Ссылка на родственные заявки
Настоящая заявка основана и испрашивает приоритет по китайской патентной заявке № CN201711394677.8, поданной 21 декабря 2017 года, полное содержание которой включено в данный документ посредством ссылки во всей своей полноте.
Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к аморфному производному пирролидина в качестве агониста PPAR и к способу его получения.
Предшествующий уровень техники настоящего изобретения
Неалкогольная жировая болезнь печени (НАЖБП) является наиболее распространенным заболеванием печени в развитых странах или регионах, это значит, что слишком большое количество жира накапливается в печени в форме триглицеридов (ткань с гепатоцитами со стеатозом >5%). Помимо слишком большого количества жира у пациентов, страдающих от НАЖБП, выявлена связь с повреждением гепатоцитов и воспалением (стеатогепатит), и в последнем случае присутствует НАСГ (неалкогольный стеатогепатит). Чистый стеатоз при НАЖБП не ассоциирован с повышением заболеваемости или смертности в краткосрочной перспективе, но будет существенно повышать риски цирроза печени, печеночной недостаточности и печеночно-клеточной карциномы (НСС) в случае, когда чистый стеатоз прогрессирует в НАСГ. Цирроз печени вследствие НАСГ является одной из причин растущей потребности в трансплантации печени. У пациентов с НАСГ как заболеваемость, так и смертность от заболеваний печени существенно возрастают и тесно связаны с повышенной заболеваемостью и смертностью от сердечно-сосудистых заболеваний. Диагностика у не проявляющих симптомы заболевания пациентов-мужчин среднего возраста показала, что 46% пациентов имеют неалкогольную болезнь печени (НАЖБП), а 12,2% имеют НАСГ. Пациентами с НАЖБП преимущественно являются мужчины, люди пожилого возраста, пациенты с гипертензией и диабетом. 60-76% пациентов с диабетом страдают от НАЖБП, и 22% из них страдают от НАСГ. Количество пациентов-детей с НАЖБП также растет с каждым годом, и 38-53% детей с ожирением страдают от НАЖБП. В Китае заболеваемость неалкогольной жировой болезнью печени значительно выросла.
В настоящее время не существует одобренных FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) лекарственных средств для лечения этого заболевания, и в клинической практике в Китае обычно применяют полиен-фосфатидилхолин, силимарин, урсодезоксихолевую кислоту, глицирризиновую кислоту и другие лекарственные средства-гепатопротекторы.
Рецептор, активируемый пролифератором пероксисом, (PPAR), член суперсемейства ядерных гормональных рецепторов, представляет собой активируемый лигандом транскрипционный фактор, который регулирует экспрессию генов, и преимущественно имеет три подтипа: PPAR-альфа экспрессируется в основном в бурой жировой ткани, печени, сердце и скелетных мышцах и играет главную роль в метаболизме желчных кислот, липидов и Сахаров; PPAR-дельта не проявляет специфичность экспрессии и может оказывать противовоспалительный эффект; а гамма-форма оказывает определенное воздействие на резистентность к инсулину. Рецептор ассоциирован с рядом болезненных состояний, в том числе с дислипидемией, гиперлипидемией, гиперхолестеринемией, атеросклерозом, атерогенезом, гипертриглицеридемией, сердечной недостаточностью, инфарктом миокарда, сосудистым заболеванием, сердечно-сосудистым заболеванием, гипертензией, ожирением, воспалением, артритом, раком, болезнью Альцгеймера, кожным заболеванием, заболеванием органов дыхания, глазным заболеванием, IBD (синдром раздраженного кишечника), язвенным колитом и болезнью Крона. Исходя из механизмов, обеспечивающих многочисленные благоприятные свойства PPAR для функционирования печени, агонисты PPAR являются одними из наиболее эффективных потенциальных лекарственных средств для лечения жировой инфильтрации печени.
Следующие соединения представляют собой соединения-агонисты PPAR, о которых сообщалось в литературе.
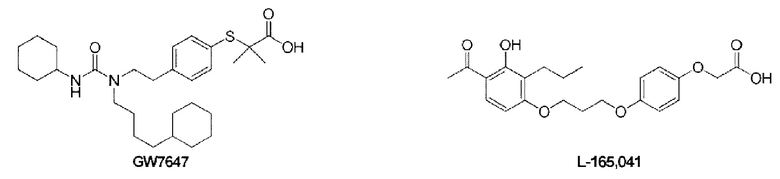
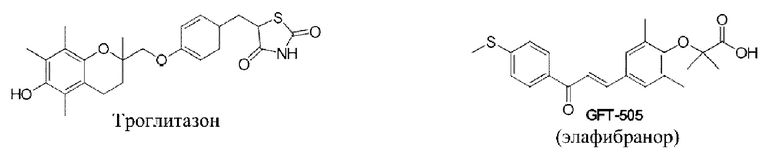
Краткое раскрытие настоящего изобретения
Для преодоления недостатков предшествующего уровня техники первая цель настоящего изобретения заключается в обеспечении аморфного соединения, представленного формулой (I), причем аморфная форма характеризуется значительной стабильностью и, следовательно, имеет определенные перспективы медицинского применения, таким образом обеспечивая подходящий вариант активного фармацевтического ингредиента для разработки соединения, представленного формулой (I), в качестве используемого в клинической практике лекарственного средства.
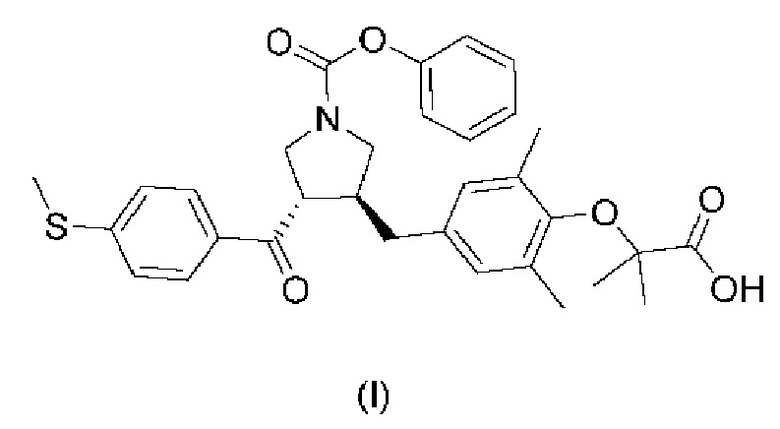
Вышеизложенную цель настоящего изобретения достигают в соответствии со следующими техническими решениями.
Предусмотрена аморфная форма соединения, представленного формулой (I), характеризующаяся тем, что порошковая рентгеновская дифрактограмма (XRPD) аморфной формы не имеет какого-либо острого дифракционного пика.
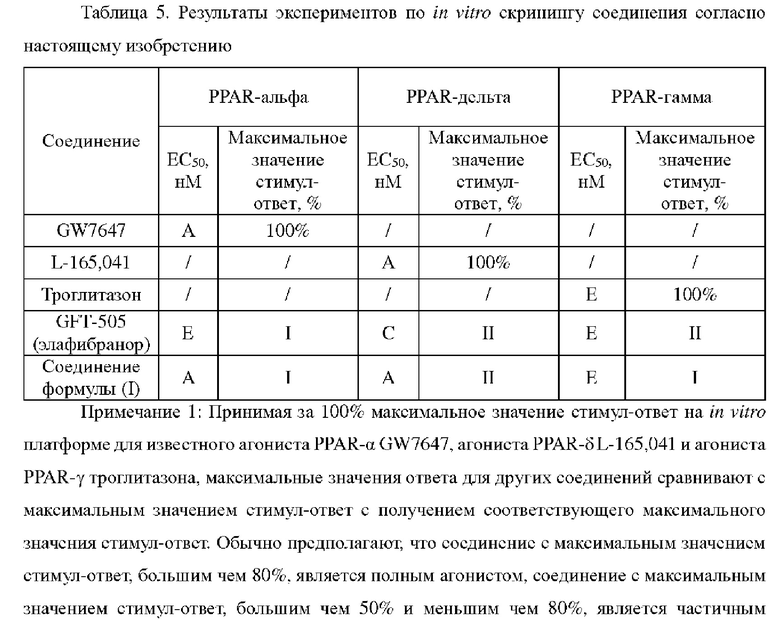
Квалифицированному специалисту в данной области техники хорошо известно, что аморфная форма является высокоэнергетическим термодинамическим состоянием, что обеспечивает термодинамически метастабильную структуру. Элементарные частицы, составляющие соединение, демонстрируют случайное расположение в трехмерном пространстве. Спектр порошковой рентгеновской дифракции является одним из наиболее наглядных средств для определения аморфной формы. В частности, когда соединение существует в аморфной форме, его порошковая рентгеновская дифрактограмма обычно не демонстрирует острого дифракционного пика, то есть оказывается, что спектр XRPD не имеет дифракционного пика или имеет один или несколько широких и плавных дифракционных пиков (часто называемых в уровне техники «пиками в виде пирожков баоцзы»). Специалисты в данной области техники могут понять, что широкие и плавные дифракционные пики в спектре XRPD аморфной формы являются относительными по сравнению с узкими и острыми дифракционными пиками в спектре XRPD у кристаллов, В целом, широкие и плавные дифракционные пики в спектре XRPD аморфной формы могут иметь значения углов 2θ с протяженностью, составляющей 5° или еще больше.
В частности, порошковая рентгеновская дифрактограмма аморфной формы соединения, представленного формулой (I), имеет широкий и плавный дифракционный пик при значении угла 2θ от 10° до 25°.
В соответствии с вариантом осуществления настоящего изобретения аморфная форма соединения, представленного формулой (I), имеет порошковую рентгеновскую дифрактограмму, представленную на фиг. 1.
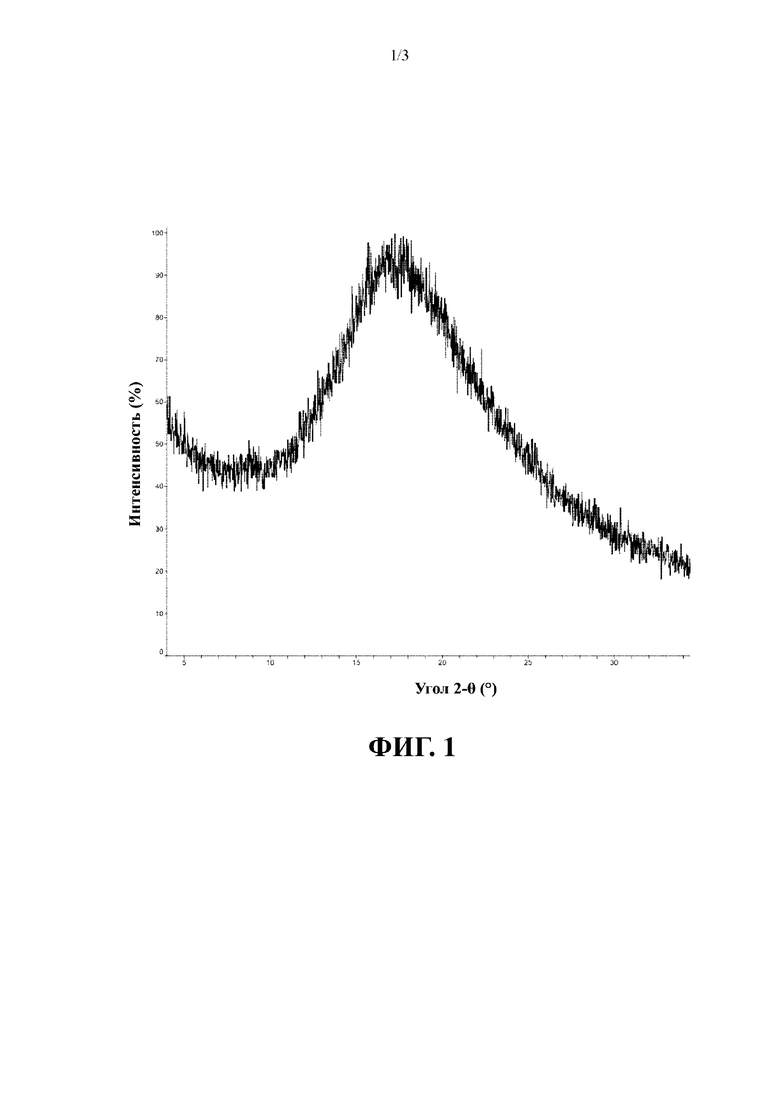
В соответствии с некоторыми вариантами осуществления вышеуказанная аморфная форма характеризуется кривой дифференциальной сканирующей калориметрии (DSC) с начальными точками двух эндотермических пиков при 69,28±3°С и 239,33±3°С.
В соответствии с вариантом осуществления настоящего изобретения вышеуказанная аморфная форма имеет график DSC, представленный на фиг. 2.
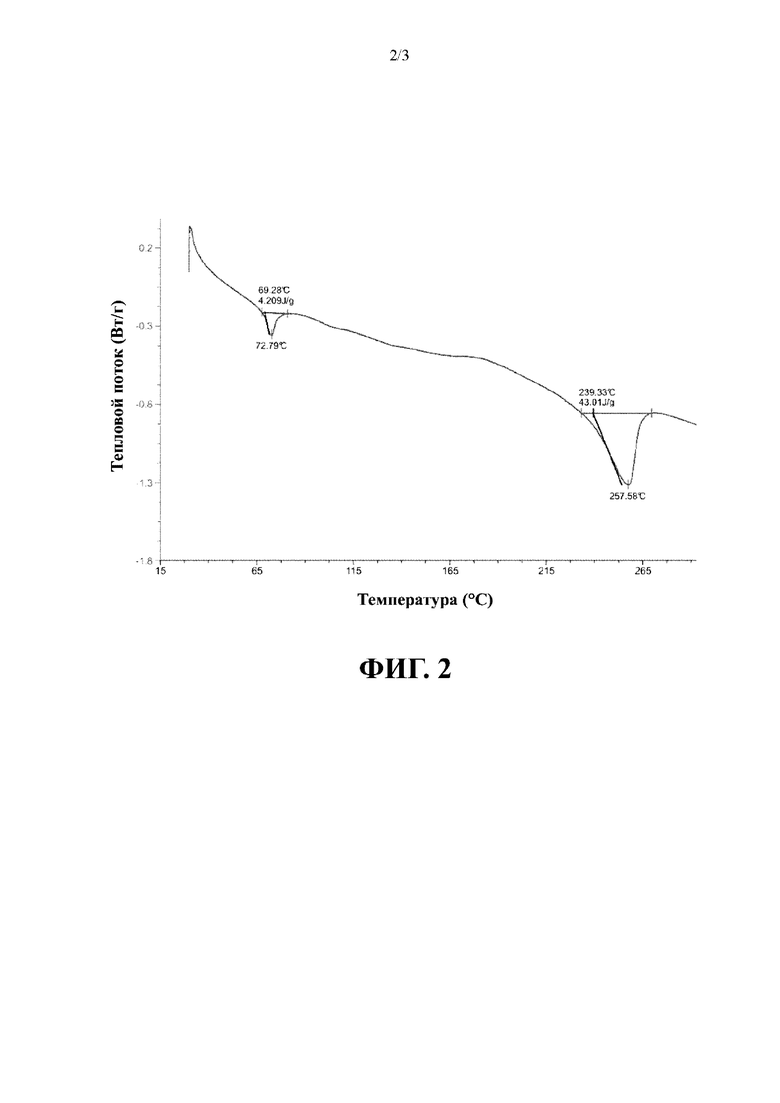
В соответствии с некоторыми вариантами осуществления вышеуказанная аморфная форма имеет кривую термогравиметрического анализа (TGA) с потерей массы, достигающей 0,9958% при 120,00±3°С.
В соответствии с вариантом осуществления настоящего изобретения вышеуказанная аморфная форма имеет график TGA, представленный на фиг. 3.
Вторая цель настоящего изобретения заключается в обеспечении способа получения аморфной формы соединения, представленного формулой (I), и способ получения включает в себя: добавление соединения формулы (I) в растворитель для нагревания с перемешиванием или перекристаллизации с получением аморфной формы; причем растворитель является выбранным из группы, состоящей из метанола, этанола, тетрагидрофурана, этилацетата и н-гептана, температура перемешивания при нагревании с перемешиванием составляет от 25°C до 45°C, период нагревания с перемешиванием (взбивания) составляет от 2 часов до 48 часов, и соотношение масса/объем соединения и растворителя составляет 1:3,5-6 г/мл в способе получения.
Этот способ предполагает стабильный процесс, мягкие условия реакции и легкодоступные сырьевые материалы, и, следовательно, его можно применять для крупномасштабного промышленного производства аморфной формы соединения формулы (I).
Технический эффект
Неожиданно, автор настоящего изобретения обнаружил, что, в отличие от традиционных аморфных соединений, которые характеризуются плохой стабильностью и плохими лечебными свойствами, аморфная форма соединения формулы (I), которая описана в настоящем изобретении, характеризуется более высокой стабильностью, которая, в частности, проявляется в том, что аморфная форма соединения формулы (I) характеризуется относительно высокой стабильностью при высокой температуре, высокой влажности и других условиях. На основании существующих данных стабильности можно прийти к выводу, что аморфная форма соединения формулы (I) имеет определенные перспективы медицинского применения.
Кроме того, аморфная форма соединения формулы (I), которая описана в настоящем изобретении, демонстрирует явный ингибирующий эффект в отношении цитокинов в ассоциированных с PPAR путях, и обнаружили, что соединение формулы (I) оказывает существенный улучшающий эффект на повреждение печени, оценку NAS и печеночный фиброз в экспериментах с индуцируемым CCU острым повреждением печени у C57BL/6 мышей и в моделях НАСГ, индуцируемого MCD рационом у db/db мышей.
Подводя итог, можно увидеть, что аморфная форма соединения, представленного формулой (I), согласно настоящему изобретению имеет лучшую стабильность и определенные перспективы медицинского применения. Таким образом, если с помощью средств выявления подтверждается, что часть соединения, представленного формулой (I), или все оно присутствует в аморфной форме в API (активный фармацевтический ингредиент) и/или продуктах-препаратах, то следует учесть, что используется аморфная форма соединения, представленного формулой (I), которая обеспечена в настоящем изобретении. Помимо порошковой рентгеновской дифракции, которая описана выше, средства выявления могут дополнительно включать в себя дифференциальную сканирующую калориметрию (DSC), инфракрасную спектроскопию (IR), спектроскопию комбинационного рассеяния света (рамановскую спектроскопию), твердотельный ядерный магнитный резонанс (SSNMR) и любые другие средства выявления, которые могут подтвердить применение аморфной формы соединения, представленного формулой (I), согласно настоящему изобретению, и для устранения эффектов, вызванных вспомогательными веществами для лекарственных средств или подобным, можно применять любой способ, обычно применяемый специалистами в данной области техники, такой как метод вычитания графиков.
Определение и разъяснение
Если не указано иное, предполагается, что следующие термины и фразы, которые используются в данном документе, имеют следующие значения. Конкретная фраза или термин не следует рассматривать как неточный или неясный без конкретного определения, и их следует понимать в их обычном значении. Если в данном документе приведено торговое название, предполагают, что оно относится к соответствующему ему продукту или его активным ингредиентам.
Промежуточные соединения согласно настоящему изобретению можно получать с помощью ряда способов синтеза, которые хорошо известны специалистам в данной области техники, в том числе с помощью конкретных вариантов осуществления, представленных ниже, с помощью вариантов осуществления, образованных комбинацией этих вариантов осуществления с другими способами химического синтеза, и с помощью эквивалентных альтернатив, которые хорошо известны специалистам в данной области техники. Предпочтительные варианты осуществления включают в себя, без ограничения, примеры согласно настоящему изобретению.
Химические реакции согласно конкретным вариантам осуществления настоящего изобретения осуществляют в соответствующем растворителе, и растворитель должен быть подходящим для химических превращений согласно настоящему изобретению, а также для реактивов и материалов, требующихся для них. Для получения соединений согласно настоящему изобретению иногда требуется, чтобы специалисты в данной области техники производили модификацию или выбор стадий синтеза или реакционных процессов, исходя из существующих вариантов осуществления.
Ниже в данном документе настоящее изобретение будет описано подробно с помощью примеров. Эти примеры не предназначены для ограничения настоящего изобретения каким-либо образом.
Все растворители, которые применяют в настоящем изобретении, являются коммерчески доступными, и их можно применять без дополнительной очистки.
Растворители, которые применяют в настоящем изобретении, являются коммерчески доступными. В настоящем изобретении используются следующие сокращения: DCM представляет собой дихлорметан; DMF представляет собой N,N-диметилформамид; DMSO представляет собой диметилсульфон; EtOH представляет собой этанол; МеОН представляет собой метанол; TFA представляет собой трифторуксусную кислоту; TsOH представляет собой пара-толуолсульфоновую кислоту; тр представляет собой температуру плавления; EtSO3H представляет собой этансульфоновую кислоту; MeSO3H представляет собой метансульфоновую кислоту; АТР представляет собой трифосаденин; HEPES представляет собой 4-гидроксиэтилпиперазинэтансульфоновую кислоту; EGTA представляет собой этилен-бис(2-аминоэтиловый эфир)тетрауксусную кислоту; MgCl2 представляет собой дихлорид магния; MnCl2 представляет собой дихлорид марганца; и DTT представляет собой дитиотреитол.
1.1 Порошковая рентгеновская дифракция (XRPD)
Модель инструмента: рентгеновский дифрактометр Brook D8 advance (Bruker D8 Advance)
Метод исследования: соответственно, 10-20 мг образца применяют для определения XRPD.
Детальные параметры XRPD являются следующими:
Рентгеновская трубка: Cu, kα, (λ=1,54056Å)
Напряжение на рентгеновской трубке: 40 кВ, ток на рентгеновской трубке: 40 мА
Щель расходимости: 0,60 мм
Щель детектора: 10,50 мм
Щель антирассеивателя: 7,10 мм
Диапазон сканирования: 4-40 градусов
Шаг: 0,02 градуса
Длина шага: 0,12 секунды
Скорость вращения диска с образцом: 15 об./мин.
1.2 Дифференциальная сканирующая калориметрия (DSC)
Модель инструмента: дифференциальный сканирующий калориметр ТА Q2000
Метод исследования: образец (~1 мг) помещали в алюминиевый тигель для DSC с целью исследования, и способ включает: нагревание образца со скоростью нагревания, составляющей 10°C/мин., от 25°C до 350°C в атмосфере потока N2 со скоростью 50 мл/мин.
1.3 Термогравиметрический анализ (TGA)
Модель инструмента: термогравиметрический анализатор ТА Q5000IR
Метод исследования: образец (2-5 мг) помещали в платиновый тигель для TGA с целью исследования, и способ включает в себя: нагревание образца со скоростью нагревания, составляющей 10°C/мин., от комнатной температуры до 350°C в атмосфере потока N2 со скоростью 25 мл/мин.
Краткое описание чертежей
Фиг. 1 представляет собой полученную при облучении Cu-Kα спектрограмму XRPD аморфной формы соединения формулы (I),
Фиг. 2 представляет собой спектрограмму DSC аморфной формы соединения формулы (I).
Фиг. 3 представляет собой спектрограмму TGA аморфной формы соединения формулы (I).
Подробное раскрытие настоящего изобретения
Ниже в данном документе настоящее изобретение подробно описано с помощью примеров, но не предполагается, что они ограничивают настоящее изобретение каким-либо неблагоприятным образом. Настоящее изобретение было подробно описано в данном документе и дополнительно раскрыто в конкретных вариантах осуществления. Квалифицированному специалисту в данной области техники будет очевидно, что ряд модификаций и улучшений можно выполнить в вариантах осуществления настоящего изобретения без отступления от принципа и объема настоящего изобретения.
ПРИМЕРЫ
Пример 1. Получение соединения формулы (I)
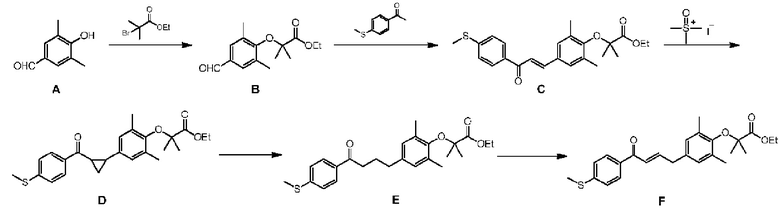
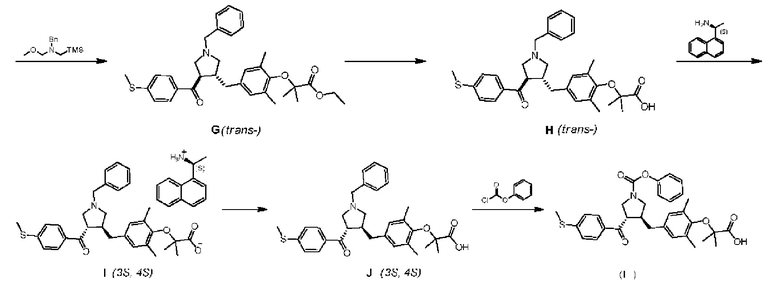
Стадия 1. Получение соединения В
При 25°C ацетонитрил (30 л) добавляли в реактор объемом 50 л, начинали перемешивание, а затем добавляли соединение А (2,00 кг, 13,32 ммоля, 1,0 экв.), этил-бромизобутират (7,79 кг, 39,95 моля, 3,0 экв.) и карбонат калия (5,52 кг, 39,95 моля, 3,0 экв.). Реакционный раствор перемешивали при 80°C в течение 16 часов. Температуру реакционной смеси снижали до 25°C, а затем ее фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток растворяли в этилацетате (5 л). Осадок на фильтре промывали этилацетатом (5 л×2) и растворы в этилацетате объединяли. Объединенные органические фазы промывали водным NaOH (1 моль/л, 5 л/раз) до тех пор, пока не становилось видно, что пятна исходного материала А не появляются в органической фазе при анализе методом TLC (тонкослойная хроматография) (петролейный эфир : этилацетат = 5:1). Органическую фазу промывали насыщенным водным раствором хлорида натрия (5 л×2), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1,51 кг соединения В, выход: 42,9%.
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 9,85 (s, 1H), 7,50 (s, 2Н), 4,31-4,23 (m, 2Н), 2,25 (s, 6Н), 1,45 (s, 6Н), 1,33 (t, J=7,2 Гц, 1H).
Стадия 2. Получение соединения С
В бане с сухим льдом и этанолом (-60°C) газообразную HCl (3,67 кг, 100,54 моля, 5,3 экв.) вводили в этанол (12 л) и температуру системы контролировали так, чтобы она была ниже 0°C. Этанол (13 л) и свежеприготовленный этанольный раствор HCl добавляли в реактор объемом 50 л. Смесь перемешивали и подогревали в естественных условиях до 25°C. Затем добавляли соединение В (5,01 кг, 18,97 моля, 1,0 экв.). После полного растворения материалов порциями добавляли пара-метилтиоацетофенон (2,83 кг, 17,07 моля, 0,9 экв.). Смесь перемешивали при 25°C в течение 16 часов. Реакционную систему подвергали фильтрованию с отсасыванием. Осадок на фильтре растворяли в этилацетате (30 л), промывали водой (10 л×2), водным NaOH (1 н, 8 л×2) и насыщенным водным раствором хлорида натрия (8 л×2), сушили над безводным сульфатом натрия (1,5 кг), фильтровали и концентрировали при пониженном давлении с получением 6,10 кг соединения С, выход: 76,8%.
MSm/z(ESI): 413,1 [М+1].
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 7,95 (d, J=8,28 Гц, 2Н), 7,71 (d, J=15,56 Гц, 1H), 7,42 (d, J=15,56 Гц, 1H), 7,31-7,28 (m, 4Н), 4,30 (q, J=7,28 Гц, 2Н), 2,54 (s, 3Н), 2,25 (s, 6Н), 1,50 (s, 6Н), 1,36 (t, J=7,15 Гц, 3Н).
Стадия 3. Получение соединения D
N,N-диметилформамид (15 л) добавляли в реактор объемом 50 л, начинали перемешивание и добавляли йодид триметилсульфоксония (3,78 кг, 16,01 моля, 1,2 экв,), затем охлаждали до 0°C и добавляли порциями трет-бутоксид калия (1,79 кг, 16,01 моля, 1,2 экв.). После того как смесь перемешивали при 0°C в течение 30 мин., медленно добавляли раствор соединения С (5,5 кг, 13,34 моля, 1,0 экв.) в N,N-диметилформамиде (15 л). Смесь перемешивали при 0°C в течение 2 часов. Реакционную жидкость медленно выливали в ледяную воду (0-5°C, 30 л), а затем экстрагировали смесью петролейный эфир/этилацетат (1:1, 10 л×3). Объединенную органическую фазу промывали водой (10 л×2) и насыщенным водным раствором хлорида натрия (10 л×2), сушили над безводным сульфатом натрия (2 кг), фильтровали и концентрировали при пониженном давлении с получением 5,48 кг соединения D, выход: 96,3%.
MS m/z (ESI): 427,2 [М+1].
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 7,91 (d, J=8,5 Гц, 2Н), 7,27 (d, J=8,5 Гц, 2Н), 6,81-6,70 (m, 1H), 6,75 (s, 1H), 4,29 (q, J=7,0 Гц, 2Н), 2,83-2,74 (m, 1H), 2,56 (m, 1H), 2,51 (s, 3Н), 2,18 (s, 6Н), 1,84 (m, 1H), 1,46 (s, 6Н), 1,35 (t, J=7,2 Гц, 3Н).
Стадия 4. Получение соединения Е
Этанол (35,0 л) добавляли в сухой реактор объемом 50 л, начинали перемешивание, а затем добавляли соединение D (5,45 кг, 12,79 моля, 1,0 экв.) и ледяную уксусную кислоту (2,30 кг, 38,37 моля, 3,0 экв.). После нагревания реакционной смеси до 80°C порциями добавляли цинковый порошок (2,45 кг, 38,37 моля, 3,0 экв.). Полученную в результате суспензию непрерывно перемешивали при 80°C в течение 16 часов. Реакционную жидкость фильтровали и осадок на фильтре промывали этилацетатом (3 л×2). Объединенную органическую фазу концентрировали при пониженном давлении. Концентрированный раствор растворяли в этилацетате (10 л), закачивали в делительную воронку объемом 50 л. Этилацетат (15 л) и воду (10 л) закачивали в делительную воронку объемом 50 л и перемешивали в течение 5 минут, затем отстаивали для разделения фаз. Органическую фазу последовательно промывали 10% водным карбонатом натрия (10 л×2) и насыщенным водным раствором хлорида натрия (10 л×1), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 5,15 кг соединения Е, выход: 92,9%.
MS m/z (ESI): 429,2 [М+1].
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 7,74 (d, J=8,5 Гц, 2Н), 7,17 (d, J=8,5 Гц, 2Н), 6,71 (s, 2Н), 4,21 (q, J=7,1 Гц, 2Н), 2,82 (t, J=7,3 Гц, 2Н), 2,51 (t, J=7,7 Гц, 2H), 2,45 (s, 3Н), 2,09 (s, 6Н), 1,94 (quin, J=7,5 Гц, 2H), 1,39 (s, 6Н), 1,28 (t, J=7,0 Гц, 3Н)
Стадия 5. Получение соединения F
Безводный дихлорметан (20 л) добавляли в реактор объемом 50 л, начинали перемешивание, затем добавляли соединение Е (5,21 кг, 12,02 моля, 1,0 экв.) и 2,6-лутидин (4,50 кг, 42,07 моля, 3,5 экв.). Смесь охлаждали до 0°C.
Триметилсилилтрифторметансульфонат (8,01 кг, 36,06 моля, 3,0 экв.) добавляли в реакционный раствор и смесь непрерывно перемешивали при 0°C в течение около 30 минут. Реакционную жидкость подвергали анализу методом TLC (тонкослойная хроматография) (петролейный эфир : этилацетат = 5:1). Холодную воду (5-10°C, 10 л) закачивали в делительную воронку объемом 50 л, а затем туда закачивали реакционную жидкость при перемешивании. После перемешивания в течение 5 минут фазы разделяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия (10 л) и концентрировали при пониженном давлении. Концентрированный раствор добавляли к смешанному раствору из метанола/воды (2:1, 30 л) и перемешивали в течение приблизительно 20 минут. Осаждались желтые твердые вещества. Смесь фильтровали, сушили при пониженном давлении с получением неочищенного продукта в виде желтого твердого вещества.
Безводный толуол (35 л) добавляли в реактор объемом 50 л, начинали перемешивание и неочищенный продукт, который сушили при пониженном давлении, добавляли в реактор. Смесь охлаждали до 0°C и дихлор-дициано-бензохинон (2,99 кг, 13,22 моля, 1,1 экв.) добавляли порциями в реактор. Смесь непрерывно перемешивали при 0°C в течение 1 часа. Реакционную жидкость подвергали анализу с помощью TLC (петролейный эфир : этилацетат = 5:1). Воду (50 л) и сульфит натрия (3,00 кг) добавляли в бочку объемом 120 л. После перемешивания до прозрачного состояния реакционную жидкость медленно выливали в раствор сульфита натрия и добавляли этилацетат (15 л). Смесь быстро перемешивали в течение 10 минут с выпадением в осадок большого количества желтого твердого вещества, а затем фильтровали с отсасыванием. Осадок на фильтре промывали смесью петролейный эфир/этилацетат (3:1, 10 л×2). Фильтраты объединяли, а затем отделяли органическую фазу. Органическую фазу промывали 5% сульфитом натрия (10 л×2) и насыщенным водным раствором хлорида натрия (10 л×2), сушили над безводным сульфатом натрия (2,00 кг), фильтровали и концентрировали при пониженном давлении (40-50°C) с получением 4,29 кг неочищенного продукта. Затем неочищенный продукт добавляли к 12 л безводного этанола, перемешивали при 25°C в течение 0,5 часа, затем фильтровали. Осадок на фильтре собирали и сушили при пониженном давлении с получением 3,50 кг соединения F, выход: 69,9%.
MS m/z (ESI): 427,2 [М+1].
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 7,85 (d, J=8,5 Гц, 2Н), 7,28-7,27 (m, 2Н), 7,22-7,14 (m, 1H), 7,02 (s, 1H), 6,83 (s, 2Н), 4,30 (s, 2Н), 3,53 (d, J=6,8 Гц, 2Н), 2,55 (s, 3Н), 2,21 (s, 6Н), 1,49 (s, 6Н), 1,38 (s, 3Н).
Стадия 6. Получение соединения G
2-Метилтетрагидрофуран (30 л) добавляли в сухой реактор объемом 50 л, начинали перемешивание и добавляли соединение F (3,0 кг, 6,50 моля, 1,0 экв.) и трифторуксусную кислоту (37,05 г, 0,33 моля, 0,05 экв.). Затем медленно добавляли N-(метоксиметил)-N-(триметилсилилметил)бензиламин (1,85 кг, 7,80 моля, 1,2 экв.). Внутреннюю температуру контролировали так, чтобы она была ниже 30°C. После добавления по каплям смесь непрерывно перемешивали при 25°C в течение 12 часов. Реакционную жидкость выплескивали в делительную воронку объемом 50 л, последовательно промывали 5% водным раствором карбоната натрия (10 л×2) и насыщенным водным раствором хлорида натрия (10 л×2), сушили над безводным сульфатом натрия (2 кг), фильтровали и концентрировали при пониженном давлении с получением 3,92 кг соединения G.
MS m/z (ESI): 560,0 [М+1].
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 7,71 (d, J=8,5 Гц, 1H), 7,31-7,20 (m, 9Н), 6,71 (s, 1H), 4,30-4,23 (m, 2Н), 3,76-3,36 (m, 6Н), 3,07-2,96 (m, 2Н), 2,68 (t, J=7,3 Гц, 2Н), 2,51 (s, 3Н), 2,10-2,03 (m, 6Н), 1,36-1,22 (m, 9Н).
Стадия 7. Получение соединения Н
Соединение G (3,92 кг, 5,04 моля, 1,0 экв.) растворяли в безводном этаноле (20 л) и раствор добавляли в реактор объемом 50 л, начинали перемешивание. NaOH (604,8 г, 15,12 моля, 3,0 экв.) растворяли в воде (6 л), а затем раствор медленно добавляли в реакционный раствор. После добавления смесь непрерывно перемешивали при 25°C в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении с удалением большей части растворителя (этанол). Концентрированный раствор выплескивали в делительную воронку объемом 50 л, перемешивали, а затем добавляли этилацетат (20 л). Смесь промывали 10% водным KHSO4 (10 л×2) и насыщенным водным раствором хлорида натрия (10 л×2), сушили над безводным сульфатом натрия (1,5 кг) и концентрировали при пониженном давлении до тех пор, пока не оставалось приблизительно 8 л растворителя и в осадок выпадало большое количество твердого вещества. Концентрирование останавливали и смесь охлаждали до 25°C. Концентрированную суспензию фильтровали и осадок на фильтре промывали этилацетатом (2 л×3), сушили с отсасыванием и сушили при пониженном давлении в вакуумной сушильной камере с получением 2,44 кг соединения Н, выход: 89,85%.
MS m/z (ESI): 532,1[М+1].
1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm (части на миллион) 7,66 (dd, J=4,0, 8,3 Гц, 2Н), 7,41-7,33 (m, 2Н), 7,23-7,08 (m, 5Н), 6,79 (d, J=2,0 Гц, 2Н), 4,12 (q, J=7,0 Гц, 2Н), 3,93-3,60 (m, 4Н), 3,48-3,32 (m, 1H), 2,97-2,84 (m, 1H), 2,67 (d, J=7,8 Гц, 2H), 2,57-2,49 (m, 3H), 2,25-2,13 (m, 6H), 1,46 (s, 6H), 1,36 (t, J=7,2 Гц, 3Н).
Стадия 8. Получение соединения I
Ацетонитрил (24 л) и изопропанол (6 л) добавляли в сухой реактор объемом 50 л, начинали перемешивание, затем добавляли соединение Н (3,04 кг, 5,65 моля, 1,0 экв.). При 80°C медленно добавляли (S)-(-)-(1-нафтил)этамин (724,79 г). После добавления смесь непрерывно перемешивали при 80°C в течение 1 часа. Нагревание останавливали. Смесь охлаждали в естественных условиях и непрерывно перемешивали при 30°C в течение 16 часов. Перемешивание останавливали. Смесь фильтровали с отсасыванием и осадок на фильтре промывали изопропанолом (2 л×2). После отсасывания до сухого состояния твердое вещество переносили в ротационный испаритель для высушивания при пониженном давлении с получением 1,63 кг соединения I.
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм, I.D. (внутренний диаметр) 3 мкм; подвижная фаза: 40% метанол (0,05% DEA) -СО2; скорость потока: 4 мл/мин.; температура колонки: 40°C.
Время удержания для соединения I: 1,604 мин.
Стадия 9. Получение соединения J
Безводный этанол (30 л) и безводный метанол (4,5 л) добавляли в сухой реактор объемом 50 л, начинали перемешивание и добавляли соединение I (2,93 кг, 4,17 моля, 1,0 экв.). Смесь нагревали до 80°C и перемешивали в течение 1 часа. Нагревание останавливали и смесь охлаждали в естественных условиях. Смесь непрерывно перемешивали при 30°C в течение 16 часов. Суспензию фильтровали с отсасыванием и осадок на фильтре промывали этанолом (2 л×2) и сушили при пониженном давлении. К остатку добавляли метанол (1,5 л) и этилацетат (15 л) и смесь перемешивали, а затем закачивали в сепаратор для жидкостей объемом 50 л. Органическую фазу промывали 10% водным KHSO4 (10 л×5), насыщенным водным раствором хлорида натрия (5 л×2), сушили над безводным сульфатом натрия (1 кг), фильтровали и концентрировали при пониженном давлении с получением 0,97 кг соединения J.
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм, I.D. (внутренний диаметр) 3 мкм; подвижная фаза: 40% метанол (0,05% DEA) - CO2; скорость потока: 4 мл/мин.; температура колонки: 40°C.
Время удержания для соединения J: 1,576 мин.
Стадия 10. Получение соединения формулы (I)
Безводный дихлорметан (7 л) добавляли в сухой реактор объемом 50 л, начинали перемешивание и добавляли соединение J (700 г, 1,31 моля, 1,0 экв.) и триэтиламин (1,33 кг, 13,1 моля, 10,0 экв.). При 0°C фенилхлорформиат (2,24 кг, 13,1 моля, 10,0 экв.) добавляли по каплям в реакционную жидкость. После завершения добавления смесь непрерывно перемешивали при 0°C в течение 1 часа. Раствор карбоната калия (543,37 г, 3,94 моля, 3,0 экв.) в чистой воде (3 л) добавляли в реакционный раствор. Смесь нагревали до 40°C и непрерывно перемешивали в течение 20 мин. Затем добавляли LiOH (165,60 г, 3,94 моля, 3,0 экв.) и смесь непрерывно перемешивали при 25°C в течение 20 минут. Реакционную смесь подвергали разделению фаз. Органическую фазу промывали насыщенным водным раствором хлорида натрия (2 л), сушили над безводным сульфатом натрия (500 г), фильтровали и концентрировали при пониженном давлении. Концентрированный раствор растворяли в этилацетате (1,5 л), затем медленно добавляли гептан (5,6 л) при перемешивании с высокой скоростью. После добавления смесь непрерывно перемешивали в течение 30 минут и фильтровали. Осадок на фильтре добавляли в смесь гептан/этил ацетат (4:1, 3,5 л×3) и перемешивали с высокой скоростью для взбивания в течение 30 минут и фильтровали. Полученный в результате осадок на фильтре растворяли в трет-бутилметиловом эфире (5 л) и последовательно промывали 5% водным KHSO4 (1,5 л×2), деионизированной водой (1 л×2), сушили над безводным сульфатом натрия (300 г), фильтровали и концентрировали при пониженном давлении. Полученное в результате твердое вещество сушили в вакуумной печи (40-45°C) с получением соединения формулы (I).
MS m/z (ESI): 584,1 [М+23].
1H ЯМР (400 МГц, MeOD-d4) δ ppm (части на миллион) 7,65 (d, J=6,8 Гц, 2Н), 7,43-7,37 (m, 2Н), 7,29-7,22 (m, 3Н), 7,16 (t, J=7,2 Гц, 2Н), 6,87 (s, 2Н), 4,13-3,92 (m, 2Н), 3,89-3,80 (m, 1H), 3,69 (dd, J=5,4, 10,7 Гц, 1H), 3,58-3,49 (m, 1H), 2,81 (d, J=14,1 Гц, 2Н), 2,71-2,63 (m, 1H), 2,55 (s, 3Н), 2,23 (s, 6Н), 1,43 (s, 6Н).
Условия хирального разделения: хиральная колонка: Chiralpak AD-3 100×4,6 мм, I.D. (внутренний диаметр) 3 мкм; подвижная фаза: 40% метанола (0,05% DEA) в СО2; скорость потока: 2,8 мл/мин.; температура колонки: 40°C.
Время удержания для соединения формулы (I): 2,018 мин.
Пример 2. Получение аморфной формы соединения формулы (I)
Температуру контролировали так, чтобы она составляла 25°C, и неочищенное соединение формулы (I) (310,5 г) в виде бледно-желтого твердого вещества добавляли в реакционную колбу объемом 3 л. Затем добавляли гептан (1500 мл). После завершения добавления реакционную смесь перемешивали при 25°C в течение 2 часов, затем фильтровали. Осадок на фильтре промывали н-гептаном (500 мл), затем фильтровали с получением неочищенного продукта. Неочищенный продукт сушили в вакуумной сушильной камере и подвергали определению его формы с помощью метода XRPD. Полученный конечный продукт находился в аморфной форме.
Полученная при облучении Cu-Kα дифрактограмма XRPD для полученного конечного продукта представлена на фиг. 1; график DSC представлен на фиг. 2; и график TGA представлен на фиг. 3.
Пример 3. Получение аморфной формы соединения формулы (I)
Соединение формулы (I) (40,0 мг) отвешивали и добавляли в стеклянный пузырек объемом 4,0 мл и 150 мкл этилацетата добавляли для образования суспензии. Суспензию перемешивали на магнитной мешалке при 40°C в течение 2 суток, затем образец центрифугировали. Супернатант помещали в вытяжной шкаф до высыхания растворителя. Затем полученное в результате твердое вещество сушили в вакуумной сушильной камере при 40°C в течение ночи. Полученный в результате конечный продукт представлял собой аморфную форму, которая являлась такой же, как аморфная форма из примера 1.
Пример 4. Получение аморфной формы соединения формулы (I)
Соединение формулы (I) (39,9 мг) отвешивали и добавляли в стеклянный пузырек объемом 4,0 мл и 150 мкл тетрагидрофурана добавляли для образования суспензии. После того как суспензию перемешивали при 40°C на магнитной мешалке в течение 2 суток, образец центрифугировали и супернатант помещали в вытяжной шкаф до высыхания растворителя. Затем полученное твердое вещество сушили в вакуумной сушильной камере при 40°C в течение ночи. Полученный в результате конечный продукт представлял собой аморфную форму, которая являлась такой же, как аморфная форма из примера 1.
Пример 5. Исследование стабильности в твердом состоянии аморфной формы соединения формулы (I) в условиях высокой температуры и высокой влажности
Образцы (каждый массой приблизительно 100 мг) аморфной формы соединения формулы (I) отвешивали в двух параллелях и помещали на дно стеклянной бутылки для проб, распределяя их в виде тонкого слоя. Горлышко стеклянной бутылки для проб запечатывали алюминиевой фольгой, которую прокалывали с образованием в ней нескольких отверстий, чтобы гарантировать, что образцы могут в достаточной степени контактировать с окружающим воздухом. Образцы помещали в камеру с постоянной температурой и влажностью в условия с температурой 40°C/влажностью 75%. Из образцов, которые хранили при вышеуказанных условиях, отбирали пробы и осуществляли анализ в День 10, 30, 60 и 90. Результаты анализа сравнивали с результатами первоначального анализа в День 0. Анализ методом HPLC представлен в таблице 1. Результаты анализа представлены в таблице 2 ниже.
Представленные выше экспериментальные данные демонстрируют, что аморфная форма соединения формулы (I), обеспеченная согласно настоящему изобретению, не проявляет значительного изменения в содержании и количестве примесей при высокой температуре и влажности и характеризуется относительно высокой стабильностью при высокой температуре и влажности.
Пример 6. Исследование физической стабильности в твердом состоянии аморфной формы соединения формулы (I) при высокой влажности
Образцы (каждый массой приблизительно 100 мг) аморфной формы соединения формулы (I) отвешивали в двух параллелях и помещали на дно стеклянной бутылки для проб, распределяя их в виде тонкого слоя. Горлышко стеклянной бутылки для проб запечатывали алюминиевой фольгой, которую прокалывали с образованием в ней нескольких отверстий, чтобы гарантировать, что образцы могут в достаточной степени контактировать с окружающим воздухом. Приготовленные образцы помещали в условия с температурой 25°C/относительной влажностью 92,5% и осуществляли определение их физической стабильности в День 10. В то же время, образец (массой приблизительно 100 мг) аморфной формы соединения формулы (I) отвешивали отдельно, помещали на дно стеклянной бутылки для образца, которую запечатывали завинчивающейся пробкой, и хранили при -20°C в качестве контроля. В День 10 все образцы доставали и возвращали в условия с комнатной температурой. Образцы подвергали визуальному осмотру в отношении изменения их внешнего вида и определяли их форму методом XRPD. Физическую стабильность в твердом состоянии соединения формулы (I) определяли посредством сравнения образца, хранившегося в условиях ускоренной деградации, с контрольным образцом. В таблице 3 ниже представлены результаты экспериментов по исследованию физической стабильности аморфной формы соединения формулы (I).
Представленные выше экспериментальные данные демонстрируют, что аморфная форма соединения формулы (I), обеспеченная согласно настоящему изобретению, не проявляет изменений формы и внешнего вида при высокой влажности и характеризуется относительно высокой стабильностью при высокой влажности.
Пример 7. Исследования стабильности аморфной формы соединения формулы (I) в различных растворителях
Множество образцов аморфной формы соединения формулы (I) (каждый массой приблизительно 20 мг) добавляли соответственно к 0,3-0,4 мл отдельных или смешанных растворителей, которые приведены в таблице ниже, и перемешивали при 40°C. После перемешивания в течение 2 суток, если образец все еще находился в состоянии раствора или в состоянии, близком к раствору, его фильтровали с последующим естественным испарением растворителя; а если образец все еще представлял собой суспензию, образец центрифугировали для сбора осадков и супернатант помещали в вытяжной шкаф до высыхания растворителя. Затем осадки и твердые вещества, полученные после испарения растворителя, помещали в вакуумный сушильный шкаф с температурой 40°C на ночь. Твердые вещества во всех образцах собирали и определяли их состояние методом XRPD. Результаты представлены в таблице 4.
Представленные выше экспериментальные данные демонстрируют, что аморфная форма соединения формулы (I), обеспеченная согласно настоящему изобретению, не проявляет какого-либо изменения формы и характеризуется относительно высокой стабильностью.
Исследование биологической активности
Экспериментальный пример 8. Оценка in vitro
Принципы in vitro анализа активности агониста PPAR
Исследование ядерного гормонального рецептора (NHR)
Тест взаимодействия с белком NHR и ядерного транспорта PathHunter применяют для выявления способности к активации ядерного гормонального рецептора в унифицированном эксперименте без визуализации. Эта технология называется комплементацией фрагментов фермента (ETC) и разработана DiscoverX.
Тест на белок NHR основан на выявлении белок-белковых взаимодействий между белком NHR стандартной длины в активированном состоянии и гибридным ядерным белком, содержащим участок пептида, коактивирующего стероидный рецептор (SRCP), и одну или несколько стандартных действующих последовательностей LXXLL.
NHR метят на компоненте ProLinkTM тест-системы EFC и участок SRCP и компонент рецептора фермента (ЕА) сливают и экспрессируют в ядре. Когда он связывается с лигандом, NHR будет транспортироваться в ядро и взаимодействовать с участком SRCP, в котором создается комплементарный эффект, таким образом образуя эквивалентное количество активированной галактозидазы (-Gal), что сопровождается генерированием хемилюминесцентных сигналов. Выгоды, ассоциированные с этим путем, включают в себя уменьшенное время инкубирования соединения, непосредственный анализ мишени-NHR, применение последовательностей человеческого NHR со стандартной длиной и подбор некоторых новых классов соединений, исходя из нарушения белок-белковых взаимодействий.
NT-тест NHR выявлял транспорт NHR между цитоплазматическим и ядерным компартментами. Рецептор метили на компоненте ProLinkTM тест-системы EFC, в то время как ЕА и ядерную последовательность сливали, таким образом ограничивая экспрессию ЕА на ядре. Транспорт в ядро приводил к комплементарному эффекту с ЕА, таким образом образуя один эквивалент активированной галактозидазы (-Gal), что сопровождалось генерированием хемилюминесцентных сигналов.
Обработка клеток:
1. Линии клеток PathHunter NHR разращивали из замороженной концентрированной суспензии в соответствии со стандартными операциями.
2. Клетки высевали на 384-луночный белый планшет для клеток в количестве 20 мкл/лунка и инкубировали при 37°C в течение соответствующего периода времени перед тестом. Среда культивирования содержала активированный уголь-декстран с фильтрованной сывороткой для снижения уровня экспрессии гормона.
Эксперименты в режиме агониста:
1. В случае измерения агонистической активности для индукции ответа требуется инкубировать клетки с соединением,
2. Соединение составляли с буферным раствором в маточный раствор, который растворяли 5-кратно.
3. 5 мкл 5-кратно разведенного раствора соединения добавляли к клеткам и инкубировали при 37°C (или при комнатной температуре) в течение 3-16 часов, обеспечивая, чтобы конечная концентрация среды составляла 1%).
Эксперименты в режиме ингибитора:
1. В случае измерения ингибирующей активности требуется инкубировать клетки с антиагонистом, а затем провоцировать агонистом в концентрации ЕС80.
2. Соединение составляли с буферным раствором в маточный раствор, который растворяли 5-кратно.
3. 5 мкл 5-кратно разведенного раствора соединения добавляли к клеткам и инкубировали при 37°C (или при комнатной температуре) в течение 60 минут, обеспечивая, чтобы конечная концентрация среды составляла 1%).
4. 5 мкл агониста в ЕС80, который 6-кратно разводили буферным раствором, добавляли к клеткам и инкубировали при 37°C (или при комнатной температуре) в течение 3-16 часов.
Выявление сигнала:
1. Сигналы в эксперименте генерировались с помощью 12,5 мкл или 15 мкл (50% об./об.) смеси реактивов для теста PathHunter, которую после добавления затем необходимо инкубировать при комнатной температуре в течение 1 часа.
2. Хемилюминесцентные сигналы, генерируемые на микропланшете, выявляли с помощью инструмента PerkinElmer Envision.
Анализ данных:
1. Активность соединения анализировали с помощью программного обеспечения для анализа данных CIBS (Chemlnnovation, Калифорния, США).
2. Для экспериментов в режиме агониста активность в процентах рассчитывают согласно следующей формуле:
% активность = 100% × (средняя RLU для анализируемого соединения - средняя фоновая RLU для среды) / (среднее значение для лиганда с максимальным контролем - средняя фоновая RLU для среды)
3. Для экспериментов в режиме антагониста активность в процентах рассчитывают согласно следующей формуле:
% ингибирование = 100% × (1 - (средняя RLU для анализируемого соединения - средняя фоновая RLU для среды) / (средняя RLU для контрольного соединения в ЕС80 - средняя фоновая RLU для среды))
4. Следует заметить, что ответ на лиганд вызывает снижение активности рецептора (оказывает воздействие, обратное агонистическому при непрерывно активной мишени). Эту активность, обратную агонистической, рассчитывают согласно следующей формуле:
% активность, обратная агонистической = 100% × ((средняя фоновая RLU для среды - средняя RLU для анализируемого соединения) / (средняя фоновая RLU для среды - средняя RLU для лиганда с максимальным контролем))
Результаты экспериментов представлены в таблице 5.

Вышеуказанные экспериментальные данные показывают, что соединение формулы (I), характеризуется значительным активирующим эффектом в отношении рецепторов PPAR-альфа и -дельта и селективным активирующим эффектом в отношении рецептора PPAR-гамма.
На основании вышеприведенных экспериментальных оценок стабильности и активности аморфной формы соединения формулы (I), можно увидеть, что: в отличие от распространенных знаний об аморфных лекарственных средствах в данной области техники, аморфная форма соединения формулы (I), обеспеченная согласно настоящему изобретению, характеризуется высокой стабильностью в плане химических свойств и физических форм. Кроме того, аморфная форма соединения формулы (I) обладает выраженным ингибирующим эффектом в отношении цитокинов, ассоциированных со связанными с PPAR путями, и оказывает значительные эффекты на улучшение повреждения печени, оценку NAS и фиброз печени. Можно увидеть, что аморфная форма соединения формулы (I) имеет хорошие перспективы медицинского применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ATR И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2832707C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| C-Анилинохиназолиновые соединения и их использование в лечении рака | 2018 |
|
RU2769694C2 |
| ТВЕРДЫЕ ФОРМЫ (2S,3R,4R,5S,6R)-2-(4-ХЛОР-3-(4-ЭТОКСИБЕНЗИЛ)ФЕНИЛ)-6-(МЕТИЛТИО)ТЕТРАГИДРО-2Н-ПИРАН-3,4,5-ТРИОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2505543C2 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
| 3,7-ДИАЗАБИЦИКЛО[3.3.1]-ПРЕПАРАТЫ КАК АНТИАРИТМИЧЕСКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2286993C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ТРИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2775753C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| 2,3-ДИГИДРО-1H-ПИРРОЛИЗИН-7-ФОРМАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2792726C2 |
Изобретение относится к аморфной форме соединения, представленного формулой (I), причем порошковая рентгеновская дифрактограмма аморфной формы не имеет какого-либо острого дифракционного пика и имеет широкий и пологий дифракционный пик при значении угла 2θ от 10° до 25°, а также к способу его получения. Технический результат: получена новая аморфная форма соединения, представленного формулой (I), которая может быть использована в качестве агониста PPAR, причем аморфная форма характеризуется значительной стабильностью. 2 н. и 5 з.п. ф-лы, 3 ил., 5 табл., 3 пр.
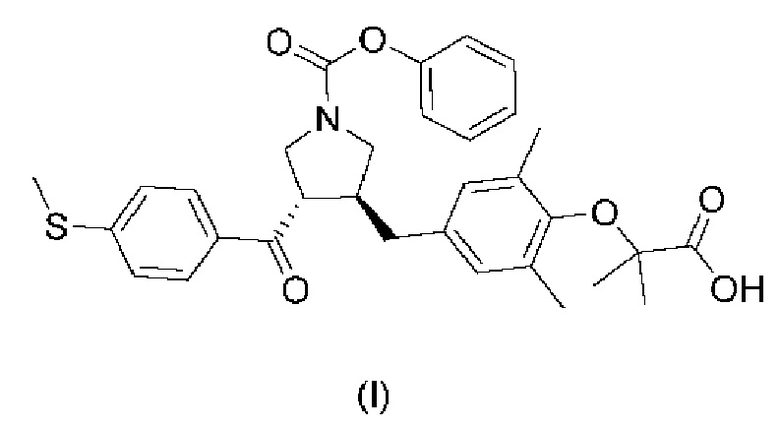
1. Аморфная форма соединения, представленного формулой (I)
причем порошковая рентгеновская дифрактограмма аморфной формы не имеет какого-либо острого дифракционного пика и имеет широкий и пологий дифракционный пик при значении угла 2θ от 10° до 25°.
2. Аморфная форма соединения, представленного формулой (I), по п. 1, причем порошковая рентгеновская дифрактограмма аморфной формы представлена на фиг. 1.
3. Аморфная форма по п. 1, причем кривая дифференциальной сканирующей калориметрии аморфной формы имеет начальные точки двух эндотермических пиков при 69,28±3°C и 239,33±3°C.
4. Аморфная форма по п. 3, причем график DSC аморфной формы представлен на фиг. 2.
5. Аморфная форма по п. 1, причем кривая термогравиметрического анализа аморфной формы характеризуется потерей массы, достигающей 0,9958% при 120,00±3°C.
6. Аморфная форма по п. 5, причем график TGA аморфной формы представлен на фиг. 3.
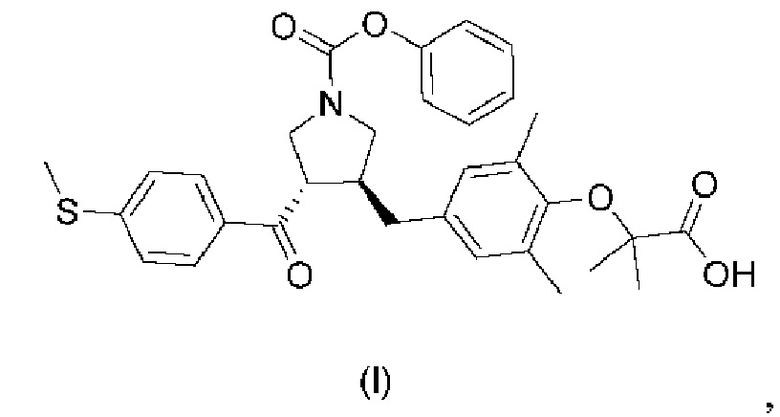
7. Способ получения аморфной формы соединения формулы (I), предусматривающий: добавление соединения формулы (I) в растворитель для нагревания с перемешиванием или перекристаллизации с получением аморфной формы
причем растворитель является выбранным из группы, состоящей из метанола, этанола, тетрагидрофурана, этилацетата и н-гептана, нагревание с перемешиванием осуществляют при температуре перемешивания, составляющей от 25°C до 45°C, период взбивания составляет от 2 часов до 48 часов, и соотношение масса/объем соединения и растворителя составляет 1:3,5-6 г/мл в способе получения.
| US 8642660 B2, 04.02.2014 | |||
| Воронка с приспособлением для очищения наливаемой жидкости | 1929 |
|
SU15106A1 |

