ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/409080, поданной 1 ноября 2010 г., №61/411829, поданной 9 ноября 2010 г., №61/412330, поданной 10 ноября 2010 г., и №61/534323, поданной 13 сентября 2011 г., содержание каждой из которых полностью включено в настоящее описание посредством ссылок.
ОБЛАСТЬ ТЕХНИКИ
[0002] Настоящее изобретение относится к соединениям, подходящим для применения в качестве мутант-селективных ингибиторов киназ рецепторов эпидермального фактора роста (EGFR). В настоящем изобретении также предложены фармацевтически приемлемые композиции, содержащие соединения согласно настоящему изобретению, и способы применения указанных композиций для лечения различных заболеваний.
УРОВЕНЬ ТЕХНИКИ
[0003] Тирозиновые протеинкиназы представляют собой класс ферментов, которые катализируют перенос фосфатной группы от АТФ или ГТФ на остаток тирозина, находящийся в белковом субстрате. Действие рецепторных тирозинкиназ состоит в передаче сигналов извне внутрь клетки путем активации вторичных эффекторов переноса посредством фосфорилирования. Указанные сигналы способствуют протеканию различных клеточных процессов, включая пролиферацию, утилизацию углеводов, синтез белков, ангиогенез, рост клеток и выживаемость клеток.
[0004] Существует серьезный прецедент вовлеченности EGFR в развитие рака у человека, поскольку более 60% всех солидных опухолей сверхэкспрессируют по меньшей мере один из указанных белков или их лигандов. Сверхэкспрессия EGFR часто наблюдается в опухолях молочной железы, легких, головы и шеи, мочевого пузыря.
[0005] Активирующие мутации в тирозинкиназном домене EGFR были обнаружены у пациентов с немелкоклеточным раком легкого (Lin N.U., Winer E.Р., Breast Cancer Res 6: 204-210, 2004). Обратимо действующие ингибиторы Тарцева (эрлотиниб) и Иресса (гефитиниб) в настоящее время представляют собой терапию первой линии для пациентов, страдающих немелкоклеточным раком легкого с активирующими мутациями. Наиболее распространенными активирующими мутациями являются L858R и deIE746-A750.
[0006] Кроме того, для большинства пациентов, которые переносили рецидив, приобретенная лекарственная устойчивость, такая как устойчивость, вызванная мутацией остатка гена-привратника Т790М, была обнаружена по меньшей мере у половины таких клинически устойчивых пациентов. Более того, Т790М может также исходно существовать, и может иметь место независимая онкогенная роль мутации Т790М. Например, существуют пациенты с мутацией L858R/T790M, которые никогда не получали лечение гефитинибом. Кроме того, генеративные мутации Т790М EGFR связаны с некоторыми видами наследственного рака легких.
[0007] Современные лекарственные средства, находящиеся на стадии разработки, включая ковалентные ингибиторы второго поколения, такие как BIBW2992, HKI-272 и PF-0299804, эффективны против мутации устойчивости Т790М, однако проявляют дозолимитирующие токсические свойства вследствие одновременного ингибирования WT EGFR. Таким образом, сохраняется необходимость в поиске мутант-селективных ингибиторов киназ EGFR, подходящих для применения в качестве терапевтических агентов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0008] В настоящем изобретении обнаружено, что соединения согласно настоящему изобретению и их фармацевтически приемлемые композиции эффективны в качестве мутант-селективных ингибиторов киназ EGFR. Такие соединения представляют собой соединения, имеющие общую формулу I:
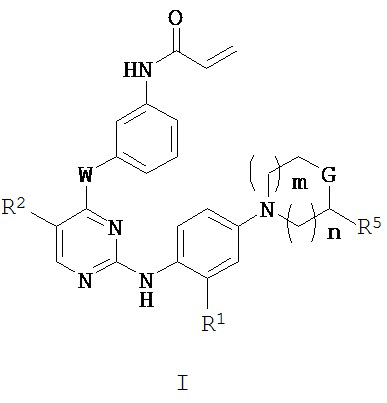 ,
,
или их фармацевтически приемлемую соль, где каждый из n, m, W, G, R1, R2 и R5 определен и описан в настоящей заявке.
[0009] Соединения согласно настоящему изобретению и их фармацевтически приемлемые композиции являются подходящими для лечения раковых заболеваний, связанных с одной или более мутациями EGFR. Такие заболевания, расстройства или состояния включают заболевания, расстройства или состояния, описанные в настоящей заявке.
[0010] Соединения, предложенные в настоящем изобретении, также являются подходящими для изучения киназ в биологических и патологических процессах, изучения путей внутриклеточной сигнальной трансдукции, опосредуемых такими киназами, и сравнительной оценки новых ингибиторов киназ.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг. 1 представлен МС-анализ, подтверждающий ковалентное модифицирование T790M/L858R EGFR соединением I-4.
ПОДРОБНОЕ ОПИСАНИЕ НЕКОТОРЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
1. Общее описание соединений согласно настоящему изобретению
[0011] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I:
или его фармацевтически приемлемая соль, где:
n составляет 0, 1 или 2;
m составляет 0, 1 или 2, причем m и n одновременно не равны 0;
W представляет собой -О- или -NH-;
R1 представляет собой -OR;
каждый R независимо представляет собой C1-4 алкил или C1-4 фторалкил;
R2 представляет собой -CF3, Cl или Br;
G представляет собой -O-, -NR3-, -S(O)2- или -CH(OR4)-;
R3 представляет собой -C(O)-R, -C(O)OR, -C(O)NHR, -SO2-R, -SO2NH2, -C(O)-C1-4 алкилен-ОН или -SO2-С1-4 алкилен-ОН;
R4 представляет собой водород, C1-4 алкил или C1-4 фторалкил и
R5 представляет собой водород или -C(O)OR.
[0012] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I:
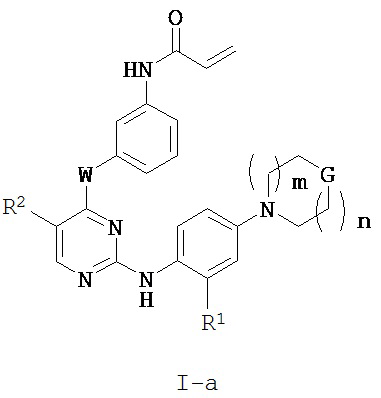
или его фармацевтически приемлемая соль, где:
n составляет 0, 1 или 2;
m составляет 0, 1 или 2, причем m и n одновременно не равны 0;
W представляет собой -О- или -NH-;
R1 представляет собой -OR;
каждый R независимо представляет собой C1-4 алкил или C1-4 фторалкил;
R2 представляет собой -CF3, Cl или Br;
G представляет собой -O-, -NR3- или -CH(OR4)-;
R3 представляет собой -C(O)-R, -C(O)OR, -C(O)NHR, -SO2-R, -SO2NH2, -C(O)-C1-4 алкилен-ОН или -SO2-С1-4 алкилен-ОН и
R4 представляет собой водород, C1-4 алкил или C1-4 фторалкил.
[0013] В настоящей заявке термин «C1-4 алкилен» означает двухвалентный насыщенный углеводородный радикал с прямой или разветвленной цепью, содержащий 1-4 атомов углерода.
[0014] В некоторых вариантах реализации настоящего изобретения n составляет 0, а G представляет собой -CH(OR4)-.
[0015] В некоторых вариантах реализации настоящего изобретения m составляет 0, а G представляет собой -CH(OR4)-.
[0016] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I или I-a, где W представляет собой -NH-.
[0017] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I или I-a, где W представляет собой -NH-, а R2 представляет собой -CF3.
[0018] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I или I-a, где W представляет собой -O-, а R2 представляет собой -Cl.
[0019] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I-a, где G представляет собой -O-, с получением таким образом соединения формулы II:
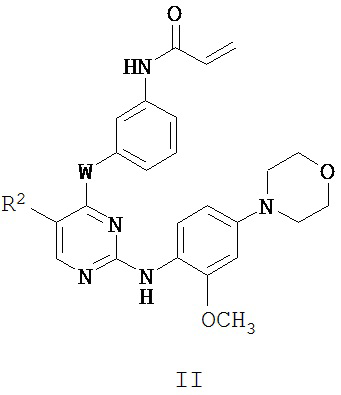
или его фармацевтически приемлемой соли, где W и R2 определены выше для формулы I и I-a.
[0020] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы II, где W представляет собой -NH-.
[0021] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы II, где W представляет собой -NH-, а R2 представляет собой -CF3.
[0022] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I, I-a или II, для которых выполняются по меньшей мере одно или оба следующих условия:
(a) W представляет собой -О- или -NH- и
(b) R2 представляет собой -CF3 или Cl.
[0023] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I, I-a или II, для которых выполняются по меньшей мере одно или оба следующих условия:
(a) W представляет собой -О- и
(b) R2 представляет собой -CF3 или Cl.
[0024] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I, I-a или II, для которых выполняются по меньшей мере одно или оба следующих условия:
(a) W представляет собой -NH- и
(b) R2 представляет собой -CF3 или Cl.
[0025] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I, где G представляет собой -NR3-, с получением таким образом соединения формулы III:
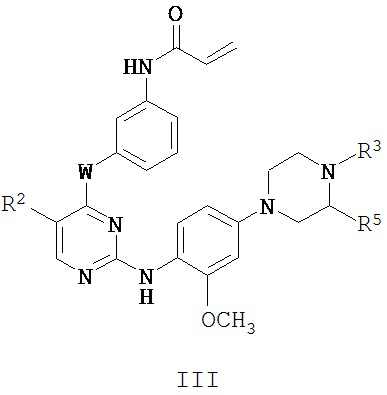
или его фармацевтически приемлемой соли, где W, R2 и R3 определены выше для формулы I.
[0026] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы I-a, где G представляет собой -NR3-, с получением таким образом соединения формулы III-a:
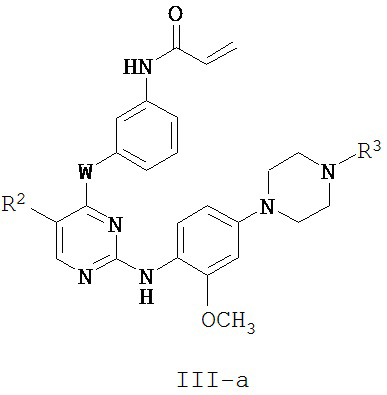
или его фармацевтически приемлемой соли, где W, R2 и R3 определены выше для формулы I.
[0027] Как определено выше, группа R3 формулы III или III-a представляет собой -C(O)-C1-4 алкил, -SO2-C1-4 алкил, -C(O)-C1-4 алкилен-ОН или -SO2-C1-4 алкилен-ОН. Специалисту в данной области будет понятно, что заместитель R3 у атома азота пиперазина превращает азот в «неосновный». Следует иметь в виду, что фрагмент, содержащий такой «неосновный» азот, «не склонен» действовать в качестве акцептора протонов, например, по сравнению с соответствующим вторичным амином или его алкилзамещенным производным.
[0028] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, где W представляет собой -NH-.
[0029] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, где W представляет собой -NH-, a R2 представляет собой -CF3.
[0030] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, где W представляет собой -O-, а R2 представляет собой -Cl.
[0031] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-а, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -О- или -NH-;
(b) R2 представляет собой -CF3 или Cl и
(c) R3 представляет собой -С(O)СН3 или -SO2CH3.
[0032] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-а, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -NH-;
(b) R2 представляет собой -CF3 или Cl и
(c) R3 представляет собой -С(O)СН3.
[0033] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-а, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -NH-;
(b) R2 представляет собой -CF3 или Cl и
(c) R3 представляет собой -SO2CH3.
[0034] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -O-;
(b) R2 представляет собой -CF3 или Cl и
(c) R3 представляет собой -С(O)СН3.
[0035] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -O-;
(b) R2 представляет собой Cl и
(c) R3 представляет собой -С(O)СН3.
[0036] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -O-;
(b) R2 представляет собой -CF3 или Cl и
(c) R3 представляет собой -SO2CH3.
[0037] В некоторых вариантах реализации настоящего изобретения предложено соединение формулы III или III-a, для которых выполняются по меньшей мере одно, по меньшей мере два или все три следующих условия:
(a) W представляет собой -O-;
(b) R2 представляет собой Cl и
(c) R3 представляет собой -SO2CH3.
[0038] Иллюстративные соединения формулы I приведены ниже в Таблице 1.
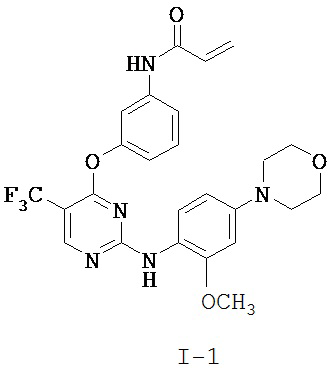
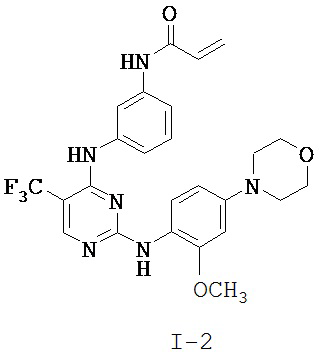
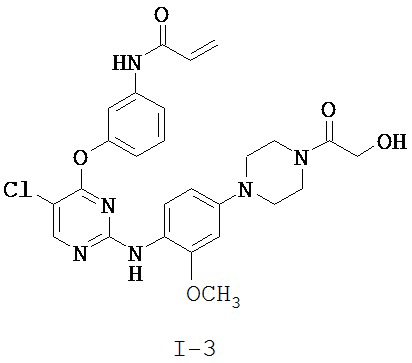
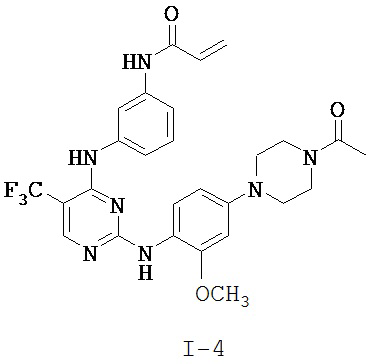
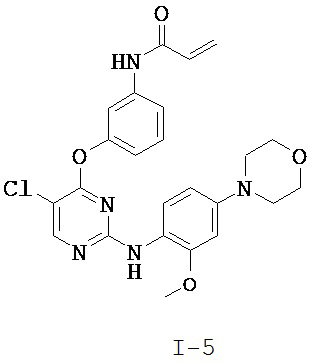
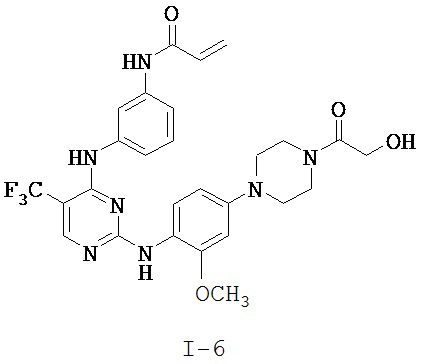
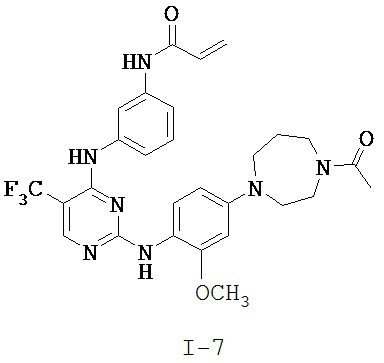
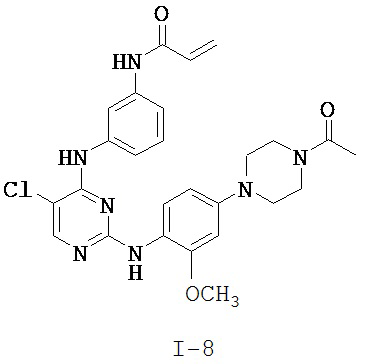
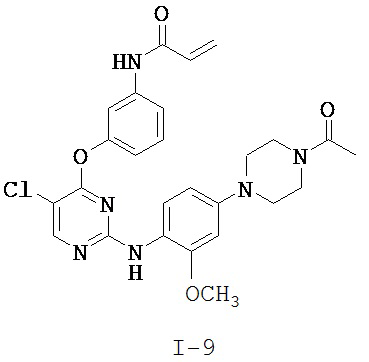
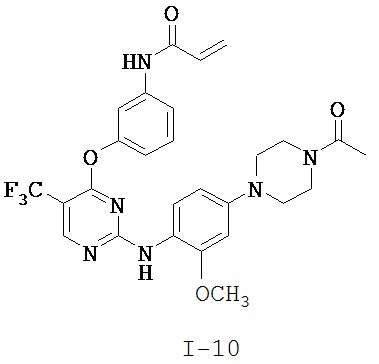

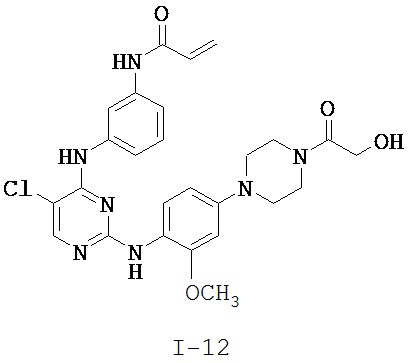
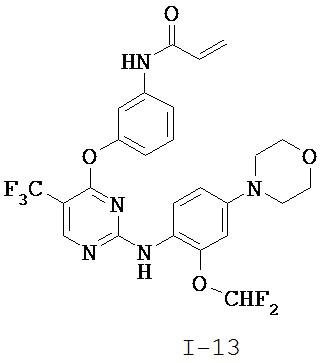
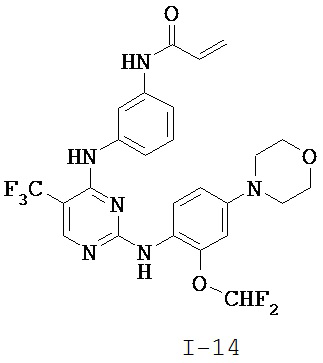
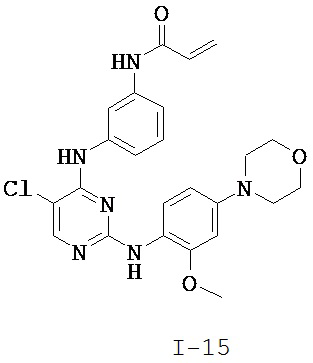
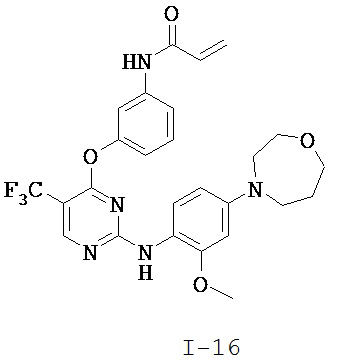
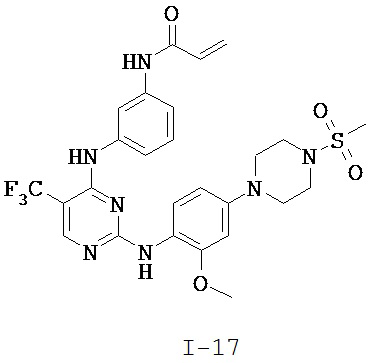
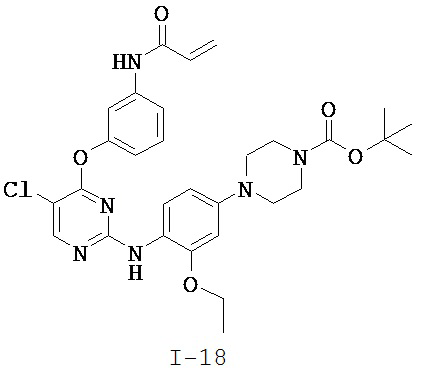
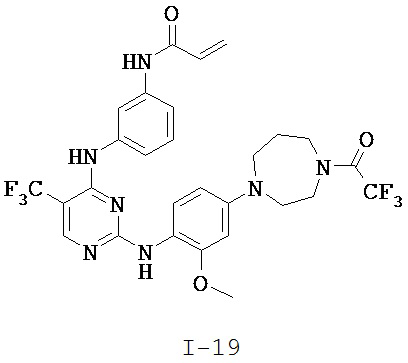
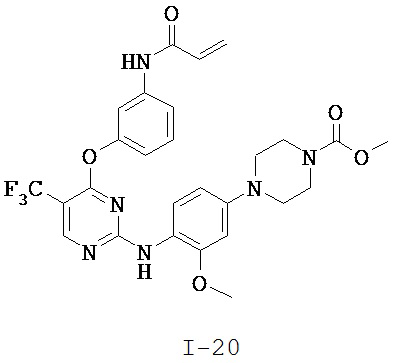
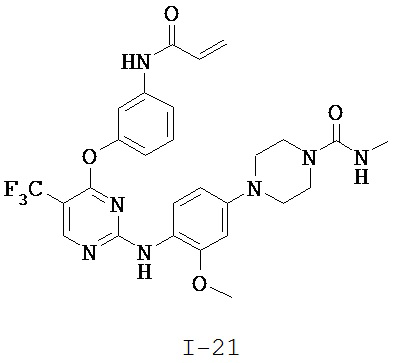
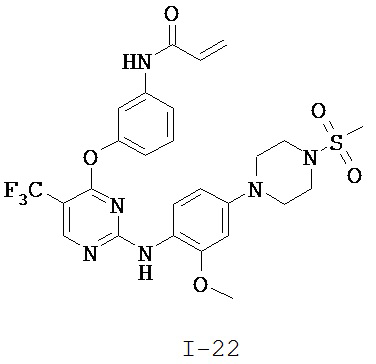
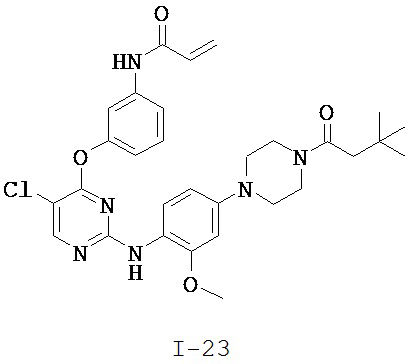
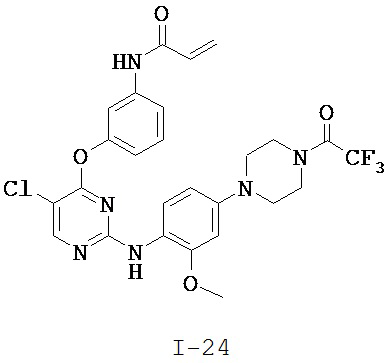
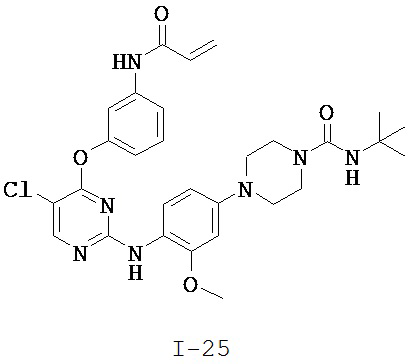
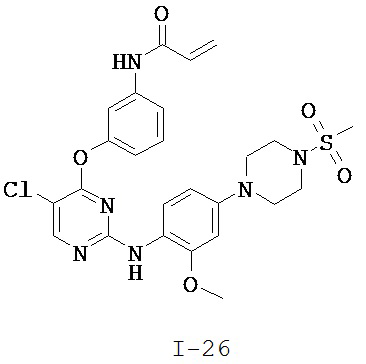
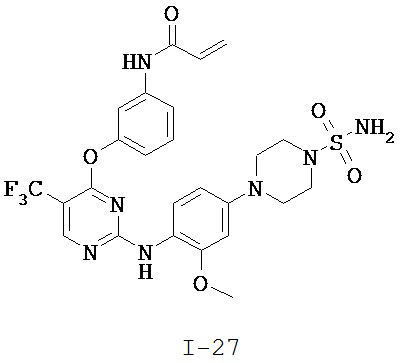
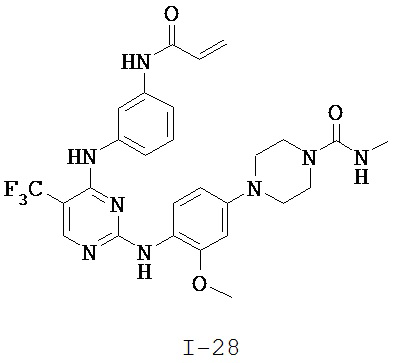
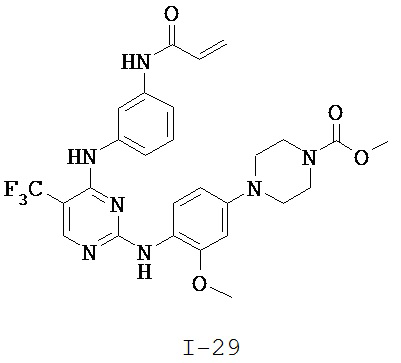
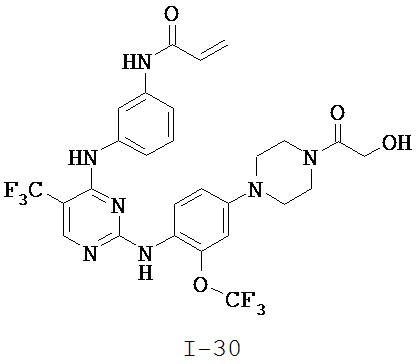
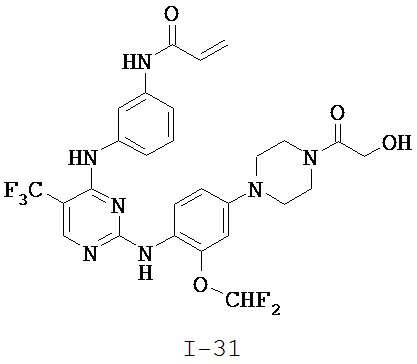
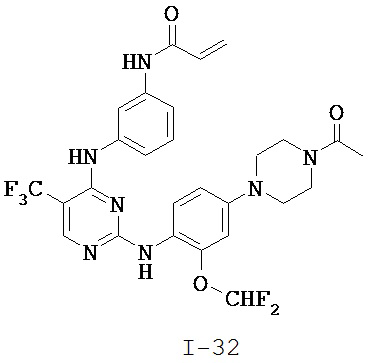
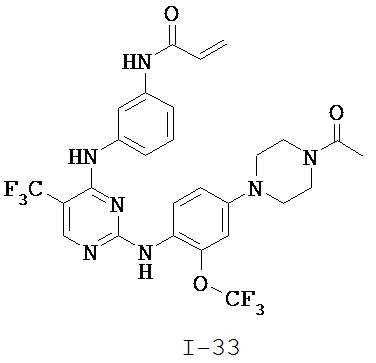
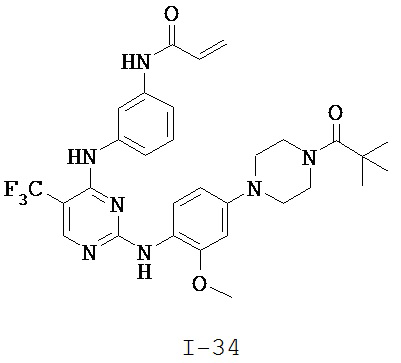
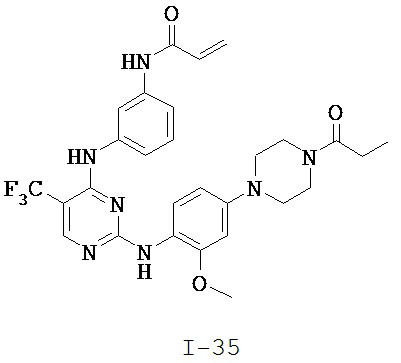
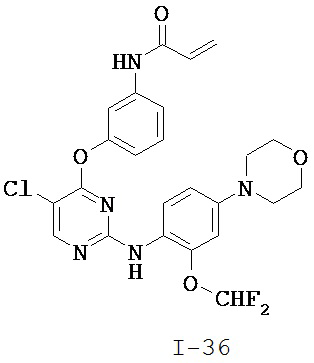
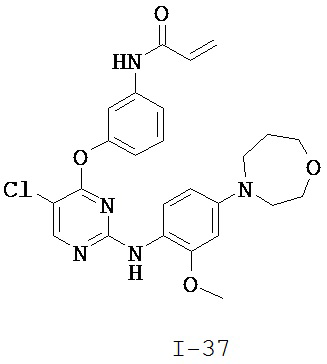
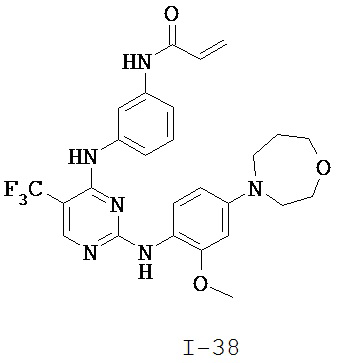
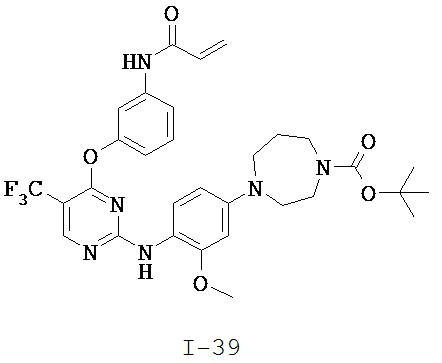
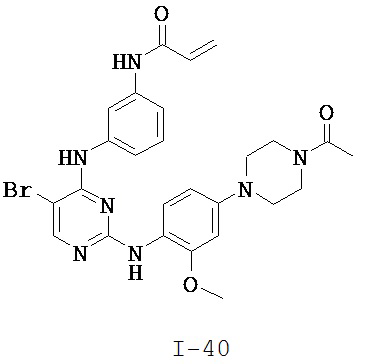
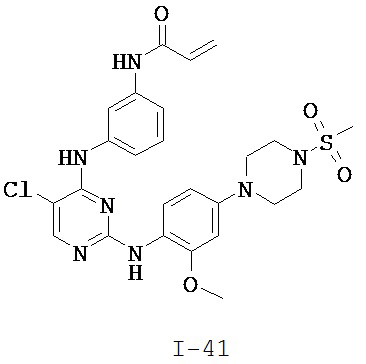
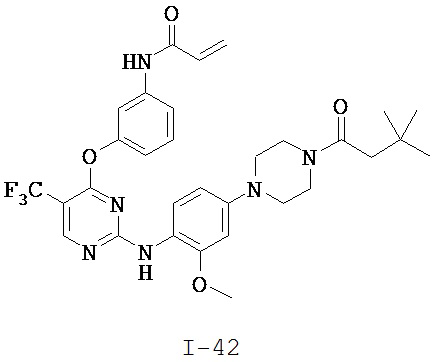
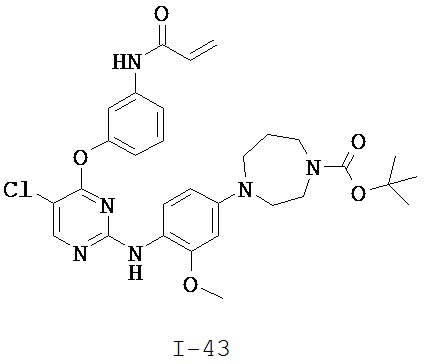
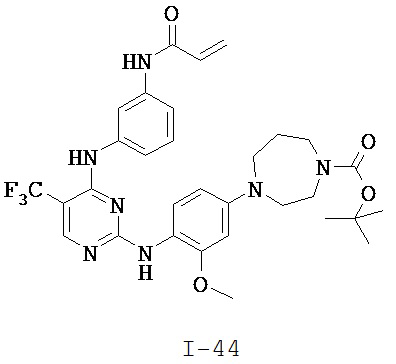
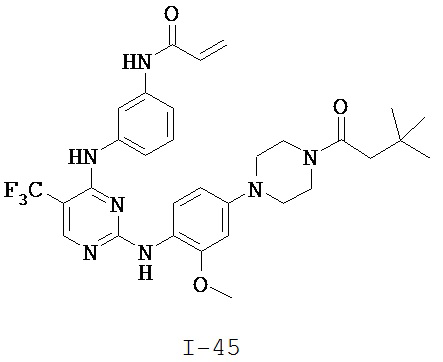
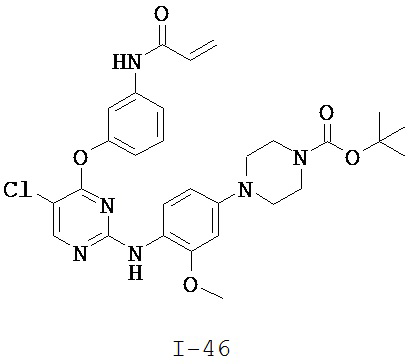
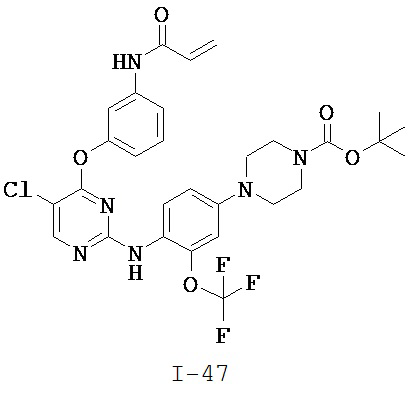
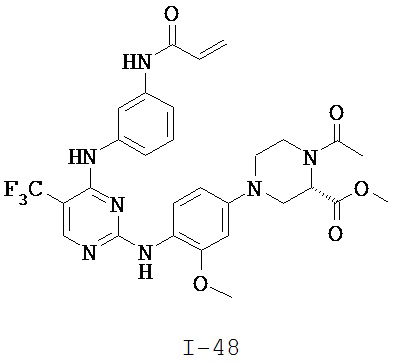
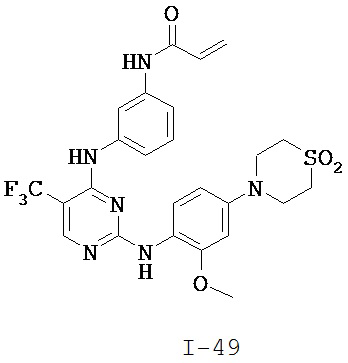
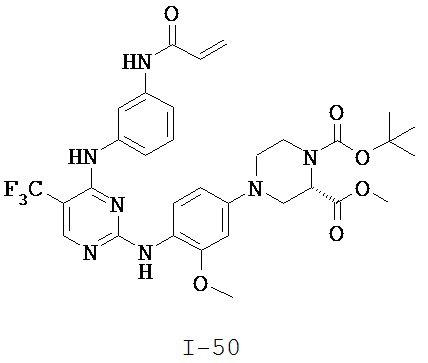
[0039] В некоторых вариантах реализации настоящего изобретения предложено соединение, приведенное выше в Таблице 1, или его фармацевтически приемлемая соль.
[0040] В некоторых вариантах реализации настоящего изобретения предложенное соединение не является соединением структуры
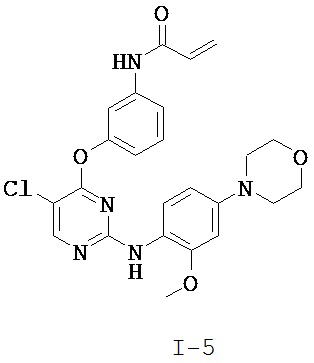 .
.
[0041] В настоящей заявке термин «фармацевтически приемлемая соль» относится к таким солям, которые по результатам тщательной медицинской оценки подходят для применения в контакте с тканями человека и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и т.п. и соответствуют разумному соотношению польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Например, S.М. Berge с соавт. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 1977, 66, 1-19, включенном в настоящее описание посредством ссылки. Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают соли, полученные из подходящих неорганических и органических кислот и оснований. Примерами фармацевтически приемлемых нетоксичных солей присоединения кислоты солей являются соли, содержащие аминогруппу, полученные с применением неорганических кислот, таких как соляная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или органических кислот, таких как уксусная кислота, щавелевая кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или другими способами, применяемыми в данной области, такими как ионный обмен. Другие фармацевтически приемлемые соли включают такие соли, как адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидройодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканоат, валерат и т.п.
[0042] Соли, полученные из подходящих оснований, включают соли щелочных металлов, щелочноземельных металлов, аммония и N+(C1-4алкил)4 соли. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и т.п. Также фармацевтически приемлемые соли при необходимости включают соли, содержащие нетоксичные катионы аммония, четвертичного аммония и амина, полученные с применением противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, низший алкилсульфонат и арилсульфонат.
2. Описание иллюстративных вариантов реализации настоящего изобретения
[0043] Как подробно описано в настоящей заявке ниже, предложенные соединения представляют собой селективные ингибиторы по меньшей мере одной мутации EGFR. Неожиданно было обнаружено, что предложенные соединения представляют собой селективные ингибиторы по меньшей мере одной мутации EGFR по сравнению с EGFR дикого типа («WT»). В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой Т790М. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой активирующую мутацию. В некоторых вариантах реализации настоящего изобретения предложенное соединение селективно ингибирует по меньшей мере одну мутацию устойчивости и по меньшей мере одну активирующую мутацию по сравнению с WT EGFR. В некоторых вариантах реализации настоящего изобретения предложенное соединение селективно ингибирует по меньшей мере одну делеционную мутацию и/или по меньшей мере одну точечную мутацию и по существу не ингибирует WT EGFR.
[0044] Мутация EGFR может быть выбрана из Т790М (мутация устойчивости или онкогенная), L858R (активирующая), deIE746-A750 (активирующая), G719S (активирующая) или их комбинации.
[0045] В настоящей заявке термин «селективно ингибирует», применяемый по сравнению с ингибированием WT EGFR, означает, что предложенное соединение ингибирует по меньшей мере одну мутацию EGFR (то есть по меньшей мере одну делеционную мутацию, по меньшей мере одну активирующую мутацию, по меньшей мере одну мутацию устойчивости или комбинацию по меньшей мере одной делеционной мутации и по меньшей мере одной точечной мутации) в ходе по меньшей мере одного анализа, описанного в настоящей заявке (например, биохимического или клеточного). В некоторых вариантах реализации настоящего изобретения термин «селективно ингибирует», применяемый по сравнению с ингибированием WT EGFR, означает, что предложенное соединение является в по меньшей мере 50 раз, по меньшей мере 45 раз, по меньшей мере 40 раз, по меньшей мере 35 раз, по меньшей мере 30 раз, по меньшей мере 25 раз или по меньшей мере 20 раз более сильным ингибитором по меньшей мере одной мутации EGFR, определенной и описанной в настоящей заявке, по сравнению с WT EGFR.
[0046] В настоящей заявке термин «по существу не ингибирует WT EGFR» означает, что селективный ингибитор по меньшей мере одной мутации EGFR, определенной и описанной выше в настоящей заявке, ингибирует EGFR при верхнем пределе обнаружения в по меньшей мере одном анализе, описанном в настоящей заявке (например, биохимическом или клеточном, подробно описанных в Примерах 56-58). В некоторых вариантах реализации настоящего изобретения термин «по существу не ингибирует WT EGFR» означает, что предложенное соединение ингибирует WT EGFR с IC50, составляющей по меньшей мере 10 мкМ, по меньшей мере 9 мкМ, по меньшей мере 8 мкМ, по меньшей мере 7 мкМ, по меньшей мере 6 мкМ, по меньшей мере 5 мкМ, по меньшей мере 3 мкМ, по меньшей мере 2 мкМ или по меньшей мере 1 мкМ.
[0047] В некоторых вариантах реализации настоящего изобретения предложенное соединение селективно ингибирует (а) по меньшей мере одну активирующую мутацию и (b) T790M и (с) по существу не ингибирует WT. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой deIE746-A750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[0048] В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой L858R и/или Т790М.
[0049] Не желая ограничиваться какой-либо конкретной теорией, полагают, что введение предложенного соединения пациенту, имеющему по меньшей мере одну активирующую мутацию, может предупредить образование мутации устойчивости Т790М. Таким образом, в некоторых вариантах реализации настоящего изобретения предложен способ ингибирования активирующей мутации у пациента, включающий введение пациенту предложенного соединения или его композиции, описанной в настоящей заявке.
[0050] Специалисту в данной области будет понятно, что некоторые пациенты имеют мутацию Т790М онкогенного типа, то есть мутация Т790М существует до введения пациенту любого ингибитора EGFR и, следовательно, является онкогенной. Таким образом, в некоторых вариантах реализации настоящего изобретения предложен способ ингибирования онкогенной Т790М у пациента, включающий введение пациенту предложенного соединения или его композиции, описанной в настоящей заявке.
[0051] Тарцева (эрлотиниб) и Иресса (гефитиниб) представляют собой лекарственные средства терапии первой линии для пациентов с активирующими мутациями, однако проявляют токсические свойства, ограничивающие дозу, вследствие одновременного ингибирования WT EGFR. Кроме того, лекарственные средства, в настоящее время находящиеся на стадии разработки, включая ковалентные ингибиторы второго поколения, такие как BIBW2992, HKI-272 и PF-0299804, эффективны против мутации устойчивости Т790М, однако проявляют токсические свойства, ограничивающие дозу, вследствие одновременного ингибирования WT EGFR.
[0052] Неожиданно было обнаружено, что предложенные соединения селективно ингибируют каждую из активирующих и делеционных мутаций EGFR. Более того, предложенные соединения по существу не ингибируют WT EGFR и не обладают обусловленными ингибированием WT EGFR токсическими свойствами, ограничивающими дозу.
[0053] Предложенные соединения с указанными свойствами составляют контаст с другими известными ингибиторами EGFR (например, BIBW2992 и HKI-272), которые являются лишь до некоторой степени эффективными против мутантов, но при этом сохраняют активность против WT EGFR, и, следовательно, их применение ограничено токсическими свойствами, обусловленными ингибированием WT EGFR. В Таблице 2 ниже приведены значения GI50 для Тарцева, BIBW2992 и HKI-272 по сравнению с предложенными соединениями I-2 и I-4 (где номера соединений соответствуют номерам соединений в Таблице 1 выше). Данные, представленные в Таблице 2, представляют собой значения GI50, полученные в результате анализа клеточной пролиферации, подробно описанного в Примере 58, где клетки А431 экспрессируют WT EGFR, HCC827 экспрессируют EGFR, обладающий делеционной мутацией deIE746-A750, а клетки Н1975 экспрессируют EGFR, обладающий двойной мутацией L858R/T790M.
[0054] В некоторых вариантах реализации настоящего изобретения предложенное соединение является в по меньшей мере 50 раз, по меньшей мере 45 раз, по меньшей мере 40 раз, по меньшей мере 35 раз, по меньшей мере 30 раз, по меньшей мере 25 раз или по меньшей мере 20 раз более сильным ингибитором по меньшей мере одной мутации EGFR по сравнению с WT EGFR, что установлено путем биохимического анализа, подробно описанного в Примере 56 ниже. В некоторых вариантах реализации настоящего изобретения предложенное соединение является в по меньшей мере 20, по меньшей мере 15 или по меньшей мере 10 раз более сильным ингибитором по меньшей мере одной мутации EGFR по сравнению с WT EGFR, что установлено путем клеточного анализа, подробно описанного в Примере 58 ниже.
[0055] В некоторых вариантах реализации настоящего изобретения предложенное соединение является по меньшей мере в 50 раз, по меньшей мере в 45 раз, по меньшей мере в 40 раз, по меньшей мере в 35 раз, по меньшей мере в 30 раз, по меньшей мере в 25 раз или по меньшей мере в 20 раз более сильным ингибитором по меньшей мере одной делеционной мутации EGFR по сравнению с WT EGFR по данным биохимического анализа, подробно описанного в Примере 56 ниже. В некоторых вариантах реализации настоящего изобретения предложенное соединение является по меньшей мере в 20, по меньшей мере в 15 или по меньшей мере в 10 раз более сильным ингибитором по меньшей мере одной делеционной мутации EGFR по сравнению с WT EGFR по данным клеточного анализа, подробно описанного в Примере 58 ниже.
[0056] В некоторых вариантах реализации настоящего изобретения предложенное соединение является по меньшей мере в 50 раз, по меньшей мере в 45 раз, по меньшей мере в 40 раз, по меньшей мере в 35 раз, по меньшей мере в 30 раз, по меньшей мере в 25 раз или по меньшей мере в 20 раз более сильным ингибитором L858R и/или Т790М мутации EGFR по сравнению с WT EGFR по данным биохимического анализа, подробно описанного в Примере 56 ниже. В некоторых вариантах реализации настоящего изобретения предложенное соединение является по меньшей мере в 20, по меньшей мере в 15 или по меньшей мере в 10 раз более сильным ингибитором L858R и/или Т790М мутации EGFR по сравнению с WT EGFR поданным клеточного анализа, подробно описанного в Примере 58 ниже.
[0057] В некоторых вариантах реализации настоящего изобретения предложенное соединение является по меньшей мере в 20, по меньшей мере в 15 или по меньшей мере в 10 раз более сильным ингибитором двойного мутанта в клетках Н1975 по сравнению с WT EGFR по данным анализа передачи сигналов, подробно описанного в Примере 57.
[0058] В некоторых вариантах реализации настоящего изобретения предложенное соединение ингибирует по меньшей мере одну мутацию EGFR селективно по сравнению с WT EGFR и по сравнению с другими протеинкиназами (например, ErbB2, ErbB4, ТЕС-киназой и/или JAK3). Следует иметь в виду, что акриламидный фрагмент, представленный в формуле I, представляет собой реакционноспособную группу, образующую ковалентную связь с ключевым остатком цистеина в связывающем домене по меньшей мере одной мутации EGFR селективно по сравнению с WT EGFR и другими протеинкиназами. Протеинкиназы, содержащие остаток цистеина в связывающем домене, известны специалисту в данной области. Такие Протеинкиназы, содержащие остаток цистеина в связывающем домене, включают Протеинкиназы семейства ТЕС (включая ТЕС, ВТК, ITK, BMX, JAK3 и RLK). В некоторых вариантах реализации настоящего изобретения остаток цистеина сохраняется у протеинкиназ всего подсемейства, таких как ErbB1 (обычно называемого EGFR), ErbB2 и ErbB4.
[0059] Не желая ограничиваться какой-либо конкретной теорией, полагают, что предложенные соединения необратимо ингибируют (то есть ковалентно модифицируют) по меньшей мере одну мутацию EGFR селективно по сравнению с WT EGFR и другими протеинкиназами. В некоторых вариантах реализации настоящего изобретения предложенное соединение необратимо ингибирует по меньшей мере одну мутацию EGFR селективно по сравнению с по меньшей мере одной протеинкиназой, выбранной из ErbB1, ErbB2, ErbB4, ТЕС, ВТК, ITK, BMX, JAK3 или RLK.
[0060] Тем не менее, в некоторых вариантах реализации настоящего изобретения предложенные соединения незначительно ингибируют, обратимо или необратимо, другие Протеинкиназы. В некоторых вариантах реализации настоящего изобретения предложенное соединение является селективным в случае ингибирования по меньшей мере одного мутанта EGFR по сравнению с протеинкиназами, не являющимися мишенями, таким образом избегая воздействия и не приобретая токсических свойств, обусловленных их ингибированием.
3. Синтез и промежуточные соединения
[0061] В некоторых вариантах реализации настоящего изобретения предложенное соединение синтезируют с применением одного или более следующих этапов и промежуточных соединений:
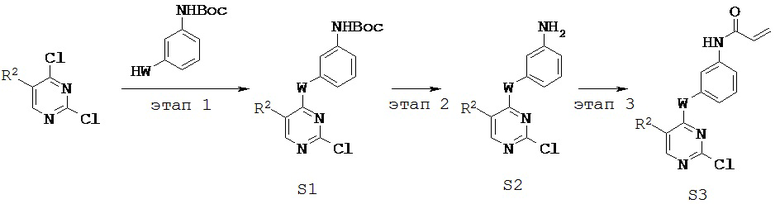 ,
,
где R2 и W определены и описаны в разделах и подразделах настоящей заявки.
[0062] На этапе 1 обеспечивают проведение реакции R2-замещенного 2,4-дихлорпиримидина, например, с Вос-защищенным 3-аминофенолом с получением промежуточного соединения S1. В некоторых вариантах реализации настоящего изобретения этап 1 проводят в щелочных условиях. В некоторых вариантах реализации настоящего изобретения этап 1 проводят в присутствии третичного амина. В некоторых вариантах реализации настоящего изобретения этап 1 проводят в присутствии основания Хунига. В некоторых вариантах реализации настоящего изобретения этап 1 проводят в протонном растворителе. В некоторых вариантах реализации настоящего изобретения этап 1 проводят в спиртовом растворителе. В некоторых вариантах реализации настоящего изобретения этап 1 проводят в н-бутаноле.
[0063] На этапе 2 с промежуточного соединения S1 снимают защитную группу с получением промежуточного соединения S2. В некоторых вариантах реализации настоящего изобретения с промежуточного соединения S1 снимают защитную группу с применением кислоты. В некоторых вариантах реализации настоящего изобретения с промежуточного соединения S1 снимают защитную группу в присутствии трифторуксусной кислоты.
[0064] На этапе 3 промежуточное соединение S2 ацилируют с применением акрилоильной группы с получением промежуточного соединения S3. В некоторых вариантах реализации настоящего изобретения ацилирующий агент представляет собой акрилоилхлорид. В некоторых вариантах реализации настоящего изобретения этап 3 осуществляют в галогенированном растворителе. В некоторых вариантах реализации настоящего изобретения этап 3 проводят в дихлорметане.
[0065] Промежуточное соединение S3 может быть подвергнут реакции с различными анилинами с получением соединений, описанных в настоящей заявке.
4. Применение, состав и введение
Фармацевтически приемлемые композиции
[0066] Согласно другому варианту реализации в настоящем изобретении предложена композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, адъювант или наполнитель. Количество соединения в композиции согласно настоящему изобретению представляет собой такое количество, которое является эффективным для поддающегося измерению ингибирования протеинкиназы, в частности для ингибирования по меньшей мере одного мутанта EGFR селективно по сравнению с WT EGFR, в биологическом образце или в организме пациента. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с мутацией Т790М. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с делеционной мутацией EGFR. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой L858R и/или Т790М.
[0067] В некоторых вариантах реализации настоящего изобретения количество соединения в предложенной композиции составляет такое количество, которое является эффективным для поддающегося измерению ингибирования по меньшей мере одной мутации EGFR селективно по сравнению с WT EGFR.
[0068] В некоторых вариантах реализации настоящего изобретения количество соединения в предложенной композиции составляет такое количество, которое является эффективным для поддающегося измерению ингибирования по меньшей мере одной мутации EGFR селективно по сравнению с WT EGFR и другими протеинкиназами (например, ErbB2, ErbB4, ТЕС-киназой и/или JAK3).
[0069] В некоторых вариантах реализации настоящего изобретения количество соединения в предложенной композиции составляет такое количество, которое является эффективным для поддающегося измерению ингибирования по меньшей мере одного мутанта EGFR селективно по сравнению с WT EGFR в биологическом образце или в организме пациента. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для введения пациенту, нуждающемуся в такой композиции. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для перорального введения пациенту.
[0070] В некоторых вариантах реализации настоящего изобретения количество соединения в предложенной композиции составляет такое количество, которое является эффективным для поддающегося измерению ингибирования по меньшей мере одного мутанта EGFR селективно по сравнению с WT EGFR в биологическом образце или в организме пациента. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для введения пациенту, нуждающемуся в такой композиции. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для перорального введения пациенту.
[0071] В некоторых вариантах реализации настоящего изобретения количество соединения в предложенной композиции составляет такое количество, которое является эффективным для поддающегося измерению ингибирования по меньшей мере одного мутанта EGFR селективно по сравнению с WT EGFR и другими протеинкиназами (например, ErbB2, ErbB4, ТЕС-киназой и/или JAK3) в биологическом образце или в организме пациента. В некоторых вариантах реализации настоящего изобретения композицию согласно настоящему изобретения приготовлена для введения пациенту, нуждающемуся в такой композиции. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для перорального введения пациенту.
[0072] В настоящей заявке термин «пациент» означает животное, предпочтительно - млекопитающее и наиболее предпочтительно - человека.
[0073] Термин «фармацевтически приемлемые носитель, адъювант или наполнитель» относится к нетоксичному носителю, адъюванту или наполнителю, который не разрушает фармакологическую активность соединения, с которым его готовят. Фармацевтически приемлемые носители, адъюванты или наполнители, которые могут быть применены в композициях согласно настоящему изобретению, включают, но не ограничиваются ими, ионообменники, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных жирных кислот растительного происхождения, воду, соли или электролиты, такие как сульфат протамина, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воски, полиэтилен-полиоксипропилен-блок-полимеры, полиэтиленгликоль и ланолин.
[0074] Композиции согласно настоящему изобретению могут быть введены перорально, парентерально, с применением спрея для ингаляций, местно, ректально, назально, буккально, вагинально или с применением имплантируемой емкости. В настоящей заявке термин «парентеральный» включает способы введения, представляющие собой подкожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, интрастернальные, интратекальные, внутрипеченочные, внутриочаговые и внутричерепные инъекции или инфузии. Предпочтительно указанные композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные формы для инъекций композиций согласно настоящему изобретению могут представлять собой водную или масляную суспензию. Указанные суспензии могут быть приготовлены способами, известными в данной области, с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Указанная стерильная форма для инъекций также может представлять собой стерильный раствор или стерильную суспензию для инъекций в нетоксичном разбавителе или растворителе, приемлемом для парентерального введения, например раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут быть применены, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно применяют стерильные жирные масла.
[0075] Для указанной цели может быть использовано любое смягчающее жирное масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота, и их производные, представляющие собой глицериды, подходят для применения в форме для инъекций, как и натуральные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, особенно в полиоксиэтилированной форме. Указанные масляные растворы или суспензии также могут содержать длинноцепочечный спирт в качестве разбавителя или диспергатора или аналогичные диспергирующие агенты, которые обычно применяют в составе фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Другие обычно применяемые поверхностно-активные вещества, такие как Tweens, Spans, и другие эмульгирующие агенты или усилители биодоступности, которые обычно применяют в изготовлении фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть применены при приготовлении.
[0076] Фармацевтически приемлемые композиции согласно настоящему изобретению могут быть перорально введены в любой лекарственной форме, приемлемой для перорального введения, включая, но не ограничиваясь ими, капсулы, таблетки, водные суспензии или растворы. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. В случае перорального введения указанных композиций в форме капсул подходящие разбавители включают лактозу и высушенный кукурузный крахмал. Если для перорального введения необходимо применять водные суспензии, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании также могут быть добавлены некоторые подсластители, вкусоароматические или красящие агенты.
[0077] В качестве альтернативы, фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены в форме суппозиториев для ректального введения. Указанные суппозитории могут быть получены путем смешивания агента с подходящим нераздражающим наполнителем, который является твердым при комнатной температуре, но при этом является жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли.
[0078] Фармацевтически приемлемые композиции согласно настоящему изобретению также могут быть введены местно, особенно если объект лечения включает участки или органы, легко доступные для местного введения, в том числе глаза, кожу или нижний отдел кишечника. Подходящие лекарственные формы для местного применения легко получают для каждого из указанных участков или органов.
[0079] Местное применение для нижнего отдела кишечника может быть осуществлено в форме ректального суппозитория (см. выше) или в форме подходящей клизмы. Также могут быть применены трансдермальные пластыри.
[0080] В случае местного применения предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или более носителях. Носители для местного введения соединений согласно настоящему изобретению включают, но не ограничиваются ими, минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, полиоксиэтилен, полиоксипропилен, эмульгирующий воск и воду. В качестве альтернативы, предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают, но не ограничиваются ими, минеральное масло, сорбитан моностеарат, полисорбат 60, воск на основе цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[0081] В случае офтальмологического применения предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде тонкоизмельченных суспензий в стерильном изотоническом физиологическом растворе с отрегулированным рН или, предпочтительно, в виде растворов в стерильном изотоническом физиологическом растворе с отрегулированным рН с применением или без применения консерванта, такого как бензалкония хлорид. В качестве альтернативы, в случае офтальмологического применения указанные фармацевтически приемлемые композиции могут быть приготовлены в виде мази, такой как вазелин.
[0082] Фармацевтически приемлемые композиции согласно настоящему изобретению также могут быть введены с применением назального аэрозоля или путем ингаляции. Такие композиции получают способами, хорошо известными в области приготовления лекарственных средств, и указанные композиции могут быть получены в виде растворов в физиологическом растворе с применением бензилового спирта или других подходящих консервантов, веществ способствующих абсорбции, для усиления биодоступности, фторуглеродов и/или других традиционных солюбилизирующих или диспергирующих агентов.
[0083] В некоторых вариантах реализации настоящего изобретения фармацевтически приемлемые композиции согласно настоящему изобретению приготовлены для перорального введения.
[0084] Количество соединений согласно настоящему изобретению, которые могут быть объединены с материалами, представляющими собой носители, с получением композиции в единичной лекарственной форме, будет варьироваться в зависимости организма, подвергаемого лечению, и конкретного способа введения. Предпочтительно предложенные композиции должны быть приготовлены таким образом, чтобы пациенту, принимающему указанные композиции, можно было вводить дозу указанного ингибитора 0,01-100 мг/кг массы тела/сутки.
[0085] Также следует понимать, что конкретная доза и курс лечения для любого конкретного пациента будут зависеть от различных факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, рацион питания, время введения, скорость выведения, комбинацию лекарственных средств, и решения лечащего врача и тяжести конкретного заболевания, подвергаемого лечению. Количество соединения согласно настоящему изобретению в указанной композиции также будет зависеть от конкретного соединения в композиции.
Применение соединений и фармацевтически приемлемых композиций
[0086] Соединения и композиции, описанные в настоящей заявке, как правило, подходят для селективного ингибирования по меньшей мере одного мутанта EGFR по сравнению с WT EGFR. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с мутацией Т790М. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с делеционной мутацией EGFR, активирующей мутацией EGFR или их комбинацией. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой L858R и/или Т790М.
[0087] В некоторых вариантах реализации настоящего изобретения предложенное соединение селективно ингибирует: (а) по меньшей мере одну активирующую мутацию, (b) T790M и (с) по существу не ингибирует WT. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой deIE746-A750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[0088] В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой L858R и/или T790M.
[0089] Активность соединения, применяемого в указанном изобретении в качестве селективного ингибитора по меньшей мере одного мутанта EGFR по сравнению с WT EGFR, может быть исследована in vitro, in vivo или с применением линии клеток. Исследования in vitro включают исследования, в ходе которых оценивают ингибирование активности фосфорилирования и/или функциональных последствий или АТФазной активности активированного EGFR (WT или мутанта). В ходе альтернативных исследований in vitro проводят количественный анализ способности указанного ингибитора связываться с EGFR (WT или мутантом). Связывание ингибитора может быть измерено путем радиоактивного мечения указанного ингибитора перед связыванием, выделения комплекса ингибитор/EGFR (WT или мутант) и определения количества связанных радиоактивных меток. В качестве альтернативы, связывание ингибитора может быть определено путем проведения эксперимента по конкурентному связыванию, в ходе которого новые ингибиторы выдерживают с EGFR (WT или мутантом), связанным с известными радиолигандами. Условия исследования соединения, применяемого в указанном изобретении в качестве ингибитора EGFR (WT или мутанта), подробно изложены в Примерах ниже.
[0090] Тирозиновые протеинкиназы представляют собой класс ферментов, которые катализируют перенос фосфатной группы от АТФ или ГТФ на остаток тирозина, находящийся в белковом субстрате. Действие рецепторных тирозинкиназ основано на передаче сигналов с внешней поверхности клеточной мембраны внутрь клетки путем активации вторичных эффекторов посредством фосфорилирования. Указанными сигналами вызваны различные клеточные процессы, включая пролиферацию, утилизацию углеводов, синтез белков, ангиогенез, рост клеток и выживаемость клеток.
[0091] В настоящей заявке термины «лечение», «лечить» и «лечащий» относятся к реверсированию, облегчению, задержке начала или ингибированию развития заболевания или расстройства или одного или более его симптомов, описанных в настоящей заявке. В некоторых вариантах реализации настоящего изобретения лечение может быть произведено после обнаружения одного или более симптомов. В других вариантах реализации настоящего изобретения лечение может быть произведено при отсутствии симптомов. Например, лечение предрасположенного к заболеванию человека может быть произведено до появления у него симптомов (например, с учетом характера развития симптомов и/или с учетом генетических или других факторов восприимчивости). Также лечение может быть продолжено после устранения симптомов, например, чтобы предотвратить или отсрочить их повторное появление.
[0092] Предложенные соединения являются ингибиторами по меньшей мере одного мутанта EGFR и, следовательно, подходят для лечения одного или более расстройств, обусловленных активностью одного или более мутантов EGFR (например, мутантов с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией). Таким образом, в некоторых вариантах реализации настоящего изобретения предложен способ лечения расстройства, медиируемого мутантом EGFR, включающий этап введения пациенту, нуждающемуся в этом, соединения согласно настоящему изобретению или его фармацевтически приемлемой композиции.
[0093] В настоящей заявке термин «расстройства или патологические состояния, опосредованные мутантом EGFR», означает любое заболевание или другое вредное для организма состояние, о котором известно, что в его возникновении играет роль по меньшей мере один мутант EGFR. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с мутацией Т790М. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с делеционной мутацией. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с активирующей мутацией. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой мутант с мутацией L858R и/или Т790М. В некоторых вариантах реализации настоящего изобретения предложенное соединение селективно ингибирует: (а) по меньшей мере одну активирующую мутацию, (b) T790M и (с) по существу не ингибирует WT. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой deIE746-A750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[0094] Соответственно, в другом варианте реализации настоящее изобретение относится к лечению или уменьшению тяжести одного или более заболеваний, о которых известно, что в их возникновении играет роль по меньшей мере один мутант EGFR. Точнее говоря, настоящее изобретение относится к способу лечения или уменьшения тяжести заболевания или патологического состояния, выбранного из пролиферативного расстройства, причем указанный способ включает введение пациенту, нуждающемуся в этом, соединение или композицию согласно настоящему изобретению.
[0095] В некоторых вариантах реализации настоящего изобретения предложен способ лечения или уменьшения тяжести одного или более расстройств, выбранных из раковых заболеваний. В некоторых вариантах реализации настоящего изобретения рак обусловлен солидной опухолью. В некоторых вариантах реализации настоящего изобретения рак представляет собой рак молочной железы, глиобластому, рак легкого, рак головы и шеи, рак прямой кишки, рак мочевого пузыря или немелкоклеточный рак легкого. В некоторых вариантах реализации настоящего изобретения предложен способ лечения или уменьшения тяжести одного или более расстройств, выбранных из плоскоклеточной карциномы, карциномы слюнных желез, карциномы яичника или рака поджелудочной железы.
[0096] В некоторых вариантах реализации настоящего изобретения предложен способ лечения или уменьшения тяжести нейрофиброматоза I типа (NF1), нейрофиброматоза II типа (NF2), новообразований из шванновских клеток (например, различных ЗООПН) или шванном.
[0097] Соединения и композиции в соответствии со способом согласно настоящему изобретению можно вводить с применением любого количества и любого пути введения, эффективного для лечения или уменьшения тяжести рака. Точное необходимое количество будет варьироваться от субъекта к субъекту в зависимости от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного агента, способа его введения и т.п. Соединения согласно настоящему изобретению предпочтительно готовят в единичной лекарственной форме для простоты введения и постоянства дозы. В настоящей заявке выражение «единичная лекарственная форма» относится к физически дискретной единице агента, подходящей для пациента, подвергаемого лечению. Однако следует понимать, что общая суточная доза соединений и композиций согласно настоящему изобретению будет определяться лечащим врачом по результатам тщательной медицинской оценки. Конкретный уровень эффективной дозы для любого конкретного пациента или организма будет зависеть от различных факторов, включая подлежащее лечению расстройство и тяжесть указанного расстройства, активность конкретного примененного соединения, конкретную примененную композицию, возраст, массу тела, общее состояние здоровья, пол и рацион пациента, время введения, способ введения и скорость выведения конкретного примененного соединения, продолжительность лечения, лекарственные средства, примененные в комбинации или одновременно с конкретным примененным соединением, и другие подобные факторы, хорошо известные в области медицины. В настоящей заявке термин «пациент» означает животное, предпочтительно - млекопитающее и наиболее предпочтительно - человека.
[0098] Фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены людям и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), буккально, в виде перорального или назального спрея или другими подобными способами в зависимости от тяжести инфекции, подлежащей лечению. В некоторых вариантах реализации настоящего изобретения соединения согласно настоящему изобретению могут быть введены перорально или парентерально в дозах от примерно 0,01 мг/кг до примерно 50 мг/кг или от примерно 1 мг/кг до примерно 25 мг/кг массы тела субъекта в сутки один или более раз в сутки с получением желаемого терапевтического эффекта.
[0099] Жидкие лекарственные формы для перорального введения включают, но не ограничиваются ими, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активных соединений жидкие лекарственные формы могут содержать инертные разбавители, обычно применяемые в данной области, например, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное масла, масло зародышей пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры сорбитана и жирных кислот и их смеси. Кроме инертных разбавителей композиции для перорального применения также могут включать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, вкусовые и ароматизирующие агенты.
[00100] Инъекционные формы, например стерильные водные или масляные суспензии для инъекций, могут быть приготовлены способами, известными из уровня техники, с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильная инъекционная форма также может представлять собой стерильный раствор, суспензию или эмульсию для инъекций в нетоксичном разбавителе или растворителе, приемлемом для парентерального введения, например раствор в 1,3-бутандиоле. Среди приемлемых наполнителей и растворителей, которые могут быть применены, можно указать воду, раствор Рингера, Фармакопея США, и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно применяют стерильные жирные масла. Для указанной цели может быть использовано любое смягчающее жирное масло, включая синтетические моно- или диглицериды. Кроме того, в форме для инъекций применяют жирные кислоты, такие как олеиновая кислота.
[00101] Формы для инъекций могут быть стерилизованы, например, посредством фильтрования через фильтр, задерживающий бактерии, терминальной (тепловой) стерилизации или стерилизации с применением ионизирующего излучения или путем введения стерилизующих агентов в виде стерильных твердых композиций, которые перед применением могут быть растворены или диспергированы в стерильной воде или другой стерильной среде для инъекций.
[00102] Для того чтобы продлить действие соединения согласно настоящему изобретению, зачастую желательно замедлить всасывание указанного соединения после подкожной или внутримышечной инъекции. Указанное продление может быть достигнуто путем применения жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. В таком случае скорость всасывания указанного соединения зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера и формы кристаллов. В качестве альтернативы, замедленное всасывание парентерально вводимой формы соединения достигается путем растворения или суспендирования указанного соединения в масляном наполнителе. Депо-формы для инъекций готовят путем получения микрокапсульных матриц указанного соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. Скорость выделения соединения можно регулировать в зависимости от отношения соединения к полимеру и природы конкретного примененного полимера. Примеры других биоразлагаемых полимеров включают сложные поли(ортоэфиры) и поли(ангидриды). Депо-формы для инъекций также получают путем включения указанного соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
[00103] Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть получены путем смешивания соединения согласно настоящему изобретению с подходящими нераздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но при этом являются жидкими при температуре тела и, следовательно, плавятся в ректальной или вагинальной полости и выделяют активное соединение.
[00104] Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивают с по меньшей мере одним инертным фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или гидроортофосфат кальция, и/или а) наполнителями или разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими, например, такими как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик, с) увлажнителями, такими как глицерин, d) разрыхляющими агентами, такими как агар, карбонат кальция, картофельный или тапиока, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами, замедляющими растворение, такими как парафин, f) ускорителями всасывания, такими как соединения четвертичного аммония, g) смачивающими агентами, например, такими как цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и их смесями. В случае капсул, таблеток и пилюль лекарственная форма также может содержать буферные агенты.
[00105] Твердые композиции аналогичного типа также могут быть применены в качестве наполнителей в мягких и твердых желатиновых капсулах с применением таких вспомогательных веществ, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Твердые лекарственные формы, представляющие собой таблетки, драже, капсулы, пилюли и гранулы, могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области разработок фармацевтических рецептур. Они, необязательно, могут содержать агенты, придающие лекарственной форме непрозрачность, а также могут представлять собой композицию, которая выделяет активный ингредиент(ы) только или преимущественно в определенной части кишечного тракта, необязательно, замедленно. Примеры покрывающих композиций, которые могут быть применены, включают полимерные вещества и воски. Твердые композиции аналогичного типа также могут быть применены в качестве наполнителей в мягких и твердых желатиновых капсулах с применением таких наполнителей, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. В некоторых вариантах реализации настоящего изобретения твердая композиция представляет собой твердую желатиновую капсулу, наполненную жидкостью, или твердую дисперсию.
[00106] Активные соединения также могут находиться в микроинкапсулированной форме с применением одного или более наполнителей, указанных выше. Твердые лекарственные формы, представляющие собой таблетки, драже, капсулы, пилюли и гранулы, могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия с регулируемым выделением активного компонента и другие покрытия, хорошо известные в области разработок фармацевтических рецептур. В таких твердых лекарственных формах активное соединение может быть смешано с по меньшей мере одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы также могут содержать, что является обычной практикой, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие вспомогательные вещества для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль указанные лекарственные формы также могут содержать буферные агенты. Они, необязательно, могут содержать агенты, придающие лекарственной форме непрозрачность, а также могут представлять собой композицию, которая выделяет активный ингредиент(ы) только или преимущественно в определенной части кишечного тракта, необязательно, замедленно. Примеры покрывающих композиций, которые могут быть применены, включают полимерные вещества и воски.
[00107] Лекарственные формы для местного или трансдермального введения соединения согласно настоящему изобретению включают мази, пасты, крема, лосьоны, гели, порошки, растворы, спреи, средства для ингаляции или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми консервантами или буферами, которые могут потребоваться. Глазные лекарственные формы, ушные капли и глазные капли также рассматриваются как входящие в рамки настоящего изобретения. Кроме того, настоящее изобретение относится к применению трансдермальных пластырей, которые имеют дополнительное преимущество, заключающееся в обеспечении контролируемой доставки соединения в организм. Такие лекарственные формы могут быть приготовлены путем растворения или диспергирования указанного соединения в подходящей среде. Для улучшения проникновение соединения через кожу также могут быть применены усилители абсорбции. Скорость можно контролировать путем обеспечения мембраны, регулирующей скорость, или путем диспергирования указанного соединения в полимерной матрице или геле.
[00108] Согласно другому варианту реализации настоящее изобретение относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, мутанта с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией) в биологическом образце, включающему этап приведения указанного биологического образца в контакт с соединением согласно настоящему изобретению или композицией, содержащей указанное соединение. В некоторых вариантах реализации настоящее изобретение относится к способу необратимого ингибирования активности по меньшей мере одного мутанта EGFR (например, мутанта с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией) в биологическом образце, включающему этап приведения указанного биологического образца с соединением согласно настоящему изобретению или композицией, содержащей указанное соединение.
[00109] В некоторых вариантах реализации настоящего изобретения предложенное соединение в биологическом образце селективно ингибирует: (а) по меньшей мере одну активирующую мутацию, (b) T790M и (с) по существу не ингибирует WT. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой deIE746-A750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[00110] В настоящей заявке термин «биологический образец» включает, без ограничения, клеточные культуры или их экстракты, биопсированный материал, полученный у млекопитающего, или его экстракты и кровь, слюну, мочу, кал, сперму, слезы или другие жидкости организма или их экстракты.
[00111] Ингибирование активности по меньшей мере одного мутанта EGFR (например, мутанта с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией) в биологическом образце подходит для различных задач, которые известны специалисту в данной области. Примеры таких задач включают, но не ограничиваются ими, переливание крови, трансплантацию органов, хранение биологических образцов и биологические анализы.
[00112] Другой вариант реализации настоящего изобретения относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, мутанта с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией) в организме пациента, включающему этап введения указанному пациенту соединения согласно настоящему изобретению или композиции, содержащей указанное соединение. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования (а) по меньшей мере одной активирующей мутации и (b) T790M в организме пациента и (с) по существу без ингибирования WT, причем указанный способ включает введение пациенту предложенное соединение или его композицию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования по меньшей мере одного мутанта EGFR в организме пациента, причем активирующая мутация представляет собой deIE746-A750. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования по меньшей мере одного мутанта EGFR в организме пациента, причем активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования по меньшей мере одного мутанта EGFR в организме пациента, причем активирующая мутация представляет собой G719S.
[00113] Согласно другому варианту реализации настоящее изобретение относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, мутанта с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией) в организме пациента, включающему этап введения указанному пациенту соединения согласно настоящему изобретению или композиции, содержащей указанное соединение. Согласно некоторым вариантам реализации настоящее изобретение относится к способу необратимого ингибирования активности по меньшей мере одного мутанта EGFR (например, мутанта с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией) в организме пациента, включающему этап введения указанному пациенту соединения согласно настоящему изобретению или композиции, содержащей указанное соединение. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования: (а) по меньшей мере одной активирующей мутации и (b) T790M в организме пациента и (с) по существу без ингибирования, причем указанный способ включает введение пациенту предложенного соединения или его композиции. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна необратимо ингибируемая активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна необратимо ингибируемая активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования по меньшей мере одного мутанта EGFR в организме пациента, причем активирующая мутация представляет собой deIE746-A750. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования по меньшей мере одного мутанта EGFR в организме пациента, причем активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования по меньшей мере одного мутанта EGFR в организме пациента, причем активирующая мутация представляет собой G719S.
[00114] В других вариантах реализации настоящего изобретения предложен способ лечения расстройства, медиируемого одним или более мутантами EGFR (например, мутантами с делеционной мутацией, активирующей мутацией, мутацией устойчивости или их комбинацией), у пациента, нуждающегося в таком лечении, включающий этап введения указанному пациенту соединения согласно настоящему изобретению или его фармацевтически приемлемую композицию. Такие расстройства подробно описаны в настоящей заявке.
[00115] В зависимости от конкретного патологического состояния или заболевания, подлежащего лечению, дополнительные терапевтические агенты, которые обычно вводят для лечения указанного состояния, также могут присутствовать в композициях согласно настоящему изобретению. В настоящей заявке дополнительные терапевтические агенты, которые обычно вводят для лечения конкретного заболевания или патологического состояния, известны как «применяемые при заболевании или патологическом состоянии, подлежащем лечению».
[00116] Например, соединения согласно настоящему изобретению или их фармацевтически приемлемую композицию вводят в комбинации с химиотерапевтическими агентами для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических агентов в числе прочих включают, но не ограничиваются ими, Адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерфероны, производные платины, таксан (например, паклитаксел), алкалоиды барвинка (например, винбластин), антрациклины (например, доксорубицин), эпиподофиллотоксины (например, этопозид), цисплатин, ингибитор mTOR (например, рапамицин), метотрексат, актиномицин D, доластатин 10, колхицин, эметин, триметрексат, метоприн, циклоспорин, даунорубицин, тенипозид, амфотерицин, алкилирующие агенты (например, хлорамбуцил), 5-фторурацил, камптотецин, цисплатин, метронидазол и Гливек™. В других вариантах реализации настоящего изобретения соединение согласно настоящему изобретению вводят в комбинации с биологическим агентом, таким как Авастин или Вектибикс.
[00117] В некоторых вариантах реализации настоящего изобретения соединения согласно настоящему изобретению или их фармацевтически приемлемую композицию вводят в комбинации с антипролиферативным или химиотерапевтическим агентом, выбранным из одного или более агентов, представляющих собой абареликс, алдеслейкин, алемтузумаб, алитретиноин, аллопуринол, алтретамин, амифостин, анастрозол, триоксид мышьяка, аспарагиназу, азацитидин, живую вакцину БЦЖ, бевацузимаб, фторурацил, бексаротен, блеомицин, бортезомиб, бусульфан, калустерон, капецитабин, камптотецин, карбоплатин, кармустин, целекоксиб, цетуксимаб, хлорамбуцил, кладрибин, клофарабин, циклофосфамид, цитарабин, дактиномицин, дарбэпоэтин альфа, даунорубицин, денилейкин, дексразоксан, доцетаксел, доксорубицин (нейтральный), доксорубицина гидрохлорид, дромостанолона пропионат, эпирубицин, эпоэтин альфа, эрлотиниб, эстрамустин, этопозида фосфат, этопозид, экземестан, филграстим, флоксуридин, флударабин, фулвестрант, гефитиниб, гемцитабин, гемтузумаб, гозерелина ацетат, гистрелина ацетат, гидроксимочевину, ибритумомаб, идарубицин, ифосфамид, иматиниба мезилат, интерферон альфа-2а, интерферон альфа-2b, иринотекан, леналидомид, летрозол, лейковорин, лейпролида ацетат, левамизол, ломустин, мегестрола ацетат, мелфалан, меркаптопурин, 6-МП, месну, метотрексат, метоксален, митомицин С, митотан, митоксантрон, нандролон, неларабин, нофетумомаб, опрелвекин, оксалиплатин, паклитаксел, палифермин, памидронат, пегадемазу, пэгаспаргазу, пэгфилграстим, пеметрексед динатрия, пентостатин, пипоброман, пликамицин, порфимер натрия, прокарбазин, хинакрин, расбуриказу, ритуксимаб, сарграмостим, сорафениб, стрептозоцин, сунитиниба малеат, тальк, тамоксифен, темозоломид, тенипозид, ВМ-26, тестолактон, тиогуанин, 6-ТГ, тиотепу, топотекан, торемифен, тозитумомаб, трастузумаб, третиноин, полностью трансретиноевая кислота (ПТРК), урамустин, валрубицин, винбластин, винкристин, винорелбин, золедронат или золедроновую кислоту.
[00118] Другие примеры агентов, с которыми также могут быть объединены ингибиторы согласно настоящему изобретению, включают, без ограничения, средства для лечения болезни Альцгеймера, такие как донепезила гидрохлорид (Арисепт®) и ривастигмин (Экселон®); средства для лечения болезни Паркинсона, такие как L-ДОФА/карбидопа, энтакапон, ропинирол, прамипексол, бромокриптин, перголид, тригексифенидил и амантадин; агенты для лечения множественный склероз (МС), такие как бета-интерферон (например, Авонекс® и Ребиф®), глатирамера ацетат (Копаксон®) и митоксантрон; средства для лечения астмы, такие как альбутерол и монтелукаст (Сингуляр®); агенты для лечения шизофрении, такие как зипрекса, рисперидал, кветиапин и галоперидол; противовоспалительные агенты, такие как кортикостероиды, блокаторы ФНО, IL-1 RA, азатиоприн, циклофосфамид и сульфасалазин; иммуномодулирующие и иммунодепрессивные агенты, такие как циклоспорин, такролимус, рапамицин, микофенолата мофетил, интерфероны, кортикостероиды, циклофосфамид, азатиоприн и сульфасалазин; нейтрофические факторы, такие как ингибиторы ацетилхолинэстеразы, ингибиторы МАО, интерфероны, антисудорожные средства, блокаторы ионных каналов, рилузол и агенты против болезни Паркинсона; агенты для лечения сердечно-сосудистого заболевания, такие как бета-блокаторы, ингибиторы АПФ, диуретики, нитраты, блокаторы кальциевых каналов и статины; агенты для лечения заболевания печени, такие как кортикостероиды, холестирамин, интерфероны и противовирусные агенты; агенты для лечения заболевания крови, такие как кортикостероиды, противолейкозные агенты и факторы роста, и агенты для лечения иммунодефицитных состояний, такие как гамма-глобулин.
[00119] В некоторых вариантах реализации настоящего изобретения соединения согласно настоящему изобретению или их фармацевтически приемлемую композицию вводят в комбинации с моноклональным антителом или терапевтическими миРНК.
[00120] Указанные дополнительные агенты могут быть введены отдельно от композиции, содержащей соединение согласно настоящему изобретению, как часть схемы с многократным дозированием. В качестве альтернативы, указанные агенты могут быть частью единой лекарственной формы, будучи смешанными с соединением согласно настоящему изобретению в единую композицию. При введении в качестве частей схемы с многократным дозированием два активных агента могут быть введены одновременно, последовательно или второй в течение некоторого периода времени после первого, обычно в течение пяти часов после первого.
[00121] В настоящей заявке термины «комбинация», «объединенный» и родственные им термины относятся к одновременному или последовательному введению терапевтических агентов согласно настоящему изобретению. Например, соединение согласно настоящему изобретению может быть введено с другим терапевтическим агентом одновременно или последовательно в отдельных единичных лекарственных формах или совместно в одной единичной лекарственной форме. Соответственно, в настоящем изобретении предложена единая единичная лекарственная форма, содержащая предложенное соединение, дополнительный терапевтический агент и фармацевтически приемлемый носитель, адъювант или наполнитель.
[00122] Общее количество соединения согласно настоящему изобретению и дополнительного терапевтического агента (в тех композициях, которые содержат дополнительный терапевтический агент, описанный выше), которые могут быть объединены с носителями с получением единой лекарственной формы, будет варьироваться в зависимости от организма, подвергаемого лечению, и конкретного способа введения. Предпочтительно, композиции согласно настоящему изобретению следует готовить так, чтобы можно было вводить дозу соединения согласно изобретению 0,01-100 мг/кг массы тела/сутки.
[00123] В указанных композициях, которые содержат дополнительный терапевтический агент, указанный дополнительный терапевтический агент и соединение согласно настоящему изобретению могут действовать синергетически. Следовательно, количество дополнительного терапевтического агента в таких композициях должно быть меньше, чем то, которое требуется для монотерапии с применением только указанного терапевтического агента. В таких композициях доза дополнительного терапевтического агента, которая может быть введена, составляет 0,01-1000 мкг/кг массы тела/сутки.
[00124] Количество дополнительного терапевтического агента, присутствующего в композициях согласно настоящему изобретению, должно быть не больше, чем количество, которое обычно вводят при введении композиции, содержащей указанный терапевтический агент в качестве единственного активного агента. Предпочтительно, количество дополнительного терапевтического агента в композициях, предложенных в настоящем изобретении, будет варьироваться от примерно 50% до 100% от количества, обычно присутствующего в композиции, содержащей указанный агент в качестве единственного терапевтически активного агента.
[00125] Соединения согласно настоящему изобретению или их фармацевтические композиции также могут быть включены в композиции для покрытия имплантируемых медицинских устройств, таких как протезы, искусственные клапаны, сосудистые трансплантанты, стенты и катереры. Например, сосудистые стенты применяют для преодоления рестеноза (повторного сужения стенки сосуда после травмы). Однако пациенты при применении стентов или других имплантируемых устройств подвергаются риску образования тромба или активации тромбоцитов. Указанные нежелательные эффекты можно предотвратить или смягчить путем предварительного нанесения на указанное устройство покрытия с применением фармацевтически приемлемой композиции, содержащей ингибитор киназы. Имплантируемые устройства, покрытые соединением согласно настоящему изобретению, являются еще одним вариантом реализации настоящего изобретения.
ПРИМЕРЫ
[00126] Как показано в Примерах ниже, в некоторых иллюстративных вариантах реализации настоящего изобретения соединения получают согласно следующим общим методикам. Следует иметь в виду, что хотя общие способы описывают синтез некоторых соединений согласно настоящему изобретению, следующие общие способы и другие способы, известные специалисту в данной области, могут быть применены ко всем соединениям и подклассам и видам каждого из указанных соединений, описанных в настоящей заявке.
[00127] Номера соединений, примененных в Примерах ниже, соответствуют номерам соединений, представленных в Таблице 1 выше.
[00128] Предложенные соединения получают способами, известными средним специалистам в данной области и включающими способы, подробно описанные в заявке на патент США 20100029610, опубликованной 4 февраля 2010 г., содержание которой полностью включено в настоящее описание посредством ссылки.
ПРИМЕР 1
Промежуточное соединение 1
Схема 1
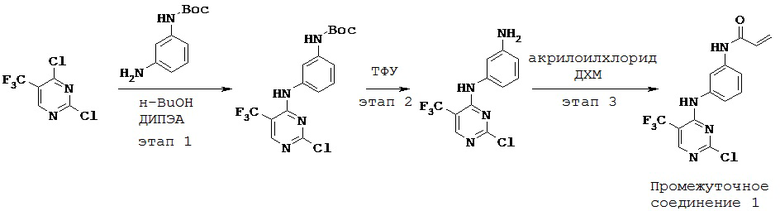
Этап 1:
[00129] В 25 мл 3-горлую круглодонную колбу предварительно снабженную магнитной мешалкой, рубашкой и предохранительной трубкой с CaCl2, загружали N-Вос-1,3-диаминобензол (0,96 г) и н-бутанол (9,00 мл). Реакционную смесь охлаждали до 0°С. К вышеуказанной реакционной смеси при 0°С по каплям добавляли 2,4-дихлор-5-трифторметилпиримидин (1,0 г). К вышеуказанной реакционной смеси при 0°С по каплям добавляли N,N-диизопропилэтиламин (ДИПЭА) (0,96 мл) и реакционную смесь перемешивали в течение 1 часа при температуре от 0°С до 5°С. Наконец, реакционную смесь оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали еще 4 часа при комнатной температуре. Окончание реакции отслеживали путем ТСХ с применением гексана: этилацетата (7:3). Твердое вещество, выпадавшее в осадок, отфильтровывали и промывали 1-бутанолом (2 мл). Твердое вещество сушили при пониженном давлении при 40°С в течение 1 часа. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,48 (с, 9Н), 7,02 (м, 1Н), 7,26 (м, 2Н), 7,58 (с, 1Н), 8,57 (с, 1Н), 9,48 (с, 1Н), 9,55 (с, 1Н).
Этап 2:
[00130] К вышеуказанному неочищенному веществу (3,1 г) в дихлорметане (ДХМ) (25 мл) медленно добавляли трифторуксусную кислоту (ТФУ) (12,4 мл) при 0°С. Реакционную смесь оставляли нагреваться до комнатной температуры. Далее реакционную смесь перемешивали еще 10 мин при комнатной температуре. Неочищенное вещество концентрировали при пониженном давлении.
Этап 3:
[00131] Концентрированное неочищенное вещество растворяли в ДИПЭА (2,0 мл) и ДХМ (25 мл), а затем охлаждали до -30°С. К реакционной смеси медленно добавляли акрилоилхлорид (0,76 г) при -30°С. Реакционную массу нагревали до комнатной температуры, после чего перемешивали при комнатной температуре в течение 1,0 ч. Ход реакции отслеживали с помощью ТСХ с применением гексана: этилацетата (7:3) в качестве подвижной фазы. Реакция завершалась через 1 ч. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 5,76 (дд, J=2,0, 10,0 Гц, 1Н), 6,24 (дд, J=2,0, 17,2 Гц, 1Н), 6,48 (м, 1Н), 7,14 (д, J=8,8 Гц, 1Н), 7,37 (т, J=8,0 Гц, 1Н), 7,94 (с, 1Н), 8,59 (с, 1Н), 9,60 (с, 1Н), 10,26 (с, 1Н).
ПРИМЕР 2
Соединение I-2 N-(3-(2-(2-метокси-4-морфолинофениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
[00132] Для получения указанного в заголовке соединения I-2 смесь промежуточного соединения 1, полученного в Примере 1, (16 мг) и 2-метокси-4-морфолиноанилина в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением указанного в заголовке соединения в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,4 (с, 1H), 9,72 (ушир., 1H), 9,18 (ушир., 1H), 8,49 (ушир., 1H), 7,83 (с, 1H), 7,59 (д, J=8,4 Гц, 1H), 7,31-7,48 (м, 2H), 7,41 (т, J=15,2 Гц, 1H), 7,12 (ушир., 1H), 6,67 (с, 1H), 6,49 (дд, J=10,0, 16,8 Гц, 1H), 6,25 (дд, J=2,0, 16,8 Гц, 1H), 5,77 (дд, J=2,0, 10,0 Гц, 1H), 3,7-3,9 (м, 7Н), 3,1 (ушир., 4H); вычисленное значение массы для C25H25F3N6O3: 514,2, полученное значение: 515,5 (M+H+).
ПРИМЕР 3
Соединение I-4 N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-метоксифениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
[00133] Указанное в заголовке соединение I-4 получали, как описано в Примере 2, с применением 2-метокси-4-(4-ацетилпиперазинил)анилина и промежуточного соединения 1, полученного в Примере 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,2 (с, 1H), 8,2 (ушир., 1H), 8,30 (с, 1H), 7,73 (ушир., 1H), 7,52 (д, J=7,8 Гц, 1H), 7,45 (д, J=7,8 Гц, 1H), 7,26 (J=8,2 Гц, 1H), 7,14 (ушир., 1H), 6,60 (с, 1H), 6,42 (дд, J=11,4, 16,9 Гц, 1H), 6,24 (д, J=16,9 Гц, 1H), 5,75 (д, J=11,4 Гц, 1H), 3,76 (с, 3H), 3,04 (ушир., 4H), 2,04 (с, 3H); вычисленное значение массы для C27H28F3N7O3: 555,2, полученное значение: 556,2 (М+Н+).
ПРИМЕР 4
Промежуточное соединение 2
Схема 2
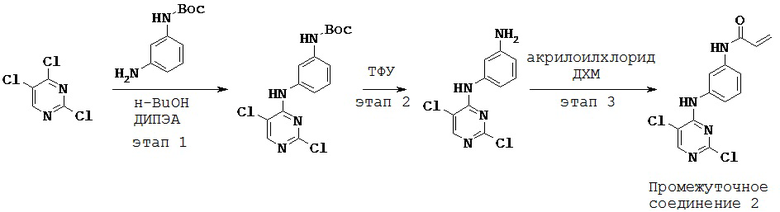
Этап 1:
[00134] Указанный в заголовке этап осуществляли согласно Этапу 1 в Схеме 1 Примера 1. 1Н-ЯМР (ДМСО-d6, 400 МГц) δ 1,48 (с, 9Н), 7,16 (д, 1H), 7,25 (м, 2H), 7,70 (с, 1H), 8,37 (с, 1H), 9,47 (с, 1H), 9,55 (с, 1H).
Этап 2:
[00135] Указанный в заголовке этап осуществляли согласно Этапу 2 в Схеме 1 Примера 1.
Этап 3:
[00136] Указанный в заголовке этап осуществляли согласно Этапу 3 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 5,76 (дд, J=1,6, 10,8, Гц 1H), 6,25 (дд, J=1,6, 16,8 Гц, 1H), 6,46 (м, 1H), 7,30 (м, 2H), 7,46 (д, J=8,0 Гц, 1H), 7,91 (с, 1H), 8,38 (с, 1H), 9,60 (с, 1H), 10,23 (с, 1H).
ПРИМЕР 5
Промежуточное соединение 3
Схема 3
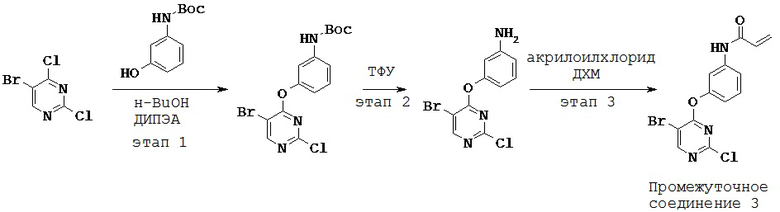
Этап 1:
[00137] Указанный в заголовке этап осуществляли согласно Этапу 1 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,47 (с, 9Н), 6,89 (д, J=7,6 Гц, 1H), 7,35 (м, 2H), 7,45 (с, 1H), 8,89 (с, 1H), 9,64 (с, 1H).
Этап 2:
[00138] Указанный в заголовке этап осуществляли согласно Этапу 2 в Схеме 1 Примера 1.
Этап 3:
[00139] Указанный в заголовке этап осуществляли согласно Этапу 3 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 5,77 (д, J=10,0 Гц, 1H), 6,25 (д, J=17,2 Гц, 1H), 6,45 (м, 1H), 7,01 (д, J=7,2 Гц, 1H), 7,53 (м, 2H), 7,73 (с, 1H), 8,98 (с, 1H), 10,40 (с, 1H).
ПРИМЕР 6
Промежуточное соединение 4
Схема 4
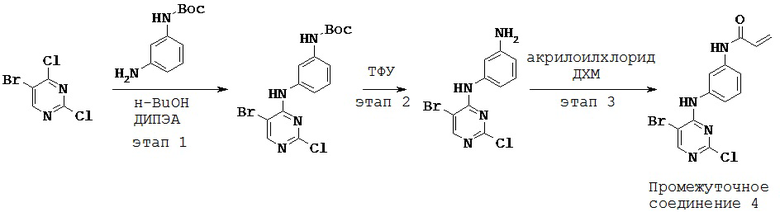
Этап 1:
[00140] Указанный в заголовке этап осуществляли согласно Этапу 1 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,50 (с, 9Н), 7,10 (д, J=6,0 Гц, 1H), 7,25 (м, 2H), 8,44 (с, 1H), 9,32 (с, 1H), 9,47 (с, 1H).
Этап 2:
[00141] Указанный в заголовке этап осуществляли согласно Этапу 2 в Схеме 1 Примера 1.
Этап 3:
[00142] Указанный в заголовке этап осуществляли согласно Этапу 3 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 5,76 (дд, J=1,6, 10,0 Гц, 1H), 6,25 (дд, J=1,6, 16,8 Гц, 1H), 6,43 (м, 1H), 7,23 (д, J=8,0 Гц, 1H), 7,35 (т, J=8,0 Гц, 1H), 7,48 (д, J=8,0 Гц, 1H), 7,80 (с, 1H), 8,38 (с, 1H), 9,36 (с, 1H), 10,23 (с, 1H).
ПРИМЕР 7
Промежуточное соединение 5
Схема 5
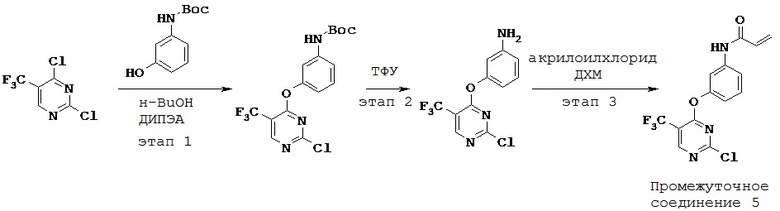
Этап 1:
[00143] Указанный в заголовке этап осуществляли согласно Этапу 1 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,47 (с, 9Н), 6,90 (д, J=6,0 Гц, 1H), 7,35 (м, 2H), 7,50 (с, 1H), 9,05 (с, 1H), 9,65 (с, 1H).
Этап 2:
[00144] Указанный в заголовке этап осуществляли согласно Этапу 2 в Схеме 1 Примера 1.
Этап 3:
[00145] Указанный в заголовке этап осуществляли согласно Этапу 3 в Схеме 1 Примера 1. 1Н-ЯМР (ДМСО-d6, 400 МГц) δ 5,76 (д, J=10,0 Гц, 1H), 6,25 (дд, J=1,6, 16,8 Гц, 1H), 6,46 (м, 2H), 7,01 (д, J=8,0 Гц, 1H), 7,08 (т, J=8,4 Гц, 1Н) 7,25 (с, 1H), 9,44 (с, 1H), 10,02 (с, 1H).
ПРИМЕР 8
Промежуточное соединение 6
Схема 6
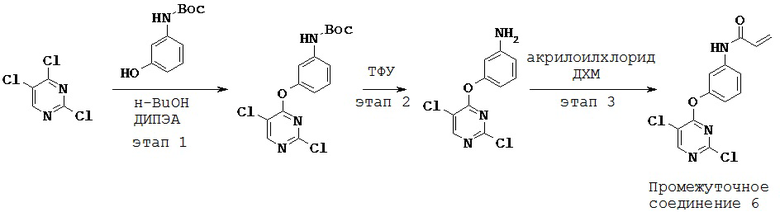
Этап 1:
[00146] Указанный в заголовке этап осуществляли согласно Этапу 1 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,47 (с, 9Н), 6,60 (с, 1H), 6,86 (д, J=8,4 Гц, 1H), 7,13 (т, J=7,6 Гц, 1H), 7,36 (м, 1H), 7,50 (с, 1H), 8,40 (с, 1H).
Этап 2:
[00147] Указанный в заголовке этап осуществляли согласно Этапу 2 в Схеме 1 Примера 1.
Этап 3:
[00148] Указанный в заголовке этап осуществляли согласно Этапу 3 в Схеме 1 Примера 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 5,78 (дд, J=2,0, 10,0 Гц, 1H), 6,25 (дд, J=2,0, 17,2 Гц, 1H), 6,40 (м, 1H), 7,02 (д, 1H), 7,50 (м, 2H), 7,71 (с, 1H), 8,40 (с, 1H), 10,35 (с, 1H).
ПРИМЕР 9
Соединение I-5 N-(3-(5-хлор-2-(2-метокси-4-морфолинофениламино)пиримидин-4-илокси)фенил)акриламид)
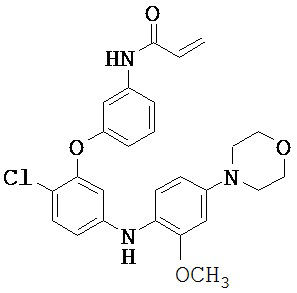
[00149] Для получения указанного в заголовке соединения смесь промежуточного соединения 6, полученного в Примере 8, и 2-метокси-4-морфолиноанилина в н-бутаноле с HCl в качестве катализатора нагревали с применением микроволнового излучения до 150°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с получением указанного в заголовке соединения. 1H-ЯМР (хлороформ-d, 400 МГц) δ 8,23 (с, 1H), 7,6-7,8 (ушир., 2H), 7,4-7,5 (м, 3H), 7,00 (дд, J=1,4, 8,2 Гц, 1H), 6,41 (м, 2H), 6,23 (м, 2H), 5,77 (дд, J=1,4, 10,1 Гц, 1H), 3,84 (м, 4H), 3,81 (с, 3H), 3,04 (м, 4H); вычисленное значение массы для C24H24ClN5O4: 481,2, полученное значение: 482,2 (M+H+).
ПРИМЕР 10
Соединение I-6 N-(3-(2-(4-(4-(2-гидроксиацетил)пиперазин-1-ил)-2-метоксифениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
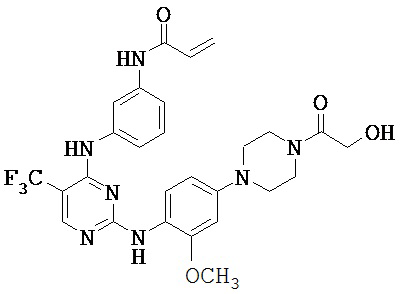
[00150] Указанное в заголовке соединение I-6 получали, как описано в Примере 2, с применением 2-метокси-4-(4-(2-гидроксиацетил)пиперазинил)анилина и промежуточного соединения 1, полученного в Примере 1. 1H-ЯМР (CDCl3, 400 МГц) δ 8,33 (с, 1H), 8,08 (ушир., 1H), 7,86 (ушир., 1H), 7,60 (ушир., 1H), 7,39 (м, 1H), 6,89 (с, 1H), 6,22-6,55 (м, 3H), 5,80 (д, J=10,0 Гц), 4,24 (с, 2H), 3,90 (с, 2H), 3,85 (с, 2H), 3,64 (с, 1H), 3,45 (с, 2H), 3,13 (с, 3H); вычисленное значение массы для C27H28F3N7O4: 571,2, полученное значение: 572,4 (M+H+).
ПРИМЕР 11
Соединение I-7 N-(3-(2-(4-(4-ацетил-1,4-диазепин-1-ил)-2-метоксифениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
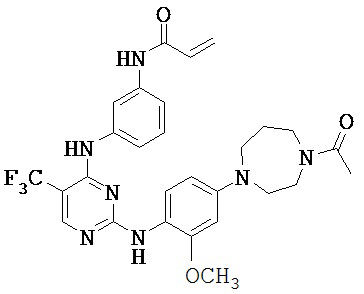
[00151] Указанное в заголовке соединение I-7 получали, как описано в Примере 2, с применением 1-(4-(4-амино-3-метоксифенил)-1,4-диазепин-1-ил)этанона и промежуточного соединения 1, полученного в Примере 1. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,2 (с, 1H), 9,2 (ушир., 1H), 8,7 (ушир., 1H), 8,4 (ушир., 1H), 7,76 (ушир., 1H), 7,49 (д, J=8,2 Гц, 1H), 7,27 (ушир., 2H), 7,1 (ушир., 1H), 6,42 (дд, J=11,0, 16,5 Гц, 1H), 6,30 (ушир., 1H), 6,24 (д, J=16,5 Гц, 1H), 5,9 (ушир., 1H), 5,74 (д, J=11,0 Гц, 1H), 3,3-3,7 (м, 4H), 1,7-1,95 (м, 5Н); вычисленное значение массы для C28H30F3N7O3: 569,2, полученное значение: 570,2 (М+Н+).
ПРИМЕР 12
Соединение I-10 N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-метоксифениламино)-5-(трифторметил)пиримидин-4-илокси)фенил)акриламид)
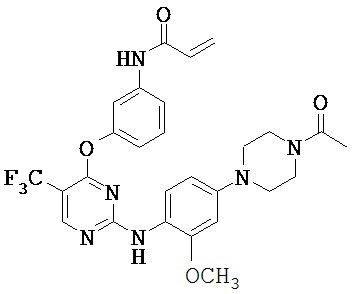
[00152] Для получения указанного в заголовке соединения смесь промежуточного соединения 5, полученного в Примере 7, и 1-(4-(4-амино-3-метоксифенил)пиперазин-1-ил)этанона (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением указанного в заголовке соединения в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,32 (с, 1H), 8,92 (с, 1H), 8,60 (с, 1H), 7,72 (т, J=2,3 Гц, 1H), 7,58 (д, J=8,7 Гц, 1H), 7,43 (м, 2H), 6,98 (м, 1H), 6,61 (д, J=2,3, 1H), 6,42 (м, 2H), 6,25 (дд, J=1,8, 16,9 Гц, 1H), 5,77 (д, J=1,8, 10,1 Гц, 1H), 3,7-4,0 (м, 4H), 3,77 (с, 3H), 3,1 (м, 4H), 1,99 (с, 3H); вычисленное значение массы для C27H27F3N6O4: 556,2, полученное значение: 557,1 (М+Н+).
ПРИМЕР 13
Соединение I-9 N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-метоксифениламино)-5-хлорпиримидин-4-илокси)фенил)акриламид)
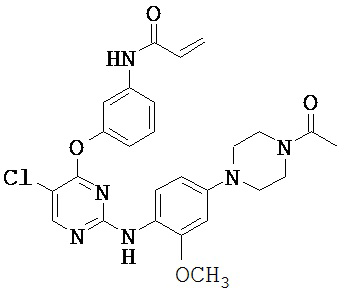
[00153] Указанное в заголовке соединение получали, как описано в Примере 9, с применением 1-(4-(4-амино-3-метоксифенил)пиперазин-1-ил)этанона и промежуточного соединения 6, полученного в Примере 8. 1H-ЯМР (хлороформ-d, 400 МГц) δ 8,26 (с, 1H), 8,08 (ушир., 1H), 7,93 (ушир., 1H), 7,68 (с, 1H), 7,57 (м, 1H), 7,45 (м, 2H), 6,96 (д, J=7,8 Гц, 1H), 6,69 (с, 1H), 6,60 (д, J=7,4 Гц, 1H), 6,41 (д, J=1,4, 17,0 Гц, 1H), 6,30 (дд, J=10,1, 16,5 Гц, 1H), 5,75 (д, J=1,4, 10,1 Гц, 1H), 3,97 (м, 2H), 3,85 (с, 3H), 3,82 (м, 2H), 3,29 (м, 2H), 3,24 (м, 2H), 2,19 (с, 3H); вычисленное значение массы для C26H27ClN6O4: 522,2, полученное значение: 523,2 (М+Н+).
ПРИМЕР 14
Соединение I-3 N-(3-(5-хлор-2-(4-(4-(2-гидроксиацетил)пиперазин-1-ил)-2-метоксифениламино)пиримидин-4-илокси)фенил)акриламид)
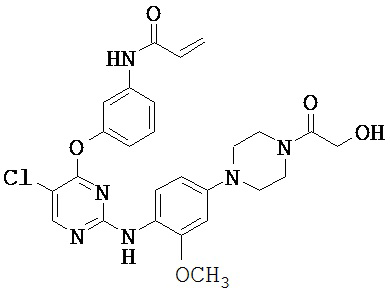
[00154] Указанное в заголовке соединение получали, как описано в Примере 9, с применением 1-(4-(4-амино-3-метоксифенил)пиперазин-1-ил)-2-гидроксиэтанона и промежуточного соединения 6, полученного в Примере 8. 1H-ЯМР (хлороформ-d, 400 МГц) δ 8,24 (с, 1H), 7,71 (ушир., 1H), 7,63 (м, 1H), 7,49 (м, 1H), 7,44 (т, J=8,2 Гц, 1H), 7,39 (д, J=6,9 Гц, 2H), 7,00 (дд, J=1,8, 7,8 Гц, 1H), 6,45 (д, J=2,8 Гц, 1H), 6,44 (дд, J=1,4, 16,9 Гц, 1H), 6,23 (дд, J=10,1, 16,9 Гц, 1H), 5,79 (дд, J=1,4, 10,1 Гц, 1H), 4,22 (с, 2H), 3,82 (с, 3H), 3,80 (с, 2H), 3,42 (м, 2H), 3,06 (м, 4H); вычисленное значение массы для C26H27ClN6O5: 538,2, полученное значение: 539,1 (М+Н+).
ПРИМЕР 15
Соединение I-8 N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-метоксифениламино)-5-хлорпиримидин-4-иламино)фенил)акриламид)
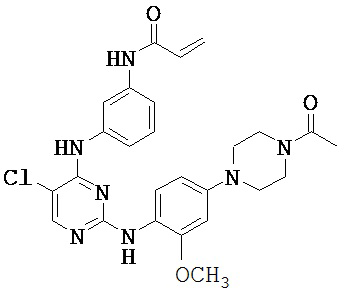
[00155] Для получения указанного в заголовке соединения смесь промежуточного соединения 2, полученного в Примере 4, и 1-(4-(4-амино-3-метоксифенил)пиперазин-1-ил)этанона в н-бутаноле с HCl в качестве катализатора нагревали с применением микроволнового излучения до 150°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с получением указанного в заголовке соединения. 1Н-ЯМР (ДМСО-d6, 400 МГц) δ 10,2 (с, 1H), 8,86 (с, 1H), 8,07 (с, 1H), 7,94 (ушир., 1H), 7,70 (с, 1H), 7,68 (с, 1H), 7,46 (д, J=7,6 Гц, 1H), 7,27 (м, 2H), 6,63 (д, J=2,4 Гц, 1H), 6,46 (дд, J=10,0, 16,8 Гц, 1H), 6,31 (ушир., 1H), 6,29 (дд, J=2,0, 16,8 Гц, 1H), 5,76 (дд, J=2,0, 10,0 Гц, 1H), 3,79 (с, 3H), 3,56 (м, 4H), 3,0-3,2 (м, 4H), 2,04 (с, 3H); вычисленное значение массы для C26H28ClN7O3: 521,2, полученное значение: 522,4 (M+H+).
ПРИМЕР 16
Соединение I-12 N-(3-(5-хлор-2-(4-(4-(2-гидроксиацетил)пиперазин-1-ил)-2-метоксифениламино)пиримидин-4-иламино)фенил)акриламид)
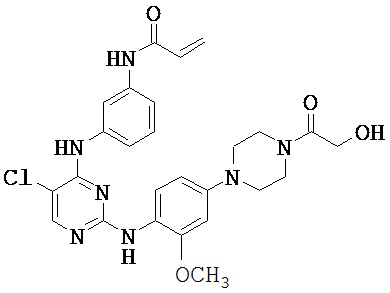
[00156] Указанное в заголовке соединение получали, как описано в Примере 15, с применением 1-(4-(4-амино-3-метоксисренил)пиперазин-1-ил)-2-гидроксиэтанона и промежуточного соединения 2, полученного в Примере 4. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,2 (с, 1H), 8,86 (с, 1H), 8,07 (с, 1H), 7,94 (ушир., 1H), 7,70 (м, 2H), 7,46 (д, J=7,6 Гц, 1H), 7,27 (м, 2H), 6,63 (д, J=2,4 Гц, 1H), 6,46 (дд, J=10,0, 16,8 Гц, 1H), 6,31 (ушир., 1H), 6,29 (дд, J=2,0, 16,8 Гц, 1H), 5,76 (дд, J=2,0, 10,0 Гц, 1H), 4,66 (т, J=5,6 Гц, 1H), 4,14 (т, J=5,6 Гц, 2H), 3,79 (с, 3H), 3,61 (ушир., 2H), 3,48 (ушир., 2H), 3,05 (м, 4H), 2,04 (с, 3H); вычисленное значение массы для C26H28ClN7O4: 537,2, полученное значение: 538,4 (М+Н+).
ПРИМЕР 17
Соединение I-11 N-(3-(2-(4-(4-(2-гидроксиацетил)пиперазин-1-ил)-2-метоксифениламино)-5-(трифторметил)пиримидин-4-илокси)фенил)акриламид)
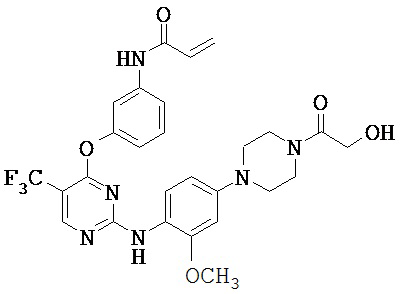
[00157] Указанное в заголовке соединение получали, как описано в Примере 12, с применением 1-(4-(4-амино-3-метоксифенил)пиперазин-1-ил)-2-гидроксиэтанона и промежуточного соединения 5, полученного в Примере 7. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,34 (с, 1H), 8,84 (с, 1H), 8,56 (ушир., 1H), 7,63 (с, 1H), 7,53 (д, J=8,0 Гц, 1H), 7,41 (м, 1H), 7,16 (м, 1H), 6,96 (ушир., 1H), 6,57 (ушир., 1H), 6,45 (ушир., 1H), 6,44 (дд, J=10,0, 16,8 Гц, 1H), 6,29 (дд, J=1,6, 16,8 Гц, 1H), 5,79 (дд, J=1,6, 10,0 Гц, 1H), 4,66 (т, J=5,6 Гц, 1H), 4,14 (д, J=5,6 Гц, 2H), 3,73 (с, 3H), 3,60 (ушир., 2H), 3,47 (ушир., 2H), 3,08 (ушир., 4H); вычисленное значение массы для C27H27F3N6O5: 572,2, полученное значение: 573,6 (M+H+).
ПРИМЕР 18
Соединение I-27, N-(3-(2-(2-метокси-4-(4-сульфамоилпиперазин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-илокси)фенил)акриламид)
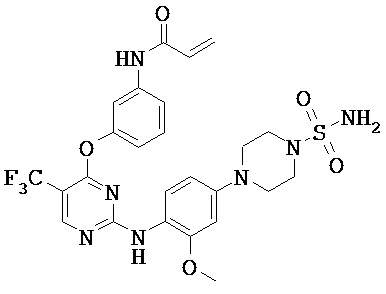
[00158] Смесь промежуточного соединения 5 (20 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N-метилморфолин (20 мкл), диоксан (0,5 мл) и сульфамид (50 мг). Реакционную смесь нагревали с применением микроволнового излучения до 90°С и поддерживали указанную температуру в течение 30 минут. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,30 (с, 1H), 8,81 (с, 1H), 8,54 (ушир., 1H), 7,62 (с, 1H), 7,52 (д, J=8,2 Гц, 1H), 7,39 (т, J=8,2 Гц, 1H), 7,16 (д, J=8,7 Гц, 1H), 6,95 (м, 1H), 6,85 (с, 2H), 6,58 (м, 1H), 6,43 (дд, J=11,4, 17,0 Гц, 1H), 6,26 (д, J=17,0 Гц, 1H), 5,78 (д, J=11,4 Гц, 1H), 3,72 (с, 3H), 3,19 (м, 4H), 3,06 (м, 4H); вычисленное значение массы для C25H26F3N7O5S: 593,2, полученное значение: 594,2 (М+Н+).
ПРИМЕР 19
Соединение I-28 (4-(4-(4-(3-акриламидофениламино)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)-N-метилпиперазин-1-карбоксамид)
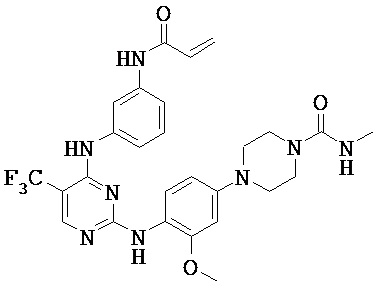
[00159] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и N-метил-N-(гидроксисукцинил)карбамат (50 мг) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C27H29F3N8O3: 570,2, полученное значение: 571,2 (M+H+).
ПРИМЕР 20
Соединение I-29 (метиловый эфир 4-(4-(4-(3-акриламидофениламино)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)пиперазин-1-карбоновой кислоты)
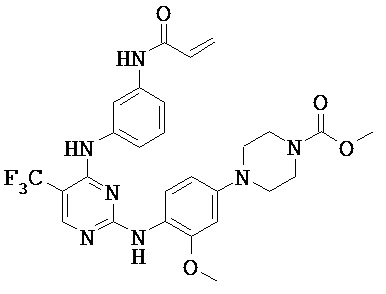
[00160] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и метиловый эфир хлормуравьиной кислоты (20 мкл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C27H28F3N7O4: 571,2, полученное значение: 572,2 (М+Н+).
ПРИМЕР 21
Соединение I-17 (N-(3-(2-(2-метокси-4-(4-(метилсульфонил)пиперазин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
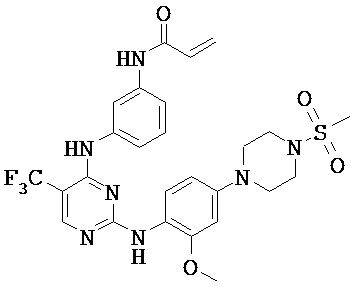
[00161] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и метансульфонилхлорид (20 мкл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,22 (с, 1H), 9,28 (ушир., 1H), 8,72 (ушир., 1H), 8,37 (ушир., 1H), 7,77 (с, 1H), 7,52 (д, J=8,0 Гц, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,30 (т, J=8,0 Гц, 1H), 7,12 (ушир., 1H), 6,62 (с, 1H), 6,43 (дд, J=10,0, 16,8 Гц, 1H), 6,24 (д, J=16,8 Гц, 1H), 6,22 (ушир., 1H), 5,76 (д, J=10,0 Гц, 1H), 3,77 (с, 3H), 3,21 (м, 4H), 3,18 (м, 4H), 2,92 (с, 3H); вычисленное значение массы для C26H28F3N7O4S: 591,2, полученное значение: 592,2 (М+Н+).
ПРИМЕР 22
Соединение I-19 (N-(3-(2-(2-метокси-4-(4-(2,2,2-трифторацетил)-1,4-диазепин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
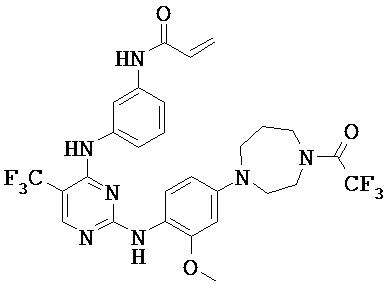
[00162] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты (21 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и трифторуксусный ангидрид (10 мкл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,2 (с, 1H), 9,1 (ушир., 1H), 8,6 (ушир., 1H), 8,3 (ушир., 1H), 7,75 (ушир., 1H), 7,50 (д, J=8,7 Гц, 1H), 7,26 (м, 2H), 7,11 (м, 1H), 6,42 (дд, J=10,1, 17,0 Гц, 1H), 6,34 (м, 1H), 6,23 (д, J=17,0 Гц, 1H), 5,74 (дд, J=1,8, 10,1 Гц,1H), 3,3-3,8 (м, 8Н), 1,88 (м, 2H); вычисленное значение массы для C28H27F6N7O3: 623,2, полученное значение: 624,2 (М+Н+).
ПРИМЕР 23
Соединение I-20 (метиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)пиперазин-1-карбоновой кислоты)
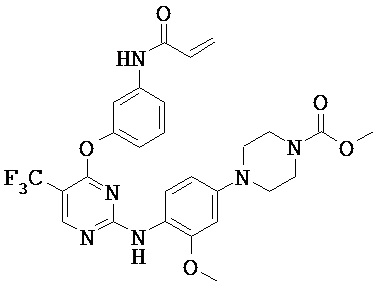
[00163] Смесь промежуточного соединения 5 (20 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и метиловый эфир хлормуравьиной кислоты (20 мкл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,30 (с, 1H), 8,80 (с, 1H), 8,53 (ушир., 1H), 7,60 (с, 1H), 7,51 (д, J=8,2 Гц, 1H), 7,40 (т, J=8,2 Гц, 1H), 7,14 (д, J=9,6 Гц, 1H), 6,93 (м, 1H), 6,55 (м, 1H), 6,40 (дд, J=10,0, 16,8 Гц, 1H), 6,24 (дд, J=1,8, 16,8 Гц, 1H), 5,76 (дд, J=1,8, 10,0 Гц, 1H), 3,70 (с, 3H), 3,58 (с, 3H), 3,49 (м, 4H), 3,05 (м, 4H); вычисленное значение массы для C27H27F3N6O5: 572,2, полученное значение: 573,2 (М+Н+).
ПРИМЕР 24
Соединение I-21 (4-(4-(4-(3-акриламидофенокси)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)-N-метилпиперазин-1-карбоксамид)
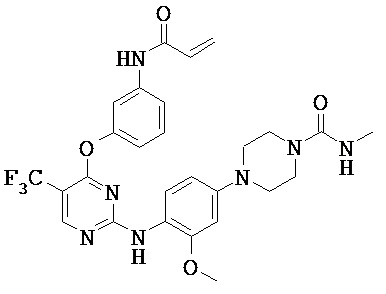
[00164] Смесь промежуточного соединения 5 (18 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и N-метил-N-(гидроксисукцинил)карбамат (50 мг) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C27H28F3N7O4: 571,2, полученное значение: 572,2 (М+Н+).
ПРИМЕР 25
Соединение I-22 (N-(3-(2-(2-метокси-4-(4-(метилсульфонил)пиперазин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-илокси)фенил)акриламид)
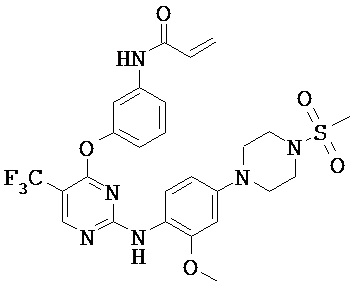
[00165] Смесь промежуточного соединения 5 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и метансульфонилхлорид (10 мкл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C26H27F3N6O5S: 592,2, полученное значение: 593,2 (М+Н+).
ПРИМЕР 26
Соединение I-23 (N-(3-(5-хлор-2-(4-(4-(3,3-диметилбутирил)пиперазин-1-ил)-2-метоксифениламино)пиримидин-4-илокси)фенил)акриламид)
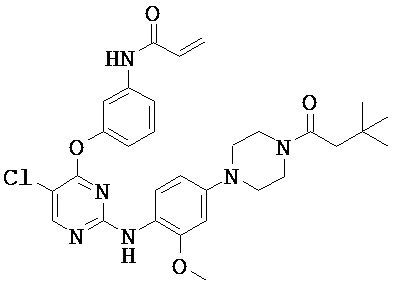
[00166] Смесь промежуточного соединения 6 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и 3,3-диметилбутирилхлорид при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,33 (с, 1H), 8,35 (с, 1H), 8,15 (с, 1H), 7,60 (с, 1H), 7,55 (д, J=8,2 Гц, 1H), 7,40 (т, J=8,2 Гц, 1H), 7,24 (д, J=8,7 Гц, 1H), 6,95 (д, J=7,8 Гц, 1H), 6,57 (с, 1H), 6,42 (дд, J=10,0, 16,8 Гц, 1H), 6,25 (д, J=16,8 Гц, 1H), 6,18 (м, 1H), 6,77 (д, J=10,0 Гц, 1H), 3,93 (с, 3H), 3,68 (м, 4H), 2,99 (м, 4H), 2,26 (с, 2H), 0,99 (с, 9Н); вычисленное значение массы для C30H35ClN6O4: 578,2, полученное значение: 579,2 (М+Н+).
ПРИМЕР 27
Соединение I-24 (N-(3-(5-хлор-2-(2-метокси-4-(4-(2,2,2-трифторацетил)пиперазин-1-ил)фениламино)пиримидин-4-илокси)фенил)акриламид)
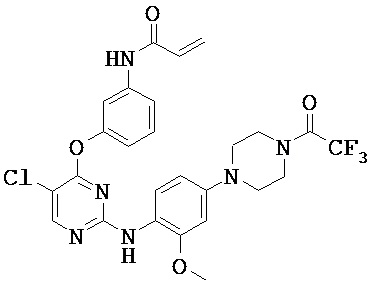
[00167] Смесь промежуточного соединения 6 (20 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и ангидрид трифторуксусной кислоты (10 мкл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,32 (с, 1H), 8,35 (с, 1H), 8,16 (с, 1H), 7,59 (с, 1H), 7,55 (д, J=8,2 Гц, 1H), 7,40 (т, J=8,2 Гц, 1H), 7,24 (д, J=8,7 Гц, 1H), 6,96 (д, J=7,8 Гц, 1H), 6,57 (д, J=2,8 Гц, 1H), 6,41 (дд, J=10,0, 16,8 Гц, 1H), 6,24 (дд, J=1,8, 16,8 Гц, 1H), 6,19 (м, 1H), 5,76 (дд, J=1,8, 10,0 Гц, 1H), 3,72 (с, 3H), 3,68 (м, 4H), 3,13 (м, 4H); вычисленное значение массы для C26H24ClF3N6O4: 576,2, полученное значение: 577,0 (М+Н+).
ПРИМЕР 28
Соединение I-25 (4-(4-(4-(3-акриламидофенокси)-5-хлорпиримидин-2-иламино)-3-метоксифенил)-(N-трет-бутил)пиперазин-1-карбоксамид)
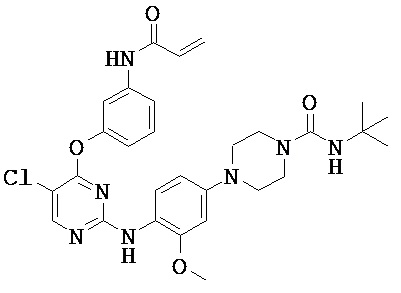
[00168] Смесь промежуточного соединения 6 (20 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и трет-бутилизоцианат при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,35 (с, 1H), 8,37 (с, 1H), 8,19 (с, 1H), 7,62 (с, 1H), 7,56 (д, J=8,2 Гц, 1H), 7,41 (т, J=8,2 Гц, 1H), 7,28 (д, J=8,2 Гц, 1H), 6,96 (д, J=8,2 Гц, 1H), 6,65 (с, 1H), 6,43 (дд, J=10,0, 16,8 Гц, 1H), 6,29 (с, 1H), 6,26 (д, J=16,8 Гц, 1H), 5,92 (с, 1H), 5,77 (д, J=10,0 Гц, 1H), 3,74 (с, 3H), 3,41 (с, 4H), 3,04 (с, 4H), 1,26 (с, 9Н); вычисленное значение массы для C29H34ClN7O4: 579,2, полученное значение: 580,2 (М+Н+).
ПРИМЕР 29
Соединение I-26 (N-(3-(5-хлор-2-(2-метокси-4-(4-(метилсульфонил)пиперазин-1-ил)фениламино)пиримидин-4-илокси)фенил)акриламид)
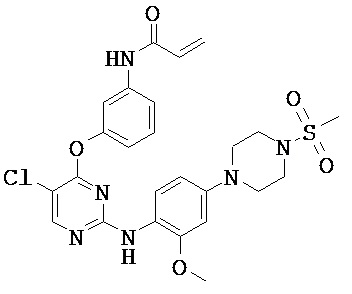
[00169] Смесь промежуточного соединения 6 (20 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и метансульфонилхлорид при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C25H27ClN6O5S: 558,2, полученное значение: 559,2 (М+Н+).
ПРИМЕР 30
Соединение I-18 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-хлорпиримидин-2-иламино)-3-этоксифенил)пиперазин-1-карбоновой кислоты)
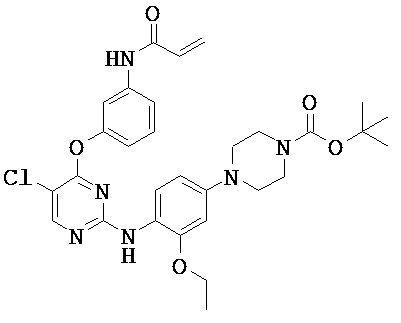
[00170] Смесь промежуточного соединения 6 (16 мг) и трет-бутилового эфира 4-(4-амино-3-этоксифенил)пиперазин-1-карбоновой кислоты (21 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C30H35ClN6O5: 594,2, полученное значение: 595,5 (M+H+).
ПРИМЕР 31
Соединение I-30 (N-(3-(2-(4-(4-(2-гидроксиацетил)пиперазин-1-ил)-2-(трифторметокси)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
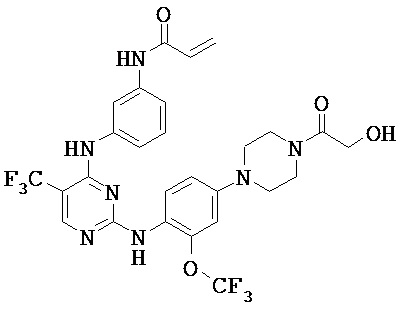
[00171] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-трифторметоксифенил)пиперазин-1-карбоновой кислоты (22 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), N,N-диметилформамид (1,0 мл), HATU и гликолевую кислоту при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,14 (с, 1H), 8,99 (с, 1H), 8,61 (с, 1H), 8,29 (с, 1H), 7,71 (с, 1H), 7,44 (с, 1H), 7,42 (с, 1H), 7,18 (м, 2H), 6,85 (с, 1H), 6,75 (м, 1H), 6,45 (дд, J=10,0, 16,8 Гц, 1H), 6,26 (д, J=16,8 Гц, 1H), 5,77 (д, J=10,0 Гц, 1H), 4,66 (т, J=5,6 Гц, 1H), 4,14 (д, J=5,6 Гц, 2H), 3,61 (ушир., 2H), 3,49 (ушир., 2H), 3,11 (ушир., 4H); вычисленное значение массы для C27H25F6N7O4: 625,2, полученное значение: 625,8 (М+Н+).
ПРИМЕР 32
Соединение I-31 (N-(3-(2-(2-(дифторметокси)-4-(4-(2-гидроксиацетил)пиперазин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
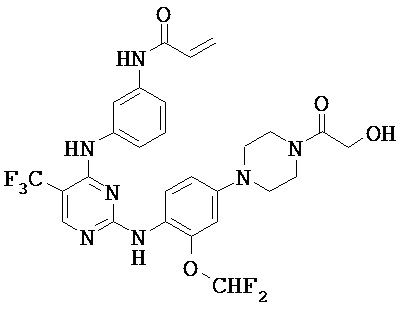
[00172] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-дифторметоксифенил)пиперазин-1-карбоновой кислоты (22 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), N,N-диметилформамид (1,0 мл), HATU и гликолевую кислоту при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C27H26F5N7O4: 607,2, полученное значение: 607,8 (M+H+).
ПРИМЕР 33
Соединение I-1 (N-(3-(2-(2-метокси-4-морфолинофениламино)-5-(трифторметил)пиримидин-4-илокси)фенил)акриламид)
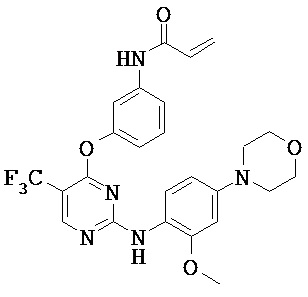
[00173] Смесь промежуточного соединения 5 (16 мг) и 2-метокси-4-морфолиноанилина (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,35 (с, 1H), 8,83 (с, 1H), 8,55 (ушир., 1H), 7,63 (с, 1H), 7,53 (д, J=8,0 Гц, 1H), 7,40 (м, 1H), 7,16 (д, J=8,8 Гц, 1H), 6,96 (ушир., 1H), 6,54 (ушир., 1H), 6,43 (м, 1H), 6,27 (дд, J=1,7, 16,8 Гц, 1H), 5,77 (дд, J=1,7, 10,4 Гц, 1H), 3,72 (ушир., 7 Н), 3,04 (ушир., 4H); вычисленное значение массы для C25H24F3N5O4: 515,2, полученное значение: 516,7 (М+Н+).
ПРИМЕР 34
Соединение I-13 (N-(3-(2-(2-(дифторметокси)-4-морфолинофениламино)-5-(трифторметил)пиримидин-4-илокси)фенил)акриламид)
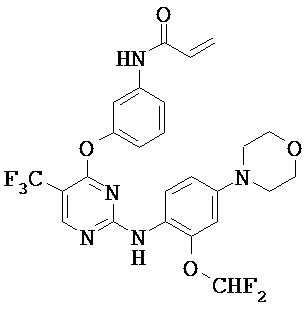
[00174] Смесь промежуточного соединения 5 (16 мг) и 2-дифторметокси-4-морфолиноанилина (22 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1Н-ЯМР (ДМСО-d6, 400 МГц) δ 10,33 (с, 1H), 9,34 (с, 1H), 8,55 (ушир., 1H), 7,63 (с, 1H), 7,51 (ушир., 1H), 7,40 (ушир., 1H), 7,19 (д, J=9,2 Гц, 1H), 6,96 (ушир., 1H), 6,65 (ушир., 1H), 6,43 (дд, J=10,0, 16,8 Гц, 1H), 6,27 (дд, J=2,0, 16,8 Гц, 1H), 5,79 (дд, J=2,0, 10,0 Гц, 1H), 3,73 (т, J=4,4 Гц, 1H), 3,06 (м, 4H); вычисленное значение массы для C25H22F5N5O4: 551,2, полученное значение: 551,7 (M+H+).
ПРИМЕР 35
Соединение I-14 (N-(3-(2-(2-(дифторметокси)-4-морфолинофениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
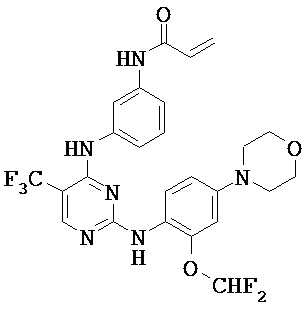
[00175] Смесь промежуточного соединения 1 (16 мг) и 2-дифторметокси-4-морфолиноанилина (22 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,14 (с, 1H), 8,63 (с, 1H), 8,28 (с, 1H), 7,71 (с, 1H), 7,46 (д, J=7,2 Гц, 1H), 7,38 (с, 1H), 7,15 (м, 2H), 6,91 (м,1H), 6,67 (м, 2H), 6,44 (дд, J=10,0, 16,8 Гц, 1H), 6,25 (д, J=16,8 Гц, 1H), 5,76 (д, J=10,0 Гц, 1H), 3,73 (т, J=4,4 Гц, 1H), 3,05 (ушир., 4H); вычисленное значение массы для C25H23F5N6O3: 550,2, полученное значение: 550,9 (М+Н+).
ПРИМЕР 36
Соединение I-15 (N-(3-(5-хлор-2-(2-метокси-4-морфолинофениламино)пиримидин-4-иламино)фенил)акриламид)
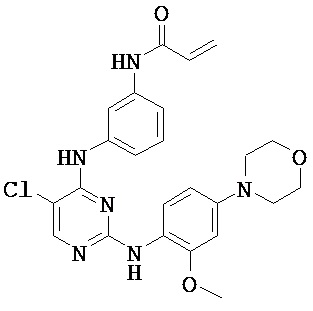
[00176] Смесь промежуточного соединения 2 (16 мг) и 2-дифторметокси-4-морфолиноанилина (20 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 150°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ С10,16 (с, 1H), 8,85 (с, 1H), 8,07 (с, 1H), 7,95 (ушир., 1H), 7,68 (м, 2H), 7,45 (д, J=7,6 Гц, 1H), 7,27 (м, 2H), 6,60 (д, J=2,4 Гц, 1H), 6,45 (дд, J=10,0, 16,8 Гц, 1H), 6,26 (м, 2H), 5,76 (дд, J=2,0, 10,0 Гц, 1H), 3,79 (с, 3H), 3,73 (м, 4H), 3,03 (м, 4H); вычисленное значение массы для C24H25ClN6O3: 480,2, полученное значение: 481,4 (М+Н+).
ПРИМЕР 37
Соединение I-32 (N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-(дифторметокси)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
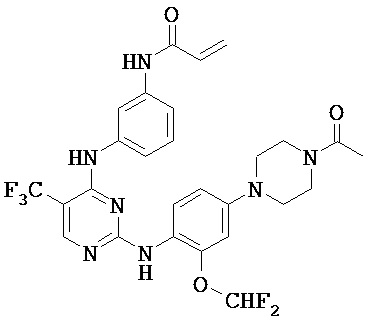
[00177] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-дифторметоксифенил)пиперазин-1-карбоновой кислоты (22 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и уксусный ангидрид (50 мкл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,13 (с, 1H), 8,63 (с, 2H), 8,29 (с, 1H), 7,71 (с, 1H), 7,47 (д, J=7,2 Гц, 1H), 7,39 (м, 1H), 7,16 (м, 2H), 6,91 (с, 1H), 6,69 (с, 1H), 9,59 (м, 1H), 6,44 (дд, J=10,0, 16,8 Гц, 1H), 6,26 (д, J=16,8 Гц, 1H), 5,77 (д, J=10,0 Гц, 1H), 3,57 (с, 4H), 3,10 (с, 2H), 3,04 (с, 2H), 2,05 (с, 3H); вычисленное значение массы для C27H26F5N7O3: 591,2, полученное значение: 591,8 (М+Н+).
ПРИМЕР 38
Соединение I-33 (N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-(трифторметокси)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
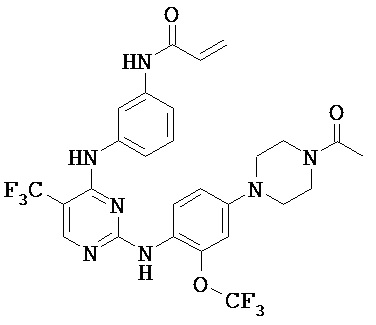
[00178] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-трифторметоксифенил)пиперазин-1-карбоновой кислоты (22 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и уксусный ангидрид (30 мкл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,15 (с, 1H), 9,00 (с, 1H), 8,62 (с, 1H), 8,29 (с, 1H), 7,71 (с, 1H), 7,43 (с, 2H), 7,18 (м, 2H), 6,84 (с, 1H), 6,75 (ушир., 1H), 6,45 (дд, J=10,0, 16,8 Гц, 1H), 6,26 (д, J=16,8 Гц, 1H), 5,77 (д, J=10,0 Гц, 1H), 3,57 (ушир., 4H), 3,13 (ушир., 2H), 3,06 (ушир., 2H), 2,05 (с, 3H); вычисленное значение массы для C27H25F6N7O3: 609,2, полученное значение: 610,0 (М+Н+).
ПРИМЕР 39
Соединение I-34 (N-(3-(2-(2-метокси-4-(4-пивалоилпиперазин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
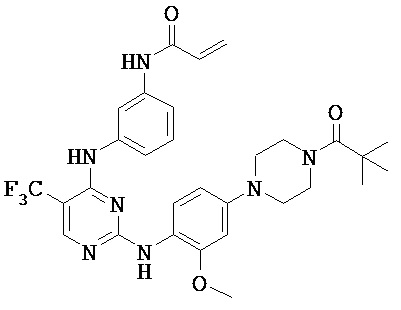
[00179] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и пивалоилхлорид (20 мкл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C30H34F3N7O3: 597,3, полученное значение: 598,3 (М+Н+).
ПРИМЕР 40
Соединение I-35 (N-(3-(2-(2-метокси-4-(4-пропионилпиперазин-1-ил)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
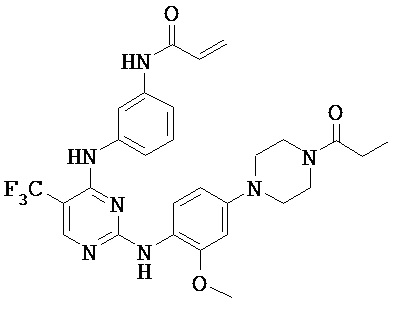
[00180] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и пропионилхлорид (10 мкл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C27H28F3N7O3: 555,2, полученное значение: 556,2 (М+Н+).
ПРИМЕР 41
Соединение I-36 (N-(3-(5-хлор-2-(2-(дифторметокси)-4-морфолинофениламино)пиримидин-4-илокси)фенил)акриламид)
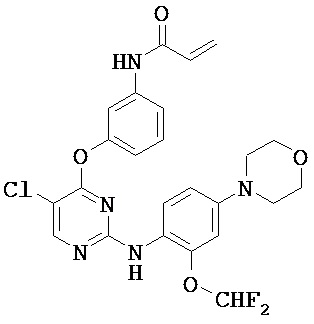
[00181] Смесь промежуточного соединения 6 (20 мг) и 2-дифторметокси-4-морфолиноанилина (22 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 150°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,33 (с, 1H), 8,71 (с, 1H), 8,35 (с, 1H), 7,62 (т, J=2,0 Гц, 1H), 7,53 (д, J=8,0 Гц, 1H), 7,40 (т, J=8,0 Гц, 1H), 7,23 (д, J=8,8 Гц, 1H), 6,97 (дд, J=64,8, 66,4 Гц, 1H), 6,95 (с, 1H), 6,64 (д, J=2,0 Гц, 1H), 6,59 (ушир., 1Н), 6,44 (дд, J=10,0, 16,8 Гц, 1H), 6,27 (дд, J=2,0, 16,8 Гц, 1H), 5,79 (дд, J=2,0, 10,0 Гц, 1H), 3,72 (м, 4H), 3,03 (м, 4H); вычисленное значение массы для C24H22ClF2N5O4: 517,1, полученное значение: 517,7 (M+H+).
ПРИМЕР 42
Соединение I-37 (N-(3-(5-хлор-2-(2-метокси-4-(1,4-оксазепин-4-ил)фениламино)пиримидин-4-илокси)фенил)акриламид)
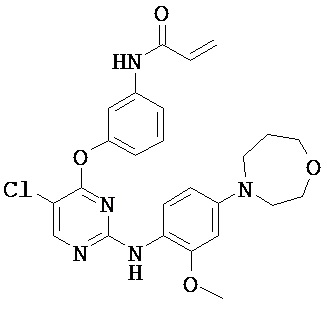
[00182] Смесь промежуточного соединения 6 (20 мг) и 2-метокси-4-(1,4-оксазепин-4-ил)анилина (21 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора с применением микроволнового излучения нагревали до 150°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C25H26ClN5O4: 495,2, полученное значение: 495,8 (М+Н+).
ПРИМЕР 43
Соединение I-38 (N-(3-(2-(2-метокси-4-(1,4-оксазепин-4-ил)фениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
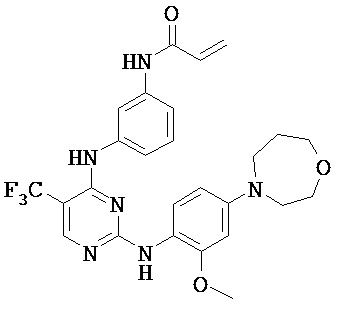
[00183] Смесь промежуточного соединения 1 (16 мг) и 2-метокси-4-(1,4-оксазепин-4-ил)анилина (21 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C26H27F3N6O3: 528,2, полученное значение: 528,8 (M+H+).
ПРИМЕР 44
Соединение I-39 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты)
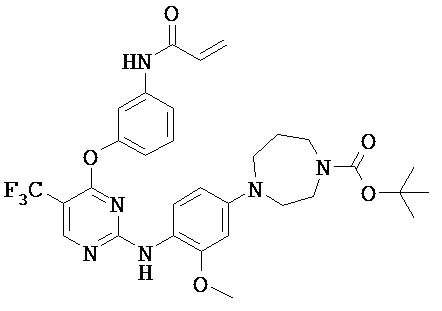
[00184] Смесь промежуточного соединения 5 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты (21 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C26H27F3N6O3: 528,2, полученное значение: 528,8 (М-Вос+Н+).
ПРИМЕР 45
Соединение I-40 (N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-метоксифениламино)-5-бромпиримидин-4-иламино)фенил)акриламид)
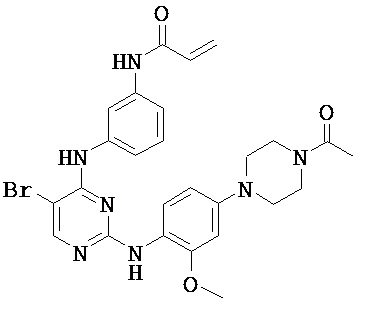
[00185] Смесь промежуточного соединения 4 и 1-(4-(4-амино-3-метоксифенил)пиперазин-1-ил)этанона (18 мг) в н-бутаноле (1 мл) с HCl в качестве катализатора нагревали с применением микроволнового излучения до 150°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с получением указанного в заголовке соединения. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,15 (с, 1H), 8,60 (ушир., 1H), 8,15 (с, 1H), 7,89 (ушир., 1H), 7,67 (м, 2H), 7,47 (д, J=6,8 Гц, 1H), 7,26 (м, 2H), 6,63 (д, J=2,4 Гц, 1H), 6,45 (дд, J=10,0, 16,8 Гц, 1H), 6,28 (м, 1H), 6,26 (дд, J=2,0, 16,8 Гц, 1H), 5,76 (дд, J=2,0, 10,0 Гц, 1H), 3,79 (с, 3H), 3,56 (м, 4H), 3,06 (м, 2H), 3,03 (м, 2H), 2,05 (с, 3H); вычисленное значение массы для C26H28BrN7O3: 565,1, полученное значение: 566,3 (M+H+).
ПРИМЕР 46
Соединение I-41 (N-(3-(5-хлор-2-(2-метокси-4-(4-(метилсульфонил)пиперазин-1-ил)фениламино)пиримидин-4-иламино)фенил)акриламид)
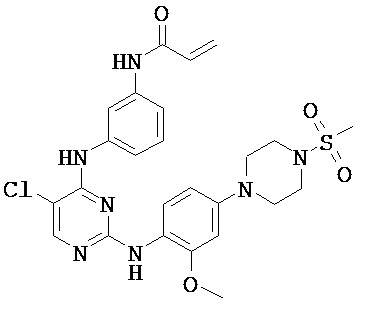
[00186] Смесь промежуточного соединения 2 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в н-бутаноле (1,0 мл) с трифторуксусной кислотой в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и метансульфонилхлорид (10 мкл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,16 (с, 1H), 8,86 (с, 1H), 8,08 (с, 1H), 7,95 (с, 1H), 7,70 (м, 2H), 7,44 (д, J=7,6 Гц, 1H), 7,28 (м, 2H), 6,64 (д, J=2,4 Гц, 1H), 6,46 (дд, J=10,0, 16,8 Гц, 1H), 6,32 (м, 1H), 6,26 (дд, J=2,0, 16,8 Гц, 1H), 5,77 (дд, J=2,0, 10,0 Гц, 1H), 3,80 (с, 3H), 3,30 (м, 4H), 3,25 (м, 2H), 3,17 (м, 2H), 2,93 (с, 3H); вычисленное значение массы для C25H28ClN7O4S: 557,2, полученное значение: 558,4 (М+Н+).
ПРИМЕР 47
Соединение I-42 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)пиперазин-1-карбоновой кислоты)
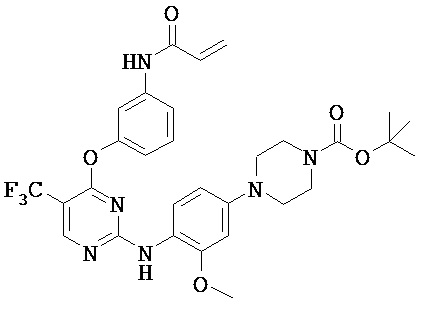
[00187] Смесь промежуточного соединения 5 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C30H33F3N6O5: 614,3, полученное значение: 615,2 (M+H+).
ПРИМЕР 48
Соединение I-43 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-хлорпиримидин-2-иламино)-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты)
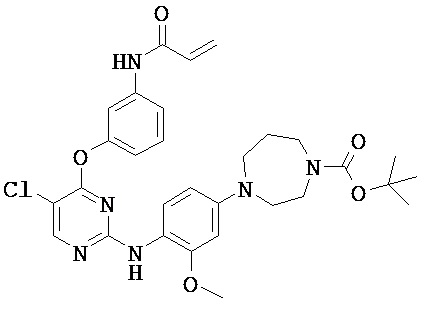
[00188] Смесь промежуточного соединения 6 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты (21 мг) в н-бутаноле (1 мл) с HCl в качестве катализатора нагревали с применением микроволнового излучения до 100°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C30H35ClN6O5: 594,2, полученное значение: 594,8 (М+Н+).
ПРИМЕР 49
Соединение I-44 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофениламино)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты)
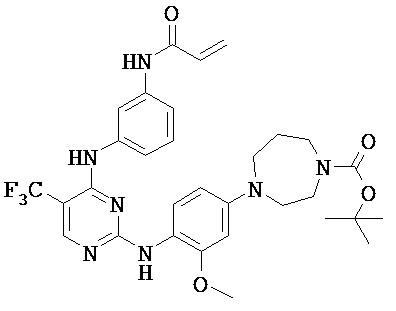
[00189] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)-1,4-диазепин-1-карбоновой кислоты (21 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C31H36F3N7O4: 627,3, полученное значение: 628,0 (M+H+).
ПРИМЕР 50
Соединение I-45 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофениламино)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)пиперазин-1-карбоновой кислоты)
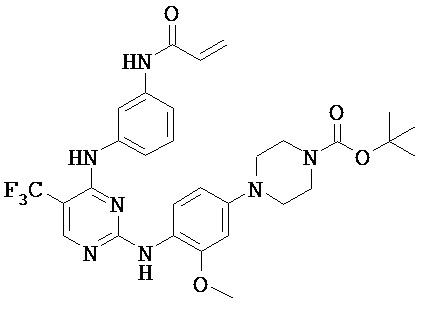
[00190] Смесь промежуточного соединения 1 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C30H34F3N7O4: 613,3, полученное значение: 614,1 (М+Н+).
ПРИМЕР 51
Соединение I-46 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-хлорпиримидин-2-иламино)-3-метоксифенил)пиперазин-1-карбоновой кислоты)
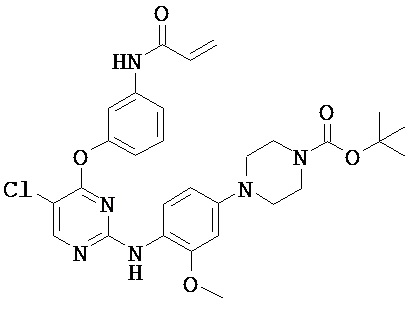
[00191] Смесь промежуточного соединения 6 (16 мг) и трет-бутилового эфира 4-(4-амино-3-метоксифенил)пиперазин-1-карбоновой кислоты (20 мг) в н-бутаноле (1 мл) с HCl в качестве катализатора нагревали с применением микроволнового излучения до 120°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C29H33ClN6O5: 580,2, полученное значение: 581,2 (M+H+).
ПРИМЕР 52
Соединение I-47 (трет-бутиловый эфир 4-(4-(4-(3-акриламидофенокси)-5-хлорпиримидин-2-иламино)-3-(трифторметокси)фенил)пиперазин-1-карбоновой кислоты)
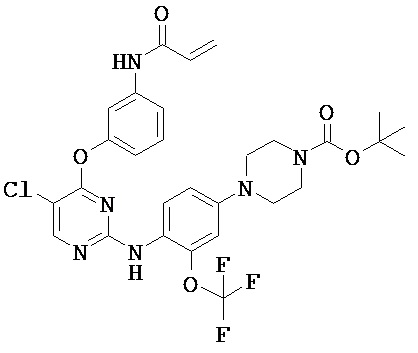
[00192] Смесь промежуточного соединения 6 (16 мг) и трет-бутилового эфира 4-(4-амино-3-трифторметоксифенил)пиперазин-1-карбоновой кислоты (22 мг) в н-бутаноле (1,0 мл) с HCl в качестве катализатора нагревали с применением микроволнового излучения до 120°С и поддерживали указанную температуру в течение 20 мин. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C29H30ClF3N6O5: 634,2, полученное значение: 635,4 (M+H+).
ПРИМЕР 53
Соединение I-48 ((S)-метиловый эфир 1-ацетил-4-(4-(4-(3-акриламидофениламино)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)пиперазин-2-карбоновой кислоты)
[00193] Смесь промежуточного соединения 1 (16 мг) и (S)-1-трет-бутил-2-метил-4-(4-амино-3-метоксифенил)пиперазин-1,2-дикарбоксилата (23 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора). Промежуточное соединение растворяли в дихлорметане (1,0 мл) и обрабатывали с применением ТФУ (0,3 мл). Через 10 минут смесь концентрировали при пониженном давлении. К остатку добавляли N,N-диэтилизопропиламин (20 мкл), дихлорметан (1,0 мл) и уксусный ангидрид (20 мкл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C29H30F3N7O5: 613,2, полученное значение: 614,2 (М+Н+).
ПРИМЕР 54
Соединение I-49 (N-(3-(2-(2-метокси-4,4-диоксо-4-тиоморфолинофениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)
[00194] Смесь промежуточного соединения 1 (18 мг) и 2-метокси-4-8,8-диоксотиоморфолиноанилина (24 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. 1H-ЯМР (ДМСО-d6, 400 МГц) δ 10,16 (с, 1H), 8,66 (ушир., 1H), 8,29 (с, 1H), 8,13 (с, 1H), 7,77 (ушир., 1H), 7,51 (с, 1H), 7,49 (с, 1H), 7,26 (т, J=8,0 Гц, 1H), 7,18 (ушир., 1H), 6,64 (д, J=2,0 Гц, 1H), 6,44 (дд, J=10,0, 16,8 Гц, 1H), 6,31 (м, 1H), 6,26 (дд, J=2,0, 16,8 Гц, 1H), 5,77 (дд, J=2,0, 10,0 Гц, 1H), 3,79 (с, 3H), 3,70 (м, 4H), 3,12 (м, 4H); вычисленное значение массы для C25H25F3N6O3S: 562,2, полученное значение: 562,8 (M+H+).
ПРИМЕР 55
Соединение I-50 ((S)-1-трет-бутил-2-метил-4-(4-(4-(3-акриламидофениламино)-5-(трифторметил)пиримидин-2-иламино)-3-метоксифенил)пиперазин-1,2-дикарбоксилат)
[00195] Смесь промежуточного соединения 1 (20 мг) и (S)-1-трет-бутил-2-метил-4-(4-амино-3-метоксифенил)пиперазин-1,2-дикарбоксилата (26 мг) в диоксане (1,0 мл) с трифторуксусной кислотой в качестве катализатора перемешивали в течение ночи при 50°С. Неочищенное вещество концентрировали при пониженном давлении и очищали с применением ВЭЖХ (с ТФУ в качестве модификатора) с получением желаемого продукта в виде соли ТФУ. Вычисленное значение массы для C32H36F3N7O6: 671,3, полученное значение: 672,3 (М+Н+).
Биологические примеры
[00196] Ниже описаны анализы, примененные для измерения биологической активности предложенных соединений в качестве селективных ингибиторов мутанта EGFR по сравнению c WT EGFR (и другими протеинкиназами).
ПРИМЕР 56
Общий протокол анализа для оценки эффективности действия против активных ферментов EGFR (WT) и EGFR (T790M/L858R)
[00197] Ниже описан протокол биохимического анализа с применением EGFR-WT и EGFR-T790M/L858R.
[00198] Принцип действия платформы для анализа лучше всего описан поставщиком (Invitrogen, Карлсбад, СА) на его интернет-сайте со следующим URL: www.invitrogen. com/content.cfm?pageid=11338 или www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/Drug-Discovery/Target-and-Lead-Identification-and-Validation/KinaseBiology/KB-Misc/Biochemical-Assays/Omnia-Kinase-Assays.html.
[00199] Вкратце, 10Х-разведенные EGFR-WT (PV3872) от Invitrogen и EGFR-T790M/L858R (40350) от BPS Bioscience, Сан-Диего, СА, 1,13Х АТФ (AS001A) и подходящие Tyr-Sox конъюгированные пептидные субстраты (KCZ1001) готовили в 1Х реакционном буферном растворе для киназы, содержащем 20 мМ Трис, рН 7,5, 5 мМ MgCl2, 1 мМ EGTA, 5 мМ β-глицерофосфата, 5% глицерина (10Х разведение, КВ002А) и 0,2 мМ ДТТ (DS001A). 5 мкл каждого фермента предварительно выдерживали в 384-луночном, белом, титрационном микропланшете с несвязывающей поверхностью Corning (#3574) (Corning, NY) в течение 30 мин при 25°С с применением 0,5 мкл 50% ДМСО и последовательно разбавляли соединения, приготовленные в 50% ДМСО. Киназные реакции запускали путем добавления 45 мкл смеси АТФ/пептидного субстрата Tyr-Sox и ход указанных реакций отслеживали каждую 71 секунду в течение 60 минут при λвозб/λэм 360/485 в ридере планшет Synergy4 от BioTek (Уинуски, VT). По завершении каждого анализа, для кривых реакции в каждой лунки определяли линейность кинетики реакции и статистические критерии согласия (R2, 95% доверительный интервал, абсолютное значение суммы квадратов). Начальную скорость (от 0 минут до ~30 минут) каждой реакции определяли по тангенсу угла наклона графика зависимости интенсивности флуоресценции в относительных единицах флуоресценции от времени (в минутах), а затем строили график зависимости от концентрации ингибитора для вычисления IC50, исходя из зависимости log[концентрации ингибитора] от ответной реакции, применяя модель переменного наклона в GraphPad Prism от GraphPad Software (Сан-Диего, СА).
[00200] [EGFR-WT]=5 нМ, [АТФ]=15 мкМ, [Y12-Sox]=5 мкМ (АТФ КМарр ~ 12 мкМ); [EGFR-T790M/L858R]=2,5 нМ, [АТФ]=20 мкМ, [Y12-Sox]=5 мкМ (АТФ КМарр ~ 20 мкМ).
[00201] В Таблице 3 представлена активность выбранных соединений согласно настоящему изобретению в анализе ингибирования EGFR, описанном выше. В Таблице 3 представлены данные для мутанта EGFR в сравнении с данными для WT EGFR и предложено отношение селективности WT к мутанту для каждого исследуемого соединения. Номера соединений соответствуют номерам соединений в Таблице 1.
ПРИМЕР 57
Культура клеток и антитела
[00202] Клетки А431 плоскоклеточной карциномы человека, Н1975 немелкоклеточного рака легкого (НМРЛ) человека и НСС827 аденокарциномы человека, представлявшей собой разновидность НМРЛ, получали из Американского центра типовых культур (Манассас, VA). Клетки А431 выращивали в среде DMEM (Invitrogen, Карлсбад, СА) с добавлением 10% ФБС (HyClone, South Logan, UT) и 1% пенициллина-стрептомицина (П/С, Lonza, Walkersville, MD). Клетки Н1975 и НСС827 выращивали в полной среде RPMI-1640 (Invitrogen) с добавлением 10% ФБС и 1% П/С. Все клетки хранили и культивировали в виде монослойных культур в инкубаторе при 37°С в увлажненной атмосфере с содержанием CO2 5%.
[00203] Все первичные антитела получали от Cell Signaling (Дэнверс, МА) и применяли в разведении 1:1000. Вторичные антитела применяли в разведении 1:10000. Козьи антимышиные IgG антитела, конъюгированные с IRDye 800CW, получали от LiCor Biosciences (Линкольн, NE), а козьи антикроличьи IgG, конъюгированные с Alexa Fluor 680, получали от Invitrogen.
Иммуноблоттинг
[00204] Клетки выращивали в 12-луночных планшетах (Corning, Коринг, NY) до 90% конфлюэнтности, а затем выдерживали в среде с низким содержанием сыворотки (0,1% ФБС) в течение 16-18 ч. Затем клетки обрабатывали 5, 1,25, 0,31, 0,078, 0,020 или 0,005 мкМ исследуемого соединение в среде с низким содержанием сыворотки (0,1% ФБС) в течение 1 часа. Далее клетки А431 стимулировали с применением 50 нг/мл EGF (Peprotech, Роки Хилл, NJ) в течение 15 мин. После обработки монослои клеток промывали охлажденным PBS (Invitrogen) и немедленно лизировали путем соскабливания в 60 мкл охлажденного буферного раствора для экстракции клеток (Invitrogen) с добавлением ингибиторов протеаз Complete (Roche, Индианаполис, IN) и ингибиторов фосфатаз PhosphoSTOP (Roche).
[00205] Концентрации белка в лизате определяли путем ВСА Assay (Pierce, Рокфорд, IL), и 50 мкг каждого лизата отделяли путем ДСН-ПААГ-электрофореза с градиентом концентрации геля 4-12% (Invitrogen), переносили на нитроцеллюлозную мембрану (Biorad, Херкулиз, СА) и исследовали с применением антител, специфичных к указанному белку. Сигналы фосфопротеинов количественно оценивали с применением Odyssey Infrared Imaging (Li-Cor Biosciences).
[00206] Для оценки передачи сигнала фосфорилированного EGFR перенесенные на мембрану лизаты исследовали с применением кроличьих антител к фосфорилированному EGFR (Y1068) и мышиных антител ко всем EGFR. Сигнал фосфорилированного EGFR нормировали по экспрессии всех EGFR для каждого образца. Результаты представлены в виде % от результатов для ДМСО контроля. Нормированные данные подбирали с применением программы анализа сигмоидальных кривых (Graph Pad Prism, версия 5) с различной величиной наклона, определяемой коэффициентом Хилла, для определения значений ЕС50.
[00207] В Таблице 4 представлены данные для мутанта EGFR в клетках Н1975 (двойная мутация L858R/T790M) и НСС827 (делеционная мутация deIE746-A750) по сравнению с данными для WT EGFR (клетки А431). Номера соединений, перечисленных в Таблице 4, соответствуют номерам соединений в Таблице 1.
ПРИМЕР 58
Пролиферация клеток
[00208] Клетки высевали на среду для роста с добавлением 5% ФБС и 1% П/С в 96-луночные планшеты для культур тканей (Corning) при плотности 3000 клетки на лунку. Клеткам позволяли оседать в течение 4 ч, а затем обрабатывали 5, 1,25, 0,31, 0,078, 0,020 или 0,005 мкМ исследуемого соединения в течение 72 ч. Жизнеспособность клеток определяли с помощью CellTiter Glo (Promega, Мэдисон, WI) и результаты преобразовывали в количество клеток с применением стандартной кривой. Значения показателя эффективности ингибирования роста (GI50) определяли с помощью Graph Pad Prism.
[00209] Результаты указанного эксперимента приведены в Таблице 5, где представлены данные селективного ингибирования мутантов в клетках H1975 (двойная мутация L858R/T790M) и HCC827 (делеционная мутация deIE746-A750), но не WT-EGFR в клетках А431.
ПРИМЕР 59
Эксперимент по вымыванию соединений из клеток Н1975, содержащих делеционную/Т790М мутацию EGFR
[00210] Клетки высевали на среду для роста с добавлением 10% ФБС и 1% П/С в 12-луночные планшеты для культур тканей при плотности 2,0×105 клеток на лунку. Клеткам позволяли оседать в течение 4 ч, а затем сохраняли в среде с низким содержанием сыворотки (0,1% ФБС) в течение ночи.
[00211] На следующее утро указанную среду удаляли и клетки обрабатывали 500 нМ исследуемого соединения в среде с низким содержанием сыворотки в течение 1 часа. Указанные клетки дважды промывали PBS (Invitrogen), смывая соединение. Одну группу клеток немедленно лизировали, как указано выше, принимая указанный момент времени за 0 ч. Остальные клетки выдерживали в полной среде для роста RPMI-1640 (10% ФБС) в течение 1, 3, 6 и 24 ч. В течение первого часа клетки дважды промывали PBS каждые 30 мин. Образцы ДМСО контроля (0,5%) собирали в моменты времени 0, 3, 6 и 24 ч.
[00212] Соединения I-2 и I-4 демонстрировали продолжительное действие после удаления соединений. Через 1 ч после удаления соединений ингибирование фосфорилирования pEGFR происходило на 80-100%. pEGFR оставался на 60-90% ингибированным в течение по меньшей мере через 8 часов после удаления соединения, но через 24 ч активность восстанавливалась до 40-60% путем синтеза новых белков.
ПРИМЕР 60
Масс-спектрометрия мутанта EGFR
[00213] Соединение I-4 модифицирует T790M/L858R EGFR по отдельности и полностью, что подтверждает МС анализ неочищенного белка. Интактный T790M/L858R EGFR (BPS, 40350) выдерживали в течение 60 мин в 10-кратном избытке соединения I-4 по отношению к белку. Аликвоты образцов объемом 5 мкл разбавляли 15 мкл 0,2% ТФУ перед нанесением с применением наконечника пипетки ZipTip, в котором осуществляется обращенно-фазовая хроматография с помощью микрослоя С4, непосредственно на мишень МАЛДИ с применением синапиновой кислоты в качестве десорбционной матрицы (10 мг/мл в 0,1% ТФУ:ацетонитрил 50:50). На Панели А представлено изображение масс-спектра интактного белка T790M/L858R EGFR (m/z составляет 88389 Да). На Панели В представлено изображение масс-спектра T790M/L858R EGFR, выдерживаемого с соединением 1-4 (молекулярная масса составляет 555,56) в течение 30 мин. Положение центра масс (m/z=88820 Да) свидетельствует о том, что произошло изменение массы на 431 Да (78%), что указывает на полную модификацию EGFR T790M/L858R соединением I-4.
[00214] Аналогично исследовали соединения I-1 и I-3 и обнаруживали ковалентную модификацию белка.
ПРИМЕР 61
Изучение опухолевых клеток Н1975 in vivo
[00215] Самкам мышей nu/nu подкожно имплантировали в бок 1×107 опухолевых клеток Н1975 в 50% Матригеле (вводимый объем 0,2 мл). Параметры опухолей фиксировали три раза в неделю. Опухоли попарно подбирали по величине, когда они достигали средней величины 100-150 мг. Размер группы составлял 10 мышей. Исследуемое соединение вводили внутрибрюшинно в количестве 25 мг/кг ежесуточно в течение 21 дня. Значения ингибирования опухолей, выраженные в процентах, определяли на 15 день, представляющий собой момент времени, в который опухоли мышей контрольной группы достигали максимального объема. Объем опухолей регистрировали до тех пор, пока опухоли не достигали объема 1500 мм3, или в течение 60 дней.
[00216] Значения ингибирования опухолей предложенными соединениями представлены в Таблице 6 ниже.
[00217] Несмотря на то, что ряд вариантов реализации настоящего изобретения описан в настоящей заявке, очевидно, что основные примеры могут быть изменены для обеспечения других вариантов реализации настоящего изобретения, в которых задействованы соединения и способы согласно настоящему изобретению. Таким образом, следует иметь в виду, что объем настоящего изобретения должен определяться прилагаемой формулой изобретения, а не конкретными вариантами реализации, которые были представлены в качестве примера.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ИНДАЗОЛ-3-КАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СИГНАЛЬНОГО ПУТИ WNT/b-КАТЕНИНА | 2012 |
|
RU2627693C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2015 |
|
RU2727700C2 |
| СОЕДИНЕНИЯ 3,5-ДИАМИНО-6-ХЛОР-N-(N-(4-ФЕНИЛБУТИЛ)КАРБАМИМИДОИЛ)ПИРАЗИН-2-КАРБОКСАМИДА | 2013 |
|
RU2671976C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ КИНАЗНОЙ АКТИВНОСТИ МУТАНТОВ EGFR | 2015 |
|
RU2822389C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ИМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2018 |
|
RU2801302C2 |
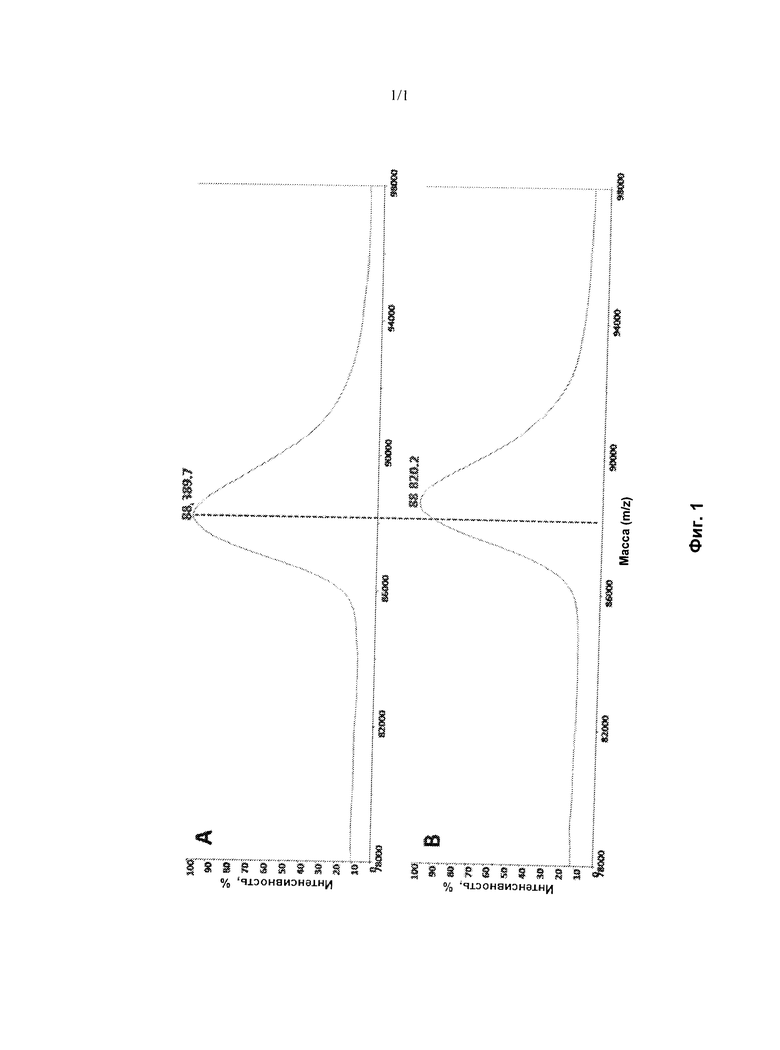
Изобретение относится к гетероциклическим пиримидиновым соединениям формулы
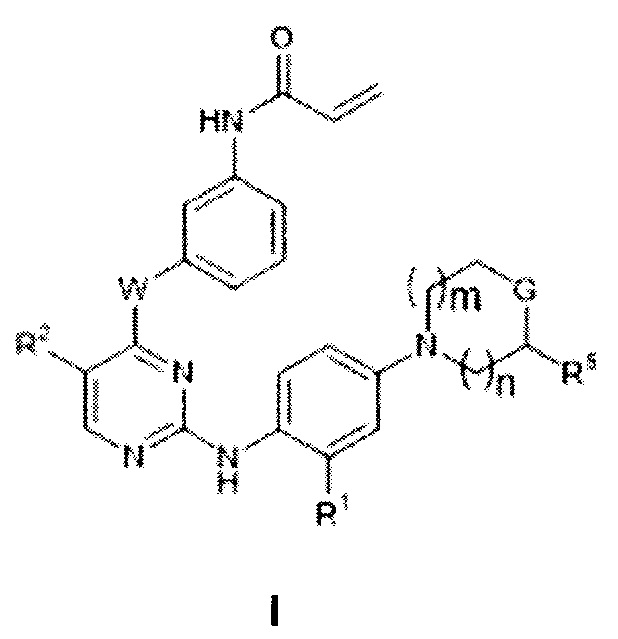 , а также к фармацевтическим композициям на их основе. Технический результат: получены новые соединения, которые модулируют мутант-селективную киназную активность рецепторов эпидермального фактора роста (EGFR) и которые применимы в способах лечения заболеваний, связанных с киназной активностью EGFR. 6 н. и 40 з.п. ф-лы, 1 ил., 6 табл., 61 пр.
, а также к фармацевтическим композициям на их основе. Технический результат: получены новые соединения, которые модулируют мутант-селективную киназную активность рецепторов эпидермального фактора роста (EGFR) и которые применимы в способах лечения заболеваний, связанных с киназной активностью EGFR. 6 н. и 40 з.п. ф-лы, 1 ил., 6 табл., 61 пр.
1. Соединение формулы I:
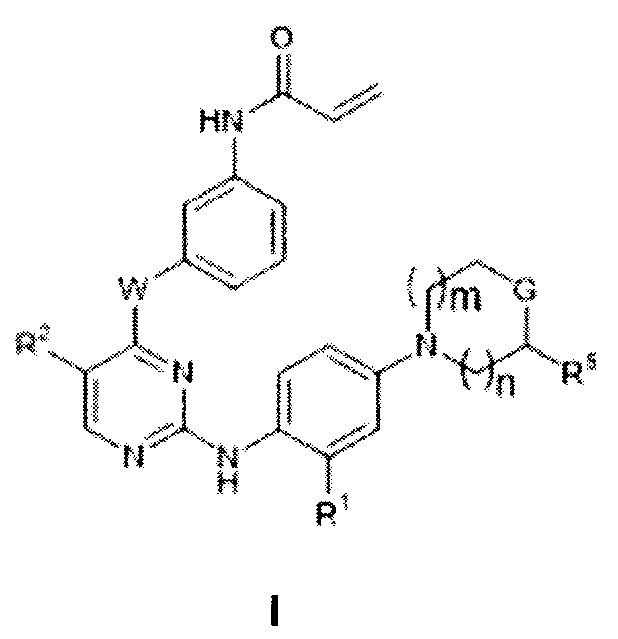
или его фармацевтически приемлемая соль, где:
n составляет 1;
m составляет 1 или 2;
W представляет собой -О- или -NH-;
R1 представляет собой -OR;
каждый R независимо представляет собой С1-4 алкил или С1-4 фторалкил;
R2 представляет собой -CF3, Cl или Br;
G представляет собой -NR3- или -S(O)2-;
R3 представляет собой -C(O)-R, -C(O)OR, -C(O)NHR, -SO2-R, -SO2NH2 или -C(O)-C1-4 алкилен-ОН; и
R5 представляет собой водород или -C(O)OR.
2. Соединение формулы I-a:
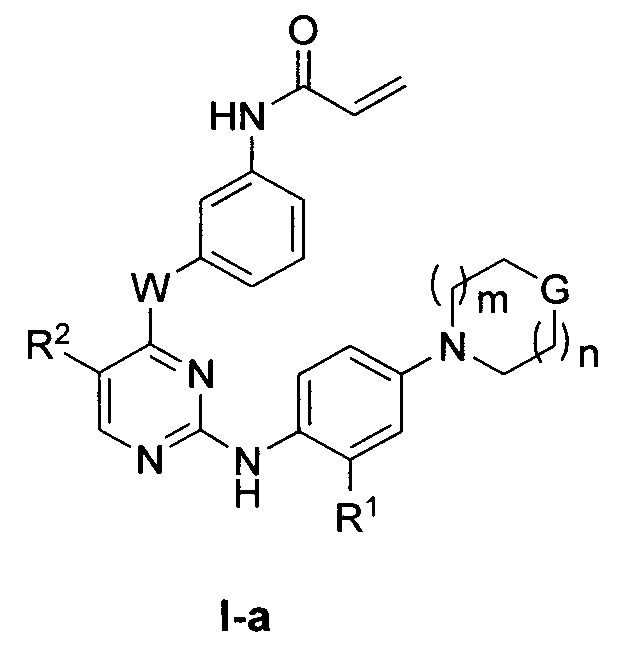
или его фармацевтически приемлемая соль, где:
n составляет 1;
m составляет 1 или 2;
W представляет собой -О- или -NH-;
R1 представляет собой -OR;
каждый R независимо представляет собой C1-4 алкил или C1-4 фторалкил;
R2 представляет собой -CF3, Cl или Br;
G представляет собой -NR3-;
R3 представляет собой -C(O)-R, -C(O)OR, -C(O)NHR, -SO2-R, -SO2NH2 или -C(O)-C1-4 алкилен-ОН.
3. Соединение по п. 1 или 2, отличающееся тем, что W представляет собой -NH-.
4. Соединение по п. 3, отличающееся тем, что R2 представляет собой -CF3.
5. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой соединение формулы III:
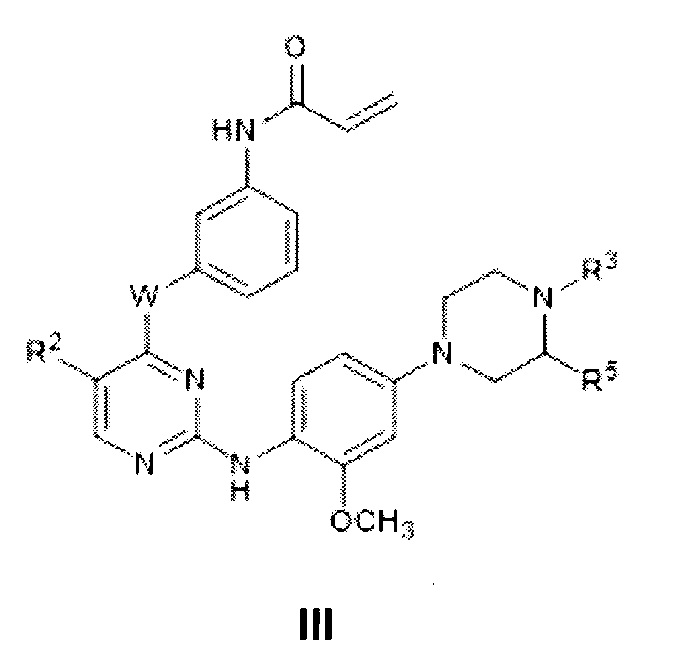
или его фармацевтически приемлемую соль.
6. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой соединение формулы III-а:
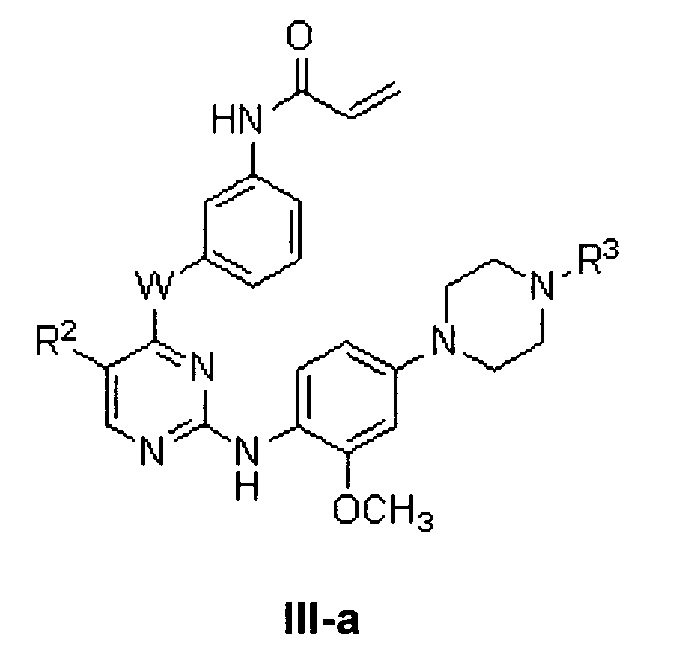
или его фармацевтически приемлемую соль.
7. Соединение по п. 5 или 6, отличающееся тем, что W представляет собой -NH-.
8. Соединение по п. 5 или 6, отличающееся тем, что R2 представляет собой -CF3.
9. Соединение по п. 5 или 6, отличающееся тем, что для него выполняется по меньшей мере одно из условий (а) или (b):
(a) R2 представляет собой -CF3 или Cl, и
(b) R3 представляет собой -С(O)СН3 или -SO2CH3.
10. Соединение по п. 9, отличающееся тем, что для него выполняются оба условия (а) и (b).
11. Соединение по п. 5 или 6, отличающееся тем, что для него выполняется по меньшей мере одно из условий (а), (b) или (с):
(a) W представляет собой -NH-;
(b) R2 представляет собой -CF3 или Cl, и
(c) R3 представляет собой -С(O)СН3.
12. Соединение по п. 11, отличающееся тем, что для него выполняются по меньшей мере два условия из (а), (b) и (с).
13. Соединение по п. 12, отличающееся тем, что для него выполняются все три условия (а), (b) и (с).
14. Соединение по п. 5 или 6, отличающееся тем, что для него выполняется по меньшей мере одно из условий (а), (b) или (с):
(a) W представляет собой -NH-;
(b) R5 представляет собой -CF3 или Cl, и
(c) R3 представляет собой -SO2CH3.
15. Соединение по п. 14, отличающееся тем, что для него выполняются по меньшей мере два условия из (а), (b) и (с).
16. Соединение по п. 15, отличающееся тем, что для него выполняются все три условия (а), (b) и (с).
17. Соединение по п. 5 или 6, отличающееся тем, что для него выполняется по меньшей мере одно из условий (а), (b) и (с):
(a) W представляет собой -О-;
(b) R2 представляет собой -CF3 или Cl, и
(c) R3 представляет собой -С(O)СН3.
18. Соединение по п. 17, отличающееся тем, что для него выполняются по меньшей мере два условия из (а), (b) и (с).
19. Соединение по п. 18, отличающееся тем, что для него выполняются все три условия (а), (b) и (с).
20. Соединение по п. 5 или 6, отличающееся тем, что для него выполняется по меньшей мере одно из условий (а), (b) и (с):
(a) W представляет собой -О-;
(b) R2 представляет собой -CF3 или Cl; и
(c) R3 представляет собой -SO2CH3.
21. Соединение по п. 20, отличающееся тем, что для него выполняются по меньшей мере два условия из (а), (b) и (с).
22. Соединение по п. 21, отличающееся тем, что для него выполняются все три условия (а), (b) и (с).
23. Соединение по п. 1, выбранное из:
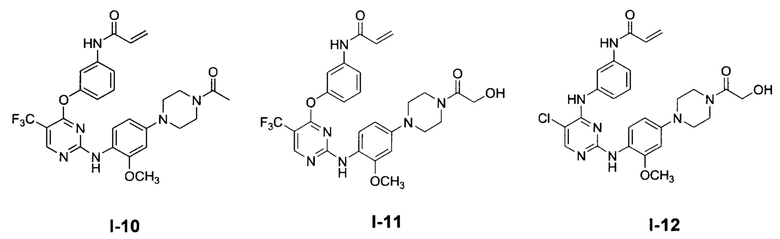
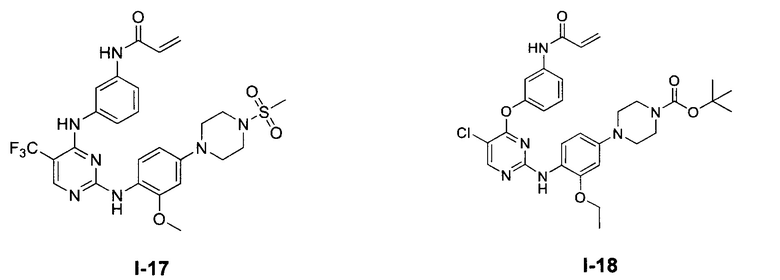
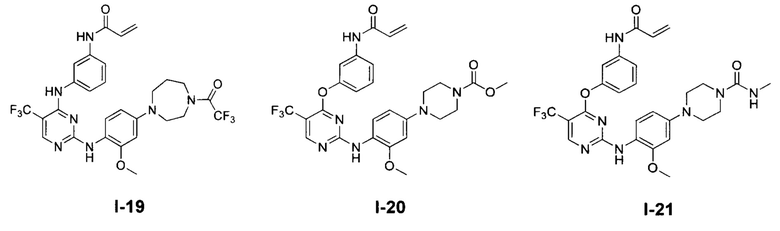

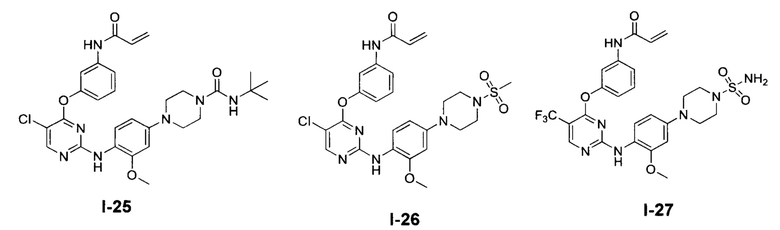
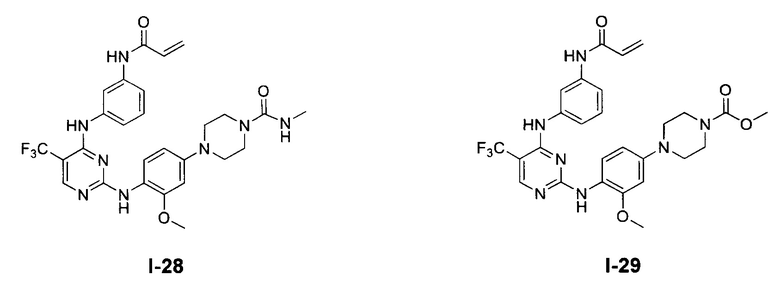
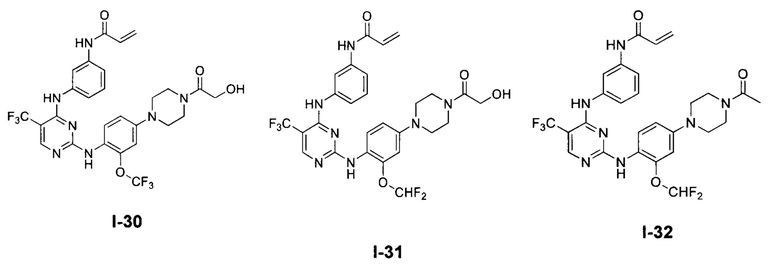
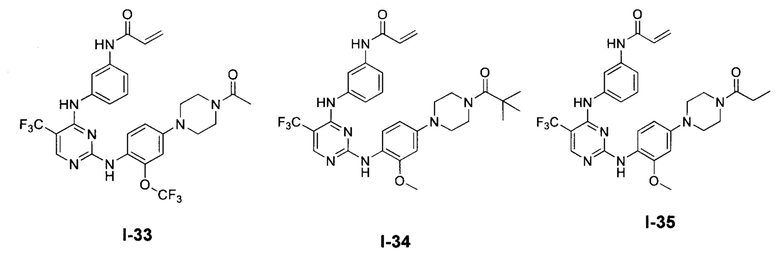
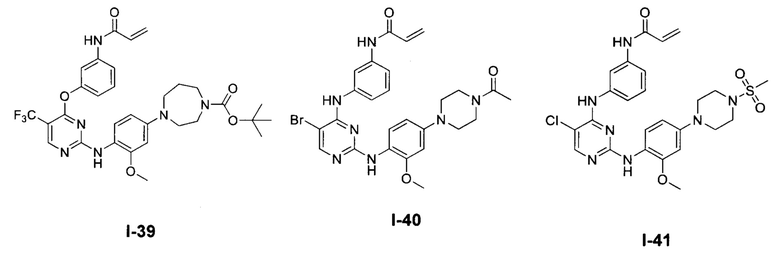
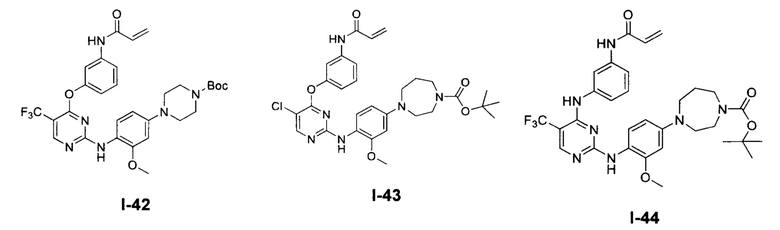
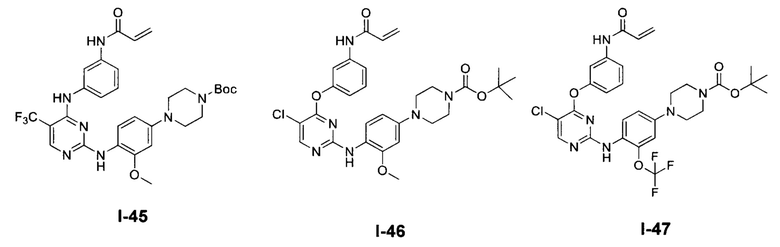
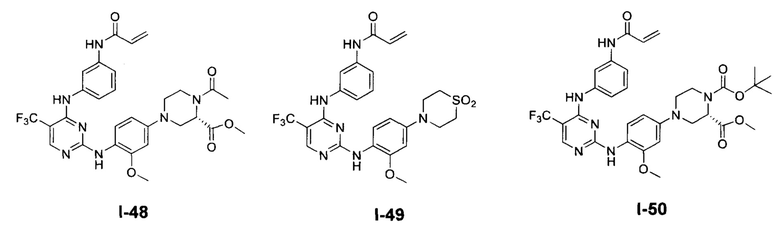
или его фармацевтически приемлемая соль.
24. Соединение по п. 5 или 6, отличающееся тем, что соединение представляет собой:
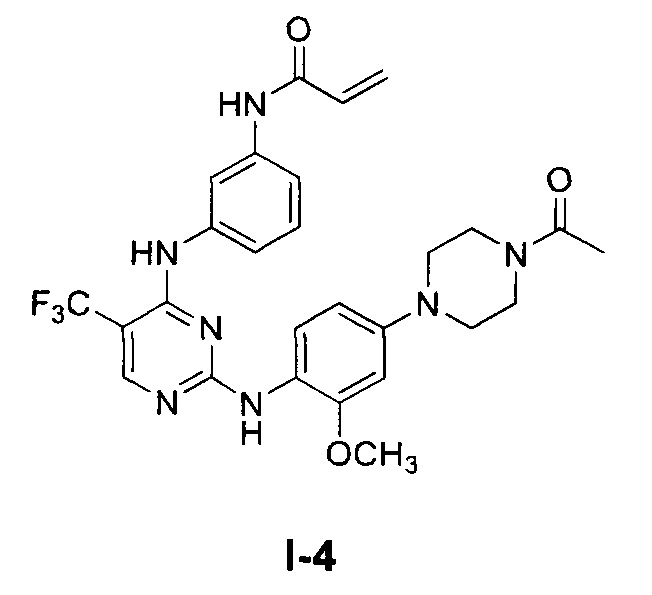
или его фармацевтически приемлемую соль.
25. Фармацевтическая композиция для применения в качестве мутант-селективного ингибитора киназы EGFR, содержащая эффективное количество соединения по любому из пп. 1-24 и фармацевтически приемлемый носитель, адъювант или наполнитель.
26. Фармацевтическая композиция для лечения раковых заболеваний, связанных с одной или более мутациями EGFR, содержащая эффективное количество соединения по любому из пп. 1-24 и фармацевтически приемлемый носитель, адъювант или наполнитель.
27. Применение соединения по любому из пп. 1-24 или фармацевтической композиции по любому из пп. 25-26 для ингибирования по меньшей мере одного мутанта EGFR селективно по сравнению с WT EGFR.
28. Применение по п. 27, отличающееся тем, что в указанном способе по существу не происходит ингибирования WT EGFR.
29. Применение по п. 27, отличающееся тем, что по меньшей мере один мутант представляет собой Т790М.
30. Применение по п. 28, отличающееся тем, что по меньшей мере один мутант представляет собой Т790М.
31. Применение по п. 27, отличающееся тем, что по меньшей мере один мутант EGFR представляет собой мутанта с активирующей мутацией.
32. Применение по п. 31, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией EGFR представляет собой мутанта с делеционной мутацией.
33. Применение по п. 31, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией EGFR представляет собой мутанта с точечной мутацией.
34. Применение по п. 32, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией представляет собой delE746-A750.
35. Применение по п. 33, отличающееся тем, что по указанный меньшей мере один мутант с активирующей мутацией представляет собой L858R.
36. Применение по п. 33, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией представляет собой G719S.
37. Применение по п. 27, отличающееся тем, что указанное соединение селективно ингибирует по меньшей мере один мутант с активирующей мутацией и Т790М.
38. Применение по п. 37, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией EGFR представляет собой мутанта с делеционной мутацией.
39. Применение по п. 37, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией EGFR представляет собой мутанта с точечной мутацией.
40. Применение по п. 38, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией представляет собой delE746-A750.
41. Применение по п. 39, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией представляет собой L858R.
42. Применение по п. 39, отличающееся тем, что указанный по меньшей мере один мутант с активирующей мутацией представляет собой G719S.
43. Применение по любому из пп. 31-42, отличающееся тем, что указанное соединение по существу не ингибирует WT EGFR.
44. Применение соединения по любому из пп. 1-24 или фармацевтической композиции по любому из пп. 25-26 для лечения опосредованного мутантом EGFR расстройства или патологического состояния.
45. Применение по п. 44, отличающееся тем, что указанное расстройство или патологическое состояние представляет собой рак.
46. Применение по п. 45, отличающееся тем, что указанный рак представляет собой немелкоклеточный рак легкого.
| US 20100016296 A1, 21.01.2010 | |||
| ZHOU et al | |||
| Способ закалки паровозных и вагонных рессор | 1920 |
|
SU790A1 |
| ВОДЯНАЯ ТУРБИНА | 1922 |
|
SU462A1 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ АВТОМАТИЧЕСКОГО ПЕРЕВОДА СТРЕЛОК | 1924 |
|
SU1070A1 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРИДИНА И ПРОИЗВОДНОЕ ПИРИМИДИНА (3) | 2006 |
|
RU2362771C1 |

