ОБЛАСТЬ ТЕХНИКИ
[1] Настоящее изобретение относится к способу очистки сырого производного бензопирана. В частности, настоящее изобретение относится к способу очистки производного бензопирана, включающему преобразование аморфного сырого производного бензопирана в его кристаллическую форму. Кроме того, настоящее изобретение относится к новой кристаллической форме производного бензопирана и способам его получения.
УРОВЕНЬ ТЕХНИКИ
[2] Производное бензопирана с формулой 1 с химическим наименованием (2R, 3R, 4S)-6-амино-4-[N-(4-хлорфенил)-N-(1Н-имидазол-2-илметил)амино]- 3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2Н-1-бензопиран известно как соединение, обладающее терапевтическим действием при раке, ревматоидном артрите и т.д. (патент Кореи № 10-0492252). Кроме того, соединение с формулой 1 может быть приготовлено в виде глазных капель на основе низкомолекулярного материала; и с пользой применено для профилактики и лечения дегенеративных процессов желтого пятна, без инъекции напрямую в пораженную область, как в случае с терапией с инъекцией антител (патентная публикация Кореи № 10-2012-0112162).
[3] Формула 1
[4] 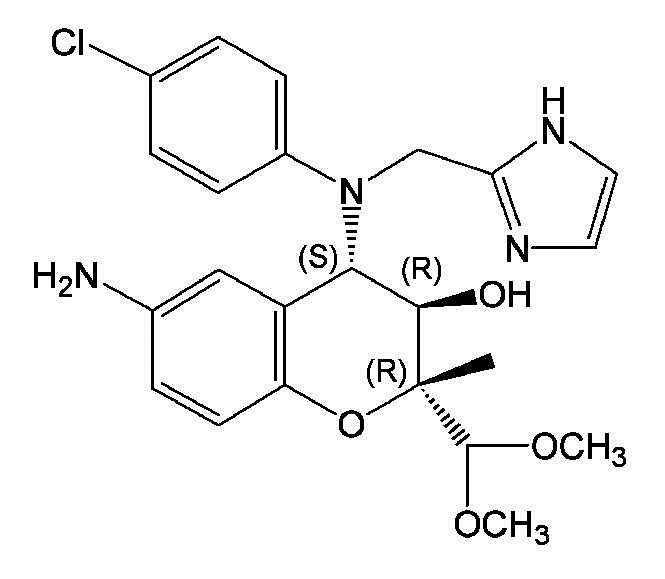
[5] Способ получения соединения с формулой 1 раскрыт в патенте Кореи № 10-0492252. В частности, как показано на следующей схеме реакции 1, способ получения соединения с формулой 1 включает преобразование олефинового соединения с формулой 4а в эпоксидное соединение с формулой 3а; реакцию эпоксидного соединения с формулой 3a с (4-хлорфенил)(1H-имидазол-2-илметил)амином с получением соединения с формулой 2a; и восстановление соединения с формулой 2a с получением соединения с формулой 1.
[6] Схема реакции 1
[7] 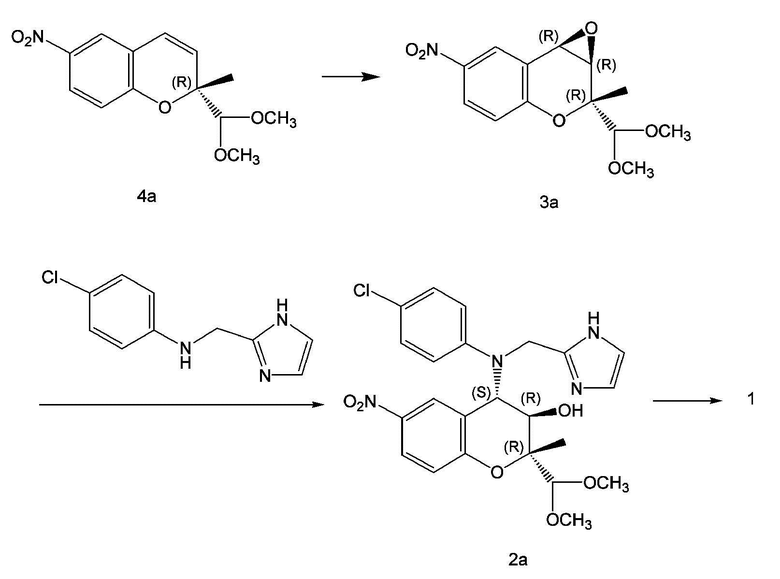
[8] Соединение с формулой 1, полученное указанным способом, выделяют путем фильтрования реакционной смеси, полученной в результате восстановления, чтобы удалить твердое вещество, увеличивая концентрацию фильтрата и затем очищая полученный остаток с использованием колоночной хроматографии на силикагеле.
[9] Авторы настоящего изобретения обнаружили, что в результате анализа соединения с формулой 1, полученного в соответствии со способом, раскрытым в патенте Кореи № 10-0492252, полученный продукт обладает низкой чистотой (менее 97 вес.% в безводной форме) и высоким содержанием воды (более 1 вес.%). В частности, соединение с формулой 1, полученное в соответствии со способом, раскрытым в патенте Кореи №10-0492252, содержит остаточные примеси (например, органические примеси, неорганические примеси, остаточные растворители и т.д.), возникающие в процессе получения или из быстро разлагающихся продуктов распада, и, таким образом, его чистота не находится в подходящем диапазоне (например, 99,0% или более) в соответствии с Положением о разрешении использования лекарственных средств Управления по контролю качества продуктов и лекарств, что вызывает проблему, состоящую в том, что полученное вещество не может быть напрямую использовано в качестве активного фармацевтического ингредиента. Кроме того, соединение с формулой 1, полученное в соответствии со способом, раскрытым в патенте Кореи № 10-0492252, демонстрирует очень высокую гигроскопичность. Например, содержание воды в нем повышается до 2,30 вес.% в течение 1 суток в условиях ускоренных испытаний; и, следовательно, требуется строгий контроль. Кроме того, сам продукт сразу же после получения уже содержит большое количество воды, что делает его непригодным для использования в качестве активного фармацевтического ингредиента.
CУЩНОСТЬ ИЗОБРЕТЕНИЯ
Техническая задача
[10] Авторы настоящего изобретения провели различные исследования для разработки способа, позволяющего принципиально решить проблемы низкой чистоты и высокого содержания воды (а также высокой гигроскопичности) производного бензопирана (т.е. сырого соединения с формулой 1), полученного в соответствии с предшествующим уровнем техники. Неожиданно было обнаружено, что продукт, полученный в соответствии со способом из предшествующего уровня техники (патент Кореи № 10-0492252), получен в аморфной форме. Кроме того, было обнаружено, что при преобразовании аморфного продукта в кристаллическую форму (например, кристаллическую форму А со специфической дебаеграммой ПРД (XRPD pattern), специфической термограммой ДСК (DSC thermogram) или специфической термограммой ТГА (TGA thermogram)), чистота продукта может быть значительно повышена, а остаточное содержание воды в полученной кристаллической форме может быть заметно уменьшено до 0,2 вес.% или менее. Кроме того, было обнаружено, что полученная кристаллическая форма по существу не проявляет свойства гигроскопичности, что может принципиально решить проблемы гигроскопичности аморфной формы.
[11] Таким образом, цель настоящего изобретения состоит в создании способа для очистки соединения с формулой 1, содержащего преобразование сырого производного бензопирана (т.е. соединения с формулой 1) в его кристаллическую форму.
[12] Кроме того, еще одна цель настоящего изобретения состоит в получении кристаллической формы соединения с формулой 1.
[13] Кроме того, еще одна цель настоящего изобретения состоит в предложении способа получения кристаллической формы соединения с формулой 1.
Техническое решение
[14] В качестве одного из объектов настоящего изобретения предложен способ очистки соединения с формулой 1, включающий преобразование сырого производного бензопирана с формулой 1 в его кристаллическую форму.
[15] Формула 1
[16]
[17] В качестве другого объекта настоящего изобретения предложена кристаллическая форма соединения с формулой 1. В одном из вариантов осуществления изобретения кристаллическая форма соединения с формулой 1 может представлять собой кристаллическую форму А с дебаеграммой ПРД с 2θ ± 0,2°2θ пиками на 12,27, 12,65, 16,07, 19,06 и 26,48.
[18] В качестве еще одного объекта настоящего изобретения предложен способ получения кристаллической формы соединения с формулой 1, включающий растворение аморфного соединения с формулой 1 в органическом растворителе для получения раствора; перемешивание, дистилляцию или охлаждение раствора с образованием твердого вещества или дистиллята, последующее охлаждение раствора с образованием твердого вещества; и выделение твердого вещества.
[19] В качестве еще одного объекта настоящего изобретения предложен способ получения кристаллической формы соединения с формулой 1, включающий растворение аморфного соединения с формулой 1 в органическом растворителе для получения раствора; добавление раствора в антирастворитель для получения твердого вещества или добавление антирастворителя в раствор для получения твердого вещества и выделение твердого вещества.
[20] В качестве еще одного объекта настоящего изобретения предложен способ получения кристаллической формы соединения с формулой 1, включающий растворение аморфного соединения с формулой 1 в воде посредством добавления в нее кислоты для получения раствора; добавление основания в раствор для получения твердого вещества и выделение твердого вещества.
ТЕХНИЧЕСКИЕ РЕЗУЛЬТАТЫ ИЗОБРЕТЕНИЯ
[21] В соответствии с настоящим изобретением обнаружено, что соединение с формулой 1, полученное способом из предшествующего уровня техники (т.е. способ, раскрытый в патенте Кореи № 10-0492252), получено в аморфной форме с низкой чистотой и высоким содержанием воды (а также с высокой гигроскопичностью). Способ очистки в соответствии с настоящим изобретением может обеспечить получение соединения с формулой 1 в кристаллической форме с высокой чистотой и уменьшенным содержанием воды. Способ очистки обладает преимуществом, состоящим в том, что этот способ может быть с легкостью применен для промышленного массового производства. Кроме того, кристаллическая форма (например, кристаллическая форма А соединения с формулой 1) со специфической дебаеграммой ПРД, специфической термограммой ДСК или специфической термограммой ТГА обладает превосходными начальными свойствами (т.е. высокая чистота и уменьшенное содержание воды). В частности, кристаллическая форма А соединения с формулой 1 по существу не демонстрирует гигроскопичность; и может сохранять стабильную форму, без изменения кристаллизации даже при нагревании и в условиях ускоренных испытаний. Следовательно, кристаллическая форма А соединения с формулой 1 обладает свойствами, подходящими для составления рецептур терапевтических лекарственных форм; и, таким образом, обеспечивает преимущества, позволяя получать эффективные рецептуры без потери активного фармацевтического ингредиента и с длительным сроком хранения.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
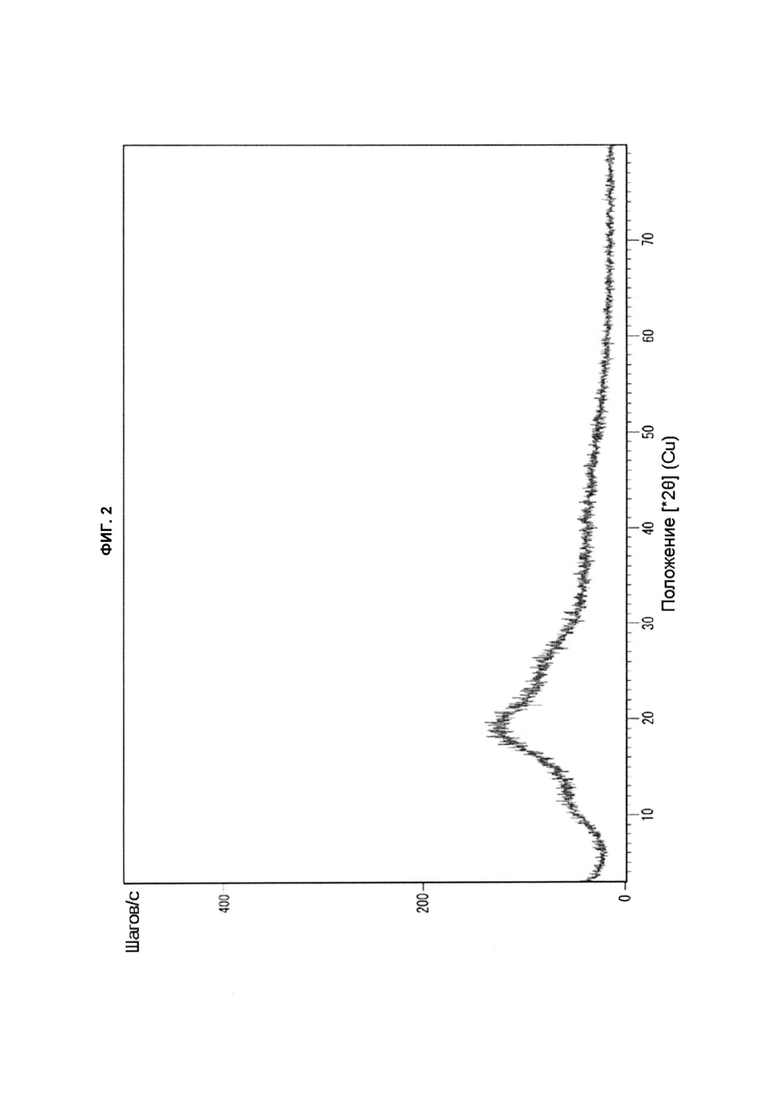
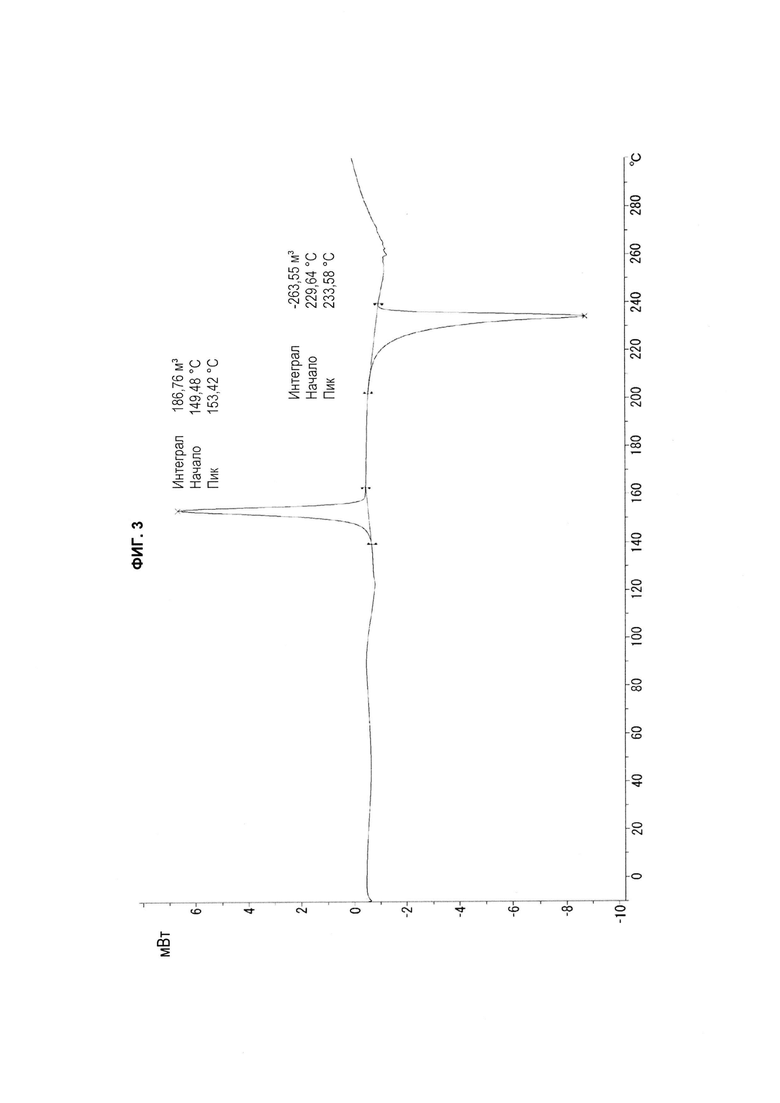
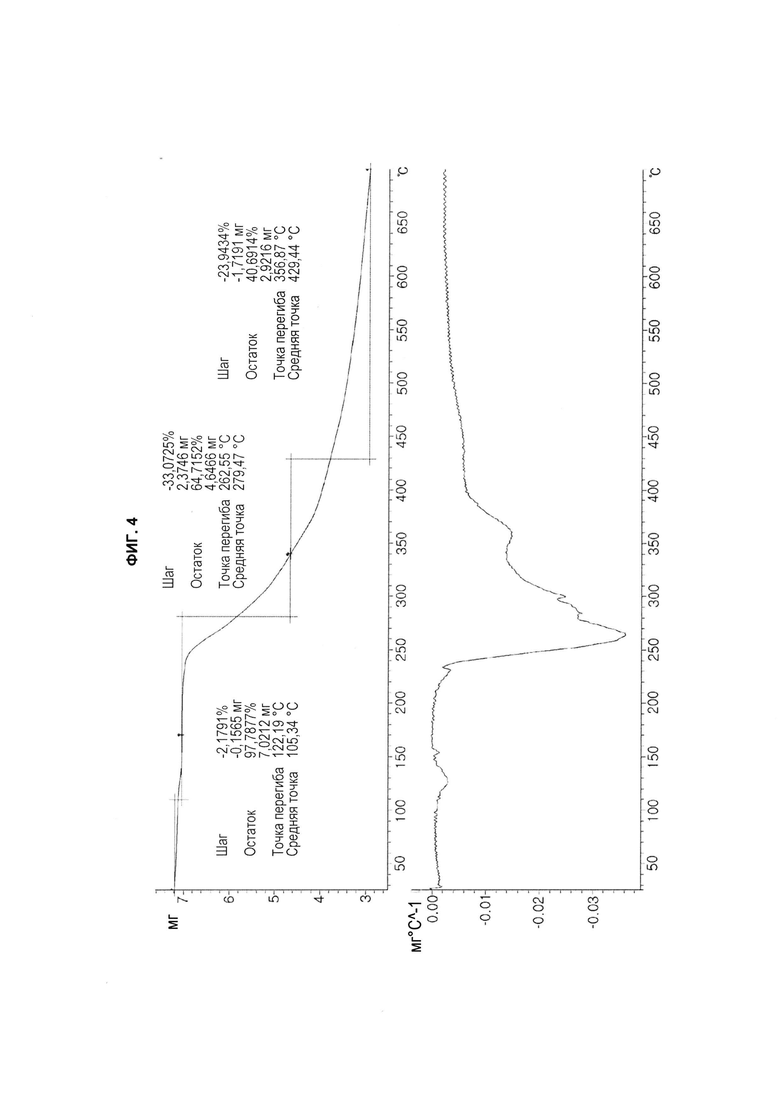
[22] На ФИГ. 1-4 показаны соответственно 1H спектр ЯМР (ФИГ. 1), спектр ПРД (ФИГ. 2), термограмма ДСК (ФИГ. 3) и термограмма ТГА (ФИГ. 4) производного бензопирана (т.е. соединение с формулой 1), полученного в соответствии со способом, раскрытым в патенте Кореи № 10-0492252.
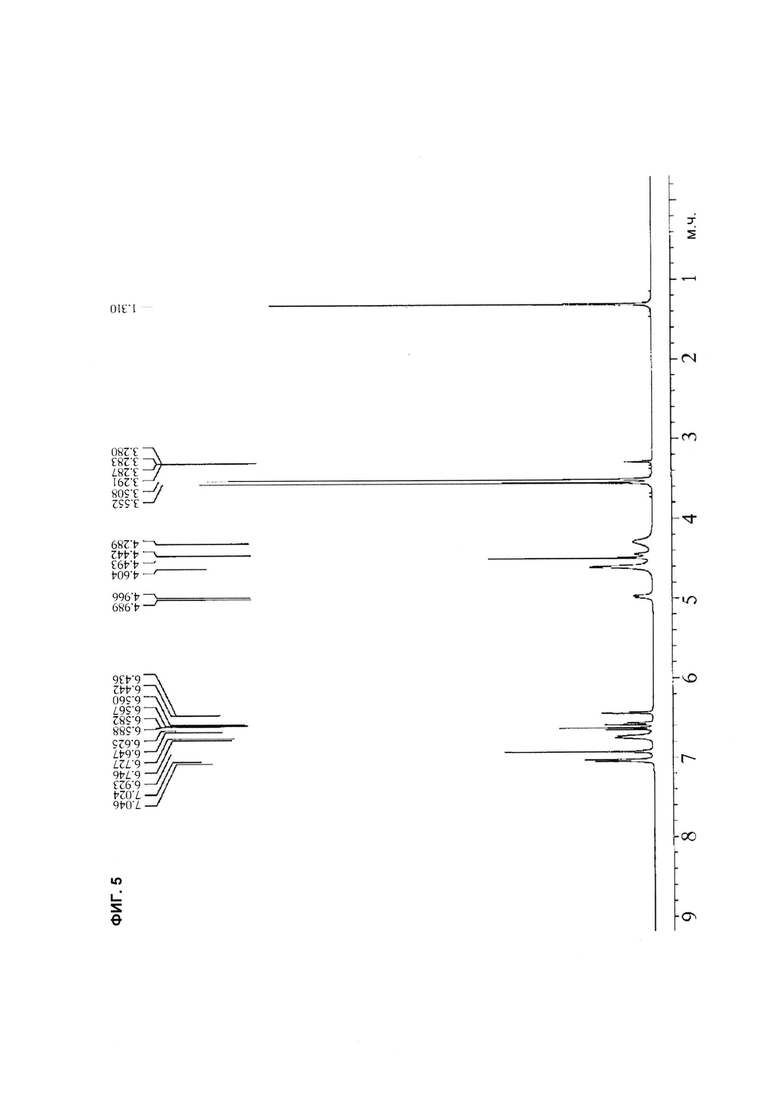
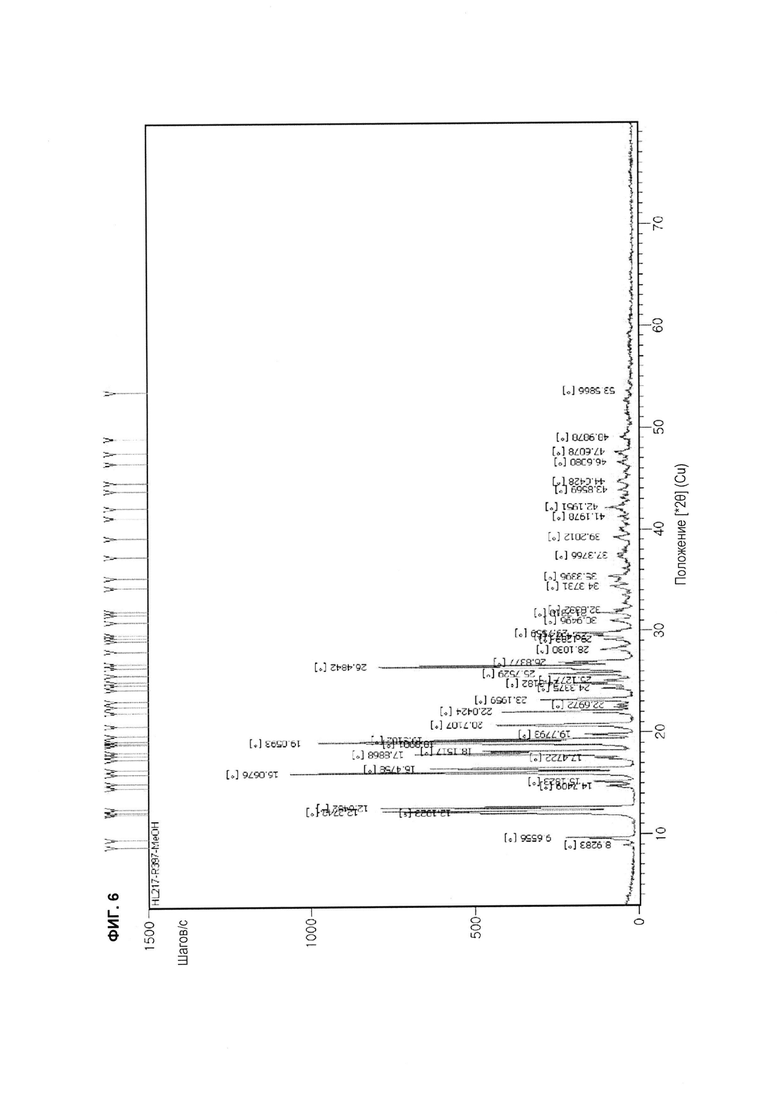
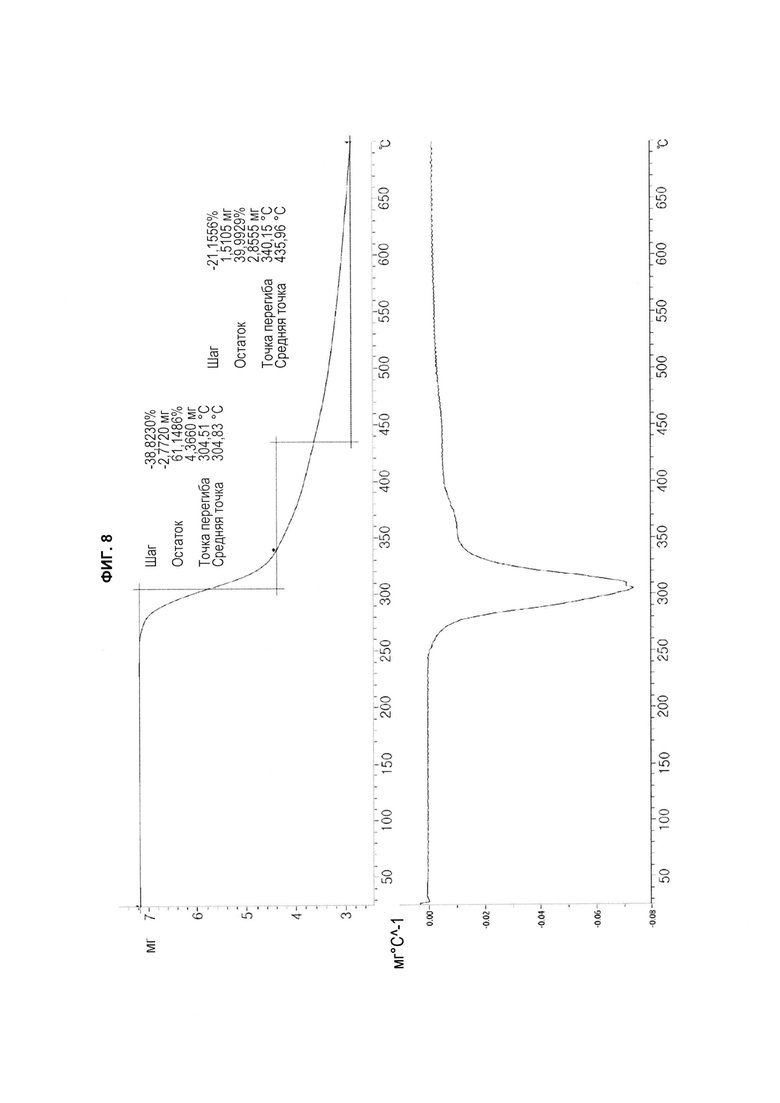
[23] На ФИГ. 5-8 показаны соответственно 1H спектр ЯМР (ФИГ. 5), спектр ПРД (ФИГ. 6), термограмма ДСК (ФИГ. 7) и термограмма ТГА (ФИГ. 8) кристаллической формы А соединения с формулой 1, полученного в соответствии с настоящим изобретением.
ЛУЧШИЕ ПРИМЕРЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[24] В настоящем изобретении предложен способ очистки соединения с формулой 1, содержащий преобразование сырого соединения с формулой 1 в его кристаллическую форму.
[25] Формула 1
[26]
[27] Используемый в данном случае термин «сырое соединение с формулой 1» относится к соединению, в котором содержание соединения с формулой 1 составляет 97 вес.% или менее, предпочтительно менее 98 вес.%, в безводной форме. Например, сырое соединение с формулой 1 может быть соединением, полученным способом, раскрытым в патенте Кореи № 10-0492252. В одном из вариантов осуществления изобретения сырое соединение с формулой 1 может быть аморфным соединением с формулой 1, полученным способом, раскрытым в патенте Кореи № 10-0492252.
[28] В соответствии с настоящим изобретением обнаружено, что соединение с формулой 1, полученное способом из предшествующего уровня техники (т.е. способом, раскрытым в патенте Кореи № 10-0492252), получено в аморфной форме с низкой чистотой и высоким содержанием воды (а также с высокой гигроскопичностью). Способ очистки в соответствии с настоящим изобретением может обеспечить получение соединения с формулой 1 в кристаллической форме с высокой чистотой и уменьшенным содержанием воды. Способ очистки обладает преимуществом, состоящим в том, что этот способ может быть с легкостью применен для промышленного массового производства. Используемый в данном случае термин «сырое соединение с формулой 1 с высокой чистотой» относится к соединению с формулой 1, в котором содержание соединения с формулой 1 составляет 98 вес.% или более, в предпочтительном варианте 99 вес.% или более, в безводной форме. Кроме того, термин «соединение с формулой 1 с уменьшенным содержанием воды» относится к соединению с формулой 1, в котором содержание воды составляет 0,5 вес.% или менее, в предпочтительном варианте 0,3 вес.% или менее, в более предпочтительном варианте 0,2 вес.% или менее.
[29] В способе очистки по настоящему изобретению кристаллическая форма может представлять собой кристаллическую форму А; и дебаеграмма ПРД (порошковая рентгеновская дифракция) кристаллической формы А может иметь характеристические пики на 12,27, 12,65, 16,07, 19,06 и 26,48°2θ ± 0,2°2θ. Предпочтительно дебаеграмма ПРД кристаллической формы А соединения с формулой 1 может иметь пики на 12,27, 12,65, 16,07, 16,48, 17,89, 18,89, 19,06, 19,31 и 26,48°2θ ± 0,2°2θ. Более предпочтительно дебаеграмма ПРД кристаллической формы А соединения с формулой 1 имеет вид, показанный на ФИГ. 6.
[30] Кроме того, термограмма ДСК (дифференциальная сканирующая калориметрия) кристаллическая форма А соединения с формулой 1 может иметь термограмму ДСК (дифференциальная сканирующая калориметрия) с эндотермическим пиком между 240°С и 250°С, например, термограмму ДСК, показанную на ФИГ. 7.
[31] Кроме того, кристаллическая форма А соединения с формулой 1 может иметь термограмму ТГА (термогравиметрический анализ), показывающую потерю веса между 300°С и 310°С, например, термограмму ТГА, показанную на ФИГ. 8.
[32] В настоящем изобретении предложена кристаллическая форма соединения с формулой 1.
[33] Формула 1
[34]
[35] Кристаллическая форма А соединения с формулой 1 обладает превосходными начальными свойствами (т.е. имеет высокую чистоту и уменьшенное содержание воды). В частности, кристаллическая форма А соединения с формулой 1 по существу не демонстрирует гигроскопичность; и может сохранять стабильную форму, не меняя упорядоченности даже при нагревании и в условиях ускоренных испытаний. Следовательно, кристаллическая форма А соединения с формулой 1 обладает свойствами, подходящими для составления рецептур терапевтических лекарственных форм; и, таким образом, обеспечивает преимущества, позволяя получать эффективные рецептуры без потери активного фармацевтического ингредиента и с длительным сроком хранения.
[36] В данном случае термин «соединение, по существу не демонстрирующее гигроскопичность» относится к соединению, демонстрирующему изменение содержания воды 0,05 вес.% или менее, предпочтительно 0,03 вес.% или менее, более предпочтительно 0,02 вес.% или менее при хранении в условиях ускоренных испытаний (40°С, относительная влажность 75%) в течение 2 недель (изменение содержания воды = содержание воды при хранении в течение 2 недель минус начальное содержание воды); или к соединению, демонстрирующему изменение содержания воды 0,05 вес.% или менее при хранении в нагретом состоянии (100°С) в течение 2 недель (изменение содержания воды = содержание воды при хранении в течение 2 недель минус начальное содержание воды); или к соединению, демонстрирующему изменение содержания воды 0,3 вес.% или менее, предпочтительно 0,2 вес.% или менее при хранении в условиях влажности (25°С), относительная влажность 98%) в течение 2 недель (изменение содержания воды = содержание воды при хранении в течение 2 недель минус начальное содержание воды).
[37] Кристаллическая форма соединения с формулой 1 может представлять собой кристаллическую форму А; дебаеграмма ПРД кристаллической формы А может иметь пики на 12,27, 12,65, 16,07, 19,06 и 26,48°2θ ± 0,2°2θ. Предпочтительно, кристаллическая форма соединения с формулой 1 может иметь дебаеграмму ПРД с пиками на 12,27, 12,65, 16,07, 16,48, 17,89, 18,89, 19,06, 19,31 и 26,48°2θ ± 0,2°2θ. Более предпочтительно, кристаллическая форма соединения с формулой 1 может иметь дебаеграмму ПРД, показанную на ФИГ. 6.
[38] Кроме того, кристаллическая форма А соединения с формулой 1 может иметь термограмму ДСК (дифференциальная сканирующая калориметрия), демонстрирующую эндотермический пик между 240°С и 250°С, например, термограмму ДСК, показанную на ФИГ. 7.
[39] Кроме того, кристаллическая форма А соединения с формулой 1 может иметь термограмму ТГА (термогравиметрический анализ), показывающую потерю веса между 300°С и 310°С, например, термограмму ТГА, показанную на ФИГ. 8.
[40] Настоящее изобретение относится к способу получения кристаллической формы соединения с формулой 1, который может быть с легкостью применен для промышленного массового производства.
[41] Формула 1
[42]
[43] В способе получения кристаллической формы соединения с формулой 1 по настоящему изобретению используется аморфное соединение с формулой 1 в качестве начального вещества, которое может быть получено в соответствии со способом, раскрытым в патенте Кореи № 10-0492252.
[44] В качестве одного из вариантов осуществления настоящего изобретения предложен способ получения кристаллической формы соединения с формулой 1, включающий растворение аморфного соединения с формулой 1 в органическом растворителе для получения раствора; перемешивание, дистилляцию или охлаждение раствора с образованием твердого вещества или дистиллята, а затем охлаждение раствора с образованием твердого вещества; и выделение твердого вещества (т.е. способ посредством рекристаллизации). Органическим растворителем может быть любой растворитель, который может растворять аморфное соединение с формулой 1, и может быть использован один органический растворитель или сочетание из двух или более органических растворителей. Например, органическим растворителем может быть один или несколько растворителей, выбранных из группы, состоящей из метанола, этанола, изопропанола, ацетона, метилэтилкетона, ацетонитрила, этилацетата, дихлорметана, тетрагидрофурана, диметилсульфоксида, диметилформамида и N-метил-2-пирролидона. Предпочтительно, органический растворитель может быть одним растворителем или смесью органических растворителей, выбранных из группы, состоящей из метанола, этанола, изопропанола, ацетона, ацетонитрила, дихлорметана, этилацетата и метилэтилкетона. Растворение может быть выполнено при температуре в диапазоне от комнатной температуры до температуры кипения используемых растворителей. Формирование твердого вещества может быть осуществлено путем перемешивания, дистилляции или охлаждения раствора; или путем дистилляции раствора для уменьшения количества растворителя с последующим охлаждением полученного раствора. Выделение твердого вещества (т.е. кристаллической формы) может быть выполнено обычной фильтрацией (например, фильтрация в условиях пониженного давления), сушкой (например, сушка при температуре около 50°С) и т.д.
[45] В другом варианте осуществления настоящего изобретения предложен способ получения кристаллической формы соединения с формулой 1, включающий растворение аморфного соединения с формулой 1 в органическом растворителе для получения раствора; добавление раствора в антирастворитель для получения твердого вещества или добавление антирастворителя в раствор для получения твердого вещества и выделение твердого вещества (т.е. способ с растворителем / антирастворителем). Органическим растворителем может быть любой растворитель, который может растворять аморфное соединение с формулой 1, и может быть использован один органический растворитель или сочетание из двух или более органических растворителей. Например, органическим растворителем может быть один или несколько растворителей, выбранных из группы, состоящей из метанола, этанола, изопропанола, ацетона, метилэтилкетона, ацетонитрила, этилацетата, дихлорметана, тетрагидрофурана, диметилсульфоксида, диметилформамида и N-метил-2-пирролидона. Растворение может быть выполнено при температуре от комнатной температуры до температуры кипения используемых растворителей. Антирастворитель может быть одним или несколькими антирастворителями, выбранными из группы, состоящей из воды, гексана, гептана, диэтилового эфира, изопропилового эфира, ди-н-бутилового эфира и толуола, но не ограничивается перечисленным. Выделение твердого вещества (т.е. кристаллической формы) может быть выполнено обычной фильтрацией (например, фильтрация в условиях пониженного давления), сушкой (например, сушка при температуре около 50°С) и т.д.
[46] В другом варианте осуществления настоящего изобретения в настоящем изобретении предложен способ получения кристаллической формы соединения с формулой 1, содержащий растворение аморфного соединения с формулой 1 в воде посредством добавления в него кислоты для получения раствора; добавление основания в раствор для получения твердого вещества; и выделение твердого вещества (т.е. способ посредством кристаллизации с контролем уровня рН). Кислотой может быть любая кислота с кислотным рН. Например, кислота может быть одной или несколькими кислотами, выбранными из группы, состоящей из хлористоводородной кислоты, уксусной кислоты и муравьиной кислоты, без ограничения. Основанием может быть любое основание, которое может нейтрализовать использованную кислоту с образованием твердого вещества. Например, основание может быть одним или несколькими основаниями, выбранными из группы, состоящей из гидроксида натрия, гидроксида калия, бикарбоната натрия и карбоната натрия, без ограничения. Кислота и/или основание могут быть использованы в виде водного раствора.
[47] Кристаллическая форма, полученная в соответствии с указанными способами для получения кристаллической формы соединения с формулой 1 по настоящему изобретению, получена в кристаллической форме А. Дебаеграмма ПРД кристаллической формы А может быть с характеристические пики на 12,27, 12,65, 16,07, 19,06 и 26,48°2θ ± 0,2°2θ. Предпочтительно дебаеграмма ПРД кристаллической формы А соединения с формулой 1 может быть с пиками на 12,27, 12,65, 16,07, 16,48, 17,89, 18,89, 19,06, 19,31 и 26,48°2θ ± 0,2°2θ. Более предпочтительно, дебаеграмма ПРД кристаллической формы А соединения с формулой 1 имеет вид, показанный на ФИГ. 6. Кроме того, термограмма ДСК (дифференциальная сканирующая калориметрия) кристаллической формы А соединения с формулой 1 может быть с эндотермическим пиком между 240°С и 250°С, например, термограмма ДСК, показанная на ФИГ. 7. Кроме того, термограмма ТГА (термогравиметрический анализ) кристаллической формы А соединения с формулой 1 может показывать потерю веса между 300°С и 310°С, например, термограмма ТГА, показанная на ФИГ. 8.
[48] Настоящее изобретение будет более подробно раскрыто со ссылкой на следующие примеры и экспериментальные примеры. Эти примеры и экспериментальные примеры приведены только для наглядности и не предназначены для ограничения объема настоящего изобретения.
[49] В следующих примерах и экспериментальных примерах выполнен анализ с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ) при следующих условиях:
[50] - Аналитическая колонка: C18, 4,6 × 250 мм, 5 мкм
[51] - Мобильная фаза: буферный раствор / ацетонитрил = 40 / 60 (об.)
[52] - Буферный раствор: формиат аммония (0,657 г), добавлен в мерную колбу объемом 1 л. Вода добавлена до метки так, чтобы растворить формиат аммония, а затем рН полученного раствора доведен до рН 5,5 ± 0,2 разбавленной муравьиной кислотой.
[53] - Длина волны: 254 нм
[54] - Температура колонки: 30°С
[55] - Расход: 1,0 мл/мин.
[56] - Объем впрыска: 10 мкл
[57] Анализ с использованием порошковой рентгеновской дифракции (ПРД) был выполнен на рентгеновском дифрактометре X-pert Pro компании PANalytical. Измерения значений 2θ под углами в диапазоне от 3 до 80° были выполнены со скоростью сканирования 3° в секунду с использованием излучения CuKα1 (λα1 = 1,54060 Å), генерируемого в условиях 40 мА и 40 кВ.
[58] Дифференциальная сканирующая калориметрия (ДСК) была выполнена на дифференциальном сканирующем калориметре DSC 823e компании Mettler Toledo в следующих условиях: начальная температура 10°С, конечная температура 300°С, скорость нагрева 10°С/мин, расход очищенного газообразного азота 50 мл/мин.
[59] Термогравиметрический анализ (ТГА) был выполнен на термогравиметрическом анализаторе TGA/SDTA 851 компании Mettler Toledo в следующих условиях: начальная температура 25°С, конечная температура 700°С, скорость нагрева 10°С/мин.
[60]
[61] Пример получения (2R, 3R, 4S)-6-амино-4-[N-(4-хлорфенил)-N- (1H-имидазол-2-илметил)амино]-3-гидрокси-2-метил-2-диметоксиметил- 3,4-дигидро-2Н-1-бензопирана (соединение с формулой 1)
[62] Согласно известному способу (пример 23 в патенте Кореи № 10-0492252) нитросоединение (52,10 г, 106,56 ммоль) растворили в метаноле (300 мл); и затем добавляли к нему 10% Pd/C (5,0 г). Смесь была гидрирована под давлением 3 атмосферы H2 в течение 12 ч. Реакционную смесь профильтровали через целитовую прокладку для удаления твердого вещества; а фильтрат сконцентрировали. Полученный остаток очистили колоночной хроматографией на силикагеле (метанол : дихлорметан = 5:95 (об.) с получением указанного в заголовке соединения 36,52 г (выход: 75%). Температура плавления, чистота (содержание соединения с формулой 1 (в безводной форме) в продукте), содержание воды и 1H спектр ЯМР (ФИГ. 1) полученного продукта следующие:
[63] Температура плавления: 191-195°С
[64] Чистота: 96,96 вес.%
[65] Содержание воды: 1,05 вес.%
[66] 1H ЯМР (400 МГц, CD3OD) δ (м. д.): 1,309(s. 3H), 3,509(s. 3H), 3,554(s. 3H), 4,288(m. 2H), 4,443-4,492(d. 2H), 4,964-4,988(d. 1H), 6,437-6,442(d. 1H), 6,562-6,591(m, 1H), 6,626-6,647(d, 1H), 6,727-6,746(d, 2H), 6,925(s, 2H), 7,025-7,047(d, 2H).
[67] Также спектр ПРД, термограмма ДСК и термограмма ТГА полученного продукта показаны на ФИГ. 2-4 соответственно. Не наблюдалось никаких характеристических пиков, показывающих угол дифракции, расстояние между слоями кристалла и относительную интенсивность в измеренном спектре ПРД; и поэтому полученный продукт представляет собой аморфное соединение. Кроме того, термограмма ДСК полученного продукта показала экзотермический пик при температуре от приблизительно 148°С до приблизительно 158°С, и эндотермический пик при температуре от приблизительно 229°С до приблизительно 239°С (ФИГ. 3); кроме того, термограмма ТГА показала характеристическую потерю веса при температуре от приблизительно 100°С до приблизительно 110°С, и от приблизительно 274°С до приблизительно 284°С (ФИГ. 4).
[68]
[69] Пример 1: Очистка соединения с формулой 1 посредством рекристаллизации и его характеристики
[70] Соединение с формулой 1, полученное в Примере получения (5,00 г), растворили в метаноле (50 мл) с обратным холодильником. Полученный раствор перегоняли до образования твердого вещества, охладили до комнатной температуры и затем профильтровали в условиях пониженного давления. Полученное твердое вещество сушили при разрежении при 50°С в течение 18 ч, получив 3,53 г соединения с формулой 1 (выход: 70,60%). Температура плавления, чистота (содержание соединения с формулой 1 (в безводной форме) в продукте), содержание воды и 1H спектр ЯМР (ФИГ. 5) полученного продукта следующие:
[71] Температура плавления: 227-231°С
[72] Чистота: 99,43 вес.%
[73] Содержание воды: 0,16 вес.%
[74] 1H ЯМР (400 МГц, CD3OD) δ (м. д.): 1,310(s. 3H), 3,508(s. 3H), 3,552(s. 3H), 4,289(m. 2H), 4,442-4,493(d. 2H), 4,966-4,989(d. 1H), 6,436-6,442(d. 1H), 6,560-6,588(m, 1H), 6,625-6,647(d, 1H), 6,727-6,746(d, 2H), 6,923(s, 2H), 7,024-7,046(d, 2H).
[75] Также спектр ПРД, термограмма ДСК и термограмма ТГА полученного продукта показаны на ФИГ. 6-8 соответственно. Углы дифракции (°2θ), расстояния между кристаллическими слоями (d) и относительные интенсивности (отношение интенсивности каждого пика (I) к интенсивности наибольшего пика (I0), I/I0) в измеренном спектре ПРД показаны в таблице 1 ниже.
[76] Таблица 1
[77]
[78] Поскольку кристаллическая схема, демонстрирующая характерные пики, подтверждена по результатам таблицы 1, форма продукта - кристаллическая. Кристаллическая форма представляет собой «кристаллическую форму А соединения с формулой 1».
[79]
[80] Примеры 2-8
[81] Соединение с формулой 1 очистили в соответствии с теми же процедурами, что и в Примере 1, за исключением использования других растворителей в соответствии с условиями, показанными в таблице 2 ниже. Выход и чистота (содержание соединения с формулой 1 (в безводной форме) в продукте) показаны в таблице 2. Кроме того, поскольку для всех продуктов были получены по существу такие же спектры ПРД, как показанный на ФИГ. 6, все полученные продукты представляли собой кристаллическую форму А.
[82] Таблица 2
[84]
[85] Пример 9: Очистка соединения с формулой 1 с использованием растворителя / антирастворителя и его характеристики
[86] Соединение с формулой 1, полученное в Примере получения (5,00 г), растворили в метаноле (50 мл) с обратным холодильником. К полученному раствору добавили очищенную воду (30 мл). Смесь охладили до комнатной температуры и затем профильтровали в условиях пониженного давления. Полученное твердое вещество сушили при разрежении при 50°С в течение 18 ч, получив 4,42 г соединения с формулой 1 (выход: 88,40%). Чистота (содержание соединения с формулой 1 (в безводной форме) в продукте) составила 99,61 вес.%. Кроме того, поскольку по продукту был получен по существу такой же спектр ПРД, как показанный на ФИГ. 6, полученный продукт представлял собой кристаллическую форму А.
[87]
[88] Примеры 10-18
[89] Соединение с формулой 1 очистили в соответствии с теми же процедурами, что и в Примере 9, за исключением использования других растворителей / антирастворителей в соответствии с условиями, показанными в таблице 3 ниже. Выход и чистота (содержание соединения с формулой 1 (в безводной форме) в продукте) показаны в таблице 3. Кроме того, поскольку по всем продуктам были получены по существу такие же спектры ПРД, как показанный на ФИГ. 6, все полученные продукты представляли собой кристаллическую форму А.
[90] Таблица 3
[91]
растворитель
20 мл
20 мл
пирролидон
6 мл
50 мл
50 мл
150 мл
75 мл
40 мл
40 мл
150 мл
50 мл
100 мл
[92]
[93[ Пример 19: Очистка соединения с формулой 1 посредством кристаллизации с контролем рН и его характеристики
[94] Соединение с формулой 1, полученное в Примере получения (3,00 г), добавили к очищенной воде; и затем растворили в ней с контролем рН на уровне 1,0 1Н раствором хлористоводородной кислоты. рН полученного раствора довели до 7,0 1Н раствором гидроксида натрия, чтобы получить твердое вещество. Смесь профильтровали в условиях пониженного давления. Полученное твердое вещество сушили при разрежении при 50°С в течение 18 ч, получив 2,81 г соединения с формулой 1 (выход: 93,67%). Чистота (содержание соединения с формулой 1 (в безводной форме) в продукте) составила 99,55 вес.%. Кроме того, поскольку по продукту был получен по существу такой же спектр ПРД, как показанный на ФИГ. 6, полученный продукт представлял собой кристаллическую форму А.
[95]
[96] Пример 20: Очистка соединения по Формуле 1 с использованием растворителя / антирастворителя и его характеристики
[97] Соединение с формулой 1, полученное в Примере получения (3,00 г), растворили в дихлорметане (30 мл). Полученный раствор порциями добавили к гексану (300 мл) и затем профильтровали в условиях пониженного давления. Полученное твердое вещество сушили при разрежении при 50°С в течение 18 ч, получив 2,93 г соединения с формулой 1 (выход 97,67 %). Чистота (содержание соединения с формулой 1 (в безводной форме) в продукте) составила 99,47 вес.%. Кроме того, поскольку по продукту был получен по существу такой же спектр ПРД, как показанный на ФИГ. 6, полученный продукт представлял собой кристаллическую форму А.
[98]
[99] Примеры 21-24
[100] Соединение с формулой 1 очистили в соответствии с теми же процедурами, что и в Примере 20, за исключением использования других растворителей / антирастворителей в соответствии с условиями, показанными в таблице 4 ниже. Выход и чистота (содержание безводного соединения с формулой 1 в продукте) показаны в таблице 4. Кроме того, поскольку по всем продуктам были получены по существу такие же спектры ПРД, как показанный на ФИГ. 6, все полученные продукты представляли собой кристаллическую форму А.
[101] Таблица 4
[102]
тель
30 мл
30 мл
300 мл
[103]
[104] Экспериментальный пример 1: Ускоренное испытание стабильности
[105] Кристаллическую форму А соединения с формулой 1, полученную в Примере 1, и аморфную форму соединения с формулой 1, полученную в Примере получения, хранили в условиях ускоренных испытаний (40°С, относительная влажность 75%) в течение 2 недель, чтобы оценить ее стабильность. Результаты показаны ниже в таблицах 5 и 6.
[106] Таблица 5
[107] Ускоренное испытание стабильности кристаллической формы соединения с формулой 1
ство
ческий порошок белого или бледно-
желтого цвета
ческий порошок
ческий порошок
ческий порошок
ческий порошок
ты распада
ные продукты распада: 0,10% или менее
но
ческая форма A
* Des-Cl: 2R,3R,4S)-6-амино-4-[N-фенил-N-(1H-имидазол-2-илметил)амино]-
3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопиран.
[108]
[109] Таблица 6
[110] Ускоренное испытание стабильности аморфной формы соединения с формулой 1
ческий порошок белого или бледно-
желтого цвета
ние воды
омер
(в безводной форме)
* Des-Cl: 2R,3R,4S)-6-амино-4-[N-фенил-N-(1H-имидазол-2-илметил)амино]-
3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопиран.
[111] Как показано в таблице 5 выше, кристаллическая форма А соединения с формулой 1 стабильно сохранена в виде белого кристаллического порошка без каких-либо изменений внешнего вида в условиях ускоренных испытаний. Содержание воды сохранено в количестве 0,16-0,18%, без значительного увеличения; содержание продуктов распада сохранено в количестве 0,030-0,041%, без значительного увеличения. Кроме того, во время испытаний не обнаружен энантиомер. Чистота 99,43-99,34% в пределах критериев пригодности (т.е. 98,5-101,0%), наблюдалось только снижение уровня погрешности эксперимента. В анализах ПРД сохранена та же самая кристаллическая форма А.
[112] Однако, как показано в таблице 6 выше, аморфная форма соединения с формулой 1 показывает более высокое начальное содержание воды (т.е. 1,05%), чем кристаллическая форма; и содержание воды увеличилось до 2,64% в зависимости от времени хранения, проявляя очень высокую гигроскопичность. Хотя начальные значения внешнего вида, содержания продуктов распада и энантиомера сохранены, начальная чистота в безводной форме (96,96%) не соответствовала критериям, без изменения в зависимости от продолжительности хранения. Поэтому видно, что аморфная форма соединения с формулой 1 демонстрирует очень высокую гигроскопичность в ускоренном состоянии в течение 2 недель.
[113]
[114] Экспериментальный пример 2: Испытание тепловой стабильности
[115] Кристаллическую форму А соединения с формулой 1, полученную в Примере 1, и аморфную форму соединения с формулой 1, полученную в Примере получения, хранили в условиях температуры (100 °С) в течение 2 недель, чтобы оценить ее стабильность. Результаты показаны ниже в таблице 7 и 8.
[116] Таблица 7
[117] Испытание тепловой стабильности кристаллической формы соединения с формулой 1
ский порошок белого или бледно-
желтого цвета
лический порошок
лический порошок
лический порошок
лический порошок бледно-
желтого цвета
лический порошок бледно-
желтого цвета
(в безводной форме)
ческая форма A
* Des-Cl: (2R,3R,4S)-6-амино-4-[N-фенил-N-(1H-имидазол-2-илметил)амино]-
3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопиран.
[118]
[119] Таблица 8
[120] Испытание тепловой стабильности аморфной формы соединения с формулой 1
ное состояние
ский порошок белого или бледно-желтого цвета
невый порошок
невый порошок
невый порошок
невый порошок
жено
(в безводной форме)
* Des-Cl: (2R,3R,4S)-6-амино-4-[N-фенил-N-(1H-имидазол-2-илметил)амино]-
3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопиран.
[121] Как показано в таблице 7 выше, внешний вид кристаллической формы А соединения с формулой 1 изменил цвет на бледно-желтый с первой недели при указанных температурных условиях (100°С). Содержание воды сохранено в количестве 0,11-0,16%, без значительного увеличения; содержание продуктов распада сохранено в количестве 0,031-0,055%, без значительного увеличения. Кроме того, во время испытаний не обнаружен энантиомер. Чистота 99,43-99,25% соответствует критериям пригодности (т.е. 98,5-101,0%), наблюдалось только снижение уровня погрешности эксперимента. В анализах ПРД сохранена та же самая кристаллическая форма А.
[122] Однако, как показано в таблице 8 выше, аморфная форма соединения с формулой 1 показывает более высокое начальное содержание воды (т.е. 1,05%), чем кристаллическая форма; и содержание воды уменьшилось до 0,34% в зависимости от времени хранения. Внешний вид также изменил цвет на коричневый с первого дня и, следовательно, не соответствует критериям. Содержание продуктов распада увеличено с начального 0,047% до 1,811%, при этом в течение периода испытаний не был обнаружен энантиомер. Начальная чистота в безводной форме (96,96%) не соответствовала критериям и была уменьшена до 92,34% и, следовательно, не соответствует критериям. Таким образом, аморфная форма соединения с формулой 1 демонстрирует ухудшенные свойства, особенно в части внешнего вида, содержания продуктов распада и чистоты при тепловом воздействии в течение 2 недель.
[123]
[124] Экспериментальный пример 3: Испытание стабильности при воздействии влажности
[125] Кристаллическую форму А соединения с формулой 1, полученную в Примере 1, и аморфную форму соединения с формулой 1, полученную в Примере получения, хранили в условиях влажности (25°С, относительная влажность 98%) в течение 2 недель, чтобы оценить ее стабильность. Результаты показаны ниже в таблицах 9 и 10.
[126] Таблица 9
[127] Испытание стабильности кристаллической формы соединения с формулой 1 в условиях влажности
ное состо- яние
ческий порошок белого или бледно-жел-
того цвета
лический порошок
лический порошок
лический порошок
желтого цвета
желтого цвета
жено
жено
(в безводной форме)
* Des-Cl: (2R,3R,4S)-6-амино-4-[N-фенил-N-(1H-имидазол-2-илметил)амино]-
3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопиран.
[128]
[129] Таблица 10
[130] Испытание стабильности аморфной формы соединения с формулой 1 в условиях влажности
ное состо-
яние
вид
ский порошок белого или бледно-желтого цвета
жено
(в безводной форме)
ная форма
* Des-Cl: (2R,3R,4S)-6-амино-4-[N-фенил-N-(1H-имидазол-2-илметил)амино]-
3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопиран.
[131] Как показано в таблице 9 выше, кристаллическая форма А соединения с формулой 1 стабильно сохранялась в виде белого кристаллического порошка без каких-либо изменений внешнего вида в условиях влажности (25°С, относительная влажность 98%). Содержание воды немного увеличено до 0,16-0,29%, без значительного увеличения; содержание продуктов распада сохранялось в количестве 0,029-0,036%, без значительного увеличения. Кроме того, во время испытаний не обнаружен энантиомер. Чистота 99,43-99,32% соответствует критериям пригодности (т.е. 98,5-101,0%), наблюдалось только снижение уровня погрешности эксперимента. В анализах ПРД видно, что сохранена та же самая кристаллическая форма А.
[132] Однако, как показано в таблице 10 выше, аморфная форма соединения с формулой 1 показала более высокое начальное содержание воды (т.е. 1,05%), чем кристаллическая форма; и содержание воды увеличилось до 3,90% в зависимости от времени хранения, проявляя очень высокую гигроскопичность. Хотя внешний вид, почти белый порошок, не изменился, содержание продуктов распада увеличилось с начального 0,047% до 0,090%. Энантиомер не обнаружен во время испытаний, но начальная чистота в безводной форме (96,96%) не соответствовала критериям, без изменений в зависимости от продолжительности хранения. Таким образом, аморфная форма соединения с формулой 1 демонстрирует гигроскопичность в условиях влажности в течение 2 недель.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СПОСОБ ЕЕ ОЧИСТКИ | 2011 |
|
RU2604734C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА TLR7/TLR8 | 2019 |
|
RU2792005C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА LTA4H | 2019 |
|
RU2808992C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА MAGL | 2017 |
|
RU2799564C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА BTK | 2020 |
|
RU2828460C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2654855C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА ЯНУС-КИНАЗЫ | 2017 |
|
RU2838992C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 9-АМИНОМЕТИЛ-ЗАМЕЩЕННОГО СОЕДИНЕНИЯ ТЕТРАЦИКЛИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2017 |
|
RU2764723C2 |


Изобретение относится к способу очистки соединения формулы 1 и/или улучшения гигроскопичности соединения формулы 1, включающий преобразование сырого соединения формулы 1 в аморфной форме в кристаллическую форму А соединения формулы 1 с дебаеграммой ПРД с пиками 2θ ± 0,2°2θ на 12,27, 12,65, 16,07, 19,06 и 26,48°, причем сырое соединение формулы 1 является соединением, в котором содержание соединения с формулой 1 составляет 97 вес.% или менее, в безводной форме. Технический результат - разработан новый способ очистки аморфного (2R,3R,4S)-6-амино-4-[N-(4-хлорфенил)-N-(1H-имидазол-2-илметил)амино]-3-гидрокси-2-метил-2-диметоксиметил-3,4-дигидро-2H-1-бензопирана, который в свою очередь может найти применение в медицине для лечения таких заболеваний, как рак, ревматоидный артрит, дегенеративные процессы желтого пятна. 3 з.п. ф-лы, 10 табл., 24 пр., 8 ил.
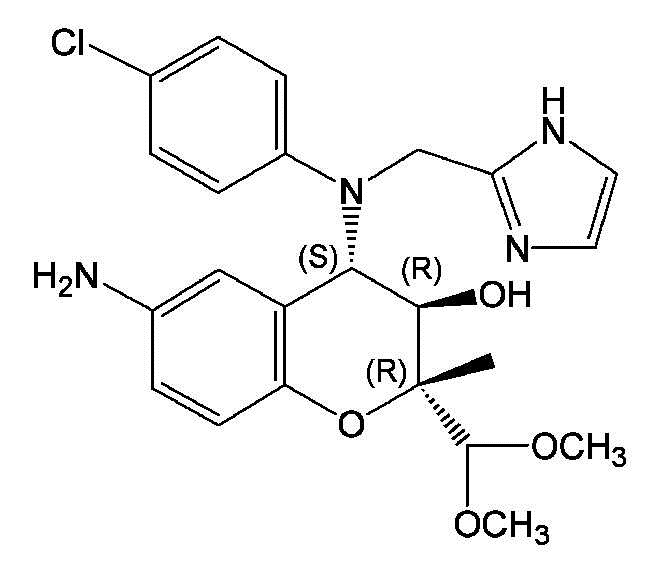
Формула 1
1. Способ очистки соединения формулы 1 и/или улучшения гигроскопичности соединения формулы 1, включающий преобразование сырого соединения формулы 1 в аморфной форме в кристаллическую форму А соединения формулы 1 с дебаеграммой ПРД с пиками 2θ ± 0,2°2θ на 12,27, 12,65, 16,07, 19,06 и 26,48°, причем сырое соединение формулы 1 является соединением, в котором содержание соединения с формулой 1 составляет 97 вес.% или менее, в безводной форме,
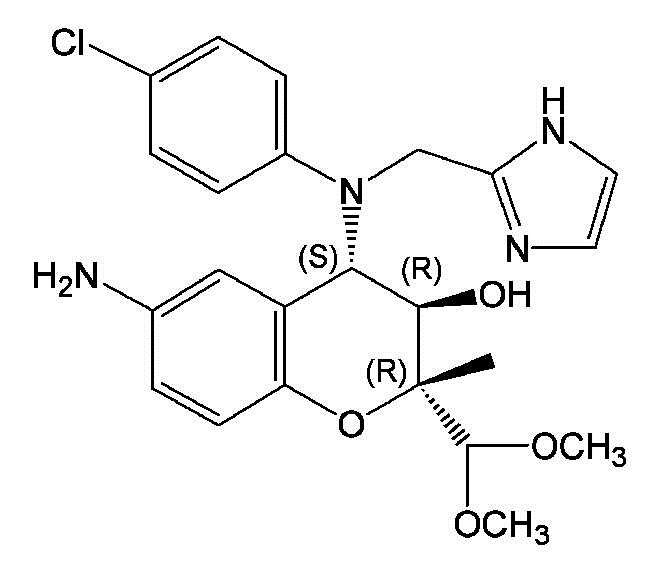
Формула 1,
при этом упомянутое преобразование включает:
(i) растворение путем кипячения с обратным холодильником сырого соединения формулы 1 в одном или более растворителей, выбранных из группы, состоящей из метанола, этанола, изопропанола, ацетона, метилэтилкетона, ацетонитрила, этилацетата и дихлорметана,
(ii) перемешивание, дистилляцию или охлаждение раствора с образованием твердого вещества или дистиллята, а затем охлаждение раствора с образованием твердого вещества,
(iii) выделение твердого вещества,
или
упомянутое преобразование включает:
(i') растворение путем кипячения с обратным холодильником сырого соединения формулы 1 в одном или более растворителей, выбранных из группы, состоящей из метанола, ацетона, ацетонитрила, этилацетата, дихлорметана, тетрагидрофурана, диметилсульфоксида, диметилформамида и N-метил-2-пирролидона,
(ii') добавление раствора в антирастворитель для получения твердого вещества или добавление антирастворителя в раствор для получения твердого вещества, при этом антирастворитель является одним или несколькими антирастворителями, выбранными из группы, состоящей из воды, гексана, гептана, диэтилового эфира, изопропилового эфира, ди-н-бутилового эфира, циклогексана и толуола, и
(iii') выделение твердого вещества,
или
упомянутое преобразование включает:
(i'') растворение сырого соединения формулы 1 в воде посредством добавления в него кислоты для получения раствора, при этом кислота является одной или несколькими кислотами, выбранными из группы, состоящей из хлористоводородной кислоты, уксусной кислоты и муравьиной кислоты,
(ii'') добавление основания в раствор для получения твердого вещества, при этом основание является одним или несколькими основаниями, выбранными из группы, состоящей из гидроксида натрия, гидроксида калия, бикарбоната натрия и карбоната натрия, и
(iii') выделение твердого вещества.
2. Способ по п. 1, в котором кристаллическая форма А соединения формулы 1 имеет дебаеграмму ПРД с пиками 2θ ± 0,2°2θ на 12,27, 12,65, 16,07, 16,48, 17,89, 18,89, 19,06, 19,31 и 26,48°.
3. Способ по п. 1, в котором кристаллическая форма А соединения формулы 1 имеет термограмму дифференциальной сканирующей калориметрии (ДСК) с эндотермическим пиком между 240°С и 250°С.
4. Способ по п. 1, в котором кристаллическая форма А соединения формулы 1 имеет термограмму термогравиметрического анализа (ТГА) с потерей веса между 300°С и 310°С.
| АЭРОПОЕЗД С ПИТАНИЕМ ОТ ТРОЛЛЕЯ (ВАРИАНТЫ) | 2018 |
|
RU2692345C1 |
| WO 2004014898 A1, 19.02.2004 | |||
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА, ЗАМЕЩЕННЫЕ ВТОРИЧНЫМИ АМИНАМИ, ВКЛЮЧАЮЩИМИ В СЕБЯ ТЕТРАЗОЛ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2003 |
|
RU2283312C2 |
| КРИСТАЛЛИЧЕСКИЕ СОЛИ СИТАГЛИПТИНА | 2009 |
|
RU2519717C2 |

