ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому содержащему медь молекулярному ситу SAPO, к процессу его синтеза, а также к его использованию в реакции денитрирования.
УРОВЕНЬ ТЕХНИКИ
NOx, как одни из главных загрязнителей атмосферы, могут вызвать множество экологических проблем, таких как кислотный дождь и фотохимический смог, и представляют серьезную опасность для здоровья человека. Загрязнение оксидами азота вызывается главным образом выхлопами транспортных средств и выбросами отходящего газа из стационарных источников. Селективное каталитическое восстановление NOx с помощью NH3, мочевины или углеводорода в качестве восстановителя является главным способом управления загрязнением, и NOx преобразуется в безвредный азот. Традиционный катализатор денитрирования представляет собой главным образом каталитическую систему V–Ti–W. Однако с широким распространением технологии экономичного сгорания в производстве двигателей температура выхлопного газа стала снижаться, и узкая температурная область применения катализатора системы V–Ti–W перестала удовлетворять требованиям. Кроме того, потенциальная вредность катализатора V–W–Ti в плане загрязнения окружающей среды также ограничивает его применение. Каталитические системы на основе молекулярных сит постепенно становились горячей точкой исследований. Среди каталитических систем на основе молекулярных сит двумя представительными системами являются катализатор на основе меди и катализатор на основе железа. Катализаторы на основе меди показывают превосходную низкотемпературную активность, но чрезмерно высокая нагрузка вызывает сильную реакцию окисления NH3 в высокотемпературной секции. Катализаторы на основе железа обладают превосходной высокотемпературной активностью, но их более низкий коэффициент преобразования в низкотемпературном диапазоне ограничивает их применение в некоторых областях.
В 1986 Iwamoto и др. впервые сообщили, что обменивающийся Cu2+ ZSM–5 способен катализировать прямое разложение NO на N2 и O2. Впоследствии исследователи сосредоточились на реакции SCR (селективного каталитического восстановления) с углеводородом в качестве восстановителя, и Fe–ZSM–5 постепенно стал следующей горячей точкой исследований. По сравнению с оксидным катализатором катализатор из системы молекулярного сита обладает преимуществами более широкого окна температуры реакции, хорошей тепловой стойкости и хорошей устойчивости к отравлению серой при высокой температуре, но он также имеет и некоторые проблемы, такие как недостаточная высокотемпературная гидротермальная стабильность, недостаточная низкотемпературная устойчивость к отравлению серой и т.п.
Молекулярные сита с малыми порами, такие как SSZ–13 и SAPO–34 в качестве носителей, могут эффективно улучшать высокотемпературную гидротермальную стабильность катализатора, а при загрузке медью в качестве активного металла они имеют высокую активность преобразования NO и высокую селективность по N2 в широком диапазоне температур. Хотя у них имеются некоторые проблемы, такие как чувствительность к сере, они постепенно решаются с усовершенствованием качества топлива.
Как правило, синтез молекулярных сит SAPO требует органического амина/аммония в качестве структурообразующего агента, который синтезируется гидротермическими или сольвотермическими способами. Новшества в способах синтеза и в выборе агентов сборки по шаблону оказывают критическое влияние на управление структурой и характеристики продукта. Cu–SAPO–18 и Cu–SAPO–34 могут быть синтезированы в одну стадию путем использования комплексного соединения амина меди в качестве шаблона. Катализатор Cu–SAPO, синтезируемый этим одностадийным процессом, упрощает процесс подготовки катализатора и имеет большое значение. Кроме того, катализатор типа Cu–SAPO, синтезированный с помощью этого одностадийного процесса, показывает превосходную каталитическую активность NH3–SCR, а также настраиваемое денатурирование состава, и имеет определенную промышленную прикладную перспективу.
Настоящее изобретение предлагает одностадийный способ синтеза катализатора молекулярного сита Cu–SAPO с управляемым содержанием Cu, который показывает превосходную денитрирующую каталитическую активность, и имеет потенциальную прикладную ценность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является предложить молекулярное сито Cu–SAPO с эвтектической структурой GME и CHA.
Инновационное молекулярное сито, синтезируемое с помощью настоящего изобретения, обладает характеристикой сосуществования широких пиков и узких пиков, и его дифракционные рисунки XRD подобны спектрам кремниево–алюминиевого цеолита со структурой сосуществующих фаз GME/CHA, описанной в литературе (Microporous and Mesoporous Materials, 30 (1999) 335–346; официальный сайт Международной цеолитной ассоциации, http://www.iza–structure.org/databases/Catalog/ABC_6.pdf). Было установлено, что этот тип материала является новым молекулярным ситом SAPO со структурой сосуществующих фаз GME/CHA.
В соответствии с одним вариантом осуществления настоящего изобретения предлагается молекулярное сито Cu–SAPO с сосуществующими кристаллическими фазами CHA и GME, отличающееся тем, что дифракционная рентгенограмма этого молекулярного сита содержит по меньшей мере следующие дифракционные пики:
Таблица 1
Неорганический каркас этого молекулярного сита имеет следующий химический состав: wCu–(SixAlyPz)O2, где x, y и z соответственно представляют мольные доли Si, Al и P, диапазоны их мольных долей соответственно составляют x=0,01~0,28, y=0,35~0,55 и z=0,28~0,50, причем x+y+z=1, w является количеством молей Cu на моль (SixAlyPz)O2, и w=0,001~0,124.
Безводный химический состав этого молекулярного сита, содержащего агент сборки по шаблону, выражается как wCu·mR1·nR3·(SixAlyPz)O2, где R1 – диизопропаноламин или диэтаноламин, R3 – триметиламин; m – количество молей агента сборки по шаблону R1 на моль (SixAlyPz)O2, n – количество молей агента сборки по шаблону R3 на моль (SixAlyPz)O2, m=0,01~0,20, n=0,01~0,10; x, y и z соответственно представляют мольные доли Si, Al и P, и диапазоны их мольных долей соответственно составляют x=0,01~0,28, y=0,35~0,55 и z=0,28~0,50, причем x+y+z=1; w – количество молей Cu на моль (SixAlyPz)O2, w=0,001~0,124. В некоторых вариантах осуществления m может быть равно 0,02–0,15; n может быть равно 0,01~0,09; x может быть равно 0,05~0,28; y может быть равно 0,40~0,50; z может быть равно 0,30~0,50; и w может быть равно 0,005~0,100.
Другой задачей настоящего изобретения является предложить способ синтеза вышеупомянутого молекулярного сита Cu–SAPO, который содержит стадии:
a) смешивания источника меди, деионизированной воды, агентов сборки по шаблону R1 и R2, источника кремния, источник алюминия и источника фосфора в такой пропорции, чтобы получить начальную гелевую смесь, имеющую следующие молярные отношения:
Cu/Al2O3=0,01~ 0,25;
SiO2/Al2O3=0,05~2,0;
P2O5/Al2O3=0,5~1,5;
H2O/Al2O3=8~40;
R1/Al2O3=5~20;
R2/Al2O3=0,1~1,5;
где R1 – диизопропаноламин (DIPA) или диэтаноламин (DEOA); R2 – любой из триметиламина (TMA), бензилтриметиламмонийхлорида (BTACl), бензилтриметиламмонийгидроксида (BTAOH) или их смеси;
опционально порядок конкретного процесса смешивания может быть следующим: сначала источник меди смешивается с водой, затем добавляются R1 и R2, все это перемешивается при комнатной температуре в течение 0,5~5 час, затем к смешанному раствору добавляются источник алюминия, источник кремния и источник фосфора, и смешанный гель перемешивается при комнатной температуре в течение 1~5 час;
b) подачи начальной гелевой смеси, полученной на стадии a), в реактор синтеза высокого давления, закрытия реактора, нагревания до 160~220°C и выполнения кристаллизации в течение 5–72 час; и
c) после завершения кристаллизации отделения твердого продукта, его промывки и сушки для того, чтобы получить молекулярное сито.
На стадии a) источник кремния выбирается из одного или более из золя кремнезема, активированного кремнезема, ортосиликата и метакаолина; источник алюминия выбирается из одного или более из соли алюминия, активированного глинозема, псевдобемита, алкоксиалюминия и метакаолина; источник фосфора выбирается из одного или более из ортофосфорной кислоты, гидрофосфата аммония, дигидрофосфата аммония, органических фосфорсодержащих соединений и фосфорных оксидов; источник меди выбирается из одной или более неорганических солей из Cu(OAc)2, CuSO4, Cu(NO3)2 и CuCl2.
На стадии b) кристаллизация выполняется в статическом или динамическом состоянии.
Предпочтительно на стадии a) начальная гелевая смесь имеет соотношение SiO2/Al2O3=0,20~1,8.
Предпочтительно на стадии a) начальная гелевая смесь имеет соотношение P2O5/Al2O3=0,8~1,5.
Предпочтительно на стадии a) начальная гелевая смесь имеет соотношение R1/Al2O3=5,0~10.
Предпочтительно на стадии a) начальная гелевая смесь имеет соотношение R2/Al2O3=0,25~1,0.
Органический агент сборки по шаблону бензилтриметиламмонийхлорид (BTACl) и бензилтриметиламмонийгидроксид (BTAOH) в R2 разлагается при синтезе молекулярного сита, образуя триметиламин, и входит в клетку поры молекулярного сита.
В вышеописанном способе синтеза молекулярных сит, когда R1 является диэтаноламином, молярное отношение R1/R2 предпочтительно составляет 16–60; а когда R1 является диизопропаноламином, предпочтительная температура кристаллизации составляет 195–220°C.
Другой задачей настоящего изобретения является предложить катализатор для селективного каталитического восстановления NOx с помощью NH3, который получается путем обжига на воздухе при температуре 550–700°C вышеописанного молекулярного сита и/или молекулярного сита, синтезированного в соответствии с вышеописанным способом.
Выгоды настоящего изобретения включают в себя:
(1) инновационное молекулярное сито Cu–SAPO.
(2) это молекулярное сито может использоваться в качестве катализатора для каталитического удаления оксидов азота и показывает хорошую каталитическую эффективность.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
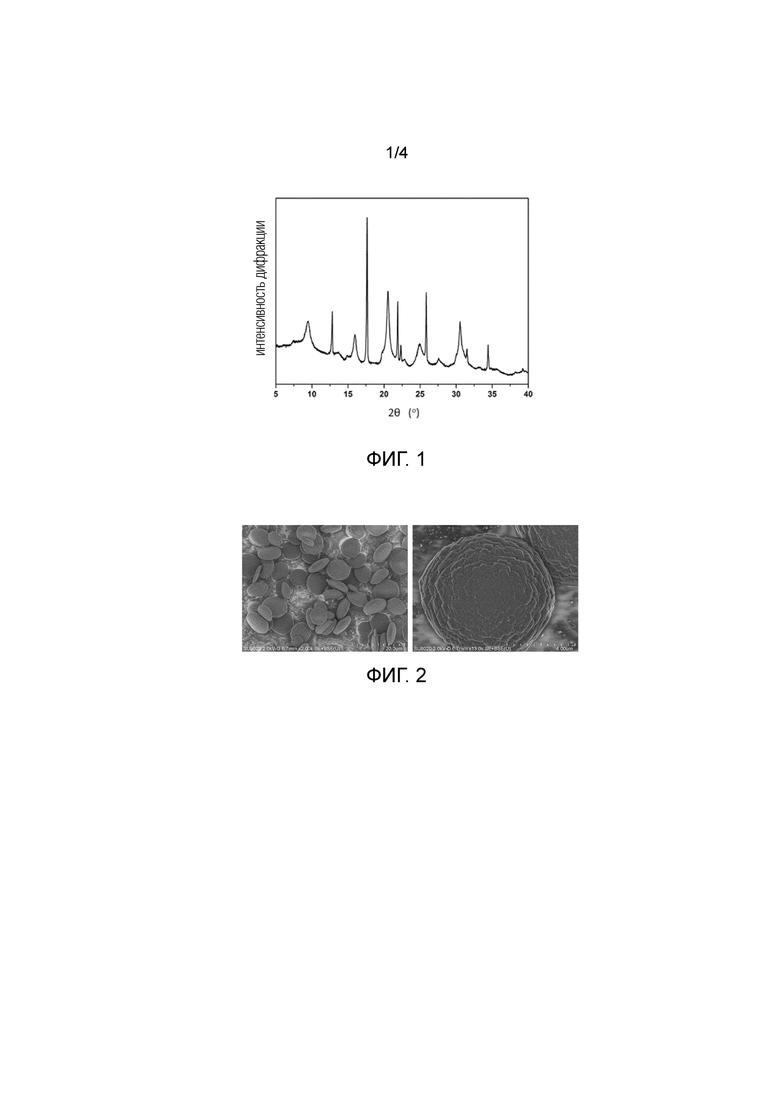
Фиг. 1 представляет собой рентгенограмму XRD продукта, синтезированного в Примере 1.
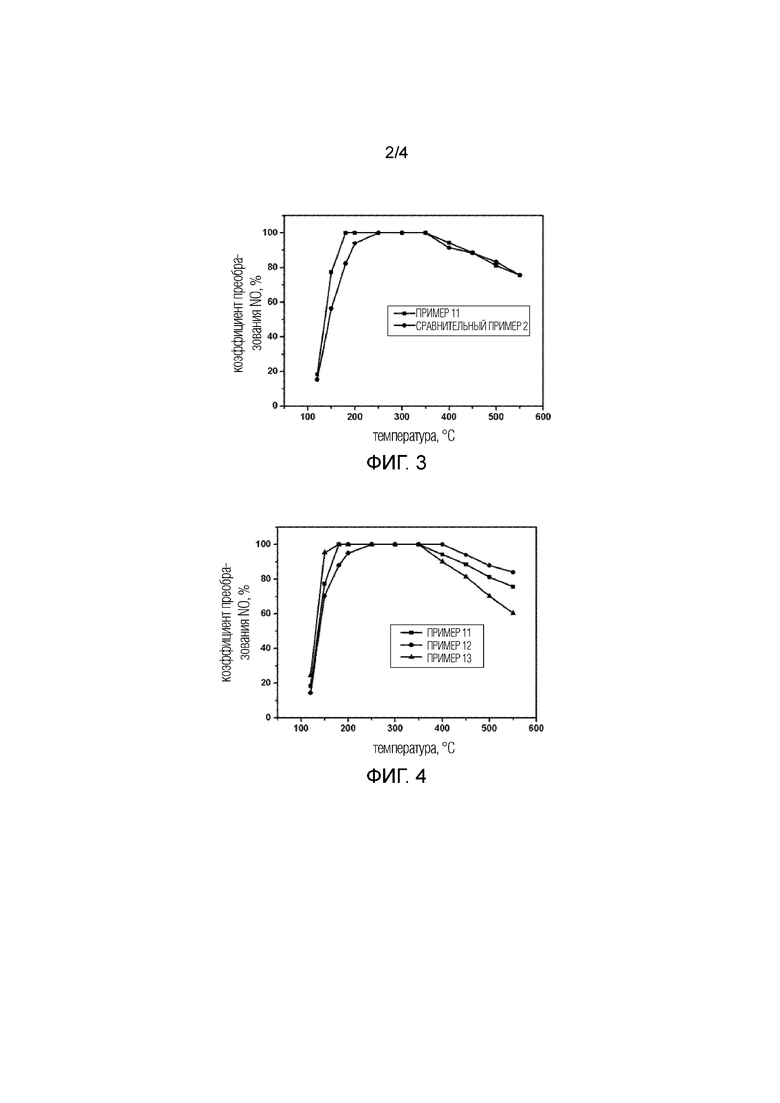
Фиг. 2 представляет собой полученное с помощью сканирующего электронного микроскопа (SEM) изображение синтетического продукта в Примере 1.
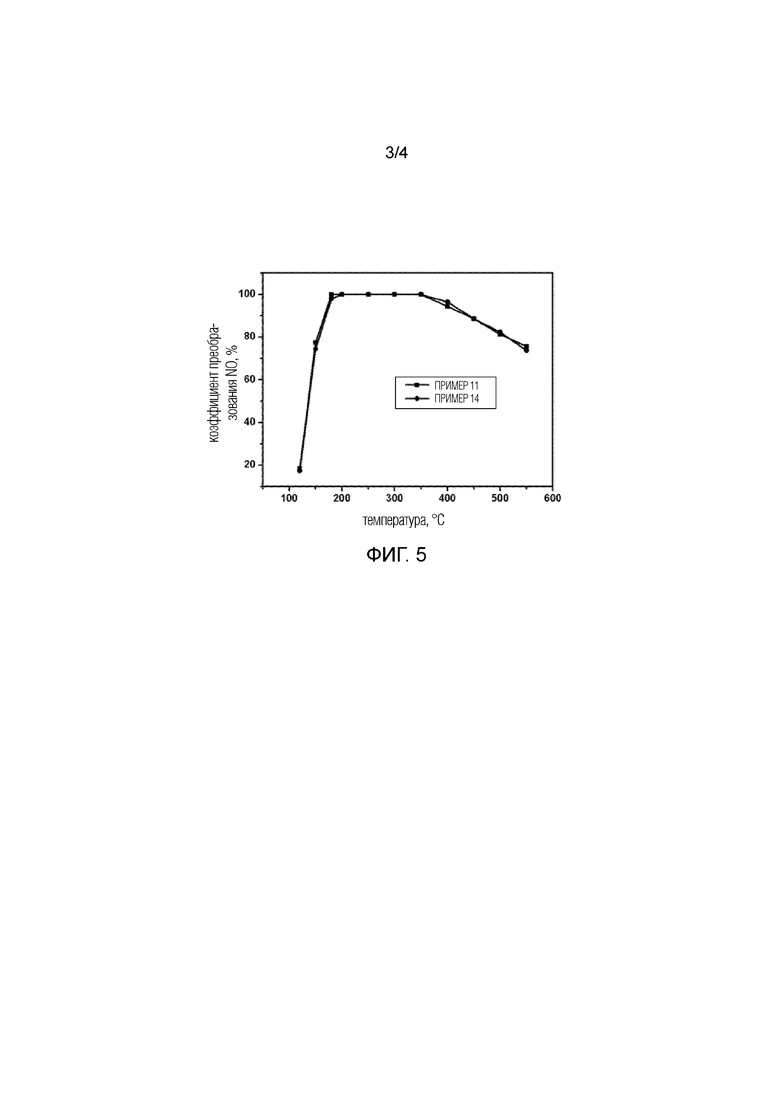
Фиг. 3 показывает результат оценки реакции NH3–SCR Примера 11 и Сравнительного примера 2.
Фиг. 4 показывает результат оценки реакции NH3–SCR катализаторов с различными содержаниями меди (Примеры 11–13).
Фиг. 5 показывает сравнение результатов оценки реакции NH3–SCR до и после высокотемпературной гидротермальной обработки для образца Примера 1 (Примера 11 и Примера 14).
Фиг. 6 показывает результат XRD образцов, соответствующих Сравнительным примерам 3–8.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТА ОСУЩЕСТВЛЕНИЯ
Далее настоящее изобретение будет более конкретно описано со ссылкой на примеры. Следует понимать, что эти примеры являются просто иллюстративными для настоящей патентной заявки и не ограничивают область охвата настоящего изобретения. Экспериментальные способы, которые не определяют особые условия в следующих примерах, как правило выполняются согласно обычным условиям или согласно условиям, рекомендуемым изготовителем. Если явно не указано иное, сырье, используемое в настоящей заявке, приобреталось коммерчески и использовалось напрямую без специальной обработки.
Если явно не указано иное, условия теста, использованные в настоящей патентной заявке, были следующими:
Элементный состав измерялся с использованием рентгеновского флуоресцентного анализатора (XRF) Magix 2424 производства Philips.
Данные FT–IR получались с использованием немецкого прибора BRUKER TENSOR 27.
Рентгеновский порошковый дифракционный фазовый анализ (XRD) выполнялся на рентгеновском дифрактометре X'Pert PRO производства PANalytical Corporation, Нидерланды, с использованием мишени из Cu, источника излучения Kα (λ = 0,15418 нм), напряжения 40 кВ и тока 40 мА.
Удельная площадь поверхности и распределение размера пор образцов определялись с использованием физического адсорбера Micromeritics ASAP Model 2020. Перед анализом образец вакуумировался и нагревался при 350°C для предварительной обработки в течение 6 час, и свободный объем образца измерялся с помощью гелия в качестве среды. Когда образец анализировался, измерения физической адсорбции и десорбции выполнялись при температуре жидкого азота (77 K) с использованием азота в качестве адсорбционного газа. Удельная площадь поверхности материала определялась с использованием формулы BET; полный объем порового пространства материала вычислялся с использованием адсорбированного количества N2 при относительном давлении (P/P0), равном 0,99. Площадь поверхности микропор и объем микропор вычислялись способом t–графика. При вычислении площадь поперечного сечения молекулы N2 составляла 0,162 нм2.
Анализ морфологии с помощью SEM выполнялся с использованием сканирующего электронного микроскопа типа Hitachi (SU8020).
Спектроскоп ядерного магнитного резонанса Infinity plus 400WB производства Varian Corporation, США, использовался для анализа ядерного магнитного резонанса углерода (13C MAS NMR) с зондом BBO MAS и рабочей напряженностью магнитного поля 9,4 Тл.
Элементный анализ CHN выполнялся с использованием элементного анализатора Vario EL Cube немецкого производства.
Далее настоящее изобретение будет подробно описано посредством примеров, но настоящее изобретение не ограничивается этими примерами.
ПРИМЕР 1
Молярная доля каждого исходного материала и условия кристаллизации показаны в Таблице 2. Конкретный процесс смешивания был следующим: сначала источник меди смешивался и растворялся в воде, затем добавлялись R1 и R2 и перемешивались при комнатной температуре в течение 2 час. После этого источник алюминия, источник кремния и источник фосфора последовательно добавлялись к смешанному раствору, и смешанный гель размешивался при комнатной температуре в течение 5 час для того, чтобы сформировать гель. Этот гель затем помещался в реактор из нержавеющей стали. После того, как реактор был помещен в сушильный шкаф, он нагревался со скоростью 2°C/мин до 200°C для кристаллизации при вращении в течение 36 час. После завершения кристаллизации твердый продукт центрифугировался, промывался и сушился на воздухе при 100°C для того, чтобы получить образец исходного порошка молекулярного сита. Этот образец подвергался анализу XRD, и дифрактограмма показывала характеристики широких пиков и узких пиков. Дифрактограмма XRD показана на Фиг. 1, а дифракционные данные XRD показаны в Таблице 3. После того, как образец был прокален, и шаблон был удален, были измерены удельная площадь поверхности и объем порового пространства. Образец имел высокую удельную площадь поверхности по BET, равную 602 м2/г, большой объем порового пространства, равный 0,27 см3/г, в которых удельная площадь поверхности пор и объем микропор, вычисленный согласно способу t–графика, составили 533 м2/г и 0,26 см3/г соответственно.
Полученный с помощью SEM микроснимок полученного образца показан на Фиг. 2. Можно заметить, что морфология полученного образца имеет круглую листовую форму, которая является слоистой и уложенной в стопку, и диапазон размера частиц составляет 3–5 мкм.
Таблица 2: Ингредиенты синтеза молекулярного сита и условия кристаллизации
моль
моль
моль
0,10 моль
моль
моль
моль
0,1 моль
моль
моль
моль
Таблица 3: Результаты XRD образца в Примере 1
ПРИМЕР 2
Конкретная пропорция ингредиентов и условий кристаллизации показаны в Таблице 2, и конкретный процесс смешивания был тем же самым, что и в Примере 1.
Синтезированные образцы были подвергнуты анализу XRD, и результаты показаны в Таблице 4.
Полученные с помощью сканирующего электронного микроскопа микроснимки показали, что морфология полученного образца была подобна морфологии образца Примера 1.
Таблица 4: Результаты XRD образца Примера 2
ПРИМЕР 3
Конкретная пропорция ингредиентов и условий кристаллизации показаны в Таблице 2, и конкретный процесс смешивания был тем же самым, что и в Примере 1.
Синтезированные образцы были подвергнуты анализу XRD, и результаты показаны в Таблице 5.
Полученные с помощью сканирующего электронного микроскопа микроснимки показали, что морфология полученного образца была подобна морфологии образца Примера 1.
Таблица 5: Результаты XRD образца в Примере 3
ПРИМЕРЫ 4–9
Конкретная пропорция ингредиентов и условий кристаллизации показаны в Таблице 2, и конкретный процесс смешивания был тем же самым, что и в Примере 1.
Синтезированные образцы были подвергнуты анализу XRD. Результаты анализа XRD Примеров 4 и 9 были подобны приведенным в Таблице 3. Результаты анализа XRD Примеров 5 и 6 были подобны приведенным в Таблице 4, а результаты анализа XRD Примеров 7 и 8 были близки к Таблице 5.
При сравнении с дифракционным спектром различных соотношений симбиотической кремниево–алюминиевой кристаллической фазы цеолита GME/CHA, приведенным на официальном сайте Международной цеолитной ассоциации, было установлено, что содержание кристаллической фазы CHA в кремниево–фосфорно–алюминиевом молекулярном сите, полученном в Примерах 1–9, является значительно более высоким, чем содержание кристаллической фазы GEM.
ПРИМЕР 10
Был выполнен анализ 13C MAS NMR порошковых образцов Примеров 1–9. При сравнении со стандартными спектрами 13C MAS NMR диизопропаноламина, диэтаноламина и триметиламина было найдено, что образцы, синтезированные с использованием диизопропаноламина в качестве растворителя, имели резонансные пики диизопропаноламина и триметиламина, а образцы, синтезированные с использованием диэтаноламина в качестве растворителя, имели резонансные пики диэтаноламина и триметиламина. Количественный анализ был выполнен на основе конкретных несовпадающих пиков NMR этих двух веществ для того, чтобы определить их соотношение.
Общий элементный состав продукта молекулярного сита был выполнен с помощью анализа XRF, и элементный анализ CHN также проводился для сырого порошка Примеров 1–9. Состав сырого порошка молекулярного сита, полученный с помощью всестороннего элементного анализа CHN, XRF и 13C MAS NMR, показан в Таблице 6.
Таблица 6: Состав образцов сырого порошка в Примерах 1–9
Образцы сырого порошка Примеров 1–9 были отдельно размолоты и спрессованы с бромистым калием, а затем подвергнуты исследованию FT–IR. Все они показали характерный пик вибрационного поглощения, приписываемый двойному шестичленному кольцу, на 637 см–1, показывающий присутствие двойного шестичленного кольца в образце.
ПРИМЕР 11
Образец, полученный в Примере 1, был прокален при высокой температуре, равной 650°C, в течение 2 час. После того, как агент сборки по шаблону был удален, была оценена каталитическая эффективность для селективного каталитического восстановления NOx с помощью NH3. Конкретные процедуры и условия эксперимента были следующими: после обжига образец таблетировался и просеивался. 0,1 г образца фракции 60–80 меш было взвешено и смешано с 0,4 г кварца (фракции 60–80 меш), и загружено в реактор неподвижного слоя. Катализаторы были продуты азотом при 600°C в течение 40 мин, затем температура была понижена до 120°C для начала оценки, и температура постепенно повышалась до 550°C. Компоненты сырого газа были следующими: NO: 500 частей на миллион, NH3: 500 частей на миллион, O2: 5%, H2O: 5%, с остатком из N2, и скорость потока газа составляла 300 мл/мин. Отходящий после реакции газ анализировался в реальном масштабе времени с помощью FTIR с использованием прибора Bruker Tensor 27. Результаты показаны на Фиг. 3 и на Фиг. 4. Можно заметить, что преобразование NO составило 77% при 150°C, и преобразование NO было выше 90% в широком диапазоне температур, составляющем 180–450°C. Аналогичным образом образцы, полученные в Примерах 2–8, показали превосходную каталитическую эффективность для селективного восстановления NOx после той же самой обработки, что и для образца Примера 1.
ПРИМЕР 12
Образец, полученный в Примере 3, был прокален при высокой температуре, равной 650°C, в течение 2 час. После того, как агент сборки по шаблону был удален, была оценена каталитическая эффективность для селективного каталитического восстановления NOx с помощью NH3. Конкретные процедуры и условия эксперимента были следующими: после обжига образец таблетировался и просеивался. 0,1 г образца фракции 60–80 меш было взвешено и смешано с 0,4 г кварца (фракции 60–80 меш), и загружено в реактор неподвижного слоя. Катализаторы были продуты азотом при 600°C в течение 40 мин, затем температура была понижена до 120°C для начала оценки, и температура постепенно повышалась до 550°C. Компоненты сырого газа были следующими: NO: 500 частей на миллион, NH3: 500 частей на миллион, O2: 5%, H2O: 5%, с остатком из N2, и скорость потока газа составляла 300 мл/мин. Отходящий после реакции газ анализировался в реальном масштабе времени с помощью FTIR с использованием прибора Bruker Tensor 27. Результаты реакции показаны на Фиг. 4.
ПРИМЕР 13
Образец, полученный в Примере 8, был прокален при высокой температуре, равной 650°C, в течение 2 час. После того, как агент сборки по шаблону был удален, была оценена каталитическая эффективность для селективного каталитического восстановления NOx с помощью NH3. Конкретные процедуры и условия эксперимента были следующими: после обжига образец таблетировался и просеивался. 0,1 г образца фракции 60–80 меш было взвешено и смешано с 0,4 г кварца (фракции 60–80 меш), и загружено в реактор неподвижного слоя. Катализаторы были продуты азотом при 600°C в течение 40 мин, затем температура была понижена до 120°C для начала оценки, и температура постепенно повышалась до 550°C. Компоненты сырого газа были следующими: NO: 500 частей на миллион, NH3: 500 частей на миллион, O2: 5%, H2O: 5%, с остатком из N2, и скорость потока газа составляла 300 мл/мин. Отходящий после реакции газ анализировался в реальном масштабе времени с помощью FTIR с использованием прибора Bruker Tensor 27. Результаты реакции показаны на Фиг. 4.
ПРИМЕР 14
Образец, полученный в Примере 1, был прокален при высокой температуре, равной 650°C, в течение 2 час. После того, как агент сборки по шаблону был удален, гидротермальная обработка старения была выполнена при 800°C, содержание водяного пара составляло 100%, а время обработки – 24 час. После обработки образец был высушен при 100°C.
Относительная кристалличность образца была определена способом XRD, и кристалличность образца составила 95% по сравнению с Примером 1, что означает, что образец, приготовленный в Примере 1, имел высокую гидротермальную стабильность, и целостность структуры образца может быть хорошо сохранена после водной обработки.
Была оценена каталитическая эффективность для селективного каталитического восстановления NOx с помощью NH3. Конкретные процедуры и условия эксперимента были следующими: после обжига образец таблетировался и просеивался. 0,1 г образца фракции 60–80 меш было взвешено и смешано с 0,4 г кварца (фракции 60–80 меш), и загружено в реактор неподвижного слоя. Катализаторы были продуты азотом при 600°C в течение 40 мин, затем температура была понижена до 120°C для начала оценки, и температура постепенно повышалась до 550°C. Компоненты сырого газа были следующими: NO: 500 частей на миллион, NH3: 500 частей на миллион, O2: 5%, H2O: 5%, с остатком из N2, и скорость потока газа составляла 300 мл/мин. Отходящий после реакции газ анализировался в реальном масштабе времени с помощью FTIR с использованием прибора Bruker Tensor 27. Результаты реакции показаны на Фиг. 5.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 1
10 г образца сырого порошка молекулярного сита, полученного в Примере 9, использовались в качестве прекурсора и были нагреты со скоростью 2°C/мин до постоянной температуры 600°C на 4 час, чтобы удалить органический агент сборки по шаблону и воду, содержавшуюся в нем.
Прокаленный образец был помещен в водный раствор азотнокислого аммония с концентрацией 3,66 моль/л в соотношении твердое–жидкость (в массовом соотношении) 1:10, перемешивался 5 мин, а затем был нагрет до 80°C для ионообмена в течение 2 час. После центрифугирования и промывки три раза деионизированной водой и сушки при 80°C было получено молекулярное сито NH4+.
7 г молекулярного сита NH4+ были помещены в раствор Cu(CH3COO)2·H2O с концентрацией 0,03 моль/л в соотношении твердое–жидкость 1:25. После перемешивания в течение 5 мин эта смесь была нагрета до 50°C для ионообмена в течение 4 час. После центрифугирования и промывки три раза деионизированной водой и сушки при 80°C полученный образец был обозначен как Cu–9/T. Элементный анализ XRF показал, что продукт имеет содержание оксида меди 3,2%, что близко к Примеру 1. Удельная площадь поверхности и объем порового пространства прокаленного образца Примера 9 и образца Cu–9/T были измерены с помощью физической адсорбции N2, и удельная площадь поверхности пор и объем порового пространства были вычислены способом t–графика. Удельная площадь поверхности микропор и объем микропор образца Примера 9 составили 559 м2/г и 0,28 см3/г, соответственно. Удельная площадь поверхности микропор и объем микропор образца Cu–9/T составили 520 м2/г и 0,25 см3/г, соответственно. Эти результаты показывают, что катализатор, приготовленный согласно способу Примера 1, может лучше поддерживать регулярность структуры каркаса образца.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 2
Образец, полученный в Сравнительном примере 1, был прокален при высокой температуре, равной 650°C, в течение 2 час, и использовался в качестве катализатора для реакции селективного восстановления NOx с помощью NH3. Конкретные процедуры и условия эксперимента были следующими: после обжига образец таблетировался и просеивался. 0,1 г образца фракции 60–80 меш было взвешено и смешано с 0,4 г кварца (фракции 60–80 меш), и загружено в реактор неподвижного слоя. Катализаторы были продуты азотом при 600°C в течение 40 мин, затем температура была понижена до 120°C для начала оценки, и температура постепенно повышалась до 550°C. Компоненты сырого газа были следующими: NO: 500 частей на миллион, NH3: 500 частей на миллион, O2: 5%, H2O: 5%, с остатком из N2, и полная скорость потока газа составляла 300 мл/мин. Полная объемная скорость реакции составляла GHSV=180000 час–1. Отходящий после реакции газ анализировался в реальном масштабе времени с помощью FTIR с использованием прибора Bruker Tensor 27. Конкретные результаты показаны на Фиг. 3.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 3
Молярное соотношение конкретных ингредиентов, исходные материалы и условия кристаллизации были теми же самыми, что и в Примере 1, за исключением того, что диэтаноламин в исходном материале был заменен на триэтиламин. Синтезированный образец представлял собой молекулярное сито SAPO–34, и результаты анализа XRD показаны на Фиг. 6.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 4
Молярное соотношение конкретных ингредиентов, исходные материалы и условия кристаллизации были теми же самыми, что и в Примере 2, за исключением того, что бензилтриметиламмонийгидроксид в исходном материале был заменен на 1,6–гександиамин. Синтезированный образец представлял собой ламеллярную фазу, и результаты XRD показаны на Фиг. 6.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 5
Молярное соотношение конкретных ингредиентов, исходные материалы и условия кристаллизации были теми же самыми, что и в Примере 3, за исключением того, что добавление триметиламина в исходный материал не выполнялось. Синтезированный образец представлял собой физическую смесь SAPO–34 и SAPO–5, и результаты XRD показаны на Фиг. 6.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 6
Молярное соотношение конкретных ингредиентов, исходные материалы и условия кристаллизации были теми же самыми, что и в Примере 4, за исключением того, что диэтаноламин в исходном материале был заменен на диэтиламин. Синтезированный образец представлял собой физическую смесь DNL–6 (молекулярное сито SAPO со структурой RHO) с небольшим количеством SAPO–34, и результаты XRD показаны на Фиг. 6.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 7
Молярное соотношение конкретных ингредиентов, исходные материалы и условия кристаллизации были теми же самыми, что и в Примере 5, за исключением того, что триметиламин в исходном материале был заменен на триэтаноламин. Синтезированный образец представлял собой физическую смесь SAPO–5 и SAPO–34, и результаты XRD показаны на Фиг. 6.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 8
Молярное соотношение конкретных ингредиентов, исходные материалы и условия кристаллизации были теми же самыми, что и в Примере 5, за исключением того, что добавление триметиламина в исходный материал не выполнялось. Синтезированный образец был аморфным, и результаты анализа XRD показаны на Фиг. 6.
Результаты синтеза Сравнительных примеров 3–8 показали, что молекулярное сито Cu–SAPO с сосуществующими кристаллическими фазами CHA и GME настоящей патентной заявки может быть получено только при конкретной комбинации агентов сборки по шаблону и подходящих условиях кристаллизации.
В то время как настоящая патентная заявка была описана выше со ссылкой на предпочтительные варианты осуществления, эти варианты осуществления не предназначены для ограничения формулы изобретения. Специалисты в данной области техники будут в состоянии, не отступая от духа настоящей патентной заявки, сделать несколько возможных вариаций и модификаций, и таким образом область защиты настоящего изобретения должна определяться областью его охвата, определяемой формулой изобретения.

Раскрыты молекулярное сито Cu–SAPO с сосуществующими кристаллическими фазами CHA и GME, процесс его синтеза и его использование в реакции денитрирования. Дифракционный рисунок XRD молекулярного сита Cu–SAPO имеет сосуществующие широкие пики и узкие пики. Это синтезируемое молекулярное сито может использоваться в качестве катализатора для селективного восстановления NOx. Способ для синтеза молекулярного сита содержит стадии: a) смешивания источника меди, деионизированной воды, агентов сборки по шаблону R1 и R2, источника кремния, источника алюминия и источника фосфора в такой пропорции, чтобы получить начальную гелевую смесь, имеющую следующие молярные отношения: Cu/Al2O3=0,01-0,25; SiO2/Al2O3=0,05-2,0; P2O5/Al2O3=0,5-1,5; H2O/Al2O3=8-40; R1/Al2O3=5-20; R2/Al2O3=0,1-1,5; где R1 – диизопропаноламин (DIPA) или диэтаноламин (DEOA); R2 – любой из триметиламина (TMA), бензилтриметиламмонийхлорида (BTACl), бензилтриметиламмонийгидроксида (BTAOH) или их смеси; b) подачи начальной гелевой смеси, полученной на стадии a), в реактор синтеза высокого давления, закрытия реактора, нагревания до 160-220°C и выполнения кристаллизации в течение 5–72 час; и c) после завершения кристаллизации отделения твердого продукта, его промывки и сушки для того, чтобы получить молекулярное сито. Технический результат – новый цеолит показывает превосходную денитрирующую каталитическую активность и имеет потенциальную прикладную ценность. 3 н. и 7 з.п. ф-лы, 6 ил., 6 табл., 14 пр.
1. Содержащее медь силикоалюмофосфатное (SAPO) молекулярное сито с сосуществующими кристаллическими фазами CHA и GME, в котором дифракционная рентгенограмма молекулярного сита содержит по меньшей мере следующие дифракционные пики:
2. Молекулярное сито по п. 1, в котором неорганический каркас молекулярного сита имеет следующий химический состав: wCu–(SixAlyPz)O2, где x, y и z соответственно представляют мольные доли Si, Al и P, диапазоны их мольных долей соответственно составляют x=0,01-0,28, y=0,35-0,55 и z=0,28-0,50, причем x+y+z=1, w является количеством молей Cu на моль (SixAlyPz)O2, и w=0,001-0,124.
3. Молекулярное сито по п. 1, в котором безводный химический состав молекулярного сита, содержащего агент сборки по шаблону, выражается как wCu·mR1·nR3·(SixAlyPz)O2, где R1 – диизопропаноламин или диэтаноламин, R3 – триметиламин; m – количество молей агента сборки по шаблону R1 на моль (SixAlyPz)O2, n – количество молей агента сборки по шаблону R3 на моль (SixAlyPz)O2, m=0,01-0,20, n=0,01-0,10; x, y и z соответственно представляют мольные доли Si, Al и P, и диапазоны этих мольных долей соответственно составляют x=0,01-0,28, y=0,35-0,55 и z=0,28-0,50, причем x+y+z=1; w – количество молей Cu на моль (SixAlyPz)O2, w=0,001-0,124.
4. Способ для синтеза молекулярного сита по любому из пп. 1–3, содержащий стадии:
a) смешивания источника меди, деионизированной воды, агентов сборки по шаблону R1 и R2, источника кремния, источника алюминия и источника фосфора в такой пропорции, чтобы получить начальную гелевую смесь, имеющую следующие молярные отношения:
Cu/Al2O3=0,01-0,25;
SiO2/Al2O3=0,05-2,0;
P2O5/Al2O3=0,5-1,5;
H2O/Al2O3=8-40;
R1/Al2O3=5-20;
R2/Al2O3=0,1-1,5;
где R1 – диизопропаноламин (DIPA) или диэтаноламин (DEOA); R2 – любой из триметиламина (TMA), бензилтриметиламмонийхлорида (BTACl), бензилтриметиламмонийгидроксида (BTAOH) или их смеси;
b) подачи начальной гелевой смеси, полученной на стадии a), в реактор синтеза высокого давления, закрытия реактора, нагревания до 160-220°C и выполнения кристаллизации в течение 5–72 час; и
c) после завершения кристаллизации отделения твердого продукта, его промывки и сушки для того, чтобы получить молекулярное сито.
5. Способ по п. 4, в котором на стадии a) процесс смешивания является следующим: сначала источник меди смешивают с водой, затем добавляют R1 и R2, перемешивают при комнатной температуре в течение 0,5-5 час, затем к смешанному раствору добавляют источник алюминия, источник кремния и источник фосфора и смешанный гель перемешивается при комнатной температуре в течение 1-5 час.
6. Способ по п. 4, в котором на стадии a) источник кремния выбирают из одного или более из золя кремнезема, активированного кремнезема, ортосиликата и метакаолина; источник алюминия выбирают из одного или более из соли алюминия, активированного глинозема, псевдобемита, алкоксиалюминия и метакаолина; источник фосфора выбирают из одного или более из ортофосфорной кислоты, гидрофосфата аммония, дигидрофосфата аммония, органических фосфорсодержащих соединений и фосфорных оксидов; источник меди выбирают из одного или более из Cu(OAc)2, CuSO4, Cu(NO3)2 и CuCl2.
7. Способ по п. 4, в котором на стадии b) кристаллизацию выполняют в статическом или динамическом состоянии.
8. Способ по п. 4, в котором на стадии a) начальная гелевая смесь имеет R1/Al2O3=5,0-10.
9. Способ по п. 4, в котором на стадии a) начальная гелевая смесь имеет R2/Al2O3=0,25-1,0.
10. Катализатор для селективного каталитического восстановления NOx, который получают путем обжига на воздухе при температуре 550-700°C молекулярного сита по любому из пп. 1-3 или молекулярного сита, синтезированного в соответствии со способом по любому из пп. 4–9.
| CN 102869427 B, 09.11.2016 | |||
| НОСИТЕЛИ НА ОСНОВЕ НЕУПОРЯДОЧЕННЫХ МОЛЕКУЛЯРНЫХ СИТ ДЛЯ СЕЛЕКТИВНОГО КАТАЛИТИЧЕСКОГО ВОССТАНОВЛЕНИЯ NOx | 2011 |
|
RU2593989C2 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Alessandro Turrina, Raquel Garcia et al | |||
| Retrosynthetic Co-Templating Method for the Preparation of Silicoaluminophosphate Molecular Sieves | |||
| Chem | |||
| Mater | |||
| Токарный резец | 1924 |
|
SU2016A1 |

