[1] Данная заявка испрашивает приоритет по заявке на патент Китая № CN 2018109690068, поданной 23 августа 2018 г., содержание которой включено в данный документ во всей своей полноте.
Область техники
[2] Настоящее изобретение относится к кристаллической форме А и кристаллической форме В соединения, представленного формулой (I), и их применению в получении лекарственного препарата для лечения HBV.
Уровни техники
[3] Гепатит В представляет собой воспалительный ответ, обусловленный проникновением вируса гепатита В, который может привести к ряду проблем, таких как боль в области печени, гепатоспленомегалия, фиброз печени, и цирроз, и даже рак печени в тяжелых случаях. Статистически в мире насчитывается 350-400 миллионов носителей вируса гепатита В, 1/3 из которых находится в Китае, а число смертей, обусловленных гепатитом В, в Китае составляет до 500000 каждый год.
[4] На данном этапе в мире нет конкретных лекарственных средств для лечения гепатита В. Лекарственные средства первой линии для лечения гепатита В в Китае в основном представляют собой нуклеозидные лекарственные средства, интерфероны и средства традиционной китайской медицины. Однако существуют такие проблемы, как высокая стоимость и подверженность рецидивам, и поэтому крайне необходимо разработать новый тип лекарственного средства для лечения гепатита.
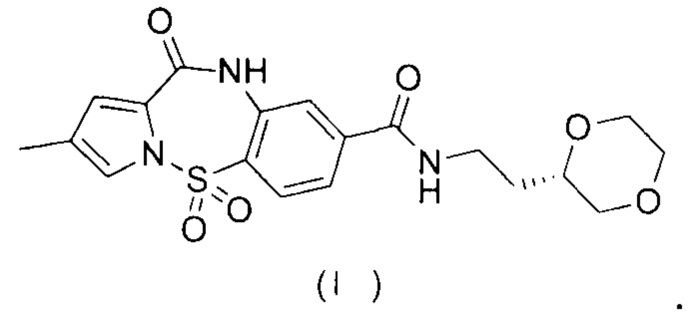
[5] В WO 2008154817 A1 раскрыта следующая структура GLS4:

Содержание изобретения
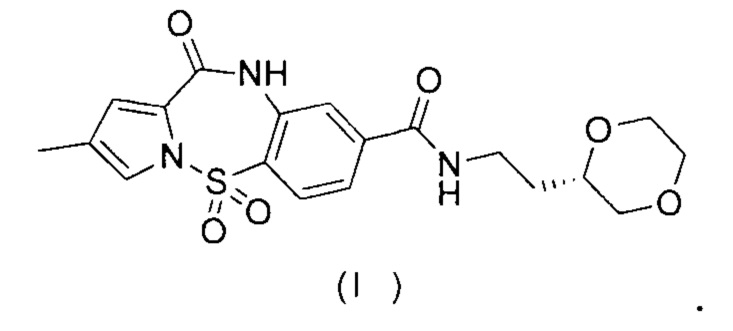
[6] В настоящем изобретении предусмотрена кристаллическая форма А соединения, представленного формулой (I), при этом ее порошковая рентгеновская дифрактограмма (XRPD) содержит характеристические дифракционные пики при следующих значениях угла 2θ: 5,56±0,2°, 10,84±0,2°, 15,56±0,2°, 16,17±0,2°, 22,14±0,2°, 22,70±0,2°, 27,76±0,2° и 28,44±0,2°,

[7] В некоторых вариантах осуществления настоящего изобретения дифрактограмма XRPD кристаллической формы А, как определено выше, представлена на фигуре 1.
[8] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы А, как определено выше, представлены в таблице 1.
[9] Таблица 1. Аналитические данные дифрактограммы XRPD кристаллической формы А
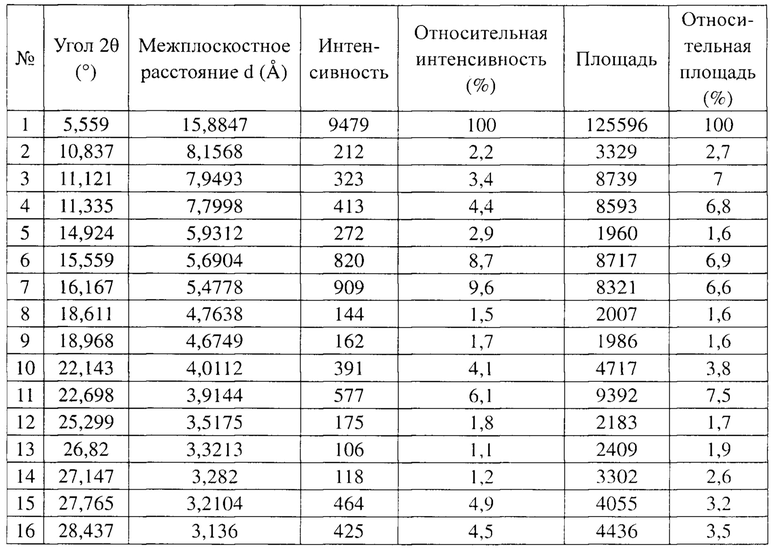
[10] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма А, как определено выше, проявляет эндотермический пик с началом при 229,95°С, как измерено с помощью кривой дифференциальной сканирующей калориметрии (DSC).
[11] В некоторых вариантах осуществления настоящего изобретения DSC кристаллической формы А, как определено выше, показана на фигуре 2.
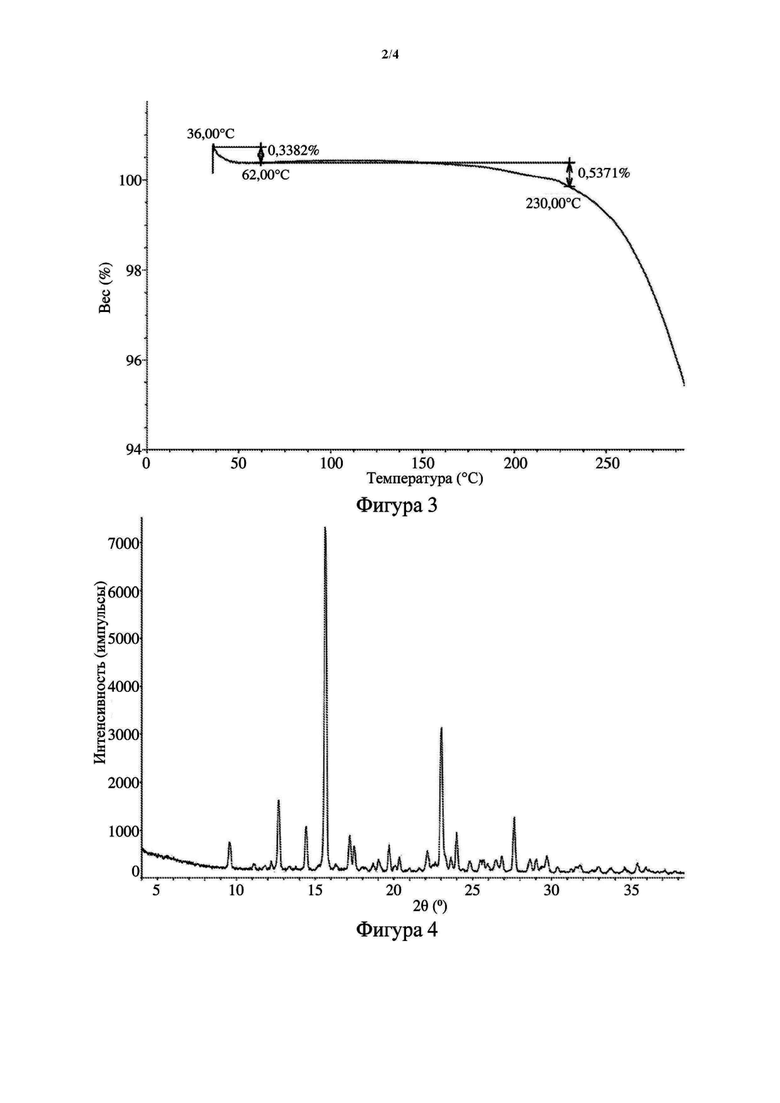
[12] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма А, как определено выше, имеет кривую термогравиметрического анализа (TGA) с потерей веса 0,3382% при 62±3°С и потерей веса 0,8753% при 230±3°С.
[13] В некоторых вариантах осуществления настоящего изобретения TGA кристаллической формы А, как определено выше, показана на фигуре 3.
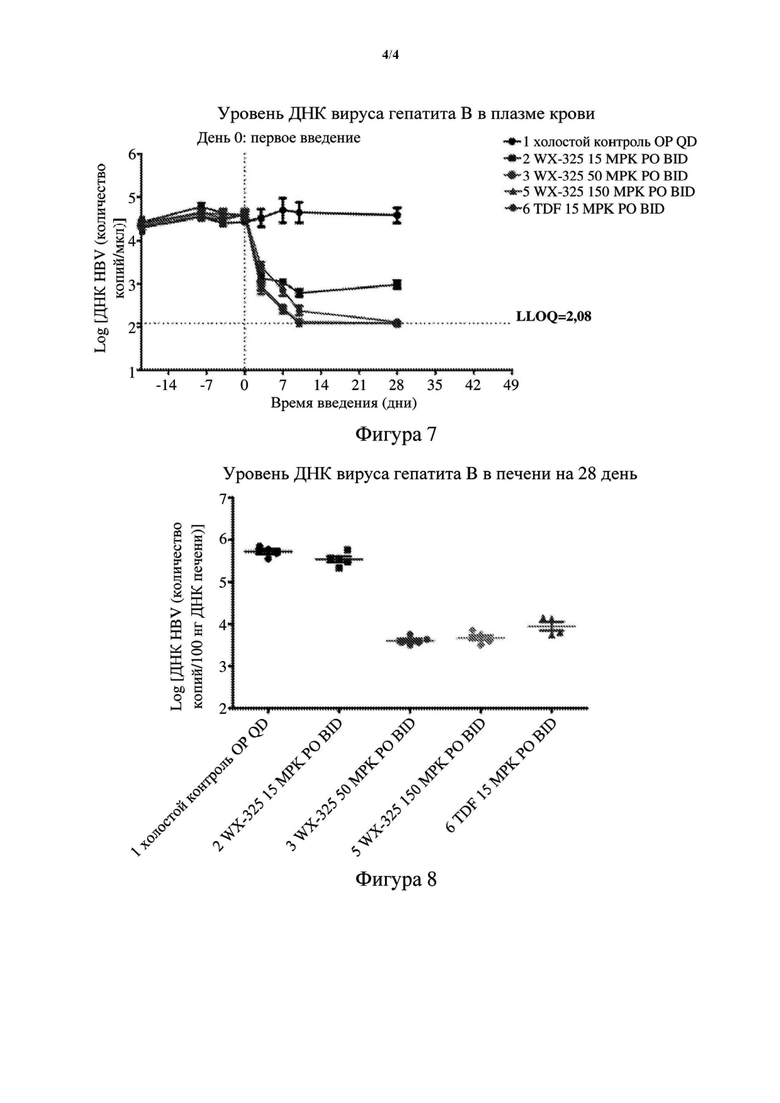
[14] В настоящем изобретении также предусмотрена кристаллическая форма В соединения, представленного формулой (I), при этом ее порошковая рентгеновская дифрактограмма содержит характеристические дифракционные пики при следующих значениях угла 2θ: 9,56±0,2°, 12,70±0,2°, 14,41±0,2°, 15,64±0,2°, 19,70±0,2°, 23,03±0,2°, 23,98±0,2° и 27,65±0,2°.
[15] В некоторых вариантах осуществления настоящего изобретения дифрактограмма XRPD кристаллической формы В, как определено выше, представлена на фигуре 4.
[16] В некоторых вариантах осуществления настоящего изобретения данные анализа из дифрактограммы XRPD кристаллической формы В, как определено выше, представлены в таблице 2.
[17] Таблица 2. Аналитические данные дифрактограммы XRPD кристаллической формы В

[18] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма В, как определено выше, проявляет эндотермический пик с началом при 233,59°С, как измерено с помощью кривой дифференциальной сканирующей калориметрии.
[19] В некоторых вариантах осуществления настоящего изобретения DSC кристаллической формы В, как определено выше, показана на фигуре 5.
[20] В некоторых вариантах осуществления настоящего изобретения кристаллическая форма В, как определено выше, имеет кривую термогравиметрического анализа с потерей веса 0,04890% при 120±3°С.
[21] В некоторых вариантах осуществления настоящего изобретения TGA кристаллической формы В, как определено выше, показана на фигуре 6.
[22] В настоящем изобретении также предусмотрено применение кристаллической формы А или кристаллической формы В, как определено выше, в получении лекарственного препарата для лечения HBV.
[23] Технические эффекты
[24] Кристаллическая форма А и кристаллическая форма В соединения, представленного формулой (I), по настоящему изобретению, являются устойчивыми и меньше подвержены влиянию тепла и влажности с хорошей эффективностью при введении in vivo и имеют широкие перспективы в получении лекарственных препаратов.
[25] Определения и объяснения
[26] Если не указано иное, предполагается, что следующие термины и выражения, используемые в настоящем документе, имеют следующие значения. Конкретный термин или выражение при отсутствии точного определения не следует считать неопределенным или неясным, а следует понимать в соответствии с традиционным значением. Если в данном документе встречается торговое название, то предполагается, что оно относится к соответствующему продукту или его активному ингредиенту.
[27] Промежуточные соединения по настоящему изобретению могут быть получены с помощью различных способов синтеза, известных специалисту в данной области техники, в том числе вариантов осуществления, описанных ниже, вариантов осуществления, образованных путем объединения вариантов осуществления, описанных ниже, с другими способами химического синтеза, и эквивалентных альтернатив, общеизвестных специалисту в данной области техники. Предпочтительные варианты осуществления включают без ограничения варианты осуществления настоящего изобретения.
[28] Химические реакции в вариантах осуществления настоящего изобретения осуществляют в подходящем растворителе, и растворитель должен быть подходящим для химического изменения, и с применением необходимых реагентов и материалов в соответствии с настоящим изобретением. С целью получения соединения по настоящему изобретению, специалистам в данной области техники иногда необходимо модифицировать или выбирать стадии синтеза или схемы реакций на основе существующих вариантов осуществления.
[29] Настоящее изобретение будет конкретно описано ниже с помощью вариантов осуществления, но объем настоящего изобретения ими не ограничивается.
[30] Все растворители, применяемые в настоящем изобретении, являются коммерчески доступными и могут непосредственно применяться без дополнительной очистки.
[31] Применяемые в настоящем изобретении растворители являются коммерчески доступными. В настоящем изобретении используются следующие сокращения: ЕЮН обозначает этанол; МеОН обозначает метанол; TFA обозначает трифторуксусную кислоту; TsOH обозначает п-толуолсульфоновую кислоту; т. пл. обозначает точку плавления; EtSO3H обозначает этансульфоновую кислоту; MeSO3H обозначает метансульфоновую кислоту; THF обозначает тетрагидрофуран; EtOAc обозначает этилацетат; THF обозначает тетрагидрофуран; ЕА обозначает этилацетат; DMAP обозначает 4-диметиламинопиридин; DCM обозначает дихлорметан; DIPEA обозначает N,N-диизопропилэтиламин.
[32] Порошковая рентгеновская дифракция (XRPD)
[33] Применяли приблизительно 10-20 мг образца для обнаружения с помощью XRPD.
[34] Подробные параметры XRPD являются следующими:
[35] рентгеновская трубка: Cu, kα, (λ = 1,54056 Å);
[36] напряжение на рентгеновской трубке: 40 кВ; сила тока на рентгеновской трубке: 40 мА;
[37] щель расходимости: 0,60 мм;
[38] щель детектора: 10,50 мм;
[39] антирассеивающая щель: 7,10 мм;
[40] диапазон сканирования: 3-40 град, или 4-40 град.;
[41] размер шага: 0,02 град.;
[42] время шага: 0,12 секунды;
[43] скорость вращения лотка для образцов: 15 об./мин.
[44] Дифференциальная сканирующая калориметрия (DSC)
[45] 0,5-1 мг образца помещали в алюминиевый тигель для DSC для проведения испытания путем повышения температуры образца от 30°С до 300°С со скоростью 10°С/ мин. при 50 мл/мин. N2.
[46] Термогравиметрический анализ (TGA)
[47] 2-5 мг образца помещали в платиновый тигель для TGA для проведения испытания и нагревали со скоростью 10°С/мин. при 25 мл/мин. N2 с повышением температуры образца от комнатной температуры до 300°С или до потери 20% веса образца.
[48] Анализ динамической сорбции пара (DVS)
[49] Условия обнаружения: 10-15 мг образца помещали в лоток для образцов для обнаружения с помощью DVS.
[50] Подробные параметры DVS указаны ниже:
[51] температура: 25°С;
[52] равновесие: dm/dt=0,01%/MHH.: (наименьшее: 10 мин., наибольшее: 180 мин.);
[53] высушивание: относительная влажность 0%, 120 мин.;
[54] градиент относительной влажности (%) для проведения испытания: 10%;
[55] диапазон градиента относительной влажности (%) для проведения испытания: 0%-90%-0%.
[56] Ниже приведены критерии для оценки.

Краткое описание графических материалов
[58] На фигуре 1 представлена дифрактограмма XRPD кристаллической формы А соединения, представленного формулой (I), с применением излучения Cu-Kα.
[59] На фигуре 2 представлена термограмма DSC кристаллической формы А соединения, представленного формулой (I).
[60] На фигуре 3 представлена термограмма TGA кристаллической формы А соединения, представленного формулой (I).
[61] На фигуре 4 представлена дифрактограмма XRPD кристаллической формы В соединения, представленного формулой (I), с применением излучения Cu-Kα.
[62] На фигуре 5 представлена термограмма DSC кристаллической формы В соединения, представленного формулой (I).
[63] На фигуре 6 представлена термограмма TGA кристаллической формы В соединения, представленного формулой (I).
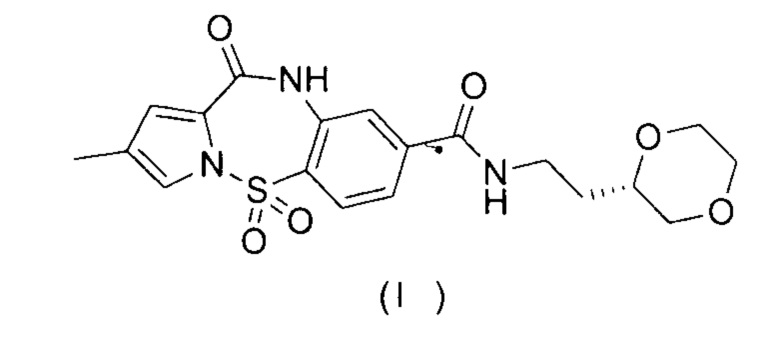
[64] Фигура 7. Уровень ДНК вируса гепатита В в плазме крови; ломаная линия 1 обозначает 10% водный раствор Solutol в качестве холостого контроля, который вводили один раз в сутки (QD), перорально (РО); ломаная линия 2 обозначает, что WX-325 вводили в дозе 15 мг/кг два раза в сутки (BID) в интервалом 8 часов, перорально (РО); ломаная линия 3 обозначает, что WX-325 вводили в дозе 50 мг/кг два раза в сутки (BID) с ин тервалом 8 часов, перорально (РО); ломаная линия 5 обозначает, что WX-325 вводили в дозе 150 мг/кг два раза в сутки (BID) с интервалом 8 часов, перорально (РО); ломаная линия 6 обозначает, что тенофовир (TDF) в качестве соединения, являющегося положительным контролем, вводили в дозе 15 мг/кг два раза в сутки (BID) с интервалом 8 часов, перорально (РО); LLOQ обозначает нижний предел обнаружения; примечание: WX-325 на фигуре 7 обозначает соединение, представленное формулой (I), по настоящему изобретению.
[65] Фигура 8. Уровень ДНК вируса гепатита В в печени на 28-й день; примечание: QD обозначает один раз в сутки; BID обозначает два раза в сутки; МРК обозначает мг/кг; носитель обозначает раствор в качестве холостого контроля; примечание: WX-325 на фигуре 8 обозначает соединение, представленное формулой (I), по настоящему изобретению.
Подробное описание предпочтительного варианта осуществления
[66] С целью лучшего понимания содержания настоящего изобретения следующие варианты осуществления дополнительно иллюстрируют настоящее изобретение, но настоящее изобретение не ограничивается ими.
[67] Пример 1. Получение соединения, представленного формулой (I)

[68] Путь синтеза
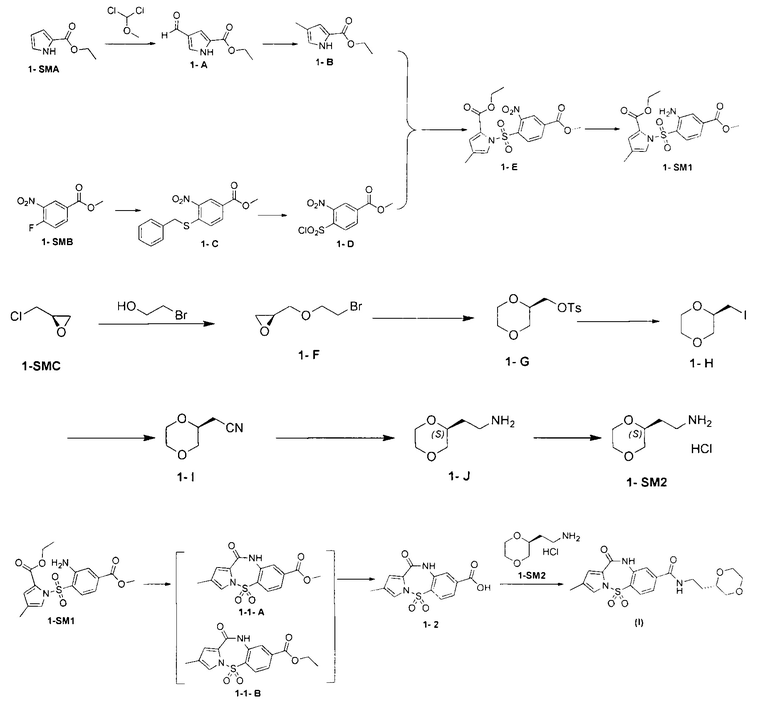
[69] Стадия 1. Синтез соединения 1-А
[70] Безводный дихлорметан (5 л) добавляли в сухую трехгорлую колбу (10 л) и перемешивали, затем в трехгорлую колбу последовательно добавляли соединение 1-SMA (500,00 г) и нитрометаы с получением смеси. Смесь помещали на баню с сухим льдом и этанолом и охлаждали до -10°С. Температуру контролировали в пределах от -10°С до 0°С. Трихлорид алюминия (1,15 кг) медленно добавляли в реакционную колбу и температуру контролировали до менее 0°С. Затем в реакционный сосуд медленно добавляли α,α-дихлордиметилметиловый эфир (495,00 г) с получением реакционного раствора, который медленно нагревали до комнатной температуры и перемешивали в течение 18 часов. С помощью мониторинга посредством TLC (РЕ:ЕА 3:1) выявили исчезновение пятна исходного материала и появление нового пятна с высокой полярностью. Раствор бисульфата калия (3 л) медленно добавляли по каплям к экстрагированному реакционному раствору до концентрации 10%, перемешивали в течение 20 минут, при этом добавляли измельченный лед, чтобы предотвратить перегрев. Смешанный раствор переносили в делительную воронку объемом 25 л и оставляли отстояться для расслоения, чтобы отделить слой дихлорметана, и экстрагировали водную фазу дихлорметаном (2 л*2). После промывания 10% раствором бисульфата калия (5 л*2) органическую фазу отделяли и высушивали над безводным сульфатом натрия (1 кг). Органическую фазу концентрировали при пониженном давлении с получением соединения 1-А в виде темно-зеленого твердого вещества.
[71] 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 9,97 (br s, 1Н), 9,87-9,82 (m, 1Н), 7,58 (dd, 7=1,5, 3,3 Гц, 1Н), 7,36-7,29 (m, 1Н), 4,37 (q, 7-7,1 Гц, 2Н), 1,38 (t, 7=7,2 Гц, 3Н).
[72] Стадия 2. Синтез соединения 1-В
[73] Добавляли п-толуолсульфонилгидразид (2,23 кг, 11,96 моль) к раствору THF (20 л), содержащему соединение 1-А (2 кг, 11,96 моль). Раствор перемешивали при 20°С в течение приблизительно 1 ч. При наблюдении за исчезновением исходного материала с помощью ТГС реакционную систему нагревали до 60°С и добавляли порциями цианоборогидрид натрия (902 г, 14,36 моль). После добавления реакционный раствор нагревали до 70°С и перемешивали в течение 3 ч. Затем нагревание прекращали и обеспечивали охлаждение до комнатной температуры, добавляли 5 л воды для гашения реакции с последующим удалением большей части THF при пониженном давлении и остаток экстрагировали большим количеством ЕА (1,5 л*3). Органические фазы объединяли, промывали с помощью насыщенного хлорида натрия и высушивали над безводным сульфатом натрия. Затем органические фазы фильтровали и растворитель удаляли при пониженном давлении. Наконец, неочищенный продукт подвергали колоночной хроматографии с получением соединения 1-В в виде бледно-желтого твердого вещества.
[74] Стадия 3. Синтез соединения 1-С
[75] Добавляли метанол (32 л) в реактор с рубашкой объемом 50 л и перемешивали, затем добавляли последовательно соединение 1-SMB (4000,00 г) и диизопропилэтиламин (5,25 л) и внутреннюю температуру понижали до 5-10°С. Медленно добавляли по каплям бензилмеркаптан (2490,00 г) и внутреннюю температуру поддерживали при 5-15°С. После завершения добавления систему охлаждения отключали для обеспечения повышения температуры естественным образом и продолжали перемешивание в течение 2,5 ч. Затем перемешивание останавливали, скорость доводили до 100 об./мин. и реакционную жидкость высвобождали и фильтровали через настольный фильтр с получением осадка на фильтре. Осадок на фильтре промывали водой трижды (5 л), затем один раз с помощью EtOH (3 л). Осадок на фильтре фильтровали путем фильтрования с отсасыванием до тех пор, пока он не переставал быть вязким, с получением соединения 1-С в виде бледно-желтого твердого вещества.
[76] Стадия 4. Синтез соединения 1-D
[77] Дихлорметан (7,5 л) добавляли в реактор объемом 50 л и перемешивали, затем добавляли соединение 1-С (1500 г). Внутреннюю температуру снижали до 0-10°С и добавляли раствор HCl (6 М, 4,12 л). Добавляли по каплям раствор гипохлорита натрия (коммерчески доступный 8% раствор, 23,0 кг) при 0-10°С с открытой крышкой. После добавления по каплям отключали систему охлаждения и продолжали перемешивание в течение приблизительно 17 часов с открытой крышкой. Затем для теплоемкости добавляли бисульфит натрия (1000 г, 5 л водного раствора) и использовали йодокрахмальную реактивную бумагу, чтобы определить, не осталось ли окислителя в водной фазе. Затем перемешивание останавливали, раствор оставляли отстояться для расслоения. Слой дихлорметана собирали при экстрагировании водного слоя дихлорметаном (2,5 л) и слои дихлорметана объединяли. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали, затем растворитель удаляли при пониженном давлении с получением соединения 1-D в виде белого твердого вещества.
[78] 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 8,50-8,43 (m, 2Н), 8,34 (d, 7=8,2 Гц, 1Н), 4,04 (s, 3Н).
[79] Стадия 5. Синтез соединения 1-Е
[80] Тетрагидрофуран (10 л) добавляли в сухой реактор с рубашкой объемом 50 л и перемешивали, затем добавляли соединение 1-В (2000 г). Внутреннюю температуру снижали до 0-10°С. Температуру поддерживали при 0-15°С в течение приблизительно 1,5 часов и добавляли трет-бутоксид калия (1 М раствор THF, 15,67 л). После добавления температуру повышали до приблизительно 20°С и продолжали перемешивание в течение 1 часа. Затем температуру снижали до 0-10°С и медленно добавляли раствор THF (10 л), содержащий соединение 1-D (4380 г). После добавления температуру медленно повышали до 15°С и перемешивание продолжали в течение приблизительно 16 часов. Добавляли этилацетат (10 л) для экстракции и дважды промывали органическую фазу насыщенным раствором хлоридом натрия (10 л). Водные фазы объединяли и экстрагировали с помощью ЕА (5 л) и органические фазы объединяли. Наконец, растворитель удаляли из органической фазы при пониженном давлении с получением соединения 1-Е в виде бледно-желтого твердого вещества.
[81] 1Н ЯМР (400 МГц, DMSO-d6) δ = 8,55 (d, 7=1,4 Гц, 1Н), 8,37 (dd, 7=1,5, 8,3 Гц, 1Н), 7,91 (d, 7=8,3 Гц, 1Н), 7,60 (s, 1Н), 7,13 (d, 7=1,8 Гц, 1Н), 4,02 (q, 7=7,0 Гц, 2Н), 3,93 (s, 3Н), 2,10 (s. 3Н), 1,08 (t, 7=7,1 Гц, 3Н).
[82] Стадия 6. Синтез соединения 1-SM1
[83] Соединение 1-Е (1000,0 г) добавляли в сухую трехгорлую колбу объемом 10 л и перемешивали, затем добавляли ледяную уксусную кислоту (5 л) и внутреннюю температуру реакции контролировали при 25-30°С. Медленно добавляли порошок железа (1 экв., 140,9 г). После перемешивания в течение 30 минут медленно добавляли вторую партию порошка железа (0,5 экв., 70,44 г). После перемешивания, продолжающегося в течение 30 минут, добавляли третью партию порошка железа (0,5 экв., 70,44 г). Через еще 30 мин. перемешивания добавляли четвертую партию порошка железа (0,5 экв. 70,44 г) и реакционную смесь продолжали перемешивать с мониторингом исчезновения исходного материала и появления новой точки с высокой полярностью. Затем перемешивание останавливали и реакционный раствор переносили в дозатор объемом 25 л для разделения жидкости. Добавляли 10 л этилацетата к раствору, который затем дважды промывали с помощью 5 л насыщенного водного раствора бисульфата натрия. После отделения жидкости водную фазу подвергали обратной экстракции с помощью 5 л этилацетата. Органические фазы затем объединяли и промывали с помощью 10% водного раствора NaOH до рН>8 и органические фазы собирали путем разделения жидкостей. Органическую фазу концентрировали при пониженном давлении с получением соединения 1-SM1 в виде белого твердого вещества.
[84] 1Н ЯМР (400 МГц, DMSO-d6) δ = 7,79-7,71 (m, 2Н), 7,50 (d, 7=1,8 Гц, 1Н), 7,14 (dd, 7=1,7, 8,5 Гц, 1Н), 6,96 (d, 7=2,0 Гц, 1Н), 6,42 (s, 2Н), 4,11 (q, 7=7,2 Гц, 2Н), 3,84 (s, 3Н), 2,04 (s, 3Н), 1,16 (t, 7=7,1 Гц, 3Н).
[85] Стадия 7. Синтез соединения 1-F
[86] Добавляли толуол (12 л) в сухой реактор с рубашкой объемом 50 л и перемешивали, затем добавляли 2-бромэтанол (9930 г) с последующим добавлением эфира трифторида бора (268 г). Реакционную смесь нагревали до 30-35°С. Медленно добавляли по каплям соединение 1-SMC (3500 г) и завершали добавление через приблизительно 1,5 часа. После добавления повышали внутреннюю температуру реакции до приблизительно 55-65°С и регулировали настройки температуры нагревателя до 60°С для поддержания внутренней температуры при 55-65°С в течение 1 часа. Затем внутреннюю температуру системы снижали до приблизительно 10°С и в реакционную систему медленно добавляли водный раствор гидроксида натрия (3783 г, 17,5 л воды) при приблизительно 20°С при поддержании внутренней температуры при 10-20°С. После добавления раствора NaOH контроль температуры нагревателя отключали и реакционную смесь продолжали перемешивать в течение приблизительно 16 ч. Затем перемешивание останавливали для стационарного разделения реакционного раствора. Водный слой экстрагировали 2-метилтетрагидрофураном (10 л) и органические фазы объединяли, промывали с помощью воды (10 л) и оставляли отстояться для расслоения для сбора органической фазы. Наконец, органическую фазу концентрировали при пониженном давлении с получением соединения 1-F в виде бесцветного масла.
[87] 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 3,87-3,71 (m, 4Н), 3,66-3,59 (m, 3Н), 3,42 (dd, J=6,0, 11,7 Гц, 1Н), 3,20-3,13 (m, 1Н), 2,79 (t. 7=4,6 Гц, 1Н), 2,65-2,59 (m, 1Н).
[88] Стадия 8. Синтез соединения 1-G
[89] Водный раствор гидроксида натрия (3240 г, 15 л воды) добавляли в реактор с рубашкой объемом 50 л, затем добавляли соединение 1-F (4430 г) и включали нагревание. После нагревания реакционной смеси до 90°С продолжали перемешивание в течение 1 ч. Включали охлаждение для снижения температуры реакции до приблизительно 15°С, затем добавляли раствор THF (6180 г, 15 л тетрагидрофурана), содержащий раствор п-толуолсульфонилхлорида. Контроль температуры нагревателя отключали и реакционную смесь дополнительно перемешивали при приблизительно 15°С в течение приблизительно 16 ч. Затем перемешивание останавливали для обеспечения отстаивания реакционной смеси для расслоения. Водную фазу экстрагировали 2-метилтетрагидрофураном (10 л) и фазу 2-метилтетрагидрофурана (представляющую собой белое нерастворимое вещество, которое исчезало после промывки) промывали водой (5 л) и органические фазы объединяли. DMAP (500 г) и триэтиламин (2,5 л) добавляли к органической фазе с последующим перемешиванием органической фазы в течение 30 минут. Затем органическую фазу промывали насыщенным раствором хлорида натрия (10 л) и оставляли отстояться для расслоения и водную фазу отбрасывали. Органическую фазу последовательно промывали раствором гидросульфата калия (3800 г, 15 л воды) и насыщенным раствором хлорида натрия (5 л, дважды), а затем оставляли отстояться для расслоения и собирали органическую фазу. Органическую фазу наконец концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта в виде соединения 1-G.
[90] 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 7,77 (d, J=8,3 Гц, 2Н), 7,34 (d, J=8,2 Гц, 2Н), 4,03-3,91 (m, 2Н), 3,80-3,49 (m, 8Н), 3,33 (dd,.7=9,9, 11,4 Гц, 1H), 2,43 (s, 3Н).
[91] Стадия 9. Синтез соединения 1-Н
[92] Ацетон (30 л) добавляли в чистый реактор с рубашкой объемом 50 л, затем добавляли соединение 1-G (4500 г) с последующим добавлением йодида натрия (6190 г). Включали нагревание и после нагревания реакционной смеси до 75°С продолжали перемешивание в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении при 50°С. Добавляли этилацетат (15 л) и воду (10 л) в концентрированный неочищенный продукт и смесь перемешивали, а затем оставляли отстояться для расслоения. Органическую фазу промывали 0,5 М тиосульфатом натрия (10 л). Водную фазу и раствор тиосульфата натрия объединяли и экстрагировали с помощью EtOAc (5 л). Органические фазы объединяли и промывали с помощью насыщенного раствора хлорида натрия (10 л), затем оставляли отстояться для расслоения и собирали органическую фазу. Органическую фазу наконец концентрировали при пониженном давлении для удаления растворителя с получением неочищенного продукта в виде соединения 1-Н.
[93] 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 3,90-3,83 (m, 2Н), 3,81-3,75 (m, 2Н), 3,74-3,65 (m, 5Н), 3,63-3,49 (m, 7Н), 3,31-3,18 (m, 3Н), 3,06-3,04 (m, 2Н).
[94] Стадия 10. Синтез соединения 1-1
[95] DMSO (20 л) добавляли в чистый реактор с рубашкой объемом 50 л, затем добавляли соединение 1-Н (4700 г) и температуру повышали до 35°С с последующим добавлением цианида натрия (1010 г). Внутреннюю температуру реакции повышали до приблизительно 60°С в течение 20 минут, затем температуру постепенно понижали до 35°С и продолжали перемешивание в течение приблизительно 16 часов. Раствор бикарбоната натрия (2000 г бикарбоната натрия, 10 л воды) добавляли в реакционную систему, которую затем перемешивали в течение приблизительно 5 минут. Добавляли EtOAc: МеОН (20 л, 2 л) и реакционную систему дополнительно перемешивали в течение 2 минут с последующим отстаиванием в течение приблизительно 1 часа. Затем реакционную систему разделяли и отделяли приблизительно 30 л нижнего слоя раствора. Нижний слой раствора экстрагировали дважды с помощью ЕЮАс : МеОН (15 л: 1,5 л для первого раза и 5 л: 0,5 л для второго раза). После экстракции верхнюю органическую фазу и верхний слой оставшейся реакционной жидкости объединяли, трижды промывали насыщенным раствором хлорида натрия (каждый по 10 л) и оставляли отстояться для расслоения. Водную фазу отбрасывали, в то время как органическую фазу собирали. Наконец, удаляли растворитель из органической фазы при пониженном давлении и неочищенный продукт подвергали колоночной хроматографии с получением соединения 1 -I в виде бесцветного масла.
[96] 1Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 3,84-3,65 (m, 6Н), 3,61-3,53 (m, 2Н), 3,35 (t, J=10,5 Гц, 1Н), 2,49-2,44 (m, 2Н).
[97] Стадия 11. Синтез соединения 1-J
[98] В защитной атмосфере аргона добавляли никель Ренея (10,00 г, 116,73 ммоль) и EtOH (150 мл) в сухую колбу для гидрогенизации, а затем добавляли 1-I (20 г, 157,31 ммоль) и NH3⋅H2O (13,65 г, 97,36 ммоль, 15,00 мл, чистота 25%) с последующей заменой и реакционную смесь перемешивали при 50 фунтах/кв. дюйм и 50°С в течение 3,5 ч. Реакционный раствор фильтровали с применением диатомита и фильтрат концентрировали при пониженном давлении с получением соединения 1-J в виде желтого масла.
[99] 'Н ЯМР (400 МГц, дейтерированный хлороформ) δ = 3,82-3,57 (m, 6Н), 3,34-3,18 (m, 1Н), 2,86-2,72 (m, 2Н), 1,60-1,38 (m, 2Н).
[100] Стадия 12. Синтез соединения 1-SM2
[101] Добавляли 1-J (800,00 г) в трехгорлую колбу объемом 5 л и перемешивали, затем добавляли этилацетат (800 мл) в течение 0,5 ч. и медленно добавляли по каплям 4М HCl/EtOAc (1,6 л) до тех пор, пока значение рН системы не стало меньше 5, при этом внутреннюю температуру поддерживали при 5-15°С. Затем систему охлаждения отключали и реакционную смесь нагревали до комнатной температуры и дополнительно перемешивали в течение 1 часа. После прекращения перемешивания реакционную смесь фильтровали через настольный фильтр с получением осадка на фильтре, который затем концентрировали при пониженном давлении (40-45°С) с получением неочищенного продукта. Ацетонитрил (2 мл/г) добавляли к предыдущему продукту и смесь суспендировали в течение 1 часа. После суспендирования продукт фильтровали, осадок на фильтре собирали по отдельности и органический раствор удаляли при пониженном давлении с получением белого твердого вещества соединения 1-SM2.
[102] 1Н ЯМР (399 МГц, МЕТАНОЛ-d4) δ = 3,88-3,72 (m, 5Н), 3,67-3,59 (m, 1Н), 3,36-3,31 (m, 1Н), 3,14 (t, J=6,7 Гц, 2Н), 1,87-1,67 (m, 2Н).
[103] Стадия 13. Синтез соединения 1-1-А и соединения 1-1-В
[104] Добавляли толуол (20 л) в сухой реактор с рубашкой объемом 50 л и перемешивали, затем добавляли соединение 1-SM1 (2500 г) и внутреннюю температуру повышали до 30-35°С.Среду инертного газа в реакторе поддерживали продувкой азотом. Затем добавляли по каплям триметилалюминий (3,0 л, температуру в реакторе медленно повышали добавлением Al(СН3)3). После добавления отключали продувку азотом. Температуру повышали до 80-85°С и реакционную смесь дополнительно перемешивали в течение приблизительно 16 часов. Затем отключали охлаждение для снижения температуры реакционной смеси до 20-30°С.Половину реакционного раствора (приблизительно 12 л), к которому добавляли EtOAc (10 л), переносили и хорошо перемешивали. Смешанный раствор добавляли к раствору 10% KHSO4 (10 мл) при перемешивании, перемешивали в течение 2 минут, а затем оставляли отстояться для расслоения. Органический слой промывали с помощью 10% раствора KHSO4 (10 л), водные фазы объединяли и экстрагировали дважды с помощью DCM (каждый раз по 7,5 л). Переносили другую половину реакционного раствора (приблизительно 12 л) и обрабатывали таким же образом, как определено выше. Затем органические фазы объединяли и концентрировали при пониженном давлении с получением неочищенного продукта. Добавляли двукратный объем н-гептана и взбивали в течение 1 часа с образованием суспензии. Суспензию фильтровали и высушивали в вакууме в течение > 12 часов при 40°С, Р≤-0,1МРа. Смесь соединения 1-1-А и соединения 1-1-В получали в виде белого твердого вещества.
[105] Стадия 14. Синтез соединения 1-2
[106] Тетрагидрофуран (3840 мл) добавляли в трехгорлую колбу объемом 10 л и перемешивали и медленно добавляли смесь соединения 1-1-А и соединения 1-1-В (480,00 г), затем медленно добавляли по каплям раствор LiOH⋅H2O (118,84) в Н2О (960 мл). После добавления температуру повышали до 60°С и реакционную смесь перемешивали в течение 1 часа. Затем добавляли концентрированную HCl к реакционному раствору для доведения рН системы до 2 и останавливали перемешивание. Затем раствор оставляли отстаиваться для расслоения жидкости. Водную фазу экстрагировали дважды с помощью THF (600 мл) и органические фазы объединяли и концентрировали при пониженном давлении (40-45°С). Твердое вещество суспендировали в чистой воде (2 мл/г) в течение 0,5 часа, и фильтровали, и осадок на фильтре высушивали в вакууме в течение 12 часов при 40°С и Р≤-0,1МРа с получением соединения 1-2 в виде бледно-желтого твердого вещества.
[107] 1Н ЯМР (399 МГц, DMSO-d6) δ = 11,19 (s, 1Н). 8,09 (d, J=8,2 Гц, 1H), 8,00 (d, J=1,6 Гц, 1H), 7,88 (dd, J=1,5, 8,3 Гц, 1H), 7,39 (dd, J=1,2, 2,0 Гц, 1H), 7,01 (d, J=2,0 Гц, 1H), 2,05 (s, 3H).
[108] Стадия 15. Синтез соединения, представленного формулой (1)
[109] DMF (2,25 л) добавляли в трехгорлую колбу объемом 5 л и перемешивали, затем последовательно добавляли соединение 1-2 (400,00 г) и HATU (744,83 г) и перемешивали в течение 30 мин. с последующим добавлением соединения 1-SM2 (229,86 г). Медленно добавляли по каплям DIPEA (568,68 мл) при комнатной температуре в течение 1 часа. После добавления реакционную смесь дополнительно перемешивали при комнатной температуре в течение 16 часов. Затем реакционный раствор переносили в делительную воронку, добавляли этилацетат (2 л) и чистую воду (1 л), и раствор перемешивали в течение 2 мин., а затем оставляли отстаиваться для расслоения с отделением водной фазы. Затем добавляли чистую воду (1 л) для промывки водной фазы, которую затем перемешивали и оставляли отстояться для расслоения. Объединенную водную фазу экстрагировали три раза с помощью EtOAc (500 мл) и органические фазы объединяли. Органическую фазу последовательно промывали дважды с помощью раствора карбоната натрия (1,5 л), дважды с помощью раствора гидросульфата калия (1 л) и дважды чистой водой (1 л). Органическую фазу концентрировали при пониженном давлении (40-45°С) с получением неочищенного продукта. Этилацетат (2 мл/г) добавляли к неочищенному продукту и смесь суспендировали в течение 1 часа. Наконец, суспендированную смесь фильтровали и собирали осадок на фильтре с получением соединения, представленного формулой (I).
[110] 1Н ЯМР (400 МГц, DMSO-d6) δ = 11,13 (br s, 1Н), 8,73 (br t, J=5,5 Гц, 1H), 8,05 (d, J=8,2 Гц, 1H), 7,83 (d, J=1,3 Гц, 1H), 7,74 (dd, J=1,5, 8,4 Гц, 1H), 7,36 (s, 1H), 6,98 (d, J=2,0 Гц, 1H), 3,71-3,48 (m, 5H), 3,45-3,31 (m, 1H), 3,45-3,30 (m, 1H), 3,27-3,21 (m, 1HJ, 3,14 (dd, J=9,9, 11,2 Гц, 1H), 2,03 (s, 3H), 1,53 (q, J=7,0 Гц, 2H).
[111] Пример 2. Получение кристаллической формы А соединения, представленного формулой (I)
[112] Последовательно добавляли 35 мг соединения, представленного формулой (I), и 400 мкл трет-бутилметилового эфира во флакон с жидкой фазой объемом 1,5 мл и затем смешивали или растворяли с помощью ультразвука. Образец суспензии перемешивали (с защитой от света) в течение 3 дней в шейкере с постоянной температурой (40°С). Затем раствор образца быстро центрифугировали и центрифугированное твердое вещество высушивали в вакуумном сушильном шкафу при 30°С в течение 5 часов. Полученный сухой образец подвергали XRPD с подтверждением того, что была получена кристаллическая форма А соединения, представленного формулой (1).
[113] Пример 3. Получение кристаллической формы В соединения, представленного формулой (I)
[114] Последовательно добавляли 35 мг соединения, представленного формулой (I), и 400 мкл ацетона во флакон с жидкой фазой объемом 1,5 мл и затем смешивали или растворяли с помощью ультразвука. Образец суспензии перемешивали (с защитой от света) в течение 3 дней в шейкере с постоянной температурой (40°С). Затем раствор образца подвергали быстрому центрифугированию и твердое вещество после центрифугирования высушивали в вакуумном сушильном шкафу при 30°С в течение 5 часов. Полученный сухой образец подвергали XRPD с подтверждением того, что была получена кристаллическая форма В соединения, представленного формулой (I).
[115] Пример 4. Испытание в отношении предварительной стабильности кристаллической формы В соединения, представленного формулой (I)
[116] Таблица 3. Условия исследования и испытуемый параметр

[117] Примечание:
[118] X указывает на испытуемые параметры, в том числе XRPD, содержание и сопутствующие вещества, образцы 0 дня хранили при -20°С;
[119] ICH указывает на условия освещения, которые определяют размещение образцов, то есть освещение (общая освещенность = 1,2×106 люкс⋅ч./ближняя ультрафиолетовая область = 200 Вт⋅ч./м2, с открытой крышкой).
[120] Стадии эксперимента.
[121] Приблизительно 10 мг кристаллической формы В соединения, представленного формулой (I), взвешивали и помещали на дно стеклянного флакона объемом 40 мл и распределяли тонким слоем. Горло флакона оборачивали алюминиевой фольгой (за исключением освещаемых образцов) и проделывали несколько небольших отверстий в алюминиевой фольге для того, чтобы убедиться, что обеспечивается полный контакт образцов с воздухом окружающей среды. Освещаемый образец помещали на дно стеклянного флакона объемом 40 мл, который помещали вертикально в освещенный бокс с открытой крышкой; контрольный образец помещали на дно стеклянных флаконов, которые помещали вертикально с открытой крышкой и оборачивали фольгой для защиты от света. При каждом условии 2 аликвоты образца взвешивали параллельно в каждый момент времени и отбирали дополнительное подходящее количество образца (не взвешивали) для обнаружения с помощью XRPD. Полученные образцы помещали в различные условия, показанные в следующей таблице, и образцы соответственно анализировали посредством HPLC после достижения моментов времени. Способ анализа показан в таблице 4, а экспериментальные результаты показаны в таблице 5.
[122] Анализ посредством HPLC.
[123] 1.1 Получение разбавителя и подвижной фазы
[124] Разбавитель: ацетонитрил: вода (2:1).
[125] Пример: равномерно смешивали 1000 мл ацетонитрила и 500 мл воды, подвергали ультразвуковой дегазации и охлаждали до комнатной температуры.
[126] Подвижная фаза А: 0,1% водный раствор трифторуксусной кислоты.
[127] Пример: добавляли 2,0 мл трифторуксусной кислоты в 2 л воды и хорошо смешивали, подвергали ультразвуковой дегазации и охлаждали до комнатной температуры.
[128] Подвижная фаза В: 100% ацетонитрил.
[129] 1.2 Получение контрольного образца и растворов образца
[130] Кристаллическую форму В соединения, представленного формулой (I), применяли в качестве контроля. Кристаллическую форму В соединения, представленного формулой (I), помещали в стеклянный флакон, затем во флакон добавляли 5 мл разбавителя до концентрации 1 мг/мл и раствор хорошо перемешивали с помощью ультразвука. Затем смесь разбавляли в 5 раз с получением раствора контрольного образца STD1. STD1 разбавляли в 1, 2, 4, 20 и 200 раз с получением 1% контрольного раствора.
[131] 1.3 Получение раствора тестируемого образца
[132] Добавляли разбавитель к каждому тестируемому образцу до концентрации 10 мл, затем образцы растворяли с помощью ультразвука. Анализ посредством HPLC проводили после охлаждения до комнатной температуры.
[133] В то же время раствор образца разбавляли в 5 раз и хорошо смешивали, а затем анализировали посредством HPLC. Способ анализа показан в таблице 4.
[134] Таблица 4. Способ анализа посредством HPLC

[135] Таблица 5. Результаты анализа посредством HPLC из испытания стабильности твердого вещества для кристаллической формы В соединения, представленного формулой (I)
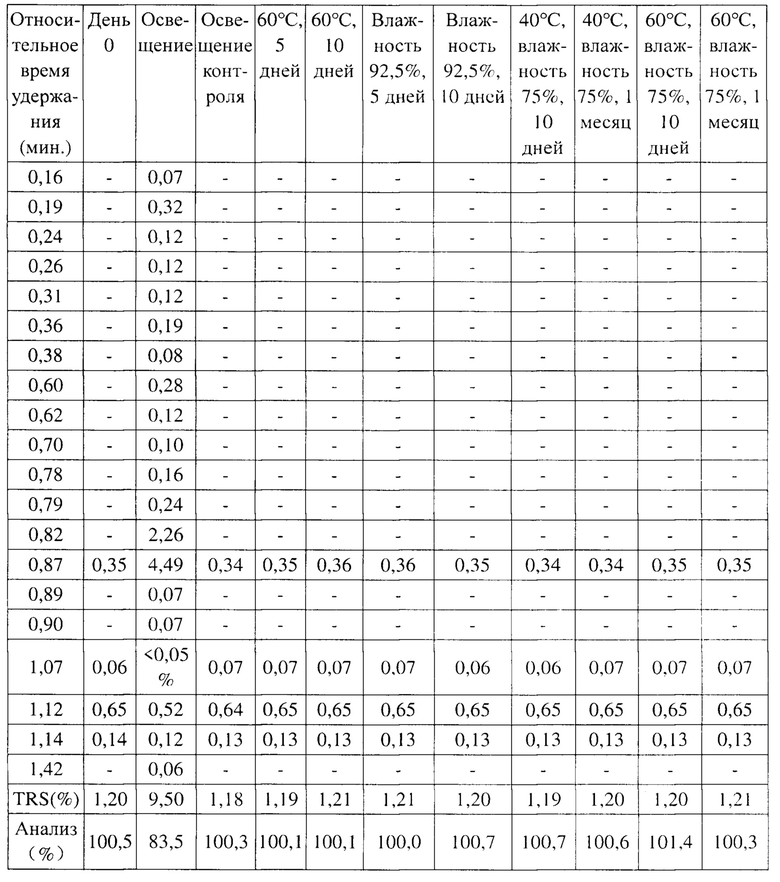
[136] Примечание: «-» означает «не определено»;
[137] TRS относится к сумме оставшихся примесей после удаления основного пика.
[138] Анализ относится к содержанию образца.
[139] Заключение: кристаллическая форма В соединения, представленного формулой (I), является стабильной в условиях высокой температуры и высокой влажности.
[140] Пример 5. Тест в отношении HBV in vitro: количественный qPCR-тест
[141] 1 Цели эксперимента
[142] Содержание ДНК HBV в клетках HepG2.2.15 определяли с помощью количественного qPCR-анализа в реальном времени, а значение ЕС50 соединений применяли в качестве индикатора для оценки ингибирующего действия соединений на HBV.
[143] 2 Экспериментальные материалы
[144] 2.1 Клеточная линия: клетки HepG2.2.15.
[145] Культуральная среда для клеток HepG2.2.15 (DMEM/F12, Invitrogen-11330057; 10% сыворотка, Invitrogen-10099141; 100 единиц/мл пенициллина и 10 мкг/мл стрептомицина, Invitrogen-15140122; 1% заменимых аминокислот, Invitrogen-11140076; 2 мМ L-глутамина, Invitrogen-25030081; 300 мкг/мл генетицина, Invitrogen-10131027).
[146] 2.2 Реагенты
[147] Панкреатин (Invitrogen-25300062);
[148] DPBS (Hyclone-SH30028.01B);
[149] DMSO(Sigma-D2650-100ML);
[150] набор для очистки ДНК с высокой пропускной способностью (QIAamp 96 DNA Blood Kit, Qiagen-51162);
[151] универсальный реагент для зонда FastStart для количественного определения (FastStart Universal Probe Master, Roche-04914058001);
[152] 2.3 Расходные материалы и оборудование
[153] 96-луночный планшет для культивирования клеток (Corning-3599);
[154] СО2-инкубатор (HERA-CELL-240);
[155] оптически прозрачная герметизирующая пленка (ABI-4311971);
[156] 96-луночный планшет для количественной ПЦР (Applied Biosystems-4306737);
[157] оборудование для количественной ПЦР с флуоресцентной детекцией (Applied Biosystems-7500 Real Time PCR System).
[158] 3. Экспериментальные стадии и способы
[159] 3.1 Клетки HepG2.2.15 (4×104 клеток/лунка) помещали в 96-луночный планшет и культивировали в течение ночи при 37°С, 5% СО2.
[160] 3.2 На 2 день соединение разбавляли до в общей сложности 8 концентраций с 3-кратным градиентным разбавлением. Соединения в разных концентрациях добавляли в лунки с культурой в двух повторностях. Конечная концентрация DMSO в культуральной среде составляла 1%. 1 мкМ GLS4 применяли в качестве контроля со 100% ингибированием; 1% DMSO применяли в качестве контроля с 0% ингибированием.
[161] 3.3 На 5 день культуральную среду заменяли свежей культуральной средой, содержащей соединение.
[162] 3.4 На 8 день собирали культуральную среду в лунке с культурой и выделяли ДНК с помощью набора для очистки ДНК с высокой пропускной способностью (Qiagen-51162). Для получения информации о конкретных стадиях обращаться к руководству по применению продукта.
[163] 3.5 Получение реакционного раствора для ПЦР показано в таблице 6.
[164] Таблица 6. Получение реакционного раствора для ПЦР

[165] Последовательность прямого праймера: GTGTCTGCGGCGTTTTATCA (SEQ ID N0.1)
[166] Последовательность обратного праймера:
GACAAACGGGCAACATACCTT (SEQ ID NO. 2)
[167] Последовательность зонда: 5'+FAM+CCTCTKCATCCTGCTGCTATGCCTCATC(SEQ ID NO.3)+TAMRA -3'
[168] 3.6 15 мкл реакционной смеси добавляли в каждую лунку 96-луночного планшета для ПЦР, а затем в каждую лунку добавляли 10 мкл образца ДНК или стандартных ДНК HBV.
[169] 3.7 Условия реакции для ПЦР: нагревание при 95°С в течение 10 минут; затем денатурация при 95°С в течение 15 секунд и удлинение при 60°С в течение 1 минуты, всего 40 циклов.
[170] 3.8 Анализ данных
[171] 3.8.1 Расчет процента ингибирования % инг. = [1 - (количество копий ДНК в образце - количество 1 мкМ копий ДНК в СЕ84)/(количество копий ДНК в контроле DMSO - количество 1 мкМ копий ДНК в GLS4)] × 100.
[172] 3.8.2 Расчет ЕС50 Значение 50% ингибирующей концентрации (ЕС50) соединения в отношении HBV рассчитывали с помощью программного обеспечения GraphPad Prism.
[173] 4. Результаты эксперимента показаны в таблице 7.
[174] Таблица 7. Результаты испытания в отношении ЕС50 из эксперимента с qPCR

[175] Заключение: соединение, представленное формулой (I). обладает значительным ингибирующим эффектом в отношении HBV.
[176] Пример 6. Ингибирование изофермента цитохрома Р450
[177] Цели эксперимента. Определение ингибирующего эффекта тестируемого соединения в отношении активности изоферментов микросомального цитохрома Р450 печени человека (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4).
[178] Процедура эксперимента. Сначала тестируемое соединение (10 мМ) подвергали градиентному разбавлению с получением рабочих растворов (100× конечная концентрация). Концентрации рабочих растворов составляли 5, 1,5. 0,5, 0,15, 0,05, 0,015 и 0,005 мМ. В то же время также получали рабочие растворы смеси (5 в 1) положительных ингибиторов каждого изофермента Р450 (CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4) и специфического субстрата изоферментов; при этом микросомы печени человека, замороженные в холодильнике при -80°С, размораживали на льду. После полного растворения микросом печени человека их разбавляли с помощью РВ с получением определенной концентрации (0,253 мг/мл) рабочего раствора; добавляли 20 мкл смеси субстратов в реакционный планшет (20 мкл РВ добавляли в холостую лунку), а также 158 мкл рабочего раствора микросом печени человека, затем реакционный планшет помещали на лед и отставляли; в это время в соответствующую лунку добавляли 2 мкл каждой концентрации тестируемого соединения (N=1) и специфический ингибитор (N=2), добавляли группу без ингибитора (тестируемое соединение или положительный ингибитор) с соответствующим органическим растворителем в качестве контрольного образца (контрольный образец тестируемого соединения представлял собой смесь 1:1 DMSO: МеОН, и положительный контрольный образец представлял собой смесь 1:9 DMSO: МеОН); после предварительной инкубации на водяной бане при 37°С в течение 10 мин. в реакционный планшет добавляли 20 мкл раствора фактора кофермента (NADPH) и воду, выдержанную при 37°С в течение 10 мин.; добавляли 400 мкл холодного раствора ацетонитрила (внутренний стандарт составлял 200 нг/мл толбутамида и лабеталола) для прекращения реакции; затем реакционный планшет помещали на шейкер и встряхивали в течение 10 мин. с последующим центрифугированием при 4000 об./мин. в течение 20 мин.; 200 мкл надосадочной жидкости добавляли в 100 мкл воды для разбавления образца; наконец, планшет герметично закрывали, хорошо встряхивали и подвергали обнаружению с помощью LC/MS/MS. Экспериментальные результаты показаны в таблице 8.
[179] Таблица 8. Результаты ингибирующего эффекта тестируемого соединения в отношении активности изоферментов микросомального цитохрома Р450 печени человека

[180] Заключение: соединение, представленное формулой (I), не оказывает явного ингибирующего эффекта в отношении CYP1A2, CYP2C9, CYP2C19, CYP2D6 и CYP3A4.
[181] Пример 7. Исследование степени связывания белка в плазме крови
[182] Цели эксперимента: определить степень связывания тестируемого соединения с белками в плазме крови человека и мыши CD-1.
[183] Процедура эксперимента. Отбирали 796 мкл контрольных образцов плазмы крови у человека и мышей CD-1 и добавляли 4 мкл рабочего раствора тестируемого соединения (400 мкМ) или рабочего раствора варфарина (400 мкМ) с получением конечных концентраций обоих тестируемых соединений и варфарина в образце плазмы крови 2 мкМ. Образец тщательно перемешивали. Конечная концентрация DMSO в органической фазе составляла 0,5%; 50 мкл образцов плазмы тестируемого соединения и варфарина переносили в планшет для приема образцов (в трех повторностях) и сразу же добавляли соответствующий объем контрольных образцов плазмы крови или буфера с получением конечного объема каждого образца 100 мкл и объемного соотношения плазмы крови и буфера для диализа 1:1. Затем к этим образцам добавляли 400 мкл останавливающего раствора. Эти образцы будут использоваться в качестве образцов ТО для определения восстановления и стабильности. Образцы Т0 хранили при 2-8°С и ожидали последующей обработки с другими диализованными образцами; 150 мкл тестируемых образцов соединения и варфарина в плазме крови добавляли в часть каждой лунки для диализа, предназначенную для введения дозы, и 150 мкл холостого диализного буфера в принимающую часть лунки каждой лунки для диализа. Затем планшет для диализа накрывали газопроницаемой мембраной и инкубировали в инкубаторе с увлажненной атмосферой при 5% СО2 и 37°С со встряхиванием при приблизительно 100 об./мин. в течение 4 часов. После диализа 50 мкл диализированного образца буфера и диализированного образца плазмы крови вносили пипеткой в новый планшет для приема образцов. Соответствующий объем контрольных образцов плазмы крови или буфера добавляли к образцам так, чтобы конечный объем каждого образца в лунке составлял 100 мкл, и объемное соотношение плазма : буфер для диализа составляло 1:1. Все образцы анализировали после осаждения белка с помощью LC/MS/MS и рассчитывали степень связывания белка и степень восстановления по формуле: % диссоциации (степень диссоциации) - 100 * FC/TC; % связывания (степень связывания)=100 - % диссоциации; % извлечения (степень извлечения) = 100 * (FC + ТС)/Т0. Результаты эксперимента показаны в таблице 9.
[184] Таблица 9. Степень связывания тестируемого соединения с белком в плазме крови человека и мыши CD-1.

[185] Заключение: соединение, представленное формулой (I), проявляет более низкую скорость связывания с белками как в плазме человека, так и в плазме мыши CD-1.
[186] Пример 8. Фармакокинетическое исследование in vivo
[187] 1. Исследование фармакокинетики перорального введения и внутривенной инъекции соединения, представленного формулой (I), на мышах Balb/c.
[188] Соединение, представленное формулой (I), смешивали с 5% DMSO/55% полиэтиленгликоля 400/40% водного раствора, встряхивали и обрабатывали с помощью ультразвука с получением 1 мг/мл практически прозрачного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Выбирали самок мышей Balb/c возрастом 7-10 недель и вводили внутривенно раствор соединения-кандидата в дозе 1 мг/кг. Соединение, представленное формулой (I), смешивали с 10% водным раствором солютола (полиэтиленгликоль-15-гидроксистеарат), встряхивали и обрабатывали с помощью ультразвука с получением 1 мг/мл практически прозрачного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Выбирали самок мышей Balb/c возрастом 7-10 недель и вводили перорально раствор соединения-кандидата в дозе 10 мг/кг.
[189] В определенный момент времени собирали цельную кровь с получением плазмы, анализировали концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[190] 2. Исследование фармакокинетики перорального введения и внутривенной инъекции соединения, представленного формулой (I), на крысах SD.
[191] Соединение, представленное формулой (I), смешивали с 5% DMSO/55% полиэтиленгликоля 400/40% водного раствора, встряхивали и обрабатывали с помощью ультразвука с получением 1 мг/мл практически прозрачного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Выбирали самцов крыс SD возрастом 7-10 недель и вводили внутривенно раствор соединения-кандидата в дозе 1 мг/кг.
[192] Соединение, представленное формулой (I), смешивали с 10% водным раствором солютола, встряхивали и обрабатывали с помощью ультразвука с получением 1 мг/мл практически прозрачного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Выбирали самцов крыс SD возрастом 7-10 недель и вводили перорально раствор соединения-кандидата в дозе 10 мг/кг.
[193] В определенный момент времени собирали цельную кровь с получением плазмы, анализировали концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[194] 3. Исследование фармакокинетики перорального введения и внутривенной инъекции соединения, представленного формулой (1), на собаках породы бигль.
[195] Соединение, представленное формулой (I), смешивали с 5% DMSO/55% полиэтиленгликоля 400/40% водного раствора, встряхивали и обрабатывали с помощью ультразвука с получением 1 мг/мл практически прозрачного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Выбирали самцов собак породы бигль весом приблизительно 10 кг и им внутривенно вводили раствор соединения-кандидата в дозе 1 мг/кг.
[196] Соединение, представленное формулой (I), смешивали с 10% водным раствором солютола, встряхивали и обрабатывали с помощью ультразвука с получением 2 мг/мл практически прозрачного раствора, который затем фильтровали через микропористую мембрану для последующего применения. Выбирали самцов собак породы бигль весом приблизительно 10 кг и им перорально вводили раствор соединения-кандидата в дозе 10 мг/кг.
[197] В определенный момент времени собирали цельную кровь с получением плазмы, анализировали концентрацию лекарственного средства с помощью способа LC-MS/MS и рассчитывали фармакокинетические параметры с помощью программного обеспечения Phoenix WinNonlin (Pharsight, США).
[198] Экспериментальные результаты показаны в таблице 10.
[199] Таблица 10. Результаты фармакокинетики тестируемого соединения

[200] Примечание: Т1/2 означает период полувыведения; Vdss означает кажущийся объем распределения; Cl означает почечный клиренс; AUC0-last означает площадь под кривой лекарственное средство-время; Tmax означает время достижения пика; Cmax означает пиковую концентрацию; F% означает биодоступность при пероральном введении; iv означает внутривенную инъекцию; РО означает пероральное введение; mpk означает мг/кг.
[201] Заключение: соединение, представленное формулой (I), имеет хороший единичный или частичный показатель фармакокинетики собак породы бигль.
[202] Пример 9. Исследование эффективности лекарственного средства in vivo
[203] Модель AAV/HBV
[204] Цели эксперимента. Выявление эффекта соединения в отношении вируса гепатита В in vivo с применением мышиной модели AAV/HBV.
[205] Процедура эксперимента. День осуществления первого введения обозначали как день 0, день перед введением обозначали как день -1, день после введения обозначали как день 1 и т.д. На 28 день перед введением всем животным внутривенно вводили в хвостовую вену l*1011v.g. вирус rAAV8-1.3HBV, 200 мкл для каждого животного. За 14 дней до введения и за 7 дней до введения у всех мышей, которым через поднижнечелюстную вену вводили вирус rAAV8-1.3HBV, собирали сыворотку крови. Собранные образцы крови помещали при температуре 37°С в течение приблизительно 30 минут, затем центрифугировали при 13200 g при 4°С в течение 3 минут и собирали надосадочную жидкость. Содержание ДНК HBV, HBeAg и HBsAg определяли с помощью сыворотки крови. Мыши с более низкими уровнями содержания ДНК HBV DNA, HBsAg и HBeAg и меньшим весом, вероятно, будут исключены из эксперимента. Выбранные 25 мышей равномерно распределяли в каждую группу и обеспечивали отсутствие статистического расхождения между уровнями содержания ДНК HBV, HBsAg и HBeAg и весом тела для мышей в каждой группе с обработкой соединением на 21 день после введения вируса по сравнению с таковыми в группе с растворителем (Р>0,05). Соединение, представленное формулой (I) (WX-325), смешивали с 10% водным раствором солютола, встряхивали и обрабатывали с помощью ультразвука с получением гомогенной суспензии, которую фильтровали посредством микропористой мембраны для последующего применения. Тенофовир в качестве соединения, являющегося положительным контролем, растворяли в физиологическом растворе, подвергали воздействию ультразвука и перемешивали до растворения, затем получали в виде 0,1 мг/мл мастер-микса, который разбавляли до 0,01 мг/мл с помощью физиологического раствора и хранили при 4°С до применения. Соединение, представленное формулой (I) (WX-325), вводили перорально (РО) и два раза в сутки (BID) с интервалом в 8 часов. Эталонное соединение тенофовир вводили перорально и два раза в сутки. Оба лекарственных средства вводили в течение 28 дней и образцы крови отбирали на 3, 7, 10 и 28 дни после введения и уровень ДНК HBV в плазме определяли с помощью qPCR. На 28 день мышей умерщвляли ингаляцией СО2, их печень собирали и измеряли уровень ДНК HBV с помощью qPCR. Результаты эксперимента показаны на фигуре 7 и фигуре 8.
[206] Заключение: соединение по настоящему изобретению проявляет хорошую эффективность in vivo и дозозависимый эффект.
[207] Специалистам в данной области должно быть понятно, что вышеуказанное описание конкретных вариантов осуществления предназначено исключительно для иллюстрации, и различные изменения или модификации будут очевидны специалистам в данной области техники без отступления от принципа и сущности настоящего изобретения. Следовательно, настоящее изобретение предполагается как не ограниченное, за исключением четко изложенного в следующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ ИНГИБИТОРА ПОВЕРХНОСТНОГО АНТИГЕНА ВИРУСА ГЕПАТИТА В | 2020 |
|
RU2823673C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ИНГИБИТОРА ATR И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2832707C2 |
| Соединение ингибитора FGFR в твердой форме и способ его получения | 2020 |
|
RU2810067C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПРОИЗВОДНОГО ТИОФЕНА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2022 |
|
RU2830948C1 |
| КРИСТАЛЛИЧЕСКАЯ ИЛИ АМОРФНАЯ ФОРМА АГОНИСТОВ FXR, ПРЕДСТАВЛЯЮЩИХ СОБОЙ ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2800751C2 |
| ТАРТРАТ 3-((1R,3R)-1-(2,6-ДИФТОР-4-((1-(3-ФТОРПРОПИЛ)АЗЕТИДИН-3-ИЛ)АМИНО)ФЕНИЛ)-3-МЕТИЛ-1,3,4,9-ТЕТРАГИДРО-2H-ПИРИДО[3,4-b]ИНДОЛ-2-ИЛ)-2,2-ДИФТОРПРОПАН-1-ОЛА, ЕГО ТВЕРДЫЕ ФОРМЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2809220C2 |
| АНАЛОГ ПИРИДО[1,2-A]ПИРИМИДОНА, ЕГО КРИСТАЛЛИЧЕСКАЯ ФОРМА, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2016 |
|
RU2753696C2 |
| СПОСОБ ПОЛУЧЕНИЯ NO-ДОНОРНЫХ СОЕДИНЕНИЙ, ТАКИХ КАК NO-ДОНОРНЫЙ ДИКЛОФЕНАК | 2003 |
|
RU2322434C2 |
| АМОРФНОЕ ПРОИЗВОДНОЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТА PPAR И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2018 |
|
RU2749056C1 |
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ТИМОДЕПРЕССИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2536685C2 |

Изобретение относится к кристаллической форме А и кристаллической форме В соединения, представленного формулой (I), и их применению в получении лекарственных препаратов для лечения HBV. Технический результат: получены новые кристаллические формы соединения, представленного формулой (I), которые могут быть применимы в получении лекарственных препаратов для лечения HBV. 3 н. и 10 з.п. ф-лы, 10 табл., 9 пр., 8 ил.
1. Кристаллическая форма А соединения, представленного формулой (I), характеризующаяся порошковой рентгеновской дифрактограммой (XRPD), содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 5,56±0,2°, 10,84±0,2°, 15,56±0,2°, 16,17±0,2°, 22,14±0,2°, 22,70±0,2°, 27,76±0,2° и 28,44±0,2°,
2. Кристаллическая форма А по п. 1, где дифрактограмма XRPD показана на фигуре 1.
3. Кристаллическая форма А по п. 1 или 2, проявляющая эндотермический пик с началом при 229,95°С, как измерено с помощью кривой дифференциальной сканирующей калориметрии (DSC).
4. Кристаллическая форма А по п. 3, где DSC показана на фигуре 2.
5. Кристаллическая форма А по п. 1 или 2, характеризующаяся кривой термогравиметрического анализа (TGA) с потерей веса 0,3382% при 62±3°С и потерей веса 0,8753% при 230±3°С.
6. Кристаллическая форма А по п. 5, где TGA показана на фигуре 3.
7. Кристаллическая форма В соединения, представленного формулой (I), характеризующаяся порошковой рентгеновской дифрактограммой (XRPD), содержащей характеристические дифракционные пики при следующих значениях угла 2θ: 9,56±0,2°, 12,70±0,2°, 14,41±0,2°, 15,64±0,2°, 19,70±0,2°, 23,03±0,2°, 23,98±0,2° и 27,65±0,2°,
8. Кристаллическая форма В по п. 7, где дифрактограмма XRPD показана на фигуре 4.
9. Кристаллическая форма В по п. 7 или 8, проявляющая эндотермический пик с началом при 233,59°С, как измерено с помощью кривой дифференциальной сканирующей калориметрии (DSC).
10. Кристаллическая форма В по п. 9, где DSC показана на фигуре 5.
11. Кристаллическая форма В по п. 7 или 8, характеризующаяся кривой термогравиметрического анализа (TGA) с потерей веса 0,04890% при 120±3°С.
12. Кристаллическая форма В по п. 11, где TGA показана на фигуре 6.
13. Применение кристаллической формы А по любому из пп. 1-6 или кристаллической формы В по любому из пп. 7-12 в получении лекарственного препарата для лечения HBV.
| CN 106413402 A, 15.02.2017 | |||
| WO 2017069279 A1, 27.04.2017 | |||
| БРОМ-ФЕНИЛ ЗАМЕЩЕННЫЕ ТИАЗОЛИЛДИГИДРОПИРИМИДИНЫ | 2008 |
|
RU2443703C2 |
| НЕКОТОРЫЕ ХИМИЧЕСКИЕ СТРУКТУРЫ, КОМПОЗИЦИИ И СПОСОБЫ | 2009 |
|
RU2537549C2 |

