ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает преимущество предварительной заявки на патент США № 62/513211, поданной 31 мая 2017 г., и предварительной заявки на патент США № 62/581919, поданной 6 ноября 2017 г.; каждая из которых включена посредством ссылки в данный документ во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, которые обладают антипаразитарной активностью в отношении простейших класса кинетопластид.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Лейшманиоз представляет собой заболевание, вызываемое паразитами, представляющими собой простейшие, которые относятся к роду Leishmania, и передаваемое при укусах некоторых видов москитов. Существует четыре основных формы лейшманиоза. Кожный лейшманиоз является наиболее распространенной формой лейшманиоза. Висцеральный лейшманиоз - наиболее опасная форма, при которой паразиты мигрируют в жизненно важные органы, вызывается паразитом Leishmania donovani и является потенциально смертельным при отсутствии лечения. Он поражает до 12 миллионов людей по всему миру, при этом каждый год регистрируют 1,5-2 миллиона новых случаев. Частота возникновения висцеральной формы лейшманиоза по оценкам составляет 500000 новых случаев, и она является причиной 60000 смертей ежегодно.
Двумя основными средствами терапии висцерального лейшманиоза являются производные сурьмы стибоглюконат натрия (Pentostam®) и меглюмина антимонат (Glucantim®). Стибоглюконат натрия применяют в течение приблизительно 70 лет, и все более значимой проблемой является устойчивость к данному лекарственному средству. Кроме того, лечение является относительно длительным и болезненным и может обуславливать нежелательные побочные эффекты. На данный момент предпочтительным средством лечения является амфотерицин (AmBisome®). Другими альтернативными средством лечения являются милтефосин (Impavido®) и паромомицин.
Амфотерицин (AmBisome) является дорогостоящим и предусматривает внутривенное введение. Паромомицин предусматривает внутримышечные инъекции в течение 3 недель; большую проблему представляет собой соблюдение режима лечения. Милтефосин является лекарственным средством, предназначенным для перорального применения, и было показано, что он является более эффективным и лучше переносимым, чем другие лекарственные средства. Однако существуют проблемы, связанные с применением милтефосина, которые обуславливаются его тератогенностью и фармакокинетикой. Было показано, что милтефосин намного более медленно выводится из организма, и его все еще можно было обнаружить через пять месяцев после окончания лечения. Присутствие субтерапевтических концентраций милтефосина в крови через более чем пять месяцев после лечения может способствовать отбору устойчивых паразитов; более того, необходимо пересматривать меры в отношении предотвращения тератогенных рисков милтефосина. В настоящее время нет вакцин, которые бы применяли в обычном порядке.
Болезнь Шагаса, также называемая американским трипаносомозом, представляет собой тропическое паразитарное заболевание, вызываемое простейшим класса жгутиковые Trypanosoma cruzi. T. cruzi обычно передается людям и другим млекопитающим кровососущими "поцелуйными клопами" из подсемейства Triatominae (семейство Reduviida). Ежегодно предположительно от 10 до 15 миллионов людей по всему миру инфицируются болезнью Шагаса, большинство из которых не знают, что они инфицированы. Ежегодно 14000 людей умирают вследствие заболевания. В Центральной и Южной Америке от болезни Шагаса умирает больше людей, чем от любого другого заболевания, вызванного паразитами, в том числе от малярии. По оценкам CDC в Соединенных Штатах Америки живет более 300000 человек, инфицированных Trypanosoma cruzi.
Симптомы болезни Шагаса изменяются в ходе протекания инфекции. На ранней, острой стадии симптомы являются слабовыраженными и обычно вызывают не более чем местное набухание в месте инфекции. Начальная острая фаза является восприимчивой к антипаразитарным средствам лечения с частотой выздоровления 60-90%. Через 4-8 недель у индивидуумов с активной инфекцией болезнь Шагаса переходит в хроническую фазу, которая является бессимптомной для 60-80% индивидуумов с хронической инфекцией в течение их жизни. Однако у оставшихся 20-40% инфицированных людей будут развиваться тяжело протекающие и иногда угрожающие жизни медицинские проблемы на протяжении их жизни.
Лечение болезни Шагаса сосредоточено на уничтожении паразита и на сдерживании признаков и симптомов. Доступными в настоящее время лекарственными средствами, которые применяют для лечения во время острой фазы болезни Шагаса, являются бензнидазол и нифуртимокс. После того, как болезнь Шагаса переходит в хроническую фазу, лекарственные препараты перестают быть эффективными для излечения заболевания. Вместо этого лечение зависит от конкретных признаков и симптомов. Однако проблемы таких видов терапии, применяемых в настоящее время, включают их различные побочные эффекты, длительность лечения и необходимость в медицинском наблюдении во время лечения. Уже существует устойчивость к двум лекарственным средствам первой линии. Противогрибковое средство амфотерицин b было предложено в качестве лекарственного средства второй линии, но оно является дорогостоящим и относительно токсичным. Вакцины против болезни Шагаса не существует.
Африканский трипаносомоз человека (HAT), также известный как африканская сонная болезнь, представляет собой трансмиссивное паразитарное заболевание, вызываемое простейшим Trypanosoma brucei. Существует два подвида, которые инфицируют людей: T.b. gambiense и T.b. rhodesiense, при этом первый является причиной более чем 95% известных случаев, и второй является причиной оставшихся известных случаев. Паразиты передаются людям через укусы мухи цеце (род Glossina), которая получила инфекцию от человека или от животных, являющихся переносчиками паразитов, патогенных для человека. Приблизительно 48000 людей умерло от сонной болезни в 2008 г. В результате усилий по предупреждению и ликвидации популяции мухи цеце общественные организации здравоохранения добились успеха в отношении контроля распространения заболевания; было зарегистрировано менее 10000 новых случаев в 2009 г. в соответствии с данными ВОЗ, что представляет собой значительное уменьшение по сравнению с предполагаемыми 300000 новых случаев в 1998 г.
Симптомы африканского трипаносомоза возникают в две стадии. На первой стадии, известной как гемолимфатическая фаза, трипаносомы размножаются в подкожной ткани, крови и лимфе. Гемолимфатическая фаза характеризуется приступами лихорадки, головных болей, болей в суставах и зуда. На второй стадии, неврологической фазе, паразиты проходят через гематоэнцефалический барьер и инфицируют центральную нервную систему. На данной стадии появляются более очевидные признаки и симптомы заболевания (например, изменения поведения, спутанность, сенсорные нарушения и нарушенная координация). Нарушение цикла сна, из-за которого заболевание получило свое название, является важной особенностью второй стадии заболевания. Без лечения заболевание является неизбежно смертельным, при этом прогрессирующее психическое нарушение приводит к коме, системной органной недостаточности и смерти.
Для лечения сонной болезни зарегистрированы четыре лекарственных средства. Протокол зависит от стадии заболевания. В настоящее время стандартное лечение заболевания на первой стадии предусматривает внутривенное или внутримышечное введение пентамидина (в случае T.b. gambiense), или внутривенное введение сурамина (в случае T.b. rhodesiense). В настоящее время стандартное лечение заболевания на второй стадии предусматривает внутривенное введение меларсопрола или внутривенное введение меларсопрола в комбинации с пероральным введением нифуртимокса, внутривенным введением отдельно эфлорнитина или эфлорнитина в комбинации с нифуртимоксом. Все лекарственные средства обладают нежелательными или потенциально опасными побочными эффектами. Например, у 3-10% пациентов, которым вводили посредством инъекции меларсопрол (Arsobal®), развивалась реактивная энцефалопатия (конвульсии, прогрессирующая кома или психотические реакции), и 10-70% таких случаев приводят к смерти.
Таким образом, все еще остается необходимость в новых и лучших средствах лечения и средствах терапии для лейшманиоза, болезни Шагаса и HAT.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
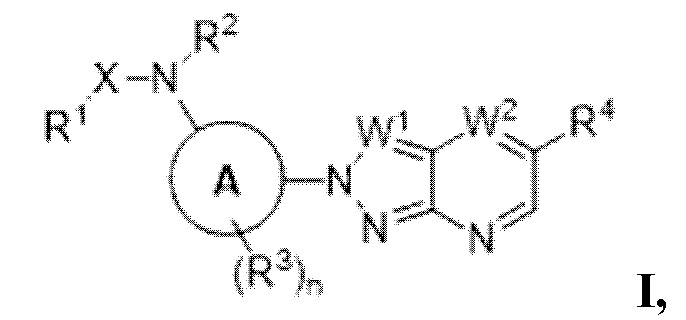
В определенных аспектах в данном документе представлено соединение формулы (I) или ее подформул:
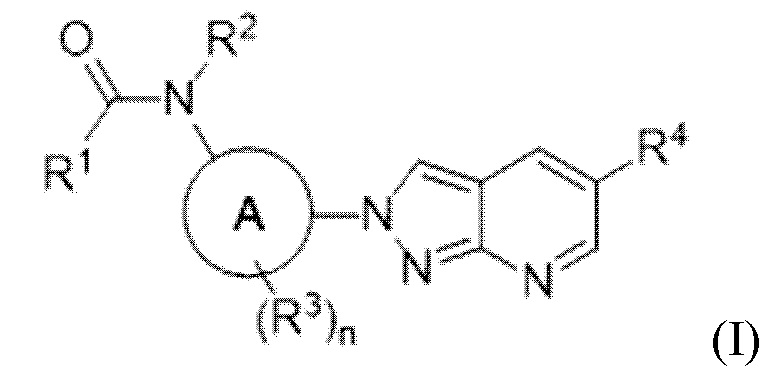 ,
,
или его фармацевтически приемлемая соль или стереоизомер; где
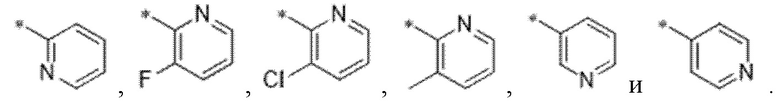
кольцо A представляет собой фенил или пиридинил;
R1 выбран из:
(a) C1-6алкила, который не замещен или замещен 1-3 заместителями, независимо выбранными из галогена и C3-6циклоалкила; и при этом указанный C3-6циклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила;
(b) C1-4алкокси, который не замещен или замещен C1-4галогеналкилом;
(c) -NR5aR5b, где R5a и R5b независимо представляют собой водород, C1-4алкил или C1-4галогеналкил; или R5a и R5b вместе с атомом азота, к которому они оба присоединены, образуют 4-7-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-7-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, C1-4алкила и C1-4алкокси; или два заместителя при одном и том же или различных атомах кольца 4-7-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо B, присоединенное к 4-7-членному гетероциклоалкилу;
где кольцо B представляет собой C3-6циклоалкил или 3-7-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца;
(d) моноциклического C3-6циклоалкила, C3-6циклоалкенила или спиропентила; каждый из которых не замещен или замещен 1-3 заместителями, независимо выбранными из галогена, циано, C1-4алкила, и C1-4галогеналкила, и C1-4алкокси;
(e) фенила или 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца; каждый из которых не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкоксиC1-4алкила, C1-4гидроксиалкила, C1-4алкокси и C3-6циклоалкила;
R2 и R7 независимо представляют собой водород или C1-4алкил;
R3 представляет собой водород или галоген, и n равняется 0 или 1; и
R4 выбран из
(a) водорода;
(b) галогена;
(c) C1-6галогеналкила или C1-6алкила, который не замещен или замещен C3-6циклоалкилом; и при этом указанный C3-6циклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила;
(d) -NR6aR6b, где R6a представляет собой водород или C1-4алкил; R6b представляет собой водород, C1-4алкоксикарбонил, C1-4галогеналкил, C3-6циклоалкил или C1-4алкил, который не замещен или замещен C1-4алкокси; или
R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-7-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-7-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси, оксо, 1,1-диоксо, -C(O)-OR7 или 4-6-членного гетероциклоалкила, содержащего 1-2-гетероатома, независимо выбранных из N, O и S; или два заместителя при одном и том же или различных атомах кольца 4-7-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-7-членному гетероциклоалкилу;
где кольцо C выбрано из C3-6циклоалкила и 3-7-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и при этом оно независимо не замещено или замещено 1-2 заместителями, независимо выбранными из галогена и оксо;
(e) C3-6циклоалкила;
(f) 4-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца; и при этом он не замещен или замещен -C(O)OR8,
-C(O)R8, где R8 представляет собой C1-4алкил, и арил-C1-4алкилом, который не замещен или замещен 1-2 заместителями, представляющими собой галоген; и
(g) 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца; который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4гидроксиалкила и C3-6циклоалкила.
В другом аспекте в настоящем изобретении представлена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или ее подформул или его фармацевтически приемлемой соли или стереоизомера и один или несколько фармацевтически приемлемых носителей.
В еще одном аспекте в настоящем изобретении представлена комбинация, в частности, фармацевтическая комбинация, содержащая терапевтически эффективное количество соединения формулы (I) или ее подформул или его фармацевтически приемлемой соли или стереоизомера и одно или несколько терапевтически активных средств.
Соединения формулы (I) или ее подформул в свободной форме или в форме фармацевтически приемлемой соли могут быть пригодными в качестве средства терапии для заболевания или состояния, при которых может наблюдаться благоприятный эффект в результате подавления роста и пролиферации паразитов класса кинетопластид. В одном аспекте в настоящем изобретении представлено соединение формулы (I) или ее подформул, его фармацевтически приемлемая соль или его стереоизомер для применения при лечении, предупреждении, ингибировании, уменьшении интенсивности или устранении патологии и/или совокупности симптомов заболевания, вызванного паразитом класса кинетопластид. В другом аспекте в настоящем изобретении представлено применение соединения, выбранного из формулы (I) или ее подформул, или его фармацевтически приемлемой соли или стереоизомера в изготовлении лекарственного препарата для лечения у субъекта заболевания, вызываемого паразитом класса кинетопластид. Паразиты класса кинетопластид включают без ограничения паразитов рода Leishmania, например, Leishmania donovani, Leishmania infantum, Leishmania braziliensis, Leishmania panamensis, Leishmania guayanensis, Leishmania amazonensis, Leishmania mexicana, Leishmania tropica, Leishmania major; или паразитов рода Trypanosoma, например, Trypanosoma cruzi и Trypanosoma brucei. Соответственно, соединения по настоящему изобретению могут быть пригодными в лечении показания, выбранного из: лейшманиоза, болезни Шагаса (также называемой американским трипаносомозом), более конкретно, болезни Шагаса, вызванной простейшими Trypanosoma cruzi; и африканского трипаносомоза человека, более конкретно, африканского трипаносомоза человека, вызванного простейшими Trypanosoma brucei.
Если не указано иное, термин "соединения по настоящему изобретению" означает соединения формулы I и ее подформул, соли соединения, гидраты или сольваты соединений, соли, а также все стереоизомеры (в том числе диастереоизомеры и энантиомеры), таутомеры и изотопно меченные соединения (в том числе замещенные дейтерием). Соединения по настоящему изобретению дополнительно предусматривают полиморфы соединений формулы (I) (или ее подформул) и их солей.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Используемые в данном документе термины в форме единственного числа и подобные термины, используемые в контексте настоящего изобретения (в частности, в контексте формулы изобретения), следует истолковывать как охватывающие как форму единственного числа, так и форму множественного числа, если в данном документе не указано иное или нет явного противоречия с контекстом.
Используемый в данном документе термин "C1-6алкил" означает углеводородный радикал с прямой или разветвленной цепью, состоящий только из атомов углерода и водорода, не содержащий ненасыщенных связей, содержащий от одного до шести атомов углерода, и который присоединен к остальной части молекулы посредством одинарной связи. Термин "C1-4алкил" следует истолковывать соответствующим образом. Используемый в данном документе термин "н-алкил" означает алкильный радикал с прямой цепью (неразветвленный), определенный в данном документе. Примеры C1-6алкила включают без ограничения метил, этил, н-пропил, изопропил, н-бутил, изобутил (-CH2CH(CH3)2), втор-бутил (-CH(CH3)CH2CH3), трет-бутил (-C(CH3)3), н-пентил, изопентил (-(CH2)2CH(CH3)2), неопентил (-CH2C(CH3)3), трет-пентил (-C(CH3)2CH2CH3), 2-пентанил (-CH(CH3)(CH2)2CH3), н-гексил и т. п.
Используемый в данном документе термин "C1-4алкиламино" означает радикал формулы -NH-Ra, где Ra представляет собой C1-4алкильный радикал, определенный выше.
Используемый в данном документе термин "ди-(C1-4алкил)амино" означает радикал формулы -N(Ra)-Ra, где каждый Ra представляет собой C1-4алкильный радикал, который может быть одинаковым или отличающимся, как определено выше.
Используемый в данном документе термин "C1-6алкокси" означает радикал формулы -ORa, где Ra представляет собой C1-6алкильный радикал, в целом определенный выше. Примеры C1-6алкокси включают без ограничения метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, пентокси и гексокси.
Используемый в данном документе термин "C1-4алкоксиC1-4алкил" означает радикал формулы -Ra-O-Ra, где каждый Ra независимо представляет собой C1-6алкильный радикал, определенный выше. Атом кислорода может быть связан с любым атомом углерода в любом алкильном радикале. Примеры C1-4алкоксиC1-4алкила включают без ограничения метоксиметил, метоксиэтил, этоксиэтил, 1-этоксипропил и 2-метоксибутил.
Используемый в данном документе термин "C1-6алкоксикарбонил" означает радикал формулы -C(=O)-O-Ra, где Ra представляет собой C1-6алкильный радикал, определенный выше.
"Арил", используемый в данном документе, означает 6-14-членную моноциклическую или полициклическую ароматическую сборку колец, где все атомы кольца представляют собой атомы углерода. Как правило, арил представляет собой 6-членную моноциклическую, 10-12-членную бициклическую или 14-членную конденсированную трициклическую ароматическую кольцевую систему. CXарил и CX-Yарил, используемые в данном документе, описывают арильную группу, где X и Y показывают количество атомов углерода в кольцевой системе. C6-14арилы включают без ограничения фенил, бифенил, нафтил, азуленил и антраценил.
"Кольцо с внутренним мостиком" или "мостиковые кольца", используемые в данном документе, означают полициклическую кольцевую систему, где атомы кольца, которые являются общими для двух колец, не являются непосредственно связанными друг с другом. Одно или несколько колец кольцевой системы могут включать C3-6циклоалкил или 4-6-членные гетероциклические кольца, содержащие гетероатомы, выбранные из N, O и S, в качестве атомов кольца. Неограничивающие примеры колец с внутренним мостиком включают адамантанил, азабицикло[3.2.1]окт-3-ен-3-ил,
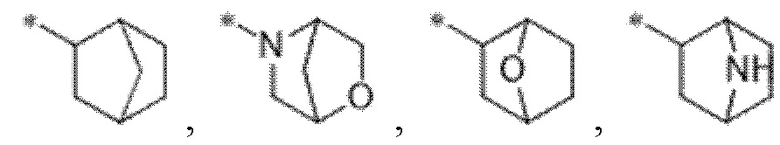 и т. п.
и т. п.
Используемый в данном документе термин "циано" означает радикал  .
.
Термин "циклоалкил" означает неароматическое карбоциклическое кольцо, которое представляет собой полностью гидрогенизированное кольцо, в том числе моно-, би- или полициклические, конденсированные, мостиковые или спирокольцевые системы. Примеры циклоалкильных групп включают без ограничения циклопропил, циклобутил, циклопентил, бицикло[1,1,1]пентанил, циклогексил, норборнил и кубанил.
Термин "конденсированное кольцо", используемый в данном документе, означает сборку нескольких колец, где кольца, составляющие сборку колец, связаны так, что атомы кольца, которые являются общими для двух колец, непосредственно связаны друг с другом. Сборки конденсированных колец могут быть насыщенными, частично насыщенными, ароматическими, карбоциклическими, гетероциклическими и т. п. Неограничивающие примеры обычных конденсированных колец включают декалин, нафталин, антрацен, фенантрен, индол, бензофуран, пурин, хинолин и т. п.
"Галоген" или "галогенид" означает бром, хлор, фтор или йод; предпочтительно фтор, хлор или бром.
Используемый в данном документе "C1-6галогеналкил" означает C1-6алкильный радикал, определенный выше, который замещен одним или несколькими представляющими собой атом галогена радикалами, определенными выше. Примеры C1-6галогеналкила включают без ограничения трифторметил, дифторметил, фторметил, трихлорметил, 2,2,2-трифторэтил, 1,3-дибромпропан-2-ил, 3-бром-2-фторпропил и 1,4,4-трифторбутан-2-ил.
Термин "гетероарил" означает ароматические фрагменты, содержащие по меньшей мере один гетероатом (например, кислород, серу, азот или их комбинации) в 5-10-членной ароматической кольцевой системе. Примеры гетероарила включают без ограничения пирролил, пиразолил, индолил, индазолил, тиенил, фуранил, бензофуранил, оксазолил, изоксазолил, имидазолил, триазолил, тетразолил, триазинил, пиридил, пиримидинил, пиразинил, тиазолил, пуринил, бензимидазолил, хинолинил, изохинолинил, хиноксалинил, бензопиранил, бензотиофенил, бензоимидазолил, бензоксазолил и 1H-бензо[d][1,2,3]триазолил. Гетероароматический фрагмент может состоять из одного кольца или конденсированной кольцевой системы. Типичное одиночное гетероарильное кольцо представляет собой 5-6-членное кольцо, содержащее от одного до четырех гетероатомов, независимо выбранных из N, O и S, и типичная конденсированная гетероарильная кольцевая система представляет собой 9-10-членную кольцевую систему, содержащую от одного до четырех гетероатомов, независимо выбранных из N, O и S. Конденсированная гетероарильная кольцевая система может состоять из двух гетероарильных колец, конденсированных вместе, или гетероарила, конденсированного с арилом (например, с фенилом).
Используемый в данном документе термин "гетероатомы" означает атомы азота (N), кислорода (O) или серы (S). Если не указано иное, предполагается, что любой гетероатом с незаполненными валентностями имеет атомы водорода в достаточном количестве для заполнения валентностей, и если гетероатом представляет собой атом серы, он может быть неокисленным (S) или окисленным до S(O) либо S(O)2.
Термин "гидроксил" или "гидрокси", используемый в данном документе, означает радикал -OH.
"Гетероциклоалкил" означает циклоалкил, определенный в данной заявке, при условии, что один или несколько указанных атомов углерода в кольце заменены на фрагмент, выбранный из -O-, -N=, -NH-, -S-, -S(O)- и -S(O)2-. Примеры 3-8-членного гетероциклоалкила включают без ограничения оксиранил, азиридинил, азетидинил, имидазолидинил, пиразолидинил, тетрагидрофуранил, тетрагидротиенил, тетрагидротиенил-1,1-диоксид, оксазолидинил, тиазолидинил, пирролидинил, пирролидинил-2-он, морфолинил, пиперазинил, пиперидинил, пиразолидинил, гексагидропиримидинил, 1,4-диокса-8-азаспиро[4.5]дец-8-ил, тиоморфолинил, сульфаноморфолинил, сульфонoморфолинил и октагидропирроло[3,2-b]пирролил.
Термин "оксо", используемый в данном документе, означает двухвалентный радикал =O.
Термин "спиро" используемый в данном документе, включает C3-6циклоалкил или 4-6-членные гетероциклические кольца, содержащие один или два гетероатома, выбранных из N, O и S в качестве членов кольца, где спирокольцо конденсировано с одним атомом углерода неароматического кольца, что делает атом углерода, общий для обоих колец, спироциклическим центром. Спирокольцо необязательно может быть замещено, как определено, например, галогеном, гидроксилом или C1-4алкилом. Иллюстративными примерами спирогрупп являются:
пунктирные связи в каждой структуре представляют собой связи неароматического кольца, с которым у спироциклической группы есть один общий атом.
Упоминаемое в данном документе выражение "замещенный" означает, что по меньшей мере один атом водорода заменен группой, отличной от водорода, при условии, что поддерживаются нормальные валентности, и что замещение приводит к образованию стабильного соединения. Если заместитель представляет собой оксо (т. е. =O), то 2 атома водорода при атоме заменены. В случаях, если в соединениях по настоящему изобретению присутствуют атомы азота (например, амины), они могут быть превращены в N-оксиды посредством обработки окисляющим средством (например, mCPBA и/или пероксидами водорода) с получением других соединений по настоящему изобретению.
Специалисту в данной области техники будет понятно, что, например, кетонная группа (-CH-C(=O)-) в молекуле может в результате таутомеризации переходить в ее енольную форму (-C=C(OH)-). Таким образом, подразумевается, что данное изобретение охватывает все возможные таутомеры, даже если структура изображает лишь один из них.
Если какая-либо переменная встречается более одного раза в любой составляющей или формуле соединения, ее определение в каждом случае является независимым от ее определения в каждом другом случае. Таким образом, например, если показано, что группа замещена 0-3 группами R, то указанная группа может быть незамещенной или замещенной не более чем тремя группами R, и в каждом случае R выбран независимо от определения R.
Используемое в данном документе обозначение " " обозначает точку присоединения X к другой части молекулы.
" обозначает точку присоединения X к другой части молекулы.
Термин "IC50", используемый в данном документе, означает молярную концентрацию ингибитора, которая обеспечивает 50% ингибирующего эффекта.
Термин "EC50", используемый в данном документе, означает молярную концентрацию ингибитора или модулятора, который обеспечивает эффективность 50%.
Используемый в данном документе термин "фармацевтический композиция" означает соединение по настоящему изобретению или его фармацевтически приемлемую соль вместе с по меньшей мере одним фармацевтически приемлемым носителем в подходящей для местного или парентерального введения форме.
Используемый в данном документе термин "фармацевтически приемлемый носитель" означает вещество, пригодное в получении или применении фармацевтической композиции, и включает, например, подходящие разбавители, растворители, дисперсионные среды, поверхностно-активные вещества, антиоксиданты, консерванты, изотонические средства, буферные средства, эмульгаторы, средства, замедляющие абсорбцию, соли, стабилизаторы лекарственных средств, связующие, вспомогательные вещества, разрыхляющие средства, смазывающие средства, смачивающие средства, подсластители, ароматизирующие средства, красители и их комбинации, как будет известно специалистам в данной области техники (см., например, Remington The Science and Practice of Pharmacy, 22nd Ed. Pharmaceutical Press, 2013, pp. 1049-1070).
Используемые в данном документе термины "ингибировать", "ингибирование" или "ингибирующий" означают снижение или ослабление данного состояния, симптома, или нарушения, или заболевания или значительное снижение исходной активности в отношении биологической активности или процесса.
Используемый в данном документе термин "предупреждать", "осуществление предупреждения" или "предупреждение" любого заболевания или нарушения означает профилактическое лечение заболевания или нарушения или задержку возникновения или прогрессирования заболевания или нарушения.
Используемый в данном документе термин "субъект" означает приматов (например, людей, мужчин или женщин), собак, кроликов, морских свинок, свиней, крыс и мышей. В определенных вариантах осуществления субъектом является примат. В других вариантах осуществления субъектом является человек.
Термин "терапевтически эффективное количество" соединения по настоящему изобретению означает количество соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ у субъекта, например, снижение или подавление активности фермента или белка, или уменьшать тяжесть симптомов, облегчать состояния, замедлять или сдерживать прогрессирование заболевания, или предупреждать развитие заболевания и т. д. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое при введении субъекту, является эффективным в отношении (1) по меньшей мере частичного облегчения, подавления, предупреждения и/или снижения тяжести состояния, или нарушения, или заболевания, обусловленного пролиферацией паразита класса кинетопластид; или (2) снижения или подавления пролиферации паразита класса кинетопластид.
Используемый в данном документе термин "лечить", "осуществление лечения" или "лечение" любого заболевания или нарушения означает облегчение или снижение тяжести заболевания или нарушения (т. е. замедление или остановку развития заболевания или по меньшей мере одного из его клинических симптомов); или снижение или уменьшение по меньшей мере одного физического параметра или биомаркера, ассоциированного с заболеванием или нарушением, в том числе таких, которые могут не ощущаться пациентом.
Варианты осуществления изобретения
Настоящее изобретение относится к соединениям, которые обладают антипаразитарной активностью в отношении простейших класса кинетопластид. В частности, оно относится к соединениям, которые подавляют рост клеток паразита класса кинетопластид путем ингибирования паразитической протеасомы и, следовательно, являются применимыми в качестве средства терапии для лейшманиоза, болезни Шагаса и африканской сонной болезни.
Различные (перечисленные) варианты осуществления настоящего изобретения описаны в данном документе. Признаки, описанные в каждом варианте осуществления, можно объединять с другими описанными признаками с получением дополнительных вариантов осуществления по настоящему изобретению.
Вариант осуществления 1. Соединение формулы (I) или его фармацевтически приемлемая соль, описанные в разделе "Краткое описание изобретения".
Вариант осуществления 2. Соединение в соответствии с вариантом осуществления 1 или его фармацевтически приемлемая соль, где кольцо A представляет собой фенил.
Вариант осуществления 3. Соединение в соответствии с вариантом осуществления 1 или его фармацевтически приемлемая соль, где кольцо A представляет собой пиридинил.
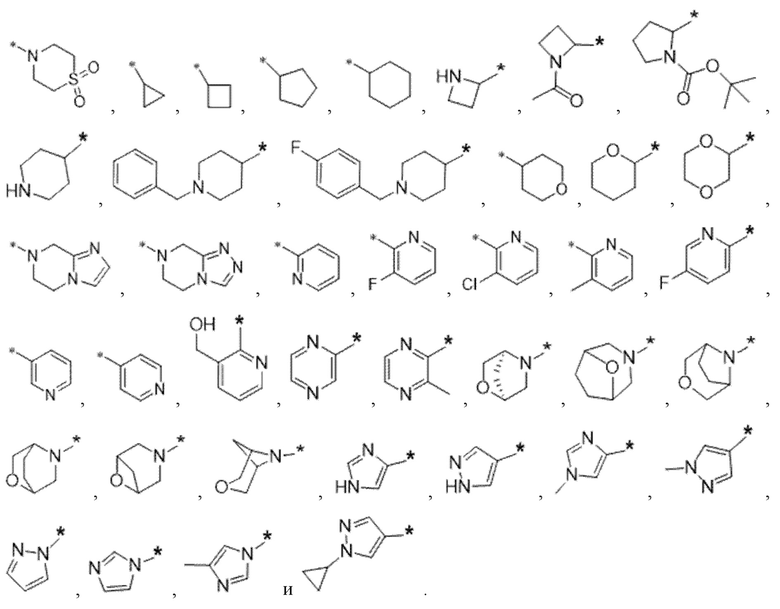
Вариант осуществления 4. Соединение в соответствии с любым из вариантов осуществления 1-3 или его фармацевтически приемлемая соль, где R1 выбран из -(CH2)1-3CF3, -(CH2)-CH(CH3)-CF3, -(CH2)-C(CH3)3, -O(CH2)2CF3, -(CH2)0-2-циклопропила, -(CH2)0-2-циклобутила, -NHCH3, -N(CH3)2, -N(CD3)2, -N(CH3)(CH2CH3), -N(CH3)(CH2CF3),
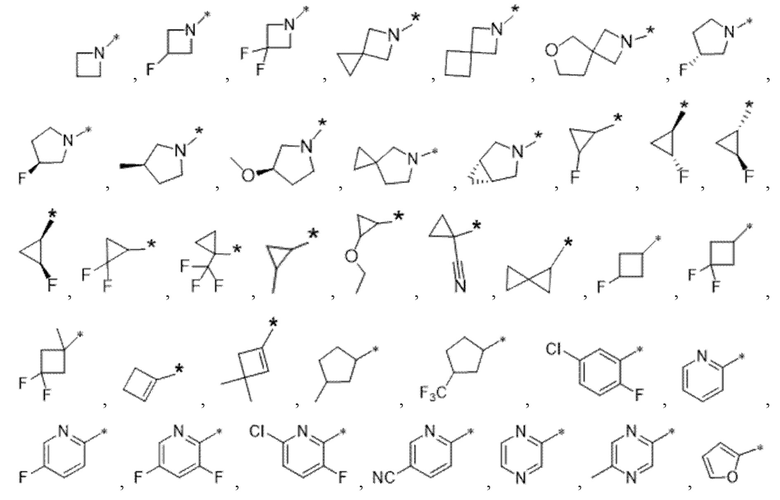
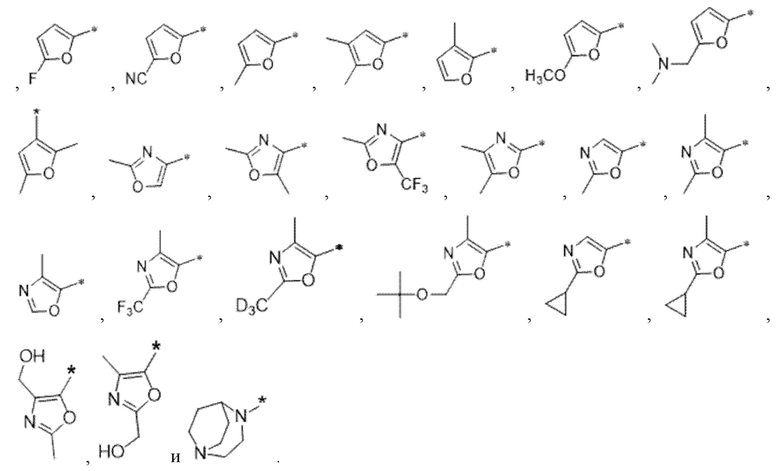
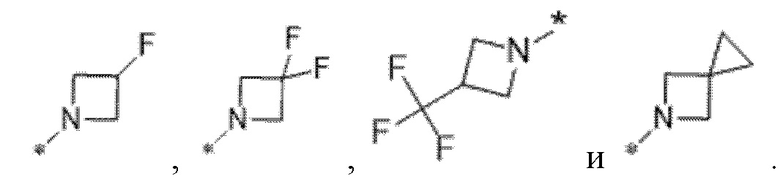
Вариант осуществления 5. Соединение в соответствии с любым из вариантов осуществления 1-3 или его фармацевтически приемлемая соль, где R1 представляет собой азетидинил, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила; или два заместителя при одном и том же атоме кольца азетидинила вместе с атомом кольца, к которому они оба присоединены, образуют спироциклопропил или спиротетрагидрофуранил, присоединенный к азетидинильному кольцу.
Вариант осуществления 6. Соединение в соответствии с любым из вариантов осуществления 1-3 или его фармацевтически приемлемая соль, где R1 выбран из
Вариант осуществления 7. Соединение в соответствии с любым из вариантов осуществления 1-6 или его фармацевтически приемлемая соль, где R3 представляет собой галоген, и n равняется 1.
Вариант осуществления 8. Соединение в соответствии с вариантом осуществления 7 или его фармацевтически приемлемая соль, где R3 представляет собой фтор.
Вариант осуществления 9. Соединение в соответствии с любым из вариантов осуществления 1-8 или его фармацевтически приемлемая соль, где R4 выбран из водорода, хлора, брома, метила, изопропила, -(CH2)1-2CH(CH3)2, -(CH2)0-1C(CH3)3, -C(CH3)2CH2CH3,
-CH(CH3)(CH2)1-2CH3, -CH2-циклобутила, -(CH2)0-1CF3, -NH-(CH2)0-1CH3, -N-(CD3)2, -N(CH3)2, -NH-CH-(CH3)2, -NH-(CH2)-CH-(CH3)2, -NHC(O)OCH(CH3)2, -NH(CH2)2OCH3,
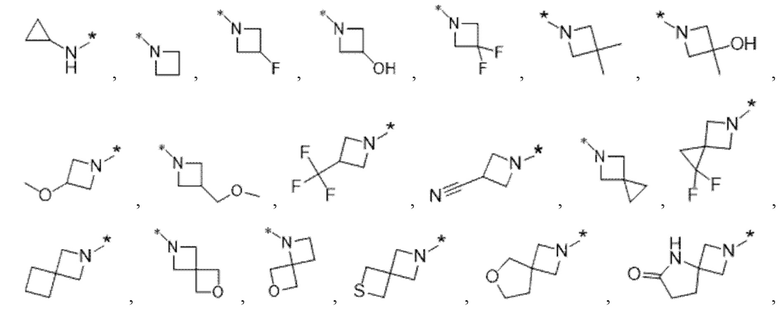
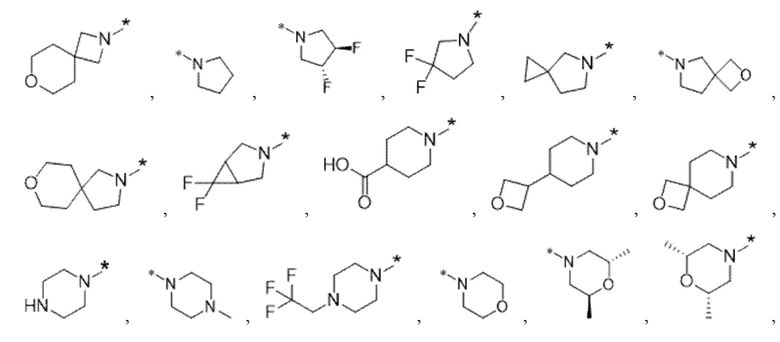
Вариант осуществления 10. Соединение в соответствии с любым из вариантов осуществления 1-8 или его фармацевтически приемлемая соль, где R4 представляет собой -NR6aR6b;
R6a представляет собой водород или C1-4алкил;
R6b представляет собой водород, C1-4алкоксикарбонил или C1-4алкил, который не замещен или замещен C1-4алкокси; или R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-6-членный гетероциклоалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-6-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси и оксо; или два заместителя при одном и том же или различных атомах кольца 4-6-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-6-членному гетероциклоалкилу;
где кольцо C выбрано из C3-6циклоалкила и 3-5-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и при этом кольцо C независимо не замещено или замещено 1-2 заместителями, независимо выбранными из галогена и оксо.
Вариант осуществления 11. Соединение в соответствии с любым из вариантов осуществления 1-8 или его фармацевтически приемлемая соль, где R4 выбран из
Вариант осуществления 12. Соединение в соответствии с любым из вариантов осуществления 1-3 или его фармацевтически приемлемая соль;
где R1 представляет собой азетидинил, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила; или два заместителя при одном и том же атоме кольца азетидинила вместе с атомом кольца, к которому они оба присоединены, образуют спироциклопропил или спиротетрагидрофуранил, присоединенный к азетидинильному кольцу.
R2 представляет собой водород или C1-4алкил;
R3 выбран из галогена, и n представляет собой 1; и
R4 представляет собой -NR6aR6b, где R6a представляет собой водород или C1-4алкил; R6b представляет собой водород, C1-4алкоксикарбонил или C1-4алкил, который не замещен или замещен C1-4алкокси; или R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-6-членный гетероциклоалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-6-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси и оксо; или два заместителя при одном и том же или различных атомах кольца 4-6-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-6-членному гетероциклоалкилу;
где кольцо C выбрано из C3-6циклоалкила; 3-5-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и при этом кольцо C независимо не замещено или замещено 1-2 заместителями, независимо выбранными из галогена и оксо.
Вариант осуществления 13. Соединение в соответствии с любым из вариантов осуществления 1-12 или его фармацевтически приемлемая соль, где R4 выбран из 
Вариант осуществления 14. Соединение в соответствии с любым из вариантов осуществления 1-12 или его фармацевтически приемлемая соль, где R4 выбран из 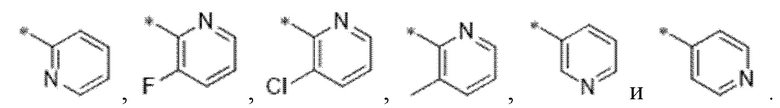
Вариант осуществления 15. Соединение формулы (I) или его фармацевтически приемлемая соль:
или его фармацевтически приемлемая соль или стереоизомер; где
каждый из W1 и W2 независимо представляет собой CH или N;
кольцо A представляет собой фенил или пиридил;
X представляет собой -C(O)- или -S(O)2-;
R1 выбран из
(a) C1-4алкила, который не замещен или замещен 1-3 заместителями, независимо выбранными из галогена и C3-6циклоалкила; где заместитель, представляющий собой C3-6циклоалкил, не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила;
(b) C1-4алкокси, который не замещен или замещен C1-4галогеналкилом;
(c) -NR5aR5b, где
(1) каждый из R5a и R5b независимо представляет собой водород или C1-4алкил; или
(2) R5a и R5b вместе с атомом азота, к которому они оба присоединены, образуют 4-6-членный гетероциклоалкил, необязательно содержащий 1-3 дополнительных гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца, при этом
4-6-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4 алкокси и оксо; или
два заместителя при одном и том же или различных атомах кольца 4-6-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо B, присоединенное к 4-6-членному гетероциклоалкилу; где кольцо B выбрано из
(i) C3-6циклоалкила,
(ii) 3-5-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и
(iii) 5-6-членного гетероарила, содержащего 1-3 гетероатома, независимо выбранных из N или O, в качестве атомов кольца;
при этом кольцо B из (i) - (iii) в каждом случае не замещено или замещено 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила и оксо;
(d) моноциклического C3-6циклоалкила и полициклического C5-9циклоалкила, каждый из которых не замещен или замещен 1-3 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила и C1-4алкоксикарбонила;
(e) 4-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкокси, оксо и C1-4алкилкарбонила;
(f) фенила, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкиламино-C1-4алкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкокси и C3-6циклоалкила;
(g) моноциклического 5-6-членного гетероарила и полициклического 7-10-членного гетероарила, каждый из которых содержит 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца, и каждый из которых не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, C1-4алкиламино-C1-4алкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкоксиC1-4алкила, C1-4алкокси и C3-6циклоалкила;
R2 представляет собой водород или C1-4алкил;
R3 представляет собой галоген или C1-4алкил, и n равняется 0, 1 или 2;
R4 выбран из
(a) галогена;
(b) C1-6алкила, который не замещен или замещен 1-3 заместителями, независимо выбранными из галогена и C3-6циклоалкила; где заместитель, представляющий собой C3-6циклоалкил, не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила;
(c) -NR6aR6b, где
(1) R6a представляет собой водород или C1-4алкил;
(2) R6b представляет собой водород или C1-4алкил, который не замещен или замещен C1-4алкокси или C1-4алкоксикарбонилом;
(3) или R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-6-членный гетероциклоалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца, при этом
4-6-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси и оксо; или
два заместителя при одном и том же или различных атомах кольца 4-6-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-6-членному гетероциклоалкилу; где кольцо C выбрано из
(i) C3-6циклоалкила,
(ii) 3-5-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и
(iii) 5-6-членного гетероарила, содержащего 1-3 гетероатома, независимо выбранных из N или O, в качестве атомов кольца;
при этом кольцо C из (i) - (iii) в каждом случае не замещено или замещено 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила и оксо;
(d) C3-6циклоалкила, который не замещен или замещен 1-2 заместителями, выбранными из галогена, циано, C1-4алкила и C1-4галогеналкила;
(e) 4-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила и оксо;
(f) фенила, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкиламино-C1-4алкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкокси и C3-6циклоалкила;
(g) 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, C1-4алкиламино-C1-4алкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкоксиC1-4алкила, C1-4алкокси и C3-6циклоалкила.
Вариант осуществления 16. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 15, где соединение характеризуется формулой (I-B):
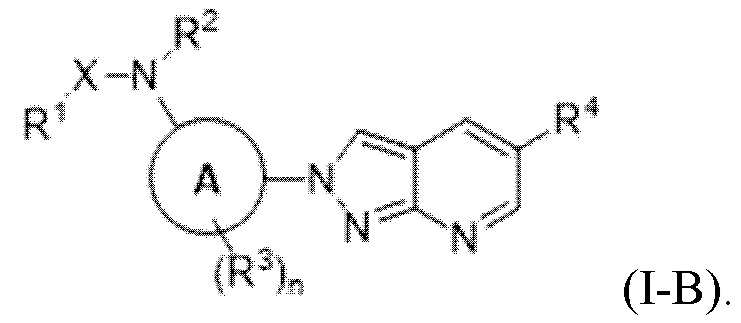
Вариант осуществления 17. Соединение или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 15 или 16, где кольцо A представляет собой фенил.
Вариант осуществления 18. Соединение или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 15 или 16, где кольцо A представляет собой пиридинил.
Вариант осуществления 19. Соединение или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 15-18, где X представляет собой -C(O)-.
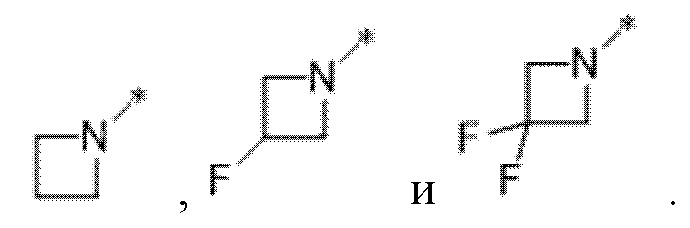
Вариант осуществления 20. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 15-19, где R1 представляет собой азетидинил, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила; или два заместителя при одном и том же атоме кольца азетидинила вместе с атомом кольца, к которому они оба присоединены, образуют спироциклопропил или спиротетрагидрофуранил, присоединенный к азетидинильному кольцу.
Вариант осуществления 21. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 20, где R1 представляет собой 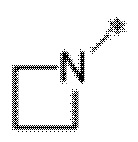 .
.
Вариант осуществления 22. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 20, где R1 представляет собой 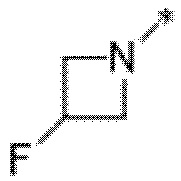 .
.
Вариант осуществления 23. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 20, где R1 представляет собой 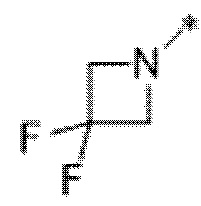 .
.
Вариант осуществления 24. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 15-23, где R3 выбран из галогена или C1-4алкила, и n равняется 1.
Вариант осуществления 25. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 24, где R3 представляет собой галоген.
Вариант осуществления 26. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 25, где R3 представляет собой фтор.
Вариант осуществления 27. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 15-26, где R4 представляет собой -NR6aR6b; где R6a представляет собой водород или C1-4алкил; и R6b представляет собой водород или C1-4алкил, который не замещен или замещен C1-4алкокси или C1-4алкоксикарбонилом; или где R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-6-членный гетероциклоалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-6-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси и оксо; или два заместителя при одном и том же или различных атомах кольца 4-6-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-6-членному гетероциклоалкилу;
где кольцо C выбрано из C3-6циклоалкила; 3-5-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и 5-членного гетероарила, содержащего 1-3 гетероатома, независимо выбранных из N или O в качестве атомов кольца; и кольцо C не замещено или замещено 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила и оксо.
Вариант осуществления 28. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 15-27, где R4 выбран из
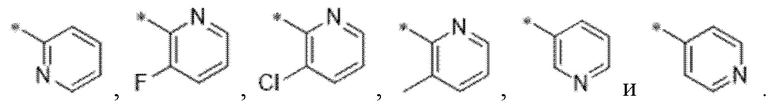
Вариант осуществления 29. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 15, где соединение характеризуется формулой (I)D:
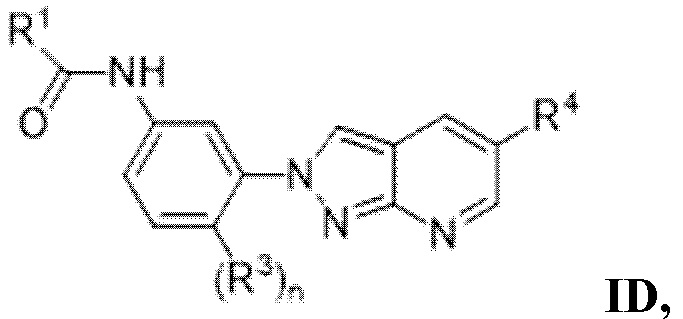
где
R1 выбран из
(a) -NR5aR5b, где R5a и R5b вместе с атомом азота, к которому они оба присоединены, образуют 4-5-членный гетероциклоалкил, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила; и
(b) моноциклического 5-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N и O, в качестве атомов кольца, и каждый из которых не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила;
R3 представляет собой галоген или C1-4алкил, и n равняется 0 или 1;
R4 выбран из
(a) -NR6aR6b, где R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4- или 5-членный гетероциклоалкил, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, гидроксила и C1-4алкила; или
2 заместителя при одном и том же или различных атомах кольца 4- или 5-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спирокольцо C, присоединенное к 4-5-членному гетероциклоалкилу, где кольцо C представляет собой C3-6циклоалкил; и
(b) 6-членного гетероарила, содержащего 1-2 атома азота в качестве атомов кольца, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила.
Вариант осуществления 30. Соединение в соответствии с вариантом осуществления 29, где R1 выбран из 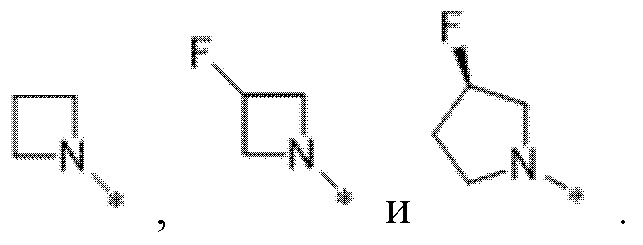
Вариант осуществления 31. Соединение формулы (I)D или его фармацевтически приемлемая соль в соответствии с одним из вариантов осуществления 29 или 30, где R3 представляет собой фтор или хлор, и n равняется 1.
Вариант осуществления 32. Соединение формулы (I)D или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 29-31, где R4 выбран из
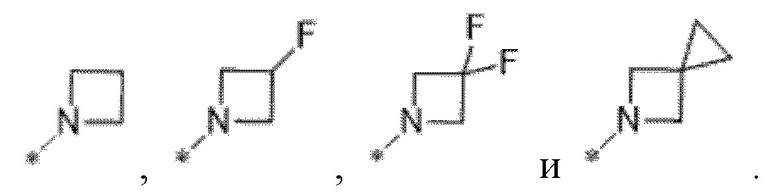
Вариант осуществления 33. Соединение формулы (I)D или его фармацевтически приемлемая соль в соответствии с любым из вариантов осуществления 29-31, где R4 выбран из
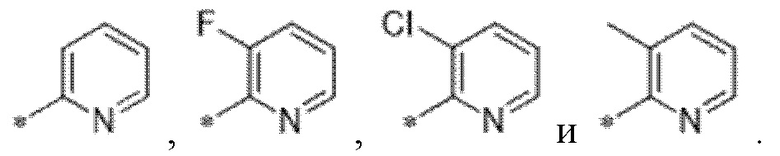
Вариант осуществления 34. Соединение формулы (I) или его фармацевтически приемлемая соль, выбранные из таблицы 1.
Таблица 1
Вариант осуществления 35. Соединение формулы (I) или его фармацевтически приемлемая соль в соответствии с вариантом осуществления 34, где указанное соединение выбрано из соединений 1-76, 78-79, 81-87, 89-115, 117-118 и 121-353 из таблицы 1.
Вариант осуществления 36. Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в соответствии с любым из вариантов осуществления 1-35 и по меньшей мере одно вспомогательное вещество.
Вариант осуществления 37. Комбинация, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в соответствии с любым из вариантов осуществления 1-35 или фармацевтическую композицию на их основе в соответствии с вариантом осуществления 36 и одно или несколько терапевтически активных средств.
Вариант осуществления 38. Соединение в соответствии с любым из вариантов осуществления 1-35 или фармацевтическая композиция в соответствии с вариантом осуществления 36 для применения в терапии.
Вариант осуществления 39. Соединение в соответствии с любым из вариантов осуществления 1-35 или фармацевтическая композиция в соответствии с вариантом осуществления 36 для применения в лечении заболевания, выбранного из лейшманиоза, форм болезни Шагаса и африканского трипаносомоза человека.
Вариант осуществления 40. Применение соединения в соответствии с любым из вариантов осуществления 1-35 или фармацевтической композиции в соответствии с вариантом осуществления 36 в изготовлении лекарственного препарата для лечения заболевания, выбранного из лейшманиоза, форм болезни Шагаса и африканского трипаносомоза человека.
Вариант осуществления 41. Способ подавления роста и пролиферации паразита класса кинетопластид у субъекта, предусматривающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в соответствии с любым из вариантов осуществления 1-35.
Вариант осуществления 42. Способ лечения нарушения или заболевания, вызванных паразитом класса кинетопластид, предусматривающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в соответствии с любым из вариантов осуществления 1-35 и необязательно в комбинации со вторым средством; где заболевание выбрано из лейшманиоза, форм болезни Шагаса и африканского трипаносомоза человека.
Вариант осуществления 43. Способ в соответствии с вариантом осуществления 42, где указанное заболевание представляет собой лейшманиоз, выбранный из висцерального лейшманиоза и кожного лейшманиоза.
Вариант осуществления 44. Способ в соответствии с вариантом осуществления 43, где указанное второе средство выбрано из стибоглюконата, меглюмина антимоната, амфотерицина, милтефосина и паромомицина.
Вариант осуществления 45. Способ в соответствии с вариантом осуществления 42, где указанное заболевание представляет собой болезнь Шагаса; и при этом указанное второе средство выбрано из бензнидазола, нифуртимокса и амфотерицина.
Вариант осуществления 46. Способ в соответствии с вариантом осуществления 42, где указанное заболевание представляет собой африканский трипаносомоз человека; и при этом указанное второе средство представляет собой пентамидин, сурамин, меларсопрол, эфлорнитин и нифуртимокс.
В зависимости от выбора исходных материалов и процедур соединения могут находиться в форме одного из возможных стереоизомеров или в виде их смесей, например, в виде чистых оптических изомеров, или в виде смеси стереоизомеров, таких как рацематы и смеси диастереоизомеров, в зависимости от числа асимметрических атомов углерода. В настоящем изобретении подразумевается включение всех таких возможных стереоизомеров, в том числе рацемических смесей, смесей диастереоизомеров и оптически чистых форм. Оптически активные (R)- и (S)-стереоизомеры можно получать с использованием хиральных синтонов или хиральных реагентов или выделять с применением традиционных методик. Если соединение содержит двойную связь, заместитель может иметь E- или Z-конфигурацию. Если соединение содержит двузамещенный циклоалкил, циклоалкильный заместитель может иметь цис- или транс-конфигурацию. Также подразумевается включение всех таутомерных форм.
Используемые в данном документе термины "соль" или "соли" означают соль присоединения кислоты или присоединения основания соединения по настоящему изобретению. "Соли" включают, в частности, "фармацевтически приемлемые соли". Термин "фармацевтически приемлемые соли" означает соли, которые сохраняют биологические эффективность и свойства соединений по настоящему изобретению и которые, как правило, не являются биологически или иным образом нежелательными. Во многих случаях соединения по настоящему изобретению способны образовывать кислотные и/или основные соли вследствие присутствия амино- и/или карбоксильных групп или подобных им групп.
Фармацевтически приемлемые соли присоединения кислоты могут быть образованы с помощью неорганических кислот и органических кислот. Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т. п. Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т. п.
Фармацевтически приемлемые соли присоединения основания могут быть образованы неорганическими и органическими основаниями. Неорганические основания, из которых могут быть получены соли, включают, например, соли аммония и металлов из групп I-XII периодической таблицы элементов. В определенных вариантах осуществления соли получены из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; в частности, подходящие соли включают аммониевые, калиевые, натриевые, кальциевые и магниевые соли. Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, в том числе встречающиеся в природе замещенные амины, циклические амины, основные ионообменные смолы и т. п. Определенные органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглюмин, пиперазин и трометамин.
В другом аспекте в настоящем изобретении представлены соединения формулы (I) или ее подформул в форме соли, представляющей собой ацетат, аскорбат, адипат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, капрат, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, глутамат, глутарат, гликолят, гиппурат, гидройодид/йодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, мукат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, себацат, стеарат, сукцинат, сульфосалицилат, сульфат, тартрат, тозилат-трифенатат, трифторацетат или ксинафоат.
В другом аспекте в настоящем изобретении представлены соединения формулы (I) или ее подформул в форме соли натрия, калия, аммония, кальция, магния, железа, серебра, цинка, меди, изопропиламина, бензатина, холината, диэтаноламина, диэтиламина, лизина, меглюмина, пиперазина или трометамина.
Любая формула, приведенная в данном документе, подразумевает немеченные формы, а также меченные изотопом формы соединений. Меченные изотопом соединения имеют структуры, изображенные с помощью формул, приведенных в данном документе, за исключением того, что один или несколько атомов заменены атомом, характеризующимся выбранными атомной массой или массовым числом. Изотопы, которые можно включать в соединения по настоящему изобретению, включают, например, изотопы водорода.
Кроме того, включение определенных изотопов, в частности дейтерия (т. е., 2H или D), может давать определенные терапевтические преимущества, обусловленные более высокой метаболической устойчивостью, например, увеличенным периодом полувыведения in vivo, или снижением требующейся дозировки, или улучшением терапевтического индекса или переносимости. Понятно, что дейтерий в данном контексте рассматривается в качестве заместителя соединения формулы (I). Концентрацию дейтерия можно определить по коэффициенту изотопного обогащения. Используемый в данном документе термин "коэффициент изотопного обогащения" означает соотношение распространенности изотопа и природной распространенности указанного изотопа. Если заместитель в соединении по настоящему изобретению указан как дейтерий, такое соединение характеризуется коэффициентом изотопного обогащения для каждого обозначенного атома дейтерия, составляющим по меньшей мере 3500 (введение 52,5% дейтерия при каждом обозначенном атоме дейтерия), по меньшей мере 4000 (введение 60% дейтерия), по меньшей мере 4500 (введение 67,5% дейтерия), по меньшей мере 5000 (введение 75% дейтерия), по меньшей мере 5500 (введение 82,5% дейтерия), по меньшей мере 6000 (введение 90% дейтерия), по меньшей мере 6333,3 (введение 95% дейтерия), по меньшей мере 6466,7 (введение 97% дейтерия), по меньшей мере 6600 (введение 99% дейтерия) или по меньшей мере 6633,3 (введение 99,5% дейтерия). Следует понимать, что термин "коэффициент изотопного обогащения" может использоваться в отношении любого изотопа таким же образом, как это описано для дейтерия.
Другие примеры изотопов, которые можно включать в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36Cl,123I, 124I, 125I соответственно. Однако следует понимать, что настоящее изобретение включает соединения, в которые включены один или несколько любых из вышеуказанных изотопов, включая, например, радиоактивные изотопы, такие как 3H и 14C, или соединения, в которых присутствуют нерадиоактивные изотопы, такие как 2H и 13C. Такие изотопно-меченные соединения применимы в метаболических исследованиях (с использованием 14C), исследованиях кинетических параметров реакций (например, с использованием 2H или 3H), методиках обнаружения или визуализации, таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая анализы распределения лекарственного средства или субстрата в тканях, или в лучевой терапии пациентов. В частности, 18F или меченое соединение может быть особенно востребованными для исследований с помощью PET или SPECT. Изотопно-меченые соединения формулы (I), как правило, можно получать с помощью традиционных методик, известных специалистам в данной области техники, или посредством способов, аналогичных описанным в сопутствующих примерах и способах получения, с использованием подходящего изотопно-меченого реагента вместо немеченого реагента, используемого ранее.
Любой асимметрический атом (например, углерод или подобный) соединения(соединений) по настоящему изобретению может находиться в рацемической или энантиомерно обогащенной форме, например, в (R)-, (S)- или (R, S)-конфигурации. В определенных вариантах осуществления каждый асимметрический атом характеризуется по меньшей мере 50% энантиомерным избытком, по меньшей мере 60% энантиомерным избытком, по меньшей мере 70% энантиомерным избытком, по меньшей мере 80% энантиомерным избытком, по меньшей мере 90% энантиомерным избытком, по меньшей мере 95% энантиомерным избытком или по меньшей мере 99% энантиомерным избытком в (R)- или (S)-конфигурации. Заместители при атомах с ненасыщенными двойными связями могут, если это возможно, находиться в цис- (Z)- или транс- (E)-форме.
Соответственно, как используется в данном документе, соединение по настоящему изобретению может находиться в форме одного из возможных стереоизомеров, ротамеров, атропоизомеров, таутомеров или их смесей, например, в виде по сути чистых геометрических (цис- или транс-) стереоизомеров, диастереоизомеров, оптических изомеров (антиподов), рацематов или их смесей.
Любые полученные смеси стереоизомеров могут быть разделены на основе физико-химических отличий составляющих компонентов на чистые или практически чистые геометрические или оптические изомеры, диастереоизомеры, рацематы, например, посредством хроматографии и/или фракционной кристаллизации.
Любые полученные рацематы конечных продуктов или промежуточных соединений могут быть разделены на оптические антиподы посредством известных способов, например посредством разделения их диастереомерных солей, полученных с помощью оптически активных кислоты или основания, и выделения оптически активного кислотного или основного соединения. В частности, основный фрагмент таким образом может быть использован для разделения соединений по настоящему изобретению на их оптические антиподы, например, путем фракционной кристаллизации соли, образованной с помощью оптически активной кислоты, например, винной кислоты, дибензоилвинной кислоты, диацетилвинной кислоты, ди-O, O'-п-толуолвинной кислоты, миндальной кислоты, яблочной кислоты или камфор-10-сульфоновой кислоты. Рацемические продукты также могут быть разделены посредством хиральной хроматографии, например жидкостной хроматографии высокого давления (HPLC) с применением хирального адсорбента.
Фармакология и применимость
В одном аспекте настоящее изобретение относится к низкомолекулярным соединениям формулы (I), которые целенаправленно воздействуют на протеасому паразитов класса кинетопластид и являются применимыми в качестве средства терапии; в частности, для предупреждения и лечения заболеваний и состояний, обусловленных инфекцией паразитами класса кинетопластид. Активность соединения по настоящему изобретению можно оценить с помощью способов in vitro и in vivo, описанных в разделе "Биологические анализы" ниже. Очевидно, что анализы иллюстрируют настоящее изобретение без ограничения каким-либо образом объема настоящего изобретения.
Таким образом, в качестве дополнительного аспекта, в настоящем изобретении представлено применение соединения формулы (I) или ее подформул или его фармацевтически приемлемой соли или его стереоизомера в качестве средства терапии для заболевания или состояния, при которых может наблюдаться благоприятный эффект в результате подавления роста и пролиферации паразитов класса кинетопластид; и в изготовлении лекарственного препарата, например, для лечения заболевания, обусловленного ростом и пролиферацией паразита класса кинетопластид. В одном варианте осуществления заболевание, подлежащее лечению, выбрано из лейшманиоза, болезни Шагаса и африканского трипаносомоза человека. В другом варианте осуществления заболевание, подлежащее лечению, представляет собой лейшманиоз, вызванный паразитом, в том числе без ограничения Leishmania donovani, Leishmania infantum, Leishmania braziliensis, Leishmania panamensis, Leishmania guayanensis, Leishmania amazonensis, Leishmania mexicana, Leishmania tropica, Leishmania major. В одном варианте осуществления заболевание, подлежащее лечению, представляет собой висцеральный лейшманиоз. В другом варианте осуществления заболевание, подлежащее лечению, представляет собой кожный лейшманиоз. В другом варианте осуществления заболевание, подлежащее лечению, представляет собой болезнь Шагаса, вызванную Trypanosoma cruzi. В еще одном варианте осуществления заболевание, подлежащее лечению, представляет собой африканский трипаносомоз человека, вызванный Trypanosoma brucei, Trypanosoma brucei gambiense или Trypanosoma brucei rhodesiense.
В соответствии с вышеуказанным, в настоящем изобретении дополнительно представлен способ предупреждения или лечения лейшманиоза, болезни Шагаса или африканского трипаносомоза человека у субъекта, нуждающегося в таком лечении, который предусматривает введение указанному субъекту терапевтически эффективного количества соединения, выбранного из формулы (I) или ее подформул, его фармацевтически приемлемой соли или его стереоизомера. Необходимая доза будет варьироваться в зависимости от пути введения, конкретного состояния, подлежащего лечению, и требуемого эффекта.
Фармацевтические композиции, дозировка и введение
В другом аспекте в настоящем изобретении представлена фармацевтическая композиция, содержащая соединение по настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В дополнительном варианте осуществления композиция содержит по меньшей мере два таких фармацевтически приемлемых носителя, которые описаны в данном документе. Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение (например, путем инъекции, инфузии, трансдермального или местного введения), и ректальное введение. Местное введение может также относиться к ингаляционному или интраназальному применению. Фармацевтические композиции по настоящему изобретению можно изготовлять в твердой форме (включая без ограничения капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включающей без ограничения растворы, суспензии или эмульсии). Таблетки могут быть либо покрыты оболочкой, либо покрыты энтеросолюбильным покрытием в соответствии со способами, известными из уровня техники. Как правило, фармацевтические композиции представляют собой таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с одним или несколькими из
a) разбавителей, например, лактозы, декстрозы, сахарозы, маннита, сорбита, целлюлозы и/или глицина;
b) смазывающих средств, например, диоксида кремния, талька, стеариновой кислоты, ее магниевой или кальциевой соли и/или полиэтиленгликоля; в случае таблеток также
c) связующих, например, алюмосиликата магния, крахмальной пасты, желатина, трагаканта, метилцеллюлозы, натрий-карбоксиметилцеллюлозы и/или поливинилпирролидона; при необходимости
d) разрыхлителей, например, видов крахмала, агара, альгиновой кислоты или ее натриевой соли или шипучих смесей; и
e) абсорбентов, красителей, ароматизаторов и подсластителей.
Фармацевтическая композиция или комбинация по настоящему изобретению может быть представлена в однократной дозировке, составляющей приблизительно 1-1000 мг активного(активных) ингредиента(ингредиентов) для субъекта весом приблизительно 50-70 кг, или приблизительно 1-500 мг, или приблизительно 1-250 мг, или приблизительно 1-150 мг, или приблизительно 0,5-100 мг, или приблизительно 1-50 мг активных ингредиентов. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинаций зависит от вида субъекта, веса тела, возраста и индивидуального состояния, нарушения или заболевания, лечение которых осуществляют, или их тяжести. Квалифицированный лечащий врач, клиницист или ветеринар может легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или подавления прогрессирования нарушения или заболевания.
Вышеупомянутые параметры дозировки являются очевидными в тестах in vitro и in vivo с применением преимущественно млекопитающих, например, мышей, крыс, собак, нечеловекообразных обезьян или выделенных органов, тканей и их препаратов. Соединения по настоящему изобретению можно применять in vitro в виде растворов, например водных растворов, и in vivo либо энтерально, либо парентерально, преимущественно внутривенно, например, в виде суспензии или водного раствора. Дозировка in vitro может находиться в диапазоне от приблизительно 10−3 молярной до 10−9 молярной концентрации. Терапевтически эффективное количество in vivo в зависимости от пути введения может находиться в диапазоне приблизительно 0,1-500 мг/кг или приблизительно 1-100 мг/кг.
Соединения по настоящему изобретению можно вводить в терапевтически эффективных количествах в комбинации с одним или несколькими терапевтическими средствами (фармацевтические комбинации). Соединение по настоящему изобретению можно вводить либо одновременно с одним или несколькими другими терапевтическими средствами, либо до, либо после их введения. Соединение по настоящему изобретению можно вводить отдельно от других средств, посредством такого же или иного пути введения, или же вместе с ними в одной и той же фармацевтической композиции. Терапевтическое средство представляет собой, например, химическое соединение, пептид, антитело, фрагмент антитела или нуклеиновую кислоту, которые являются терапевтически активными или повышают терапевтическую активность при введении пациенту в комбинации с соединением по настоящему изобретению.
Продукты, представленные в виде комбинированного препарата, включают композицию, содержащую соединение формулы (I) или ее подформул, его фармацевтически приемлемую соль или его стереоизомер и другое(другие) терапевтическое(терапевтические) средство(средства) вместе в той же фармацевтической композиции, или соединение формулы (I) и другое(другие) терапевтическое(терапевтические) средство(средства) в отдельной форме, например, в виде набора. В одном варианте осуществления в настоящем изобретении представлена фармацевтическая композиция, содержащая соединение формулы (I) или ее подформул, его фармацевтически приемлемую соль или его стереоизомер и другое(другие) терапевтическое(терапевтические) средство(средства). Необязательно фармацевтическая композиция может содержать фармацевтически приемлемый носитель, описанный выше. В другом варианте осуществления в настоящем изобретении представлен продукт, содержащий соединение формулы (I) и по меньшей мере одно другое терапевтическое средство в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии.
В одном варианте осуществления в настоящем изобретении представлен набор, содержащий две или более отдельных фармацевтических композиций, по меньшей мере одна из которых содержит соединение формулы (I) или ее подформул, его фармацевтически приемлемую соль или его стереоизомер. В одном варианте осуществления набор содержит средства для раздельного содержания указанных композиций, такие как контейнер, разделенная бутылка или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, как правило, применяемая для упаковки таблеток, капсул и т. п. Набор по настоящему изобретению можно применять для введения различных лекарственных форм, например, для перорального и парентерального применения, для введения отдельных композиций с различными интервалами между введениями доз или для титрования отдельных композиций относительно друг друга. В целях содействия соблюдению режима лечения набор по настоящему изобретению, как правило, содержит инструкции по введению.
В видах комбинированной терапии по настоящему изобретению соединение по настоящему изобретению и другое терапевтическое средство могут быть изготовлены и/или составлены одним и тем же или разными производителями. Более того, соединение по настоящему изобретению и другое терапевтическое средство можно объединять в комбинированной терапии (i) до того, как комбинированный продукт попадает к врачам (например, в случае набора, содержащего соединение по настоящему изобретению и другое терапевтическое средство); (ii) самими врачами (или под наблюдением врача) незадолго до введения; (iii) в самих пациентах, например, во время последовательного введения соединения по настоящему изобретению и другого терапевтического средства.
Соответственно, в настоящем изобретении представлено применение соединения формулы (I) для лечения заболевания или состояния, обусловленного ростом и пролиферацией паразита класса кинетопластид, где лекарственный препарат получен для введения с другим терапевтическим средством. В настоящем изобретении также представлено применение другого терапевтического средства для лечения заболевания или состояния, обусловленного ростом и пролиферацией паразита класса кинетопластид, где лекарственный препарат вводят с соединением формулы (I).
В настоящем изобретении также представлено применение соединения формулы (I) для лечения заболевания или состояния, обусловленного ростом и пролиферацией паразит класса кинетопластид, где пациент ранее (например, в течение 24 часов) подвергался лечению с помощью другого терапевтического средства. В настоящем изобретении также представлено применение другого терапевтического средства для лечения заболевания или состояния, обусловленного ростом и пролиферацией паразита класса кинетопластид, где пациент ранее (например, в течение 24 часов) подвергался лечению с помощью соединения формулы (I).
В одном варианте осуществления другое терапевтическое средство для лечения лейшманиоза выбрано из стибоглюконата, меглюмина антимоната, амфотерицина, милтефосина и паромомицина. В одном варианте осуществления другое терапевтическое средство для лечения болезни Шагаса выбрано из бензнидазола, нифуртимокса и/или амфотерицина. В другом варианте осуществления другое терапевтическое средство для лечения африканского трипаносомоза человека выбрано из пентамидина, сурамина, меларсопрола, эфлорнитина и/или нифуртимокса. Если соединения по настоящему изобретению вводят в сочетании с другими видами терапии, дозировки совместно вводимых соединений, разумеется, будут варьировать в зависимости от типа применяемого дополнительного лекарственного средства, от конкретного применяемого лекарственного средства, от состояния, подвергаемого лечению, и т. д.
ПОЛУЧЕНИЕ СОЕДИНЕНИЙ
Соединения по настоящему изобретению можно получить с помощью ряда способов, известных специалисту в области органического синтеза, с учетом способов, схем реакций и примеров, представленных в данном документе. Для иллюстративных целей схемы реакций, изображенные ниже, обеспечивают возможные пути синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Для более подробного описания отдельных стадий реакции см. раздел "Примеры" ниже. Специалистам в данной области техники будет понятно, что могут применяться другие пути синтеза для синтеза соединений в соответствии с настоящим изобретением. Хотя конкретные исходные материалы и реагенты изображены на схемах и рассмотрены ниже, они могут быть заменены другими исходными материалами и реагентами для обеспечения различных производных и/или условий реакции. Кроме того, многие из соединений, полученных посредством описанных ниже способов, можно дополнительно модифицировать с учетом настоящего изобретения с применением традиционных химических методик, широко известных специалистам в данной области техники. Все способы, описанные в данном документе, могут выполняться в любом подходящем порядке, если не указано иное, или это явно не противоречит контексту.
Соединения формулы (I) можно получать, как в общем проиллюстрировано на схемах 1-3 ниже, где R1, R2, R3, R4 и n определены в разделе "Краткое описание изобретения".
Соединения формулы (I) можно получать, как в общем проиллюстрировано на схема 1-3 ниже, где X представляет собой хлор, бром или трифторметил; и R1, R2, R3, R4 и n определены в разделе "Краткое описание изобретения".
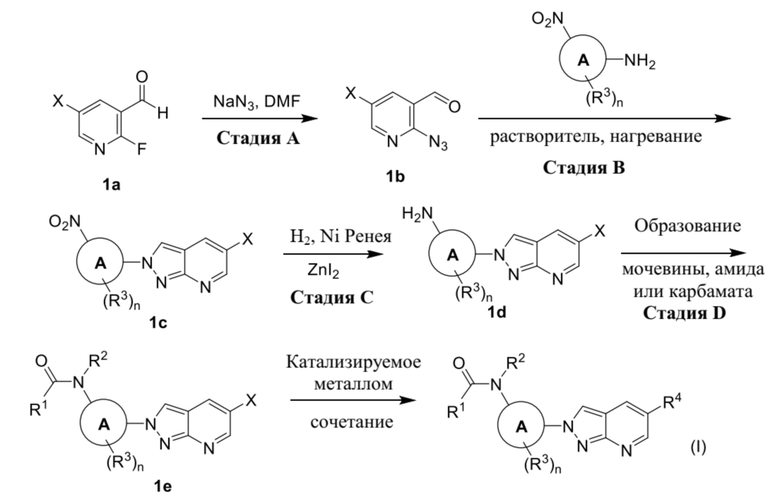
Схема 1
В качестве альтернативы, соединение формулы (I) можно получать из промежуточного соединения (1d) в соответствии с процедурами на схеме 2.
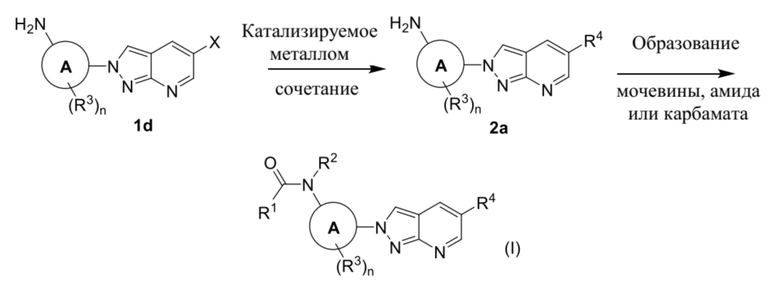
Схема 2
В качестве альтернативы, соединение формулы (I) можно получать из промежуточного соединения (1e) в соответствии с процедурами на схеме 3.
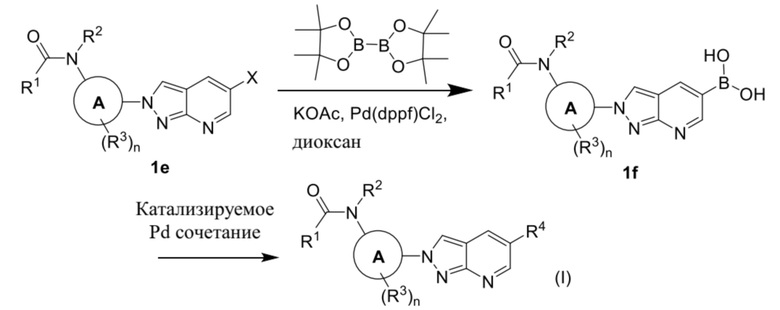
Схема 3
Настоящее изобретение дополнительно включает любой вариант способов по настоящему изобретению, в котором промежуточный продукт, получаемый на любой его стадии, применяют в качестве исходного материала и проводят остальные стадии, или в котором исходные вещества получают in situ при условиях реакции, или в котором компоненты реакционной смеси применяют в форме их солей или оптически чистого вещества. Соединения по настоящему изобретению и промежуточные соединения также можно превращать друг в друга в соответствии со способами, общеизвестными специалистам в данной области техники.
Примеры
Раздел "Примеры" в данном документе всего лишь разъясняет настоящее изобретение и не ограничивает объем настоящего изобретения, заявленный в той или иной форме.
Значения температуры приведены в градусах Цельсия. Если не указано иное, все операции выпаривания осуществляют при пониженном давлении, как правило, от приблизительно 15 мм рт. ст. до 100 мм рт. ст. (= 20-133 мбар). Структура конечных продуктов, промежуточных соединений и исходных материалов подтверждена с помощью стандартных аналитических способов, например микроанализа и спектроскопических характеристик, например, MS, IR, ЯМР.
Все исходные материалы, структурные элементы, реагенты, кислоты, основания, дегидрирующие средства, растворители и катализаторы, используемые для синтеза соединений по настоящему изобретению, либо являются коммерчески доступными, либо их можно получить посредством способов органического синтеза, известных специалисту обычной квалификации в данной области техники (Houben-Weyl 4th Ed. 1952, Methods of Organic Synthesis, Thieme, Volume 21). Также соединения по настоящему изобретению могут быть получены посредством способов органического синтеза, известных специалисту обычной квалификации в данной области техники, как показано в следующих примерах. При необходимости применяют традиционные защитные группы для защиты реакционноспособных функциональных групп в соответствии со стандартной практикой, например, см. у T. W. Greene и P.G.M. Wuts в "Protecting Groups in Organic Synthesis", John Wiley and Sons, 1991.
Если не указано иное, то исходные материалы обычно доступны из коммерческих источников, которые не являются исключительными, таких как Aldrich Chemicals (Милуоки, Висконсин), TCI Fine Chemicals (Япония), Shanghai Chemhere Co., Ltd. (Шанхай, Китай), Aurora Fine Chemicals LLC (Сан-Диего, Калифорния), FCH Group (Украина), Lancaster Synthesis, Inc. (Виндхем, Нью-Гемпшир), Acros Organics (Фейр-Лоун, Нью-Джерси), Maybridge Chemical Company, Ltd. (Корнуолл, Англия), Tyger Scientific (Принстон, Нью-Джерси), AstraZeneca Pharmaceuticals (Лондон, Англия), Chembridge Corporation (США), Matrix Scientific (США), Conier Chem & Pharm Co., Ltd (Китай), Enamine Ltd (Украина), Combi-Blocks, Inc. (Сан-Диего, США), Oakwood Products, Inc. (США), Apollo Scientific Ltd. (Великобритания), Allichem LLC. (США), Rieke Metals (США), Silicycle Inc (Канада) и Ukrorgsyntez Ltd (Латвия); или являются легко получаемыми с применением способов, общеизвестных специалистам в данной области техники (например, получаемыми с помощью способов, в общем описанных в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.), Larock, R.C., Comprehensive Organic Transformations, 2nd-ed., Wiley-VCH Weinheim, Germany (1999) или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения (также доступные с помощью онлайн-базы данных Beilstein).
Сокращения
Сокращения, используемые в данном документе, определены следующим образом: "1x" означает один раз, "2x" означает дважды, "3x" означает трижды, "C" означает градусы Цельсия, "водн." означает водный раствор, "Col" означает колонку, "экв." означает эквивалент или эквиваленты, "г" означает грамм или граммы, "мг" означает миллиграмм или миллиграммы, "л" означает литр или литры, "мл" означает миллилитр или миллилитры, "мкл" означает микролитр или микролитры, "н." означает нормальный, "М" означает молярный, "мкМ" означает микромолярный, "нМ" означает наномолярный, "моль" означает моль или моли, "ммоль" означает миллимоль или миллимоли, "мин." означает минуту или минуты, "ч." означает час или часы, "к. т." означает комнатную температуру, "ON" означает в течение ночи, "атм." означает атмосферу, "фунтов/кв. дюйм" означает фунтов на квадратный дюйм, "конц." означает концентрированный, "водн." означает водный раствор, "нас." или "насыщ." означает насыщенный, "MW" означает молекулярную массу, "mw" или "μwave" означает микроволновое излучение, "т. пл." означает точку плавления, "Wt" означает вес, "MS" или "Mass Spec" означает масс-спектрометрию, "ESI" означает масс-спектрометрию с ионизацией электрораспылением, "HR" означает высокое разрешение, "HRMS" означает масс-спектрометрию высокого разрешения, "LCMS" означает жидкостную хроматографию с масс-спектрометрией, "HPLC" означает жидкостную хроматографию высокого давления, "RP HPLC" означает HPLC с обращенной фазой, "TLC" или "tlc" означает тонкослойную хроматографию, "ЯМР" означает ядерную магнитно-резонансную спектроскопию, "nOe" означает спектроскопию ядерного эффекта Оверхаузера, "1H" означает протон, "δ " означает дельта, "s" означает синглет, "d" означает дуплет, "t" означает триплет, "q" означает квартет, "m" означает мультиплет, "br" означает широкий, "Гц" означает герц, "ee" означает "энантиомерный избыток", и "α", "β", "R", "r", "S", "s", "E", и "Z" являются стереохимическими обозначениями, известными специалисту в данной области техники.
Следующие сокращения, применяемые в данном документе ниже, имеют соответствующие значения:
AIBN азобисизобутиронитрил
Bn бензил
Boc трет-бутоксикарбонил
Boc2O ди-трет-бутилдикарбонат
Bu бутил
Cs2CO3 безводный карбонат цезия
CHCl3 хлороформ
DAST трифторид диэтиламиносеры
DBA дибензилиденацетон
DBU 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин
DCM дихлорметан
DIEA N, N-диизопропилэтиламин
DMAP 4-диметиламинопиридин
DMF диметилформамид
DMSO диметилсульфоксид
DPP дифенилфосфин
DPPA дифенилфосфорилазид
Dppf 1,1'-бис(дифенилфосфино)ферроцен
EA этилацетат
Экв. эквивалент
Et этил
Et2O диэтиловый эфир
EtOH этанол
EtOAc этилацетат
FA муравьиная кислота
HATU 2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат
HCl хлористоводородная кислота
i-Bu изобутил
i-Pr изопропил
KOAc ацетат калия
LiAlH4 алюмогидрид лития
Me метил
mCPBA 3-хлорпероксибензойная кислота
MeCN ацетонитрил
MnO2 диоксид марганца
N2 азот
NaBH4 борогидрид натрия
NaHCO3 бикарбонат натрия
Na2SO4 сульфат натрия
NBS N-бромсукцинимид
Ph фенил
PPh3 трифенилфосфин
Ph3P=O трифенилфосфиноксид
Rf фактор удерживания
к. т. комнатная температура (C)
SFC сверхкритическая флюидная хроматография
t-Bu или But трет-бутил
T3P® ангидрид пропанфосфоновой кислоты
TEA триэтиламин
TFA трифторуксусная кислота
THF тетрагидрофуран
Troc 2,2,2-трихлорэтил
LCMS
Способ 1 :
Система для UPLC Acquity Waters:
-Система управления бинарным градиентом Acquity с дегазатором
-Отделение колонки Acquity, установленное на 50°C
-Диодно-матричный детектор Acquity
Автоматический дозатор HTS Pal Leap Technologies
Хемилюминесцентный детектор на азот (CLND) Antek
Масс-спектрометр Waters 3100
Колонка для HPLC: Syncronis C18 30×2,1 мм Thermo
Подвижная фаза: (A) 95% H2O/5% MeOH/IPA (75/25, об./об.) + 0,05% муравьиной кислоты, (B) MeOH/IPA (75/25, об./об.) + 0,035% муравьиной кислоты
Градиент: 0,4 мл/минута, исходный 2% B в течение 1,0 минуты, подъем до 95% B за 2,5 минуты, до момента времени 4,0 минуты, возврат к 2% B до момента времени 4,25 минуты и до окончания цикла в момент времени 5,0.
Сканирование при MS: от 150 до 1000 а.е.м. за 1 секунду
Диодно-матричный детектор: 190 нм - 400 нм
Способ 2 :
Система для UPLC Acquity Waters:
-Система управления бинарным градиентом Acquity с дегазатором
-Отделение колонки Acquity, установленное на 50°C
-Диодно-матричный детектор Acquity
-Acquity ELSD
Автоматический дозатор HTS Pal Leap Technologies
Масс-детектор Waters Qda
Колонка для HPLC: Acquity C18 1,7 мкм, 2,1 × 30 мм, Waters
Подвижная фаза: (A) H2O+0,05% TFA и (B) ацетонитрил+0,05% TFA
Градиент: 1 мл/минута, исходный 5% B в течение 0,1 минуты, подъем до 95% B в течение 1,5 минуты, выдерживание до момента времени 1,6 минуты, затем до 100% B в момент времени 1,7 и возврат к 5% B до момента времени 1,9 минуты и до окончания цикла в момент времени 2,25.
Сканирование при MS: от 160 до 1000 а.е.м. за 0,4 секунды
Диодно-матричный детектор: 214 нм - 400 нм
ЯМР. Протонные спектры регистрировали на Bruker AVANCE II 400 МГц с 5 мм криозондом QNP или Bruker AVANCE III 500 МГц с 5 мм зондом QNP, если не указано иное. Химические сдвиги приведены в ppm относительно диметилсульфоксида (δ 2,50), хлороформа (δ 7,26), метанола (δ 3,34) или дихлорметана (δ 5,32). Небольшое количество сухого образца (2-5 мг) растворяли в подходящем дейтерированном растворителе (1 мл).
Промежуточное соединение 1: 3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторанилин (I-1)
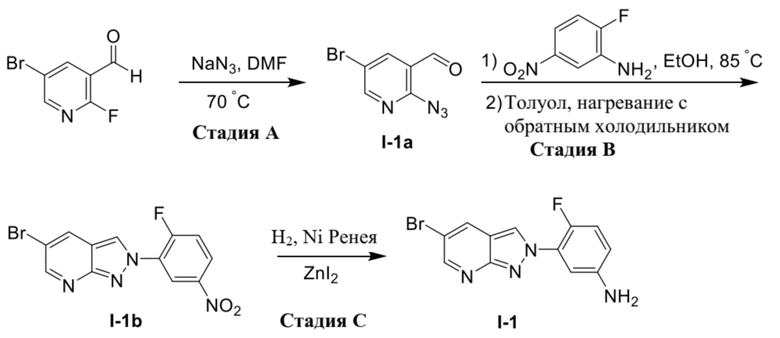
Стадия A. Перемешивали реакционную смесь NaN3 (8 г, 122,5 ммоль, 1 экв.), 5-бром-2-фторпиридин-3-карбальдегида (25 г, 122,5 ммоль, 1 экв.) в DMF (250 мл) при 70°C в течение 1 часа. Реакционную смесь разбавляли этилацетатом (500 мл) и выливали в ледяную воду (500 мл). Водный слой отделяли и экстрагировали этилацетатом (500 мл × 6). Объединенные органические слои промывали солевым раствором (300 мл × 3), высушивали над сульфатом натрия и концентрировали. Остаток растирали с Et2O (100 мл) с получением соединения I-1a в виде желтого твердого вещества. 1H ЯМР: (400 МГц, DMSO-d6) δ ppm 10,26-10,46 (m, 1 H), 10,12 (d, J=1,51 Гц, 1 H), 8,64 (d, J=1,51 Гц, 1 H). С целью безопасного обращения с низкомолекулярными азидами данную реакцию проводили в нескольких партиях и объединяли для применения на стадии B.
Стадия B. Перемешивали реакционную смесь соединения 1-1a (30 г, 132,1 ммоль, 1 экв.) и 2-фтор-5-нитроанилина (20,6 г, 132,1 ммоль, 1 экв.) в EtOH (500 мл) при 85°C в течение 4 часов в атмосфере N2. Реакционную смесь концентрировали in vacuo. Добавляли толуол (700 мл) в неочищенный материал и реакционную смесь нагревали с обратным холодильником в течение 16 ч. Реакционную смесь концентрировали и материал очищали с помощью хроматографии на силикагеле с элюированием от 0 до 50% EtOAc в DCM. Полученный материал растирали с EtOAc (100 мл) с получением соединения 1-1b в виде желтого твердого вещества. 1H ЯМР: (400 МГц, DMSO-d6) δ ppm 9,04 (d, J=2,26 Гц, 1 H), 8,85 (dd, J=6,53, 3,01 Гц, 1 H), 8,79 (d, J=2,26 Гц, 1 H), 8,60-8,71 (m, 1 H), 8,47 (dt, J=9,03, 3,39 Гц, 1 H), 7,92 (dd, J=10,54, 9,29 Гц, 1 H).
Стадия C. Перемешивали реакционную смесь соединения 1-1b (25 г, 74,2 ммоль, 1 экв.), дийодцинка (9,5 г, 29,7 ммоль, 0,4 экв.) и Ni Ренея (50 г, 583,6 ммоль, 7,9 экв.) в THF (500 мл) при 20°C в течение 12 часов в атмосфере H2 при давлении 15 фунтов/кв. дюйм. Реакционную смесь разбавляли с помощью MeOH (450 мл), фильтровали и осадок на фильтре промывали с помощью MeOH (200 мл × 3). Фильтрат концентрировали, и растирали с водой (500 мл), и высушивали in vacuo с получением соединения I-1 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,81 (br d, J=1,51 Гц, 1 H), 8,71 (br d, J=1,76 Гц, 1 H), 8,58 (br d, J=1,76 Гц, 1 H), 7,10-7,41 (m, 2 H), 6,56-6,85 (m, 1 H), 5,45 (br s, 2 H).
Промежуточное соединение 2: 2,2,2-трихлорэтил-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)карбамат (I-2) 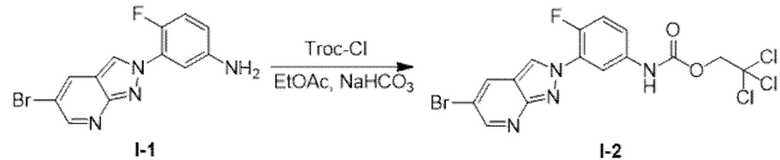
В раствор соединения I-1 (5 г, 16,3 ммоль, 1 экв.) в EtOAc (150 мл) добавляли насыщенный водный раствор NaHCO3 (150 мл). Реакционную смесь охлаждали до 0°C с последующим добавлением по каплям 2,2,2-трихлорэтилкарбонилхлорида (6,9 г, 32,6 ммоль, 2 экв.). Смесь перемешивали при 0°C в течение 2 ч. и затем перемешивали при 25°C в течение 20 ч. Твердое вещество собирали посредством фильтрования и промывали водой (150 мл) и этилацетатом (20 мл) с получением соединения I-2 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 10,57 (br s, 1 H), 8,92 (d, J=2,51 Гц, 1 H), 8,76 (d, J=2,26 Гц, 1 H), 8,63 (d, J=2,51 Гц, 1 H), 8,27 (br d, J=4,52 Гц, 1 H), 7,62-7,67 (m, 1 H), 7,55-7,61 (m, 1 H), 4,99 (s, 2 H).
Промежуточное соединение 3: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-3,3-дифторазетидин-1-карбоксамид (I-3) (также соединение 125)
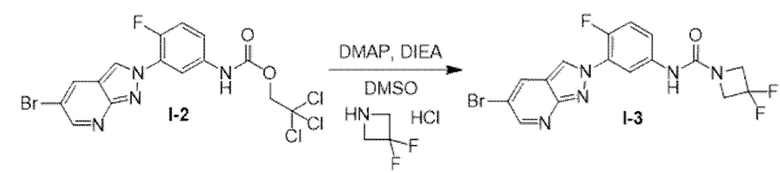
В раствор соединения I-2 (2 г, 4,14 ммоль, 1 экв.) и 3,3-дифторазетидина-HCl (1,1 г, 8,3 ммоль, 2 экв.) в DMSO (15 мл) добавляли DMAP (25,3 мг, 207 мкмоль, 0,05 экв.) и DIEA (1,1 г, 8,3 ммоль, 1,45 мл, 2 экв.). Смесь перемешивали при 60°C в течение 15 ч. Продукт осаждали путем добавления 60 мл воды и 10 мл этилацетата и собирали посредством фильтрования. Осадок на фильтре промывали с помощью 40 мл воды и 10 мл этилацетата и полученное твердое вещество высушивали при пониженном давлении с получением соединения I-3 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 9,25 (s, 1 H), 8,91 (br s, 1 H), 8,76 (br s, 1 H), 8,63 (br s, 1 H), 8,25 (br s, 1 H), 7,69 (br s, 1 H), 7,51 (br t, J=10,16 Гц, 1 H), 4,41 (br t, J=12,67 Гц, 4 H).
Промежуточное соединение 4: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-3-фторазетидин-1-карбоксамид (I-4) (также соединение 126)
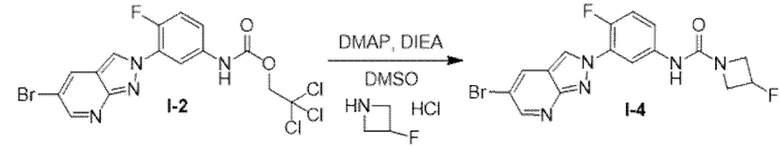
В раствор соединения I-2 (2 г, 4,14 ммоль, 1 экв.) и 3-фторазетидина-HCl (924 мг, 8,28 ммоль, 2 экв.) в DMSO (15 мл) добавляли DMAP (25,3 мг, 207 мкмоль, 0,05 экв.) и DIEA (1,07 г, 8,28 ммоль, 1,45 мл, 2 экв.). Смесь перемешивали при 60°C в течение 15 ч. Продукт осаждали путем добавления 60 мл воды и 10 мл этилацетата и собирали посредством фильтрования. Осадок на фильтре промывали с помощью 40 мл воды и 10 мл этилацетата и полученное твердое вещество высушивали при пониженном давлении с получением соединения I-4 в виде твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 8,98 (s, 1 H), 8,90 (d, J=2,51 Гц, 1 H), 8,76 (d, J=2,51 Гц, 1 H), 8,63 (d, J=2,51 Гц, 1 H), 8,24 (dd, J=7,03, 2,76 Гц, 1 H), 7,67-7,74 (m, 1 H), 7,49 (dd, J=11,17, 9,16 Гц, 1 H), 5,29-5,51 (m, 1 H), 4,25-4,39 (m, 2 H), 3,96-4,10 (m, 2 H).
Промежуточное соединение 5: (R)-N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-3-фторпирролидин-1-карбоксамид (I-5) (также соединение 127)
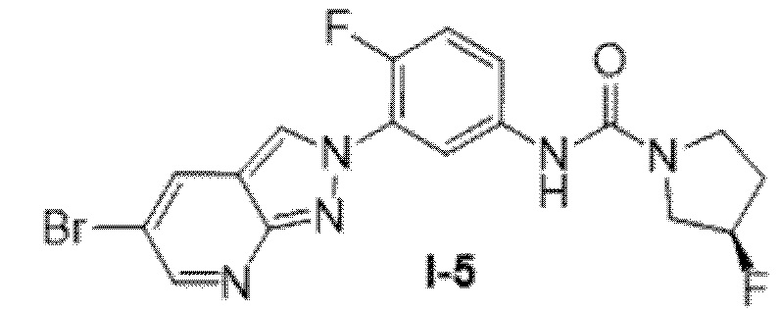
Промежуточное соединение I-5 получали подобным образом, как промежуточное соединение I-3, с применением (R)-3-фторпирролидина-HCl в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 8,90 (d, J=2,26 Гц, 1 H), 8,76 (d, J=2,51 Гц, 1 H), 8,66 (s, 1 H), 8,63 (d, J=2,26 Гц, 1 H), 8,27 (dd, J=7,03, 2,76 Гц, 1 H), 7,71-7,77 (m, 1 H), 7,48 (dd, J=11,17, 9,16 Гц, 1 H), 5,28-5,48 (m, 1 H), 3,40-3,78 (m, 4 H), 2,01-2,27 (m, 2 H).
Промежуточное соединение 6: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)азетидин-1-карбоксамид I-6 (также соединение 129)
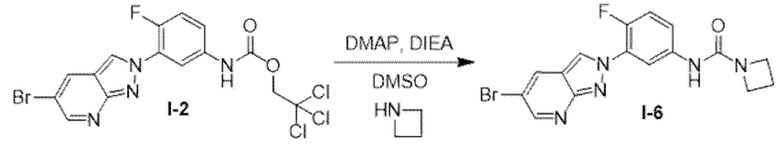
В раствор соединения I-2 (1,6 г, 3,32 ммоль, 1 экв.) и азетидина (379 мг, 6,63 ммоль, 2 экв.) в DMSO (15 мл) добавляли DMAP (20,3 мг, 166 мкмоль, 0,05 экв.) и DIEA (857 мг, 6,63 ммоль, 2 экв.). Смесь перемешивали при 60°C в течение 15 ч. Продукт осаждали путем добавления 60 мл воды и 10 мл этилацетата и собирали посредством фильтрования. Осадок на фильтре промывали с помощью 40 мл воды и 10 мл этилацетата и полученное твердое вещество высушивали при пониженном давлении. Полученное твердое вещество растирали с 10 мл этилацетата с получением соединения I-6 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 8,89 (d, J=2,01 Гц, 1 H), 8,70-8,80 (m, 2 H), 8,62 (d, J=2,26 Гц, 1 H), 8,25 (dd, J=6,90, 2,38 Гц, 1 H), 7,72 (br d, J=9,03 Гц, 1 H), 7,46 (br t, J=10,16 Гц, 1 H), 3,98 (br t, J=7,53 Гц, 4 H), 2,20 (квинт., J=7,59 Гц, 2 H).
Промежуточное соединение 7: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-2,4-диметилоксазол-5-карбоксамид (I-7) (также соединение 128)
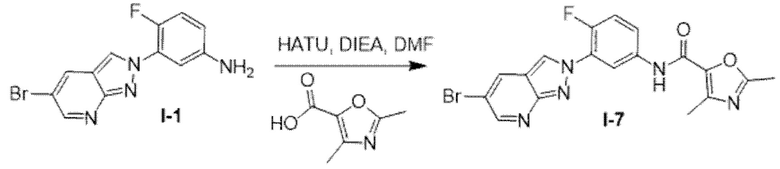
В раствор соединения I-1 (1 г, 3,3 ммоль, 1 экв.) и 2,4-диметилоксазол-5-карбоновой кислоты (552,1 мг, 3,9 ммоль, 1,2 экв.) в DMF (15 мл) добавляли HATU (1,5 г, 3,9 ммоль, 1,2 экв.) и DIEA (632 мг, 4,9 ммоль, 854 мкл, 1,50 экв.). Смесь перемешивали при 25°C в течение 13 часов. Добавляли насыщенный водный раствор NaHCO3 (40 мл) в реакционную смесь и собирали твердое вещество посредством фильтрования. Осадок на фильтре промывали водой (30 мл) и этилацетатом (10 мл). Полученное твердое вещество растирали в воде (20 мл) с получением соединения I-7 в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 10,53 (s, 1 H), 8,94 (d, J=2,51 Гц, 1 H), 8,77 (d, J=2,26 Гц, 1 H), 8,64 (d, J=2,26 Гц, 1 H), 8,58 (dd, J=7,15, 2,64 Гц, 1 H), 7,89-7,97 (m, 1 H), 7,59 (dd, J=11,17, 9,16 Гц, 1 H), 2,52-2,53 (s, 3 H), 2,40 (s, 3 H).
Промежуточное соединение 8: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)фуран-2-карбоксамид (I-8) (также соединение 130)
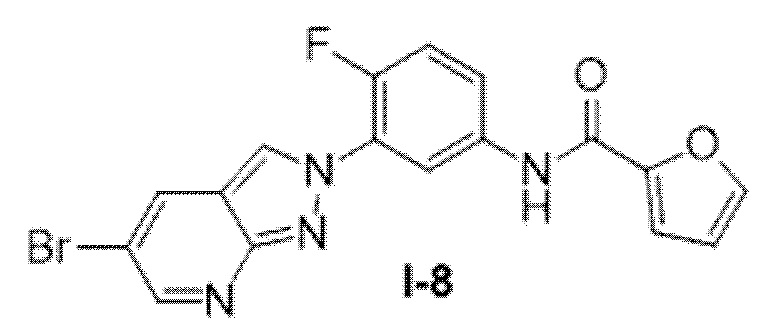
Промежуточное соединение I-8 получали подобным образом, как промежуточное соединение I-7, с применением фуран-2-карбоновой кислоты в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 10,58 (br s, 1 H), 8,94 (d, J=2,26 Гц, 1 H), 8,77 (d, J=2,26 Гц, 1 H), 8,64 (d, J=2,26 Гц, 1 H), 8,53 (dd, J=7,03, 2,51 Гц, 1 H), 7,92-8,02 (m, 2 H), 7,60 (dd, J=11,04, 9,29 Гц, 1 H), 7,40 (d, J=3,26 Гц, 1 H), 6,74 (dd, J=3,26, 1,51 Гц, 1 H).
Промежуточное соединение 9: 4-фтор-3-(5-изопропил-2H-пиразоло[3,4-b]пиридин-2-ил)анилин (I-9)
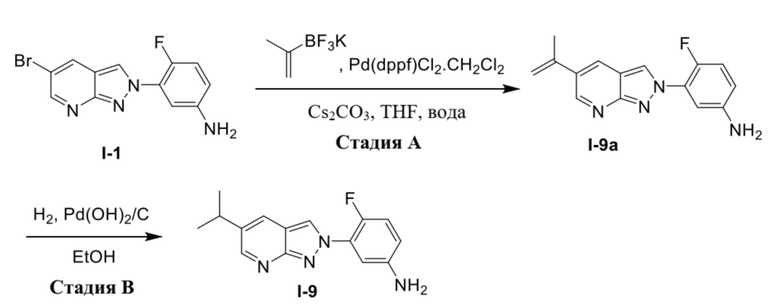
Стадия A. В раствор промежуточного соединения I-1 (4 г, 13,0 ммоль, 1 экв.) в THF (60 мл) и H2O (9 мл) добавляли Cs2CO3 (12,7 г, 39,1 ммоль, 3 экв.), Pd(dppf)Cl2.CH2Cl2 (531,8 мг, 651,2 мкмоль, 0,05 экв.) и трифтор(проп-1-ен-2-ил)борат калия (2,2 г, 14,7 ммоль, 1,1 экв.). Смесь перемешивали при 80°C в течение 16 ч. Реакционную смесь экстрагировали с помощью EtOAc (100 мл × 2). Объединенные органические слои промывали солевым раствором (100 мл), высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью хроматографии на силикагеле с элюированием петролейным эфиром: EtOAc=1:2) с получением соединения I-9a в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,00 (d, J=2,26 Гц, 1 H), 8,78 (d, J=2,38 Гц, 1 H), 8,23 (d, J=2,26 Гц, 1 H), 7,09-7,29 (m, 2 H), 6,68 (dt, J=8,88, 3,34 Гц, 1 H), 5,64 (s, 1 H), 5,44 (s, 2 H), 5,22 (s, 1 H), 2,18 (s, 3 H), M+H=269,1.
Стадия B. В раствор промежуточного соединения I-9a (3,4 г, 12,7 ммоль, 1 экв.) в EtOH (216 мл) добавляли 20% Pd(OH)2/C (5,3 г, 7,60 ммоль, 0,6 экв.) и смесь перемешивали при 25°C в течение 4 ч. в атмосфере H2 при давлении 15 фунтов/кв. дюйм. Реакционную смесь фильтровали через целит и осадок на фильтре промывали этанолом (30 мл). Фильтрат концентрировали, полученный остаток растворяли в EtOAc (10 мл) и добавляли 4 М HCl в EtOAc (2 мл). Твердое вещество собирали посредством фильтрования и высушивали в вакууме с получением HCl-соли соединения I-9 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,10 (d, J=2,26 Гц, 1 H), 8,90 (d, J=2,01 Гц, 1 H), 8,47 (br d, J=1,63 Гц, 1 H), 8,07 (br dd, J=6,46, 2,45 Гц, 1 H), 7,72 (br dd, J=10,85, 9,10 Гц, 1 H), 7,58 (dt, J=8,50, 3,28 Гц, 1 H), 3,07-3,25 (m, 1 H), 1,32 (d, J=6,78 Гц, 6 H), M+H=271,1.
Промежуточное соединение 10: (2-(2-фтор-5-(3-фторазетидин-1-карбоксамидо)фенил)-2H-пиразоло[3,4-b]пиридин-5-ил)бороновая кислота (I-10)
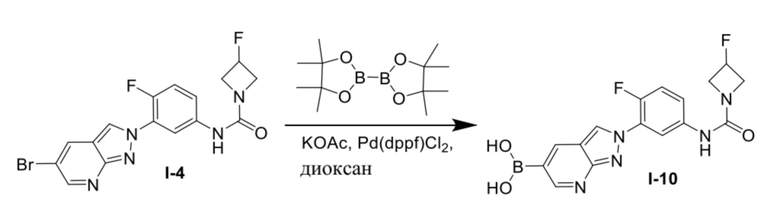
В раствор промежуточного соединения I-4 (300 мг, 734,9 мкмоль, 1 экв.) в диоксане (18 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (223,9 мг, 881,9 мкмоль, 1,2 экв.), Pd(dppf)Cl2 (53,8 мг, 73,5 мкмоль, 0,1 экв.) и KOAc (144,3 мг, 1,5 ммоль, 2 экв.). Смесь перемешивали при 100°C в течение 16 ч. Реакционную смесь концентрировали и остаток промывали с помощью EtOAc (5 мл), воды (5 мл) и высушивали при пониженном давлении с получением соединения I-10 в виде черного твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,00 (br s, 2 H), 8,70 (br s, 1 H), 8,45 (br s, 1 H), 8,24 (br s, 1 H), 7,69 (br s, 1 H), 7,56 (br s, 1 H), 7,50 (br s, 1 H), 6,59 (br s, 1 H), 5,25-5,56 (m, 1 H), 4,33 (br s, 2 H), 3,90-4,13 (m, 2 H), M+H=374,2.
Промежуточное соединение 11: 4-фтор-3-(5-(пиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)анилин I-11
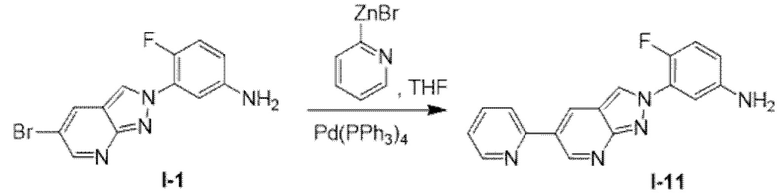
В колбе объединяли соединение I-1 (947 мг, 3,1 ммоль) и Pd(Ph3P)4 (178 мг, 0,15 ммоль) и помещали в атмосферу азота. Добавляли 0,5 М бромид пиридин-2-илцинка(II) в THF (24,7 мл, 12,3 ммоль) и реакционную смесь нагревали до 65°C в течение 4 ч. Реакционную смесь гасили водой и концентрировали. Неочищенный материал очищали с помощью флэш-хроматографии на колонке C18 с элюированием от 10 до 80% ацетонитрила в воде с получением соединения I-11 в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 9,45 (d, J=2,3 Гц, 1H), 8,93 (dd, J=3,4, 2,4 Гц, 2H), 8,73 (ddd, J=4,8, 1,8, 0,9 Гц, 1H), 8,12 (dt, J=8,0, 1,0 Гц, 1H), 7,95 (td, J=7,7, 1,8 Гц, 1H), 7,46-7,40 (m, 1H), 7,26-7,16 (m, 2H), 6,69 (ddd, J=8,9, 3,9, 2,9 Гц, 1H), 5,46 (s, 2H).
Промежуточное соединение 12: 2-(5-амино-2-фторфенил)-N, N-диметил-2H-пиразоло[3,4-b]пиридин-5-амин (I-12)
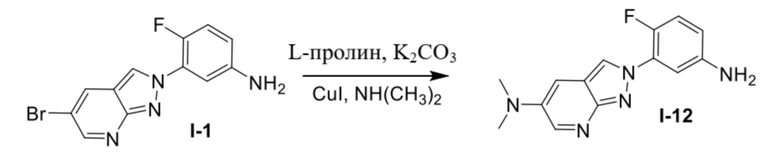
Перемешивали реакционную смесь N-метилметанамина (4,4 г, 98,7 ммоль, 5 мл, 50,5 экв.), I-1 (0,6 г, 1,9 ммоль, 1 экв.), L-пролина (22,5 мг, 195,4 мкмоль, 0,1 экв.), CuI (37,2 мг, 195,4 мкмоль, 0,1 экв.) и K2CO3 (540 мг, 3,9 ммоль, 2 экв.) в DMSO (5 мл) при 90°C в течение 12 ч. Реакционную смесь гасили 10% водным раствором NH4OH (20 мл) и экстрагировали этилацетатом (25 мл × 4). Объединенные слои на основе этилацетата промывали солевым раствором (15 мл × 3), высушивали над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием с помощью 20% этилацетата и 20% DCM в петролейном эфире с получением I-12 в виде желтого твердого вещества. 1H ЯМР: (400 МГц, DMSO-d6) δ ppm 8,68 (br s, 1 H), 8,48 (br s, 1 H), 7,16 (br s, 3 H), 6,63 (br s, 1 H), 5,38 (br s, 2 H), 2,95 (br s, 6 H). M+H=391,1.
Промежуточное соединение 13: 2,2,2-трихлорэтил-(3-(5-(диметиламино)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)карбамат (I-13)
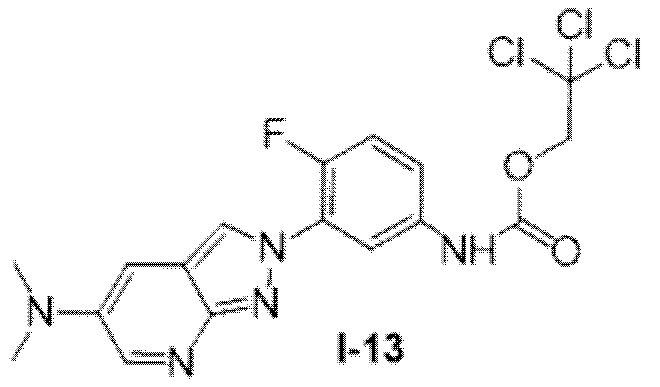
Синтез I-13 описан в примере 9.
Промежуточное соединение 14: (R)-N-(3-(5-(3,6-дигидро-2H-пиран-4-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-3-фторпирролидин-1-карбоксамид (I-14)
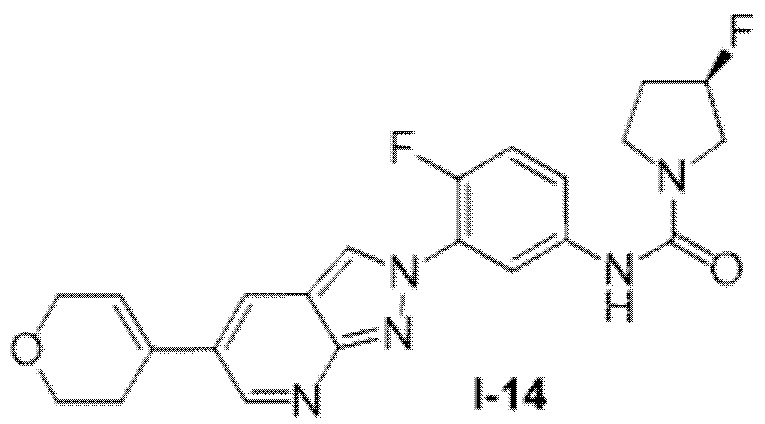
Синтез I-14 описан в примере 18.
Промежуточное соединение 15 : N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-5-азаспиро[2.3]гексан-5-карбоксамид I-15 (также соединение 254)
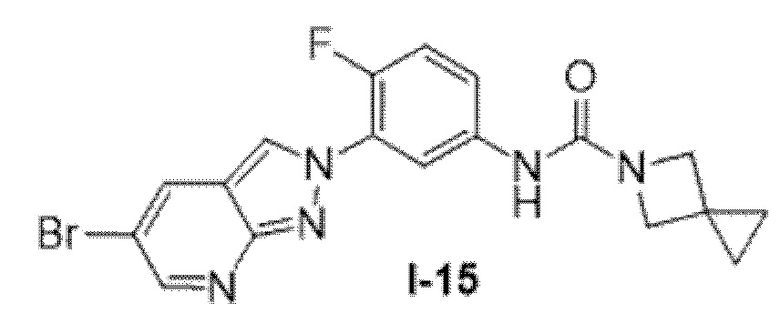
Промежуточное соединение I-15 (соединение 254) получали подобным образом, как промежуточное соединение I-3, с применением полуоксалата 5-азаспиро[2.3]гексана в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 8,90 (d, J=2,45 Гц, 1 H), 8,85 (s, 1 H), 8,74-8,78 (m, 1 H), 8,61-8,65 (m, 1 H), 8,26 (dd, J=7,09, 2,69 Гц, 1 H), 7,69-7,76 (m, 1 H), 7,47 (dd, J=11,25, 9,29 Гц, 1 H), 4,05 (s, 4 H), 0,66 (s, 4 H). Способ 1: RT=2,95 мин.; M+H=416,0, 418,0.
Промежуточное соединение 16 : 3-(5-хлор-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторанилин (I-16)
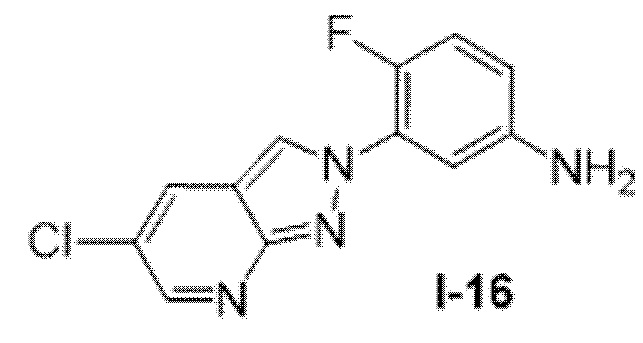
Промежуточное соединение I-16 получали подобным образом, как промежуточное соединение I-1, с применением 5-хлор-2-фторникотинальдегида в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,82 (d, J=2,26 Гц, 1 H), 8,67 (d, J=2,51 Гц, 1 H), 8,44 (d, J=2,51 Гц, 1 H), 7,12-7,28 (m, 2 H), 6,61-6,75 (m, 1 H), 5,44 (br s, 2 H).
Промежуточное соединение 17 : 2-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)пиридин-4-амин (I-17)
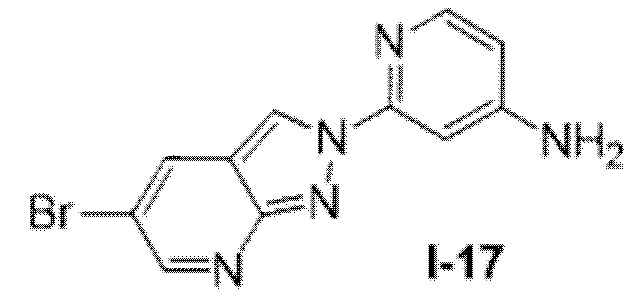
Промежуточное соединение I-17 получали подобным образом, как промежуточное соединение I-1, с применением 4-нитропиридин-2-амина в качестве исходного материала на стадии B. 1H ЯМР (400 МГц, DMSO) δ ppm 9,19 (br s, 1H), 8,73 (br s, 1H), 8,59 (br s, 1H), 8,01 (br d, J=5,27 Гц, 1H), 7,42 (br s, 1H), 6,54-6,74 (m, 3H).
Промежуточное соединение 18 : 3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-хлоранилин (I-18)
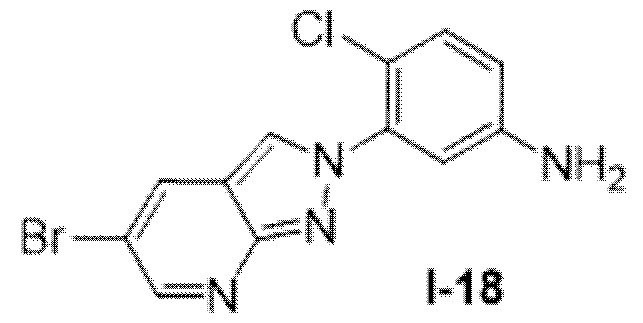
Промежуточное соединение I-18 получали подобным образом, как промежуточное соединение I-1, с применением 2-хлор-5-нитроанилина в качестве исходного материала на стадии B. M+H=325,0.
Промежуточное соединение 19 : 3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)анилин (I-19)
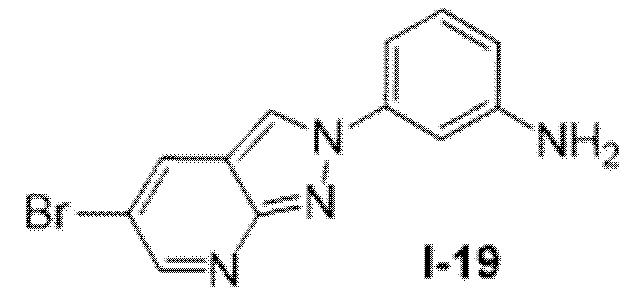
Промежуточное соединение I-19 получали подобным образом, как промежуточное соединение I-1, с применением 3-нитроанилина в качестве исходного материала на стадии B. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,99 (s, 1H), 8,69 (d, J=2,44 Гц, 1H), 8,56 (d, J=2,45 Гц, 1H), 7,31 (t, J=2,08 Гц, 1H), 7,12-7,26 (m, 2H), 6,61-6,72 (m, 1H), 5,56 (br s, 2H).
Промежуточное соединение 20 : 2,2,2-трихлорэтил-(2-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)пиридин-4-ил)карбамат (I-20)
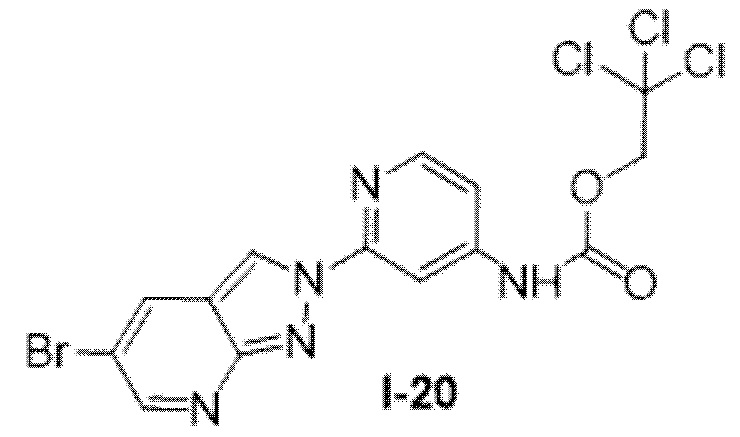
Синтез I-20 описан в примере 21.
Промежуточное соединение 21 : 3-(2-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)пиридин-4-ил)-1,1-диметилмочевина (I-21)
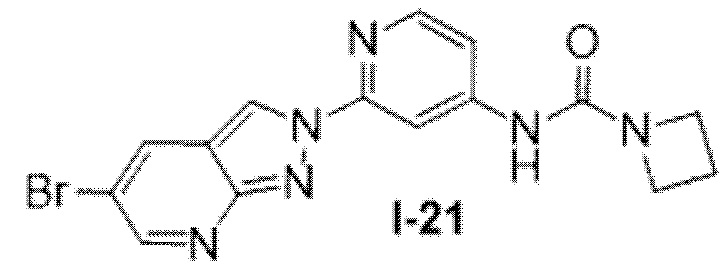
Синтез I-21 описан в примере 21.
Промежуточное соединение 22 : N-(2-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)пиридин-4-ил)-3,3-дифторазетидин-1-карбоксамид (I-22)
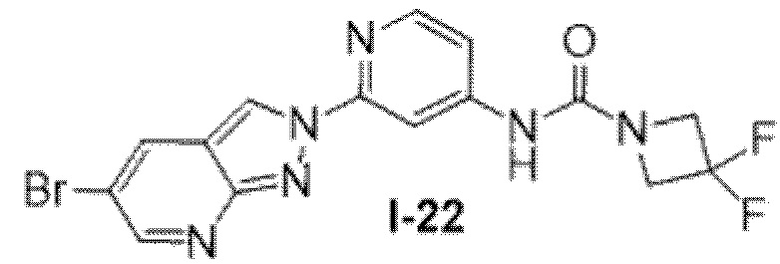
Синтез I-22 описан в примере 22.
Промежуточное соединение 23 : 4-фтор-3-(5-(трифторметил)-2H-пиразоло[3,4-b]пиридин-2-ил)анилин (I-23)
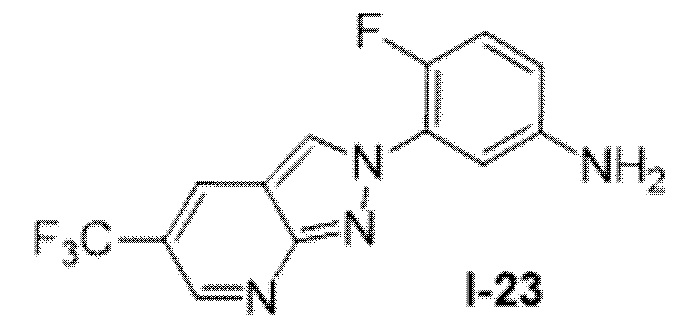
Промежуточное соединение I-23 получали подобным образом, как промежуточное соединение I-1, с применением 2-фтор-5-(трифторметил)никотинальдегида в качестве исходного материала. 1H ЯМР (400 МГц, DMSO), δ ppm, 9,06 (d, J=2,01 Гц, 1 H), 8,98 (d, J=2,26 Гц, 1 H), 8,85 (s, 1 H), 7,16-7,30 (m, 2 H), 6,72 (dt, J=8,91, 3,33 Гц, 1 H), 5,48 (s, 2 H).
Промежуточное соединение 24: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)циклопропанкарбоксамид (I-24)
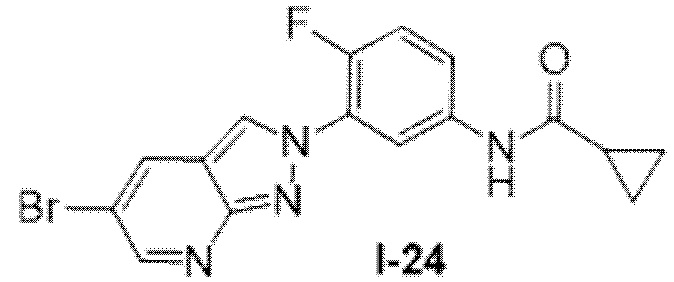
Промежуточное соединение I-24 получали подобным образом, как промежуточное соединение I-7, с применением циклопропанкарбоновой кислоты в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,90 (d, J=2,38 Гц, 1 H), 8,75 (d, J=2,38 Гц, 1 H), 8,62 (d, J=2,38 Гц, 1 H), 8,38 (dd, J=7,03, 2,51 Гц, 1 H), 8,25-8,30 (m, 1H), 7,66-7,75 (m, 1 H), 7,53 (dd, J=11,17, 9,16 Гц, 1 H), 1,72-1,83 (m, 1 H), 0,83 (d, J=6,15 Гц, 4 H).
Промежуточное соединение 25: 3-(5-(1H-пиразол-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторанилин (I-25)
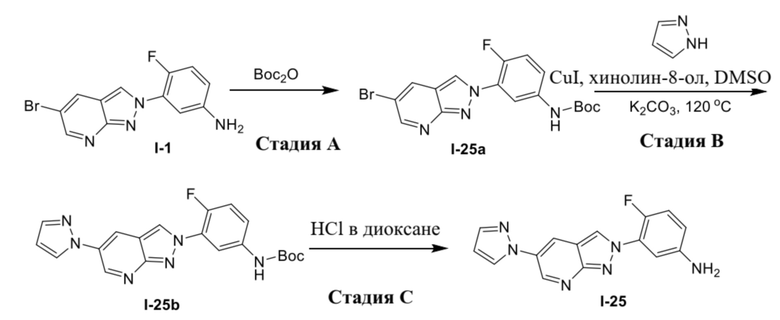
Стадия A. Перемешивали реакционную смесь I-1 (10 г, 32,6 ммоль, 1 экв.) и Boc2O (21,3 г, 97,7 ммоль, 22,4 мл, 3 экв.) в THF (100 мл) при 76°C в течение 12 ч. Реакционную смесь концентрировали и остаток растирали с петролейным эфиром (100 мл) с получением I-25a в виде коричневого твердого вещества. M+H=408,9.
Стадия B. Перемешивали реакционную смесь I-25a (0,5 г, 1,2 ммоль, 1 экв.), 1H-пиразола (126 мг, 1,8 ммоль, 1,5 экв.), CuI (35 мг, 184,5 мкмоль, 0,15 экв.), хинолин-8-ола (27 мг, 184,5 мкмоль, 31,9 мкл, 0,15 экв.) и K2CO3 (221 мг, 1,6 ммоль, 1,3 экв.) в DMSO (5 мл) при 120°C в течение 12 ч. Реакционную смесь выливали в 20 мл воды, экстрагировали с помощью DCM (20 мл × 3). Объединенные органические слои промывали солевым раствором (10 мл), высушивали над Na2SO4 и концентрировали с получением I-25b в виде черного твердого вещества. M+H=395,3.
Стадия C. Перемешивали реакционную смесь I-25b (0,4 г, 1,0 ммоль, 1 экв.) в HCl/диоксане (10 мл) при 25°C в течение 12 ч. Реакционную смесь концентрировали и очищали с помощью препаративной HPLC (Phenomenex Gemini 150 × 25 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 18% до 48%, 12 мин.) с получением I-25 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ ppm 9,28 (d, J=2,69 Гц, 1 H), 8,90 (d, J=2,45 Гц, 1 H), 8,61 (dd, J=10,64, 2,57 Гц, 2 H), 7,84 (d, J=1,47 Гц, 1 H), 7,14-7,31 (m, 2 H), 6,66-6,75 (m, 1 H), 6,57-6,65 (m, 1 H), 5,45 (s, 2 H).
Промежуточное соединение 26: 4-фтор-3-(5-(4-метил-1H-имидазол-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)анилин (I-26)
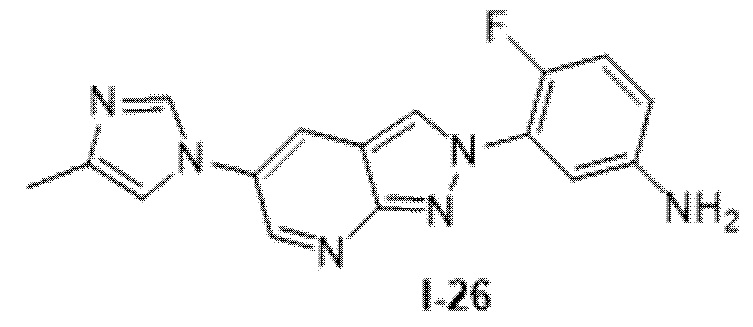
Промежуточное соединение I-26 получали подобным образом, как промежуточное соединение I-25, с применением 4-метил-1H-имидазола в качестве исходного материала на стадии B. 1H ЯМР: (400 МГц, DMSO) δ ppm 9,00 (d, J=2,69 Гц, 1 H), 8,89 (d, J=2,20 Гц, 1 H), 8,37-8,49 (m, 1 H), 8,20 (s, 1 H), 7,53 (s, 1 H), 7,13-7,30 (m, 3 H), 6,61-6,80 (m, 1 H), 5,45 (s, 2 H), 2,21 (s, 3 H).
Промежуточное соединение 27: N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)пирролидин-1-карбоксамид (I-27)
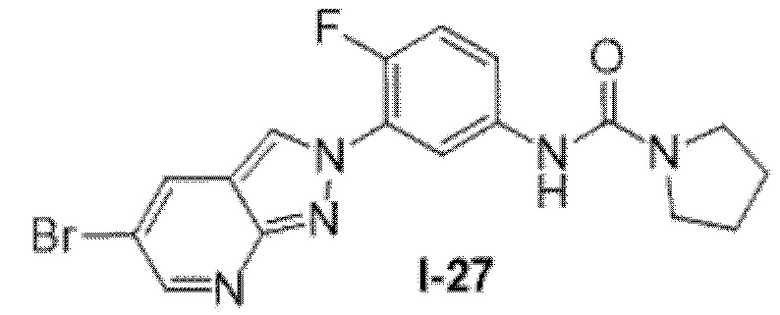
Промежуточное соединение I-27 получали подобным образом, как промежуточное соединение I-3, с применением пирролидина в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 8,89 (d, J=2,26 Гц, 1 H), 8,75 (d, J=2,26 Гц, 1 H), 8,62 (d, J=2,51 Гц, 1 H), 8,51 (s, 1 H), 8,28 (dd, J=7,03, 2,51 Гц, 1 H), 7,74 (dt, J=9,10, 3,48 Гц, 1 H), 7,46 (dd, J=11,29, 9,29 Гц, 1 H), 3,39 (br s, 4 H), 1,87 (br t, J=6,40 Гц, 4 H).
Промежуточное соединение 28: 4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)анилин I-28
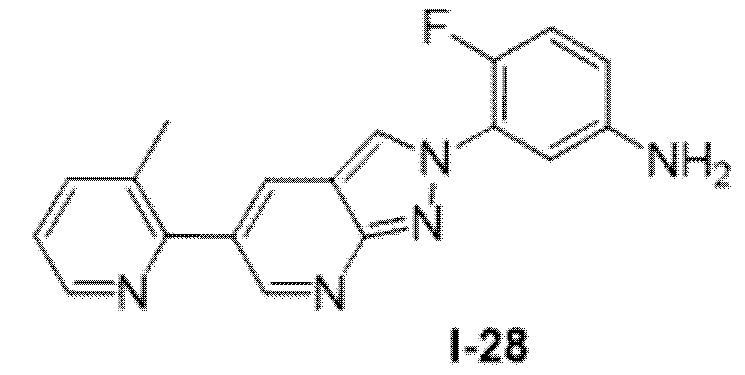
Промежуточное соединение I-28 получали подобным образом, как промежуточное соединение I-11, с применением бромида (3-метилпиридин-2-ил)цинка(II) в качестве исходного материала. 1H ЯМР (400 МГц, DMSO-d6) 8,89 (dd, J=8,2, 2,4 Гц, 2H), 8,56 (dd, J=4,6, 1,7 Гц, 1H), 8,47 (d, J=2,2 Гц, 1H), 7,80 (ddd, J=7,7, 1,7, 0,8 Гц, 1H), 7,40-7,32 (m, 1H), 7,24-7,14 (m, 2H), 6,69 (ddd, J=9,0, 3,9, 2,8 Гц, 1H), 5,46 (s, 2H), 2,44 (s, 3H).
Промежуточное соединение 29: 4-фтор-3-(5-(3-(трифторметил)азетидин-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)анилин I-29
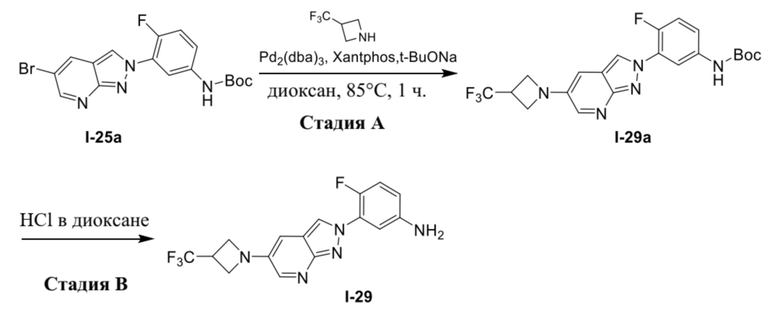
Стадия A. Дегазировали смесь I-25a (100 мг, 245,6 мкмоль, 1 экв.), 3-(трифторметил)азетидина (48 мг, 294,7 мкмоль, 1,2 экв., HCl), NaOBu-t (2 М, 368,3 мкл, 3 экв.), XantPhos (28 мг, 49,11 мкмоль, 0,2 экв.) и Pd2(dba)3 (22 мг, 24,6 мкмоль, 0,1 экв.) в диоксане (2 мл) и затем перемешивали при 80°C в течение 1 часа в атмосфере N2. Смесь гасили и регулировали pH до 7 с помощью 1 М HOAc. Смесь очищали с помощью хроматографии на силикагеле (петролейный эфир/этилацетат=1/1) с получением I-29a в виде желтого твердого вещества. M+H=452,3.
Стадия B. Данную стадию проводили подобным образом, как для I-25, стадия C, с получением I-29. 1H ЯМР (400 МГц, DMSO) δ ppm 8,49 (d, J=2,45 Гц, 1 H), 8,29 (d, J=2,81 Гц, 1 H), 7,09-7,22 (m, 2 H), 7,02 (d, J=2,69 Гц, 1 H), 6,63 (dt, J=8,74, 3,33 Гц, 1 H), 5,38 (s, 2 H), 4,07-4,23 (m, 2 H), 3,98 (dd, J=7,95, 5,38 Гц, 2 H), 3,66-3,83 (m, 1 H).
Промежуточное соединение 30 : трет-бутил-2-(2-(2-фтор-5-(3-фторазетидин-1-карбоксамидо)фенил)-2H-пиразоло[3,4-b]пиридин-5-ил)азетидин-1-карбоксилат (I-30)
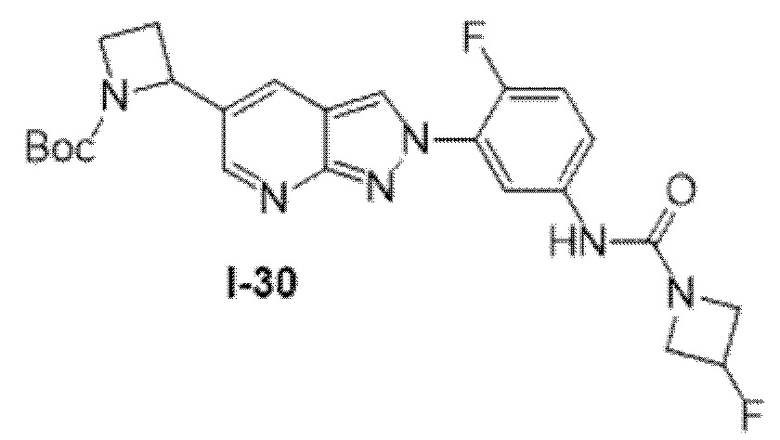
Синтез I-30 описан в примере 24.
Промежуточное соединение 31
: транс-N-(3-(5-бром-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-2-фторциклопропан-1-карбоксамид (I-31) 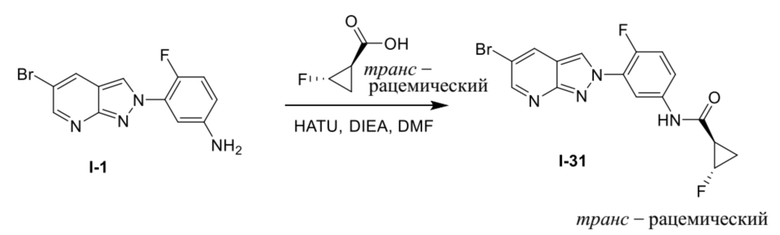
В раствор I-1 (2,5 г, 8,1 ммоль, 1 экв.) и транс-2-фторциклопропанкарбоновой кислоты (1,0 г, 9,8 ммоль, 1,2 экв.) в DMF (20 мл) добавляли HATU (3,7 г, 9,8 ммоль, 1,2 экв.) и DIEA (1,6 г, 12,2 ммоль, 2,1 мл, 1,5 экв.). Смесь перемешивали при 25°C в течение 16 ч. Реакционную смесь разбавляли этилацетатом (150 мл) и промывали насыщенным раствором NaHCO3 (50 мл) и с помощью 100 мл солевого раствора (50 мл × 2), высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток растирали в трет-бутилметиловом эфире (20 мл) и собирали посредством фильтрования с получением I-31 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,74 (s, 1 H), 8,91 (d, J=2,51 Гц, 1 H), 8,75 (d, J=2,26 Гц, 1 H), 8,62 (d, J=2,26 Гц, 1 H), 8,36 (dd, J=7,03, 2,51 Гц, 1 H), 7,60-7,74 (m, 1 H), 7,55 (dd, J=11,17, 9,16 Гц, 1 H), 4,70-5,12 (m, 1 H), 2,89 (s, 1 H), 2,23-2,41 (m, 1 H), 1,46-1,66 (m, 1 H).
Промежуточное соединение 32: 4-(метоксиметил)-2-метилоксазол-5-карбоновая кислота (I-32)
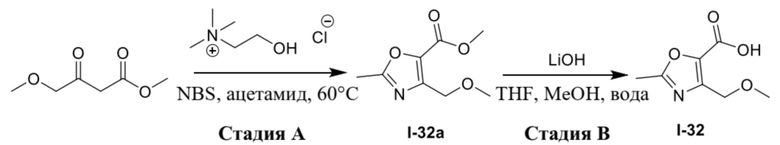
Стадия A. Объединяли ацетамид (1,7 г, 28,6 ммоль, 4 экв.) и 2-гидрокси-N, N, N-триметилэтан-1-аминия хлорид (1 г, 7,2 ммоль, 1 экв.) и нагревали до 60°C в течение 30 мин. В реакционную смесь добавляли NBS (2,5 г, 14,3 ммоль, 2 экв.) и метил-4-метокси-3-оксобутаноат (2,1 г, 14,3 ммоль, 2 экв.) и нагревали до 60°C в течение 1 ч. Реакционную смесь очищали с помощью препаративной TLC с элюированием с помощью 50% этилацетата в петролейном эфире с получением I-32a в виде светло-желтого масла. 1H ЯМР: (400 МГц, CDCl3) δ ppm 4,66 (s, 2H), 3,94 (s, 3H), 3,47 (s, 3H), 2,55 (s, 3H).
Стадия B. Перемешивали смесь I-32a (60 мг, 324,01 мкмоль, 1 экв.) и LiOH·H2O (27,2 мг, 648,0 мкмоль, 2 экв.) в THF (1 мл), MeOH (1 мл) и H2O (0,5 мл) при 25°C в течение 16 ч. в атмосфере N2. Реакционную смесь концентрировали в вакууме с получением 60 мг литиевой соли I-32 в виде желтого твердого вещества.
Промежуточное соединение 33: N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)-4-(метоксиметил)-2-метилоксазол-5-карбоксамид (I-33)
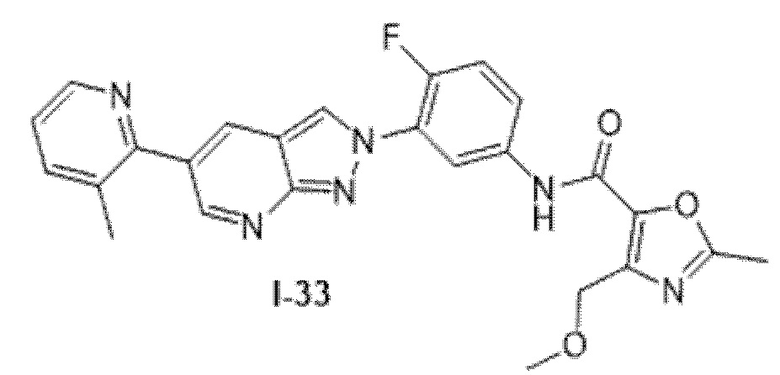
Синтез I-33 описан в примере 28.
Промежуточное соединение 34: 4-метил-2-(метил-d3)оксазол-5-карбоновая кислота (I-34)
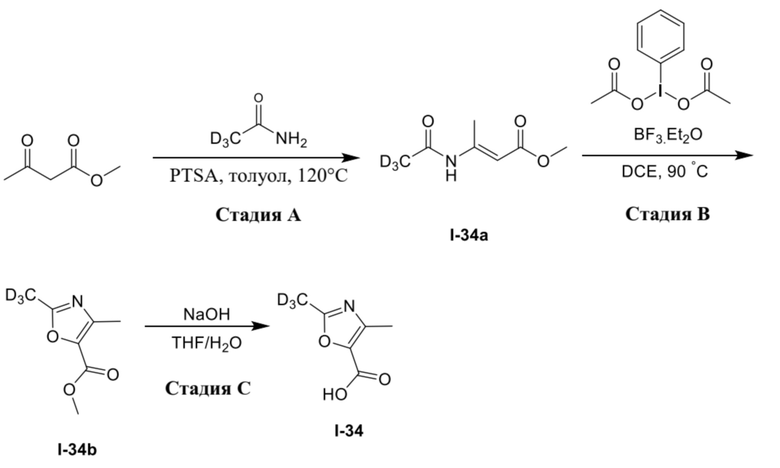
Стадия A. В раствор метил-3-оксобутаноата (900 мг, 7,7 ммоль, 833 мкл, 1 экв.) в толуоле (15 мл) добавляли 2,2,2-тридейтероацетамид (962 мг, 15,5 ммоль, 2 экв.) и TsOH (13,3 мг, 77,5 мкмоль, 0,01 экв.). Смесь перемешивали при 120°C в течение 12 ч. Смесь концентрировали в вакууме с получением остатка, который очищали с помощью хроматографии на силикагеле с элюированием смесью петролейный эфир:этилацетат с градиентом от 30:1 до 10:1 с получением I-34a в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,88 (br s, 1 H), 5,02 (s, 1 H), 3,63 (s, 3 H), 2,29 (s, 3 H).
Стадия B. В раствор I-34a (250 мг, 1,6 ммоль, 1 экв.) в DCE (8 мл) добавляли (диацетоксийод)бензол (654 мг, 2,0 ммоль, 1,3 экв.) и BF3·Et2O (443 мг, 3,1 ммоль, 385 мкл, 2 экв.). Смесь перемешивали при 90°C в течение 2 ч. Смесь концентрировали в вакууме с получением остатка, который очищали с помощью хроматографии на силикагеле с элюированием смесью петролейный эфир:этилацетат с градиентом от 30:1 до 10:1 с получением I-34b в виде белого твердого вещества. 1H ЯМР: (400 МГц, DMSO-d6) δ ppm 3,81 (s, 3 H), 2,33 (s, 3 H).
Стадия C. В раствор I-34b (100 мг, 632 мкмоль, 1 экв.) в THF (1 мл) и H2O (1 мл) добавляли 15% водный раствор NaOH (1 мл). Смесь перемешивали при 70°C в течение 2 ч. Регулировали pH до 5 путем добавления водного раствора HCl (1 М), и образовывалось белое твердое вещество. Суспензию фильтровали и осадок на фильтре промывали с помощью H2O (5 мл). Осадок на фильтре высушивали при пониженном давлении с получением I-34 в виде белого твердого вещества. 1H ЯМР: (400 МГц, DMSO-d6) δ ppm 13,30 (br s, 1 H), 2,32 (s, 3 H).
Промежуточное соединение 35: 2-(трет-бутоксиметил)-N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)-4-метилоксазол-5-карбоксамид (I-35)
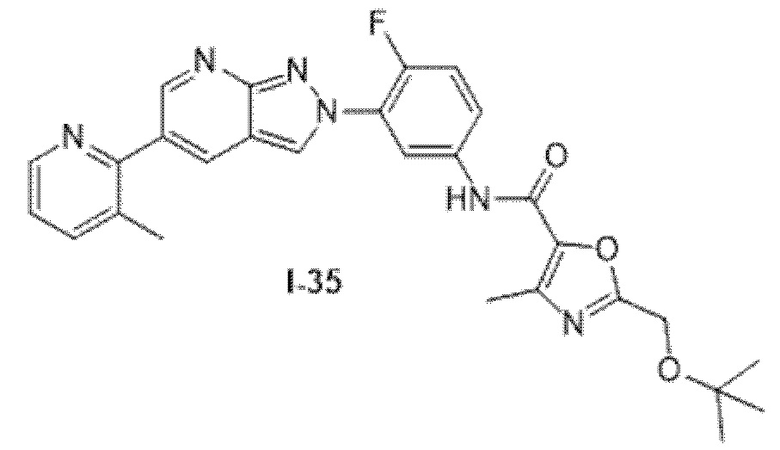
Синтез I-35 описан в примере 29.
Промежуточное соединение 36: 4-фтор-3-(5-фенил-2H-пиразоло[3,4-b]пиридин-2-ил)анилин (I-36)
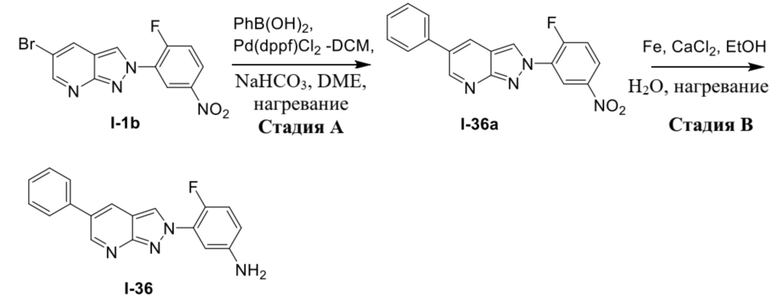
Стадия A. Закрытую пробирку объемом 100 мл, содержащую I-1b (500 мг, 1,483 ммоль) и фенилбороновую кислоту (181 мг, 1,483 ммоль) в DME:H2O (8:2) (15 мл), продували газообразным аргоном в течение 15 мин. Добавляли NaHCO3 (249 мг, 2,966 ммоль) и Pd(dppf)Cl2·DCM (61 мг, 0,074 ммоль) и реакционную смесь перемешивали при 80°C в течение 1 ч. Реакционную смесь разбавляли водой, экстрагировали с помощью EtOAc (100 мл × 2). Объединенные органические слои промывали 1x водой (150 мл) и 1x солевым раствором (100 мл) и затем высушивали над сульфатом натрия и концентрировали in vacuo. Неочищенное соединение очищали с помощью хроматографии на силикагеле с элюированием с помощью 25% EtOAc в н-гексане с получением I-36a. 1H ЯМР (300 МГц, CDCl3): δ 9,32-9,29 (m, 1H), 9,10 (s, 1H), 8,69 (s, 1H), 8,34-8,29 (m, 1H), 8,23 (s, 1H), 7,65 (d, J=6,9 Гц, 2H), 7,55-7,41 (m, 4H); M+H=335,2.
Стадия B. В колбу, содержащую I-36a (150 мг, 0,449 ммоль) в EtOH:H2O (8:2) (20 мл), добавляли порошок железа (370 мг, 6,730 ммоль) и CaCl2 (995 мг, 8,973 ммоль) и реакционную смесь перемешивали при 65°C в течение 2 ч. Реакционную смесь разбавляли водой и экстрагировали с помощью EtOAc (100 мл × 2). Объединенные органические слои промывали 1x водой (100 мл) и 1x солевым раствором (100 мл) и затем высушивали над сульфатом натрия и концентрировали in vacuo. Неочищенное соединение очищали путем промываний н-пентаном с получением I-36. 1H ЯМР (300 МГц, CDCl3): δ 9,04 (s, 1H), 8,61 (s, 1H), 8,22 (s, 1H), 7,70-7,40 (m, 6H), 7,20-7,00 (m, 1H), 6,75-6,60 (m, 1H); M+H=305,30.
Пример 1: N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)-2,4-диметилоксазол-5-карбоксамид (соединение 1)
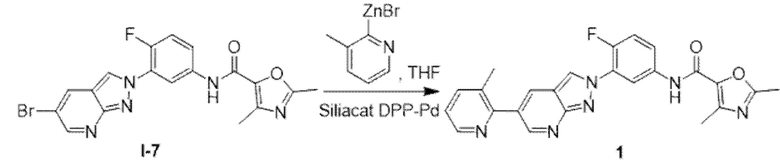
В смесь I-7 (30 мг, 0,070 ммоль) и Siliacat DPP-Pd 0,25 ммоль/г (0,014 г, 3,52 мкмоль, 0,05 экв.) добавляли 0,5 мл безводного THF. Реакционную смесь помещали в атмосферу азота и добавляли бромид (3-метилпиридин-2-ил)цинка(II) (0,5 М в THF, 0,56 мл, 0,28 ммоль, 4 экв.) и реакционную смесь перемешивали при 65°C в течение ночи. Реакционную смесь гасили с помощью 0,2 мл воды и очищали с помощью препаративной HPLC (колонка Waters Atlantis T3 C18 19 × 50 мм × 10 мкм) с элюированием от 20 до 80% ацетонитрила (0,035% TFA) в воде (0,05% TFA) с получением соединения 1. 1H ЯМР (400 МГц, Метанол-d4) δ 8,90 (dd, J=13,2, 2,3 Гц, 2H), 8,59-8,43 (m, 3H), 7,97-7,81 (m, 2H), 7,55-7,35 (m, 2H), 2,56 (s, 3H), 2,48-2,45 (m, 6H). Способ 1: RT=2,69 мин.; M+H=443,1.
Соединения 16, 17, 101, 102, 103, 104, 105, 106, 107 и 108 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов цинка(II).
Пример 2: N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)фуран-2-карбоксамид (соединение 2)
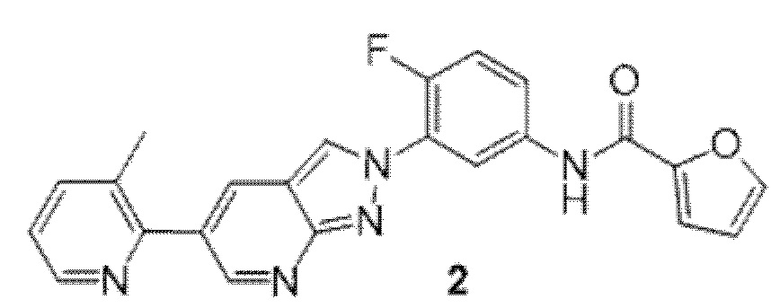
Соединение 2 получали подобным образом, как соединение 1, с использованием I-8 в качестве исходного материала. 1H ЯМР (400 МГц, Метанол-d4) δ 8,92 (d, J=2,4 Гц, 1H), 8,88 (d, J=2,2 Гц, 1H), 8,55-8,45 (m, 3H), 7,96 (m, 1H), 7,87 (m, 1H), 7,78 (m, 1H), 7,52-7,39 (m, 2H), 7,32 (m, 1H), 6,68 (m, 1H), 2,46 (s, 3H). Способ 2: RT=0,89 мин.; M+H=414,3.
Соединения 54, 55, 56, 57, 59, 60, 61, 62, 63 и 64 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов цинка(II).
Пример 3: N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 3)
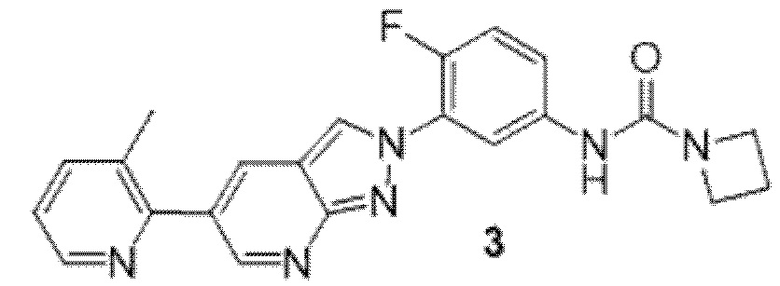
Соединение 3 получали подобным образом, как соединение 1, с использованием I-6 в качестве исходного материала. 1H ЯМР (400 МГц, Метанол-d4) δ 8,89-8,84 (m, 2H), 8,52 (m, 1H), 8,48 (m, 1H), 8,18 (m, 1H), 7,86 (m, 1H), 7,67 (m, 1H), 7,44-7,32 (m, 2H), 4,18-4,06 (m, 4H), 2,46 (s, 3H), 2,41-2,26 (m, 2H). Способ 1: RT=2,41 мин.; M+H=403,3.
Соединение 99 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующего бромида цинка(II).
Пример 4: (R)-3-фтор-N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)пирролидин-1-карбоксамид (соединение 4)
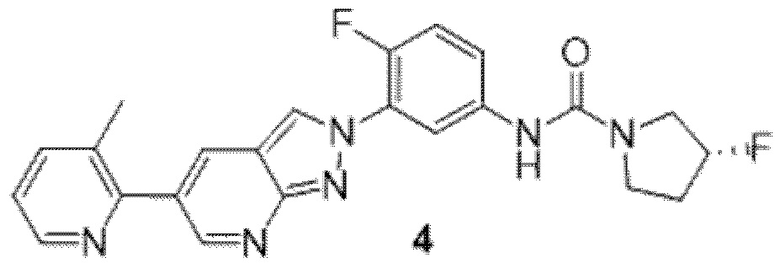
Соединение 4 получали подобным образом, как соединение 1, с использованием I-5 в качестве исходного материала. 1H ЯМР (400 МГц, Метанол-d4) δ 8,87 (dd, J=4,7, 2,3 Гц, 2H), 8,52 (m, 1H), 8,49 (m, 1H), 8,19 (m, 1H), 7,86 (m, 1H), 7,67 (m, 1H), 7,45-7,34 (m, 2H), 5,42-5,25 (m, 1H), 3,85-3,55 (m, 4H), 2,46 (s, 3H), 2,39-2,19 (m, 2H). Способ 1: RT=2,41 мин.; M+H=435,3.
Соединения 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52 и 53 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов цинка(II).
Пример 5: 3,3-дифтор-N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 5)
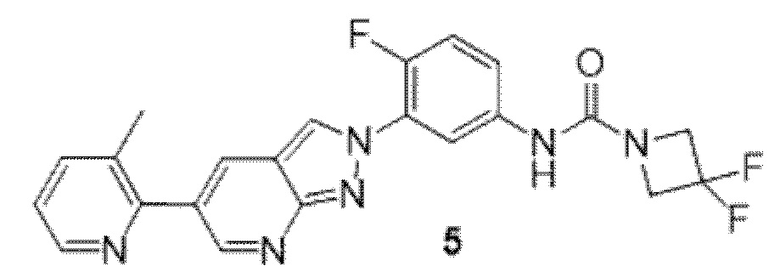
Соединение 5 получали подобным образом, как соединение 1, с использованием I-3 в качестве исходного материала. 1H ЯМР (400 МГц, Метанол-d4) δ 8,88 (dd, J=5,4, 2,3 Гц, 2H), 8,52 (m, 1H), 8,48 (m, 1H), 8,22 (m, 1H), 7,86 (m, 1H), 7,69 (m, 1H), 7,45-7,36 (m, 2H), 4,44 (m, 4H), 2,46 (s, 3H). Способ 2: RT=0,76 мин.; M+H=439,1.
Пример 6: 3-фтор-N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 6)
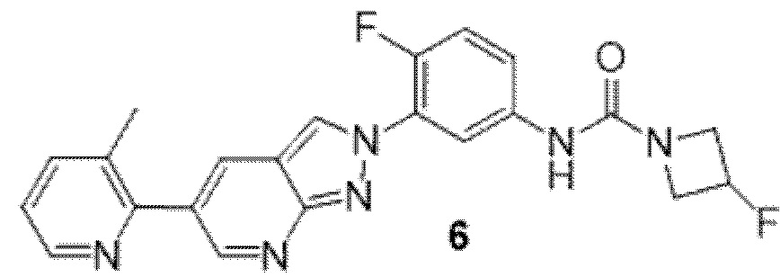
Соединение 6 получали подобным образом, как соединение 1, с использованием I-4 в качестве исходного материала. 1H ЯМР (500 МГц, DMSO-d6) δ 8,98 (s, 1H), 8,94 (dd, J=16,9, 2,3 Гц, 2H), 8,57 (ddd, J=4,8, 1,7, 0,7 Гц, 1H), 8,49 (d, J=2,3 Гц, 1H), 8,28 (dd, J=7,0, 2,7 Гц, 1H), 7,80 (ddd, J=7,7, 1,7, 0,8 Гц, 1H), 7,72 (ddd, J=9,1, 4,1, 2,7 Гц, 1H), 7,49 (dd, J=11,2, 9,1 Гц, 1H), 7,37 (dd, J=7,7, 4,7 Гц, 1H), 5,41 (dtt, J=57,5, 6,1, 3,1 Гц, 1H), 4,34 (dddd, J=21,7, 10,4, 6,0, 1,4 Гц, 2H), 4,04 (dddd, J=24,9, 10,4, 3,1, 1,4 Гц, 2H), 2,44 (s, 3H). Способ 1: RT=2,39 мин.; M+H=421,1.
Соединения 14, 15, 18, 19, 20, 21, 23, 24, 25, 26, 27 и 28 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов цинка(II).
Соединение 318 синтезировали в соответствии с протоколом, описанным выше, с применением диметилцинка.
Пример 7: N-(4-фтор-3-(5-изобутил-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 7)
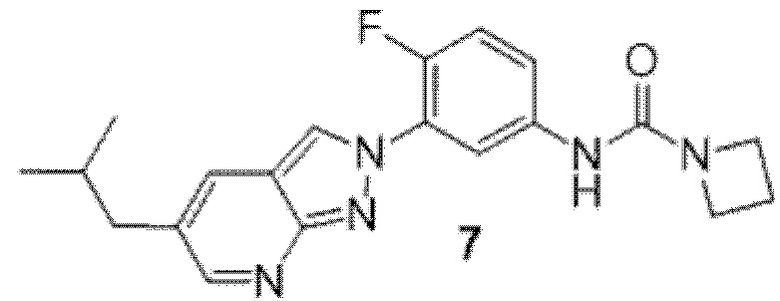
Соединение 7 получали подобным образом, как соединение 1, с использованием I-6 и бромида изобутилцинка(II) в качестве исходных материалов. 1H ЯМР (400 МГц, Метанол-d4) δ 8,66 (d, J=2,4 Гц, 1H), 8,56 (d, J=2,2 Гц, 1H), 8,12 (m, 1H), 8,07 (m, 1H), 7,64 (m, 1H), 7,34 (m, 1H), 4,19-4,01 (m, 4H), 2,67 (m, 2H), 2,41-2,24 (m, 2H), 1,97 (m, 1H), 0,99 (m, 6H). Способ 1: RT=3,11 мин.; M+H=368,3.
Соединения 91, 93, 94, 95, 96, 97 и 98 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов цинка(II).
Пример 8: 3,3-дифтор-N-(4-фтор-3-(5-изобутил-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 8)
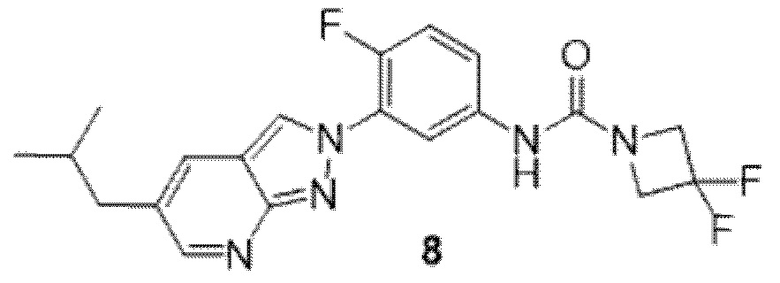
Соединение 8 получали подобным образом, как соединение 1, с использованием I-3 и бромида изобутилцинка(II) в качестве исходных материалов. 1H ЯМР (400 МГц, Метанол-d4) δ 8,59 (d, J=2,5 Гц, 1H), 8,46 (d, J=2,2 Гц, 1H), 8,06 (m, 1H), 7,97 (m, 1H), 7,56 (m, 1H), 7,30-7,24 (m, 1H), 4,33 (m, 4H), 2,57 (m, 2H), 1,92-1,81 (m, 1H), 0,89 (m, 6H). Способ 2: RT=1,09 мин. M+H=404,1.
Соединения 29, 30, 31, 32, 33, 34, 35, 36, 37 и 38 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов цинка(II).
Пример 9: N-(3-(5-(диметиламино)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-3,3-дифторазетидин-1-карбоксамид (соединение 9)
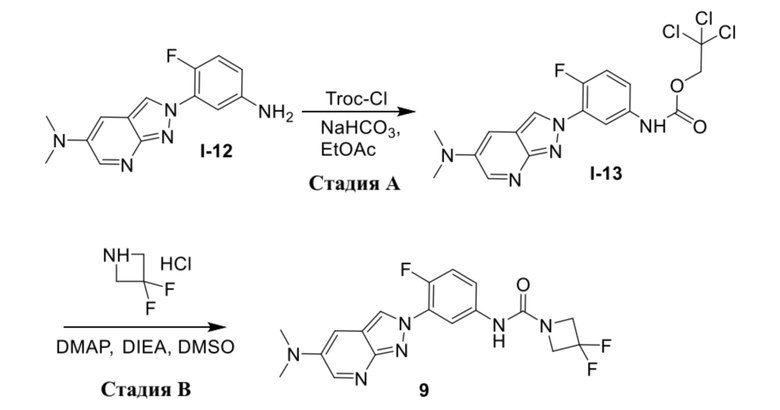
Стадия A. В раствор I-12 (0,5 г, 1,8 ммоль, 1 экв.) в этилацетате (5 мл) и насыщенном водном растворе NaHCO3 (5 мл) добавляли 2,2,2-трихлорэтилкарбонилхлорид (585,7 мг, 2,8 ммоль, 370,7 мкл, 1,5 экв.) при 0°C и реакционную смесь перемешивали при 0°C в течение 1 ч. Реакционную смесь экстрагировали с помощью EtOAc (30 мл × 3) и объединенные органические вещества высушивали над сульфатом натрия и концентрировали. Полученный остаток очищали с помощью хроматографии на силикагеле с элюированием с помощью 20% EtOAc и 20% DCM в петролейном эфире с получением I-13 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,52 (br s, 1 H), 8,71 (d, J=2,93 Гц, 1 H), 8,57 (d, J=2,69 Гц, 1 H), 8,25 (br d, J=5,14 Гц, 1 H), 7,44-7,64 (m, 2 H), 7,16 (d, J=2,69 Гц, 1 H), 4,99 (s, 2 H), 2,96 (s, 6 H).
Стадия B. Перемешивали реакционную смесь I-13 (60 мг, 134,3 мкмоль, 1 экв.), 3,3-дифторазетидина-HCl (25 мг, 268,6 мкмоль, 2 экв.), DIEA (34,6 мг, 268,6 мкмоль, 46,7 мкл, 2 экв.), DMAP (1,6 мг, 13,4 мкмоль, 0,1 экв.) в DMSO (1 мл) при 60°C в течение 12 часов и при 70°C в течение еще 12 часов. Реакционную смесь очищали с помощью препаративной HPLC, колонка: Phenomenex Gemini C18 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 25% до 55%, 12 мин., с получением 9 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,18 (s, 1 H), 8,70 (d, J=3,01 Гц, 1 H), 8,55 (d, J=2,76 Гц, 1 H), 8,20 (dd, J=6,90, 2,64 Гц, 1 H), 7,56-7,78 (m, 1 H), 7,45 (dd, J=11,42, 9,16 Гц, 1 H), 7,15 (d, J=3,01 Гц, 1 H), 4,41 (t, J=12,67 Гц, 4 H), 2,95 (s, 6 H). Способ 1: RT=2,84 мин.; M+H=391,1.
Соединения 100, 124, 133, 142, 143, 156, 167, 170 195, 219, 220, 221, 222, 223, 224, 225, 226, 227, 228, 229, 260, 276, 319, 320, 321, 322, 323, 324 и 328 синтезировали в соответствии с протоколом, описанным выше, с применением промежуточных соединений I-9, I-12, I-16, I-23, I-25 или I-26 и соответствующего амина.
Пример 10: N-(3-(5-(азетидин-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)азетидин-1-карбоксамид (соединение 10)
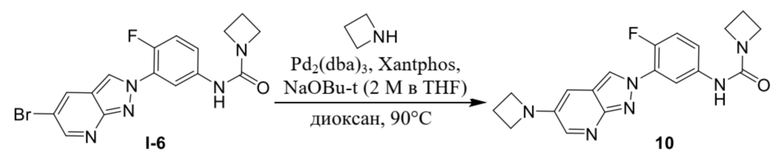
Дегазировали смесь I-6 (80 мг, 205 мкмоль, 1 экв.), азетидина (17,6 мг, 307,5 мкмоль, 20,7 мкл, 1,5 экв.), NaOt-Bu (2 М в THF, 307,5 мкл, 3 экв.), XantPhos (23,7 мг, 41 мкмоль, 0,20 экв.) и Pd2(dba)3 (18,8 мг, 20,5 мкмоль, 0,10 экв.) в диоксане (3 мл) с помощью аргона и нагревали до 90°C в течение 4 ч. Реакционную смесь гасили с помощью 1 М HOAc и очищали с помощью препаративной HPLC (колонка: Phenomenex Gemini C18 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 28% до 58%, 12 мин.) с получением 10 в виде желтого твердого вещества. 1H ЯМР (400 МГц, Метанол-d4), δ ppm, 8,42 (d, J=2,51 Гц, 1 H), 8,22 (d, J=2,76 Гц, 1 H), 8,05 (dd, J=6,78, 2,76 Гц, 1 H), 7,58-7,62 (m, 1 H), 7,30 (dd, J=11,04, 9,16 Гц, 1 H), 7,01 (d, J=2,64 Гц, 1 H), 4,10 (t, J=7,65 Гц, 4 H), 3,99 (t, J=7,22 Гц, 4 H), 2,41-2,49 (m, 2 H), 2,27-2,35 (m, 2 H). Способ 1: RT=2,80 мин.; M+H=367,1.
Соединения 40, 58, 92, 122, 131, 132, 134, 135, 137, 141, 144, 145, 147, 149, 151, 155, 157, 158, 161, 163, 171, 172, 177, 178, 179, 180, 181, 183, 184, 185, 186, 190, 191, 192, 193, 194, 197, 198, 199, 200, 201, 202, 203, 204, 205, 230, 231, 232, 234, 235, 236 237, 238, 239, 240, 241, 243, 244, 245, 248, 249, 250, 251, 252, 253, 255, 263, 266, 272 290, 291, 292, 298, 299, 300, 302, 303, 306, 307, 308, 314, 326, 327, 338 и 339 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих промежуточных соединений I-3, I-4, I-5, I-6, I-7, I-8, I-24, I-27 или I-31 и соответствующих аминов.
Соединение 242 выделяли в дополнение к соединению 241 с применением указанных выше условий реакции. Подобным образом, соединение 246 выделяли в дополнение к соединению 202 с применением указанных выше условий реакции.
Соединение 270 синтезировали с применением протокола, описанного выше, с применением Boc-пиперидина, после чего Boc-группу удаляли с применением TFA в DCM в соотношении 0,36:1 с последующей нейтрализацией до pH=7 с помощью Et3N и затем очисткой с помощью препаративной HPLC (колонка: Phenomenex Synergi C18 150 × 25 × 10 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 3% до 30%, 10 мин.) с получением соединения 270.
Пример 11: 4,4,4-трифтор-N-(4-фтор-3-(5-изопропил-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)бутанамид (соединение 173)
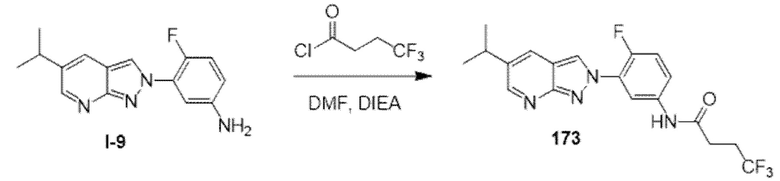
В раствор I-9 (25 мг, 0,092 ммоль) в DMF (1,5 мл) и DIEA (0,040 мл, 0,23 ммоль) добавляли 4,4,4-трифторбутаноилхлорид (14,9 мг, 0,092 ммоль) и реакционную смесь перемешивали в течение 12 ч. при комнатной температуре. Неочищенную реакционную смесь очищали непосредственно на колонке C18 15 г для флэш-хроматографии с элюированием от 20 до 90% ацетонитрила в воде с получением соединения 173 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 10,45 (s, 1H), 8,81 (d, J=2,7 Гц, 1H), 8,69 (d, J=2,3 Гц, 1H), 8,36 (dd, J=7,0, 2,7 Гц, 1H), 8,05 (dd, J=2,3, 0,8 Гц, 1H), 7,68 (ddd, J=9,1, 4,2, 2,7 Гц, 1H), 7,54 (dd, J=11,2, 9,0 Гц, 1H), 3,08 (pd, J=6,9, 0,8 Гц, 1H), 2,72-2,55 (m, 4H), 1,30 (d, J=6,9 Гц, 6H). Способ 1: RT=3,19 мин.; M+H=395,2.
Соединение 174 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующего хлорформиата вместо хлорангидрида.
Пример 12: N-(4-фтор-3-(5-изопропил-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)пиразин-2-карбоксамид (соединение 87)
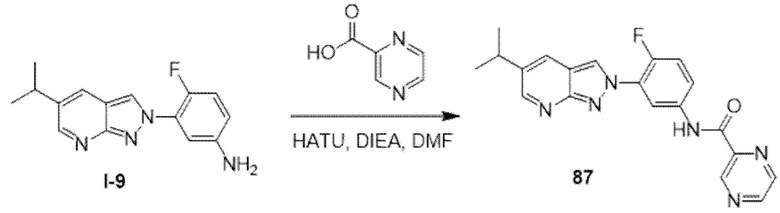
В колбу добавляли пиразин-2-карбоновую кислоту (16,5 мг, 0,13 ммоль) с последующим добавлением I-9 (30 мг, 0,11 ммоль), растворенного в 0,25 мл DMF. Добавляли раствор DIEA (0,058 мл, 0,33 ммоль) и HATU (50,6 мг, 0,13 ммоль) в 0,25 мл DMF в реакционную смесь и полученную смесь перемешивали в течение ночи. Реакционную смесь фильтровали через шприцевой фильтр 0,2 микрон и материал очищали с помощью препаративной HPLC (колонка Waters Atlantis T3 C18 19 × 50 мм × 10 мкм) с элюированием от 20 до 80% ацетонитрила (0,035% TFA) в воде (0,05% TFA) с получением соединения 87. 1H ЯМР (400 МГц, Метанол-d4) δ 9,36 (m, 1H), 8,84 (m, 1H), 8,76 (m, 1H), 8,72 (m, 1H), 8,66 (m, 1H), 8,57 (m, 1H), 8,13 (m, 1H), 7,99 (m, 1H), 7,46 (m, 1H), 3,13 (m, 1H), 1,38 (2s, 6H). Способ 1: RT=3,05 мин.; M+H=377,3.
Соединения 65, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 196, 207, 208, 209, 210, 211, 212, 213, 214, 215, 216, 217, 261, 262 и 351 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих карбоновых кислот.
Соединения 139, 140, 153, 154, 279, 330 и 352 синтезировали в соответствии с протоколом, описанным выше, с применением I-12, I-29 или I-36 и соответствующих карбоновых кислот.
Соединение 278 синтезировали в соответствии с протоколом, описанным выше, с применением I-28 и I-34 в качестве исходных материалов.
Пример 13: 3-фтор-N-(4-фтор-3-(5-(2,2,2-трифторэтил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 152)
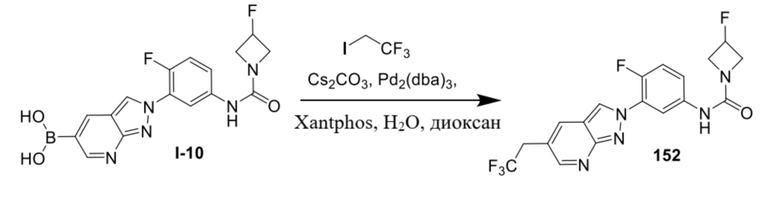
В раствор I-10 (106 мг, 285,6 мкмоль, 1 экв.) в диоксане (1 мл) добавляли 1,1,1-трифтор-2-йодэтан (119,9 мг, 571,10 мкмоль, 56,03 мкл, 2 экв.), Pd2(dba)3 (13,1 мг, 14,3 мкмоль, 0,05 экв.), XantPhos (28,1 мг, 48,5 мкмоль, 0,2 экв.) и Cs2CO3 (372,15 мг, 1,1 ммоль, 4 экв.) в атмосфере N2. Через 1 мин. добавляли воду (92,6 мг, 5,1 ммоль, 92,6 мкл, 18 экв.) и смесь перемешивали при 80°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли с помощью DCM (30 мл) и органический слой промывали водой (10 мл × 2), высушивали над Na2SO4, фильтровали и концентрировали с получением остатка. Остаток очищали с помощью препаративной TLC (SiO2, DCM: метанол=10:1) и дополнительно очищали с помощью препаративной HPLC (колонка: Phenomenex Gemini C18 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 28% до 58%, 12 мин.) с получением соединения 152 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 8,98 (s, 1 H), 8,95 (d, J=2,51 Гц, 1 H), 8,69 (d, J=2,01 Гц, 1 H), 8,33 (d, J=1,76 Гц, 1 H), 8,24 (dd, J=6,90, 2,63 Гц, 1 H), 7,67-7,74 (m, 1 H), 7,48 (dd, J=11,29, 9,29 Гц, 1 H), 5,30-5,52 (m, 1 H), 4,26-4,39 (m, 2 H), 3,97-4,09 (m, 2 H), 3,87 (q, J=11,71 Гц, 2 H). Способ 1: RT=2,76 мин.; M+H=412,2.
Соединения 162, 164, 166, 175 и 176 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующей бороновой кислоты, полученной из промежуточных соединений I-3, I-5, I-6, I-7 или I-8, полученных подобным образом, как I-10.
Пример 14: изопропил-(2-(2-фтор-5-(3-фторазетидин-1-карбоксамидо)фенил)-2H-пиразоло[3,4-b]пиридин-5-ил)карбамат (соединение 168)
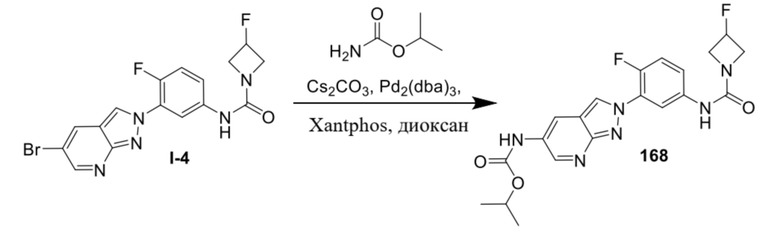
В смесь I-4 (120 мг, 294 мкмоль, 1 экв.), изопропилкарбамата (45,5 мг, 441 мкмоль, 1,5 экв.) и Cs2CO3 (287,3 мг, 881,9 мкмоль, 3 экв.) в диоксане (3 мл) добавляли XantPhos (34,0 мг, 58,8 мкмоль, 0,20 экв.) и Pd2(dba)3 (26,9 мг, 29,4 мкмоль, 0,10 экв.) в атмосфере аргона. Смесь нагревали до 90°C и перемешивали в течение 4 часов. Смесь гасили путем добавления 1 М HOAc и очищали с помощью препаративной HPLC (колонка: Boston pH-lex 150 × 25 10 мкм; подвижная фаза: [вода (0,225% муравьиной кислоты)-ацетонитрил]; B%: от 33% до 63%, 10 мин. с последующей дополнительной очисткой на колонке Phenomenex Gemini C18 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 28% до 58%, 12 мин.). Соединение 168 получали в виде желтого твердого вещества. 1H ЯМР (400 МГц, Метанол-d4), δ ppm, 8,64 (br d, J=2,26 Гц, 1 H), 8,61 (br d, J=2,64 Гц, 1 H), 8,44 (br s, 1 H), 8,10 (dd, J=6,71, 2,70 Гц, 1 H), 7,60-7,67 (m, 1 H), 7,33 (dd, J=10,92, 9,16 Гц, 1 H), 5,25-5,47 (m, 1 H), 5,01 (dt, J=12,52, 6,23 Гц, 1 H), 4,32-4,43 (m, 2 H), 4,07-4,19 (m, 2 H), 1,33 (d, J=6,27 Гц, 6 H). Способ 1: RT=2,88 мин.; M+H=431,1.
Соединения 123, 136, 138, 146 и 159 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих промежуточных соединений I-3, I-5, I-6, I-7 или I-8.
Пример 15: 3-фтор-N-(4-фтор-3-(5-(3-фторпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 187)
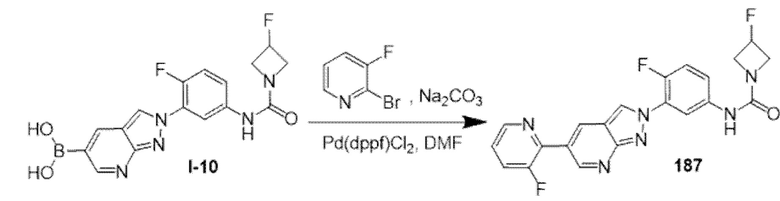
В раствор промежуточного соединения I-10 (95 мг, 254,6 мкмоль, 1 экв.) в DMF (3 мл) добавляли Pd(dppf)Cl2 (18,6 мг, 25,5 мкмоль, 0,1 экв.), Na2CO3 (81 мг, 763,8 мкмоль, 3 экв.) и 2-бром-3-фторпиридин (89,5 мг, 509,2 мкмоль, 2 экв.). Смесь перемешивали при 90°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и очищали с помощью препаративной HPLC (колонка: Boston Green ODS 150 × 30 5 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 35% до 65%, 10 мин.) с получением соединения 187 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,27 (s, 1 H), 9,00-9,09 (m, 2 H), 8,82 (s, 1 H), 8,63 (br d, J=4,39 Гц, 1 H), 8,29 (dd, J=6,90, 2,51 Гц, 1 H), 7,93 (dd, J=10,73, 8,47 Гц, 1 H), 7,72 (dt, J=8,60, 3,48 Гц, 1 H), 7,52-7,58 (m, 1 H), 7,46-7,51 (m, 1 H), 5,28-5,55 (m, 1 H), 4,25-4,44 (m, 2 H), 3,93-4,14 (m, 2 H). Способ 1: RT=2,74 мин.; M+H=425,1.
Соединения 188, 294, 295, 297, 301, 304, 312, 325, 329, 330 и 335 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих бромидов.
Пример 16: 5-фтор-N-(4-фтор-3-(5-(пиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)фуран-2-карбоксамид (соединение 11)
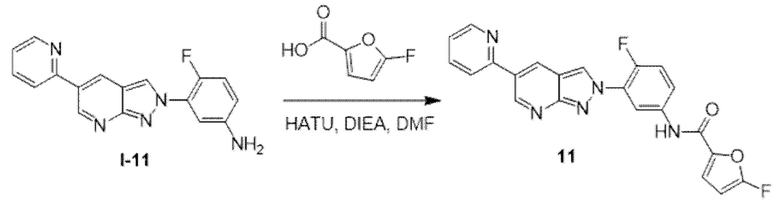
Растворяли I-11 (40 мг, 0,131 ммоль) в 2 мл DMF. В реакционную смесь добавляли 5-фторфуран-2-карбоновую кислоту (25,6 мг, 0,20 ммоль), DIEA (0,058 мл, 0,33 ммоль) и HATU (50,6 мг, 0,13 ммоль) и полученную смесь перемешивали в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и промывали 1x насыщ. водн. раствором Na2CO3 и 1x водой. Органические вещества высушивали над Na2SO4 и концентрировали. Неочищенный материал очищали с помощью хроматографии на силикагеле с элюированием от 0 до 100% смеси 3:1 EtOAc:EtOH в гептанах с получением соединения 11. 1H ЯМР (400 МГц, DMSO-d6) δ 10,53 (s, 1H), 9,49 (d, J=2,3 Гц, 1H), 9,06 (d, J=2,6 Гц, 1H), 8,96 (d, J=2,3 Гц, 1H), 8,74 (ddd, J=4,8, 1,9, 0,9 Гц, 1H), 8,53 (dd, J=7,0, 2,7 Гц, 1H), 8,14 (dt, J=8,0, 1,1 Гц, 1H), 8,01-7,91 (m, 2H), 7,62 (dd, J=11,2, 9,1 Гц, 1H), 7,50-7,38 (m, 2H), 6,14 (dd, J=7,0, 3,7 Гц, 1H). Способ 2: RT=0,91 мин.; M+H=418,1.
Соединения 12, 13, 206, 218, 345, 346, 347, 348 и 349 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующей карбоновой кислоты.
Соединение 350 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующей карбоновой кислоты и I-28.
Пример 17: 3-фтор-N-(4-фтор-3-(5-(пиридин-4-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 182)
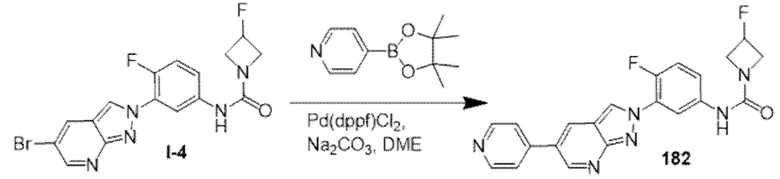
В раствор I-4 (80 мг, 196 мкмоль, 1 экв.) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (60,3 мг, 294 мкмоль, 1,5 экв.) в DME (3 мл) добавляли Pd(dppf)Cl2 (14,34 мг, 19,6 мкмоль, 0,1 экв.) и Na2CO3 (2 М, 294 мкл, 3 экв.) в атмосфере N2. Смесь перемешивали при 90°C в течение 20 ч. Реакционную смесь концентрировали и остаток разбавляли с помощью EtOAc (30 мл). Органический слой промывали водой (10 мл × 2), высушивали над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью препаративной HPLC (колонка: Phenomenex Gemini C18 250 × 50 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 18% до 48%, 12 мин.) с получением соединения 182 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), δ ppm, 9,17 (d, J=2,26 Гц, 1 H), 9,03 (d, J=2,51 Гц, 1 H), 9,00 (s, 1 H), 8,75 (d, J=2,51 Гц, 1 H), 8,67-8,73 (m, 2 H), 8,28 (dd, J=6,90, 2,64 Гц, 1 H), 7,84-7,90 (m, 2 H), 7,68-7,75 (m, 1 H), 7,50 (dd, J=11,29, 9,03 Гц, 1 H), 5,30-5,52 (m, 1 H), 4,27-4,40 (m, 2 H), 3,97-4,10 (m, 2 H). Способ 2: RT=0,58 мин.; M+H=407,1.
Соединение 189 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующего сложного эфира бороновой кислоты.
Пример 18: (R)-3-фтор-N-(4-фтор-3-(5-(тетрагидро-2H-пиран-4-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)пирролидин-1-карбоксамид (соединение 150)
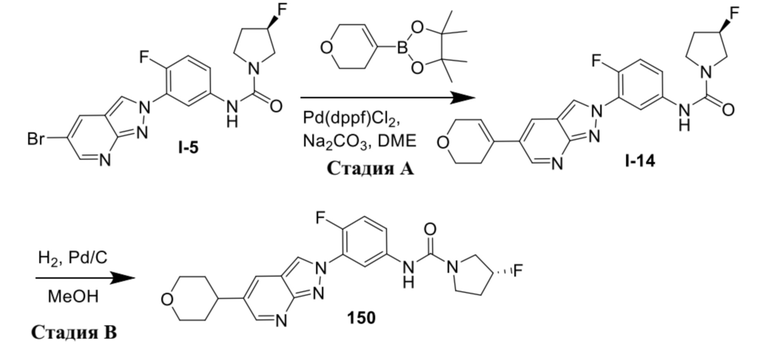
Стадия A. В смесь 2-(3,6-дигидро-2H-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (65,7 мг, 312,6 мкмоль, 1,1 экв.), I-5 (120 мг, 284,2 мкмоль, 1 экв.) и Na2CO3 (2 M, 426,3 мкл, 3 экв.) в DME (3 мл) и воде (0,5 мл) в атмосфере аргона добавляли Pd(dppf)Cl2 (20,8 мг, 28,4 мкмоль, 0,1 экв.). Смесь нагревали до 90°C в течение 4 ч. Смесь очищали с помощью препаративной TLC с элюированием с помощью 90% EtOAc в петролейном эфире с получением I-14 в виде белого твердого вещества. M+H=426,2.
Стадия B. В смесь I-14 (85 мг, 199,8 мкмоль, 1 экв.) в метаноле (10 мл) добавляли Pd/C (30 мг, чистота 10%). Суспензию дегазировали в вакууме и несколько раз продували с помощью H2. Смесь перемешивали в атмосфере H2 (15 фунтов/кв. дюйм) при 10°C в течение 1 ч. Смесь фильтровали и фильтрат концентрировали с получением остатка. Остаток очищали с помощью препаративной HPLC (колонка: Boston Green ODS 150 × 30 5 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 27% до 51%, 10 мин.) с получением соединения 150 в виде желтого твердого вещества. 1H ЯМР (400 МГц, Метанол-d4), δ ppm, 9,15 ((d, J=2,01 Гц, 1 H)), 8,98 (s, 1 H), 8,85 (d, J=1,88 Гц, 1 H), 8,33 (dd, J=6,78, 9,16 Гц, 1 H), 7,61-7,65 (m, 1 H), 7,42 (dd, J=11,04, 9,16 Гц, 1 H), 5,28-5,41 (m, 1 H), 4,11-4,14 (m, 2 H), 3,57-3,85 (m, 6 H), 3,14-3,20 (m, 1 H), 2,11-2,32 (m, 2 H), 1,91-1,97 (m, 4 H). Способ 1: RT=2,60 мин.; M+H=428,2.
Соединения 148, 160, 165, 169, 296 и 305 синтезировали в соответствии с протоколом, описанным выше, с применением промежуточных соединений I-3, I-4, I-6 или I-7 и соответствующей бороновой кислоты или сложного эфира бороновой кислоты.
Соединение 293 синтезировали с применением протокола, описанного выше, с применением I-4 и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилата, после чего Boc-группу удаляли с применением TFA в DCM в соотношении 1:5 с последующим концентрированием и затем очисткой с помощью препаративной HPLC (колонка: Phenomenex Synergi C18 150 × 25 × 10 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 2% до 22%, 10 мин.) с получением соединения 293.
Пример 19: N-(3-(5-(5-азаспиро[2.3]гексан-5-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)азетидин-1-карбоксамид (соединение 257)
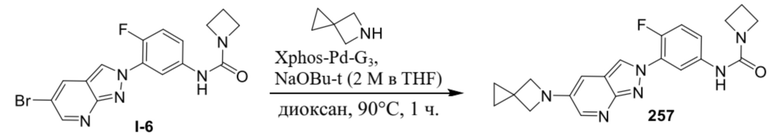
Дегазировали смесь I-6 (400 мг, 1,0 ммоль, 1 экв.), полуоксалата 5-азаспиро[2.3]гексана (128 мг, 1,0 ммоль, 1 экв.), NaOBu-t (2 М в THF, 1,5 мл, 3 экв.) и Xphos Pd G3 (87 мг, 102,5 мкмоль, 0,1 экв.) в диоксане (10 мл), помещали в атмосферу аргона и нагревали до 90°C в течение 1 часа. Реакционную смесь гасили путем добавления насыщенного раствора NH4Cl (40 мл). Полученную смесь экстрагировали дихлорметаном (30 мл × 6) и органическую фазу высушивали над Na2SO4 и фильтровали. Фильтрат концентрировали и остаток очищали с помощью хроматографии на силикагеле с элюированием с помощью 50% MeOH в DCM с последующей очисткой с помощью препаративной HPLC (колонка: Phenomenex Synergi C18 150 × 25 × 10 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 30% до 60%, 10 мин.) с получением соединения 257 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ ppm 8,69 (s, 1 H), 8,52 (d, J=2,63 Гц, 1 H), 8,28 (d, J=2,76 Гц, 1 H), 8,20 (dd, J=7,03, 2,63 Гц, 1 H), 7,62-7,69 (m, 1 H), 7,40 (dd, J=11,29, 9,16 Гц, 1 H), 6,91 (d, J=2,76 Гц, 1 H), 4,01 (s, 4 H), 3,97 (t, J=7,53 Гц, 4 H), 2,19 (квинт., J=7,56 Гц, 2 H), 0,68 (s, 4 H). Способ 1: RT=2,90 мин.; M+H=393,2.
Соединения 233, 258, 259 и 337 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих промежуточных соединений I-3, I-4, I-5 или I-27.
Пример 20: 3-фтор-N-(4-фтор-3-(5-(3-(трифторметил)азетидин-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 265)
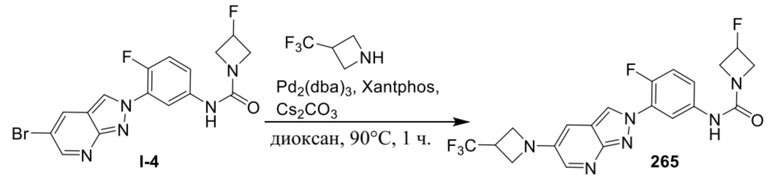
Дегазировали смесь I-4 (3,5 г, 8,6 ммоль, 1 экв.), HCl-соли 3-(трифторметил)азетидина (1,7 г, 10,3 ммоль, 1,2 экв.), Pd2(dba)3 (785 мг, 857,4 мкмоль, 0,1 экв.), XantPhos (992 мг, 1,7 ммоль, 0,2 экв.) и Cs2CO3 (8,4 г, 25,7 ммоль, 3 экв.) в диоксане (60 мл) и затем нагревали до 85°C в течение 1,2 ч. в атмосфере N2. Реакционную смесь гасили насыщ. раствором NH4Cl (400 мл) и экстрагировали с помощью EtOAc (300 мл × 2). Объединенные органические слои промывали солевым раствором (200 мл), высушивали над Na2SO4 и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием смесью MeOH/этилацетат/DCM=от 0:1:1 до 1:5:1, с получением желтого твердого вещества, которое дополнительно очищали с помощью препаративной HPLC; колонка: Phenomenex Synergi Max-RP 250 × 80 мм × 10 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 30% до 60%, 35 мин. Регулировали pH раствора до pH=7 с помощью 20% NH3 в H2O и удаляли ацетонитрил в вакууме при 30°C. Полученное желтое твердое вещество собирали посредством фильтрования, промывали водой, 100 мл × 4, и лиофилизировали с получением соединения 265 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ ppm 8,93 (s, 1 H), 8,58 (d, J=2,76 Гц, 1 H), 8,32 (d, J=2,76 Гц, 1 H), 8,20 (dd, J=7,03, 2,64 Гц, 1 H), 7,65 (dd, J=7,34, 4,58 Гц, 1 H), 7,44 (dd, J=11,17, 9,16 Гц, 1 H), 7,03 (d, J=2,89 Гц, 1 H), 5,29-5,51 (m, 1 H), 4,24-4,38 (m, 2 H), 4,18 (t, J=8,34 Гц, 2 H), 3,95-4,10 (m, 4 H), 3,65-3,85 (m, 1 H). Способ 1: RT=2,86 мин.; M+H=453,1.
Соединения 274 и 275 синтезировали в соответствии с протоколом, описанным выше, с применением соответствующих промежуточных соединений I-5 или 1-6.
Пример 21: N-(2-(5-(3,3-дифторазетидин-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)пиридин-4-ил)азетидин-1-карбоксамид (соединение 280)
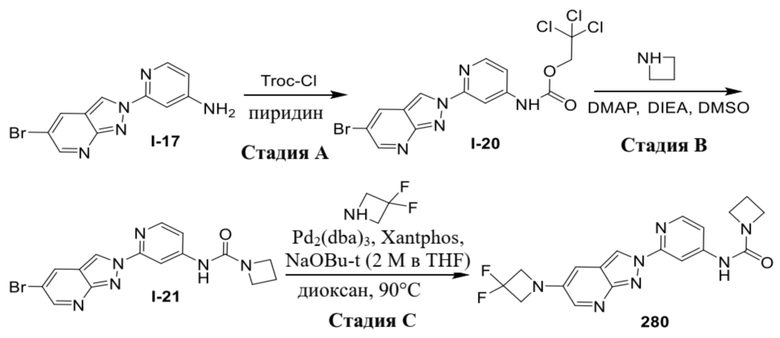
Стадия A. В раствор I-17 (2,5 г, 8,6 ммоль, 1 экв.) в пиридине (30 мл) добавляли 2,2,2-трихлорэтилкарбонилхлорид (3,6 г, 17,2 ммоль, 2,3 мл, 2 экв.). Смесь перемешивали при 30°C в течение 22 ч. Реакционную смесь осаждали путем добавления 100 мл воды и затем фильтровали. Осадок на фильтре промывали с помощью 50 мл воды и высушивали при пониженном давлении. Затем осадок растирали в трет-бутилметиловом эфире (20 мл) и собирали посредством фильтрования с получением I-20 в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ ppm 11,08 (s, 1 H), 9,32 (s, 1 H), 8,77 (d, J=1,71 Гц, 1 H), 8,62 (d, J=1,96 Гц, 1 H), 8,52 (d, J=1,71 Гц, 1 H), 8,49 (d, J=3,91 Гц, 1 H), 7,75 (dd, J=5,38, 1,71 Гц, 1 H), 5,05 (s, 2 H).
Стадия B. В раствор I-20 (1,2 г, 2,6 ммоль, 1 экв.) и гидрохлорида азетидина (482 мг, 5,2 ммоль, 2 экв.) в DMSO (12 мл) добавляли DMAP (31 мг, 257,8 мкмоль, 0,10 экв.) и DIEA (666 мг, 5,2 ммоль, 898,0 мкл, 2 экв.). Смесь перемешивали при 60°C в течение 144 ч. Реакционную смесь осаждали путем добавления 100 мл воды и затем фильтровали. Осадок на фильтре промывали с помощью 50 мл воды, высушивали при пониженном давлении с получением I-21 в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ ppm 9,24 (s, 2 H), 8,75 (d, J=2,45 Гц, 1 H), 8,60 (d, J=2,20 Гц, 1 H), 8,49 (d, J=1,71 Гц, 1 H), 8,35 (d, J=5,62 Гц, 1 H), 7,73 (dd, J=5,75, 1,83 Гц, 1 H), 4,04 (br t, J=7,58 Гц, 4 H), 2,23 (квинт., J=7,64 Гц, 2 H).
Стадия C. Перемешивали смесь I-21 (180 мг, 482,3 мкмоль, 1 экв.), 3,3-дифторазетидина (125 мг, 964,6 мкмоль, 2 экв., HCl), XantPhos (56 мг, 96,5 мкмоль, 0,20 экв.), Pd2(dba)3 (44 мг, 48,2 мкмоль, 0,10 экв.) и NaOBu-t (2 М, 964,6 мкл, 4 экв.) в диоксане (3 мл) при 90°C в течение 16 ч. в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении с удалением растворителя. Остаток очищали с помощью хроматографии на силикагеле с элюированием от 10 до 100% петролейного эфира в этилацетате с получением 65 мг остатка. Остаток растирали в MeOH (3 мл) и твердое вещество собирали посредством фильтрования с получением соединения 280 в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,18 (s, 1 H), 8,99 (s, 1 H), 8,44 (d, J=1,76 Гц, 1 H), 8,36 (d, J=2,76 Гц, 1 H), 8,31 (d, J=5,77 Гц, 1 H), 7,67 (dd, J=5,52, 2,01 Гц, 1 H), 7,14 (d, J=2,76 Гц, 1 H), 4,42 (t, J=12,17 Гц, 4 H), 4,04 (t, J=7,53 Гц, 4 H), 2,16-2,29 (m, 2 H). Способ 1: RT=2,76 мин.; M+H=386,0.
Соединения 282, 284, 286, 287 и 343 синтезировали в соответствии с протоколом, описанным выше, с применением промежуточных соединений I-17, I-18 или I-19 и соответствующих аминов.
Соединение 340 синтезировали в соответствии с протоколом, описанным на стадии A и стадии B, с применением (R)-3-фторпирролидина.
Пример 22: 3,3-дифтор-N-(2-(5-(пиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)пиридин-4-ил)азетидин-1-карбоксамид (соединение 281)
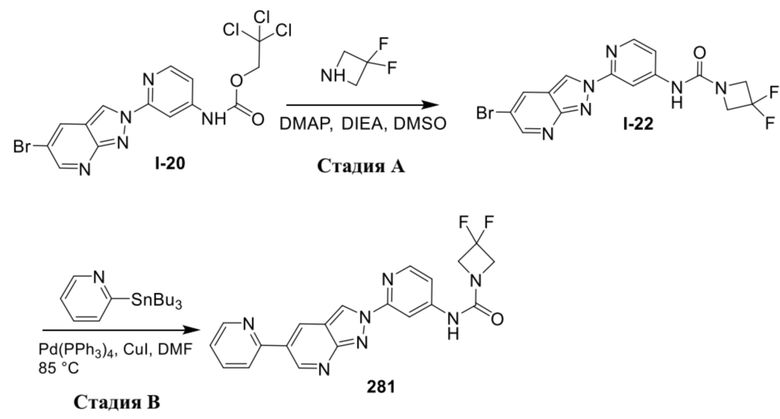
Стадия A. В раствор I-20 (800 мг, 1,7 ммоль, 1 экв.) и HCl-соли 3,3-дифторазетидина (445 мг, 3,4 ммоль, 2 экв.) в DMSO (10 мл) добавляли DMAP (21 мг, 171,8 мкмоль, 0,1 экв.) и DIEA (444 мг, 3,4 ммоль, 598,7 мкл, 2 экв.). Смесь перемешивали при 60°C в течение 320 ч. Реакционную смесь осаждали путем добавления воды (100 мл) и затем фильтровали. Осадок на фильтре промывали с помощью 50 мл воды, высушивали при пониженном давлении с получением I-22 в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,69 (s, 1 H), 9,23-9,27 (m, 1 H), 8,75 (d, J=2,45 Гц, 1 H), 8,61 (d, J=2,44 Гц, 1 H), 8,47 (d, J=1,96 Гц, 1 H), 8,41 (d, J=5,62 Гц, 1 H), 7,71 (dd, J=5,62, 1,96 Гц, 1 H), 4,48 (t, J=12,72 Гц, 4 H).
Стадия B. В раствор I-22 (460 мг, 1,12 ммоль, 1 экв.) и трибутил(2-пиридил)станнана (497 мг, 1,3 ммоль, 1,2 экв.) в DMF (10 мл) добавляли Pd(PPh3)4 (130 мг, 112,4 мкмоль, 0,1 экв.) и CuI (21 мг, 112,4 мкмоль, 0,1 экв.). Смесь перемешивали при 85°C в течение 16 ч. в атмосфере N2. Реакционную смесь концентрировали при пониженном давлении с удалением растворителя. Остаток очищали путем элюирования силикагеля от 10 до 100% петролейного эфира в этилацетате с получением твердого вещества, которое растирали в DCM (3 мл). Полученное твердое вещество собирали посредством фильтрования с получением соединения 281 в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,70 (s, 1 H), 9,47 (d, J=2,20 Гц, 1 H), 9,38 (s, 1 H), 8,95 (d, J=2,20 Гц, 1 H), 8,71-8,77 (m, 1 H), 8,49 (d, J=1,71 Гц, 1 H), 8,42 (d, J=5,62 Гц, 1 H), 8,12 (d, J=8,07 Гц, 1 H), 7,96 (td, J=7,76, 1,83 Гц, 1 H), 7,72 (dd, J=5,62, 1,96 Гц, 1 H), 7,39-7,47 (m, 1 H), 4,49 (t, J=12,72 Гц, 4 H). Способ 1: RT=2,81 мин.; M+H=408,0.
Соединения 283, 285, 309, 310, 313, 315, 317, 341, 342 и 344 синтезировали в соответствии с протоколом, описанным выше, с применением промежуточных соединений I-1, I-17, I-18 или I-19 и соответствующего амина и соответствующего оловоорганического вещества.
Соединения 256, 332, 333 и 336 синтезировали в соответствии с протоколом, описанным на стадии B, с применением I-24, I-27 или I-31 и соответствующих оловоорганических веществ.
Пример 23: 3-фтор-N-(4-фтор-3-(2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 311)
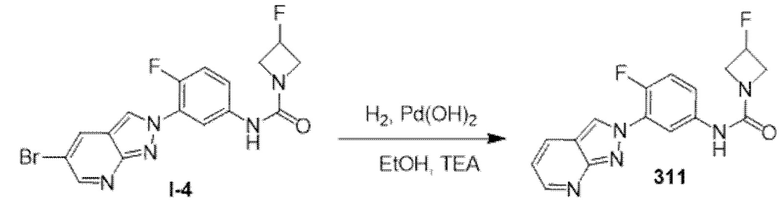
В раствор I-4 (100 мг, 245 мкмоль, 1 экв.) в EtOH (2 мл) добавляли TEA (25 мг, 245 мкмоль, 34,1 мкл, 1 экв.) и Pd(OH)2/C (34 мг, 24,5 мкмоль, чистота 10%, 0,1 экв.) в атмосфере N2. Суспензию дегазировали и 3 раза продували с помощью H2. Смесь перемешивали в атмосфере H2 (15 фунтов/кв. дюйм) при 25°C в течение 3 ч. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью препаративной HPLC (колонка: Luna C18 150 × 25 × 5 мкм; подвижная фаза: [вода (0,225% FA)-ацетонитрил]; B%: от 17% до 44%, 10 мин.) и лиофилизировали с получением соединения 311 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6), ppm, 8,98 (s, 1 H), 8,90 (d, J=2,26 Гц, 1 H), 8,72 (d, J=3,01 Гц, 1 H), 8,31 (d, J=8,28 Гц, 1 H), 8,25 (dd, J=7,03, 2,51 Гц, 1 H), 7,66-7,73 (m, 1 H), 7,43-7,51 (m, 1 H), 7,20 (dd, J=8,41, 4,14 Гц, 1 H), 5,29-5,51 (m, 1 H), 4,24-4,39 (m, 2 H), 3,96-4,10 (m, 2 H). Способ 1: RT=2,41 мин.; M+H=330,1.
Пример 24 : N-(3-(5-(1-ацетилазетидин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-3-фторазетидин-1-карбоксамид (соединение 267)
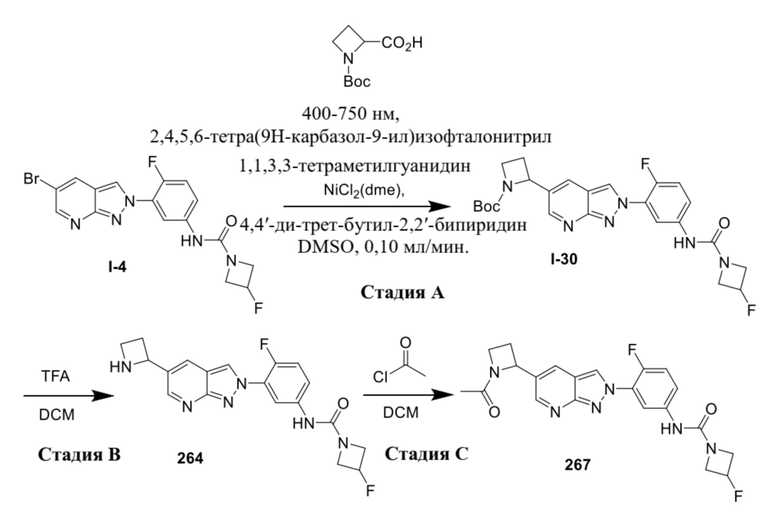
Стадия A. Растворяли 1-(трет-бутоксикарбонил)азетидин-2-карбоновую кислоту (394 мг, 2 ммоль), I-4 (400 мг, 1 ммоль), 1,1,3,3-тетраметилгуанидин (226 мг, 1,96 ммоль), NiCl2(dme) (22 мг, 0,1 ммоль), 4,4'-ди-трет-бутил-2,2'-бипиридин (26 мг, 0,1 моль) и 2,4,5,6-тетра(9H-карбазол-9-ил)изофталонитрил (23 мг, 0,03 ммоль) в DMSO (9,8 мл) при нагревании и ультразвуковой обработке. Раствор прокачивали (0,10 мл/мин.) через фотореактор (объем реактора - 0,9 мл; PFA-трубки с ID 0,04 дюйма, ID 0,0625 дюйма; облучаемая площадь 20 см2; расстояние до LED-панели 1 см; LED с длиной волны 400-750 нм; яркость 8800 лм) и собирали во флакон. Реакционную смесь очищали с помощью флэш-хроматографии на колонке C18 с элюированием от 15 до 85% ацетонитрила в воде и концентрировали. Материал дополнительно очищали с помощью хроматографии на силикагеле с элюированием от 40 до 100% EtOAc в гептане с получением I-30. 1H ЯМР (400 МГц, DMSO-d6) δ 8,98 (s, 1H), 8,94 (d, J=2,3 Гц, 1H), 8,71 (d, J=4,3 Гц, 1H), 8,22 (dd, J=7,0, 2,7 Гц, 1H), 7,70 (ddd, J=9,1, 4,1, 2,8 Гц, 1H), 7,48 (dd, J=11,1, 9,1 Гц, 1H), 7,13 (d, J=4,4 Гц, 1H), 5,55 (dd, J=9,0, 6,4 Гц, 1H), 5,48-5,30 (m, 1H), 4,32 (dddd, J=21,7, 10,4, 6,0, 1,4 Гц, 2H), 4,11-3,94 (m, 6H), 1,27 (s, 9H).
Стадия B. Растворяли I-30 (83 мг, 0,171 ммоль) в DCM (1 мл) и добавляли TFA (0,5 мл). Реакционную смесь перемешивали в течение 3 ч. и концентрировали, растворяли в MeOH и очищали с помощью флэш-хроматографии на колонке C18 с элюированием от 5 до 40% ацетонитрила в воде. В результате лиофилизации получали соединение 264 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ 9,38 (s, 1H), 9,07-8,96 (m, 2H), 8,84 (d, J=4,3 Гц, 1H), 8,30 (dd, J=7,0, 2,7 Гц, 1H), 7,72-7,62 (m, 1H), 7,52 (dd, J=11,2, 9,1 Гц, 1H), 7,33 (dd, J=4,4, 1,2 Гц, 1H), 6,04 (s, 1H), 5,41 (dtt, J=57,4, 6,1, 3,1 Гц, 1H), 4,40-4,25 (m, 2H), 4,27-4,16 (m, 1H), 4,10-3,94 (m, 2H), 3,90-3,77 (m, 1H), 3,06-2,94 (m, 1H), 2,86 (dtd, J=11,7, 8,7, 4,4 Гц, 1H). Способ 1: RT=1,69 мин.; M+H=385,2.
Стадия C. Соединение 264 (20 мг, 0,052 ммоль) растворяли в DCM (объем: 1 мл) и добавляли DIEA (0,027 мл, 0,16 ммоль) и ацетилхлорид (5,55 мкл, 0,078 ммоль) и перемешивали в течение 60 ч. Реакционную смесь очищали с помощью SFC на колонке Princeton PPU 250 × 21,2 мм с элюированием от 20 до 60% MeOH в жидком CO2 с получением соединения 267. 1H ЯМР (400 МГц, Метанол-d4) δ 8,86 (dd, J=3,4, 2,2 Гц, 1H), 8,74 (d, J=4,4 Гц, 0,5H), 8,67 (d, J=4,5 Гц, 0,5H), 8,17 (dd, J=6,8, 2,7 Гц, 0,5H), 8,13 (dd, J=6,8, 2,7 Гц, 0,5H), 7,66 (dddd, J=8,8, 4,3, 2,7, 1,7 Гц, 1H), 7,37 (ddd, J=10,9, 9,1, 3,7 Гц, 1H), 7,28 (dd, J=4,4, 0,8 Гц, 0,5H), 7,18 (dd, J=4,5, 0,9 Гц, 0,5H), 5,93 (dd, J=9,2, 5,6 Гц, 0,5H), 5,73 (dd, J=9,3, 6,1 Гц, 0,5H), 5,37 (dtt, J=57,1, 6,1, 3,2 Гц, 1H), 4,48-4,32 (m, 3H), 4,24-4,15 (m, 2H), 4,11 (ddd, J=10,3, 3,2, 1,4 Гц, 1H), 3,05-2,91 (m, 1H), 2,34-2,21 (m, 1H), 2,03 (s, 1,5H), 1,71 (s, 1,5H). Способ 2: RT=0,66 мин.; M+H=427,2.
Соединение 247 синтезировали в соответствии с протоколом, указанным выше на стадии A, с применением 1-(трет-бутоксикарбонил)пирролидин-2-карбоновой кислоты в качестве исходного материала.
Пример 25 : N-(4-фтор-3-(5-(тетрагидро-2H-пиран-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 288)
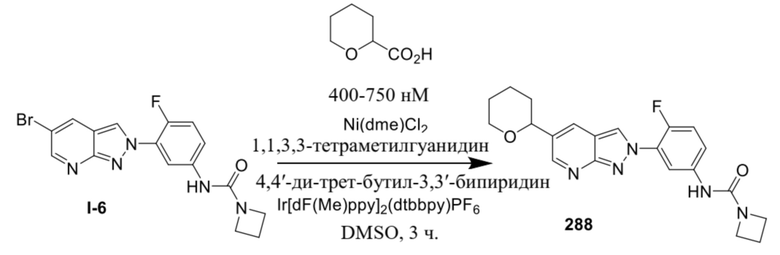
Растворяли I-6 (30 мг, 0,077 ммоль), комплекс хлорида никеля(II), этиленгликоля и диметилового эфира (1,7 мг, 7,8 мкмоль), 1,1,3,3-тетраметилгуанидин (53 мг, 0,46 ммоль), 4,4'-ди-трет-бутил-3,3'-бипиридин (2,1 мг, 7,7 мкмоль), тетрагидро-2H-пиран-2-карбоновую кислоту (60 мг, 0,46 ммоль) и [4,4'-бис(трет-бутил)-2,2'-бипиридин]бис[3,5-дифтор-2-[5-метил-2-пиридинил]фенил]иридия(III) гексафторфосфат (3,90 мг, 3,84 мкмоль) в DMSO (769 мкл) при нагревании и ультразвуковой обработке. Раствор добавляли в лунку объемом 0,3 мл 96-луночного тефлонового планшета и подвергали облучению в течение 3 часов (облучаемая площадь 20 см2; расстояние до LED-панели 10 см; LED с длиной волны 400-750 нм; яркость 7600 лм). Полученный раствор очищали с помощью препаративной HPLC (колонка Waters Atlantis T3 C18 19 × 50 мм × 10 мкм) с элюированием ацетонитрилом (0,035% TFA) в воде (0,05% TFA) с получением соединения 288. 1H ЯМР (400 МГц, Метанол-d4) δ 9,03 (d, J=2,2 Гц, 1H), 8,73 (d, J=4,9 Гц, 1H), 8,19 (dd, J=6,8, 2,7 Гц, 1H), 7,63 (ddd, J=9,1, 4,2, 2,7 Гц, 1H), 7,37 (dd, J=10,9, 9,1 Гц, 1H), 7,32 (dd, J=5,0, 0,9 Гц, 1H), 4,92 (ddt, J=2,7, 1,4, 0,7 Гц, 1H), 4,23 (d, J=10,3 Гц, 1H), 4,17-4,07 (m, 2H), 3,74 (td, J=11,6, 2,5 Гц, 1H), 3,37-3,32 (m, 2H), 2,33 (p, J=7,6 Гц, 2H), 2,15-1,98 (m, 2H), 1,92-1,57 (m, 4H). Способ 2: RT=0,89 мин.; M+H=396,0.
Соединение 289 синтезировали в соответствии с указанным выше протоколом с применением 1,4-диоксан-2-карбоновой кислоты в качестве исходного материала.
Примеры 26A и B: (1S,2R)-2-фтор-N-(4-фтор-3-(5-(пиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)циклопропан-1-карбоксамид или (1R,2S)-2-фтор-N-(4-фтор-3-(5-(пиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)циклопропан-1-карбоксамид (соединения 269 и 273)
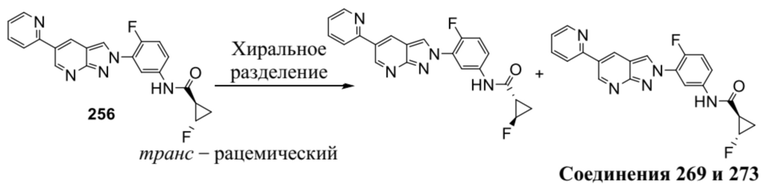
Соединение 256 (170 мг) разделяли с помощью SFC (колонка: CHIRALCEL® OJ-H (250 мм × 25 мм, 10 мкм); подвижная фаза: [CO2 - MeOH (0,1% NH3H2O)]; B%: 50%) с получением соединения 269 (пик 1, RT=5,42 мин.) в виде желтого твердого вещества и соединения 273 (пик 2, RT=6,75 мин.) в виде грязно-белого твердого вещества.
269 (пик 1): SFC RT: 5,42 мин. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,75 (s, 1 H), 9,48 (d, J=2,26 Гц, 1 H), 9,03 (d, J=2,38 Гц, 1 H), 8,95 (d, J=2,26 Гц, 1 H), 8,74 (d, J=4,14 Гц, 1 H), 8,39 (dd, J=6,96, 2,57 Гц, 1 H), 8,13 (d, J=8,03 Гц, 1 H), 7,96 (td, J=7,75, 1,69 Гц, 1 H), 7,65-7,74 (m, 1 H), 7,57 (dd, J=11,11, 9,10 Гц, 1 H), 7,42 (dd, J=7,22, 5,21 Гц, 1 H), 4,78-5,06 (m, 1 H), 2,26-2,35 (m, 1 H), 1,49-1,66 (m, 1 H), 1,24-1,33 (m, 1 H). Способ 1: RT=2,80 мин.; M+H=392,2.
273 (пик 2): SFC RT: 6,75 мин. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,75 (s, 1 H), 9,48 (d, J=2,08 Гц, 1 H), 9,03 (d, J=2,32 Гц, 1 H), 8,95 (d, J=2,20 Гц, 1 H), 8,74 (d, J=4,52 Гц, 1 H), 8,40 (dd, J=7,03, 2,51 Гц, 1 H), 8,13 (d, J=8,07 Гц, 1 H), 7,96 (td, J=7,79, 1,77 Гц, 1 H), 7,69 (dt, J=8,47, 3,53 Гц, 1 H), 7,57 (dd, J=11,00, 9,17 Гц, 1 H), 7,42 (dd, J=7,09, 5,14 Гц, 1 H), 4,80-5,05 (m, 1 H), 2,25-2,34 (m, 1 H), 1,48-1,67 (m, 1 H), 1,28 (dq, J=13,13, 6,67 Гц, 1 H). Способ 1: RT=2,82 мин.; M+H=392,2.
Примеры 27A и B: (1S,2R)-N-(3-(5-(3,3-дифторазетидин-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-2-фторциклопропан-1-карбоксамид или (1R,2S)-N-(3-(5-(3,3-дифторазетидин-1-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)-4-фторфенил)-2-фторциклопропан-1-карбоксамид (соединения 268 и 271)
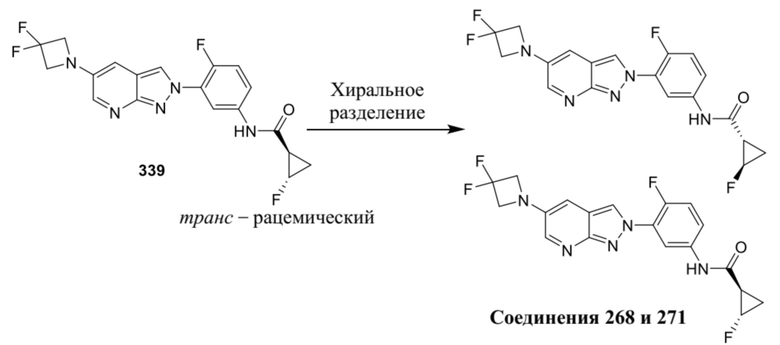
Соединение 339 (130 мг) разделяли с помощью SFC (колонка: CHIRALCEL® OD-H (250 мм × 25 мм, 10 мкм); подвижная фаза: [CO2 - MeOH (0,1% NH3H2O)]; B%: 50%) с получением соединения 271 (пик 1, RT=4,58 мин.) в виде желтого твердого вещества и соединения 268 (пик 2, RT=8,01 мин.) в виде желтого твердого вещества.
268 (пик 2): SFC RT: 8,01 мин. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,69 (s, 1 H), 8,64 (d, J=2,69 Гц, 1 H), 8,37 (d, J=2,81 Гц, 1 H), 8,32 (dd, J=7,03, 2,51 Гц, 1 H), 7,61-7,67 (m, 1 H), 7,52 (dd, J=11,19, 9,11 Гц, 1 H), 7,15 (d, J=2,81 Гц, 1 H), 4,80-5,03 (m, 1 H), 4,41 (t, J=12,29 Гц, 4 H), 2,24-2,33 (m, 1 H), 1,46-1,67 (m, 1 H), 1,27 (dd, J=13,27, 6,54 Гц, 1 H). Способ 1: RT=2,89 мин.; M+H=406,0.
271 (пик 1): SFC RT: 4,58 мин. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,75 (s, 1 H), 8,70 (d, J=2,69 Гц, 1 H), 8,43 (d, J=2,81 Гц, 1 H), 8,38 (dd, J=6,97, 2,57 Гц, 1 H), 7,66-7,77 (m, 1 H), 7,52-7,63 (m, 1 H), 7,20 (d, J=2,81 Гц, 1 H), 4,85-5,08 (m, 1 H), 4,47 (t, J=12,23 Гц, 4 H), 2,27-2,39 (m, 1 H), 1,52-1,72 (m, 1 H), 1,33 (dq, J=13,11, 6,43 Гц, 1 H). Способ 1: RT=2,89 мин.; M+H=406,0.
Пример 28: N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)-4-(гидроксиметил)-2-метилоксазол-5-карбоксамид (соединение 277)
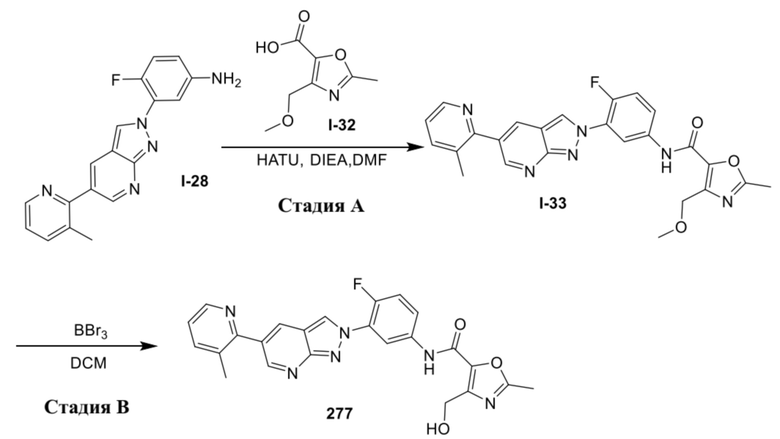
Стадия A. Промежуточное соединение I-33 получали подобным образом, как в примере 12, с применением I-28 и I-32 в качестве исходных материалов. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,70 (s, 1 H), 9,04 (d, J=2,45 Гц, 1 H), 8,95 (d, J=2,20 Гц, 1 H), 8,60-8,66 (m, 2 H), 8,55 (d, J=2,20 Гц, 1 H), 7,89-7,96 (m, 2 H), 7,62 (dd, J=11,00, 9,29 Гц, 1 H), 7,44-7,52 (m, 1 H), 4,64 (s, 2 H), 3,31 (s, 3 H), 2,57 (s, 3 H), 2,46 (s, 3 H). M+H=473,0.
Стадия B. В раствор I-33 (15 мг, 28,9 мкмоль, 1 экв., FA) в DCM (1 мл) добавляли по каплям BBr3 (14,5 мг, 57,9 мкмоль, 5,6 мкл, 2 экв.) в DCM (0,5 мл) при −78°C. Смесь перемешивали при 20°C в течение 18 ч. в атмосфере N2. Добавляли насыщенный раствор NaHCO3 (3 мл) и смесь экстрагировали с помощью DCM (10 мл). Органический слой высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью препаративной HPLC (колонка: Phenomenex Gemini 150 × 25 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 30% до 60%, 12 мин.) и лиофилизировали с получением соединения 277 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 9,00 (d, J=2,20 Гц, 1 H), 8,94 (d, J=1,96 Гц, 1 H), 8,53-8,63 (m, 2 H), 8,51 (d, J=2,20 Гц, 1 H), 7,85-7,95 (m, 1 H), 7,81 (br d, J=7,34 Гц, 1 H), 7,55-7,67 (m, 1 H), 7,37 (dd, J=7,46, 4,77 Гц, 1 H), 4,69 (s, 2 H), 2,55 (s, 3 H), 2,45 (s, 3 H). Способ 1: RT=2,50 мин.; M+H=459,0.
Пример 29: N-(4-фтор-3-(5-(3-метилпиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)-2-(гидроксиметил)-4-метилоксазол-5-карбоксамид (соединение 334)
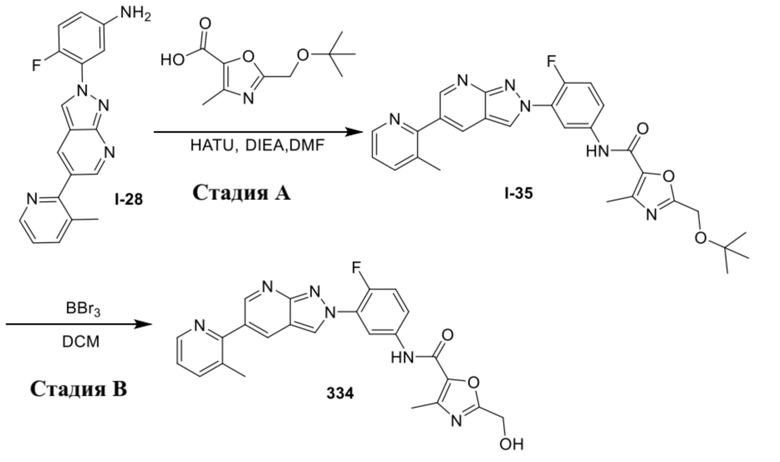
Стадия A. Промежуточное соединение I-35 получали подобным образом, как в примере 12, с применением I-28 и 2-(трет-бутоксиметил)-4-метилоксазол-5-карбоновой кислоты в качестве исходных материалов. 1H ЯМР (400 МГц, DMSO-d6) δ ppm 10,55-10,67 (m, 1 H) 9,00 (d, J=2,45 Гц, 1 H) 8,91-8,97 (m, 1 H) 8,54-8,65 (m, 2 H) 8,47-8,53 (m, 1 H) 7,96 (s, 1 H) 7,80 (dd, J=7,70, 0,86 Гц, 1 H) 7,55-7,65 (m, 1 H) 7,37 (dd, J=7,70, 4,77 Гц, 1 H) 4,54 (s, 2 H) 2,69 (s, 3 H) 2,44 (s, 3 H) 1,24 (s, 9 H).
Стадия B. В раствор I-35 (70 мг, 136,04 мкмоль, 1 экв.) в DCM (2 мл) добавляли TFA (0,5 мл, 6,75 ммоль, 49,6 экв.). Смесь перемешивали при 25°C в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении с удалением растворителя. Регулировали pH до 7 с помощью TEA. Остаток очищали с помощью препаративной HPLC (колонка: Phenomenex Gemini 150 × 25 мм × 10 мкм; подвижная фаза: [вода (0,05% гидроксида аммония об./об.)-ацетонитрил]; B%: от 20% до 50%, 12 мин.) и лиофилизировали с получением соединения 334 в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ ppm, 10,63 (s, 1 H), 9,00 (d, J=2,20 Гц, 1 H), 8,94 (d, J=2,20 Гц, 1 H), 8,62 (dd, J=7,09, 2,45 Гц, 1 H), 8,57 (d, J=3,67 Гц, 1 H), 8,51 (d, J=2,20 Гц, 1 H), 7,91-8,01 (m, 1 H), 7,81 (d, J=7,34 Гц, 1 H), 7,60 (dd, J=11,00, 9,05 Гц, 1 H), 7,37 (dd, J=7,70, 4,77 Гц, 1 H), 5,83 (t, J=5,99 Гц, 1 H), 4,60 (d, J=6,11 Гц, 2 H), 2,45 (s, 6 H). Способ 1: RT=2,57 мин.; M+H=459,0.
Пример 30: 3,3-дифтор-N-(4-фтор-3-(5-(пиридин-2-ил)-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 39)
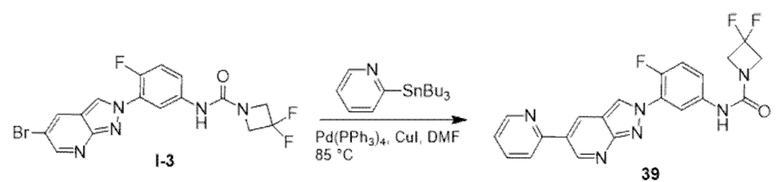
В раствор I-3 (3,00 г, 7,04 ммоль, 1 экв.) и трибутил(2-пиридил)станнана (3,4 г, 9,1 ммоль, 1,3 экв.) в DMF (40 мл) добавляли Pd(PPh3)4 (813 мг, 703,9 мкмоль, 0,1 экв.) и CuI (134 мг, 703,9 мкмоль, 0,1 экв.) в атмосфере N2. Смесь перемешивали при 85°C в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении с удалением растворителя. Остаток очищали с помощью хроматографии на силикагеле с элюированием смесью петролейный эфир/этилацетат=от 2:1 до 0:1, с получением соединения 39 в виде белого твердого вещества. 1H ЯМР (400 МГц, Метанол-d4) δ 9,36 (d, J=2,3 Гц, 1H), 9,00-8,93 (m, 2H), 8,79-8,75 (m, 1H), 8,26 (dd, J=6,8, 2,7 Гц, 1H), 8,19-8,14 (m, 2H), 7,70-7,64 (m, 1H), 7,64-7,56 (m, 1H), 7,46-7,35 (m, 1H), 4,44 (td, J=12,3, 3,0 Гц, 4H). Способ 2: RT=0,99 мин.; M+H=425,0.
Пример 31: 3,3-дифтор-N-(4-фтор-3-(5-фенил-2H-пиразоло[3,4-b]пиридин-2-ил)фенил)азетидин-1-карбоксамид (соединение 353)
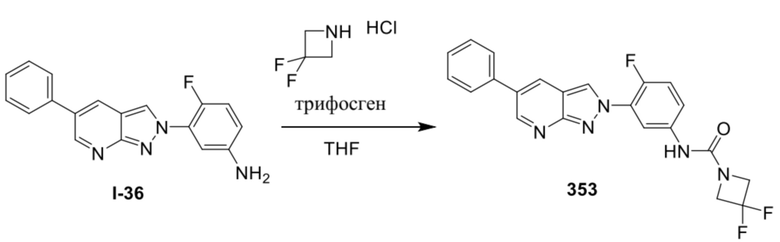
В перемешиваемый раствор трифосгена (23 мг, 0,079 ммоль) в сухом THF (1 мл) добавляли I-36 (40 мг, 0,131 ммоль) в сухом THF (2 мл) при -5°C - 0°C и перемешивали при к. т. в течение 10 мин. Добавляли по каплям гидрохлорид 3,3-дифторазетидина (34 мг, 0,263 ммоль) и DIEA (51 мг, 0,394 ммоль) в сухом THF (2 мл) при 0°C и реакционную смесь перемешивали при к. т. в течение 1 ч. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали с помощью EtOAc (50 мл × 2). Объединенные органические слои промывали водой (20 мл), солевым раствором (10 мл) и высушивали над сульфатом натрия и концентрировали in vacuo. Неочищенное соединение очищали с помощью хроматографии на силикагеле с элюированием с помощью 50% EtOAc в н-гексане с получением соединения 353 в виде грязно-белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) 9,25 (s, 1H), 9,06-9,05 (d, J=2,8, 1H), 8,96-8,95 (d, J=2,0, 1H), 8,52-8,51 (d, J=2,4, 1H), 8,27-8,25 (m, 1H), 7,80-7,78 (d, J=7,2, 2H), 7,72-7,67 (m, 1H), 7,56-7,48 (m, 3H), 7,44-7,41 (m, 1H), 4,44-4,38 (t, J=16,4, 4H).
В таблице 2 перечислены соединения, полученные в соответствии с примерами выше.
Таблица 2
соед.
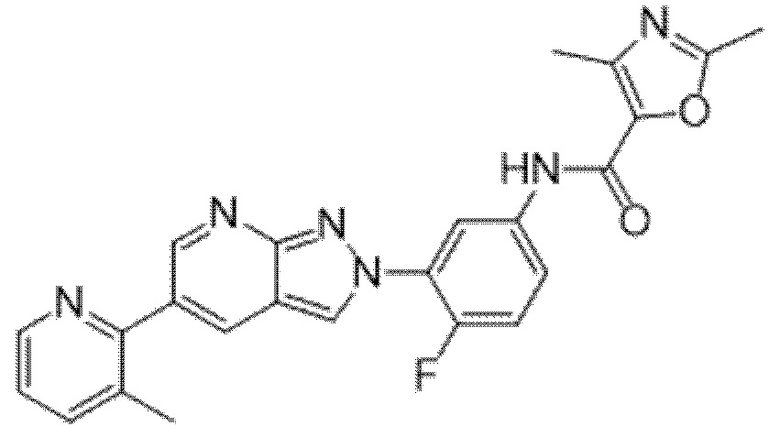
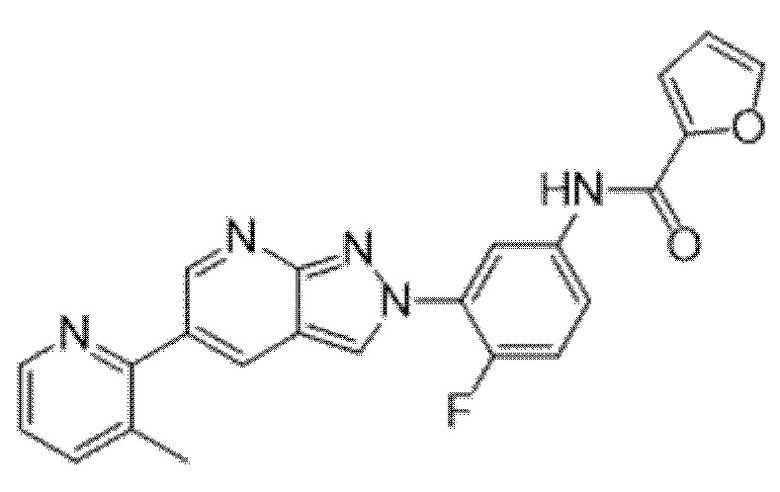
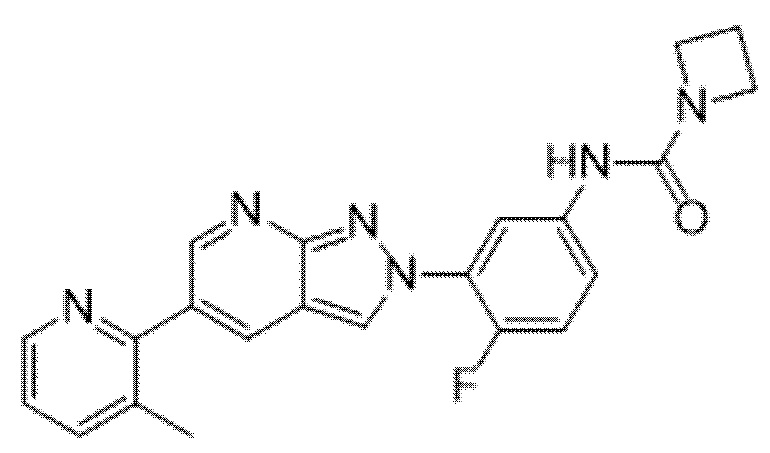
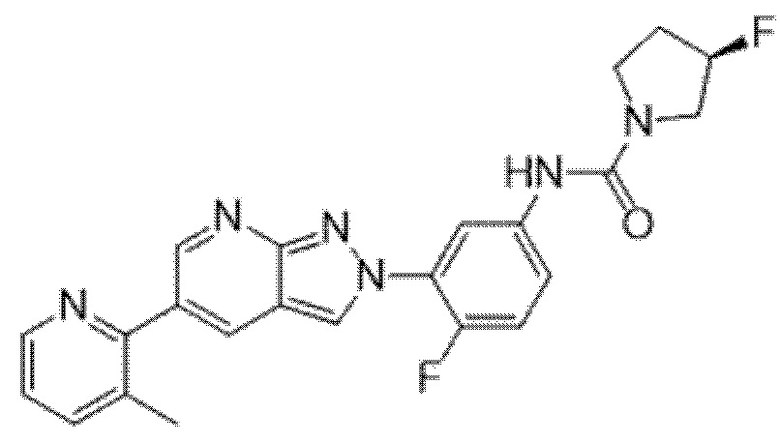
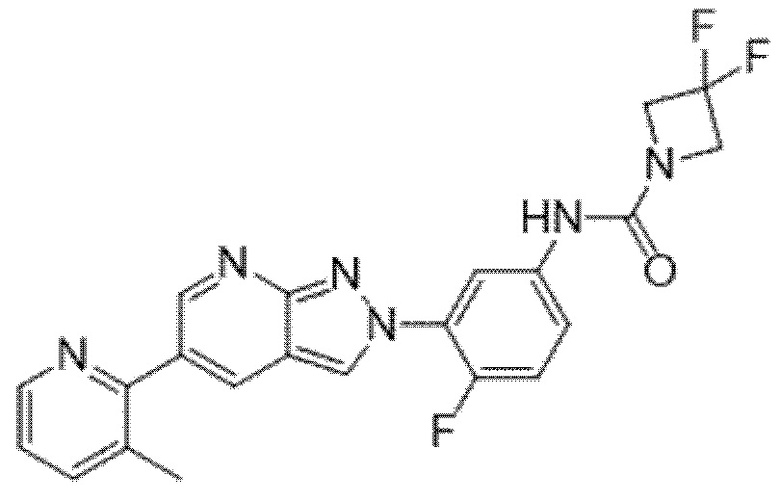
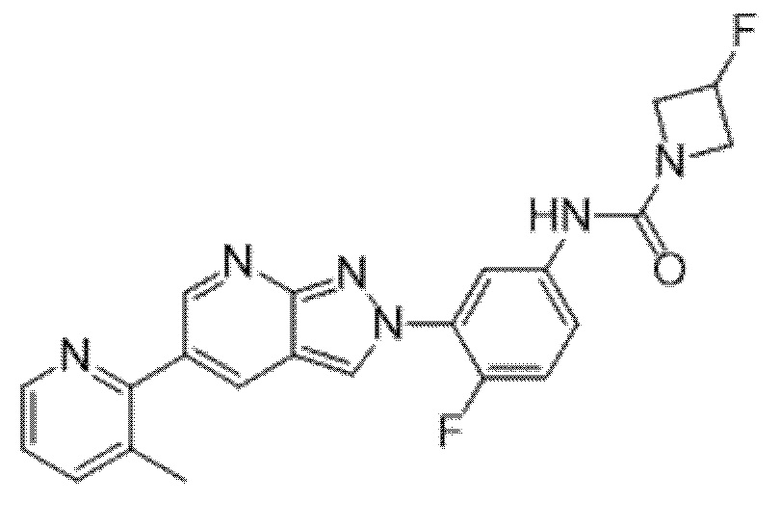
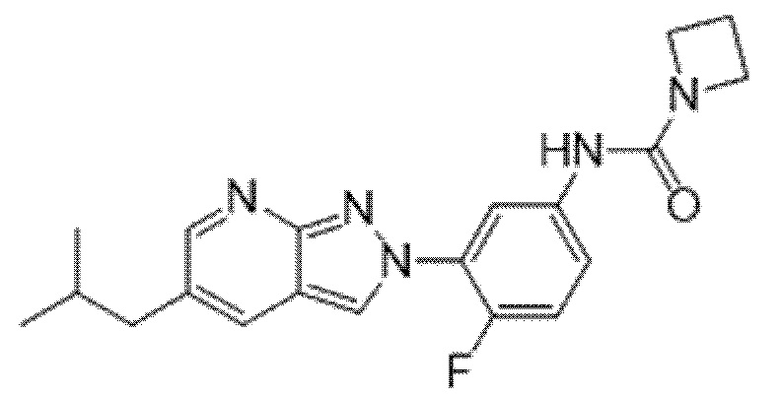
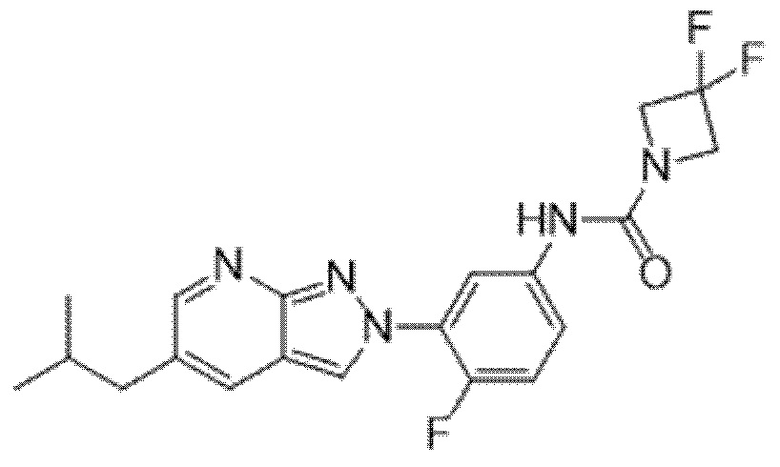
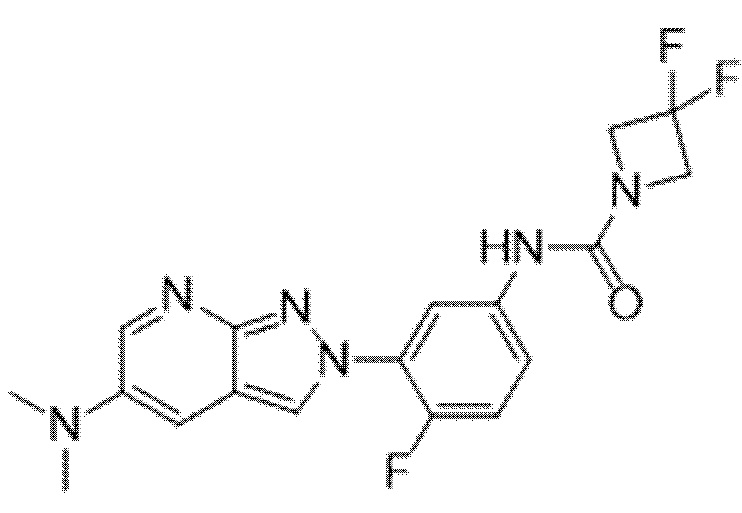
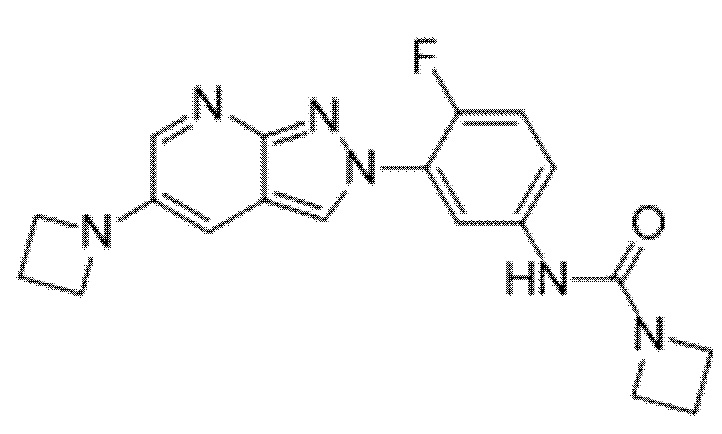
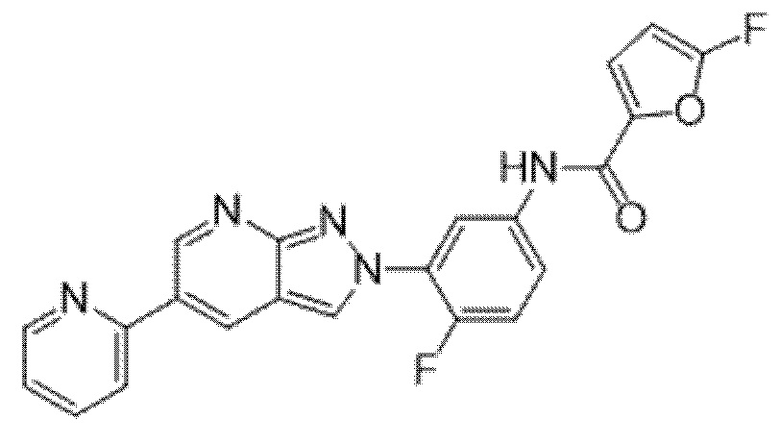
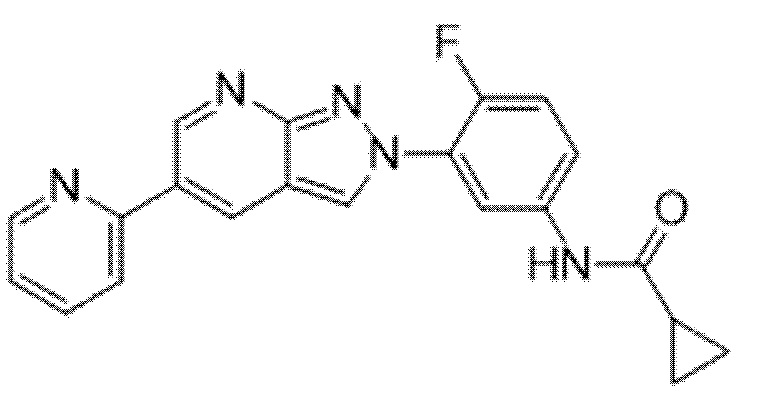
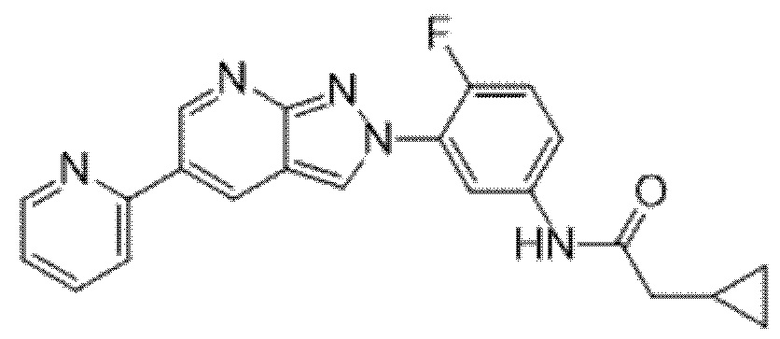
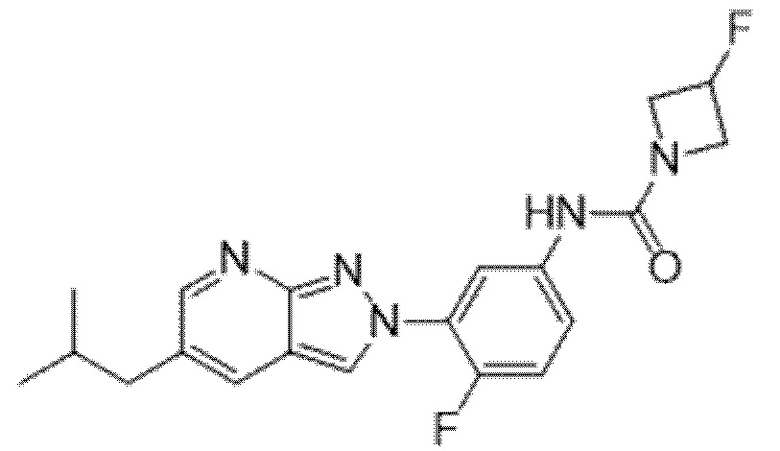
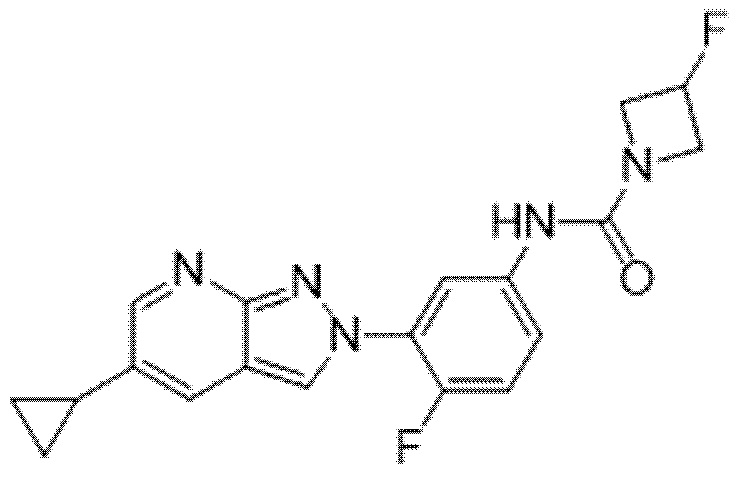
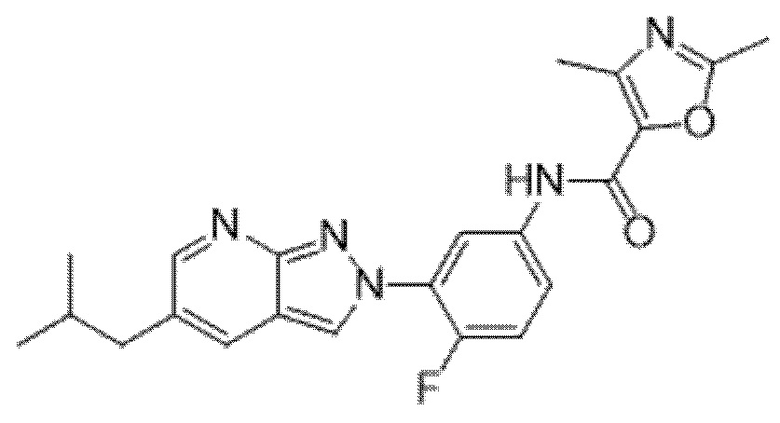
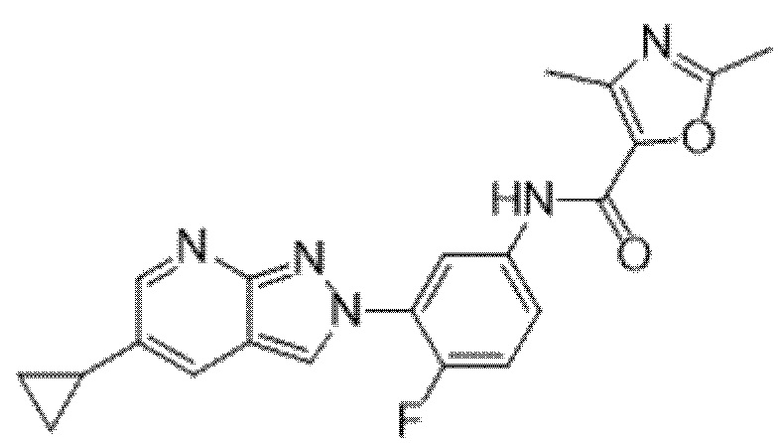
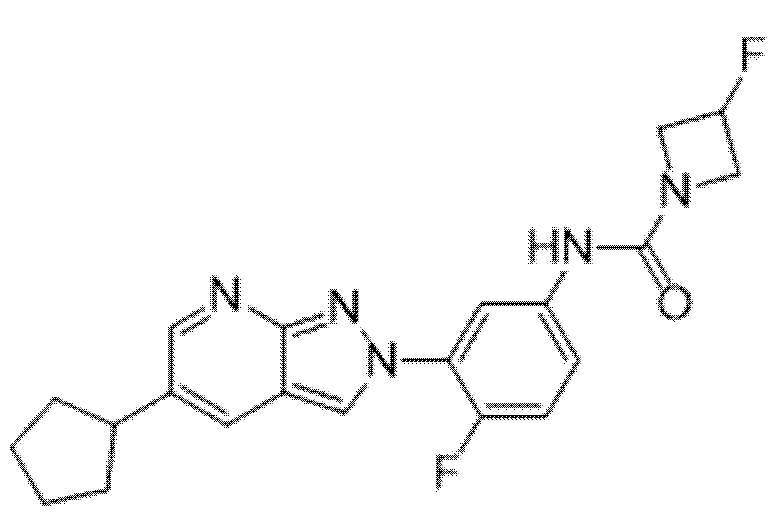
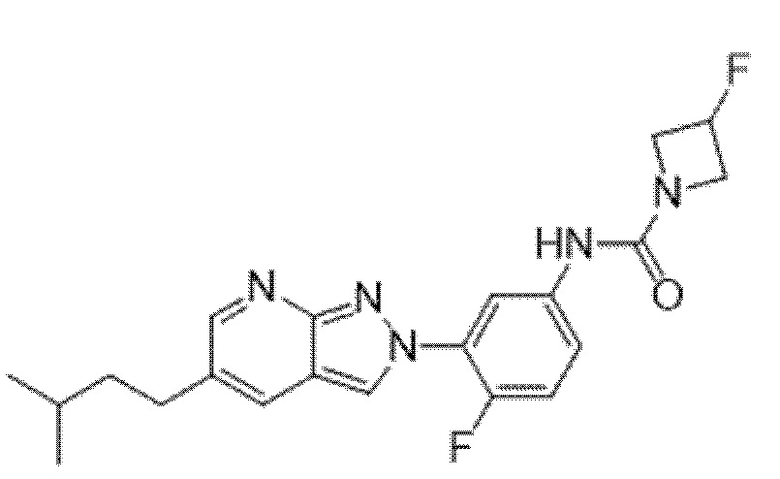
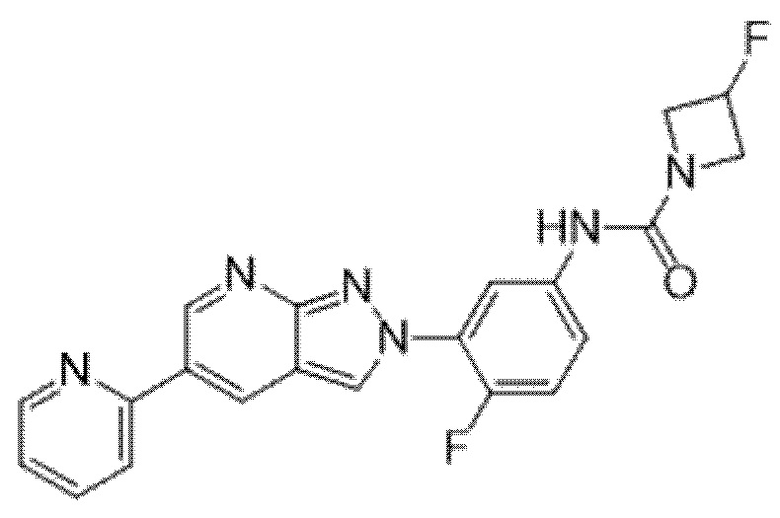
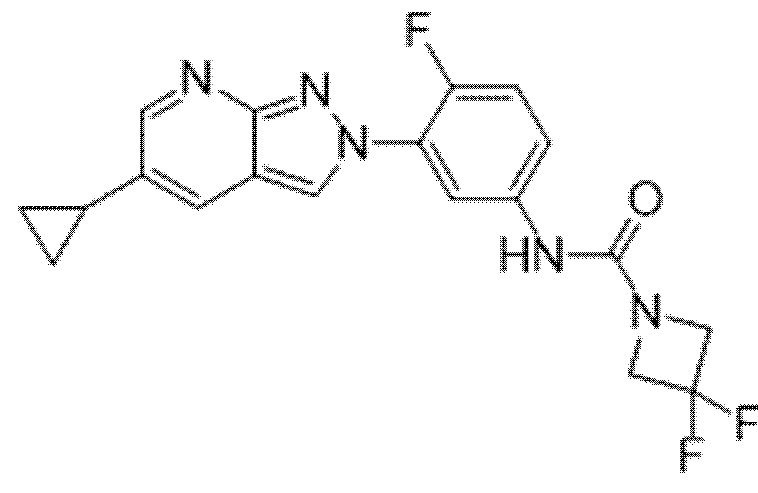
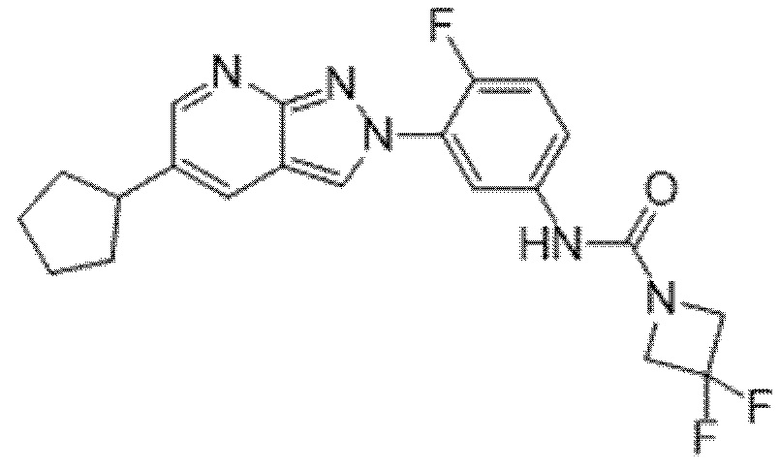
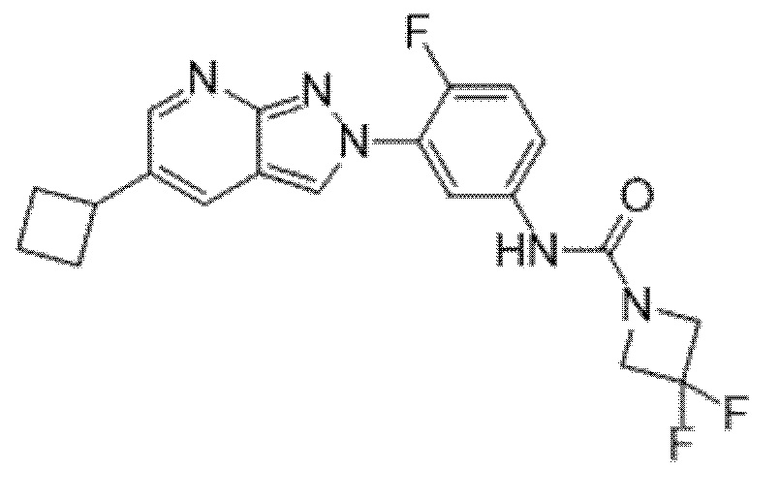
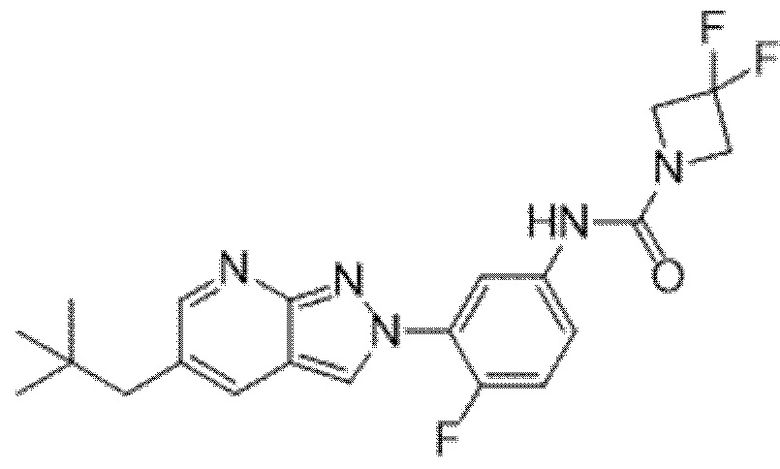
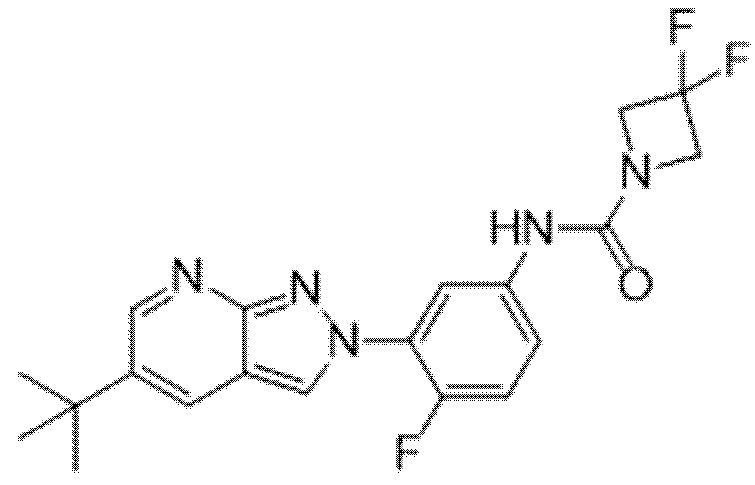
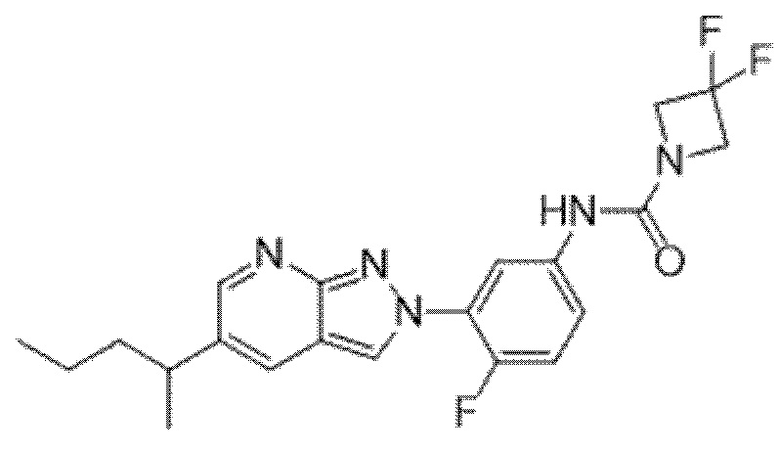
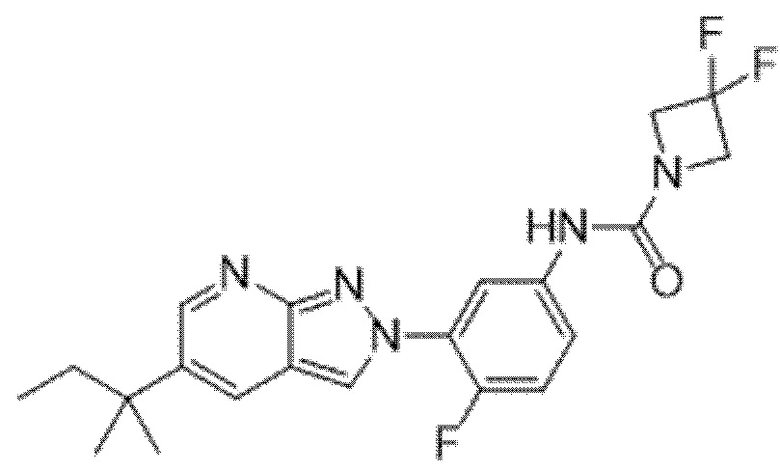
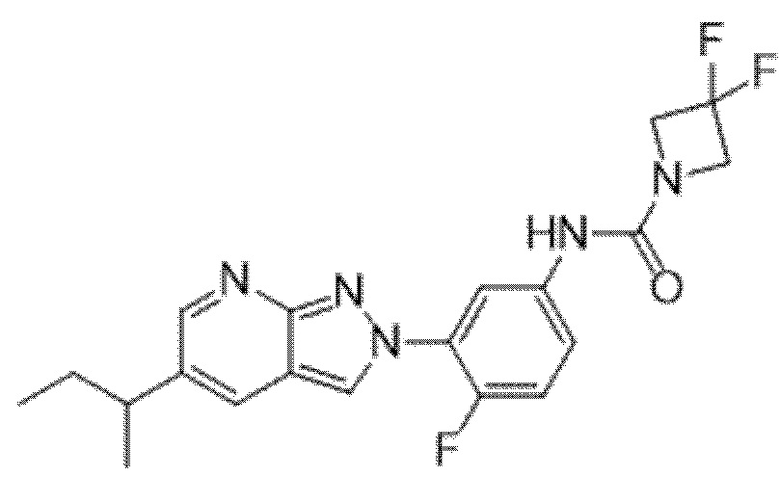
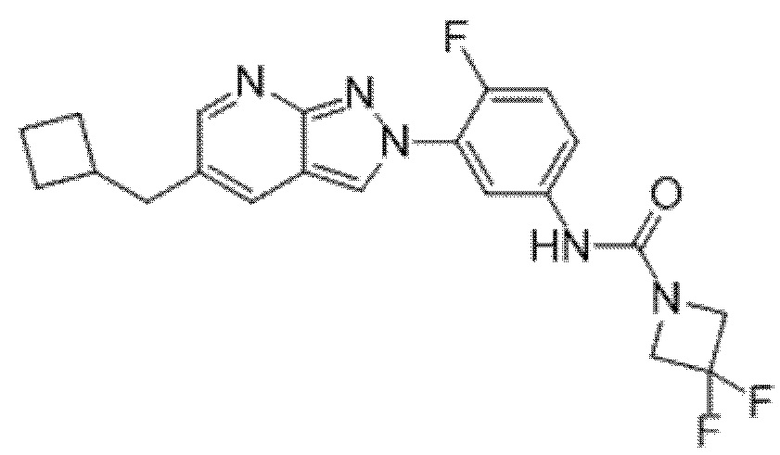
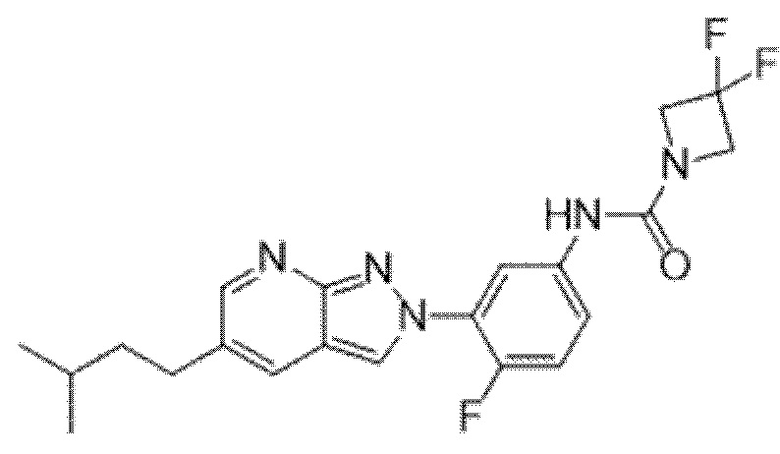
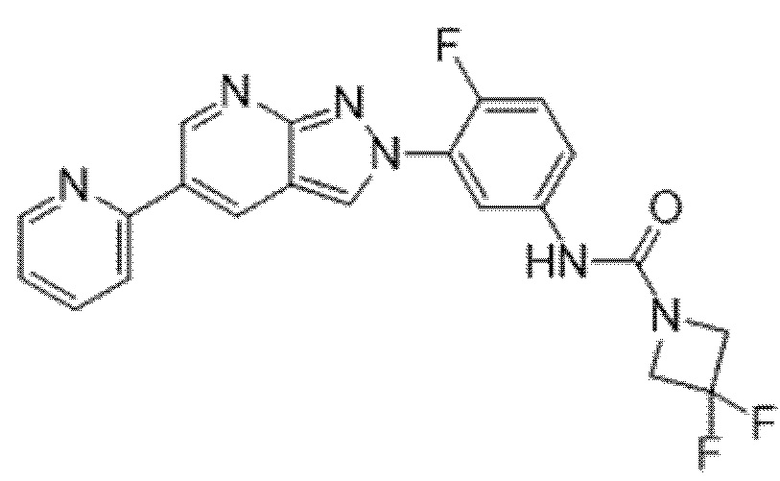
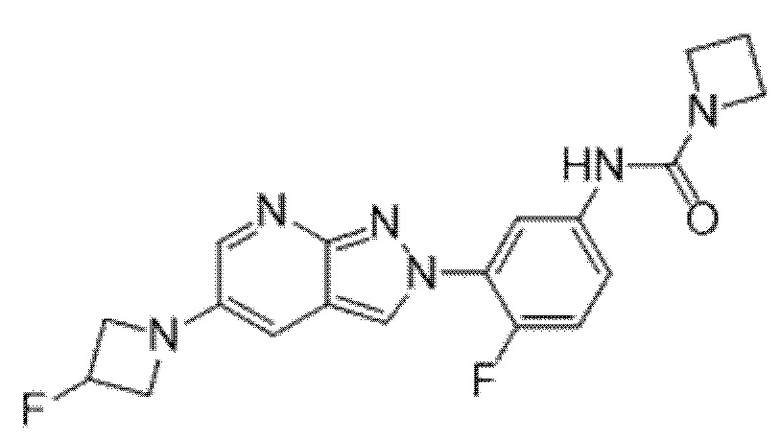
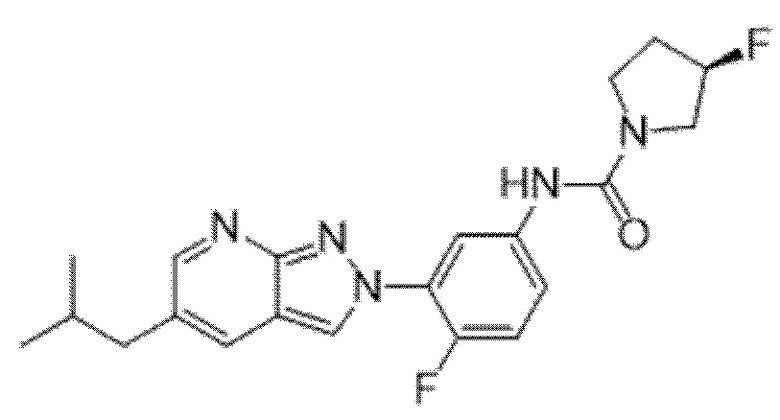
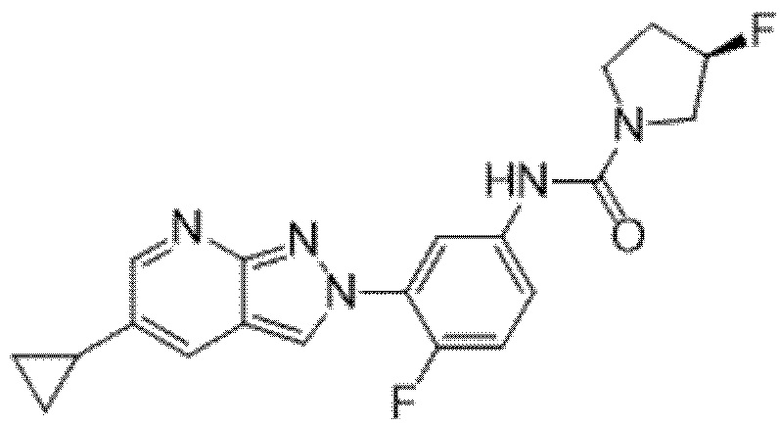
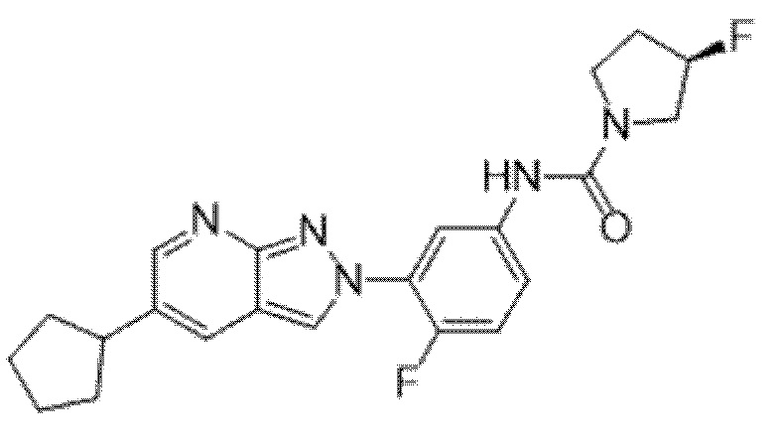
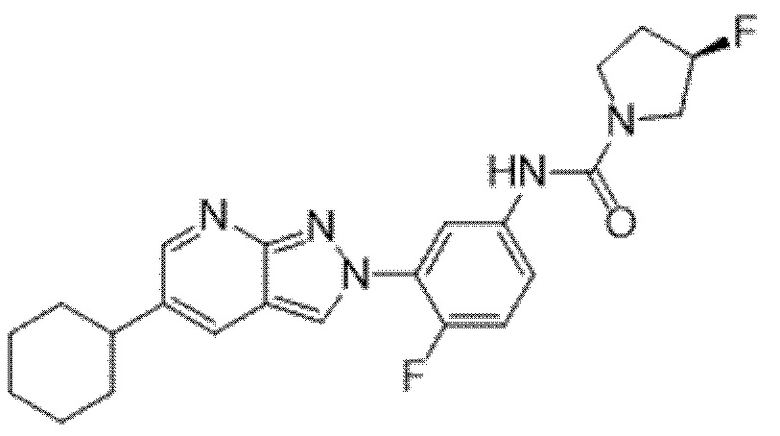
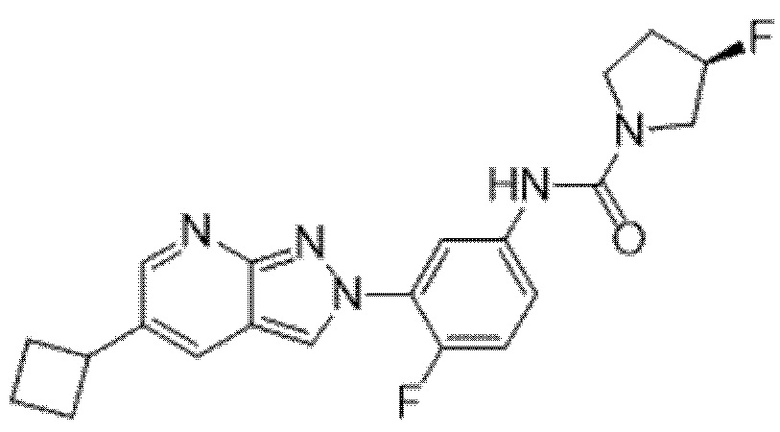
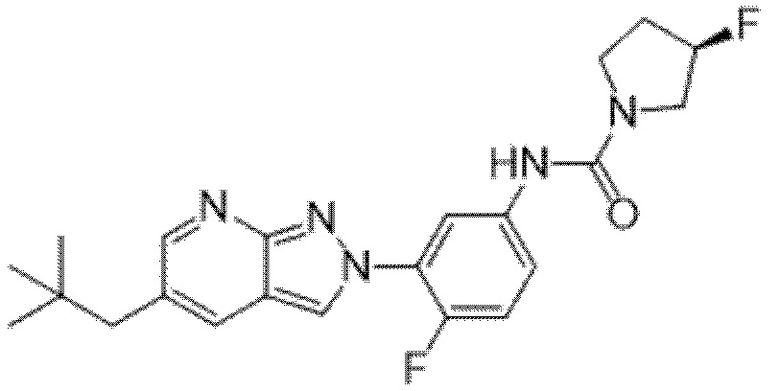
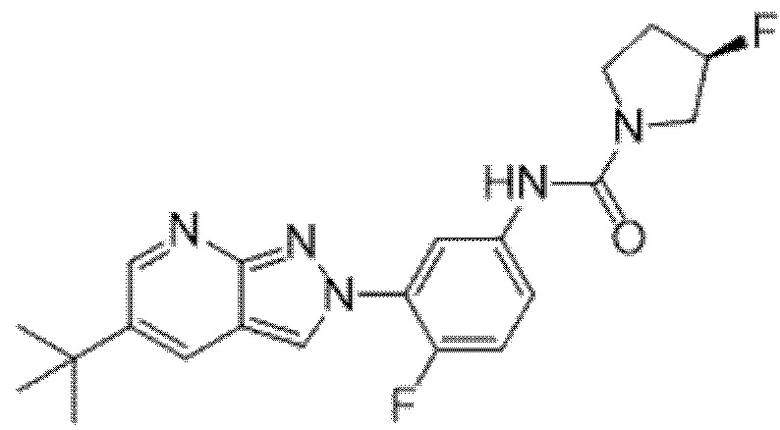
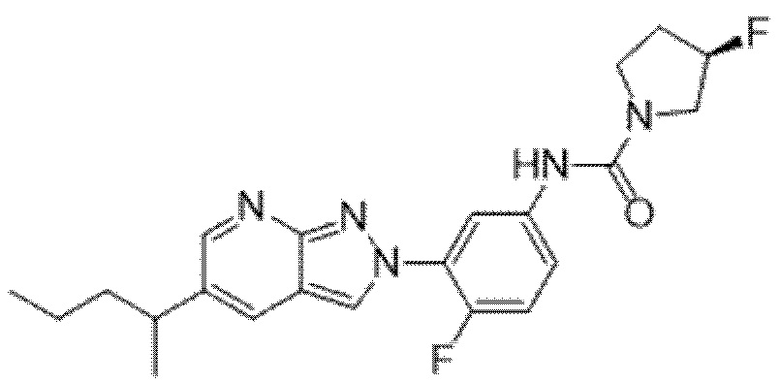
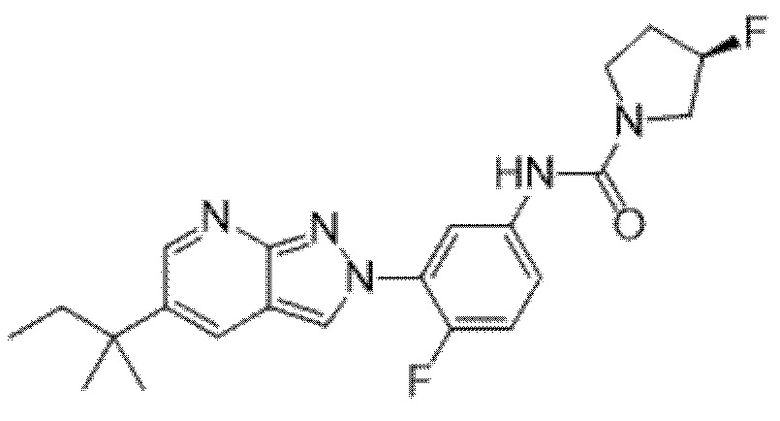
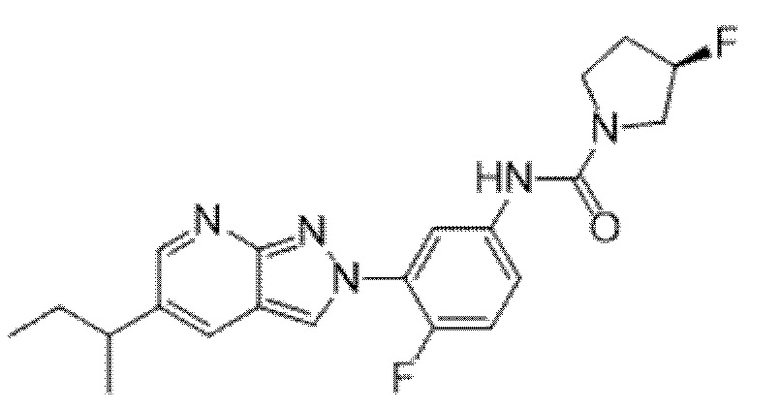
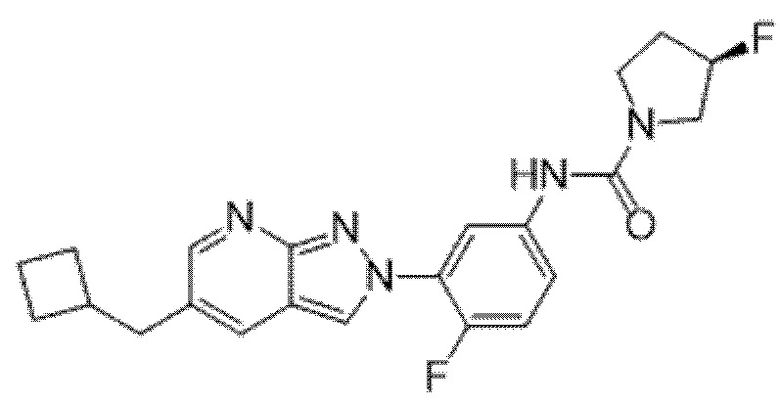
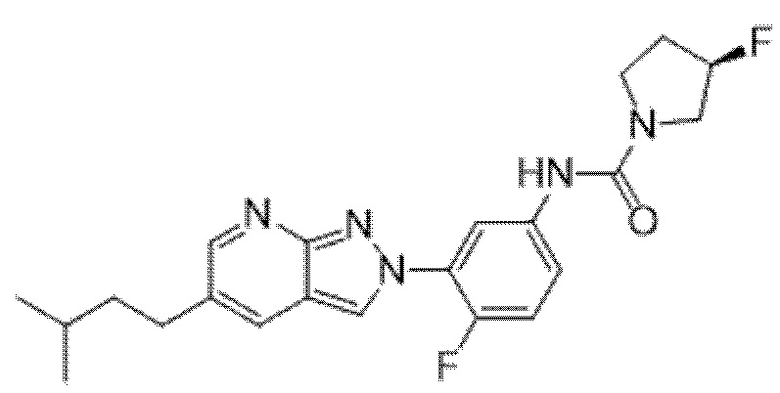
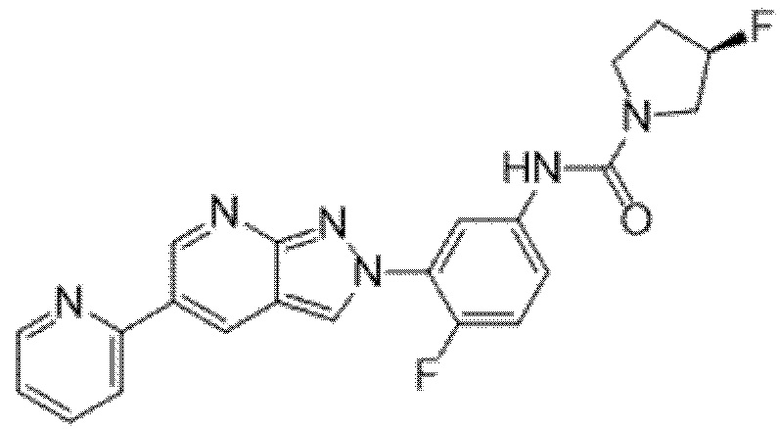
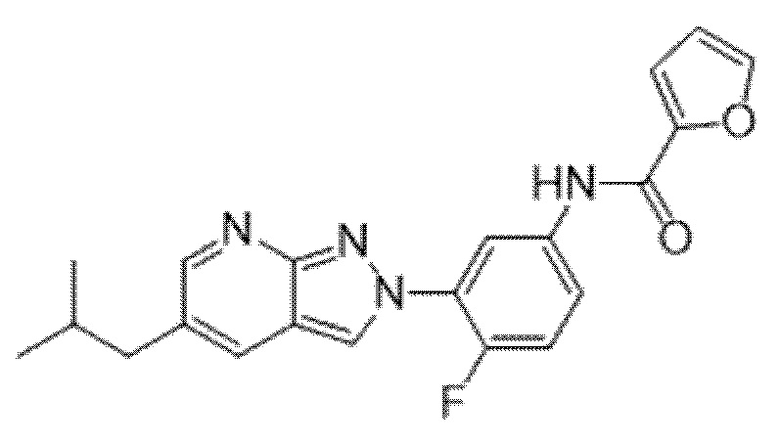
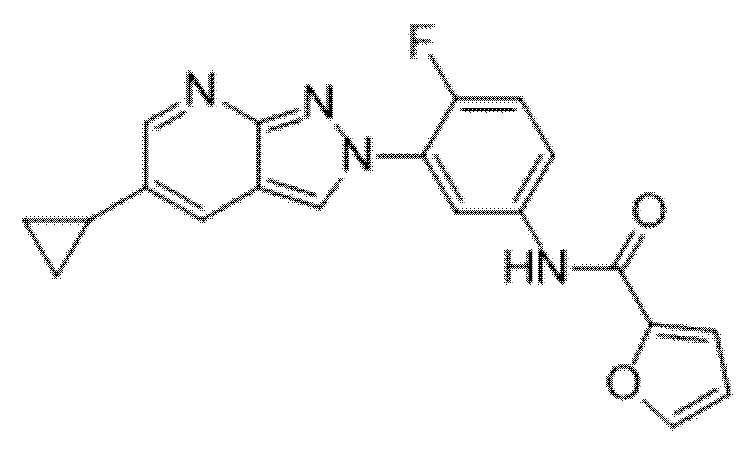
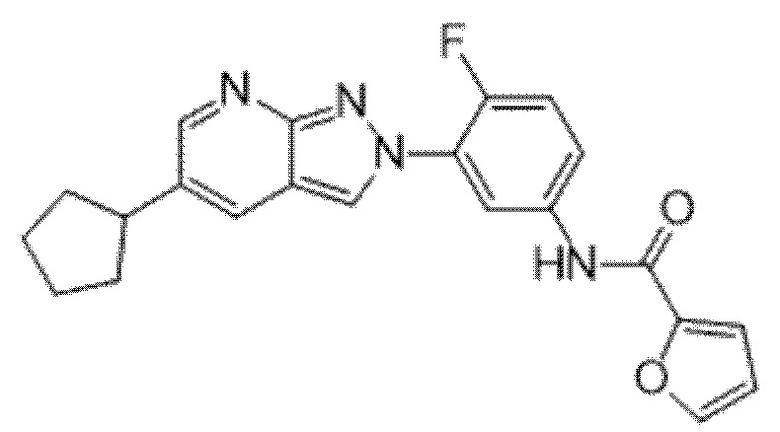
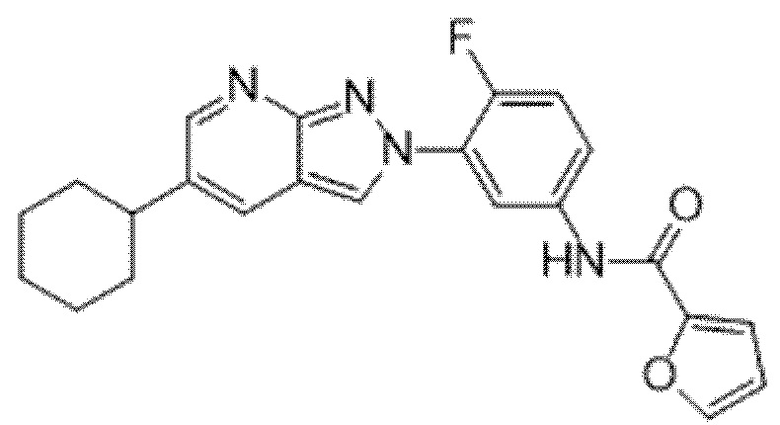
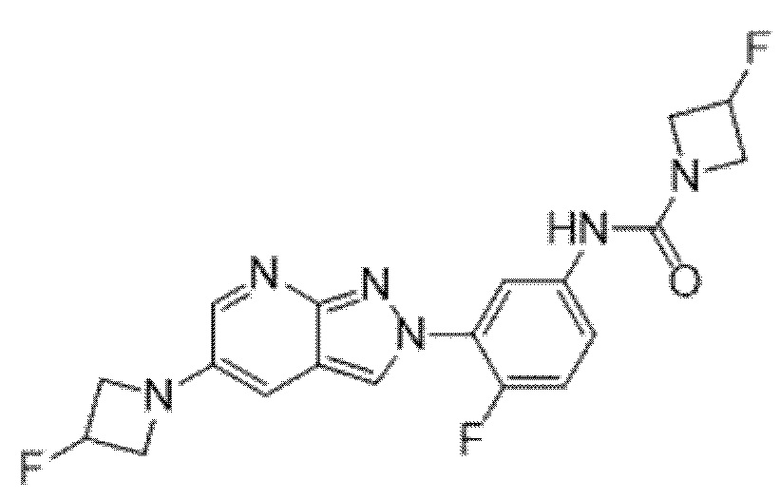
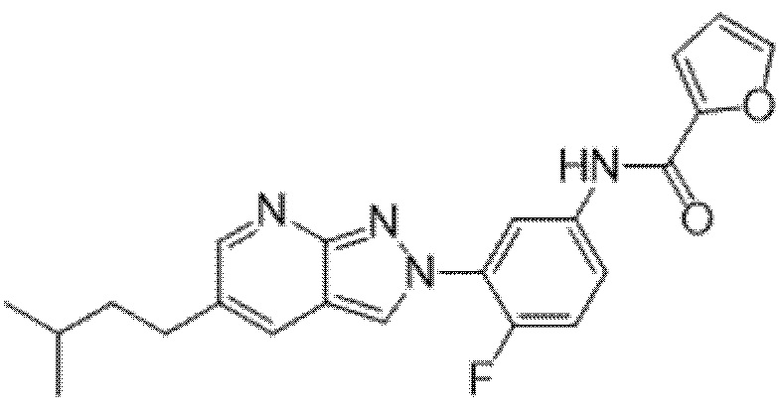
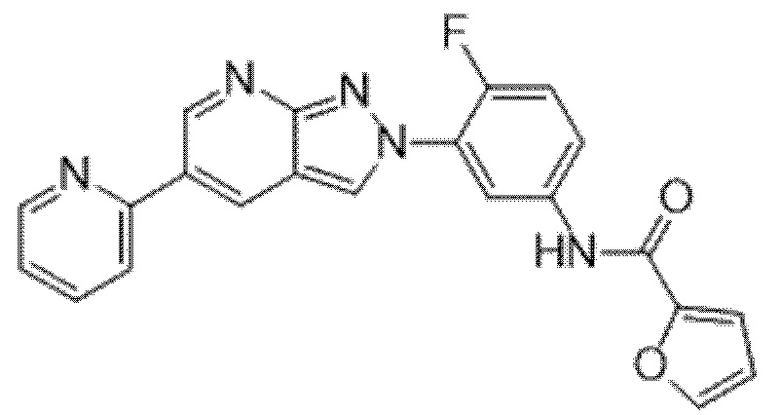
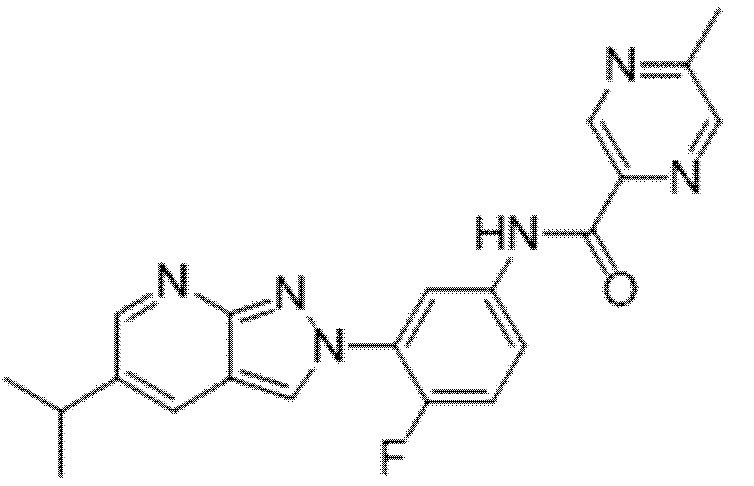
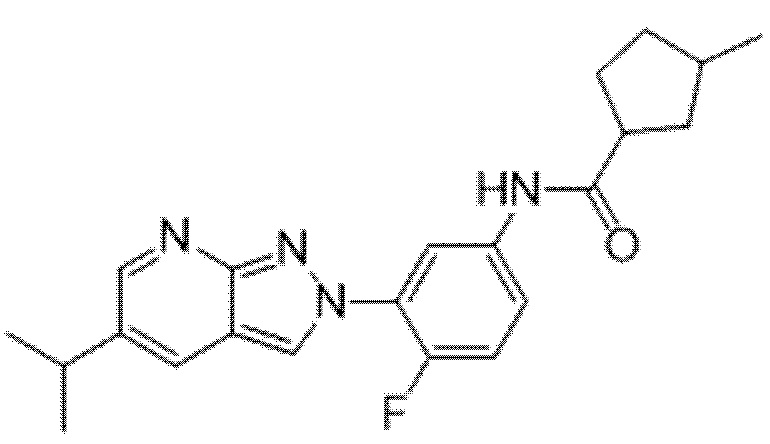
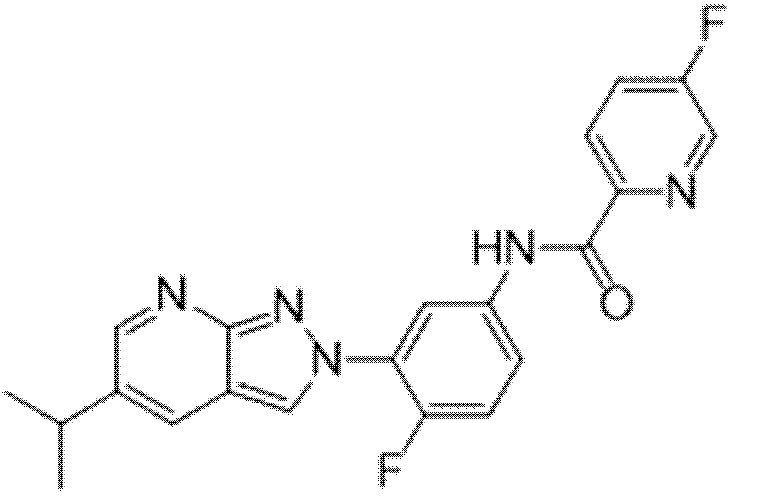
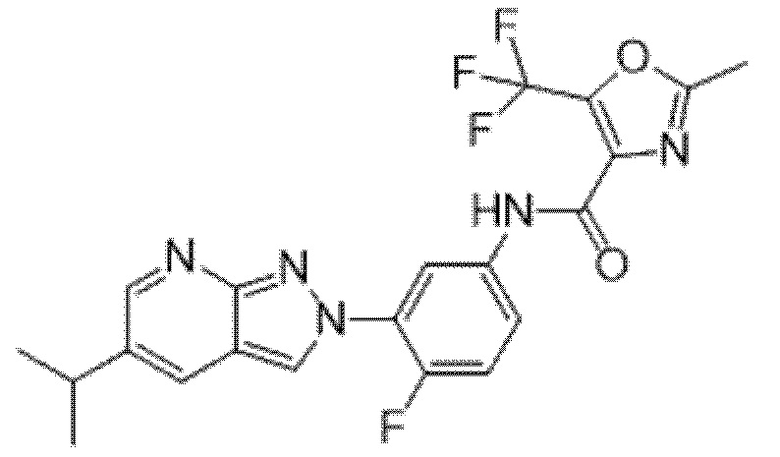
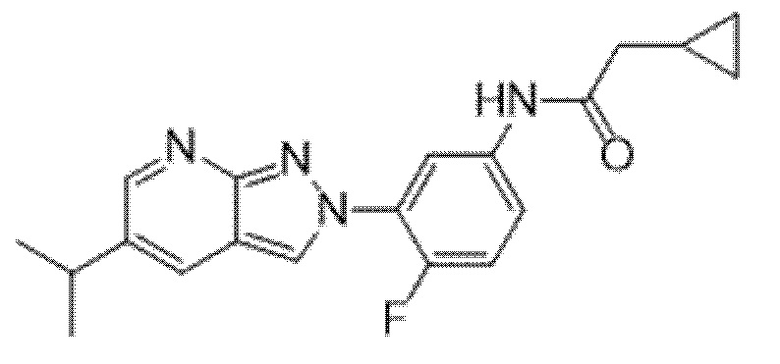
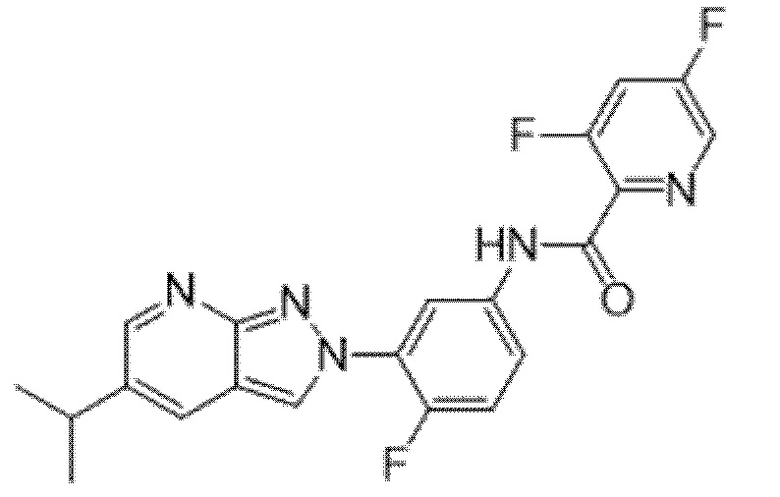
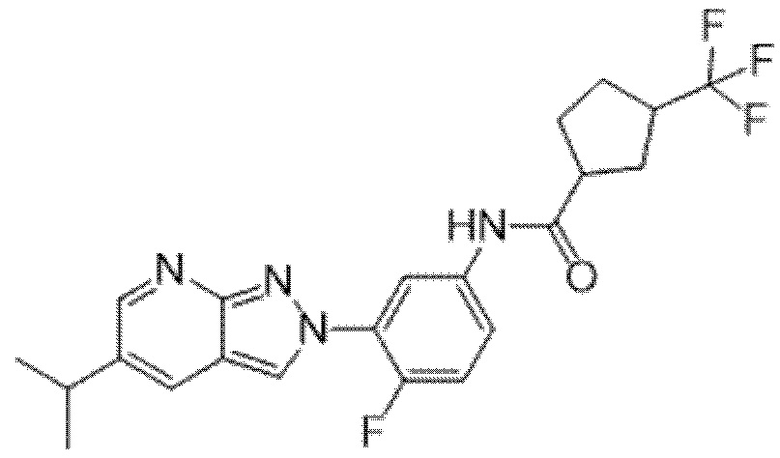
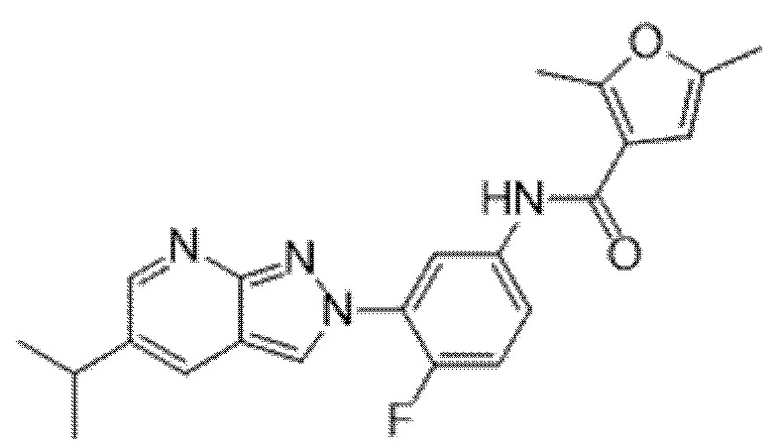
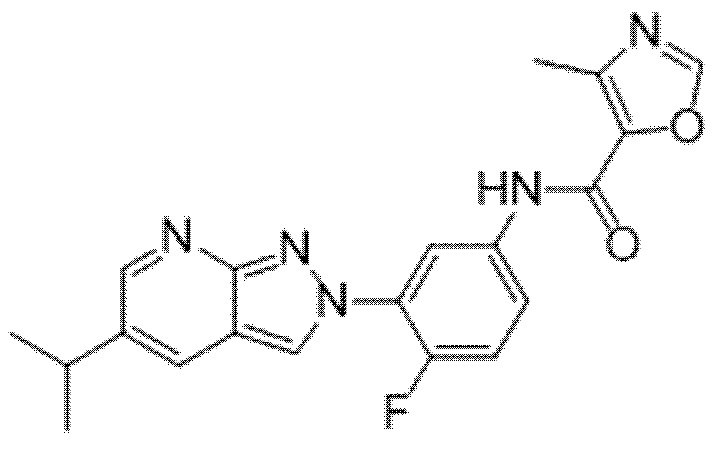
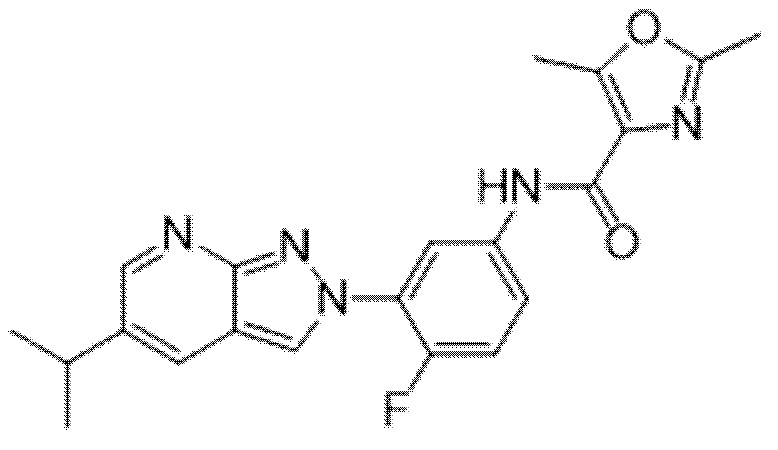
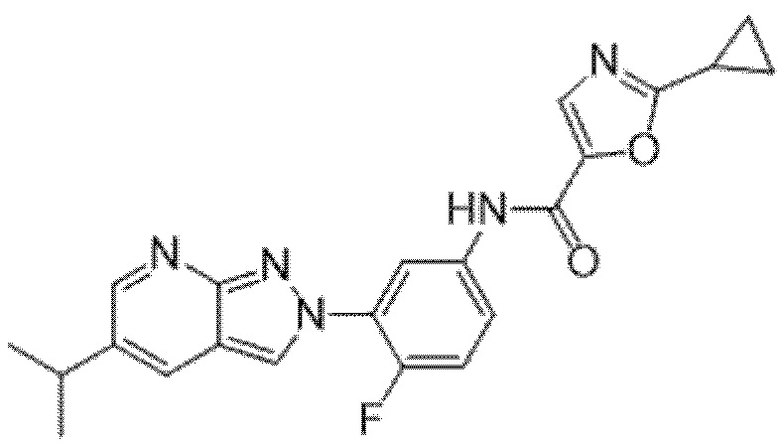
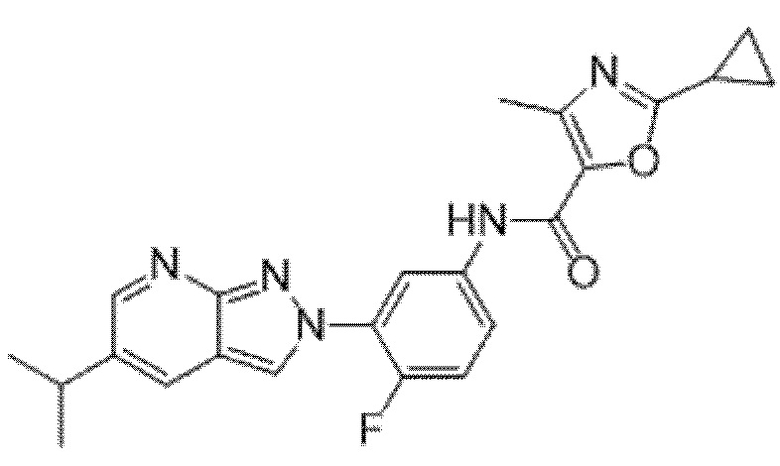
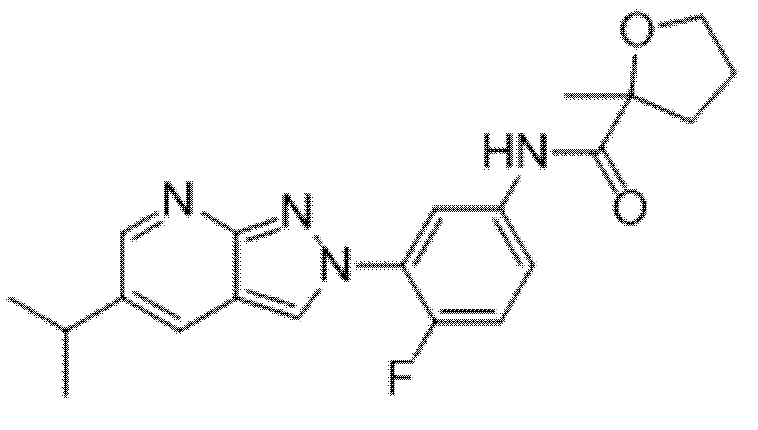
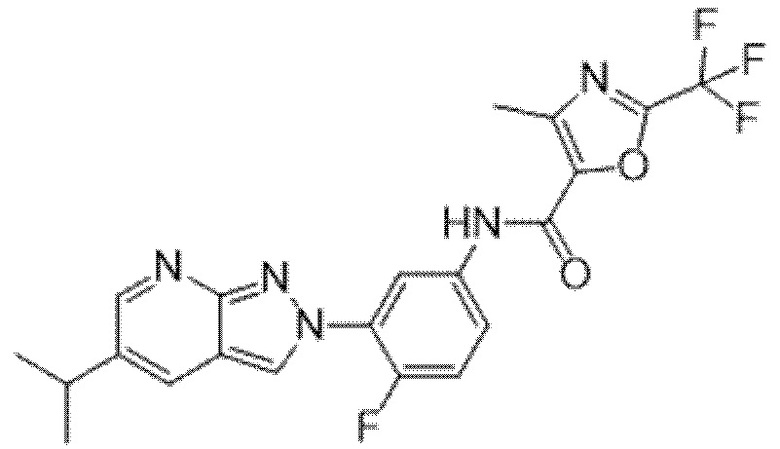
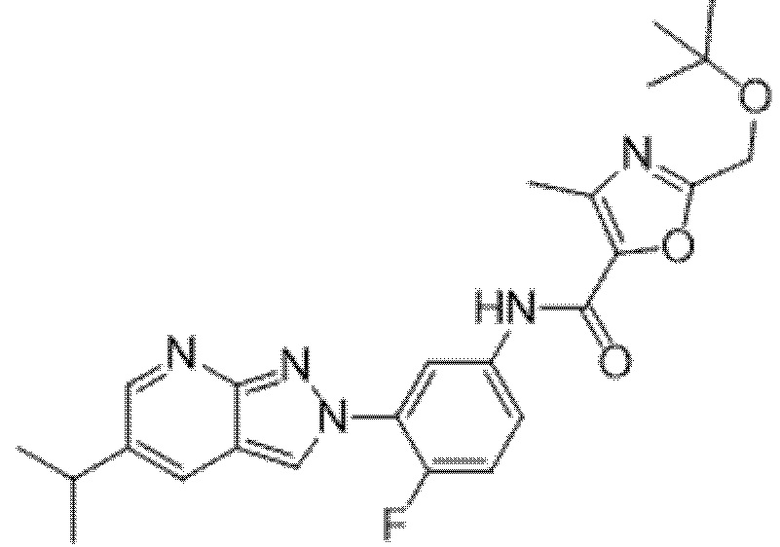
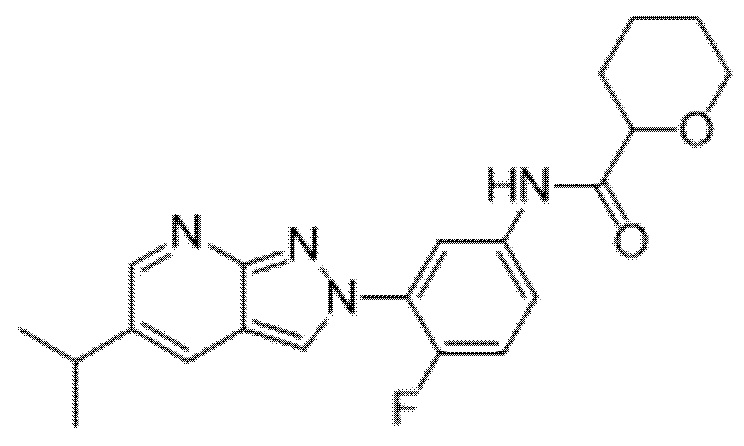
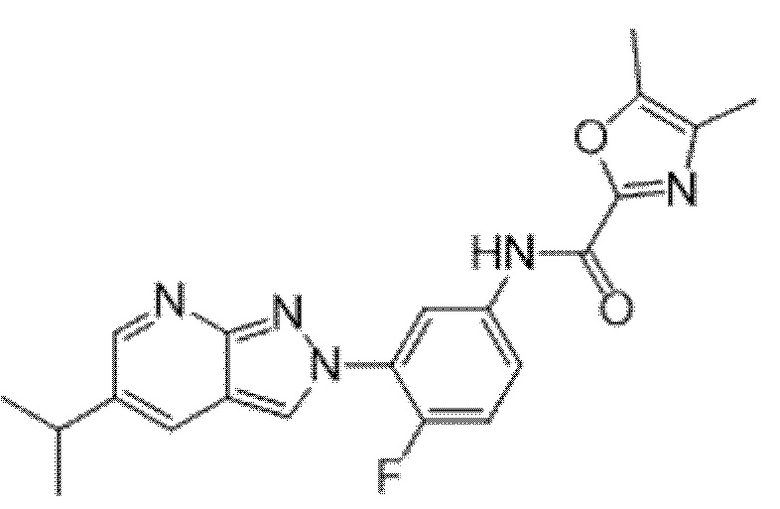
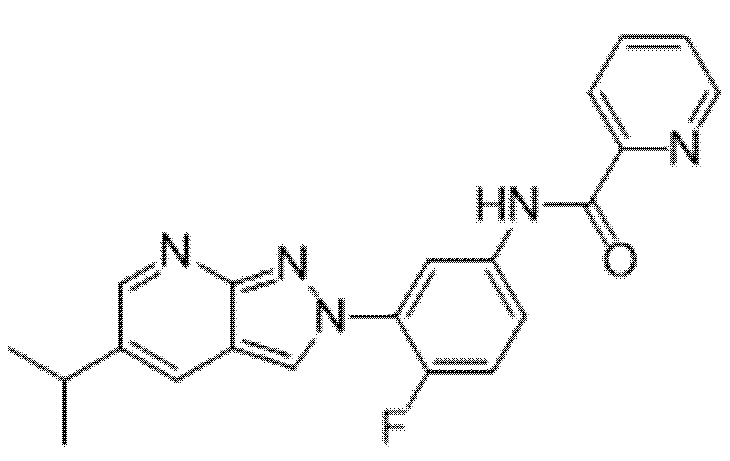
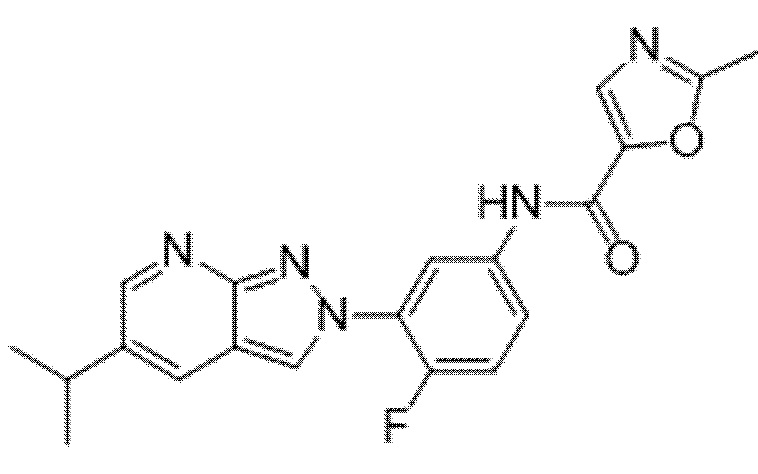
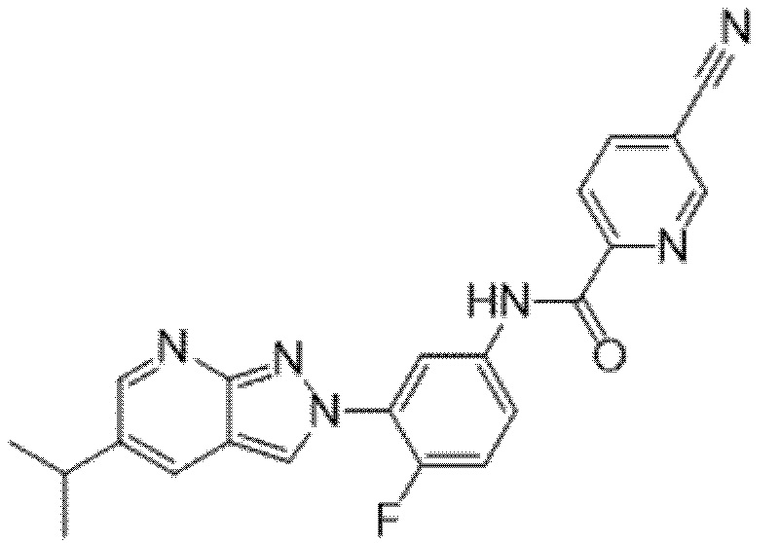
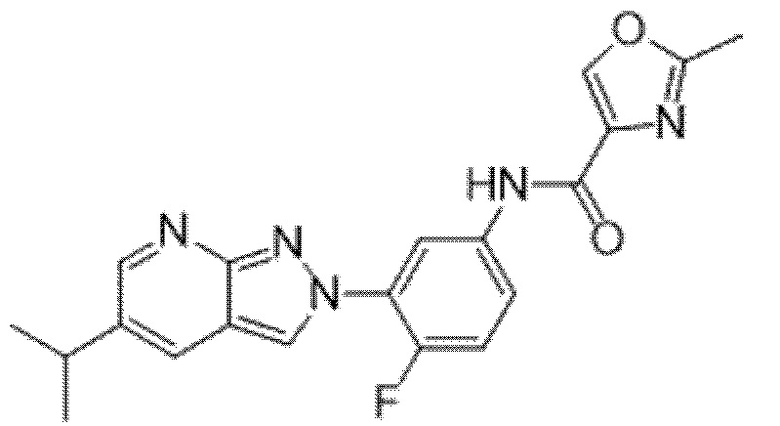
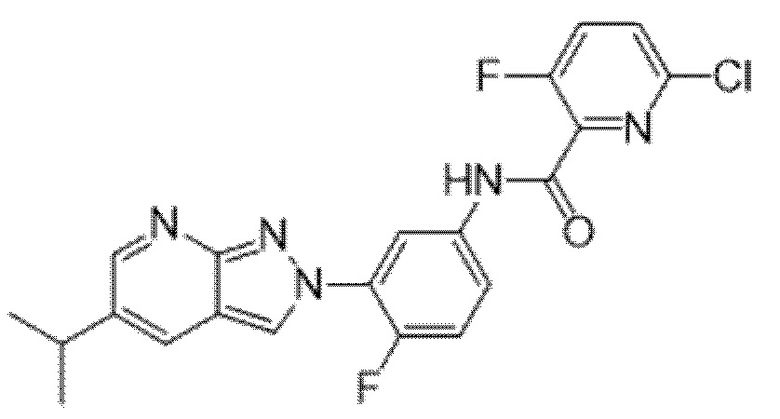
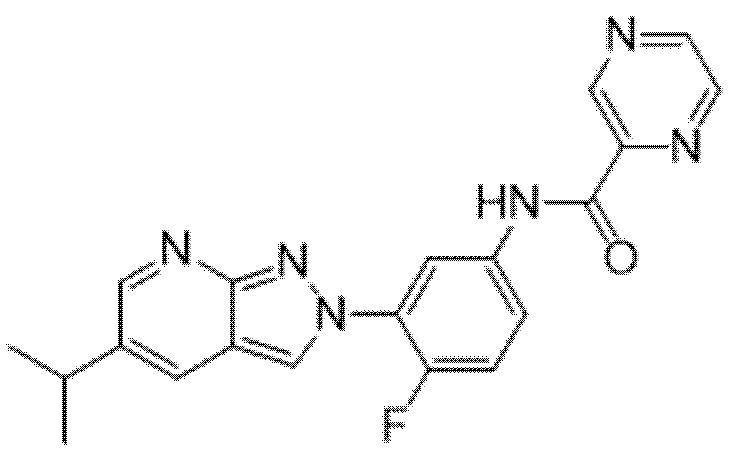
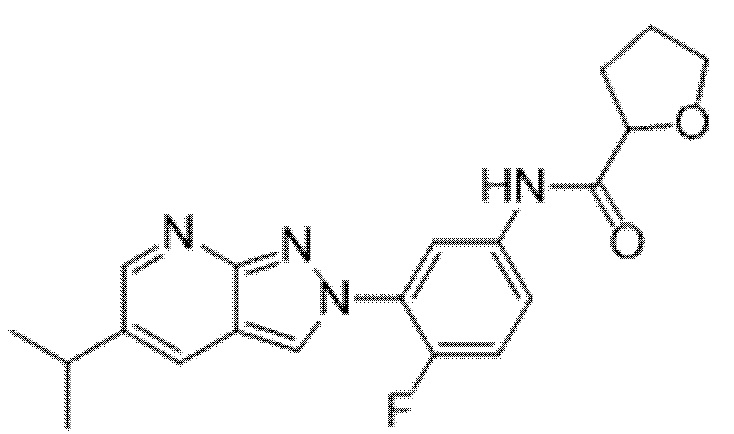
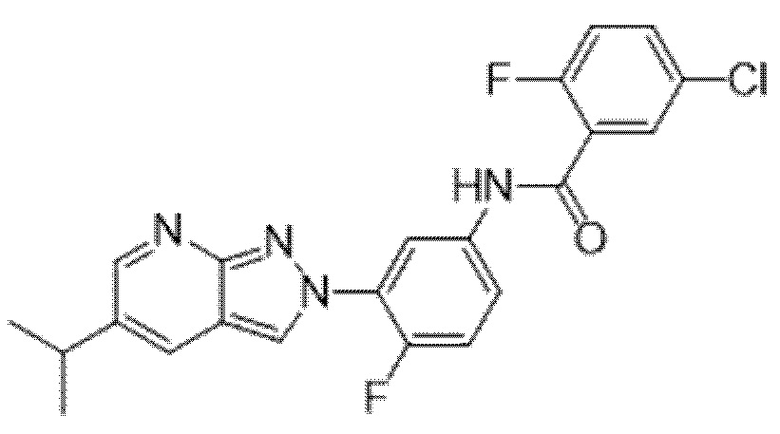
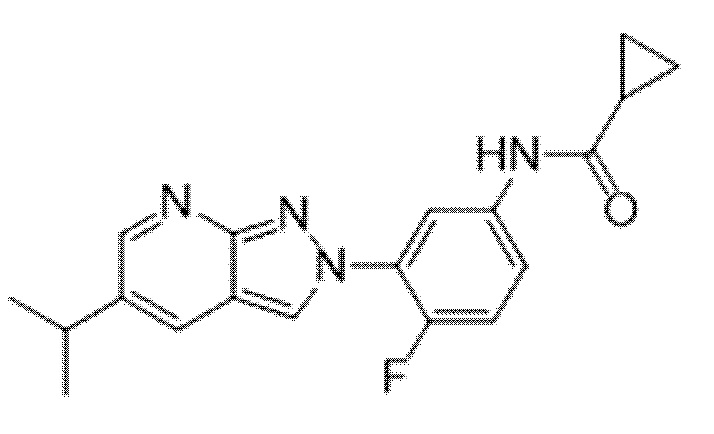
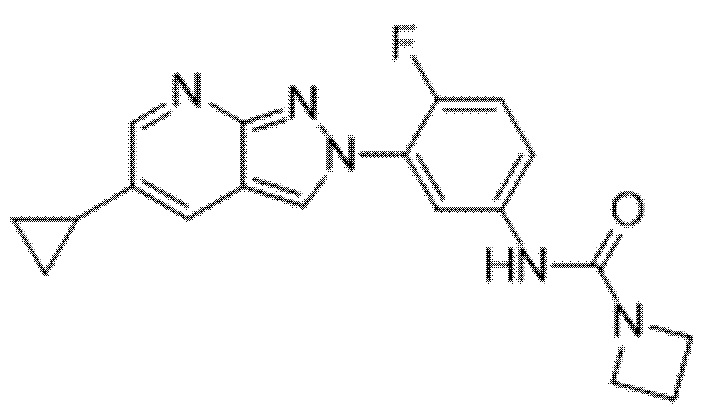
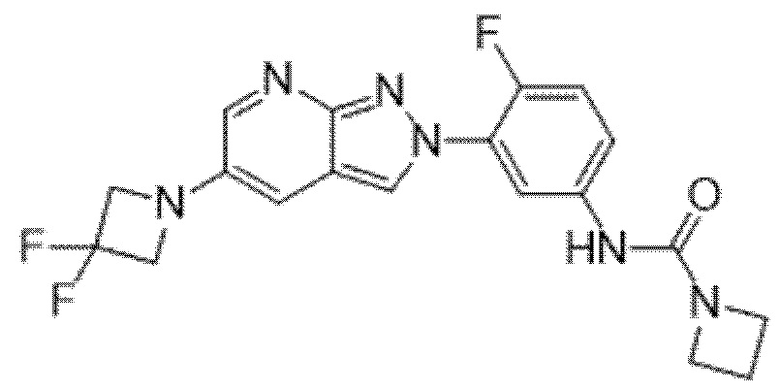
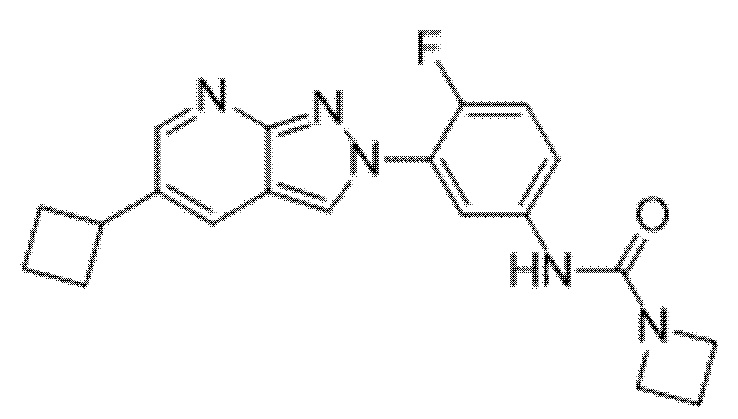
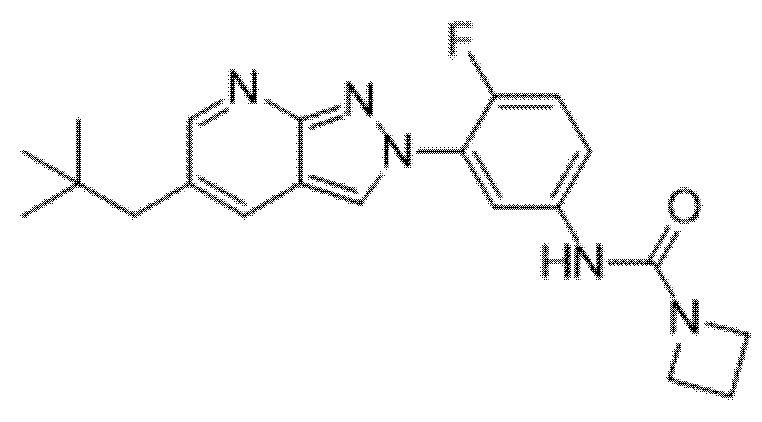
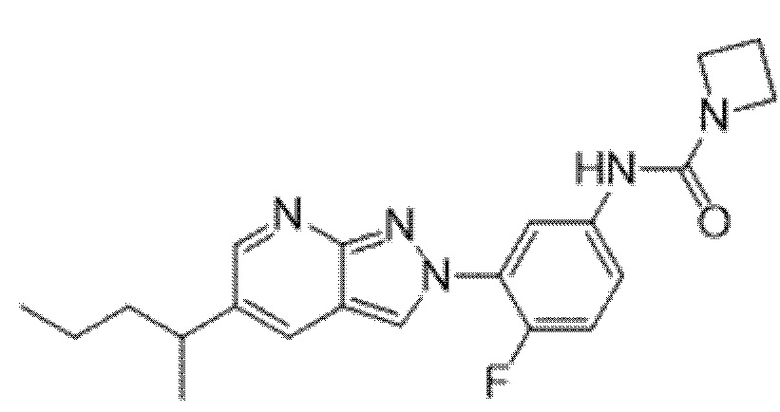
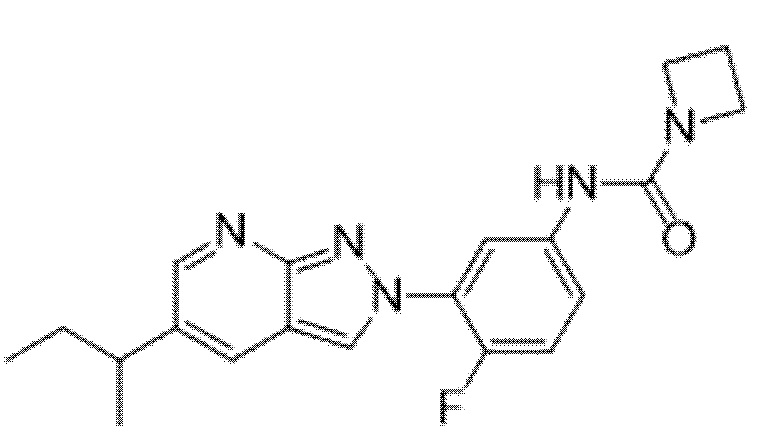
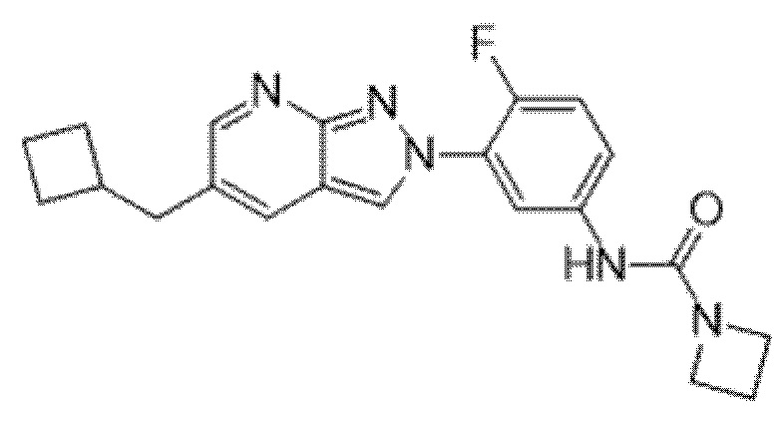
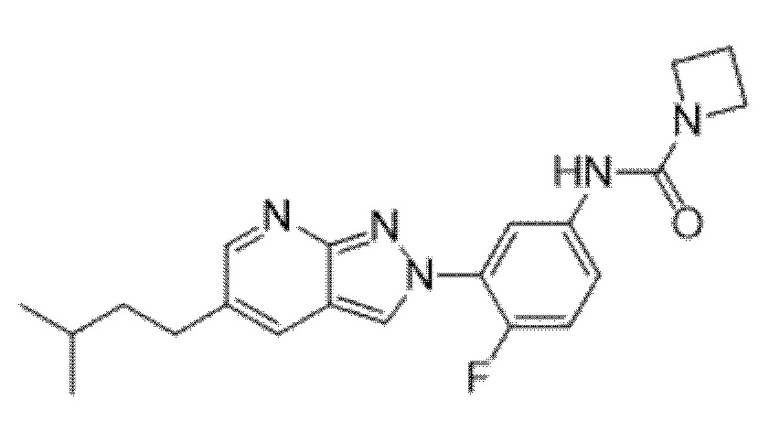
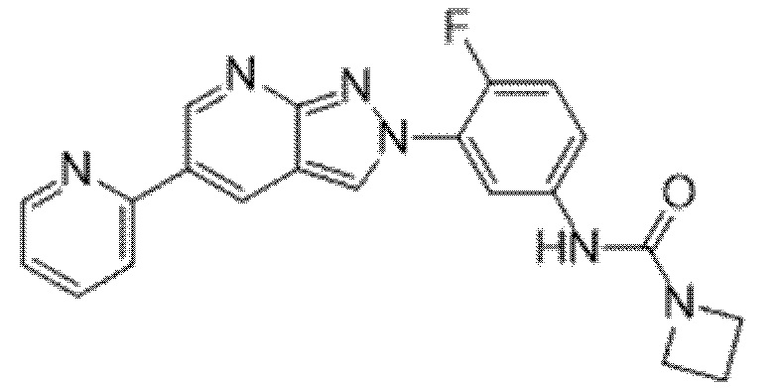
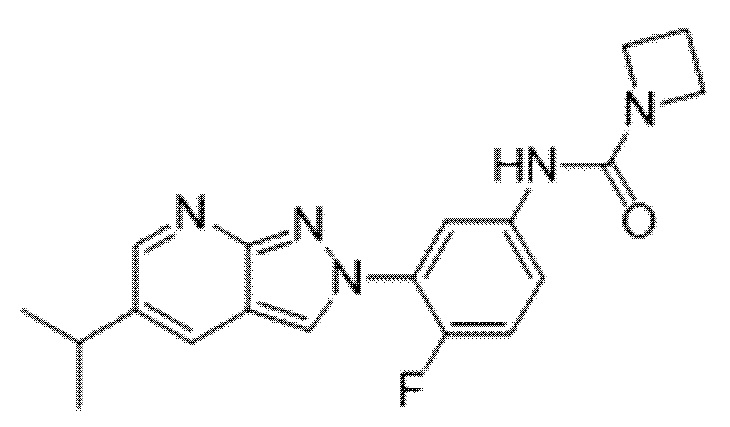
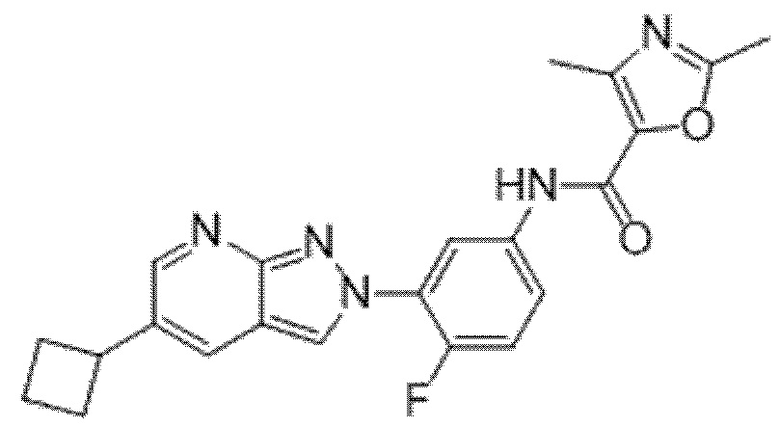
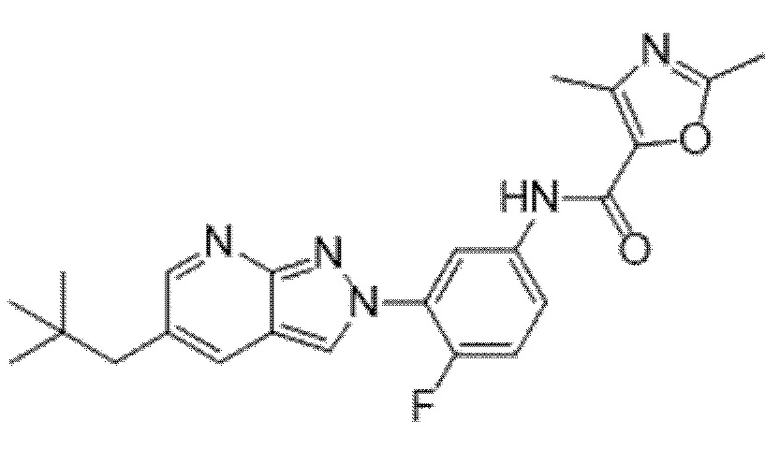
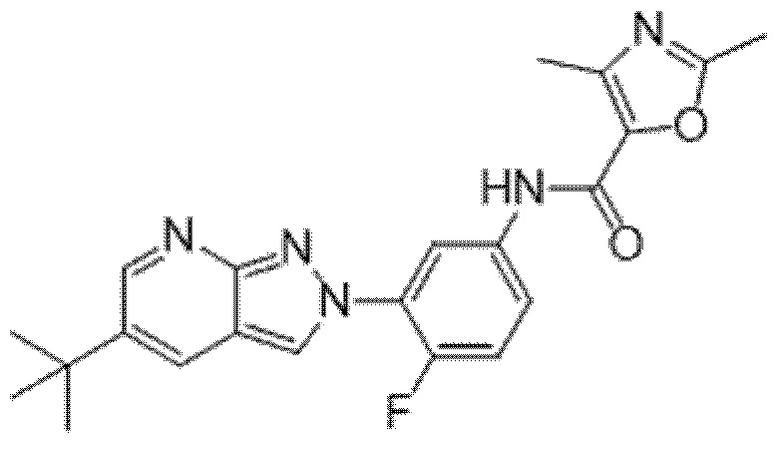
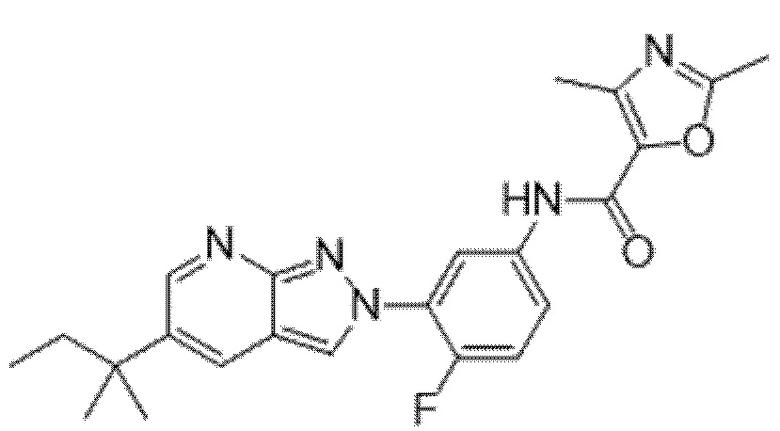
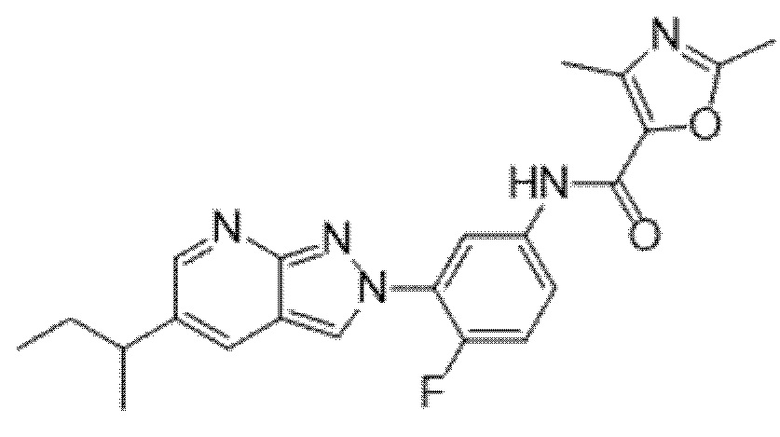
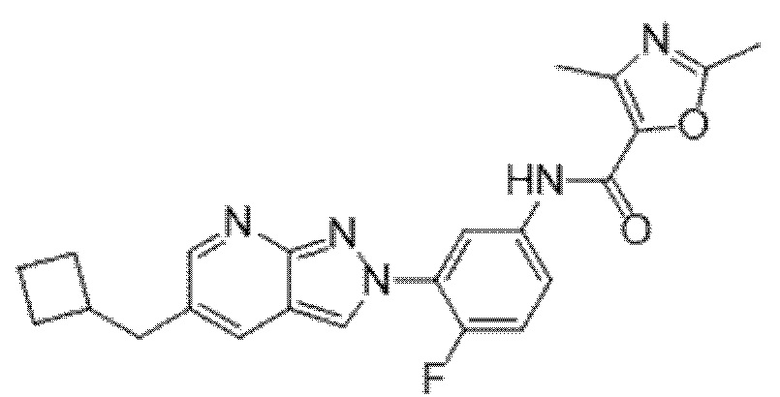
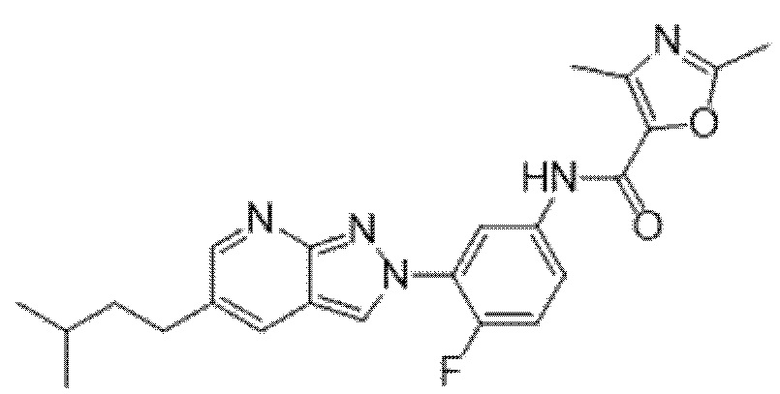
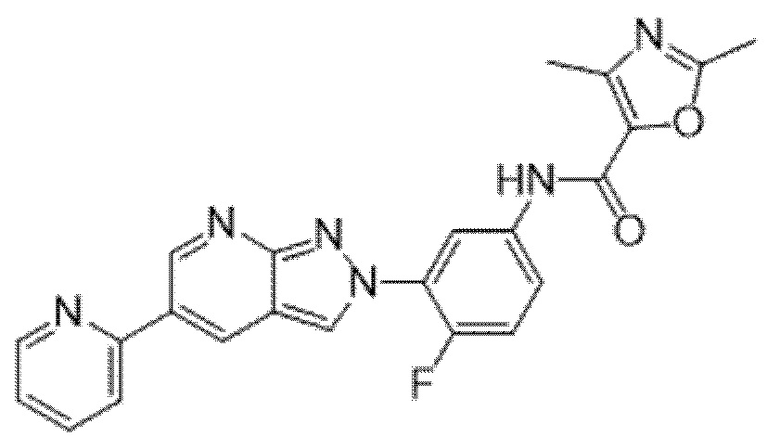
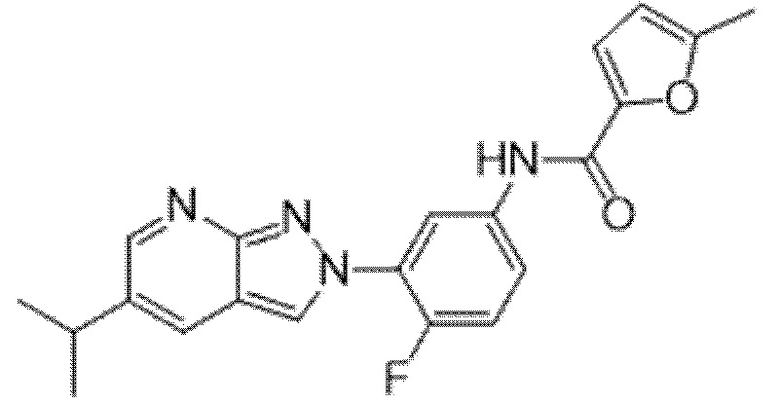
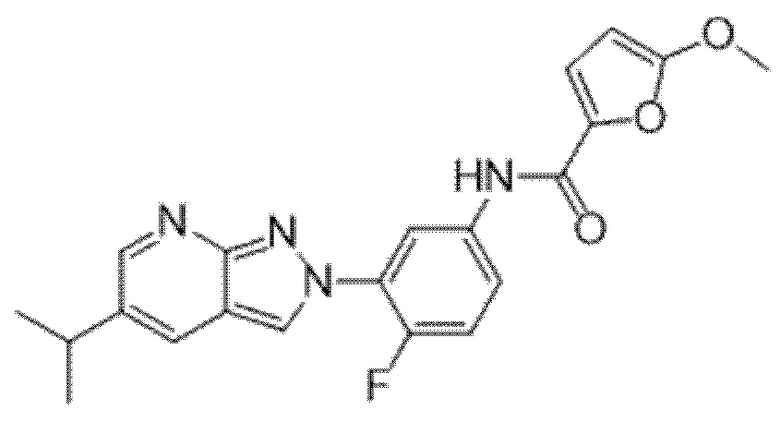
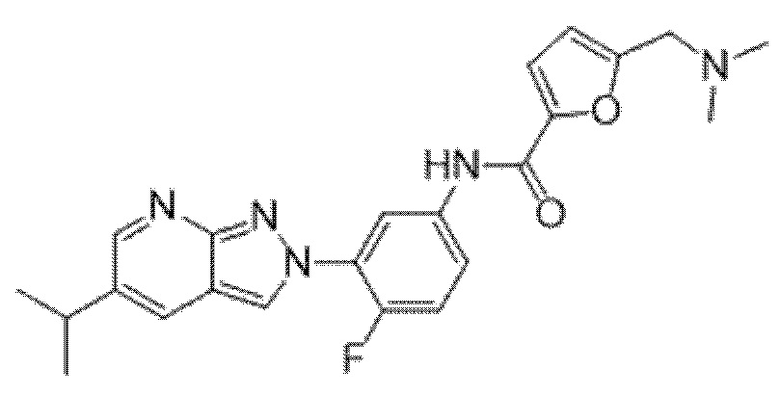
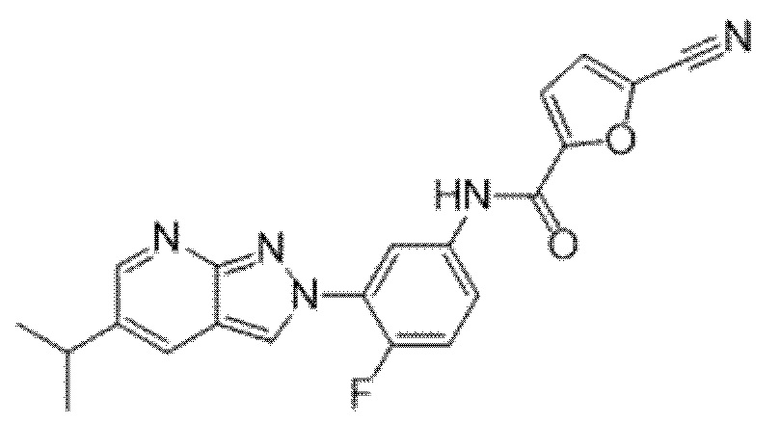
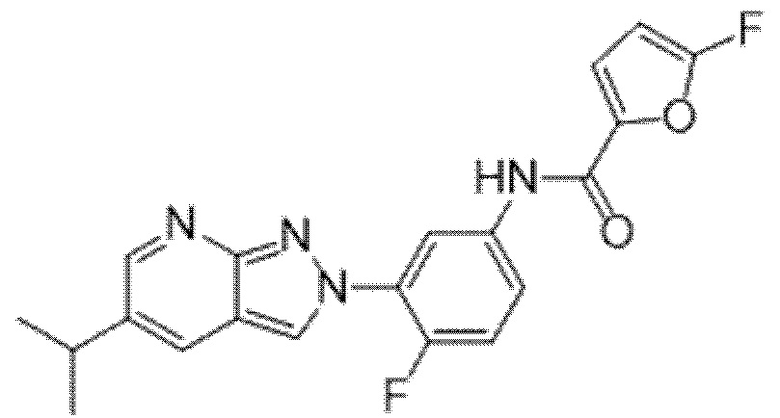
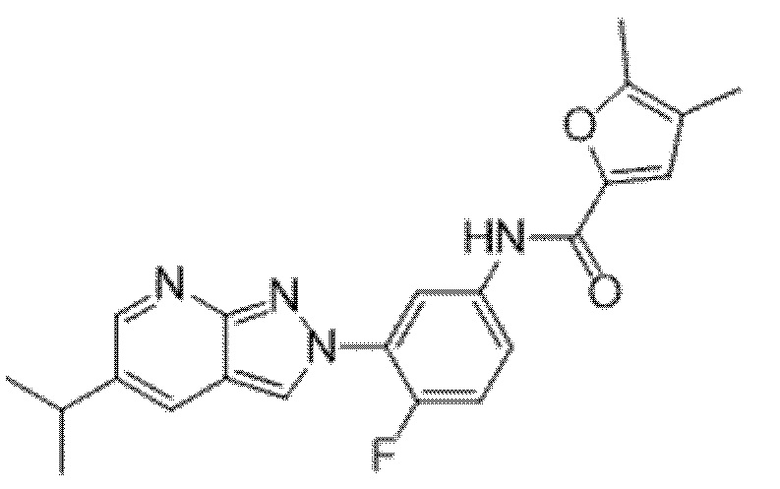
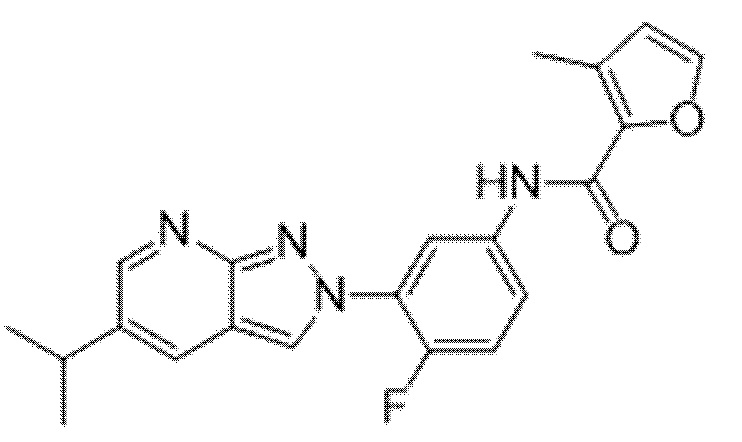
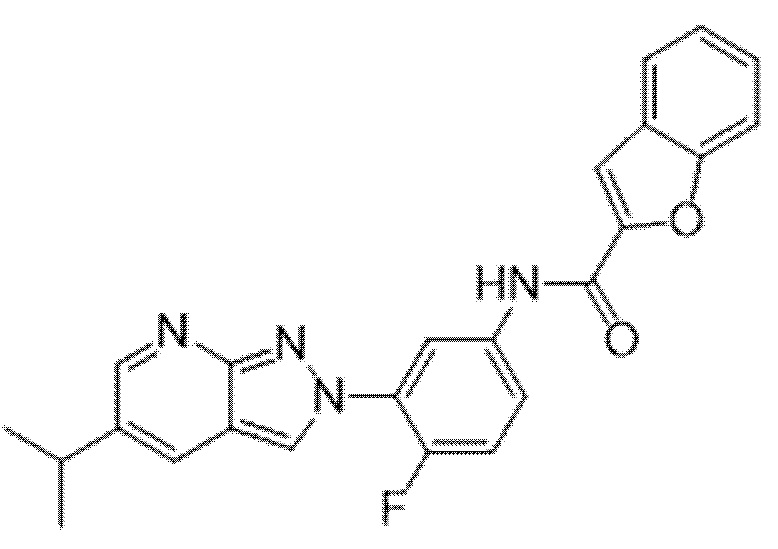
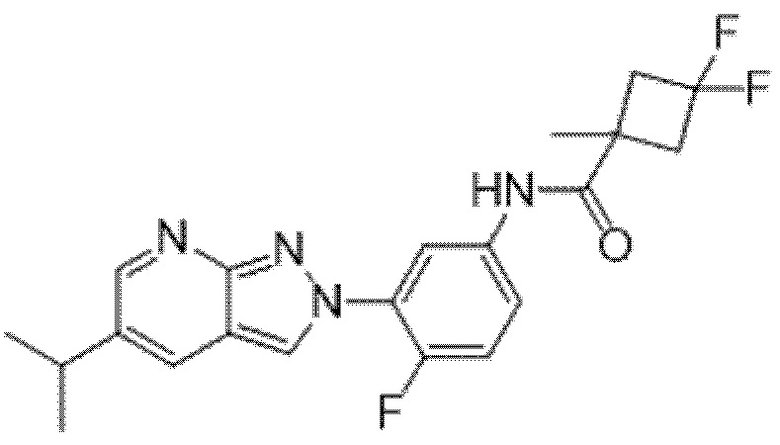
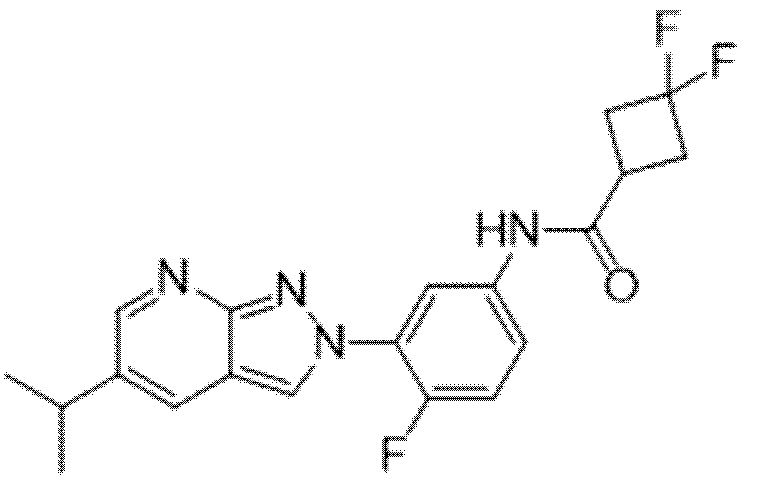
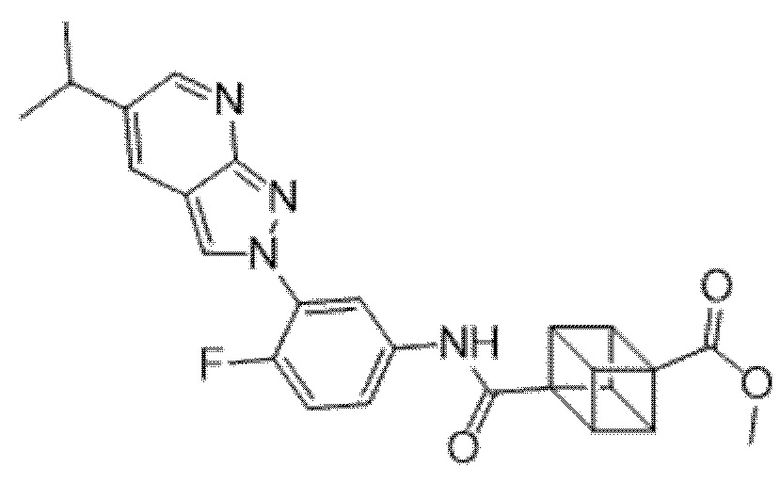
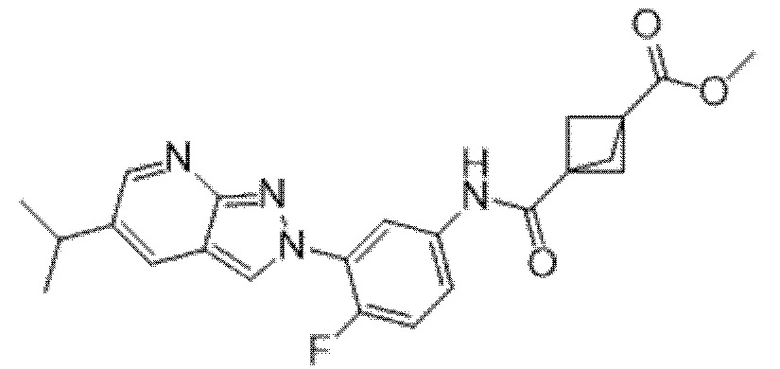
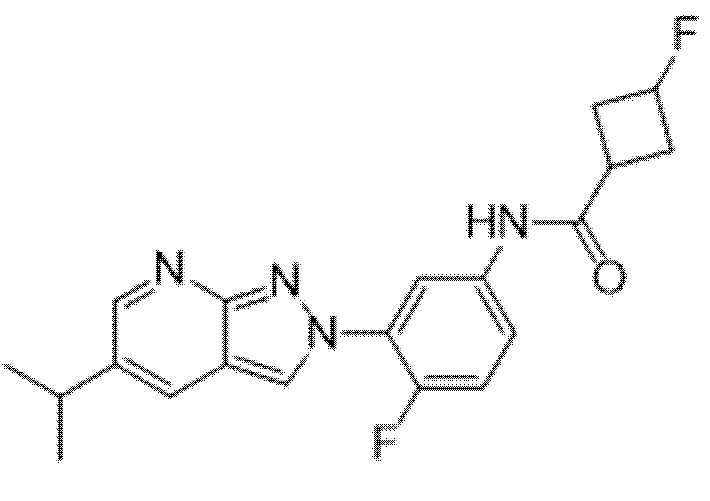
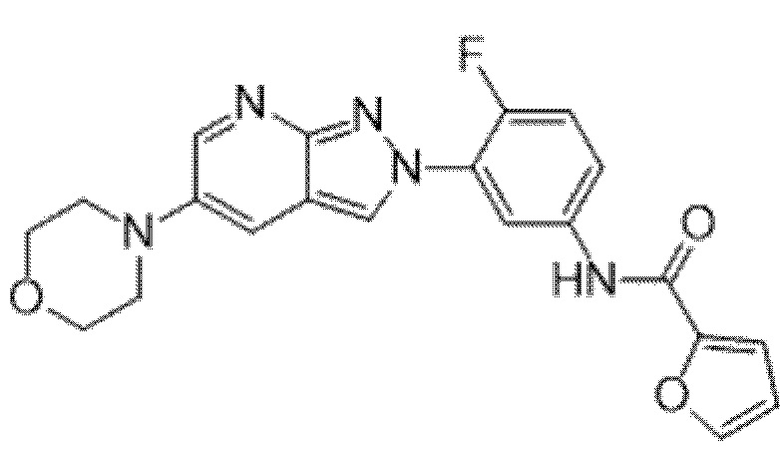
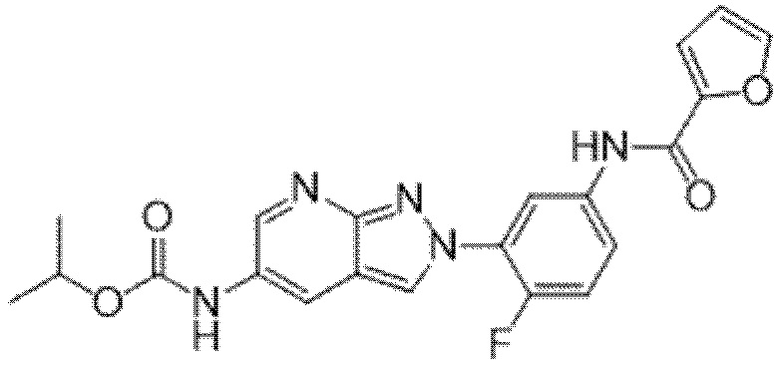
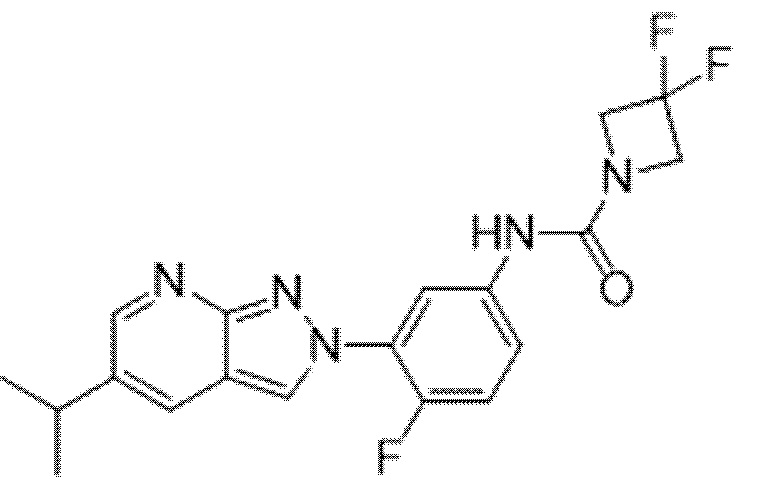
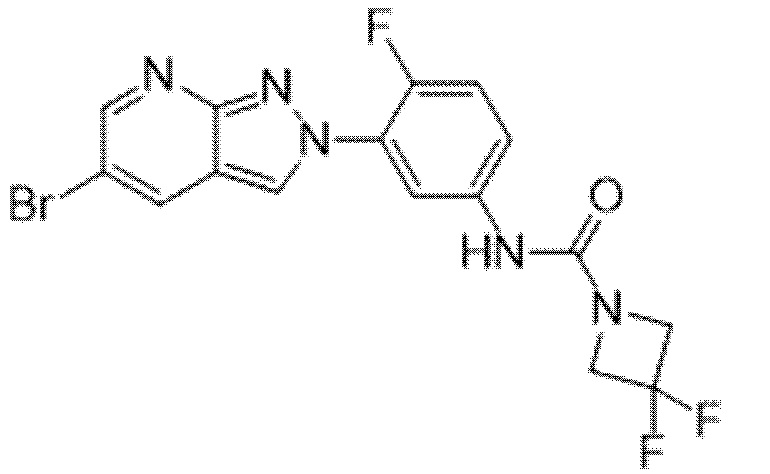
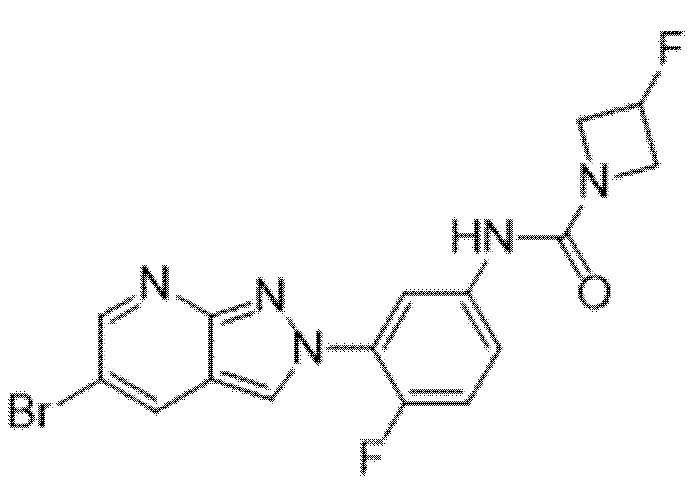
Способ 1: RT=2,82 мин.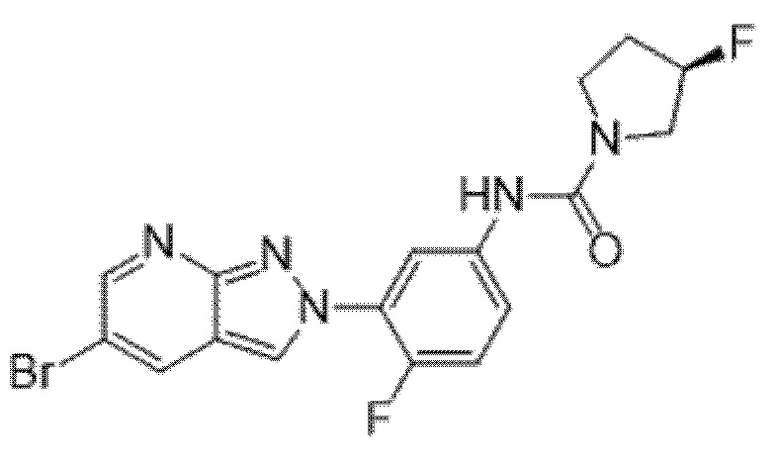
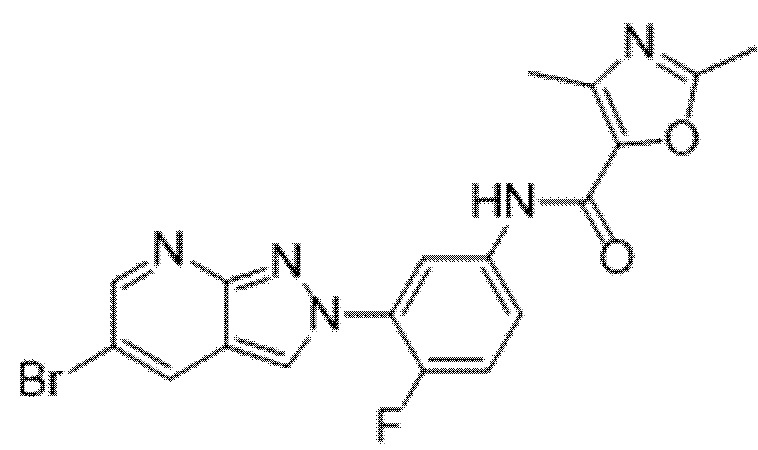
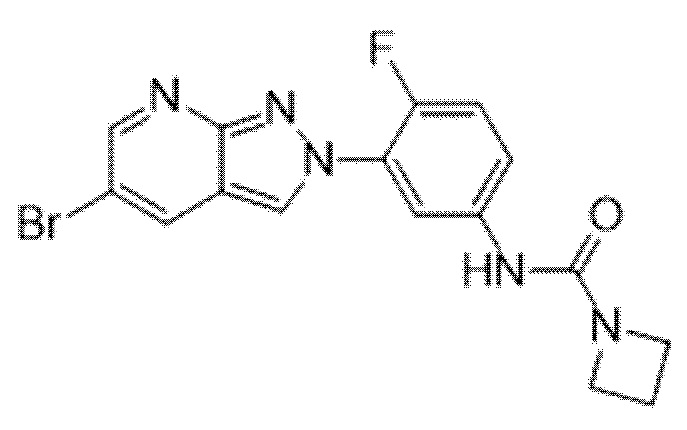
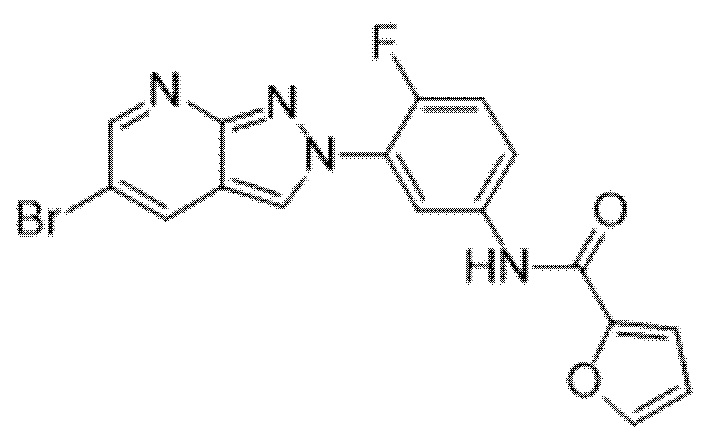
Способ 1: RT=2,98 мин.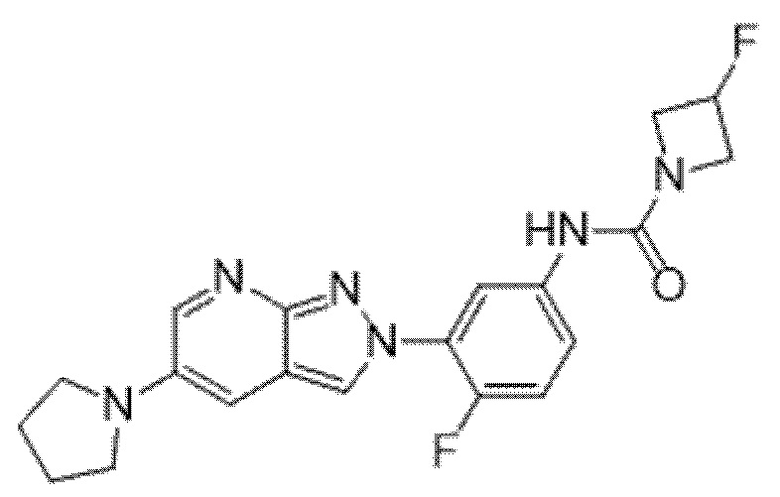
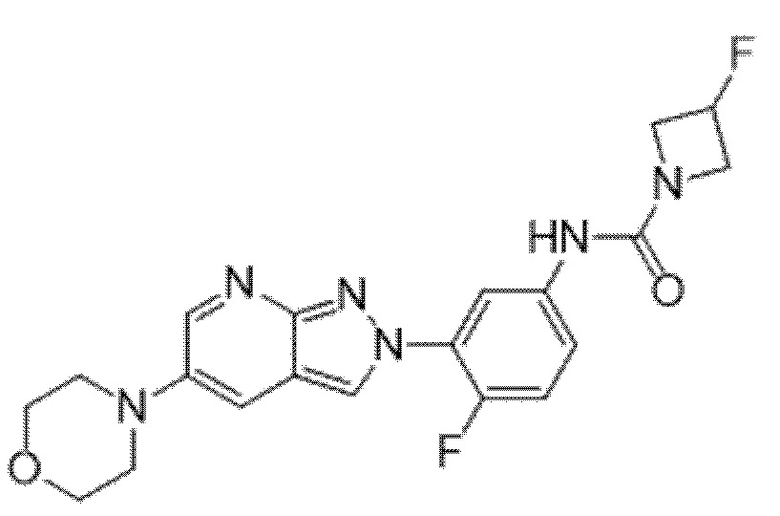
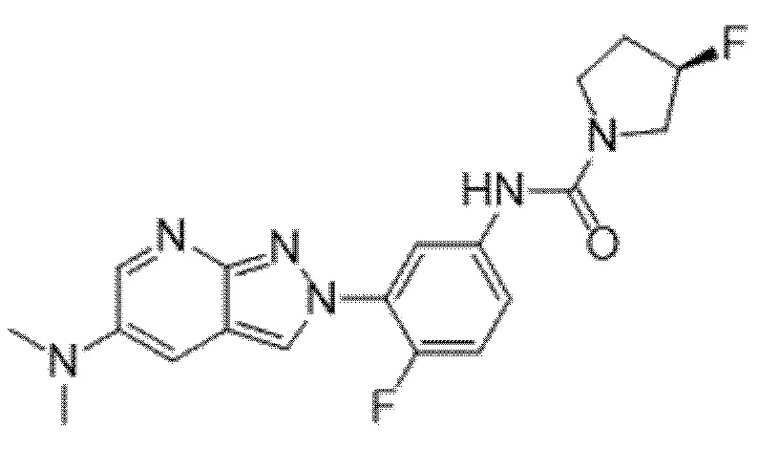
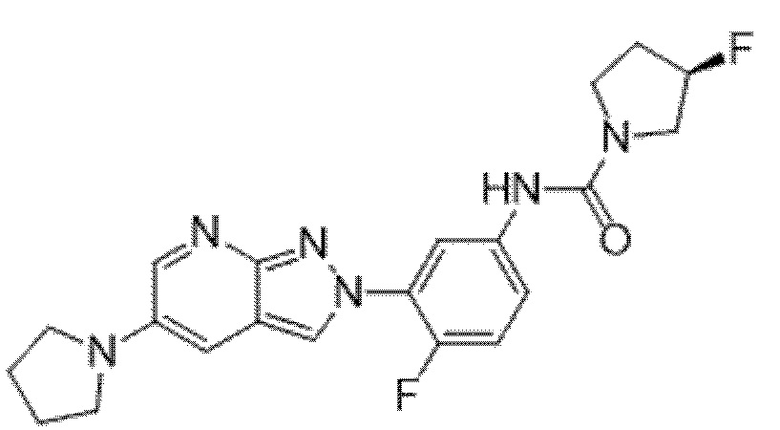
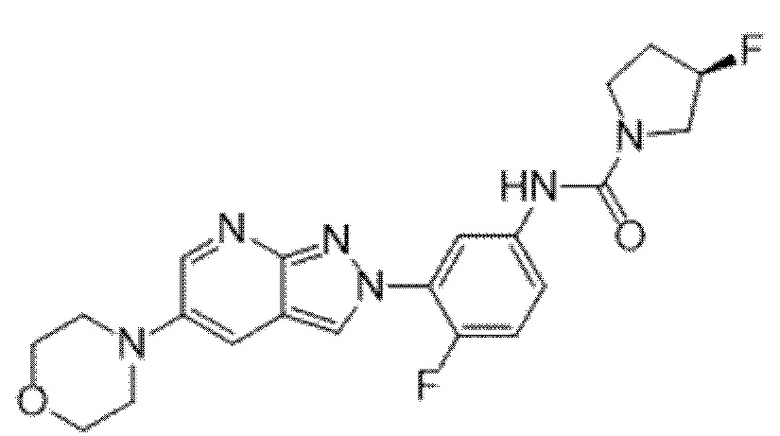
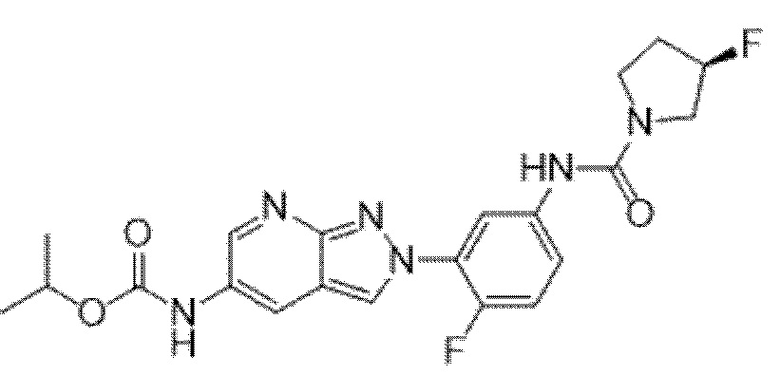
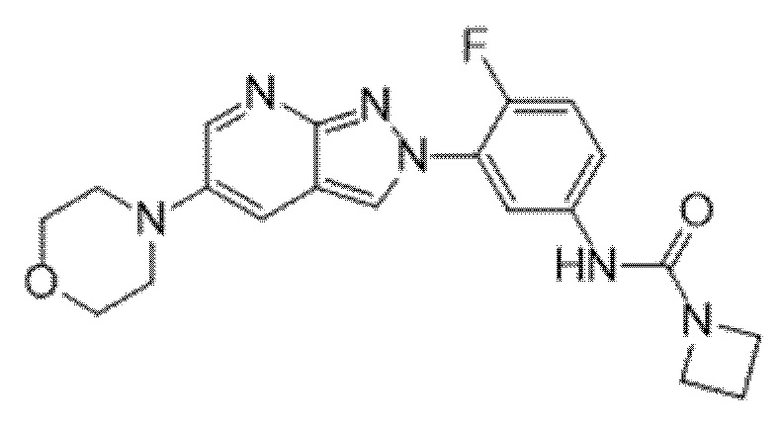
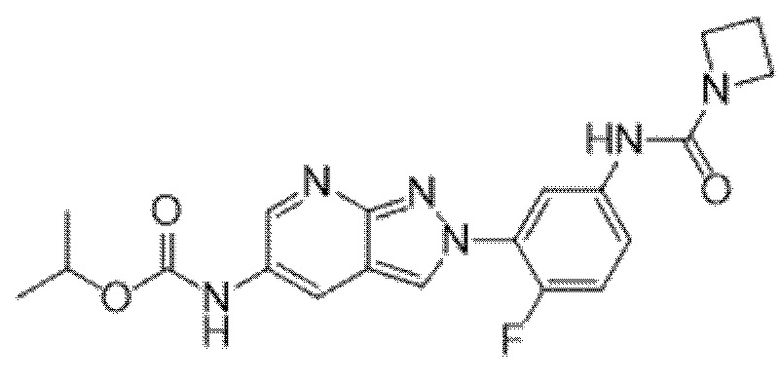
Способ 1: RT=2,87 мин.; M+H=413,1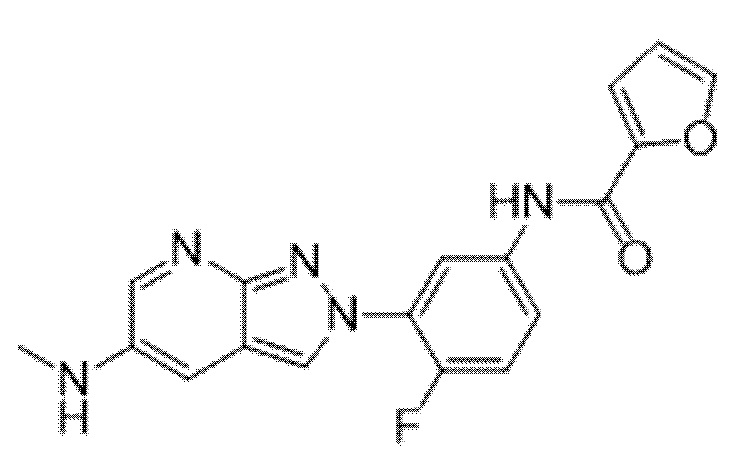
Способ 1: RT=2,67 мин.; M+H=352,0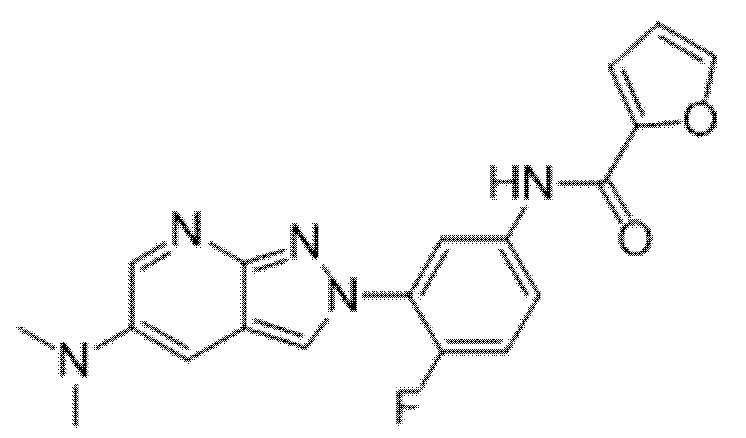
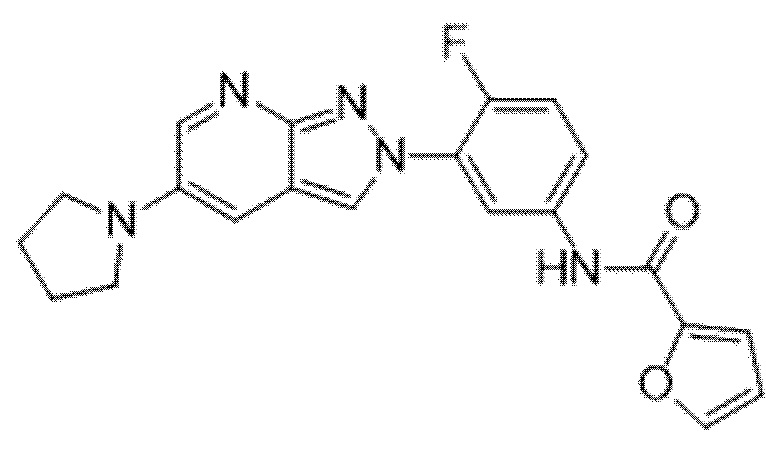
Способ 1: RT=3,18 мин.; M+H=392,2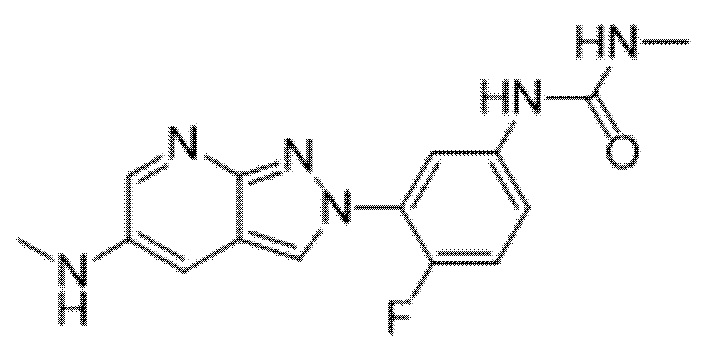
Способ 1: RT=2,56 мин.; M+H=315,1
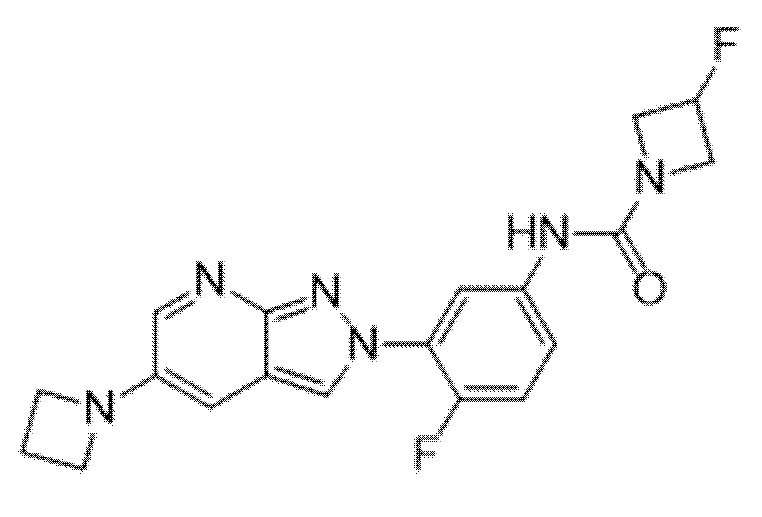
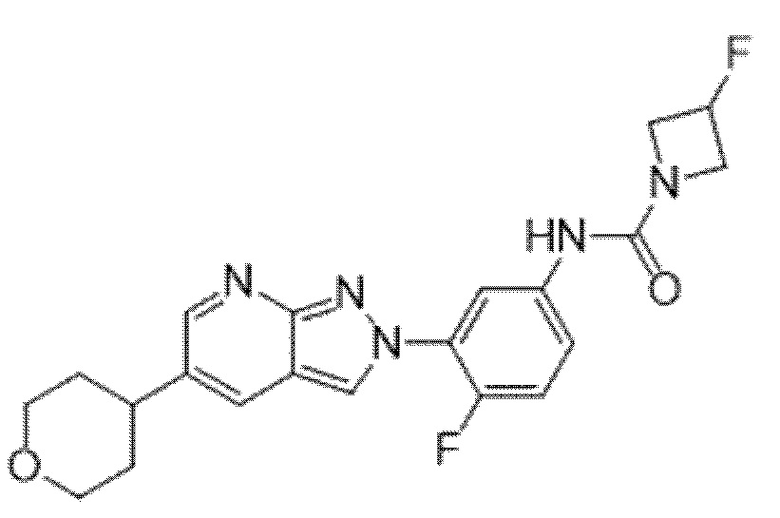
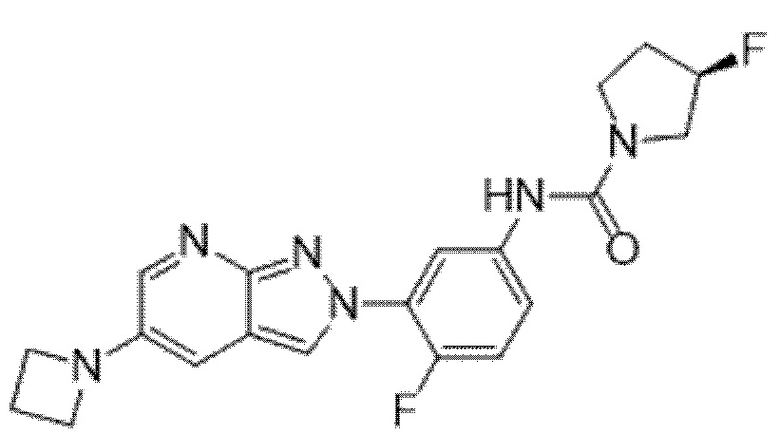
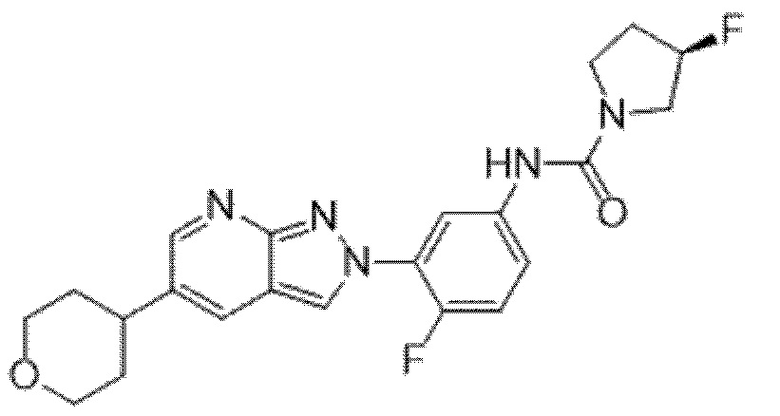
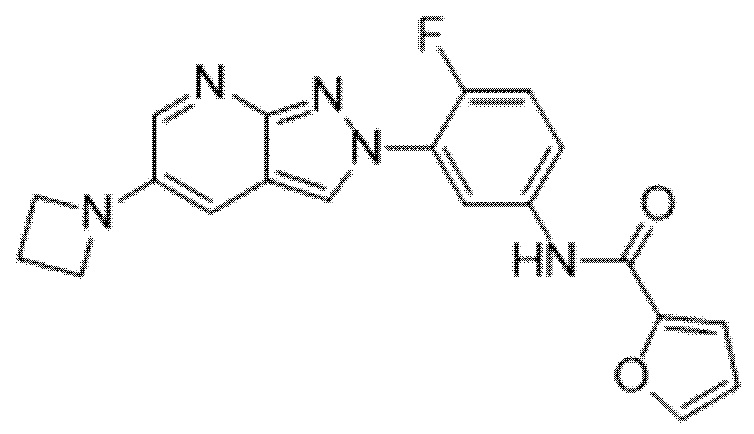
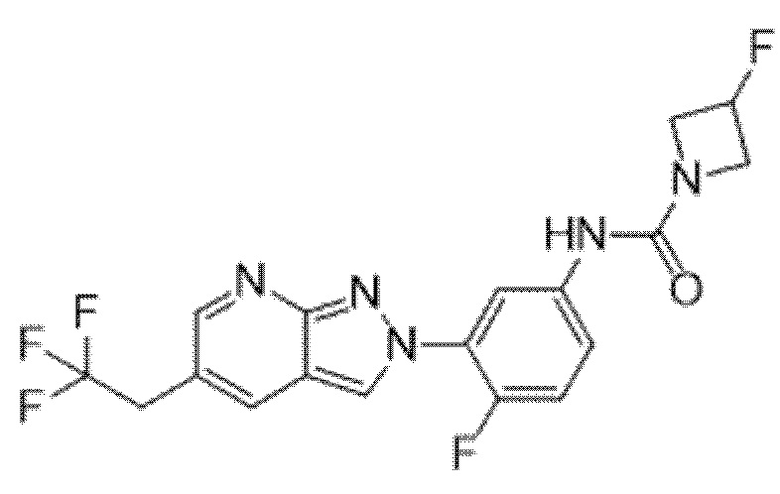
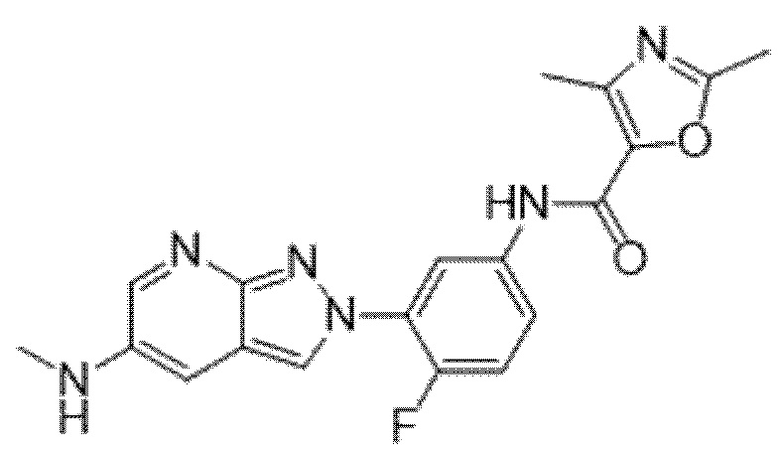
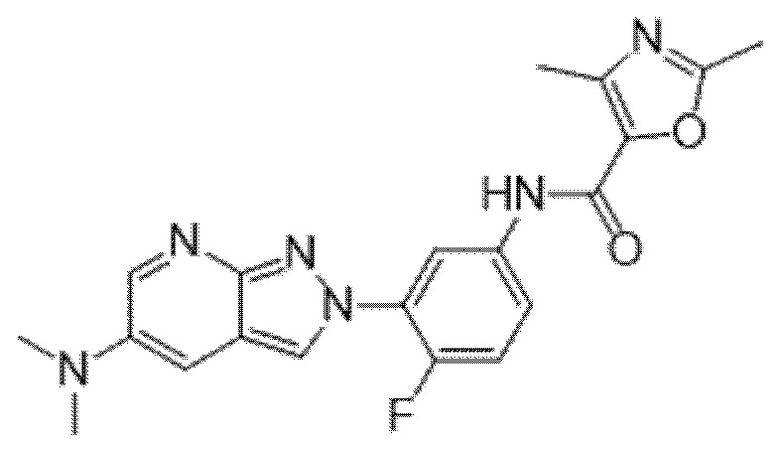
Способ 1: RT=2,84 мин.; M+H=395,1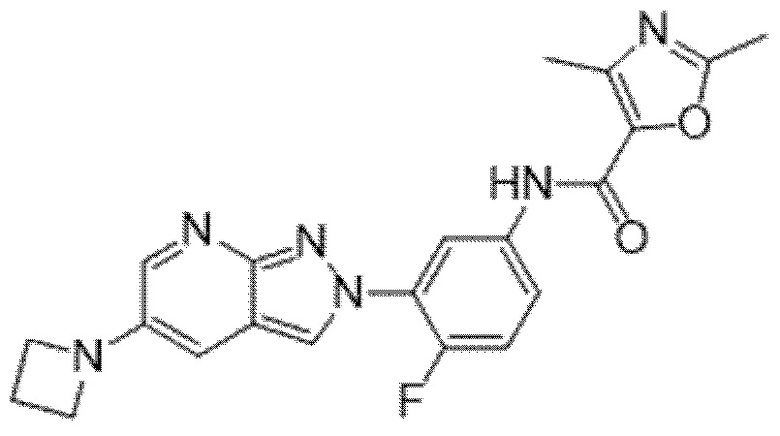
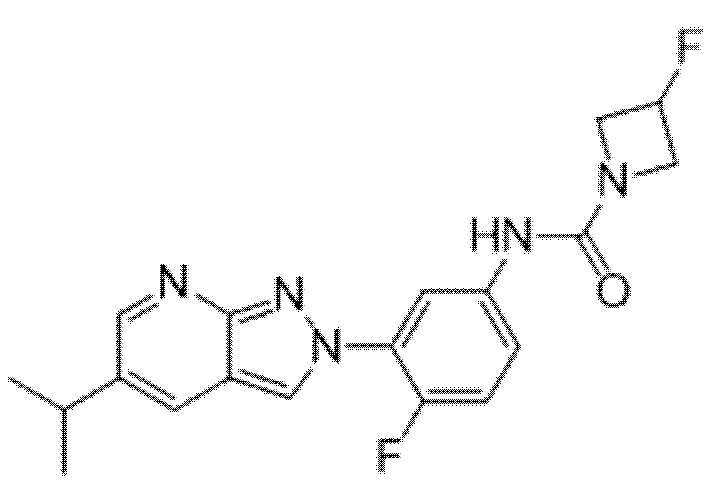
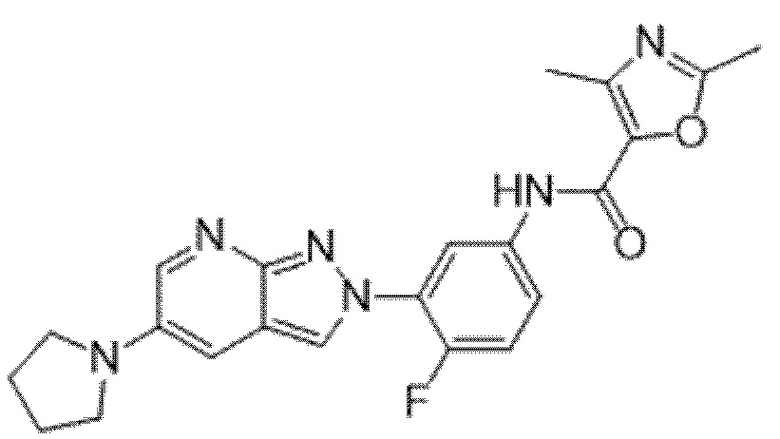
Способ 1: RT=3,12 мин.; M+H=421,1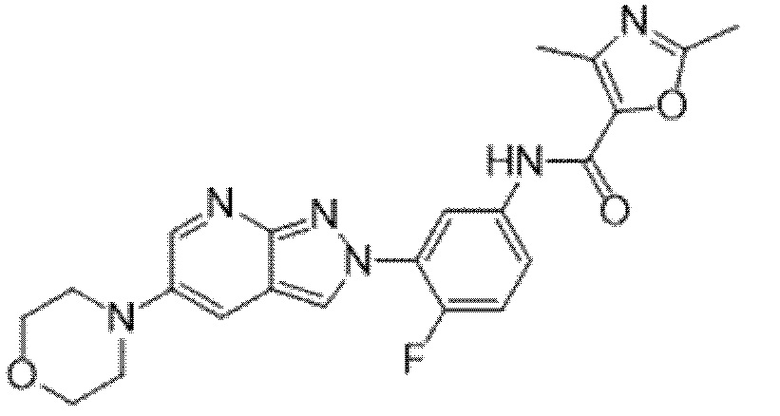
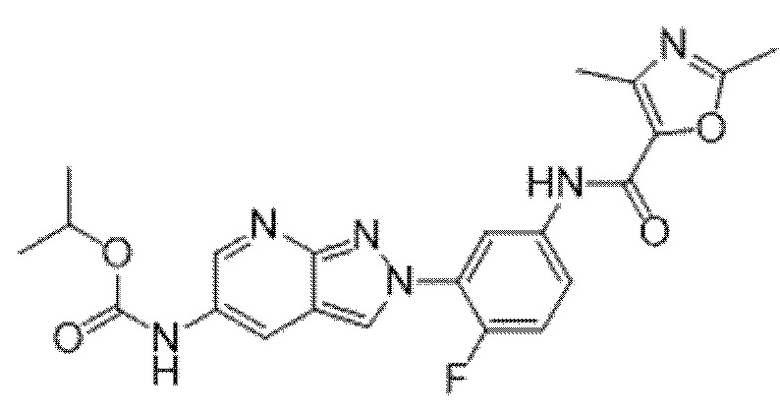
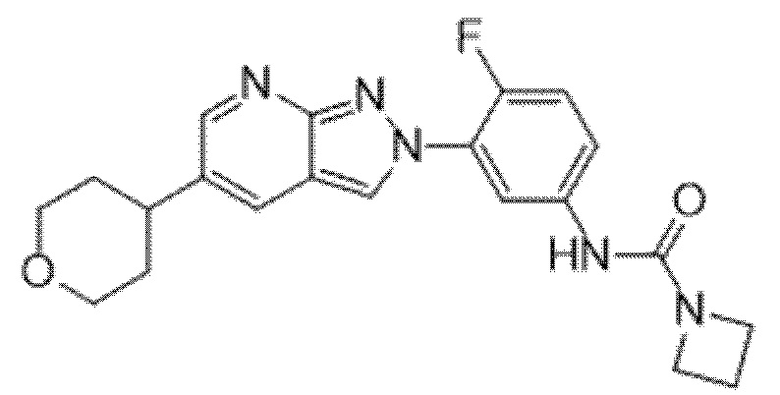
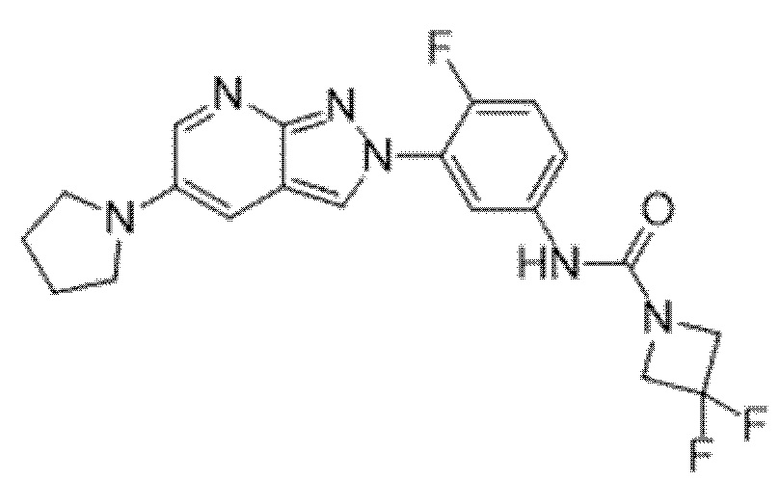
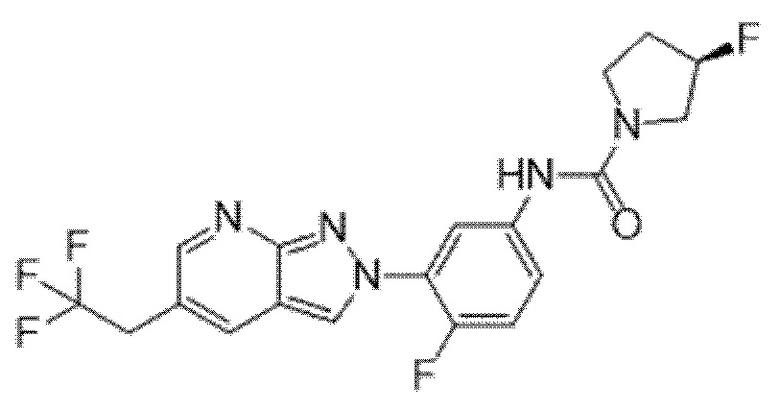
Способ 1: RT=2,82 мин.; M+H=426,2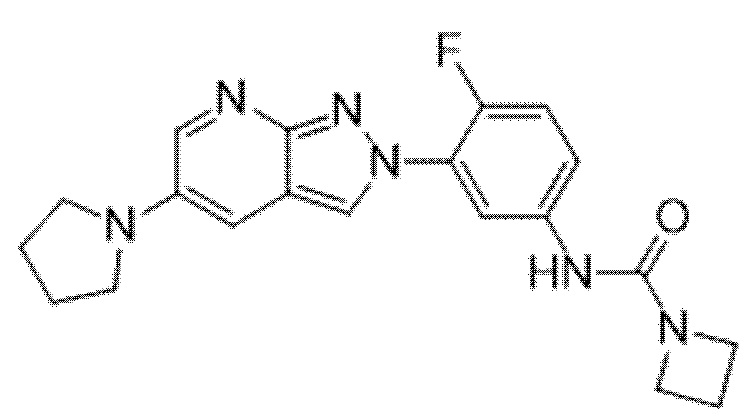
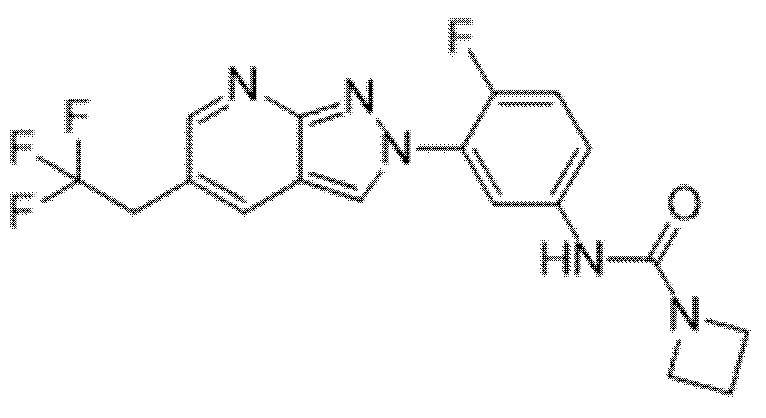
Способ 1: RT=2,75 мин.; M+H=394,2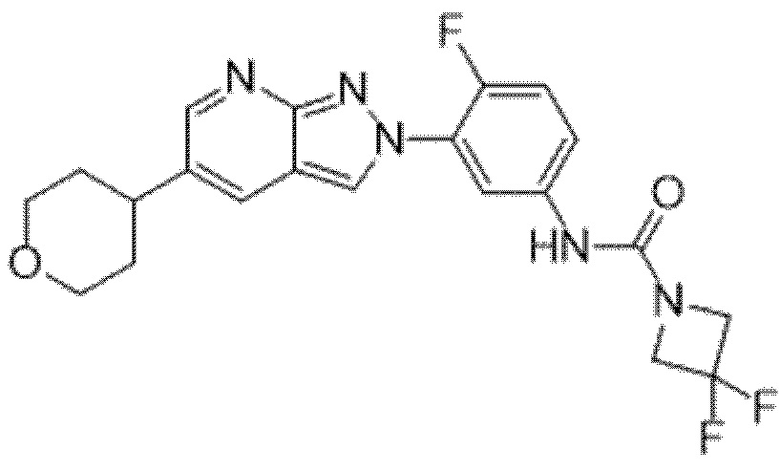
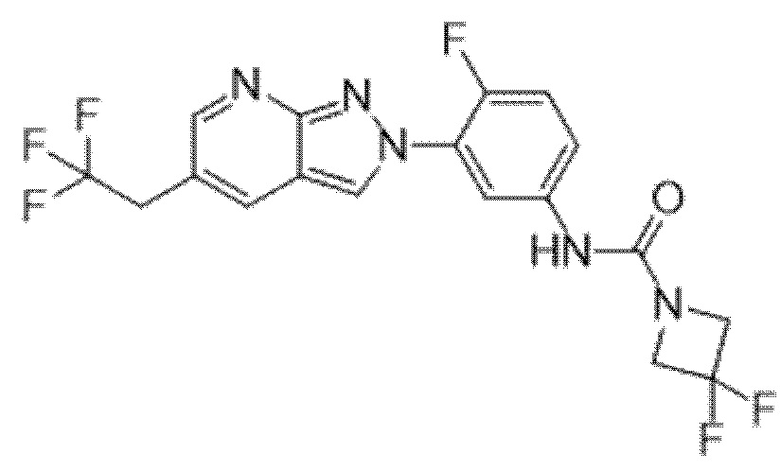
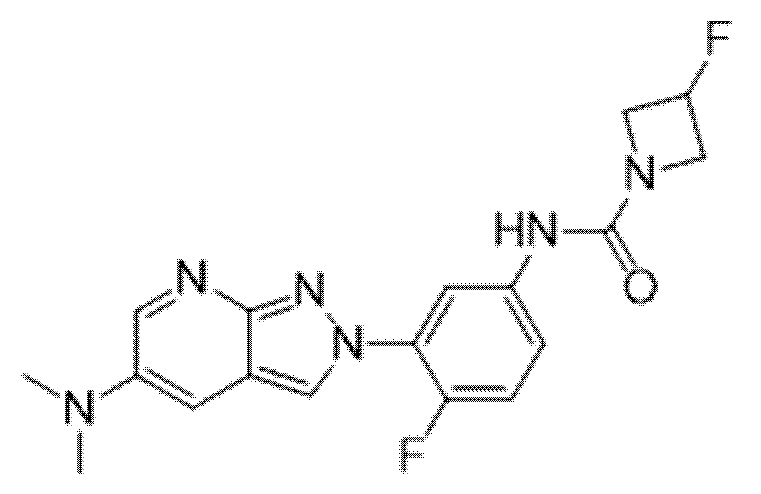
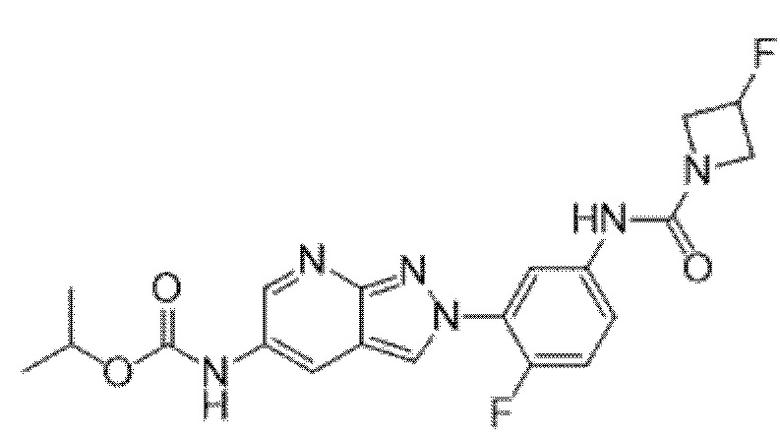
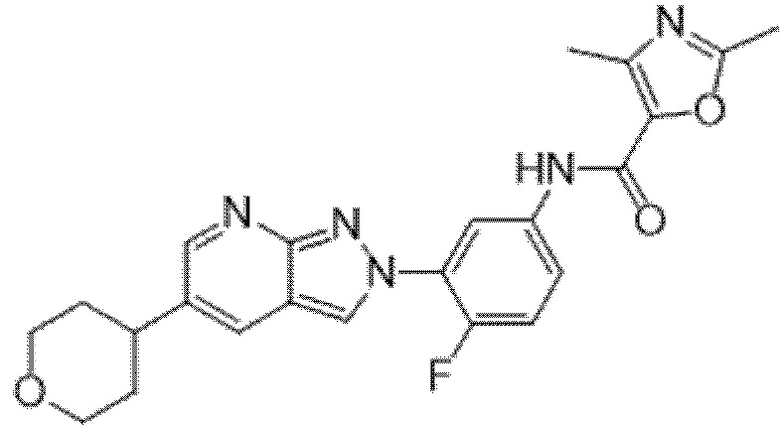
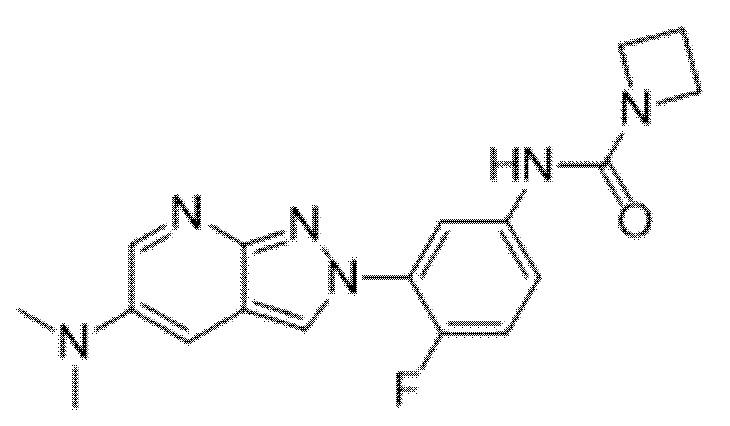
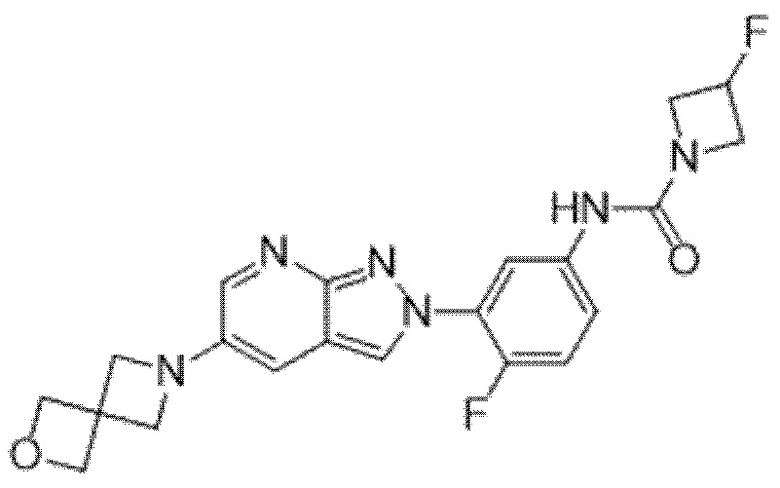
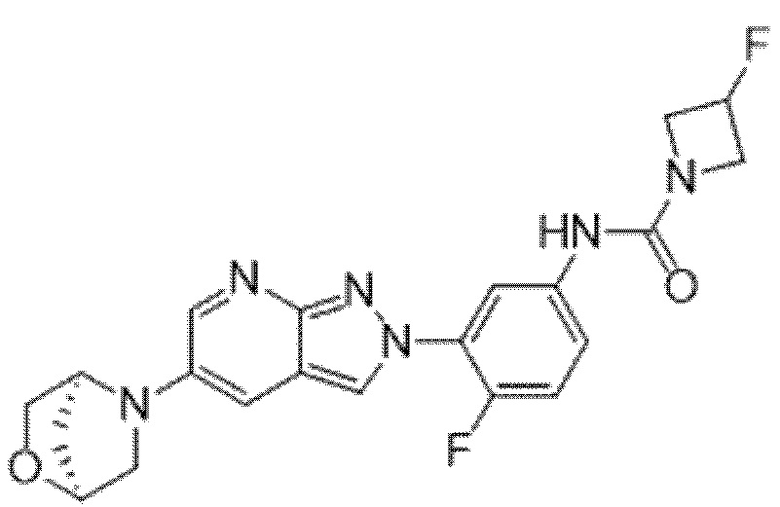
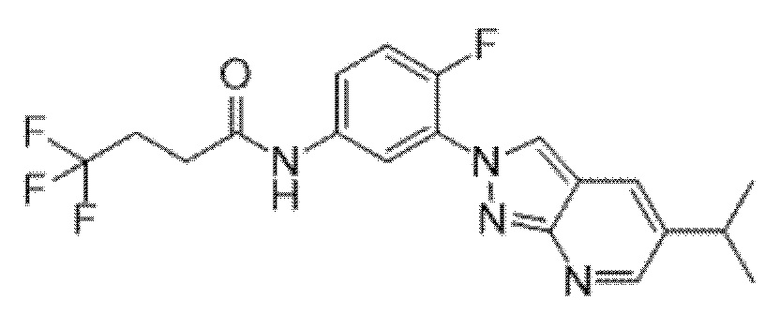
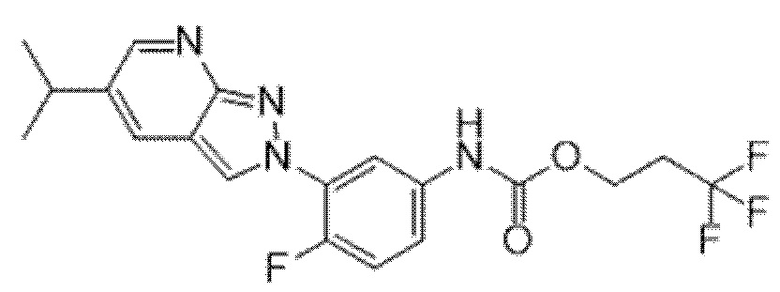
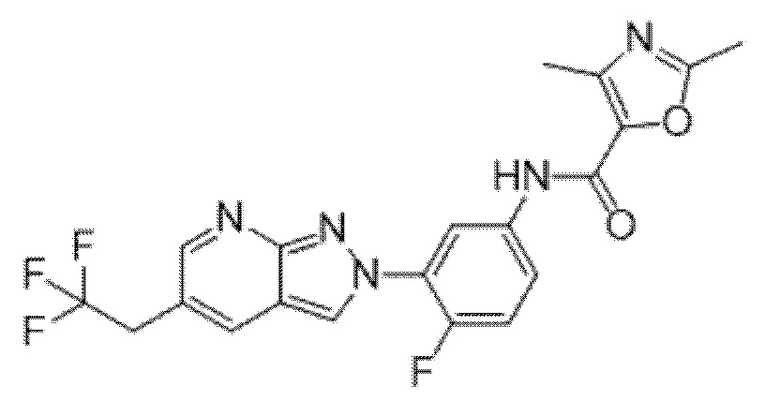
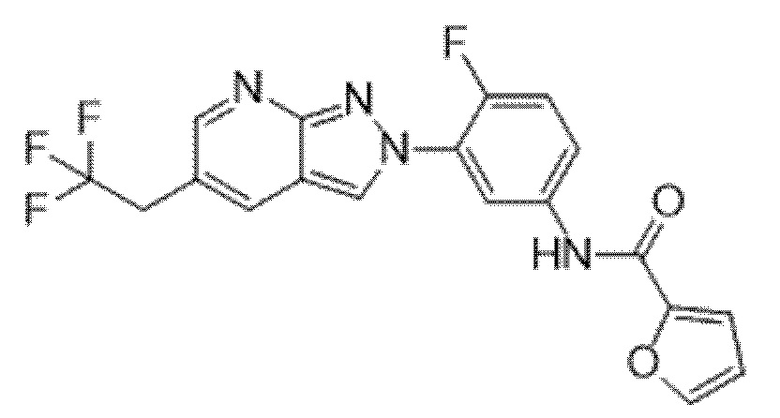
Способ 1: RT=2,86 мин.; M+H=405,1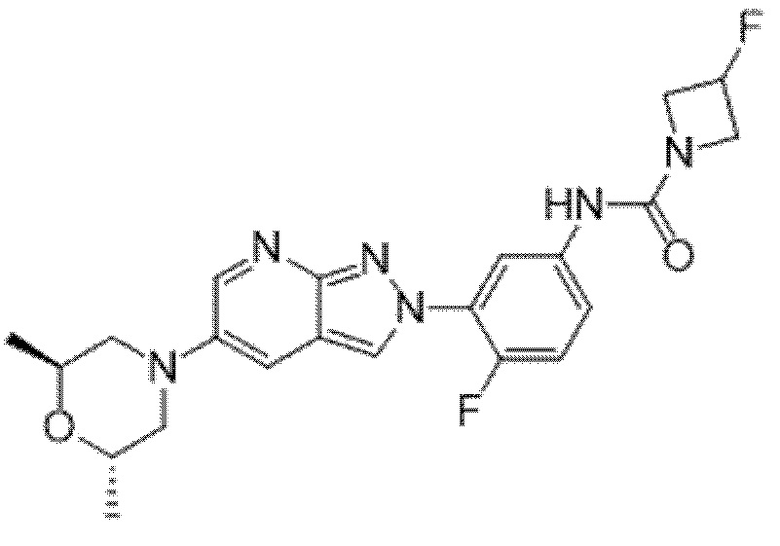
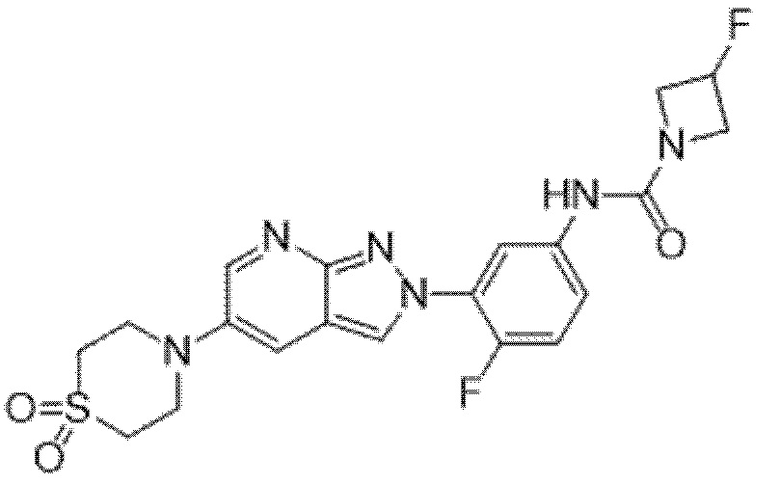
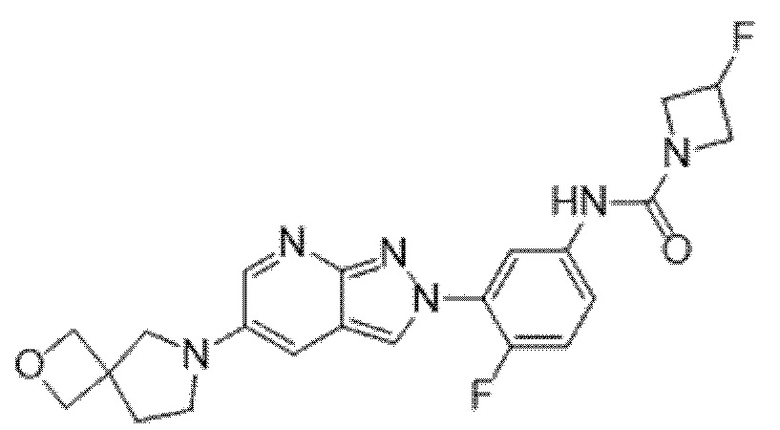
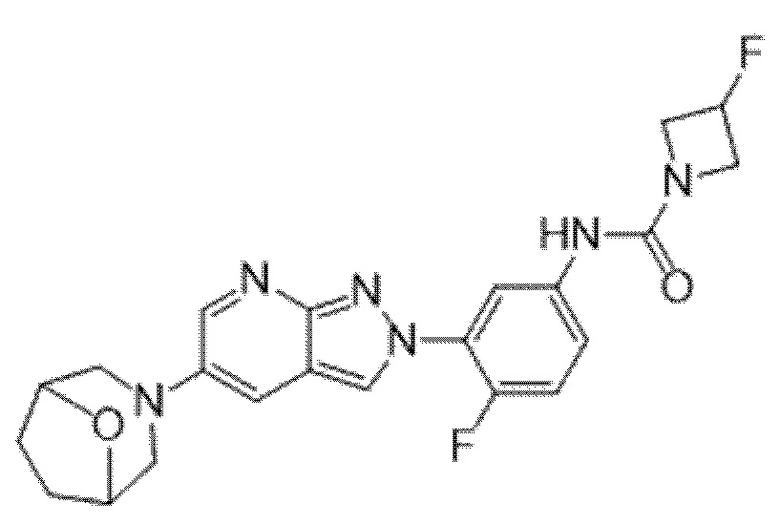
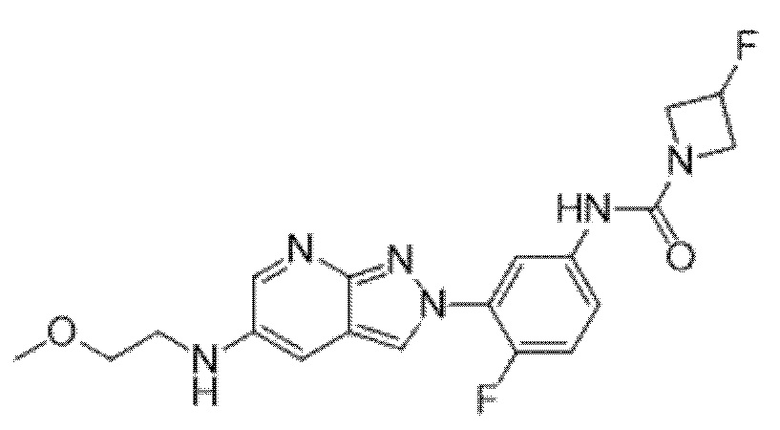
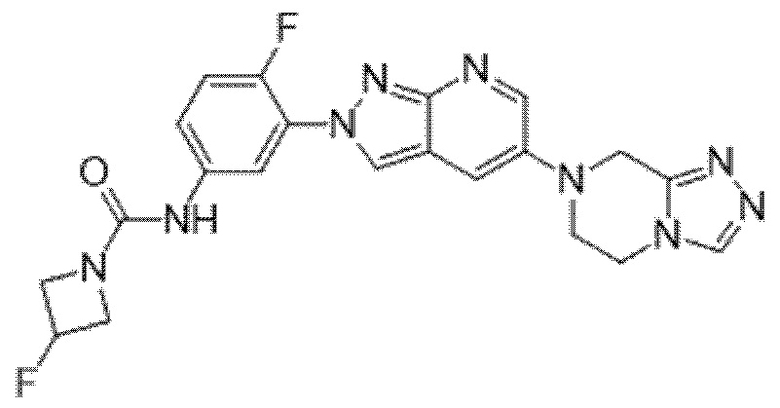
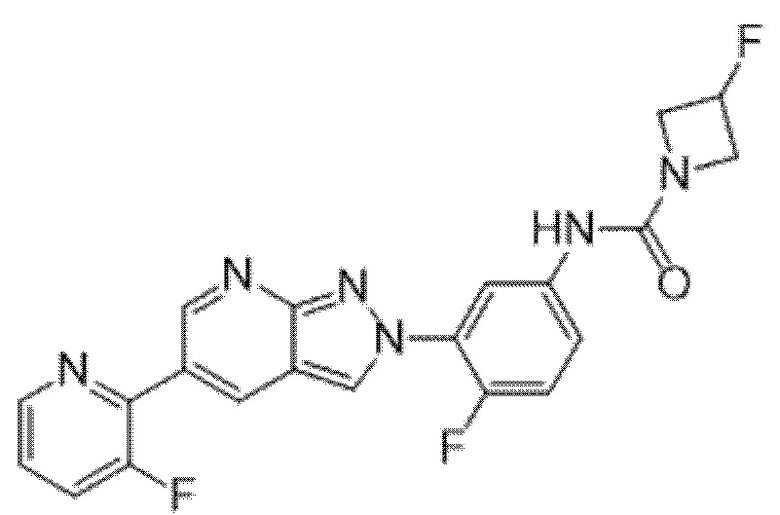
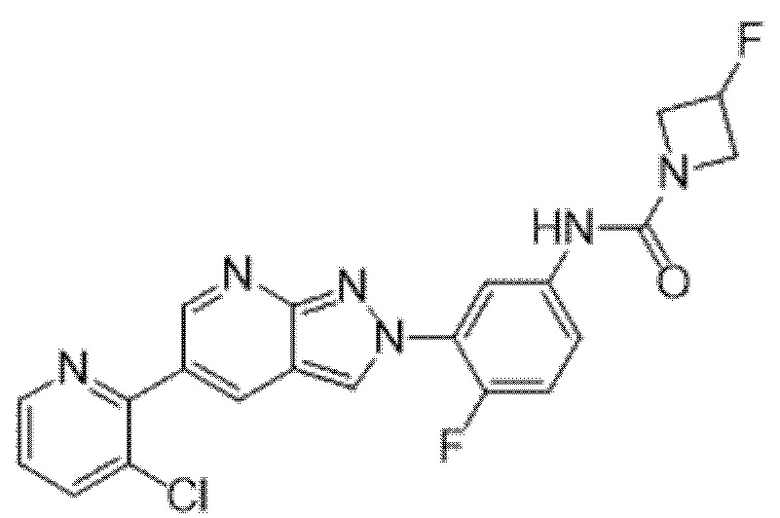
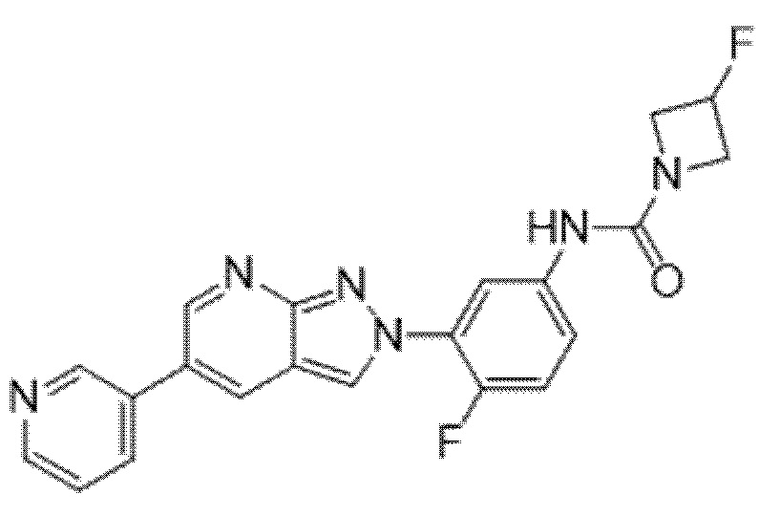
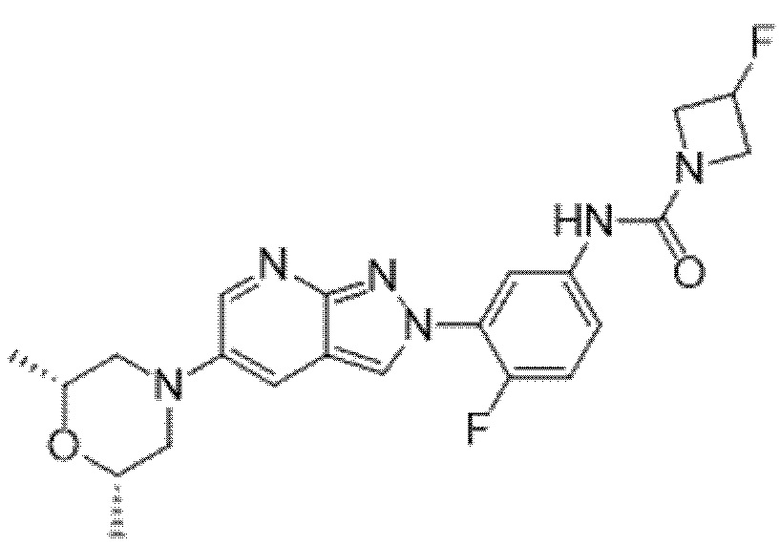
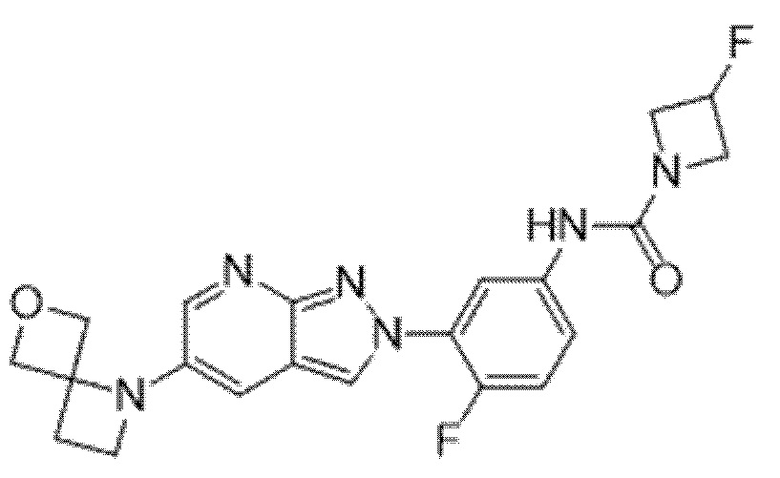
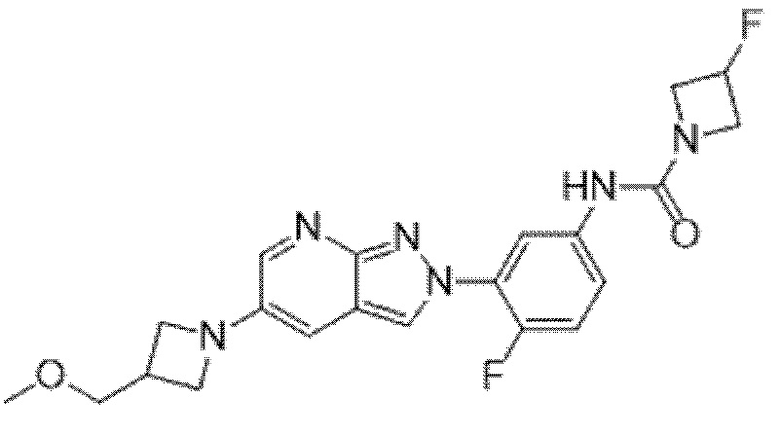
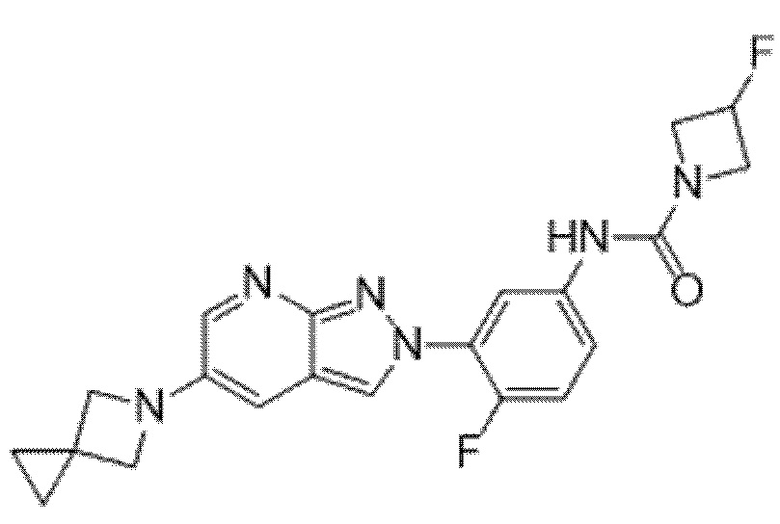
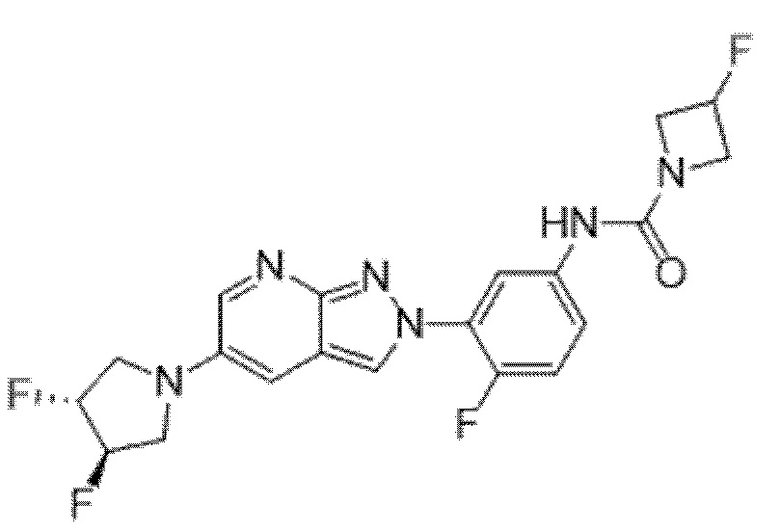
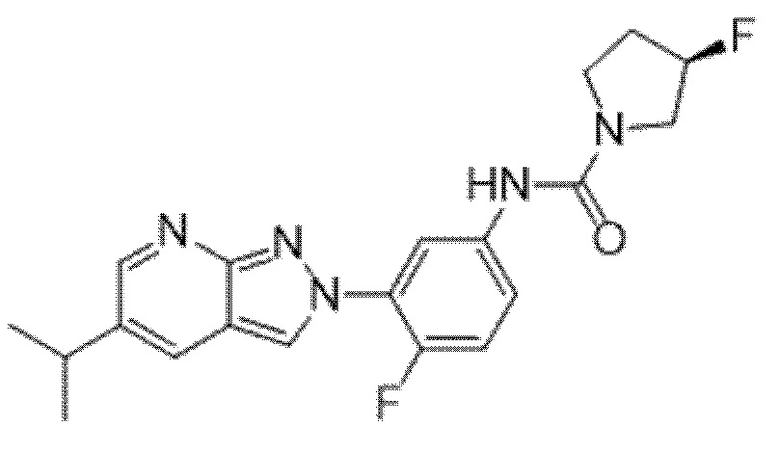
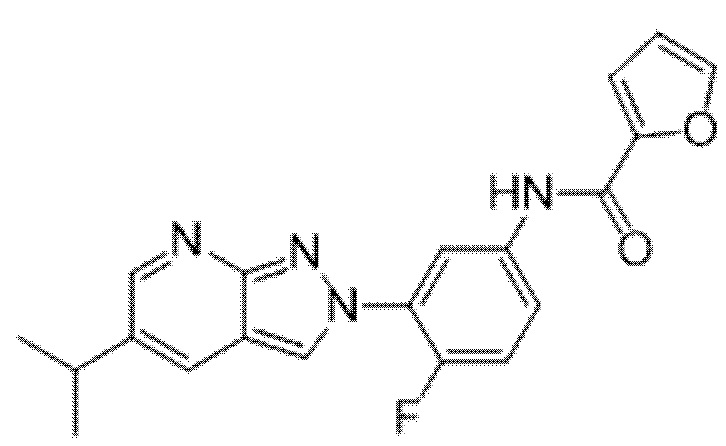
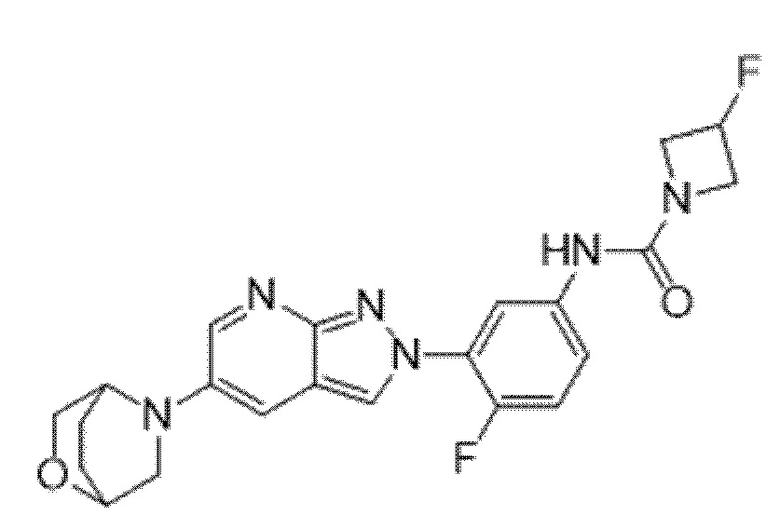
Способ 1: RT=2,69 мин.; M+H=441,3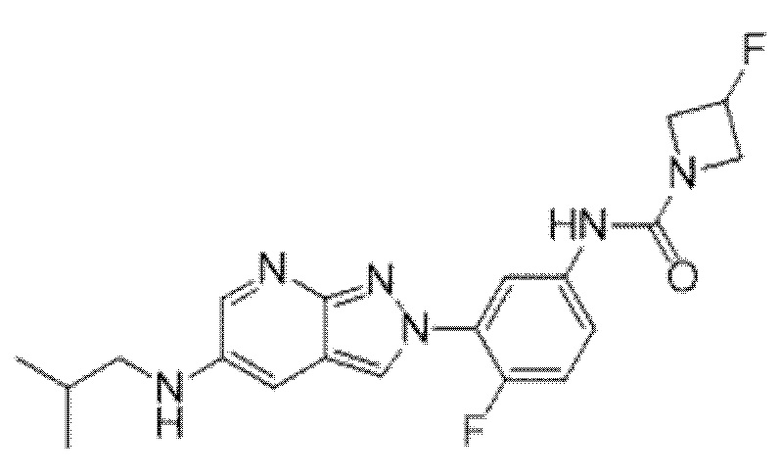
Способ 1: RT=3,00 мин.; M+H=401,4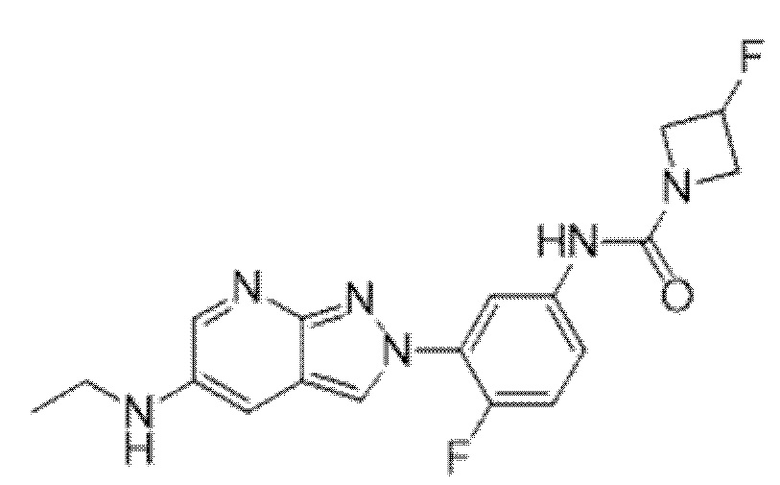
Способ 2: RT=0,80 мин.; M+H=373,4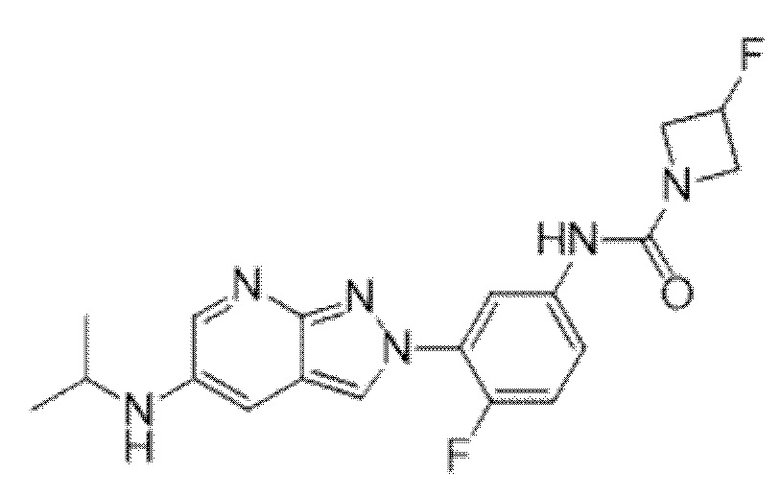
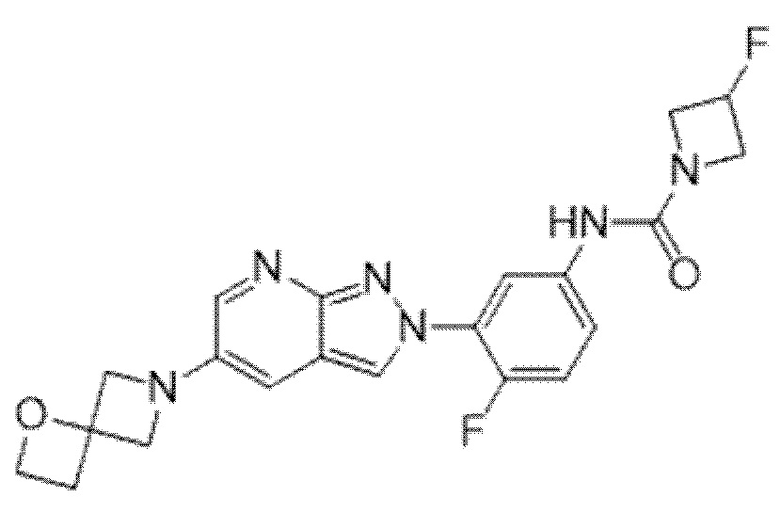
Способ 2: RT=0,78 мин.; M+H=427,3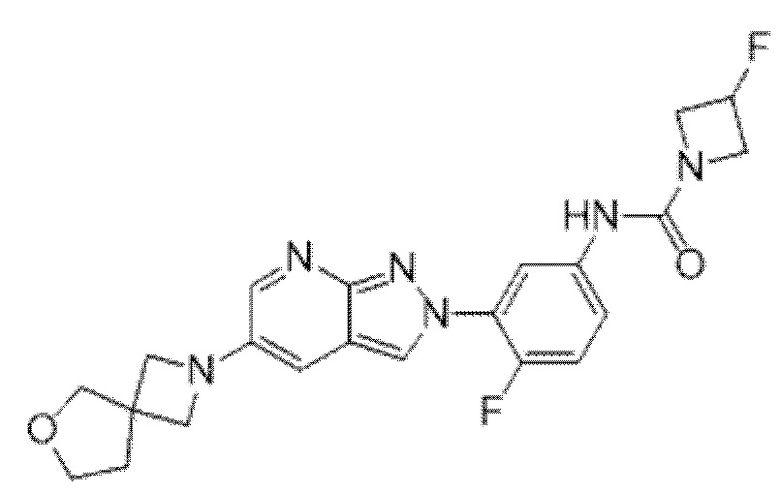
Способ 2: RT=0,80 мин.; M+H=441,3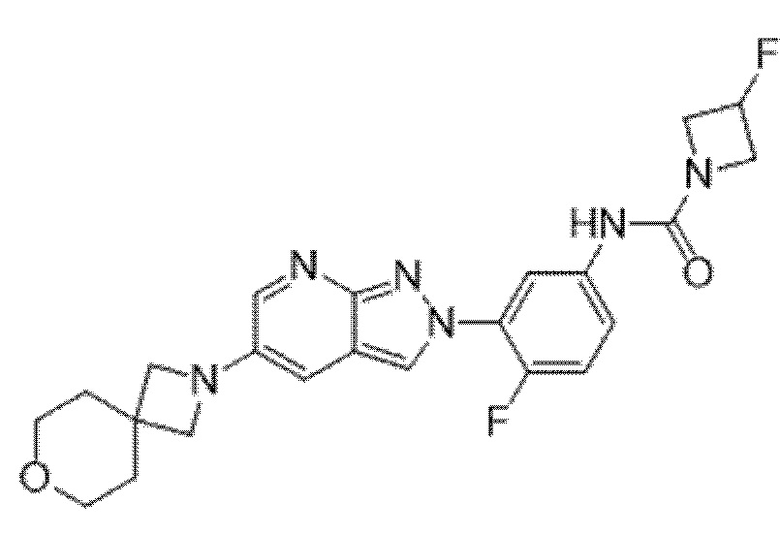
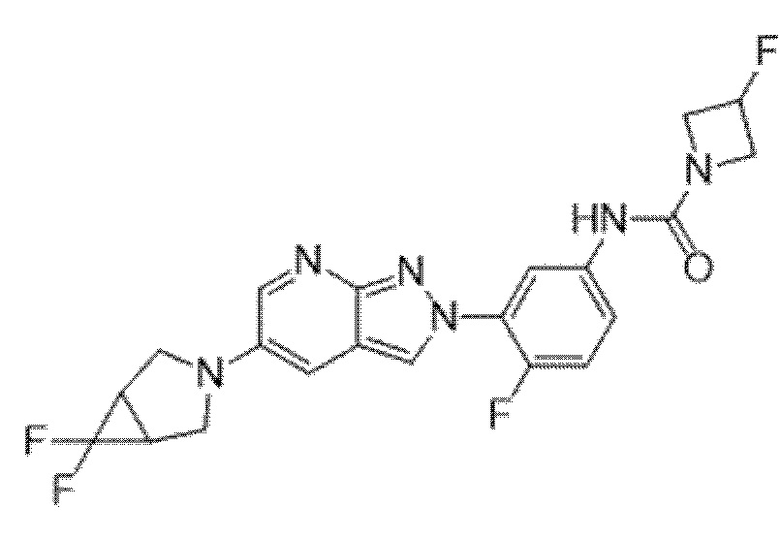
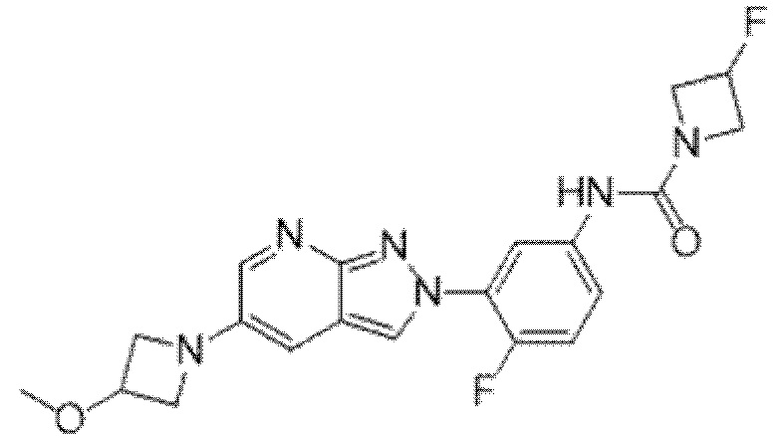
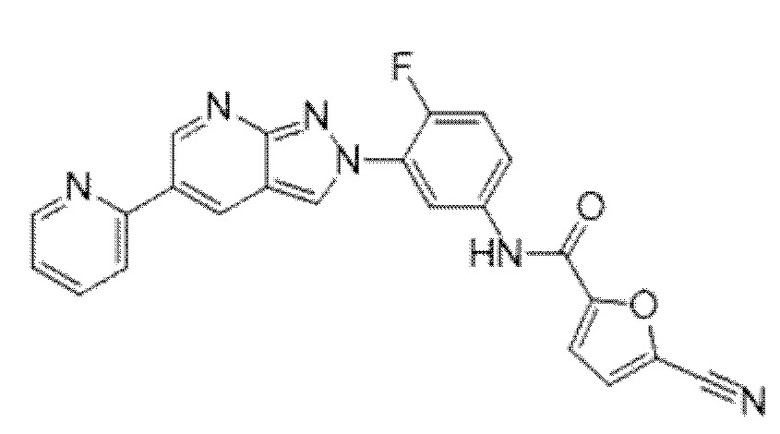
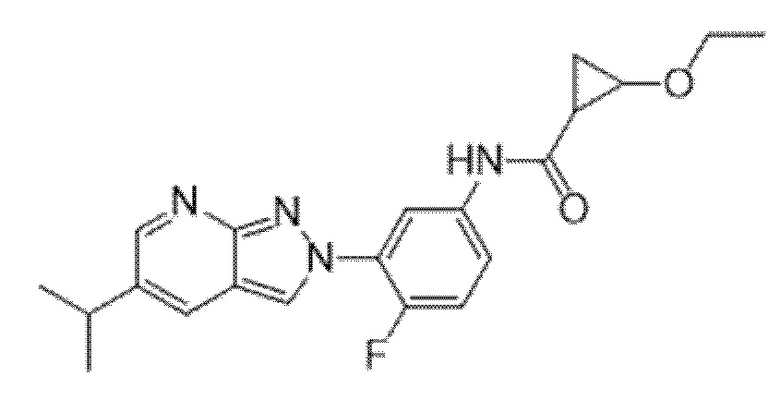
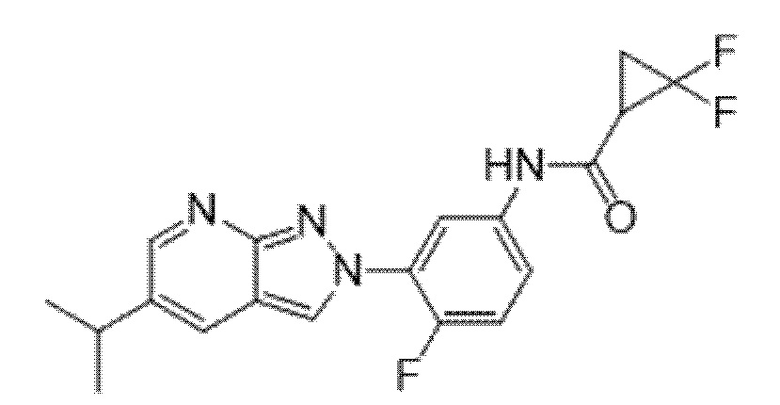
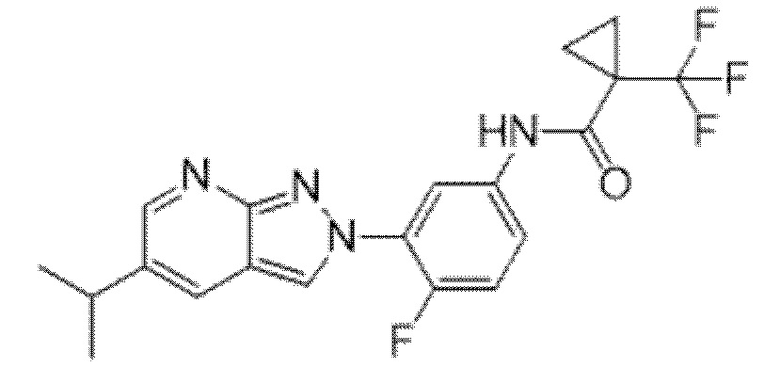
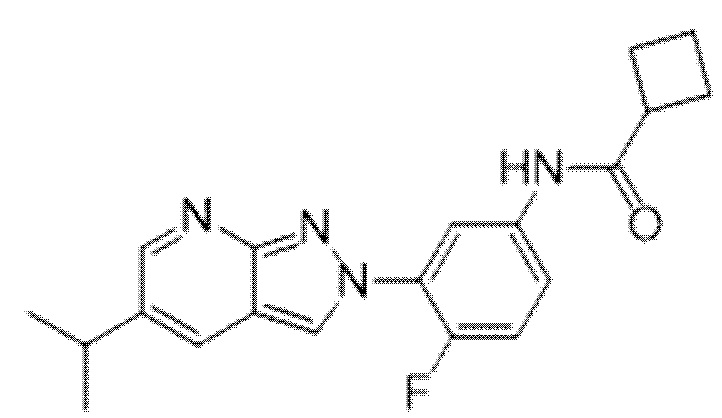
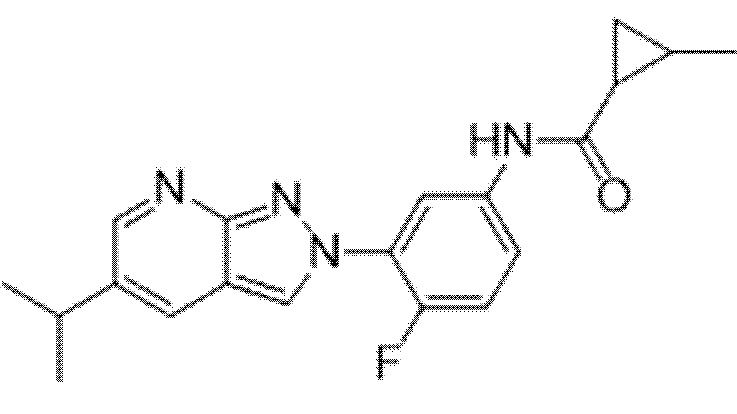
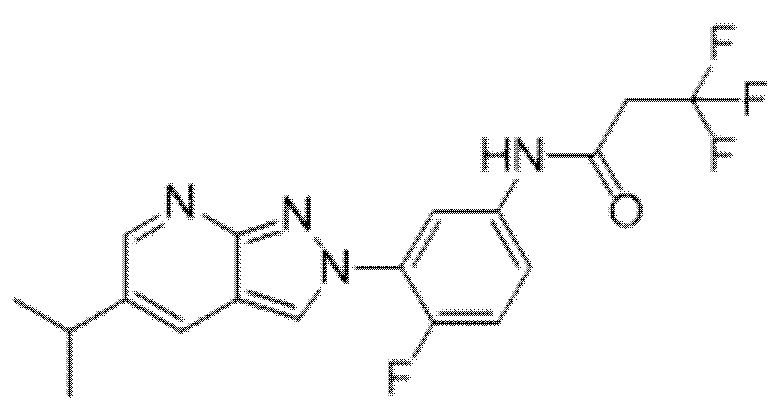
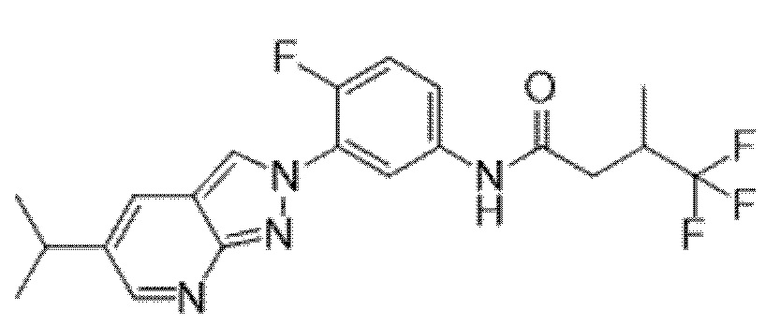
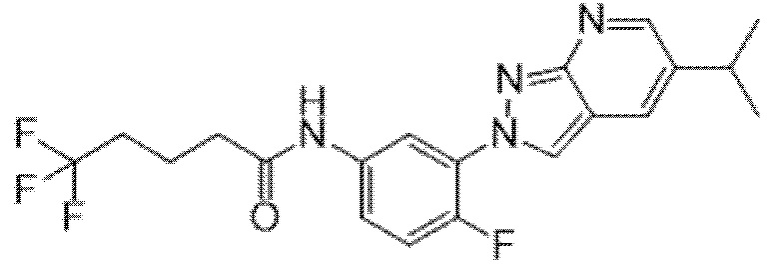
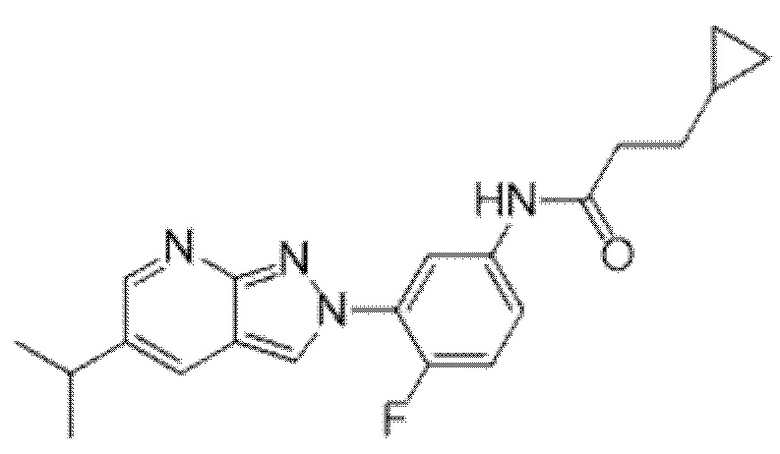
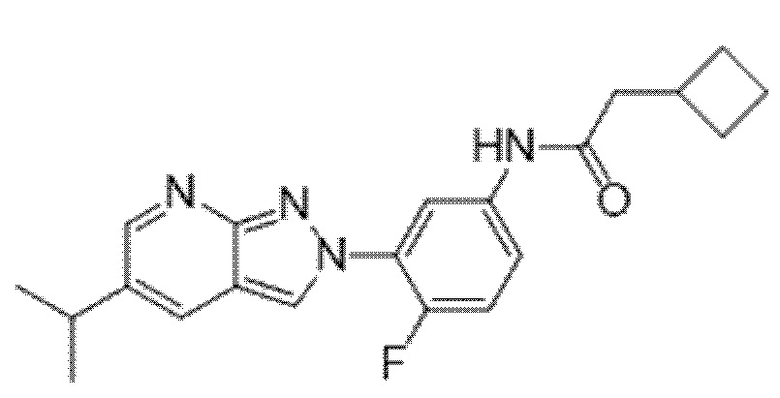
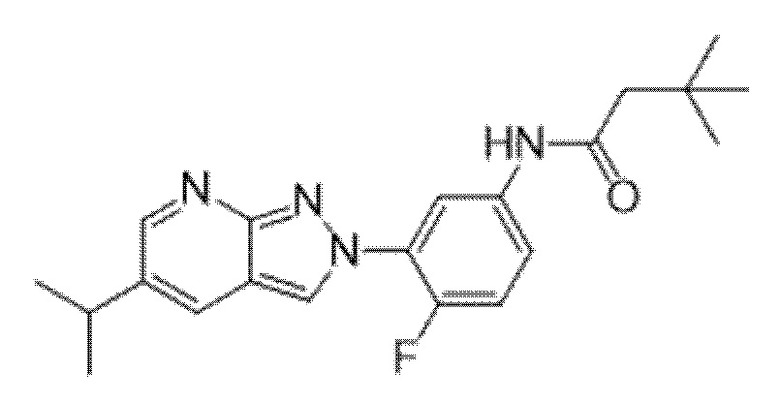
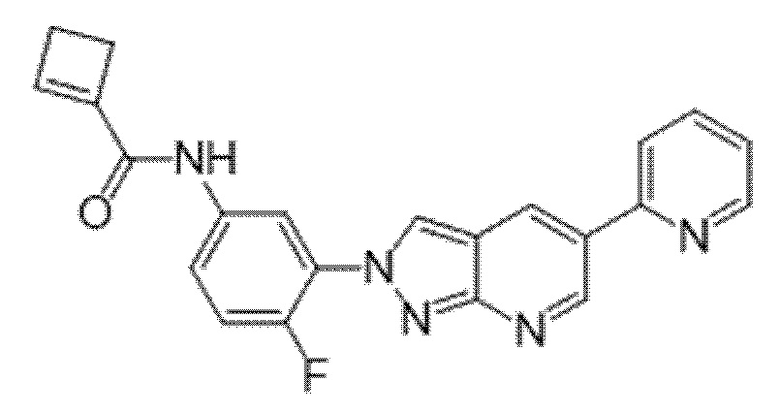
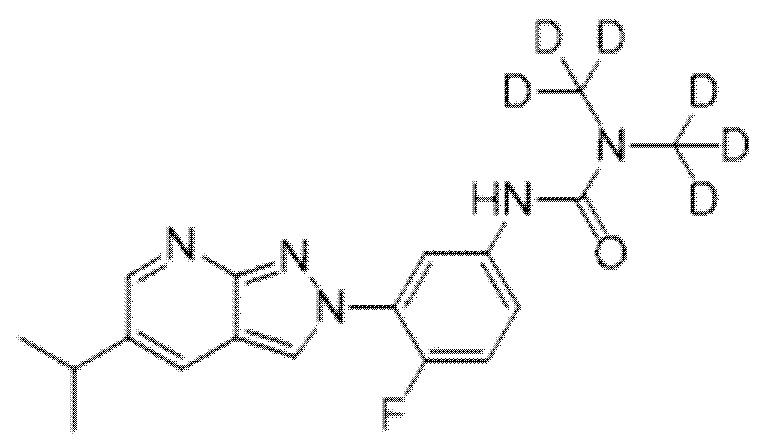
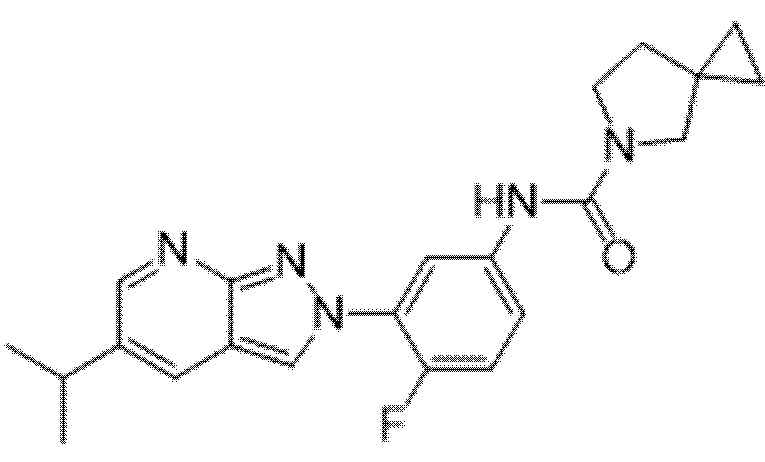
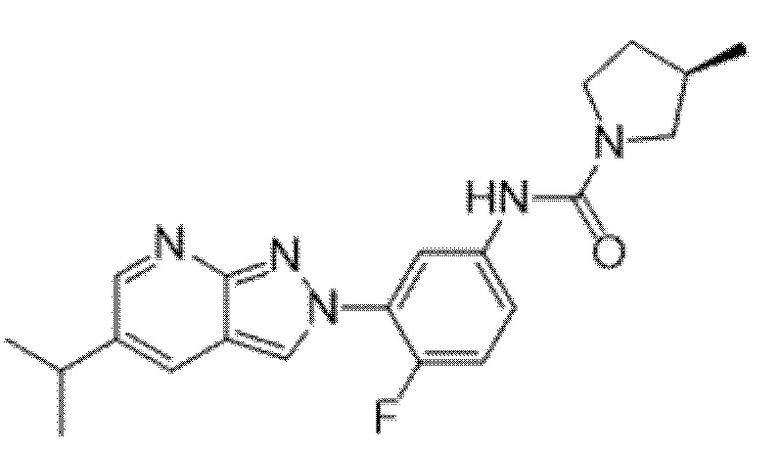
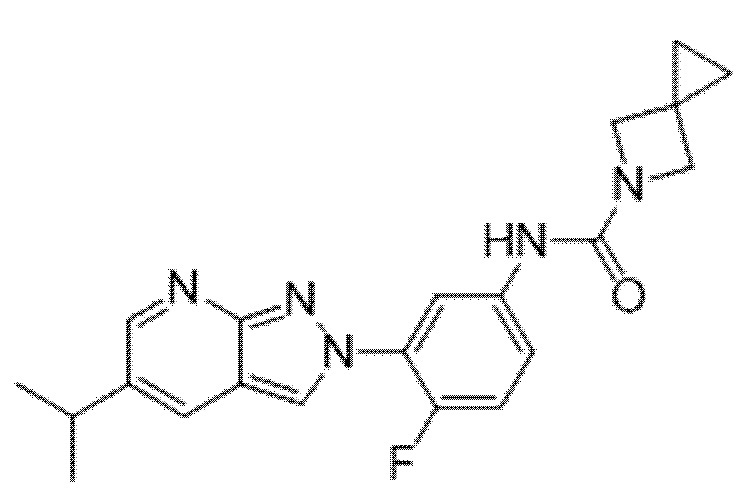
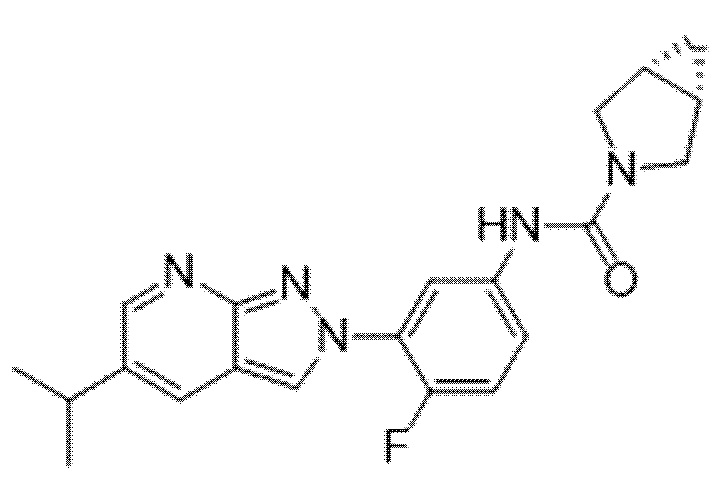
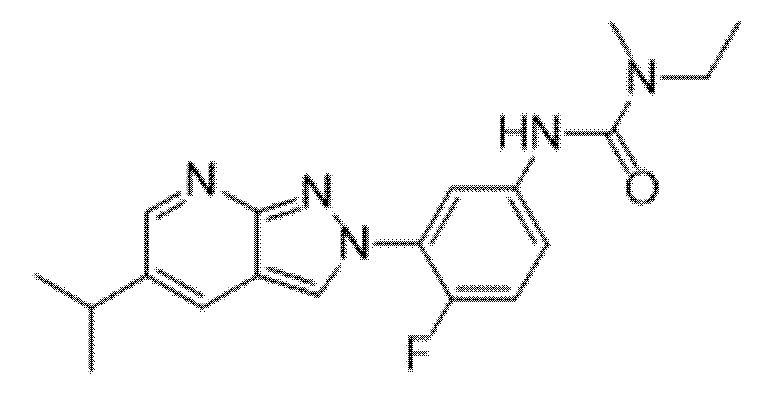
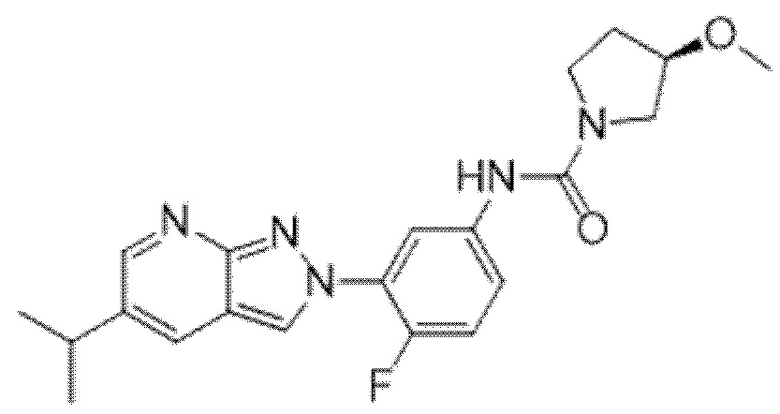
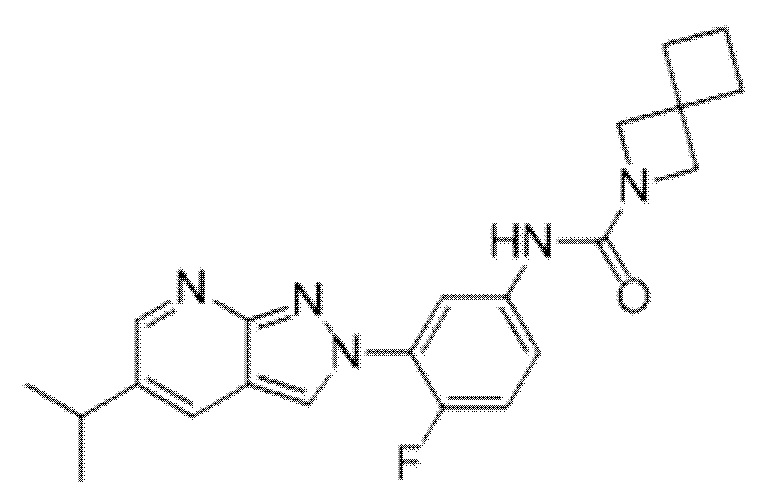
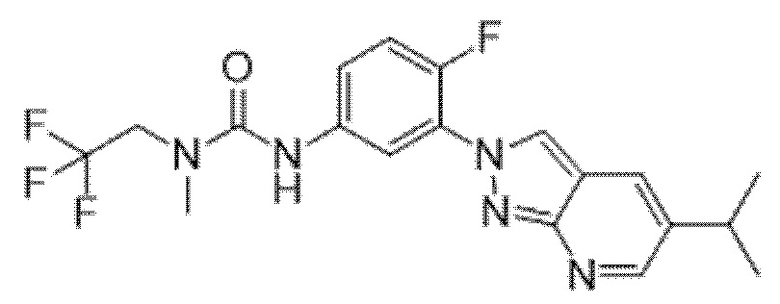
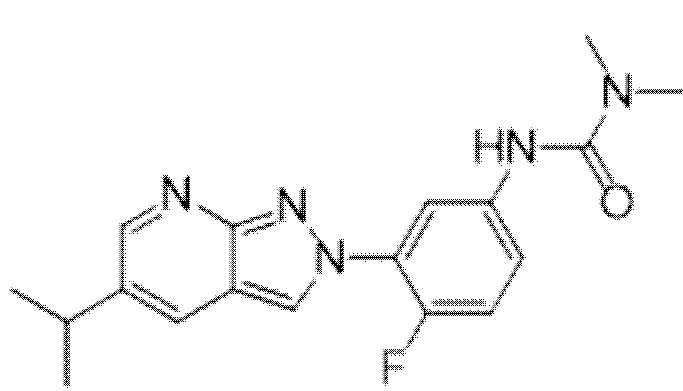
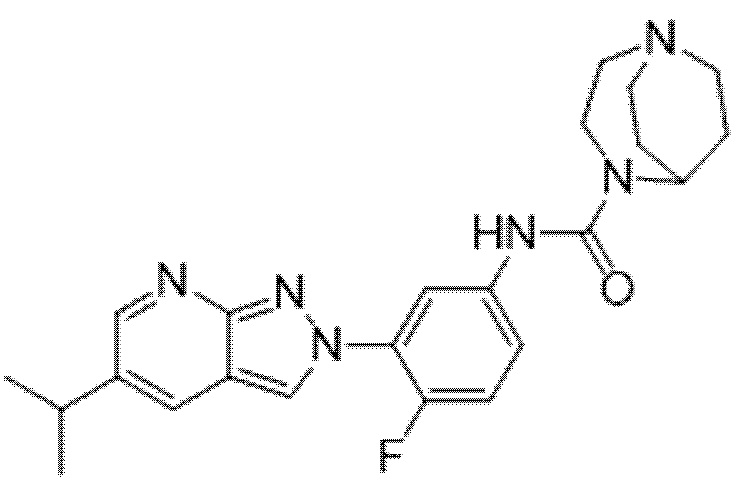
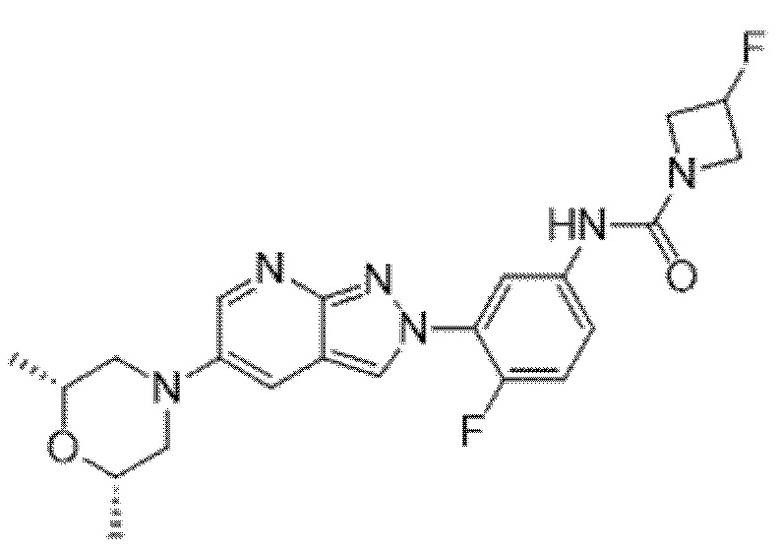
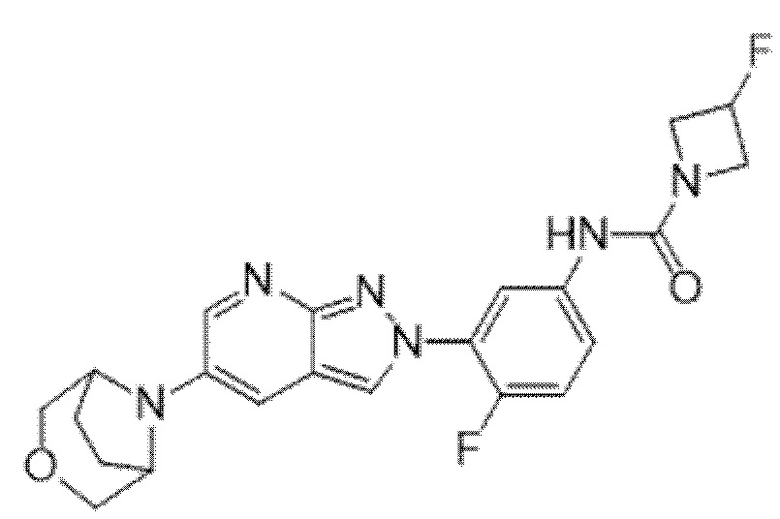
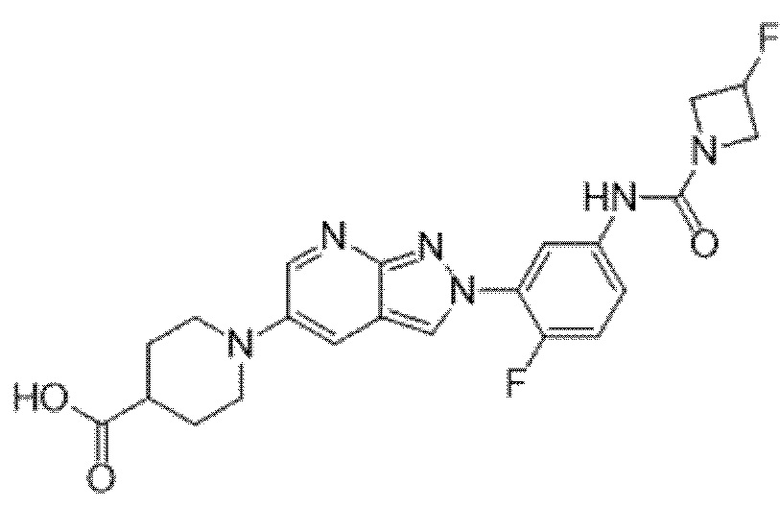
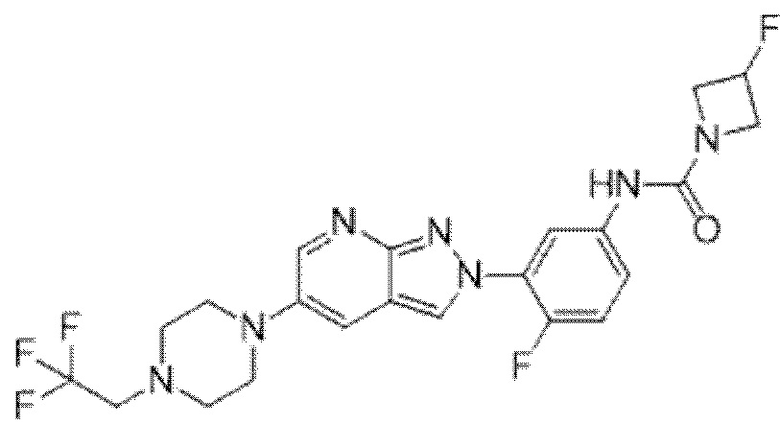
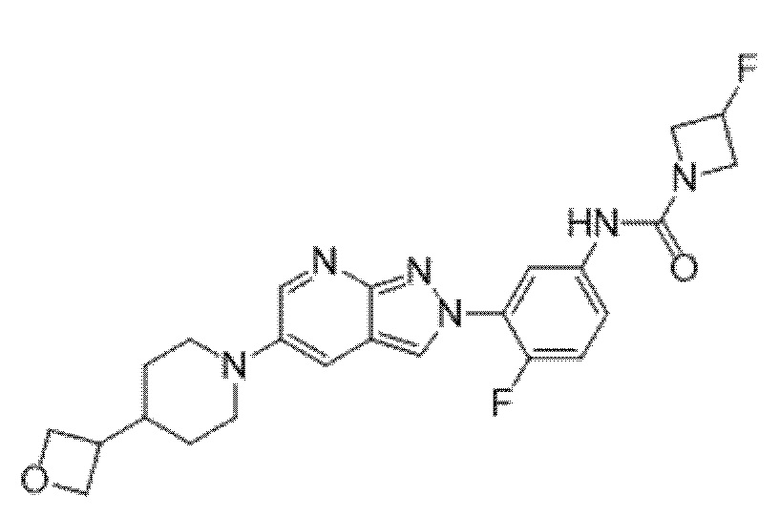
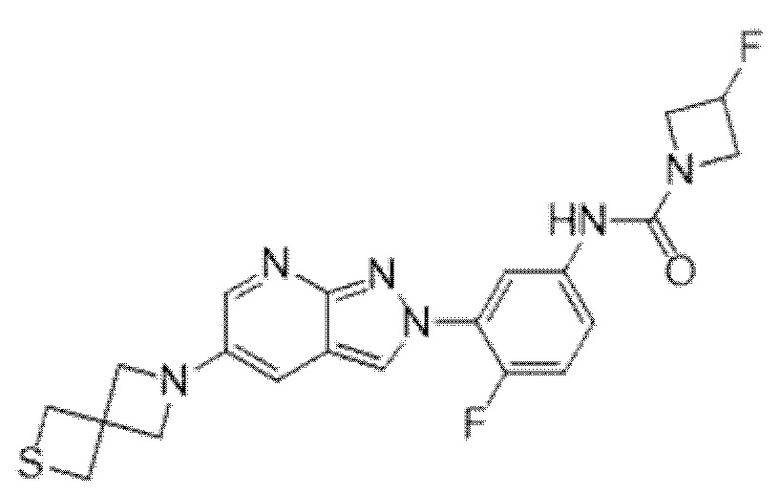
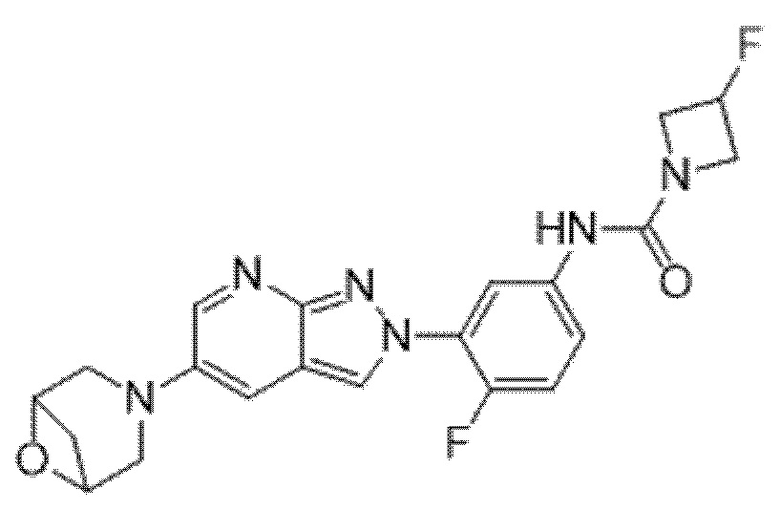
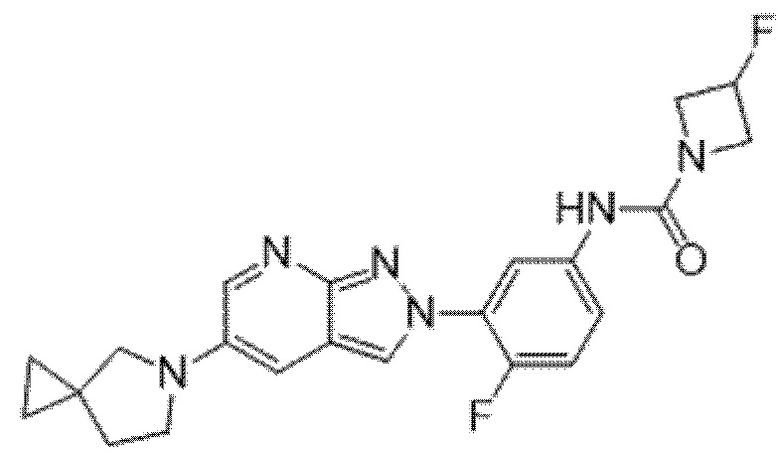
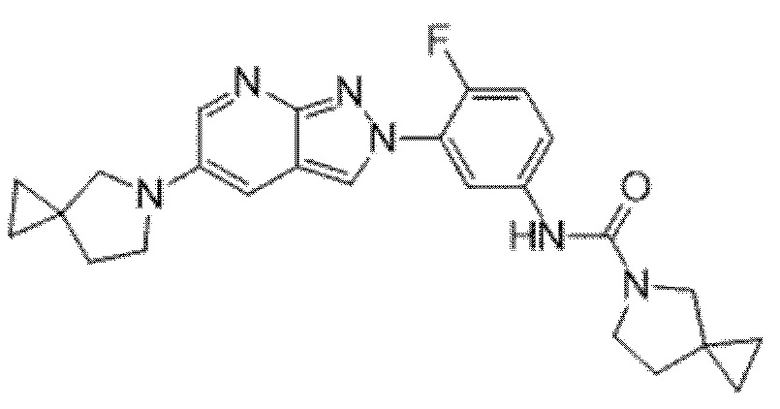
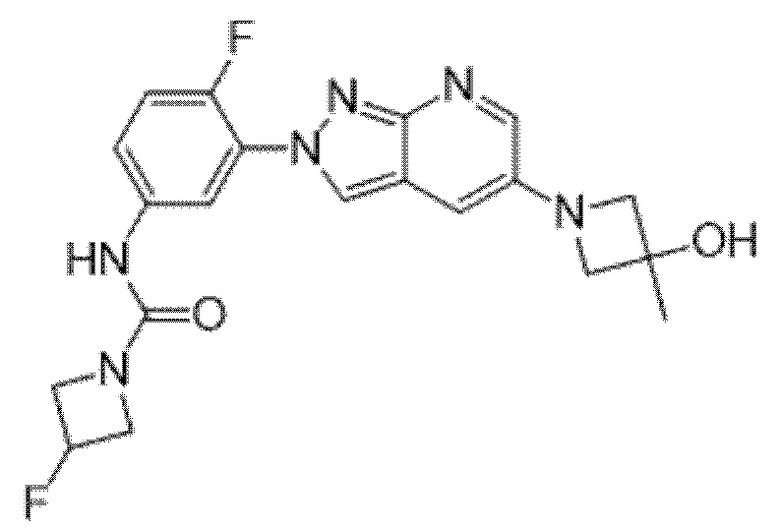
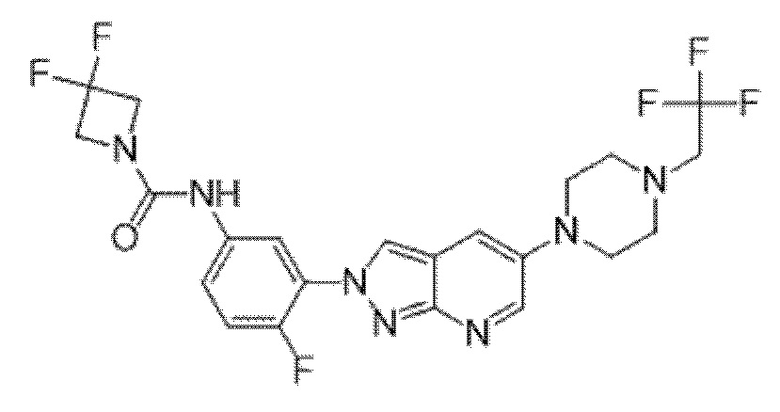
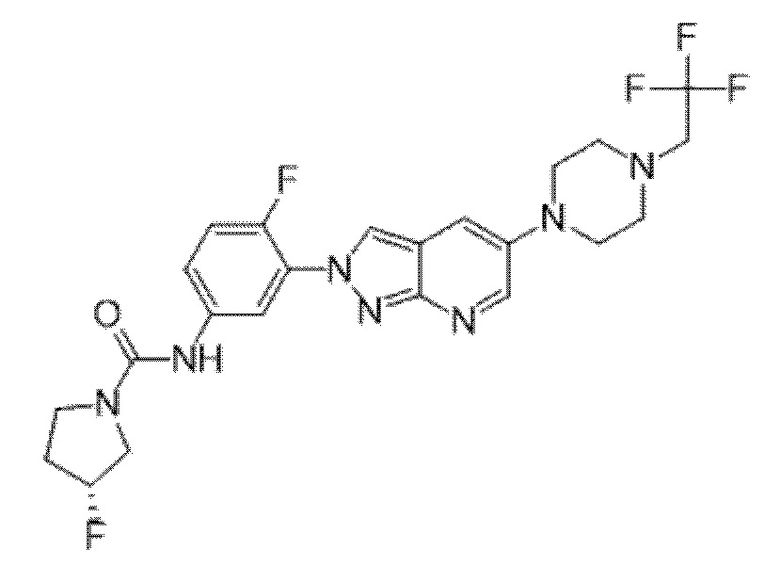
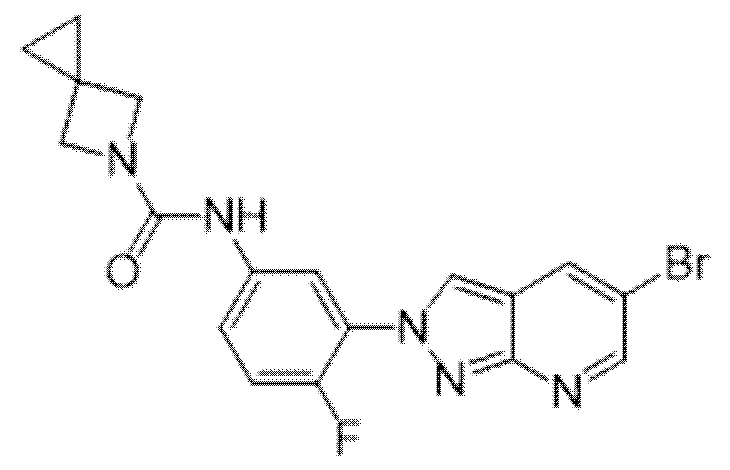
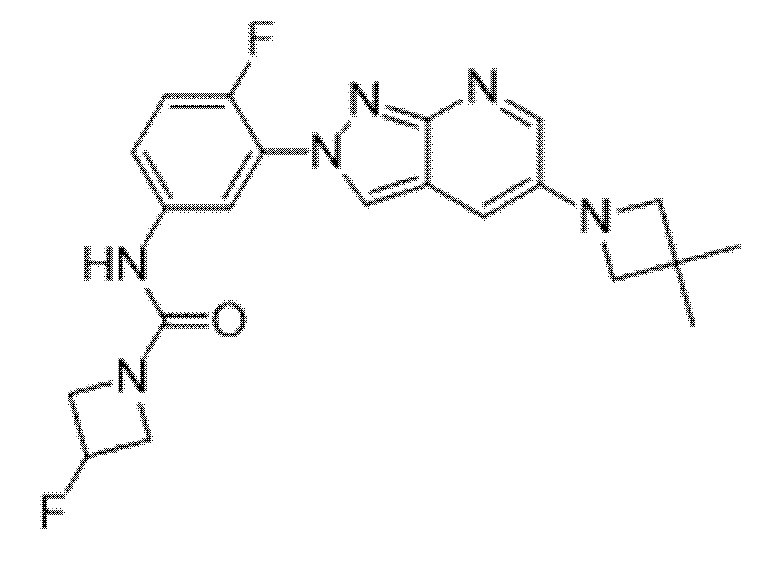
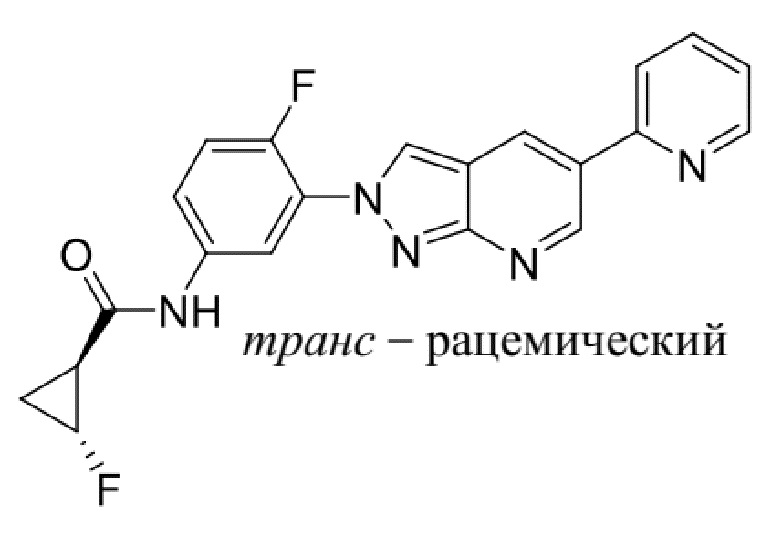
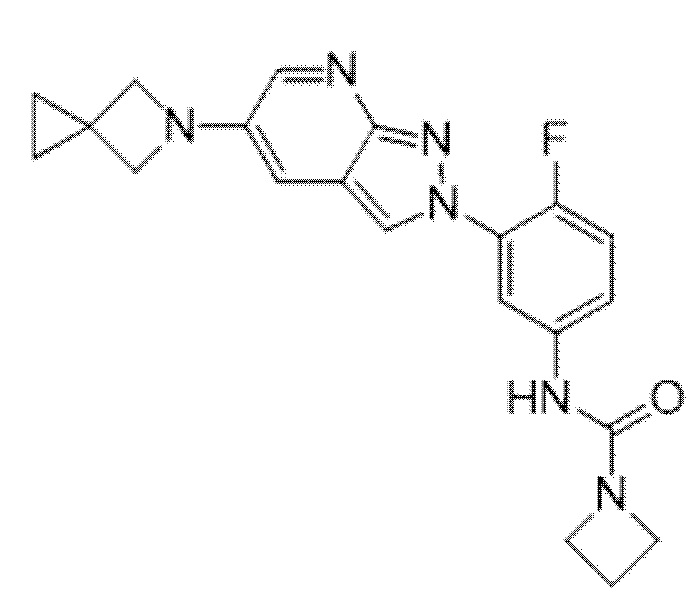
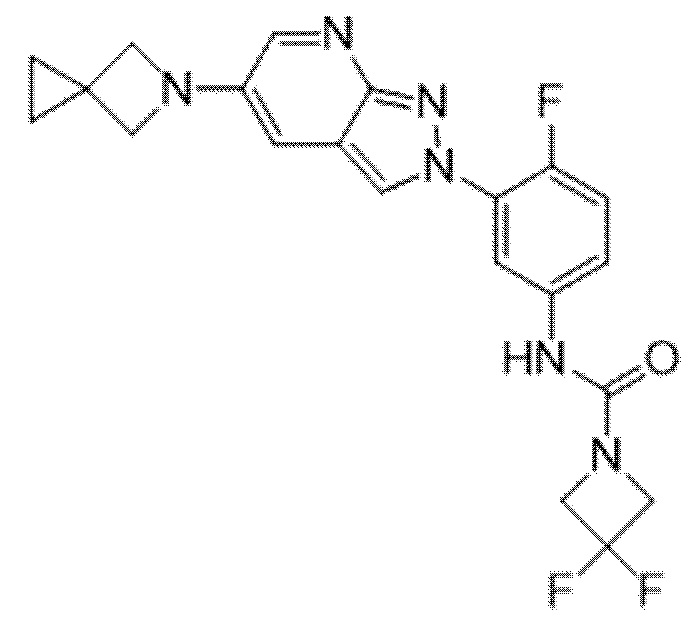
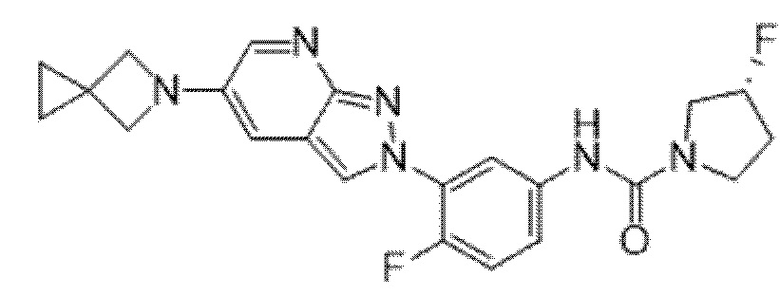
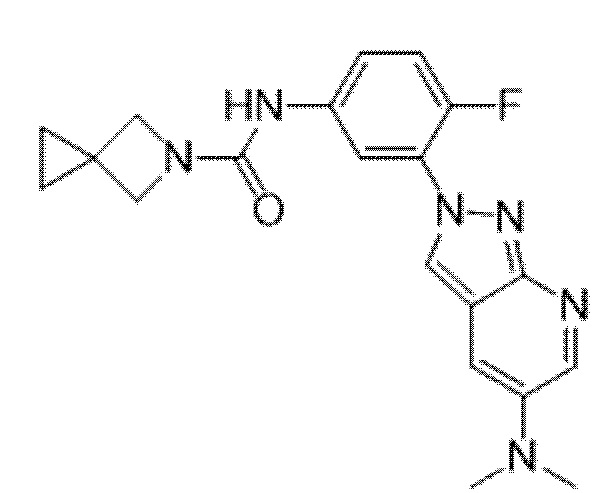
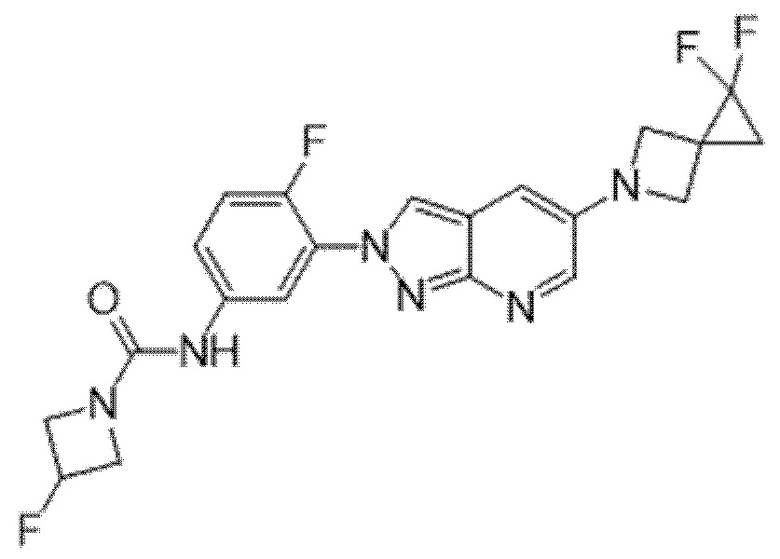
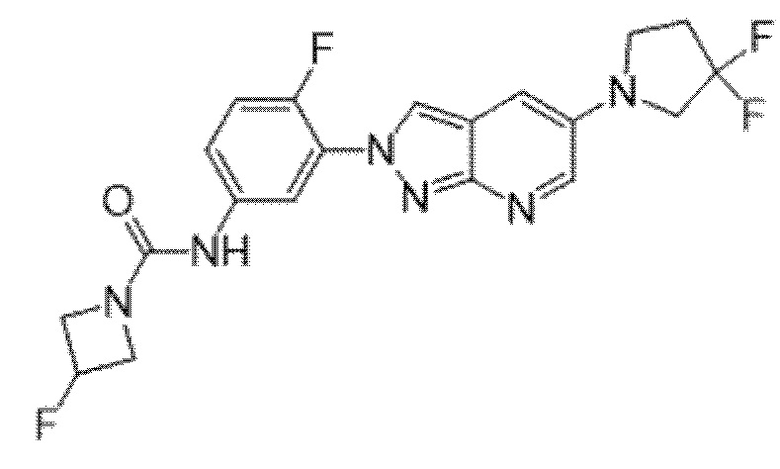
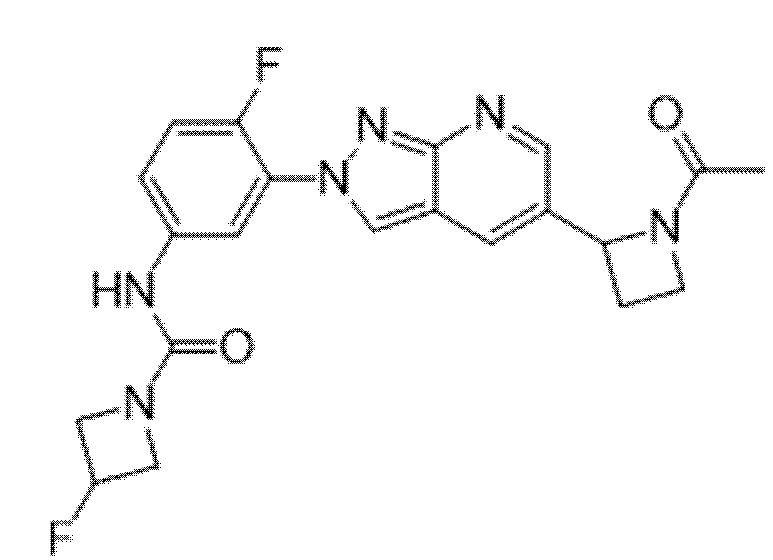
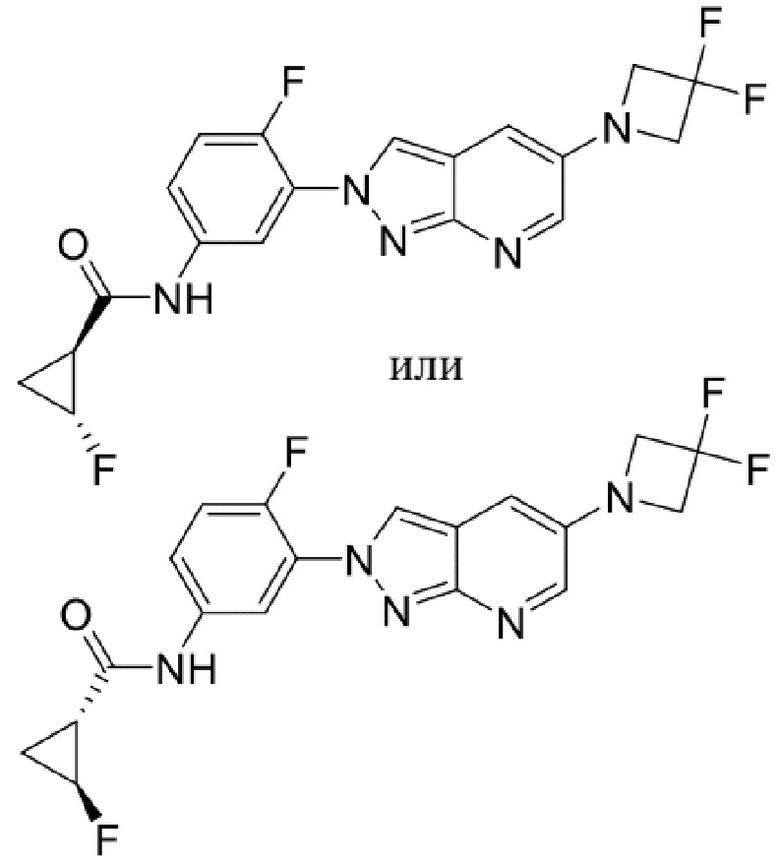
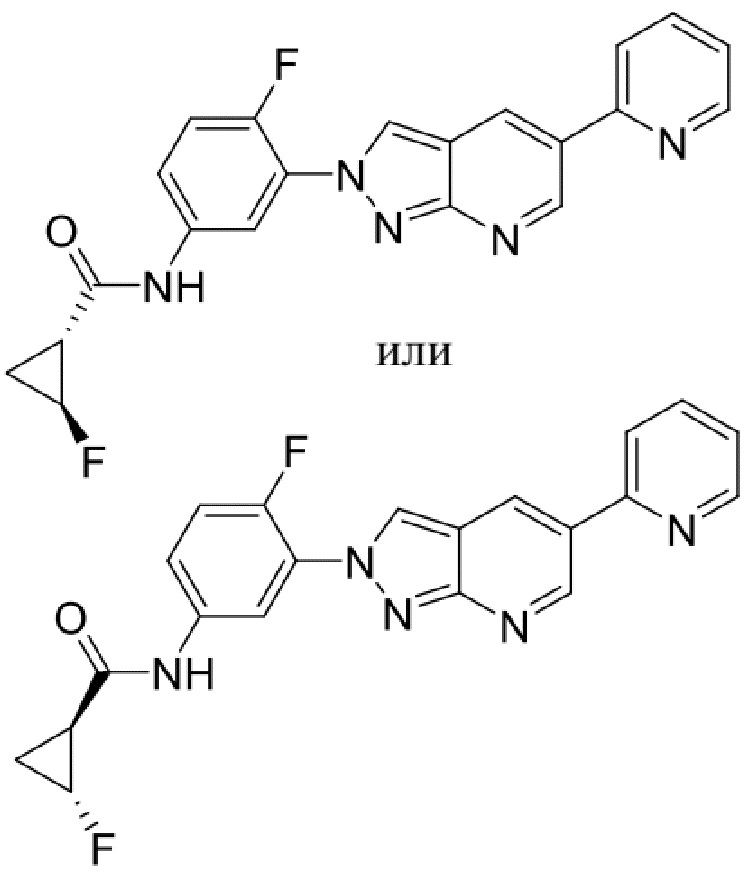
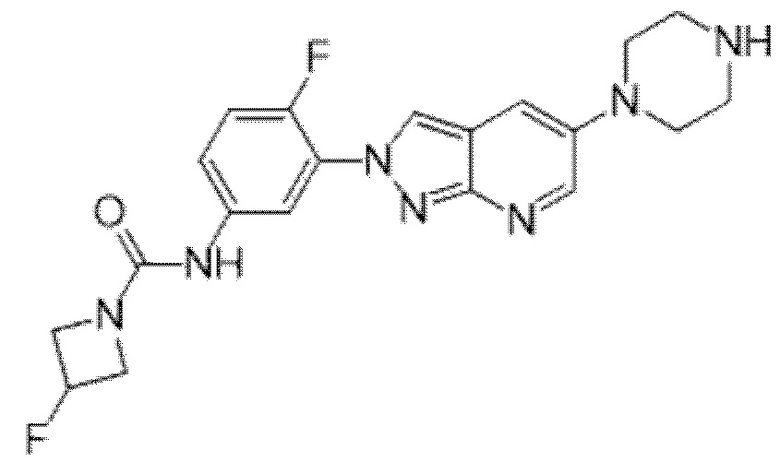
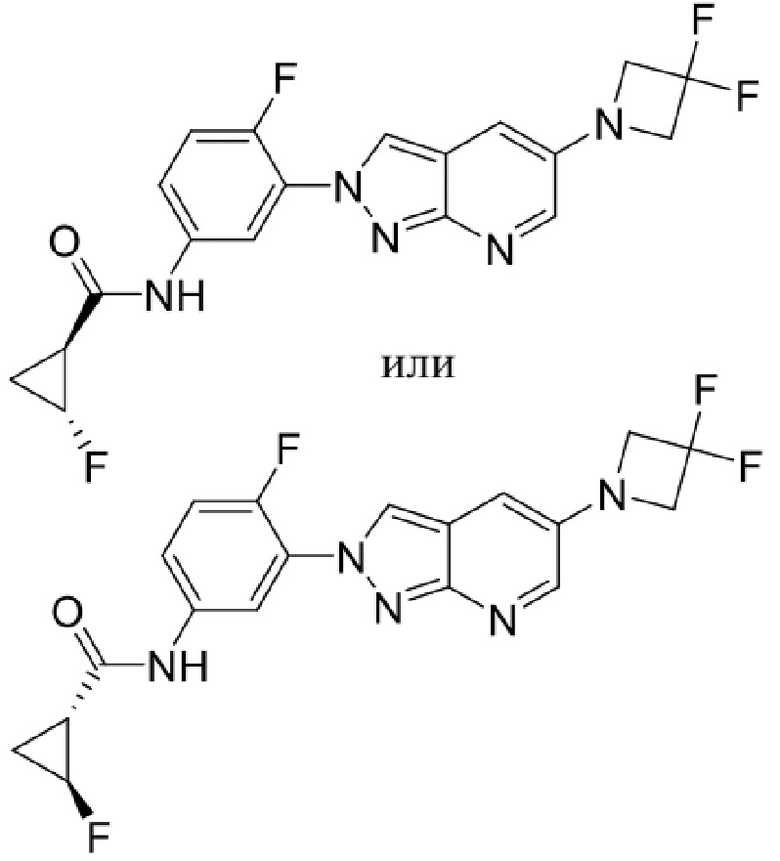
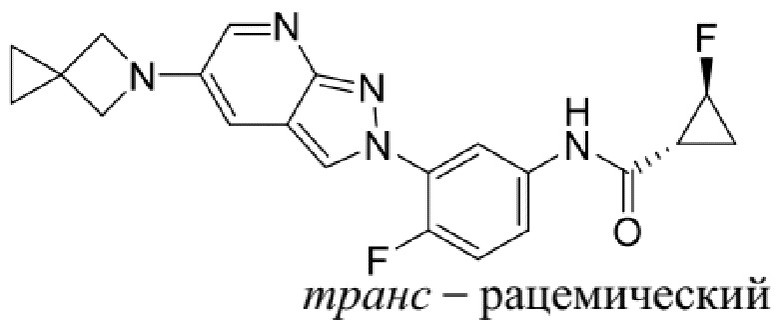
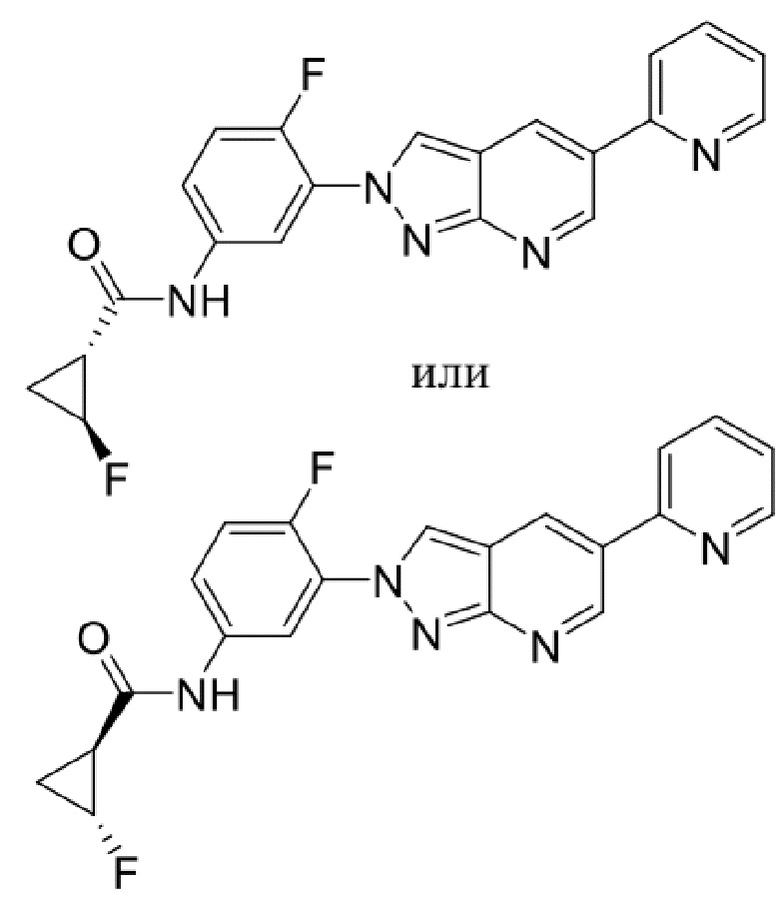
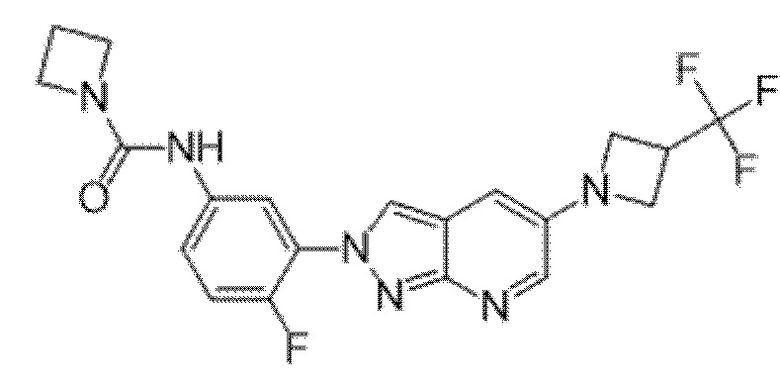
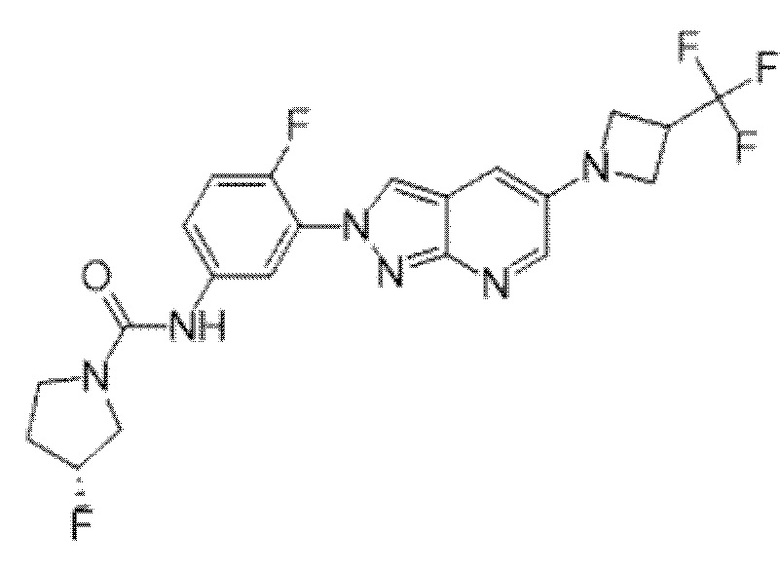
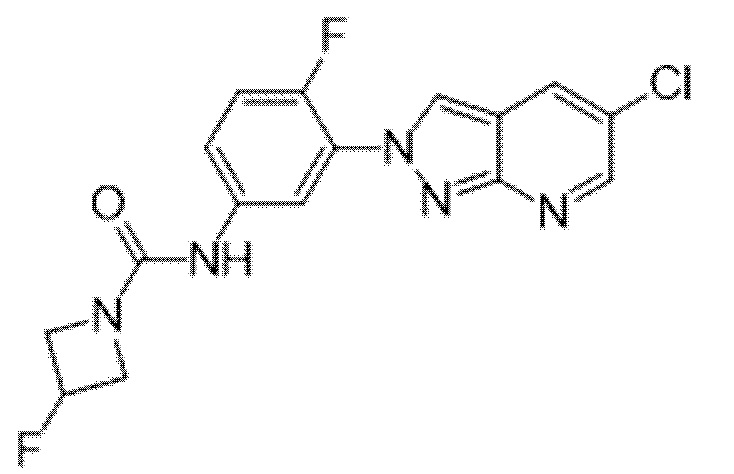
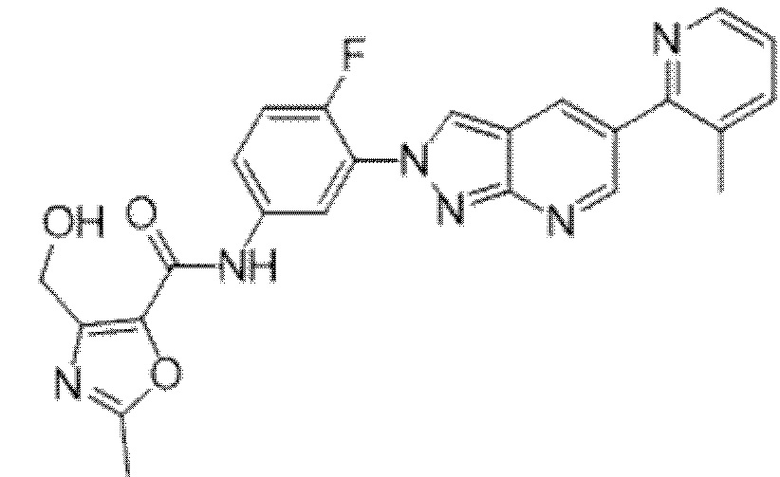
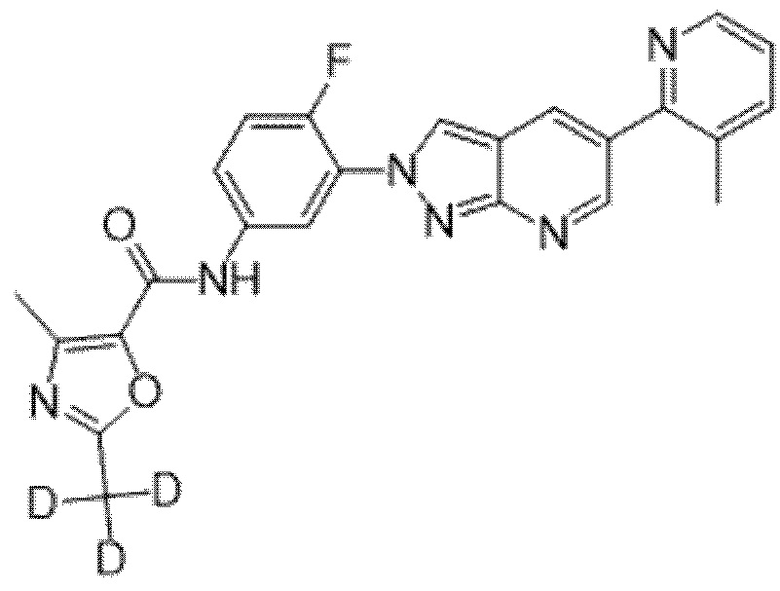
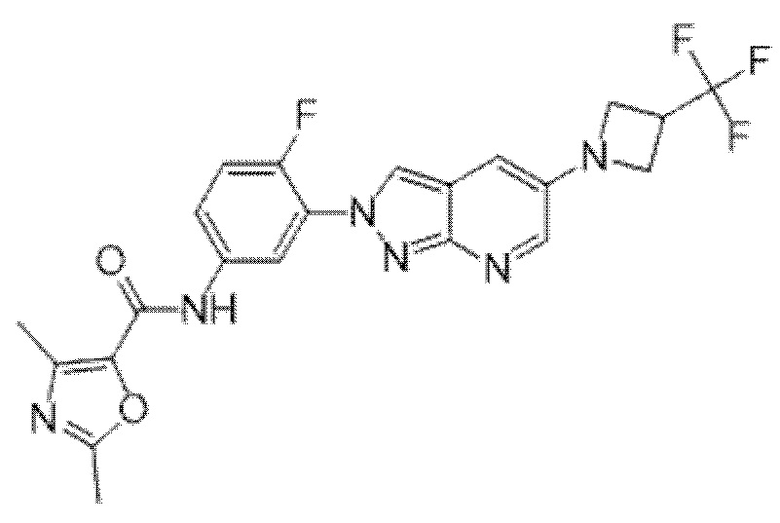
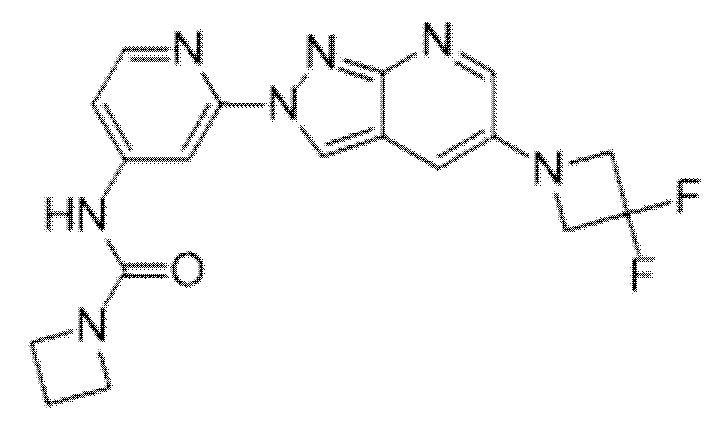
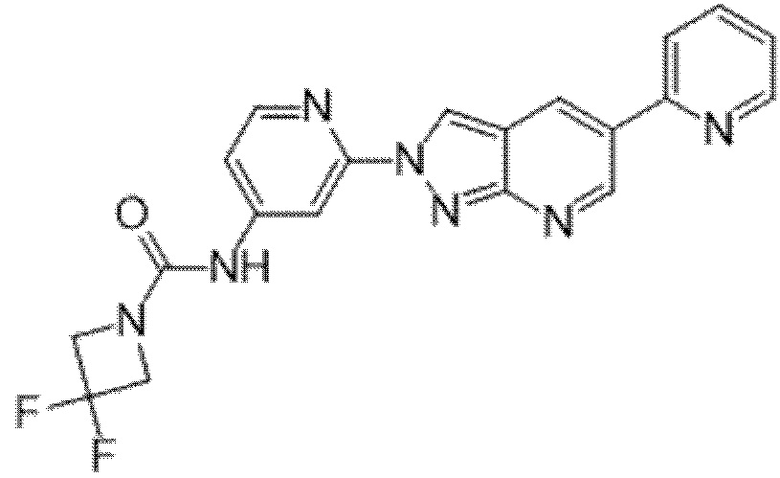
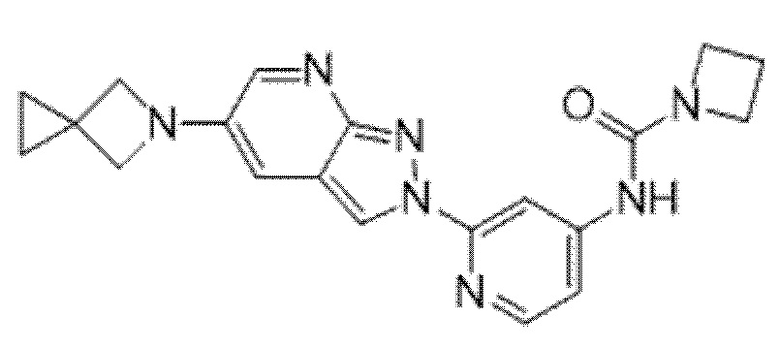
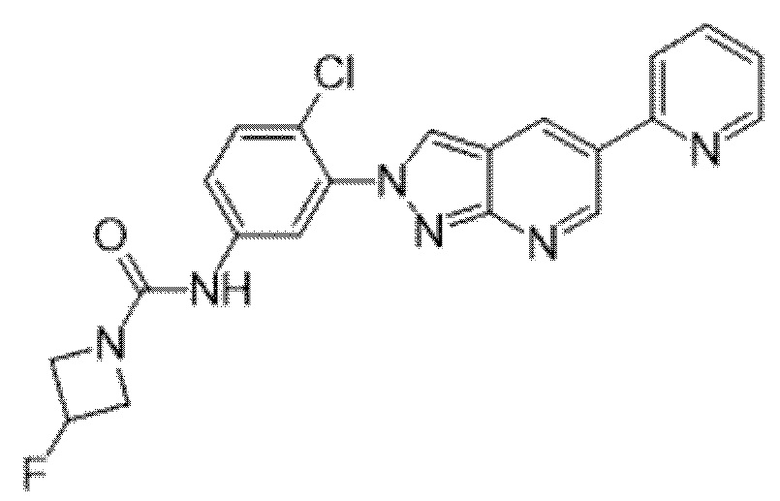
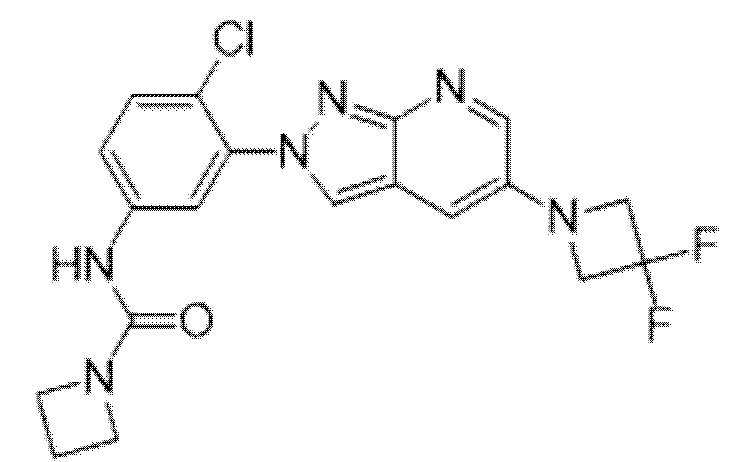
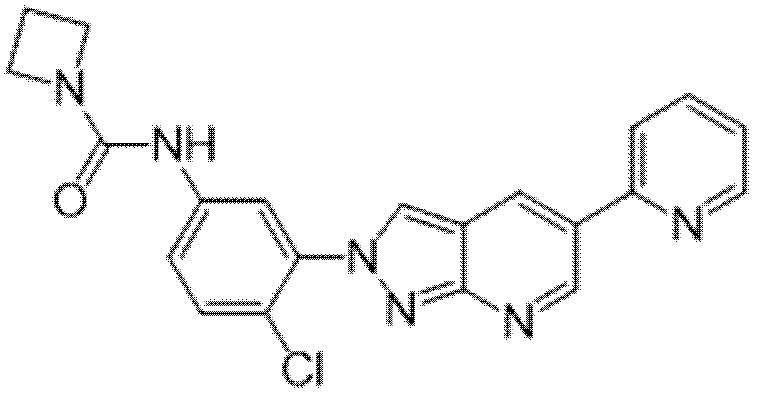
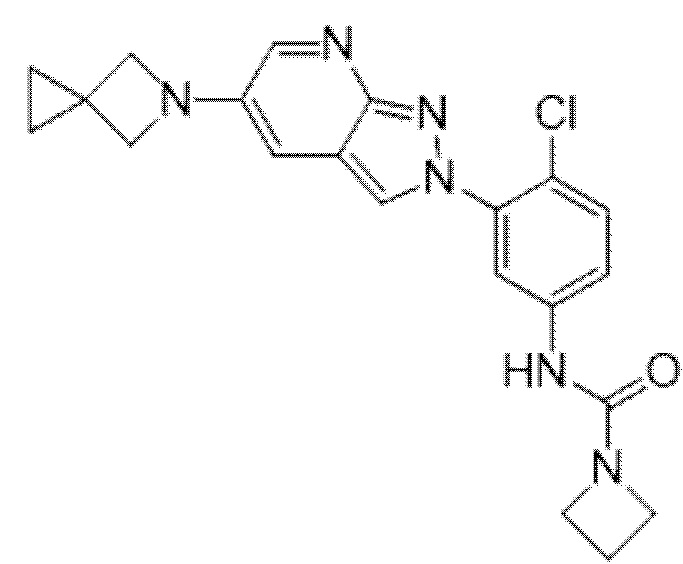
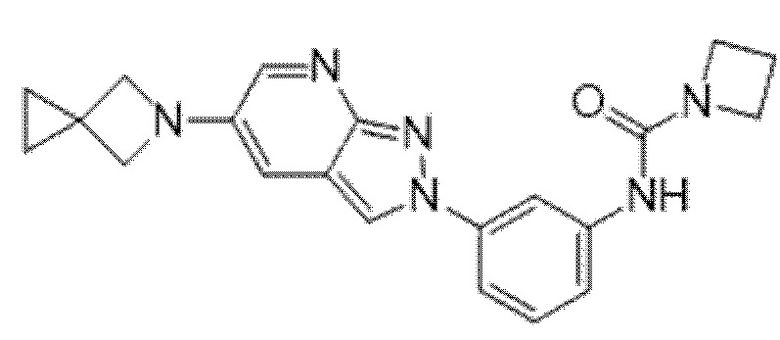
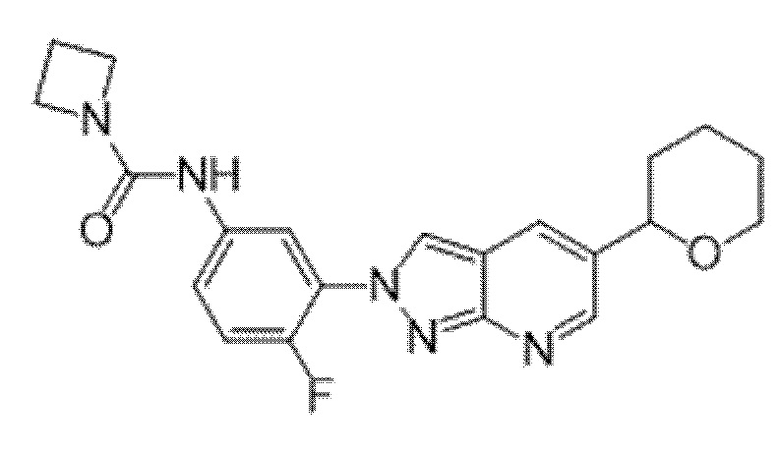
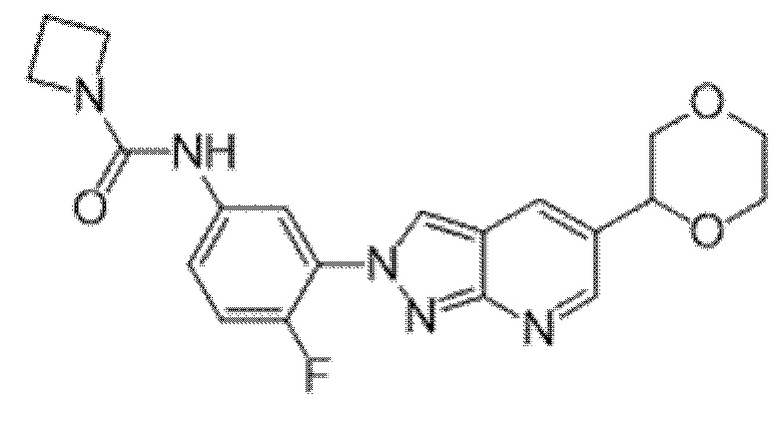
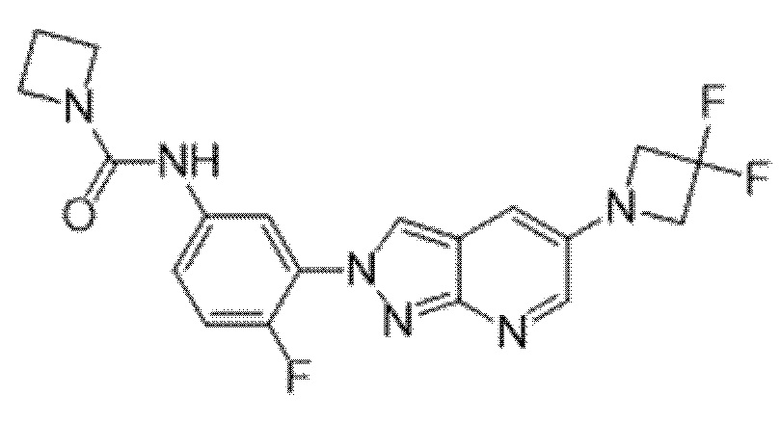
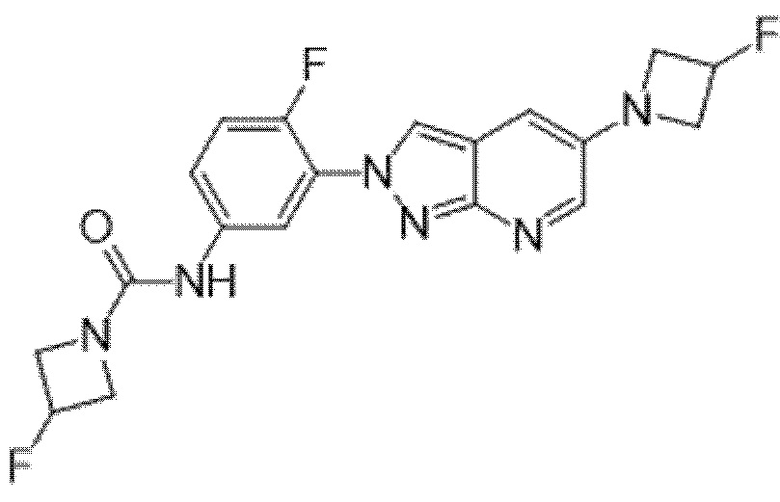
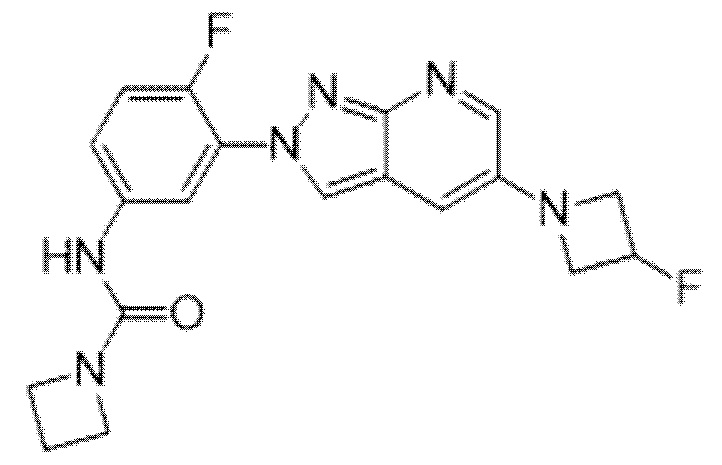
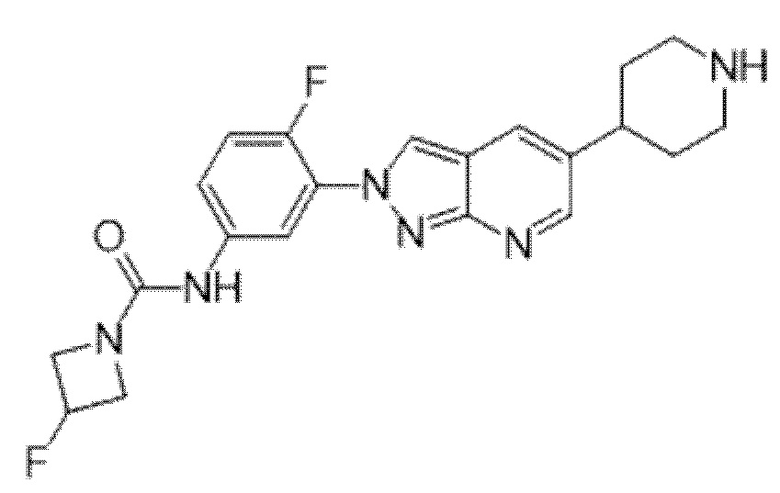
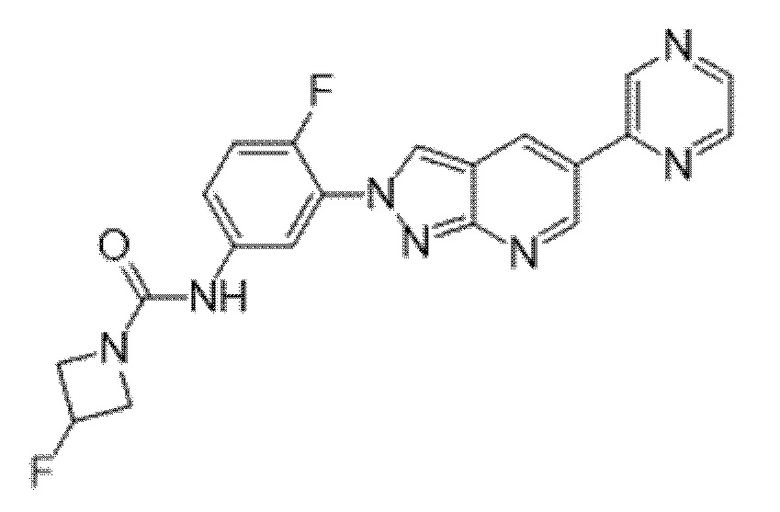
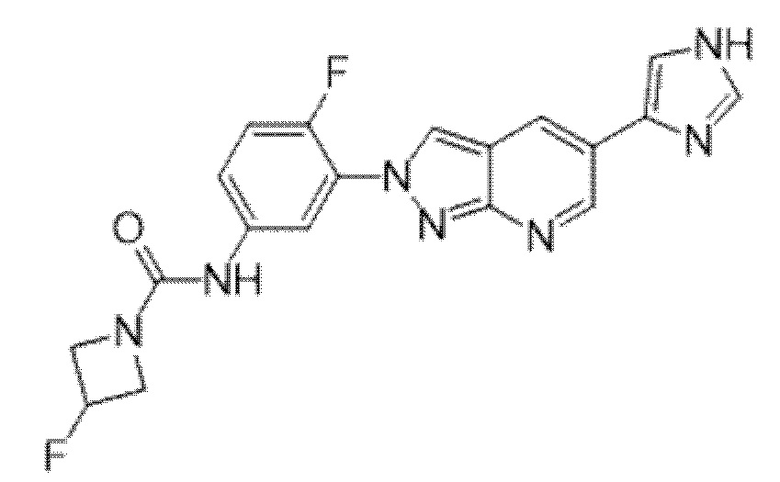
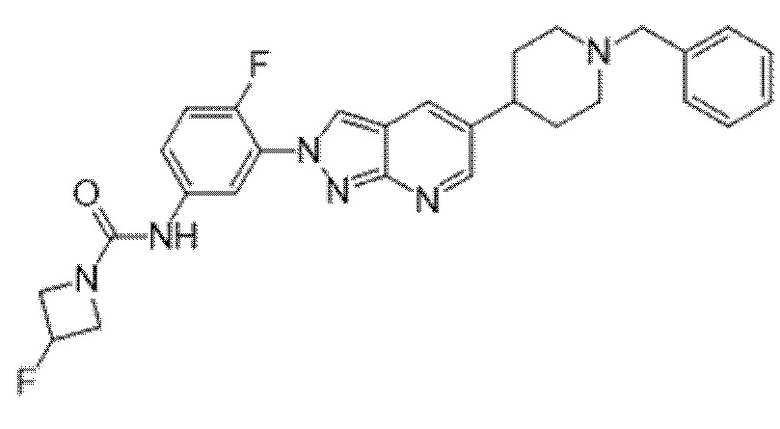
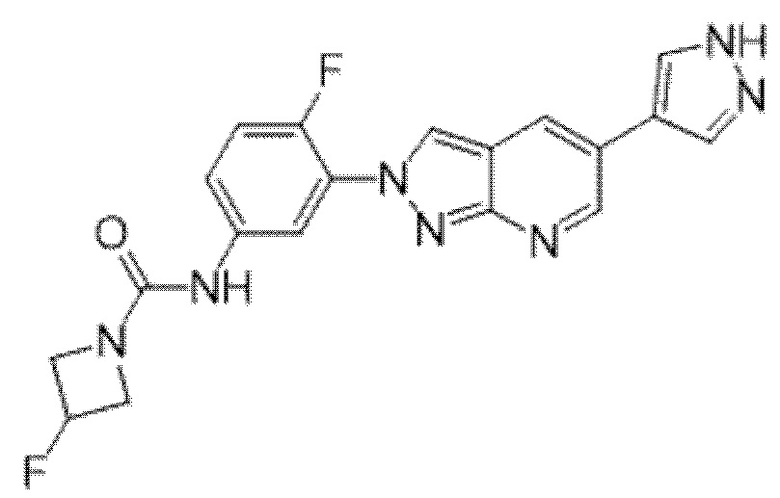
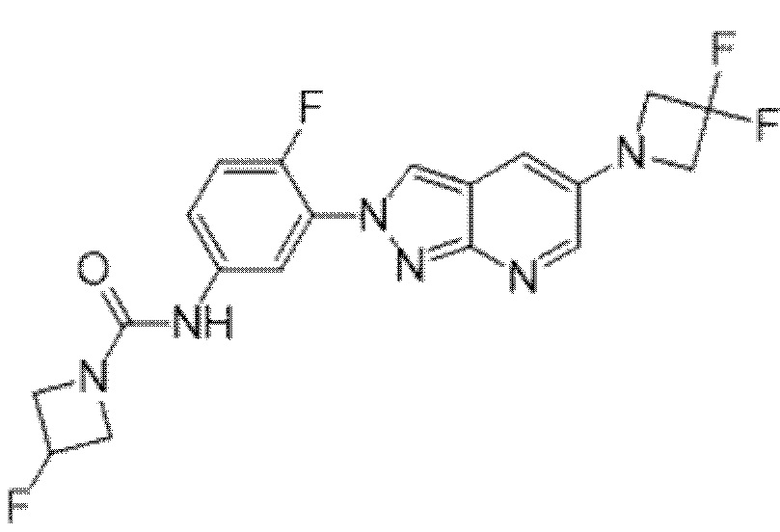
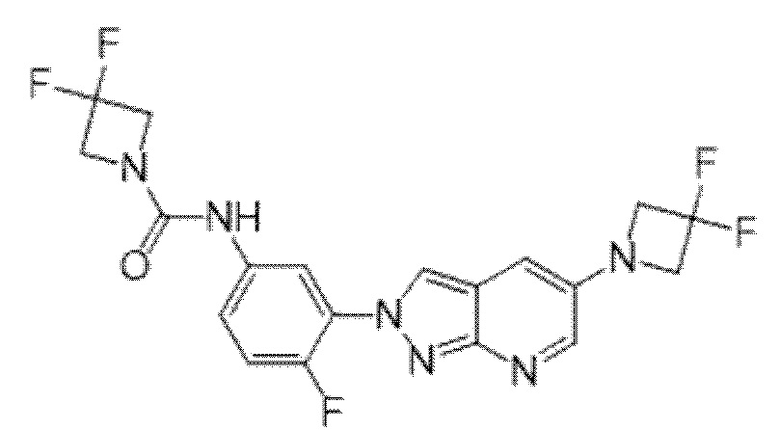
Способ 1: RT=2,90 мин.; M+H=439,2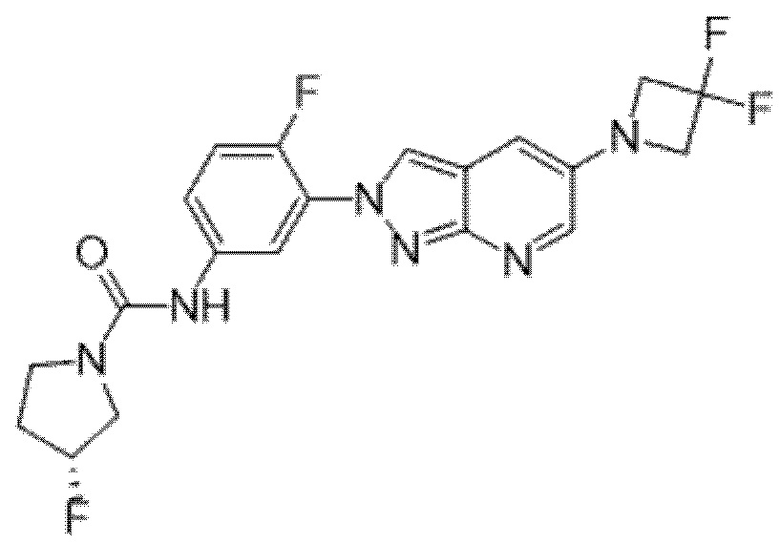
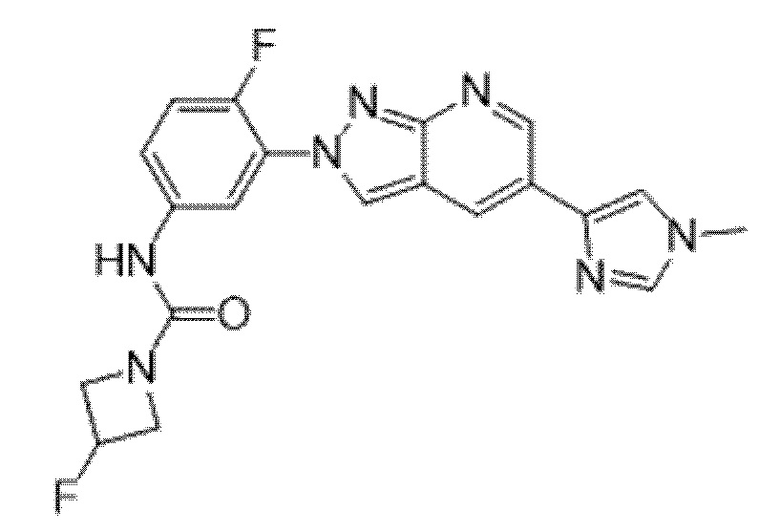
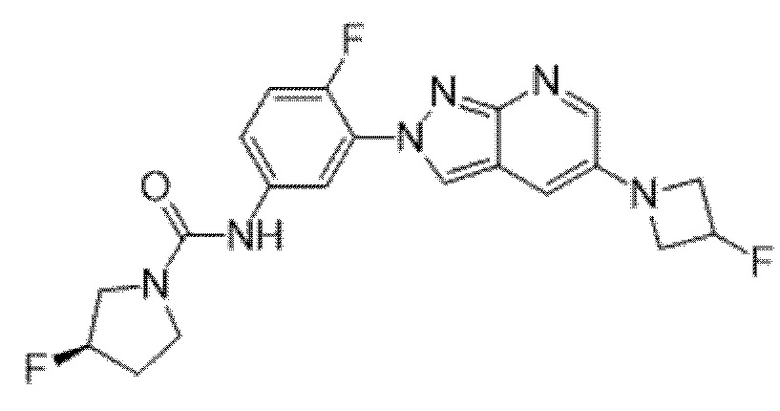
Способ 2: RT=0,83 мин.; M+H=417,1
Способ 1: RT=2,74 мин.; M+H=421,3
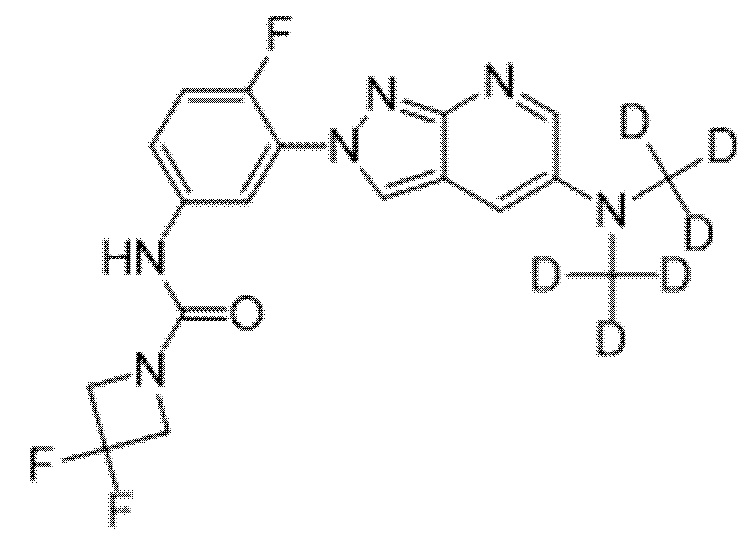
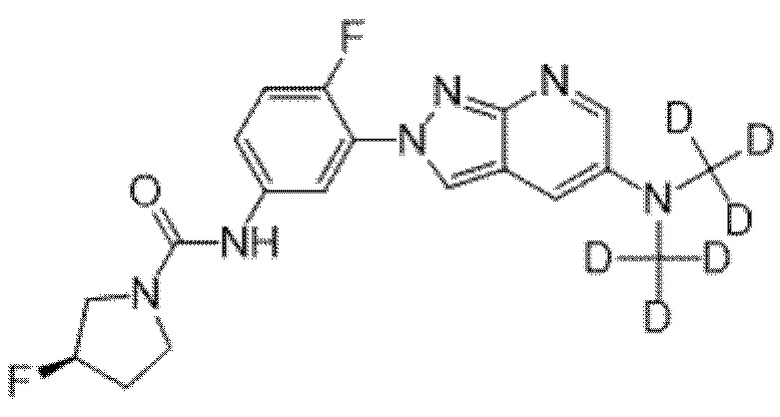
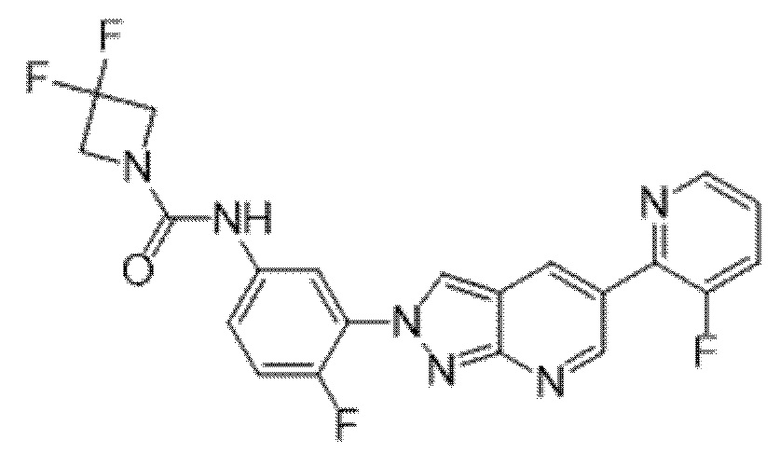
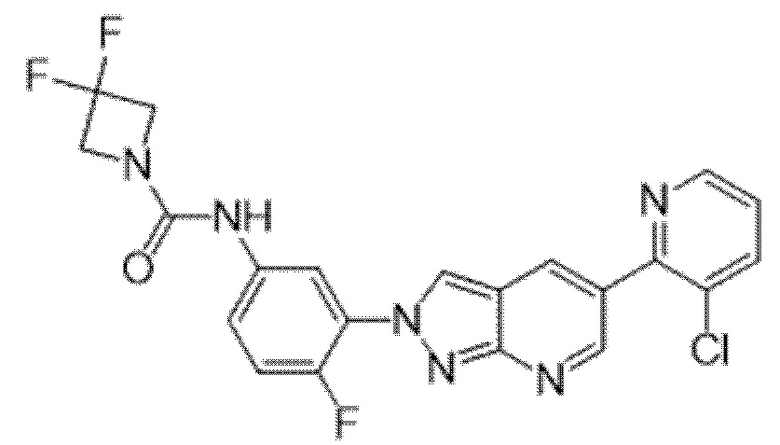
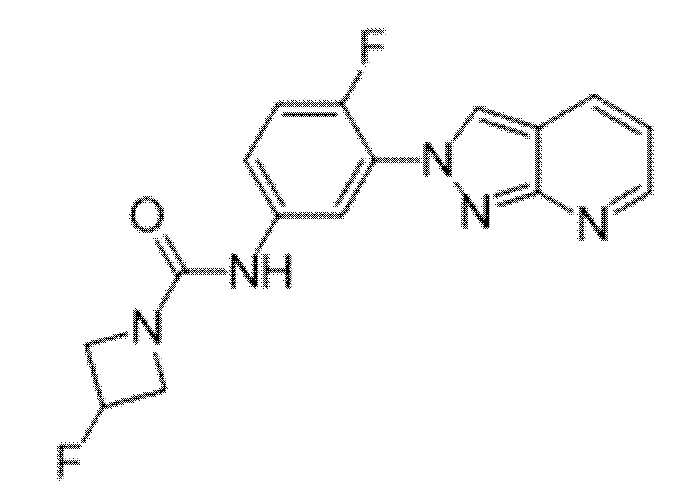
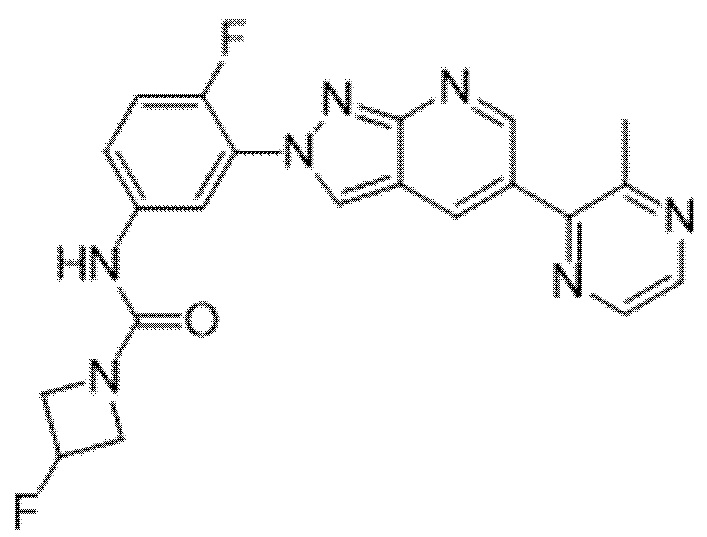
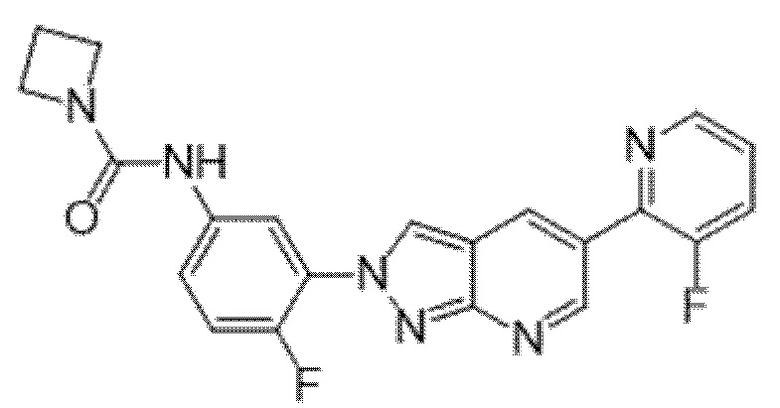
Способ 2: RT=0,85 мин.; M+H=407,1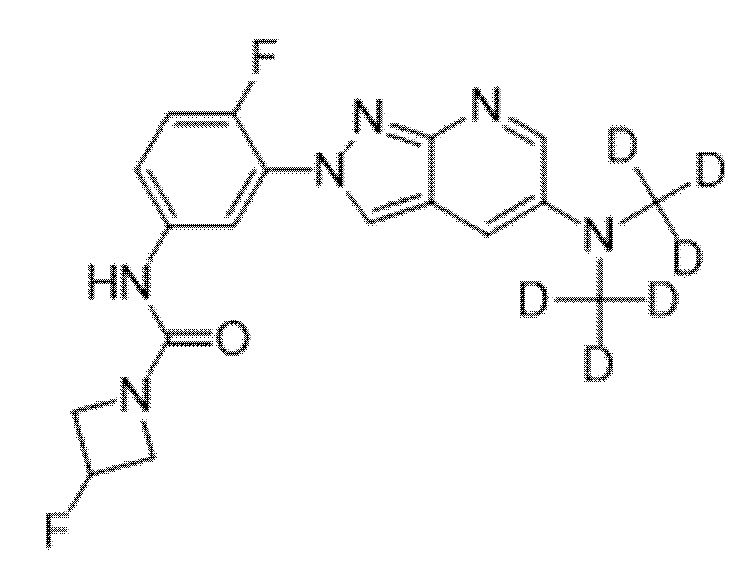
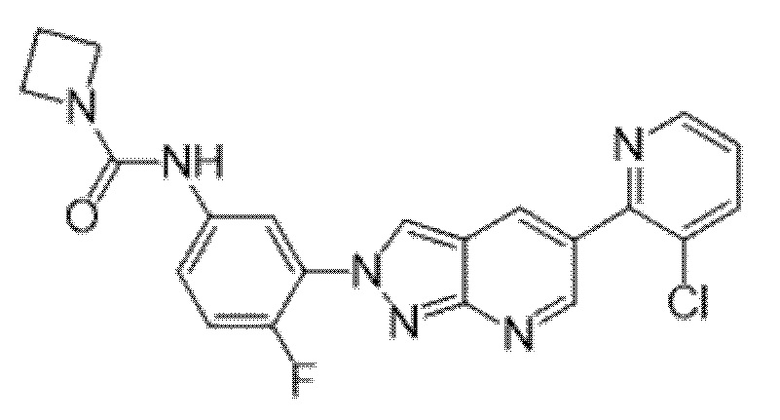
Способ 1: RT=2,73 мин.; M+H=423,1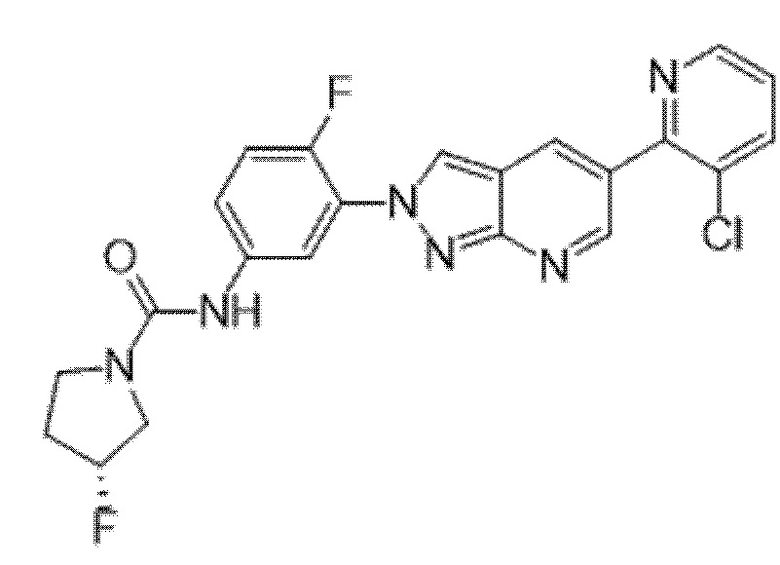
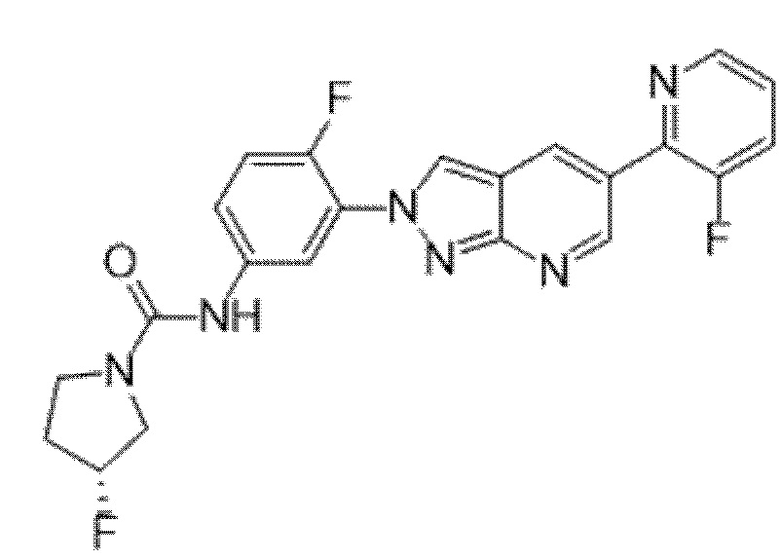
Способ 2: RT=0,87 мин.; M+H=439,1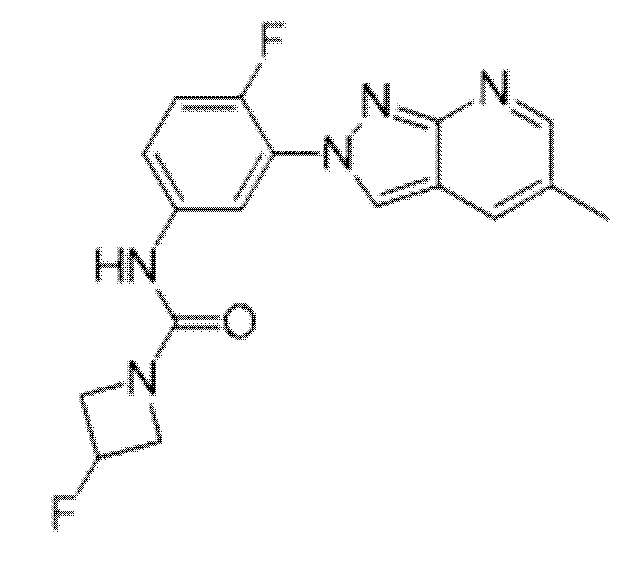
Способ 1: RT=2,62 мин.; M+H=344,1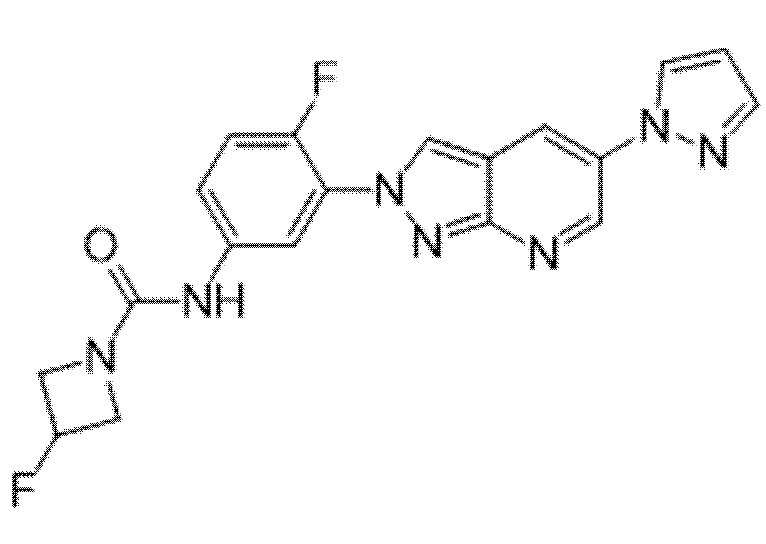
Способ 1: RT=2,59 мин.; M+H=396,1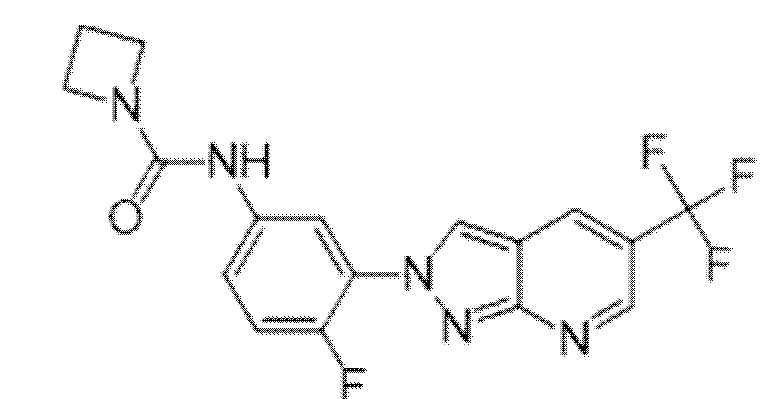
Способ 1: RT=2,82 мин.; M+H=380,1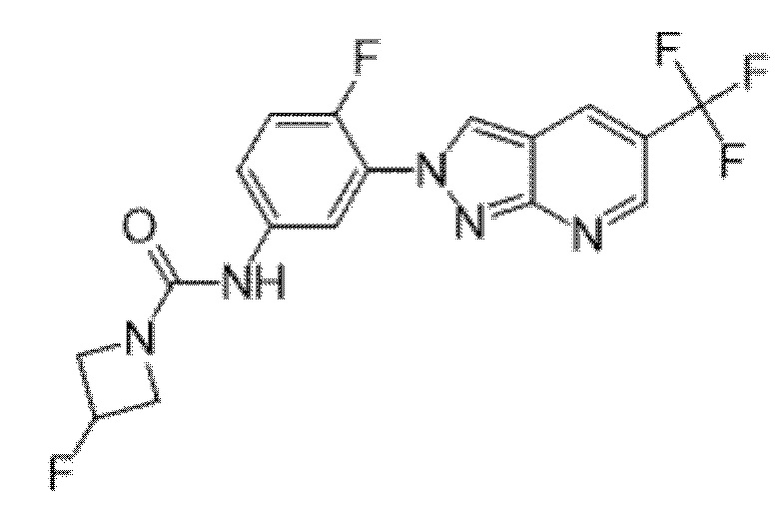
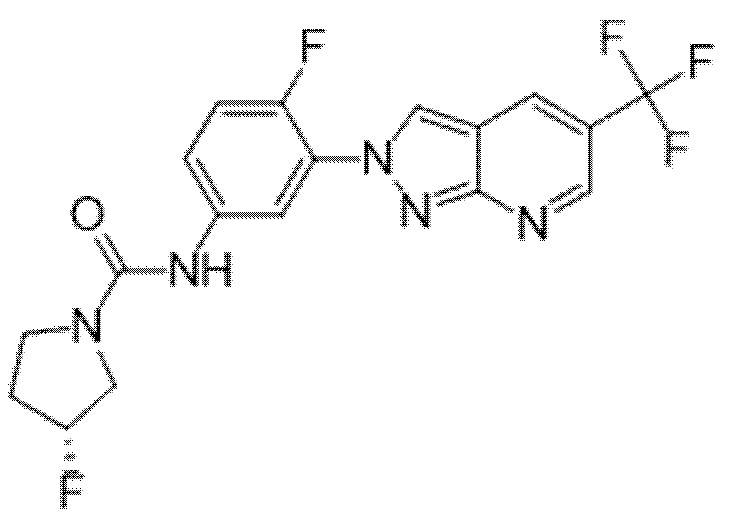
Способ 1: RT=2,83 мин.; M+H=412,1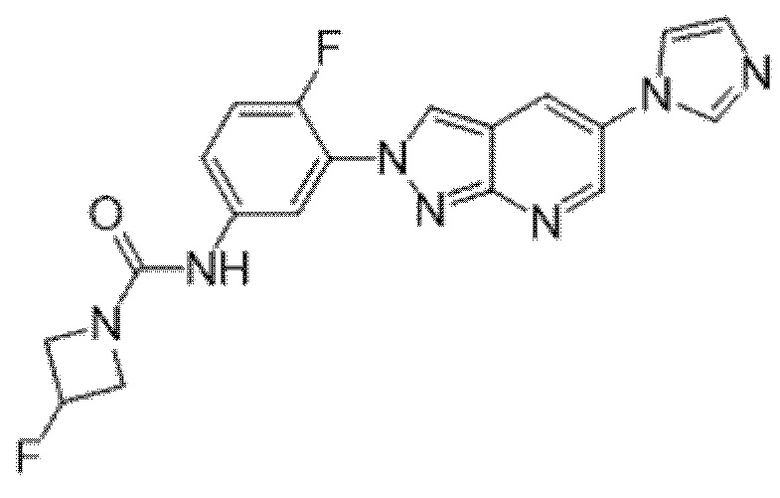
Способ 1: RT=1,83 мин.; M+H=396,1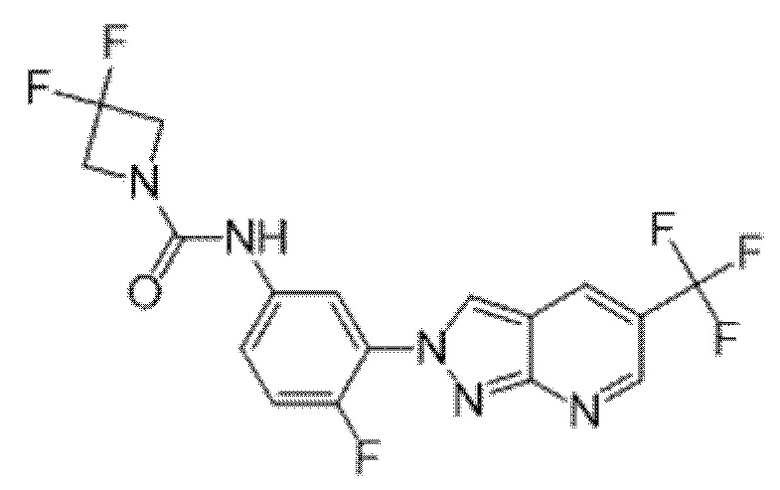
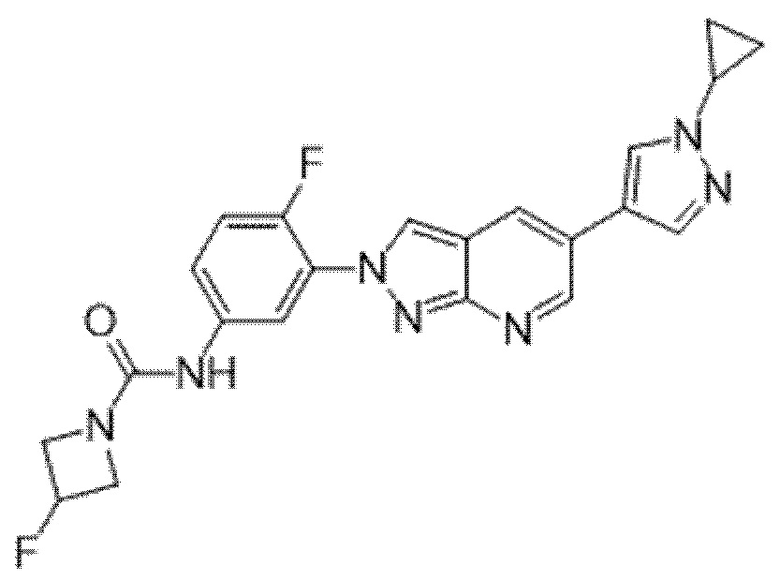
Способ 1: RT=2,73 мин.; M+H=436,1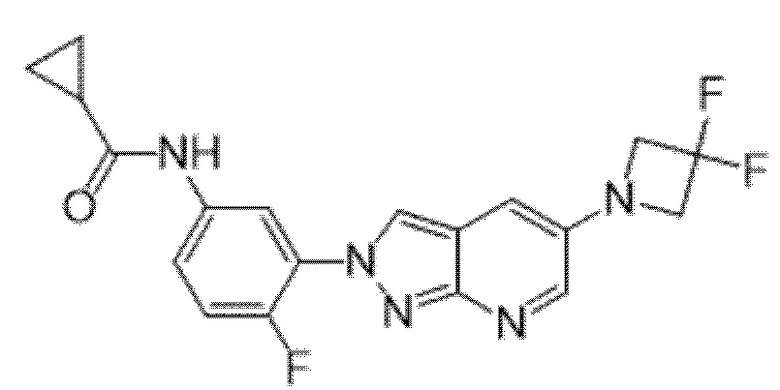
Способ 1: RT=1,87 мин.; M+H=410,1
Способ 2: RT=0,88 мин.; M+H=425,0
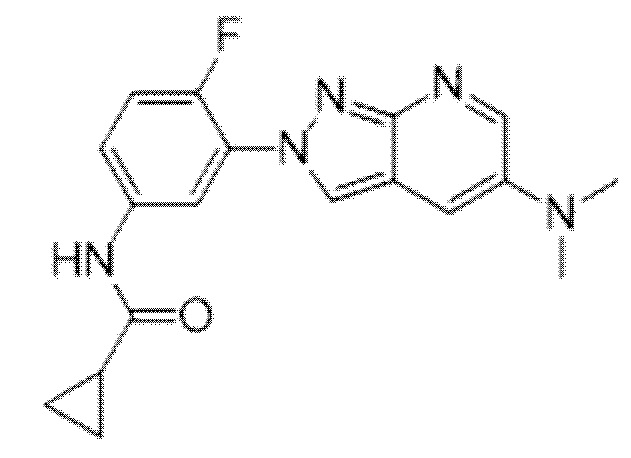
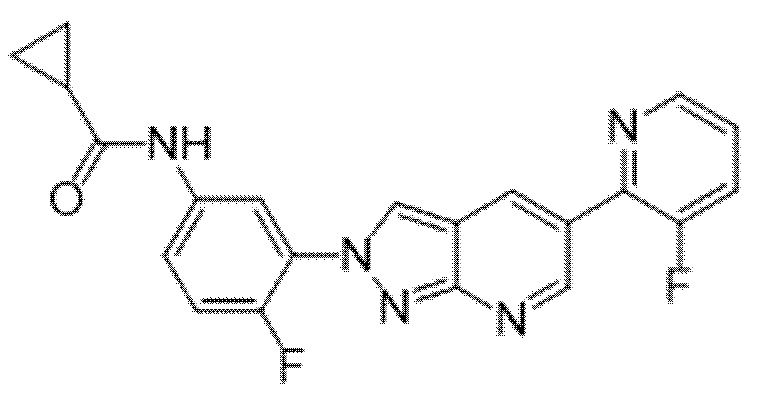
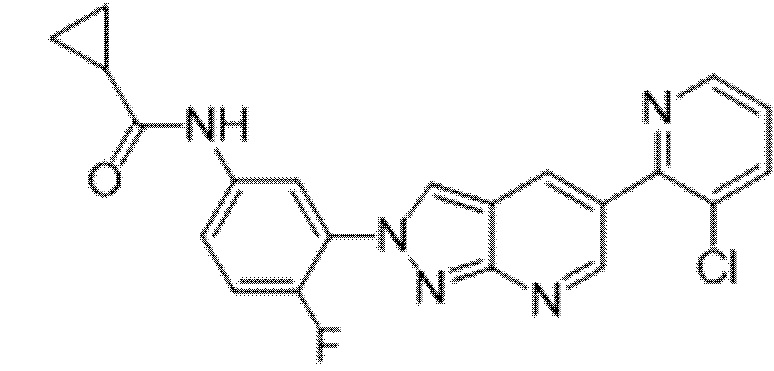
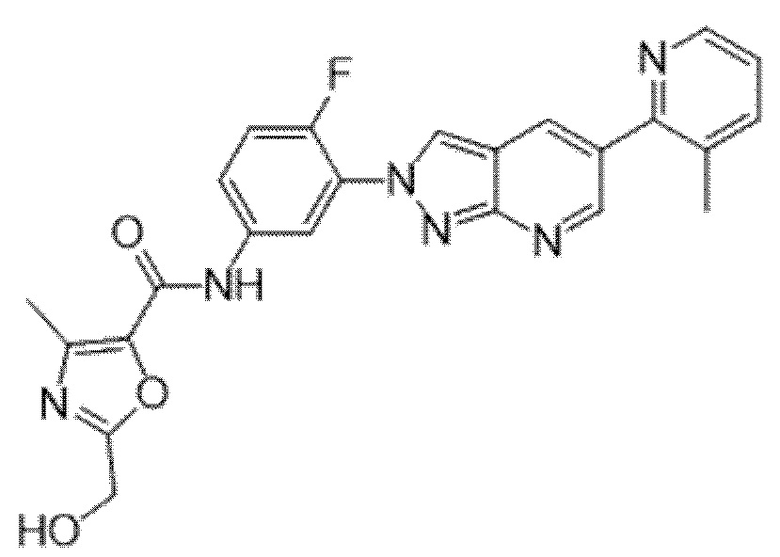
Способ 1: RT=2,57 мин.; M+H=459,0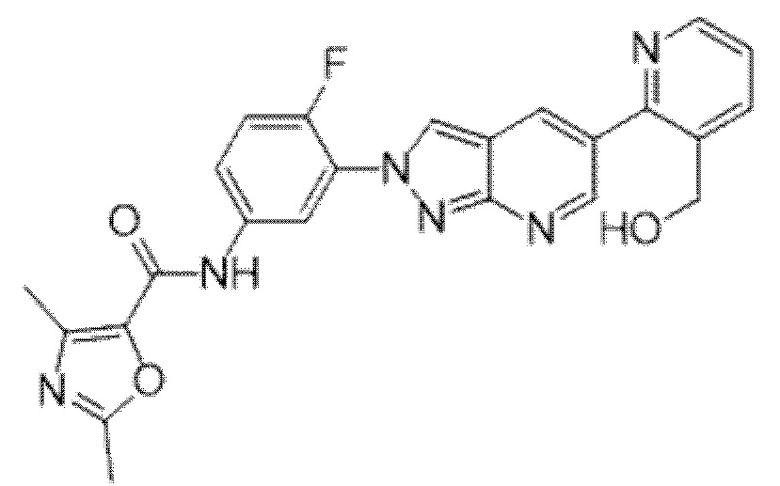
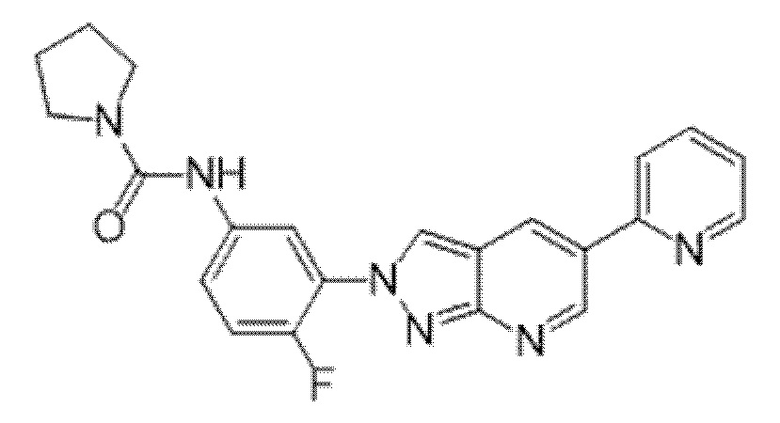
Способ 1: RT=2,67 мин.; M+H=403,3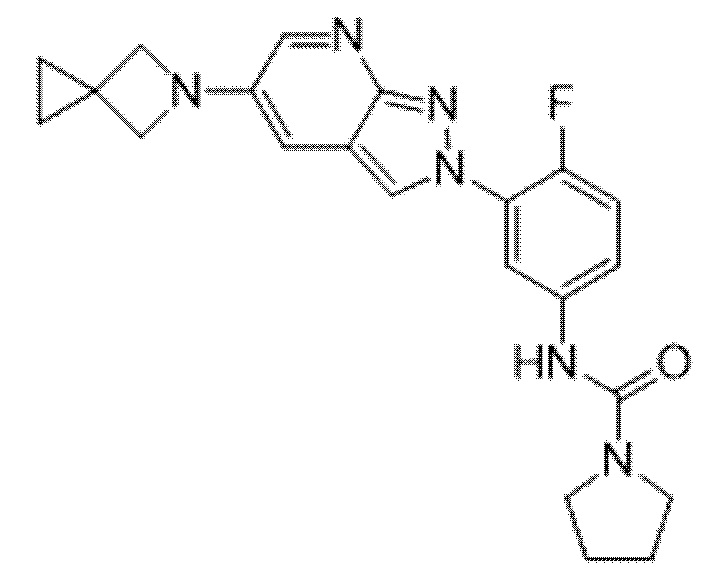
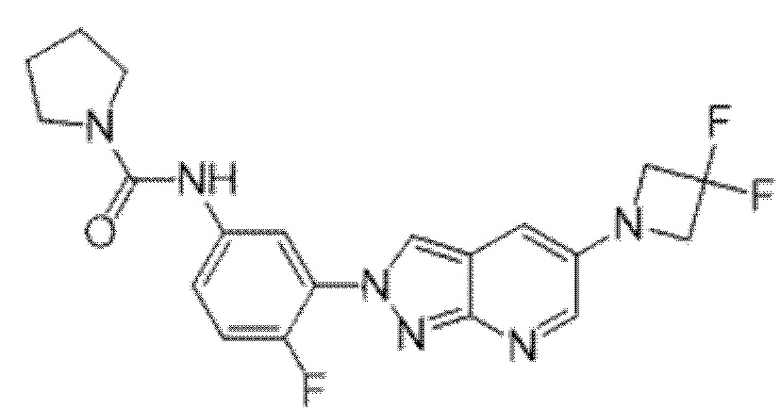
Способ 1: RT=2,86 мин.; M+H=417,0 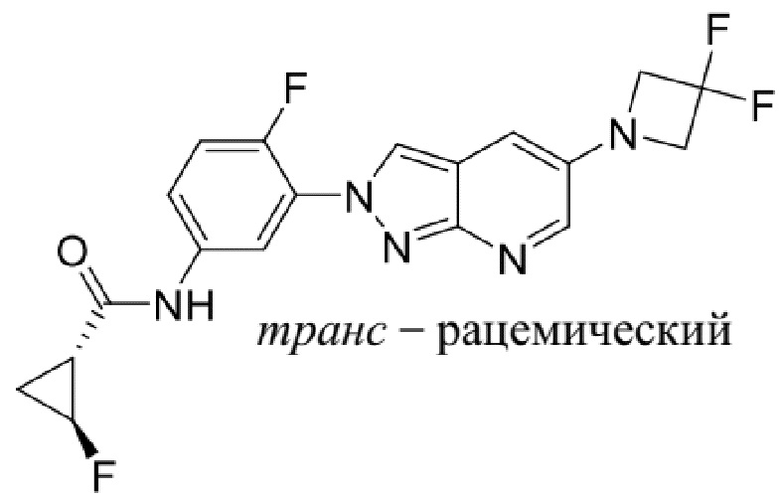
Способ 2: RT=0,97 мин.; M+H=406,2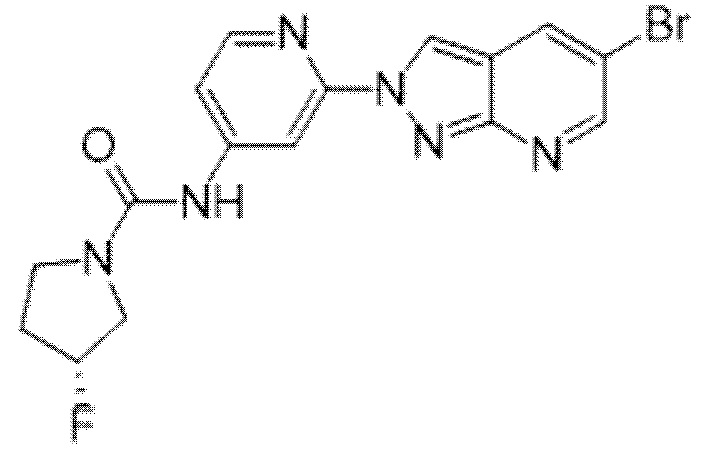
Способ 2: RT=0,90 мин.; M+H=405,1, 407,1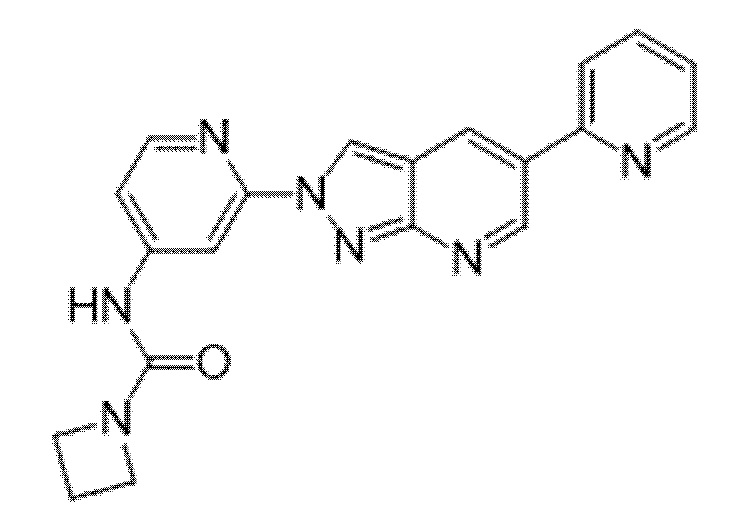
Способ 1: RT=2,67 мин.; M+H=372,0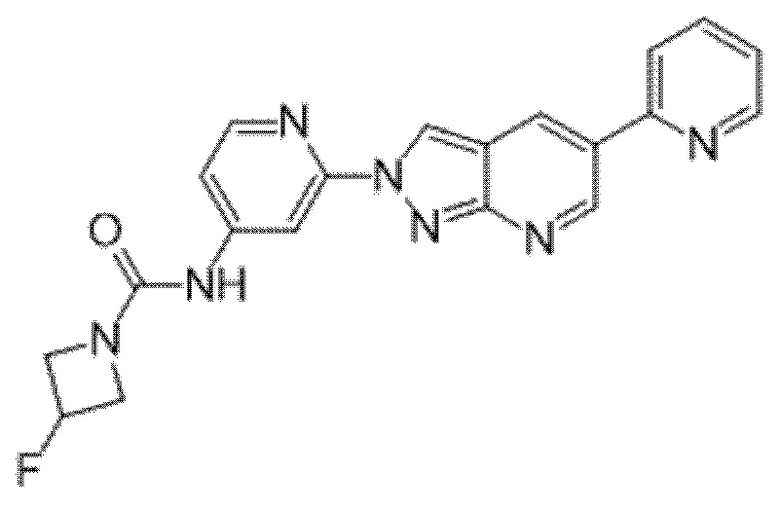
Способ 1: RT=2,67 мин.; M+H=390,0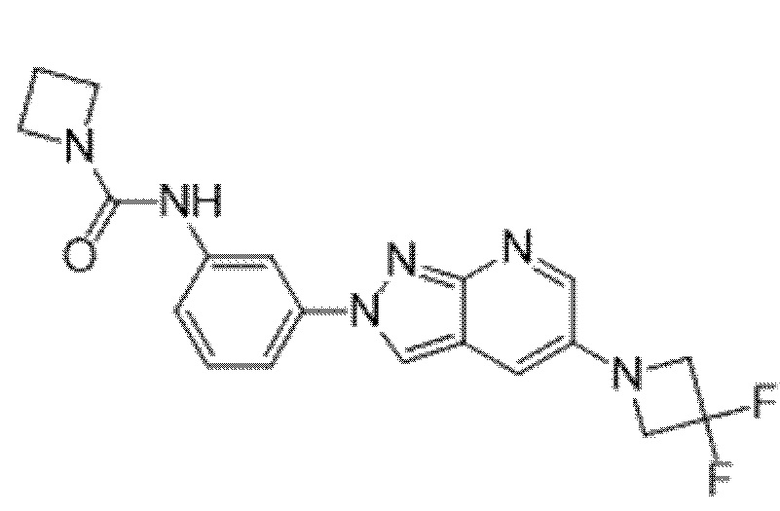
Способ 2: RT=0,84 мин.; M+H=385,2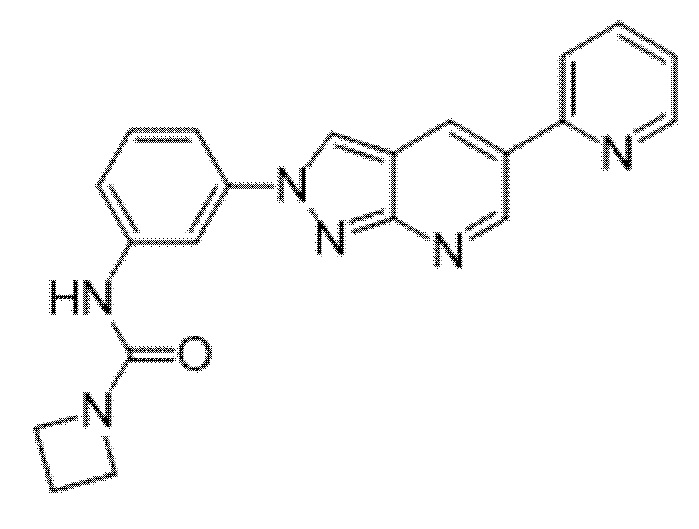
Способ 2: RT=0,74 мин.; M+H=371,0 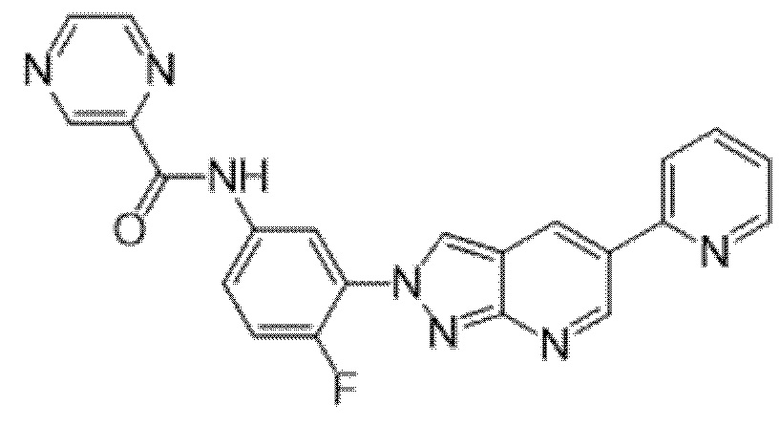
Способ 1: RT=2,71 мин.; M+H=412,10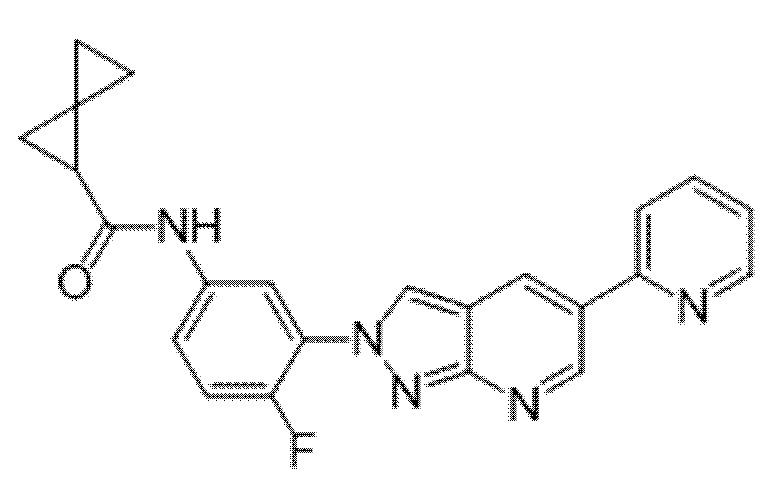
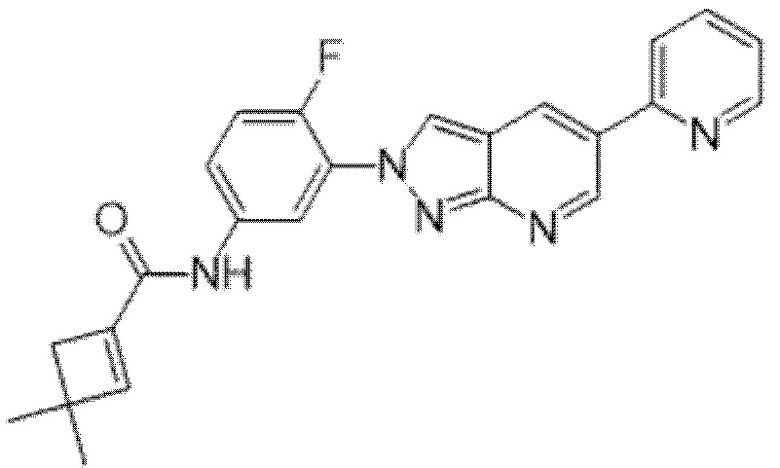
Способ 1: RT=3,08 мин.; M+H=414,1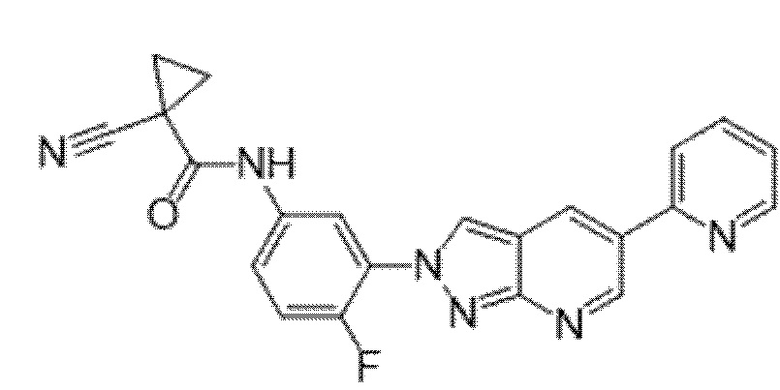
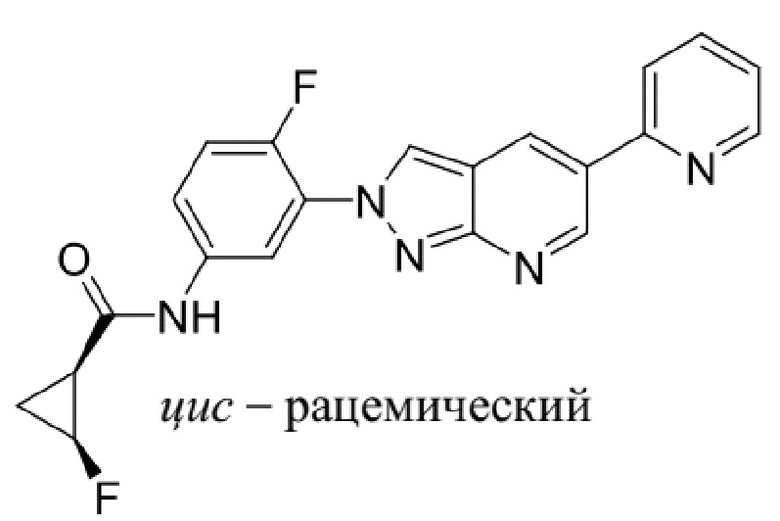
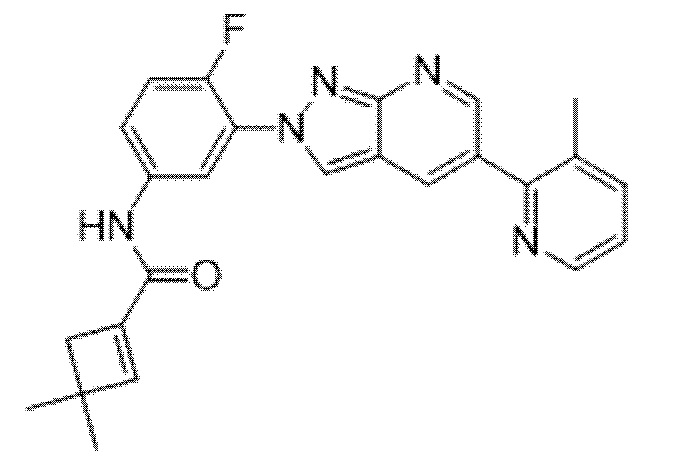
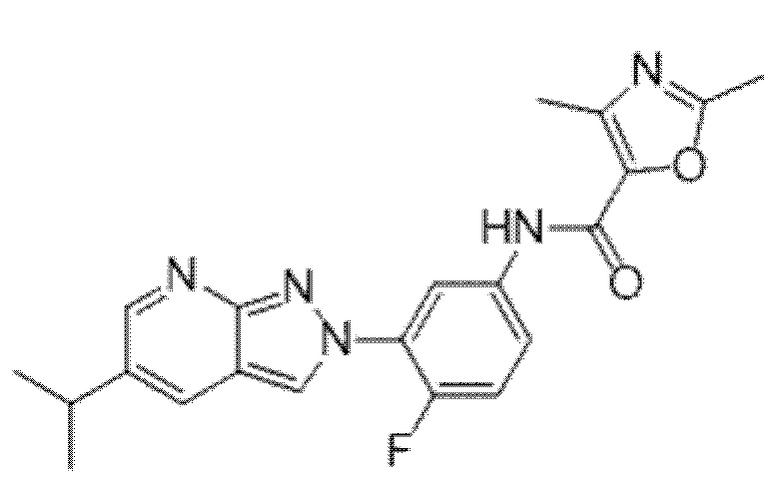
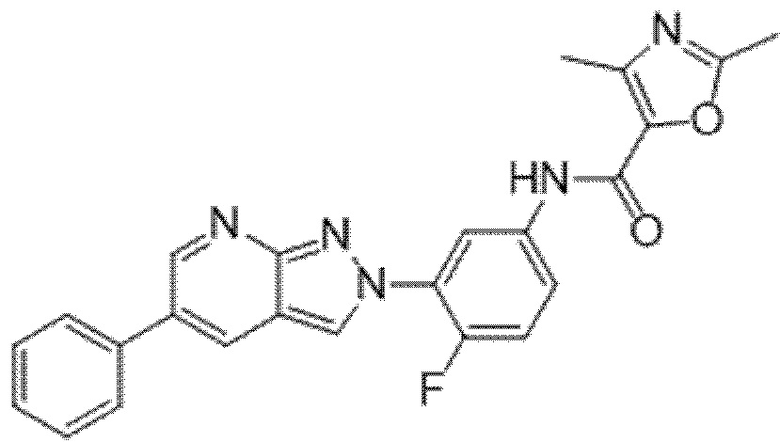
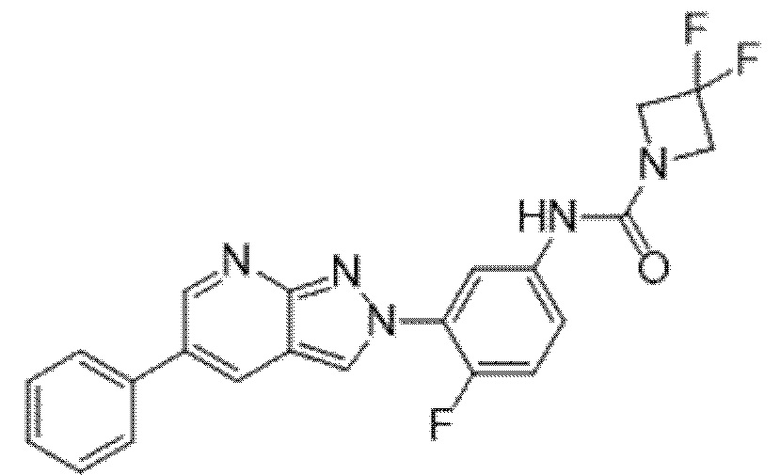
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Анализы, описанные в данном документе, иллюстрируют настоящее изобретение, но не ограничивают его объем.
Сокращения
Сокращения, используемые в данном документе ниже, имеют соответствующие значения:
Анализ in vitro подавления роста аксенической амастиготы Leishmania donovani
Аксеническую амастиготу паразитов Leishmania donovani выращивают при 37°C, 5% CO2 в среде, состоящей из RPMI 1640, 4 мМ L-глутамина, 20% термически инактивированной FBS, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина, 23 мкМ фолиевой кислоты, 100 мкМ аденозина, 22 мМ D-глюкозы, 25 мМ MES. Регулируют pH среды до 5,5 при 37°C с применением HCl. Сначала 20 мкл среды отмеривают в 384-луночные планшеты и в лунки планшетов добавляют 100 нл соединений по настоящему изобретению в DMSO. В то же время добавляют контрольные соединения и DMSO в планшеты, которые будут выполнять функцию положительного и отрицательного контролей соответственно. Затем в лунки планшетов добавляют 40 мкл культуры паразитов (9600 паразитов). Затем планшеты помещают в инкубаторы. Через два дня инкубации в лунки планшетов добавляют 20 мкл Cell TiterGlo® (Promega). Люминесцентный сигнал каждой лунки измеряют с применением ридера Envision (Perkin Elmer).
Анализ подавления паразитемии Leishmania donovani в макрофагах мышей
В анализе измеряют увеличение количества паразитов в анализируемой лунке планшета с применением красителя, включающегося в ДНК, - красителя SYBR Green I®, (INVITROGEN), для окрашивания ядер клеток Leishmania. Штамм L. donovani HU3 размножают путем инфицирования мышей линии BALB/c путем инъекции в хвостовую вену 107 паразитов Leishmania. Обеспечивали развитие у инфицированных мышей инфекции в течение 9-11 недель после инфицирования. В течение данного времени паразиты накапливаются в селезенке инфицированных мышей в большом количестве, и инфицированные мыши выполняют функцию источника паразитов для измерения in vitro эффективности соединений. Для анализа соединения в отношении антилейшманиозной активности перитонеальные макрофаги, выделенные из неинфицированных мышей линии BALB/c, высевают в 384-луночные планшеты при плотности 2 × 104 макрофагов на лунку в 25 мл среды (RPMI1640, 10% фетального сывороточного альбумина, 10 мМ HEPES, 1 мМ пирувата натрия, 1% Pen/Strep). Затем планшеты с высеянной культурой помещают в инкубатор, настроенный на поддержание температуры 37°C и атмосферы с 5% CO2.
На следующий день паразитов Leishmania выделяют из селезенок мышей, инфицированных в течение 9-11 недель, и добавляют 4 × 105 выделенных паразитов в 10 мл вышеуказанной среды в каждую лунку планшета. Затем планшеты возвращают в инкубаторы и обеспечивают развитие инфекции в течение 24 часов. После завершения инфицирования макрофагов добавляют 5 мл соединений по настоящему изобретению в вышеуказанной среде, которая также содержит 5% DMSO, в лунки планшетов, содержащие инфицированные макрофаги. В то же время добавляют контрольные соединения (милтефосин и амфотерицин B) и DMSO в планшеты, которые будут выполнять функцию положительного и отрицательного контролей соответственно. После добавления соединения планшеты возвращают в инкубатор и клетки, инфицированные паразитами, культивируют в течение 5 дней. После завершения культивирования добавляют 40 мл 8% параформальдегида в лунки планшетов и инкубируют в течение 15 мин. при комнатной температуре. После инкубации параформальдегид аспирируют из лунок планшетов и в лунки добавляют 40 мл PBS, содержащей 0,2% Triton X-100. Через 15 мин. инкубации раствор снова аспирируют из лунок и заменяют раствором красителя Sybr® Green в PBS (разбавление 1:125000). Инфицированные клетки визуализируют с помощью микроскопа для многопараметрического анализа Evotec Opera, и количество паразитов в лунке определяют путем подсчета ядер паразитов, которые являются видимыми в результате окрашивания красителем Sybr® Green.
Анализ подавления роста паразита класса кинетопластид Trypanosoma cruzi
Соединения по настоящему изобретению можно анализировать в отношении ингибирующей активности в отношении амастигот Trypanosoma cruzi, культивированных в клетках фибробластов 3T3. Анализ выполняют с применением безжгутиковой формы (амастигот) T. cruzi, которая реплицируется во внутриклеточном пространстве клеток-хозяев. Клетки-хозяева изначально инфицированы полученными из культуры трипомастиготами, которые быстро проникают в них и затем делятся в виде амастигот. Протокол предусматривает использование штамма Tulahuen T. cruzi, который был разработан для экспрессирования у E. coli гена бета-галактозидазы (Lac-Z) (Antimicr. Agents Chemoth. 40:2592, 1996). Это обеспечивает возможность считывания колориметрических данных путем применения субстрата CPRG и планшет-ридера для считывания поглощения.
Клетки фибробластов 3T3 повторно суспендируют в среде RPMI-1640 без фенолового красного, при этом среда дополнена 10% FBS (термически инактивированной), 100 мкг/мл пенициллина и 100 мкг/мл стрептомицина. Отмеривают сорок мкл суспензии (1000 клеток) в 384-луночные планшеты и инкубируют в течение ночи при температуре 37°C и в атмосфере, содержащей 5% CO2. На следующий день добавляют 100 нл соединений по настоящему изобретению в DMSO в лунки планшетов, содержащие клетки 3T3. В то же время добавляют контрольные соединения (бензнидазол и нифуртимокс) и DMSO в планшеты, которые будут выполнять функцию положительного и отрицательного контролей соответственно. После этого добавляют 10 мкл среды, содержащей 10000 трипомастигот T. cruzi, в каждую лунку планшета и планшеты помещают обратно в инкубаторы. Через 6 дней инкубации добавляют 10 мкл раствора реагентов (0,6 мМ CPRG, 0,6% NP-40 в PBS) в планшеты и инкубируют при комнатной температуре в течение 2 часов. Затем измеряют поглощение на флуориметре SpectraMax GEMINI с определением относительного количества клеток T. cruzi, присутствующих в каждой лунке планшета.
Анализ подавления роста в отношении Trypanosoma brucei brucei
Пролиферацию количественно определяют путем применения люминесцентного анализа жизнеспособности клеток Cell TiterGlo® (Promega®), с помощью которого измеряют количество жизнеспособных клеток в культуре на основе количественного определения количества клеточного ATP, который является показателем метаболически активных клеток.
Штамм Trypanosoma brucei brucei (Lister 427) выращивали в среде Hirumi 9 (HMI-9), дополненной 10% об./об. фетальной бычьей сыворотки (FBS) и 10% об./об. Serum Plus. Для измерения подавления пролиферации клеток тестируемые соединения последовательно разбавляли в три раза в двух повторностях в 384-луночные белые планшеты, в результате чего получали 10 разбавлений для каждого соединения. В каждую лунку добавляли культуру T. b. brucei в объеме 40 мкл (10000 паразитов/мл) и планшеты для анализа инкубировали при 37°C в течение 2 дней в инкубаторе с CO2. Подавление роста отслеживали путем измерения уровней ATP, который используют в качестве заменителя маркера роста. Относительные единицы люминесценции измеряли с применением TECAN® M1000 через 30 мин. после добавления 40 мкл Cell TiterGlo®. Значения IC50 определяли путем анализа данных с применением программного обеспечения HELIOS. IC50 определен как наименьшая концентрация соединения, которая обеспечивает 50% подавление роста штамма T. b. brucei дикого типа по сравнению с необработанными контролями.
В таблице 3 показана ингибирующая эффективность соединений (EC50) в отношении аксенических амастигот L. donovani in vitro (колонка 2); L. donovani в перитонеальных макрофагах мыши (колонка 3); T. cruzi (колонка 4) и T. brucei brucei (колонка 5). Соединения по настоящему изобретению характеризуются значением EC50, находящимся в диапазоне от >10 мкМ до <0,01 мкМ. В конкретных вариантах осуществления соединения по настоящему изобретению характеризуются EC50 <1 мкМ, <500 нМ, <100 нМ или < 50 нМ.
Таблица 3
EC50 (мкМ)
в макрофагах мыши
EC50 (мкМ)
T. cruzi
EC50 (мкМ)
EC50 (мкМ)
В таблице 4 сравнивается активность соединения формулы (I) с соединениями, имеющими различное ядро. Значения EC50 получены в анализах, описанных выше, если не указано иное.
Таблица 4
EC50 (нМ)
T. cruzi
EC50 (нМ)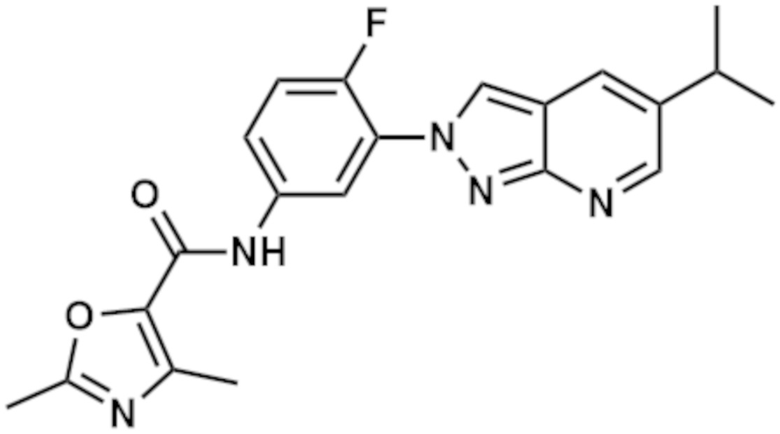
Следует понимать, что примеры и варианты осуществления, описанные в данном документе, предназначены только для иллюстративных целей, и что различные модификации или изменения в их свете будут предложены специалистам в данной области техники и должны быть включены в сущность и область действия настоящей заявки и объем прилагаемой формулы изобретения. Все публикации, патенты и заявки на патенты, цитируемые в данном документе, включены в данный документ с помощью ссылки для всех целей.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ПИРАЗОЛО[1,5-A]ПИРИМИДИНИЛКАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2017 |
|
RU2799332C2 |
| Гетероциклическое соединение в качестве ингибитора протеинкиназы | 2018 |
|
RU2783723C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ | 2016 |
|
RU2744766C2 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
Изобретение относится к соединению формулы (I) и его применению в изготовлении лекарственного препарата для лечения нарушений или заболеваний, выбранных из лейшманиоза, форм болезни Шагаса и африканского трипаносомоза человека. В формуле (I) кольцо A представляет собой фенил или пиридинил; R1 выбран из (a) C1-6алкила, который не замещен или замещен 1-3 заместителями, независимо выбранными из галогена и C3-6циклоалкила; (b) C1-4алкокси, который не замещен или замещен C1-4галогеналкилом; (c) -NR5aR5b, где R5a и R5b независимо представляют собой водород, C1-4алкил или C1-4галогеналкил или R5a и R5b вместе с атомом азота, к которому они оба присоединены, образуют 4-7-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, O и S; (d) моноциклического C3-6циклоалкила, C3-6циклоалкенила или спиропентила; каждый из которых не замещен или замещен 1-3 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила и C1-4алкокси; (e) фенила или 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S; каждый из которых не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкоксиC1-4алкила, C1-4гидроксиалкила, C1-4алкокси и C3-6циклоалкила; R2 и R7 независимо представляют собой водород или C1-4алкил; R3 представляет собой водород или галоген и n равняется 0 или 1; R4 выбран из (a) водорода; (b) галогена; (c) C1-6галогеналкила или C1-6алкила, который не замещен или замещен C3-6циклоалкилом; (d) -NR6aR6b, где R6a представляет собой водород или C1-4алкил; R6b представляет собой водород, C1-4алкоксикарбонил, C1-4галогеналкил, C3-6циклоалкил или C1-4алкил, который не замещен или замещен C1-4алкокси; или R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-7-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N, O и S; (e) C3-6циклоалкила; (f) 4-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S; при этом он не замещен или замещен -C(O)OR8, -C(O)R8, где R8 представляет собой C1-4алкил, и арилC1-4алкилом, который не замещен или замещен 1-2 заместителями, представляющими собой галоген; и (g) 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S; который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4гидроксиалкила и C3-6циклоалкила. Изобретение относится также к конкретным соединениям формулы (I). 3 н. и 15 з.п. ф-лы, 4 табл., 31 пр.
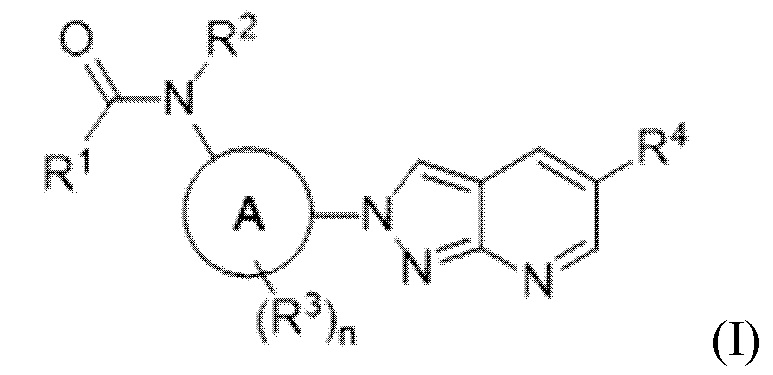
1. Соединение формулы (I):
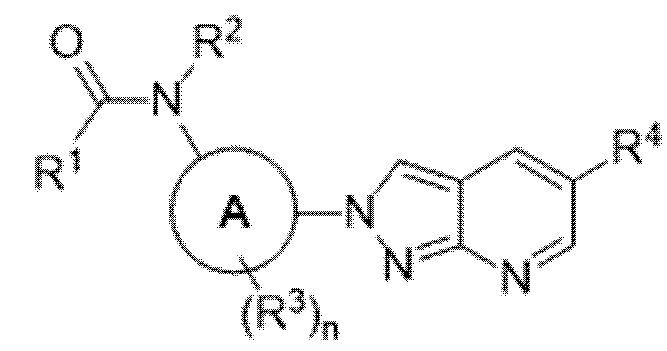 (I),
(I),
где
кольцо A представляет собой фенил или пиридинил;
R1 выбран из:
(a) C1-6алкила, который не замещен или замещен 1-3 заместителями, независимо выбранными из галогена и C3-6циклоалкила;
(b) C1-4алкокси, который не замещен или замещен C1-4галогеналкилом;
(c) -NR5aR5b, где R5a и R5b независимо представляют собой водород, C1-4алкил или C1-4галогеналкил или R5a и R5b вместе с атомом азота, к которому они оба присоединены, образуют 4-7-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-7-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, C1-4алкила и C1-4алкокси или два заместителя при одном и том же или различных атомах кольца 4-7-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо B, присоединенное к 4-7-членному гетероциклоалкилу;
где кольцо B представляет собой C3-6циклоалкил или 3-7-членный гетероциклоалкил, содержащий 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца;
(d) моноциклического C3-6циклоалкила, C3-6циклоалкенила или спиропентила; каждый из которых не замещен или замещен 1-3 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила и C1-4алкокси;
(e) фенила или 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца; каждый из которых не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, C1-4алкила, C1-4галогеналкила, ди-C1-4алкиламино-C1-4алкила, C1-4алкоксиC1-4алкила, C1-4гидроксиалкила, C1-4алкокси и C3-6циклоалкила;
R2 и R7 независимо представляют собой водород или C1-4алкил;
R3 представляет собой водород или галоген и n равняется 0 или 1;
R4 выбран из:
(a) водорода;
(b) галогена;
(c) C1-6галогеналкила или C1-6алкила, который не замещен или замещен C3-6циклоалкилом;
(d) -NR6aR6b, где R6a представляет собой водород или C1-4алкил; R6b представляет собой водород, C1-4алкоксикарбонил, C1-4галогеналкил, C3-6циклоалкил или C1-4алкил, который не замещен или замещен C1-4алкокси; или
R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-7-членный гетероциклоалкил, содержащий 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-7-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси, оксо, 1,1-диоксо, -C(O)-OR7 или 4-6-членного гетероциклоалкила, содержащего 1-2-гетероатома, независимо выбранных из N, O и S; или два заместителя при одном и том же или различных атомах кольца 4-7-членного гетероциклоалкила вместе с атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-7-членному гетероциклоалкилу;
где кольцо C выбрано из C3-6циклоалкила и 3-7-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и при этом оно независимо не замещено или замещено 1-2 заместителями, независимо выбранными из галогена и оксо;
(e) C3-6циклоалкила;
(f) 4-6-членного гетероциклоалкила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца; и при этом он не замещен или замещен -C(O)OR8, -C(O)R8, где R8 представляет собой C1-4алкил, и арилC1-4алкилом, который не замещен или замещен 1-2 заместителями, представляющими собой галоген; и
(g) 5-6-членного гетероарила, содержащего 1-2 гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца; который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, C1-4алкила, C1-4гидроксиалкила и C3-6циклоалкила.
2. Соединение по п. 1, где кольцо A представляет собой фенил.
3. Соединение по п. 1, где кольцо A представляет собой пиридинил.
4. Соединение по любому из пп. 1-3, где R1 выбран из -(CH2)1-3CF3, -(CH2)-CH(CH3)-CF3, -(CH2)-C(CH3)3, -O(CH2)2CF3, -(CH2)0-2-циклопропила, -(CH2)0-2-циклобутила, -NHCH3, -N(CH3)2, -N(CD3)2,
-N(CH3)(CH2CH3), -N(CH3)(CH2CF3),  ,
,  ,
, 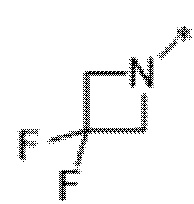 ,
,  ,
, 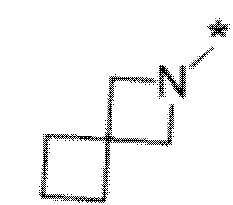 ,
,  ,
, 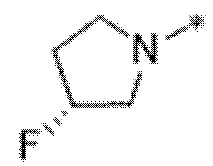 ,
, 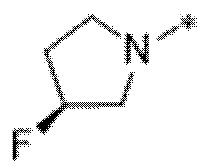 ,
, 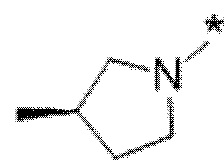 ,
, 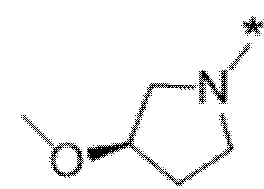 ,
,  ,
, 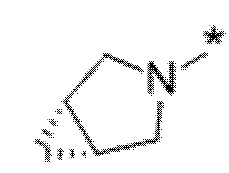 ,
,  ,
, 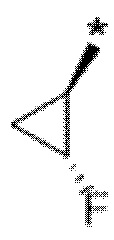 ,
, 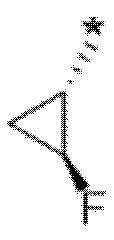 ,
,  ,
, 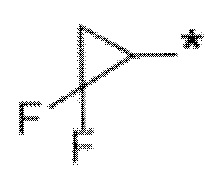 ,
, 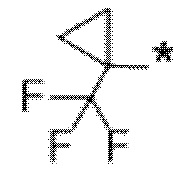 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 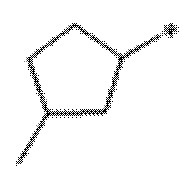 ,
, 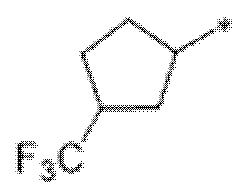 ,
, 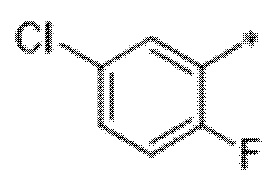 ,
, 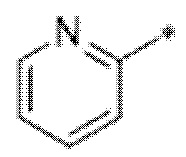 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 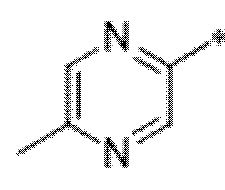 ,
, 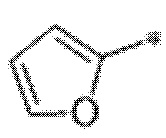 ,
, 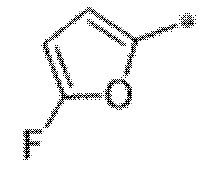 ,
, 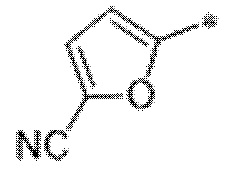 ,
,  ,
,  ,
,  ,
,  ,
, 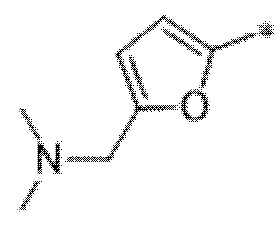 ,
, 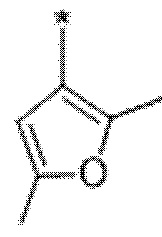 ,
, 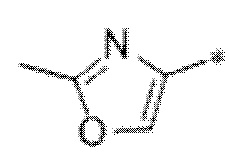 ,
, 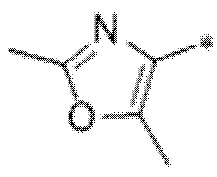 ,
, 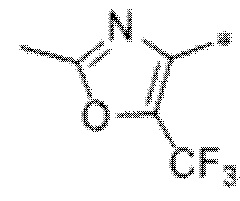 ,
, 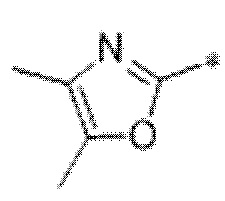 ,
, 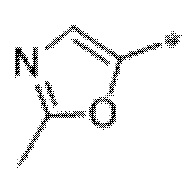 ,
, 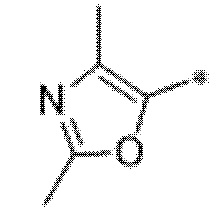 ,
,  ,
,  ,
,  ,
,  ,
, 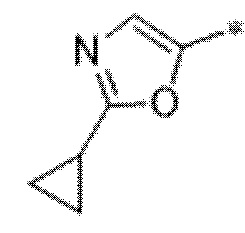 ,
, 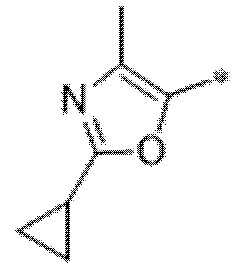 ,
,  ,
,  и
и  .
.
5. Соединение по любому из пп. 1-3, где R1 представляет собой азетидинил, который не замещен или замещен 1-2 заместителями, независимо выбранными из галогена и C1-4алкила; или два заместителя при одном и том же атоме кольца азетидинила вместе с атомом кольца, к которому они оба присоединены, образуют спироциклопропил или спиротетрагидрофуранил, присоединенный к азетидинильному кольцу.
6. Соединение по любому из пп. 1-3, где R1 выбран из
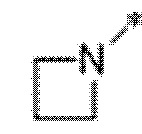 ,
, 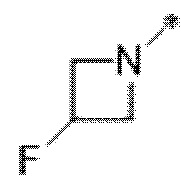 и
и 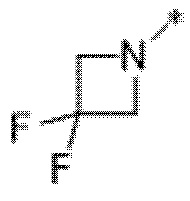 .
.
7. Соединение по любому из пп. 1-6, где R3 представляет собой галоген и n равняется 1.
8. Соединение по п. 7, где R3 представляет собой фтор.
9. Соединение по любому из пп. 1-8, где R4 выбран из водорода, хлора, брома, метила, изопропила, -(CH2)1-2CH(CH3)2, -(CH2)0-1C(CH3)3, -C(CH3)2CH2CH3, -CH(CH3)(CH2)1-2CH3, -CH2-циклобутила, -(CH2)0-1CF3, -NH-(CH2)0-1CH3, -N-(CD3)2, -N(CH3)2, -NH-CH-(CH3)2, -NH-(CH2)-CH-(CH3)2,
-NHC(O)OCH(CH3)2, -NH(CH2)2OCH3, 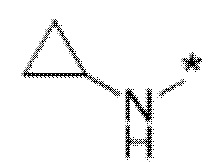 ,
, 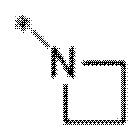 ,
, 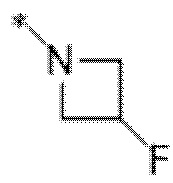 ,
, 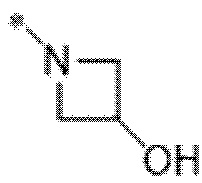 ,
,  ,
, 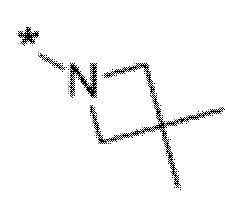 ,
, 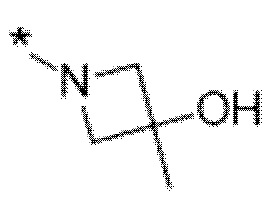 ,
, 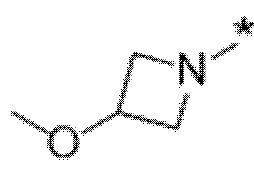 ,
, 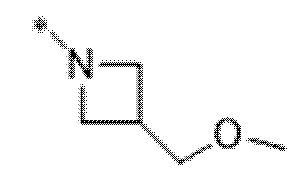 ,
, 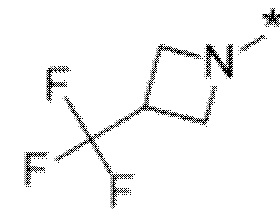 ,
, 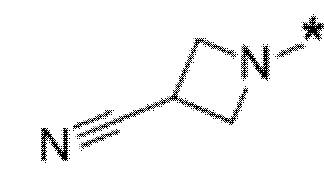 ,
, 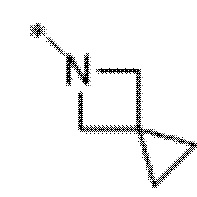 ,
, 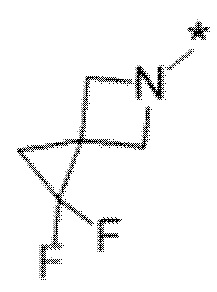 ,
, 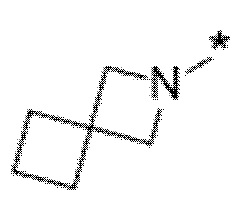 ,
, 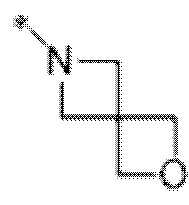 ,
, 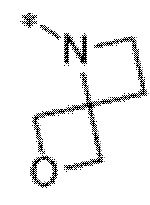 ,
,  ,
, 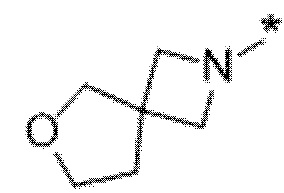 ,
,  ,
, 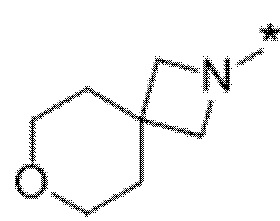 ,
, 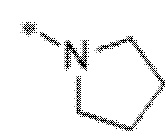 ,
, 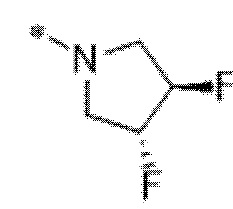 ,
,  ,
,  ,
, 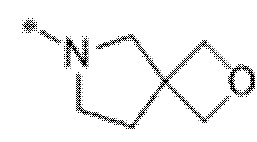 ,
, 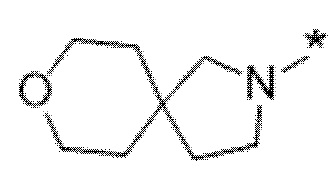 ,
,  ,
,  ,
, 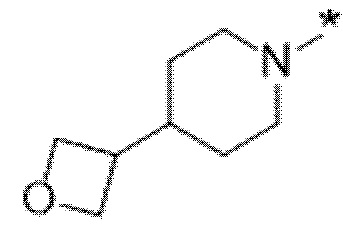 ,
, 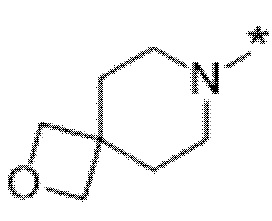 ,
,  ,
,  ,
, 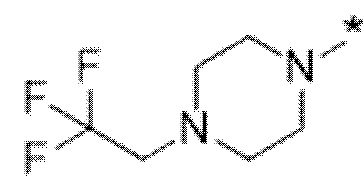 ,
, 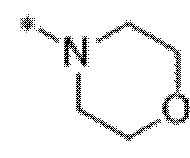 ,
, 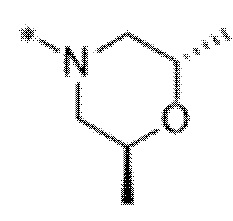 ,
, 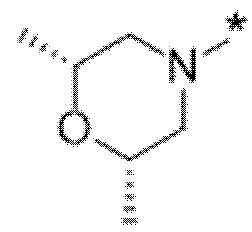 ,
, 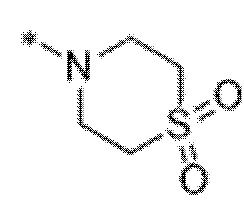 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 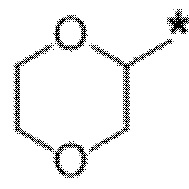 ,
, 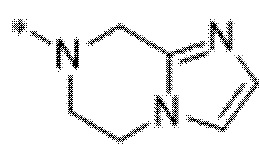 ,
, 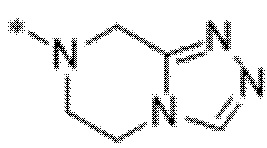 ,
,  ,
, 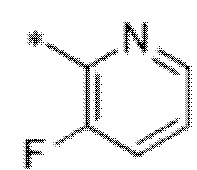 ,
, 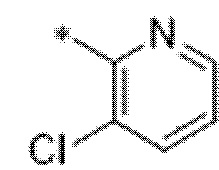 ,
, 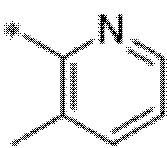 ,
, 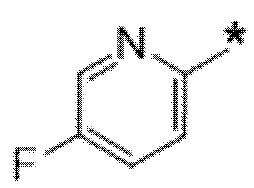 ,
, 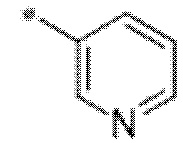 ,
, 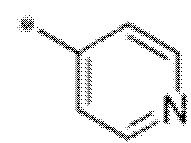 ,
, 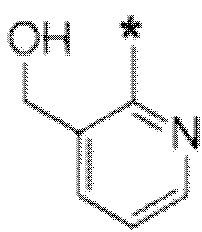 ,
, 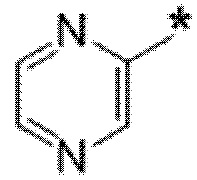 ,
, 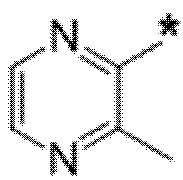 ,
, 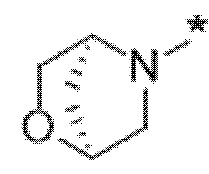 ,
, 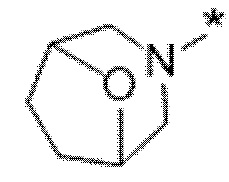 ,
, 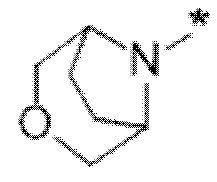 ,
, 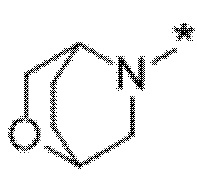 ,
, 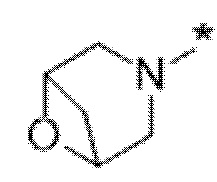 ,
,  ,
,  ,
, 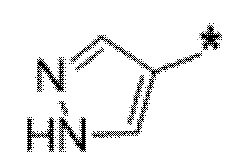 ,
, 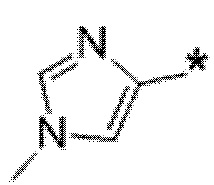 ,
, 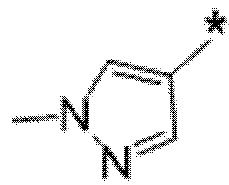 ,
, 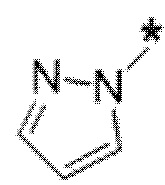 ,
, 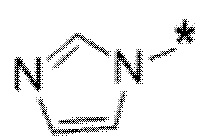 ,
, 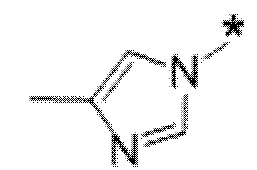 и
и 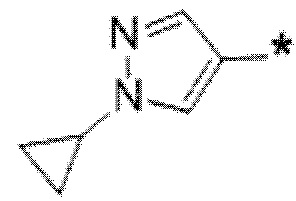 .
.
10. Соединение по любому из пп. 1-8, где R4 представляет собой -NR6aR6b;
R6a представляет собой водород или C1-4алкил;
R6b представляет собой водород, C1-4алкоксикарбонил или C1-4алкил, который не замещен или замещен C1-4алкокси; или R6a и R6b вместе с атомом азота, к которому они оба присоединены, образуют 4-6-членный гетероциклоалкил, необязательно содержащий 1-2 дополнительных гетероатома, независимо выбранных из N, O и S, в качестве атомов кольца;
при этом 4-6-членный гетероциклоалкил не замещен или замещен 1-2 заместителями, независимо выбранными из галогена, циано, гидроксила, C1-4алкила, C1-4галогеналкила, C1-4алкоксиC1-4алкила, C1-4алкокси и оксо; или два заместителя при одном и том же или различных атомах кольца 4-6-членного гетероциклоалкила вместе атомами, к которым они присоединены, образуют спиро-, мостиковое или конденсированное кольцо C, присоединенное к 4-6-членному гетероциклоалкилу;
где кольцо C выбрано из C3-6циклоалкила и 3-5-членного гетероциклоалкила, содержащего 1-3 гетероатома, независимо выбранных из N, O или S, в качестве атомов кольца; и при этом кольцо C независимо не замещено или замещено 1-2 заместителями, независимо выбранными из галогена и оксо.
11. Соединение по любому из пп. 1-8, где R4 выбран из
 ,
, 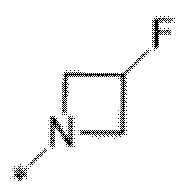 ,
, 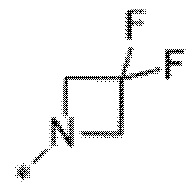 ,
, 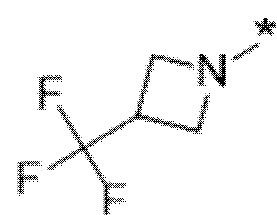 и
и 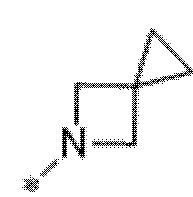 .
.
12. Соединение по любому из пп. 1-8, где R4 выбран из 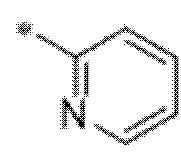 ,
, 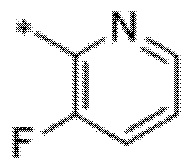 ,
, 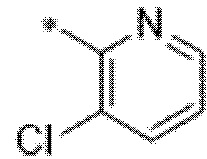 ,
, 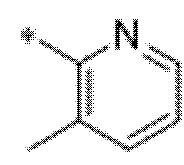 ,
,  и
и  .
.
13. Соединение, выбранное из следующих:
14. Соединение по п. 1, выбранное из следующих:
N–(3–(5–бром–2H–пиразоло[3,4–b]пиридин–2–ил)–4–фторфенил)–3–фторазетидин–1–карбоксамид (I-4),
N–(3–(5–бром–2H–пиразоло[3,4–b]пиридин–2–ил)–4–фторфенил)азетидин–1–карбоксамид (I-6),
3–(2–(5–бром–2H–пиразоло[3,4–b]пиридин–2–ил)пиридин–4–ил)–1,1–диметилмочевина (I-21).
15. Соединение по п. 1, которое представляет собой
3–фтор–N–(4–фтор–3–{5–[3–(трифторметил)азетидин–1–ил]–2H–пиразоло[3,4–b]пиридин–2–ил}фенил)азетидин–1–карбоксамид (265).
16. Соединение по п. 1, которое представляет собой
N–[3–(5–{5–азаспиро[2.3]гексан–5–ил}–2H–пиразоло[3,4–b]пиридин–2–ил)–4–фторфенил]азетидин–1–карбоксамид (257).
17. Соединение по п. 1, которое представляет собой
N–{2–[5–(3,3–дифторазетидин–1–ил)–2H–пиразоло[3,4–b]пиридин–2–ил]пиридин–4–ил}азетидин–1–карбоксамид (280).
18. Применение соединения по любому из пп. 1-17 в изготовлении лекарственного препарата для лечения нарушения или заболевания, выбранных из лейшманиоза, форм болезни Шагаса и африканского трипаносомоза человека.
| Токарный резец | 1924 |
|
SU2016A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| T | |||
| NOVINSON ET AL, Novel heterocyclic nitrofurfural hydrazones | |||
| In vivo antitrypanosomal activity, JOURNAL OF MEDICINAL CHEMISTRY, 1976, vol | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| СПОСОБ ПОЛУЧЕНИЯ ТВЕРДЫХ ПРОДУКТОВ УПЛОТНЕНИЯ ФОРМАЛЬДЕГИДА С ФЕНОЛАМИ И ДРУГИМИ ВЕЩЕСТВАМИ | 1925 |
|
SU512A1 |
| 7-АЗАИНДОЛЫ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ 4 И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2268887C2 |

