Изобретение относится к области химии, нефтехимии и нефтепереработки и может быть использовано для приготовления катализаторов, в частности, для получения синтез-газа путем углекислотной конверсии метана.
Известно, что для получения синтез-газа используется три способа конверсии метана: паровой риформинг, основанный на процессе СН4+H2O=СО+3Н2; углекислотная конверсия, основанная на процессе СН4+CO2=2СО+2Н2, и частичное окисление метана, протекающее по реакции СН4+1/2O2=СО+2Н2, из которых синтез-газ, полученный углекислотной конверсией метана с использованием приготовленного катализатора, благодаря оптимальному соотношению образующихся продуктов (Н2/СО), может быть эффективно использован для получения углеводородов, кислородсодержащих соединений (спиртов, альдегидов, эфиров и кислот) реакцией Фишера-Тропша.
Известны способы приготовления катализаторов, которые могут быть использованы для получения синтез-газа путем углекислотной конверсии метана [1-3].
Известен способ приготовления катализаторов на основе переходных металлов (Fe, Со, Ni), являющихся альтернативой дорогостоящим катализаторам на основе благородных металлов [S.М. De Lima and J.M. Assaf, "Ni-Fe catalysts based on perovskite-type oxides for dry reforming of methane to syngas," Catal. Letters, vol. 108, no. 1-2, pp. 63-70, 2006 - (1)], однако они подвержены быстрой дезактивации и отложению углерода на поверхности. Перспективными катализаторами являются оксиды со структурой перовскита (ABO3, где А - редкоземельный металл, В - переходный металл), данные оксиды могут быть модифицированы путем частичного замещения А или В, что позволяет, варьируя состав, увеличивать стабильность и каталитическую активность. Благодаря ряду структурных особенностей оксиды со структурой перовскита обладают повышенной химической и термической стабильностью, устойчивостью к каталитическим ядам. К недостаткам известного способа относится длительная высокотемпературная обработка, поскольку получение оксидов со структурой перовскита обычно включает стадию прокаливания при длительном времени и высокой температуре, что приводит к уменьшению удельной поверхности и укрупнению размера частиц, что может снижать активность катализатора.
Известен способ приготовления [CN 103933991 B 2014 Perovskite type composite oxide catalyst for producing controllable synthesis gas - (2)] композитного катализатора, состоящего из перовскита с добавлением оксида железа (III). К недостаткам данного способа следует отнести высокую температуру синтеза - 600-1200°С, длительное время прокаливания - 4-24 часа, что приводит к получению частиц большего размера, 150-250 мкм.
Известен способ приготовления оксида со структурой перовскита с размером частиц 60-100 нм [CN 102942225 2013 Preparation method of GdFeO3 nanocrystallines (3)], который может быть использован в качестве катализатора получения синтез-газа. По решаемой задаче и достигаемому техническому результату этот способ выбран в качестве прототипа. Данный способ включает следующие стадии: растворение нитратов Gd(NO3)3⋅6H2O, Fe(NO3)3⋅9H2O в этиленгликоле (1:1:40-120), выпаривание при 60-80°С в течение 2-4 часов до образования золя, сушку полученного золя при 120-160°С в течение 6-10 часов, с последующей гомогенизацией полученного порошка и трехкратное прокаливание в муфельной печи при 300-500°С с промежуточным измельчением. Прокаливание в муфельной печи при 600-900°С в течение 2-8 часов. Промывание водой и этанолом, сушку в печи при 60-80°С в течение 5-6 часов. Известный способ позволяет получать частицы размером 60-100 нм.
К недостаткам прототипа относятся большое количество повторяющихся энергозатратных стадий прокаливания, длительная термическая обработка и использование большого количества реагентов (комплексообразователя - этиленгликоля и этанола для промывки), приводящих к снижению экономической эффективности способа.
Техническим результатом заявленного изобретения является снижение энергозатрат на приготовление катализаторов за счет того, что разработанный способ позволяет уменьшить число стадий прокаливания, сократить время и температуру термической обработки, уменьшить количества используемых реагентов, что дает возможность снизить экономические затраты на приготовление катализатора.
Указанный технический результат достигается тем, что в способе приготовления катализатора для получения синтез-газа путем углекислотной конверсии метана, заключающемся в растворении солей-предшественников, добавлении комплексообразователя, выпаривании раствора, прокаливании в муфельной печи, в соответствии с заявленным изобретением, в качестве дополнительной соли-предшественника используют нитрат кобальта Co(NO3)2⋅3H2O, соли-предшественники Gd(NO3)3⋅6H2O, Fe(NO3)3⋅9H2O, Co(NO3)2⋅3H2O, взятые в мольном соотношении 1:х:(1-х), где х=0-0.9, перед добавлением комплексообразователя растворяют в деионизированной воде, в качестве комплексообразователя используют лимонную кислоту, взятую в весовом соотношении к смеси нитратов от 2:1 до 2.5:1, после полного растворения лимонной кислоты добавляют раствор аммиака до установления рН от 6 до 6.5, а полученный после выпаривания порошок прокаливают при 450°С в течение 2-х часов и 1-2 часа при температуре 600-800°С.
Общими с прототипом признаками заявленного способа приготовления катализаторов включает следующие стадии: растворение солей-предшественников Gd(NO3)3⋅6H2O и Fe(NO3)3⋅9H2O, добавление комплексообразователя, выпаривание раствора и прокаливание полученного порошка в муфельной печи.
Отличительными от прототипа признаками заявленного способа приготовления катализатора являются:
1. Использование в качестве дополнительной соли-предшественника нитрата кобальта Co(NO3)2⋅3H2O.
2. Соли-предшественники Gd(NO3)3⋅6H2O, Fe(NO3)3⋅9H2O, Co(NO3)2⋅3H2O, взятые в мольном соотношении 1:х:(1-х), где х=0-0.9, перед добавлением комплексообразователя растворяют в деионизированной воде.
3. В качестве комплексообразователя используют лимонную кислоту, взятую в весовом соотношении к смеси нитратов от 2:1 до 2.5:1.
4. После полного растворения лимонной кислоты добавляют раствор аммиака до установления рН от 6 до 6.5.
5. Полученный после выпаривания порошок прокаливают при 450°С в течение 2 часов и 1-2 часа при температуре 600-800°С.
Сущность заявленного способа приготовления катализаторов заключатся в растворении солей-предшественников Gd(NO3)3⋅6H2O, Fe(NO3)3⋅9H2O, Co(NO3)2⋅3H2O, взятых в мольном соотношении 1:х:(1-х), где х=0-0.9, в деионизированной воде добавлении комплексообразователя - лимонной кислоты, взятой в весовом соотношении к смеси нитратов от 2:1 до 2.5:1, до полного растворения лимонной кислоты, добавлении раствора аммиака при комнатной температуре, до установления рН от 6 до 6.5 и образования золя, выпаривании полученного раствора, при постоянном перемешивании при температуре 120°С до полного удаления воды, прокаливании порошка при 450°С в течение 2 часов, гомогенизации порошка с последующим таблетированием, прокаливании 1-2 часа при температуре 600-800°С.
Заявленный способ позволяет приготовить катализатор состава, соответствующего формуле GdCo1-xFexO3, где х=0-0.9.
Для приготовленных предложенным способов образцов катализатора проводили анализ фазового состава, определение морфологии и тестирование каталитической активности в реакции получения синтез-газа путем углекислотной конверсии метана.
Рентгенофазовый анализ образцов проведен при комнатной температуре, на приборе Rigaku «MiniFlex II», с СuКα излучением, интервал сканирования углов - 2θ=10-60°, скорость сканирования - 5°/мин. Для идентификации фаз использована база данных PDF2.
Морфологию образцов исследовали с использованием полевого эмиссионного сканирующего микроскопа (ZeissMerlin и ZeissSupra 40VP).
Удельная площадь поверхности была определена методом адсорбции азота при -196°С на адсорбционном анализаторе QuadrasorbSI. Перед проведением измерений адсорбции все образцы были дегазированы при 300°С в течение 5 часов для удаления остаточной влаги и других летучих веществ.
Каталитические свойства образцов, полученных описанным способом, анализировали в реакции получения синтез-газа углекислотной конверсией метана. Испытания были проведены в реакторе проточного типа, при атмосферном давлении, температура реакции - при 950°С, скорость подачи смеси - 1,0 л/ч, состав исходной смеси - CO2:СН4=1:1.
Состав реакционной смеси регистрировался после достижения катализатором стационарного состояния, о котором судили по постоянству площадей хроматографических пиков. Основными показателями протекания реакции были выбраны конверсия метана, скорости расходования метана, диоксида углерода и скорости образования монооксида углерода и водорода.
Количественная интерпретация полученных результатов была проведена методом абсолютной калибровки с использованием программы Хроматэк Аналитик 2.5.
Число молей реагентов и продуктов в анализируемой дозе рассчитывали с использованием регрессионных уравнений:

где ni - число молей i-го вещества в анализируемой дозе; Si - площадь i-го хроматографического пика, мВ⋅мин; ki - поправочный коэффициент для i-го вещества, моль/мин⋅мВ.
Скорости расходования реагентов (wi моль/ (ч⋅га.ф.)) рассчитывали по формуле:

ni - количество i-го продукта в газовой фазе, моль; n0 - исходное количество i-го продукта в реакционной смеси, моль; w - объемная скорость реакционной смеси, л/час, V - объем петли хроматографической колонки, 0.208⋅10-3 л, m - масса катализатора, г.
Содержание продуктов в газовой фазе (скорость образования моль/(ч⋅га.ф.)) определяли по формуле:

Конверсии (αi) СН4 и CO2 рассчитывали по формуле:

где ni - количество СН4 или CO2 в газовой фазе, моль; ni0 - исходное количество i-го продукта в реакционной смеси, моль.
Погрешность в определении каждого компонента не превышала 5% и складывалась из погрешности измерения температуры и погрешности измерения скорости подачи реакционной смеси.
Сущность изобретения иллюстрируется нижеприведенными примерами (1-3), таблицами (1-2) и чертежами (Фиг. 1 - Фиг. 6).
Пример 1 иллюстрирует способ приготовления катализатора состава GdCo0.1Fe0.9O3.
В качестве солей-предшественников использовали: Gd(NO3)3⋅6H2O (квалификации "ХЧ"), Co(NO3)2⋅3H2O (квалификации "Ч"), Fe(N3)3⋅9H2O (квалификации "Ч"). Нитраты, взятые в мольном соотношении 1:0.1:0.9, растворяли в деионизированной воде, затем колба помещалась на магнитную мешалку с терморегулятором. К полученному раствору нитратов при постоянном перемешивании добавляли лимонную кислоту в весовом соотношении 2:1. После полного растворения лимонной кислоты, для установления рН раствора на уровне 6, при комнатной температуре добавляли раствор аммиака и при этом наблюдали образование золя. Далее проводили выпаривание раствора при температуре ~120°С до полного удаления воды и образования порошка черного цвета. Затем температуру поднимали до 450°С и прокаливали полученный порошок в течение 2 часов. Полученный порошок гомогенизировали перетиранием в агатовой ступке с последующим таблетированием, затем прокаливали в муфельной печи при температуре 600°С в течение 2 часов.
На Фиг. 1 приведены результаты рентгенофазового анализа приготовленного катализатора. Исследование его фазового состава показывает, что указанного времени и температуры прокаливания достаточны для образования оксида со структурой перовскита.
Результаты фазового анализа и значение удельной площади поверхности приведены в таблице 1.

На Фиг. 2 приведена микрофотография поверхности приготовленного катализатора, на которой видно, что полученный образец обладает пористой морфологией с размером частиц порядка 50-60 нм.
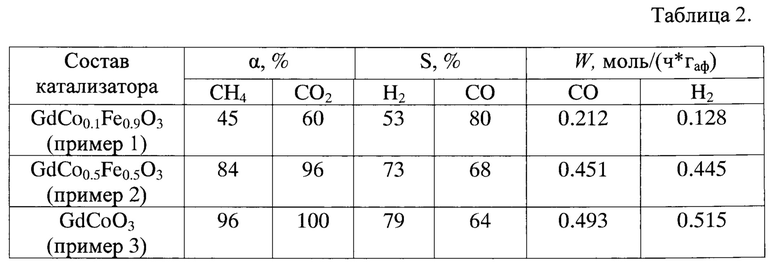
Результаты исследования каталитических свойств, а именно, конверсия (α) метана и диоксида углерода, скорости их образования (W) и селективности (S) по продуктам СО и Н2 при температуре 950°С, приведены в таблице 2.
Таким образом, применение заявленного способа, как видно из приведенного примера, позволяет существенно упростить получение катализатора на основе феррита гадолиния, а подбор температуры прокаливания обеспечивает получение нанодисперсного порошка с развитой поверхностью.
Пример 2 иллюстрирует способ приготовления катализатора состава GdCo0.5Fe0.5O3.
В качестве солей-предшественников использовали: Gd(NO3)3⋅6H2O (квалификации "ХЧ"), Co(NO3)2⋅3H2O (квалификации "Ч"), Fe(NO3)3⋅9H2O (квалификации "Ч"). Нитраты, взятые мольном соотношении 1:0.5:0.5, растворяли в деионизированной воде, затем колба помещалась на магнитную мешалку с терморегулятором. К полученному раствору солей-предшественников при постоянном перемешивании добавляли лимонную кислоту в весовом соотношении: лимонная кислота: нитраты (2.2:1). После полного растворения лимонной кислоты, при комнатной температуре добавляли раствор аммиака до установления рН раствора на уровне 6.2 и образования золя. Далее проводили выпаривание раствора при температуре ~120°С, до полного удаления воды и образования порошка черного цвета. Затем температуру поднимали до 450°С и прокаливали полученный порошок в течение 2 часов. Полученный порошок гомогенизировали в агатовой ступке и таблетировали, затем прокаливали в муфельной печи при температуре 750°С 1.5 часа.
Данные рентгенофазового анализа (Фиг. 3), подтверждают присутствие только целевого продукта со структурой перовскита и отсутствие примесных фаз.
На Фиг. 4 приведена микрофотография полученного образца катализатора, частицы которого имеет размер в диапазоне 50-200 нм и образуют пористую поверхность, удельная площадь поверхности приведена в таблице 1.
Результаты изучения каталитических свойств образца катализатора, приготовленного по описанному способу, приведены в таблице 2.
Пример 3 иллюстрирует способ приготовления катализатора состава-GdCoO3.
В качестве солей-предшественников использовали: Gd(NO3)3⋅6H2O (квалификации "ХЧ"), Co(NO3)2⋅3H2O (квалификации "Ч"). Нитраты, взятые в мольном соотношении 1:1, растворяли в деионизированной воде, затем колба помещалась на магнитную мешалку с терморегулятором. К полученному раствору солей при постоянном перемешивании добавляли лимонную кислоту в весовом соотношении: лимонная кислота: нитраты (2.5:1). После полного растворения лимонной кислоты, при комнатной температуре добавляли раствор аммиака до установления рН раствора на уровне 6.5и образования золя, после чего проводили выпаривание раствора при температуре ~120°С до полного удаления воды и образования порошка черного цвета. Затем нагревали до 450°С и прокаливали полученный порошок в течение 2 часов. Полученный порошок гомогенизировали в агатовой ступке и таблетировали, затем прокаливали в муфельной печи при температуре 800°С 1 час.
На Фиг. 5 приведены результаты рентгенофазового анализа образца катализатора. Рентгенограмма полученного порошка показала наличие однофазного GdCoO3 со структурой перовскита.
На Фиг. 6 приведена микрофотография GdCoO3, свидетельствующая о том, что приготовленный катализатор имеет частицы размером порядка 100 нм. В таблице 1 приведена удельная площадь поверхности.
Результаты изучения каталитической активности, полученного по описанному способу катализатора, приведены в таблице 2.
Приведенные выше примеры подтверждают возможность использования заявленного способа приготовления катализаторов с частицами размером 50-200 нм и высокой каталитической активностью в реакции получения синтез-газа путем углекислотной конверсии метана.
Техническим результатом заявленного изобретения является разработка способа, позволяющего уменьшить число стадий прокаливания, сократить время и снизить температуру термической обработки, уменьшить количества используемых реагентов, что дает возможность снизить экономические затраты на приготовление катализатора. Заявленный способ позволяет получать однофазные оксиды с заданными характеристиками, к которым можно отнести развитую поверхность, термическую стабильность, высокую каталитическую активность.
Использование заявленного изобретения предполагается на предприятиях нефтегазового сектора, таких как ПАО «Газпром нефть», ПАО «Лукойл», ОАО «Салаватнефтеоргсинтез», ЗАО «Промышленные катализаторы».
Источники информации
1. S.М. De Lima and J.M. Assaf, "Ni-Fe catalysts based on perovskite-type oxides for dry reforming of methane to syngas," Catal. Letters, vol. 108, no. 1-2, pp. 63-70, 2006.
2. Патент CN 103933991 B 2014 Perovskite type composite oxide catalyst for producing controllable synthesis gas.
3. Патент CN 102942225 2013 Preparation method of GdFeO3 nanocrystallines - прототип.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ приготовления катализатора для получения углеводородов реакцией Фишера-Тропша | 2019 |
|
RU2744708C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРОВСКИТОВ | 2009 |
|
RU2440292C2 |
| СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА | 2014 |
|
RU2572530C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА В ЕГО ПРИСУТСТВИИ | 2023 |
|
RU2814309C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДНЫХ КАТАЛИЗАТОРОВ С ИСПОЛЬЗОВАНИЕМ МИКРОВОЛНОВОГО ИЗЛУЧЕНИЯ (ВАРИАНТЫ) | 2006 |
|
RU2301705C1 |
| КАТАЛИЗАТОР ПОЛУЧЕНИЯ БУТАДИЕНА-1,3 И СПОСОБ ПОЛУЧЕНИЯ БУТАДИЕНА-1,3 С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА | 2014 |
|
RU2552984C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА И СПОСОБ ОЧИСТКИ ГАЗОВЫХ СМЕСЕЙ ОТ ОКСИДА УГЛЕРОДА | 2008 |
|
RU2381064C1 |
| СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА | 2014 |
|
RU2573005C1 |
| СПОСОБ АКТИВАЦИИ МЕТАЛЛОКСИДНЫХ КАТАЛИЗАТОРОВ СИНТЕЗА УГЛЕРОДНЫХ НАНОМАТЕРИАЛОВ | 2010 |
|
RU2443470C2 |
| КАТАЛИЗАТОР И СПОСОБ ПОЛУЧЕНИЯ СИНТЕЗ-ГАЗА | 2010 |
|
RU2453366C1 |






Изобретение относится к области химии, нефтехимии и нефтепереработки, в частности, к способу приготовления катализаторов для получения синтез-газа реакцией углекислотной конверсии метана. Способ приготовления катализатора заключается в растворении солей-предшественников, добавлении комплексообразователя, выпаривании раствора, прокаливании в муфельной печи, при этом в качестве дополнительной соли-предшественника используют нитрат кобальта Co(NO3)2⋅3H2O, перед добавлением комплексообразователя соли-предшественники Gd(NO3)3⋅6H2O, Fe(NO3)3⋅9H2O, Co(NO3)2⋅3H2O, взятые в мольном соотношении 1:х:(1-х), где х=0-0.9, растворяют в деионизированной воде, в качестве комплексообразователя используют лимонную кислоту, взятую в весовом соотношении к смеси нитратов от 2:1 до 2.5:1, после полного растворения лимонной кислоты добавляют раствор аммиака до установления рН от 6 до 6.5, а полученный после выпаривания порошок прокаливают при 450°С в течение 2 ч и 1-2 ч при температуре 600-800°С. Технический результат заключается в сокращении времени и снижении температуры термической обработки, уменьшении числа стадий и количества используемых реагентов, что позволяет снизить экономические затраты на приготовление катализатора. 6 ил., 3 пр.
Способ приготовления катализатора для получения синтез-газа путем углекислотной конверсии метана, заключающийся в растворении солей-предшественников, добавлении комплексообразователя, выпаривании раствора, прокаливании в муфельной печи, отличающийся тем, что в качестве дополнительной соли-предшественника используют нитрат кобальта Co(NO3)2⋅3H2O, перед добавлением комплексообразователя соли-предшественники Gd(NO3)3⋅6H2O, Fe(NO3)3⋅9H2O, Co(NO3)2⋅3H2O, взятые в мольном соотношении 1:х:(1-х), где х=0-0.9, растворяют в деионизированной воде, в качестве комплексообразователя используют лимонную кислоту, взятую в весовом соотношении к смеси нитратов от 2:1 до 2.5:1, после полного растворения лимонной кислоты добавляют раствор аммиака до установления рН от 6 до 6.5, а полученный после выпаривания порошок прокаливают при 450°С в течение 2 ч и 1-2 ч при температуре 600-800°С.
| CN 102942225 A, 27.02.2013 | |||
| CN 103933991 A, 23.07.2014 | |||
| Устройство для механической чистки топок паровозных котлов | 1928 |
|
SU17572A1 |
| CN 101185885 A, 28.05.2008 | |||
| КАТАЛИЗАТОРЫ | 2010 |
|
RU2517700C2 |

