Изобретение относится к технологии получения солей олова (IV) и может быть использовано в различных областях химической и иных видов практик, в аналитическом контроле и в научных исследованиях.
Известно образование основного нитрата олова (IV) с брутто-формулой Sn(OH)2(NO3)2 путем непосредственного взаимодействия диоксида олова с азотной кислотой в присутствии трибохимического катализатора и механической активации процесса (Агеева Л.С., Пожидаева С.Д., Иванов A.M. Глубокие и быстрые при комнатных температурах превращения олова, его оксидов, гидроксидов и солей в присутствии кислот и механической активации процессов // Материалы III Международной Российско-Казахстанской научно-практической конференции «Химические технологии функциональных материалов», 27-29 апреля 2017 г, г. Новосибирск, Изд-во НГТУ, 2017. - 363 с; С. 52-55).
Недостатками данного варианта являются:
1. Он не оформлен в качестве способа, т.е. не содержит никаких загрузочных показателей, условий проведения, фазовых характеристик по ходу протекания и в конечной реакционной смеси (PC), как выделяется целевой продукт и т.д.
2. В приведенной информации нет сведений о природе трибохимического катализатора, обеспечивающего высокий выход основного нитрата олова (IV).
Известно, что действием разбавленной азотной кислоты на олово получают смесь нитратов олова (II) и олова (IV) (Г. Реми Курс неорганической химии. - М.: Изд-во иностран. лит., Т. 1, 1963, с. 583). Нитрат брутто-формулы Sn2O(NO3)2, в отличие от Sn(NO3)2⋅20H2O, кристаллизуется без воды и может быть нагрет до 100°C без разложения. При более сильном и более быстром нагреве, а также при ударе и растирании он детонирует с сильным треском.
Здесь также речь идет о нитратах олова (II), что вряд ли как по способу, так и режимным характеристикам операций подлежит переносу на нитраты олова (IV).
Известно (А.С. СССР №356248, опубл. 23.10.1972), что при восстановлении меди из раствора нитрата меди оловом невозможно получение чистого раствора нитрата олова (II) из-за сопутствующего получения основной соли, находящейся в растворе в виде мути, причем за время длительного получения часть двухвалентного олова успевает окислиться до четырехвалентного состояния. Для исключения этого олово применяют в виде амальгамы.
Авторы данного изобретения не указывают природу накапливающейся основной соли, в том числе и степень окисления металла в ней, как и то, что окисляется легче: основной или средний нитрат олова (II).
Наиболее близким к заявляемому является получение основного нитрата олова с брутто-формулой {SnO(NO3)2} при взаимодействии нитрата олова (IV) и оксида азота (II) NO в растворе четыреххлористого углерода (Philip G. Harrison, Mutassim I. Khalil, Norman Logan A contribution to the chemistry of tin (IV) nitrate // Inorganica Chimica Acta. Volume 30, 1978, Pages 165-170). Оно предполагает медленное барботирование чистого сухого оксида азота (II) через раствор нитрата олова (IV) в четыреххлористом углероде. Реакция начинается сразу и сопровождается появлением бурой окраски газового пространства реактора, что свидетельствует о быстром накоплении оксида азота (IV) над реакционной смесью. Одновременно появляется белая твердая фаза в реакционной смеси. Когда образование продукта прекращается, процесс завершают. Избыток оксида азота (II) из реактора удаляют с помощью продувки азота, продукт выделяют путем фильтрования и сушат под вакуумом.
Недостатками данного варианта является:
1. Он не оформлен в качестве способа получения заявленной композиции основной соли олова (IV).
2. Основная соль SnO(NO3)2 не является эквивалентом основной соли Sn(OH)2(NO3)2. Поэтому никаких оснований считать, что способы их получения будут не только близкими, но даже похожими друг на друга.
3. Более того приведенная авторами формула основной соли относится не к индивидуальному соединению, а к композиции, в состав которой входят и полимерные структуры. Следовательно стехиометрическое уравнение
Sn(NO3)2+NO→{OSn(NO3)2}+3NO2
относится к сложному процессу с рядом промежуточных стадий, в которых участвуют и соединения олова (II), но которые не попадают в общее стехиометрическое уравнение (образуясь в одних стадиях и расходуясь в других), что совсем не означает, что их нет в реакционной смеси.
4. Следует отметить и то, что по прототипу «основную соль» получают из средней соли, что не может быть эквивалентным получению из оксида олова (IV) ни по реагентам, ни по типу химического взаимодействия, ни по составу продуктов, ни по используемому для этих целей растворителю объемной фазы.
Задачей предлагаемого решения является подобрать такие трибохимические блокаторы основной соли нитрата олова (IV), природу и состав объемной фазы и прочие условия проведения процесса взаимодействия оксида олова (IV) с азотной кислотой, которые при близких к комнатным температурах обеспечивали бы высокую селективность по целевому продукту при практически количественном расходовании оловосодержащего исходного реагента, а также приемлемые для практических целей скорости получения основного нитрата олова (IV) Sn(OH)2(NO3)2.
Поставленная задача достигается тем, что получение основного нитрата олова (IV) Sn(OH)2(NO3)2 ведут путем прямого взаимодействия оксида олова (IV) с азотной кислотой в растворе этилцеллозольва при комнатной температуре в условиях интенсивного механического перемешивания в присутствии стеклянного бисера как перетирающего агента и солянокислых гидроксиламина, анилина, п-нитроанилина, о-дианизидина, м-фенилендиамина и бензидина, сернокислых гидразина, анилина или бензидина в качестве стабилизатора основной соли в количестве 10-2 моль/кг, оксид олова (IV) загружают в количестве 0,1-0,6 моль/кг в мольном соотношении с кислотой (1:2,02)÷(1:11), процесс начинают с ввода интенсивного механического перемешивания, контролируют по расходованию кислоты и прекращают, когда указанное расходование приближается к удвоенной начальной мольной загрузке оксида олова (IV), после этого перемешивание прекращают, проводят отделение перетирающего агента и его отмывку от остатков реакционной смеси, отделенную реакционную смесь фильтруют, осадок на фильтре промывают промывным растворителем и сушат на воздухе до постоянного веса. При этом раствор азотной кислоты в этилцеллозольве может быть приготовлен заблаговременно без каких-либо ограничений времени хранения или его готовят непосредственно в рабочем реакторе для процесса путем соответствующих дозировок расчетных количеств растворителя и кислоты при кратковременном перемешивании перед вводом стабилизатора основной соли и оксида олова (IV); избыточная кислота может быть израсходована путем дробного ввода в реакционную смесь дополнительного количества оксида олова (IV), а фильтрат с непрореагировавшей избыточной кислотой и растворенным стабилизатором основной соли может быть возвращен в повторный процесс с возможной дополнительной подпиткой кислоты, но без дополнительной подпитки стабилизатора основной соли.
Характеристика используемого сырья:
Оксид олова (IV) ГОСТ 22516-77
Азотная кислота ГОСТ 4461-77
Этилцеллозольв ГОСТ 8313-88
Гидроксиламин солянокислый ГОСТ 5456-65
Гидразин сернокислый ГОСТ 5841-74
Бензидин солянокислый ТУ 6-09-4222-76
Бензидин сернокислый ТУ МХП 1739-48
Анилин солянокислый ГОСТ 5822-78
Анилин сернокислый ГОСТ 5818-78
n-Нитроанилин солянокислый ТУ 6-09-06-545-75
о-Дианизидин солянокислый ТУ 6-09-3347-73
м-Фенилендиамин солянокислый ТУ 6-09-11-660-75
Проведение процесса заявляемым способом следующее. В бисерную мельницу вертикального типа с высокооборотной (3000 об/мин) механической мешалкой лопастного типа из текстолита, стеклянным корпусом объемом 300 мл и диаметром входного отверстия 50 мм, вводят расчетные количества стеклянного бисера с диаметром (0,8-2,2) мм в качестве перетирающего агента, этилцеллозольва и концентрированной азотной кислоты, либо предварительно приготовленного раствора азотной кислоты в этилцеллозольве с концентрацией 1-1,25 моль/кг (срок хранения такого раствора принципиального значения не имеет) и, спустя некоторое время, если раствор кислоты готовится непосредственно в реакторе, стабилизатора основной соли и оксида олова (IV). Диоксид вводится последним, причем при работающем механическом перемешивании. В противном случае могут образоваться небольшие количества средней соли, что уменьшит селективность по основной соли. Момент ввода последнего компонента принимают за начало проводимого процесса.
Контроль за ходом процесса осуществляют методом отбора проб и определения в них непрореагировавшей кислоты. Последнее достигает определенного значения и далее перестает меняться, что свидетельствует о завершении проводимого процесса. Выключают механическое перемешивание, отсоединяют корпус реактора от крышки с мешалкой и опускают в гнезде каркасной рамы вниз, давая возможность остаткам суспензии реакционной смеси (PC) стечь. Затем корпус с PC вынимают из гнезда каркасной рамы, а его содержимое выливают в приемлемую воронку узла отделения перетирающего агента с сеткой с ячейками 0,3×0,3 мм в качестве фильтровальной перегородки. Оставшийся на сетке перетирающий агент возвращают в корпус реактора, собирают и крепят в гнезде каркасной рамы бисерную мельницу вновь, вручную проворачивают вал мешалки, вводят определенное количество растворителя, включают механическое перемешивание и проводят отмывку реактора и перетирающего агента от остатков PC. Далее проводят повторное отделение перетирающего агента от промывного растворителя, используя в дальнейшем на промывку осадка на фильтре, получаемого при фильтровании PC. Осадок после промывки аккуратно отжимают, снимают с фильтра и отправляют на воздушную сушку при комнатной температуре до постоянной массы. Определяют последнюю, делают анализ на эквивалент и рассчитывают выход основной соли в проведенном процессе.
Фильтрат и промывной растворитель анализируют на остаточную кислоту и содержание продукта. Последнее в выбранных условиях менее 10-3 моль/кг. Содержание же кислоты может варьироваться от 0,5 моль/кг и более 0,05-0,1 моль/кг. В случае значимых концентраций возможен ввод расчетного количества оксида олова (IV) и проведение очередного процесса получения основной соли. При таком подходе ввод дополнительного количества трибохимического катализатора не требуется: вполне достаточно оставшегося в фильтрате количества.
Пример №1
В бисерную мельницу вертикального типа со стеклянным корпусом емкостью 300 мл и высотой 125 мм, высокооборотной (3000 об/мин) мешалкой лопастного типа с размерами лопасти из текстолита 46×22×2,3 мм вводят 100 г стеклянного бисера, 96,13 г 1,13 моль/кг предварительно приготовленного раствора азотной кислоты в этилцеллозольве, 0,07 г солянокислого гидроксиламина и 3,8 г оксида олова (IV). Корпус реактора с загрузкой быстро помещают в предназначенное для него гнездо каркасной рамы, соединяют с крышкой с мешалкой, сальниковой коробкой и валом, соединенным с валом электромотора, а также гнездами для помещения датчика температуры и пробоотборника, прокручивают вал мешалки вручную и включают механическое перемешивание, принимая этот момент за начало эксперимента. Процесс ведут при близкой к комнатной естественно складывающейся температуре и текущем контроле за содержанием непрореагировавшей кислоты, предусматривающим отбор проб по ходу протекания без прекращения механического перемешивания в этот момент. Полученные данные указанного контроля приведены в таблице 1.

При достижении прекращения дальнейшего расходования кислоты, совпадающего с расчетным значением (для образования основной соли Sn(OH)2(NO3)2 с 0,25 моль/кг загруженного SnO2 должно прореагировать 0,5 моль/кг кислоты) процесс прекращают, для чего останавливают механическое перемешивание, отсоединяют крышку реактора от корпуса, последний опускают непосредственно в гнезде каркасной рамы таким образом, чтобы нижняя кромка лопасти мешалки оказалась над реакционной смесью в корпусе и в таком положении оставляют на 7 мин, давая возможность остаткам суспензии реакционной смеси стечь с лопасти и вала мешалки. После этого корпус с PC вынимают из гнезда каркасной рамы, а его содержимое выливают в воронку узла отделения перетирающего агента с сеткой с размерами ячеек 0,3×0,3 мм вместо фильтровальной перегородки. Суспензия PC быстро проваливается в приемную емкость, а стеклянный бисер остается на сетке. Его аккуратно снимают с сетки и возвращают в корпус реактора. Бисерную мельницу собирают в гнезде каркасной рамы вновь, добавляют 30 г этилцеллозольва, включают механическое перемешивание и в течение 12 мин проводят отмывку бисера, внутренних поверхностей корпуса и элементов реактора от остатков реакционной смеси. По истечении данного времени перемешивание прекращают и проводят повторное отделение стеклянного бисера. Последний сушат и направляют на загрузку повторного процесса.
Промывной растворитель собирают в соответствующую емкость и используют для промывки получаемого при фильтровании отделенной от перетирающего агента суспензии PC осадка на фильтре узла фильтрования. После промывки осадок аккуратно отжимают, снимают вместе с фильтром и подвергают воздушной сушке при комнатной температуре до постоянной массы. Последнюю сравнивают с теоретически рассчитываемой величиной и рассчитывают выход отделенной основной соли. В данном случае он составил 95,3%. Делают эквивалент и рентгеноструктурный анализ, подтверждающие природу полученной соли.
Фильтрат и промывной растворитель взвешивают и используют в повторном процессе. Один из вариантов такого использования приведен ниже.
Примеры №2-9
Реактор, оловосодержащий реагент, предварительно приготовленный раствор азотной кислоты, их дозировка, дозировка стабилизатора основной соли, последовательности операций при загрузке компонентов, проведении процесса, определении момента его завершения, отделении перетирающего агента, отмывки от остатков PC, выделении продукта из PC и работе с продуктом и жидкой фазой PC аналогичны описанным в примере 1. Отличаются природой стабилизатора основной соли и начальным соотношением содержаний кислоты и оксида олова (IV). Указанное различие и предопределенные им характеристики процесса сведены в таблицу 2.

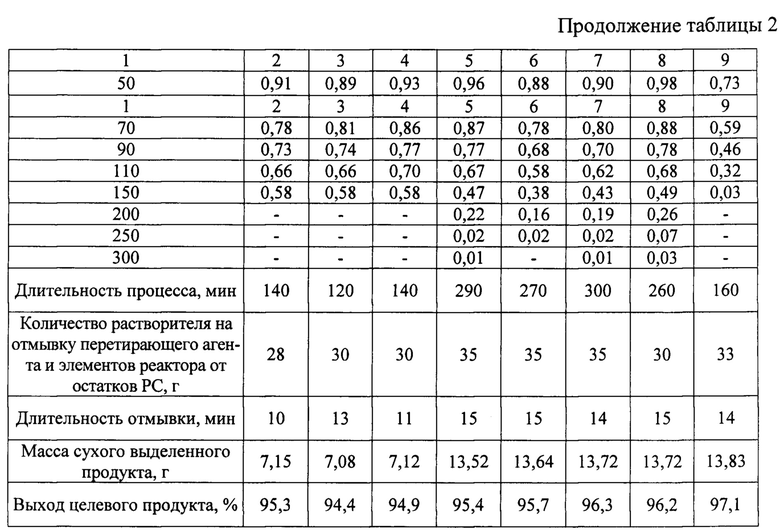
Примеры №10-18
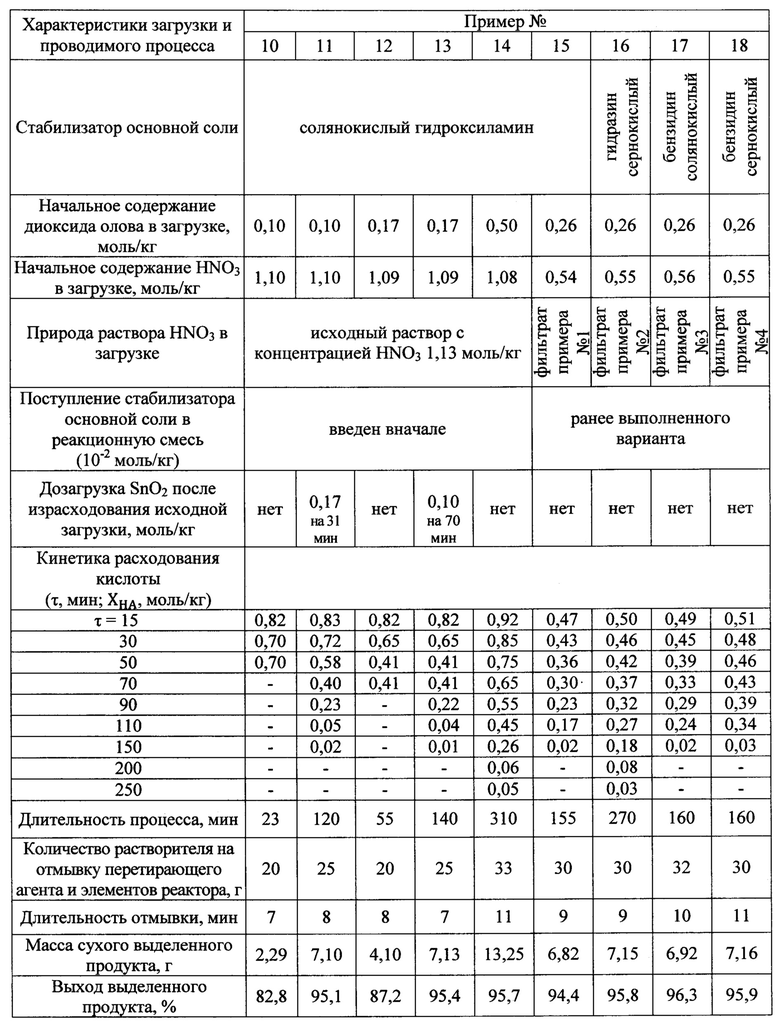
Реактор, природа используемых реагентов и растворителя объемной фазы, а также способ приготовления раствора кислоты, последовательности операций при загрузке, проведении процесса, его завершении, отделения перетирающего агента, выделении продукта, работе с ними аналогичны приведенным в примере 1. Отличаются величиной загрузки оксида олова (IV), избытком кислоты в начальной загрузке, природой стабилизатора основной соли, дробным вводом оксида олова (IV), а также использованием фильтратов после выделения целевого продукта в качестве самостоятельных объемных фаз с дозагрузкой кислоты или без нее, но в отсутствие дозагрузки стабилизатора основной соли. Указанные различия и предопределенные ими характеристики процесса сведены в таблицу 3.
Таблица 3
Примеры №19-26
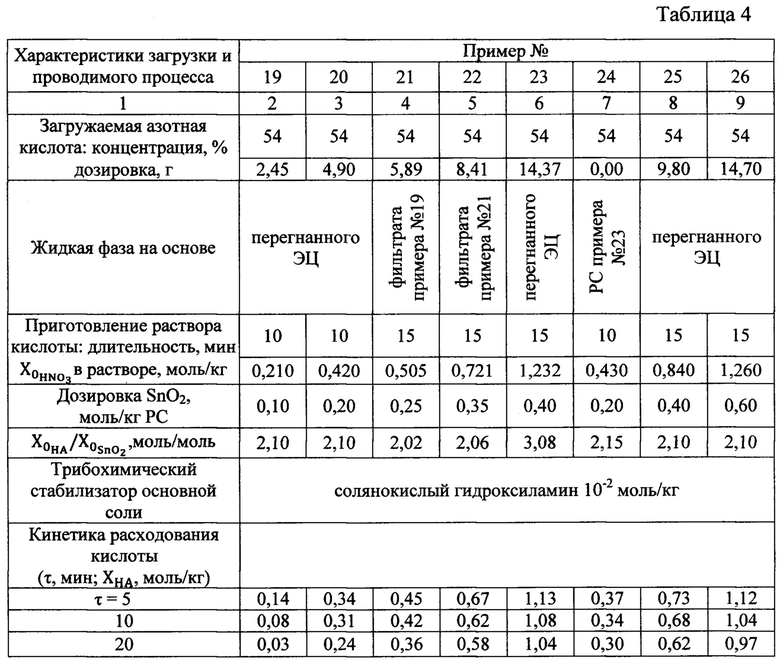
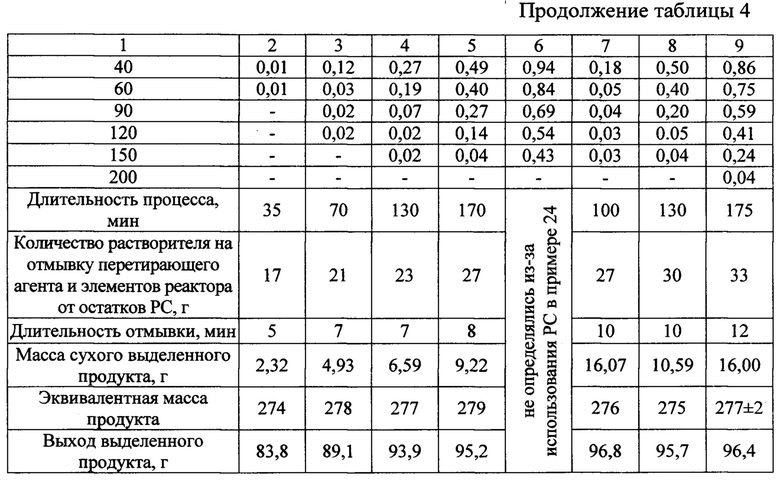
Исходные реагенты, растворитель, трибохимический стабилизатор основной соли и его количество, перетирающий агент, операции проведения процесса, контроля за ходом протекания, определения момента прекращения, отделения перетирающего агента, его подготовки к повторному использованию, выделения целевого продукта и по работе с ним и другими составными частями PC аналогичны описанным в примере 1. Отличаются приготовлением раствора кислоты непосредственно в бисерной мельнице, концентрационными факторами, избытком кислоты и вариантами использования такого избытка для получения целевого продукта. Указанные различия и предопределенные ими характеристики процесса приведены в таблице 4.
Положительный эффект предлагаемого решения состоит:
1. Получаемый продукт позволяет синтезировать соли олова (IV) с разными анионами при одном и том же катионе и таким путем существенно влиять на физические и химические свойства соли, а следовательно и на области их использования.
2. Метод прост в исполнении, не требует каких-либо специфических реагентов, подвода внешнего тепла и каких-либо существенных затрат на поддержание температурного режима по ходу протекания.
3. Отделение целевого продукта от остальной реакционной смеси проводится простым фильтрованием с промывкой осадка на фильтре с последующей воздушной сушкой. Твердая фаза продукта не склонна к захвату больших количеств жидкой фазы реакционной смеси, что существенно облегчает как отделение реакционной смеси от перетирающего агента бисерной мельницы, так и проведение всех остальных операций по выделению, промывке и сушке целевого продукта.
4. Предлагаемое решение позволяет многократное использование компонентов реакционной смеси в повторных процессах, причем в разных вариантах, в том числе без разделения, концентрирования и т.д. Это касается и трибохимического стабилизатора основной соли, ассортимент которого довольно широкий.
5. Аппаратурное оформление предлагаемого решения довольно простое и доступное. В нем нет котлонадзорного оборудования, а также оборудования, требующего индивидуального изготовления.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения нитрата олова (IV) путем окисления нитрата олова (II) | 2019 |
|
RU2717810C1 |
| Способ получения нитрата олова (IV) | 2017 |
|
RU2655142C1 |
| Двухстадийный способ получения карбоксилатов олова (II) из металла | 2017 |
|
RU2678092C1 |
| Способ получения соли олова (IV) с анионами азотной и бензойной кислот | 2020 |
|
RU2735433C1 |
| Способ получения карбоксилатов олова (II) | 2017 |
|
RU2671197C1 |
| Способ получения нитрата олова (II) при окислении металла | 2020 |
|
RU2744006C1 |
| Способ получения бензоата и замещенных бензоатов олова (IV) | 2017 |
|
RU2660905C1 |
| Способ получения бензоата и замещенных бензоатов олова (IV) из вторичного сырья | 2017 |
|
RU2673470C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ОЛОВА (IV) ИЗ СОЛЕЙ ОЛОВА (II) | 2022 |
|
RU2797089C1 |
| Способ получения основного бензоата олова (II) | 2017 |
|
RU2650893C1 |
Изобретение может быть использовано в неорганической химии. Получение основного нитрата олова (IV) Sn(OH)2(NO3)2 ведут путем прямого взаимодействия оксида олова (IV) с азотной кислотой в растворе этилцеллозольва при комнатной температуре в условиях интенсивного механического перемешивания. Перемешивание проводят в присутствии стеклянного бисера в качестве перетирающего агента и солянокислых гидроксиламина, анилина, n-нитроанилина, о-дианизидина, м-фенилендиамина, бензидина, сернокислых гидразина, анилина или бензидина в качестве стабилизатора основной соли. Оксид олова (IV) загружают в количестве 0,1-0,6 моль/кг в мольном соотношении с кислотой (1:2,02)÷(1:11). Процесс начинают с интенсивного механического перемешивания, контролируют по расходованию кислоты и прекращают, когда указанное расходование приближается к удвоенной начальной мольной загрузке оксида олова (IV). После этого перемешивание прекращают, проводят отделение перетирающего агента и его отмывку от остатков реакционной смеси. Отделенную реакционную смесь фильтруют. Осадок на фильтре промывают промывным растворителем и сушат на воздухе до постоянного веса. Изобретение позволяет упростить получение основного нитрата олова (IV) при повышении селективности по целевому продукту. 2 з.п. ф-лы, 4 табл., 26 пр.
| HARRISON PHILIP G | |||
| et al., A contribution to the chemistry of tin (IV) nitrate, Inorganica Chimica Acta, 1978, Vol | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| Устройство для отыскания металлических предметов | 1920 |
|
SU165A1 |
| СПОСОБ ПОЛУЧЕНИЯ РАСТВОРА АЗОТНОКИСЛОГО ДВУХВАЛЕНТНОГО ОЛОВА | 0 |
|
SU356248A1 |
| WO 2011124659 A1, 13.10.2011 | |||
| CN 101519561 A, 02.09.2009 | |||
| АГЕЕВА Л.С | |||
| и др., Глубокие и быстрые при комнатных температурах превращения олова, его оксидов, гидроксидов и солей в присутствии кислот и механической активации процессов, Материалы III Международной Российско-Казахстанской научно-практической конференции "Химические технологии функциональных материалов", 27-29 апреля 2017 г, Новосибирск, НГТУ, 2017, сс | |||
| Устройство для устранения мешающего действия зажигательной электрической системы двигателей внутреннего сгорания на радиоприем | 1922 |
|
SU52A1 |

