Область техники
Изобретение относится к биотехнологии, а именно, к новым ферментам Cas нуклеазам систем CRISPR-Cas, применяемым для разрезания ДНК и редактирования генома различных организмов. Данная технология может применяться в будущем для генной терапии наследственных заболеваний человека, а также для редактирования генома других организмов.
Уровень техники
Изменение последовательности ДНК – одна из актуальных задач биотехнологии на сегодняшний день. Редактирование и изменение геномов эукариотических и прокариотических организмов, а также манипуляции с ДНК in vitro, требуют направленного внесения двунитевых разрывов в последовательности ДНК.
Для решения этой задачи в настоящее время используют следующие методики: искусственные нуклеазные системы, содержащей домены типа «цинковые пальцы», TALEN-системы и бактериальные CRISPR-Cas системы. Первые два метода требуют трудозатратой оптимизации аминокислотной последовательности нуклеазы для узнавания конкретной последовательности ДНК. В отличие от них в случае CRISPR-Cas систем структурами, узнающими ДНК мишень, являются не белки, а короткие направляющие РНК. Разрезание конкретной ДНК мишени не требует синтеза нуклеазы или ее гена de novo, а обеспечивается за счет использования направляющих РНК, комплементарных целевой последовательности. Это делает CRISPR Cas системы удобными и эффективными инструментами разрезания различных ДНК-последовательностей. Методика позволяет осуществлять единовременное разрезание ДНК в нескольких участках при использовании направляющих РНК разной последовательностей. Такой подход используется в том числе для одновременного изменения нескольких генов в эукариотических организмах.
По своей природе CRISPR-Cas системы являются иммунными системами прокариот, способными высоко специфично вносить разрывы в генетический материал вирусов (Mojica F. J. M. et al. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements //Journal of molecular evolution. – 2005. – Т. 60. – №. 2. – С. 174-182). Аббревиатура CRISPR-Cas расшифровывается как “Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR associated genes” (Jansen R. et al. Identification of genes that are associated with DNA repeats in prokaryotes //Molecular microbiology. – 2002. – Т. 43. – №. 6. – С. 1565-1575), что переводе с английского обозначает “короткие палиндромные повторы, регулярно расположенные группами, и aссоциированные с ними гены”. Все CRISPR-Cas системы состоят из CRISPR кассет и генов, кодирующих различные Cas белки (Jansen R. et al. , Molecular microbiology. – 2002. – Т. 43. – №. 6. – С. 1565-1575). CRISPR кассеты состоят из последовательностей-спейсеров, каждый из которых имеет уникальную нуклеотидную последовательность, и повторяющихся палиндромных повторов (Jansen R. et al. , Molecular microbiology. – 2002. – Т. 43. – №. 6. – С. 1565-1575). В результате транскрипции CRISPR кассет и их последующего процессинга образуются направляющие крРНК, которые вместе с Cas белками формируют эффекторный комплекс (Brouns S. J. J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes //Science. – 2008. – Т. 321. – №. 5891. – С. 960-964). За счет комплементарного спаривания крРНК с целевым участком ДНК, именуемым протоспейсером, Cas-нуклеаза узнает ДНК-мишень и высоко специфично вносит в нее разрыв.
CRISPR-Cas системы, представленными одиночным белком-эффектором, разделяют на шесть различных типов (от I до VI) в зависимости от Cas белков, входящих в состав систем. Система CRISPR-Cas9 II типа отличается простотой состава и механизма работы: для ее функционирования необходимо формирование эффекторного комплекса, состоящего лишь из одного белка Cas9 и двух коротких РНК: крРНК (crRNA) и трейсерной РНК (tracrRNA). Трейсерная РНК комплементарно спаривается с участком крРНК, проиcходящим из CRISPR повтора, образуя вторичную структуру, необходимую для связывания направляющих РНК с Cas эффектором. Эффекторный белок Cas9 является РНК-зависимой ДНК эндонуклеазой с двумя нуклеазными доменами (HNH и RuvC), вносящими разрывы в комплементарные нити целевой ДНК, таким образом образуя двунитевой разрыв ДНК (Deltcheva E. et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III //Nature. – 2011. – Т. 471. – №. 7340. – С. 602).
На сегодняшний день известно несколько CRISPR-Cas нуклеаз, способных направлено и специфично вносить двунитевые разрывы в ДНК. Одной из основных характеристик, ограничивающих применение CRISPR-Cas систем является PAM последовательность, фланирующая ДНК-мишень с 3'-конца, наличие которой необходимо для правильного узнавания ДНК Cas9 нуклеазой. Различные CRISPR-Cas белки имеют разные PAM последовательности, которые ограничивают возможности применения нуклеаз на любых участках ДНК. Использование CRISPR-Cas белков с новыми разнообразными PAM последовательностями необходимо для обеспечения возможности изменения любого участка ДНК, как in vitro, так и в геноме живых организмов. Изменение эукариотических геномов также требует использования нуклеаз малого размера для обеспечения доставки CRISPR-Cas систем в клетки посредством AAV вирусов.
Несмотря на известность ряда способов разрезания ДНК и изменения последовательности геномной ДНК, на сегодняшний день сохраняется потребность в
новых эффективных инструментах для модификации ДНК в различных организмах и в строго определенных местах последовательности ДНК. Данное изобретение обладает рядом свойств, необходимых для решения этой задачи.
Сущность изобретения
Задачей настоящего изобретения является создание новых инструментов для изменения последовательности геномной ДНК одноклеточных или многоклеточных организмов на основе систем CRISPR-Cas9. Существующие в настоящее время системы имеют ограниченное применение из-за специфичной последовательности РАМ, которая должна присутствовать на 3'-конце участка ДНК, подвергающегося модификации. Поиск новых ферментов Cas9 с другими РАМ последовательностями позволит расширить арсенал имеющихся средств для образования двунитевого разрыва в необходимых, строго определенных местах в молекулах ДНК разных организмов. Для решения этой задачи авторами была охарактеризована ранее предсказанная для Defluviimonas sp. 20V17 CRISPR нуклеаза II типа DfCas9, которая может быть применена для внесения направленных изменений в геном как этого, так и других организмов. Существенными признаками, отличающими настоящее изобретение, являются: (а) короткая, отличающаяся от других известных последовательность PAM; (б) относительно малый размер охарактеризованного белка DfCas9 – 1079 аминокислотных остатков (а.о.).
Указанная задача решается путем применения белка, содержащего аминокислотную последовательность SEQ ID NO: 1, или содержащего аминокислотную последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1 и имеет отличия по сравнению с SEQ ID NO: 1 только в неконсервативных аминокислотных остатках, для образования двунитевого разрыва в молекуле ДНК, расположенного непосредственно перед нуклеотидной последовательностью 5'-NN(G/A)NA(C/T)N-3' в указанной молекуле ДНК. В некоторых вариантах изобретения данное применение характеризуется тем, что образование двунитевого разрыва в молекуле ДНК происходит при температуре от 35 оС до 37 оС и в присутствии ионов Mn2+. В предпочтительных вариантах изобретения данное применение характеризуется тем, что концентрация ионов Mn2+ составляет более 5 мM.
Указанная задача также решается путем создания способа образования двунитевого разрыва в последовательности геномной ДНК одноклеточного или многоклеточного организма, непосредственно примыкающей к последовательности 5'-NN(G/A)NA(C/T)N-3', включающего введение в по меньшей мере одну клетку этого организма эффективного количества: а) белка, содержащего аминокислотную последовательность SEQ ID NO: 1, или нуклеиновой кислоты, кодирующей белок, содержащий аминокислотную последовательность SEQ ID NO: 1, и б) направляющей РНК, содержащей последовательность, образующую дуплекс с нуклеотидной последовательностью участка геномной ДНК организма, непосредственно примыкающей к нуклеотидной последовательности 5'-NN(G/A)NA(C/T)N-3', и взаимодействующей с указанным белком после образования дуплекса, или последовательности ДНК, кодирующей указанную направляющую РНК; при этом взаимодействие указанного белка с направляющей РНК и нуклеотидной последовательностью 5’-NN(G/A)NA(C/T)N-3’ приводит к образованию двунитевого разрыва в последовательности геномной ДНК, непосредственно примыкающей к последовательности 5'-NN(G/A)NA(C/T)N-3'. В некоторых вариантах изобретения данный способ характеризуется тем, что дополнительно включающий введение экзогенной последовательности ДНК одновременно с направляющей РНК.
В качестве направляющей РНК может быть использована смесь из крРНК (crRNA) и трейсерной РНК (tracrRNA), способных образовать комплекс с участком целевой ДНК и белком DfCas9. В предпочтительных вариантах изобретения в качестве направляющей РНК может быть использована гибридная РНК, сконструированная на основе крРНК и трейсерной РНК. Методы конструирования гибридной направляющей РНК известны специалистам (Hsu PD, et al., DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013 Sep;31(9):827-32). Один из вариантов конструирования гибридной РНК раскрыт в Примерах ниже.
Изобретение может быть использовано как для разрезания целевой ДНК in vitro, так и для модификации генома какого-либо живого организма. Модификация генома может проводиться прямым способом – разрезанием генома в соответствующем сайте, а также вставкой экзогенной последовательности ДНК за счет гомологичной репарации.
В качестве экзогенной последовательности ДНК может быть использован любой участок двунитевой или однонитевой ДНК из генома организма, отличного от организма, используемого при введении (или смесь таких участков между собой и с другими фрагментами ДНК), при этом этот участок (или смесь участков) предназначен для интеграции в место двуцепочечного разрыва в целевой ДНК, образованного под действием нуклеазы DfCas9. В некоторых вариантах изобретения в качестве экзогенной последовательности ДНК может быть использован участок двуцепочечной ДНК из генома организма, используемого при введении белка DfCas9, но при этом измененный мутациями (заменой нуклеотидов), а также вставками или делециями одного или нескольких нуклеотидов.
Техническим результатом настоящего изобретения является повышение универсальности доступных систем CRISPR-Cas9, позволяющее использовать нуклеазу Cas9 для разрезания геномной или плазмидной ДНК в большем количестве специфических сайтов и специфических условий.
Краткое описание рисунков
Фиг. 1. Схема CRISPR локусов в Defluviimonas sp.
Фиг. 2. Разработка системы порезки ДНК, ограниченной NN(G/A)NA(C/T)N последовательностью.
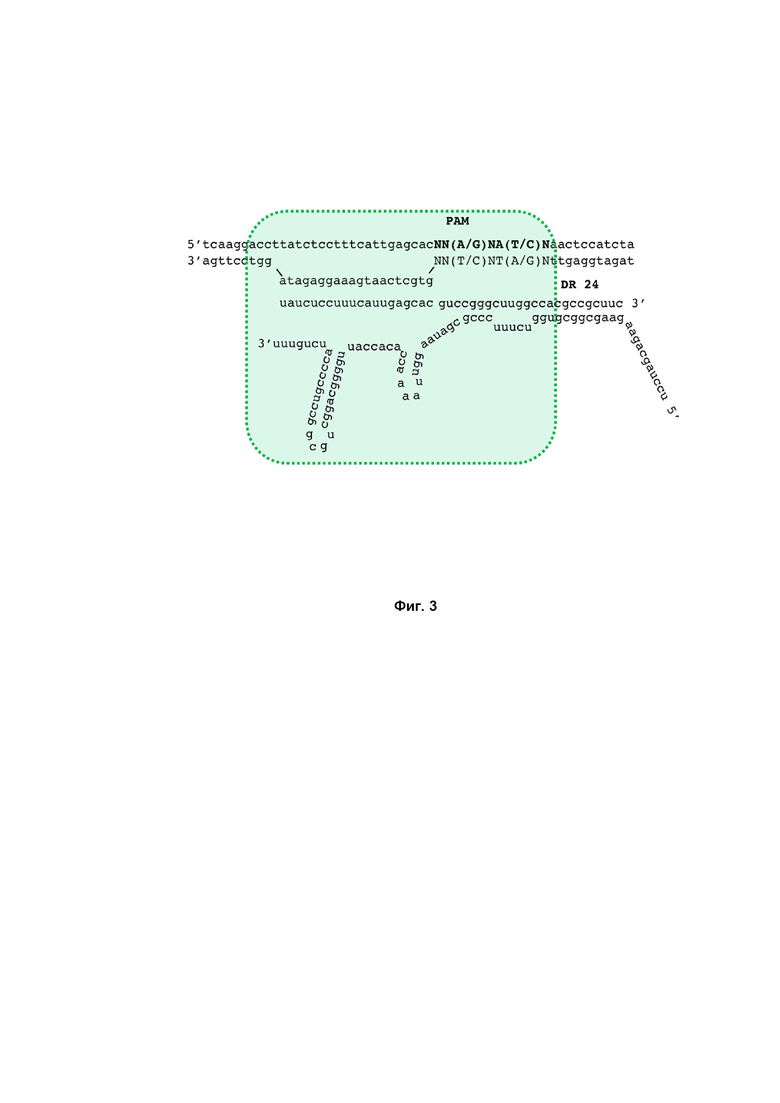
Фиг. 3. Схема взаимодействия направляющей РНК с участком целевой ДНК.
Фиг. 4. Зависимость эффективности реакции разрезания ДНК от присутствия ионов.
Фиг. 5. Проверка активности белка в разрезании различных ДНК мишеней.
Фиг. 6. Реакции in vitro разрезания ДНК фрагмента гена человека grin2b.
Фиг. 7. Влияние концентрации ионов на эффективность реакции разрезания ДНК.
Фиг. 8. Подбор последовательности sgRNA.
Фиг. 9. Выравнивание начальных частей последовательностей белков Cas9 из организмов Staphylococcus aureus (SaCas9), Campylobacter jejuni (CjCas9) и DfCas9. Подчеркнуты снизу неконсервативные участки последовательностей.
Подробное раскрытие изобретения
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из». Если не определено отдельно, технические и научные термины в данной заявке имеют стандартные значения, общепринятые в научной и технической литературе.
Используемый здесь термин «процент гомологии двух последовательностей» эквивалентен термину «процент идентичности двух последовательностей». Идентичность последовательностей определяется на основании референсной последовательности. Алгоритмы для анализа последовательности известны в данной области, такие как BLAST, описанный в Altschul et al., J. Mol. Biol., 215, pp. 403-10 (1990). Для целей настоящего изобретения для определения уровня идентичности и сходства между нуклеотидными последовательностями и аминокислотными последовательностями может быть использовано сравнение нуклеотидных и аминокислотных последовательностей, производимое с помощью пакета программ Blast, предоставляемого National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/blast) с использованием содержащего разрывы выравнивания со стандартными параметрами. Процент идентичности двух последовательностей определяется числом положений идентичных аминокислот в этих двух последовательностях с учетом числа пробелов и длины каждого пробела, которые необходимо ввести для оптимального сопоставления двух последовательностей путем выравнивания. Процент идентичности равен числу идентичных аминокислот в данных положениях с учетом выравнивания последовательностей, разделенному на общее число положений и умноженному на 100.
Термин "специфически гибридизуется" относится к ассоциации между двумя одноцепочечными молекулами нуклеиновых кислот или в достаточной степени комплементарными последовательностями, что разрешает такую гибридизацию в предопределенных условиях, обычно использующихся в данной области.
Фраза "двунитевой разрыв, расположенный непосредственно перед нуклеотидной последовательностью РАМ" означает, что двунитевой разрыв в целевой последовательности ДНК будет произведен на расстоянии от 0 до 25 нуклеотидов перед нуклеотидной последовательностью РАМ.
Под экзогенной последовательностью ДНК, вводимой одновременно с направляющей РНК, следует понимать последовательность ДНК, подготовленную специально для специфической модификации двуцепочечной целевой ДНК в месте разрыва, определяемого специфичностью направляющей РНК. Подобной модификацией может быть, например, вставка или делеция определенных нуклеотидов в месте разрыва целевой ДНК. Экзогенной ДНК может служить как участок ДНК из другого организма, так и участок ДНК из того же организма, что и целевая ДНК.
Под белком, содержащим определенную аминокислотную последовательность следует понимать белок, имеющий аминокислотную последовательность, составленную из указанной аминокислотной последовательности и, возможно, других последовательностей, соединённых пептидными связями с указанной аминокислотной последовательностью. Примером других последовательностей может служить последовательность сигнала ядерной локализации (NLS), или другие последовательности, обеспечивающие повышенную функциональность для указанной аминокислотной последовательности.
Под экзогенной последовательностью ДНК, вводимой одновременно с направляющей РНК, следует понимать последовательность ДНК, подготовленную специально для специфической модификации двуцепочечной целевой ДНК в месте разрыва, определяемого специфичностью направляющей РНК. Подобной модификацией может быть, например, вставка или делеция определенных нуклеотидов в месте разрыва целевой ДНК. Экзогенной ДНК может служить как участок ДНК из другого организма, так и участок ДНК из того же организма, что и целевая ДНК.
Под эффективным количеством вводимых в клетку белка и РНК следует понимать такое количество белка и РНК, которое при попадании в указанную клетку будет способно образовать функциональный комплекс, то есть комплекс, который будет специфически связываться с целевой ДНК и производить в ней двунитевой разрыв в месте, определяемом направляющей РНК и РАМ последовательностью на ДНК. Эффективность этого процесса может быть оценена при помощи анализа целевой ДНК, выделенной из указанной клетки с помощью стандартных методов, известных специалистам.
Доставка белка и РНК в клетку может быть осуществлена различными способами. Например, белок может быть доставлен в виде ДНК-плазмиды, которая кодирует ген этого белка, как мРНК для трансляции этого белка в цитоплазме клетки, или как рибонуклеопротеидный комплекс, включающий этот белок и направляющую РНК. Доставка может быть осуществлена различными методами, известными специалистам.
Нуклеиновая кислота, кодирующая компоненты системы, может быть введена в клетку, непосредственно или опосредованно: за счет трансфекции или трансформации клеток известными специалистам способами, за счет использования рекомбинатного вируса, за счет манипуляций с клеткой, таких как микроинъекция ДНК и т.п.
Доставка рибонуклеинового комплекса, состоящего из нуклеазы и направляющих РНК и экзогенной ДНК (при необходимости) может осуществляться путем трансфекции комплексов в клетку или за счет механического введения комплекса внутрь клетки, например, микроинъекции.
Молекула нуклеиновой кислоты, кодирующая белок, который необходимо ввести в клетку, может быть интегрирована в хромосому или может представлять собой внехромосомно реплицирующуюся ДНК. В некоторых вариантах для обеспечения эффективной экспрессии гена белка с вводимой в клетку ДНК необходимо изменить последовательность этой ДНК в соответствии с типом клетки в целях оптимизации кодонов при экспрессии, обусловленное неравномерностью частот встречаемости синонимичных кодонов в кодирующих областях генома различных организмов. Оптимизация кодонов необходима для увеличения экспрессии в клетках животных, растений, грибов или микроорганизмов.
Для функционирования белка, имеющего последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, в эукариотической клетке необходимо, чтобы этот белок оказался в ядре этой клетки. Поэтому, в некоторых вариантах изобретения, для образования двунитевых разрывов в целевой ДНК используют белок, имеющий последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1, и который дополнительно модифицирован с одного или с обоих концов добавлением одного или нескольких сигналов ядерной локализации. Например, может быть использован сигнал ядерной локализации из вируса SV40. Для эффективной доставки в ядро сигнал ядерной локализации может быть отделен от основной последовательности белка спейсерной последовательностью, например, описанной в Shen B, et al. "Generation of gene-modified mice via Cas9/RNA-mediated gene targeting", Cell Res. 2013 May;23(5):720-3. Также, в других вариантах осуществления, может быть использован другой сигнал ядерной локализации, или альтернативный метод доставки указанного белка в ядро клетки.
Настоящее изобретение охватывает применение белка из организма Defluviimonas sp. 20V17, гомологичного ранее охарактеризованным белкам Cas9, для внесения двуцепочечных разрывов в молекулы ДНК в строго определенных положениях. Использование CRISPR нуклеаз для внесения направленных изменений в геном имеет ряд преимуществ. Во-первых, специфичность действия системы определяется последовательностью крРНК, что позволяет использовать один тип нуклеазы для всех локусов-мишеней. Во-вторых, методика позволяет доставить в клетку сразу несколько направляющих РНК, комплементарных разным генам-мишеням, что позволяет осуществлять единовременное изменение сразу нескольких генов.
Для биохимической характеризации белка Cas9 из бактерии Defluviimonas sp. 20V17 локус CRISPR, кодирующий основные компоненты системы (гены белков DfCas9, cas1, cas2, а также CRISPR кассету и направляющие РНК), был клонирован в малокопийный бактериальный вектор pACYC184. Эффекторному рибонуклеиновому комплексу, состоящему из Cas9 и дуплекса крРНК (crRNA) и тракрРНК (tracrRNA), для распознавания и последующего гидролиза ДНК помимо комплементарного соответствия спейсера крРНК и протоспейсера необходимо присутствие PAM (от англ. “PAM” - protospacer adjusted motif) на ДНК мишени (Mojica F. J. M. et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system //Microbiology. – 2009. – Т. 155. – №. 3. – С. 733-740). PAM представляет собой строго определенную последовательность из нескольких нуклеотидов, расположенных в системах типа II вплотную либо в нескольких нуклеотидах от 3’-конца протоспейсера на нецелевой цепи. При отсутствии PAM гидролиза связей в ДНК с образованием двунитевого разрыва не происходит. Необходимость присутствия PAM последовательности на мишени повышает специфичность узнавания, но в то же время накладывает ограничение в выборе целевых участков ДНК, в которые необходимо внести разрыв.
Для определения последовательностей направляющей РНК системы CRISPR-Cas9 было проведено РНК-секвенирование бактерий E.coli DH5alpha, несущих созданную конструкцию DfCas9_pacyc184. Секвенирование показало, что CRSIPR кассета системы активно транскрибируется, как и трейсерная РНК (Фиг. 1). Анализ последовательности crRNA и tracrRNA позволил предположить возможное формирование ими вторичных структур, распознаваемых DfCas9 нуклеазой.
Далее авторами было произведено определение PAM последовательности белка DfCas9 при помощи бактериального PAM скрининга. Для определения PAM последовательности белка DfCas9 клетки E.coli DH5alpha, несущие плазмиду DfCas9_pacyc184, были трансформированы библиотекой плазмид, содержащих последовательность спейсера 5’- TAGACCTTCGGGATCATGTCGATCATGATC-3’ CRISPR кассеты DfCas9 системы, фланкированной рандомизированной семибуквенной последовательностью с 5’ или 3’ конца. Плазмиды, несущие последовательность, соответствующую последовательности PAM системы DfCas9, подвергались деградации под действием функционирующей CRISPR-Cas системы, в то время как остальные плазмиды библиотеки эффективно трансформировались в клетки, придавая им устойчивость к антибиотику ампициллину. После трансформации и инкубации клеток на чашках с антибиотиком колонии смывались с поверхности агара и из них экстрагировали ДНК с помощью набора Qiagen Plasmid Purification Midi. Из выделенного пула плазмид амплифицировали при помощи ПЦР участки, включающие рандомизированную PAM последовательность, которые затем подвергали высокоэффективному секвенированию на платформе Illumina. Полученные прочтения анализировали: сравнивали эффективность трансформации плазмид с уникальными PAM, входящими в библиотеку, в клетки, несущие DfCas9_pacyc184 или в контрольные клетки, несущие пустой вектор pacyc184. Полученные результаты анализировали методами биоинформатики. В результате удалось выявить PAM системы DfCas9 – трехбуквенную последовательность 5'-NN(G/A)NA(C/T)N-3' (Фиг. 2).
Для разработки системы порезки ДНК in vivo и in vitro необходимо получить все компоненты, входящие в состав эффекторного DfCas9 комплекса: направляющие РНК и DfCas9 нуклеазу. Определение РНК-секвенированием последовательности направляющих РНК позволило синтезировать in vitro молекулы crRNA и tracrRNA. Синтез осуществлялся с помощью набора NEB HiScribe T7 RNA synthesis.
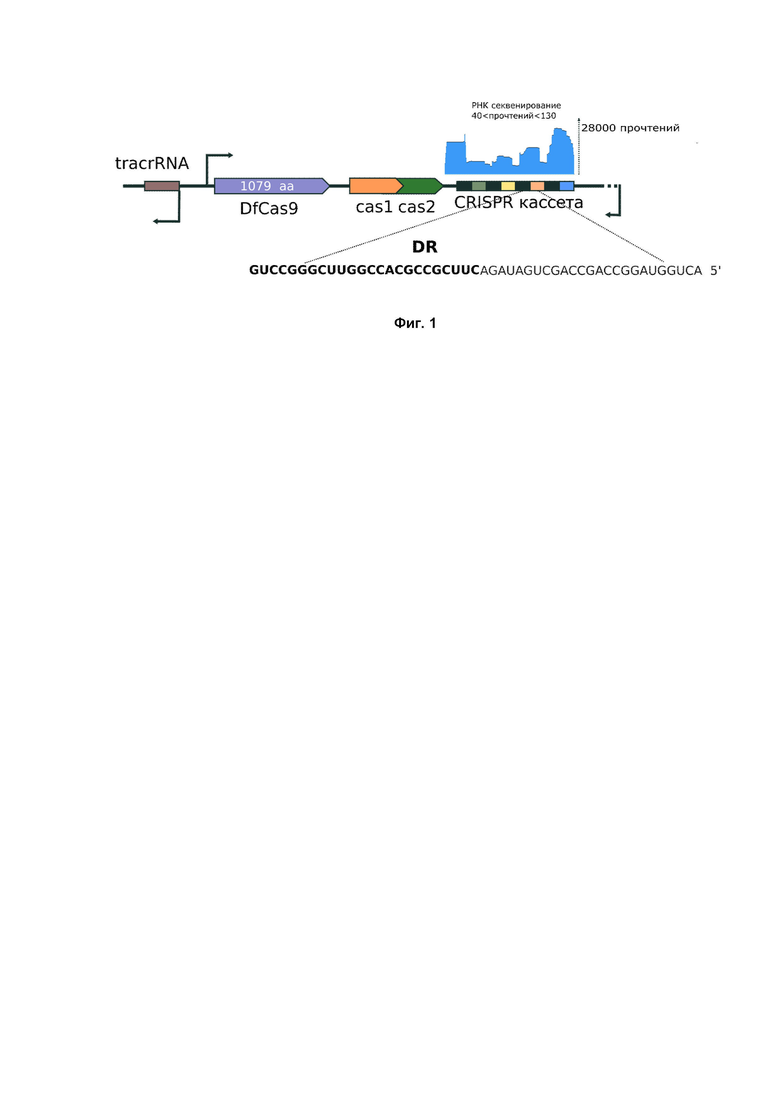
Для разрезания ДНК мишеней, ограниченных c 3’ конца NN(G/A)NA(C/T)N последовательностью использовали направляющие РНК следующей последовательности:
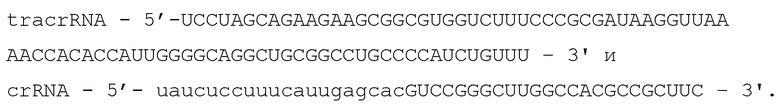
Первые 20 нуклеотидов, входящих в состав последовательности crRNA, комплементарно спариваются с соответствующей последовательностью на ДНК мишени и обеспечивают ее специфическое узнавание DfCas9 нуклеазой. В случае необходимости разрезания другой ДНК мишени, эта 20-буквенная спейсерная последовательность изменяется (Фиг. 3).
Для получения рекомбинантного белка DfCas9 его ген был заклонирован в плазмиду pET21a. Клетки E.coli Rosetta были трансформированы полученной плазмидой DfCas9_pET21a. Клетки, несущие плазмиду, наращивали до оптической плотности OD600=0.6, далее проводилась индукция экспрессии гена DfCas9 при помощи добавления IPTG до концентрации 1mM. Клетки инкубировали в течение 4 часов при 25 °С, после чего проводили их лизис. Очистку рекомбинантного белка проводили двумя этапами: с помощью аффинной хроматографии (Ni-NTA) и разделением белков по размерам на колонке Superdex 200. Полученный белок концентрировали c помощью фильтров Amicon с размером пор 30 кДа. После этого белок замораживали на минус 80 °С и использовали для проведения in vitro реакций.
В качестве ДНК мишени использовали двунитевой фрагмент ДНК длиной 378 пар нуклеотидов (п.н.), несущий последовательность протоспейсера на расстоянии около 30 п.н. c 3’ конца, ограниченную соответствующей PAM последовательностью, определенной в экспериментах на бактериях: NN(G/A)NA(C/T)N.

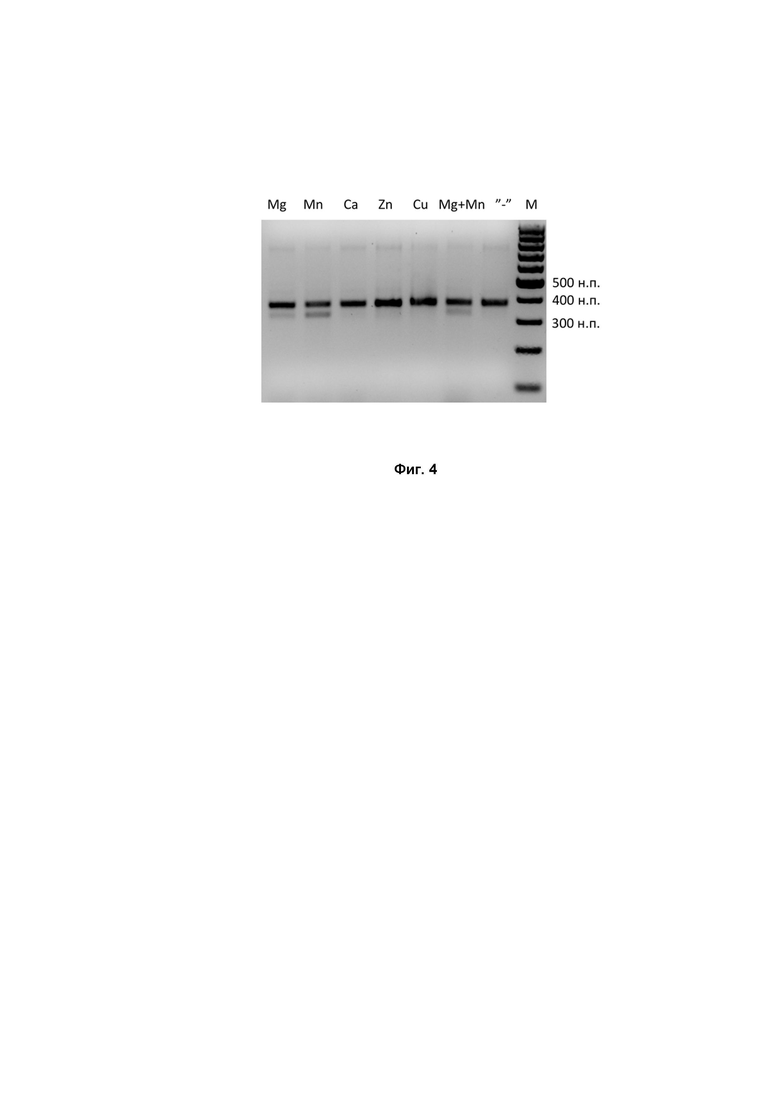
Нуклеазные домены Cas9 белков для своей активности требуют присутствия дивалентных ионов. В связи с этим проведение in vitro реакций по разрезанию ДНК-мишени проводили в присутствии солей магния, марганца, кальция и цинка. In vitro реакцию порезки ДНК-мишени проводили в следующих условиях:
1x буфер Tris-HCl, pH = 7.9 (при 25 °С)
400 нM DfCas9
20 нM ДНК-мишень
2 мкM crRNA
2 мкM tracrRNA
1 мM соли соответствующего иона
Общий объем реакции был 20 мкл, продолжительность реакции 30 минут, температура инкубации 37 °С.
Продукты реакции наносили на 1.5% агарозный гель и подвергали электрофорезу. В случае успешной порезки мишени ДНК фрагмент распадался на две части, одна из которых (длиной около 325 н.п.) образовывала хорошо различимую на геле полосу (Фиг. 4). Результаты проведенных экспериментов показали, что белок DfCas9 требует для своей работы ионы марганца, а не магния, что является необычным свойством для охарактеризованных на сегодняшний день Cas9 нуклеаз.
Для подтверждения значимости различных PAM последовательности DfCas9 были проведены эксперименты по разрезанию in vitro ДНК мишеней, аналогичным использованным ранее, но содержащим точечные (однонуклеотидные) мутации в PAM последовательности 5’- AAAAACG -3’ (Фиг. 5).
Результаты эксперимента подтвердили, что фермент DfCas9 способен вносить двунитевые разрывы в ДНК последовательности, ограниченные PAM последовательностью NN(G/A)NA(C/T)N с 3’ конца. Замены в 3, 5 и 6 позициях PAM критичны для эффекторного комплекса DfCas9 и препятствуют эффективному разрезанию целевой ДНК.
Кроме того, были проведены эксперименты по поиску оптимальной температуры работы DfCas9: она оказалась равной 35-37°С, что открывает перед DfCas9 перспективы использования в клетках человека.
Нижеследующие примеры осуществления способа приведены в целях раскрытия характеристик настоящего изобретения и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения.
Пример 1. Тестирование активности белка DfCas9 в разрезании различных ДНК мишеней.
Для того, чтобы проверить способность DfCas9 узнавать различные последовательности ДНК, фланкированные мотивом 5'-NN(G/A)NA(C/T)N-3', были проведены эксперименты по разрезанию in vitro ДНК-мишеней из последовательности гена grin2b человека (см. таблицу 1).
Таблица 1. ДНК-мишени, выделенные из гена grin2b человека.
Реакции in vitro разрезания ДНК проводили в условиях, аналогичных предыдущим экспериментам. Для каждой из целевых последовательностей была синтезирована направляющая crRNA. В качестве ДНК-мишени использовали фрагмент гена человека grin2b размером около 760 н.п.: 5- ttgtctctgcctgtagctgccaatgactatagcaatagcaccttttattgccttgttcaaggatttctgaggcttttgaaagtttcattttctctcattctgcagagcaaataccagagataagagagtaggctggtagatggagttgggtttggtgctcaatgaaaggagataaggtccttgaattgcagtatctagcctcttctaagacaggttacgtgatgtagatcctattttaacatgctctttctttgtgtttgcagggagtcgacgagttgaagatgaagcccagagcggagtgctgttctcccaagttctggttggtgttggccgtcctggccgtgtcaggcagcagagctcgttctcagaagagcccccccagcattggcattgctgtcatcctcgtgggcacttccgacgaggtggccatcaaggatgcccacgagaaagatgatttccaccatctctccgtggtaccccggg - 3’.
В качестве ДНК мишеней были выбраны последовательности, фланкированные с 3’ конца участками ДНК, соответствующими PAM последовательности 3'-NN(G/A)NA(C/T)N-5'. Из шести выбранных последовательностей только две были эффективно разрезаны белком DfCas9: порезанные эффекторным комплексом ДНК-фрагменты соответствующего размера видны на геле (Фиг. 6). Избирательность в разрезании различных мишеней может объясняться отличающейся эффективностью узнавания DfCas9 различных вторичных структур ДНК, или другими причинами. Избирательность DfCas9 в разрезании различных мишеней может повысить специфичность этого белка при разрезании им геномов эукариотических клеток.
Таким образом, DfCas9 в комплексе с направляющими РНК является новым инструментом для разрезания двунитевой ДНК, ограниченной последовательностью 3'-NN(G/A)NA(C/T)N-5' с характерным температурным диапазоном работы от 35 °С до 37 °С.
Пример 2. Влияние концентрации ионов Mn2+ на функциональность нуклеазы.
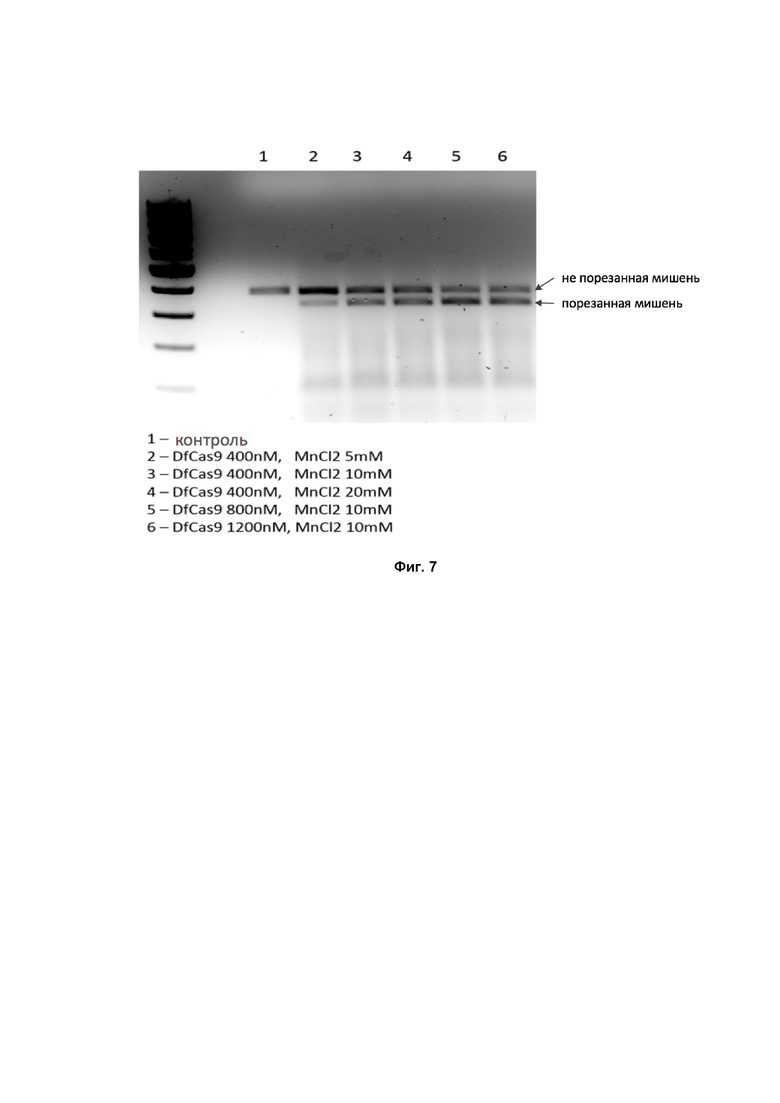
Для проверки влияния ионов Mn2+ на нуклеазную активность DfCas9 были проведены in vitro эксперименты разрезания двунитевого ДНК фрагмента, содержащего целевую ДНК последовательность, ограниченную PAM последовательностью AAAAAC, согласующуюся с консенсусом 3’- NN(G/A)NA(C/T) -5’. Для проведения реакции использовали буфер 1x CutSmart buffer (NEB), концентрацию целевой ДНК - 20 нM и концентрацию trRNA/crRNA – 2 мкM. 
На Фиг. 7 показано, что увеличение концентрации MnСl2 с 5 мM до 10 мM приводит к более эффективному разрезанию ДНК-мишени, в то время как дальнейшее увеличение концентрации дивалентных ионов не сказывается на эффективности протекания реакции. Таким образом, для эффективной работы DfCas9 необходимо присутствие ионов марганца в концентрации равной 10 или более мM.
Пример 3. Использование гибридной направляющей РНК для разрезания ДНК мишени.
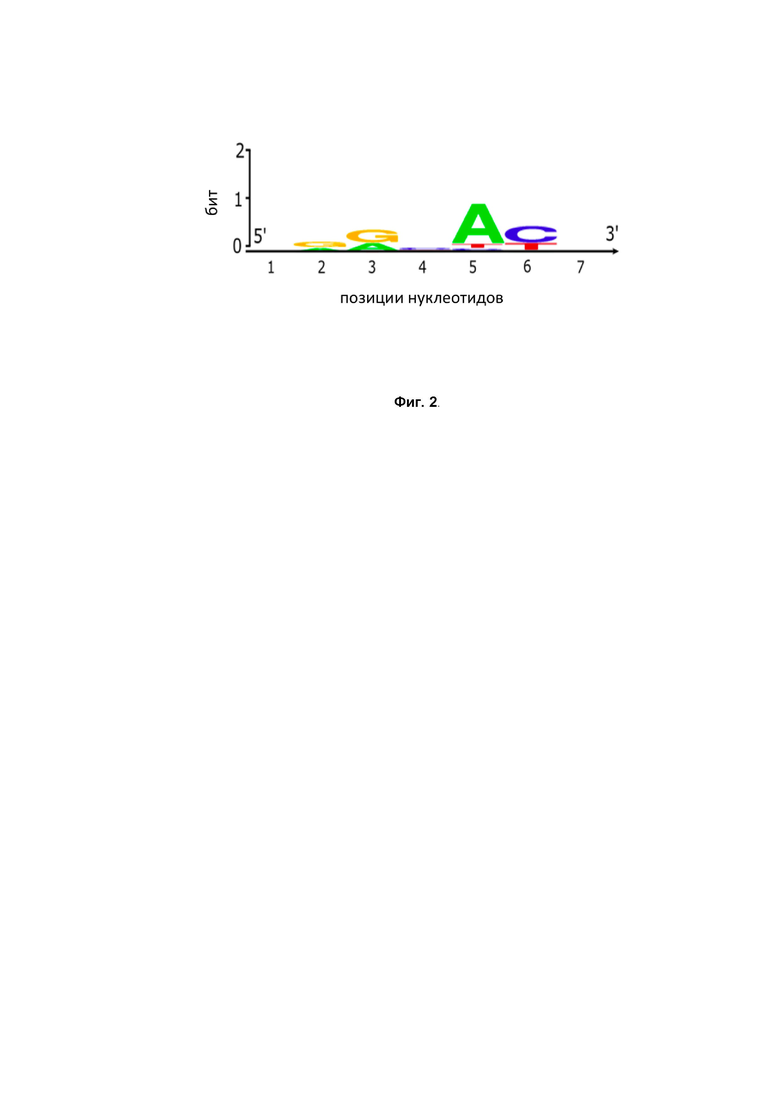
sgRNA - форма направляющих РНК, которая представляет собой слитые воедино tracrRNA (трейсерная РНК) и crRNA. Для подбора оптимальной sgRNA были сконструированы три варианта этой последовательности, отличающиеся длиной tracrRNA – crRNA дуплекса. РНК синтезировали in vitro и проводили с ними эксперименты по разрезанию ДНК -мишени (Фиг. 8 – показаны реакции разрезания DfCas9 ДНК с использованием различных вариантов sgRNA).
Подобранная sgRNA3 оказалась так же эффективна, как и нативные последовательности tracrRNA и crRNA: порезка более 50% ДНК-мишени.
Этот вариант sgRNA может быть использован для разрезания любой другой целевой ДНК при изменении последовательности, непосредственно спаривающейся с ДНК -мишенью.
В качестве гибридных РНК были использованы следующие РНК последовательности: 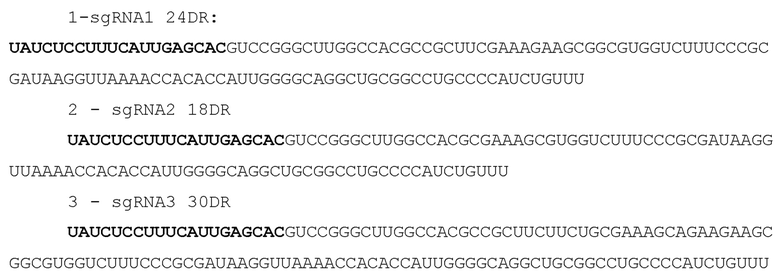
4 – контроль (без РНК)
5 – положительный контроль (порезка мишени с помощью crRNA+trRNA)
Жирным шрифтом обозначена 20-нуклеотидная последовательность, обеспечивающая спаривание с ДНК -мишенью (вариабельная часть sgRNA). В эксперименте делали контрольную пробу без РНК, а также положительный контроль - порезка мишени с помощью crRNA+trRNA.
Реакцию проводили в следующих условиях: концентрация ДНК последовательности, содержащей PAM (AAAACG) – 20 нM, концентрация белка - 400 нM, концентрация РНК - 2 мкM; время инкубирования 30 минут, температура инкубирования 37 °С.
Данный вариант гибридной РНК может быть использован для разрезания любой другой целевой ДНК при изменении последовательности, непосредственно спаривающейся с ДНК -мишенью.
Пример 4. Белки Cas9 из близкородственных организмов, относящихся к Defluviimonas sp.
На сегодняшний день в Defluviimonas не охарактеризовано ни одного фермента системы CRISPR-Cas9. Сравнимые по размерам белки Cas9 из Staphylococcus aureus идентичен DfCas9 на 21 %, Cas9 из Campylobacter jejuni на 28% (степень идентичности была рассчитана по программе BLASTp, default parameters).
Таким образом, белок DfCas9 существенно отличается по аминокислотной последовательности от других Cas9 белков, изученных на сегодняшний день.
Специалисту в области генетической инженерии очевидно, что полученный и охарактеризованный Заявителем вариант последовательности белка DfCas9 может быть изменен без изменения функции самого белка (например, направленным мутагенезом аминокислотных остатков, напрямую не влияющих на функциональную активность (Sambrook et al., Molecular Cloning: A Laboratory Manual, (1989), CSH Press, pp. 15.3-15.108)). В частности, специалисту известно, что могут быть изменены неконсервативные аминокислотные остатки, не затрагивающие остатки, определяющие функциональность белка (определяющие его функцию или структуру). Примерами таких изменений могут служить замены неконсервативных аминокислотных остатков на гомологичные. Некоторые из участков, содержащих неконсервативные аминокислотные остатки, приведены на Фиг. 9. В некоторых вариантах осуществления изобретения возможно использование белка, содержащего аминокислотную последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1 и имеет отличия по сравнению с SEQ ID NO: 1 только в неконсервативных аминокислотных остатках, для образования двунитевого разрыва в молекуле ДНК, расположенного непосредственно перед нуклеотидной последовательностью 5’-NN(G/A)NA(C/T)N-3’ в указанной молекуле ДНК. Гомологичные белки могут быть получены путем мутагенеза (например, сайт-направленного или ПЦР-опосредуемого мутагенеза) соответствующих молекул нуклеиновых кислот с последующим тестированием кодируемого модифицированного белка Cas9 на сохранение его функций в соответствии с описанными здесь функциональными анализами.
Пример 5. Описанная в настоящем изобретении система DfCas9 в комплексе с направляющими РНК может быть использована для изменения последовательности геномной ДНК многоклеточного организма, в том числе эукариотического. Для введения система DfCas9 в комплексе с направляющими РНК в клетки этого организма (во все клетки или в часть клеток) могут быть применены различные подходы, известные специалистам. Например, методы доставки CRISPR-Cas9 систем в клетки организмов раскрыты в источниках (Liu C et al., Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J Control Release. 2017 Nov 28;266:17-26; Lino CA et al., Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 2018 Nov;25(1):1234-1257), и в источниках, раскрытых внутри этих источников.
Для эффективной экспрессии нуклеазы DfCas9 в эукариотических клетках будет желательно провести оптимизацию кодонов для аминокислотной последовательности белка DfCas9 методами, известными специалистам (например, IDT codon optimization tool).
Для эффективной работы нуклеазы DfCas9 в эукариотических клетках необходимо обеспечить импорт этого белка внутрь ядра эукариотической клетки. Для этого можно использовать сигнал ядерной локализации из Т-антигена вируса SV40 (Lanford et al., Cell, 1986, 46: 575–582), соединённый с последовательностью DfCas9 с помощью спейсерной последовательности, описанной в Shen B, et al. "Generation of gene-modified mice via Cas9/RNA-mediated gene targeting", Cell Res. 2013 May;23(5):720-3 или без нее. Таким образом, полная аминокислотная последовательность нуклеазы транспортируемой внутрь ядра эукариотической клетки будет представлять собой следующую последовательность: MAPKKKRKVGIHGVPAA-DfCas9-KRPAATKKAGQAKKKK (далее DfCas9 NLS). Для доставки белка с приведенной выше аминокислотной последовательностью, могут быть использованы по меньшей мере два подхода.
Доставка в виде гена осуществляется путем создания плазмиды, несущей ген DfCas9 NLS под регуляцией промотора (например, CMV промотора) и последовательности, кодирующей направляющие РНК под регуляцией U6 промотора. В качестве ДНК- мишеней используются ДНК последовательности фланкированные 5’- NN(G/A)NA(C/T)-3’, например, последовательности гена grin2b человека:
5’-ttgcagtatctagcctcttc-3’
Таким образом, кассета для экспрессии крРНК выглядит следующим образом:

Жирным шрифтом выделена последовательность U6 промотора, далее последовательность, необходимая для узнавания целевой ДНК, а последовательность прямого повтора выделена заглавными буквами.
Кассета для экспрессии трейсерной РНК выглядит следующим образом:

Жирным шрифтом выделена последовательность U6 промотора, далее последовательность, кодирующая трейсерную РНК.
Плазмидную ДНК очищают и трансфицируют в клетки человека HEK293 c помощью реагента Lipofectamine 2000 (Thermo Fisher Scientific). Клетки инкубируют в течение 72 часов, после чего из них выделяется геномная ДНК с помощью колонок для очистки геномной ДНК (Thermo Fisher Scientific). Целевой ДНК сайт анализируется с помощью секвенирования на платформе Illumina с целью определения числа вставок-делеций в ДНК, происходящих в целевом сайте по причине направленного двунитевого разрыва и последующей его репарации.
Для амплификации целевых фрагментов используют праймеры, фланкирующие предположительное место внесения разрыва, например для указанных выше сайтов гена grin2b:
5’-GACTATAGCAATAGCAC-3’
5’TCAACTCGTCGACTCCCTG-3’
После амплификации пробы готовятся по протоколу реагента Ultra II DNA Library Prep Kit for Illumina (NEB) для подготовки образцов к высокопроизводительному секвенированию. Затем проводится секвенирование на платформе Illumina 300cycles, прямое прочтение. Результаты секвенирования анализируются биоинформатическими методами. В качестве детекции разрезания принимается вставка или делеция нескольких нуклеотидов в целевой последовательности ДНК.
Доставка в виде рибонуклеинового комплекса осуществляется путем инкубации рекомбинантной формы DfCas9 NLS c направляющими РНК в CutSmart буфере (NEB). Рекомбинантный белок получают из бактериальных клеток-продуцентов, очищая его с помощью аффинной хроматографии (NiNTA, Qiagen) разделением по размеру (Superdex 200).
Белок смешивают с РНК в соотношении 1:2:2 (DfCas9 NLS : crRNA :tracrRNA), инкубируют в течение 10 минут на комнатной температуре, затем смесь трансфицируют в клетки.
Далее проводится анализ экстрагированной из них ДНК на предмет вставок-делеций в целевом ДНК сайте (как описано выше).
Охарактеризованная в настоящем изобретении DfCas9 нуклеаза из глубоководной бактерии Defluviimonas sp. 20V17 имеет ряд преимуществ относительно ранее охарактеризованных Cas9 белков. Например, DfCas9 обладает относительно простым, трехбуквенным, отличным от других известных PAM Cas нуклеаз, необходимый для функционирования системы. Большинство известных на сегодняшний день Cas нуклеаз, способных вносить двунитевые разрывы в ДНК, имеют многобуквенные сложные PAM мотивы, ограничивающие выбор последовательностей, пригодных для разрезания. Среди изученных Cas нуклеаз, распознающих короткие PAM, только DfCas9 может распознавать последовательности, ограниченные 5'-NN(G/A)NA(C/T)N-3' нуклеотидами.
Второе преимущество DfCas9 – относительно малый размер белка (1079 а.о.). На сегодняшний день это один из немногих изученных малоразмерных белков, обладающих простой PAM последовательностью и активных при 37 оС. Необычным свойством DfCas9 является необходимость присутствия ионов марганца для успешного разрезания целевых ДНК-мишеней. Это свойство может быть использовано для регуляции работы Cas9 комплекса.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные случаи приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть, понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Средство разрезания ДНК на основе Cas9 белка из биотехнологически значимой бактерии Clostridium cellulolyticum | 2018 |
|
RU2712497C1 |
| СРЕДСТВО РАЗРЕЗАНИЯ ДНК НА ОСНОВЕ CAS9 БЕЛКА ИЗ БАКТЕРИИ DEMEQUINA SEDIMINICOLA | 2019 |
|
RU2722933C1 |
| ПРИМЕНЕНИЕ CAS9 БЕЛКА ИЗ БАКТЕРИИ PASTEURELLA PNEUMOTROPICA ДЛЯ МОДИФИКАЦИИ ГЕНОМНОЙ ДНК В КЛЕТКАХ | 2019 |
|
RU2724470C1 |
| СРЕДСТВО РАЗРЕЗАНИЯ ДНК НА ОСНОВЕ CAS9 БЕЛКА ИЗ БАКТЕРИИ PASTEURELLA PNEUMOTROPICA | 2019 |
|
RU2722934C1 |
| Средство разрезания ДНК на основе Cas9 белка из бактерии Streptococcus uberis NCTC3858 | 2022 |
|
RU2788197C1 |
| Средство разрезания ДНК на основе Cas9 белка из бактерии Capnocytophaga ochracea | 2021 |
|
RU2778156C1 |
| Средство разрезания ДНК на основе ScCas12a белка из бактерии Sedimentisphaera cyanobacteriorum | 2022 |
|
RU2791447C1 |
| Средство разрезания двунитевой ДНК с помощью Cas12d белка из Katanobacteria и гибридной РНК, полученной путем слияния направляющей CRISPR РНК и scout РНК | 2020 |
|
RU2771626C1 |
| СИСТЕМА РЕДАКТИРОВАНИЯ ГЕНОМНОЙ ДНК ЭУКАРИОТИЧЕСКОЙ КЛЕТКИ НА ОСНОВЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ, КОДИРУЮЩЕЙ БЕЛОК SUCAS9NLS | 2022 |
|
RU2804422C1 |
| Средство обнаружения нуклеиновых кислот на основе ScCas12a белка из бактерии Sedimentisphaera cyanobacteriorum | 2023 |
|
RU2820345C1 |




Изобретение относится к биотехнологии и описывает новую бактериальную нуклеазу системы CRISPR-Cas9 из бактерии Defluviimonas sp. 20V17, а также ее применение для образования строго специфичных двунитевых разрывов в молекуле ДНК. Данная нуклеаза обладает необычными свойствами и может быть использована в качестве инструмента для внесения изменений в строго определенных местах в последовательности геномной ДНК одноклеточных или многоклеточных организмов. Изобретение позволяет повысить универсальность доступных систем CRISPR-Cas9, что позволит использовать нуклеазы Cas9 из различных организмов для разрезания геномной или плазмидной ДНК в большем количестве специфических сайтов и при различных условиях. 2 н. и 3 з.п. ф-лы, 9 ил., 5 пр.
1. Применение белка, содержащего аминокислотную последовательность SEQ ID NO: 1 или содержащего аминокислотную последовательность, которая по меньшей мере на 95% идентична аминокислотной последовательности SEQ ID NO: 1 и имеет отличия по сравнению с SEQ ID NO: 1 только в неконсервативных аминокислотных остатках, для образования двунитевого разрыва в молекуле ДНК, расположенного непосредственно перед нуклеотидной последовательностью 5'-NN(G/A)NA(C/T)N-3' в указанной молекуле ДНК.
2. Применение по п. 1, характеризующееся тем, что образование двунитевого разрыва в молекуле ДНК происходит при температуре от 35 до 37°С и в присутствии ионов Mn2+.
3. Применение белка по п. 1, где белок содержит аминокислотную последовательность SEQ ID NO: 1.
4. Способ образования двунитевого разрыва в последовательности геномной ДНК одноклеточного или многоклеточного организма, непосредственно примыкающей к последовательности 5'-NN(G/A)NA(C/T)N-3', включающий введение в по меньшей мере одну клетку этого организма эффективного количества: а) белка, содержащего аминокислотную последовательность SEQ ID NO: 1, или нуклеиновой кислоты, кодирующей белок, содержащий аминокислотную последовательность SEQ ID NO: 1, и б) направляющей РНК, содержащей последовательность, образующую дуплекс с нуклеотидной последовательностью участка геномной ДНК организма, непосредственно примыкающей к нуклеотидной последовательности 5'-NN(G/A)NA(C/T)N-3', и взаимодействующей с указанным белком после образования дуплекса, или последовательности ДНК, кодирующей указанную направляющую РНК;
при этом взаимодействие указанного белка с направляющей РНК и нуклеотидной последовательностью 5'-NN(G/A)NA(C/T)N-3' приводит к образованию двунитевого разрыва в последовательности геномной ДНК, непосредственно примыкающей к последовательности 5'-NN(G/A)NA(C/T)N-3' .
5. Способ по п. 4, дополнительно включающий введение экзогенной последовательности ДНК одновременно с направляющей РНК.
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ МОДИФИКАЦИИ ЗАДАННОЙ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВОЙ КИСЛОТЫ-МИШЕНИ | 2012 |
|
RU2663354C2 |
| WO 2014093661 A9, 19.06.2014. | |||

