Настоящая заявка испрашивает приоритет по патентной заявке Китая (заявка № CN202010219601.7), поданной 25 марта 2020 года, и патентной заявке Китая (заявка № CN202110287012.7), поданной 17 марта 2021 года.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтических препаратов и, в частности, относится к фармацевтической композиции, содержащей конъюгат антитела и лекарственного средства и ее применению в качестве противоракового лекарственного препарата.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Изложенное в данном документе представляет собой лишь общую информацию, относящуюся к настоящему изобретению, и необязательно может представлять собой известный уровень техники.
Конъюгат антитела и лекарственного средства (ADC) связывает моноклональное антитело или фрагмент антитела с биологически активным цитотоксином через стабильное химическое линкерное соединение, полностью используя специфичность связывания антитела с поверхностными антигенами нормальных клеток и опухолевых клеток и высокую эффективность цитотоксина, а также устраняя недостаток первого, связанный с тем, что оно имеет плохой терапевтический эффект, недостаток последнего, связанный с тем, что оно имеет серьезные токсические побочные эффекты, и т. п. Это означает, что конъюгат антитела и лекарственного средства может более точно связываться с опухолевыми клетками и оказывает сниженное влияние на нормальные клетки по сравнению с общепринятыми химиотерапевтическими лекарственными средствами в прошлом (Mullard A, (2013) Nature Reviews Drug Discovery, 12:329-332; DiJoseph JF, Armellino DC, (2004) Blood, 103:1807-1814).
Милотарг® (гемтузумаб озогамицин, Wyeth Pharmaceutical Co.,Ltd.), первый конъюгат антитела и лекарственного средства, был одобрен FDA США (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) в 2000 году для лечения острого миелоцитарного лейкоза (Drugs of the Future (2000) 25(7):686; US4970198; US5079233; US5585089; US5606040; US5693762; US5739116; US5767285; US5773001).
Адцетрис® (брентуксимаб ведотин, Seattle Genetics) был одобрен исследованием по ускоренной процедуре, разработанным FDA США, в августе 2011 года для лечения лимфомы Ходжкина и рецидивирующей анапластической крупноклеточной лимфомы (Nat. Biotechnol (2003) 21(7):778-784; WO2004010957; WO2005001038; US7090843A; US7659241; WO2008025020). Адцетрис® представляет собой новое ADC-направленное лекарственное средство, которое может позволить лекарственному средству непосредственно воздействовать на CD30-мишень на клетках лимфомы, а затем проводить эндоцитоз, чтобы индуцировать апоптоз опухолевых клеток.
И Милотарг®, и Адцетрис® являются средствами терапии, направленными на гематологические опухоли, которые относительно просты по структуре ткани по сравнению с солидными опухолями. Кадцила® (адо-трастузумаб эмтанзин, T-DM1) был одобрен FDA США в феврале 2013 г. для лечения HER2-положительных пациентов с запущенным или метастатическим раком молочной железы и имеющих лекарственную резистентность к тратузумабу (торговое название: Герцептин) и паклитакселу (WO2005037992; US8088387). Кадцила® является первым ADC лекарственным средством, одобренным FDA США для лечения солидных опухолей.
Существует несколько классов малых молекул с цитотоксичностью для конъюгатов антитела и лекарственного средства, одним из которых являются производные камптотецина, обладающие противоопухолевым действием путем ингибирования топоизомеразы I. В WO2014057687; Clinical Cancer Research (2016) 22(20): 5097-5108; и Cancer Sci (2016) 107: 1039-1046 сообщалось, что производное камптотецина эксатекан (химическое название: (1S,9S)-1-амино-9-этил-5-фтор-2,3-дигидро-9-гидрокси-4-метил-1H,12H-бензо[де]пирано[3',4':6,7]имидазо[1,2-b]хинолин-10,13(9H,15H)-диона) применяли к конъюгатам антитела и лекарственного средства (ADC). По-прежнему существует необходимость в дальнейшей разработке ADC-лекарсвтенных средств с лучшими терапевтическими эффектами.
Однако ADC имеют более сложные гетероструктуры, чем антитела, и, следовательно, для ADC-препаратов для терапевтических целяей была поставлена более сложная задача.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложена фармацевтическая композиция, содержащая конъюгат антитела и лекарственного средства и буфер, где конъюгат антитела и лекарственного средства имеет структуру общей формулы (Pc-L-Y-D):
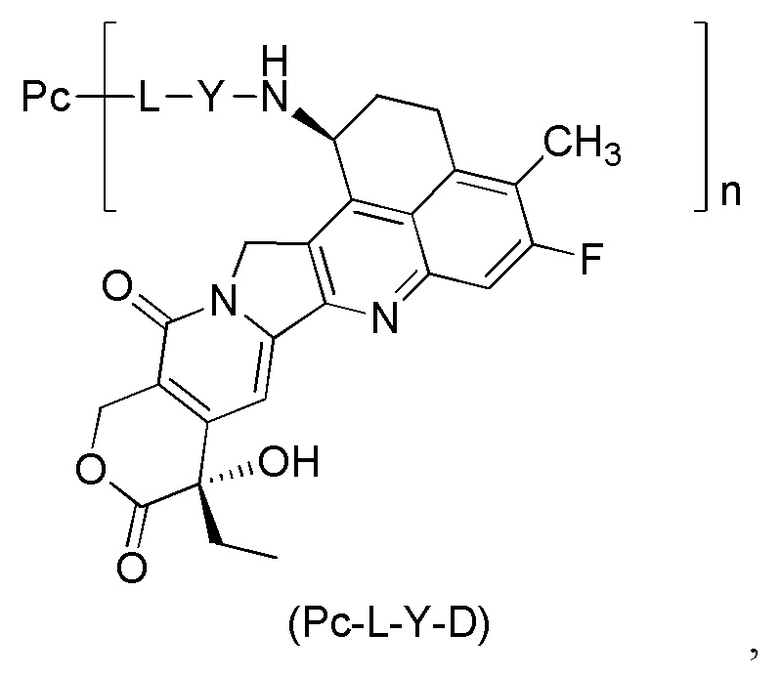
где:
Y выбран из группы, состоящей из -O-(CRaRb)m-CR1R2-C(O)-, -O-CR1R2-(CRaRb)m-, -O-CR1R2-, -NH-(CRaRb)m-CR1R2-C(O)- и -S-(CRaRb)m-CR1R2-C(O);
Ra и Rb являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, дейтерия, галогена, алкила, галогеналкила, дейтерированного алкила, алкокси, гидрокси, амино, циано, нитро, гидроксиалкила, циклоалкила и гетероциклила;
или Ra и Rb, вместе с атомами углерода, связанными с ними, образуют циклоалкил или гетероциклил;
R1 выбран из группы, состоящей из галогена, галогеналкила, дейтерированного алкила, циклоалкила, циклоалкилалкила, алкоксиалкила, гетероциклила, арила и гетероарила;
R2 выбран из группы, состоящей из водорода, галогена, галогеналкила, дейтерированного алкила, циклоалкила, циклоалкилалкила, алкоксиалкила, гетероциклила, арила и гетероарила;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют циклоалкил или гетероциклил;
или, Ra и R2, вместе с атомами углерода, присоединенными с ними, образуют циклоалкил или гетероциклил;
m представляет собой целое число от 0 до 4;
n представляет собой десятичное или целое число от 1 до 10;
L представляет собой линкерный фрагмент;
Pc представляет собой антитело или его антигенсвязывающий фрагмент;
причем указанная композиция имеет рН от около 4,5 до около 6,0, предпочтительно рН от около 4,8 до около 5,3 и более предпочтительно рН от около 5,0 до около 5,1.
В альтернативном варианте осуществления буфер в фармацевтической композиции имеет рН от около 4,5 до около 6,0; неограничивающие примеры включают около 4,5, около 4,6, около 4,7, около 4,8, около 4,9, около 5,0, около 5,1, около 5,2, около 5,3, около 5,4, около 5,5, около 5,6, около 5,7, около 5,8, около 5,9 и около 6,0; от около 4,8 до около 5,3 является предпочтительным, и от около 5,0 до около 5,1 является более предпочтительным. В некоторых вариантах осуществления фармацевтическая композиция имеет рН от 4,5 до 5,2, предпочтительно рН от 4,8 до 5,2 и более предпочтительно рН от 5,0 до 5,1. В некоторых вариантах осуществления фармацевтическая композиция имеет рН 5,0.
В альтернативном варианте осуществления фармацевтическая композиция дополнительно содержит поверхностно-активное вещество, которое может быть выбрано из группы, состоящей из полисорбата, полисорбата 20, полисорбата 80, полоксамера, тритона, додецилсульфоната натрия, лаурилсульфоната натрия, октилгликозида натрия, лаурилсульфобетаина, миристил-сульфобетаина, линолеил-сульфобетаина, стеарил-сульфобетаина, лаурил-саркозина, миристил-саркозина, линолеил-саркозина, стеарил-саркозина, линолеил-бетаина, миристил-бетаина, цетил-бетаина, лаурамидопропил-бетаина, кокарамидопропил-бетаина, линолеинамидопропил-бетаина, миристиламидопропил-бетаина, пальмитамидопропил-бетаина, изостеарамидопропил-бетаина, миристиламидопропил-диметиламина, пальмитамидопропил-диметиламина, изостеарамидопропил-диметиламина, метилкокоила натрия, метилолеилтаурата натрия, полиэтиленгликоля, полипропиленгликоля, сополимера этилена и пропиленгликоля и т.п. Поверхностно-активное вещество предпочтительно представляет собой полисорбат 80 или полисорбат 20, более предпочтительно полисорбат 80.
В альтернативном варианте осуществления поверхностно-активное вещество в фармацевтической композиции присутствует в концентрации от около 0,01 мг/мл до около 1,0 мг/мл. В альтернативном варианте осуществления поверхностно-активное вещество в фармацевтической композиции присутствует в концентрации от около 0,05 мг/мл до около 0,5 мг/мл, предпочтительно от около 0,1 мг/мл до около 0,3 мг/мл, от около 0,2 мг/мл до около 0,6 мг/мл, от около 0,2 мг/мл до около 0,5 мг/мл или от около 0,2 мг/мл до около 0,3 мг/мл, и более предпочтительно около 0,2 мг/мл; неограничивающие примеры включают 0,1 мг/мл, 0,15 мг/мл, 0,2 мг/мл, 0,25 мг/мл, 0,3 мг/мл, 0,35 мг/мл, 0,4 мг/мл, 0,45 мг/мл, 0,5 мг/мл и 0,6 мг/мл.
В альтернативном варианте осуществления вышеупомянутая фармацевтическая композиция дополнительно содержит сахарид. "Сахарид" по настоящему изобретению включает общую композицию (CH2O)n и ее производные, включая моносахариды, дисахариды, трисахариды, полисахариды, сахарные спирты, восстанавливающие сахара, невосстанавливающие сахара и т.д. Сахарид может быть выбран из группы, состоящей из глюкозы, сахарозы, трегалозы, лактозы, фруктозы, мальтозы, декстрана, глицерина, эритрита, глицерина, арабитола, силита, сорбита, маннита, меллибиозы, мелезитозы, раффинозы, маннотриозы, стахиозы, мальтозы, лактулозы, мальтулозы, глюцита, мальтита, лактита, изомальтулозы и т.д. Сахарид предпочтительно представляет собой невосстанавливающий дисахарид, более предпочтительно трегалозу или сахарозу и наиболее предпочтительно сахарозу.
В альтернативном варианте осуществления сахарид в вышеупомянутых фармацевтических композициях присутствует в концентрации от около 60 мг/мл до около 90 мг/мл; неограничивающие примеры включают 60 мг/мл, 65 мг/мл, 70 мг/мл, 75 мг/мл, 80 мг/мл, 85 мг/мл и 90 мг/мл; предпочтительно 80 мг/мл. В некоторых вариантах осуществления сахарид присутствует в концентрации от 70 мг/мл до 90 мг/мл.
В альтернативном варианте осуществления конъюгат антитела и лекарственного средства в фармацевтической композиции имеет концентрацию от около 1 мг/мл до около 100 мг/мл; неограничивающие примеры включают 1 мг/мл, 10 мг/мл, 11 мг/мл, 12 мг/мл, 13 мг/мл, 14 мг/мл, 15 мг/мл, 16 мг/мл, 17 мг/мл, 18 мг/мл, 19 мг/мл, 20 мг/мл, 21 мг/мл, 22 мг/мл, 23 мг/мл, 24 мг/мл, 25 мг/мл, 26 мг/мл, 27 мг/мл, 28 мг/мл, 29 мг/мл, 30 мг/мл, 40 мг/мл, 50 мг/мл, 60 мг/мл, 70 мг/мл, 80 мг/мл, 90 мг/мл и 100 мг/мл; предпочтительно от около 10 мг/мл до около 30 мг/мл и более предпочтительно от около 20 мг/мл до около 22 мг/мл. В частности, неограничивающие примеры включают 20,1 мг/мл, 20,2 мг/мл, 20,3 мг/мл, 20,4 мг/мл, 20,5 мг/мл, 20,6 мг/мл, 20,7 мг/мл, 20,8 мг/мл, 20,81 мг/мл, 20,82 мг/мл, 20,83 мг/мл, 20,84 мг/мл, 20,85 мг/мл, 20,86 мг/мл, 20,87 мг/мл, 20,88 мг/мл, 20,89 мг/мл, 20,9 мг/мл, 20,9 мг/мл, 20,91 мг/мл, 20,92 мг/мл, 20,93 мг/мл, 20,94 мг/мл, 20,95 мг/мл, 20,96 мг/мл, 20,97 мг/мл, 20,98 мг/мл, 20,99 мг/мл и 21 мг/мл. В альтернативном варианте осуществления конъюгат антитела и лекарственного средства в фармацевтической композиции присутствует в концентрации от около 10 мг/мл до около 30 мг/мл, предпочтительно около 20 мг/мл, на основе голого антитела (т.е. части антитела ADC).
В альтернативном варианте осуществления буфер в вышеупомянутой фармацевтической композиции выбран из группы, состоящей из гистидинового солевого буфера, сукцинатного буфера и цитратного буфера, предпочтительно сукцинатного буфера и более предпочтительно буфера янтарная кислота-сукцинат натрия.
В альтернативном варианте осуществления буфер в фармацевтической композиции присутствует в концентрации от около 5 мМ до около 50 мМ; неограничивающие примеры включают 1 мМ, 5 мМ, 6 мМ, 7 мМ, 8 мМ, 9 мМ, 10 мМ, 11 мМ, 12 мМ, 13 мМ, 14 мМ, 15 мМ, 16 мМ, 17 мМ, 18 мМ, 19 мМ, 20 мМ, 30 мМ, 40 мМ и 50 мМ; предпочтительно от около 5 мМ до около 20 мМ и наиболее предпочтительно около 10 мМ.
В альтернативном варианте осуществления содержание лекарственного средства (n) может составлять от 3 до 8, от 4 до 8, от 5 до 7, более предпочтительно от 5,3 до 6,1, 5,7 цитотоксических лекарственных средств, связывающихся с каждым антителом или его антигенсвязывающим фрагментом (Pc), n представляет собой десятичное или целое число.
В альтернативном варианте осуществления фармацевтическая композиция содержит:
(a) конъюгат антитела и лекарственного средства в концентрации от около 10 мг/мл до около 30 мг/мл, (b) полисорбат в концентрации от около 0,05 мг/мл до около 0,5 мг/мл, (c) сахарид в концентрации от около 60 мг/мл до около 90 мг/мл и (d) буфер в концентрации от около 5 мМ до около 20 мМ; причем фармацевтическая композиция имеет рН от около 4,8 до около 5,3.
В альтернативном варианте осуществления фармацевтическая композиция содержит:
(a) конъюгат антитела и лекарственного средства в концентрации от около 10 мг/мл до около 30 мг/мл, (b) полисорбат в концентрации от около 0,05 мг/мл до около 0,5 мг/мл, (c) сахарид в концентрации от около 60 мг/мл до около 90 мг/мл и (d) буфер в концентрации от около 5 мМ до около 20 мМ; причем фармацевтическая композиция имеет рН 4,8-5,2.
В альтернативном варианте осуществления фармацевтическая композиция содержит:
(a) конъюгат антитела и лекарственного средства в концентрации от около 20 мг/мл до около 22 мг/мл, (b) полисорбат 80 в концентрации около 0,2 мг/мл, (c) сахароза в концентрации 80 мг/мл и (d) сукцинатный буфер в концентрации 10 мМ; причем фармацевтическая композиция имеет рН от около 5,0 до около 5,1. В некоторых вариантах осуществления фармацевтическая композиция имеет рН 5,0-5,1.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
-Y- представляет собой -O-(CRaRb)m-CR1R2-C(O)-;
Ra и Rb являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, дейтерия, галогена и алкила;
R1 представляет собой C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
m представляет собой 0 или 1.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
-Y- представляет собой -O-(CH2)m-CR1R2-C(O)-;
R1 представляет собой C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
m представляет собой 0 или 1.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
-Y- представляет собой -O-(CH2)m-CR1R2-C(O)-;
R1 представляет собой C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 представляет собой водород;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
m представляет собой 0 или 1.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
-Y- представляет собой -O-(CH2)m-CR1R2-C(O)-;
R1 представляет собой C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 представляет собой водород;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
m представляет собой 0.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
-Y- выбран из группы, состоящей из:
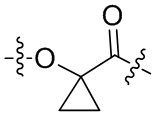 ,
, 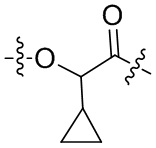 ,
, 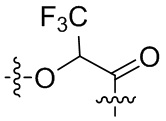 ,
, 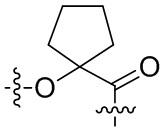 ,
, 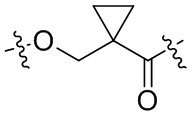 ,
, 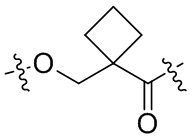 и
и 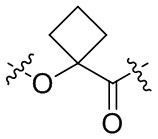 .
.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
О-конец Y присоединен к линкерному фрагменту L.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства,
-Y- выбран из группы, состоящей из:
, , , , , и .
В альтернативном варианте осуществления вышеупомянутый конъюгат антитела и лекарственного средства имеет структуру общей формулы (Pc-L-D1):
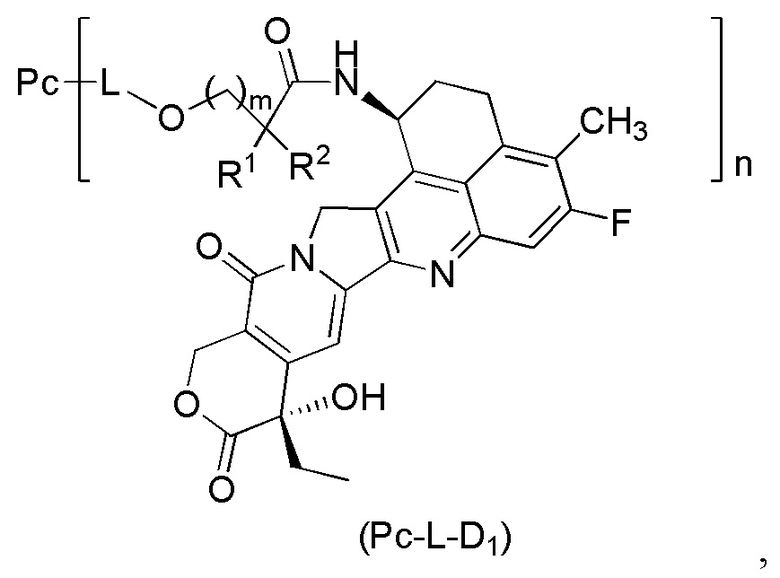
где:
R1 представляет собой циклоалкилалкил или циклоалкил, предпочтительно C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
m представляет собой 0 или 1;
n может быть целым или десятичным числом от 1 до 10;
Pc представляет собой антитело или его антигенсвязывающий фрагмент; и L представляет собой линкерный фрагмент.
В альтернативном варианте осуществления изобретения в вышеупомянутом конъюгате антитела и лекарственного средства n может быть целым или десятичным числом от 2 до 8, предпочтительно целым или десятичным числом от 3 до 8.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственноего средства линкерный фрагмент -L- представляет собой -L1-L2-L3-L4-, где:
L1 выбран из группы, состоящей из -(сукцинимидил-3-ил-N)-W-C(O)-, -CH2-C(O)-NR3-W-C(O)- и -C(O)-W-C(O)-, где W выбран из группы, состоящей из C1-8 алкила, C1-8 алкил-циклоалкила и линейного гетероалкила от 1 до 8 атомов, и гетероалкил содержит от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, O и S, где каждый из C1-8 алкила, циклоалкила и линейного гетероалкила независимо дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, алкила, хлоралкила, дейтерированного алкила, алкокси и циклоалкила;
L2 выбран из группы, состоящей из -NR4(CH2CH2O)p1CH2CH2C(O)-, -NR4(CH2CH2O)p1CH2C(O)-, -S(CH2)p1C(O)- и химической связи, где p1 представляет собой целое число от 1 до 20; L2 предпочтительно представляет собой химическую связь;
L3 представляет собой пептидный остаток, состоящий из 2-7 аминокислот, где аминокислоты необязательно дополнительно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, алкила, хлоралкила, дейтерированного алкила, алкокси и циклоалкила;
L4 выбран из группы, состоящей из -NR5(CR6R7)t-, -C(O)NR5, -C(O)NR5(CH2)t- и химической связи, где t представляет собой целое число от 1 до 6; L4 предпочтительно представляет собой -NR5(CR6R7)t-;
R3, R4 и R5 являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
R6 и R7 являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства линкерный фрагмент L1 выбран из группы, состоящей из -(сукцинимидин-3-ил-N)-(CH2)s1-C(O)-, -(сукцинимидин-3-ил-N)-CH2-циклогексил-C(O)-, -(сукцинимидин-3-ил-N)-(CH2CH2O)s2-CH2CH2-C(O)-, -CH2-C(O)-NR3-(CH2)s3-C(O)- и -C(O)-(CH2)s4C(O)-, где s1 представляет собой целое число от 2 до 8, s2 представляет собой целое число от 1 до 3, s3 представляет собой целое число от 1 до 8, и s4 представляет собой целое число от 1 до 8; s1 предпочтительно представляет собой 5.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства линкерный фрагмент L2 представляет собой -NR4(CH2CH2O)p1CH2C(O)- или химическую связь, и p1 представляет собой целое число от 6 до 12.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства L4 представляет собой -NR5(CR6R7)t-, R5 представляет собой водород или алкил, R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил, и t представляет собой 1 или 2, предпочтительно 2; L4 предпочтительно представляет собой -NR5CR6R7-, более предпочтительно -NHCH2-.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственноего средства линкерный фрагмент -L- представляет собой -L1-L2-L3-L4-, где:
L1 представляет собой 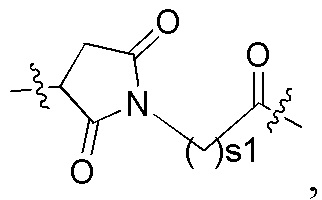 и s1 представляет собой целое число от 2 до 8;
и s1 представляет собой целое число от 2 до 8;
L2 представляет собой химическую связь;
L3 представляет собой остаток тетрапептида;
L4 представляет собой -NR5(CR6R7)t-, R5 представляет собой водород или алкил, R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил, и t представляет собой 1 или 2.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственноего средства линкерный фрагмент -L- представляет собой -L1-L2-L3-L4-, где:
L1 представляет собой -(сукцинимидин-3-ил-N)-CH2-циклогексил-C(O)-;
L2 представляет собой -NR4(CH2CH2O)9CH2C(O)-;
L3 представляет собой остаток тетрапептида;
L4 представляет собой -NR5(CR6R7)t-, R5 представляет собой водород или алкил, R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил, и t представляет собой 1 или 2.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства пептидный остаток L3 представляет собой аминокислотный остаток, образованный из одной, двух или более аминокислот, выбранных из группы, состоящей из фенилаланина (E), глицина (G), валина (V), лизина (K), цитруллина, серина (S), глутаминовой кислоты (E) и аспарагиновой кислоты (N), предпочтительно аминокислотный остаток, образованный из одной, двух или более аминокислот, выбранных из группы, состоящей из фенилаланина и глицина, более предпочтительно, тетрапептидного остатка и наиболее предпочтительно, тетрапептидного остатка GGFG (глицин-глицин-фенилаланин-глицин).
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства линкерный фрагмент -L- представляет собой -L1-L2-L3-L4-, где L1-конец связан с антителом или его антигенсвязывающим фрагментом, а L4-конец связан с Y.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства -L-Y- представляет собой:
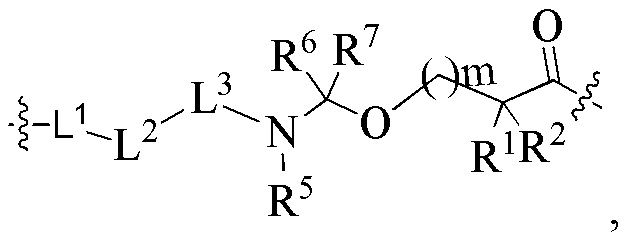
L1 представляет собой -(сукцинимидин-3-ил-N)-(CH2)s1-C(O)- или -(сукцинимидин-3-ил-N)-CH2-циклогексил-C(O)-;
L2 представляет собой -NR4(CH2CH2O)p1CH2C(O)- или химическую связь, и p1 представляет собой целое число от 6 до 12; R4 выбран из группы, состоящей из водорода, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
L3 представляет собой остаток тетрапептида GGFG;
R1 представляет собой циклоалкилалкил или циклоалкил, предпочтительно C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
R5 выбран из группы, состоящей из водорода и алкила, и R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил;
s1 представляет собой целое число от 2 до 8, предпочтительно 5;
m представляет собой целое число от 0 до 4.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства -L-Y- представляет собой:
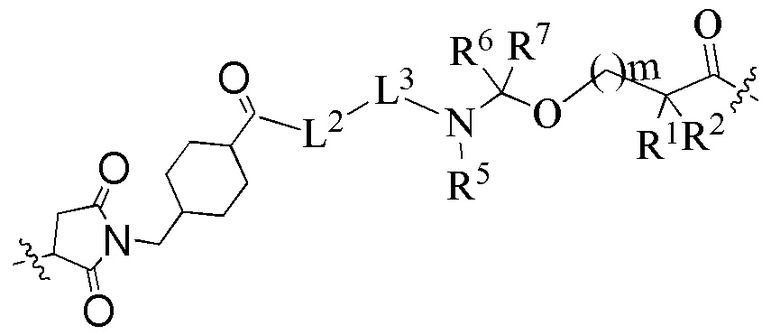
предпочтительно:
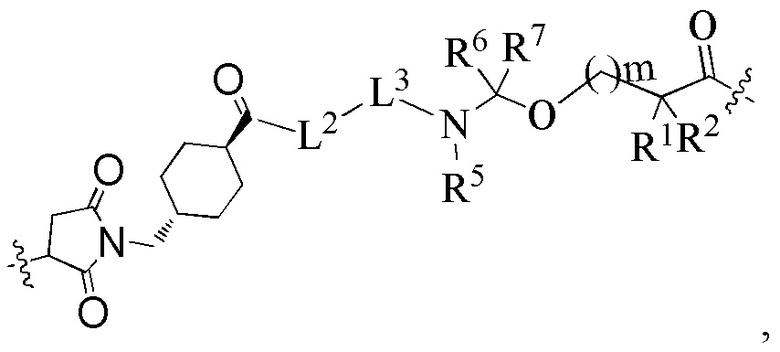
L2 представляет собой -NR4(CH2CH2O)9CH2C(O)-; R4 выбран из группы, состоящей из водорода, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
L3 представляет собой остаток тетрапептида GGFG;
R1 представляет собой циклоалкилалкил или циклоалкил, предпочтительно C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
R5 выбран из группы, состоящей из водорода и алкила, и R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил;
m представляет собой целое число от 0 до 4.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства -L-Y- представляет собой:
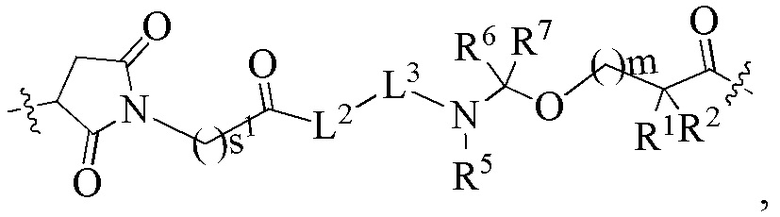
L2 представляет собой химическую связь;
L3 представляет собой остаток тетрапептида GGFG;
R1 представляет собой циклоалкилалкил или циклоалкил, предпочтительно C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
R5 выбран из группы, состоящей из водорода и алкила, и R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил;
s1 представляет собой целое число от 2 до 8, предпочтительно 5;
m представляет собой целое число от 0 до 4.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства -L-Y- представляет собой:
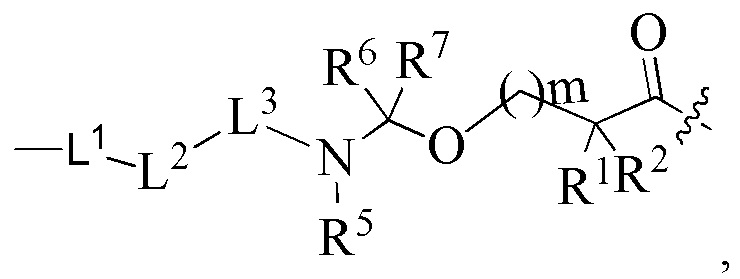
где:
L1 представляет собой -(сукцинимидин-3-ил-N)-(CH2)s1-C(O)- или -(сукцинимидин-3-ил-N)-CH2-циклогексил-C(O)-;
L2 представляет собой -NR4(CH2CH2O)p1CH2C(O)- или химическую связь, и p1 представляет собой целое число от 1 до 20; R4 выбран из группы, состоящей из водорода, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
L3 представляет собой остаток тетрапептида GGFG;
R1 представляет собой циклоалкилалкил или циклоалкил, предпочтительно C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
R5, R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил;
s1 представляет собой целое число от 2 до 8;
m представляет собой целое число от 0 до 4.
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства -L-Y- представляет собой:
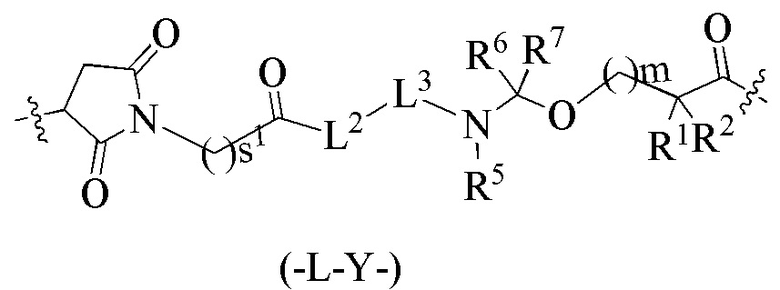 ,
,
где:
L2 представляет собой химическую связь;
L3 представляет собой остаток тетрапептида GGFG;
R1 представляет собой циклоалкилалкил или циклоалкил, предпочтительно C3-7 циклоалкилалкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, галогеналкила и C3-7 циклоалкила, предпочтительно водорода;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
R5 выбран из группы, состоящей из водорода и алкила, и R6 и R7 являются одинаковыми или различными и каждый независимо представляет собой водород или алкил;
s1 представляет собой целое число от 2 до 8;
m представляет собой целое число от 0 до 4.
В альтернативном варианте осуществления вышеупомянутый конъюгат антитела и лекарственного средства имеет структуру общей формулы (Pc-La-Y-D):
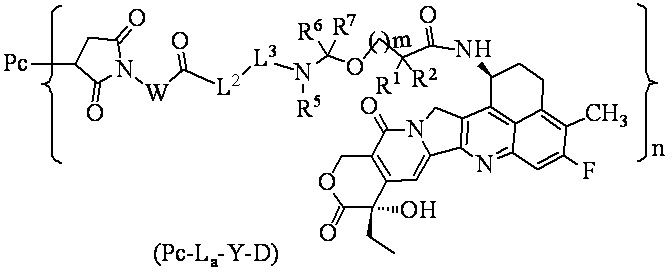 ,
,
где
W выбран из группы, состоящей из C1-8 алкила, C1-8 алкил-C3-7 циклоалкила и линейного гетероалкила, содержащего от 1 до 8 атомов, и гетероалкил содержит от 1 до 3 гетероатомов, выбранных из группы, состоящей из N, O и S, где каждый из C1-8 алкила, C3-7 циклоалкила и линейного гетероалкила независимо дополнительно необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, C1-6 алкила, C1-6 хлоралкила, дейтерированного C1-6 алкила, C1-6 алкокси и C3-7 циклоалкила;
L2 выбран из группы, состоящей из -NR4(CH2CH2O)p1CH2CH2C(O)-, -NR4(CH2CH2O)p1CH2C(O)-, -S(CH2)p1C(O)- и химической связи, где p1 представляет собой целое число от 1 до 20; R4 выбран из группы, состоящей из водорода, алкила, галогеналкила, дейтерированного алкила и гидроксиалкила;
L3 представляет собой пептидный остаток, состоящий из 2-7 аминокислотных остатков, где аминокислотные остатки выбраны из группы, состоящей из аминокислотных остатков, образованных из аминокислот фенилаланина (F), глицина (G), валина (V), лизина (K), цитруллина, серина (S), глутаминовой кислоты (Q) и аспарагиновой кислоты (D), и необязательно дополнительно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, амино, C1-6 алкила, C1-6 хлоралкила, дейтерированного C1-6 алкила, C1-6 алкокси и C3-7 циклоалкила;
R1 представляет собой C1-6 галогеналкил или C3-7 циклоалкил;
R2 выбран из группы, состоящей из водорода, C1-6 галогеналкила и C3-7 циклоалкила;
или, R1 и R2, вместе с атомами углерода, связанными с ними, образуют C3-7 циклоалкил;
R5 выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила, дейтерированного C1-6 алкила и гидрокси C1-6 алкила;
R6 и R7 являются одинаковыми или различными и каждый независимо выбран из группы, состоящей из водорода, галогена, C1-6 алкила, C1-6 галогеналкила, дейтерированного C1-6 алкила и гидрокси C1-6 алкила;
m представляет собой 0 или 1;
n представляет собой десятичное или целое число от 3 до 8;
Pc представляет собой антитело или его антигенсвязывающий фрагмент.
В альтернативном варианте осуществления вышеупомянутый конъюгат антитела и лекарственного средства имеет структуру общей формулы (Pc-Lb-Y-D):
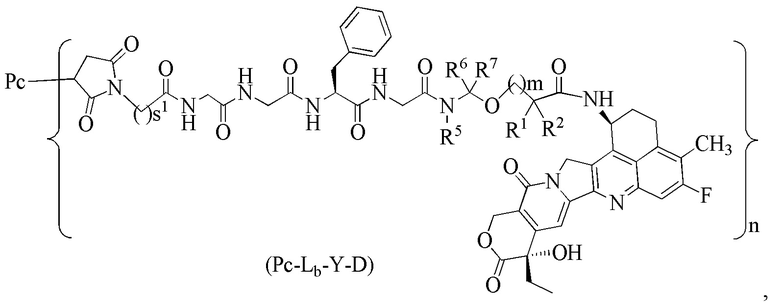
где:
s1 представляет собой целое число от 2 до 8;
Pc, R1, R2, R5, R6, R7, m и n являются такими, как определено в общей формуле (Pc-La-Y-D).
В альтернативном варианте осуществления в вышеупомянутом конъюгате антитела и лекарственного средства -L-Y- включает, но не ограничивается ими:
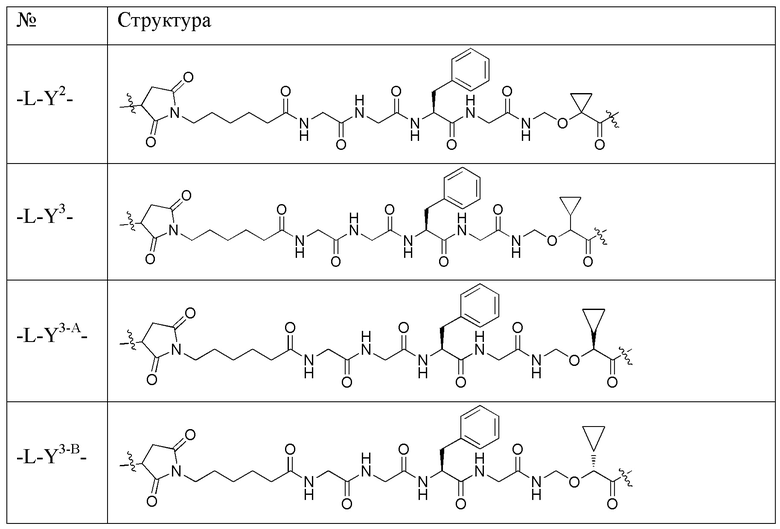
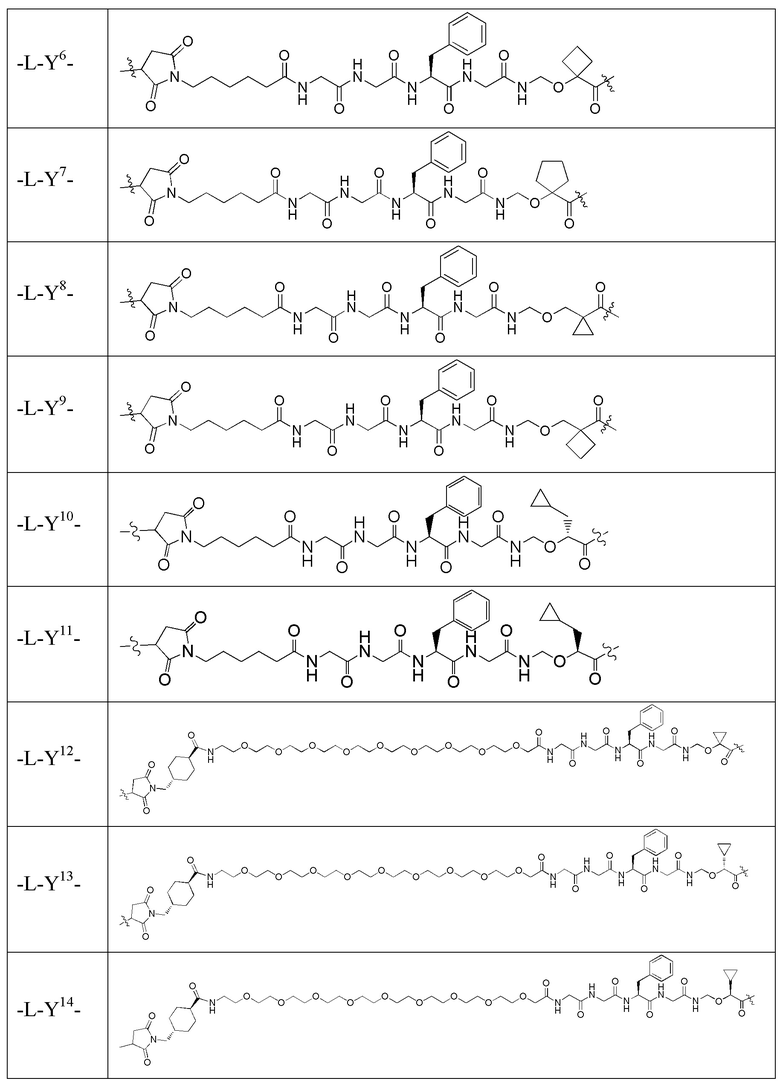
В альтернативном варианте осуществления вышеупомянутый конъюгат антитела и лекарственного средства имеет следующую структуру:
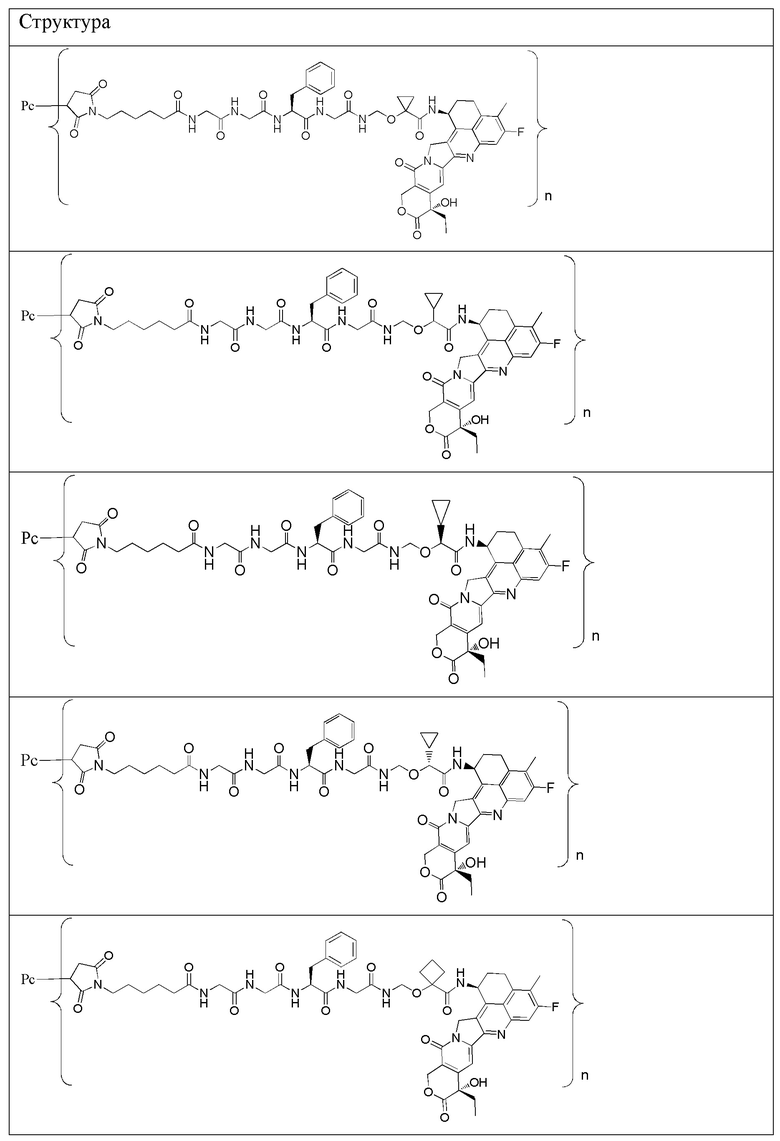
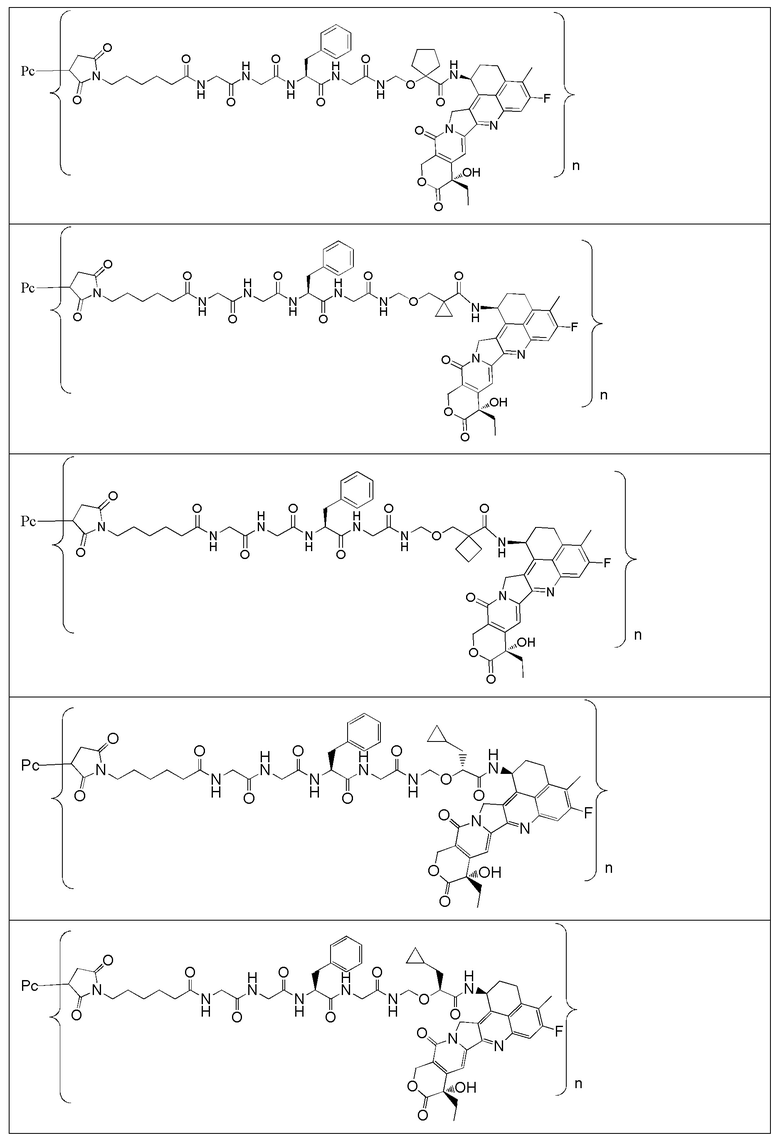
где Pc и n являются такими, как определено в общей формуле (Pc-La-Y-D).
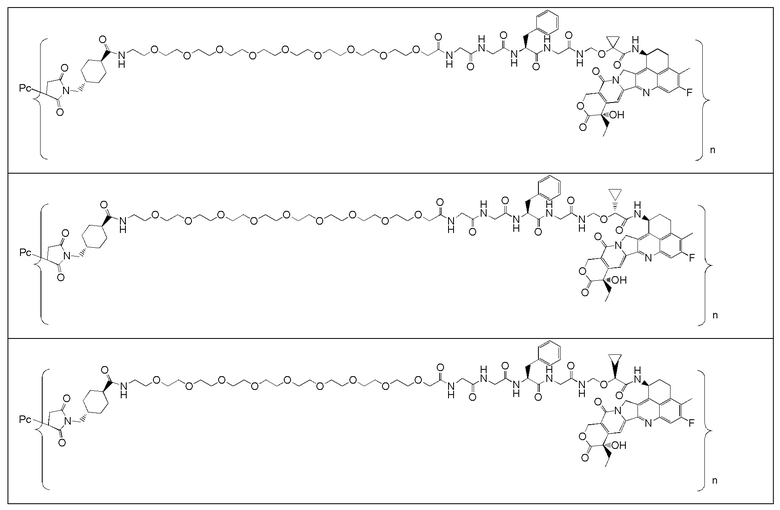
В альтернативном варианте осуществления конъюгат антитела и лекарственного средства имеет следующую структуру:
 ,
,
где:
n представляет собой десятичное или целое число от 3 до 8;
Pc представляет собой антитело или его антигенсвязывающий фрагмент.
В альтернативном варианте осуществления Pc представляет собой антитело или его антигенсвязывающий фрагмент, где антитело выбрано из группы, состоящей из химерного антитела, гуманизированного антитела или полностью человеческого антитела, предпочтительно моноклонального антитела.
В альтернативном варианте осуществления Pc выбрано из группы, состоящей из антитела против HER2 (рецептор эпидермального фактора роста 2) (ErbB2), антитела против EGFR (рецептор эпидермального фактора роста 1), антитела против B7-H3 (белок гомолог 3 B7), антитела против c-Met (рецептор фактора роста гепатоцитов), антитела против HER3 (рецептор эпидермального фактора роста 3) (ErbB3), антитела против HER4 (рецептор эпидермального фактора роста 4) (ErbB4), антитела против CD20, антитела против CD22, антитела против CD30, антитела против CD33, антитела против CD44, антитела против CD56, антитела против CD70, антитела против CD73, антитела против CD105, антитела против CEA, антитела против A33, антитела против Cripto (Крипто), антитела против EphA2, антитела против G250, антитела против MUCl, антитела против Льюис Y, антитела против VEGFR (фактор роста эндотелия сосудов), антитела против GPNMB (гликопротеин B неметастатической меланомы), антитела против интегрина, антитела против PSMA (простатспецифический мембранный антиген), антитела против тенасцина-C, антитела против SLC44A4 (белок 4, подобный переносчику холина), антитела против мезотелина или их антигенсвязывающих фрагментов.
В альтернативном варианте осуществления антитело или его антигенсвязывающий фрагмент в конъюгате антитела и лекарственного средства выбрано из группы, состоящей из трастузумаба, пертузумаба, нимотузумаба, эноблитузумаба, эмибетузумаба, инотузумаба, пинатузумаба, брентуксимаба, гемтузумаба, биватузумаба, лорвотузумаба, cBR96 и глематумамаба или их антигенсвязывающих фрагментов.
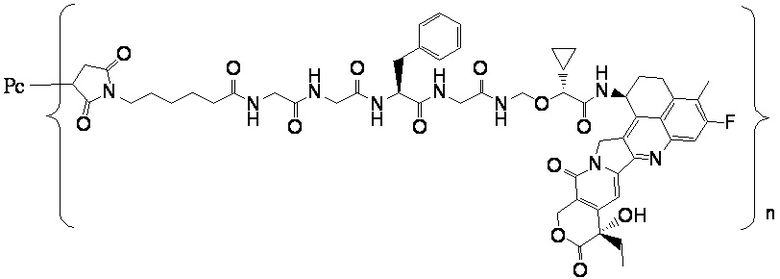
В альтернативном варианте осуществления конъюгат антитела и лекарственного средства имеет следующую структуру:
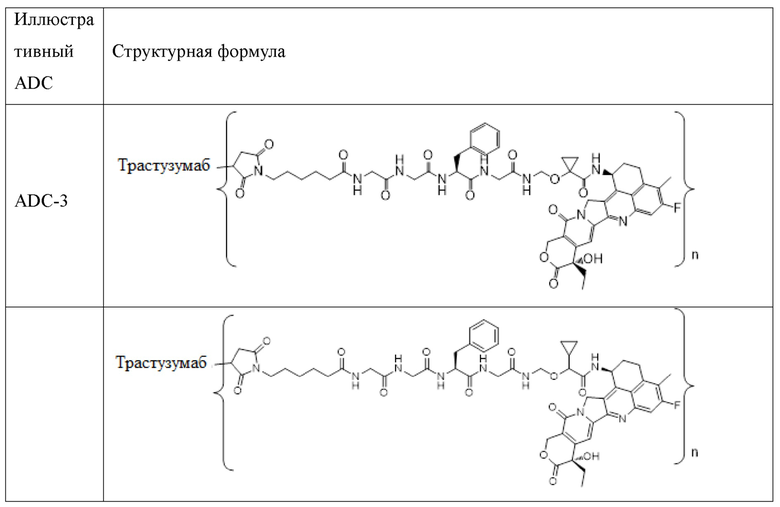
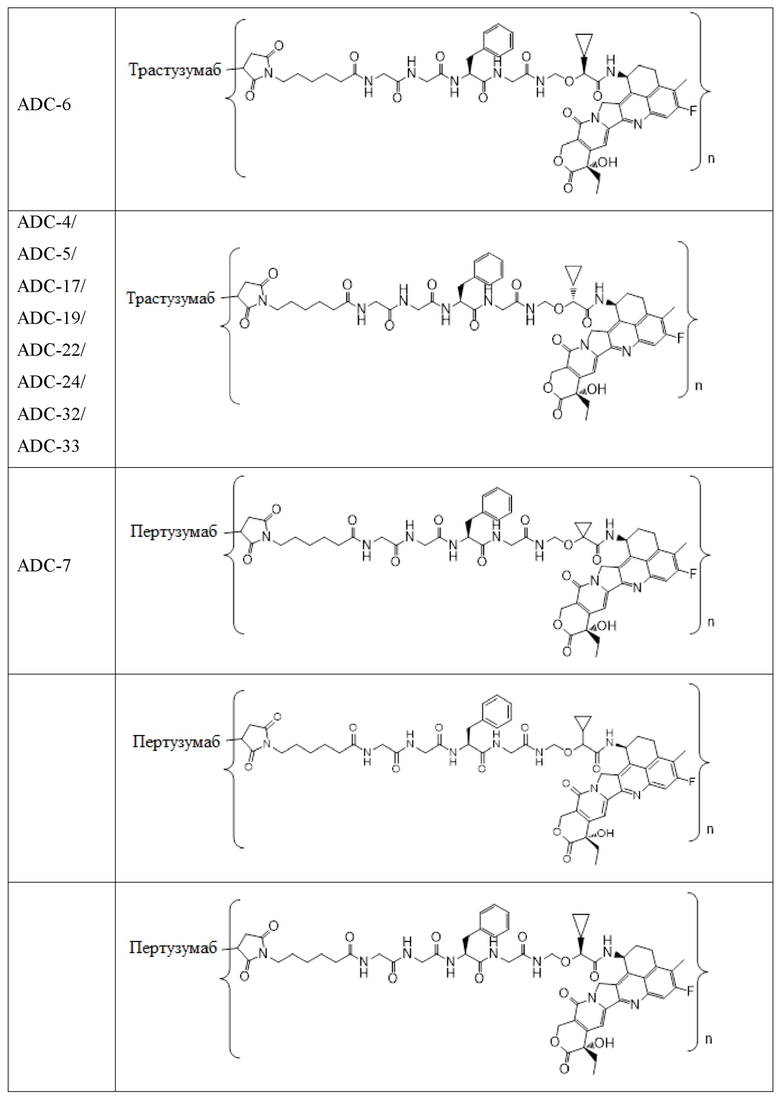
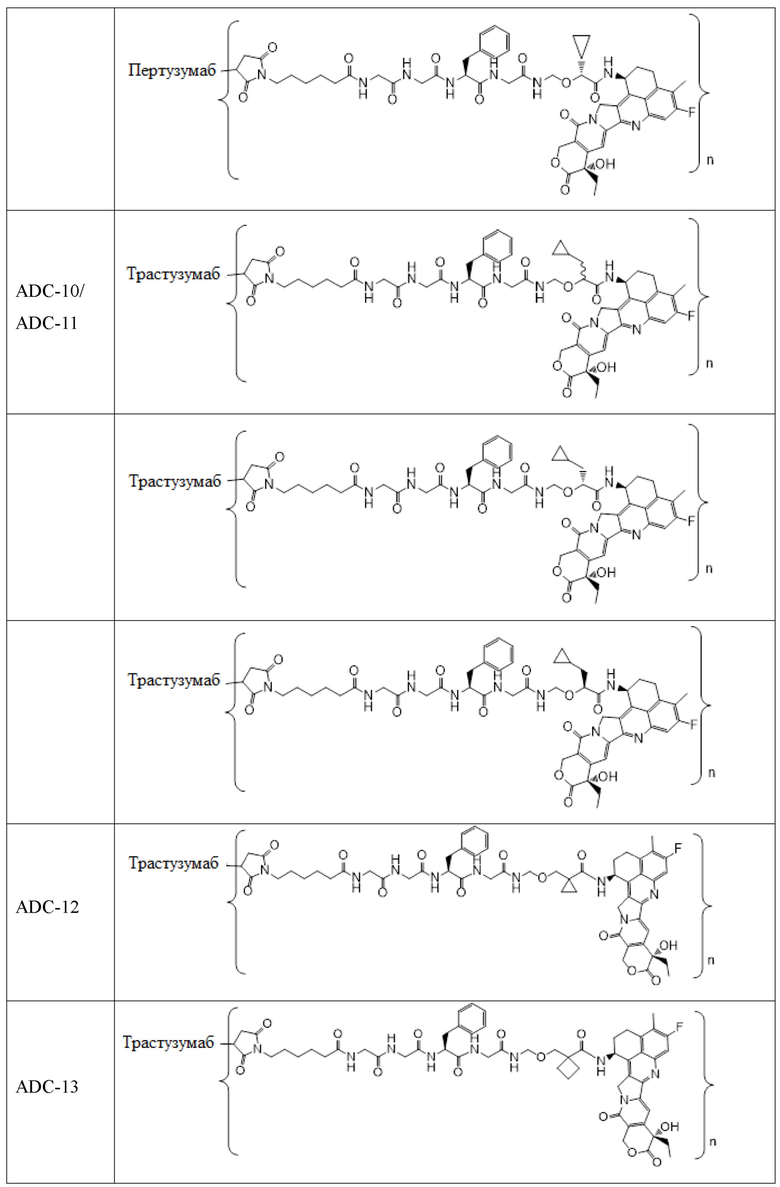
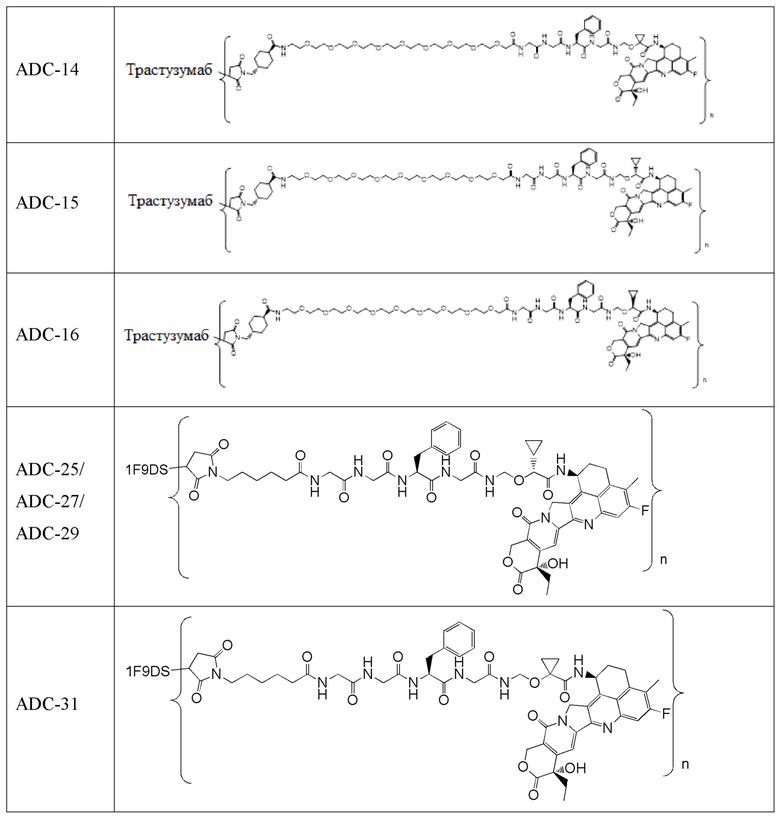
где n представляет собой целое или десятичное число от 0 до 10, не равное нулю, предпочтительно целое или десятичное число от 1 до 10, более предпочтительно целое или десятичное число от 2 до 8, и наиболее предпочтительно целое или десятичное число от 3 до 8.
В альтернативном варианте осуществления конъюгат антитела и лекарственного средства в фармацевтической композиции имеет следующую структуру:
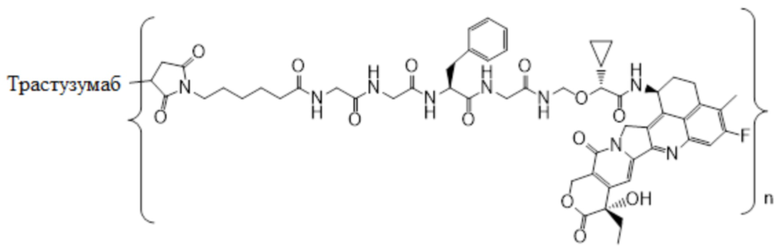 ,
,
где n представляет собой десятичное или целое число от 3 до 8.
В настоящем изобретении предложена фармацевтическая композиция, содержащая: (a) конъюгат антитела и лекарственного средства в концентрации от около 10 мг/мл до около 30 мг/мл, (b) полисорбат в концентрации от около 0,05 мг/мл до около 0,5 мг/мл, (c) сахарид в концентрации от около 60 мг/мл до около 90 мг/мл и (d) буфер в концентрации от около 5 мМ до около 20 мМ; причем композиция имеет рН 4,8-5,2,
где конъюгат антитела и лекарственного средства имеет следующую структуру:
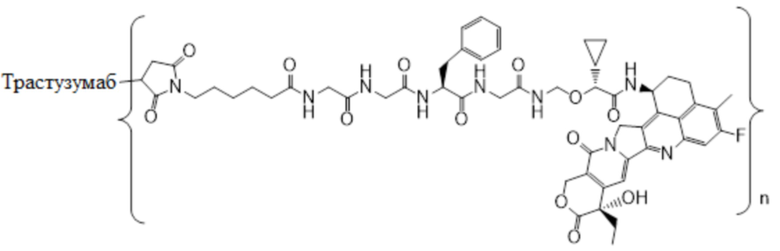 ,
,
где n представляет собой десятичное или целое число от 3 до 8.
В настоящем изобретении предложена фармацевтическая композиция, содержащая (a) конъюгат антитела и лекарственного средства в концентрации от около 20 мг/мл до около 22 мг/мл, (b) полисорбат 80 в концентрации около 0,2 мг/мл, (c) сахароза в концентрации около 80 мг/мл и (d) сукцинатный буфер в концентрации около 10 мМ; причем фармацевтическая композиция имеет рН 5,0-5,1,
где конъюгат антитела и лекарственного средства имеет следующую структуру:
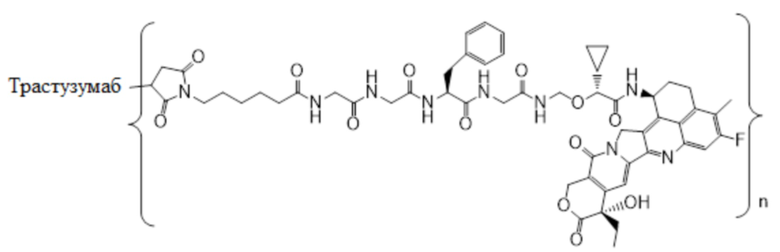 ,
,
где n представляет собой десятичное или целое число от 3 до 8.
В настоящем описании также предложен лиофилизированный препарат, содержащий конъюгат антитела и лекарственного средства, где препарат может быть восстановлен с образованием фармацевтической композиции, описанной выше.
В настоящем изобретении также предложен способ получения лиофилизированного препарата, содержащего конъюгат антитела и лекарственного средства, включающий стадию лиофилизации фармацевтической композиции, описанной выше.
В альтернативном варианте осуществления лиофилизация в способе получения лиофилизированного препарата, содержащего конъюгат антитела и лекарственного средства, включает стадии предварительного замораживания, первичной сушки и вторичной сушки. Лиофилизацию проводили путем замораживания препарата и последующего сублимирования воды при температуре, подходящей для первичной сушки. В этих условиях продукт находится при температуре ниже точки эвтектики или температуры разрушения препарата. Как правило, температура для первичной сушки составляет от около -30 °С до 25 °С (при условии, что продукт остается замороженным во время первичной сушки). Препарат, размер и тип контейнера (например, стеклянный флакон), содержащего образец, и объем жидкости определяют время, необходимое для сушки, которое может составлять от нескольких часов до нескольких дней (например, 40-60 часов). Вторичная сушка может быть проведена при температуре около 0-40 °C, в зависимости в первую очередь от типа и размера контейнера и типа используемого белка. Время, необходимое для вторичной сушки, определяется желаемым остаточным содержанием воды в продукте и обычно занимает по меньшей мере около 5 часов. Как правило, содержание воды в препарате, лиофилизированном при низком давлении, составляет менее чем около 5%, предпочтительно менее чем около 3%. Давление может быть таким же, как и на стадии первичной сушки; предпочтительно, давление вторичной сушки ниже, чем давление первичной сушки. Условия лиофилизации могут варьироваться в зависимости от состава и размера флакона.
В альтернативном варианте осуществления настоящего изобретения 5 мл исходного раствора фармацевтической композиции лиофилизировали, при этом программа лиофилизации выглядит следующим образом: для предварительного замораживания температура составляет -5 °С или -45 °С; для первичной сушки температура составляет -20 °С, а степень вакуума составляет 10 Па; для вторичной сушки температура составляет 25 °С, а степень вакуума составляет 1 Па.
В некоторых вариантах осуществления лиофилизированный препарат стабилен при 2-8 °C в течение по меньшей мере 3 месяцев, по меньшей мере 6 месяцев, по меньшей мере 12 месяцев, по меньшей мере 18 месяцев или по меньшей мере 24 месяцев. В некоторых вариантах осуществления лиофилизированный препарат стабилен при 40 °С в течение по меньшей мере 7 дней, по меньшей мере 14 дней или по меньшей мере 28 дней.
В настоящем изобретении также предложен лиофилизированный препарат, содержащий конъюгат антитела и лекарственного средства, полученный путем лиофилизации фармацевтической композиции, содержащей конъюгат антитела против HER2 и лекарственного средства, описанного выше.
В настоящем изобретении также предложен восстановленный раствор, содержащий конъюгат антитела и лекарственного средства, полученный путем восстановления лиофилизированного препарата, описанного выше.
В настоящем изобретении также предложен способ получения восстановленного раствора, описанного выше, включающий стадию восстановления вышеупомянутого лиофилизированного препарата раствором, выбранным из группы, состоящей из, но не ограничиваясь этим, воды для инъекций, физиологического раствора или раствора глюкозы.
В альтернативном варианте осуществления восстановленный раствор содержит следующие компоненты:
(a) конъюгат антитела и лекарственного средства в концентрации от около 10 мг/мл до около 30 мг/мл, (b) полисорбат в концентрации от около 0,05 мг/мл до около 0,5 мг/мл, (c) сахарид в концентрации от около 60 мг/мл до около 90 мг/мл и (d) буфер в концентрации от около 5 мМ до около 20 мМ; причем восстановленный раствор имеет рН 4,8-5,2.
В альтернативном варианте осуществления восстановленный раствор содержит следующие компоненты:
(a) конъюгат антитела и лекарственного средства в концентрации от около 20 мг/мл до 22 мг/мл, (b) полисорбат 80 в концентрации около 0,2 мг/мл, (c) сахароза в концентрации около 80 мг/мл и (d) сукцинатный буфер в концентрации около 10 мМ; причем восстановленный раствор имеет рН 5,0-5,1.
В настоящем изобретении также предложено изделие, содержащее контейнер, включающий фармацевтическую композицию, лиофилизированный препарат или восстановленный раствор, описанный выше. В некоторых вариантах осуществления контейнер представляет собой трубчатый флакон для инъекций, изготовленный из нейтрального боросиликатного стекла.
В изобретении также предложено применение вышеупомянутой фармацевтической композиции, или лиофилизированного препарата, или восстановленного раствора, или изделия для получения лекарственного препарата для лечения или предотвращения опухолей.
В настоящем изобретении также предложен способ лечения заболевания, включающий обеспечение вышеупомянутой фармацевтической композиции или лиофилизированного преперата или восстановленного раствора или изделия.
В настоящем изобретении также предложена вышеупомянутая фармацевтическая композиция или лиофилизированный препарат, или восстановленный раствор, или изделие в качестве лекарственного препарата, предпочтительно для лечения или предотвращения опухолевого заболевания.
В альтернативном варианте осуществления заболевание или опухоль представляет собой рак, связанный с экспрессией HER2, HER3, B7H3 или EGFR.
В альтернативном варианте осуществления рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака шейки матки, рака матки, рака предстательной железы, рака почки, рака мочевыводящих путей, рака мочевого пузыря, рака печени, рака желудка, рака эндометрия, карциномы слюнной железы, рака пищевода, меланомы, нейроглиомы, нейробластомы, саркомы, рака легкого, рака толстой кишки, рака прямой кишки, колоректального рака, лейкоза, рака кости, рака кожи, рака щитовидной железы, рака поджелудочной железы и лимфомы.
Как хорошо известно специалистам в данной области техники, один, некоторые или все признаки различных вариантов осуществления, описанных в настоящем описании, могут быть дополнительно объединены с образованием других вариантов осуществления настоящего изобретения. Вышеуказанные варианты осуществления настоящего изобретения и другие варианты осуществления, полученные комбинацией, дополнительно проиллюстрированы следующим подробным описанием.
В настоящем изобретении предложена фармацевтическая композиция, которая способствует получению и введению и является стабильной по свойствам. В частности, фармацевтическая композиция, описанная в настоящем описании, содержит конъюгат антитела и лекарственного средства и буфер.
Термины
Чтобы облегчить понимание настоящего изобретения, некоторые технические и научные термины конкретно определены ниже. Если иное четко не определено в данном документе, все другие технические и научные термины, используемые в данном описании, имеют значения, обычно понятные специалистам в области техники, к которой относится настоящее изобретение.
Заявка PCT/CN2019/107873 (WO2020/063676) полностью включена в настоящий документ посредством ссылки.
«Конъюгат антитела и лекарственного средства (ADC)» получали путем соединения антитела или фрагмента антитела с цитотоксином с биологической активностью или низкомолекулярным лекарственным средством с активностью уничтожения клеток стабильным химическим линкерным соединением, в этом случае полностью реализуются специфичность связывания антитела с антигенами, специфичными для опухолевых клеток или высокоэкспрессируемыми антигенами и высокую эффективность цитотоксина, и избегаются токсические побочные эффекты на нормальные клетки. Конъюгат антитела и лекарственного средства может точно связываться с опухолевыми клетками и оказывать сниженное влияние на нормальные клетки по сравнению с обычными химиотерапевтическими препаратами, используемыми ранее.
«Буфер» относится к буферу, который противостоит изменениям рН за счет действия своих кислотно-основных сопряженных компонентов. Примеры буферов, которые контролируют рН в соответствующем диапазоне, включают ацетат, сукцинат, глюконат, гистидиновую соль, оксалат, лактат, фосфат, цитрат, тартрат, фумарат, глицилглицин и другие органические кислотные буферы.
"Буфер на основе соли гистидина" представляет собой буфер, содержащий ионы гистидина. Примеры буферов на основе соли гистидина включают гистидин-гидрохлоридный буфер, гистидин-ацетатный буфер, гистидин-фосфатный буфер, гистидин-сульфатный буфер и т. п., и гистидин-ацетатный буфер является предпочтительным. Гистидин-ацетатный буфер получали из гистидина и уксусной кислоты, а гистидин-гидрохлоридный буфер получали из гистидина и соляной кислоты.
"Цитратный буфер" представляет собой буфер, содержащий цитратные ионы. Примеры цитратных буферов включают лимонную кислоту-цитрат натрия, лимонную кислоту-цитрат калия, лимонную кислоту-цитрат кальция, лимонную кислоту-цитрата магния и т.п. Предпочтительно цитратный буфер представляет собой лимонную кислоту-цитрат натрия.
"Сукцинатный буфер" представляет собой буфер, содержащий сукцинатные ионы. Примеры сукцинатных буферов включают янтарную кислоту-сукцинат натрия, янтарную кислоту-сукцинат калия, янтарную кислоту-сукцинат кальция и т.п. Предпочтительным сукцинатным буфером является янтарная кислота-сукцинат натрия. В качестве примера, буфер янтарная кислота-сукцинат натрия может быть получен из янтарной кислоты и гидроксида натрия или из янтарной кислоты и сукцината натрия.
"Фосфатный буфер" представляет собой буфер, содержащий фосфат-ионы. Примеры фосфатных буферов включают гидрофосфат динатрия-дигидрофосфат натрия, гидрофосфат динатрия-дигидрофосфат калия, гидрофосфат динатрия-лимонная кислота и т.п. Предпочтительным фосфатным буфером является гидрофосфат динатрия-дигидрофосфат натрия.
"Ацетатный буфер" представляет собой буфер, содержащий ацетатные ионы. Примеры ацетатных буферов включают уксусную кислоту-ацетат натрия, уксусную кислоту-соль гистидина, уксусную кислоту-ацетат калия, уксусную кислоту-ацетат кальция, уксусную кислоту-ацетат магния и т.п. Предпочтительно ацетатным буфером является уксусная кислота-ацетат натрия.
"Фармацевтическая композиция" относится к смеси, содержащей один или более конъюгатов антитела и лекарственного средства, описанных в данном документе, или их физиологически/фармацевтически приемлемые соли или пролекарства и другие химические компоненты, а другие компоненты представляют собой, например, физиологически/фармацевтически приемлемые носители и эксципиенты. Назначением фармацевтической композиции является поддержание стабильности активного ингредиента антитела и содействие введению в организм, что облегчает абсорбцию активного ингредиента, проявляя таким образом свою биологическую активность.
Используемые здесь термины «фармацевтическая композиция» и «препарат» не исключают друг друга.
Фармацевтическая композиция, описанная в настоящем изобретении, находится в форме раствора, и если не указано иное, растворитель представляет собой воду.
"Лиофилизированный препарат" относится к препарату или фармацевтической композиции, полученной путем вакуумной лиофилизации фармацевтической композиции или препарата в жидкой или растворенной форме.
Термины «около» и «приблизительно», используемые в данном документе, означают, что числовое значение находится в пределах допустимого диапазона погрешности для конкретного значения, определенного специалистом в данной области техники, и числовое значение частично зависит от того, как измеряется или определяется значение (т.е. пределы измерительной системы). Например, "около" может означать нахождение в пределах 1 или более чем 1 стандартного отклонения согласно практике в данной области. Или «около» или «по существу содержащий» может означать диапазон до 20%. Кроме того, в частности, для биологических систем или процессов, этот термин может означать до порядка величины или до 5-кратного числового значения. Когда конкретное значение представлено в настоящей заявке и формуле изобретения, если не указано иное, значение «около» или «по существу содержащий» следует понимать как находящееся в пределах допустимого диапазона погрешности для этого конкретного значения.
Фармацевтическая композиция по настоящему изобретению может достигать стабильного эффекта: фармацевтическая композиция, в которой конъюгат антитела и лекарственного средства по существу сохраняет свою физическую и/или химическую стабильность и/или биологическую активность после хранения; предпочтительно фармацевтическая композиция по существу сохраняет свою физическую и химическую стабильность, а также свою биологическую активность после хранения. Период хранения, как правило, выбирали на основании заранее определенного срока годности фармацевтической композиции. В настоящее время существует множество аналитических методик, доступных для измерения стабильности белка, и стабильность после хранения в течение выбранного периода времени при выбранной температуре может быть измерена.
Стабильный препарат представляет собой препарат, в котором не наблюдается значительных изменений при следующих условиях: хранение при температуре охлаждения (2-8 °C) в течение по меньшей мере 3 месяцев, предпочтительно 6 месяцев, более предпочтительно 1 года и еще более предпочтительно до 2 лет. Кроме того, стабильные жидкие препараты включают жидкие препараты, которые проявляют желательные признаки после хранения при температурах, включая 25 °C, в течение периодов, включающих 1 месяц, 3 месяца и 6 месяцев. Типичными примерами стабильности являются следующие: как правило, не более чем около 10%, предпочтительно не более чем около 5% мономеров антител агрегируют или разлагаются, как измерено с помощью эксклюзионной SEC-HPLC (эксклюзионная высокоэффективная жидкостная хроматография). Препарат представляет собой бледно-желтую, почти бесцветную и прозрачную жидкость, или бесцветную, или прозрачную, или слегка опалесцирующую, при визуальном анализе. Концентрация, рН и осмотическое давление препарата изменяются не более чем на ±10%. Как правило, наблюдается снижение не более чем около 10%, предпочтительно не более чем около 5%. Как правило, образуется не более чем около 10%, предпочтительно не более чем около 5% агрегации.
Конъюгат антитела и лекарственного средства "сохраняет свою физическую стабильность" в фармацевтическом препарате, если оно не демонстрирует значительного усиления агрегации, осаждения и/или денатурации при визуальном осмотре цвета и/или прозрачности, или при измерении с помощью УФ-рассеяния света (UV, УФ - ультрафиолетовый), эксклюзионной хроматографии (SEC) и динамического рассеяния света (DLS). Изменения конформации белка могут быть оценены с помощью флуоресцентной спектроскопии (которая определяет третичную структуру белка) и спектроскопии FTIR (инфракрасная спектроскопия на основе преобразования Фурье) (которая определяет вторичную структуру белка).
Конъюгат антитела и лекарственного средства "сохраняет свою химическую стабильность" в фармацевтическом препарате, если оно не демонстрирует существенных химических изменений. Химическая стабильность может быть оценена путем обнаружения и количественного определения химически измененных форм белка. Процессы деградации, которые часто изменяют химическую структуру белков, включают гидролиз или клипирование (оценивается с помощью таких методов, как эксклюзионная хроматография и CE-SDS (капиллярный электрофорез в присутствии додецилсульфата натрия)), окисление (оценивается с помощью таких методов, как пептидное картирование в сочетании с масс-спектроскопией или MALDI/TOF/MS (времяпролетная масс-спектрометрия с матрично-активированной лазерной десорбцией/ионизацией), дезамидирование (оценивается с помощью таких методов, как ионообменная хроматография, капиллярная изоэлектрическая фокусировка, пептидное картирование и измерение изоаспарагиновой кислоты) и изомеризацию (оценивается путем измерения содержания изоаспарагиновой кислоты, пептидного картирования и т. д.).
Конъюгат антитела и лекарственного средства "сохраняет свою биологическую активность" в фармацевтическом препарате, если биологическая активность конъюгата антитела и лекарственного средства в данный момент времени находится в пределах заданного диапазона биологической активности, проявленной во время получения фармацевтического препарата.
Трехбуквенные и однобуквенные коды аминокислот, используемые в данном документе, описаны в J. Biol. Chem., 243, с. 3558 (1968).
"Антитело", описанное в данном документе, относится к иммуноглобулину, и интактное антитело представляет собой структуру тетрапептидной цепи, образованную соединением между двумя идентичными тяжелыми цепями и двумя идентичными легкими цепями посредством межцепочечных дисульфидных связей. Константные области тяжелой цепи иммуноглобулина отличаются по своему аминокислотному составу и расположению, и, таким образом, по своей антигенности. Соответственно, иммуноглобулины можно разделить на пять классов, иначе называемых изотипами иммуноглобулинов, а именно IgM, IgD, IgG, IgA и IgE, при этом их соответствующие тяжелые цепи представляют собой μ-цепь, δ-цепь, γ-цепь, α-цепь и ε-цепь, соответственно. Ig одного класса может быть разделен на различные подклассы в зависимости от различий в аминокислотном составе шарнирных областей и количества и положения дисульфидных связей тяжелых цепей; например, IgG может быть разделен на IgG1, IgG2, IgG3 и IgG4. Легкие цепи классифицируются на κ- или λ-цепи по различиям в константных областях. Каждый из пяти классов Ig может иметь κ-цепь или λ-цепь. Антитело, описанное в настоящем изобретении, предпочтительно представляет собой специфические антитела против антигенов клеточной поверхности на клетках-мишенях, неограничивающие примеры антител, представляющих собой одно или более из антитела против HER2 (ErbB2), антитела против EGFR, антитела против B7-H3, антитела против c-Met, антитела против HER3 (ErbB3), антитела против HER4 (ErbB4), антитела против CD20, антитела против CD22, антитела против CD30, антитела против CD33, антитела против CD44, антитела против CD56, антитела против CD70, антитела против CD73, антитела против CD105, антитела против CEA, антитела против A33, антитела против Cripto, антитела против EphA2, антитела против G250, антитела против MUCl, антитела против Льюис-Y, антитела против VEGFR, антитела против GPNMB, антитела против интегрина, антитела против PSMA, антитела против тенасцина-C, антитела против SLC44A4 и антитела против мезотелина, предпочтительно трастузумаба (под торговым названием Herceptin), пертузумаба (также известный как 2C4, под торговым названием Perjeta), нимотузумаба (под торговым названием Taixinsheng), эноблитузумаба, эмибетузумаба, инотузумаба, пинатузумаба, брентуксимаба, гемтузумаба, биватузумаба, лорвотузумаба, cBR96 и глематумамаба.
В тяжелых и легких цепях антитела последовательности около 110 аминокислот вблизи N-конца значительно варьируются и, таким образом, называются вариабельными областями (Fv-области); остальные аминокислотные последовательности вблизи С-конца являются относительно стабильными и, таким образом, называются константными областями. Вариабельная область включает 3 гипервариабельные области (HVR) и 4 каркасные области (FR) с относительно консервативными последовательностями. Три гипервариабельные области определяют специфичность антитела и таким образом также известны как определяющие комплементарность области (CDR). Каждая вариабельная область легкой цепи (LCVR) или вариабельная область тяжелой цепи (HCVR) состоит из 3 CDR и 4 FR, расположенных от аминоконца к карбоксиконцу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3 и FR4. Три области CDR легкой цепи обозначаются как LCDR1, LCDR2 и LCDR3, три области CDR тяжелой цепи обозначаются как HCDR1, HCDR2 и HCDR3. Аминокислотные остатки CDR областей LCVR и HCVR антител или антигенсвязывающих фрагментов, описанных в настоящем описании, соответствуют известной схеме нумерации по Kabat (LCDR 1-3, HCDR 1-3) по количеству и положениям.
В настоящем изобретении легкая цепь антитела по настоящему изобретению может дополнительно содержать константную область легкой цепи, содержащую человеческие или мышиные κ- и λ-цепи или их варианты.
В настоящем изобретении тяжелая цепь антитела по настоящему изобретению может дополнительно содержать константную область тяжелой цепи, содержащую IgG1, IgG2, IgG3 и IgG4 человека или мыши, или их варианты.
Антитело согласно настоящему изобретению включает мышиное антитело, химерное антитело и гуманизированное антитело, и предпочтительно гуманизированное антитело.
Термин "мышиное антитело", используемый в данном документе, относится к антителу, полученному из мыши в соответствии со знаниями и опытом в данной области техники. Во время получения испытуемому субъекту вводят антиген, а затем выделяют гибридомы, экспрессирующие антитела с желаемыми последовательностями или функциональными свойствами.
Термин "химерное антитело" относится к антителу, полученному путем слияния вариабельной области мышиного антитела и константной области человеческого антитела, которое может снижать иммунный ответ, индуцированный мышиным антителом. Химерное антитело создают, сначала создавая гибридому, секретирующую мышиное специфическое моноклональное антитело, затем клонируя ген вариабельной области из клеток гибридомы мыши, клонируя ген константной области человеческого антитела по мере необходимости, соединяя ген вариабельной области мыши и ген константной области человека в химерный ген, вставляя химерный ген в человеческий вектор и, наконец, экспрессируя молекулы химерного антитела в эукариотической промышленной системе или прокариотической промышленной системе.
Термин «гуманизированное антитело», также известное как CDR-привитое антитело, относится к антителу, полученному путем прививания последовательностей мышиных CDR в каркас вариабельной области человеческого антитела, т. е. другой тип каркасной последовательности антитела человеческого вида. Такое антитело может преодолеть сильную гетерогенную реакцию, индуцированную химерным антителом, из-за переноса большого количества белковых компонентов мыши. Такие каркасные последовательности можно получить из общедоступных баз данных ДНК или опубликованных ссылок, которые включают последовательности генов антител зародышевой линии. Например, последовательности ДНК зародышевой линии генов вариабельных областей тяжелой и легкой цепи человека можно найти в базе данных последовательностей видов человека «VBase», а также в Kabat, E. A. и др., 1991 Sequences of Proteins of Immunological Interest, 5-е издание. Во избежание снижения активности, вызванного снижением иммуногенности, последовательность FR (каркасная область) в вариабельной области антитела человека может быть подвергнута минимальной реверсивной или обратной мутации для поддержания активности. Гуманизированное антитело по настоящему изобретению также включает гуманизированные антитела, которые дополнительно подвергались созреванию аффинности CDR с помощью фагового дисплея.
Термин «голое антитело» относится к антителу, которое не конъюгировано с гетерологичным модулем (например, цитотоксическим модулем) или радиоактивной меткой.
"Антигенсвязывающий фрагмент антитела", описанный в настоящем документе, может относиться к фрагменту Fab, фрагменту Fab', фрагменту F(ab')2 или фрагменту scFv, фрагменту Fv, который связывается с антигеном, обладающим антигенсвязывающей активностью. Фрагмент Fv содержит вариабельную область тяжелой цепи и вариабельную область легкой цепи антитела, но без константной области, и имеет наименьший фрагмент антитела из всех антигенсвязывающих сайтов. Обычно Fv антитела также содержит полипептидный линкер между доменами VH (вариабельный домен тяжелой цепи) и VL (вариабельный домен легкой цепи) и способно образовывать структуру, необходимую для связывания антигена. Две вариабельные области антитела также могут быть связаны в одну полипептидную цепь с использованием различных линкеров, известных как одноцепочечное антитело или одноцепочечный fv (sFv).
Термин "антигенсвязывающий сайт", раскрытый в данном документе относится к непрерывному или прерывистому трехмерному пространственному сайту на антигене, который распознается антителом или антигенсвязывающим фрагментом по настоящему изобретению.
"ADCC", т.е. антителозависимая клеточно-опосредованная цитотоксичность, описанная в данном документе, означает, что клетки, экспрессирующие Fc-рецептор, непосредственно убивают клетки-мишени, покрытые антителом, путем распознавания Fc-сегмента антитела. Эффекторная функция ADCC антитела может быть снижена или устранена путем модификации Fc-сегмента IgG. Модификация относится к мутации в константной области тяжелой цепи антитела, такой как мутация, выбранная из группы, состоящей из N297A, L234A и L235A IgG1; химеры IgG2/4 и L234A/E235A IgG4.
"Мутации" в мутантных последовательностях, описанных в настоящем документе, включают, но не ограничиваются ими, "обратную мутацию", "консервативную модификацию" или "консервативную замену или замещение". «Консервативная модификация» или «консервативная замена или замещение», описанные в данном документе, относятся к замене аминокислот в белке другими аминокислотами, имеющими сходные характеристики (например, зарядом, размером боковой цепи, гидрофобностью/гидрофильностью или конформацией и жесткостью каркаса), так что изменения часто могут быть сделаны без изменения биологической активности белка. Специалистам в данной области техники известно, что, как правило, замена одной аминокислоты в несущественной области полипептида существенно не изменяет биологическую активность (см., например, Watson и др. (1987) Molecular Biology of the Gene, The Benjamin/Cummings Pub. Co., с. 224, (4-е издание)). Кроме того, замена аминокислот с аналогичной структурой или функцией вряд ли нарушит биологическую активность.
"Мутантная последовательность", описанная в данном документе, относится к нуклеотидной последовательности и/или аминокислотной последовательности, имеющей различную степень процентной идентичности последовательности с нуклеотидной последовательностью и/или аминокислотной последовательностью по настоящему изобретению, когда в нуклеотидную последовательность и/или аминокислотную последовательность по настоящему изобретению соответствующим образом вносятся мутационные модификации, такие как замены, вставки или делеции. Идентичность последовательности, описанной в данном документе, может составлять по меньшей мере 85%, 90% или 95% и предпочтительно по меньшей мере 95%. Неограничивающие примеры включают: 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% и 100%. Сравнение последовательностей и определение процента идентичности между двумя последовательностями могут быть выполнены с помощью настроек по умолчанию или алгоритма BLASTN/BLASTP, доступных на веб-сайте Национального центра биотехнологического института.
Термин «линкерная единица» или «линкерный фрагмент» относится к фрагменту химической структуры или связи, который связан с антителом или его антигенсвязывающим фрагментом на одном конце и с лекарственным средством на другом конце, а также может быть связан с антителом или лекарственным средством после того, как он связан с другим линкером. В предпочтительных вариантах осуществления настоящего изобретения они обозначены как L и L1-L4, где L1 конец связан с антителом, а L4 конец связан со структурной единицей Y, а затем с соединением или токсином.
Линкер содержать растягивающие звенья, спейсерные звенья и аминокислотные звенья и может быть синтезирован с использованием способов, известных в данной области техники, таких как описанные в US2005-0238649A1. Линкер может представлять собой "расщепляемый линкер", благоприятствующий высвобождению лекарственных средств в клетках. Например, могут быть использованы кислотолабильные линкеры (например, гидразоны), чувствительные к протеазе (например, чувствительные к пептидазе) линкеры, фотолабильные линкеры, диметильные линкеры или дисульфидсодержащие линкеры (Chari и др., Cancer Research 52: 127-131(1992); патент США № 5208020).
Сконструированное антитело или антигенсвязывающий фрагмент в настоящем изобретении можно получить и очистить общепринятыми методами. Например, последовательности кДНК (комплементарная ДНК), кодирующие тяжелую и легкую цепи, можно клонировать и рекомбинировать в экспрессионный вектор GS. Векторы экспрессии рекомбинантного иммуноглобулина могут быть стабильно трансфицированы в клетки CHO (клетки яичника китайского хомяка). В качестве более рекомендуемых в предшествующем уровне техники системы экспрессии млекопитающих могут привести к гликозилированию антител, в частности, в высококонсервативном N-концевом сайте области Fc. Положительные клоны культивировали в бессывороточной среде биореактора для получения антител. Культура с секретируемым антителом может быть очищена и собрана с использованием обычных методик. Например, очистку проводили на колонке A или G Sepharose FF, содержащей скорректированный буфер. Неспецифически связанные фракции вымывали. Связанное антитело элюировали методом градиента рН, и фрагменты антител детектировали с помощью SDS-PAGE (электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия) и собирали. Антитело может быть отфильтровано и концентрировано общепринятыми методами. Растворимые смеси и полимеры также могут быть удалены общепринятыми способами, такими как молекулярные сита и ионный обмен. Полученный продукт должен быть немедленно заморожен, например, при -70 °C, или лиофилизирован.
Термин "алкил" относится к насыщенной алифатической углеводородной группе, которая представляет собой линейную или разветвленную группу, содержащую от 1 до 20 атомов углерода, предпочтительно алкилу, содержащему от 1 до 12 атомов углерода, более предпочтительно алкилу, содержащему от 1 до 10 атомов углерода, и наиболее предпочтительно алкилу, содержащему от 1 до 6 атомов углерода. Неограничивающие примеры алкила включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил, различные разветвленные изомеры и т.д. Более предпочтительно низший алкил содержит от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкил может быть замещенным или незамещенным. При замещении заместитель может быть замещен в любом доступном месте соединения, где заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин "гетероалкил" относится к алкилу, содержащему один или более гетероатомов, выбранных из группы, состоящей из N, O и S, где алкил является таким, как определено выше.
Термин "алкилен" относится к насыщенной линейной или разветвленной алифатической углеводородной группе, имеющей остатки, полученные из исходного алкана путем удаления двух атомов водорода из одного и того же атома углерода или двух разных атомов углерода. Это линейная или разветвленная группа, содержащая от 1 до 20 атомов углерода, предпочтительно алкилен, содержащий от 1 до 12 атомов углерода, более предпочтительно алкилен, содержащий от 1 до 6 атомов углерода. Неограничивающие примеры алкилена включают, но не ограничиваются ими, метилен(-CH2-), 1,1-этилиден(-CH(CH3)-), 1,2-этилиден(-CH2CH2)-, 1,1-пропилиден(-CH(CH2CH3)-), 1,2-пропилиден(-CH2CH(CH3)-), 1,3-пропилиден(-CH2CH2CH2-), 1,4-бутилиден(-CH2CH2CH2CH2-), 1,5-бутилиден(-CH2CH2CH2CH2CH2-) и т. д. Алкилен может быть замещен или незамещен. При замещении заместитель может быть замещен в любом доступном месте соединения одним или более заместителями, предпочтительно независимо необязательно выбранными из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин "алкокси" относится к группе -O-(алкил) и -O-(незамещенный циклоалкил), где алкил или циклоалкил является таким, как определено выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропокси, циклобутокси, циклопентилокси и циклогексилокси. Алкокси может быть необязательно замещен или незамещен, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин "циклоалкил" относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю. Циклоалкильное кольцо содержит от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода и наиболее предпочтительно от 3 до 7 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т. п. Полициклический циклоалкил включает спироциклоалкил, конденсированный циклоалкил и мостиковый циклоалкил.
Термин "гетероциклил" относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю, содержащему от 3 до 20 кольцевых атомов, где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), за исключением циклической части -O-O-, -O-S- или -S-S-, и остальные кольцевые атомы представляют собой атомы углерода. Гетероциклоалкил предпочтительно содержит от 3 до 12 кольцевых атомов, из которых от 1 до 4 представляют собой гетероатомы; более предпочтительно циклоалкильное кольцо содержит от 3 до 10 кольцевых атомов. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.д. Неограничивающие примеры полициклического гетероциклила включают спирогетероциклил, конденсированный гетероциклил и мостиковый гетероциклил.
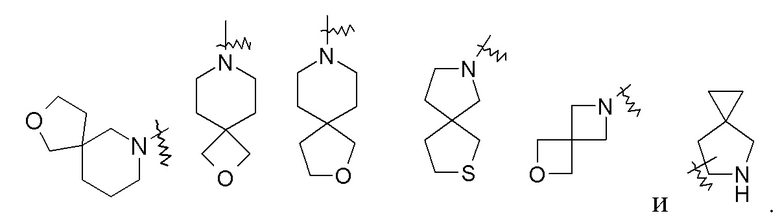
Термин "спирогетероциклил" относится к 5-20-членной полициклической гетероциклильной группе, в которой моноциклические кольца имеют общий один атом (называемый спироатомом), где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), а остальные кольцевые атомы представляют собой атомы углерода. Эти кольца могут содержать одну или более двойных связей, но ни одно из них не имеет полностью сопряженной π-электронной системы. Предпочтительно спирогетероциклил является 6-14-членным и более предпочтительно 7-10-членным. В соответствии с количеством спироатомов, являющихся общими между кольцами, спирогетероциклил может являться моноспирогетероциклилом, биспирогетероциклилом или полиспирогетероциклилом, предпочтительно моноспирогетероциклилом и биспирогетероциклилом и более предпочтительно 4-членным/4-членным, 4-членным/5-членным, 4-членным/6-членным, 5-членным/5-членным или 5-членным/6-членным моноспирогетероциклилом. Неограничивающие примеры спирогетероциклила включают:
Термин "конденсированный гетероциклил" относится к 5-20-членной полициклической гетероциклльной группе, в которой каждое кольцо имеет общую пару соседних атомов с другими кольцами в системе, где одно или более колец могут содержать одну или более двойных связей, но ни одно из них не имеет полностью сопряженной π-электронной системы, где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), а остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно конденсированный гетероциклил является 6-14-членным и более предпочтительно 7-10-членным. В соответствии с количеством образованных колец, конденсированный гетероциклил может быть бициклическим, трициклическим, тетрациклическим или полициклическим конденсированным гетероциклилом, предпочтительно бициклическим или трициклическим конденсированным гетероциклилом и более предпочтительно 5-членным/5-членным или 5-членным/6-членным бициклическим конденсированным гетероциклилом. Неограничивающие примеры конденсированного гетероциклила включают:
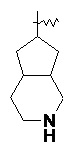
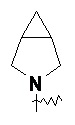
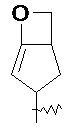
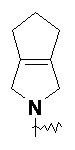
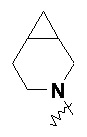
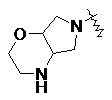
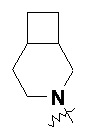
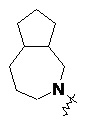
 и
и  .
.
Термин "мостиковый гетероциклил" относится к 5-14-членной полициклической гетероциклильной группе, в которой любые два кольца имеют общих два атома углерода, которые непосредственно не присоединены друг к другу, где эти кольца могут содержать одну или более двойных связей, но ни одно из них не имеет полностью сопряженной π-электронной системы, причем один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое от 0 до 2), а остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно мостиковый гетероциклил является 6-14-членным и более предпочтительно 7-10-членным. В соответствии с количеством образованных колец, мостиковый гетероциклил может быть бициклическим, трициклическим, тетрациклическим или полициклическим мостиковым гетероциклилом, предпочтительно бициклическим, трициклическим или тетрациклическим мостиковым гетероциклилом и более предпочтительно бициклическим или трициклическим мостиковым гетероциклилом. Неограничивающие примеры мостикового гетероциклила включают:
 .
.
Гетероциклильное кольцо может быть конденсировано с арильным, гетероарильным или циклоалкильным кольцом, где кольцо, соединенное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры гетероциклильного кольца включают:
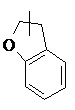
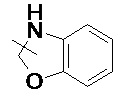
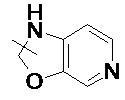
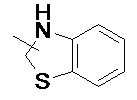 , и т. д.
, и т. д.
Гетероциклил может быть необязательно замещенным или незамещенным, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклоалкилтио и оксо.
Термин "арил" относится к 6-14-членной, предпочтительно 6-10-членной углеродной моноциклической или конденсированной полициклической (т.е., кольца, имеющие общую пару соседних атомов углерода) группе, имеющей сопряженную π-электронную систему, такой как фенил и нафтил, предпочтительно фенил. Арильное кольцо может быть конденсировано с гетероарильным, гетероциклильным или циклоалкильным кольцом, где кольцо, соединенное с исходной структурой, представляет собой арильное кольцо. Неограничивающие примеры арильного кольца включают:
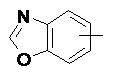
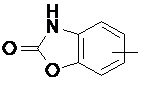
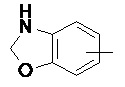
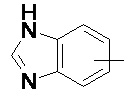
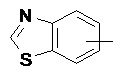
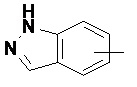
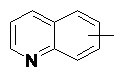
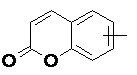 и
и  .
.
Арил может быть замещен или незамещен, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин "гетероарил" относится к гетероароматической системе, содержащей от 1 до 4 гетероатомов и от 5 до 14 кольцевых атомов, где гетероатомы выбраны из группы, состоящей из кислорода, серы и азота. Гетероарил предпочтительно представляет собой 5-10-членный, более предпочтительно 5- или 6-членный, такой как фуранил, тиенил, пиридинил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил и тетразолил. Гетероарильное кольцо может быть конденсировано с арильным, гетероциклильным или циклоалкильным кольцом, где кольцо, соединенное с исходной структурой, представляет собой гетероарильное кольцо. Неограничивающие примеры гетероарильного кольца включают:
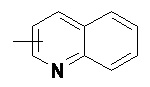
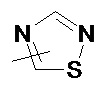 и
и 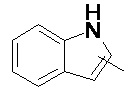 .
.
Гетероарил может быть необязательно замещен или незамещен, и когда он замещен, заместитель предпочтительно представляет собой одну или более из следующих групп, независимо выбранных из группы, состоящей из: алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, меркапто, гидрокси, нитро, циано, циклоалкила, гетероциклоалкила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио и гетероциклоалкилтио.
Термин "аминозащитная группа" относится к группе, которая может быть легко удалена и предназначена для защиты аминогруппы от изменения, когда реакция проводится в другом месте молекулы. Неограничивающие примеры аминозащитных групп включают 9-флуоренилметоксикарбонил, трет-бутоксикарбонил, ацетил, бензил, аллил, п-метоксибензил и т.д. Эти группы могут быть необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из галогена, алкокси и нитро. Аминозащитная группа предпочтительно представляет собой 9-флуоренилметоксикарбонил.
Термин "циклоалкилалкил" относится к алкилу, замещенному одной или более циклоалкильными группами, предпочтительно одной циклоалкильной группой, где алкил является таким, как определено выше, и циклоалкил является таким, как определено выше.
Термин "галогеналкил" относится к алкилу, замещенному одним или более галогенами, где алкил является таким, как определено выше.
Термин "дейтерированный алкил" относится к алкилу, замещенному одним или более атомами дейтерия, где алкил является таким, как определено выше.
Термин «гидрокси» относится к группе -ОН.
Термин «галоген» относится к фтору, хлору, брому или йоду.
Термин "амино" относится к -NH2.
Термин "нитро" относится к -NO2.
Термин "необязательный" или "необязательно" означает, что событие или обстоятельство, описанное впоследствии, может, но не обязательно, иметь место, и что описание включает случаи, когда событие или обстоятельство происходит или не происходит. Например, "необязательно содержащий 1-3 вариабельные области тяжелой цепи антитела" означает, что вариабельная область тяжелой цепи антитела конкретной последовательности может, но не обязательно, присутствовать.
Термин "замещен" означает, что один или более, предпочтительно вплоть до 5, более предпочтительно от 1 до 3 атомов водорода в группе независимо замещены соответствующим количеством заместителей. Само собой разумеется, что заместитель находится только в своем возможном химическом положении, и специалисты в данной области техники смогут определить (экспериментально или теоретически) возможное или невозможное замещение без особых усилий. Например, оно может быть нестабильным, когда амино или гидрокси, имеющая свободный водород, связана с атомом углерода, имеющим ненасыщенную (например, олефиновую) связь.
Термин «нагрузка лекарственным средством» относится к среднему количеству цитотоксических лекарственных средств, нагруженных на каждое антитело или его антигенсвязывающий фрагмент в молекулах конъюгата антитела и лекарственного средства, и он также может быть выражен в виде отношения количества лекарственных средств к количеству антител. Нагрузка лекарственным средством может составлять от 0 до 12, предпочтительно от 1 до 10, более предпочтительно от 3 до 8 и наиболее предпочтительно от 5,3 до 6,1 цитотоксических лекарственных средств, связанных с каждым антителом или его антигенсвязывающим фрагментом (Pc). В вариантах осуществления настоящего изобретения нагрузка лекарственным средством представлена в n, которое также может быть обозначено как значение DAR (соотношение лекарственного средства и антитела), и приведенные в качестве примера значения могут быть средними значениями 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10. Среднее количество лекарственных средств на молекулу ADC при реакциях связывания может быть охарактеризовано обычными методами, такими как спектроскопия в видимой и ультрафиолетовой областях спектра (UV-Vis), гидрофобная хроматография с масс-спектрометрией (HIC), анализы ELISA (иммуноферментный анализ) и HPLC.
Нагрузка цитотоксического лекарственного средства может контролироваться с помощью следующих неограничивающих способов, включая:
(1) контроль молярного соотношения линкерного реагента к моноклональному антителу,
(2) контроль времени и температуры реакции, и
(3) выбор различных реагентов.
Для получения общепринятых фармацевтических композиций делается ссылка на Китайскую фармакопею.
Термин «носитель» для лекарственного средства по настоящему изобретению относится к системе, которая может изменять способ попадания лекарственного средства в организм человека и распределение лекарственного средства в организме человека, контролировать скорость высвобождения лекарственного средства и доставлять лекарственное средство в орган-мишень. Система высвобождения и нацеливания на основе носителя лекарственного средства может уменьшить деградацию и потерю лекарственного средства, уменьшить побочные эффекты и улучшить биодоступность. Например, полимерные поверхностно-активные вещества, которые могут быть использованы в качестве носителей, могут самостоятельно собираться благодаря своим уникальным амфифильным структурам с образованием различных форм агрегатов, таких как мицеллы, микроэмульсии, гели, жидкие кристаллы и везикулы, в качестве предпочтительных примеров. Агрегаты имеют способность инкапсулировать молекулы лекарственного средства и имеют хорошую проницаемость для мембран, и поэтому могут быть использованы как превосходные носители лекарственного средства.
Термины "лечить" и "лечение", когда они применяются к животным, людям, экспериментальным субъектам, клеткам, тканям, органам или биологическим жидкостям, относятся к приведению в контакт экзогенного лекарственного средства, терапевтического агента, диагностического агента или композиции с животными, людьми, субъектами, клетками, тканями, органами или биологическими жидкостями. Термины "лечить" и "лечение" могут относиться, например, к терапевтическим, фармакокинетическим, диагностическим, исследовательским и экспериментальным способам. Лечение клеток включает приведение в контакт реагента с клетками и приведение в контакт реагента с жидкостью. Термины "лечить" и "лечение" также относятся к лечению, например, клеток реагентами, диагностике, связывающими композициями или другой клеткой in vitro и ex vivo. «Лечение» применительно к людям, ветеринарным или исследовательским субъектам, относится к терапевтическому лечению, предупредительной или профилактической мерам, и исследовательским и диагностическим применениям.
«Лечить» или «лечение» относятся к введению терапевтического агента, такого как композиция, содержащая любое из соединений конъюгации по настоящему изобретению, либо внутри, либо снаружи пациенту с одним или более симптомами заболевания, в отношении которого известно, что терапевтический агент оказывает терапевтический эффект. Как правило, терапевтический агент вводили в количестве, эффективном для облегчения одного или более симптомов заболевания у пациента или популяции, получавших лечение, для индуцирования регрессии таких симптомов или для ингибирования развития таких симптомов в любой клинически измеримой степени. Количество терапевтического агента, эффективное для облегчения симптомов любого конкретного заболевания (также называемое "терапевтически эффективным количеством"), может варьироваться в зависимости от множества факторов, как состояние болезни, возраст и вес пациента, а также от способности лекарственного средства производить желаемый терапевтический эффект у пациента. Облегчение симптома заболевания может быть оценено с помощью любых методов клинического тестирования, обычно используемых врачами или другими специалистами в области здравоохранения для оценки тяжести или прогрессирования симптома. Хотя варианты осуществления настоящего изобретения (например, способы лечения или продукты) могут быть неэффективными в облегчении симптомов каждого интересующего заболевания, они должны уменьшать симптомы интересующего заболевания у статистически значимого числа пациентов, как определено в соответствии с любыми статистическими методами тестирования, известными в данной области техники, такими как t-критерий Стьюдента, критерий хи-квадрат, U-критерий Манна и Уитни, критерий Крускала-Уоллиса (H-критерий), критерий Джонкера-Терпстра и критерий Уилкоксона.
"Эффективное количество" включает количество, достаточное для облегчения или предотвращения появления симптома или состояния медицинского заболевания. Эффективное количество также относится к количеству, достаточному для обеспечения или облегчения диагностики. Эффективное количество для конкретного пациента или ветеринарного субъекта может варьироваться в зависимости от таких факторов, как состояние, подлежащее лечению, общее состояние здоровья пациента, способ, и путь, и дозировка введения, а также тяжесть побочных эффектов. Эффективное количество может представлять собой максимальную дозу или режим введения, чтобы избежать значительных побочных эффектов или токсических эффектов.
"Обмен" относится к обмену системы растворителей, которая солюбилизирует белок антитела. Например, систему с высоким содержанием солей или гипертонических растворителей, содержащую белок антитела, заменяется физическими операциями буферной системой стабильного препарата, так что белок антитела присутствует в стабильном препарате. Физические операции включают, но не ограничиваются ими, ультрафильтрацию, диализ или восстановление после центрифугирования.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1A показаны результаты теста стабильности в плазме для ADC-19 по настоящему изобретению.
На фиг. 1B показаны результаты теста стабильности в плазме для ADC-18 по настоящему изобретению.
На фиг. 1C показаны результаты теста стабильности в плазме для ADC-20 по настоящему изобретению.
На фиг. 2 показана оценка эффективности ADC-21 и ADC-24 по настоящему изобретению у мышей с опухолью JIMT-1.
На фиг. 3 показана оценка терапевтического эффекта ADC на голых мышах с ксенотрансплантатом клеток SK-BR-3 рака молочной железы человека.
На фиг. 4 показаны результаты теста стабильности в плазме для ADC-25 по настоящему изобретению.
На фиг. 5 показана эффективность ADC по настоящему изобретению против опухоли астроцитомы головного мозга человека U87MG, ксенотрансплантированной голым мышам.
На фиг. 6 показана эффективность ADC по настоящему изобретению против раковых клеток опухоли метастатического рака глотки человека Detroit 562 с плевральным выпотом, ксенотрансплантированных голым мышам.
На фиг. 7 показана эффективность ADC по настоящему изобретению против опухоли глиобластомы головного мозга человека U87MG, ксенотрансплантированной голым мышам.
На фиг. 8 показаны подогнанные графики тенденции для эксперимента по скринингу формулы, где PS80 находится в единицах 10-4 г/мл, а концентрация белка ADC-32 (на основе голого антитела) находится в единицах мг/мл.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение дополнительно описано ниже со ссылкой на примеры, которые, однако, не предназначены для ограничения объема настоящего изобретения. Экспериментальные способы в примерах по настоящему изобретению, в которых конкретные условия не указаны, как правило, выполняют в обычных условиях, например, со ссылкой на Antibodies: A Laboratory Manual («Антитела: лабораторное руководство») и Molecular Cloning: A Laboratory Manual («Молекулярное клонирование: лабораторное руководство») от Cold Spring Harbor Laboratory («Лаборатория Колд-Спринг-Харбор»), или в условиях, рекомендованных производителем сырья или товаров. Реагенты без указания конкретного происхождения являются коммерчески доступными общепринятыми реагентами.
I. Конъюгаты антитела и лекарственного средства
Структуры соединений идентифицировали с помощью ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (MS). Спектры NMR измеряли с использованием прибора ядерного магнитного резонанса Bruker AVANCE-400, с дейтерированным диметилсульфоксидом (DMSO-d6 (диметилсульфоксид)), дейтерированным хлороформом (CDCl3) и дейтерированным метанолом (CD3OD) в качестве растворителей и тетраметилсиланом (ТМС) в качестве внутреннего стандарта. Химические сдвиги приведены в единицах измерения 10-6 (млн-1).
MS-анализ проводили с использованием масс-спектрометра FINNIGAN LCQAd (ESI) (производитель: Thermo, модель: Finnigan LCQ advantage MAX).
Анализ UPLC (сверхэффективная жидкостная хроматография) проводили с использованием системы жидкостной хроматографии-масс-спектрометрии Waters Acquity UPLC SQD.
Анализ UPLC проводили с использованием жидкостного хроматографа Agilent 1200DAD высокого давления (хроматографическая колонка Sunfire C18 150×4,6 мм) и жидкостного хроматографа Waters 2695-2996 высокого давления (хроматографическая колонка Gimini C18 150×4,6 мм).
Анализ UV-HPLC (высокоэффективная жидкостная хроматография с использованием UV детектора) проводили с использованием спектрофотометра в УФ-спектре Thermo nanodrop2000.
Степень ингибирования пролиферации и значения IC50 (концентрация полумаксимального ингибирования) измеряли с помощью ридера для микропланшетов PHERA starFS (BMG, Германия).
Силикагелевые пластины Yantai Huanghai HSGF254 или Qingdao GF254 со спецификациями от 0,15 мм до 0,2 мм применяли для тонкослойной хроматографии (TLC) и от 0,4 мм до 0,5 мм для разделения и очистки с помощью TLC.
Силикагель Yantai Huanghai 200-300 меш обычно использовали в качестве носителя в колоночной хроматографии.
Известные исходные материалы по настоящему изобретению могут быть синтезированы с использованием или в соответствии со способами, известными в данной области техники, или могут быть приобретены у ABCR GmbH & Co.KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc, Chembee Chemicals и т.д.
В примерах все реакции проводили в атмосфере аргона или в атмосфере азота, если не указано иное.
Атмосфера аргона или атмосфера азота означает, что реакционная колба соединена с баллоном, содержащим около 1 л аргона или азота.
Атмосфера водорода означает, что реакционная колба соединена с баллоном, содержащим около 1 л водорода.
Гидрогенизатор Parr 3916EKX, гидрогенизатор Qinglan QL-500 или гидрогенизатор HC2-SS использовали в реакциях гидрирования под давлением.
Реакции гидрирования обычно включают 3 цикла вакуумирования и продувки водородом.
В реакциях, проводимых под воздействием микроволнового излучения, использовали микроволновый реактор CEM Discover-S 908860.
В примерах раствор в реакции относится к водному раствору, если не указано иное.
В примерах температура реакции представляет собой комнатную температуру, если не указано иное.
Комнатная температура является оптимальной температурой реакции, которая находится в диапазоне от 20 до 30 °C.
Получение буфера PBS (фосфатно-солевой буферный раствор) при рН 6,5 в примерах: 8,5 г KH2PO4, 8,56 г K2HPO4.3H2O, 5,85 г NaCl и 1,5 г EDTA (этилендиаминтетрауксусная кислота) добавляли в колбу, и объем доводили до 2 л. Все добавления растворяли ультразвуком, и раствор хорошо перемешивали путем встряхивания с получением требуемого буфера.
Элюирующая система для колоночной хроматографии и система проявляющего растворителя для тонкослойной хроматографии, используемая для очистки соединения, включают: A: дихлорметанольную и изопропанольную систему, B: дихлорметанольную и метанольную систему и C: петролейноэфирную и этилацетатную систему. Объемное соотношение растворителей корректировали в соответствии с полярностью соединения или добавляли небольшое количество триэтиламина и кислотного или основного реагента.
Некоторые из соединений по настоящему изобретению характеризовали с помощью анализа Q-TOF LC/MS (жидкостная хроматография с масс-спектрометрией). Анализа Q-TOF LC/MS выполняли квадрупольный времяпролетный масс-спектрометр Agilent 6530 с точной массой и сверхвысокоэффективным жидкостным хроматографом Agilent 1290-Infinity (хроматографическая колонка Agilent Poroshell 300SB-C8 5 мкм, 2,1×75 мм).
Пример 1
N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-1-гидроксициклопропан-1-карбоксамид 1
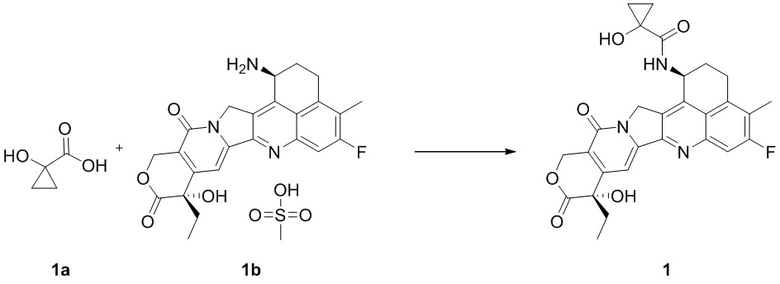
К экзатекан мезилату 1b (2,0 мг, 3,76 мкмоль, полученному, как описано в патентной заявке "EP0737686A1") добавляли 1 мл N,N-диметилформамида. Смесь охлаждали до 0-5 °C на ледяной водяной бане и добавляли каплю триэтиламина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 1-гидроксициклопропилкарбоновую кислоту 1а (1,4 мг, 3,7 мкмоль, полученную известным способом "Tetrahedron Letters, 25(12), 1269-72; 1984") и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (3,8 мг, 13,7 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 2 часов, гасили 5 мл воды и экстрагировали этилацетатом (8 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 1 (1,6 мг, выход 82,1 %).
MS m/z (ESI (ионизация электроспреем)): 520,2 [M+1].
1H ЯМР (400 МГц, CDCl3): δ 7,90-7,84 (m, 1H), 7,80-7,68 (m, 1H), 5,80-5,70 (m, 1H), 5,62-5,54 (m, 2H), 5,44-5,32 (m, 2H), 5,28-5,10 (m, 2H), 3,40-3,15 (m, 3H), 2,44 (s, 3H), 2,23 (t, 1H), 2,06-1,75 (m, 2H), 1,68-1,56 (m, 1H), 1,22-1,18 (m, 2H), 1,04-0,98 (m, 2H), 0,89 (t, 3H).
Пример 1-2.
(S)-2-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксиацетамид 2-A
(R)-2-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксиацетамид 2-B
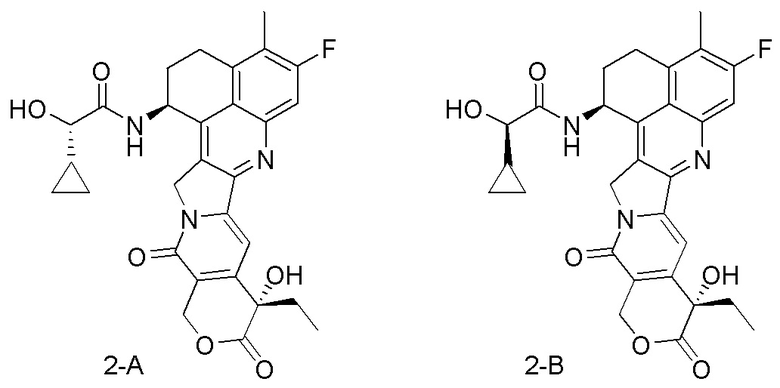
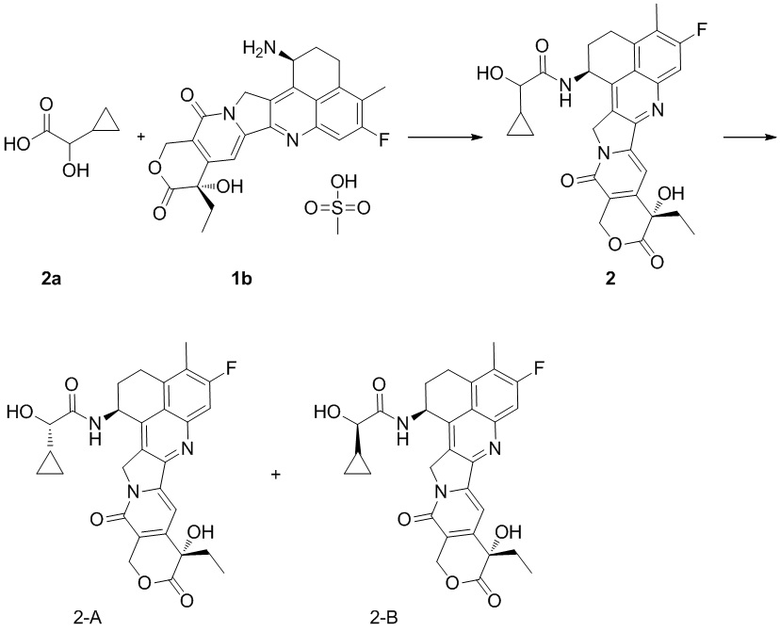
К соединению 1b (4 мг, 7,53 мкмоль) добавляли 2 мл этанола и 0,4 мл N,N-диметилформамида. Систему трижды продували аргоном, и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением по каплям 0,3 мл N-метилморфолина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 2-циклопропил-2-гидроксиуксусную кислоту 2а (2,3 мг, 19,8 мкмоль, полученную, как описано в патентной заявке "WO2013106717"), 1-гидроксибензотриазол (3 мг, 22,4 мкмоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (4,3 мг, 22,4 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 1 часа. Ледяную водяную баню удаляли, и реакционную смесь нагревали до 30 °С, перемешивали в течение 2 ч и концентрировали при пониженном давлении. Полученное неочищенное соединение 2 очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: хроматографическая колонка: XBridge Prep C18 OBD 5 мкм 19 × 250 мм; подвижная фаза: A - вода (10 ммоль NH4OAc), B - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин), и соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продуктов (2-A: 1,5 мг, 2-B: 1,5 мг).
MS m/z (ESI): 534,0 [M+1].
Соединение 2-B с единственной конфигурацией (с более коротким временем удерживания):
Анализ UPLC: время удерживания: 1,06 мин; чистота: 88 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 8,37 (d, 1H), 7,76 (d, 1H), 7,30 (s, 1H), 6,51 (s, 1H), 5,58-5,56 (m, 1H), 5,48 (d, 1H), 5,41 (s, 2H), 5,32-5,29 (m, 2H), 3,60 (t, 1H), 3,19-3,13 (m, 1H), 2,38 (s, 3H), 2,20-2,14 (m, 1H), 1,98 (q, 2H), 1,87-1,83 (m, 1H), 1,50-1,40 (m, 1H), 1,34-1,28 (m, 1H), 0,86 (t, 3H), 0,50-0,39 (m, 4H).
Соединение с единственной конфигурацией 2-A (с более длительным временем удерживания):
Анализ UPLC: время удерживания: 1,10 мин; чистота: 86 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР(400 МГц, DMSO-d6): δ 8,35 (d, 1H), 7,78 (d, 1H), 7,31 (s, 1H), 6,52 (s, 1H), 5,58-5,53 (m, 1H), 5,42 (s, 2H), 5,37 (d, 1H), 5,32 (t, 1H), 3,62 (t, 1H), 3,20-3,15 (m, 2H), 2,40 (s, 3H), 2,25-2,16 (m, 1H), 1,98 (q, 2H), 1,87-1,82 (m, 1H), 1,50-1,40 (m, 1H), 1,21-1,14 (m, 1H), 0,87 (t, 3H), 0,47-0,35 (m, 4H).
Пример 1-3
(S)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-3,3,3-трифтор-2-гидроксипропионамид 3-A
(R)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-3,3,3-трифтор-2-гидроксипропионамид 3-B
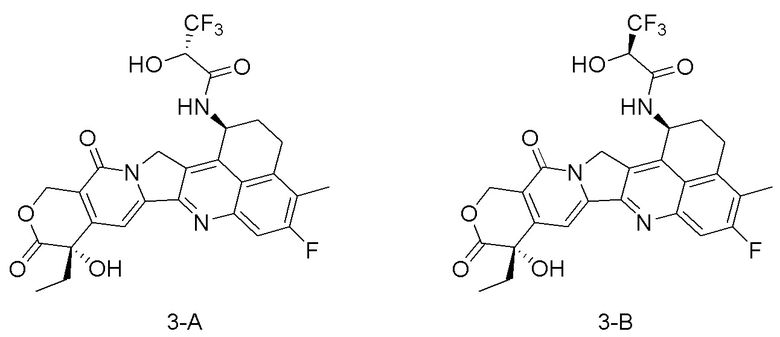
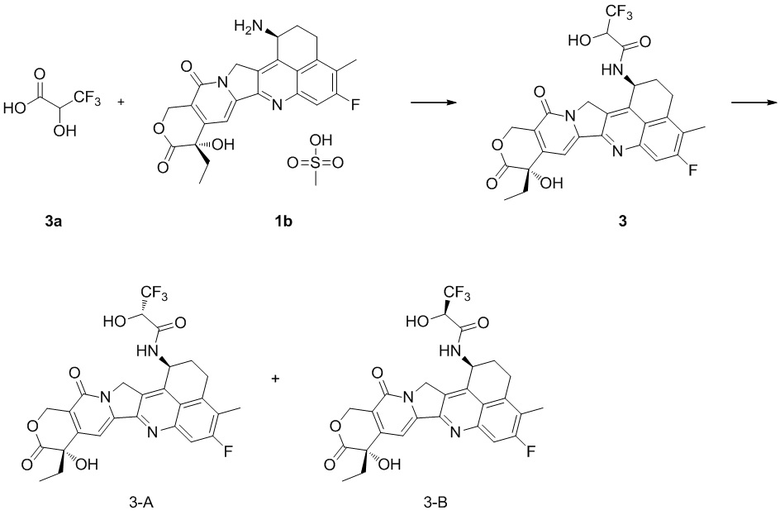
К соединению 1b (5,0 мг, 9,41 мкмоль) добавляли 2 мл этанола и 0,4 мл N,N-диметилформамида, и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением по каплям 0,3 мл N-метилморфолина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 3,3,3-трифтор-2-гидроксипропионовую кислоту 3a (4,1 мг, 28,4 мкмоль, поставляемый Alfa), 1-гидроксибензотриазол (3,8 мг, 28,1 мкмоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (5,4 мг, 28,2 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 10 минут. Ледяную водяную баню удаляли, и реакционную смесь нагревали до 30 °C, перемешивали в течение 8 часов и концентрировали при пониженном давлении. Полученное неочищенное соединение 3 очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: хроматографическая колонка: XBridge Prep C18 OBD 5 мкм 19 × 250 мм; подвижная фаза: A - вода (10 ммоль NH4OAc), B - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин), и соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продуктов 1,5 мг, 1,5 мг).
MS m/z (ESI): 561,9 [M+1].
Соединение с единственной конфигурацией (с более коротким временем удерживания):
Анализ UPLC: время удерживания: 1,11 мин; чистота: 88 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 8,94 (d, 1H), 7,80 (d, 1H), 7,32 (s, 1H), 7,20 (d, 1H), 6,53 (s, 1H), 5,61-5,55 (m, 1H), 5,45-5,23 (m, 3H), 5,15-5,06 (m, 1H), 4,66-4,57 (m, 1H), 3,18-3,12 (m, 1H), 2,40 (s, 3H), 2,26-2,20 (m, 1H), 2,16-2,08 (m, 1H), 2,02-1,94 (m, 1H), 1,89-1,82 (m, 1H), 1,50-1,40 (m, 1H), 0,87 (t, 3H).
Соединение с единственной конфигурацией (с более длительным временем удерживания):
Анализ UPLC: время удерживания: 1,19 мин; чистота: 90 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 8,97 (d, 1H), 7,80 (d, 1H), 7,31 (s, 1H), 7,16 (d, 1H), 6,53 (s, 1H), 5,63-5,55 (m, 1H), 5,45-5,20 (m, 3H), 5,16-5,07 (m, 1H), 4,66-4,57 (m, 1H), 3,18-3,12 (m, 1H), 2,40 (s, 3H), 2,22-2,14 (m, 1H), 2,04-1,95 (m, 2H), 1,89-1,82 (m, 1H), 1,50-1,40 (m, 1H), 0,87 (t, 3H).
Пример 1-4
N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-1-гидроксициклопентан-1-карбоксамид 4
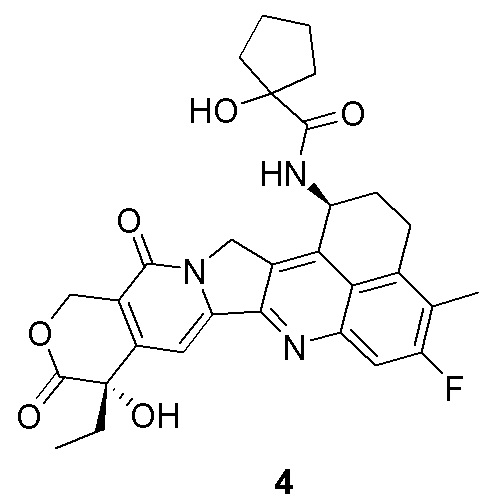
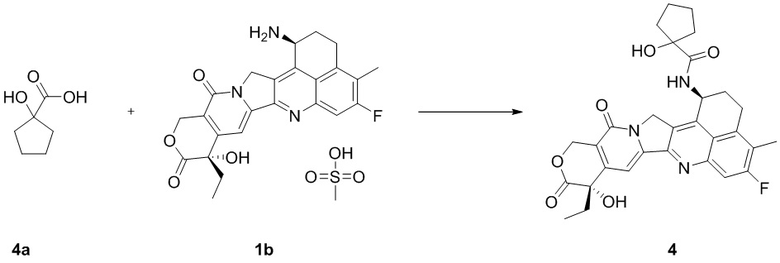
К соединению 1b (3,0 мг, 5,64 мкмоль) добавляли 1 мл N,N-диметилформамида. Смесь охлаждали до 0-5 °C на ледяной водяной бане и добавляли каплю триэтиламина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 1-гидрокси-циклопентанкарбоновую кислоту 4a (2,2 мг, 16,9 мкмоль, полученную, как описано в патентной заявке "WO2013106717") и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиний хлорид (4,7 мг, 16,9 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 1 часа, гасили 5 мл воды и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 4 (2,5 мг, выход 80,9 %).
MS m/z (ESI): 548,0 [M+1].
1H ЯМР (400 МГц, CDCl3): δ 7,73-7,62 (м, 2H), 5,75-5,62 (м, 1H), 5,46-5,32 (м, 2H), 5,26-5,10 (м, 1H), 3,30-3,10 (м, 1H), 2,43 (с, 3H), 2,28-2,20 (м, 2H), 2,08-1,84 (м, 8H), 1,69-1,58 (м, 2H), 1,04-1,00 (м, 2H), 0,89 (т, 3H).
Пример 1-5
N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-1-(гидроксиметил)циклопропан-1-карбоксамид 5
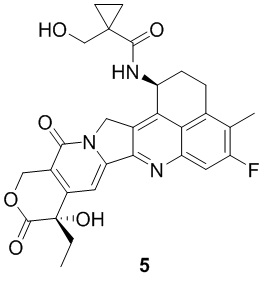
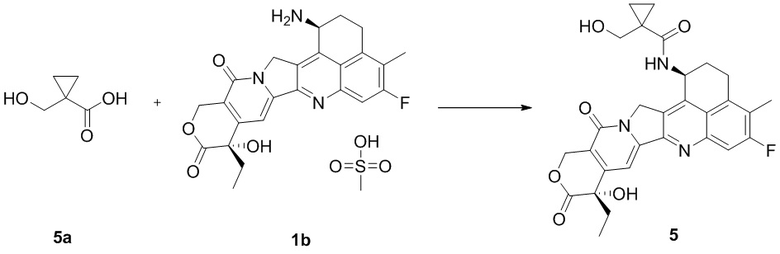
К соединению 1b (2,0 мг, 3,76 мкмоль) добавляли 1 мл N,N-диметилформамида. Смесь охлаждали до 0-5 °C на ледяной водяной бане и добавляли каплю триэтиламина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 1-(гидроксиметил)-циклопентанкарбоновую кислоту 5a (0,87 мг, 7,5 мкмоль, полученную, как описано в патентной заявке "WO201396771") и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (2 мг, 7,24 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 2 часов, гасили 5 мл воды и экстрагировали этилацетатом (8 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 5 (1,0 мг, выход 50 %).
MS m/z (ESI): 533,9 [M+1].
1H ЯМР (400 МГц, CDCl3): δ 8,07 (s, 1H), 7,23-7,18 (m, 2H), 6,71-6,64 (m, 1H), 6,55-6,51 (m, 1H), 5,36-5,27 (m, 2H), 4,67-4,61 (m, 2H), 3,53-3,48 (m, 1H), 3,30-3,22 (m, 2H), 3,18-3,13 (m, 1H), 2,71-2,61 (m, 2H), 2,35-2,28 (m, 1H), 2,04-1,91 (m, 4H), 1,53-1,40 (m, 3H), 0,91-0,75 (m, 4H).
Пример 1-6
N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-1-(гидроксиметил)циклобутан-1-карбоксамид 6
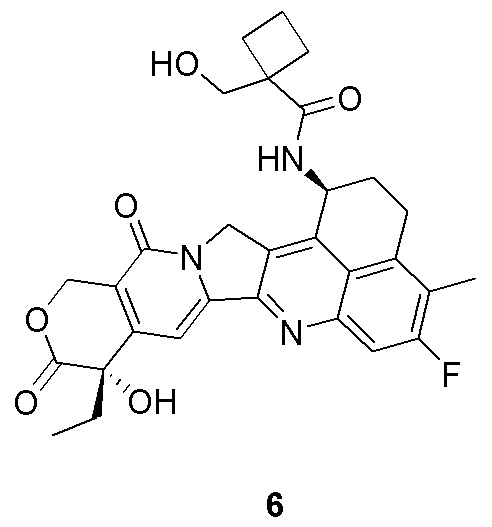
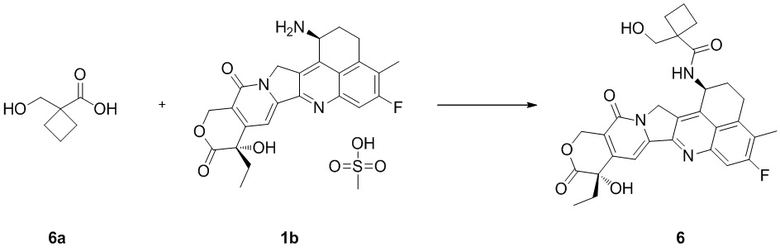
К соединению 1b (3,0 мг, 5,64 мкмоль) добавляли 1 мл N,N-диметилформамида. Смесь охлаждали до 0-5 °C на ледяной водяной бане и добавляли каплю триэтиламина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 1-(гидроксиметил)циклобутан-1-карбоновую кислоту 6a (2,2 мг, 16,9 мкмоль, полученную, как описано в "Journal of the American Chemical Society, 2014, vol.136, #22, с. 8138-8142") и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (4,7 мг, 16,9 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 1 часа, гасили 5 мл воды и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 6 (2,1 мг, выход 67,9 %).
MS m/z (ESI): 548,0 [M+1].
1H ЯМР (400 MГц, DMSO-d6): δ 7,85-7,62 (м, 1H), 6,88 (бр, 1H), 5,87-5,48 (м, 2H), 5,47-5,33 (м, 1H), 5,31-5,06 (м, 1H), 4,25-3,91 (м, 2H), 3,25 (бр, 1H), 2,60-2,32 (м, 3H), 2,23 (т, 1H), 2,15-1,95 (м, 3H), 1,70-1,56 (м, 2H), 1,41-1,17 (м, 9H), 1,03 (с, 1H), 0,95-0,80 (м, 2H).
Пример 1-7
N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-1-гидроксициклобутан-1-карбоксамид 7
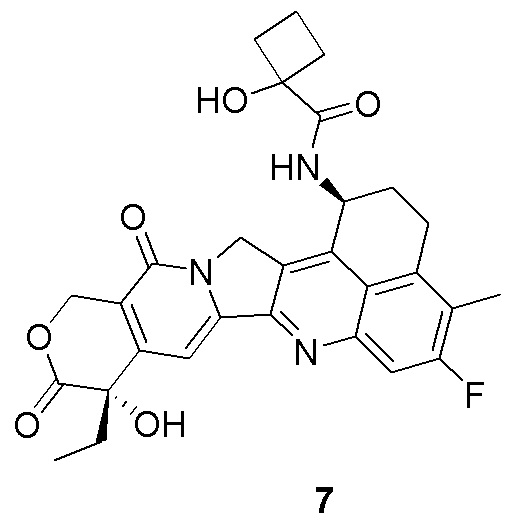
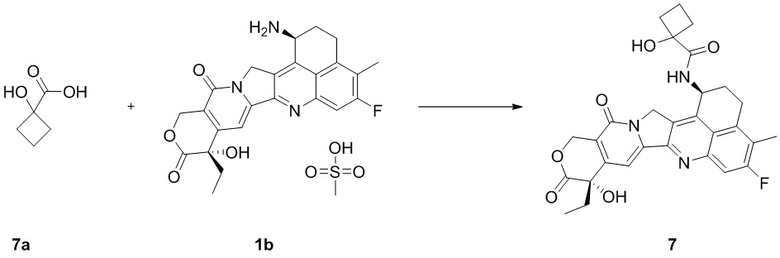
К соединению 1b (3,0 мг, 5,64 мкмоль) добавляли 2 мл этанола и 0,4 мл N,N-диметилформамида, и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением по каплям 0,3 мл N-метилморфолина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли 1-гидроксициклобутанкарбоновую кислоту 7а (2,0 мг, 17,22 мкмоль, поставляемый PharmaBlock), 1-гидроксибензотриазол (2,3 мг, 17,0 мкмоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (3,2 мг, 16,7 мкмоль). После добавления реакционную смесь перемешивали при 0-5 °C в течение 10 минут. Ледяную водяную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 2 ч, и концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 7 (2,5 мг, выход 83,1 %).
MS m/z (ESI): 534,0 [M+1].
1H ЯМР (400 MГц, DMSO-d6): δ 8,28 (d, 1H), 7,75 (d, 1H), 7,29 (s, 1H), 6,51 (s, 1H), 6,12 (s, 1H), 5,59-5,51 (m, 1H), 5,41 (s, 2H), 5,20-5,01 (m, 2H), 3,27-3,17 (m, 1H), 3,15-3,05 (m, 1H), 2,71-2,63 (m, 1H), 2,37 (s, 3H), 2,12-2,05 (m, 1H), 2,03-1,94 (m, 2H), 1,92-1,78 (m, 4H), 1,50-1,42 (m, 1H), 0,90-0,83 (m, 4H).
Пример 1-8
1-(((S)-7-бензил-20-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-3,6,9,12,15-пентаоксо-2,5,8,11,14-пентаазеикозил)окси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклопропан-1-карбоксамид 8
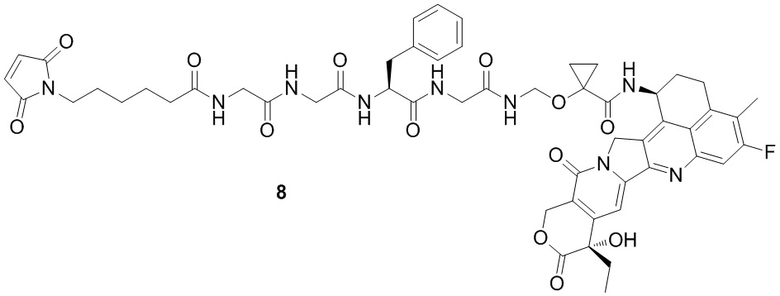
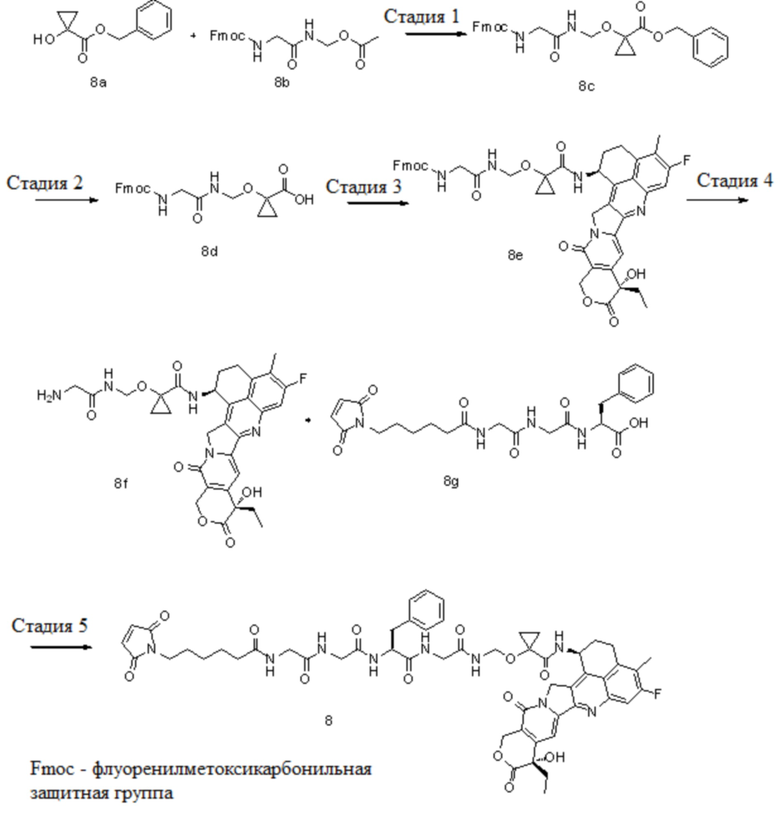
Стадия 1
Бензил-1-((2-((((9H-флуорен-9-ил)метокси)карбонил)амино)ацетиламино)метокси)циклопропан-1-карбоксилат 8c
Бензил-1-гидроксициклопропан-1-карбоксилат 8a (104 мг, 0,54 ммоль; полученный, как описано в патентной заявке "US2005/20645") и 2-((((9H-флуорен-9-ил)метокси)карбонил)амино)ацетиламино)метилацетат 8b (100 мг, 0,27 ммоль; полученный, как описано в патентной заявке "CN105829346A") добавляли в реакционную колбу и добавляли 5 мл тетрагидрофурана. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида калия (61 мг, 0,54 ммоль). Ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 10 минут с последующим добавление 20 мл ледяной воды и экстракцией этилацетатом (5 мл×2) и хлороформом (5 мл×5). Органические фазы объединяли и концентрировали. Полученный остаток растворяли в 3 мл 1,4-диоксана и добавляли 0,6 мл воды, бикарбоната натрия (27 мг, 0,32 ммоль) и 9-фторенилметилхлорформиата (70 мг, 0,27 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 20 мл воды с последующей экстракцией этилацетатом (8 мл×3). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 8c (100 мг, выход 73,6 %).
MS m/z (ESI): 501,0 [M+1].
Стадия 2
1-((2-((((9H-флуорен-9-ил)метокси)карбонил)амино)ацетиламино)метокси)циклопропан-1-карбоновая кислота 8d
Соединение 8c (50 мг, 0,10 ммоль) растворяли в 3 мл смеси растворителей тетрагидрофурана и этилацетата (V:V = 2:1) и добавляли палладий на угле (25 мг, нагрузка 10 %). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали через целит, а осадок на фильтре промывали тетрагидрофураном. Фильтрат концентрировали с получением указанного в заголовке продукта 8d (41 мг, выход 100 %).
MS m/z (ESI): 411,0 [M+1].
Стадия 3
(9H-флуорен-9-ил)метил(2-(((1-((1S,9S) -9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклопропокси)метил)амино)-2-оксоэтил)карбамат 8e
Соединение 1b (7 мг, 0,013 ммоль) добавляли в реакционную колбу и добавляли 1 мл N,N-диметилформамида. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина, раствора соединения 8d (7 мг, 0,017 ммоль) в 0,5 мл N,N-диметилформамида и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (7 мг, 0,026 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 35 мин. Добавляли 10 мл воды с последующей экстракцией этилацетатом (5 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 8e (8,5 мг, выход 78,0 %).
MS m/z (ESI): 828,0 [M+1].
Стадия 4
1-((2-аминоацетиламино)метокси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклопропан-1-карбоксамид 8f
Соединение 8e (4 мг, 4,84 мкмоль) растворяли в 0,2 мл дихлорметана и добавляли 0,1 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 8f (2,9 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 606,0 [M+1].
Стадия 5
1-(((S)-7-бензил-20-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-3,6,9,12,15-пентаоксо-2,5,8,11,14-пентаазеикозил)окси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклопропан-1-карбоксамид 8
Неочищенный 8f (2,9 мг, 4,84 мкмоль) растворяли в 0,5 мл N,N-диметилформамида. Систему трижды продували аргоном, и раствор охлаждали до 0-5 °C на ледяной водяной бане. Добавляли раствор (S)-2(-2-(-2-(6-(2,5-диоксо-1H-пиррол-1-ил)гексанамидо)ацетиламино)ацетиламино)-3-фенилпропионовой кислоты 8 г (2,7 мг, 5,80 мкмоль, полученный, как описано в патентной заявке "EP2907824") в 0,3 мл N,N-диметилформамида с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиний хлорида (2,7 мг, 9,67 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 30 мин. Затем ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры, перемешивали в течение 15 мин и очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: хроматографическая колонка: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: A - вода (10 ммоль NH4OAc), B - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 8 (2 мг, выход 39,0 %).
MS m/z (ESI): 1060,0 [M+1].
1H ЯМР (400 MHz, DMSO-d6): δ 9,01 (d, 1H), 8,77 (t, 1H), 8,21 (t, 1H), 8,08-7,92 (m, 2H), 7,73 (d, 1H), 7,28 (s, 1H), 7,24-7,07 (m, 4H), 6,98 (s, 1H), 6,50 (s, 1H), 5,61 (q, 1H), 5,40 (s, 2H), 5,32 (t, 1H), 5,12 (q, 2H), 4,62 (t, 1H), 4,52 (t, 1H), 4,40-4,32 (m, 1H), 3,73-3,47 (m, 8H), 3,16-3,04 (m, 2H), 2,89 (dd, 1H), 2,69-2,55 (m, 2H), 2,37-2,23 (m, 4H), 2,12-1,93 (m, 4H), 1,90-1,74 (m, 2H), 1,52-1,38 (m, 4H), 1,33-1,11 (m, 5H), 0,91-0,81 (m, 4H).
Пример 1-9
N-((2R,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 9-A
N-((2S,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 9-B
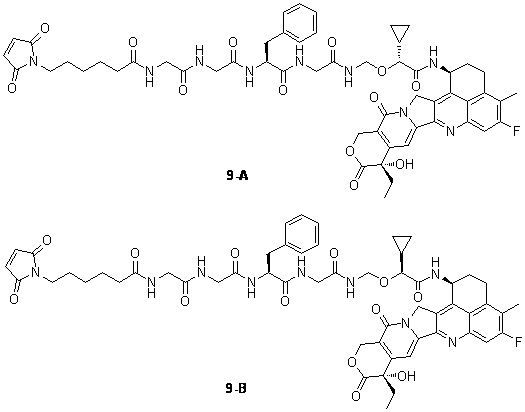
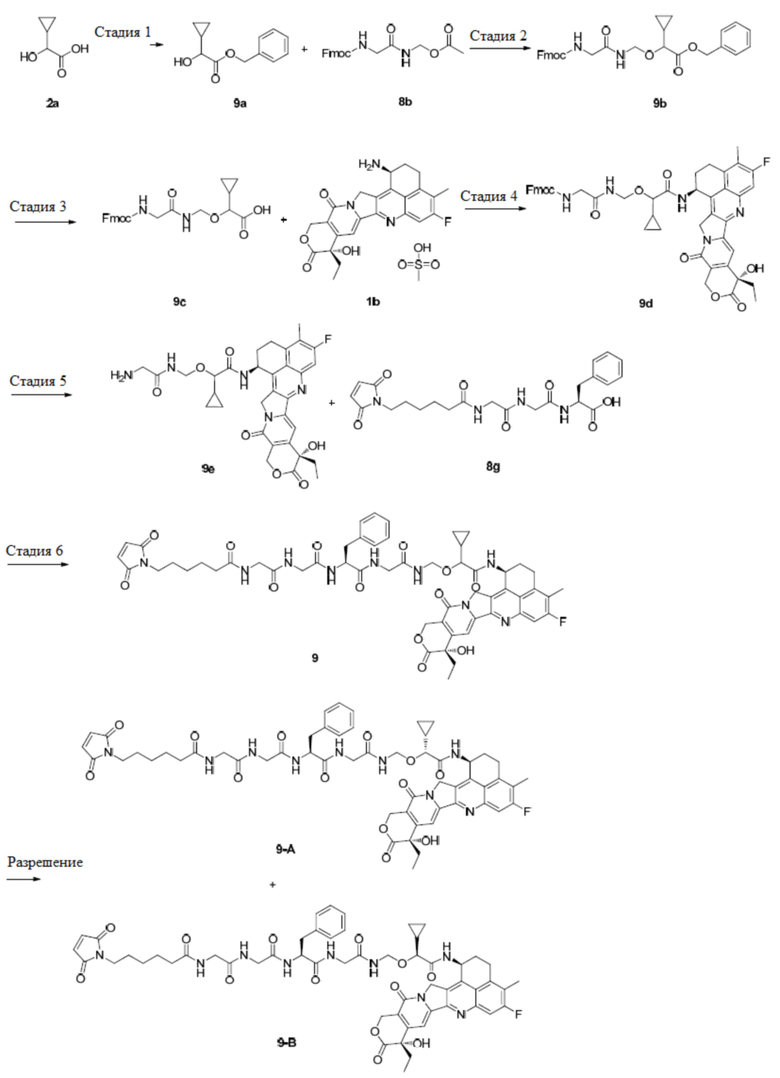
Стадия 1
Бензил-2-циклопропил-2-гидроксиацетат 9a
Соединение 2a (1,3 г, 11,2 ммоль; полученный, как описано в патентной заявке "WO2013/106717") растворяли в 50 мл ацетонитрила и последовательно добавляли карбонат калия (6,18 г, 44,8 ммоль), бензилбромид (1,33 мл, 11,2 ммоль) и тетрабутиламмонийиодид (413 мг, 1,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов и фильтровали через целит, и осадок на фильтре промывали этилацетатом (10 мл). Фильтраты объединяли и концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя C с получением указанного в заголовке продукта 9a (2 г, 86,9 % выхода).
Стадия 2
Бензил-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оат 9b
Соединение 9a (120,9 мг, 0,586 ммоль) и 8b (180 мг, 0,489 ммоль) добавляли в реакционную колбу и добавляли 4 мл тетрагидрофурана. Систему трижды продували аргоном и охлаждали реакционную смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида калия (109 мг, 0,98 ммоль). Ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 40 минут с последующим добавлением 10 мл ледяной воды и экстракцией этилацетатом (20 мл×2) и хлороформом (10 мл×5). Органические фазы объединяли и концентрировали. Полученный остаток растворяли в 4 мл диоксана и добавляли 2 мл воды, бикарбонат натрия (49,2 мг, 0,586 ммоль) и 9-флуоренилметилхлорформиат (126 мг, 0,49 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли 20 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя C с получением указанного в заголовке продукта 9b (48 мг, выход 19 %).
MS m/z (ESI): 515,0 [M+1].
Стадия 3
10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оевая кислота 9c
Соединение 9b (20 мг, 0,038 ммоль) растворяли в 4,5 мл смеси растворителей тетрагидрофурана и этилацетата (V:V = 2:1) и добавляли палладий на угле (12 мг, нагрузка 10%, сухое вещество). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали через целит, а осадок на фильтре промывали этилацетатом. Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 9c (13 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 424,9 [M+1].
Стадия 4
(9H-флуорен-9-ил)метил(2-(((1-циклопропил-2-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-2-оксоэтокси)метил)амино)-2-оксоэтил)карбамат 9d
Соединение 1b (10 мг, 18,8 мкмоль) добавляли в реакционную колбу и добавляли 1 мл N,N-диметилформамида. Систему трижды продували аргоном и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением капли неочищенного триэтиламина 9c (13 мг, 30,6 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (16,9 мг, 61,2 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 40 мин. Добавляли 10 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 9d (19 мг, выход 73,6 %).
MS m/z (ESI): 842,1 [M+1].
Стадия 5
2-((2-аминоацетиламино)метокси)-2-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)ацетамид 9e
Соединение 9d (19 мг, 22,6 мкмоль) растворяли в 2 мл дихлорметана и добавляли 1 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Добавляли 1 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и оставляли стоять. Затем супернатант удаляли и оставляли твердое вещество. Твердый остаток концентрировали при пониженном давлении и сушили с помощью масляного насоса для получения неочищенного указанного в заголовке продукта 9e (17 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 638,0 [М+18].
Стадия 6
N-((2R,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 9-A
N-((2S,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 9-B
Неочищенный 9e (13,9 мг, 22,4 мкмоль) растворяли в 0,6 мл N,N-диметилформамида. Систему трижды продували аргоном, и раствор охлаждали до 0-5 °C на ледяной водяной бане. Добавляли раствор 8 г (21,2 мг, 44,8 мкмоль) в 0,3 мл N,N-диметилформамида с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (18,5 мг, 67,3 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 10 мин. Затем ледяную баню удаляли и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа с получением соединения 9. Реакционную смесь очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке продуктов (9-A: 2,4 мг, 9-B: 1,7 мг).
MS m/z (ESI): 1074,4 [M+1].
Соединение 9-А с единственной конфигурацией (с более коротким временем удерживания):
Анализ UPLC: время удерживания: 1,14 мин; чистота: 85 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 MHz, DMSO-d6): δ 8,60 (t, 1H), 8,51-8,49 (d, 1H), 8,32-8,24 (m, 1H), 8,13-8,02 (m, 2H), 8,02-7,96 (m, 1H), 7,82-7,75 (m, 1H), 7,31 (s, 1H), 7,26-7,15 (m, 4H), 6,99 (s, 1H), 6,55-6,48 (m, 1H), 5,65-5,54 (m, 1H), 5,41 (s, 2H), 5,35-5,15 (m, 3H), 4,74-4,62 (m, 1H), 4,54-4,40 (m, 2H), 3,76-3,64 (m, 4H), 3,62-3,48 (m, 2H), 3,20-3,07 (m, 2H), 3,04-2,94 (m, 1H), 2,80-2,62 (m, 1H), 2,45-2,30 (m, 3H), 2,25-2,15 (m, 2H), 2,04-2,15 (m, 2H), 1,93-1,78 (m, 2H), 1,52-1,39 (m, 3H), 1,34-1,12 (m, 5H), 0,87 (t, 3H), 0,64-0,38 (m, 4H).
Соединение с единственной конфигурацией 9-B (с более длительным временем удерживания):
Анализ UPLC: время удерживания: 1,16 мин; чистота: 89 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 MHz, DMSO-d6): δ 8,68-8,60 (m, 1H), 8,58-8,50 (m, 1H), 8,32-8,24 (m, 1H), 8,13-8,02 (m, 2H), 8,02-7,94 (m, 1H), 7,82-7,75 (m, 1H), 7,31 (s, 1H), 7,26-7,13 (m, 3H), 6,99 (s, 1H), 6,55-6,48 (m, 1H), 5,60-5,50 (m, 1H), 5,41 (s, 2H), 5,35-5,15 (m, 2H), 4,78-4,68 (m, 1H), 4,60-4,40 (m, 2H), 3,76-3,58 (m, 4H), 3,58-3,48 (m, 1H), 3,20-3,10 (m, 2H), 3,08-2,97 (m, 2H), 2,80-2,72 (m, 2H), 2,45-2,30 (m, 3H), 2,25-2,13 (m, 2H), 2,13-2,04 (m, 2H), 2,03-1,94 (m, 2H), 1,91-1,78 (m, 2H), 1,52-1,39 (m, 3H), 1,34-1,12 (m, 4H), 0,91-0,79 (m, 3H), 0,53-0,34 (m, 4H).
Пример 1-10
N-((2S,10S)-10-бензил-2-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)-1,1,1-трифтор-6,9,12,15-тетраоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 10-A
N-((2R,10S)-10-бензил-2-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)-1,1,1-трифтор-6,9,12,15-тетраоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 10-B
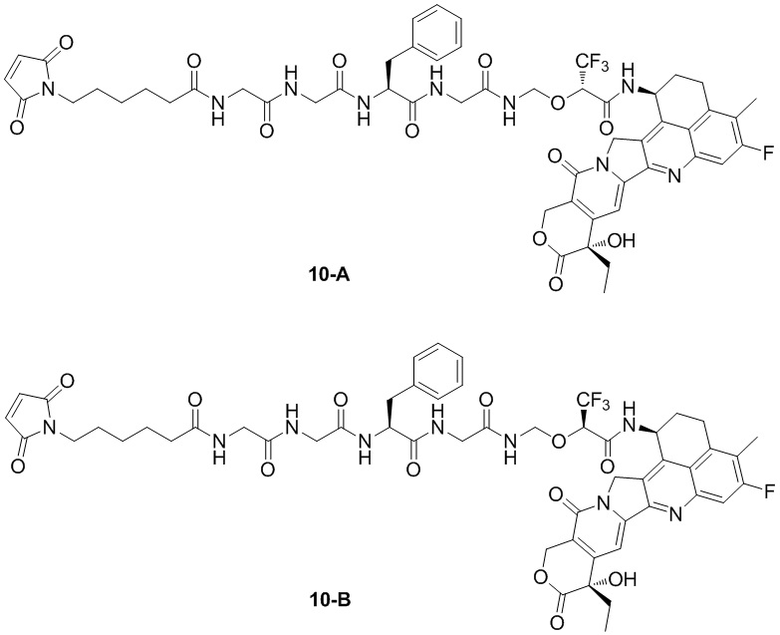
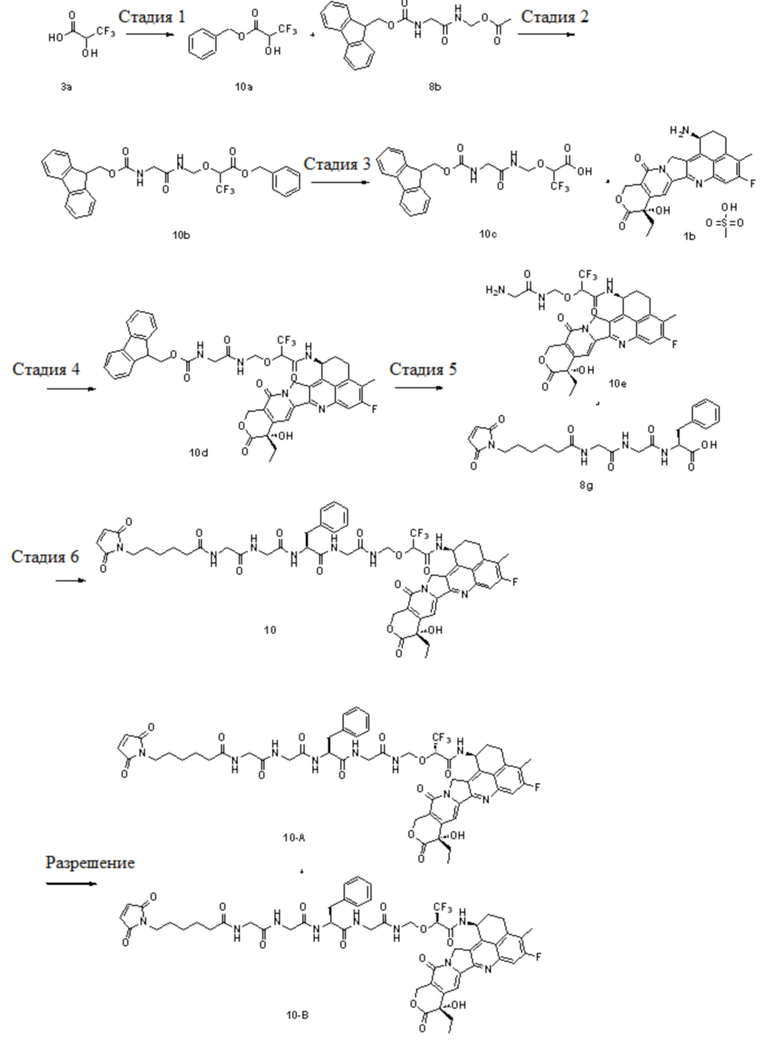
Стадия 1
Бензил 3,3,3-трифтор-2-гидроксипропионат 10A
Соединение 3a (1,80 г, 12,5 ммоль) растворяли в 100 мл ацетонитрила и последовательно добавляли карбонат калия (5,17 г, 37,5 ммоль), бензилбромид (4,48 мл, 37,5 ммоль) и йодид тетрабутиламмония (231 мг, 0,63 ммоль). Реакционную смесь нагревали до 60 °C, перемешивали в течение 5 ч, охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы C проявляющего растворителя с получением указанного в заголовке продукта 10a (980 мг, выход 33,5 %).
1H ЯМР (400 МГц, CDCl3): δ 7,43-7,36 (m, 5H), 5,34 (s, 2H), 4,53 (s, 1H), 3,44 (s, 1H).
Стадия 2
Бензил 1-(9H-флуорен-9-ил)-3,6-диоксо-10-(трифторметил)-2,9-диокса-4,7-диазаундекан-11-оат 10b
Соединение 8b (63 мг, 0,17 ммоль) и 10a (80 мг, 0,34 ммоль) добавляли в реакционную колбу и добавляли 3 мл тетрагидрофурана. Систему трижды продували аргоном и охлаждали реакционную смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида калия (38 мг, 0,34 ммоль). Ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 20 минут с последующим добавлением 10 мл ледяной воды и экстракцией этилацетатом (20 мл×2) и хлороформом (10 мл×5). Органические фазы объединяли и концентрировали. Полученный остаток растворяли в 2 мл диоксана и добавляли 0,4 мл воды, бикарбоната натрия (19 мг, 0,23 ммоль) и 9-флуоренилметилхлорформиата (49 мг, 0,19 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 20 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы C проявляющего растворителя с получением указанного в заголовке продукта 10b (51 мг, выход 55,3 %).
MS m/z (ESI): 559,9 [M+18].
Стадия 3
1-(9H-флуорен-9-ил)-3,6-диоксо-10-(трифторметил)-2,9-диокса-4,7-диазаундекан-11-овая кислота 10c
Соединение 10b (15 мг, 0,28 ммоль) растворяли в 3 мл смеси растворителей тетрагидрофурана и этилацетата (V:V = 2:1) и добавляли палладий на угле (15 мг, нагрузка 10 %). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали через целит, а осадок на фильтре промывали тетрагидрофураном. Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 10C (13 мг).
MS m/z (ESI): 452,9 [M+1].
Стадия 4
(9H-флуорен-9-ил)метил(2-((((3-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,1,1-трифтор-3-оксопроп-2-ил)окси)метил)амино)-2-оксоэтил)карбамат 10d
Соединение 1b (10 мг, 18,8 мкмоль) добавляли в реакционную колбу и добавляли 1 мл N,N-диметилформамида. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина, раствора 10c (13 мг, 28,7 мкмоль) в 0,5 мл N,N-диметилформамида и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (11 мг, 39,7 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 30 мин. Добавляли 10 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 10d (16 мг, выход 97,8 %).
MS m/z (ESI): 870,0 [M+1].
Стадия 5
2-((2-аминоацетиламино)метокси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-3,3,3-трифторпропионамид 10e
Соединение 10d (16 мг, 18,4 мкмоль) растворяли в 0,6 мл дихлорметана и добавляли 0,3 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и оставляли стоять. Затем супернатант удаляли и твердое вещество выдерживали; процедуры повторяли три раза. Твердый остаток концентрировали при пониженном давлении и сушили с помощью масляного насоса для получения неочищенного указанного в заголовке продукта 10e (12 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 647,9 [M+1].
Стадия 6
N-((2S,10S)-10-бензил-2-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)-1,1,1-трифтор-6,9,12,15-тетраоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 10-A
N-((2R,10S)-10-бензил-2-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)-1,1,1-трофтор-6,9,12,15-тетраоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 10-B
Неочищенный 10e (12 мг, 18,5 мкмоль) растворяли в 1,0 мл N,N-диметилформамида. Систему трижды продували аргоном, и раствор охлаждали до 0-5 °C на ледяной водяной бане. Добавляли раствор 8 г (14 мг, 29,6 мкмоль) в 0,3 мл N,N-диметилформамида с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (15 мг, 54,2 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 30 мин. Затем ледяную баню удаляли и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа с получением соединения 10. Реакционную смесь очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке продуктов (2,7 мг, 2,6 мг).
MS m/z (ESI): 1102,0 [M+1].
Соединение с единственной конфигурацией (с более коротким временем удерживания):
Анализ UPLC: время удерживания: 1,18 мин; чистота: 91 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 8,97 (d, 1H), 8,85-8,76 (m, 1H), 8,37-8,27 (m, 1H), 8,12-8,02 (m, 1H), 8,02-7,95 (m, 1H), 7,80 (d, 1H), 7,31 (s, 1H), 7,26-7,10 (m, 4H), 6,99 (s, 1H), 6,66 (br, 1H), 6,52 (s, 1H), 5,65-5,54 (m, 1H), 5,41 (s, 1H), 5,37-5,25 (m, 3H), 5,23-5,13 (m, 1H), 4,81-4,68 (m, 2H), 4,51-4,41 (m, 1H), 3,78-3,45 (m, 6H), 3,21-3,13 (m, 1H), 3,02-2,93 (m, 1H), 2,77-2,63 (m, 2H), 2,45-2,29 (m, 3H), 2,24-2,05 (m, 3H), 2,04-1,93 (m, 5H), 1,90-1,75 (m, 2H), 1,52-1,38 (m, 4H), 0,90-0,78 (m, 5H).
Соединение с единственной конфигурацией (с более длительным временем удерживания):
Анализ UPLC: время удерживания: 1,23 мин; чистота: 90 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 9,05 (d, 1H), 8,97-8,88 (m, 1H), 8,35-8,27 (m, 1H), 8,11-8,03 (m, 1H), 8,02-7,95 (m, 1H), 7,80 (d, 1H), 7,34 (s, 1H), 7,29-7,13 (m, 4H), 6,99 (s, 1H), 6,66 (br, 1H), 6,54 (s, 1H), 5,64-5,55 (m, 1H), 5,43 (s, 1H), 5,36-5,20 (m, 3H), 4,92-4,85 (m, 1H), 4,82-4,72 (m, 2H), 4,52-4,42 (m, 1H), 3,77-3,48 (m, 6H), 3,21-3,14 (m, 1H), 3,03-2,95 (m, 1H), 2,79-2,65 (m, 2H), 2,47-2,28 (m, 3H), 2,25-2,05 (m, 3H), 2,05-1,94 (m, 5H), 1,91-1,76 (m, 2H), 1,52-1,37 (m, 4H), 0,92-0,77 (m, 5H).
Пример 1-11
1-(((S)-7-бензил-20-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-3,6,9,12,15-пентаоксо-2,5,8,11,14-пентаазеикозил)окси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклобутан-1-карбоксамид 11
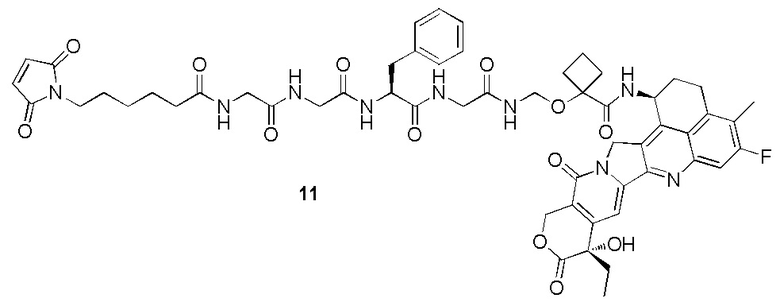
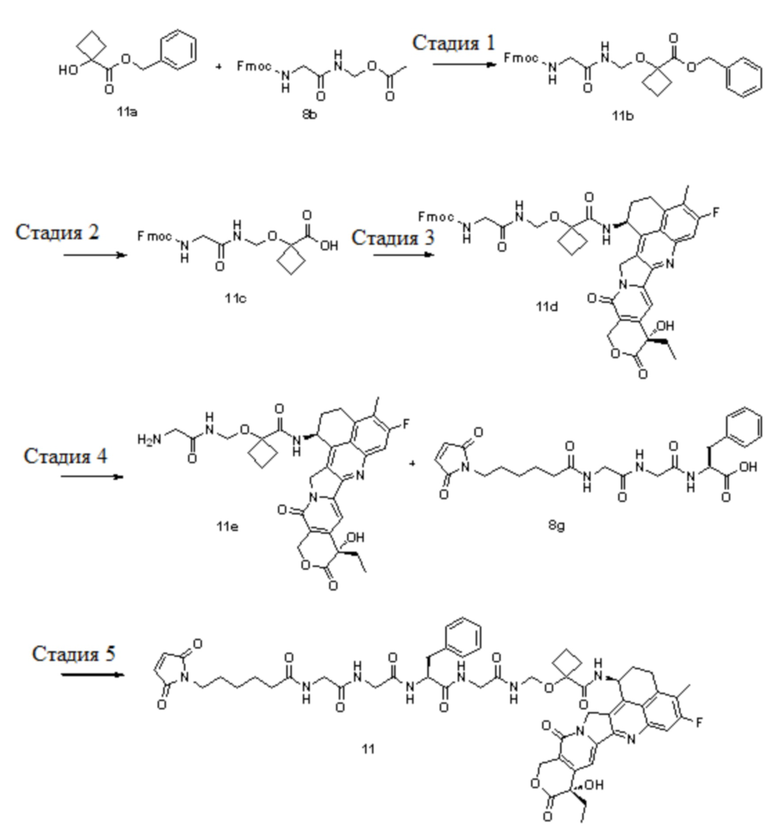
Стадия 1
Бензил-1-((2-((((9H-флуорен-9-ил)метокси)карбонил)амино)ацетиламино)метокси)циклобутан-1-карбоксилат 11b
В реакционную колбу добавляли бензил-1-гидроксициклобутан-карбоксилат 11a (167 мг, 0,81 ммоль, полученный, как описано в "Journal of Medicinal Chemistry, 2013, vol. 56, #13, с. 5541-5552") и 8b (150 мг, 0,41 ммоль) и добавляли 5 мл тетрагидрофурана. Систему трижды продували аргоном и охлаждали реакционную смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида калия (92 мг, 0,82 ммоль). Ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 10 минут с последующим добавление 20 мл ледяной воды и экстракцией этилацетатом (5 мл×2) и хлороформом (5 мл×5). Органические фазы объединяли и концентрировали. Полученный остаток растворяли в 3 мл диоксана и добавляли 0,6 мл воды, бикарбоната натрия (41 мг, 0,48 ммоль) и 9-фторенилметилхлорформиата (105 мг, 0,41 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 20 мл воды с последующей экстракцией этилацетатом (8 мл×3). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы C проявляющего растворителя с получением указанного в заголовке продукта 11b (37 мг, выход 17,6 %).
MS m/z (ESI): 514,6 [M+1].
Стадия 2
1-((2-((((9H-флуорен-9-ил)метокси)карбонил)амино)ацетиламино)метокси)циклобутан-1-карбоновая кислота 11c
Соединение 11b (37 мг, 71,9 мкмоль) растворяли в 3 мл смеси растворителей тетрагидрофурана и этилацетата (V:V = 2:1) и добавляли палладий на угле (15 мг, нагрузка 10 %). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь фильтровали через целит, а осадок на фильтре промывали тетрагидрофураном. Фильтрат концентрировали с получением указанного в заголовке продукта 11C (35 мг, выход 82 %), который непосредственно использовали на следующей стадии.
Стадия 3
(9H-флуорен-9-ил)метил(2-(((1-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклобутокси)метил)амино)-2-оксоэтил)карбамат 11d
Соединение 1b (10 мг, 0,018 ммоль) добавляли в реакционную колбу и добавляли 1 мл N,N-диметилформамида. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина, раствора соединения 11c (13 мг, 0,031 ммоль) в 0,5 мл N,N-диметилформамида и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (25 мг, 0,091 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 40 мин. Добавляли 8 мл воды с последующей экстракцией этилацетатом (5 мл×3). Органическую фазу промывали насыщенным раствором хлорида натрия (8 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы A проявляющего растворителя с получением указанного в заголовке продукта 11d (19 мг, выход 73,9 %).
MS m/z (ESI): 842,3 [M+1].
Стадия 4
1-((2-аминоацетиламино)метокси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклобутан-1-карбоксамид 11e
Соединение 11d (19 мг, 22,6 мкмоль) растворяли в 2 мл дихлорметана и добавляли 1 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и концентрировали при пониженном давлении. Добавляли 1 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 4 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 11e (15 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 5
1-(((S)-7-бензил-20-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-3,6,9,12,15-пентаоксо-2,5,8,11,14-пентаазеикозил)окси)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклобутан-1-карбоксамид 11
Неочищенный 11e (2 мг, 3,22 мкмоль) растворяли в 0,5 мл N,N-диметилформамида. Систему трижды продували аргоном, и раствор охлаждали до 0-5 °C на ледяной водяной бане. Добавляли раствор 8 г (1,5 мг, 3,17 мкмоль) в 0,3 мл N,N-диметилформамида с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (2,7 мг, 9,67 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и концентрировали досуха масляным насосом для удаления DMF (N,N-диметилформамид), и остаток растворяли в DCM (дихлорметан) и дважды очищали непосредственно тонкослойной хроматографией (полярность проявляющего растворителя: DCM/MeOH (MeOH - метанол) = 10/1) с получением указанного в заголовке продукта 11 (1 мг, выход 28,8%).
MS m/z (ESI): 1073,6 [M+1].
1H ЯМР (400 МГц, CDCl3): δ 8,70-8,60 (m, 1H), 8,28-8,19 (m, 1H), 8,13-7,91 (m, 3H), 7,79-7,71 (d, 1H), 7,29 (s, 1H), 7,25-7,09 (m, 4H), 6,98 (s, 1H), 6,71-6,62 (m, 1H), 6,55-6,47 (m, 1H), 5,64-5,54 (m, 2H), 5,40 (s, 1H), 5,35-5,27 (t, 2H), 5,17-5,10 (m, 2H), 4,60-4,51 (m, 1H), 4,51-4,35 (m, 2H), 3,93-3,78 (m, 3H), 3,71-3,59 (m, 3H), 3,01-2,88 (m, 3H), 2,70-2,64 (m, 2H), 2,44-2,30 (m, 3H), 2,28-2,14 (m, 3H), 2,11-1,92 (m, 6H), 1,90-1,76 (m, 3H), 1,51-1,39 (m, 4H), 0,92-0,75 (m, 6H).
Пример 1-12
(S)-3-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксипропионамид 12-A
(R)-3-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксипропионамид 12-B
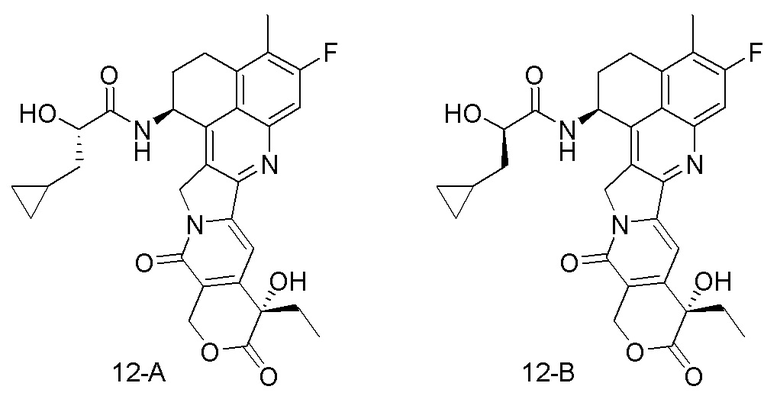
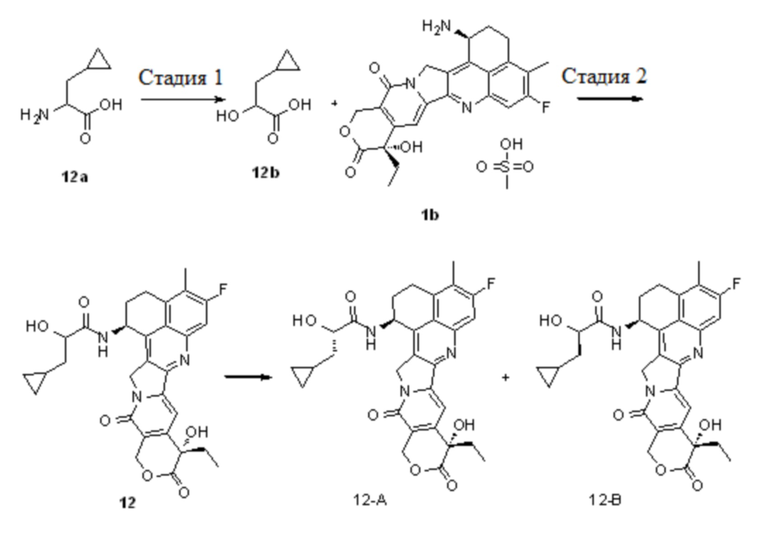
Стадия 1
3-циклопропил-2-гидроксипропионовая кислота 12b
Соединение 12a (0,5 г, 3,87 ммоль, поставляемое Adamas) растворяли в 35 мл смеси растворителей воды и уксусной кислоты (V:V = 4:1) и охлаждали раствор до 0-5 °C на водяной бане со льдом с последующим добавлением по каплям 2 М водного раствора нитрита натрия (0,53 г, 7,74 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. К реакционной смеси добавляли твердый хлорид натрия для насыщения водной фазы. Реакционную смесь экстрагировали этилацетатом (8 мл×8), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали с получением указанного в заголовке продукта 12b (0,45 мг, выход 89,3 %).
Стадия 2
(S)-3-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксипропионамид 12-A
(R)-3-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксипропионамид 12-B
К соединению 1b (45 мг, 0,085 ммоль) добавляли 1,5 мл этанола и 1,5 мл N,N-диметилформамида. Систему трижды продували аргоном и по каплям добавляли 0,1 мл N-метилморфолина. Реакционную смесь перемешивали, пока она не становилась чистой. К реакционной смеси последовательно добавляли соединение 12b (90 мг, 0,691 ммоль), 1-гидроксибензотриазол (34 мг, 0,251 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (49 мг, 0,256 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 3 ч и концентрировали при пониженном давлении, и полученное неочищенное соединение 12 очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка для хроматографии: Sharpsil-T C18 5 мкм 21,2×250 мм; подвижная фаза: A-вода (10 ммоль NH4OAc), B-ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин) с получением указанных в заголовке продуктов (7 мг, 15 мг).
MS m/z (ESI): 547,9 [M+1].
Соединение с единственной конфигурацией (с более коротким временем удерживания):
Анализ UPLC: время удерживания: 1,345 мин; чистота: 72 % (хроматографическая колонка: ZORBAX Ecliphase Plus 18 1,8 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 8,42 (d, 1H), 7,78 (d, 1H), 7,30 (s, 1H), 6,51 (s, 1H), 5,60-5,50 (m, 2H), 5,42 (s, 1H), 5,19 (q, 2H), 4,02-4,00 (m, 1H), 3,21-3,11 (m, 2H), 2,39 (s, 3H), 2,21-2,07 (m, 2H), 2,05-1,95 (m, 1H), 1,92-1,68 (m, 4H), 1,53-1,41 (m, 1H), 0,87 (t, 3H), 0,48-0,34 (m, 2H), 0,14-0,01 (m, 2H).
Соединение с единственной конфигурацией (с более длительным временем удерживания):
Анализ UPLC: время удерживания: 1,399 мин; чистота: 88 % (хроматографическая колонка: ZORBAX Ecliphase Plus 18 1,8 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
1H ЯМР (400 МГц, DMSO-d6): δ 8,36 (d, 1H), 7,77 (d, 1H), 7,31 (s, 1H), 6,51 (s, 1H), 5,58-5,51 (m, 1H), 5,48 (d, 1H), 5,42 (s, 1H), 5,20 (q, 2H), 4,09-4,02 (m, 1H), 3,22-3,11 (m, 2H), 2,39 (s, 3H), 2,27-2,06 (m, 2H), 2,05-1,95 (m, 1H), 1,93-1,81 (m, 2H), 1,65-1,43 (m, 2H), 1,32-1,21 (m, 1H), 0,87 (t, 3H), 0,48-0,33 (m, 2H), 0,14-0,01 (m, 2H).
Пример 1-13 (эталонный пример)
N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)-2-гидроксиацетамид
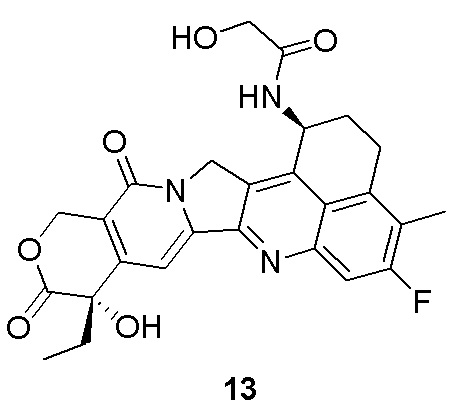
Указанное в заголовке соединение 13 получали таким образом, как описано в "Примере 76 на стр. 147 описания патента EP2907824A1".
Пример 1-14
N-((2R,10S)-10-бензил-2-(циклопропилметил)-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 14-A
N-((2S,10S)-10-бензил-2-(циклопропилметил)-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 14-B
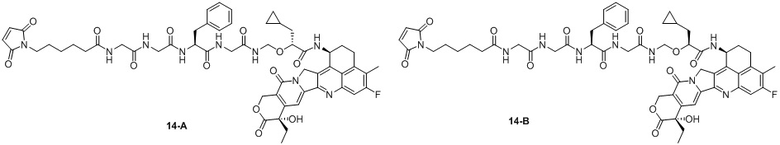
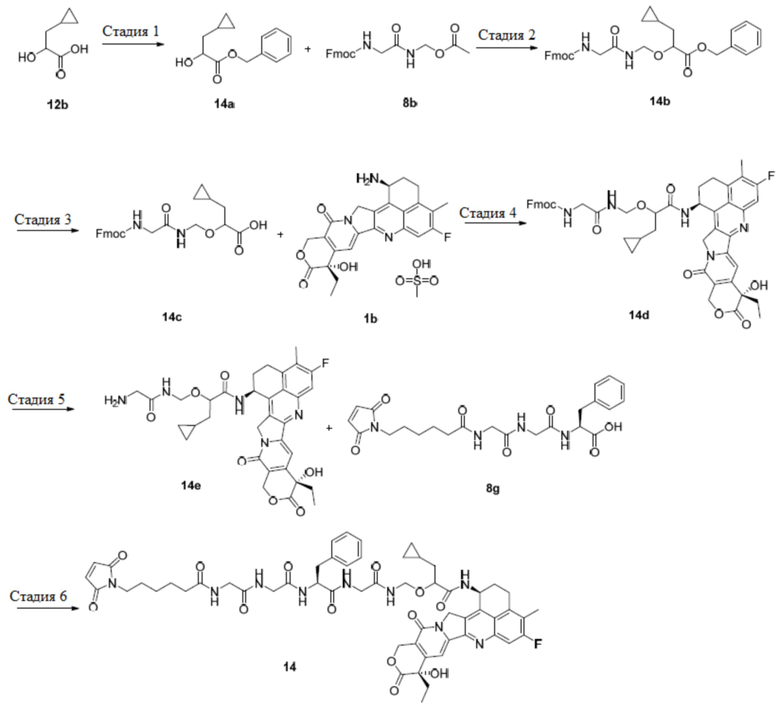
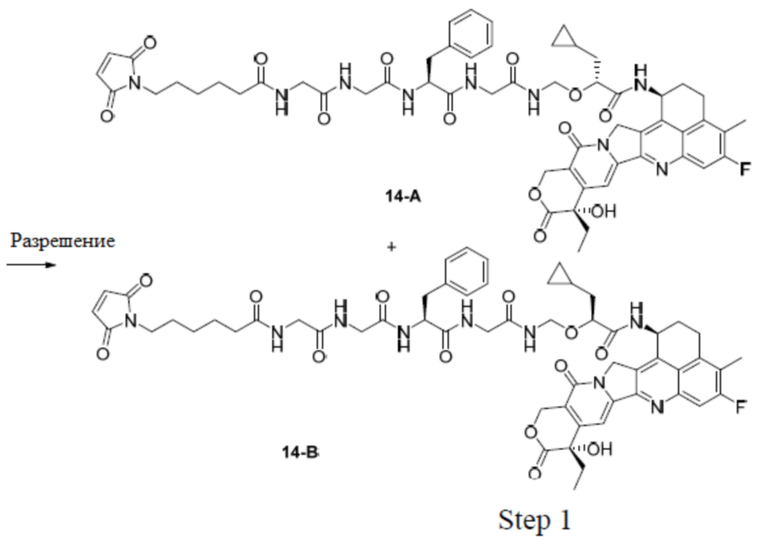
Стадия 1
Бензил-3-циклопропил-2-гидроксипропионат 14a
Соединение 12b (200 мг, 1,54 ммоль) растворяли в 20 мл ацетонитрила и последовательно добавляли карбонат калия (1,06 г, 7,68 ммоль), бензилбромид (0,16 мл, 1,34 ммоль) и йодид тетрабутиламмония (28 мг, 0,07 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов и фильтровали через целит, и осадок на фильтре промывали этилацетатом (10 мл). Фильтраты объединяли и концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя C с получением указанного в заголовке продукта 14a (140 г, 41,3 % выхода).
Стадия 2
Бензил-10-(циклопропилметил)-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оат 14b
Соединение 14a (94 мг, 0,427 ммоль) и 8b (130 мг, 0,353 ммоль) добавляли в реакционную колбу и добавляли 10 мл тетрагидрофурана. Систему трижды продували аргоном и охлаждали реакционную смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида калия (79 мг, 0,704 ммоль). Ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 10 минут с последующим добавление 20 мл ледяной воды и экстракцией этилацетатом (10 мл×4). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя C с получением указанного в заголовке продукта 14b (50 мг, выход 26,8 %).
MS m/z (ESI): 529,2 [M+1].
Стадия 3
10-(циклопропилметил)-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оевая кислота 14c
Соединение 14B (27 мг, 0,051 ммоль) растворяли в 3 мл этилацетата и добавляли палладий на угле (7 мг, нагрузка 10%, сухая основа). Систему продували водородом три раза. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и фильтровали через целит, и осадок на фильтре промывали этилацетатом. Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 14c (23 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 439,1 [M+1].
Стадия 4
(9H-флуорен-9-ил)метил(2-((((3-циклопроил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1-оксопропан-2-ил)окси)метил)амино)-2-оксоэтил)карбамат 14d
Соединение 1b (22 мг, 42,38 мкмоль) добавляли в реакционную колбу и добавляли 3 мл N,N-диметилформамида. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением по каплям триэтиламина (4,3 мг, 42,49 мкмоль) и добавлением неочищенного 14c (23 мг, 51,1 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (17,6 мг, 63,6 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 40 мин. Добавляли 15 мл воды с последующей экстракцией этилацетатом (8 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 14d (29 мг, выход 79,9 %).
MS m/z (ESI): 856,1 [M+1].
Стадия 5
2-((2-аминоацетиламино)метокси)-3-циклопропил-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)пропанамид 14e
Соединение 14d (29 мг, 33,9 мкмоль) растворяли в 0,8 мл дихлорметана и добавляли 0,4 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и концентрировали при пониженном давлении. Добавляли 1 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и оставляли стоять. Затем супернатант удаляли; и процедуры повторяли три раза. Остаток концентрировали при пониженном давлении и сушили с помощью масляного насоса для получения неочищенного указанного в заголовке продукта 14e (22 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 634,1 [M+1].
Стадия 6
N-((2R,10S)-10-бензил-2-(циклопропилметил)-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 14-A
N-((2S,10S)-10-бензил-2-(циклопропилметил)-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)-6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексанамид 14-B
Неочищенный 14e (22 мг, 33,9 мкмоль) растворяли в 2,5 мл N,N-диметилформамида. Систему трижды продували аргоном, и раствор охлаждали до 0-5 °C на ледяной водяной бане, и последовательно добавляли 8 г (24 мг, 50,8 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (14 мг, 50,6 мкмоль). Затем ледяную баню удаляли и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа с получением соединения 14. Реакционную смесь очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин) с получением указанных в заголовке продуктов (2 мг, 2 мг).
MS m/z (ESI): 1088,4 [M+1].
Соединение с единственной конфигурацией (с более коротким временем удерживания):
Анализ UPLC: время удерживания: 1,18 мин; чистота: 88 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
Соединение с единственной конфигурацией (с более длительным временем удерживания):
Анализ UPLC: время удерживания: 1,23 мин; чистота: 96 % (хроматографическая колонка: ACQUITY UPLC BEHC18 1,7 мкм 2,1×50 мм; подвижная фаза: A - вода (5 ммоль NH4OAc), B - ацетонитрил).
Пример 1-15
1-((S)-9-бензил-22-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-5,8,11,14,17-пентаоксо-2-окса-4,7,10,13,16-пентаазадокозил)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3,4':6,7]индолизино[1,2-b]хинолин-1-ил)циклопропан-1-карбоксамид 15
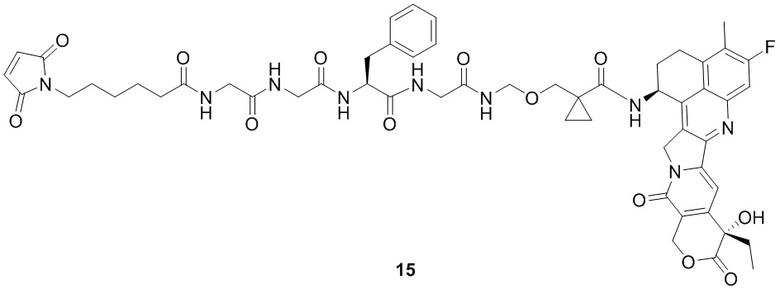
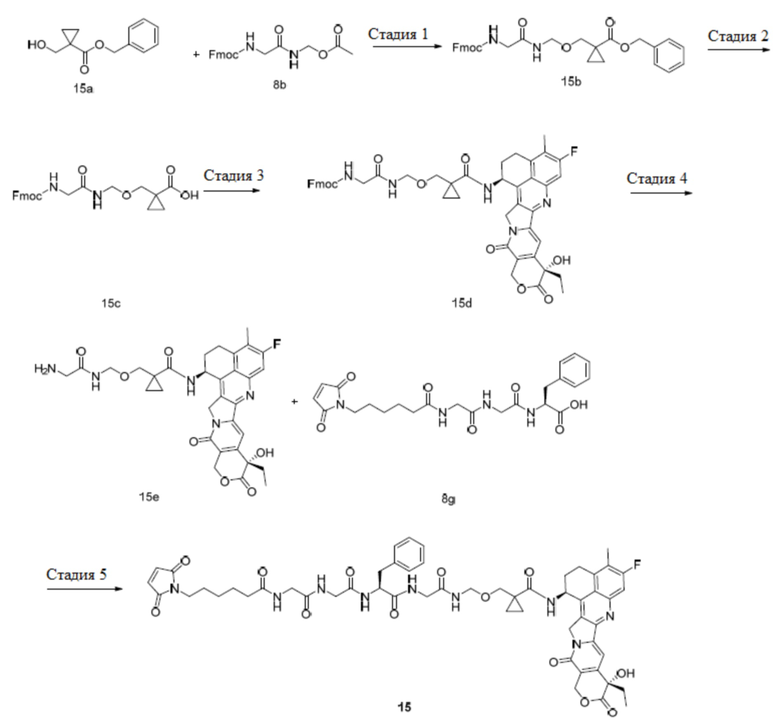
Стадия 1
Бензил 1-(10-(9H-флуорен-9-ил)-5,8-диоксо-2,9-диокса-4,7-диазадецил)циклопропан-1-карбоксилат 15b
Соединение 8b (500 мг, 1,35 ммоль) добавляли в реакционную колбу с последующим добавлением 6 мл тетрагидрофурана, бензил-1-гидроксиметилциклопропан-1-карбоксилата 15A (233 мг, 1,13 ммоль; полученного, как описано в "Примере 22-2 на стр. 262 описания патентной заявки EP2862856A1"). Систему трижды продували аргоном и охлаждали реакционную смесь до 0-5 °C на ледяной водяной бане с последующим добавлением гидрида натрия (54 мг, 1,35 ммоль). Затем ледяную баню удаляли, и реакционную смесь нагревали до комнатной температуры, перемешивали в течение 40 минут и остужали до 0 °C, с последующим добавлением 20 мл ледяной воды и экстракцией этилацетатом (5 мл×2) и хлороформом (5 мл×5). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя B с получением указанного в заголовке продукта 15b (15 мг, выход 2,5 %).
MS m/z (ESI): 515,2 [M+1].
Стадия 2
1-(10-(9H-флуорен-9-ил)-5,8-диоксо-2,9-диокса-4,7-диазадецил)циклопропан-1-карбоновая кислота 15c
Соединение 15b (15 мг, 0,029 ммоль) растворяли в 2 мл этилацетата и добавляли палладий на угле (3 мг, нагрузка 10 %, сухая основа). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 4,5 часов и осадок на фильтре промывали этилацетатом. Фильтрат концентрировали с получением указанного в заголовке продукта 15c (11 мг, выход 89 %).
MS m/z (ESI): 425,2 [M+1].
Стадия 3
(9H-флуорен-9-ил)метил(2-((((1-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклопропил)метокси)метил)амино)2-оксоэтил)карбамат 15d
Соединение 1b (10 мг, 0,021 ммоль) добавляли в реакционную колбу и добавляли 1 мл N,N-диметилформамида. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина, соединения 15c (11 мг, 0,026 ммоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (10,7 мг, 0,039 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 60 мин. Добавляли 10 мл воды с последующей экстракцией этилацетатом (5 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 15d (19 мг, выход 87,0 %).
MS m/z (ESI): 842,2 [M+1].
Стадия 4
1-(((2-аминоацетиламино)метокси)метил)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклопропан-1-карбоксамид 15e
Соединение 15d (19 мг, 22,56 мкмоль) растворяли в 2 мл дихлорметана и добавляли 1 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и концентрировали при 0 °C и пониженном давлении. Добавляли 1 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 15e (13,9 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 620,1 [M+1].
Стадия 5
1-((S)-9-бензил-22-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-5,8,11,14,17-пентаоксо-2-окса-4,7,10,13,16-пентаазадокозил)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклопропан-1-карбоксамид 15
Неочищенный 15e (13,9 мг, 22,4 мкмоль) растворяли в 1 мл N,N-диметилформамида. Систему трижды продували аргоном, и раствор охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением 8 г (15,8 мг, 33,4 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (9,3 мг, 33,6 мкмоль). Реакционную смесь нагревали до комнатной температуры, перемешивали 60 мин и очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 15 (2,5 мг, выход 10,3 %).
MS m/z (ESI): 1074,2 [M+1].
1H ЯМР (400 МГц, DMSO-d6): δ 8,51-8,37 (m, 1H), 8,22 (t, 1H), 8,14-8,02 (m, 2H), 8,011-7,94 (m, 1H), 7,82-7,73 (m, 1H), 7,29 (s, 1H), 7,26-7,10 (m, 3H), 6,98 (s, 1H), 6,53-6,47 (m, 1H), 5,62-5,50 (m, 1H), 5,45-5,36 (m, 1H), 5,35-5,23 (m, 2H), 5,13-5,02 (m, 2H), 4,61-4,50 (m, 2H), 4,42-4,28 (m, 2H), 3,76-3,61 (m, 3H), 3,60-3,45 (m, 3H), 3,27-3,23 (m, 1H), 3,20-2,81 (m,7H), 2,75-2,61 (m, 3H), 241-2,28 (m, 3H), 2,23-2,13 (m, 2H), 2,11-2,01 (m, 1H), 2,03-1,94 (m, 1H), 1,90 (s, 1H), 1,87-1,74 (m, 2H), 1,53-1,36 (m, 3H), 1,29-1,08 (m, 4H), 0,90-0,68 (m, 4H).
Пример 1-16
1-((S)-9-бензил-22-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-5,8,11,14,17-пентаоксо-2-окса-4,7,10,13,16-пентаазадокозил)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклобутан-1-карбоксамид 16
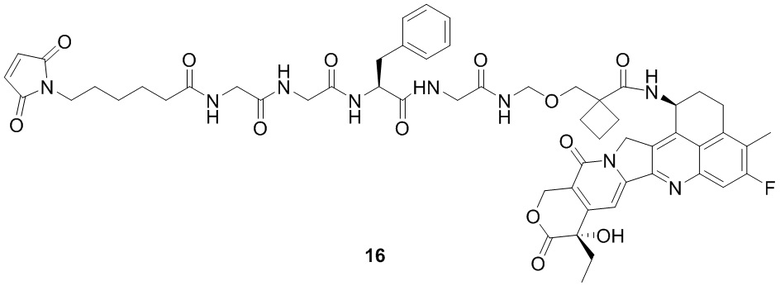
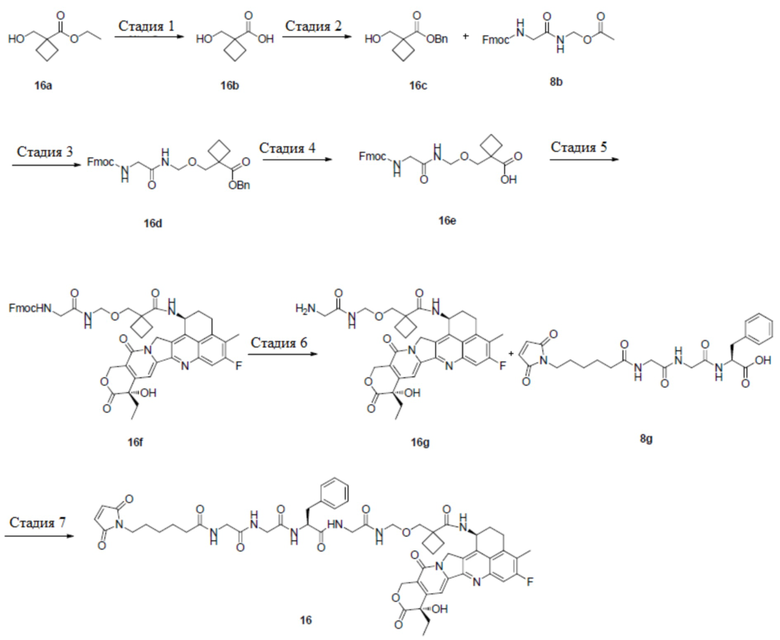
Стадия 1
1-(гидроксиметил)циклобутан-1-карбоновая кислота 16b
Этил-1-(гидроксиметил)циклобутанкарбоксилат 16a (250 мг, 1,58 ммоль, поставляемый Alfa) растворяли в метаноле (2 мл) и воде (1 мл) и добавляли гидроксид натрия (126 мг, 3,15 ммоль). Реакционную смесь нагревали до 40 °C, перемешивали в течение 3 часов, охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления органического растворителя. Реакционную смесь обратно экстрагировали простым диэтиловым эфиром (10 мл) для сбора водной фазы. Водную фазу доводили до pH 3-4 6 N водной соляной кислотой и концентрировали при пониженном давлении с получением твердого вещества. Добавляли 3 мл толуола с последующим концентрированием досуха при пониженном давлении; процедуры повторяли трижды. Остаток сушили с помощью масляного насоса для получения неочищенного указанного в заголовке продукта 16b (206 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI, NEG (масс-спектрометрии отрицательных ионов)):129,2 [M-1].
Стадия 2
Бензил-1-(гидроксиметил)циклобутан-1-карбоксилат 16C
Неочищенный 16b (206 мг, 1,58 ммоль) растворяли в ацетонитриле (15 мл) и добавляли безводный карбонат калия (1,09 г, 7,90 ммоль), йодид тетрабутиламмония (29 мг, 78,51 ммоль) и бензилбромид (216 мг, 1,26 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы C проявляющего растворителя с получением указанного в заголовке продукта 16c (112 мг, выход 32,1 %).
MS m/z (ESI): 221,1 [M+1].
Стадия 3
Бензил-1-(10-(9H-флуорен-9-ил)-5,8-диоксо-2,9-диокса-4,7-диазадецил)циклобутан-1-карбоксилат 16d
Соединение 16c (77 мг, 0,35 ммоль) и 8b (100 мг, 0,27 ммоль) добавляли в реакционную колбу и добавляли 3 мл тетрагидрофурана. Систему трижды продували аргоном и охлаждали реакционную смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида калия (61 мг, 0,54 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 10 мин. Добавляли 20 мл ледяной воды с последующей экстракцией этилацетатом (5 мл) и хлороформом (5 мл×5). Органические фазы объединяли и концентрировали. Полученный остаток растворяли в 3 мл 1,4-диоксана и добавляли 0,5 мл воды, бикарбоната натрия (27 мг, 0,32 ммоль) и 9-фторенилметилхлорформиата (71 мг, 0,27 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 20 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы C проявляющего растворителя с получением указанного в заголовке продукта 16d (24 мг, выход 16,7 %).
MS m/z (ESI): 551,3 [M+23].
Стадия 4
1-(10-(9H-флуорен-9-ил)-5,8-диоксо-2,9-диокса-4,7-диазадецил)циклобутан-1-карбоновая кислота 16e
Соединение 16d (12 мг, 22,7 мкмоль) растворяли в 1,5 мл смеси растворителей тетрагидрофурана и этилацетата (V:V = 2:1) и добавляли палладий на угле (5 мг, нагрузка 10 %). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь фильтровали через целит, а осадок на фильтре промывали этилацетатом. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 16e (10 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 5
(9H-флуорен-9-ил)метил(2-((((1-((1S,9S) -9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано [3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклобутил)метокси)метил)амино)-2-оксоэтил)карбамат 16f
Соединение 1b (7,5 мг, 0,014 ммоль) добавляли в реакционную колбу и добавляли 1 мл N,N-диметилформамида. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина, раствора неочищенного 16e (10 мг) в 0,5 мл N,N-диметилформамида и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (6 мг, 0,026 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 30 мин. Добавляли 10 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью тонкослойной хроматографии с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 16f (10,6 мг, выход 87,8 %).
MS m/z (ESI): 856,2 [M+1].
Стадия 6
1-(((2-аминоацетиламино)метокси)метил)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклобутан-1-карбоксамид 16g
Соединение 16f (10,6 мг, 12,4 мкмоль) растворяли в 0,6 мл дихлорметана и добавляли 0,3 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Остаток концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 16g (8 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 634,1 [M+1].
Стадия 7
1-((S)-9-бензил-22-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-5,8,11,14,17-пентаоксо-2-окса-4,7,10,13,16-пентаазадокозил)-N-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)циклобутан-1-карбоксамид 16
Неочищенный 16g (8 мг) растворяли в 1 мл N,N-диметилформамида и добавляли 8g (8,8 мг, 18,6 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (5,2 мг, 18,8 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин) с получением указанного в заголовке продукта 16 (1,0 мг, выход 7,2 %).
MS m/z (ESI): 1088,0 [M+1].
Пример 1-17
(1r,4r)-N-((S)-7-бензил-1-(1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклопропокси)-3,6,9,12,15-пентаоксо-17,20,23,26,29,32,35,38,41-нонаокса-2,5,8,11,14-пентаазатетратридек-43-ил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоксамид 17
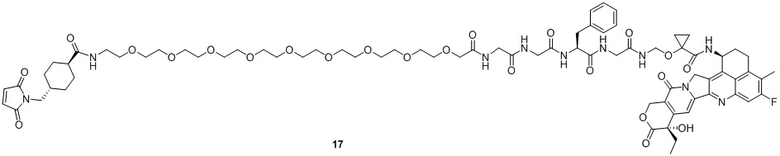
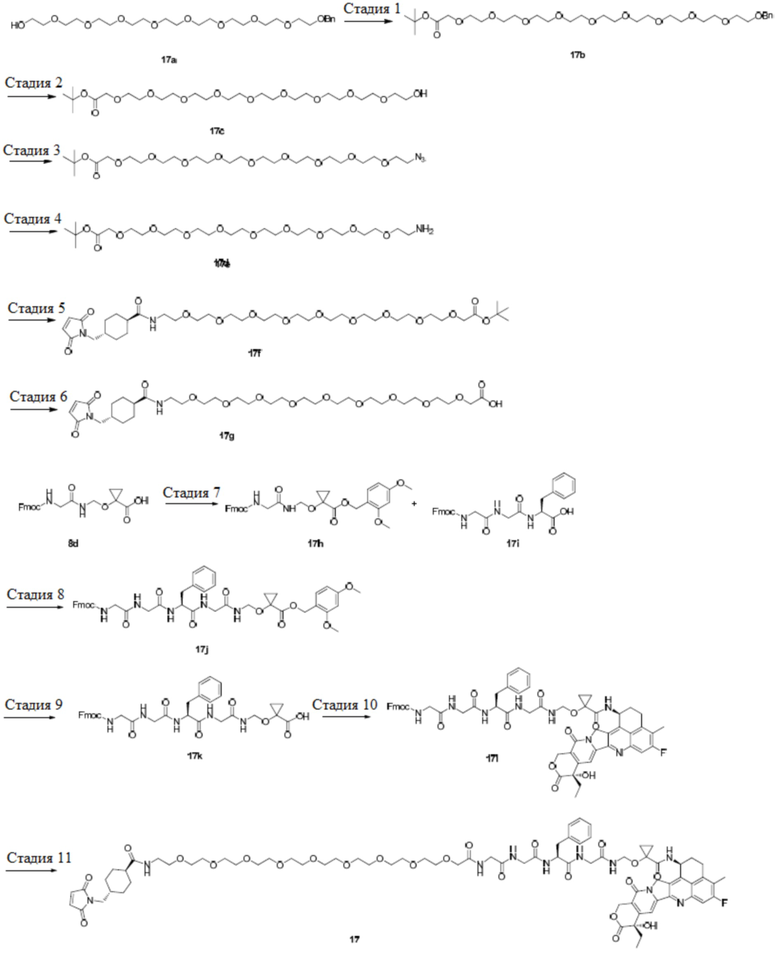
Стадия 1
Трет-бутил-1-фенил-2,5,8,11,14,17,20,23,26,29-декаоксаентриаконтан-31-оат 17b
1-фенил-2,5,8,11,14,17,20,23,26-нонаоксаоктакозан-28-ол 17a (0,34 г, 0,67 ммоль, поставляется Bide) растворяли в 10 мл дихлорметана и последовательно добавляли оксид серебра (0,24 г, 1,01 ммоль), трет-бутилбромацетат (0,16 г, 0,81 ммоль) и йодид калия (0,07 г, 0,40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и фильровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с помощью системы B проявляющего растворителя с получением указанного в заголовке продукта 17b (0,42 мг, выход 100 %).
MS m/z (ESI): 636,3 [M+18].
Стадия 2
Трет-бутил-29-гидрокси-3,6,9,12,15,18,21,24,27-ноноксанонакозан-1-оат 17c
Соединение 17b (417 мг, 0,67 ммоль) растворяли в 15 мл тетрагидрофурана и добавляли палладий на угле (110 мг, нагрузка 10%, сухая основа). Систему трижды продували водородом, и реакционную смесь нагревали при 60 °C, перемешивали в течение 3 часов, фильтровали через целит, а осадок на фильтре промывали тетрагидрофураном. Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 17c (357 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 546,2 [M+18].
Стадия 3
Трет-бутил-29-азидо-3,6,9,12,15,18,21,24,27-ноноксанонакозан-1-оат 17d
Соединение 17c (357 мг, 0,675 ммоль) растворяли в 10 мл толуола и добавляли дифенилфосфорилазид (279 мг, 1,014 ммоль) и 1,8-диазабициклоундек-7-ен (206 мг, 1,353 ммоль). Систему трижды продували аргоном, и реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем нагревали до 105 °С, подвергали реакции в течение 19 ч, охлаждали до комнатной температуры и концентрировали. Добавляли 20 мл воды с последующей экстракцией этилацетатом (10 мл×4). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя B с получением неочищенного указанного в заголовке продукта 17d (412 мг).
MS m/z (ESI): 571,3 [M+18].
Стадия 4
Трет-бутил-29-амино-3,6,9,12,15,18,21,24,27-ноноксанонакозан-1-оат 17e
Соединение 17d (230 мг, 0,415 ммоль) растворяли в 8 мл тетрагидрофурана и добавляли палладий на угле (58 мг, нагрузка 10%, сухая основа). Систему продували водородом три раза. Реакционную смесь перемешивали при комнатной температуре в течение 2 часа и фильтровали через целит, и осадок на фильтре промывали тетрагидрофураном. Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 17e (220 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 528,2 [M+1].
Стадия 5
Трет-бутил-1-((1r,4r)-4-((2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексил)-1-оксо-5,8,11,14,17,20,23,26,29-нонаокса-2-азатриундек-31-оат 17f
(1r,4r)-4-((2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоновую кислоту (98,5 мг, 0,415 ммоль) растворяли в 10 мл дихлорметана и добавляли 2-(7-бензотриазол оксид)-N,N,N',N'-тетраметилурония гексафторфосфат (190 мг, 0,500 ммоль) и N,N-диизопропилэтиламин (162 мг, 1,253 ммоль). Систему трижды продували аргоном и добавляли неочищенный 17e (220 мг, 0,417 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 15 мл воды с последующей экстракцией дихлорметаном (8 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя B с получением указанного в заголовке продукта 17f (122 мг, выход 39,2 %).
MS m/z (ESI): 747,2 [M+1].
Стадия 6
1-((1r,4r)-4-((2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексил)-1-оксо-5,8,11,14,17,20,23,26,29-нонаокса-2-азатриундек-31-овая кислота 17g
Соединение 17f (122 мг, 0,163 ммоль) растворяли в 0,8 мл дихлорметана и добавляли 0,4 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, разбавляли 15 мл дихлорметана и концентрировали при пониженном давлении; добавляли 10 мл н-гексана, затем концентрировали при пониженном давлении; процедуры повторяли дважды. Затем добавляли 10 мл толуола, и полученную смесь концентрировали при пониженном давлении, трижды суспендировали с 10 мл смеси растворителей н-гексан:простой диэтиловый эфир составляет 5:1 до тех пор, пока pH не достиг 7, концентрировали и сушили масляным насосом с получением указанного в заголовке продукта 17g (98 мг, выход 86,8%).
MS m/z (ESI): 691,2 [M+1].
Стадия 7
2,4-диметоксибензил-1-((2-((((9H-флуорен-9-ил)метокси)карбонил)амино)ацетиламино)метокси)циклопропил-1-карбоксилат 17h
Соединение 8d (164 мг, 0,40 ммоль) растворяли в дихлорметане (5 мл) и последовательно добавляли 2,4-диметоксибензиловый спирт (81 мг, 0,48 ммоль), 1-этил-(3-диметиламинопропил)карбонилдиимина гидрохлорид (115 мг, 0,60 ммоль) и 4-диметиламинопиридин (5 мг, 0,041 ммоль). После добавления реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 20 мл воды и после встряхивания образовывались слои. Водную фазу экстрагировали дихлорметаном (8 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя C с получением указанного в заголовке продукта 17h (124 мг, выход 55,4 %).
MS m/z (ESI): 583,1 [M+23].
Стадия 8
2,4-диметоксибензил-(S)-1-((11-бензил-1-(9H-флуорен-9-ил)-3,6,9,12,15-пентаоксо-2-окса-4,7,10,13,16-пентаазаптадекан-17-ил)окси)циклопропил-1-карбоксилат 17j
Соединение 17h (39 мг, 69,6 мкмоль) растворяли в 0,6 мл дихлорметана и добавляли 0,3 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза с последующим концентрированием при пониженном давлении. Полученный неочищенный продукт растворяли в 2 мл N,N-диметилформамида и добавляли (((9H-флуорен-9-ил)метокси)карбонил)глицил-L-фенилаланин 17i (35 мг, 69,8 мкмоль, полученный, как описано в "Примере 7-12 на странице 13 описания патентной заявки CN108853514A") и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (23 мг, 83,1 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 10 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 17j (48 мг, выход 83,9 %).
MS m/z (ESI): 822,0 [M+1].
Стадия 9
(S)-1-((11-бензил-1-(9H-флуорен-9-ил)-3,6,9,12,15-пентаоксо-2-окса-4,7,10,13,16-пентаазаптадекан-17-ил)окси)циклопропан-1-карбоновая кислота 17k
Соединение 17j (48 мг, 58,4 мкмоль) растворяли в 1,4 мл 3% (об./об.) раствора дихлоруксусной кислоты в дихлорметане и охлаждали раствор до 0-5 °C на ледяной водяной бане с последующим добавлением триэтилсилана (21 мг, 180,6 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении на ледяной бане для удаления половины органического растворителя. Добавляли 5 мл простого диэтилового эфира, и полученную смесь нагревали до комнатной температуры естественным образом и суспендировали для осаждения белого твердого вещества и фильтровали. Осадок на фильтре собирали и сушили с помощью масляного насоса с получением указанного в заголовке продукта 17k (33 мг, 84,1% выхода).
Стадия 10
(9H-флуорен-9-ил)метил((S)-7-бензил-1-(1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклопропокси)-3,6,9,12-тетраоксо-2,5,8,11-тетраазатридекан-13-ил)карбамат 17l
Соединение 1b (20 мг, 42,4 мкмоль) добавляли в реакционную колбу и добавляли 1 мл 10% (об./об.) раствора метанола в дихлорметане. Систему трижды продували аргоном, и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина. Смесь перемешивали до растворения соединения 1b. Соединение 17k (33 мг, 49,1 мкмоль) растворяли в 1 мл 10% (об./об.) раствора метанола в дихлорметане, а затем по каплям добавляли вышеуказанную реакционную смесь с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (17,6 мг, 63,6 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Добавляли 10 мл дихлорметана и 5 мл воды, и полученную смесь перемешивали в течение 5 минут и оставляли стоять с образованием слоев. Органическую фазу собирали, и водную фазу экстрагировали дихлорметаном (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 17l (37 мг, выход 80,2 %).
MS m/z (ESI): 1090,1 [M+1].
Стадия 11
(1r,4r)-N-((S)-7-бензил-1-(1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)аминокарбонил)циклопропокси)-3,6,9,12,15-пентаоксо-17,20,23,26,29,32,35,38,41-нонаокса-2,5,8,11,14-пентаазатетратридек-43-ил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоксамид 17
Соединение 17l (15,5 мг, 14,23 мкмоль) растворяли в 0,6 мл дихлорметана и добавляли 0,3 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Реакционную смесь концентрировали при пониженном давлении и затем сушили с помощью масляного насоса. Полученный неочищенный продукт растворяли в 1 мл N,N-диметилформамида и добавляли соединение 17g (11 мг, 15,92 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (6,0 мг, 21,68 мкмоль). Систему трижды продували аргоном и реакционную смесь перемешивали при комнатной температуре, перемешивали 30 мин и очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 17 (6 мг, выход 27,4 %).
MS m/z (ESI): 1556,4 [M+18].
1H ЯМР (400 МГц, DMSO-d6): δ 8,98 (d, 1H), 8,76 (s, 1H), 8,20 (br, 1H), 8,12-7,95 (m, 3H), 7,93-7,76 (m, 2H), 7,75-7,66 (m, 2H), 7,24 (s, 1H), 7,20-7,05 (m, 6H), 6,97 (s, 1H), 6,64 (br, 1H), 6,55 (d, 1H), 6,47 (s, 1H), 5,61-5,52 (m, 2H), 5,37 (s, 1H), 5,33-5,23 (m, 2H), 5,18 (s, 1H), 5,13 (s, 1H), 5,05 (s, 1H), 5,00 (s, 1H), 4,65-4,55 (m, 2H), 4,53-4,45 (m, 1H), 4,38-4,28 (m, 2H), 3,84 (s, 2H), 3,67 (d, 3H), 3,60-3,40 (m, 33H), 3,18 (d, 1H), 3,15-3,08 (m, 3H), 2,28 (s, 3H), 2,00-1,92 (m, 3H), 1,85 (s, 2H), 1,82-1,73 (m, 2H), 1,68-1,52 (m, 4H), 1,29-1,15 (m, 3H), 0,86-0,76 (m, 5H).
Пример 1-18
(1r,4r)-N-((2R,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15,18-гексаоксо-3,20,23,26,29,32,35,38,41,44-декаокса-5,8,11,14,17-пентааза-тетрагексадек-46-ил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоксамид 18
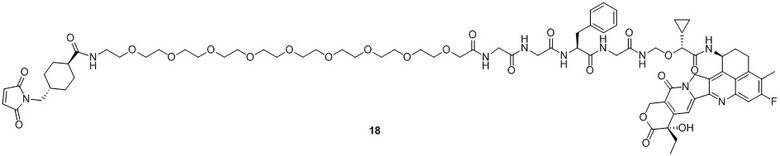
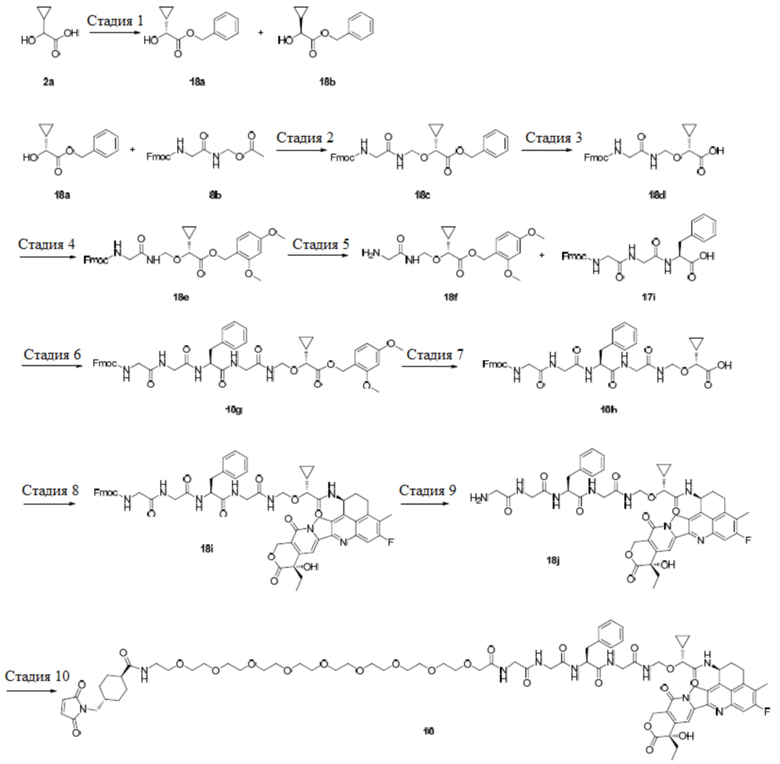
Стадия 1
Бензил (R)-2-циклопропил-2-гидроксиацетат 18a
Бензил (S)-2-циклопропил-2-гидроксиацетат 18b
Соединение 2a (7,4 г, 63,7 ммоль) растворяли в 200 мл ацетонитрила и последовательно добавляли карбонат калия (35 г, 253,6 ммоль), бензилбромид (9,3 г, 54,4 ммоль) и тетрабутиламмония йодид (500 мг, 1,36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и фильтровали через целит, и осадок на фильтре промывали этилацетатом (10 мл). Фильтраты объединяли и концентрировали при пониженном давлении, и полученный остаток (4,1 г) очищали с помощью колоночной хроматографии на силикагеле с проявляющей системой растворителей C и далее с помощью хирального разрешения с получением указанных в заголовке продуктов 18a (1,1 г) и 18b (1,2 г).
Стадия 2
Бензил-(R)-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оат 18c
Соединение 8b (3,1 г, 8,41 ммоль) растворяли в тетрагидрофуране (55 мл) и добавляли соединение 18a (2,0 г, 9,70 ммоль). Смесь охлаждали до 0-5 °C на водяной бане со льдом и добавляли трет-бутоксид калия (1,89 г, 16,84 ммоль). Смесь перемешивали на ледяной водяной бане в течение 10 мин. Добавляли этилацетат (30 мл) и воду (20 мл), и полученную смесь оставляли стоять с образованием слоев. Водную фазу экстрагировали хлороформом (30 мл×5), органические фазы объединяли и концентрировали при пониженном давлении. Полученный остаток растворяли в 1,4-диоксане (32 мл) и воде (8 мл) и добавляли карбонат натрия (1,78 г, 16,79 ммоль) и 9-фторенилметилхлорформиат (2,18 г, 8,42 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли воду (30 мл) с последующей экстракцией этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии с помощью системы растворителей C с получением указанного в заголовке продукта 18c (1,3 г, выход 30,0%).
MS m/z (ESI): 515,2 [M+1].
Стадия 3
(R)-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оевая кислота 18d
Соединение 18c (1,29 мг, 2,51 ммоль) растворяли в этилацетате (15 мл) и добавляли палладий на угле (260 мг, нагрузка 10 %, сухая основа). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 5 часов и осадок на фильтре промывали этилацетатом (20 мл) и метанолом (20 мл). Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 18d (980 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 425,1 [M+1].
Стадия 4
2,4-диметоксибензил(R)-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-сложный эфир 18E
Неочищенный 18d (980 мг, 2,31 ммоль) растворяли в дихлорметане (15 мл) и добавляли 2,4-диметоксибензиловый спирт (777 мг, 4,62 ммоль), 1-этил-(3-диметиламинопропил)карбонилдиимина гидрохлорид (664 мг, 3,46 ммоль) и 4-диметиламинопиридин (28 мг, 0,23 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали при пониженном давлении для удаления органического растворителя. Добавляли 20 мл воды с последующей экстракцией этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии с системой проявляющего растворителя C с получением указанного в заголовке продукта 8e (810 мг, выход 61,1 %).
MS m/z (ESI): 575,0 [M+1].
Стадия 5
2,4-диметоксибензил(R)-2-((2-аминоацетиламино)метокси)-2-циклопропилацетат 18f
Соединение 18e (33 мг, 57,4 мкмоль) растворяли в 0,6 мл дихлорметана и добавляли 0,3 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 18f (21 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 6
2,4-диметоксибензил(11S,19R)-11-бензил-19-циклопропил-1-(9H-флуорен-9-ил)-3,6,9,12,15-пентаоксо-2,18-диокса-4,7,10,13,16-пентаазаикозан-20-оат 18g
Неочищенный 18f (21 мг, 57,4 мкмоль) растворяли в 3 мл N,N-диметилформамида и добавляли соединение 17i (29 мг, 57,8 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (19 мг, 68,7 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 10 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой проявляющего растворителя В с получением указанного в заголовке продукта 18g (37 мг, выход 77,1 %).
MS m/z (ESI): 853,0 [М+18].
Стадия 7
(11S,19R)-11-бензил-19-циклопропил-1-(9H-флуорен-9-ил)-3,6,9,12,15-пентаоксо-2,18-диокса-4,7,10,13,16-пентаазаикозан-20-оевая кислота 18h
Соединение 18g (37 мг, 44,3 мкмоль) растворяли в 1,4 мл 3% (об./об.) раствора дихлоруксусной кислоты в дихлорметане и охлаждали раствор до 0-5 °C на ледяной водяной бане с последующим добавлением триэтилсилана (15,4 мг, 132,4 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении на ледяной бане для удаления половины органического растворителя и добавляли 5 мл простого диэтилового эфира. Полученную смесь нагревали до комнатной температуры естественным образом и суспендировали для осаждения белого твердого вещества и фильтровали. Осадок на фильтре собирали и сушили с помощью масляного насоса с получением указанного в заголовке продукта 18h (24 мг, выход 79,1%).
MS m/z (ESI): 708,2 [M+23].
Стадия 8
(9H-флуорен-9-ил)метил((2R,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)карбамат 18i
Соединение 1b (30 мг, 63,6 мкмоль) добавляли в реакционную колбу и добавляли 1 мл 10% (об./об.) раствора метанола в дихлорметане. Систему трижды продували аргоном, и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина. Смесь перемешивали до растворения соединения 1b. Соединение 18H (65 мг, 94,8 мкмоль) растворяли в 1 мл 10% (об./об.) раствора метанола в дихлорметане, а затем по каплям добавляли вышеуказанную реакционную смесь с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (27 мг, 97,6 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Добавляли 10 мл дихлорметана и 5 мл воды, и полученную смесь перемешивали в течение 5 минут и оставляли стоять с образованием слоев. Органическую фазу собирали, и водную фазу экстрагировали дихлорметаном (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 18i (25 мг, выход 35,6 %).
MS m/z (ESI): 1104,4 [M+1].
Стадия 9
(S)-2-(2-(2-аминоацетиламино)ацетиламино)-N-(2-((((R)-1-циклопропил-2-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизин[1,2-b]хинолин-1-ил)амино)-2-оксоэтокси)метил)амино)-2-оксоэтокси)-3-фенилпропанамид 18j
Соединение 18i (12 мг, 10,9 мкмоль) растворяли в 0,6 мл дихлорметана и добавляли 0,3 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 18j (10 мг), который непосредственно использовали на следующей стадии без очистки.
MS m/z (ESI): 881,0 [M+1].
Стадия 10
(1r,4r)-N-((2R,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15,18-гексаоксо-3,20,23,26,29,32,35,38,41,44-декаокса-5,8,11,14,17-пентааза-тетрагексадек-46-ил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоксамид 18
Неочищенный 18j (10 мг) растворяли в 1 мл N,N-диметилформамида и добавляли соединение 17g (8,5 мг, 12,3 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (4,6 мг, 16,6 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и фильтровали. Фильтрат очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 18 (9,5 мг, выход 56,2 %).
MS m/z (ESI): 1570,2 [M+18].
1H ЯМР (400 МГц, DMSO-d6): δ 8,77 (d, 1H), 8,59-8,55 (m, 1H), 8,42 (d, 1H), 8,37-8,28 (m, 1H), 8,25-8,06 (m, 2H), 7,96-7,86 (m, 1H), 7,86-7,70 (m, 2H), 7,32-7,28 (m, 1H), 7,25-7,14 (m, 3H), 6,67 (m, 1H), 5,96 (s, 1H), 5,80-5,72 (m, 1H), 5,62-5,52 (m, 2H), 5,43-5,30 (m, 3H), 5,28-5,17 (m, 2H), 5,12-5,08 (m, 1H), 4,72-4,35 (m, 8H), 3,95-3,70 (m, 13H), 3,35-3,22 (m, 14H), 2,42-2,32 (m, 3H), 2,05-1,98 (m, 4H), 1,88-1,82 (m, 12H), 1,47-1,39 (m, 3H), 1,32-1,18 (m, 11H), 0,90-0,80 (m, 4H), 0,52-0,37 (m, 3H), 0,32-0,18 (m, 2H).
Пример 1-19
(1r,4r)-N-((2S,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15,18-гексаоксо-3,20,23,26,29,32,35,38,41,44-декаокса-5,8,11,14,17-пентааза-тетрагексадек-46-ил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоксамид 19
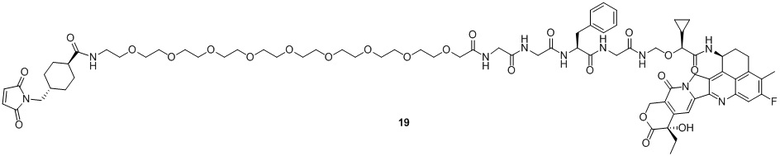
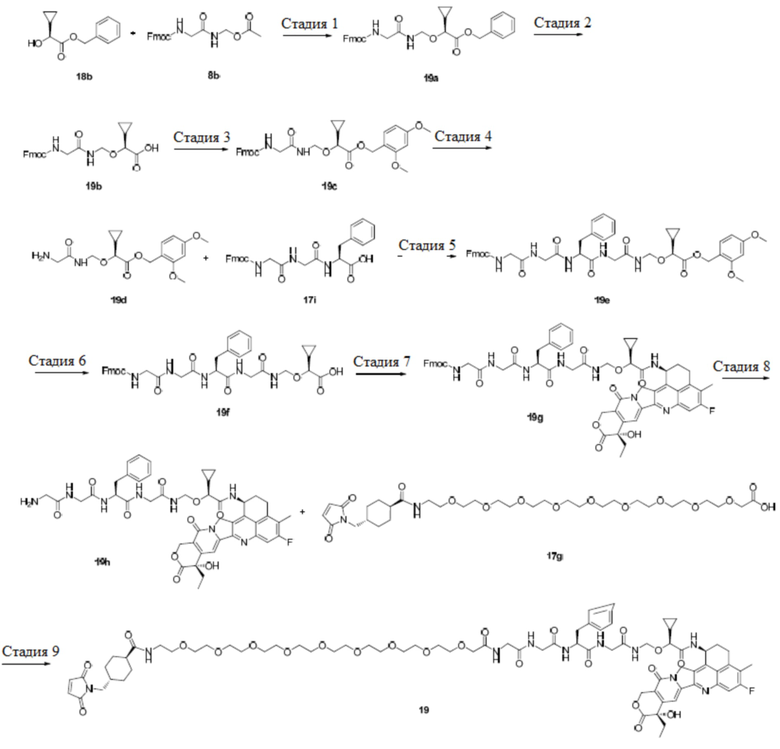
Стадия 1
Бензил-(S)-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оат 19a
Соединение 18b (252 мг, 1,22 ммоль) добавляли в реакционную колбу и добавляли 4 мл дихлорметана. Систему трижды продували аргоном и охлаждали смесь до 0-5 °C на ледяной водяной бане с последующим добавлением трет-бутоксида лития (98 мг, 1,22 ммоль). Реакционную смесь перемешивали на водяной бане со льдом в течение 15 мин она становилась чистой, и добавляли 8b (300 мг, 814,3 мкмоль). Реакционную смесь перемешивали на ледяной водяной бане в течение 2,5 ч. Добавляли воду (10 мл) для отделения жидкости. Водную фазу экстрагировали дихлорметаном (8 мл×2), а органические фазы объединяли и промывали водой (10 мл×1) и насыщенным солевым раствором (10 мл×2), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с системой проявляющего растворителя C с получением указанного в заголовке продукта 19a (282 мг, выход 67,2%).
Стадия 2
(S)-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оевая кислота 19b
Соединение 19a (280 мг, 0,554 ммоль) растворяли в 8 мл этилацетата и добавляли палладий на угле (84 мг, нагрузка 10 %, сухая основа). Систему трижды продували водородом, и реакционную смесь перемешивали при комнатной температуре в течение 3 часов и осадок на фильтре промывали этилацетатом. Фильтрат концентрировали с получением неочищенного указанного в заголовке продукта 19b (230 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 3
2,4-диметоксибензил(S)-10-циклопропил-1-(9H-флуорен-9-ил)-3,6-диоксо-2,9-диокса-4,7-диазаундекан-11-оат 19c
Неочищенный 19b (230 мг, 541,8 мкмоль) растворяли в 7 мл дихлорметана и последовательно добавляли 2,4-диметоксибензиловый спирт (136,7 мг, 812,7 мкмоль), 1-этил-(3-диметиламинопропил)карбонилдиимина гидрохлорид (155 мг, 808,5 мкмоль) и 4-диметиламинопиридин (6,6 мг, 53,5 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли 10 мл дихлорметана, промывали водой (10 мл×1) и насыщенным солевым раствором (10 мл×2), сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта. Полученный остаток очищали с помощью тонкослойной хроматографии с системой проявляющего растворителя В с получением указанного в заголовке продукта 19c (159 мг, выход 51,0 %).
Стадия 4
2,4-диметоксибензил(S)-2-((2-аминоацетиламино)метокси)-2-циклопропилацетат 19d
Соединение 19c (60 мг, 104,4 мкмоль) растворяли в 1 мл дихлорметана и добавляли 0,5 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 19d (21 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 5
2,4-диметоксибензил(11S,19S)-11-бензил-19-циклопропил-1-(9H-флуорен-9-ил)-3,6,9,12,15-пентаоксо-2,18-диокса-4,7,10,13,16-пентаазеикоза-20-оат 19e
Неочищенный 19d (36 мг, 102,2 мкмоль) растворяли в 4 мл N,N-диметилформамида и добавляли соединение 17i (52 мг, 103,6 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (34,6 мг, 125,0 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 10 мл воды с последующей экстракцией этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 19e (70 мг, выход 80,2 %).
Стадия 6
(11S,19S)-11-бензил-19-циклопропил-1-(9H-флуорен-9-ил)-3,6,9,12,15-пентаоксо-2,18-диокса-4,7,10,13,16-пентаазеикоза-20-оевая кислота 19f
Соединение 19e (70 мг, 83,7 мкмоль) растворяли в 2,5 мл 3% (об./об.) раствора дихлоруксусной кислоты в дихлорметане и охлаждали раствор до 0-5 °C на ледяной водяной бане с последующим добавлением триэтилсилана (29 мг, 249,4 мкмоль). Реакционную смесь перемешивали на ледяной бане в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении на ледяной бане для удаления половины органического растворителя и добавляли 5 мл простого диэтилового эфира. Полученную смесь нагревали до комнатной температуры естественным образом и суспендировали для осаждения белого твердого вещества и фильтровали. Осадок на фильтре собирали и сушили с помощью масляного насоса с получением указанного в заголовке продукта 19f (57 мг, выход 99,2%).
Стадия 7
(9H-флуорен-9-ил)метил((2S,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15-пентаоксо-3-окса-5,8,11,14-тетраазагексадек-16-ил)карбамат 19g
Соединение 1b (30 мг, 63,6 мкмоль) добавляли в реакционную колбу и добавляли 1 мл 10% (об./об.) раствора метанола в дихлорметане. Систему трижды продували аргоном, и смесь охлаждали до 0-5 °C на ледяной водяной бане с последующим добавлением капли триэтиламина. Смесь перемешивали до растворения соединения 1b. Соединение 19f (57 мг, 83,1 мкмоль) растворяли в 1 мл 10% (об./об.) раствора метанола в дихлорметане, а затем по каплям добавляли вышеуказанную реакционную смесь с последующим добавлением 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (26 мг, 93,9 мкмоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Добавляли 10 мл дихлорметана и 5 мл воды, и полученную смесь перемешивали в течение 5 минут и оставляли стоять с образованием слоев. Органическую фазу собирали, и водную фазу экстрагировали дихлорметаном (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью тонкослойной хроматографии с системой В проявляющего растворителя с получением указанного в заголовке продукта 19g (56 мг, выход 79,8 %).
MS m/z (ESI): 1103,1 [M+1].
Стадия 8
(S)-2-(2-(2-аминоацетиламино)ацетиламино)-N-(2-((((S)-1-циклопропил-2-((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-2-оксоэтокси)метил)амино)-2-оксоэтил)-3-фенилпропанамид 19h
Соединение 19g (4,6 мг, 4,16 мкмоль) растворяли в 1,5 мл дихлорметана и добавляли 0,75 мл диэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,6 ч и концентрировали при пониженном давлении. Добавляли 2 мл толуола с последующим концентрированием при пониженном давлении; процедуры повторяли дважды. Остаток суспендировали с 3 мл н-гексана и удаляли верхний слой н-гексана; процедуры повторяли три раза. Суспензию концентрировали при пониженном давлении с получением указанного в заголовке неочищенного продукта 19h (4,0 мг), который непосредственно использовали на следующей стадии без очистки.
Стадия 9
(1r,4r)-N-((2S,10S)-10-бензил-2-циклопропил-1-(((1S,9S)-9-этил-5-фтор-9-гидрокси-4-метил-10,13-диоксо-2,3,9,10,13,15-гексагидро-1H,12H-бензо[де]пирано[3',4':6,7]индолизино[1,2-b]хинолин-1-ил)амино)-1,6,9,12,15,18-гексаоксо-3,20,23,26,29,32,35,38,41,44-декаокса-5,8,11,14,17-пентааза-тетрагексадек-46-ил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)метил)циклогексан-1-карбоксамид 19
Неочищенный 19h (4,0 мг) растворяли в 1 мл N,N-диметилформамида и добавляли соединение 17g (2,9 мг, 4,2 мкмоль) и 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид (1,5 мг, 5,4 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 40 минут и фильтровали. Фильтрат очищали высокоэффективной жидкостной хроматографией (условия разделения: колонка для хроматографии: XBridge Prep C18 OBD 5 мкм 19×250 мм; подвижная фаза: А - вода (10 ммоль NH4OAc), В - ацетонитрил, градиентное элюирование, скорость потока: 18 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 19 (2,1 мг, выход 32,4 %).
1H ЯМР (400 МГц, DMSO-d6): δ 8,71-8,62 (m, 1H), 8,59-8,51 (m, 1H), 8,34-8,26 (m, 1H), 8,14-8,02 (m, 2H), 7,95-7,86 (m, 1H), 7,83-7,69 (m, 2H), 7,35-7,31 (m, 1H), 7,29-7,11 (m, 3H), 7,01 (s, 1H), 6,72-6,50 (m, 3H), 5,59-5,50 (m, 2H), 5,42 (s, 2H), 5,38-5,18 (m, 3H), 4,79-4,69 (m, 2H), 4,61-4,42 (m, 3H), 3,91 (s, 2H), 3,79-3,65 (m, 4H), 3,63-3,44 (m, 13H), 3,41-3,30 (m, 2H), 3,26-3,09 (m, 5H), 3,08-2,84 (m, 4H), 2,81-2,64 (m, 3H), 2,42-2,28 (m, 3H), 2,24-2,12 (m, 2H), 2,05-1,93 (m, 4H), 1,89-1,77 (m, 2H), 1,72-1,56 (m, 3H), 1,53-1,38 (m, 3H), 1,34-1,10 (m, 11H), 0,94-0,78 (m, 5H), 0,52-0,35 (m, 3H).
Пример 1-20 (эталонный пример)
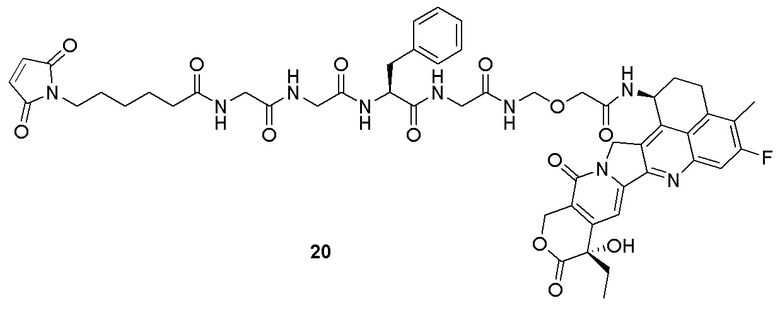
Указанное в заголовке соединение 20 получали таким образом, как описано в "Примере 58 на странице 163 описания патента CN104755494A".
Следующие антитела могут быть получены с использованием общепринятого способа получения антител и могут быть получены, например, векторной конструкцией, трансфекцией эукариотических клеток, таких как клетки HEK293 (Life Technologies Cat. № 11625019), и очистку экспрессии.
Ниже представлена последовательность трастузумаба:
Легкая цепь
DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
SEQ ID NO: 1
Тяжелая цепь
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
SEQ ID NO: 2
Ниже представлена последовательность пертузумаба:
Легкая цепь
DIQMTQSPSSLSASVGDRVTITCKASQDVSIGVAWYQQKPGKAPKLLIYSASYRYTGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQYYIYPYTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
SEQ ID NO: 3
Тяжелая цепь
EVQLVESGGGLVQPGGSLRLSCAASGFTFTDYTMDWVRQAPGKGLEWVADVNPNSGGSIYNQRFKGRFTLSVDRSKNTLYLQMNSLRAEDTAVYYCARNLGPSFYFDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
SEQ ID NO: 4
Ниже представлена последовательность B7H3 антитела 1F9DS:
Легкая цепь
DTVVTQEPSFSVSPGGTVTLTCGLSSGSVSTSHYPSWYQQTPGQAPRMLIYNTNTRSSGVPDRFSGSILGNKAALTITGAQADDESDYYCAIHVDRDIWVFGGGTKLTVLGQPKANPTVTLFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASSYLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTEC
SEQ ID NO: 5
Тяжелая цепь
QVQLVQSGGGVVQPGTSLRLSCAASGFIFSSSAMHWVRQAPGKGLEWVAVISYDGSNKYYVDSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARSARLYASFDYWGQGALVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
SEQ ID NO: 6
Пример 1-21. ADC-1
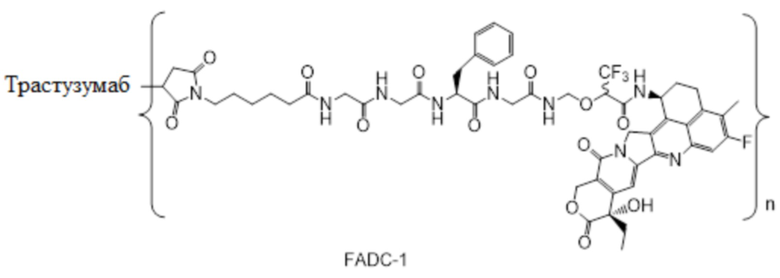
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 2,5 мл, 9,96 мг/мл, 0,168 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,082 мкмл, 0,82 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 5,0 мг/мл, и собирали 2,0 мл раствора для использования на следующем этапе.
Соединение 10 - соединение с более коротким временем удерживания (2,1 мг, 2,02 мкмоль) растворяли в 0,10 мл DMSO и раствор добавляли к вышеуказанным 2,0 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-1 FADC-1 общей формулы в буфере PBS (5,0 мг/мл, 1,1 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 5,09.
Пример 1-22. ADC-2
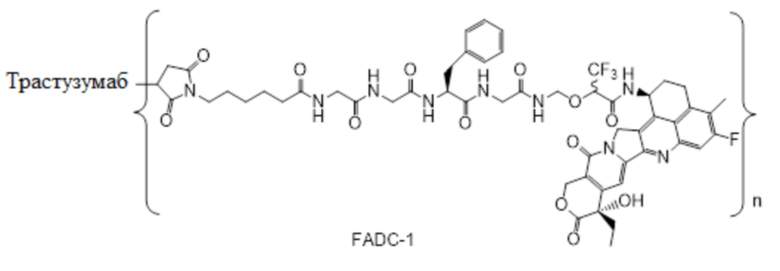
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 2,5 мл, 9,96 мг/мл, 0,168 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,082 мл, 0,82 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 5,0 мг/мл, и собирали 2,0 мл раствора для использования на следующем этапе.
Соединение 10 с более длительным временем удерживания (2,1 мг, 2,02 мкмоль) растворяли в 0,10 мл DMSO и раствор добавляли к вышеуказанным 2,0 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-2 FADC-1 общей формулы в буфере PBS (4,95 мг/мл, 1,1 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 7,39.
Пример 1-23. ADC-3
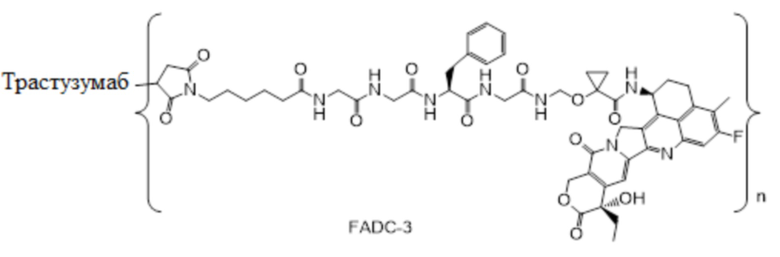
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 2,5 мл, 9,96 мг/мл, 0,168 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,082 мл, 0,82 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 5,0 мг/мл, и собирали 2,0 мл раствора для использования на следующем этапе.
Соединение 8 (2,1 мг, 2,02 мкмоль) растворяли в 0,10 мл DMSO и раствор добавляли к вышеуказанным 2,0 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-3 FADC-3 общей формулы в буфере PBS (5,24 мг/мл, 1,1 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 7,36.
Пример 1-24. ADC-4
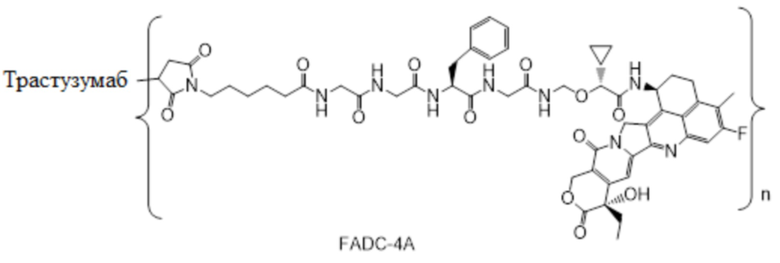
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 3,74 мл, 13,38 мг/мл, 0,338 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,173 мл, 1,73 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 6,7 мг/мл, и собирали 1,3 мл раствора для использования на следующем этапе.
Соединение 9 - соединение 9-A с более коротким временем удерживания (1,0 мг, 0,93 мкмоль) растворяли в 0,10 мл DMSO и раствор добавляли к вышеуказанным 1,3 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-4 FADC-4A общей формулы в буфере PBS (1,72 мг/мл, 2,36 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 7,39.
Пример 1-25. ADC-5
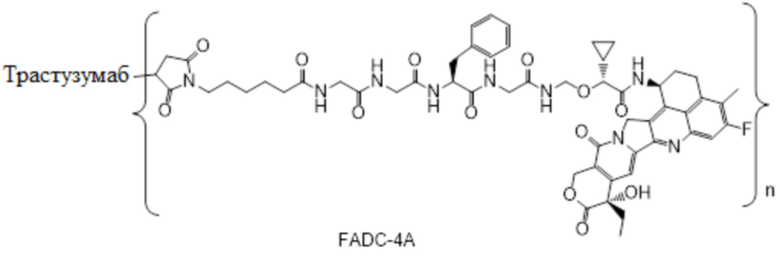
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 3,0 мл, 6,70 мг/мл, 0,136 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,067 мл, 0,67 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и собирали 0,614 мл раствора для использования на следующем этапе.
Соединение 9 - соединение 9-A с более коротким временем удерживания (0,5 мг, 0,42 мкмоль) растворяли в 0,031 мл DMSO и раствор добавляли к вышеуказанным 0,614 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-5 FADC-4A общей формулы в буфере PBS (3,08 мг/мл, 0,82 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 3,16.
Пример 1-26. ADC-6
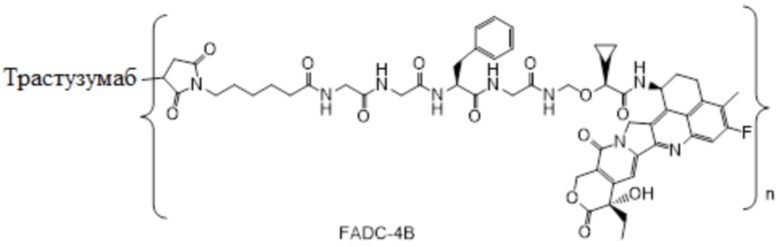
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 3,74 мл, 13,38 мг/мл, 0,338 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,173 мл, 1,73 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 6,7 мг/мл, и собирали 0,75 мл раствора для использования на следующем этапе.
Соединение 9 - соединение 9-B с более длительным временем удерживания (0,68 мг, 0,63 мкмоль) растворяли в 0,10 мл DMSO и раствор добавляли к вышеуказанным 0,75 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-6 FADC-4B общей формулы в буфере PBS (1,78 мг/мл, 1,78 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 3,94.
Пример 1-27. ADC-7
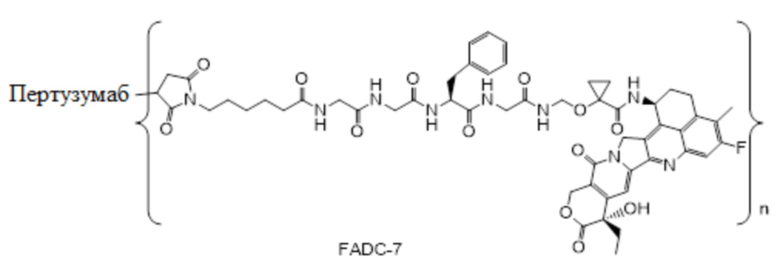
К антителу пертузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 5,0 мл, 10 мг/мл, 0,338 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,173 мл, 1,73 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 5,0 мг/мл, и собирали 1,0 мл раствора для использования на следующем этапе.
Соединение 8 (0,65 мг, 0,6 мкмоль) растворяли в 0,1 мл DMSO и раствор добавляли к вышеуказанным 1,0 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-7 FADC-7 общей формулы в буфере PBS (1,42 мг/мл, 2,15 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 6,91.
Пример 1-28. ADC-8
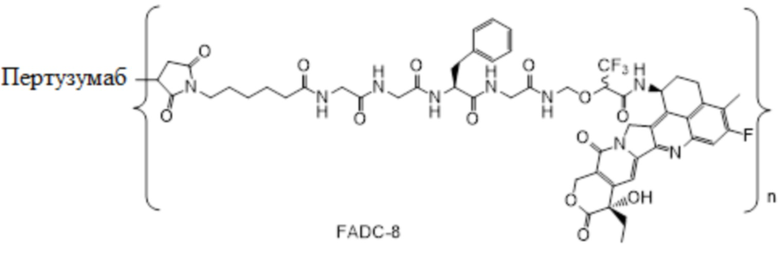
К антителу пертузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 5,0 мл, 10 мг/мл, 0,338 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,173 мл, 1,73 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 5,0 мг/мл, и собирали 1,6 мл раствора для использования на следующем этапе.
Соединение 10 - соединение с более коротким временем удерживания (1,04 мг, 1,0 мкмоль) растворяли в 0,1 мл DMSO и раствор добавляли к вышеуказанным 1,6 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-8 FADC-8 общей формулы в буфере PBS (2,14 мг/мл, 2,31 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 6,58.
Пример 1-29. ADC-9
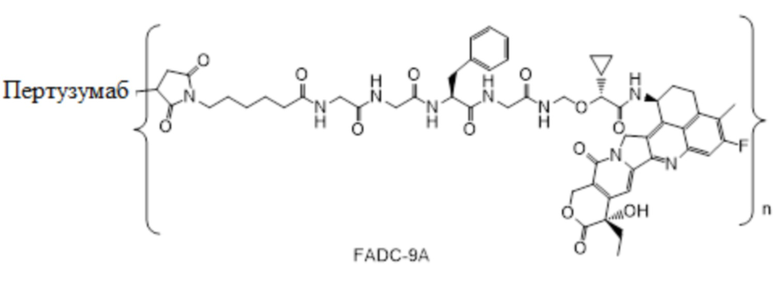
К антителу пертузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 5,0 мл, 10 мг/мл, 0,338 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (10 мМ, 0,173 мл, 1,73 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане и разбавляли до 5,0 мг/мл, и собирали 0,8 мл раствора для использования на следующем этапе.
Соединение 9 - соединение 9-A с более коротким временем удерживания (0,55 мг, 0,5 мкмоль) растворяли в 0,1 мл DMSO и раствор добавляли к вышеуказанным 0,8 мл раствора. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-9 FADC-9A общей формулы в буфере PBS (2,27 мг/мл, 1,11 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное методом UV-HPLC: n составляет 3,16.
Пример 1-30. ADC-10
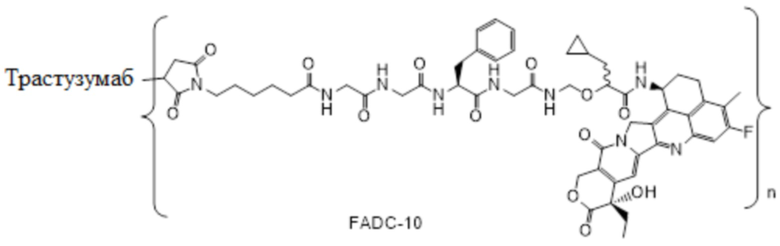
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,574 мл, 38,78 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 19,76 мкл, 197,6 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 14 - соединение с более коротким временем удерживания (0,64 мг, 588 нмоль) растворяли в 40 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-10 FADC-10 общей формулы в буфере PBS (5,48 мг/мл, 1,03 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,25.
Пример 1-31. ADC-11
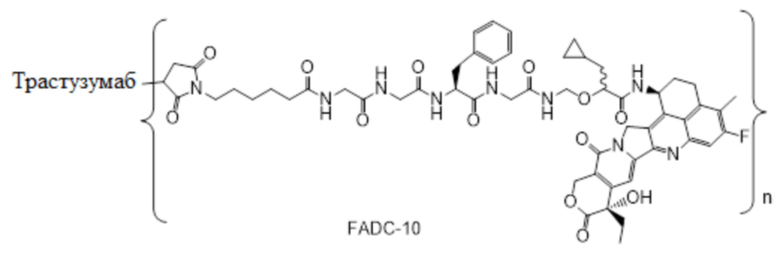
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,646 мл, 43,64 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 22,24 мкл, 222,4 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 14 с более длительным временем удерживания (0,72 мг, 662 нмоль) растворяли в 40 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-11 FADC-10 общей формулы в буфере PBS (2,13 мг/мл, 1,87 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,03.
Пример 1-32. ADC-12
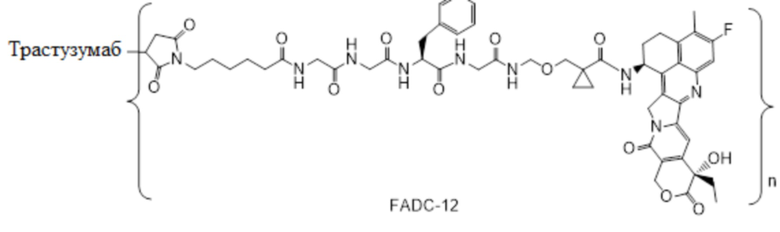
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,726 мл, 49,05 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 25,0 мкл, 250,0 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 15 (0,81 мг, 754 нмоль) растворяли в 40 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-12 FADC-12 общей формулы в буфере PBS (3,34 мг/мл, 1,45 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,93.
Пример 1-33. ADC-13
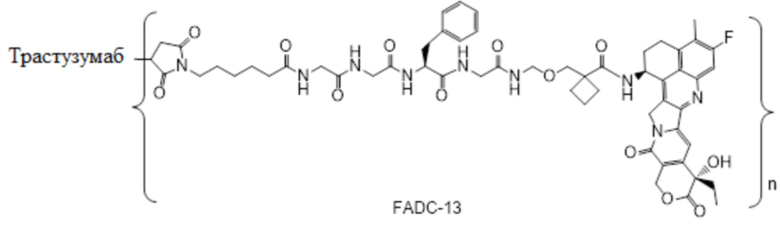
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,287 мл, 19,39 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 9,88 мкл, 98,8 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 16 (0,32 мг, 294 нмоль) растворяли в 20 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-13 FADC-13 общей формулы в буфере PBS (2,37 мг/мл, 0,88 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,53.
Пример 1-34. ADC-14
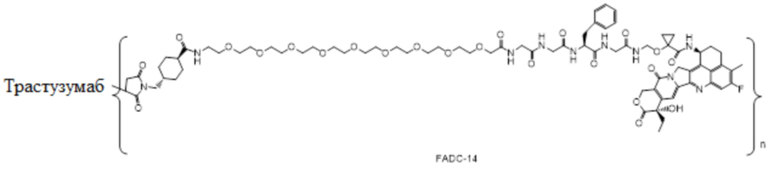
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,592 мл, 40,0 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 20,38 мкл, 203,8 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 17 (0,92 мг, 598 нмоль) растворяли в 40 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-14 FADC-14 общей формулы в буфере PBS (0,30 мг/мл, 12,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,61.
Пример 1-35. ADC-15
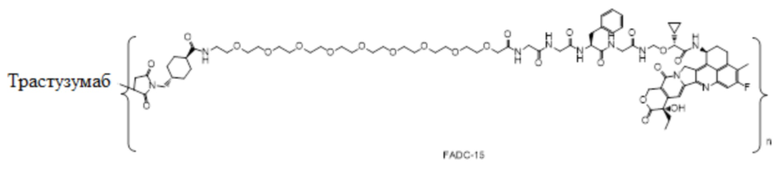
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,592 мл, 40,0 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 20,38 мкл, 203,8 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 18 (0,93 мг, 599 нмоль) растворяли в 40 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-15 FADC-15 общей формулы в буфере PBS (0,32 мг/мл, 11,8 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,89.
Пример 1-36. ADC-16
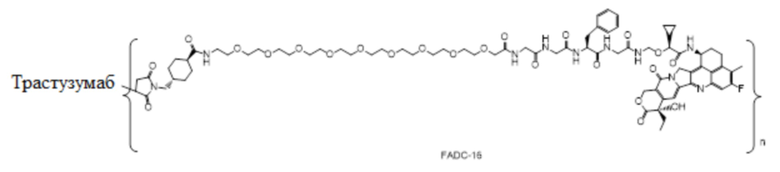
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,53 мл, 35,8 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 18,25 мкл, 182,5 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 19 (0,83 мг, 534 нмоль) растворяли в 35 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-16 FADC-16 общей формулы в буфере PBS (0,32 мг/мл, 12,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,43.
Пример 1-37. ADC-17
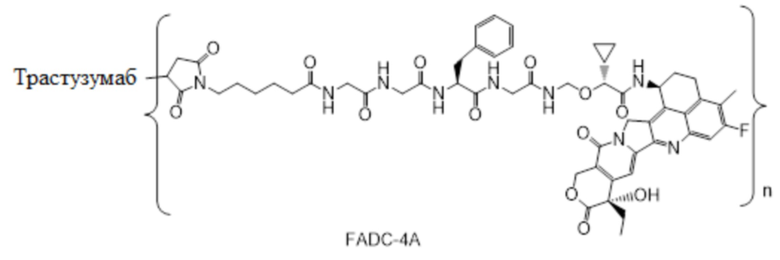
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 2,0 мл, 135,12 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 43,2 мкл, 432 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9-A с более коротким временем удерживания (2,22 мг, 2067 нмоль) растворяли в 175 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-17 FADC-4A общей формулы в буфере PBS (1,32 мг/мл, 12,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 5,42.
Пример 1-38. ADC-18 (эталонный пример)
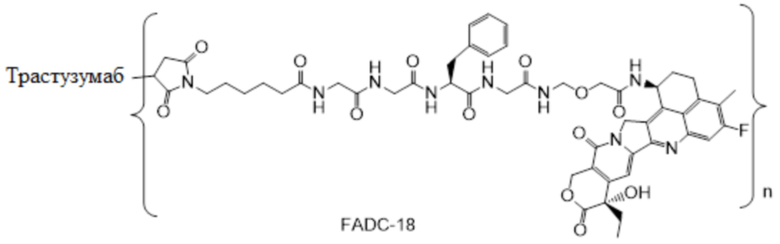
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 1,5 мл, 101,3 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 51,7 мкл, 517 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 20 (2,0 мг, 1934 нмоль) растворяли в 100 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-18 FADC-18 общей формулы в буфере PBS (0,79 мг/мл, 13,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,23.
Пример 1-39. ADC-19
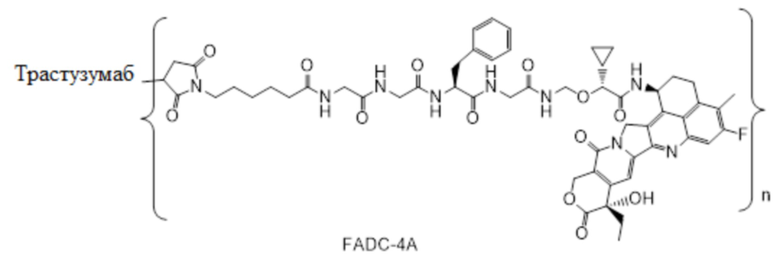
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 1,36 мл, 91,9 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 46,9 мкл, 469 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9-A с более коротким временем удерживания (2,0 мг, 1862 нмоль) растворяли в 100 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-19 FADC-4A общей формулы в буфере PBS (0,73 мг/мл, 13,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,26.
Пример 1-40. ADC-20
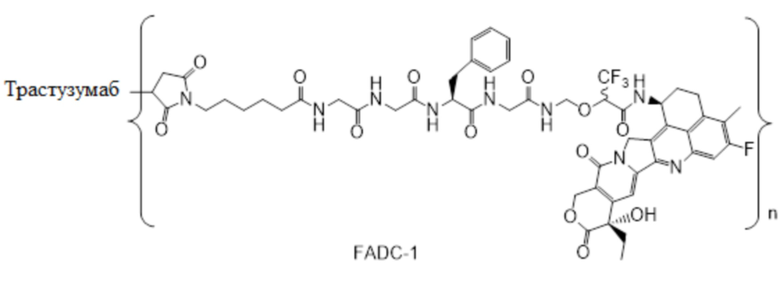
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 1,5 мл, 101,3 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 51,7 мкл, 517 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 10 с более длительным временем удерживания (2,0 мг, 1815 нмоль) растворяли в 100 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-20 FADC-1 общей формулы в буфере PBS (0,73 мг/мл, 13,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,43.
Пример 1-41. ADC-21 (эталонный пример)
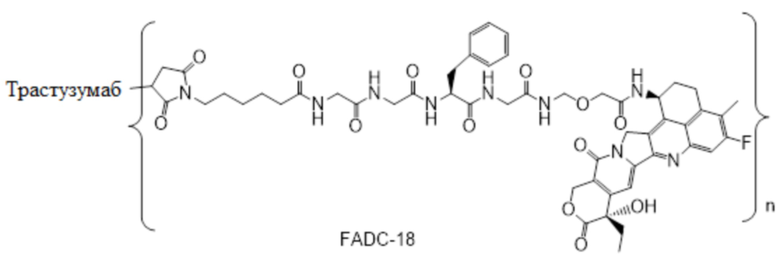
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 1,86 мл, 125,4 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 63,9 мкл, 639 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 20 (2,07 мг, 2001 нмоль) растворяли в 150 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-21 FADC-18 общей формулы в буфере PBS (2,91 мг/мл, 4,44 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,23.
Пример 1-42. ADC-22
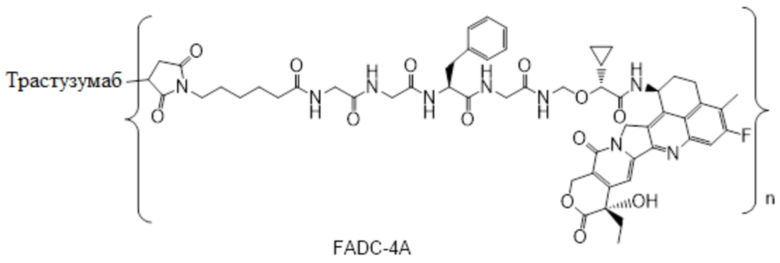
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 1,88 мл, 127,2 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 64,9 мкл, 649 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9-A с более коротким временем удерживания (2,1 мг, 1955 нмоль) растворяли в 150 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-22 FADC-4A общей формулы в буфере PBS (3,56 мг/мл, 3,98 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,79.
Пример 1-43. ADC-23 (эталонный пример)
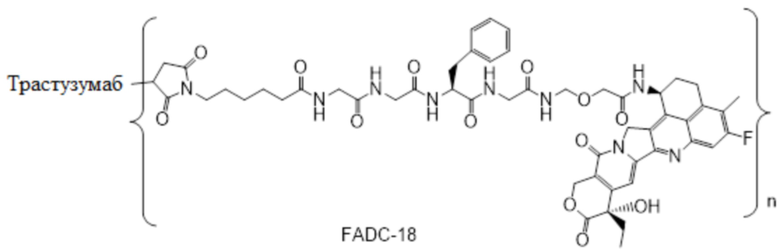
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 345 мл, 23,31 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 11,89 мкл, 118,9 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3,5 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 20 (362 мг, 350 мкмоль) растворяли в 7,12 мл MeCN (ацетонитрил) и 3,56 мл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь очищали путем обессоливания через ультрафильтрационные мембраны с помощью 2% (об./об.) MeCN и 1% (об./об.) DMSO-содержащего водного буфера PBS (0,05 M водного буфера PBS при pH 6,5) и водного буфера янтарной кислоты (0,01 M водного буфера янтарной кислоты при pH 5,3) последовательно. Затем добавляли сахарозу в концентрации 60 мг/мл и tween (твин) 20 в концентрации 0,2 мг/мл. Смесь разливали в бутылки и лиофилизировали с получением лиофилизированного порошкового образца иллюстративного продукта ADC-23 общей формулы FADC-18, который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,05.
Пример 1-44. ADC-24
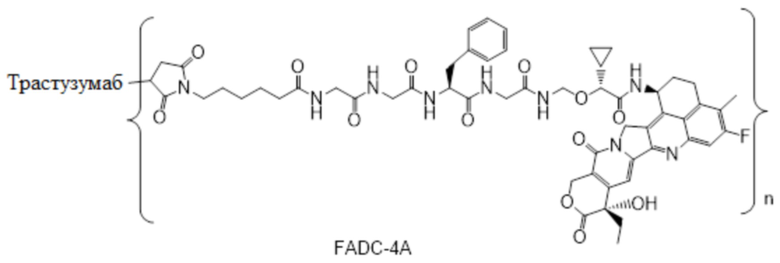
К антителу трастузумаб в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 332 мл, 22,43 мкмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 11,44 мл, 114,4 мкмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3,5 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9A c более коротким временем удерживания (241 мг, 224 мкмоль) растворяли в 13,76 мл MeCN и 6,88 мл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь очищали путем обессоливания через ультрафильтрационные мембраны с помощью 4% (об./об.) MeCN и 2% (об./об.) DMSO-содержащего водного буфера PBS (0,05 M водного буфера PBS при pH 6,5) и водного буфера янтарной кислоты (0,01 M водного буфера янтарной кислоты при pH 5,3) последовательно. Затем добавляли сахарозу в концентрации 60 мг/мл и tween 20 в концентрации 0,2 мг/мл. Смесь разливали в бутылки и лиофилизировали с получением лиофилизированного порошкового образца иллюстративного продукта ADC-24 общей формулы FADC-4A, который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,07.
Пример 1-45. ADC-25
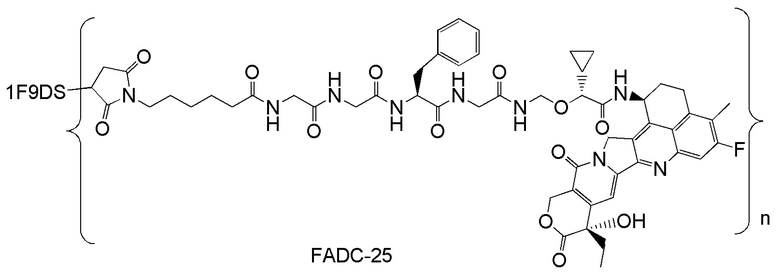
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 2,14 мл, 144,60 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 73,7 мкл, 740 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9-A с более коротким временем удерживания (3,0 мг, 2793 нмоль) растворяли в 150 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-25 FADC-25 общей формулы в буфере PBS (1,28 мг/мл, 13,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,87.
Пример 1-46. ADC-26 (эталонный пример)
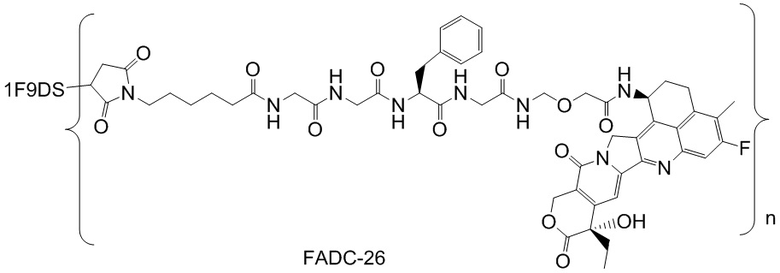
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,89 мл, 60,14 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 30,1 мкл, 300 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 20 (1,0 мг, 967 нмоль) растворяли в 100 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-26 FADC-26 общей формулы в буфере PBS (1,61 мг/мл, 4,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,15.
Пример 1-47. ADC-27
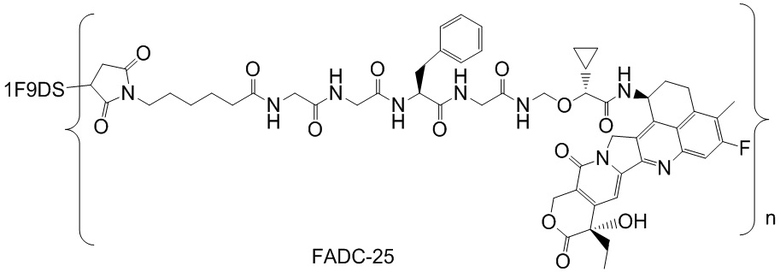
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,89 мл, 60,14 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 30,1 мкл, 300 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9-A с более коротким временем удерживания (1,02 мг, 950 нмоль) растворяли в 100 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-27 FADC-25 общей формулы в буфере PBS (1,94 мг/мл, 3,5 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,11.
Пример 1-48. ADC-28 (эталонный пример)
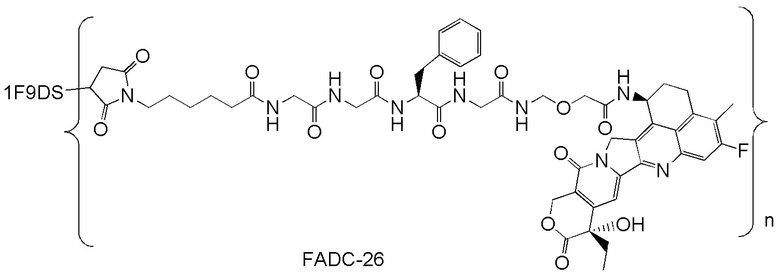
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 2,36 мл, 159,47 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 81,3 мкл, 810 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 20 (3,0 мг, 2901 нмоль) растворяли в 150 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-28 FADC-26 общей формулы в буфере PBS (1,29 мг/мл, 13,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,46.
Пример 1-49. ADC-29
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,80 мл, 50,06 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 28,6 мкл, 290 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 9 - соединение 9-A с более коротким временем удерживания (1,29 мг, 1201 нмоль) растворяли в 100 мкл DMSO, и раствор добавляли к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-29 FADC-25 общей формулы в буфере PBS (2,63 мг/мл, 2,4 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 7,24.
Пример 1-50. ADC-30 (эталонный пример)
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,86 мл, 58,4 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 29,1 мкл, 290 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 20 (1,0 мг, 967 нмоль) растворяли в 100 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-30 FADC-26 общей формулы в буфере PBS (1,61 мг/мл, 4,0 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,15.
Пример 1-51. ADC-31
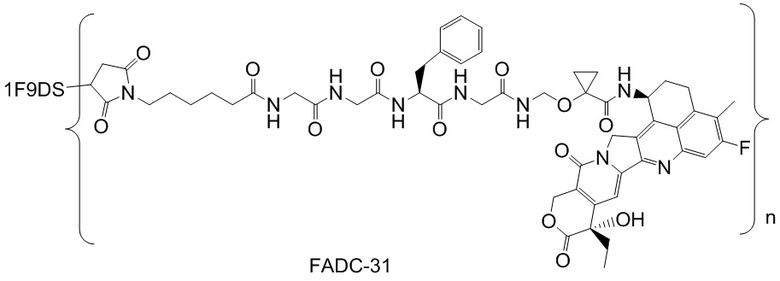
К антителу B7H3 антитело 1F9DS в водном буфере PBS (0,05 М водный буфер PBS при рН 6,5, 10,0 мг/мл, 0,89 мл, 60,14 нмоль) добавляли при 37 °C полученный водный раствор трис(2-карбоксиэтил)фосфина (TCEP) (10 мМ, 30,1 мкл, 300 нмоль). Реакционную смесь встряхивали на водяной бане-шейкере при 37 °С в течение 3 часов перед прекращением реакции. Реакционную смесь охлаждали до 25 °C на водяной бане.
Соединение 8 (1,0 мг, 943 нмоль) растворяли в 100 мкл DMSO и добавляли раствор к вышеуказанной реакционной смеси. Реакционную смесь встряхивали на водяной бане-шейкере при 25 °С в течение 3 часов перед прекращением реакции. Реакционную смесь обессоливали и очищали через колонку с гелем Sephadex G25 (фаза элюирования: 0,05 М водный буфер PBS при рН 6,5, содержащий 0,001 М EDTA) с получением иллюстративного продукта ADC-31 FADC-31 общей формулы в буфере PBS (1,47 мг/мл, 4,5 мл), который затем хранили при 4 °C.
Среднее значение, рассчитанное по методу UV-Vis: n составляет 6,33.
Пример 1-52. ADC-32
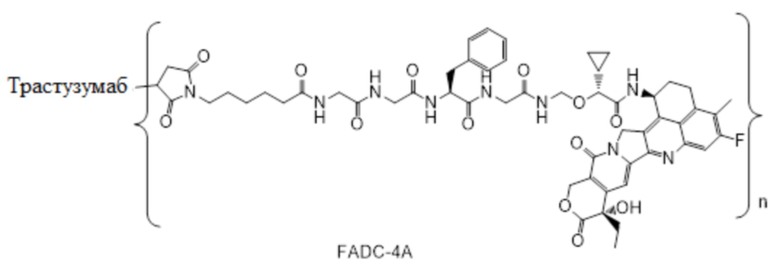
При 25 °C, в буфере, содержащем 20 мМ гистидин-соляную кислоту и 2,5 мМ EDTA (pH 5,6), 5,0 г исходного раствора трастузумаба (34,44 мкмоль; трастузумаб разбавляли 20 мМ гистидин-соляной кислотой до конечной концентрации 15 мг/мл) и 34,64 мг трис(2-карбоксиэтил)фосфина гидрохлорида (восстановитель TCEP, Sigma, 120,84 мкмоль) перемешивали на водяной бане при постоянной температуре в течение 3 часов с получением раствора промежуточного соединения I.
Соединение 9 - соединение 9-A с более коротким временем удерживания (406,2 мг, 378,17 мкмоль) растворяли в 9,98 мл DMSO с получением раствора соединения 9-A в DMSO. 23,42 мл DMSO предварительно добавляли к вышеуказанному раствору промежуточного соединения I, а затем вышеуказанный раствор соединения 9-A в DMSO добавляли к раствору промежуточного соединения I, к которому DMSO добавляли заранее. Реакционную смесь перемешивали на водяной бане при 25 °C в течение 1 часа, гасили цистеином и фильтровали. При 25 °С реакционную смесь подвергали буферному обмену путем 10-кратной и 16-кратной объемной равнообъемной ультрафильтрации через мембрану для ультрафильтрации (30 кДа) с 10% (об./об.) DMSO-содержащим водным буфером, содержащим 20 мМ гистидин-соляную кислоту и 2,5 мМ EDTA (pH = 6,0), и водным буфером, содержащим 10 мМ гистидин-соляную кислоту (pH составляет 5,5), последовательно для удаления малых молекул и остаточных растворителей, и получали типовой продукт ADC-32 общей формулы FADC-4A.
Среднее значение, рассчитанное по HIC: n составляет 6,0.
Пример 1-53. ADC-33
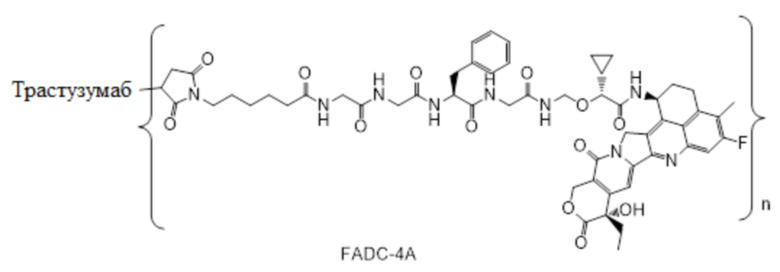
При 37 °C, в буфере, содержащем 20 мМ гистидин-соляную кислоту и 2,5 мМ EDTA (pH 6,0), 8,0 г исходного раствора трастузумаба (55,11 мкмоль; трастузумаб разбавляли 20 мМ гистидин-соляной кислотой до конечной концентрации 15 мг/мл) и 123,95 мг трис(2-карбоксиэтил)фосфина гидрохлорида (восстановитель TCEP, Sigma, 432,41 мкмоль) перемешивали на водяной бане при постоянной температуре в течение 5 часов с получением раствора промежуточного соединения I.
Соединение 9 - соединение 9-A с более коротким временем удерживания (754,7 мг, 702,63 мкмоль) растворяли в 15,99 мл DMSO с получением раствора соединения 9-A в DMSO. 37,34 мл DMSO предварительно добавляли к вышеуказанному раствору промежуточного соединения I, а затем вышеуказанный раствор соединения 9-A в DMSO добавляли к раствору промежуточного соединения I, к которому DMSO добавляли заранее. Реакционную смесь перемешивали на водяной бане при 37 °C в течение 1 часа, после чего реакцию останавливали, и реакционную смесь фильтровали. При 25 °С реакционную смесь подвергали буферному обмену путем 8-кратной и 16-кратной объемной равнообъемной ультрафильтрации через мембрану для ультрафильтрации (30 кДа) с 10% (об./об.) DMSO-содержащим водным буфером, содержащим 20 мМ гистидин-соляную кислоту и 2,5 мМ EDTA (pH = 6,0), и водным буфером, содержащим 10 мМ гистидин-соляную кислоту (pH составляет 6,0), последовательно для удаления малых молекул и остаточных растворителей, и получали иллюстративный продукт ADC-33 общей формулы FADC-4A.
Среднее значение, рассчитанное по HIC: n составляет 7,15.
Анализ нагрузки лекарственным средством исходного раствора ADC
Цель и принцип эксперимента
ADC представляет собой сшитое лекарственное средство с антителом. Его механизм лечения заболеваний заключается в переносе молекул токсина в клетки в зависимости от нацеливающей способности антитела и уничтожении клеток. Нагрузка лекарственного средства играет решающую роль в эффективности лекарственного средства. Нагрузку лекарственного средства в исходный раствор ADC определяли с помощью спектрофотометрии в видимом и ультрафиолетовом спектре (UV-Vis) или хроматографии с гидрофобным взаимодействием (HIC).
1. Спектроскопия в видимой и ультрафиолетовой областях спектра
Кюветы, содержащие буфер сукцинат натрия, помещали в эталонную ячейку и ячейку образца, и вычитали абсорбцию холостого растворителя. Затем в ячейку с образцом помещали кювету, содержащую тестовый раствор, и определяли значения абсорбции при 280 нм и 370 нм.
Расчет: нагрузку исходного раствора ADC определяли с помощью спектроскопии в видимой и ультрафиолетовой областях спектра (прибор: спектрофотометр в ультрафиолетовой области спектра Thermo nanodrop2000), исходя из принципа, что суммарное значение абсорбции исходного раствора ADC при определенной длине волны представляет собой сумму значений абсорбции цитотоксического лекарственного средства и моноклонального антитела при этой длине волны, а именно:
(1) A280 нм = εmab-280bCmab + εDrug-280bCDrug (mab - моноклональное антитело, Drug - лекарственное средство)
εDrug-280: средний молярный коэффициент экстинкции лекарственного средства при 280 нм составил 5100;
CDrug: концентрация лекарственного средства;
εmab-280: средний молярный коэффициент экстинкции исходного раствора трастузумаба или исходного раствора пертузумаба при 280 нм составлял 214 600;
Cmab: концентрация исходного раствора моноклональных антител трастузумаба или пертузумаба;
b: оптическая длина пути составляет 1 см.
Аналогично, уравнение для значения общей абсорбции образца при 370 нм может быть представлено как:
(2) A370 нм = εmab-370bCmab + εDrug-370bCDrug
εDrug-370: средний молярный коэффициент экстинкции лекарственного средства при 370 нм составил 19000;
CDrug: концентрация лекарственного средства;
εmab-370: коэффициент экстинкции исходного раствора моноклональных антител трастузумаба или пертузумаба при 370 нм составил 0;
Cmab: концентрация исходного раствора моноклональных антител трастузумаба;
b: оптическая длина пути составляет 1 см.
Нагрузка лекарственного средства может быть рассчитана с использованием как уравнения (1), так и (2), а также коэффициентов экстинкции моноклонального антитела и лекарственного средства при обеих длинах волн и их концентрациях.
Нагрузка лекарственного средства n = CDrug/Cmab.
2. Хроматография с гидрофобным взаимодействием
2.1 Анализ HPLC проводили в следующих условиях измерения:
Система HPLC: система высокоэффективного жидкостного хроматографа Agilent HPLC
Детектор: детектор DAD (детектор с фотодиодной матрицей) (длина волны измерения: 280 нм)
Колонка для хроматографии: TSKgel Butyl-NPR (4,6 мм×100 мм, 2,5 мкм)
Температура колонки: 30 °C
[Скорость потока]: 0,5 мл/мин
Температура камеры для образцов: 4 °C
Подвижная фаза A: водный раствор с pH 7,00, содержащий 1,5 М сульфата аммония ((NH4)2SO4) и 20 мМ динатрия фосфата (Na2HPO4).
Подвижная фаза B: смешанный раствор с pH 7,00, содержащий 75% 20 мМ динатрия фосфата (Na2HPO4) и 25% изопропанола.
Градиентная программа: от 0,0% B до 0,0% B (от 0 мин до 3,0 мин), от 0,0% B до 100,0% B (от 3,0 мин до 30,0 мин), от 100,0% B до
100,0% B (от 30,0 мин до 33,0 мин), от 0,0% B до 0,0% B (от 33,1 мин до 40,0 мин),
Объем вводимой пробы: 50 мкг
2.2 Анализ данных
Исходя из текущих данных, из-за особенностей колонки, исходя из разницы в концентрациях солей, конъюгат антитела и лекарственного средства элюировали в порядке возрастания в соответствии с количеством связанных лекарственных средств. Поэтому распределение количества конъюгированных лекарственных препаратов получали путем измерения площади каждого пика. В соответствии с порядком элюирования пики составляли D0 (антитело без связывания с лекарственным линкером), D2, D4, D6 и D8. Нагрузка препарата DAR (n) была рассчитана следующим образом:
Нагрузка препарата n равно Σ (площади пиков нагрузки)/100
Биологическая оценка
Тестовый пример 1-1: Тест на ингибирование in vitro пролиферации опухолевых клеток соединениями, описанными в данном документе
I. Назначение
Этот эксперимент предназначен для определения ингибирующей активности фармацевтических соединений по настоящему изобретению в отношении пролиферации in vitro клеток U87MG (Банк клеток, Китайская академия наук, кат. № TCHu138) и опухолевых клеток SK-BR-3 (клетки рака молочной железы человека, ATCC (Американская коллекция типовых культур), кат. № HTB-30). Клетки обрабатывали in vitro соединением в различных концентрациях. Через 6 дней культивирования определяли пролиферацию клеток с использованием реагента CTG (CellTiter-Glo® Luminescent Cell Viability Assay, Promega, кат. № G7573) и оценивали активность соединения in vitro в соответствии со значением IC50.
II. Способ
Способ тестирования ингибирования in vitro пролиферации клеток U87MG был описан ниже в качестве примера способа анализа ингибирующей активности соединений по настоящему изобретению в отношении in vitro пролиферации опухолевых клеток. Способ также применим, но не ограничивается этим, к испытаниям на ингибирующую активность в отношении in vitro пролиферации других опухолевых клеток.
1. Культура клеток: Клетки U87MG и SK-BR-3 культивировали в 10% среде EMEM, содержащей FBS (GE, кат. № SH30024.01), и 10% среде 5A McCoy, содержащей FBS (Gibco, кат. № 16600-108), соответственно.
2. Получение клеток: клетки U87MG и SK-BR-3, растущие в логарифмической фазе, собирали, промывали один раз PBS (фосфатно-солевым буфером, Shanghai BasalMedia Technologies Co., Ltd.), а затем расщепляли 2-3 мл трипсина (0,25% трипсин-EDTA (1×), Gibico, Life Technologies) в течение 2-3 мин; после полного расщепления клетки элюировали 10-15 мл клеточной культуральной среды; элюат центрифугировали при 1000 об/мин в течение 5 мин, и супернатант отбрасывали; затем клетки ресуспендировали в 10-20 мл клеточной культуральной среды с образованием одноклеточных суспензий.
3. Посев клеток: суспензии отдельных клеток U87MG и SK-BR-3 хорошо перемешивали, а их плотность клеток доводили средой для культивирования клеток до 2,75×103 клеток/мл и 8,25×103 клеток/мл, соответственно; суспензии клеток с поправкой на плотность хорошо перемешивали и добавляли в 96-луночный планшет для культивирования клеток при 180 мкл/лунку. В каждую из периферийных лунок 96-луночных планшетов добавляли только 200 мкл среды. Планшеты инкубировали в инкубаторе (37 °C, 5% CO2) в течение 24 часов.
4. Соединение растворяли в DMSO (диметилсульфоксид, Shanghai Titan Scientific Co., Ltd.) для получения холостого раствора при начальной концентрации 10 мМ.
Низкомолекулярные соединения были в начальной концентрации 500 нМ. Лекарственную форму получали следующим образом.
Различные тестовые образцы в концентрации 100 мкМ (30 мкл) добавляли в первый вертикальный ряд 96-луночного планшета с U-образным дном, и 20 мкл DMSO добавляли в каждую лунку второго вертикального ряда по одиннадцатый вертикальный ряд. Образцы в первом ряду (10 мкл) добавляли к 20 мкл DMSO во втором вертикальном ряду, и смеси хорошо перемешивали. Смеси (10 мкл) добавляли в третий вертикальный ряд и так далее до десятого вертикального ряда. Лекарственные средства в планшете (5 мкл на лунку) переносили в среду EMEM (95 мкл) и смеси хорошо перемешивали для последующего использования.
ADC получали при начальной концентрации 10 нМ или 500 нМ следующим образом.
Различные тестовые образцы в концентрации 100 нМ или 5 мкМ (100 мкл) добавляли в первый вертикальный ряд 96-луночного планшета и 100 мкл PBS добавляли в каждую лунку второго вертикального ряда по одиннадцатый вертикальный ряд. Образцы в первом вертикальном ряду (50 мкл) добавляли к 100 мкл PBS во втором вертикальном ряду и смеси хорошо перемешивали. Смеси (50 мкл) добавляли в третий вертикальный ряд и так далее, 3-кратным разведением, до десятого вертикального ряда.
5. Тестовые образцы, полученные в различных концентрациях (20 мкл), добавляли в планшет для культур, с двумя дублирующими лунками, установленными для каждого образца. Планшет инкубировали в инкубаторе (37 °C, 5% CO2) в течение 6 дней.
6. Цветопроявление: извлекали 96-луночный планшет для культивирования клеток и добавляли 90 мкл раствора CTG в каждую лунку с последующей 10-минутной инкубацией при комнатной температуре.
7. Считывание планшета: 96-луночный планшет для культивирования клеток извлекали и тестировали на хемилюминесцентном ридере для микропланшетов (BMG labtech, PHERAstar FS).
III. Анализ данных
Данные обрабатывали и анализировали с помощью Microsoft Excel и Graphpad Prism 5. Результаты эксперимента приведены в таблице 2 ниже.
Вывод: Низкомолекулярные фрагменты по настоящему изобретению обладают значительной ингибирующей активностью в отношении пролиферации клеток SK-BR-3 и клеток U87, и хиральные центры оказывают определенное влияние на ингибирующую активность соединений.
Тестовый пример 1-2: Тест на ингибирование in vitro пролиферации опухолевых клеток, нацеленных на HER2, с помощью конъюгатов антитела и лекарственного средства по настоящему изобретению
Этот эксперимент был предназначен для определения ингибирующей активности конъюгатов HER2-таргетирующего антитела с лекарственным средством по настоящему изобретению против пролиферации in vitro SK-BR-3 (клетки рака молочной железы человека, ATCC, кат. № HTB-30) и MDA-MB-468 (клетки рака молочной железы человека, ATCC, кат. № HTB-132). Клетки обрабатывали in vitro соединением в различных концентрациях. Через 6 дней культивирования определяли пролиферацию клеток с использованием реагентов CTG и оценивали активность соединения in vitro в соответствии со значением IC50.
Был использован метод испытания из тестового примера 1. Клетки SK-BR-3 и MDA-MB-468 использовали в испытаниях, а питательные среды для культур клеток представляли собой 10% FBS-содержащую среду McCoy 5A (Gibco, кат. № 16600-108), 10% FBS-содержащую среду EMEM (GE, кат. № SH30024.01) и 10% FBS-содержащую среду L-15 (ThermoFisher, кат. № 11415-114). Плотность клеток трех штаммов клеток доводили до 8,33×103 клеток/мл, 8,33×103 клеток/мл и 1,39×104 клеток/мл со средой для культивирования клеток, соответственно, и клеточные суспензии, скорректированные по плотности, хорошо перемешивали и добавляли в 96-луночные планшеты для культивирования клеток при 180 мкл/лунку. Соответствующие соединения были протестированы, и результаты показаны в таблице 3 ниже.
Вывод: конъюгаты антитела и лекарственного средства, нацеленное на HER2, по настоящему изобретению показали значительную ингибирующую активность в отношении пролиферации HER2-положительных клеток SK-BR-3, в то время как показали слабую ингибирующую активность в отношении пролиферации HER2-негативных клеток MDA-MB-468, демонстрируя хорошую селективность.
Тестовый пример 1-3: Испытание стабильности Her2-ADC в плазме
Образцы ADC-19, ADC-18 и ADC-20, плазму человека, плазму обезьяны (Shanghai Medicilon Inc.) и раствор 1% BSA (бычий сывороточный альбумин) (Sigma) в PBS (Sangon Biotech (Shanghai)) фильтровали через фильтр 0,22 мкм для стерилизации. Каждый из ADC-19, ADC-18 и ADC-20 добавляли к стерильной плазме или 1% раствору BSA в PBS в конечной концентрации 200 мкг/мл. Полученную смесь инкубировали в клеточном инкубаторе при 37 °C. День инкубации определяли как день 0. Образцы отбирали на 7, 14 и 21 день и тестировали на свободный токсин.
25 мкл образцов выносили на 96-луночный планшет; добавляли 50 мкл рабочего раствора внутреннего стандарта (100 нг/мл раствора камптотецина в ацетонитриле) и 150 мкл ацетонитрила; смесь перемешивали на вортексе в течение 5 мин и центрифугировали в течение 10 мин (4000 об/мин), и 5 мкл полученных образцов использовали для анализа LC/MS/MS (жидкостная хроматография и тандемной масс-спектрометрии) (Applied Biosystems, США).
Результаты показали, что: ADC-19 был достаточно стабильным как в плазме человека, так и в плазме обезьяны, а также в растворе 1% BSA в PBS, со скоростью высвобождения свободного токсина не более 2,1% на самом высоком уровне, и, как правило, был стабильным на 14-й день, см. фиг. 1А.
ADC-18 был плохо стабилен в плазме человека и обезьяны с частотой высвобождения свободного токсина 14,5% и 8,10% на самом высоком уровне, соответственно. Он был относительно стабильным в 1% растворе BSA в PBS, см. фиг. 1B.
ADC-20 был плохо стабилен в плазме человека, плазме обезьяны и растворе 1% BSA в PBS, при этом скорость высвобождения свободного токсина составляла 21,7%, 29,7% и 21,7% на самом высоком уровне, соответственно. Он находился в деградированном состоянии в растворе 1% BSA в PBS, см. фиг. 1C.
Тестовый пример 1-4: Оценка эффективности лекарственного средства на мышах с опухолью JIMT-1
I. Назначение
При использовании nu/nu голых мышей в качестве подопытных животных антитела Her2-ADC T-DM1, ADC-21 и ADC-24 вводили путем внутрибрюшинной инъекции, а затем оценивали их эффективность на голых мышах с ксенотрансплантатом опухоли линии JIMT-1 рака молочной железы человека, устойчивых к трастузумабу (герцептину).
II. Испытуемые лекарственные средства и материалы
1. Испытуемые лекарственные средства
T-DM1 (полученно с отылкой на US20050169933)
ADC-21: 3 мг/кг
ADC-21: 10 мг/кг
ADC-24: 3 мг/кг
ADC-24: 10 мг/кг
Холостой контроль (бланк): PBS
2. Способ получения: все получены путем разведения в PBS.
3. Испытуемые животные
nu/nu голые мыши, приобретенные у Beijing Vital River.
III. Способ
Мышам подкожно инокулировали в правый бок клетки JIMT-1 (Nanjing Cobioer) (5×106 клеток/мышь, с 50% искусственной базальной мембраной). Через 8 дней опухоли выросли до размера 203,09±11,94 мм3, и животных рандомизировали в 6 групп по 8 (d1).
Препарат вводили путем внутрибрюшинной инъекции в общей сложности 2 раза. Объемы опухоли и массу тела измеряли два раза в неделю, а результаты записывали.
Использовали статистическое программное обеспечение Excel 2003. Средние значения рассчитывали как avg; значения SD (стандартное отклонение) рассчитывали как STDEV (квадратный корень дисперсии выборки); значения SEM (стандартная ошибка среднего) рассчитывали как STDEV/SQRT (квадратный корень); и разность значения P между группами рассчитывали как TTEST.
Объем опухоли (V) рассчитывали как: V = 1/2 × Lдлина × Lширина2
Относительный объем (RTV) = VT/V0
Степень ингибирования опухоли (%) = (CRTV - TRTV)/CRTV (%)
Где V0 и VT представляют собой объемы опухоли в начале и в конце эксперимента, соответственно. CRTV и TRTV представляют собой относительные объемы опухоли холостой группы (носитель, PBS) и экспериментальной групп, соответственно, в конце эксперимента.
IV. Результаты
Экспериментальные результаты показаны на фиг. 2. Эксперимент завершился после двух внутрибрюшинных инъекций, а затем наблюдение проводили в течение 34 дней. T-DM1 (10 мг/кг) не оказывал ингибирующего действия на опухоли; 3 мг/кг ADC-21 имел степень ингибирования опухоли 46,22% (P < 0,01); 10 мг/кг ADC-21 имел степень ингибирования опухоли 56,77% (P < 0,001); 3 мг/кг ADC-24 имел степень ингибирования опухоли 62,77% (P < 0,001); 10 мг/кг ADC-24 имел степень ингибирования опухоли 76,32% (P < 0,001). В условиях той же дозировки эффект ингибирования опухоли ADC-24 был значительно лучше, чем у ADC-21.
Пример испытания 1-5: Оценка эффективности лекарственного средства на мышах с опухолью SK-BR-3
I. Назначение
При использовании nu/nu голых мышей в качестве подопытных животных Her2-ADC антитела ADC-21 и ADC-22 вводили путем внутрибрюшинной инъекции, а затем оценивали их эффективность на голых мышах с ксенотрансплантатом клеток SK-BR-3 рака молочной железы человека.
II. Испытуемые лекарственные средства и материалы
1. Испытуемые лекарственные средства
ADC-21: 1 мг/кг
ADC-21: 6 мг/кг
ADC-22: 1 мг/кг
ADC-22: 6 мг/кг
Холостой контроль (бланк): PBS.
2. Способ получения: все получены путем разведения в PBS.
3. Испытуемые животные
nu/nu голые мыши, приобретенные у Beijing Vital River.
III. Способ
Мышам подкожно инокулировали в правый бок клетки SK-BR-3 (ATCC) (5×106 клеток/мышь, с 50% искусственной базальной мембраной). Через 20 дней опухоли выросли до размера 153,34±11,73 мм3, и животных рандомизировали в 5 групп по 8 (d0).
Препарат вводили путем внутрибрюшинной инъекции в общей сложности 1 раз. Объемы опухоли и массу тела измеряли два раза в неделю, а результаты записывали.
Использовали статистическое программное обеспечение Excel 2003. Средние значения рассчитывали как avg; значения SD рассчитывали как STDEV; значения SEM рассчитывали как STDEV/SQRT; и разность значения P между группами рассчитывали как TTEST.
Объем опухоли (V) рассчитывали как: V = 1/2 × Lдлина × Lширина2
Относительный объем (RTV) = VT/V0
Степень ингибирования опухоли (%) = (CRTV - TRTV)/CRTV (%)
Где V0 и VT представляют собой объемы опухоли в начале и в конце эксперимента, соответственно. CRTV и TRTV представляют собой относительные объемы опухоли холостой контрольной и экспериментальной групп, соответственно, в конце эксперимента.
IV. Результаты
Экспериментальные результаты показаны на фиг. 3. Эксперимент завершился после одной внутрибрюшинной инъекции, а затем наблюдение проводили в течение 28 дней. 1 мг/кг ADC-21 имел степень ингибирования опухоли 15,01%; 6 мг/кг ADC-21 имел степень ингибирования опухоли 77,4% и имел существенное различие по сравнению с холостым контролем (P < 0,001). 1 мг/кг ADC-22 имел степень ингибирования опухоли 19,82%; 6 мг/кг ADC-22 имел степень ингибирования опухоли 98,38% (P < 0,001). В условиях той же дозировки, 6 мг/кг, эффект ингибирования опухоли ADC-22 было значительно лучше, чем у ADC-21.
Пример испытания 1-6: Стабильность в плазме
Образец ADC-25 хорошо перемешивали с каждой из плазмы человека, плазмы обезьяны и 1% раствора BSA в PBS в конечной концентрации 100 мкг/мл, а затем смеси стерилизовали фильтрацией и инкубировали на водяной бане при 37 °C. День инкубации определяли как день 0. Образцы отбирали на 7, 14 и 21 день и тестировали на свободный токсин.
Образцы в разные моменты времени отбирали, оставляли остывать до комнатной температуры и хорошо перемешивали с помощью вихревой мешалки. 25 мкл каждого из образцов добавляли в 96-луночный планшет с последующим добавлением 50 мкл рабочего раствора внутреннего стандарта (100 нг/мл раствора камптотецина в ацетонитриле) и 150 мкл ацетонитрила. Полученные смеси перемешивали на вортексе в течение 5 мин и центрифугировали при 4000 об/мин в течение 10 мин. 5 мкл супернатанта собирали для анализа LC/MS/MS.
Результаты показали, что: ADC-25 был достаточно стабильным как в плазме человека, так и в плазме обезьяны, а также в растворе 1 % BSA в PBS, со скоростью высвобождения свободного токсина не более 2 % на самом высоком уровне, и, как правило, был стабильным на 14-й день, см. фиг. 4.
Тестовый пример 1-7: Оценка эффективности ADC против опухоли астроцитомы головного мозга человека U87MG, ксенотрансплантантированной голым мышам.
I. Назначение
При использовании голых мышей BALB/cA в качестве подопытных животных в этом эксперименте оценивали эффективность соединений ADC по настоящему изобретению против опухоли астроцитомы головного мозга человека U87MG, ксенотрансплантированной голым мышам.
II. Испытуемые лекарственные средства и материалы
1. Испытуемые лекарственные средства
ADC-27 (3 мг/кг)
ADC-26 (3 мг/кг)
Холостой контроль (бланк): буфер PBS с рН 7,4.
2. Способ получения: буфер PBS с рН 7,4.
3. Испытуемые животные
Голые мыши BALB/cA: приобретенные у Shanghai Jiesijie Laboratory Animal Co., Ltd.
III. Способ
Шести-семинедельным лабораторным самкам голых мышей BALB/cA подкожно инокулировали клетки U87MG астроцитомы головного мозга человека (астроцитома головного мозга человека, Банк клеток, Китайская академия наук, кат. № TCHu138). На десятый день после инокуляции клеток животных случайным образом сгруппировали (D0) по 8 животных на группу, вводили внутрибрюшинно один раз в неделю в общей сложности 3 раза, а объемы опухоли и массу тела измеряли 2-3 раза в неделю и регистрировали данные. Объем опухоли (V) рассчитывали следующим образом:
V = 1/2 × a × b2
где a и b представляют собой соответственно длину и ширину.
Относительный объем (RTV) = VT/V0
Степень ингибирования опухоли (%) = (CRTV - TRTV)/CRTV (%)
Где V0 и VT представляют собой объемы опухоли в начале и в конце эксперимента, соответственно. CRTV и TRTV представляют собой относительные объемы опухоли контрольной (холостой) и экспериментальной групп, соответственно, в конце эксперимента.
IV. Результаты
Внутрибрюшинное введение (в/б) осуществляли один раз в неделю в общей сложности 3 раза; после наблюдения в течение 22 дней 3 мг/кг ADC-27 имел степень ингибирования опухоли 63,3% (P < 0,0001), а 3 мг/кг ADC-26 имел степень ингибирования 49,1%. ADC-27 продемонстрировал более высокую противоопухолевую эффективность, чем ADC-26.
Масса всех животных в каждой группе была нормальной в процессе введения, и ADC не имели очевидного токсического или побочного эффекта. Результаты представлены в таблице 4 и фиг. 5. Обнаруженные антитела могут эффективно ингибировать рост опухоли ксенотрансплантата U87MG у голых мышей с опухолью и демонстрировать дозозависимость.
Пример испытания 1-8: Оценка эффективности ADC против клеток опухоли метастатического рака глотки человека Detroit 562 с плевральным выпотом, ксенотрансплантированных голым мышам.
I. Назначение
В этом эксперименте оценивали эффективность соединений ADC по настоящему изобретению против клеток опухоли метастатического рака глотки человека Detroit 562 с плевральным выпотом, ксенотрансплантированной голой мыши.
II. Испытуемые лекарственные средства и материалы
1. Испытуемые лекарственные средства
ADC-29 (3 мг/кг)
ADC-28 (3 мг/кг)
Отрицательного контроль ADC (3 мг/кг): конъюгат антитела и лекарственного средства, образованный путем связывания антител, не нацеленных на B7H3, с соединением 20.
2. Способ получения: все получены путем разведения в PBS.
3. Испытуемые животные
Голые мыши BALB/cA-nude: приобретенные у Changzhou Cavens Laboratory Animal Co., Ltd.
III. Способ
Шести-семинедельным лабораторным самкам голых мышей BALB/cA инокулировали подкожно клетки метастатического рака глотки человека Detroit 562 с плевральным выпотом (ATCC, кат. № ATCC® CCL-138™). На десятый день после инокуляции клеток животных случайным образом сгруппировали (D0) по 8 животных на группу, вводили внутрибрюшинно один раз в неделю в общей сложности 3 раза, а объемы опухоли и массу тела измеряли 2-3 раза в неделю и регистрировали данные. Объем опухоли (V) рассчитывали следующим образом:
V = 1/2 × a × b2
где a и b представляют собой соответственно длину и ширину.
Относительный объем (RTV) = VT/V0
Степень ингибирования опухоли (%) = (CRTV - TRTV)/CRTV (%)
Где V0 и VT представляют собой объемы опухоли в начале и в конце эксперимента, соответственно. CRTV и TRTV представляют собой относительные объемы опухоли контрольной (отрицательной) и экспериментальной групп, соответственно, в конце эксперимента.
IV. Результаты
Препараты вводили путем внутрибрюшинной инъекции один раз в неделю, и в общей сложности осуществляли 3 инъекции. На 28 день наблюдения степени ингибирования опухоли тестируемых ADC ADC-29 3 мг/кг (3 мг/кг) и ADC-28 3 мг/кг мг/кг (3 мг/кг) достигли 72,27% (P < 0,001) и 56,2% (P < 0,001), соответственно. ADC-29 продемонстрировал более высокую противоопухолевую эффективность, чем ADC-28.
Масса всех животных в каждой группе была нормальной в процессе введения, и ADC не имели очевидного токсического или побочного эффекта. Результаты представлены в таблице 5 и фиг. 6. Обнаруженные антитела могут эффективно ингибировать рост опухоли ксенотрансплантата Detroit 562 у голых мышей с опухолью и демонстрировать дозозависимость.
День 28
Тестовый пример 1-9: Оценка эффективности лекарственного средства на мышах с опухолью U87-MG
I. Назначение
При использовании голых мышей Balb/c в качестве подопытных животных эффективность конъюгата антитела B7H3 и лекарственного средства после внутрибрюшинной инъекции оценивали на модели клеток опухоли глиобластомы человека U87MG, ксенотрансплантированных мышам.
II. Испытуемые лекарственные средства и материалы
1. Испытуемые лекарственные средства
ADC-30 1 мг/кг
ADC-30 3 мг/кг
ADC-31 1 мг/кг
ADC-31 3 мг/кг
Холостой контроль (бланк): PBS
2. Способ получения: все получены путем разведения в PBS.
3. Испытуемые животные
Голые мыши BALB/cA: приобретенные у Shanghai Slac Laboratory Animal Co., Ltd.
III. Способ
Мышам подкожно инокулировали в правый бок клетки U87MG (астроцитома головного мозга человека, банк клеток, Китайская академия наук, кат. № TCHu138) (2,5×106 клеток/мышь). Через 14 дней опухоли выросли до размера 167,49 мм3, и животных рандомизировали в 5 групп по 8 (d1).
Препарат вводили путем внутрибрюшинной инъекции один раз в неделю, и в общей сложности было осуществлено 3 инъекции. Объемы опухоли и массу тела измеряли два раза в неделю, а результаты записывали.
Использовали статистическое программное обеспечение Excel 2003. Средние значения рассчитывали как avg; значения SD рассчитывали как STDEV; значения SEM рассчитывали как STDEV/SQRT; и разность значения P между группами рассчитывали как TTEST.
Объем опухоли (V) рассчитывали как: V = 1/2 × Lдлина × Lширина2
Относительный объем (RTV) = VT/V0
Степень ингибирования опухоли (%) = (CRTV - TRTV)/CRTV (%)
Где V0 и VT представляют собой объемы опухоли в начале и в конце эксперимента, соответственно. CRTV и TRTV представляют собой относительные объемы опухоли холостой контрольной группы (носитель) и экспериментальной групп, соответственно, в конце эксперимента.
IV. Результаты
Результаты эксперимента показаны на фиг. 7. Препараты вводили путем внутрибрюшинной инъекции один раз в неделю, и в общей сложности осуществляли 3 инъекции. На 18-й день наблюдения степени ингибирования опухоли тестируемых ADC ADC-30 1 мг/кг, ADC-30 3 мг/кг, ADC-31 1 мг/кг и ADC-31 3 мг/кг достигли 0,31%, 45,23% (P < 0,0001), 39,22% (P < 0,01) и 80,24% (P < 0,0001), соответственно. В условиях той же дозировки эффект ингибирования опухоли ADC-31 был значительно лучше, чем у ADC-30.
II. Составы
Оборудование, используемое при получении и определении состава, и метод расчета результатов показаны ниже.
SEC молекулярная эксклюзионная хроматография:
Это метод анализа разделения растворенного вещества по относительной зависимости между размером пор геля и размером клубка молекул образца полимера.
SEC% (SEC процент содержания мономера) = A мономера/A общая × 100% (A мономер представляет собой площадь пика основного пика мономера в образце, а A общая представляет собой сумму площадей всех пиков).
Прибор для SEC измерения: Agilent 1260; колонка: вода, XBrige BEH200Å SEC (300 × 7,8 мм 3,5 мкм).
Капиллярный гель-электрофорез CE:
Это способ перемещения геля в капилляр в качестве поддерживающей среды для электрофореза и разделения в соответствии с молекулярной массой образца при определенном напряжении.
Сниженный процент чистоты CE = A основной пик/A общая × 100% (A основной пик представляет собой площадь основного пика легкой цепи + площадь основного пика тяжелой цепи, а A общая представляет собой сумму всех площадей пика).
Прибор для измерения CE: модель Beckman plus800
Измерение мутности:
Степень, в которой свет не проходит через слой воды, указывает на способность слоя воды рассеивать и поглощать свет. Она связана не только с содержанием взвешенных веществ, но и с составом, размером и формой частиц, а также с характеристиками отражения их поверхности. При сравнении значений поглощения одного и того же образца белка при той же концентрации на той же длине волны (в пределах ближних ультрафиолетовых и видимых областей длины волны), чем больше значение поглощения, тем больше мутность, тем больше молекул белка в образце склонны к агрегации. Прибор для измерения представлял собой многофункциональный считыватель микропланшетов (Molecular Devices M5). Образцы в том же объеме добавляли в 96-луночный планшет и считывали значения оптической плотности.
Измерение осмотического давления:
Для измерения осмотического давления использовали метод точки замерзания. Точку замерзания раствора измеряли с помощью высокочувствительного термочувствительного элемента на основе пропорционального соотношения между величиной понижения температуры замерзания и молярной концентрацией раствора, а затем преобразовывали в осмотическое давление через электрическое количество. Производитель приборов: Loser, модель: OM815.
Определение концентрации белка:
Поскольку токсин в конъюгате антитела и лекарственного средства поглощал свет при 280 нм, концентрацию белка корректировали с использованием следующих формул:
A280 = Cd × ε280d + Cmab × ε280mab
A370 = Cd × ε370d,
где Cd представляет собой концентрацию токсина, Cmab представляет собой концентрацию белка, ε280d представляет собой коэффициент экстинкции токсина при 280 нм, ε280mab представляет собой коэффициент экстинкции белка при 280 нм, ε370d представляет собой коэффициент экстинкции токсина при 370 нм, ε280mab = 1,49 мг-1*см-1*мл, ε280d = 5000 (молярный коэффициент экстинкции токсина при 280 нм)/1074,13 (молекулярная масса токсина) = 4,65 мг-1*см-1*мл, и ε370d = 19000 (молярный коэффициент экстинкции токсина при 370 нм)/1074,13 (молекулярная масса токсина) = 17,69 мг-1*см-1*мл; указанные коэффициенты экстинкции представляют собой коэффициенты экстинкции массы.
Прибор для измерения концентрации белка: спектрофотометр, видимый в ультрафиолетовом диапазоне; модель: Nano Drop oneC; длина оптического пути: 1 мм.
В качестве примера, молекулярная масса голого антитела составляет 145,181 кДа, а молекулярная масса токсина составляет 1074 Да; если среднее значение DAR составляет 5,7, расчетная молекулярная масса ADC составит 151,317 кДа, что в 1,042 раза больше, чем у голого антитела. В качестве примера, если значение ADC DAR составляет 5,7, а концентрация белка открытого антитела составляет 20,00 мг/мл, концентрация ADC будет составлять 20,84 мг/мл.
Пример 2-1 Скрининг буферных систем и значений pH для препаратов
20 мг/мл (концентрация белка на основе голого антитела) препарата на основе ADC-33 (не содержащие сахарида, поверхностно-активного вещества и других буферов, за исключением следующих буферных систем) были получены с использованием следующих буферных систем:
1) 10 мМ гистидина-ацетата натрия, pH 5,0
2) 10 мМ гистидина-ацетата натрия, pH 5,5
3) 10 мМ гистидина-ацетата натрия, pH 6,0
4) 10 мМ гистидина-ацетата натрия, pH 6,5
5) 10 мМ гистидина-гидрохлорида гистидина, pH 5,5
6) 10 мМ янтарной кислоты-сукцината натрия, pH 5,5
7) 10 мМ лимонной кислоты-цитрата натрия, pH 5,5
Образцы были взяты для высокотемпературного исследования стабильности (40 °C) и исследования встряхиванием (25 °C, 300 об/мин). Встряхиваемый образец содержал 0,1 мг/мл полисорбата 80 (PS80), а остальные образцы содержали 0,4 мг/мл PS80. Каждый препарат фильтровали, и контейнер заполняли им, закупоривали и закрывали крышкой. Образцы подвергали экспериментам принудительной деградации, описанным выше, и исследовали на внешний вид, SEC и мутность.
Результаты представлены в таблице 6. Результаты показывают, что там, где в качестве буфера использовали гистидин-ацетат натрия, внешний вид образцов постепенно ухудшался, мутность увеличивалась, а пропорция мономера SEC уменьшалась, поскольку значение рН увеличивалось в пределах рН 5,0-6,5. При рН 5,5 образец, содержащий 10 мМ гистидин-ацетата натрия, и образец, содержащий 10 мМ гистидина-гидрохлорид, были схожими по внешнему виду и SEC% и превосходили образцы, содержащие 10 мМ янтарной кислоты-сукцината натрия или 10 мМ лимонной кислоты-цитрата натрия. Неожиданно, препарат, содержащий 10 мМ янтарной кислоты-сукцината натрия, значительно превосходил по мутности другие препараты после исследования встряхиванием.
Примечание: в таблице «D» обозначает день; например, D12 обозначает 12 день, а D0 обозначает начало эксперимента; то же самое применимо ниже.
Пример 2-2. Скрининг видов поверхностно-активных веществ
Были получены 20 мг/мл препаратов (концентрация белка на основе голого антитела) ADC-32, содержащих различные поверхностно-активные вещества полисорбата, и каждый из которых содержал буфер 10 мМ янтарной кислоты-сукцината натрия (pH 5,0) и 80 мг/мл сахарозы. Каждый препарат фильтровали, и контейнер заполняли им, закупоривали и закрывали крышкой. Образцы подвергали исследованию стабильности при высокой температуре (40 °C) и исследованию встряхиванием (25 °C, 300 об/мин) и исследованию внешнего вида, SEC и восстановленную CE.
Результаты приведены в таблице 7. Между препаратами, содержащими PS20 (полисорбат 20), и препаратами, содержащими PS80, в различных условиях не было значительных различий во внешнем виде, SEC и восстановленном CE, что указывает на то, что как PS20, так и PS80 были способны эффективно стабилизировать ADC-32.
0-0,2 мг/мл
0-0,2 мг/мл
Примеры 2-3. Скрининг концентрации поверхностно-активных веществ
Получали образцы, содержащие 0, 0,1, 0,2, 0,4 и 0,6 мг/мл PS80. Каждый образец также содержал 10 мМ янтарной кислоты-сукцината натрия (pH 5,0), 80 мг/мл сахарозы и 20 мг/мл (концентрация белка на основе голого антитела) ADC-32. Образцы фильтровали для удаления бактерий и частиц и разбавляли 0,9% NaCl таким образом, что концентрация ADC-32 составляла 0,2 мг/мл. Разведения исследовали на внешний вид и наличие нерастворимых микрочастиц.
Результаты приведены в таблице 8. Если концентрация PS80 в препарате составляла 0-0,2 мг/мл, по мере увеличения концентрации увеличение количества нерастворимых микрочастиц в разведении становилось меньше, а видимых частиц было все меньше и меньше. Там, где концентрация составляла 0,2-0,6 мг/мл, не было различий в нерастворимых микрочастицах между разведениями и внешний вид был хорошим.
Примеры 2-4. Скрининг видов сахара
Были получены различные образцы, содержащие 80 мг/мл сахарозы, трегалозы и маннита. Каждый образец также содержал 10 мМ янтарной кислоты-сукцината натрия (pH 5,0), 80 мг/мл сахарозы и 20 мг/мл (концентрация белка на основе голого антитела) ADC-32. Образцы фильтровали для удаления бактерий и частиц. Каждый препарат фильтровали, и контейнер заполняли им, закупоривали и закрывали крышкой. Образцы подвергали исследованию стабильности при высокой температуре (40 °C) и исследованию цикла замораживания/оттаивания при -35 °C/4 °C и исследованию внешнего вида, SEC и восстановленную CE.
Результаты приведены в таблице 9. В условиях замораживания/размораживания сахароза превосходила трегалозу и маннит по внешнему виду. Сахароза и трегалоза превосходили маннит по результатам SEC. При 40 °C сахароза слегка превосходила трегалозу по снижению результатов CE.
оттаивания
оттаивания
оттаивания
Примеры 2-5. Скрининг рН, концентраций конъюгата антитела и лекарственного средства и полисорбатов
Содержит 10 мМ янтарной кислоты-сукцината натрия в качестве буфера и 80 мг/мл сахарозы в качестве стабилизатора, формулы были разработаны в отношении рН, концентрации белка ADC-32 (на основе голого антитела) и концентрации полисорбата, как показано в таблице 10. Было проведено исследование стабильности при высокой температуре (40 °C), исследование при освещении (4 °C, 4500 лк) и исследование цикла замораживания/оттаивания при -35 °C/4 °C, и результаты были статистически проанализированы с использованием метода наименьших квадратов с SEC и сниженным CE в качестве оценочных индексов.
Результаты приведены в таблице 11 и фиг. 8. Показано, что препараты по настоящему изобретению сильно зависят от рН. В условиях освещения, встряхивания и 40 °C, при увеличении рН, наблюдалось большее снижение SEC; аналогичным образом, в условиях освещения, при увеличении рН, уменьшенное CE увеличивалось, и наблюдалось большее снижение пика мономера, что предполагает, что препараты по настоящему изобретению должны иметь низкие значения рН, чтобы иметь улучшенную стабильность. В условиях встряхивания при увеличении концентрации PS80 наблюдалась понижательная тенденция к SEC. Различий в воздействии PS80 на SEC и снижении CE при других условиях не наблюдалось. По мере увеличения концентрации белка ADC-32 наблюдалось большее снижение пика мономера SEC при 40 °C. Различий в воздействии концентрации белка ADC-32 на SEC и снижение CE в других условиях не наблюдалось.
(10-4 г/мл)
Примеры 2-6. Разработка процесса лиофилизации
Получали 20 мг/мл (концентрация белка на основе голого антитела) исходного раствора ADC-33, содержащего 10 мМ янтарной кислоты-сукцината натрия с pH 5,0, 80 мг/мл сахарозы и 0,2 мг/мл PS80, фильтровали, разливали по бутылкам и затем лиофилизировали в соответствии с параметрами процесса лиофилизации 1 (см. таблицу 12). Образец для лиофилизации был в виде белого порошка с плоской поверхностью и содержал 1,05% воды. Нижняя часть порошка была слегка вдавлена в центр. Образец восстанавливали водой для инъекций и измеряли значение рН. Результаты представлены в таблице 13. Ионнная сила (10 мМ или более) была достаточно высокой для буферизации. Формула включала 80 мг/мл сахарозы, которая может соответствовать изоосмотическому требованию.
Получали 20 мг/мл (концентрация белка на основе голого антитела) исходного раствора ADC-33, содержащего 10 мМ янтарной кислоты-сукцината натрия с pH 5,0, 80 мг/мл сахарозы и 0,2 мг/мл PS80, фильтровали, разливали по бутылкам и затем лиофилизировали в соответствии с параметрами процесса лиофилизации 2 (см. таблицу 14). Поверхность порошковой таблетки была плоской и не имела морщин и углублений. Содержание воды составило 0,89%. Процесс лиофилизации может соответствовать требованиям к качеству продукта.
Получали 20 мг/мл (концентрация белка на основе голого антитела) исходного раствора ADC-32, содержащего 10 мМ янтарной кислоты-сукцината натрия с pH 5,0, 80 мг/мл сахарозы и 0,2 мг/мл PS80, фильтровали, разливали по бутылкам и затем лиофилизировали в соответствии с параметрами процесса лиофилизации 3 (см. таблицу 15). Поверхность порошковой таблетки была плоской и не имела углублений. Нижний край порошковой таблетки слегка сжался. Содержание воды составило 1,17%. Процесс лиофилизации по существу может соответствовать требованиям к качеству продукта.
Примеры 2-7. Данные испытаний на долгосрочную стабильность
Получали, фильтровали и разливали 20 мг/мл (концентрация белка на основе голого антитела) исходного раствора ADC-32, содержащего 10 мМ янтарной кислоты-сукцината натрия с pH 5,0, 80 мг/мл сахарозы и 0,2 мг/мл PS80, а затем готовые серии исходного раствора составляли в соответствии с параметрами 2 процесса лиофилизации, подвергали долгосрочному хранению при 2-8 °C, восстанавливали и затем испытывали. Результаты представлены в таблице 16. По сравнению с продуктами на D0, те в условиях 2-8 °C M3 (месяц 3) показали SEC, пониженные уровни CE и свободного токсина в пределах приемлемых диапазонов (SEC% - 93% или более; пониженный CE% - 95% или более; уровень свободного токсина 330 млн-1 или менее). SEC слегка снизилась на 0,5%, в то время как пониженный уровень CE и свободного токсина остался неизменным.
Примечание: в таблице "M" означает месяц; например, "M3" означает 3 месяца.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> ЦЗЯНСУ ХЭНЖУЙ ФАРМАСЬЮТИКАЛС КО., ЛТД.
ШАНХАЙ ХЭНЖУЙ ФАРМАСЬЮТИКАЛ КО., ЛТД.
<120> ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ КОНЪЮГАТ
АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА, И ЕЕ ПРИМЕНЕНИЕ
<130> 721021CPCT
<150> CN202010219601.7
<151> 2020-03-25
<150> CN202110287012.7
<151> 2021-03-17
<160> 6
<170> SIPOSequenceListing 1.0
<210> 1
<211> 214
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> ЦЕПЬ
<223> Легкая цепь трастузумаба
<400> 1
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Asp Val Asn Thr Ala
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Phe Leu Tyr Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Arg Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln His Tyr Thr Thr Pro Pro
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 2
<211> 450
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> ЦЕПЬ
<223> Тяжелая цепь трастузумаба
<400> 2
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Asn Ile Lys Asp Thr
20 25 30
Tyr Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Arg Ile Tyr Pro Thr Asn Gly Tyr Thr Arg Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn Thr Ala Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ser Arg Trp Gly Gly Asp Gly Phe Tyr Ala Met Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile
245 250 255
Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly Lys
450
<210> 3
<211> 214
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> ЦЕПЬ
<223> Легкая цепь пертузумаба
<400> 3
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Ile Gly
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Tyr Tyr Ile Tyr Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 4
<211> 449
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> ЦЕПЬ
<223> Тяжелая цепь пертузумаба
<400> 4
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Arg Phe Thr Leu Ser Val Asp Arg Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
Lys
<210> 5
<211> 215
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> ЦЕПЬ
<223> Легкая цепь 1F9DS антитела B7H3
<400> 5
Asp Thr Val Val Thr Gln Glu Pro Ser Phe Ser Val Ser Pro Gly Gly
1 5 10 15
Thr Val Thr Leu Thr Cys Gly Leu Ser Ser Gly Ser Val Ser Thr Ser
20 25 30
His Tyr Pro Ser Trp Tyr Gln Gln Thr Pro Gly Gln Ala Pro Arg Met
35 40 45
Leu Ile Tyr Asn Thr Asn Thr Arg Ser Ser Gly Val Pro Asp Arg Phe
50 55 60
Ser Gly Ser Ile Leu Gly Asn Lys Ala Ala Leu Thr Ile Thr Gly Ala
65 70 75 80
Gln Ala Asp Asp Glu Ser Asp Tyr Tyr Cys Ala Ile His Val Asp Arg
85 90 95
Asp Ile Trp Val Phe Gly Gly Gly Thr Lys Leu Thr Val Leu Gly Gln
100 105 110
Pro Lys Ala Asn Pro Thr Val Thr Leu Phe Pro Pro Ser Ser Glu Glu
115 120 125
Leu Gln Ala Asn Lys Ala Thr Leu Val Cys Leu Ile Ser Asp Phe Tyr
130 135 140
Pro Gly Ala Val Thr Val Ala Trp Lys Ala Asp Gly Ser Pro Val Lys
145 150 155 160
Ala Gly Val Glu Thr Thr Lys Pro Ser Lys Gln Ser Asn Asn Lys Tyr
165 170 175
Ala Ala Ser Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His
180 185 190
Arg Ser Tyr Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys
195 200 205
Thr Val Ala Pro Thr Glu Cys
210 215
<210> 6
<211> 449
<212> ПРТ
<213> Искусственная последовательность
<220>
<221> ЦЕПЬ
<223> Тяжелая цепь 1F9DS антитела B7H3
<400> 6
Gln Val Gln Leu Val Gln Ser Gly Gly Gly Val Val Gln Pro Gly Thr
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Ile Phe Ser Ser Ser
20 25 30
Ala Met His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Val Ile Ser Tyr Asp Gly Ser Asn Lys Tyr Tyr Val Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Ser Ala Arg Leu Tyr Ala Ser Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Ala Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Asp Glu Leu Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
Lys
<---
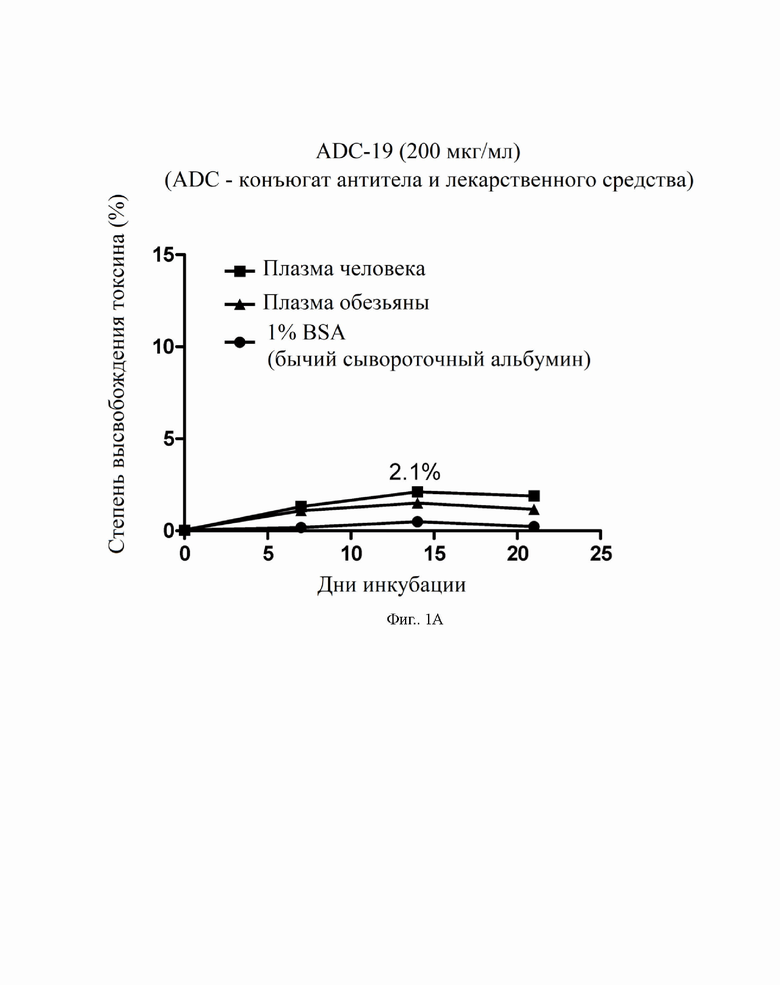
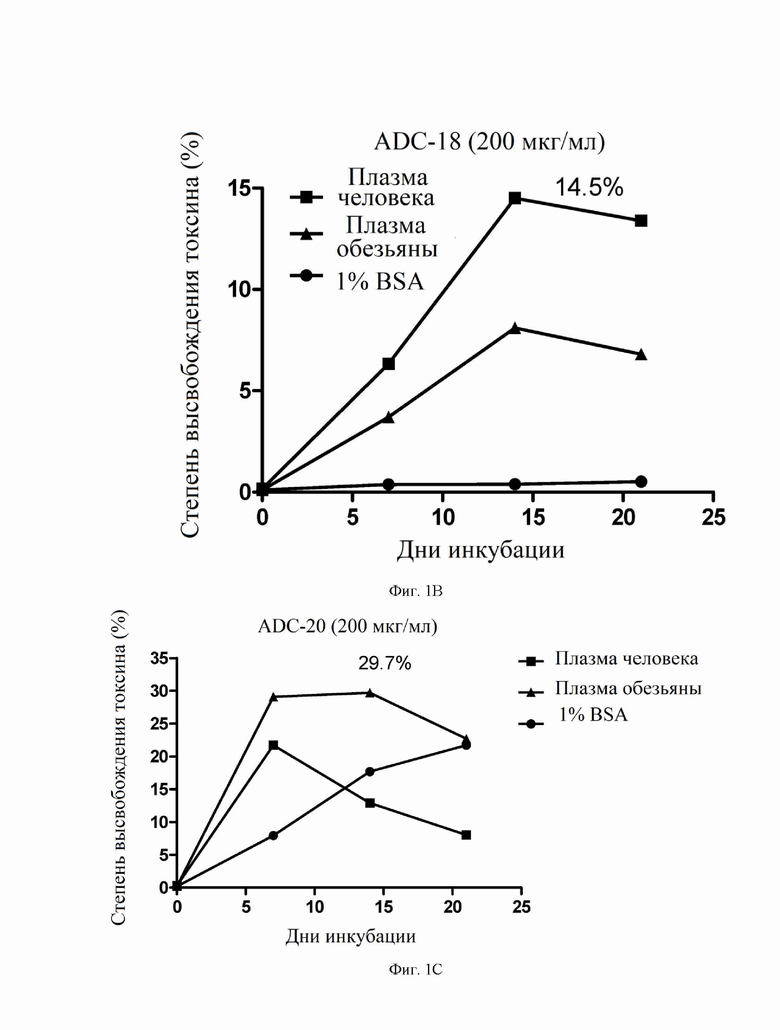
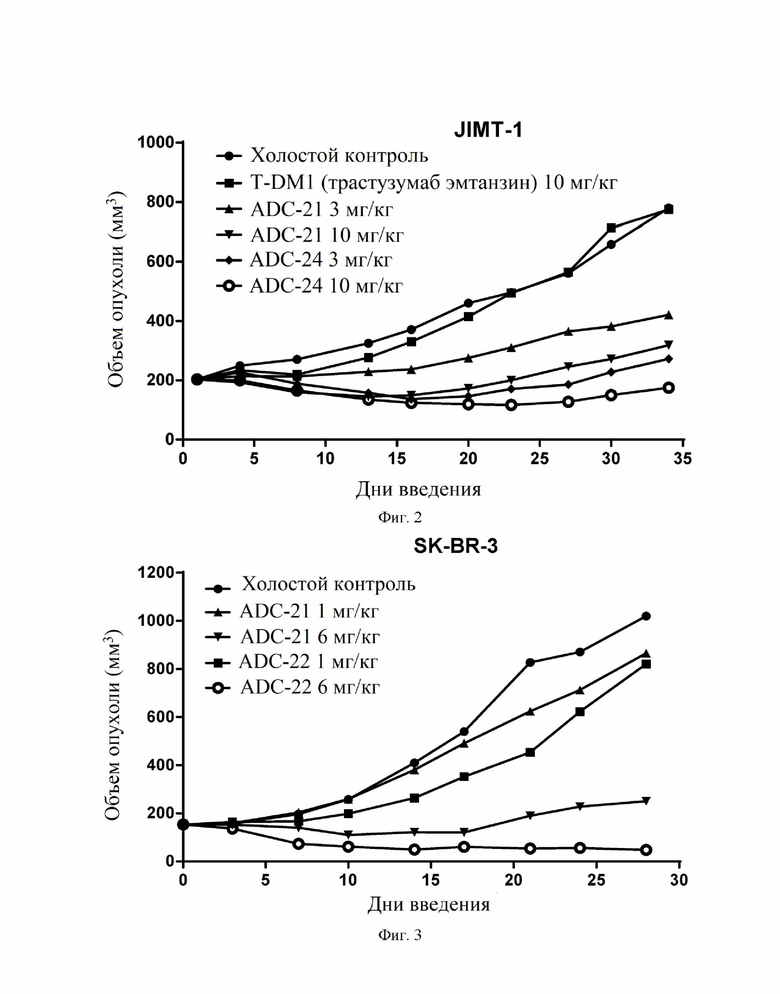
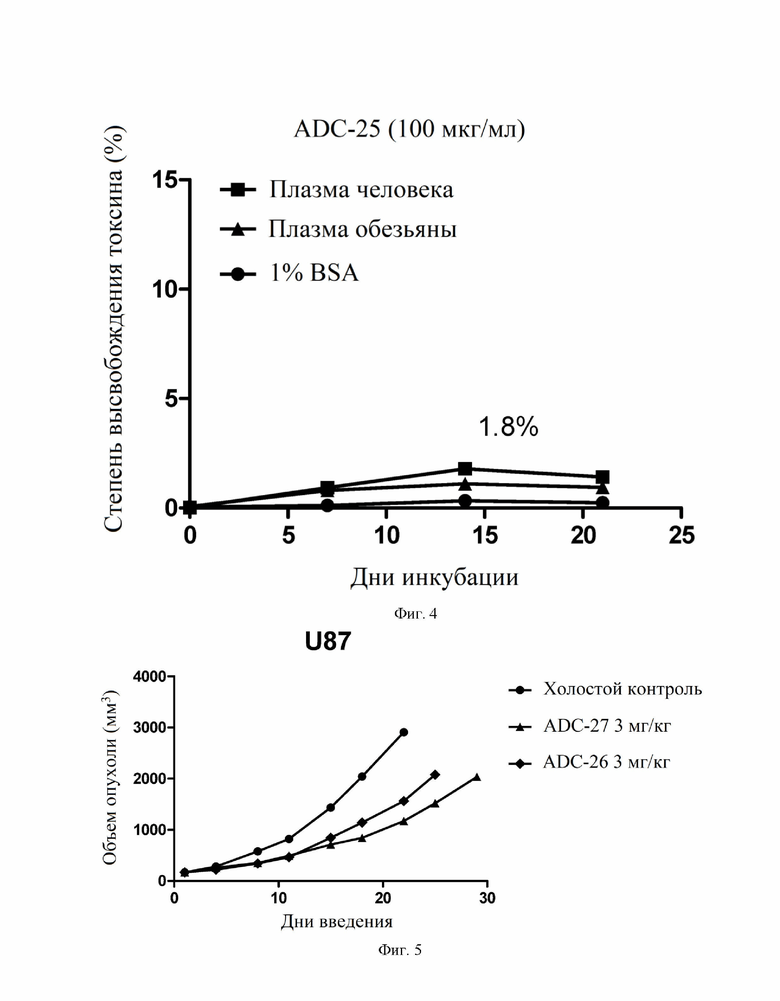
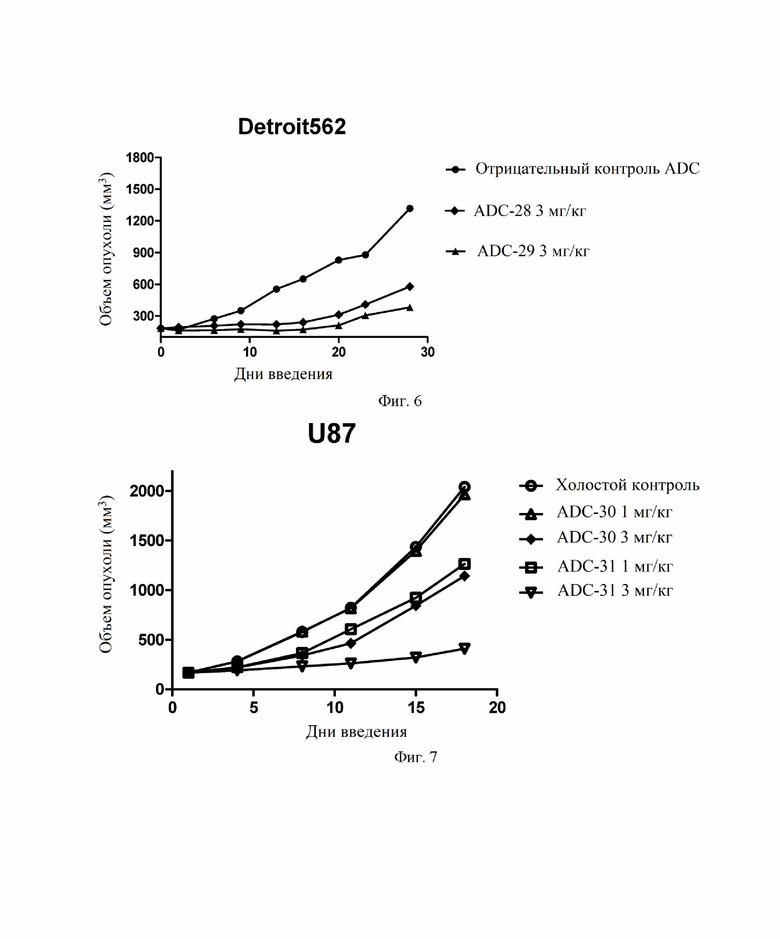
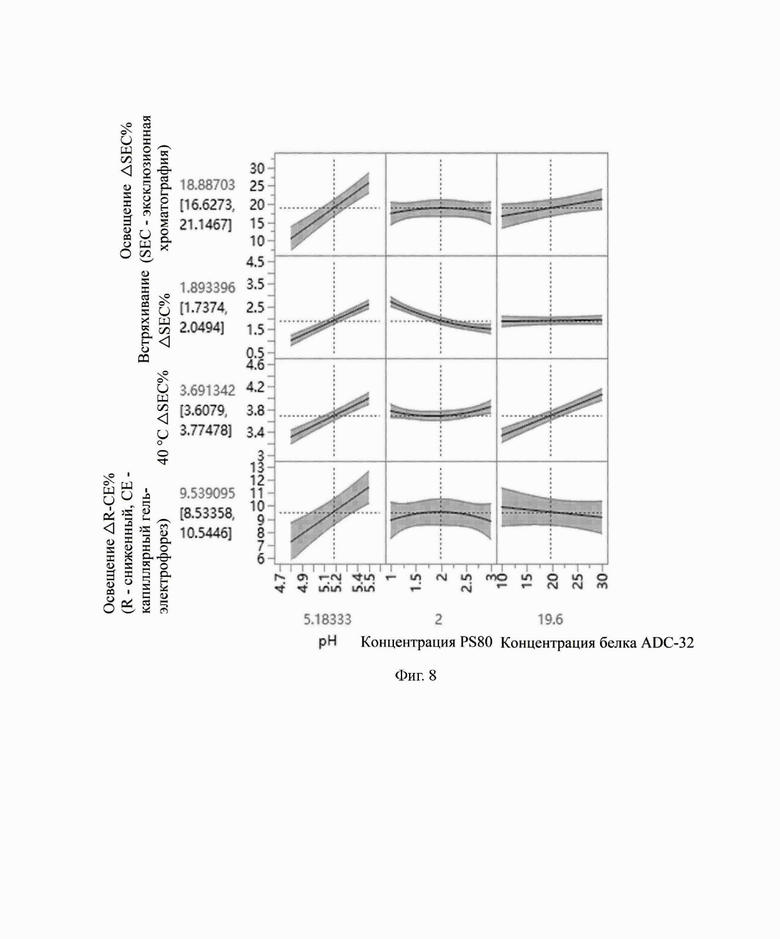
Группа изобретений относится к области фармацевтических препаратов, содержащих конъюгат антитела и лекарственного средства, и их применению в качестве противоракового лекарственного препарата. Предложены фармацевтические композиции для лечения рака, связанного с экспрессией HER2, содержащие от 10 мг/мл до 30 мг/мл конъюгата антитела и лекарственного средства следующей структуры: 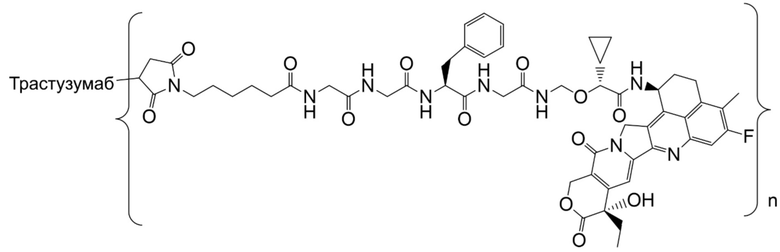 ,
,
где n представляет собой десятичное или целое число от 1 до 10, полисорбат 80 в концентрации от 0,05 мг/мл до 0,5 мг/мл, сахарозу в концентрации от 60 мг/мл до 90 мг/мл и буфер янтарная кислота-сукцинат натрия в концентрации от 5 мМ до 20 мМ. Фармацевтические композиции имеют рН от 4,5 до 5,2. Также предложены способ получения лиофилизированного препарата и способ лечения рака, связанного с экспрессией HER2. Изобретения обеспечивают получение стабильного препарата конъюгата, в котором не наблюдается значительных изменений при хранении при температурах 2-8°C и 25°C в течение по меньшей мере 3 месяцев. 4 н. и 8 з.п. ф-лы, 8 ил., 16 табл., 69 пр.
1. Фармацевтическая композиция для лечения рака, связанного с экспрессией HER2, содержащая конъюгат антитела и лекарственного средства следующей структуры:
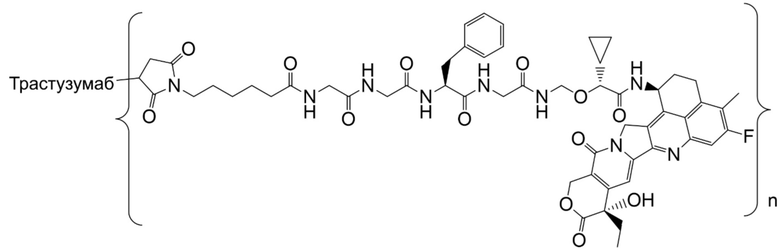
где n представляет собой десятичное или целое число от 1 до 10;
причем фармацевтическая композиция содержит:
(a) конъюгат антитела и лекарственного средства в концентрации от 10 мг/мл до 30 мг/мл, (b) полисорбат 80 в концентрации от 0,05 мг/мл до 0,5 мг/мл, (c) сахарозу в концентрации от 60 мг/мл до 90 мг/мл и (d) буфер янтарная кислота-сукцинат натрия в концентрации от 5 мМ до 20 мМ;
фармацевтическая композиция имеет рН от 4,5 до 5,2.
2. Фармацевтическая композиция по п. 1, которая имеет рН от 4,8 до 5,2.
3. Фармацевтическая композиция по п. 1 или 2, которая имеет рН от 5,0 до 5,1.
4. Фармацевтическая композиция по любому из пп. 1-3, где полисорбат 80 присутствует в концентрации 0,2 мг/мл.
5. Фармацевтическая композиция по любому из пп. 1-4, где сахароза присутствует в концентрации 80 мг/мл.
6. Фармацевтическая композиция по любому из пп. 1-5, где конъюгат антитела и лекарственного средства присутствует в концентрации от 20 мг/мл до 22 мг/мл.
7. Фармацевтическая композиция по любому из пп. 1-6, где буфер янтарная кислота-сукцинат натрия присутствует в концентрации 10 мМ.
8. Фармацевтическая композиция по любому из пп. 1-7, содержащая следующие компоненты:
(a) конъюгат антитела и лекарственного средства в концентрации от 20 мг/мл до 22 мг/мл, (b) полисорбат 80 в концентрации 0,2 мг/мл, (c) сахароза в концентрации 80 мг/мл и (d) буфер янтарная кислота-сукцинат натрия в концентрации 10 мМ; причем композиция имеет рН 5,0-5,1.
9. Фармацевтическая композиция по любому из пп. 1-8, где конъюгат антитела имеет следующую структуру:
где n представляет собой десятичное или целое число от 3 до 8.
10. Фармацевтическая композиция для лечения рака, связанного с экспрессией HER2, содержащая конъюгат антитела и лекарственного средства, имеющий следующую структуру:
где n представляет собой десятичное или целое число от 3 до 8;
фармацевтическая композиция содержит (a) конъюгат антитела и лекарственного средства в концентрации от 20 мг/мл до 22 мг/мл, (b) полисорбат 80 в концентрации 0,2 мг/мл, (c) сахарозу в концентрации 80 мг/мл и (d) буфер янтарная кислота-сукцинат натрия в концентрации 10 мМ; причем фармацевтическая композиция имеет рН 5,0-5,1.
11. Способ получения лиофилизированного препарата, содержащего конъюгат антитела и лекарственного средства, включающий стадию лиофилизации фармацевтической композиции по любому из пп. 1-10.
12. Способ лечения рака, связанного с экспрессией HER2, включающий введение пациенту эффективного количества фармацевтической композиции по любому из пп. 1-10, где рак выбран из группы, состоящей из рака молочной железы, рака яичника, рака шейки матки, рака матки, рака предстательной железы, рака почки, рака мочевыводящих путей, рака мочевого пузыря, рака печени, рака желудка, рака эндометрия, карциномы слюнных желез, рака пищевода, меланомы, нейроглиомы, нейробластомы, саркомы, рака легкого, рака толстой кишки, рака прямой кишки, колоректального рака, лейкоза, рака костей, рака кожи, рака щитовидной железы, рака поджелудочной железы и лимфомы.
| EP 3130608 B1, 04.09.2019 | |||
| CA 3073383 A1, 28.02.2019 | |||
| EP 3101032 B1, 07.12.2016 | |||
| EP 2968588 B1, 20.01.2016 | |||
| EP 3342785 B1, 04.07.2018 | |||
| YUSUKE OGITANI ET AL | |||
| Электрический контакт | 1927 |
|
SU8201A1 |

