Перекрестная Ссылка на Родственные Заявки
Данная заявка заявляет приоритет согласно § 119(e) Раздела 35 Кодекса Законов США по предварительной заявке №62/447359, поданной 17 января 2017 г., полное раскрытие которой включено в данный документ в полном объеме посредством ссылки.
Список последовательностей Данная заявка содержит список последовательностей, который был подан в электронном виде в формате ASCII и полностью включен в данный документ посредством ссылки. Указанная копия в формате ASCII, созданная 12 января 2018 года, имеет название P34027-WO_SL.txt и имеет размер 32675 байт.
ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к препаратам антител против HER2 с фиксированной дозой для подкожного введения и их применению для лечения онкологических заболеваний. В частности, данное изобретение относится к препаратам пертузумаба с фиксированной дозой, препаратам для подкожного введения, содержащим пертузумаб и трастузумаб, и их применению для лечения онкологических заболеваний.
УРОВЕНЬ ТЕХНИКИ
Антитела против HER2
Представители семейства рецепторных тирозинкиназ HER являются важными медиаторами роста, дифференциации и выживания клеток. Семейство рецепторов включает четыре отдельных представителя, включая рецептор эпидермального фактора роста (EGFR, ErbB1 или HER1), HER2 (ErbH2 или р185neu), HER3 (ErbB3) и HER4 (ErbB или tyro2). Представители семейства рецепторов были задействованы при различных типах злокачественных опухолей человека.
Рекомбинантная гуманизированная версия мышиного анти-HER2 антитела 4D5 (huMAb4D5-8, rhuMAb HER2, трастузумаб или HERCEPTIN®; патент США №5,821,337) клинически активна у пациентов со сверхэкспрессирующим HER2 метастатическим раком молочной железы, которые ранее подвергались обширной противораковой терапии (Baselga et al., J. Clin. Oncol. 14:737-744(1996)).
Трастузумаб получил разрешение на продажу от Управления по Контролю за Продуктами и Лекарствами (FDA) 25 сентября 1998 года для лечения пациентов с метастатическим раком молочной железы, опухоли которого сверхэкспрессируют белок HER2. В данное время трастузумаб одобрен для использования в качестве отдельного средства или в сочетании с химиотерапией или гормональной терапией в условиях метастазирования, и в качестве отдельного средства или в сочетании с химиотерапией в качестве вспомогательного средства для пациентов с ранней стадией HER2-положительного рака молочной железы. В данное время терапия на основе трастузумаба является рекомендованным лечением для пациентов с HER2-положительным раком молочной железы на ранней стадии, у которых нет противопоказаний для его применения (инструкция по применению препарата Herceptin®; Руководство NCCN, версия 2.2011). Трастузумаб с доцетакселом (или паклитакселом) является зарегистрированным стандартом медицинской помощи первой линии в условиях лечения метастатического рака молочной железы (МРМЖ) (Slamon et al., N Engl J Med. 2001;344(11):783-792.; Marty et al., J Clin Oncol. 2005; 23(19):4265-4274).
Пациенты, получавшие антитело против HER2 трастузумаб, отбираются для терапии на основе экспрессии HER2. См., например, WO 99/31140 (Paton et al.), US 2003/0170234A1 (Hellmann S.) и US 2003/0147884 (Paton et al.); атакже WO 01/89566, US 2002/0064785 и US 2003/0134344 (Mass et al.). См. также патент США №6573043, патент США №6905830 и US 2003/0152987, Cohen et al., касающиеся иммуногистохимии (IHC) и флуоресцентной гибридизации in situ (FISH) для выявления сверхэкспрессии и амплификации HER2. Таким образом, оптимальное лечение метастатического рака молочной железы в настоящее время учитывает не только общее состояние пациента, историю болезни и статус рецептора, но также статус HER2.
Пертузумаб (также известный как рекомбинантное гуманизированное моноклональное антитело 2С4 (rhuMAb 2С4); Genentechh, Inc, Саус-Сан-Франциско) представляет собой первый в новом классе агентов, известных как ингибиторы димеризации HER (HDI), и действует для подавления способности HER2 формировать активные гетеродимеры или гомодимеры с другими рецепторами HER (такими как EGFR/HER1, HER2, HER3 и HER4). См., например, Harari and Yarden Oncogene 19:6102-14 (2000); Yarden and Sliwkowski. Nat Rev Mol Cell Biol 2:127-37 (2001); Sliwkowski Nat Struct Biol 10:158-9 (2003); Cho et al. Nature 421:756-60 (2003); и Malik et al., Pro Am Soc Cancer Res 44:176-7 (2003).
Было показано, что блокада пертузумабом образования гетеродимеров HER2-HER3 в опухолевых клетках ингибирует критическую передачу сигналов клеток, что приводит к снижению пролиферации и выживаемости опухоли (Agus et al., Cancer Cell 2:127-37 (2002)).
Пертузумаб прошел испытания в качестве отдельного агента при клинических испытаниях фазы Ia у пациентов с прогрессирующим онкологическим заболеванием и испытаниях фазы II у пациентов с раком яичников и раком молочной железы, а также раком легких и простаты. В испытаниях фазы I пациенты с неизлечимыми, локально прогрессирующими, рецидивирующими или метастатическими солидными опухолями, которые прогрессировали во время или после стандартной терапии, получали пертузумаб, вводимый внутривенно каждые 3 недели. Пертузумаб в целом хорошо переносился. Регрессия опухоли была достигнута у 3 из 20 пациентов, которая оценивалась по ответу. У двух пациентов подтвердились частичные ответы. Стабильное заболевание, продолжающееся более 2,5 месяцев, наблюдалось у 6 из 21 пациента (Agus et al., Pro Am Soc Clin Oncol 22:192 (2003)). В дозах 2,0-15 мг/кг фармакокинетика пертузумаба была линейной, а средний клиренс составлял от 2,69 до 3,74 мл/день/кг, а средний терминальный период полувыведения колебался от 15,3 до 27,6 дней. Антитела к пертузумабу не были обнаружены (Allison et al., Pro Am Soc Clin Oncol 22:197 (2003)).
В US 2006/0034842 описаны способы лечения рака, экспрессирующего ErbB, комбинациями антител против ErbB2. В US 2008/0102069 описано применение трастузумаба и пертузумаба в лечении HER2-положительного метастатического рака, такого как рак молочной железы. В Baselga et al., J Clin Oncol, 2007 ASCO Annual Meeting Proceedings Part I, Col. 25, No. 18S (дополнение от 20 июня), 2007:1004 сообщается о лечении пациентов, ранее проходивших лечение HER2-положительного рака молочной железы, который прогрессировал во время лечения трастузумабом, комбинацией трастузумаба и пертузумаба. В Portera et al., J Clin Oncol, 2007 ASCO Annual Meeting Proceedings Part I. Vol.25, No. 18S (дополнение от 20 июня), 2007:1028 оценивается эффективность и безопасность комбинированной терапии трастузумаб+пертузумаб у пациентов с HER2-позположительным раком молочной железы, у которых прогрессировало заболевание при терапии на основе трастузумаба. Авторы пришли к выводу, что необходима дальнейшая оценка эффективности комбинированного лечения, чтобы определить общий риск и пользу от этой схемы лечения.
Пертузумаб оценивался в испытаниях фазы II в сочетании с трастузумабом у пациентов с HER2-положительным метастатическим раком молочной железы, которые ранее получали трастузумаб при лечении метастатического заболевания. Одно исследование, проведенное Национальным Институтом Рака (NCI), включало 11 пациентов, которые ранее проходили лечение HER2-положительного метастатического рака молочной железы. У двух из 11 пациентов был частичный ответ (PR) (Baselga et al., J Clin Oncol 2007 ASCO Annual Meeting Proceedings; 25:18S (дополнение от 20 июня): 1004).
Результаты неоадъювантного испытания фазы II, оценивающего эффект новой комбинированной схемы введения пертузумаба и трастузумаба плюс химиотерапия (доцетаксел) у женщин с ранней стадией HER2-положительного рака молочной железы, представленные на Симпозиуме о раке молочной железы в Сан-Антонио CTRC-AACR (SABCS), 8-12 декабря 2010 г., показали, что два антитела против HER2 плюс доцетаксел, применяемые в качестве неоадъювантной терапии перед операцией, значительно увеличили частоту полного исчезновения опухоли (частота патологического полного ответа, pCR, 45,8%) в молочной железе более чем на половину по сравнению с трастузумабом плюс доцетаксел (pCR 29,0%), р=0,014.
Клиническая оценка пертузумаба и трастузумаба (CLEOPATRA) фазы II клинического испытания оценила эффективность и безопасность пертузумаба плюс трастузумаб плюс доцетаксел по сравнению с плацебо плюс трастузумаб плюс доцетаксел, в качестве лечения первой линии у пациентов с местно-рецидивирующим, неоперабельным или метастатическим HER2-положительным раком молочной железы. Комбинация пертузумаб плюс трастузумаб плюс доцетаксел, по сравнению с плацебо плюс трастузумаб плюс доцетаксел, при использовании в качестве лечения первой линии HER2-положительного метастатического рака молочной железы, значительно продлевала выживаемость без прогрессирования заболевания без увеличения токсических эффектов на сердце. (Baselga et al., NEng JMed 2012 366:2, 109-119).
В фазе II клинического испытания NeoSphere была оценена эффективность и безопасность неоадъювантного введения пертузумаба и трастузумаба у женщин, не получавших лечение (пациентов, которые ранее не получали противораковую терапию) с операбельным, местно-прогрессирующим и воспалительным раком молочной железы. Пациенты, получавшие пертузумаб и трастузумаб плюс доцетаксел, продемонстрировали значительно увеличенную частоту патологического полного ответа по сравнению с пациентами, получавшими трастузумаб плюс доцетаксел, без существенных различий в переносимости (Gianni et al., Lancet Oncol 2012 13(1):25-32). Результаты 5-летнего наблюдения представлены в Gianni et al., Lancet Oncol 2016 17(6):791-800).
Патентные публикации, относящиеся к антителам против HER2, включают: патенты США №5677171; 5720937; 5720954; 5725856; 5770195; 5772997; 6165464; 6387371; 6399063; 6015567; 6333169; 4968603; 5821337; 6054297; 6407213; 6639055;6719971; 6800738; 5648237; 7018809; 6267958; 6695940; 6821515; 7060268; 7682609; 7371376; 6127526; 6333398; 6797814; 6339142; 6417335; 6489447; 7074404; 7531645; 7846441; 7892549; 6573043; 6905830; 7129840; 7344840; 7468252; 7674589; 6949245; 7485302; 7498030; 7501122; 7537931; 7618631; 7862817; 7041292; 6627196; 7371379; 6632979; 7097840; 7575748; 6984494; 7279287; 7811773; 7993834; 7435797; 7850966; 7485704; 7807799; 7560111; 7879325; 7449184; 7700299; и US 2010/0016556; US 2005/0244929; US 2001/0014326; US 2003/0202972; US 2006/0099201; US 2010/0158899; US 2011/0236383; US 2011/0033460; US 2005/0063972; US 2006/018739; US 2009/0220492; US 2003/0147884; US 2004/0037823; US 2005/0002928; US 2007/0292419; US 2008/0187533; US 2003/0152987; US 2005/0100944; US 2006/0183150; US 2008/0050748; US 2010/0120053; US 2005/0244417; US 2007/0026001; US 2008/0160026; US 2008/0241146; US 2005/0208043; US 2005/0238640; US 2006/0034842; US 2006/0073143; US 2006/0193854; US 2006/0198843; US 2011/0129464; US 2007/0184055; US 2007/0269429; US 2008/0050373; US 2006/0083739; US 2009/0087432; US 2006/0210561; US 2002/0035736; US 2002/0001587; US 2008/0226659; US 2002/0090662; US 2006/0046270; US 2008/0108096; US 007/0166753; US 2008/0112958; US 2009/0239236; US 2004/008204; US 2009/0187007; US 2004/0106161; US 2011/0117096; US 2004/048525; US 2004/0258685; US 2009/0148401; US 2011/0117097; US 2006/0034840; US 2011/0064737; US 2005/0276812; US 2008/0171040; US 2009/0202536; US 2006/0013819; US 2006/0018899; US 2009/0285837; US 2011/0117097; US 2006/0088523; US 2010/0015157; US 2006/0121044; US 2008/0317753; US2006/0165702; US 2009/0081223; US 2006/0188509; US 2009/0155259; US 2011/0165157; US 2006/0204505; US 2006/0212956; US 2006/0275305; US 2007/0009976; US 2007/0020261; US 2007/0037228; US 2010/0112603; US 2006/0067930; US 2007/0224203; US 2008/0038271; US 2008/0050385; 2010/0285010; US 2008/0102069; US 2010/0008975; US 2011/0027190; US 2010/0298156; US 2009/0098135; US 2009/0148435; US 2009/0202546; US 2009/0226455; US 2009/0317387; и US 2011/0044977.
Ферменты-Гиалуронидазы
Гиалуронидазы представляют собой группу обычно нейтрально- или кислото-активных ферментов, встречающихся в животном мире. Гиалуронидазы варьируют в зависимости от субстратной специфичности и механизма действия (WO 2004/078140). Существует три основных класса гиалуронидаз: 1. Гиалуронидазы, характерные для млекопитающих (ЕС 3.2.1.35), которые представляют собой эндо-β-N-ацетилгексозаминидазы с тетрасахаридами и гексасахаридами в качестве основных конечных продуктов. Они обладают как гидролитической, так и трансгликозидазной активностью и могут разлагать гиалуронан и хондроитинсульфаты (CS), обычно C4-S и C6-S. 2. Бактериальные гиалуронидазы (ЕС 4.2.99.1) разлагают гиалуронан и, в разной степени, CS и DS. Они представляют собой эндо-β-N-ацетилгексозаминидазы, которые действуют с помощью реакции бета-элиминации, которая приводит к получению в основном дисахаридных конечных продуктов. 3. Гиалуронидазы (ЕС 3.2.1.36) из пиявок, других паразитов и ракообразных являются эндо-бета-глюкуронидазами, которые приводят к получению тетрасахаридов и гексасахаридов в качестве конечных продуктов посредством гидролиза связи β1-3.
Гиалуронидазы млекопитающих можно в свою очередь разделить на две группы: нейтрально-активные и кислото-активные ферменты. В геноме человека имеется шесть гиалуронидазоподобных генов: HYAL1, HYAL2, HYAL3, HYAL4, HYALP1 и PH20/SPAM1. HYALP1 представляет собой псевдоген, и было показано, что HYAL3 не обладает ферментативной активностью в отношении каких-либо известных субстратов. HYAL4 представляет собой хондроитиназу и проявляет небольшую активность по отношению к гиалуронану. HYAL1 представляет собой прототипный кислотно-активный фермент, а РН20 представляет собой прототипный нейтрально-активный фермент. Кислото-активные гиалуронидазы, такие как HYAL1 и HYAL2, обычно не обладают каталитической активностью при нейтральном рН (то есть рН 7). Например, HYAL1 имеет небольшую каталитическую активность in vitro при рН 4,5 [Frost I. G. and Stern, R., "A microtiter-based assay for hyaluronidase activity not requiring specialized reagents", Anal. Biochemistry, 1997; 251:263-269]. HYAL2 представляет собой кислото-активный фермент с очень низкой удельной активностью in vitro.
Гиалуронидазоподобные ферменты также могут быть охарактеризованы как ферменты, которые обычно связываются с плазматической мембраной через гликозилфосфатидилинозитный якорь, такие как HYAL2 человека и РН20 человека [Danilkovitch-Miagkova et al., Proc. Natl. Acad. Sci. USA, 2003; 100(8):4580-4585; Phelps et al., Science 1988; 240(4860): 1780-1782], и такие, которые обычно растворимы, такие как HYAL1 человека [Frost, I. G. et al., "Purification, cloning, and expression of human plasma hyaluronidase", Biochem. Biophys. Res. Commun. 1997; 236(1): 10-15]. Однако существуют различия между видами: например, бычий РН20 очень слабо прикреплен к плазматической мембране и не закреплен с помощью чувствительного к фосфолипазе якоря [Lalancette et al., Biol. Reprod., 2001; 65(2):628-36]. Эта уникальная особенность бычьей гиалуронидазы позволила использовать растворимый фермент гиалуронидазу из бычьих тестикул в качестве экстракта для клинического применения (Wydase™, Hyalase™). Другие виды РН20 представляют собой закрепленные на липидах ферменты, которые обычно не растворимы без использования детергентов или липаз. Например, РН20 человека прикрепляется к плазматической мембране через GPI-якорь. Попытки создать человеческие ДНК-конструкции РН20, которые не вводили бы липидный якорь в полипептид, приводили либо к получению каталитически неактивного фермента, либо к получению нерастворимого фермента [Arming et al., Eur. J. Biochem., 1997; 247(3):810-4]. Встречающаяся в природе гиалуронидаза из спермы макака встречается как в растворимой, так и в мембраносвязанной форме. В то время как мембраносвязанная форма массой 64 кДа обладает энзиматической активностью при рН 7,0, форма массой 54 кДа активна только при рН 4,0 [Cherr et al., Dev. Biol., 1996; 10; 175(1): 142-53]. Таким образом, растворимые формы РН20 часто не имеют ферментативной активности при нейтральных условиях.
В WO 2006/091871 описано, что небольшие количества растворимых гликопротеинов гиалуронидазы (sHASEGP) могут быть введены в препарат для облегчения введения терапевтического лекарственного средства в гиподерму. Быстро деполимеризуя ГК во внеклеточном пространстве, sHASEGP снижает вязкость интерстициальной ткани, увеличивая тем самым гидропроницаемость и позволяя безопасно и комфортно вводить большие объемы в подкожную ткань. Повышенная гидропроницаемость, индуцируемая sHASEGP за счет снижения межклеточной вязкости, обеспечивает большую дисперсию, потенциально увеличивая системную биодоступность терапевтического лекарственного средства, вводимого подкожно (п/к).
При введении в гиподерму деполимеризация ГК с помощью sHASEGP локализуется в месте инъекции в подкожной ткани. Экспериментальные данные показывают, что sHASEGP инактивируется локально в интерстициальном пространстве с периодом полураспада у мышей от 13 до 20 минут, без заметного системного поглощения в кровь после однократного внутривенного введения у мышей CD-I. В сосудистом компартменте sHASEGP демонстрирует период полураспада 2,3 и 5 минут у мышей и обезьян Cynomolgus, соответственно, с дозами до 0,5 мг/кг. Быстрый клиренс sHASEGP в сочетании с постоянным синтезом субстрата ГК в подкожной ткани приводит к кратковременному и локально активному усилению проницаемости для других коинъецированных молекул, эффекты которых полностью обратимы в течение 24-48 часов после введения [Bywaters G. L., et al., "Reconstitution of the dermal barrier to dye spread after Hyaluronidase injection", Br. Med. J., 1951; 2 (4741): 1178-1183].
В дополнение к его влиянию на локальную дисперсию жидкости, sHASEGP также действует как усилитель абсорбции. Макромолекулы размером более 16 килодальтон (кДа) в значительной степени не проявляют абсорбцию через капилляры посредством диффузии и в основном абсорбируются через дренирующие лимфатические узлы. Следовательно, подкожно вводимая макромолекула, такая как, например, терапевтическое антитело (молекулярная масса около 150 кДа), должна пройти через интерстициальный матрикс, прежде чем достигнет дренирующих лимфатических сосудов для последующего всасывания в сосудистый компартмент. Увеличивая локальную дисперсию, sHASEGP увеличивает скорость (Ka) поглощения многих макромолекул. Это приводит к повышению пиковых уровней в крови (Cmax) и, возможно, к увеличению биодоступности по сравнению с введением п/к в отсутствие sHASEGP [Bookbinder L. Н., et al., "A recombinant human enzyme for enhanced interstitial transport of therapeutics", J. Control. Release 2006; 114: 230-241].
Продукты гиалуронидазы животного происхождения использовались в клинических условиях в течение более 60 лет, главным образом, для увеличения дисперсии и абсорбции других совместно вводимых лекарственных средств и для гиподермоклиза (п/к инъекция/инфузия жидкости в большом объеме) [Frost G. I., "Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration", Expert Opinion on Drug Delivery, 2007; 4: 427-440]. Детали механизма действия гиалуронидаз подробно описаны в следующих публикациях: Duran-Reynolds F., "A spreading factor in certain snake venoms and its relation to their mode of action", CR Soc Biol Paris, 1938; 69-81; Chain E., "A mucolytic enzyme in testes extracts", Nature 1939; 977-978; Weissmann В., "The transglycosylative action oftesticular hyaluronidase", J. Biol. Chem., 1955; 216: 783-94; Tammi, R., Saamanen, A. M., Maibach, H. I., Tammi M., "Degradation of newly synthesized high molecular mass hyaluronan in the epidermal and dermal compartments of human skin in organ culture", J. Invest. Dermatol. 1991; 97:126-130; Laurent, U. B. G., Dahl, L. В., Reed, R. K., "Catabolism of hyaluronan in rabbit skin takes place locally, in lymph nodes and liver", Exp. Physiol. 1991; 76: 695-703; Laurent, T. C. and Fraser, J. R. E., "Degradation of Bioactive Substances: Physiology and Pathophysiology", Henriksen, J. H. (Ed) CRC Press, Boca Raton, Fla.; 1991. pp.249-265; Harris, E. N., et al., "Endocytic function, glycosaminoglycan specificity, and antibody sensitivity of the recombinant human 190-kDa hyaluronan receptor for endocytosis (HARE)", J. Biol. Chem. 2004; 279:36201-36209; Frost, G. I., "Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration", Expert Opinion on Drug Delivery, 2007; 4: 427-440. Продукты гиалуронидазы, одобренные в странах ЕС, включают Hylase® «Dessau» и Hyalase®. Продукты гиалуронидазы животного происхождения, одобренные в США, включают Vitrase™, Hydase™, and Amphadase™.
Стабильные препараты лиофилизированных антител, содержащие лиопротектор, буфер и поверхностно-активное вещество, были описаны Andya et al. (WO 97/04801 и патенты США №6,267,958, 6,685,940, 6,821,151, 7,060,268). В WO 2006/044908 представлены препараты антител, содержащие моноклональные антитела, приготовленные в гистидинацетатном буфере, рН от 5,5 до 6,5, предпочтительно от 5,8 до 6,2. Препараты антител против HER2 раскрыты в патенте США №8,372,396; 9,017,671. Препараты антител против HER2 для подкожного введения и их применение описаны в патенте США №9,345,661. Внутривенное введение пертузумаба с фиксированной дозой описано в патенте США №7,449,184 и 8,404,234.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте данное изобретение относится к изделию, содержащему флакон с однократной дозой, содержащий одну фиксированную дозу антитела против HER2, содержащего аминокислотные последовательности вариабельной области легкой цепи и вариабельной области тяжелой цепи SEQ ID NO: 7 и 8 соответственно, при этом фиксированная доза составляет около 600 мг или около 1200 мг. Предпочтительно, антитело против HER2 представляет собой пертузумаб.
В одном варианте реализации изобретения изделие содержит два флакона с однократной дозой, причем первый флакон содержит одну фиксированную дозу около 1200 мг пертузумаба, а второй флакон содержит одну фиксированную дозу около 600 мг пертузумаба.
Во втором варианте реализации изобретения изделие содержит два флакона с однократной дозой, причем первый флакон содержит одну фиксированную дозу около 600 мг пертузумаба, а второй флакон содержит одну фиксированную дозу около 600 мг трастузумаба.
В третьем варианте реализации изобретения изделие содержит два флакона с однократной дозой, причем первый флакон содержит одну фиксированную дозу около 1200 мг пертузумаба, а второй флакон содержит одну фиксированную дозу 600 мг трастузумаба.
Во всех вариантах реализации изобретения по меньшей мере один из флаконов с однократной дозой может содержать фиксированную дозу(ы) в жидком препарате для подкожного введения.
Во всех вариантах реализации изобретения жидкий препарат для подкожного введения может дополнительно содержать фермент гиалуронидазу, такой как рекомбинантная гиалуронидаза человека (rHuPH20). rHuPH20 может присутствовать в количестве, достаточном, чтобы привести к увеличению дисперсии пертузумаба или трастузумаба, содержащихся в том же жидком препарате, во время подкожного введения.
rHuPH2 может присутствовать в жидком препарате, содержащем трастузумаб, например, в концентрации от около 150 Ед/мл до 16000 Ед/мл или в концентрации от около 600 Ед/мл до около 16000 Ед/мл, или в концентрация от около 1000 Ед/мл до около 2000 Ед/мл, например, в концентрации около 2000 Ед/мл или в концентрации, по меньшей мере, около 600 Ед/мл.
rHuPH20 может присутствовать в жидком препарате, содержащем пертузумаб, в концентрации от около 600 до около 2000 Ед/мл, например, в концентрации около 600 Ед/мл или в концентрации около 667 Ед/мл, или в концентрации около 1000 Ед/мл, или в концентрации около 2000 Ед/мл.
В другом варианте реализации изобретения флакон с однократной дозой, присутствующий в изделии, дополнительно содержит однократную фиксированную дозу трастузумаба.
В одном варианте реализации изобретения однократная фиксированная доза пертузумаба и однократная фиксированная доза трастузумаба содержатся в одном жидком препарате для подкожного введения, при этом жидкий препарат может, например, содержать однократную фиксированную дозу около 600 мг пертузумаба и однократную фиксированную дозу около 600 мг трастузумаба или однократную фиксированную дозу около 1200 мг пертузумаба и однократную фиксированную дозу около 600 мг трастузумаба.
Жидкий препарат, содержащий фиксированную дозу пертузумаба и фиксированную дозу пертузумаба, может дополнительно содержать фермент гиалуронидазу, такую как рекомбинантная гиалуронидаза человека (rHuPH20), которая может присутствовать в указанном жидком препарате в количестве, достаточном для увеличения дисперсии пертузумаба и трастузумаба, содержащихся в одном и том же жидком препарате во время подкожного введения, например, в концентрации, по меньшей мере, около 600 Ед/мл или в концентрации от около 600 Ед/мл до около 2000 Ед/мл, например, в концентрация около 1000 Ед/мл.
В некоторых вариантах реализации изобретения изделия согласно данного документа дополнительно содержат листок-вкладыш, инструктирующий пользователя вводить фиксированную дозу (дозы) подкожно пациенту с HER2-положительным онкологическим заболеванием.
В одном варианте реализации изобретения листок-вкладыш инструктирует пользователя вводить фиксированные дозы пертузумаба и трастузумаба подкожно пациенту с HER2-положительным онкологическим заболеванием.
В другом варианте реализации изобретения листок-вкладыш инструктирует пользователя совместно вводить фиксированную дозу пертузумаба и фиксированную дозу трастузумаба подкожно в виде двух отдельных подкожных инъекций.
В дополнительном варианте реализации изобретения листок-вкладыш инструктирует пользователя вводить фиксированную дозу пертузумаба, смешанного с фиксированной дозой трастузумаба, в виде одной подкожной инъекции.
В еще одном варианте реализации изобретения листок-вкладыш инструктирует пользователя вводить фиксированные дозы пертузумаба и трастузумаба подкожно пациенту с HER2-положительным онкологическим заболеванием.
Онкологическое заболевание может представлять собой, например, рак молочной железы, перитонеальный рак, рак маточной трубы, рак легких, колоректальный рак, рак желчных путей или рак мочевого пузыря, такое как ранний рак молочной железы (РРМЖ) или метастатический рак молочной железы (МРМЖ).
В другом аспекте изобретение относится к изделию, содержащему флакон объемом 10 мл или 20 мл, содержащий одну фиксированную дозу антитела против HER2, содержащего вариабельные аминокислотные последовательности легкой и тяжелой цепей, представленные в SEQ ID NO: 7 и 8 соответственно, при этом фиксированная доза составляет около 600 мг или около 1200 мг антитела против HER2, и листок-вкладыш, инструктирующий пользователя подкожно вводить фиксированную дозу пациенту с HER2-положительным онкологическим заболеванием.
В одном варианте реализации изобретения антитело против HER2 представляет собой пертузумаб.
В другом варианте реализации изобретения фиксированная доза пертузумаба содержится в жидком препарате для подкожного введения, причем жидкий препарат может, например, содержать пертузумаб в концентрации около 100-150 мг/мл, например, в концентрации около 120 мг/мл.
В различных вариантах реализации изобретения жидкий препарат, присутствующий в изделии, дополнительно содержит рекомбинантную гиалуронидазу человека (rHuPH20) в количестве, достаточном для того, чтобы привести к увеличению дисперсии пертузумаба при подкожном введении, например, в концентрации около 2000 Ед/мл или в концентрации около 1000 Ед/мл.
Изделие может дополнительно содержать один из более эксципиентов, выбранных из группы, состоящей из буферных агентов, стабилизаторов и поверхностно-активных веществ.
В одном варианте реализации изобретения буферный агент подходит для доведения рН до от около 5,0 до около 6,0, например, от 5,5 до 5,7, например 5,5. Типовой буфер представляет собой гистидиновый буфер, такой как ацетат L-гистидиновый.
Стабилизатор может содержать сахарозу и, необязательно, метионин и/или трегалозу.
Предпочтительно, поверхностно-активное вещество представляет собой полисорбат 20.
В дополнительном аспекте изобретение относится к водному препарату для подкожного введения, содержащему пертузумаб в концентрации около 120 мг/мл, rHuPH20 в концентрации около 1000-2000 Ед/мл, L-гистидиновый буфер для доведения рН до значений около 5,5-5,7, сахарозу, метионин и полисорбат 20.
В одном варианте реализации изобретения rHuPH20 присутствует в концентрации около 1000 Ед/мл.
В другом варианте реализации изобретения rHuPH20 присутствует в концентрации около 2000 Ед/мл.
В дополнительном варианте реализации изобретения водный раствор имеет рН 5,7.
Изобретение, кроме того, относится к жидкой фармацевтической композиции для подкожного введения, содержащей фиксированную дозу пертузумаба и фиксированную дозу трастузумаба, совместно приготовленных в водном растворе, дополнительно содержащем rHuPH20, буферный агент, подходящий для доведения рН до значения около 5,0-6,0, стабилизатор и поверхностно-активное вещество.
В одном варианте реализации изобретения буферный агент представляет собой гистидиновый буфер.
В другом варианте реализации изобретения буферный агент представляет собой ацетат L-гистидина.
В еще одном варианте реализации изобретения рН составляет 5,5-5,7, например 5,5.
В других вариантах реализации изобретения жидкая фармацевтическая композиция содержит сахарозу в качестве стабилизатора и может дополнительно содержать метионин и/или трегалозу в качестве стабилизатора.
В одном конкретном аспекте жидкая фармацевтическая композиция содержит 600 мг пертузумаба в концентрации 60 мг/мл, 600 мг трастузумаба в концентрации 60 мг/мл, 1000 Ед/мл rHuPH20, 20 мМ His-HCl рН 5,5, 105 мМ трегалозы, 100 мМ сахарозы, 0,04% полисорбата 20, 10 мМ метионина и стерильную воду для инъекций до общего объема 10 мл, которая, например, должна содержаться во флаконе объемом 15 мл.
В другом конкретном аспекте жидкая фармацевтическая композиция содержит 1200 мг пертузумаба в концентрации 80 мг/мл, 600 мг трастузумаба в концентрации 40 мг/мл, 1000 Ед/мл rHuPH20, 20 мМ His-HCl рН 5,5, 70 мМ трегалозы, 133 мМ сахарозы, 0,04% полисорбата 20, 10 мМ метионина и стерильную воду для инъекций до общего объема 15 мл, которая может содержаться во флаконе объемом 20 мл.
Вышеуказанные изделия могут дополнительно содержать листок-вкладыш с указаниями для подкожного введения содержащейся в изделии жидкой фармацевтической композиции человеку с HER2-положительным онкологическим заболеванием, таким как, например, рак молочной железы, перитонеальный рак, рак маточной трубы, рак легких, колоректальный рак, рак желчных путей или рак мочевого пузыря, таким как ранний рак молочной железы (РРМЖ) или метастатический рак молочной железы (МРМЖ).
В дополнительном аспекте изобретение относится к способу лечения онкологического заболевания, включающему подкожное введение субъекту, представляющему собой человека с HER2-положительным онкологическим заболеванием, одной или более фиксированных доз(ы) антитела против HER2, содержащего вариабельные аминокислотные последовательности легкой и тяжелой цепей, представленные в SEQ ID NO: 7 и 8 соответственно в количестве, эффективном для лечения онкологического заболевания, при этом фиксированная доза составляет около 600 мг и/или около 1200 мг.
Антитело против HER2 предпочтительно представляет собой пертузумаб.
В одном варианте реализации изобретения способ включает введение субъекту, представляющему собой человека, пертузумаба в фиксированной нагрузочной дозе около 1200 мг с последующей по меньшей мере одной поддерживающей дозой около 600 мг.
Во втором варианте реализации изобретения введение нагрузочной дозы сопровождается введением нескольких поддерживающих доз.
В третьем варианте реализации изобретения первую поддерживающую дозу пертузумаба вводят человеку спустя около две недели или около три недели после введения нагрузочной дозы пертузумаба.
В других вариантах реализации изобретения фиксированные дозы пертузумаба вводят человеку приблизительно каждые 2 недели или приблизительно каждые 3 недели.
Онкологическое заболевание может представлять собой HER2-положительное онкологическое заболевание, такое как рак молочной железы, перитонеальный рак, рак маточной трубы, рак легких, колоректальный рак, рак желчных путей или рак мочевого пузыря, такое как ранний рак молочной железы (РРМЖ) или метастатический рак молочной железы (МРМЖ).
Необязательно, способ может дополнительно включать введение пациенту второго терапевтического агента, такого как другое антитело против HER2, например трастузумаб, или химиотерапевтического агента.
В одном варианте реализации изобретения фиксированную дозу пертузумаба вводят подкожно в комбинации с подкожно вводимым трастузумабом.
В другом варианте реализации изобретения фиксированную дозу пертузумаба и трастузумаба вводят совместно подкожно в виде двух отдельных подкожных инъекций.
В еще одном варианте реализации изобретения фиксированную дозу пертузумаба смешивают с фиксированной дозой трастузумаба и вводят в виде единой подкожной инъекции.
В дополнительном варианте реализации изобретения фиксированную дозу пертузумаба и фиксированную дозу трастузумаба вводят в виде одного совместного препарата для подкожного введения, такого как любой из совместных препаратов, описанных выше и в данном раскрытии.
Химиотерапевтический агент, если он вводится, может, например, представлять собой таксан и/или антрациклин, такой как паклитаксел, доцетаксел, даунорубицин, доксорубицин и/или эпирубицин.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 изображена схема структуры белка HER2 и аминокислотные последовательности доменов I-IV (SEQ ID NO: 1-4, соответственно) его внеклеточного домена.
На Фиг. 2А и 2В изображены выравнивания аминокислотных последовательностей вариабельных доменов легкой (VL) (Фиг. 2А) и тяжелой (Vh) (Фиг. 2В) цепей мышиного моноклонального антитела 2С4 (SEQ ID NO: 5 и 6 соответственно); домены VL и VH варианта 574/пертузумаб (SEQ ID NO: 7 и 8, соответственно), и консенсусные каркасы VL и VH человека (hum κ1, каппа-подгруппа I легкой цепи; humIII, подгруппа III тяжелой цепи) (SEQ ID NO: 9 и 10 соответственно). Звездочками обозначены различия между вариабельными доменами пертузумаба и мышиного моноклонального антитела 2С4 или между вариабельными доменами пертузумаба и каркасной последовательностью человека. Области, определяющие комплементарность (CDR) указаны в скобках.
На Фиг. 3А и 3В изображены аминокислотные последовательности легкой цепи пертузумаба (Фиг. 3А; SEQ ID NO: 11) и тяжелой цепи (Фиг. 3В; SEQ ID NO: 12). CDR выделены жирным шрифтом. Расчетная молекулярная масса легкой цепи и тяжелой цепи составляет 23 526,22 Да и 49 216,56 Да (цистеины в восстановленной форме). Углеводный фрагмент присоединен к Asn 299 тяжелой цепи.
На Фиг. 4А и 4В изображены аминокислотные последовательности легкой цепи трастузумаба (Фиг. 4А; SEQ ID NO: 13) и тяжелой цепи (Фиг. 4В; SEQ ID NO: 14), соответственно. Границы вариабельных доменов легкой и тяжелой цепей отмечены стрелками.
На Фиг. 5А и 5В изображен вариант последовательности легкой цепи пертузумаба (Фиг. 5А; SEQ ID NO: 15) и вариант последовательности тяжелой цепи пертузумаба (Фиг. 5В; SEQ ID NO: 16), соответственно.
На Фиг. 6 изображена схема исследования для определения дозы для подкожного введения пертузумаба отдельно и в комбинации с трастузумабом.
На Фиг. 7 изображена диаграмма решений.
На Фиг. 8 изображена общая схема исследования.
На Фиг. 9 изображены нормализованные концентрации (мкг/мл) пертузумаба, вводимого подкожно, с трастузумабом и без него, в виде функции времени (дни).
На Фиг. 10 изображены нормированные по дозе концентрации (мкг/мл) пертузумаба в виде функции времени (дни) с различными концентрациями rHuPH20.
На Фиг. 11 изображены оценки параметров с использованием моделей пертузумаба и исторической популяционной ФК (попФК) в/в в сравнении.
На Фиг. 12 изображено демографическое и возрастное распределение.
На Фиг. 13 представлен обзор нежелательных явлений, Часть 1.
На Фиг. 14 представлен обзор нежелательных явлений, Часть 1, число субъектов.
На Фиг. 15 представлены наиболее распространенные нежелательные явления (все классы) - частота ≥5% во всем исследовании, число субъектов.
На Фиг. 16 изображена EGFR-опосредованная токсичность.
На Фиг. 17 изображены реакции, связанные с инъекцией, и реакции в месте инъекции. На Фиг. 18 изображены оценки ФВЛЖ (Эхокардиография).
На Фиг. 19 изображены композиции для подкожного введения лекарственных веществ пертузумаба, трастузумаба и rHuPH20 (SC DS), используемых при приготовлении препаратов с фиксированной дозой пертузумаба-трастузумаба.
На Фиг. 20 изображено количество (%) высокомолекулярных соединений (HMWS) в различных композициях пертузумаба и трастузумаба для подкожного введения, а также в совместных препаратах пертузумаба/трастузумаба при температуре 5°С и 25°С, соответственно.
На Фиг. 21 изображено среднее значение концентрации сывороточного пертузумаба в зависимости от времени для разных когорт.
На Фиг. 22 изображена среднегеометрическая нормализованная по дозе сывороточная концентрация пертузумаба в зависимости от времени, с сопутствующим герцептином и без него.
На Фиг. 23 изображена среднегеометрическая сывороточная концентрация пертузумаба в зависимости от времени при 667 Ед/мл или 2000 Ед/мл rHuPH20 (3МД)
На Фиг. 24 изображена среднегеометрическая сывороточная концентрация трастузумаба в зависимости от времени при 667 Ед/мл или 2000 Ед/мл rHuPH20 (3МД)
На Фиг. 25 изображена среднегеометрическая сывороточная концентрации пертузумаба в зависимости от времени после введения доз перьета (Perjeta) 600 мг подкожно и перьета 420 мг в/в.
На Фиг. 26 изображена среднегеометрическая сывороточная концентрации пертузумаба в зависимости от времени у ЗМД (Здоровых Мужчин-Добровольцев) или пациентов с РРМЖ.
На Фиг. 27 изображена среднегеометрическая нормализованная по дозе сывороточная концентрация пертузумаба в зависимости от времени при 667 Ед/мл, 1000 Ед/мл или 2000 Ед/мл rHuPH20.
На Фиг. 28 изображена среднегеометрическая сывороточная концентрация трастузумаба в зависимости от времени при 667 Ед/мл или 1000 Ед/мл, или 2000 Ед/мл rHuPH20.
На Фиг. 29 изображено исследование лекарственного вещества пертузумаба на стабильность (Drug Substance Stability Scratch & Sprinkle Test): Данные эксклюзионной хроматографии.
На Фиг. 30 изображены различия в препаратах FDC - мутность.
На Фиг. 31 изображены различия в препаратах FDC - SEC/HMWS
Подробное описание предпочтительных вариантов реализации изобретения
I. Определения
Термин «фармацевтический препарат» относится к препарату, который находится в такой форме, которая обеспечивает реализацию биологической активности активного ингредиента, и который не содержит дополнительных компонентов, которые являются неприемлемо токсичными для субъекта, которому будет вводиться композиция. Такие препараты являются стерильными.
«Стерильный» препарат является асептическим или не содержит никаких живых микроорганизмов и их спор.
«Стабильный» препарат представляет собой препарат, в котором содержащийся в нем белок по существу сохраняет свою физическую стабильность и/или химическую стабильность и/или биологическую активность при хранении. Предпочтительно, препарат в основном сохраняет свою физическую и химическую стабильность, а также свою биологическую активность при хранении. Период хранения обычно выбирается на основе предполагаемого срока годности препарата. Различные аналитические способы для измерения стабильности белка доступны в данной области техники и рассматриваются, например, в Peptide and Protein. Drug Delivery, 247-301, Vincent Lee Ed., Marcel Dekker, Inc., New York, N.Y., Pubs. (1991) и Jones, A. Adv. Drug Delivery Rev. 10: 29-90 (1993). Стабильность может быть измерена при выбранной температуре в течение выбранного периода времени. Предпочтительно, препарат стабилен при температуре около 40°С в течение по меньшей мере около 2-4 недель и/или стабилен при температуре около 5,0 и/или 15°С в течение как минимум 3 месяцев и/или стабилен при температуре около -20°С в течение не менее 3 месяцев или не менее 1 года. Кроме того, препарат, предпочтительно, стабилен после замораживания (например, до -70°С) и оттаивания препарата, например, после 1, 2 или 3 циклов замораживания и оттаивания. Стабильность может быть оценена качественно и/или количественно различными способами, включая оценку образования агрегатов (например, с использованием эксклюзионной хроматографии по размеру, путем измерения мутности и/или с помощью визуального контроля); оценку неоднородности заряда с помощью катионообменной хроматографии или капиллярного зонного электрофореза; анализ аминоконцевой или карбоксиконцевой последовательности; масс-спектрометрический анализ; анализ ДСН-ПААГ для сравнения восстановленного и интактного антитела; анализ пептидной карты (например, триптический или LYS-C); оценку биологической активности или антигенсвязывающей функции антитела; и т.д. Нестабильность может включать один или более из следующих факторов: агрегацию, дезамидирование (например, дезамидирование Asn), окисление (например, окисление Met), изомеризацию (например, изомеризацию Asp), укорочение/гидролиз/фрагментацию (например, фрагментацию шарнирной области), образование сукцинимида, непарный цистеин (цистеины), удлинение N-конца, процессинг С-конца, различия гликозилирования и т.д.
Антитело, которое «подвержено дезамидированию», представляет собой антитело, содержащее один или более остатков, которые, как было обнаружено, склонны к дезамидированию.
Антитело, которое «подвержено агрегации», представляет собой антитело, которое, как было установлено, агрегирует с другой молекулой антитела, особенно при замораживании и/или перемешивании.
Антитело, которое «подвержено фрагментации», представляет собой антитело, которое, как было обнаружено, расщепляется на два или более фрагмента, например, в его шарнирной области.
Под «уменьшением дезамидирования, агрегации или фрагментации» подразумевается предотвращение или уменьшение уровня дезамидирования, агрегации или фрагментации по отношению к моноклональному антителу, приготовленному при другом рН или в другом буфере.
В контексте данного документа «биологическая активность» моноклонального антитела относится к способности антитела связываться с антигеном и приводить к измеримому биологическому ответу, который может быть измерен in vitro или in vivo. В случае пертузумаба в одном варианте реализации изобретения биологическая активность относится к способности приготовленного антитела ингибировать пролиферацию клеточной линии рака молочной железы человека MDA-MB-175-VII.
Под «изотоническим» подразумевается, что представляющий интерес препарат имеет по существу такое же осмотическое давление, что и кровь человека. Изотонические препараты обычно будут иметь осмотическое давление от 250 до 350 мОсм. Изотоничность может быть измерена, например, с использованием парового осмометра или осмометра по точке замерзания.
Используемый в контексте данного документа термин «буфер» относится к буферному раствору, который препятствует изменениям рН путем влияния компонентов его кислотно-основного конъюгата. Буфер согласно данного изобретения предпочтительно имеет рН в диапазоне от около 5,0 до около 7,0, предпочтительно от около 5,5 до около 6,5, например от около 5,5 до около 6,2, такой как, например, 5,5 или 5,7. Примеры буферов, которые будут контролировать рН в этом диапазоне, включают ацетатный, сукцинатный, глюконатный, гистидиновыей, цитратный, глицилглициновый буферы и буферы на основе других органических кислот. Предпочтительный буфер согласно данному документу представляет собой гистидиновый буфер.
«Гистидиновый буфер» представляет собой буфер, содержащий ионы гистидина. Примеры гистидиновых буферов включают гистидин-хлоридный, гистидин-ацетатный, гистидин-фосфатный, гистидин-сульфатный. Было обнаружено, что предпочтительным гистидиновым буфером, определенным в приведенных в данном документе примерах, является ацетат гистидина. В предпочтительном варианте реализации изобретения гистидин-ацетатный буфер получают путем титрования L-гистидина (свободное основание, твердое вещество) уксусной кислотой (жидкость). Предпочтительно, гистидиновый буфер или гистидин-ацетатный буфер имеют рН от 5,5 до 6,5 или рН от 5,7 до 6,2, например рН 5,7.
«Сахарид» в контексте данного документа включает общую композицию (СН20)n и ее производные, включая моносахариды, дисахариды, трисахариды, полисахариды, сахарные спирты, восстанавливающие сахара, невосстанавливающие сахара и т.д. Примеры сахаридов в данном документе включают глюкозу, сахарозу, трегалозу, лактозу, фруктозу, мальтозу, декстран, глицерин, эритрит, глицерол, арабит, силитол, сорбит, маннит, меллибиозу, мелезитозу, раффинозу, маннотритозу, стахиозу, лактулозу, мальтулозу, глюцитол, мальтит, лактит, изо-мальтулозу и т.д. Предпочтительным сахаридом в данном документе является невосстанавливающий дисахарид, такой как трегалоза или сахароза.
В контексте данного документа «поверхностно-активное вещество» относится к поверхностно-активному агенту, предпочтительно неионогенному поверхностно-активному веществу. Примеры поверхностно-активных веществ в данном документе включают полисорбат (например, полисорбат 20 и полисорбат 80); полоксамер (например, полоксамер 188); тритон; додецилсульфат натрия (SDS); лаурил сульфат натрия; октилгликозид натрия; лаурил-, миристил-, линолеил- или стеарилсульфобетаин; лаурил-, миристил-, линолеил- или стеарил-саркозин; линолеил-, миристил- или цетил-бетаин; лауроамидопропил-, кокамидопропил-, линолеамидопропил-, миристамидопропил-, пальмидопропил- или изостеарамидопропил-бетаин (например, лауроамидопропил); миристамидопропил-, пальмидопропил- или изостеарамидопропил-диметиламин; метилкокоил- или динатрийметил-олеилтаурат натрия; и серию MONAQUAT™ (Mona Industries, Inc., Патерсон, Нью-Джерси); полиэтиленгликоль, полипропилгликоль и сополимеры этилена и пропиленгликоля (например, Pluronics, PF68 и т.д.) и т.д. Предпочтительным поверхностно-активным веществом в данном документе является полисорбат 20.
«Рецептор HER» представляет собой рецепторную протеинтирозинкиназу, которая принадлежит к семейству рецепторов HER и включает рецепторы EGFR, HER2, HER3 и HER4. Рецептор HER обычно содержит внеклеточный домен, который может связывать лиганд HER и/или димеризоваться с другой молекулой рецептора HER; липофильный трансмембранный домен; консервативный внутриклеточный домен тирозинкиназы; и карбоксильный концевой сигнальный домен, содержащий несколько остатков тирозина, которые могут быть фосфорилированы. Рецептор HER может представлять собой рецептор HER «с нативной последовательностью» или его «вариант аминокислотной последовательности». Предпочтительно, рецептор HER представляет собой рецептор HER человека с нативной последовательностью.
Фразы «ErbB2» и «HER2» используются в данном документе взаимозаменяемо и относятся к белку HER2 человека, описанному, например, в Semba et al, PNAS (USA) 82:6497-6501 (1985) и Yamamoto et al., Nature 319: 230-234 (1986) (номер доступа Genebank X03363). Термин «erbB2» относится к гену, кодирующему ErbB2 человека, а «neu» относится к гену, кодирующему p185neu крысы. Предпочтительный HER2 представляет собой нативную последовательность HER2 человека.
В контексте данного документа «внеклеточный домен HER2» или «ECD HER2» относится к домену HER2, который находится вне клетки, либо прикреплен к клеточной мембране, либо находится в кровотоке, включая его фрагменты. Аминокислотная последовательность HER2 изображена на Фиг. 1. В одном варианте реализации изобретения внеклеточный домен HER2 может содержать четыре домена: «Домен I» (аминокислотные остатки от около 1 до около 195; SEQ ID NO: 1), «Домен II» (аминокислотные остатки от около 196 до около 319; SEQ ID NO: 2), «Домен III» (аминокислотные остатки от около 320 до около 488: SEQ ID NO: 3) и «Домен IV» (аминокислотные остатки от около 489 до около 630; SEQ ID NO: 4) (нумерация остатков без учета сигнального пептида). Смотри Garrett et al. Mol. Cell. 11: 495-505 (2003), Cho et al., Nature 421: 756-760 (2003), Franklin et al. Cancer Cell 5:317-328 (2004), и Plowman et al. Proc. Natl. Acad. Set. 90:1746-1750 (1993), а также Фиг. 1 в данном документе.
«HER3» или «ErbB3» в контексте данного документа относятся к рецептору, описанному, например, в патентах США №5183884 и 5480968, а также в Kraus et al. PNAS (USA) 86:9193-9197 (1989).
Онкологическое заболевание с низким уровнем HER3 представляет собой онкологическое заболевание, при котором HER3 экспрессируется на уровне, меньшем, чем средний уровень экспрессии HER3 при данном типе онкологического заболевания. В одном варианте реализации изобретения онкологическое заболевание с низким уровнем HER3 представляет собой рак эпителия яичников, перитонеальный рак или рак маточной трубы. Уровень ДНК, белка и/или мРНК HER3 при онкологическом заболевании можно оценить для определения является ли онкологическое заболевание таковым с низким уровнем HER3. См., например, патент США №7981418 для получения дополнительной информации об онкологическом заболевании с низким уровнем HER3. Необязательно, проводится анализ экспрессии мРНК HER3, чтобы определить, является ли онкологическое заболевание таковым с низким уровнем HER3. В одном варианте реализации изобретения уровень мРНК HER3 при раке оценивают, например, с использованием полимеразной цепной реакции (ПНР), такой как количественная ПНР с обратной транскрипцией (ОТ-ПЦР). Необязательно, при онкологическом заболевании HER3 экспрессируется в соотношении концентраций, равном или меньшем, чем около 2,81, при оценке с помощью ОТ-ПЦР, например, с использованием прибора COBAS z480®.
«Димер HER» в контексте данного документа представляет собой нековалентно связанный димер, содержащий по меньшей мере два рецептора HER. Такие комплексы могут формироваться в случае, когда клетка, экспрессирующая два или более рецепторов HER, подвергается воздействию лиганда HER, и может быть выделена путем иммунопреципитации и проанализирована с помощью SDS-PAGE, как описано, например, в Sliwkowski et al., J. Biol. Chem., 269(20): 14661-14665 (1994). Другие белки, такие как субъединица рецептора цитокина (например, gp130), могут быть ассоциированы с димером. Предпочтительно, димер HER содержит HER2.
«Гетеродимер HER» в контексте данного документа представляет собой нековалентно связанный гетеродимер, содержащий по меньшей мере два разных рецептора HER, такой как гетеродимеры EGFR-HER2, HER2-HER3 или HER2-HER4.
«Антитело против HER» представляет собой антитело, которое связывается с рецептором HER. Необязательно, антитело против HER дополнительно препятствует активации или функции HER. Предпочтительно, антитело против HER связывается с рецептором HER2. Представляющие интерес антитела против HER2 представляют собой пертузумаб и трастузумаб.
«Активация HER» относится к активации или фосфорилированию любого одного или более рецепторов HER. Как правило, активация HER приводит к передаче сигнала (например, вызванной внутриклеточным доменом киназы рецептора HER, фосфорилирующего остатки тирозина в рецепторе HER или субстратном полипептиде). Активация HER может быть опосредована связыванием лиганда HER с димером HER, содержащим представляющий интерес рецептор HER. Связывание лиганда HER с димером HER может активировать киназный домен одного или более рецепторов HER в димере и тем самым приводит к фосфорилированию остатков тирозина в одном или более рецепторах HER и/или фосфорилированию остатков тирозина в дополнительном субстратном полипептиде(ах), таком как внутриклеточные киназы Akt или МАРК.
«Фосфорилирование» относится к добавлению одной или более фосфатных групп к белку, такому как рецептор HER или его субстрат.
Антитело, которое «ингибирует димеризацию HER», представляет собой антитело, которое ингибирует или препятствует образованию димера HER. Предпочтительно, такое антитело связывается с HER2 на своем гетеродимерном сайте связывания. Наиболее предпочтительное антитело, ингибирующее димеризацию, представляет собой пертузумаб или MAb 2С4. Другие примеры антител, которые ингибируют димеризацию HER, включают антитела, которые связываются с EGFR и ингибируют его димеризацию с одним или более другими рецепторами HER (например, моноклональное антитело EGFR 806, Mab 806, которое связывается с активированным или «не связанным» EGFR; см. Johns et al., J. Biol. Chem. 279 (29): 30375-30384 (2004)); антитела, которые связываются с HER3 и ингибируют его димеризацию с одним или более другими рецепторами HER; и антитела, которые связываются с HER4 и ингибируют его димеризацию с одним или более другими рецепторами HER.
«Ингибитор димеризации HER2» представляет собой агент, который ингибирует образование димера или гетеродимера, содержащего HER2.
«Гетеродимерный сайт связывания» на HER2 относится к области во внеклеточном домене HER2, которая контактирует или взаимодействует с областью во внеклеточном домене EGFR, HER3 или HER4 при образовании с ними димера. Эта область находится в домене II HER2 (SEQ ID NO: 15). Franklin et al., Cancer Cell 5:317-328 (2004).
Антитело против HER2, которое «связывается с гетеродимерным сайтом связывания» HER2, связывается с остатками в Домене II (SEQ ID NO: 2) и, необязательно, также связывается с остатками в других доменах внеклеточного домена HER2, таких как домены I и III, (SEQ ID NO: 1 и 3) и могут стерически препятствовать, по меньшей мере, до некоторой степени, формированию гетеродимера HER2-EGFR, HER2-HER3 или HER2-HER4. В Franklin et al., Cancer Cell 5:317-328 (2004) охарактеризована кристаллическая структура HER2-пертузумаб, и внесена в RCSB Protein Data Bank (ID Code IS78), иллюстрируя типовое антитело, которое связывается с гетеродимерным сайтом связывания HER2.
Антитело, которое «связывается с доменом II» HER2, связывается с остатками в домене II (SEQ ID NO: 2) и, необязательно, с остатками в другом домене (доменах) HER2, таком как домены I и III (SEQ ID NO: 1 и 3, соответственно). Предпочтительно, антитело, которое связывается с доменом II, связывается с соединением между доменами HER2 I, II и III.
В контексте данного документа термины «пертузумаб» и «rhuMAb 2С4», используемые взаимозаменяемо, относятся к антителу, содержащему вариабельные аминокислотные последовательности легкой и тяжелой цепи, представленные в SEQ ID NO: 7 и 8, соответственно. Когда пертузумаб представляет собой интактное антитело, он предпочтительно содержит антитело IgG1; в одном варианте реализации изобретения содержит аминокислотную последовательность легкой цепи, представленную в SEQ ID NO: 11 или 15, и аминокислотную последовательность тяжелой цепи, представленную в SEQ ID NO: 12 или 16. Антитело необязательно продуцируется рекомбинантными клетками яичника китайского хомячка (СНО). Термины «пертузумаб» и «rhuMAb 2С4» в контексте данного документа охватывают биоподобные версии препарата согласно принятому в США названию (United States Adopted Name, USAN) или международному непатентованному наименованию (INN): пертузумаб.
В контексте данного документа термины «трастузумаб» и «rhuMAb4D5», используемые взаимозаменяемо, относятся к антителу, содержащему вариабельные аминокислотные последовательности легкой и тяжелой цепи согласно SEQ ID NO: 13 и 14, соответственно. Когда трастузумаб представляет собой интактное антитело, он предпочтительно содержит антитело IgG1; в одном варианте реализации изобретения содержит аминокислотную последовательность легкой цепи, представленную в SEQ ID NO: 13 и аминокислотную последовательность тяжелой цепи, представленную в SEQ ID NO: 14. Антитело необязательно продуцируется клетками яичника китайского хомячка (СНО). Термины «трастузумаб» и «rhuMAb4D5» в контексте данного документа охватывают биоподобные версии препарата согласно принятому в США названию (United States Adopted Name, USAN) или международному непатентованному наименованию (INN): трастузумаб.
Термин «антитело» в контексте данного документа используется в самом широком смысле и, в частности, охватывает моноклональные антитела, поликлональные антитела, полиспецифические антитела (например, биспецифичные антитела) и фрагменты антител, при условии, что они проявляют желаемую биологическую активность.
«Гуманизированные» формы антител, не относящихся к человеку (например, грызунов) представляют собой химерные антитела, которые содержат минимальную последовательность, полученную из иммуноглобулина, не относящегося к человеку. По большей части гуманизированные антитела представляют собой человеческие иммуноглобулины (антитело реципиент), в которых остатки из гипервариабельной области реципиента заменены остатками из гипервариабельной области вида, не относящегося к человеку (донорского антитела), такого как мышь, крыса, кролик или примат, не относящийся к человеку, имеющие желаемую специфичность, аффинность и активность. В некоторых случаях остатки каркасной области (FR) человеческого иммуноглобулина заменены соответствующими остатками, не относящимися к человеку. Кроме того, гуманизированные антитела могут содержать остатки, которые не обнаружены в реципиентном антителе или в донорном антителе. Эти модификации сделаны для дальнейшего улучшения характеристик антител. В общем, гуманизированное антитело будет содержать, по существу, все из по меньшей мере, одного, и, как правило, двух вариабельных доменов, в которых все или, по существу, все гипервариабельные петли соответствуют таковым у иммуноглобулина, не относящегося к человеку, и все или, по существу, все FR представляют собой таковые из последовательности иммуноглобулина человека. Гуманизированное антитело необязательно также будет содержать, по меньшей мере, часть константной области иммуноглобулина (Fc), обычно константной области иммуноглобулина человека. Для получения дополнительной информации см. Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature 332:323-329 (1988); и Presta, Curr. Op.Struct. Biol. 2:593-596 (1992). Гуманизированные антитела против HER2, в частности, включают трастузумаб (HERCEPTIN®), как описано в Таблице 3 патента США №5,821,337, специально включенном в данный документ посредством ссылки и определенно в данном документе; и гуманизированные антитела 2С4, такие как пертузумаб, как описано и определено в данном документе.
«Интактное антитело» в контексте данного документа представляет собой антитело, которое содержит две антигенсвязывающие области и область Fc. Предпочтительно, интактное антитело имеет функциональную область Fc.
«Фрагменты антител» содержат часть интактного антитела, предпочтительно содержащую его антигенсвязывающую область. Примеры фрагментов антител включают фрагменты Fab, Fab', F (ab')2 и Fv; димеры; линеаризированные антитела; молекулы одноцепочечных антител; и мультиспецифичные антитела, образованные из фрагмента(ов) антител.
«Нативные антитела» обычно представляют собой гетеротетрамерные гликопротеины массой около 150000 дальтон, состоящие из двух идентичных легких (L) цепей и двух идентичных тяжелых (Н) цепей. Каждая легкая цепь связана с тяжелой цепью одной ковалентной дисульфидной связью, в то время как число дисульфидных связей варьирует среди тяжелых цепей разных изотипов иммуноглобулина. Каждая тяжелая и легкая цепь также имеет регулярно расположенные внутрицепочечные дисульфидные мостики. Каждая тяжелая цепь имеет на одном конце вариабельный домен (VH), за которым следует ряд константных доменов. Каждая легкая цепь имеет вариабельный домен на одном конце (VL) и константный домен на другом конце. Константный домен легкой цепи выровнен с первым константным доменом тяжелой цепи, а вариабельный домен легкой цепи выровнен с вариабельным доменом тяжелой цепи. Считается, что конкретные аминокислотные остатки формируют поверхность взаимодействия между вариабельными доменами легкой цепи и тяжелой цепи.
Термин «гипервариабельная область» при использовании в контексте данного документа относится к аминокислотным остаткам антитела, которые отвечают за связывание антигена. Гипервариабельная область обычно содержит аминокислотные остатки из «определяющей комплементарность области» или «CDR» (например, остатки 24-34 (L1), 50-56 (L2) и 89-97 (L3) в вариабельном домене легкой цепи и 31-35 (H1), 50-65 (Н2) и 95-102 (Н3) в вариабельном домене тяжелой цепи; Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD. (1991)) и/или эти остатки из «гипервариабельной петли» (например, остатки 26-32 (L1), 50-52 (L2) и 91-96 (L3) в вариабельном домене легкой цепи и 26-32 (H1), 53-55 (Н2) и 96-101 (Н3) в вариабельном домене тяжелой цепи; Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)). Остатки «каркасной области» или «FR» представляют собой остатки вариабельного домена, отличные от остатков гипервариабельной области, как определено в данном документе.
Термин «область Fc» в контексте данного документа используется для определения С-концевой области тяжелой цепи иммуноглобулина, включая области Fc с нативной последовательностью и варианты областей Fc. Хотя границы области Fc тяжелой цепи иммуноглобулина могут варьировать, область Fc тяжелой цепи IgG человека обычно определена как область от аминокислотного остатка в положении Cys226 или от Pro230 до его карбоксильного конца. С-концевой лизин (остаток 447 согласно системе нумерации EU) Fc-области может быть удален, например, во время продуцирования или очистки антитела или путем рекомбинантного конструирования нуклеиновой кислоты, кодирующей тяжелую цепь антитела. Соответственно, композиция интактных антител может содержать популяции антител со всеми удаленными остатками K447, популяции антител без удаленных остатков K447 и популяции антител, имеющие смесь антител с остатком K447 и без него.
Если не указано иначе, в данном документе нумерация остатков в тяжелой цепи иммуноглобулина соответствует нумерации индекса EU, как в Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991), специально включенной в данный документ посредством ссылки. «Индекс EU согласно Kabat» относится к нумерации остатков антитела IgG1 EU человека.
«Функциональная область Fc» обладает «эффекторной функцией» области Fc нативной последовательности. Типовые «эффекторные функции» включают связывание Clq; комплемент-зависимую цитотоксичность; Связывание с Fc-рецептором; антитело-зависимую клеточно-опосредованную цитотоксичность (ADCC); фагоцитоз; подавление рецепторов клеточной поверхности (например, В-клеточного рецептора; BCR) и т.д. Такие эффекторные функции обычно требуют, чтобы область Fc сочеталась с доменом связывания (например, вариабельным доменом антитела), и может оцениваться с использованием различных анализов, как описано, например, в данном документе.
«Область Fc с нативной последовательностью» содержит аминокислотную последовательность, идентичную аминокислотной последовательности области Fc, встречающейся в природе. Области Fc человека с нативной последовательностью содержат область Fc IgG1 человека с нативной последовательностью (аллотипы А и не-А); область Fc IgG2 человека с нативной последовательностью; область Fc IgG3 человека с нативной последовательностью; и область Fc IgG4 человека с нативной последовательностью, а также их встречающиеся в природе варианты.
«Вариант области Fc» содержит аминокислотную последовательность, которая отличается от последовательности области Fc с нативной последовательностью по меньшей мере одной модификацией аминокислоты, предпочтительно одной или более аминокислотным заменам. Предпочтительно, вариант области Fc имеет, по меньшей мере, одну аминокислотную замену по сравнению с областью Fc нативной последовательности или областью Fc исходного полипептида, например, от около одной до около десяти аминокислотных замен и, предпочтительно, от около одной до около пяти аминокислотных замен в области Fc нативной последовательности или в области Fc исходного полипептида. Вариант области Fc в данном документе предпочтительно будет обладать гомологией, по меньшей мере, около 80% с областью Fc нативной последовательности и/или с областью Fc исходного полипептида, и, наиболее предпочтительно, гомологией, по меньшей мере, около 90% с указанной последовательностью, более предпочтительно, гомологией, по меньшей мере, около 95% с указанной последовательностью.
В зависимости от аминокислотной последовательности константного домена их тяжелых цепей интактные антитела можно отнести к разным «классам». Существует пять основных классов интактных антител: IgA, IgD, IgE, IgG и IgM, и некоторые из них могут быть дополнительно разделены на подклассы (изотипы), например, IgG1, IgG2, IgG3, IgG4, IgA и IgA2. Константные домены тяжелой цепи, которые соответствуют различным классам антител, называются α, δ, ε, γ и μ соответственно. Структуры субъединиц и трехмерные конфигурации различных классов иммуноглобулинов хорошо известны.
«Голое антитело» представляет собой антитело, которое не конъюгировано с гетерологичной молекулой, такой как цитотоксическая часть или радиоактивная метка.
«Аффинно-зрелое» антитело представляет собой антитело с одним или более изменениями в одной или более его гипервариабельных областях, которые приводят к увеличению аффинности антитела к антигену по сравнению с родительским антителом, которое не обладает этим изменением (изменениями). Предпочтительные аффинно-зрелые антитела будут иметь наномолярные или даже пикомолярные уровни аффинности к антигену-мишени. Аффинно-зрелые антитела получают способами, известными в данной области техники. В Marks et al., Bio/Technology 10:779-783 (1992) описано созревание аффинности путем перетасовки доменов VH и VL. Случайный мутагенез остатков CDR и/или каркаса описан в: Barbas et al., Proc Nat. Acad. Sci, USA 91:3809-3813 (1994); Schier et al., Gene 169:147-155 (1995); Yelton et al. J. Immunol. 155:1994-2004 (1995); Jackson et al., J. Immunol. 154(7):3310-9 (1995); и Hawkins et al., J. Mol. Biol. 226:889-896 (1992).
«Дезамидированное» антитело представляет собой антитело, в котором один или более его аспарагиновых остатков были дериватизированы, например, в аспарагиновую кислоту, сукцинимид или изоаспарагиновую кислоту.
Термины «онкологическое заболевание» и «раковый» относятся или описывают физиологическое состояние у млекопитающих, которое обычно характеризуется нерегулируемым ростом клеток.
«Прогрессирующее» онкологическое заболевание представляет собой онкологическое заболевание, которое распространилось за пределы места или органа происхождения, либо путем локальной инвазии («местнораспространенное»), либо метастазирования («метастатическое»). Соответственно, термин «прогрессирующее» онкологическое заболевание включает как местнораспространенное, так и метастатическое заболевание.
«Метастатическое» онкологическое заболевание относится к онкологическому заболеванию, которое распространилось из одной части тела (например, молочной железы) в другую часть тела.
«Резистентное» онкологическое заболевание представляет собой онкологическое заболевание, которое прогрессирует, не смотря на то, что противоопухолевый агент, такой как химиотерапия или биологическая терапия, такая как иммунотерапия, проводится больному онкологическим заболеванием. Типовое резистентное онкологическое заболевание представляет собой онкологическое заболевание, устойчивое к платине.
«Рецидивирующее» онкологическое заболевание представляет собой онкологическое заболевание, которое возникло заново либо в начальном, либо в отдаленном месте после ответа на начальную терапию, такую как операция.
«Местнорецидивирующее» онкологическое заболевание представляет собой онкологическое заболевание, которое возникает заново после лечения в том же месте, что и ранее леченное онкологическое заболевание.
«Нерезектабельный» или «неоперабельный» рак не может быть удален (резецирован) хирургическим путем.
«Рак молочной железы на ранней стадии» в контексте данного документа относится к раку молочной железы, который не распространился за пределы груди или подмышечных лимфатических узлов. Такое онкологическое заболевание обычно лечат с помощью неоадъювантной или адъювантной терапии.
«Неоадъювантная терапия» или «неоадъювантное лечение» или «неоадъювантное введение» относится к системной терапии, проводимой до операции.
«Адъювантная терапия» или «адъювантное лечение» или «адъювантное введение» относится к системной терапии, проводимой после операции.
В контексте данного документа «пациент» или «субъект» представляет собой пациента-человека. Пациент может быть «болен онкологическим заболеванием», то есть быть тем, кто страдает или подвержен риску влияния одного или более симптомов онкологического заболевания, в частности рака молочной железы.
Термин «популяция пациентов» относится к группе больных онкологическим заболеванием. Такие популяции можно использовать для демонстрации статистически значимой эффективности и/или безопасности лекарственного средства, такого как пертузумаб и/или трастузумаб.
Пациент «с рецидивом» представляет собой такового, у которого есть признаки или симптомы онкологического заболевания после ремиссии. Необязательно, у пациента возникает рецидив после адъювантной или неоадъювантной терапии.
Онкологическое заболевание или биологический образец, которые «проявляют экспрессию, амплификацию или активацию HER», представляют собой таковые, которые в диагностическом тесте экспрессируют (включая сверхэкспрессию) рецептор HER, имеют амплифицированный ген HER и/или иным образом демонстрируют активацию или фосфорилирование рецептора HER.
Онкологическое заболевание или биологический образец, которые «проявляют активацию HER», представляет собой таковые, которые в диагностическом тесте демонстрируют активацию или фосфорилирование рецептора HER. Такая активация может быть определена напрямую (например, путем измерения фосфорилирования HER с помощью ИФА) или косвенно (например, путем профилирования экспрессии генов или путем детектирвоания гетеродимеров HER, как описано в данном документе).
Раковая клетка со «сверхэкспрессией или амплификацией рецептора HER» представляет собой клетку, которая имеет значительно более высокие уровни белка или гена рецептора HER по сравнению с нераковой клеткой того же типа ткани. Такая сверхэкспрессия может быть вызвана амплификацией генов или усилением транскрипции или трансляции. Сверхэкспрессию или амплификацию рецептора HER можно определять в диагностическом или прогностическом анализе путем оценки повышенных уровней белка HER, присутствующего на поверхности клетки (например, с помощью иммуногистохимического анализа; IHC). Альтернативно или дополнительно, можно измерять уровни нуклеиновой кислоты, кодирующей HER, в клетке, например, посредством гибридизации in situ (ISH), включая флуоресцентную гибридизацию in situ (FISH; см. WO 98/45479, опубликованную в октябре 1998 г. ) и хромогенную гибридизацию in situ. (CISH; см., например, Tanner et al., Am.J. Pathol. 157(5): 1467-1472 (2000); Bella et al., J. Clin. Oncol. 26: (May 20 Suppl; abstr 22147) (2008)), способов саузерн-блоттинга или полимеразной цепной реакции (ПЦР), такой как количественная ПЦР в режиме реального времени (количественная ПЦР). Можно также изучить сверхэкспрессию или амплификацию рецептора HER путем измерения отделенного антигена (например, внеклеточного домена HER) в биологической жидкости, такой как сыворотка (см., например, патент США №4,933,294, выданный 12 июня 1990 г.; WO 91/05264, опубликованную 18 апреля, 1991; патент США №5,401,638, выданный 28 марта 1995 г., и Sias et al., J. Immunol. Methods 132: 73-80 (1990)). Помимо вышеуказанных исследований, для квалифицированного практикующего специалиста доступны различные исследования in vivo. Например, можно подвергать клетки в теле пациента воздействию антитела, которое необязательно помечено детектируемой меткой, например радиоактивной меткой in situ для придания радиоактивности, или путем исследования образца биопсии, взятого у пациента, ранее подвергавшегося воздействию антитела.
«HER2-положительный» рак содержит раковые клетки, у которых уровень HER2 выше нормального. Необязательно, HER2-положительный рак имеет показатель иммуногистохимии (IHC) 2+ или 3+ и/или положительную гибридизацию in situ (ISH), флуоресцентную гибридизацию in situ (FISH) или хромогенную гибридизацию in situ (CISH), например, имеет коэффициент амплификации ISH/FISH/CISH ≥ 2,0.
«HER2-мутированный» рак содержит раковые клетки с HER2-активирующей мутацией, включая мутации киназного домена, которые можно, например, идентифицировать с помощью секвенирования следующего поколения (NGS) или полимеразной цепной реакции в режиме реального времени (количественная ПЦР). «HER2-мутированный» рак, в частности, включает рак, характеризующийся инсерциями в экзоне 20 HER2, делециями вокруг аминокислотных остатков 755-759 HER2, любой из мутаций G309A, G309E, S310F, D769H, D769Y, V777L, P780-Y781insGSP, V842I, R896C (Bose et al., Cancer Discov 2013; 3:1-14), а также ранее сообщалось об идентичных несинонимичных предполагаемых активирующих мутациях (или инсерционно-делеционных мутациях) в базе данных COSMIC, обнаруженных в двух или более уникальных экземплярах. Для получения дополнительной информации см., например, Stephens et al., Nature 2004; 431:525-6; Shigematsu et al., Cancer Res 2005; 65:1642-6; Buttitta et al., Int J Cancer 2006; 119:2586-91; Li et al., Oncogene 2008; 27:4702-11; Sequist et al., JClin Oncol 2010; 28:3076-83; Arcila et al., Clin Cancer Res 2012; 18:4910-8; Greulich et al., Proc Natl Acad Sci USA 2012; 109:14476-81; и Herter-Sprie et al., Front Oncol 2013;3:1-10.
В контексте данного документа «противоопухолевый агент» относится к лекарственному средству, используемому для лечения онкологического заболевания. Неограничивающие примеры противоопухолевых агентов в данном документе включают химиотерапевтические агенты, ингибиторы димеризации HER, антитела против HER, антитела, нацеленные против антигенов, ассоциированных с опухолью, антигормональные соединения, цитокины, лекарственные средства, нацеленные на EGFR, антиангиогенные агенты, ингибиторы тирозинкиназы, ингибирующие рост ангенты и антитела, цитотоксические агенты, антитела, индуцирующие апоптоз, ингибиторы СОХ, ингибиторы фарнезилтрансферазы, антитела, связывающиеся с онкофетальным белком СА 125, HER2-вакцины, ингибиторы Raf или ras, липосомальный доксорубицин, топотекан, таксан, двойные ингибиторы тирозинкиназы, TLK286, EMD-7200, пертузумаб, трастузумаб, эрлотиниб и бевацизумаб.
«Эпитоп 2С4» представляет собой область во внеклеточном домене HER2, с которой связывается антитело 2С4. Для скрининга антител, которые связываются по существу с эпитопом 2С4, может быть проведено обычное перекрестное конкурентное исследование, такое как описанное в Antibodies, A Laboratory Manual, Cold Spring Harbor Laboratory, Ed Harlow and David Lane (1988). Предпочтительно, антитело блокирует связывание 2С4 с HER2 на около 50% или более. Альтернативно, может быть выполнено картирование эпитопа для оценки того, связывается ли антитело по существу с эпитопом 2С4 HER2. Эпитоп 2С4 содержит остатки из домена II (SEQ ID NO: 2) во внеклеточном домене HER2. 2С4 и пертузумаб связываются с внеклеточным доменом HER2 на стыке доменов I, II и III (SEQ ID NO: 1, 2 и 3, соответственно). Franklin et al., Cancer Cell 5:317-328 (2004).
«Эпитоп 4D5» представляет собой область во внеклеточном домене HER2, с которой связываются антитело 4D5 (АТСС CRL 10463) и трастузумаб. Этот эпитоп близок к трансмембранному домену HER2 и находится в домене IV HER2 (SEQ ID NO: 4). Для скрининга антител, которые связываются по существу с эпитопом 4D5, может быть проведено обычное перекрестное конкурентное исследование, такое как описанное в Antibodies, A Laboratory Manual, Cold Spring Harbor Laboratory, Ed Harlow and David Lane (1988). Альтернативно, может быть выполнено картирование эпитопа для оценки того, связывается ли антитело по существу с эпитопом 4D5 HER2 (например, с любым одним или более из остатков в области от около остатка 529 до около остатка 625, включая ECD HER2, при нумерации остатков, включающей сигнальный пептид).
«Лечение» относится как к терапевтическому лечению, так и к профилактическим или предотвращающим мерам. К числу тех, кто нуждается в лечении, относятся те, у кого уже есть онкологическое заболевание, а также те, у кого онкологическое заболевание необходимо предотвратить. Следовательно, пациент, которого лечат в контексте данного документа, может быть диагностирован как имеющий онкологическое заболевание или может быть предрасположен или подвержен онкологическому заболеванию.
Термин «эффективное количество» относится к количеству лекарственного средства, эффективному для лечения онкологического заболевания у пациента. Эффективное количество препарата может снизить количество раковых клеток; уменьшить размер опухоли; ингибировать (т.е. замедлять до некоторой степени и, предпочтительно, останавливать) инфильтрацию раковых клеток в периферические органы; ингибировать (т.е. замедлять до некоторой степени и, предпочтительно, останавливать) метастазирование опухоли; ингибировать до некоторой степени рост опухоли; и/или ослабить до некоторой степени один или более симптомов, ассоциированных с онкологическим заболеванием. В той степени, в которой лекарственное средство может предотвращать рост и/или убивать существующие раковые клетки, оно может быть цитостатическим и/или цитотоксическим. Эффективное количество может продлить выживаемость без прогрессирования (например, при измерении критериями оценки ответа для солидных опухолей, RECIST или СА-125), привести к объективному ответу (включая частичный ответ, PR или полный ответ, CR), увеличению общего времени выживания и/или улучшению относительно одного или более симптомов онкологического заболевания (например, при оценке FOSI).
Используемый в контексте данного документа термин «цитотоксический агент» относится к веществу, которое ингибирует или предотвращает функцию клеток и/или приводит к разрушению клеток. Предполагается, что этот термин включает радиоактивные изотопы (например, At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32 и радиоактивные изотопы Lu), химиотерапевтические агенты и токсины, такие как низкомолекулярные токсины или ферментативно активные токсины бактериального, грибного, растительного или животного происхождения, включая их фрагменты и/или варианты.
«Химиотерапия» представляет собой использование химического соединения, полезного для лечения онкологического заболевания. Примеры химиотерапевтических агентов, используемых в химиотерапии, включают алкилирующие агенты, такие как тиотепа и циклофосфамид CYTOXAN®; алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, включая алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилоломеламин; TLK 286 (TELCYTA™); ацетогенины (особенно булатацин и булатацинон); дельта-9-тетрагидроканнабинол (дронабинол, MARINOL®); бета-лапакон; лапахол; колхицины; бетулиновую кислоту; камптотецин (включая синтетический аналог топотекана (HYCAMTIN®), СРТ-11 (иринотекан, CAMPTOSAR®), ацетилкамптотецин, скополектин и 9-аминокамптотецин); бриостатин; каллистатин; СС-1065 (включая его синтетические аналоги адозелезина, карзелезина и бизелезина); подофиллотоксин; подофиллиновую кислоту; тенипозид; криптофицины (в частности криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги KW-2189 и СВ1-ТМ1); элейтеробин; панкратистатин; саркодиктин; спонгистатин; азотные иприты, такие как хлорамбуцил, хлорнафазин, холофосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид оксидмехлорэтамина, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урациловый иприт; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин и ранимнустин; бисфосфонаты, такие как клодронат; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, особенно калихеамицин гамма-1I и калихеамицин-омега-1I (см., например, Agnew, Chem Intl. Ed. Engl, 33: 183-186 (1994)) и антрациклины, такие как аннамицин, AD 32, алкарубицин, даунорубицин, доксорубицин, дексразоксан, DX-52-1, эпирубицин, GPX-100, идарубицин, валрубицин, KRN5500, меногарил, динемицин, включая динемицин, эсперамицин, неокарциностатиновый хромофор и родственные хромопротеиновые энедииновые антибитические хромофоры, аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карзинофилин, хромомуцины, дактиномицин, деторубицин, 6-диазо-5-оксо-b-норлейцин, доксорубицин ADRIAMYCIN®(включая морфолино-доксорубицин, цианоморфолино доксорубицин, 2-пирролинодоксорубицин, липосомальный доксорубицин и дезоксидоксорубицин), эсорубицин, марцеломицин, митомицины, такие как митомицин С, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, потфиромицин, пуромицин, хеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, и зорубицин; аналоги фолиевой кислоты, такие как деноптерин, птероптерин и триметрексат; аналоги пурина, такие как флударабин, 6-меркаптопурин, тиамиприн и тиогуанин; аналоги пиримидина, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин и флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан и тестолактон; антиадреналовые средства, такие как аминоглутетимид, митотан и трилостан; пополнитель фолиевой кислоты, такой как фолиновая кислота (лейковорин); ацеглатон; антифолатные противоопухолевые агенты, такие как ALIMTA®, пеметрексед LY231514, ингибиторы дигидрофолатредуктазы, такие как метотрексат, антиметаболиты, такие как 5-фторурацил (5-FU) и его пролекарственные формы, такие как UFT, S-1 и капецитабин, и ингибиторы тимидилатсинтазы и ингибиторы глицинамид рибонуклеотидформилтрансферазы, такие как ралтитрексед (TOMUDEXRM, TDX); ингибиторы дигидропиримидиндегидрогеназы, такие как энилурацил; альдофосфамидный гликозид; аминолевулиновая кислота; амсакрин; бестрабуцил; бизантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфорнитин; ацетат эллиптиния; эпотилон; этоглюцид; нитрат галлия; гидроксимочевина; лентинан; лонидаинин; мейтансиноиды, такие как мейтанзин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраерин; пентостатин; фенамет; пирарубицин; лозоксантрон; 2-этилгидразид; прокарбазин; Полисахаридный комплекс PSK7 (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2''-трихлортриэтиламин; трихотецены (особенно токсин Т-2, верракурин А, роридин А и ангидин); уретан; виндезин (ELDISINE®, FILDESIN®); дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-С"); циклофосфамид; тиотепа; таксаны; хлоранбуцил; гемцитабин (GEMZAR®); 6-тиогуанин; меркаптопурин; платину; аналоги платины или аналоги на основе платины, такие как цисплатин, оксалиплатин и карбоплатин; винбластин (VELBAN®); этопозид (VP-16); ифосфамид; митоксантрон; винкристин (ONCOVIN®); алкалоид барвинка; винорелбин (NAVELBINE®); новантрон; эдатрексат; дауномицин; аминоптерин; кселода; ибандронат; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; фармацевтически приемлемые соли, кислоты или производные любого из вышеперечисленного; а также комбинации двух или более из вышеперечисленного, такие как CHOP, аббревиатура для комбинированной терапии циклофосфамидом, доксорубицином, винкристином и преднизолоном, и FOLFOX, аббревиатура для программа лечения оксалиплатином (ELOXATIN™) в сочетании с 5-FU и лейковорином.
В это определение также включены антигормональные агенты, которые действуют для регулирования или ингибирования действия гормонов на опухоли, такие как антиэстрогены и селективные модуляторы рецепторов эстрогена (SERMs), включая, например, тамоксифен (включая NOLVADEX® тамоксифен), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и FARESTON® торемифен; ингибиторы ароматазы; и антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин; а также троксацитабин (1,3-диоксолан нуклеозид, аналог цитозина); антисмысловые олигонуклеотиды, в частности, ингибирующие экспрессию генов в пути передачи сигнала, задействованного в пролиферации аберрантных клеток, такие как, например, PKC-альфа, Raf, H-Ras и рецептор эпидермального фактора роста (EGF-R); вакцины, такие как вакцины для генной терапии, например, вакцина ALLOVECTIN®, вакцина LEUVECTIN® и вакцина VAXID®; PROLEUKIN® rIL-2; LURTOTECAN® ингибитор топоизомеразы 1; ABARELFX® rmRH; и фармацевтически приемлемые соли, кислоты или производные любого из вышеуказанных соединений.
«Таксан» относится к химиотерапии, которая ингибирует митоз и влияет на микротрубочки. Примеры таксанов включают в себя Паклитаксел (TAXOL®; Bristol-Myers Squibb Oncology, Princeton, N.J.); без кремофора, разработанная композиция из наночастиц паклитаксела с добавленным альбумином или наб-паклитаксела (ABRAXANE™; American Pharmaceutical Partners, Schaumberg, Illinois); и Доцетаксел (TAXOTERE®; Rhone-Poulenc Rorer, Antony, France).
«Антациклин» представляет собой тип антибиотика, который получают из гриба Streptococcus peucetius, примеры включают: Даунорубицин, Доксорубицин, Эпирубицин и любые другие антрациклиновые химиотерапевтические агенты, в том числе перечисленные ранее.
«Химиотерапия на основе антрациклина» относится к режиму химиотерапии, который состоит из или включает один или более антрациклинов. Примеры включают, но не ограничиваются этим, 5-FU, эпирубицин и циклофосфамид (FEC); 5-FU, доксорубицин и циклофосфамид (FAC); доксорубицин и циклофосфамид (АС); эпирубицин и циклофосфамид (ЕС); уплотненное теравпевтическое воздействие доксорубицина и циклофосфамида (ddAC) и тому подобное.
В контексте данного документа «химиотерапия на основе карбоплатина» относится к режиму химиотерапии, который состоит из или включает один или более Карбоплатинов. Примером является ТСН (доцетаксел/TAXOL®, Карбоплатин и трастузумаб/HERCEPTIN®).
«Ингибитор ароматазы» ингибирует фермент ароматазу, которая регулирует выработку эстрогена в надпочечниках. Примеры ингибиторов ароматазы включают: 4(5)-имидазолы, аминоглутетимид, MEGASE® мегестрола ацетат, AROMASIN® экземестан, форместан, фадрозол, RIVISOR® ворозол, FEMARA® летрозол и ARIMIDEX® анастрозол. В одном варианте реализации данного изобретения ингибитор ароматазы представляет собой летрозол или анастрозол.
«Антиметаболитная химиотерапия» представляет собой использование агента, который структурно похож на метаболит, но не может продуктивно использоваться организмом. Многие антиметаболитные химиотерапевтические препараты мешают выработке нуклеиновых кислот, РНК и ДНК. Примеры антиметаболитных химиотерапевтических агентов включают гемцитабин (GEMZAR®), 5-фторурацил (5-FU), капецитабин (XELODA™), 6-меркаптопурин, метотрексат, 6-тиогуанин, пеметрексед, ралтитрексед, арабинозилцитозин ARA-C цитарабин (CYTOSAR-U®), дакарбазин (DTIC-DOME®), азоцитозин, дезоксицитозин, пиридмиден, флударабин (FLUDARA®), кладрабин, 2-дезокси-D-глюкоза и др.
Под «резистентным к химиотерапии» онкологическим заболеванием подразумевается, что состояние онкологического больного ухудшалось во время применения режима химиотерапии (т.е. пациент является «рефрактерным к химиотерапии»), или состояние пациента ухудшалось в течение 12 месяцев (например, в течение 6 месяцев) после завершения режима химиотерапии.
Термин «платин» используется в контексте данного документа для обозначения химиотерапии на основе платины, включая, но не ограничиваясь этим, цисплатин, карбоплатин и оксалиплатин.
Термин «фторпиримидин» используется в контексте данного документа для обозначения антиметаболитной химиотерапии, включая, но не ограничиваясь этим, капецитабин, флоксуридин и фторурацил (5-FU).
«Фиксированная» или «базовая» доза терапевтического агента в контексте данного документа относится к дозе, которую вводят пациенту-человеку без учета веса (WT) или площади поверхности тела (BSA) пациента. Поэтому фиксированная или базовая доза предоставляется не в виде дозы мг/кг или дозы мг/м2, а в виде абсолютного количества терапевтического агента.
«Нагрузочная» доза в контексте данного документа, как правило, включает начальную дозу терапевтического агента, вводимого пациенту, и сопровождается одной или более поддерживающими дозами. Как правило, вводят однократную нагрузочную дозу, но в контексте данного документа предусмотрены множественные нагрузочные дозы. Обычно количество вводимой нагрузочной дозы (доз) превышает количество вводимой поддерживающей дозы (доз) и/или нагрузочную дозу (дозы) вводят чаще, чем поддерживающую дозу (дозы), чтобы достичь желаемой постоянной концентрации терапевтического агента раньше, чем это может быть достигнуто при поддерживающей дозе (дозах).
«Поддерживающая» доза в контексте данного документа относится к одной или более дозам терапевтического агента, вводимого пациенту в течение периода лечения. Обычно поддерживающие дозы вводят через определенные интервалы лечения, например, приблизительно каждую неделю, приблизительно каждые 2 недели, приблизительно каждые 3 недели или приблизительно каждые 4 недели, предпочтительно каждые 3 недели.
«Флакон» представляет собой контейнер, подходящий для хранения жидкого или лиофилизированного препарата. В одном варианте реализации изобретения флакон представляет собой одноразовый флакон, например, одноразовый флакон объемом 10 мл или 20 мл с пробкой, такой как стеклянный флакон одноразового использования объемом 10 мл с 20 мм пробкой.
«Листок-вкладыш» представляет собой инструкцию по применению, которая по заказу Управления по Контролю за качеством Пищевых продуктов и Лекарственных препаратов (FDA) или другого Регулирующего Органа должна быть помещена в упаковку каждого отпускаемого по рецепту лекарства. Инструкция, как правило, содержит указание товарного знака препарата, его общее название и механизм действия; указываются его показания, противопоказания, предупреждения, меры предосторожности, побочные эффекты и лекарственные формы; и содержит инструкции относительно рекомендуемой дозы, времени и пути введения.
Выражение «данные по безопасности» относится к данным, полученным в контролируемом клиническом испытании, которые показывают распространенность и тяжесть нежелательных явлений, чтобы сориентировать пользователя относительно безопасности лекарственного средства, включая руководство о том, как контролировать и предупреждать побочные реакции на лекарственное средство.
«Данные по эффективности» относятся к данным, полученным в контролируемых клинических испытаниях, показывающим, что лекарственное средство эффективно лечит заболевание, такое как онкологическое заболевание.
Под «стабильной смесью», когда речь идет о смеси двух или более лекарств, таких как пертузумаб и трастузумаб, подразумевается, что каждое из лекарств в смеси по существу сохраняет свою физическую и химическую стабильность в смеси, при оценке одним или более аналитическими исследованиями. Типовые аналитические исследования для этой цели включают: цвет, внешний вид и прозрачность (САС), анализ концентрации и мутности, анализ частиц, эксклюзионную хроматографию (SEC), ионообменную хроматографию (IEC), капиллярный зонный электрофорез (CZE), капиллярное изоэлектрическое фокусирование с детекцией в капилляре (iCIEF) и анализ активности. В одном варианте реализации изобретения было показано, что смесь является стабильной вплоть до 24 часов при температуре 5°С или 30°С.
Введение «в комбинации» включает комбинированное введение и раздельное введение, и в этом случае введение одного терапевтического агента может происходить до, одновременно и/или после введения другого терапевтического агента. Таким образом, введение в комбинации пертузумаба и трастузумаба (или совместное введение пертузумаба и трастузумаба) охватывает комбинированное введение и раздельное введение в любом порядке.
Лекарственное средство, которое вводят «одновременно» с одним или более другими лекарственными средствами, вводят в течение того же цикла лечения, в тот же день лечения, что и одно или более других лекарств, и, необязательно, в то же время, что и одно или более других лекарств. Например, для лечения онкологического заболевания, проводимого каждые 3 недели, одновременно вводимые лекарства вводят каждое в день 1 трехнедельного цикла.
Термин «совместное введение» используется в контексте данного документа для обозначения отдельного введения, включая, например, введение пертузумаба и трастузумаба в виде двух отдельных подкожных (п/к) инъекций.
Термин «введение смеси» используется в контексте данного документа для обозначения одновременного введения в виде однократной инъекции, включая, например, введение пертузумаба и трастузумаба в виде одной подкожной (п/к) инъекции, приготовленной медицинским работником непосредственно на месте, непосредственно перед введением п/к путем смешивания отдельных препаратов пертузумаба и трастузумаба.
Термин «совместный препарат» используется в контексте данного документа для обозначения одного готового к применению фармацевтического препарата, содержащего два или более активных ингредиента, включая, например, один готовый к применению фармацевтический препарат, содержащий пертузумаб и трастузумаб, приготовленные вместе для подкожного (п/к) введения.
II. Композиции антител и химиотерапевтических препаратов
(i) Антитела против HER2
Антиген HER2, который следует использовать для продуцирования антител, может представлять собой, например, растворимую форму внеклеточного домена рецептора HER2 или его часть, содержащую желаемый эпитоп. Альтернативно, клетки, экспрессирующие HER2 на своей клеточной поверхности {например, клетки NIH-3T3, трансформированные для сверхэкспрессии HER2; или клеточная линия карциномы, такая как клетки SK-BR-3, см. Stancovski et al. PNAS (USA) 88:8691-8695 (1991)) можно использовать для генерации антител. Другие формы рецептора HER2, подходящие для получения антител, будут очевидны для специалистов в данной области.
Различные способы получения моноклональных антител в контексте данного документа известны в данной области техники. Например, моноклональные антитела могут быть получены с использованием гибридомного способа, впервые описанного Kohler et al., Nature, 256:495 (1975), посредством способов рекомбинантной ДНК (патент США №4,816,567).
Антитела против HER2, используемые в соответствии с данным изобретением, пертузумаб и трастузумаб, являются коммерчески доступными.
Патент США №6,949,245 описывает получение типовых гуманизированных антител против HER2, которые связываются с HER2 и блокируют лигандную активацию рецептора HER.
Гуманизированные антитела против HER2, в частности, включают трастузумаб, как описано в Таблице 3 патента США №5,821,337, непосредственно включенного в данный документ посредством ссылки и как определено в данном документе; и гуманизированные антитела 2С4, такие как пертузумаб, как описано и определено в данном документе.
Гуманизированные антитела в контексте данного документа могут, например, содержать остатки гипервариабельной области, не принадлежащей человеку, которые включены в вариабельный домен тяжелой цепи человека, и могут дополнительно содержать замену в каркасной области (FR) в положении, выбранном из группы, состоящей из 69Н, 71Н и 73Н с использованием системы нумерации вариабельного домена, изложенной у Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD (1991). В одном варианте реализации изобретения гуманизированное антитело содержит замены FR в двух или всех положениях 69Н, 71Н и 73Н.
Типовое представляющее интерес гуманизированное антитело в данном документе содержит остатки, определяющие комплементарность вариабельного домена тяжелой цепи 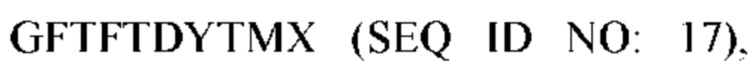 где X предпочтительно представляет собой D или S;
где X предпочтительно представляет собой D или S; 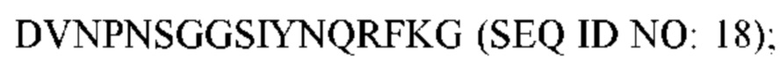 и/или
и/или  необязательно содержит аминокислотные модификации этих остатков CDR, например, когда модификации по существу поддерживают или улучшают аффинность антитела. Например, вариант антитела для использования в способах согласно данному изобретению может иметь от около одной до около семи или около пяти аминокислотных замен в вышеуказанных вариабельных последовательностях CDR тяжелой цепи. Такие варианты антител могут быть получены путем созревания аффинности, например, как описано ниже.
необязательно содержит аминокислотные модификации этих остатков CDR, например, когда модификации по существу поддерживают или улучшают аффинность антитела. Например, вариант антитела для использования в способах согласно данному изобретению может иметь от около одной до около семи или около пяти аминокислотных замен в вышеуказанных вариабельных последовательностях CDR тяжелой цепи. Такие варианты антител могут быть получены путем созревания аффинности, например, как описано ниже.
Гуманизированное антитело может содержать остатки, определяющие комплементарность вариабельного домена легкой цепи 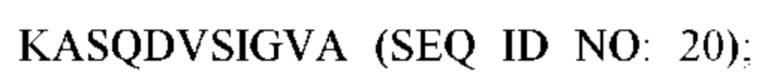 SASYX1X2X3, где X1 предпочтительно представляет собой R или L, X2 предпочтительно представляет собой Y или Е, и X3 предпочтительно представляет собой Т или S (SEQ ID NO: 21); и/или
SASYX1X2X3, где X1 предпочтительно представляет собой R или L, X2 предпочтительно представляет собой Y или Е, и X3 предпочтительно представляет собой Т или S (SEQ ID NO: 21); и/или 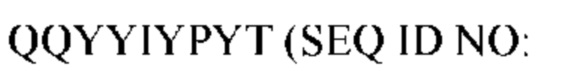
 например, в дополнение к тем остаткам вариабельной последовательности CDR тяжелой цепи в предыдущем абзаце. Такие гуманизированные антитела необязательно содержат аминокислотные модификации вышеуказанных остатков CDR, например, модификации, которые по существу поддерживают или улучшают аффинность антитела. Например, представляющий интерес вариант антитела может иметь от около одной до около семи или около пяти аминокислотных замен в вышеуказанных вариабельных последовательностях CDR легкой цепи. Такие варианты антител могут быть получены путем созревания аффинности, например, как описано ниже.
например, в дополнение к тем остаткам вариабельной последовательности CDR тяжелой цепи в предыдущем абзаце. Такие гуманизированные антитела необязательно содержат аминокислотные модификации вышеуказанных остатков CDR, например, модификации, которые по существу поддерживают или улучшают аффинность антитела. Например, представляющий интерес вариант антитела может иметь от около одной до около семи или около пяти аминокислотных замен в вышеуказанных вариабельных последовательностях CDR легкой цепи. Такие варианты антител могут быть получены путем созревания аффинности, например, как описано ниже.
Данная заявка также относится к аффинно зрелым антителам, которые связываются с HER2. Исходное антитело может представлять собой антитело человека или гуманизированное антитело, например, антитело, содержащее вариабельные последовательности легкой цепи и/или вариабельные последовательности тяжелой цепи из SEQ ID NO: 7 и 8, соответственно (т.e. содержащее VL и/или VH пертузумаба). Аффинно-зрелый вариант пертузумаба предпочтительно связывается с рецептором HER2 с аффинностью, лучшей, чем мышиное 2С4 или пертузумаб {например, лучшей от около в два или около в четыре раза, до около в 100 раз или около 1000 раз, например, по измерениям с использованием внеклеточного домена (ECD) HER2 (ELISA)). Типовые остатки вариабельной последовательности CDR тяжелой цепи для замены включают Н28, Н30, Н34, Н35, Н64, Н96, Н99, или комбинацию двух или более (например, двух, трех, четырех, пяти, шести, или семи этих остатков). Примеры остатков вариабельной последовательности CDR легкой цепи, которые могут быть изменены, включают L28, L50, L53, L56, L91, L92, L93, L94, L96, L97 или комбинацию двух или более (например, от двух до трех, четырех, пяти или вплоть до около десяти этих остатков).
Гуманизация мышиного антитела 4D5 для создания его гуманизированных вариантов, в том числе трастузумаба, описана в патентах США №: 5,821,337, 6,054,297, 6,407,213, 6,639,055, 6,719,971, и 6,800,738, а также Carter et al. PNAS (USA), 89:4285-4289 (1992). HuMAb4D5-8 (трастузумаб) связывалось с антигеном HER2 в три раза сильнее, чем мышиное антитело 4D5 и имело вторичную иммунную функцию (ADCC), которая дает возможность для направленной цитотоксической активности гуманизированного антитела в присутствии эффекторных клеток человека. HuMAb4D5-8 содержало остатки вариабельной последовательности CDR легкой цепи (VL) вставленные в консенсусную последовательность каркаса VL подгруппы I, и остатки вариабельной последовательности CDR тяжелой цепи (VH), вставленные в консенсусную последовательность каркаса VH подгруппы III. Антитело дополнительно содержало замены в каркасной области (FR) в положениях: 71, 73, 78 и 93 в VH (нумерация остатков FR согласно Kabat; и замены FR в положении 66 в VL (нумерация остатков FR согласно Kabat). Трастузумаб содержит Fc-область γ-1 человека аллотипа не-А.
Рассматриваются различные формы гуманизированного антитела или аффинно зрелого антитела. Например, гуманизированное антитело или аффинно зрелое антитело могут представлять собой фрагмент антитела. Альтернативно, гуманизированное антитело или аффинно зрелое антитело может представлять собой интактное антитело, такое как интактное антитело IgG1.
(ii) Композиции пертузумаба
В одном варианте реализации композиции антитела против HER2, композиция содержит смесь основных видов антитела пертузумаба и одного или более его вариантов. Предпочтительный вариант реализации в данном изобретении основных видов антитела пертузумаба представляет собой антитело, содержащее аминокислотные последовательности вариабельной легкой и тяжелой цепей из SEQ ID NO: 7 и 8, и наиболее предпочтительно, содержащее аминокислотную последовательность легкой цепи из SEQ ID NO: 11, и аминокислотную последовательность тяжелой цепи из SEQ ID NO: 12 (включая дезаминированные и/или окисленные варианты таких последовательностей). В одном варианте реализации изобретения композиция содержит смесь основных видов антитела пертузумаб и аминокислотную последовательность его варианта, содержащую удлинение аминоконцевой лидирующей последовательности. Предпочтительно, удлинение ами ноконцевой лидирующей последовательности находится на легкой цепи вариантов антител (например, на одной или двух легких цепях вариантов антител). Основные виды антител против HER2 или вариантов антител могут представлять собой полноразмерное антитело или фрагмент антитела (например, Fab из фрагментов F(ab=)2), но, предпочтительно, оба являются полноразмерными антителами. Вариант антитела в данном документе может содержать удлинение аминоконцевой лидирующей последовательности на любой одной или более из его тяжелых или легких цепей. Предпочтительно, удлинение аминоконцевой лидирующей последовательности находится на одной или двух легких цепях антитела. Удлинение аминоконцевой лидирующей последовательности предпочтительно содержит или состоит из VHS-. Присутствие удлинения аминоконцевой лидирующей последовательности в композиции можно выявлять при помощи различных аналитических способов, включая, но не ограничиваясь этим, анализ N-концевой последовательности, анализ гетерогенности зарядов (например, катионообменная хроматография или капиллярный зонный электрофорез), масс-спектрометрию и т.д. Количество вариантов антител в композиции, в основном, находится в диапазоне от количества, составляющего предел детекции любого анализа (предпочтительно, анализа N-концевой последовательности), применяемого для детекции варианта, до количества, меньшего чем количество основных видов антитела. В основном, около 20% или меньше (например, от около 1% до около 15%, например, от 5% до около 15%) молекул антитела в композиции содержат удлинение аминоконцевой лидирующей последовательности. Такие процентные количества предпочтительно определяют с использованием количественного анализа N-концевой последовательности или катионообменного анализа (предпочтительно с использованием высокоразрешающей колонки со слабой катионообменной смолой, такой как катионообменная колонка PROPAC WCX-10™). Помимо варианта с удлинением аминоконцевой лидирующей последовательности, рассматриваются дополнительные изменения аминокислотной последовательности основных видов антител и/или вариантов, включая, но не ограничиваясь этим, антитело, содержащее С-концевой остаток лизина на одной или обеих тяжелых цепях, дезаминированный вариант антитела и т.д.
Кроме того, основные виды антитела или вариант могут дополнительно содержать изменения гликозилирования, неограничивающие примеры которых включают антитело, содержащее олигосахаридную структуру G1 или G2, присоединенную к Fc-области, антитело, содержащее углеводную группу, присоединенную к легкой цепи (например, одну или две углеводных группы, такие как глюкоза или галактоза, присоединенные к одной или двум легким цепям антитела, например, присоединенные к одному или более остаткам лизина), антитело, содержащее одну или две негликозилированные тяжелые цепи, или антитело, содержащее сиалированный олигосахарид, присоединенный к одной или двум тяжелым цепям и т.д.
Композиция может быть получена из генно-инженерной клеточной линии, например, клеточной линии китайского хомячка (СНО), экспрессирующей антитело против HER2, или может быть получена путем пептидного синтеза.
Для дальнейшей информации относительно примеров композиций пертузумаба см. патенты США №7,560,111 и 7,879,325, а также US 2009/0202546А1.
(iii) Композиции трастузумаба
Композиция трастузумаба, в основном, содержит смесь основных видов антител (содержащих последовательности легкой и тяжелой цепи, представленные в SEQ ID NO: 13 и 14, соответственно), и их вариантные формы, в частности кислые варианты (включая дезаминированные варианты). Предпочтительно, количество таких кислых вариантов в композиции составляет менее чем около 25%, или менее чем около 20%, или менее чем около 15%. См., патент США №6,339,142. См. также, Harris et al., J. Chromatography, В 752:233-245 (2001) относительно форм трастузумаба, разделяемых при помощи катионообменной хроматографии, включая Пик А (Asn30, дезаминированный до Asp в обеих легких цепях); Пик В (Asn55, дезаминированный до isoAsp в одной тяжелой цепи); Пик 1 (Asn30, дезаминированный до Asp в одной легкой цепи); Пик 2 (Asn30, дезаминированный до Asp в одной легкой цепи, и Asp 102, изомеризованный до isoAsp в одной тяжелой цепи); Пик 3 (основная пиковая форма, или основные виды антитела); Пик 4 (Asp 102, изомеризованный до isoAsp в одной тяжелой цепи); и Пик С (Asp 102 сукцинимид (Asu) в одной тяжелой цепи). Такие вариантные формы и композиции включены в изобретение, представленное в данном документе.
(iv) Подкожные препараты, содержащие фермент гиалуронидазу
Фермент гиалуронидаза действует главным образом как усилитель проницаемости, увеличивая дисперсию и абсорбцию других лекарств, вводимых совместно. Гиалуронидаза временно гидролизует гиалуронан, компонент подкожного матрикса, что приводит к снижению вязкости внеклеточного матрикса гиподермы и, таким образом, к улучшению доставки подкожно вводимых лекарств в системный кровоток.
Растворимые гликопротеины гиалоронидазы (sHASEGP), способ их получения и их применение в фармацевтических композициях были описаны в WO 2004/078140. Использование растворимых гликопротеинов Гиалуронидазы в сочетании с различными типовыми антителами, такими как, например, трастузумаб, упоминалось в WO 2006/091871.
Фермент гиалуронидаза в препаратах согласно данному изобретению усиливает доставку антитела или антител против HER2 (например, пертузумаба и/или трастузумаба) в системную циркуляцию, например, путем увеличения абсорбции активного вещества (он действует как усилитель проницаемости). Фермент гиалуронидаза также усиливает доставку терапевтического антитела или антител против HER2 (например, пертузумаба и/или трастузумаба) в системный кровоток при подкожном введении путем обратимого гидролиза гиалуронана, внеклеточного компонента межклеточной подкожной ткани. Гидролиз гиалуронана в гиподерме временно открывает каналы в интерстициальном пространстве подкожной ткани и тем самым улучшает доставку терапевтического антитела против HER2 в системный кровоток. Кроме того, при введении наблюдается уменьшение боли у людей и уменьшение объемного отека подкожной ткани.
Гиалуронидаза при местном введении оказывает локальный эффект. Другими словами, гиалуронидаза инактивируется и метаболизируется локально за считанные минуты и не имеет системных или долгосрочных эффектов. Быстрая инактивация гиалуронидазы в течение нескольких минут, когда она попадает в кровоток, препятствует реалистичной возможности проводить сопоставимые исследования биораспределения между различными продуктами гиалуронидазы. Это свойство также сводит к минимуму любые потенциальные системные проблемы безопасности, потому что продукт гиалуронидазы не может действовать в отдаленных местах.
Объединяющей чертой всех ферментов гиалуронидазы является их способность деполимеризовать гиалуронан независимо от различий в химической структуре, от принадлежности к виду, от типа ткани или от партий лекарственного продукта, полученного из одних и тех же видов и тканей. Они необычны тем, что их активность одинакова (за исключением эффективности), несмотря на наличие разных структур.
Эксципиент гиалуронидазного фермента в соответствии с препаратом согласно данному изобретению характеризуется отсутствием нежелательного влияния на молекулярную целостность антитела или антител против HER2 в стабильных фармацевтических препаратах, описанных в данном документе. Кроме того, фермент гиалуронидаза просто модифицирует доставку антител против HER2 или антител против HER2 в системный кровоток, но не обладает какими-либо свойствами, которые могли бы обеспечивать или способствовать терапевтическим эффектам системно абсорбированного антитела или антител против HER2. Фермент гиалуронидаза не является системно биодоступным и не оказывает вредного влияния на молекулярную целостность антитела или антител против HER2 при рекомендуемых условиях хранения стабильного фармацевтического препарата в соответствии с изобретением.
Ряд подходящих ферментов гиалуронидазы в соответствии с данным изобретением известен в данной области техники. Предпочтительным ферментом является фермент гиалуронидаза человека, наиболее предпочтительно, рекомбинантный фермент гиалуронидаза человека, известный как rHuPH20. rHuPH20 является членом семейства нейтральных и кислотно-активных β-1,4-гликозилгидролаз, которые деполимеризуют гиалуронан путем гидролиза β-1,4-связи между положением C1 N-ацетилглюкозамина и положением С4 глюкуроновой кислоты. Гиалуронан представляет собой полисахарид, обнаруженный во внутриклеточном основном веществе соединительной ткани, такой как подкожная интерстициальная ткань, и в некоторых специализированных тканях, таких как пуповина и стекловидное тело. Гидролиз гиалуронана временно уменьшает вязкость межклеточной ткани и способствует дисперсии вводимых жидкостей или локализованных транссудатов или экссудатов, способствуя тем самым их абсорбции. Эффекты гиалуронидазы носят локальный и обратимый характер с полным восстановлением гиалуронана в тканях в течение 24-48 часов (Frost, G. I., "Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration", Expert Opinion on Drug Delivery, 2007; 4:427-440). Увеличение проницаемости соединительной ткани посредством гидролиза гиалуронана коррелирует с эффективностью гиалуронидазы в отношении их способности увеличивать дисперсию и абсорбцию совместно вводимых молекул.
Геном человека содержит несколько генов гиалуронидазы. Только продукт гена РН20 обладает эффективной гиалуронидазной активностью в физиологических внеклеточных условиях и действует как распространяющий агент, тогда как кислые активные гиалуронидазы не обладают этим свойством.
rHuPH20 является первым и единственным рекомбинантным ферментом гиалуронидазы человека, доступным в настоящее время для терапевтического применения. Природный человеческий белок РН20 имеет липидный якорь, присоединенный к карбоксиконцевой аминокислоте, которая прикрепляет его к плазматической мембране. Фермент rHuPH20, разработанный Halozyme, представляет собой усеченный делеционный вариант, в котором отсутствуют такие аминокислоты на карбоксиконце, ответственные за прикрепление липидов. Это приводит к образованию растворимого, активного при нейтральном рН фермента, подобного белку, обнаруженному в препаратах бычьих семенников. Белок rHuPH20 синтезируется с сигнальным пептидом из 35 аминокислот, который удаляется с N-конца в процессе секреции. Зрелый белок rHuPH20 содержит аутентичную N-концевую аминокислотную последовательность, ортологичную той, которая обнаружена в некоторых препаратах бычьей гиалуронидазы.
Гиалуронидазы РН20, включая РН20 животного происхождения и рекомбинантную человеческую rHuPH20, деполимеризуют гиалуронан путем гидролиза связи (3-1,4 между положением C1 N-ацетилглюкозамина и положением С4 глюкуроновой кислоты. Тетрасахарид является наименьшим продуктом расщепления (Weissmann, В., "The transglycosylative action of testicular hyaluronidase", J. Biol. Chem., 1955; 216: 783-94). Эта структура N-ацетил глюкозаминовой/глюкуроновой кислоты не обнаружена в N-связанных гликанах рекомбинантных биологических продуктов, и, следовательно, rHuPH20 не будет влиять на гликозилирование антител, с которыми он находится в смеси, таких как, например, пертузумаб и/или трастузумаб. Сам фермент rHuPH20 содержит шесть N-связанных гликанов на молекулу с основными структурами, подобными тем, которые обнаружены в моноклональных антителах. Как и предполагалось, эти N-связанные структуры не изменяются со временем, подтверждая отсутствие ферментативной активности rHuPH20 на этих N-связанных гликановых структурах. Короткий период полураспада rHuPH20 и постоянный синтез гиалуронана приводят к короткому и локальному действию фермента на ткани.
Фермент гиалуронидаза, присутствующий в подкожном препарате в соответствии с данным изобретением, может быть получен с использованием технологии рекомбинантных ДНК. Таким образом обеспечивается постоянное получение одного и того же белка (идентичной аминокислотной последовательности) и предотвращение аллергической реакции, например, вызванной загрязнением белков, совместно очищенных во время экстракции из ткани. Фермент гиалуронидаза, используемый в препарате, представленном в качестве примера в данном документе, представляет собой человеческий фермент, а именно, viz. rHuPH20.
Аминокислотная последовательность rHuPH20 (HYLENEX™) хорошо известна и доступна под регистрационным номером CAS Registry No. 75971-58-7. Приблизительная молекулярная масса составляет 61 кДа.
В то время как безопасность и эффективность продуктов гиалуронидазы была установлена, есть только два моноклональных антитела ((Herceptin® и MabThera®), которые были одобрены для подкожного введения, используя препараты, содержащие гиалуронидазу. Не существует известного подкожного препарата, содержащего гиалуронидазу, содержащего два антитела в одном и том же препарате (совместное приготовление двух антител).
Концентрация фермента гиалуронидазы зависит от фактического фермента гиалуронидазы, используемого при приготовлении препарата в соответствии с изобретением. Специалист в данной области техники может легко определить эффективное количество фермента гиалуронидазы на основании описания, приведенного ниже.
Фермент гиалуронидаза должен быть предоставленн в достаточном количестве, чтобы привести к увеличению дисперсии и абсорбции совместно вводимого антитела против HER2 или антител, таких как пертузумаб и/или трастузумаб. Минимальное количество фермента гиалуронидазы составляет, по меньшей мере, около 150 Ед/мл. Конкретнее, эффективное количество фермента гиалуронидазы составляет от около 150 Ед/мл до около 16,000 Ед/ мл, или от около 600 Ед/мл до около 16,000 мл, или от около 1,000 до 16,000 Ед/мл, причем последнее соответствует от около 0,01 мг до около 0,16 мг белка в расчете на предполагаемую удельную активность 100,000 Ед/мг. Альтернативно, концентрация фермента гиалуронидазы составляет от около 1,500 до 12,000 Ед/мл или конкретнее, около 2,000 Ед/мл или около 12,000 Ед/мл. Указанные количества соответствуют количеству фермента гиалуронидазы, первоначально добавленного в препарат. Концентрации гиалуронидазного фермента, измеренные в конечном препарате, могут варьировать в определенных пределах. Соотношение (мас./мас.) фермента гиалуронидазы к антителу или антителам против HER2 обычно находится в диапазоне от 1:1000 до 1:8000 или в диапазоне от 1:4000 до 1:5000 или около 1:6000.
Фермент гиалуронидаза может быть получен из животных, образцов человека или изготовлен на основе технологии рекомбинантной ДНК, как описано далее ниже.
В некоторых вариантах реализации изобретения, препараты подкожных антител против HER2, представленные в данном документе, содержат рекомбинантную человеческую гиалуронидазу (rHuPH20) в концентрации от около 600 Ед/мл до около 16,000 Ед/мл или от около 1,000 Ед/мл до около 16,000 Ед/мл или от около 1,000 до около 2,000 Ед/мл или в концентрации около 600 Ед/мл, или около 667 Ед/мл, или около 1,000 Ед/мл, или около 2,000 Ед/мл, предпочтительно около 1,000 Ед/мл.
В некоторых вариантах реализации изобретения высококонцентрированные стабильные препараты пертузумаба согласно данному изобретению содержат фиксированную дозу 600 мг или 1200 мг пертузумаба и рекомбинантную человеческую гиалуронидазу (rHuPH20) в концентрации 1,000 Ед/мл.
Как отмечено выше, растворимый гликопротеин гиалуронидазы может считаться еще одним вспомогательным веществом в препарате против HER2. Растворимый гликопротеин гиалуронидазы может быть добавлен к препарату против HER2 во время изготовления препарата против HER2 или может быть добавлен незадолго до инъекции. Альтернативно, растворимый гликопротеин гиалуронидазы может быть предоставлен в виде отдельной инъекции. В последнем случае растворимый гликопротеин гиалуронидазы может быть предоставлен в отдельном флаконе либо в лиофилизированной форме, которая должна быть восстановлена подходящими разбавляющим веществом до того, как произойдет подкожная инъекция, либо может быть предоставлена производителем в виде жидкого препарата. Препарат против HER2 и растворимый гликопротеин гиалуронидазы могут быть приобретены в виде отдельных компонентов или также могут быть предоставлены в виде наборов, содержащих как компоненты для инъекций, так и подходящие инструкции для их подкожного введения. Также могут быть предоставлены подходящие инструкции для восстановления и/или введения одного или обоих препаратов.
В дополнение к ферменту гиалуронидазы, такому как rHuPH20, препараты для подкожного введения согласно данному изобретению содержат один или более дополнительных эксципиентов, таких как один или более буферных агентов, один или более стабилизаторов и/или одно или более поверхностно-активное вещество.
Буфер, используемый в препаратах в соответствии с данным изобретением, имеет рН в диапазоне от около 5,0 до около 7,0, или от около 5,0 до около 6,0, или от около 5,3 до около 5,8, или от около 5,5 до около 5,7.
Для подкожных (п/к) препаратов пертузумаба было установлено, что наиболее подходящим является рН около 5,7. Предпочтительный рН подкожного (п/к) препарата трастузумаба составляет около 5,5.
Примеры буферных агентов, которые будут контролировать рН в этом диапазоне, включают ацетатный, сукцинатный, глюконатный, гистидиновый, цитратный, глицилглициновый и другие органические кислотные буферы. Наиболее подходящим буфером в соответствии с данным изобретением является гистидиновый буфер, такой как, например, гистидинхлорид, гистидинацетат, гистидинфосфат, гистидинсульфат, предпочтительно гистидинхлоридный буфер. Буфер гистидинхлорида может быть получен титрованием L-гистидина (свободное основание, твердое вещество) разбавленной соляной кислотой. В частности, гистидиновый буфер или гистидинхлоридный буфер представляет собой L-гистидиновый буфер при рН 5,5±0,6, конкретнее при рН от около 5,3 до около 5,8, в частности рН 5,5 или 5,7.
Стабилизатор может представлять собой, например, сахарид или комбинацию сахаридов, включая моносахариды, дисахариды, трисахариды, полисахариды, сахарные спирты, восстанавливающие сахара, невосстанавливающие сахара и т.д. Примеры сахаридов в данном документе включают глюкозу, сахарозу, трегалозу, лактозу, фруктозу, мальтозу, декстран, глицерин, эритрит, глицерол, арабит, силитол, сорбит, маннит, меллибиозу, мелезитозу, рафинозу, манотриозу, стахиозу, мальтозу, лактулозу, мальтулозу, глюцитол, мальтитол, лактитол и изо-мальтулозу. Особенно подходящим сахаридом для применения в подкожных препаратах трастузумаба является трегалоза, а особенно подходящим сахаридом для применения в подкожных препаратах пертузумаба является сахароза.
Поверхностно-активное вещество предпочтительно представляет собой неионогенное поверхностно-активное вещество. Примеры поверхностно-активных веществ в данном документе включают полисорбат; полоксамер (например, полоксамер 188); тритон; додецилсульфат натрия (SDS); лаурил сульфат натрия; октилгликозид натрия; лаурил-, миристил-, линолеил- или стеарилсульфобетаин; лаурил-, миристил-, линолеил- или стеарил-саркозин; линолеил-, миристил-или цетил-бетаин; лауроамидопропил-, кокамидопропил-, линолеамидопропил-, миристамидопропил-, пальмидопропил- или изостеарамидопропил-бетаин (например, лауроамидопропил); миристамидопропил-, пальмидопропил- или изостеарамидопропил-диметиламин; метилкокоил- или динатрийметил-олеилтаурат натрия; и серию MONAQU AT™ (Mona Industries, Inc., Paterson, N.J.); полиэтиленгликоль, полипропилгликоль и сополимеры этилена и пропиленгликоля (например, Pluronics, PF68 и т.д.) и т.д. Полисорбат 20 (PS20) и Полисорбат 80 (PS80), соответственно, особенно подходят для использования в препаратах, описанных в данном документе.
III. Отбор пациентов для терапии
Для отбора пациентов для лечения в соответствии с данным изобретением можно использовать детекцию экспрессии или амплификации HER2. Некоторые одобренные FDA коммерческие анализы способны выявлять пациентов с HER2-положительным, HER2-экспрессирующим, HER2-сверхэкспрессирующим или HER2-амплифицированным онкологическим заболеванием. Эти способы включают HERCEPTEST® (Dako) и PATHWAY® HER2 (иммуногистохимические (IHC) анализы), и PathVysion®h HER2 FISH pharmDx™ (FISH анализы). Пользователям следует обратить внимание на листки-вкладыши в упаковки наборов для конкретного анализа для получения информации о валидации и проведении каждого анализа.
Например, экспрессия или сверхэкспрессия HER2 могут быть проанализированы с помощью IHC, например, с использованием HERCEPTEST® (Dako). Погруженные в парафин тканевые срезы из опухолевых биоптатов можно подвергать анализу IHC и согласовывать с критериями интенсивности окрашивания белка HER2 следующим образом:
Показатель 0 наблюдается отсутствие окрашивания или наблюдают окрашивание мембран менее чем у 10% опухолевых клеток.
Показатель 1+ выявлется слабое/едва заметное окрашивание мембран более чем у 10% опухолевых клеток. Клетки окрашиваются только в части их мембраны.
Показатель 2+ наблюдаются полное окрашивание мембран от слабого до умеренного более чем у 10% опухолевых клеток.
Показатель 3+ наблюдается полное окрашивание мембран от умеренного до сильного более чем у 10% опухолевых клеток.
Опухоли с показателями для оценки сверхэкспрессии HER2 0 или 1+ могут быть охарактеризованы как НЕК-негативные, в то время как опухоли с показателями 2+ или 3+ могут быть охарактеризованы как HER2-положительные.
Опухоли, сверхэкспрессирующие HER2, могут быть оценены по иммуногистохимическим показателям, соответствующим числу копий молекул HER2, экспрессирующимся на клетку, и могут быть определены биохимически:
0 = 0-10,000 копий/клетку,
1+ = по меньшей мере, около 200,000 копий/клетку
2+ = по меньшей мере, около 500,000 копий/клетку
3+ = по меньшей мере, около 2,000,000 копий/клетку.
Сверхэкспрессия HER2 на уровне 3+, которая приводит к лиганд-независимой активации тирозинкиназы (Hudziak et al., Proc. Natl. Acad. Sci. USA, 84: 7159-7163 (1987)), встречается приблизительно в 30% случаев рака молочной железы, и у этих пациентов, безрецидивная выживаемость и общая выживаемость снижены (Slamon et al., Science, 244:707-712 (1989); Slamon et al., Science, 235:177-182 (1987)).
Наличие сверхэкспрессии белка HER2 и амплификация гена сильно коррелируют, таким образом, альтернативно, или дополнительно, для выявления амплификации гена можно также использовать анализ гибридизации in situ гибридизация (ISH), например, флуоресцентной гибридизации in situ (FISH), для отбора пациентов, подходящих для лечения в соответствии с данным изобретением. Анализы FISH, такие как the INFORM™ (продается у Ventana, Arizona) или PathVysion® (Vysis, Illinois) можно проводить на фиксированной формалином, погруженной в парафин опухолевой ткани для определения степени (при наличии) амплификации HER2 в опухоли.
Чаще всего НЕР2-положительный статус подтверждают с использованием архивного, погруженного в парафин, образца опухолевой ткани при помощи любого из указанных выше способов.
Предпочтительно, HER2-положительные пациенты, имеющие показатель IHC 2+ или 3+ или которые являются FISH или ISH-положительными, выбираются для лечения в соответствии с данным изобретением. Пациенты с показателем IHC 3+ и которые являются FISH/ISH-положительными, в частности, подходят для лечения в соответствии с данным изобретением.
Также были идентифицированы мутации HER2, связанные с чувствительностью к HER2-направленной терапии. Такие мутации включают, но не ограничиваются этим, вставки в экзон 20 HER2, делеции вокруг аминокислотных остатков 755-759 HER2, любую из мутаций G309A, G309E, S310F, D769H, D769Y, V777L, P780-Y781insGSP, V842I, R896C (Bose et al., Cancer Discov 2013; 3:1-14), а также ранее сообщалось об идентичных не синонимичных предполагаемых активирующих мутациях (или инделах) в базе данных COSMIC, обнаруженной в двух или более уникальных образцах.
См. также Примеры и патент США №7,981,418 для альтернативных анализов для отбора пациентов для терапии пертузумабом.
IV. Фармацевтические Препараты
Терапевтические препараты антител против HER2, применяемых в соответствии с данным изобретением, получают для хранения путем смешивания антитела необходимой степени чистоты с произвольными фармацевтически приемлемыми носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980)), в основном в форме лиофилизированных препаратов или водных растворов. Рассматриваются также кристаллы антитела (см. патентную заявку США 2002/0136719). Приемлемые носители, эксципиенты, или стабилизаторы нетоксичны для реципиентов в используемых дозировках и концентрациях, и включают буферы, такие как фосфатный, цитратный, и другие органические кислоты; антиоксиданты, в том числе аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензил хлорид аммония; гексаметоний хлорид; хлорид бензалкония, хлорид бензетония; фенол, бутил или бензиновый спирт; алкилпарабены, такие как метил или пропилпарабен; катехол; резорцинол; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее чем около 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин, или иммуноглобулины; гидрофильные полимеры, так как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин, или лизин; моносахариды, дисахариды, и другие углеводы, включая глюкозу, маннозу, или декстрины; хелатирующие агенты, так как ЭДТА; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексные соединения с металлами (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Лиофилизированные препараты антител описаны в WO 97/04801, специально включенной в данный документ в полном объеме посредством ссылки.
Лиофилизированные препараты антител описаны в патентах США №6,267,958, 6,685,940 и 6,821,515, специально включенных в данный документ в полном объеме посредством ссылки. Предпочтительно, препарат HERCEPTIN® (трастузумаб) является стерильным, лиофилизированным порошком от белого до бледно-желтого цвета без консервантов, предназначенным для внутривенного (в/в) введения, содержащим 440 мг трастузумаба, 400 мг альфа-а,а-трегалозы дегидрата, 9,9 мг L-гистидин-HCl, 6,4 мг L-гистидина и 1,8 мг полисорбата 20, USP. Восстановление в 20 мл бактериостатической воды для инъекций (БВДИ), содержащей 1,1% бензилового спирта в качестве консерванта, дает многодозовый раствор, содержащий 21 мг/мл трастузумаба, при рН приблизительно 6,0. Для получения более подробной информации смотрите информацию по применению трастузумаба.
Предпочтительно, препарат пертузумаба для терапевтического использования содержит 30 мг/мл пертузумаба в 20 мМ гистидин ацетата, 120 мМ сахарозы, 0,02% полисорбата 20, при рН 6,0.
Альтернативный препарат пертузумаба содержит 25 мг/мл пертузумаба, 10 мМ гистидин-HCl буфера, 240 мМ сахарозы, 0,02% полисорбата 20, рН 6,0.
Препарат плацебо, использованный в клинических испытаниях, описанных в примерах, является эквивалентным пертузумабу без активного агента.
Препарат в контексте данного документа может также содержать более чем одно активное соединение по мере необходимости для лечения конкретного состояния, предпочтительно, активные соединения с дополняющими действиями, которые не оказывают нежелательного влияния друг на друга. Различные лекарственные средства, которые можно комбинировать с ингибитором димеризации HER, описаны в разделе Способов ниже. Такие молекулы предпочтительно присутствуют в комбинации в количествах, которые эффективны для намеченного использования.
Препараты, которые будут использованы для введения in vivo, должны быть стерильными. Это легко достигается путем фильтрации через стерильные мембраны для фильтрации.
Типовые специфические препараты, подходящие для использования в способах согласно данному изобретению, являются следующими:
Пертузумаб в/в: концентрат пертузумаба 420 мг/14 мл для внутривенной инфузии представляет собой стерильную прозрачную или слегка опалесцирующую жидкость от бесцветного до бледно-коричневого цвета, поставляемую в одноразовых стеклянных флаконах по 20 мл с пробками 20 мм. Каждый одноразовый флакон содержит 420 мг пертузумаба в концентрации 30 мг/мл в 20 мМ L-гистидин ацетата (рН 6,0), 120 мМ сахарозы и 0,02% полисорбата 20.
Пертузумаб п/к с rHuPH20: пертузумаб 600 мг/5 мл с раствором rHuPH20 для подкожных инъекций представляет собой стерильную, не содержащую консервантов, бесцветную или слегка коричневатую жидкость, поставляемую в одноразовых флаконах по 10 мл с пробками 20 мм. Флаконы заполнены, чтобы обеспечить доставку и передачу 5,0 мл исследуемого лекарственного препарата (заполненного около 5,4 мл лекарственного препарата). Каждый флакон вмещает препараты, содержащие 120 мг/мл R04368451 в L-гистидинацетатном буфере, содержащем эксципиенты сахарозу, полисорбат 20, метионин и rHuPH20 (2000 Ед/мл) при рН 5,7.
Специфический препарат пертузумаба п/к с rHuPH20 имеет следующие ингредиенты:
120 мг/мл Пертузумаба
240 мМ Сахарозы
0,02% Полисорбата 20
10 мМ Метионина
2000 Ед/мл rhuPH20
20 мМ Гистидина/Ацетата
рН5,7
Пертузумаб п/к без rHuPH20: раствор пертузумаба 500 мг/5 мл для подкожных инъекций представляет собой стерильную, не содержащую консервантов, бесцветную или слегка коричневатую жидкость, поставляемую в одноразовых стеклянных флаконах по 10 мл с пробками 20 мм. Флаконы заполнены, чтобы обеспечить доставку и передачу 5,0 мл исследуемого лекарственного препарата. Каждый флакон вмещает препарат, содержащий 120 мг/мл пертузумаба в L-гистидинацетатном буфере, содержащего эксципиенты сахарозу, полисорбат 20 и метионин при рН 5,7.
Трастузумаб п/к: трастузумаб для подкожного введения обычно содержит следующие ингредиенты: рекомбинантную гиалуронидазу человека (rHuPH20); L-гистидин; моногидрат гидрохлорида L-гистидина; α, α-трегалоза дегидрат; L-метионин; Полисорбат 20; воду для инъекций, трастузумаб 600 мг/5 мл. Раствор трастузумаба для подкожной инъекции представляет собой стерильную, не содержащую консервантов, бесцветную или слегка коричневатую жидкость, поставляемую в стеклянных флаконах одноразового использования объемом 6 мл с пробками по 20 мм. Каждый флакон вмещает препарат, содержащий 120 мг/мл трастузумаба в L-гистидин-HCl-буфере, содержащего эксципиенты трегалозу, полисорбат 20, метионин и rHuPH20 (2000 Ед/мл) при рН5,5.
Специфический препарат трастузумаба п/к имеет следующие ингредиенты:
120 мг/мл Трастузумаба
210 мМ Трегалозы
0,04% Полисорбата 20
10 мМ Метионина
2,000 Ед/мл rhuPH20
20 мМ Гистидин-НС1
рН5,5
Специфическая комбинация фиксированных доз (FDC) пертузумаба-трастузумаба п/к в форме Нагрузочной дозы имеет следующий состав: трастузумаб 600 мг и пертузумаб 1,200 мг в 15 мл раствора для подкожной инъекции представляет собой стерильную, не содержащую консервантов, бесцветную или слегка коричневатую жидкость, поставляемую в одноразовых стеклянных флаконах по 20 мл с пробками 20 мм. Каждый флакон вмещает препарат, содержащий 40 мг/мл трастузумаба и 80 мг/мл пертузумаба в L-гистидин-HCl буфере, содержащем эксципиенты трегалозу, сахарозу, полисорбат 20, метионин и rHuPH20 (1,000 Ед/мл) при рН 5,5.
Специфическая комбинация фиксированных доз (FDC) пертузумаба-трастузумаба п/к в форме Поддерживающей дозы имеет следующий состав: трастузумаб 600 мг и пертузумаб 600 мг в 10 мл раствора для подкожной инъекции представляет собой стерильную, не содержащую консервантов, от бесцветного до слегка коричневатого цвета жидкость, поставляемую в одноразовых стеклянных флаконах по 15 мл с пробками 20 мм. Каждый флакон вмещает препарат, содержащий 60 мг/мл трастузумаба и 60 мг/мл пертузумаба в L-гистидин-HCl буфере, содержащем эксципиенты трегалозу, сахарозу, полисорбат 20, метионин и rHuPH20 (1,000 Ед/мл) при рН 5,5.
V. Способы лечения
Для внутривенного введения пертузумаб и трастузумаб вводят в соответствии с информацией по применению препарата.
Пертузумаб обычно вводят каждые три недели путем внутривенной инфузии, начиная с первой инфузии 840 мг, вводимой в течение 60 минут, с последующей второй и любой последующей внутривенной инфузией 420 мг, вводимой в течение от 30 до 60 минут. Более подробная информация о подходящих графиках введения дана в информации по применению трастузумаба.
Трастузумаб обычно вводят каждые три недели путем внутривенной инфузии, начиная с первой нагрузочной дозы 8 мг/кг в течение 90 минут, с последующей второй и любыми последующими внутривенными инфузиями поддерживающих доз 6 мг/кг вводимых в течение от 30 до 60 минут. Более подробная информация о подходящих графиках введения дана в информации по применению трастузумаба.
Пертузумаб и трастузумаб можно вводить во время одного и того же посещения в любом порядке.
Согласно данному изобретению пертузумаб или пертузумаб+трастузумаб вводят подкожно.
Пертузумаб п/к обычно вводят каждые три недели в виде подкожной инъекции, начиная с фиксированной нагрузочной дозы около 1200 мг, с последующей второй и любыми последующими фиксированными поддерживающими дозами около 600 мг, как описано выше и описано в Примерах. Место инъекции должно чередоваться между левым и правым бедром. Новые инъекции следует делать на расстоянии не менее 2,5 см от старого участка на здоровой коже, а не в местах, где кожа покраснела, ушиблена, чувствительна или тверда.
Трастузумаб п/к обычно вводят в виде подкожных инъекций в дозе 600 мг в течение 2-5 минут каждые три недели. Место инъекции должно чередоваться между левым и правым бедром. Новые инъекции следует делать на расстоянии не менее 2,5 см от старого участка на здоровой коже, а не в местах, где кожа покраснела, ушиблена, чувствительна или тверда.
Совмесный препарат пертузумаб/трастузумаб п/к вводят аналогичным образом.
Для совместного подкожного введения пертузумаба и трастузумаба необходимо смешать конечную смесь в шприцах, используя коннектор для шприца. В итоге, подкожная инъекция осуществляется с помощью одноразового пластикового шприца и иглы из нержавеющей стали.
VI. Изделия
В другом варианте реализации изобретения изделие содержит материалы, полезные для лечения онкологического заболевания. Изделие содержит флакон с фиксированной дозой пертузумаба для подкожного введения, при этом фиксированная доза составляет приблизительно 600 мг или приблизительно 1200 пертузумаба. Изделие предпочтительно дополнительно содержит листок-вкладыш. Листок-вкладыш может содержать инструкции по введению фиксированной дозы пациенту с HER2-экспрессирующим, например HER2-позитивным, HER2-амплифицированным или HER2-мутированным онкологическим заболеванием подкожно, отдельно или в сочетании с подкожным введением трастузумаба, при этом введение в комбинации включает, но не ограничивается этим, совместное введение, введение смеси и введение совместного препарата, как выше определено и описано, и как описано в Примерах. В некоторых вариантах реализации изобретения онкологическое заболевание представляет собой рак молочной железы, рак яичников, рак брюшины, рак маточной трубы, рак легких, рак предстательной железы, колоректальный рак, рак желчных путей и рак мочевого пузыря. В других вариантах реализации изобретения онкологическое заболевание представляет собой рак молочной железы, рак брюшины, рак маточной трубы, рак легких, колоректальный рак, рак желчных путей и рак мочевого пузыря. В конкретном варианте реализации изобретения онкологическое заболевание представляет собой рак молочной железы, такой как ранний рак молочной железы или метастатический рак молочной железы.
В одном варианте реализации изобретения изделие представляет собой стеклянный одноразовый флакон, оборудованный пробкой, который содержит препарат для введения.
Другой формой изделия является шприц, содержащий препарат для введения, который может быть присоединен к игле для подкожных инъекций из нержавеющей стали для подкожного введения.
В одном варианте реализации изобретения изделие содержит два флакона, при этом первый флакон содержит фиксированную дозу приблизительно 1200 мг пертузумаба, а второй флакон содержит фиксированную дозу приблизительно 600 мг пертузумаба.
В другом варианте реализации изобретения изделие содержит два флакона, при этом первый флакон содержит фиксированную дозу приблизительно 600 мг пертузумаба, а второй флакон содержит фиксированную дозу приблизительно 600 мг трастузумаба.
В другом варианте реализации изобретения изделие содержит флакон с однократной дозой, содержащей около 600 мг пертузумаба.
IV. Хранение Биологических Материалов
Следующие гибридомные клеточные линии были сданы на хранение в Американскую Коллекцию Типовых Культур (American Type Culture Collection), 10801 University Boulevard. Manassas, VA 20110-2209, USA (ATCC):


Более подробно изобретение проиллюстрировано следующими неограничивающими Примерами. Раскрытие всех ссылок в описании включено в данный документ посредством ссылки.



ПРИМЕР 1
Фаза I Исследование по подбору дозы подкожной инъекции пертузумаба в комбинации с трастузумабом
Фаза I представляет собой открытое, двухэтапное комплексное клиническое исследование по подбору дозы подкожной инъекции пертузумаба в комбинации с трастузумабом у здоровых мужчин-добровольцев и женщин-пациентов с ранним раком молочной железы.
В исследовании была определена безопасность и ФК пертузумаба п/к для лечения Q3W путем применения подхода, основанного на ФК, для сравнения состава препарата п/к с апробированным препаратом в/в. В этом исследовании по подбору дозы предполагается определить дозу п/к, которая сопоставима с дозой в/в по отношению к сывороточной концентрации. Концентрации Ctrough Q3W сыворотки пертузумаба п/к неизвестны.
В исследовании будут оцениваться различные типы инъекций пертузумаба п/к:
 Раздельное введение пертузумаба п/к с или без трастузумаба п/к в виде отдельных инъекций (совместное введение)
Раздельное введение пертузумаба п/к с или без трастузумаба п/к в виде отдельных инъекций (совместное введение)
 Одновременное введение пертузумаба п/к и трастузумаба п/к в виде однократной инъекции (введение смеси)
Одновременное введение пертузумаба п/к и трастузумаба п/к в виде однократной инъекции (введение смеси)
 Введение пертузумаба и трастузумаба в виде подкожного введения в виде однократной инъекции (совместный препарат) Первоначально, в Части 1, здоровые мужчины-добровольцы (ЗМД) получат однократную дозу пертузумаба в/в или п/к (с трастузумабом или без него), чтобы выбрать дозу (дозы) пертузумаба, которая, как ожидается, приведет к сывороточной концентрации, сопоставимой с пертузумабом в/в, как при совместном введении или введении смеси. Доза(ы) пертузумаба п/к будет затем подтверждена у пациентов с РРМЖ.
Введение пертузумаба и трастузумаба в виде подкожного введения в виде однократной инъекции (совместный препарат) Первоначально, в Части 1, здоровые мужчины-добровольцы (ЗМД) получат однократную дозу пертузумаба в/в или п/к (с трастузумабом или без него), чтобы выбрать дозу (дозы) пертузумаба, которая, как ожидается, приведет к сывороточной концентрации, сопоставимой с пертузумабом в/в, как при совместном введении или введении смеси. Доза(ы) пертузумаба п/к будет затем подтверждена у пациентов с РРМЖ.
На основании данных ФК в Части 1 (здоровые группы добровольцев) модель попФК пертузумаба будет использоваться для определения целевой дозы (доз) для Части 2. (См., Garg et al., Cancer Chemother Pharmacol (2014) 74:819-829.)
После выбора целевой дозы и на основании информации о допустимости FDC пациенты с ранним раком молочной железы (РРМЖ), которые завершили свое стандартное лечение, будут включены в Часть 2 для получения пертузумаба п/к в дозе (дозах), определенной в Части 1. Эта идентифицированная доза пертузумаба будет либо вводиться совместно с трастузумабом п/к, либо смешиваться с трастузумабом п/к, либо в виде совместного препарата с трастузумабом п/к в комбинации фиксированных доз (FDC). Часть 2 будет включать подтверждение дозы пертузумаба п/к, а также сравнение ФК в результате введения смеси и FDC.
В этом исследовании, в котором два моноклональных антитела (mAb) будут вводиться в объеме до приблизительно 15 мл, также будет оцениваться концентрация rHuPH20. Ранее 2000 Ед/мл усилителя абсорбции использовали с трастузумабом п/к и ритуксимабом п/к, однако это отдельные антитела и в объемах, меньших, чем комбинация пертузумаб/трастузумаб, изученная в данном документе.
Чтобы определить, приведет ли меньшее количество rHuPH20 к адекватной абсорбции mAb, исследование было разработано для проверки концентрации фермента 2000 Ед/мл, когда даны оба антитела (объем 15 мл) и концентрации фермента 667 Ед/мл (с использованием пертузумаба, который не содержит rHuPH20), когда даны оба антитела (объем 15 мл). Если параметры ФК приблизительно эквивалентны, то уменьшенное количество rHuPH20 может быть потенциально использовано при разработке FDC совместного препарата.
ЗАДАЧИ И КОНЕЧНЫЕ ТОЧКИ (ИЗМЕРЯЕМЫЕ ПАРАМЕТРЫ) ОСНОВНЫЕ ЗАДАЧИ
Часть 1 (Нахождение дозы)
Основными задачами для Части 1 этого исследования являются следующие:
▪ Подобрать подкожную (п/к) нагрузочную и поддерживающую дозу пертузумаба, которая приводит к сопоставимому воздействию пертузумаба для внутривенного введения (в/в), когда пертузумаб п/к вводится в виде инъекций единственного агента (для последующего использования при совместном введении с трастузумабом п/к).
▪ Подобрать нагрузочную и поддерживающую дозу пертузумаба п/к, которая приводит к сопоставимому воздействию пертузумаба в/в, когда пертузумаб п/к назначают в комбинации с трастузумабом п/к в виде однократной инъекции (введение смеси).
▪ Оценить, необходим ли дополнительный rHuPH20, когда пертузумаб п/к и трастузумаб п/к являются смешанными п/к.
Часть 2 (Подтверждение Дозы)
Основные задачи для Части 2 этого исследования заключаются в следующем:
▪ Подтвердить поддерживающую дозу пертузумаба п/к при введении в виде инъекции единственного агента как части совместного введения с трастузумабом п/к или
▪ Подтвердить поддерживающую дозу пертузумаба п/к при введении в смеси с трастузумабом п/к в однократной инъекции (введение смеси) или в сочетании с трастузумабом п/к в готовой к применению однократной инъекции (комбинация фиксированных доз (FDC)).
ВТОРИЧНЫЕ ЗАДАЧИ
Вторичные задачи этого исследования заключаются в следующем:
▪ Оценить безопасность и переносимость пертузумаба п/к при отдельном назначении или в комбинации с трастузумабом п/к (введение смеси или FDC) у здоровых мужчин-добровольцев (ЗМД) и женщин с ранним раком молочной железы (РРМЖ), которые завершили стандартную терапию рака молочной железы, на основе следующих конечных точек:
- Уровня, характера и степени тяжести нежелательных явлений, которые оцениваются в соответствии с Общей Терминологией Критериев Нежелательных Явлений Национального Института Рака (NCI СТСАЕ) v4.03;
- Изменения показателей жизненно важных функций, фракции выброса левого желудочка (ФВЛЖ) и параметров электрокардиограммы (ЭКГ);
- Изменения в клинических лабораторных результатах;
- Частоты ответа на антитерапевтические антитела (АТА).
ПЛАН ИССЛЕДОВАНИЯ
ОПИСАНИЕ ИССЛЕДОВАНИЯ
Обзор Плана Исследования
Это открытое, состоящее из двух частей, многоцентровое исследование пертузумаба п/к.
Часть 1 исследования - это определение дозы, при котором нагрузочная и поддерживающая доза пертузумаба п/к будет определяться у ЗМД. Будут оцениваться два типа инъекций пертузумаба п/к: пертузумаб, вводимый как инъекция единственного агента (для последующего использования при совместном введении с инъекцией единственного агента трастузумаба п/к), и пертузумаб для покожного введения, смешанный с трастузумабом п/к в однократной инъекции.
В Части 2 исследования будет подтвеждена доза (дозы) пертузумаба для пациентов с РРМЖ, которые завершили стандартную терапию рака молочной железы. Доза пертузумаба п/к в Части 2 будет вводиться совместно с трастузумабом п/к, в смеси с трастузумабом п/к или в виде совместного препарата с трастузумабом п/к как FDC. Часть 2 будет включать подтверждение дозы пертузумаба п/к, а также сравнение ФК из смеси и FDC перед Фазой III исследования.
См. Фиг. 6 для изучения схемы. Безопасность будет контролироваться, и образцы крови для оценки ФК будут отбираться в соответствии с графиком исследований.
Часть 1 (Нахождение дозы)
ЗМД были включены в Когорты с 1 по 8 (6 субъектов на группу). Каждый субъект получил однократную инъекцию. Когорты 2-4 оценивали разные дозы пертузумаба п/к. Когорты с 5 по 8 оценивали дозы смеси пертузумаба + трастузумаба. Дозы, оцениваемые в каждой когорте, следующие:
• Когорта 1: 420 мг пертузумаба в/в (контроль)
• Когорта 2: 400 мг пертузумаба п/к
• Когорта 3: 600 мг пертузумаба п/к
• Когорта 4: 1200 мг пертузумаба п/к
• Когорта 5: 600 мг трастузумаба п/к (контроль)
• Когорта 6: 400 мг пертузумаба п/к плюс 600 мг трастузумаба п/к (в смеси)
• Когорта 7: 1200 мг пертузумаба п/к плюс 600 мг трастузумаба п/к (в смеси)
• Когорта 8: 1200 мг пертузумаба п/к (без rHuPH20) плюс 600 мг трастузумаба п/к (в смеси).
Различные дозы пертузумаба вводили, регулируя объем вводимой дозы. Концентрация пертузумаба и трастузумаба составляла 120 мг/мл, a rHuPH20 составляла 2000 Ед/мл в дозе растворов вводимой подкожно.
Когорты 6 и 7 получали пертузумаб п/к и трастузумаб п/к, и тот и другой содержали rhuPH20 в концентрации 2000 Ед/мл, в то время как ЗМД в Когорте 8 получали пертузумаб п/к, не содержащий rHuPH20, смешанный с трастузумабом п/к, содержащим rhuPH20 в концентрации 2000 Ед/мл, следовательно, общая концентрация rhuPH20, полученная Когортой 8, составила приблизительно 667 Ед/мл. Когорта 8 была запланирована для оценки воздействия более низкой концентрации rHuPH20 на ФК пертузумаба и трастузумаба при введении в смеси.
Время Наблюдения
Для обеспечения безопасности во время испытания первый здоровый доброволец в Когорте 2 тщательно наблюдался на предмет безопасности и переносимости препарата после лечения пертузумабом п/к и до конца Дня 3. Трехдневный мониторинг был завершен до расширения Когорты 2 и до начала введения дозы в Когорте 3 или 4.
Если доза пертузумаба п/к считалась безопасной и переносимой у первого здорового добровольца в Когорте 2, то последующих здоровых добровольцев лечили параллельно в Когортах 2, 3 и 4 без добавления 3-дневного периода времени наблюдения.
Точно так же 3 здоровых добровольца в Когорте 6 получали пертузумаб и трастузумаб п/к и тщательно наблюдались на предмет безопасности и переносимости препарата в течение 3 дней после введения дозы, до расширения Когорты 6 и до начала введения дозы в Когорте 7 или 8.
Если доза пертузумаба и трастузумаба считалась безопасной и переносимой у первых 3 здоровых добровольцев в Когорте 6, последующие здоровые добровольцы будут проходить параллельное лечение в Когорте 6 без добавления 3-дневного периода времени наблюдения.
Когорты 7 и 8 были открыты одновременно. Трое ЗМД получали пертузумаб и трастузумаб п/к и тщательно наблюдались на предмет безопасности и переносимости препарата в течение 3 дней после введения дозы, перед расширением соответствующих когорт.
Если доза пертузумаба и трастузумаба п/к считалась безопасной и переносимой у первых 3 здоровых добровольцев в Когортах 7 и 8, последующие здоровые добровольцы будут проходить параллельное лечение в этих Когортах без добавления 3-дневного периода времени наблюдения.
Здоровые добровольцы в Когортах 1 и 5 могут получать дозы параллельно и могут быть включены в исследование до первого здорового добровольца в Когорте 2.
Выбор Дозы для Части 2
Выбор доз пертузумаба п/к в Части 1 (Когорты 2, 3 и 4) основан на модели популяционной фармакокинетики пертузумаба в/в попФК) с включенными значениями параметров ФК трастузумаба п/к. Как только достаточный объем данных в Части 1 позволит оценить фиксированные параметры ФК (т.е. Ctrough, AUCO-inf, максимальная концентрация в сыворотке [Cmax], время максимальной концентрации в сыворотке [Tmax]), (поддерживающая) доза (дозы) пертузумаба п/к) будет выбрана для Части 2. Эта доза п/к будет рассчитана, чтобы обеспечить воздействие пертузумаба, аналогичное воздействию пертузумаба в/в в дозе 420 мг.Точно так же, основываясь на параметрах ФК, будет рассчитана одна (нагрузочная) доза пертузумаба п/к, чтобы обеспечить воздействие пертузумаба, аналогичное воздействию пертузумаба в/в в дозе 840 мг.Модель попФК пертузумаба в/в будет обновлена с помощью параметров пертузумаба п/к с использованием данных Части 1 и будет использоваться для правильной идентификации поддерживающих и нагрузочных доз п/к.
Доза трастузумаба п/к в 600 мг была определена в Фазе 1b исследования ВР22023 по выявлению дозы и подтверждена в Фазе III исследования HannaH.
Дополнительные когорты для определения дозы могут быть открыты, если дозы от запланированных когорт приводят к тому, что воздействие пертузумаба отличается от целевой концентрации или если вариабельность фармакокинетики слишком высока, чтобы определить дозу для Части 2 исследования.
Используя данные Части 1, была рассчитана доза пертузумаба п/к, чтобы обеспечить воздействие пертузумаба, аналогичное воздействию пертузумаба в/в в дозе 420 мг (поддерживающая доза) и 840 мг (нагрузочная доза). Выбранная концентрация rHuPH20 для введения смеси пертузумаба и трастузумаба в Когорте В была основана на сопоставимости безопасности и воздействия пертузумаба в Когортах 7 и 8.
Часть 2 (Подтверждение Дозы)
Женщины с РРМЖ, которые завершили стандартную (нео)адъювантную терапию рака молочной железы, были включены в Часть 2.
Если допустимость FDC препарата подтверждена, то только Когорты В и С (не Когорта А; совместное введени) могут быть включены в исследование в Части 2. Это может позволить подтвердить дозу, указанную в Части 1, и продемонстрировать сопоставимость введения смеси и FDC препарата. Если между пертузумабом и трастузумабом существует ФК-взаимодействие при смешивании или если разработка FDC невозможна, то только Когорта А может быть включена в исследование в Части 2, что позволит подтвердить дозу Части 1. Этот план исследования позволит выбрать дозу пертузумаба п/к и вариант препарата для Фазы III исследования при включении в исследование наименьшего числа пациентов. Таким образом, общая схема для Части 2 должна быть только для Когорты А или для Когорты В и Когорты С (см. 185Н Фиг. 2). Каждая когорта будет включать 20 пациентов, и каждый пациент получит одну дозу пертузумаба и трастузумаба.
Каждая Когорта включала 20 пациентов, и каждый пациент получил одну дозу пертузумаба и трастузумаба.
• Когорта А: пертузумаб п/к (доза, определенная в Части 1) с 600 мг трастузумаба п/к; каждый агент вводится отдельно (совместное введение) или
• Когорта В: пертузумаб п/к (доза, определенная в Части 1) с 600 мг трастузумаба п/к; оба агента вводятся в одной инъекции (введение смеси) и
• Когорта С: пертузумаб п/к (доза, определенная в Части 1) с 600 мг трастузумаба п/к; оба агента приготовлены вместе и вводятся в виде одной инъекции (FDC)
Максимальная запланированная доза пертузумаба п/к, вводимая в Части 1 и Части 2, не будет превышать 1200 мг.
Замечания: Дозы пертузумаба и трастузумаба будут изменены путем регулирования объема вводимой дозы. Концентрация пертузумаба и трастузумаба составляет 120 мг/мл, a rHuPH20 (если присутствует) составляет 2000 Ед/мл в дозе растворов вводимой подкожно. Диаграмма решений показана на Фиг. 7.
Если было ФК-взаимодействие между пертузумабом и трастузумабом, которые вводили в смеси, или если разработка FDC было невозможным, то только Когорта А (совместное введение) должна была быть включена в исследование в Части 2. Этот план исследования позволил выбрать дозу и вариант препарата пертузумаба п/к для дальнейшей оценки в Фазе III исследовании при включении наименьшего числа пациентов. Таким образом, общая схема для Части 2 была только для Когорты А или Когорты В и Когорты С.
Критерии Продолжения или Прекращения Дозирования
Безопасность, переносимость и данные ФК будут оцениваться непрерывно и до расширения когорт или (при необходимости) добавления когорт. Начальные дозы будут составлять 420 мг пертузумаба в/в и 400, 600 и 1200 мг п/к.
Чтобы принять обоснованные решения относительно дозирования здоровых добровольцев в когортах с подкожным введением препарата, соответствующие данные о безопасности и переносимости определенных субъектов будут рассмотрены через 3 дня, до введения следующему субъекту в когорте или открытия других когорт.
Решение о продолжении дозирования будет принято исследователем и Медицинским Наблюдателем Roche и любым другим лицом, которое, по мнению исследователя или Медицинского Наблюдателя, необходимо для принятия этого решения.
Доза больше не будет вводиться любому другому здоровому добровольцу или пациенту с РРМЖ, если переносимость или безопасность предшествующего здорового мужчины-добровольца или пациента с РРМЖ не приемлемы по мнению Исследователя и Медицинского Наблюдателя. Не следует дополнительно вводить дозу любому другому здоровому добровольцу или пациенту с РРМЖ, если происходит какое-либо из событий, перечисленных ниже, если только не очевидно, что возникновение не связано с назначением лечения:
• Тяжелые нежелательные явления, связанные с лекарственным препаратом
• Реакции гиперчувствительности по NCI СТСАЕ (Степени с 3 до 5)
• Снижение ФВЛЖ > 10% или до < 50% (для ЗМД)
• Снижение ФВЛЖ > 10% или до < 50% (для ЕВС)
• Повторная оценка должна быть проведена в течение 3 недель после первого зарегистрированного снижения, и случай должен быть рассмотрен кардиологом. Застойная сердечная недостаточность (ЗСН) II класса или выше согласно класссификации Нью-Йоркской Ассоциации Кардиологов (NYHA) должна быть подтверждена кардиологом. Следует четко указать, что это только рекомендации, и Исследователь вместе с Медицинским Наблюдателем может сделать исключение. Однако, когда такое исключение сделано, причины этого должны быть четко задокументированы в электронной истории болезни (eCRF).
КОНЕЦ И ПРОДОЛЖИТЕЛЬНОСТЬ ИССЛЕДОВАНИЯ
Конец этого исследования определяется как дата, когда произошел последний визит последнего пациента (LPLV). Ожидается, что LPLV произойдет через 7 месяцев после регистрации последнего пациента. Ожидается, что общая продолжительность исследования, от скрининга первого пациента до конца исследования, составит приблизительно от 16 до 24 месяцев. От скрининга до последующего наблюдения у здоровых добровольцев/пациентов будет максимум 34 недели (до 4 недель на период скрининга и 30 недель на проведение исследования и последующее наблюдение).
Для каждого участника исследования (пациенты с ЗМД и ЕВС) период скрининга составлял до 4 недель, и последующее наблюдение проводилось приблизительно через 7 месяцев после введения исследуемого лекарственного препарата.
МАТЕРИАЛЫ И СПОСОБЫ
ПОПУЛЯЦИЯ ИССЛЕДОВАНИЯ
Часть 1 Критерии Включения
Для включения в исследование ЗМД должны соответствовать следующим критериям:
• Подписанная Форма Информированного Согласия
• Здоровые мужчины в возрасте от 18 до 45 лет включительно
• Способность соблюдать протокол исследования согласно мнению исследователя
• ФВЛЖ ≥ 55%, измеренная с помощью эхокардиографии (ЭХО) или радионуклидной ангиографии (MUGA)
• Индекс массы тела (ИМТ) от 18 до 32 кг/м2 включительно
• Согласие воздерживаться от ведения половой жизни (воздерживаться от гетеросексуальных контактов) или использовать противозачаточные средства и согласие воздерживаться от донорства спермы, как определено ниже:
• В течение периода лечения и в течение не менее 7 месяцев после введения пертузумаба и/или трастузумаба мужчины должны восдерживаться от ведения половой жизни с женщинами-партнерами, имеющими детородный потенциал, или использовать презерватив плюс дополнительный способ контрацепции, что в совокупности приводит к частоте беременности < 1% в год. Надежность полового воздержания следует оценивать в зависимости от продолжительности клинического испытания и предпочтительного и обычного образа жизни пациента. Периодическое воздержание (например, календарный, овуляционный, симптотермальный или постовуляционные способы) и прерваный половой акт не являются приемлемыми способами контрацепции.
• Мужчины должны воздерживаться от сдачи спермы в течение этого же периода.
• С беременными женщинами-партнерами мужчины должны воздерживаться от ведения половой жизни или использовать презерватив во время периода лечения и в течение по крайней мере 7 месяцев после введения пертузумаба и/или трастузумаба, чтобы избежать воздействия на эмбрион.
• Отсутствие противопоказаний в подробном медицинском и хирургическом анамнезе и физически неповрежденная нормальная кожа без татуировок, пигментации или повреждений, которые находятся в потенциальной области предполагаемой инъекции в бедра.
Часть 1 Критерии Исключения
• ЗМД, которые соответствуют любому из следующих критериев, будут исключены из исследования:
• Положительный анализ мочи на наркотики в соответствии с местным стандартом
• Положительный результат теста на вирус гепатита В (ВГВ), вируса гепатита С (ВГС) или вируса иммунодефицита человека (ВИЧ) 1 или 2.
• История контактирования с ВГВ, ВГС или ВИЧ
• Активная вирусная инфекция гепатита (гепатит В или С) или ВИЧ-инфекция
• Систолическое артериальное давление (АД) ≥ 140 мм рт. ст. или < 90 мм рт. ст., или диастолическое давление > 0 мм рт.ст. или < 50 мм рт.
• Использование запрещенных медикаментов или лекарственных средств растительного происхождения в течение 10 дней или 5-кратного периода полувыведения (в зависимости от того, что дольше) до введения исследуемого лекарственного препарата
• Клинически значимые отклонения в результатах лабораторных исследований (в том числе анализах фунции печени и почек, общем анализе крови, развернутом биохимическом анализе крови и анализе мочи)
• Клинически значимые отклонения ЭКГ при скрининге или исходной ЭКГ, включая, но не ограничиваясь этим, следующее:
• Интервал QTc (QTcB > 450 мсек)
• Выраженная тахикардия в покое (ЧСС > 100 ударов в минуту)
• Разница между самым высоким и самым низким значением любого базового QTc в определенный момент времени >30 мс
• Показатель отклонений параметров интервала QT (например, плоские зубцы Т, аритмии и т.д.)
• Признаки фибрилляции предсердий, трепетания предсердий, блокады правой или левой ножки пучка Гиса, синдрома Вольфа-Паркинсона-Уайта или наличие кардиостимулятора
• Любое другое существенное отклонение от нормы
• В анамнезе любое сердечное заболевания или ФВЛЖ < 55%
• Участие в исследовании экспериментального препарата или устройства в течение 90 дней до отбора
• Донорство крови>500 мл в течение 3 месяцев до отбора
• Обнаруженная аллергия на гиалуронидазу, пчелиный яд или яд перепончатокрылых или любой другой ингредиент в составе rHuPH20 (рекомбинантный Hylenex® [инъекция гиалуронидазы человека])
• Обнаруженная повышенная чувствительность к любому из исследуемых препаратов или к вспомогательным веществам рекомбинантных человеческих или гуманизированных антител.
• В анамнезе гиперчувствительность или значительные аллергические реакции, спонтанные или после любого предшествующего введения лекарственного препарата
• Очевидный клинически значимый семейный анамнез гиперчувствительности, аллергии или тяжелых заболеваний сердца
• Отек или патология нижних конечностей (например, целлюлит, лимфатическое расстройство или предшествующая операция, ранее существовавший болевой синдром, предыдущее расслоение лимфатического узла и т.д.), которые могут помешать любой указанной в протоколе оценке результатов
 Любой клинически значимый анамнез системного заболевания (например, злокачественная опухоль, сахарный диабет, болезни желудочно-кишечного тракта, почечная недостаточность, болезнь печени, сердечно-сосудистое, ревматическое или легочное заболевание)
Любой клинически значимый анамнез системного заболевания (например, злокачественная опухоль, сахарный диабет, болезни желудочно-кишечного тракта, почечная недостаточность, болезнь печени, сердечно-сосудистое, ревматическое или легочное заболевание)
• В анамнезе рак молочной железы, лечение рака молочной железы или лечение антрациклинами или другими кардиотоксическими лекарственными препаратами
• Текущие заболевания или состояния, которые могут помешать, или для которых лечение может помешать, проведению исследования или которые, по мнению исследователя, представляют неприемлемый риск для субъекта в этом исследовании
• Текущее хроническое ежедневное лечение (непрерывно в течение >3 месяцев) кортикостероидами (доза ≥ 10 мг/день метилпреднизолона), за исключение ингаляционных кортикостероидов
•Прием внутривенных антибиотиков при лечении инфекции в течение 7 дней до включения в исследование.
• Критерии включения в исследование: Часть 2 (Пациенты с Ранним Раком Молочной Железы)
Часть 2 Критерии Включения
• Пациенты должны соответствовать следующим критериям для включения в исследование:
• Подписанная Форма Информированного Согласия
• Женщины в возрасте ≥18 лет
• Способность соблюдать протокол исследования согласно мнению исследователя
•Общее состояние 0 по шкале Восточной Объединенной Группы Онкологов
•Текущая неметастатическая аденокарцинома молочной железы, соответствующая следующим критериям:
a) Получает лечение с помощью адекватной хирургической процедуры
b) Завершено стандартное противоопухолевое (нео)адъювантное лечение (химиотерапия/биологическое) > за 7 месяцев до введения исследуемого лекарственного препарата
c) Получает лечение лучевой терапией, при определенных условиях
•Исходная ФВЛЖ ≥ 55%, измеренная с помощью ЭхоКГ или сканированием MUGA
• Отрицательный тест на беременность у женщин с детородным потенциалом, которые находятся в пременопаузе или у которых аменорея менее 12 месяцев в пост-менопаузе, и которые не подвергались хирургической стерилизации.
• Для женщин с детородным потенциалом: в течение периода лечения и в течение не менее 7 месяцев после приема пертузумаба и трастузумаба согласие воздерживаться от ведения половой жизни (воздерживаться от гетеросексуальных контактов) или использовать негормональные способы контрацепции, которые приводят к частоте беременности <1% в год.
Женщина считается имеющей детородный потенциал, если она находится в постменархиальном периоде, не достигла постменопаузального состояния (≥ 12 непрерывных месяцев аменореи без идентифицированной причины, кроме менопаузы), и не подверглась хирургической стерилизации (удаление яичников и/или матки). Примеры способов контрацепции с частотой беременности < 1% в год включают двустороннюю перевязку маточных труб, стерилизацию мужчин и медь-содержащие внутриматочные средства (ВМС).
• Надежность полового воздержания следует оценивать в зависимости от продолжительности клинического испытания и предпочтительного и обычного образа жизни пациента. Периодическое воздержание (например, календарный, овуляционный, симптотермальный или постовуляционные способы) и прерваный половой акт не являются приемлемыми способами.
Часть 2 Критерии Исключения
• Пациенты, которые соответствуют любому из следующих критериев, будут исключены из исследования:
• Одновременное другое злокачественное новообразование, требующее терапии любого типа, которая может помешать проведению исследований ФК или привести к неожиданной токсичности
• Максимальная кумулятивная доза доксорубицина >360 мг/м2 или максимальная кумулятивная доза эпирубицина >720 мг/м2 или любых предыдущих антрациклинов, не связанных с текущим раком молочной железы
• Серьезное, неконтролируемое сопутствующее заболевание, при котором противопоказано использование любого из экспериментальных лекарственных препаратов, используемых в этом исследовании, или которое подвергнет пациента высокому риску осложнений, связанных с лечением.
• В анамнезе другое злокачественных новообразование в течение 5 лет до отбора, за исключением рака шейки матки in situ, который подвергся лечению надлежащим образом, немеланомного рака кожи или рака матки I стадии
•Пациенты, в настоящее время участвующие в других исследованиях экспериментальных агентов, если это не согласовано исследователем и Спонсором
• Серьезные заболевания сердца или медицинские показания
• Любое предыдущее или сопутствующее состояние, предполагающее склонность к гиперчувствительности или аллергическим реакциям. Пациенты с легкой или сезонной аллергией могут быть включены после обсуждения между исследователем и Спонсором.
• Тяжелые инфузионные реакции (IRR), возникавшие во время любой предыдущей терапии пертузумабом или трастузумабом
• Обнаруженная аллергия на гиалуронидазу, пчелиный яд или яд перепончатокрылых или любой другой ингредиент в составе Hylenex®
• Любой из следующих лабораторных тестов, который отклоняется от нормы, в День -1 перед лечением трастузумабом:
Общий билирубин сыворотки > 1,25 х верхнего предела нормы (ВГН; за исключением синдрома Гилберта)
Аланинаминотрансфераза (АЛТ) или аспартатаминотрансфераза (ACT) > 1,25 × ВГН;
Альбумин < 25 г/л
Щелочная фосфатаза (ЩФ) > 2.5 х ВГН
Креатинин сыворотки > 1,5 х ВГН
Общее количество лейкоцитов (WBC) < 2500 клеток/мм3
Абсолютное количество нейтрофилов < 1500 клеток/мм3
Тромбоциты<100,000 клеток/мм3
• Беременные или кормящие женщины, или женщины, намеревающиеся забеременеть во время исследования
• Женщины с детородным потенциалом или менее 1 года после менопаузы (если не хирургически стерильны), которые не могут или не хотят применять адекватные меры контрацепции во время лечение в рамках исследования и в течение 7 месяцев после введения исследуемого лекарственного препарата
• Остаточная токсичность в результате предыдущей терапии (например, гематологическая, сердечно-сосудистая или неврологическая, Степень ≥2). Алопеция допускается.
• Неконтролируемая артериальная гипертензия (систолическое АД >150 мм рт.ст. и/или диастолическое АД >100 мм рт.ст.)
• Клинически значимое (т.е. активное) сердечно-сосудистое заболевание, включая, но не ограничиваясь этим, цереброваскулярная катастрофа/инсульт или инфаркт миокарда в течение 6 месяцев до первого лечения в рамках исследования; нестабильная стенокардия; ЗСН класса II или выше по NYHA; серьезная сердечная аритмия, требующая лечения; или другие сердечно-сосудистые проблемы, которые не контролируются или в настоящее время контролируются с помощью лекарств
• Положительный результат теста на ВГВ, ВГС или ВИЧ 1 или 2
• История контактирования с ВГВ, ВГС или ВИЧ
• Активная вирусная инфекция гепатита (гепатит В или С) или ВИЧ-инфекция
• Прием внутривенных (в/в) антибиотиков при лечении инфекции в течение 7 дней до включения в исследование.
• Текущее хроническое ежедневное лечение (непрерывно в течение > 3 месяцев) кортикостероидами (доза, эквивалентная или превышающая 10 мг/день метилпреднизолона), за исключением ингаляционных стероидов.
• Обнаруженная повышенная чувствительность к любому из исследуемых препаратов или к вспомогательным веществам рекомбинантных человеческих или гуманизированных антител.
СПОСОБ РАСПРЕДЕЛЕНИЯ В ГРУППЫ ЛЕЧЕНИЯ
Здоровые добровольцы и пациенты будут идентифицированы для возможного пополнения с использованием предварительной проверки регистрационных журналов, одобренной Советом по надзору (ЭСО)/Комитетом по Этике (КЭ) рекламы в газетах и на радио, а также списков рассылки до получения согласия на участие в исследовании.
Часть 1 (Здоровые Добровольцы)
Приблизительно 48 здоровых добровольцев будут набраны первоначально для Части 1. Номера пациентов будут распределяться последовательно в том порядке, в котором они были зарегистрированы. При необходимости могут быть открыты дополнительные когорты по определению дозы.
Часть 2 (Пациенты с РРМЖ)
Приблизительно 40 пациентов с РРМЖ будут набраны для Части 2. Номера пациентов будут распределяться последовательно в том порядке, в котором они были зарегистрированы.
ИССЛЕДУЕМЫЙ ПРЕПАРАТ
Исследуемыми лекарственными средствами (IMP) для этого исследования являются пертузумаб и трастузумаб.
Препарат, Упаковка и Правила Использования
Упаковка исследуемого лекарственного средства будет контролироваться отделом снабжения клинических испытаний Roche и иметь маркировку с идентификацией, требуемой местным законодательством, номером протокола, а также идентификацией и дозировкой лекарственного средства. Упаковка и маркировка исследуемого лекарственного препарата будут соответствовать стандарту Roche и местным нормам. После доставки IMP в исследовательский центр, персонал центра должен проверить их на наличие повреждений и проверить правильность идентификации, количество, целостность пломб и температурные условия, а также сообщать о любых отклонениях или жалобах на препарат наблюдателю клинических исследований при обнаружении. Квалифицированный специалист, ответственный за выдачу исследуемого лекарственного препарата, подготовит правильную дозу в соответствии с графиком. Этот специалист запишет дату выдачи, дату введения и номер пациента и инициалы, в зависимости от случая, на этикетке флакона с исследуемым лекарственным препаратом и/или в Отчете об Учете Препарата. Этот специалист также запишет партию исследуемого лекарственного препарата или номер партии, полученного каждым пациентом во время исследования. Пертузумаб
Были использованы три препарата пертузумаба:
Препарат I пертузумаба представляет собой стерильный, бесцветный или слегка коричневатый концентрат для раствора для инфузии, предоставленный в виде препарата для единоразового внутривенного введения (в\в), содержащего 30 мг/мл пертузумаба в L-гистидин ацетатном буфере, содержащем эксципиенты сахарозу и полисорбат 20. Каждый 20-мл флакон содержит 420 мг пертузумаба (14,0 мл/флакон).
Препарат 2 пертузумаба представляет собой стерильный, бесцветный или слегка коричневатый раствор для инъекций, предоставленный в виде препарата для единоразового подкожного введения, содержащего 120 мг/мл пертузумаба в L-гистидинацетатном буфере, содержащем эксципиенты сахарозу, полисорбат 20, метионин и rHuPh20 (2000 Ед/мл). Каждый 10-мл флакон содержит 600 мг пертузумаба (5,0 мл/флакон).
Препарат 3 пертузумаба представляет собой стерильный, бесцветный или слегка коричневатый раствор для инъекций, предоставленный в виде препарата для единоразового подкожного введения, содержащего 120 мг/мл пертузумаба в L-гистидинацетатном буфере, содержащем эксципиенты сахарозу, полисорбат 20 и метионин. Каждый 10-мл флакон содержит 600 мг пертузумаба (5,0 мл/флакон).
С пертузумабом не используется консервант, поскольку флаконы предназначены только для одноразового использования. Рекомендуемые условия хранения лекарственного препарата: от 2°С до 8°С, в защищенном от света месте. Лекарственный препарат не должен быть заморожен.
Трастузумаб
Препарат трастузумаба представляет собой стерильный, бесцветный или слегка коричневатый концентратный раствор для инъекций, содержащий 120 мг/мл трастузумаба в буфере L-гистидин/гистидин-HCl, содержащий эксципиенты трегалозу, полисорбат 20, метионин и rHuPh20 (2000 Ед/Мл). Каждый 5-мл флакон содержит 600 мг RO0452317 (5,0 мл/флакон).
С трастузумабом не используется консервант, поскольку флаконы предназначены только для одноразового использования. Рекомендуемые условия хранения лекарственного препарата: от 2°С до 8°С, в защищенном от света месте. Лекарственный препарат не должен быть заморожен.
Дозировка, Введение и Соблюдение инструкций
Пертузумаб и трастузумаб п/к
Квалифицированный специалист, ответственный за выдачу исследуемого лекарственного препарата, подготовит правильную дозу. Этот специалист запишет дату выдачи, номер субъекта и инициалы на этикетке флакона исследуемого лекарственного препарата и в Отчете об Учете Препарата. Этот специалист также запишет партию исследуемого лекарственного препарата или номер партии, полученного каждым субъектом во время исследования.
ЗМД получат однократную дозу пертузумаба в/в, пертузумаба п/к, трастузумаба п/к или смешанных пертузумаба п/к и трастузумаба п/к, вместе (введение смеси). Пациенты будут получать одну дозу пертузумаба и трастузумаба в виде двух инъекций одного агента (совместное введение) или одной инъекции смешанных пертузумаба и трастузумаба, (введение смеси) или пертузумаб, совместно с трастузумабом, в виде одной инъекции FDC.
Здоровым добровольцам и пациентам также может быть назначена медикаментозная подготовка (например, ацетаминофен [парацетамол] и/или прометазин) перед введением пертузумаба и/или трастузумаба п/к по усмотрению исследователя, чтобы снизить риск инфузионной или инъекционной реакции.
Любая передозировка или неправильное введение исследуемого лекарственного препарата должны быть отмечены в eCRF Управления по учету исследуемого лекарственного препарата. Нежелательные явления, связанные с передозировкой или неправильным введением исследуемого препарата, должны регистрироваться в eCRF «Нежелательные Явления».
Введение (пертузумаб в/в)
Здоровые добровольцы, получавшие пертузумаб в/в (Когорта 1 - контроль), получали дозу
420 мг.
Дозу пертузумаба вводили в течение 60 (±10) минут, и здоровых добровольцев наблюдали еще в течение 60 минут. Инфузия должна быть замедлена или прервана, если пациент испытывает симптомы, связанные с инфузией.
Вводимые дозы (пертузумаб п/к и трастузумаб п/к)
Здоровые добровольцы и пациенты, получавшие пертузумаб п/к (Когорты 2-8, А и В), получали дозы от 400 до 1200 мг. Здоровым добровольцам и пациентам, получавшим трастузумаб п/к (Когорты 5-8, А и В), давали дозу 600 мг (см. Таблицу 3).

Инъекции п/к вводили в переднюю область бедра. Пациенты в Когорте А получат две инъекции совместного введения в противоположные бедра, причем вторая инъекция будет проведена сразу после первой.
Соответствующее количество раствора должно быть изъято из флаконов. Обратитесь к фармацевтическому руководству за инструкциями.
Инъекционная игла 27-го калибра вводится с помощью стерильного оборудования в подкожную ткань бедра. Игла должна быть полностью введена, следя за тем, чтобы кончик иглы был глубже дермы, но не так глубоко, как нижележащая мышца. Угол размещения и глубина введения иглы должны быть таковыми, чтобы обеспечить одинаковое размещения в подкожной ткани каждого пациента. Исследуемый лекарственный препарат не следует вводить в родинки, шрамы или синяки. Кожу необходимо сдавить и ввести иглу до того, как кожа вернется в прежнее состояние, и на шприц может быть оказано давление.
Инъекцию следует вводить вручную при скорости потока не более 2 мл/мин, поэтому введение должно занимать приблизительно 2-8 минуты в зависимости от вводимой дозы. Если субъект просит прервать инъекцию, давление на шприц должно быть сначала ослаблено, чтобы облегчить боль. Если боль не уменьшается, то инъекцию следует прекратить и субъекта следует спросить, когда ему удобно возобновить инъекцию.
Расписание Питания, Физической Активности и Процедур
Питание было одинаковым по составу и времени приема во всех когортах. Потребление продуктов и напитков, содержащих кофеин (например, чай, кофе, шоколад и безалкогольные напитки) или алкоголя, не разрешается с Дня -1 по День 2. Курение табака не разрешается во время клинической части исследования.
Легкая амбулаторная активность будет разрешена, при этом уровень активности будет сохраняться максимально одинаковым во все дни в отделении клинических исследований.
СОПУТСТВУЮЩАЯ ТЕРАПИЯ, ЗАПРЕЩЕННАЯ ПИЩА И ДОПОЛНИТЕЛЬНЫЕ ОГРАНИЧЕНИЯ
Сопутствующая терапия включает любые медицинские препараты (например, отпускаемые по рецепту лекарства, лекарства, отпускаемые без рецепта, вакцины, растительные или гомеопатические средства, пищевые добавки), используемые здоровым добровольцем/пациентом в течение 30 дней скрининга исследования. Все такие медицинские препараты должны сообщаться исследователю и записываться в eCRF «Сопутствующие Медицинские Препараты».
Разрешенная Терапия
Для здоровых добровольцев не допускаются никакие сопутствующие медицинские препараты, за исключением медицинских препаратов для лечения нежелательных явлений, если только причина исключения не обсуждена между исследователем и Медицинским наблюдателем и четко не задокументирована.
Для пациентов с РРМЖ во время исследования разрешены следующие средства для лечения:
Приемлемые способы контрацепции должны использоваться, когда пациентка или мужчина-партнер не стерилизованы хирургическим путем или пациентка не соответствует определению исследования в постменопаузе (≥12 месяцев аменореи)
Антагонисты H1 и H2 (например, дифенгидрамин, циметидин)
Сердечно-сосудистые медицинские препараты: ингибиторы ангиотензинпревращающего фермента (АПФ), блокаторы рецепторов ангиотензина, β-блокаторы, блокаторы кальциевых каналов и диуретики (для лечения артериальной гипертонии с целью снижения артериального давления до < 140/90 мм рт. ст.), β-блокаторы, кальций блокаторы и дигоксин (для контроля сердечного ритма) и ингибиторы агрегации тромбоцитов
Анальгетики/противовоспалительные средства (например, парацетамол/ацетаминофен, меперидин, опиоиды)
Краткосрочное использование кортикостероидов для лечения или профилактики аллергических или инфузионных реакций
Противорвотные средства (одобренные профилактические антагонисты серотонина, бензодиазепины, антагонисты дофамина и др.)
Препарат для лечения диареи (например, лоперамид)
Антагонисты рецепторов эстрогена (например, тамоксифен), ингибиторы ароматазы (например, анастразол, экземестан) и агонисты гонадотропин-рилизинг-гормона (например, бусерелин, трипторелин) после операции в соответствии с местной практикой и рекомендациями
Подавление функции яичников (аналог лютеинизирующего гормона, высвобождающего гормон [LHRH])
Бисфосфонаты (для использования в соответствии с утвержденными маркированными показаниями и/или национально признанными руководящими нормами лечения)
По усмотрению исследователя здоровым добровольцам и пациентам также может быть назначено предварительное лечение (например, ацетаминофен [парацетамол] и/или прометазин) перед введением пертузумаба и/или трастузумаба п/к для снижения риска IRR или инъекционных реакций.
Запрещенная Терапия
Использование следующих способов лечения запрещено во время исследования и не менее чем за 10 дней до начала лечения:
• Противораковые терапии, отличные от тех, которые назначаются в данном исследовании или перечислены в разрешенных способах лечения, указанных выше, включая цитотоксическую химиотерапию, лучевую терапию, иммунотерапию и биологическую противораковую терапию.
• Любая целевая терапия, кроме тех, которые используются в этом исследовании
• Любой исследовательский агент, кроме тех, которые используются для этого исследования
• Прием растительных лекарственных средств: Растительные лекарственные средства, прием которых начался до начала исследования и продолжается во время исследования, не допускаются и должны быть указаны в соответствующей eCRF.
• Любой системно-активный, оральный, инъецированный или имплантированный гормональный способ контрацепции, за исключением покрытых прогестероном ВМС, которые были ранее имплантированы
• Эстроген-заместительная терапия (гормон-заместительная терапия)
• Никакие отпускаемые по рецепту лекарства, отпускаемые без рецепта лекарства или растительные лекарственные средства не допускаются в течение по крайней мере 10 дней до введения исследуемой дозы препарата и до конца исследования, если иное не согласовано с врачом-исследователем. Запрещенные Пищевые Продукты
Потребление продуктов и напитков, содержащих кофеин (например, чай, кофе, шоколад и безалкогольные напитки) или алкоголя, не разрешается с Дня -1 по День 2. Дополнительные Ограничения
Питание будет одинаковым по составу и времени приема во всех когортах. Курение табака не разрешается во время клинической части исследования. Легкая амбулаторная активность будет разрешена, при этом уровень активности будет сохраняться максимально одинаковым во все дни в отделении клинических исследований.
ОЦЕНКА ИССЛЕДОВАНИЯ
Часть 1 (Здоровые Мужчины-Добровольцы)
Здоровые добровольцы прибудут в отделение в День -1 для оценки перед введением дозы и останутся на ночь (в течении 3 ночей) в отделении. Здоровые добровольцы могут быть выписаны утром в День 2 по усмотрению исследователя и вернуться в клинику на День 3.
В День 1 здоровым добровольцам введут пертузумаб путем в/в инфузии или п/к инъекции пертузумаба, трастузумаба п/к или пертузумаба и трастузумаба п/к (в смеси) в переднюю область бедра. Места инъекции будут фотографироваться цифровым способом после п/к инъекции, если в месте инъекции будет наблюдаться серьезная нежелательная реакция.
Оценки безопасности и фармакокинетики будут проводиться через регулярные промежутки времени во время исследования в соответствии с графиком исследования. Здоровые добровольцы будут оставаться в отделении до тех пор, пока не завершится 48-часовая оценка фармакокинетики. Они вернутся для оценки ФК и безопасности в указанные дни после этого.
Последующее посещение будет выполнено через 7 месяцев после введения исследуемого препарата. Здоровые добровольцы будут исключены из исследования ответственным врачом после завершения контрольного посещения.
Часть 2 (Пациентки с РРМЖ)
Пациенты прибудут в отделение в День -1 для оценки перед введением дозы. Пациенты вернутся в отделение в День 1 и получат пертузумаб и трастузумаб в виде п/к инъекции в переднюю область бедра. Пациенты в Когорте А получат 2 инъекции в противоположные бедра, причем вторая инъекция будет введена сразу после первой. Места инъекции будут фотографироваться цифровым способом после п/к инъекции, если будет наблюдаться серьезная нежелательная реакция в месте инъекции.
Оценки безопасности и ФK будут проводиться через регулярные промежутки времени во время исследования в соответствии с графиком исследования. Пациенты будут оставаться в отделении до 12 часов после введения дозы. Они вернутся для оценки фармакокинетики и безопасности в указанные дни после этого.
Последующее посещение будет выполнено через 7 месяцев после введения исследуемого препарата. Пациенты будут исключены из исследования ответственным врачом после завершения контрольного посещения.
Контрольное посещение
Для ЗМД или пациентов с РРМЖ с текущими нежелательными явлениями со стороны сердечно-сосудистой системы (независимо от причины) или с нежелательными явлениями, связанными с лечением в рамках исследования, серьезными нежелательными явлениями или явлениями, представляющими особый интерес на День 85, или нежелательными явлениями, серьезными нежелательными явлениями или явлениями, представляющими особый интерес и возникающими между Днем 85 и контрольным посещением, в контрольном посещении будут выполнены все оценки и взяты образцы ФK/АТА.
Для ЗМД без нежелательных явлений со стороны сердечно-сосудистой системы (независимо от причины) или нежелательных явлений, связанных с лечением в рамках исследования, серьезных нежелательных явлений или нежелательных явлений, представляющих особый интерес и продолжающихся на День 85, и если ничего не произошло между Днем 85 и контрольным посещением, требуется только наблюдение за беременностью партнеров-женщин. Это посещение может быть выполнено в телефонном режиме.
Для пациентов с РРМЖ без нежелательных явлений со стороны сердечно-сосудистой системы (независимо от причины) или нежелательных явлений, связанных с лечением в рамках исследования, серьезных нежелательных явлений или нежелательных явлений, представляющих особый интерес и продолжающихся на День 85, и если ничего не происходит между Днем 85 и контрольным посещением, при этом посещении требуется только тест на беременность (для пациентов с детородным потенциалом).
Для пациентов с РРМЖ в постменопаузе (≥12 месяцев аменореи) без нежелательных явлений со стороны сердечно-сосудистой системы (независимо от причины) или нежелательных явлений, связанных с лечением в рамках исследования, серьезных нежелательных явлений и нежелательных явлений, представляющих особый интерес и продолжающихся в День 85, и если ничего не происходит между Днем 85 и контрольным посещением, контрольное посещение может быть выполнено телефонном режиме.
ПАРАМЕТРЫ БЕЗОПАСНОСТИ И НЕЖЕЛАТЕЛЬНЫЕ ЯВЛЕНИЯ
Оценки безопасности будут состоять из наблюдения и регистрации нежелательных явлений, включая серьезные нежелательные явления и нежелательные явления, представляющие особый интерес, проведения лабораторных оценок безопасности, определенных протоколом, измерения показателей жизнедеятельности, определенных протоколом, и проведения других тестов, определенных протоколом, которые считаются критическими для оценки безопасности исследования.
Нежелательные Явления
Согласно Правилам проведения качественных клинических исследований, нежелательным явлением является любое нежелательное медицинское явление у субъекта клинического исследования, которому вводили фармацевтический препарат, независимо от причинно-следственной связи. Следовательно, нежелательным явлением может быть любое из следующего:
• Любое неблагоприятное и непредусмотренное проявление (включая отклоняющиеся от нормы данные лабораторного исследования), симптом или заболевание, временно связанне с использованием лекарственного препарата, независимо от того, считаются ли они связанными с лекарственным препаратом или нет.
• Любое новое заболевание или обострение существующего заболевания (ухудшение в характере, частоте или тяжести известного состояния).
• Рецидив перемежающегося медицинского состояния (например, головная боль), отсутствующего в начале исследования.
• Любое ухудшение лабораторных показателей или других клинических испытаний (например, ЭКГ, рентгенограммы), которые связаны с симптомами или приводят к изменению лечения в рамках исследования или сопутствующего лечения или прекращения приема исследуемого лекарственного препарата.
• Нежелательные явления, связанные с вмешательством, допускаемым протоколом, в том числе те, которые происходят до назначения лечения в рамках исследования (например, скрининг инвазивных процедур, таких как биопсия).
Серьезные Нежелательные Явления (Немедленно Сообщаемые Спонсору)
Серьезное нежелательное явление представляет собой любое нежелательное явление, которое соответствует любому из следующих критериев:
• Является смертельным (то есть нежелательное явление фактически вызывает или приводит к смерти)
• Опасно для жизни (то есть, по мнению исследователя, нежелательное явление подвергает пациента непосредственному риску смерти). Это не включает любое нежелательное явление, если бы оно произошло в более тяжелой форме или если при его продолжающемся проявлении могло привести к смерти.
• Требует или продлевает госпитализацию.
• Приводит к стойкой или значительной утрате трудоспособности или инвалидность (т.е. нежелательное явление приводит к существенному нарушению способности пациента выполнять нормальные жизненные функции.
• Представляет собой врожденную аномалию/врожденный дефект у новорожденного/младенца первого года жизни, рожденного матерью, подвергшейся воздействию исследуемого лекарственного препарата.
• Представляет собой значимое медицинское явление согласно мнению исследователя (например, может поставить под угрозу пациента или может потребоваться медицинское/хирургическое вмешательство для предотвращения одного из результатов, перечисленных выше). Термины «тяжелый» и «серьезный» не являются синонимами. Степень тяжести относится к интенсивности нежелательного явления (например, оценивается как легкое, умеренное или тяжелое, или в соответствии с NCI СТСАЕ v4.03; само явление может иметь относительно незначительное медицинское значение (например, сильная головная боль без каких-либо дополнительных проявлений).

Нежелательные Явления, Представляющие Особый Интерес (Немедленно Сообщаемые Спонсору)
Нежелательные явления, представляющие особый интерес, должны сообщаться Спонсору немедленно (т.е. не позднее чем через 24 часа после изучения явления). Нежелательные явления, представляющие особый интерес для данного исследования, включают следующее:
• Случаи потенциального лекарственного повреждения печени, которые включают повышенную АЛТ или ACT в сочетании с повышенным билирубином или клинической желтухой, согласно определению закона Хая.
• Предполагаемая передача инфекционного агента исследуемым лекарственным средством, согласно определению: Любой организм, вирус или инфекционная частица (например, прионный белок, передающий трансмиссивную губчатообразную энцефалопатию), патогенные или непатогенные, считаются инфекционным агентом. Передача инфекционного агента может быть заподозрена по клиническим симптомам или лабораторным данным, которые указывают на инфекцию у пациента, подвергшегося воздействию лекарственного препарата. Этот термин применяется только в случае,если есть подозрения на загрязнение исследуемого лекарственного препарата.
• Бессимптомное снижение ФВЛЖ, требующее лечения. Замечания: В общем, бессимптомное снижение ФВЛЖ не следует сообщать как нежелательные явления, так как данные ФВЛЖ собираются отдельно по eCRF. Исключения из этого правила следующие:
• Бессимптомное снижение ФВЛЖ до значения на 10 процентов ниже исходного уровня или ниже и <50% должно быть зарегистрировано как нежелательное явление.
• Об бессимптомном снижении ФВЛЖ, которое требует лечения или которое приводит к прекращению лечения в рамках исследования, следует сообщать в ускоренном порядке с использованием eCRF поля Нежелательные Явления и классифицировать явление как несерьезное явление, представляющее особый интерес, о котором немедленно сообщается.
Отдельные Нежелательные Явления
Сердечная Недостаточность
Симптоматическую LVSD (называемую сердечной недостаточностью) следует сообщать как серьезное нежелательное явление. Если поставлен диагноз сердечная недостаточность, то о ней следует сообщать как о таковой, а не как об отдельных признаках и симптомах сердечной недостаточности. Признаки и симптомы должны быть записаны в eCRF. Консультация у кардиолога рекомендуется для пациентов, у которых развивается симптоматическая LVSD (сердечная недостаточность). Сердечная недостаточность должна оцениваться в соответствии с NCI СТСАЕ v4.03 (Степень 2, 3, 4 или 5), а также в соответствии с классификацией NYHA (класс II, III и IV). Систолическая дисфункция левого желудочка не должна использоваться для описания симптоматической дисфункции, согласно NCI СТСАЕ v4.03.
О сердечной недостаточности, возникающей во время исследования и до 5 лет после последнего включения пациента, следует сообщать независимо от причинно-следственной связи и прослеживать до тех пор, пока не произойдет одно из следующих событий: нормализация или улучшение исходного состояния, дальнейшее улучшение не ожидается или смерть.
Бессимптомное Снижение Фракции Выброса Левого Желудочка
Бессимптомное снижение ФВЛЖ не следует сообщать как нежелательное явление, поскольку данные ФВЛЖ собираются отдельно по eCRF. Исключения из этого правила следующие:
• Бессимптомное снижение ФВЛЖ ≥ 10 процентов от исходного уровня до ФВЛЖ < 50% должно быть зарегистрировано как нежелательное явление с выводом уменьшенная фракция выброса, согласно NCI СТСАЕ v4.03. Кроме того, комментарий в поле комментариев нежелательных явлений должен подтвердить, что явление было бессимптомным.
• Также следует сообщать о бессимптомном снижении ФВЛЖ, требующем лечения или ведущем к прекращению применения пертузумаба и трастузумаба. Это нежелательное явление также следует регистрировать как несерьезное явление, представляющее особый интерес в форме серьезного нежелательного явления, и в поле для комментариев нежелательных явлений следует добавить комментарий, подтверждающий, что явление было бессимптомным.
В Таблице 5 показана классификация Нью-Йоркской Ассоциации Кардиологов и Систолическая Дисфункция Левого Желудочка согласно Общей Терминологии Критериев Нежелательных Явлений Национального Института Рака, Версии 4.03.

Weatherall DJ, Lendingham JGG, editors. Oxford Rextbook of Medicine. Third Edition. New York: Oxford University Press, 1996.
Таблица 6 суммирует соглашения об отчетности по LVSD и сердечной недостаточности:

Шкала оценки серьезности нежелательных явлений по NCI СТСАЕ (v4.03) будет использоваться для оценки серьезности нежелательного явления. См. Таблицу 4 выше для оценки тяжести нежелательных явлений, которые конкретно не перечислены в NCI СТСАЕ.
Оценка Достоверности Причинно-Следственной Связи Нежелательных Явлений
Исследователи должны использовать свои собственные знания о пациенте, обстоятельствах, в которых происходило явление, и оценку любых возможных альтернативных причин, чтобы определить, относится ли нежелательное явление к исследуемому лекарственному препарату, указав соответственно «да» или «нет». Следующее руководство должно быть принято во внимание:
 Временная связь наступления явления с началом исследования лекарственного препарата
Временная связь наступления явления с началом исследования лекарственного препарата
 Развитие явления, особенно учитывая результаты снижения дозы, отмены исследуемого лекарственного препарата или повторного введения исследуемого лекарственного препарата (в зависимости от обстоятель0 ств)\
Развитие явления, особенно учитывая результаты снижения дозы, отмены исследуемого лекарственного препарата или повторного введения исследуемого лекарственного препарата (в зависимости от обстоятель0 ств)\
 Известная связь явления с исследуемым лекарственным препаратом или с аналогичным лечением
Известная связь явления с исследуемым лекарственным препаратом или с аналогичным лечением
 Известная связь явления с изучаемой болезнью
Известная связь явления с изучаемой болезнью
 Наличие факторов риска у пациента или использование сопутствующих лекарственных средств, которые, как известно, увеличивают возникновение явления
Наличие факторов риска у пациента или использование сопутствующих лекарственных средств, которые, как известно, увеличивают возникновение явления
 Наличие факторов, не связанных с лечением, которые, как известно, связаны с возникновением явления
Наличие факторов, не связанных с лечением, которые, как известно, связаны с возникновением явления
Для пациентов, получающих комбинированную терапию, причинно-следственная связь будет оцениваться индивидуально для каждой назначенной протоколом терапии.
Инфузионные Реакции, Инъекционные Реакции и Локальные Реакции в Месте Инъекции
Нежелательные явления, которые происходят в течение или в пределах 24 часов после введения исследуемого лекарственного препарата и которые, как считается, связаны с инфузией или инъекцией исследуемого лекарственного препарата, должны регистрироваться как диагноз (например, «инфузионная реакция», «инъекционная реакция», «реакция в месте инъекции») в поле eCRF Нежелательные Явления. Если возможно, то необходимо избегать двусмысленных терминов, таких как «системная реакция».
Связанные признаки и симптомы должны быть записаны в специальном поле eCRF Инфузионная Реакция, eCRF Инъекционная Реакция или eCRF Реакция в Месте Инъекции. Если пациент испытывает как локальную, так и системную реакцию на одну и ту же дозу исследуемого лекарственного препарата, каждую реакцию следует регистрировать отдельно в eCRF Побочные Явления, а признаки и симптомы также записывать отдельно в выделенном поле eCRF Инфузионная Реакция, eCRF Инъекционная Реакция или eCRF Реакция в Месте Инъекции.
Нежелательные Явления, которые являются Вторичными по отношению к Другим Явлениям
Как правило, нежелательные явления, которые являются вторичными по отношению к другим явлениям (например, каскадные явления или клинические осложнения), следует идентифицировать по их основной причине, за исключением тяжелых или серьезных вторичных явлений. Второстепенное нежелательное явление с медицинской точки зрения, которое по времени отделено от исходного явления, должно быть зарегистрировано как независимое явление в поле eCRF Нежелательное Явление. Например:
 Если рвота приводит к легкому обезвоживанию без дополнительного лечения у здорового взрослого человека, в eCRF следует сообщать только о рвоте
Если рвота приводит к легкому обезвоживанию без дополнительного лечения у здорового взрослого человека, в eCRF следует сообщать только о рвоте
 Если рвота приводит к сильному обезвоживанию, оба явления должны быть зарегистрированы отдельно в eCRF
Если рвота приводит к сильному обезвоживанию, оба явления должны быть зарегистрированы отдельно в eCRF
 Если тяжелое желудочно-кишечное кровотечение приводит к почечной недостаточности, оба явления должны быть зарегистрированы отдельно в eCRF
Если тяжелое желудочно-кишечное кровотечение приводит к почечной недостаточности, оба явления должны быть зарегистрированы отдельно в eCRF
 Если головокружение приводит к падению и последующему перелому, все три явления должны быть зарегистрированы отдельно в eCRF.
Если головокружение приводит к падению и последующему перелому, все три явления должны быть зарегистрированы отдельно в eCRF.
 Если нейтропения сопровождается инфекцией, оба явления должны сообщаться отдельно в eCRF.
Если нейтропения сопровождается инфекцией, оба явления должны сообщаться отдельно в eCRF.
Все нежелательные явления должны регистрироваться отдельно в поле eCRF «Нежелательное Явление», если неясно, связаны ли эти явления.
Постоянно Рецидивирующие Нежелательные Явления
Постоянное нежелательное явление представляет собой явление, которое непрерывно распространяется без нормализации между временными точками состояния пациента. Такие явления должны быть записаны только один раз в поле eCRF Нежелательное Явление. Первоначальная тяжесть (интенсивность или степень) явления будет записана во время первого сообщения о явлении. Если постоянное нежелательное явление становится более серьезным, наиболее серьезную степень тяжести следует также зарегистрировать в поле eCRF Нежелательное Явление. Если явление становится серьезным, об этом следует немедленно сообщить Спонсору (т.е. не позднее чем через 24 часа после получения информации о том, что явление стало серьезным). Поле eCRF Нежелательное Явление следует обновить, изменив явление с «несерьезного» на «серьезное», указав дату, когда явление стало серьезным, и заполнив все поля данных, связанные с серьезными нежелательными явлениями.
Рецидивирующим нежелательным явлением является то, которое находится в стадии ремиссии между контрольными точками мониторинга состояния пациента и впоследствии повторяется. Каждое повторение нежелательного явления должно регистрироваться как отдельное явление в поле eCRF Нежелательное Явление.
Отклоняющиеся от нормы Лабораторные Значения
Не каждое лабораторное отклонение квалифицируется как нежелательное явление. Результаты лабораторных испытаний должны быть представлены как нежелательное явление, если оно соответствует одному из следующих критериев:
 Сопровождается клиническими симптомами
Сопровождается клиническими симптомами
 Приводит к изменению лечения в рамках исследования (например, изменение дозировки, прерывание лечения или прекращение лечения)
Приводит к изменению лечения в рамках исследования (например, изменение дозировки, прерывание лечения или прекращение лечения)
 Результатом является медицинское вмешательство (например, добавление калия при гипокалиемии) или изменение сопутствующей терапии
Результатом является медицинское вмешательство (например, добавление калия при гипокалиемии) или изменение сопутствующей терапии
 Является клинически значимым по мнению исследователя. Замечания: При исследовании онкологии определенные отклоняющиеся от нормы значения могут не квалифицироваться как нежелательные явления. Исследователь обязан проверить все лабораторные данные. Медицинское и научное суждение должно приниматься во внимание при принятии решения о том, следует ли классифицировать отдельное отклонение лабораторных показателей от нормы как нежелательное явление.
Является клинически значимым по мнению исследователя. Замечания: При исследовании онкологии определенные отклоняющиеся от нормы значения могут не квалифицироваться как нежелательные явления. Исследователь обязан проверить все лабораторные данные. Медицинское и научное суждение должно приниматься во внимание при принятии решения о том, следует ли классифицировать отдельное отклонение лабораторных показателей от нормы как нежелательное явление.
Если клинически значимое лабораторное отклонение является признаком заболевания или синдрома (например, ЩФ и билирубин 5 х ВГН, связанные с холестазом), только диагноз (т.е. холестаз) должен быть записан в поле eCRF Нежелательное Явление.
Если клинически значимое лабораторное отклонение является признаком заболевания или синдрома (например, ЩФ и билирубин 5 х ВГН, связанные с холестазом), только диагноз (т.е. холестаз) должен быть записан в поле eCRF Нежелательное Явление. Если лабораторное отклонение может быть охарактеризовано точным клиническим термином в соответствии со стандартными определениями, клинический термин должен быть зарегистрирован как нежелательное явление. Например, повышенный уровень калия в сыворотке крови на 7,0 мэкв/л следует регистрировать как «гиперкалиемия».
Наблюдения за подобным клинически значимым лабораторным отклонением от посещения к посещению должны регистрироваться только один раз в поле eCRF Нежелательное Явление.
Отклоняющиеся Показатели Жизненно Важных Функций
Не каждое нарушение жизненно важных функций квалифицируется как нежелательное явление. Результат измерения жизненно важных функций должен быть отмечен как нежелательное явление, если он соответствует одному из следующих критериев:
 Сопровождается клиническими симптомами
Сопровождается клиническими симптомами
 Приводит к изменению лечения в рамках исследования (например, изменение дозировки, прерывание лечения или прекращение лечения)
Приводит к изменению лечения в рамках исследования (например, изменение дозировки, прерывание лечения или прекращение лечения)
 Приводит к медицинскому вмешательству или изменению сопутствующей терапии
Приводит к медицинскому вмешательству или изменению сопутствующей терапии
 Является клинически значимым по мнению исследователя Исследователь обязан проверить все данные по показателям жизненно важных функций. Медицинское и научное суждение должно приниматься во внимание при принятии решения о том, следует ли классифицировать отдельное нарушение жизненно важных функций как нежелательное явление.
Является клинически значимым по мнению исследователя Исследователь обязан проверить все данные по показателям жизненно важных функций. Медицинское и научное суждение должно приниматься во внимание при принятии решения о том, следует ли классифицировать отдельное нарушение жизненно важных функций как нежелательное явление.
Если клинически значимое нарушение жизненно важных функций является признаком заболевания или синдрома (например, высокого АД), должен быть отмечен только диагноз (например, гипертония) в поле eCRF Нежелательное Явление.
Наблюдения за подобным клинически значимым отклонением жизненно важных функций от посещения к посещению должны регистрироваться только один раз в поле eCRF нежелательное Явление.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЙ ФК И ВЫБОР ДОЗЫ
Часть 1 Анализы п/к ФК и Выбор Дозы
Выбор доз пертузумаба п/к в Части 1 (Когорты 2, 3 и 4) был основан на модели попФК пертузумаба в/в с включенными значениями параметров ФК трастузумаба п/к. Для большей точности в/в попФК из исторических оценок параметров модели также использовался в анализах выбора дозы п/к. После оценки фиксированных параметров ФК (то есть Ctrough, AUCO-inf, максимальной концентрации в сыворотке [Cmax], времени максимальной концентрации в сыворотке [Tmax]), дозу (дозы) пертузумаба п/к (поддерживающую) выбирали для Части 2. Эта доза п/к была рассчитана, чтобы обеспечить воздействие пертузумаба, аналогичное воздействию пертузумаба в/в в дозе 420 мг.Точно так же, основываясь на параметрах ФК, была рассчитана одна доза пертузумаба п/к (нагрузочная), чтобы обеспечить воздействие пертузумаба, аналогичное воздействию пертузумаба в/в в дозе 840 мг. Модель попФК пертузумаба в/в была обновлена с помощью параметров ФК пертузумаба п/к с использованием данных Части 1 и использовалась для правильной идентификации поддерживающих и нагрузочных п/к доз.
На Фиг. 8 изображен обзор исследования, включая дозы антител, объемы инъекций, а также концентрации и количества rHuPH20 (Halozyme) для когорт 1-8.
На Фиг. 9 изображены нормализованные концентрации (мкг/мл) пертузумаба, вводимого подкожно, с трастузумабом и без него, в виде функции времени (дни). Данные показывают, что при пероральном введении ФК-взаимодействие между пертузумабом п/к и трастузумабом п/к отсутствует.Не было выявлено различий между ФК трастузумаба п/к, вводимого в виде монотерапии, или с пертузумабом п/к.
На Фиг. 10 изображены нормированные концентрации (мкг/мл) пертузумаба в виде функции времени (дни). Не было никаких существенных различий в ФК пертузумаба или ФК трастузумаба (не показаны) при введении с 2000 Ед/мл или 667 Ед/мл rHuPH20.
На Фиг. 11 изображены оценки параметров с использованием моделей пертузумаба и исторической популяционной ФК (попФК) в/в при разных концентрациях пертузумаба вводимых в/в или п/к, с трастузумабом и без него.
Характеристика Фармакокинетики Пертузумаба - Часть 1
Профили средней концентрации пертузумаба в зависимости от времени для Когорт 1-4 и 6-8 изображены на Фиг. 24. Концентрации пертузумаба после внутривенного введения дозы 420 мг соответствовали двухфазной схеме с четкой фазой распределения и элиминации. Подкожно вводимый пертузумаб приводил к достижению максимальной концентрации (Tmax) между 4-7 днями и дозозависимому увеличению воздействия. Наблюдалась изменчивость в некоторых Когортах п/к 1200 мг, вероятно, из-за небольшого размера выборки.
Средние геометрические нормализованные по дозе концентрации пертузумаба, при введении с трастузумабом и без него сравнивали для оценки потенциального влияния трастузумаба на ФК пертузумаба. Как изображено на Фиг. 22, не было очевидного влияния трастузумаба на ФК пертузумаба, когда два антитела доставлялись п/в в виде смеси. Это согласуется с данными ФК из предыдущих исследований, где два антитела вводились последовательно.
Средние геометрические концентрации пертузумаба при введении с 667 Ед/мл или 2000 Ед/мл rHuPH20 сравнивали для оценки потенциального влияния фермента, усиливающего всасывание, rhuPH20 на ФК пертузумаба. Не было очевидного влияния снижения концентрации rHuPH20 с 2000 Ед/мл до 667 Ед/мл на ФК пертузумаба или трастузумаба, как изображено на Фиг. 20 и 21, соответственно.
Характеристика ФК пертузумаба показала, что трастузумаб (в результате одновременного введения трастузумаба п/к) не оказывает видимого влияния на пертузумаб. ФК пертузумаба оказалась в основном без изменений при снижении концентрации rHuPH20 с 2000 Ед/мл до 667 Ед/мл, однако, поскольку только небольшое количество субъектов подверглось воздействию каждой концентрации (n=6 на когорту), было трудно исключить возможность различий, в частности в конечной фазе и Ctrough. Эти наблюдения подтверждают дальнейшие исследования по определению дозы пертузумаба п/к, которая является не менее эффективной, чем доза пертузумаба в/в 420 мг.
Разработка Модели Популяционной ФК для Выбора Дозы Пертузумаба п/к
Нагрузочная доза пертузумаба п/к 1200 была выбрана на основе пропорциональности дозы пертузумаба и линейной фармакокинетики, как было определено с помощью разработанной модели попФК. Расчеты на основе модели подтвердили сопоставимые воздействия пертузумаба 1200 п/к и 840 мг в/в. Наблюдаемые воздействия пертузумаба в когортах п/к 1200 мг из Части 1 дополнительно подтвердили выбор нагрузочной дозы по сравнению с воздействиями в/в, наблюдаемыми в исторических исследованиях.
Характеристика Фармакокинетики Пертузумаба - Часть 2
Учитывая отсутствие явного фармакокинетического (ФК) лекарственного взаимодействия (DDI) между пертузумабом и трастузумабом при введении смеси в Части 1, и то, что техническая разработка препарата с комбинацией фиксированных доз (FDC) была возможна, Когорта А (совместное введение 600 мг пертузумаба с 600 мг трастузумаба) не была включена в Часть 2. В Когорте В исследовалась инъекция смеси пертузумаба п/к и трастузумаба п/к с 1,000 Ед/мл rHuPH20, чтобы подтвердить дозу пертузумаба, выбранную в Части 1 исследования. Смешанный материал в Когорте В является заменителем для FDC (описано в Примере 2), который будет испытан в Когорте С.
Некомпартментный и популяционный ФК анализы данных ФК в Части 2 были проведены, чтобы:
подтвердить отсутствие ФК лекарственного взаимодействия (DDI) между пертузумабом
и трастузумабом, когда и тот и другой вводили в виде смеси п/к,
изучить влияние 1,000 Ед/мл rHuPH20 на ФК пертузумаба и трастузумаба, и подтвердить, что поддерживающая доза пертузумаба п/к в 600 мг является не менее эффективной, чем доза пертузумаба в/в 420 мг (вводимая ЗМД) на основании равновесных Ctrough у 20 пациентов с РРМЖ.
Средние геометрические концентрации пертузумаба после введение дозы смеси пертузумаба п/к 500 мг, трастузумаба п/к 600 мг и rHuPH20 1,000 Ед/мл у 20 пациентов с РРМЖ в Части 2 Когорты В сравнивали со средними геометрическими концентрациями пертузумаба после дозы пертузумаба в/в 420 мг у 6 ЗМД в Части 1, Когорте 1. Как изображено на Фиг. 22, воздействия пертузумаба (Ctrough и AUC) аналогичны между 600 мг п/к (пациенты с РРМЖ) и 420 мг в/в (ЗМД).
Средние геометрические нормализированные по дозе концентрации пертузумаба после введения дозы смеси пертузумаба п/к 600 мг, трастузумаба п/к 600 мг и rHuPH20 1,000 Ед/мл для 20 пациентов с РРМЖ в Части 2 Когорте В также сравнивали со средними геометрическими концентрациями пертузумаба после дозы пертузумаба п/к 600 мг и rHuPH20 2,000 Ед/мл у 6 ЗМД в Части 1, Когорте 3. Как изображено на Фиг. 23, профили ФК пертузумаба очень похожи между пациентами с РРМЖ (Часть 2, Когорта В) и ЗМД после введения дозы 600 мг партузумаба п/к (Часть 1, Когорта 3).
Сравнение средних геометрических нормализированных по дозе концентраций пертузумаба и трастузумаба при введении с 667 Ед/мл, 1,000 Ед/мл или 2000 Ед/мл rHuPH20 может оценить потенциальное влияние фермента, усиливающего всасывание rHuPH20, на ФК пертузумаба и трастузумаба. Как изображено на Фиг. 24, не было очевидного влияния снижения концентрации rHuPH20 с 2,000 Ед/мл до 1,000 Ед/мл или 667 Ед/мл на ФК пертузумаба. Сравнение ФК данных пертузумаба от пациентов с РРМЖ и ЗМД дополнительно подтверждает отсутствие взаимодействия между пертузумабом и трастузумабом, когда пертузумаб и трастузумаб вводят подкожно в виде смеси.
Как изображено на Фиг. 25, не было очевидных различий в ФК трастузумаба при введении с 667 Ед/мл, 2,000 Ед/мл или 2,000 Ед/мл rHuPH20. Характеристика ФК пертузумаба в Части 2 Когорты В подтвердила результаты исследования в Части 1. В Части 2, Когорты В было обнаружено, что трастузумаб (получаемый в результате единовременного введения трастузумаба п/к) не оказывал видимого влияния на ФК пертузумаба, и ФК пертузумаба и трастузумаба аналогичны при концентрациях rhuPH20 от 2,000 Ед/мл до 1,000 Ед/мл.
Наблюдаемые данные ФК, модельные расчеты с использованием модели попФК, обновленной дополнительными данными ФК, собранными в Части 2 Когорте В, и результирующие вероятности в Части 2 Когорте В были практически идентичны данным, полученным в Части 1. Эти данные были дополнительно подтверждены после того, как была построена вторая модель попФК с использованием данных п/к из Части 1 и 2 Фазы I текущего исследования и данных в/в из исторической модели попФК пертузумаба в/в (Garg et al., Cancer Chemother Pharmacol. 2014; 74:819-829). Добавление надежного набора данных пертузумаба в/в у пациентов в сочетании с п/к данными у ЗМД подтвердило оценки и расчеты параметров ФК и подтвердило выбор нагрузочной и поддерживающей дозы пертузумаба в 1200 мг и 600 мг соответственно.
Фармакокинетика rHuPH20
Концентрации rHuPH20 в плазме измеряют перед введением и через 0,5 и 24 часа после введения для пациентов в Когортах 2-8 Части 1 и Когорте В Части 2. Для измерения концентраций rHuPH20 в плазме использовали утвержденный «сэндвич» метод иммунологического анализа с использованием показаний электролюминесценции (ECL). Минимальная количественно определяемая концентрация составляла 0,061444 нг/мл.
Концентрации rHuPH20 в плазме были ниже предела количественного определения для всех точек времени отбора проб, что указывает на отсутствие количественного системного воздействия фермента при дозах rHuPH20, используемых в данном исследовании.
Выводы
ФК подкожно вводимого пертузумаба соответствовала ФК подкожно вводимого трастузумаба. Испытанные низкие и более высокие количества/концентрации rHuPH20 не продемонстрировали влияния на ФК подкожно вводимого пертузумаба. Таким образом, обе из испытанных концентраций rHuPH20 (667 Ед/мл и 1,000 Ед/мл) подходят для использования в способах, описанных в данном документе.
При использовании rHUPH20 в концентрации 2,000, 1,000 или 667 Ед/мл наблюдались аналогичные ФК пертузумаба и трастузумаба. Профили безопасности были сопоставимы в смешанных когортах, получающих 1,000, 1,000 или 667 Ед/мл rHuPH20. Однако, поскольку различные концентрации rHuPH20 были оценены в двух группах популяций (ЗМД и пациенты с РРМЖ), в каждой из которых было небольшое количество субъектов, потенциальное влияние более низкой концентрации (например, 1000 Ед/мл) на ФК пертузумаба и/или безопасность в этих различных категориях пациентов нельзя исключать, и рекомендуемая концентрация rHuPH20 в FDC (2000 Ед/мл) была определена по совокупности других доступных клинических испытаний.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ БЕЗОПАСНОСТИ
Часть 1 Данные по Безопасности
Демографическое и возрастное распределение исследуемой популяции изображено на Фиг. 12.
На Фиг. 13 представлен обзор Нежелательных Явлений в Части 1 исследования. Общее количество нежелательных явлений составило 145, и % нежелательных явлений Степени 1, Степени 2 и ≥ Степени 3 для различных когорт указаны в скобках. Наблюдаемое одно НЯ Степени 3 в виде диареи на 32-й день представляло собой несвязанное сопутствующее заболевание, возможную вирусную инфекцию, оцениваемую как сопутствующая инфекция верхних дыхательных путей (ОРВИ). Не было выявлено Серьезного Нежелательного Явления (СНЯ), Нежелательного Явления Особого Интереса (НЯОИ) или Нежелательного Явления (НЯ), приводящего к прекращению участия в исследовании, или НЯ, приводящего к смерти. На момент оценки некоторые НЯ продолжались, поэтому окончательная оценка Степени Тяжести не может быть определена.
На Фиг. 14 представлен обзор Нежелательных Явлений в Части 1 исследования, с перечислением количества субъектов для каждой когорты и нежелательного явления.
Фиг. 15 представляет собой таблицу наиболее распространенных Нежелательных Явлений (все степени) с общей частотой ≥ 5% у субъектов, проходивших исследование в Когорте 18
На Фиг. 16 представлена токсичность, связанная с EGF, т.е. диарея, воспаление слизистой оболочки полости рта и сыпь, связанная с EGFR.
На Фиг. 17 представлены инъекционные реакции и реакции в месте инъекции, включая системные и местные реакции, для Когорт с 1 по 8. В Когорте 7 была выявлена одна реакция в месте инъекции (1200 мг пертузумаба (П) п/к + 600 мг трастузумаба (Г) п/к с rHuPH20). Симптомами были дискомфорт, боль, уплотнение и онемение в месте инъекции. Системные реакции, связанные с инъекцией, включали лихорадку, озноб, тошноту, ригидность, головокружение, чувствительность кожи, светочувствительность, колебания температуры и головную боль.
На Фиг. 18 представлены результаты оценок ФВЛЖ - Эхокардиографии в когортах 1-8. В когорте 3 (600 мг пертузумаба (П) п/к) у одного здорового мужчины добровольца (ЗМД) обнаружено падение >10% с 67% в начале исследования до 56% в День 22. Уровень Фракции Выброса (ФВ) в день 85 составил 60%. Кардиолог подтвердил отсутствие признаков кардиотоксичности. Предполагалось, что падение было вызвано изменчивостью способа визуализации.
Общие Нежелательные Явления
В части 1 (Когорты 1-8) было зарегистрировано в общей сложности 148 НЯ у 44 из 48 (91,7%) здоровых мужчин-добровольцев (ЗМД). Сообщалось, что большинство НЯ были низкой интенсивности (Степень 1 или 2). Наиболее часто наблюдаемыми SOC были инфекции и инвазии, у 22 (45,8%) ЗМД, испытывающих в общей сложности 33 НЯ в этой категории, из которых большинство явлений рассматривались исследователем как не связанные с исследуемым препаратом. Наиболее часто наблюдаемыми НЯ (согласно РТ) в разных когортах были: инфекция верхних дыхательных путей (у 13 ЗМД [27,1%]), головная боль (у 9 ЗМД [18,8%]), лекарственный дерматит (у 9 ЗМД [18,8%]) и диарея (у 9 ЗМД [18,8%]). Среди этих распространенных НЯ, НЯ, связанные с исследуемым лекарственным препаратом (оцененные исследователем) были зарегистрированы у 4 (8,3%), 8 (18,8%) и 7 (14,6%) ЗМД, соответственно. Все НЯ в Части 1 были устранены к концу Части 1.
В Когорте 1 (контроль), в которой ЗМД получали однократную в/в инъекцию 420 мг пертузумаба, наиболее часто наблюдаемые НЯ (согласно сведений пациента) включали: диарею (у 3 из 6 ЗМД [50%]), инфекцию верхних дыхательных путей (у 2 ЗМД [33,3%]) и ангулярный стоматит (у 2 ЗМД [33,3%]).
В Когортах 2-4, в которых ЗМД получали однократную п/к инъекцию пертузумаба в дозах 400, 600 и 1200 мг соответственно, наиболее часто наблюдаемыми НЯ (согласно сведений пациента) были инфекция верхних дыхательных путей, сыпь и диарея, каждое из которых возникало у 4 из 18 (22,2%) ЗМД.
В Когорте 5 (контроль), в которой ЗМД получали однократную п/к инъекцию трастузумаба в дозе 600 мг, наиболее часто наблюдаемыми НЯ (согласно сведений пациента) была боль в конечностях (на ягодицах и в верхней части бедер, не в месте инъекции) (у 2 из 6 HMVs [33,3%]). Все другие НЯ были зарегистрированы только у 1 ЗМД.
В Когортах 6-8, в которых ЗМД получали п/к инъекцию 600 мг трастузумаба, смешанного с пертузумабом, в дозе 400 мг, 1200 мг (с 2,000 Ед/мл rHuPH20) и 1,200 мг (с 667 Ед/мл rHuPH20), соответственно, частота возникновения НЯ была сходна с частотой возникновения у ЗМД, получавших только пертузумаб (Когорты 2-4). Наиболее часто наблюдаемыми НЯ были инфекция верхних дыхательных путей и лекарственный дерматит, каждое из которых встречалось у 7 из 18 (38,9%) ЗМД. Другими распространенными НЯ, о которых сообщалось по крайней мере в 20% случаев ЗМД, были ангулярный стоматит (4 ЗМД [22,2%]) и головная боль (4 ЗМД [22,2%]).
Частота всех НЯ была одинаковой у ЗМД, получавшими концентрацию rHuPH20 2,000 Ед/мл (Когорта 7), и теми, кто получал концентрацию rHuPH20 667 Ед/мл (Когорта 8) как часть инъекции смеси пертузумаба и трастузумаба.
Касательно дополнительных целей проверки безопасности, то:
 Не было зарегистрировано никаких значительных общих изменений артериального давления (АД), частоты сердечных сокращений (ЧСС) или интервалов между ударами (интервал RR)
Не было зарегистрировано никаких значительных общих изменений артериального давления (АД), частоты сердечных сокращений (ЧСС) или интервалов между ударами (интервал RR)
 У четырех субъектов было повышение температуры после введения дозы в День 1, связанное с реакцией на инъекцию
У четырех субъектов было повышение температуры после введения дозы в День 1, связанное с реакцией на инъекцию
 Не зарегистрировано клинически значимых изменений ЭКГ
Не зарегистрировано клинически значимых изменений ЭКГ
Относительно лабораторных изменений:
 Значительных лабораторных отклонений НЯ небыло зарегистрировано
Значительных лабораторных отклонений НЯ небыло зарегистрировано
 У одного субъекта отмечалась гиперурикемия Степени 4 (День 22), которая была подтверждена исследовательским центром как клинически не значимое явление и, вероятно, связанное с физической нагрузкой, уровень нормализовался на День 85.
У одного субъекта отмечалась гиперурикемия Степени 4 (День 22), которая была подтверждена исследовательским центром как клинически не значимое явление и, вероятно, связанное с физической нагрузкой, уровень нормализовался на День 85.
 У четырех субъектов отмечалась гиперурикемия Степени 3, которая у всех была подтверждена исследовательским центром как не являющеяся клинически значимым событием и относилась к Степени 1.
У четырех субъектов отмечалась гиперурикемия Степени 3, которая у всех была подтверждена исследовательским центром как не являющеяся клинически значимым событием и относилась к Степени 1.
Часть 2 Данные по исследованию Безопасности
Во второй части все 20 [1005] пациенток с РРМЖ получили п/к инъекцию смеси 600 мг пертузумаба и 600 мг трастузумаба (с 1,000 Ед/мл rHuPH20). Все пациенты испытывали, по крайней мере, одно НЯ, в общей сложности зарегистрировано 102 НЯ к моменту прекращения клинического испытания. Сообщалось, что большинство НЯ были низкой интенсивности (Степень 1 или 2).
SOC, в которых встречались наиболее распространенные НЯ (зарегистрированные у>50% пациентов), включали следующее:
Расстройства нервной системы (у 14 пациентов [70%])
Желудочно-кишечные расстройства (у 10 [50%]), НЯ связанные с исследуемым лекарственным препаратом (у 10 [50%])
Болезни опорно-двигательного аппарата и заболевания соединительной ткани (у 10 [50%])
Наиболее часто наблюдаемыми НЯ (согласно показаниям пациентам), по меньшей мере, у 20% пациентов, были: головная боль (у 13 пациентов [65%]), миалгия (у 7 пациентов [35%]), диарея (у 6 пациентов [30%]), реакция в месте инъекции (у 6 пациентов [30%]) и тошнота (у 4 пациента [20%]). Среди этих распространенных НЯ, НЯ связанные с исследуемым лекарственным препаратом (оцененные исследователем) были зарегистрированы у 9 (45%), 6 (30%), 4 (20%), 6 (30%) и 1 (5%) пациентов, соответственно.
И когорта В (пациенты с РРМЖ) Части 2 и Когорты 6-8 (ЗМД) Части 1 получали инъекцию смеси пертузумаба и трастузумаба, а типы НЯ, которые регистрировались (согласно показаниям пациентов) у пациентов с РРМЖ, получавших пертузумаб п/к и трастузумаб п/к соответствуют известным рискам, связанным с комбинированной терапией. Реакция в месте инъекции, во всех случаях Степени 1 или 2, происходила с большей частотой у пациентов с РРМЖ по сравнению с ЗМД (у 6 [30%] в сравнении с 1 [5,6%]).
Выводы
В Части 1 все нежелательные явления (НЯ) в когортах с подкожным введением доз относились к Степени 1 или Степени 2.
Не было обнаружено Серьезных Нежелательных Явлений (СНЯ), Нежелательных Явлений Особого Интереса (НЯОИ), или НЯ>Степени 3, приводящих к прекращению исследования, или фатальных явлений.
Не наблюдалось никаких значимых кардиологических явлений.
В большинстве когорт с введение П п/к и П+Г п/к было онаружено более высокое количество НЯ по сравнению с контрольной когортой (П в/в, Г п/к), однако не наблюдалось никакой закономерности с увеличением дозы или добавлением трастузумаба (Г).
Наиболее распространенные НЯ (встречающиеся у>5% субъектов) представляли собой инфекции верхних дыхательных путей, диарею, головную боль и лекарственный дерматит.Не было различий между Когортами 7 и 8 (с более низкой концентрацией rHuPH20).
Наблюдались четыре реакции на инъекцию (1 в Г п/к) и одна реакцию в месте инъекции (Когорта 7). Все реакции относились к Степени 1/2 и были сопоставимы с показателями при подкожном введении трастузумаба (Г п/к).
В заключение следует отметить, что показатели безопасности подкожно вводимого пертузумаба (П) и результаты в целом соответствовали известным показателям безопасности при внутривенном введении пертузумаба и подкожном введении трастузумаба. Соответственно, было безопасно продолжить исследование и перейти к Части 2.
Пертузумаб п/к, вводимый в виде нагрузочной дозы 1200 мг и поддерживающей дозы 600 мг, обеспечивает те же Ctrough и AUC, что и пертузумаб в/в 840 мг и 420 мг соответственно, как было определено у ЗМД. Доза пертузумаба п/к 600 мг у пациентов с РРМЖ обеспечивает аналогичные Ctrough и AUC с когортами с введением 420 мг в/в и 600 мг п/к у ЗМД в Части 1, а пропорциональность ose при линейности ФК подтверждает нагрузочную дозу пертузумаба п/к 1200. Таким образом, дозы пертузумаба п/к (нагрузочная 1200 мг, поддерживающая 600 мг) подтверждены у пациентов с РРМЖ.
В целом, показатели безопасности пертузумаба п/к согласуются с известными показателями безопасности пертузумаба в/в и он хорошо переносится при применении в комбинации с трастузумабом п/к. Не было обнаружено никаких новых сообщений о проблемах безопасности. Большинство ЗМД в Части 1 и все пациенты с РРМЖ в Части 2 Когорты В испытывали, по крайней мере, одно НЯ. Во время исследования было обнаружено 3 НЯ Степени 2 в Части 1 и 3 НЯ Степени 1 в Части 2. Сообщалось, что большинство НЯ были низкой интенсивности (Степень 1 или 2). Во время исследования не было зарегистрировано никаких СНЯ, смертельных случаев или СНЯ, приводящих к прекращению исследования. После п/к инъекции смеси пертузумаба и трастузумаба у пациенток с РРМЖ наблюдалась более высокая частота реакций в месте инъекции по сравнению с ЗМД.
Принимая во внимание результаты исследований ФК и безопасности в текущем исследовании (Части 1 и 2), безопасность, переносимость и результаты ФК этого исследования Фазы I подтверждают возможность продолжения исследования и набора Когорты С для приема комбинированного препарата с фиксированной дозой пертузумаб + трастузумаб (FDC).
ПРИМЕР 2
Стабильные Подкожные Комбинированные Препараты (SC FDC) с Фиксированной Дозой Пертузумаба и Трастузумаба
Стабильные комбинированные препараты с фиксированной дозой (FDC) пертузумаба и трастузумаба были разработаны для подкожного (п/к) введения.
В исследованиях комбинированого препарата использовали п/к композиции лекарственного препарата (DS) пертузумаба и трастузумаба и композицию rHuPH20, представленную на Фиг. 19.
Количество (%) высокомолекулярных частиц (HMWS) в различных подкожных композициях пертузумаба и трастузумаба и в композициях пертузумаба/трастузумаба, содержащих трегалозу и/или сахарозу в качестве стабилизатора при температуре 5°С и 25°С, представлено на Фиг. 23.
Было обнаружено, что следующие нагрузочные и поддерживающие дозы препаратов SC FDS являются стабильными и пригодными для подкожного введения единого комбинированного препарата пертузумаба и трастузумаба пациентам-людям:
Нагрузочная Доза Пертузумаб
Доза: 1,200 мг
Концентрация: 80 мг/мл
Трастузумаб
Доза: 600 мг
Концентрация: 40 мг/мл
rHuPH20
Концентрация: 1,000 Ед/мл или 2,000 Ед/мл
рН: 5,5
20 мМ L-Гистидин/HCl
Трегалоза: 70 мМ
Сахароза: 133 мМ
Полисорбат 20 (PS20): 0,04%; 0,4 мг/мл
10 мМ Метионина
Номинальный объем препарата: 15 мл
Флакон: 20 мл/20 мм
Поддерживающая доза
Пертузумаб
Доза: 600 мг
Концентрация: 60 мг/мл
Трастузумаб
Доза: 600 мг
Концентрация: 60 мг/мл
rHuPH20
Концентрация: 1,000 Ед/мл или 2,000 Ед/мл
рН: 5,5
20 мМ L-Гистидин/HCl
Трегалоза: 105 мМ
Сахароза: 100 мМ
Полисорбат PS20: 0,04%; 0,4 мг/мл
10 мМ метионина
Номинальный объем препарата: 10 мл
Флакон: 15 мл/20 мм
Стабильность Лекарственного Препарата Пертузумаба
Тест Стабильности (Scratch & Sprinkle)
Агрегация белка может происходить вследствие кристаллизации эксципиента (сахарозы) в замороженном состоянии в условиях хранения лекарственного вещества. В тесте «Scratch & Sprinkle» флаконы с лекарственным препаратом замораживают и добавляют немного сахара (трегалозы или сахарозы) в верхнюю часть замороженного препарата, а затем царапают металлическим шпателем для ускорения любой возможной кристаллизации сахара в замороженном препарате. В заранее определенные моменты времени препараты размораживают и анализируют способом эксклюзионной хроматографии (SEC).
Данные SEC, представленные на Фиг. 29 демонстрируют, что сахароза является наиболее предпочтительным эксципиентом для лекарственного препарата пертузумаба, который хранится при температуре -20°С.
Было изучено влияние различий препаратов в препаратах следующих комбинаций фиксированных доз пертузумаба и трастузумаба (FDC) на мутность и количество высокомолекулярных частиц (HMWS).
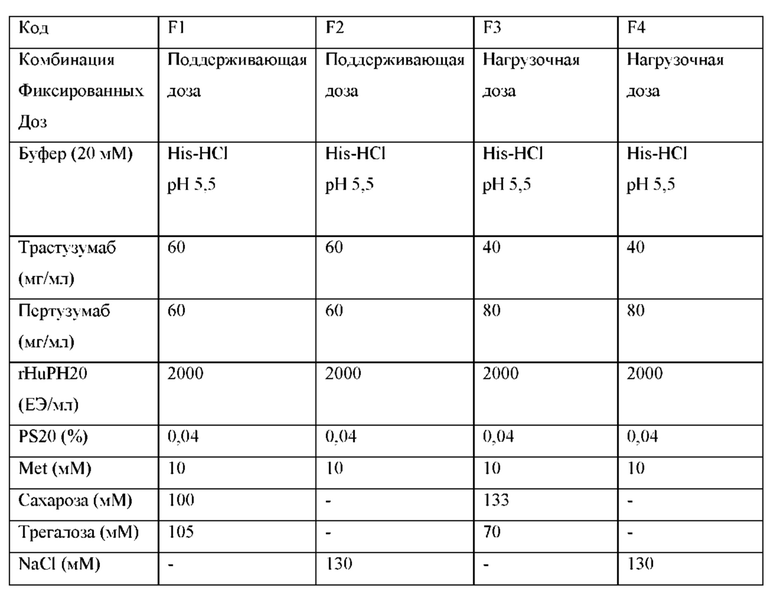
Данные, представленые на Фиг. 30, демонстрируют, что NaCl в качестве эксципиента приводит к высокой мутности в комбинации с фиксированными дозами пертузумаба-трастузумаба п/к (FDC). Аналогично, данные, представленные на Фиг. 31 демонстрируют, что NaCl в качестве эксципиента приводит к более высоким количествам высокомолекулярных частиц (HMW). Соответственно, сахароза и трегалоза являются наиболее предпочтительными эксципиентами для FDC.
Хотя некоторые варианты реализации данного изобретения были показаны и описаны в данном документе, специалистам в данной области техники будет понятно, что такие варианты реализации данного изобретения представлены только в качестве примера. Многочисленные вариации, изменения и замены в пределах сущности изобретения будут очевидны специалистам в данной области техники. Следует понимать, что различные альтернативы вариантам реализации данного изобретения, описанные в данном документе, могут быть использованы при практическом применении изобретения. Предполагается, что представленная ниже формула изобретения определяет объем изобретения и то, какие способы и структуры в пределах объема данной формулы изобретения и их эквиваленты должны быть охвачены ею.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> GENENTECH, INC.
F. HOFFMANN-LA ROCHE AG
<120> ПРЕПАРАТЫ АНТИТЕЛ ПРОТИВ HER2 ДЛЯ ПОДКОЖНОГО ВВЕДЕНИЯ
<130> P34027-WO
<140>
<141>
<150> 62/447,359
<151> 2017-01-17
<160> 22
<170> PatentIn версия 3.5
<210> 1
<211> 195
<212> БЕЛОК
<213> Homo sapiens
<400> 1
Thr Gln Val Cys Thr Gly Thr Asp Met Lys Leu Arg Leu Pro Ala Ser
1 5 10 15
Pro Glu Thr His Leu Asp Met Leu Arg His Leu Tyr Gln Gly Cys Gln
20 25 30
Val Val Gln Gly Asn Leu Glu Leu Thr Tyr Leu Pro Thr Asn Ala Ser
35 40 45
Leu Ser Phe Leu Gln Asp Ile Gln Glu Val Gln Gly Tyr Val Leu Ile
50 55 60
Ala His Asn Gln Val Arg Gln Val Pro Leu Gln Arg Leu Arg Ile Val
65 70 75 80
Arg Gly Thr Gln Leu Phe Glu Asp Asn Tyr Ala Leu Ala Val Leu Asp
85 90 95
Asn Gly Asp Pro Leu Asn Asn Thr Thr Pro Val Thr Gly Ala Ser Pro
100 105 110
Gly Gly Leu Arg Glu Leu Gln Leu Arg Ser Leu Thr Glu Ile Leu Lys
115 120 125
Gly Gly Val Leu Ile Gln Arg Asn Pro Gln Leu Cys Tyr Gln Asp Thr
130 135 140
Ile Leu Trp Lys Asp Ile Phe His Lys Asn Asn Gln Leu Ala Leu Thr
145 150 155 160
Leu Ile Asp Thr Asn Arg Ser Arg Ala Cys His Pro Cys Ser Pro Met
165 170 175
Cys Lys Gly Ser Arg Cys Trp Gly Glu Ser Ser Glu Asp Cys Gln Ser
180 185 190
Leu Thr Arg
195
<210> 2
<211> 124
<212> БЕЛОК
<213> Homo sapiens
<400> 2
Thr Val Cys Ala Gly Gly Cys Ala Arg Cys Lys Gly Pro Leu Pro Thr
1 5 10 15
Asp Cys Cys His Glu Gln Cys Ala Ala Gly Cys Thr Gly Pro Lys His
20 25 30
Ser Asp Cys Leu Ala Cys Leu His Phe Asn His Ser Gly Ile Cys Glu
35 40 45
Leu His Cys Pro Ala Leu Val Thr Tyr Asn Thr Asp Thr Phe Glu Ser
50 55 60
Met Pro Asn Pro Glu Gly Arg Tyr Thr Phe Gly Ala Ser Cys Val Thr
65 70 75 80
Ala Cys Pro Tyr Asn Tyr Leu Ser Thr Asp Val Gly Ser Cys Thr Leu
85 90 95
Val Cys Pro Leu His Asn Gln Glu Val Thr Ala Glu Asp Gly Thr Gln
100 105 110
Arg Cys Glu Lys Cys Ser Lys Pro Cys Ala Arg Val
115 120
<210> 3
<211> 169
<212> БЕЛОК
<213> Homo sapiens
<400> 3
Cys Tyr Gly Leu Gly Met Glu His Leu Arg Glu Val Arg Ala Val Thr
1 5 10 15
Ser Ala Asn Ile Gln Glu Phe Ala Gly Cys Lys Lys Ile Phe Gly Ser
20 25 30
Leu Ala Phe Leu Pro Glu Ser Phe Asp Gly Asp Pro Ala Ser Asn Thr
35 40 45
Ala Pro Leu Gln Pro Glu Gln Leu Gln Val Phe Glu Thr Leu Glu Glu
50 55 60
Ile Thr Gly Tyr Leu Tyr Ile Ser Ala Trp Pro Asp Ser Leu Pro Asp
65 70 75 80
Leu Ser Val Phe Gln Asn Leu Gln Val Ile Arg Gly Arg Ile Leu His
85 90 95
Asn Gly Ala Tyr Ser Leu Thr Leu Gln Gly Leu Gly Ile Ser Trp Leu
100 105 110
Gly Leu Arg Ser Leu Arg Glu Leu Gly Ser Gly Leu Ala Leu Ile His
115 120 125
His Asn Thr His Leu Cys Phe Val His Thr Val Pro Trp Asp Gln Leu
130 135 140
Phe Arg Asn Pro His Gln Ala Leu Leu His Thr Ala Asn Arg Pro Glu
145 150 155 160
Asp Glu Cys Val Gly Glu Gly Leu Ala
165
<210> 4
<211> 142
<212> БЕЛОК
<213> Homo sapiens
<400> 4
Cys His Gln Leu Cys Ala Arg Gly His Cys Trp Gly Pro Gly Pro Thr
1 5 10 15
Gln Cys Val Asn Cys Ser Gln Phe Leu Arg Gly Gln Glu Cys Val Glu
20 25 30
Glu Cys Arg Val Leu Gln Gly Leu Pro Arg Glu Tyr Val Asn Ala Arg
35 40 45
His Cys Leu Pro Cys His Pro Glu Cys Gln Pro Gln Asn Gly Ser Val
50 55 60
Thr Cys Phe Gly Pro Glu Ala Asp Gln Cys Val Ala Cys Ala His Tyr
65 70 75 80
Lys Asp Pro Pro Phe Cys Val Ala Arg Cys Pro Ser Gly Val Lys Pro
85 90 95
Asp Leu Ser Tyr Met Pro Ile Trp Lys Phe Pro Asp Glu Glu Gly Ala
100 105 110
Cys Gln Pro Cys Pro Ile Asn Cys Thr His Ser Cys Val Asp Leu Asp
115 120 125
Asp Lys Gly Cys Pro Ala Glu Gln Arg Ala Ser Pro Leu Thr
130 135 140
<210> 5
<211> 107
<212> БЕЛОК
<213> Mus musculus
<400> 5
Asp Thr Val Met Thr Gln Ser His Lys Ile Met Ser Thr Ser Val Gly
1 5 10 15
Asp Arg Val Ser Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Ile Gly
20 25 30
Val Ala Trp Tyr Gln Gln Arg Pro Gly Gln Ser Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Asp Arg Phe Thr Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Phe Thr Ile Ser Ser Val Gln Ala
65 70 75 80
Glu Asp Leu Ala Val Tyr Tyr Cys Gln Gln Tyr Tyr Ile Tyr Pro Tyr
85 90 95
Thr Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys
100 105
<210> 6
<211> 119
<212> БЕЛОК
<213> Mus musculus
<400> 6
Glu Val Gln Leu Gln Gln Ser Gly Pro Glu Leu Val Lys Pro Gly Thr
1 5 10 15
Ser Val Lys Ile Ser Cys Lys Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Lys Gln Ser His Gly Lys Ser Leu Glu Trp Ile
35 40 45
Gly Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Lys Ala Ser Leu Thr Val Asp Arg Ser Ser Arg Ile Val Tyr
65 70 75 80
Met Glu Leu Arg Ser Leu Thr Phe Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Thr Leu Thr Val Ser Ser
115
<210> 7
<211> 107
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 7
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Ile Gly
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Tyr Tyr Ile Tyr Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys
100 105
<210> 8
<211> 119
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 8
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Arg Phe Thr Leu Ser Val Asp Arg Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser
115
<210> 9
<211> 107
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 9
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Ser Ile Ser Asn Tyr
20 25 30
Leu Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ala Ala Ser Ser Leu Glu Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Tyr Asn Ser Leu Pro Trp
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys
100 105
<210> 10
<211> 119
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 10
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr
20 25 30
Ala Met Ser Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Val Ile Ser Gly Asp Gly Gly Ser Thr Tyr Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Gly Arg Val Gly Tyr Ser Leu Tyr Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser
115
<210> 11
<211> 214
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 11
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Lys Ala Ser Gln Asp Val Ser Ile Gly
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Tyr Tyr Ile Tyr Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 12
<211> 448
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 12
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Arg Phe Thr Leu Ser Val Asp Arg Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
<210> 13
<211> 214
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 13
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Asp Val Asn Thr Ala
20 25 30
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ser Ala Ser Phe Leu Tyr Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Arg Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln His Tyr Thr Thr Pro Pro
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 14
<211> 449
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 14
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Asn Ile Lys Asp Thr
20 25 30
Tyr Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Arg Ile Tyr Pro Thr Asn Gly Tyr Thr Arg Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn Thr Ala Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ser Arg Trp Gly Gly Asp Gly Phe Tyr Ala Met Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp
210 215 220
Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly
225 230 235 240
Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile
245 250 255
Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu
260 265 270
Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His
275 280 285
Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg
290 295 300
Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys
305 310 315 320
Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu
325 330 335
Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr
340 345 350
Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu
355 360 365
Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp
370 375 380
Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val
385 390 395 400
Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp
405 410 415
Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His
420 425 430
Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro
435 440 445
Gly
<210> 15
<211> 217
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 15
Val His Ser Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala
1 5 10 15
Ser Val Gly Asp Arg Val Thr Ile Thr Cys Lys Ala Ser Gln Asp Val
20 25 30
Ser Ile Gly Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys
35 40 45
Leu Leu Ile Tyr Ser Ala Ser Tyr Arg Tyr Thr Gly Val Pro Ser Arg
50 55 60
Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser
65 70 75 80
Leu Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Gln Tyr Tyr Ile
85 90 95
Tyr Pro Tyr Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr
100 105 110
Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu
115 120 125
Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro
130 135 140
Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly
145 150 155 160
Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr
165 170 175
Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His
180 185 190
Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val
195 200 205
Thr Lys Ser Phe Asn Arg Gly Glu Cys
210 215
<210> 16
<211> 449
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Полипептид"
<400> 16
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Thr Asp Tyr
20 25 30
Thr Met Asp Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe
50 55 60
Lys Gly Arg Phe Thr Leu Ser Val Asp Arg Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr Trp Gly Gln Gly
100 105 110
Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe
115 120 125
Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu
130 135 140
Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp
145 150 155 160
Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu
165 170 175
Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser
180 185 190
Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro
195 200 205
Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp Lys
210 215 220
Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro
225 230 235 240
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser
245 250 255
Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp
260 265 270
Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn
275 280 285
Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val
290 295 300
Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu
305 310 315 320
Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys
325 330 335
Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr
340 345 350
Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr
355 360 365
Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu
370 375 380
Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu
385 390 395 400
Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys
405 410 415
Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu
420 425 430
Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly
435 440 445
Lys
<210> 17
<211> 10
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Пептид"
<220>
<221> ВАРИАНТ
<222> (10)..(10)
<223> /замена="Ser"
<220>
<221> MISC_FEATURE
<222> (10)..(10)
<223> /примечание=" Остаток, указанный в последовательности, не имеет
предпочтений по сравнению с остатком, указанным в аннотации для
указанной позиции"
<400> 17
Gly Phe Thr Phe Thr Asp Tyr Thr Met Asp
1 5 10
<210> 18
<211> 17
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечания="Описание Искусственной Последовательности:
Синтетический Пептид"
<400> 18
Asp Val Asn Pro Asn Ser Gly Gly Ser Ile Tyr Asn Gln Arg Phe Lys
1 5 10 15
Gly
<210> 19
<211> 10
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечание="Описание Искусственной Последовательности:
Синтетический Пептид"
<400> 19
Asn Leu Gly Pro Ser Phe Tyr Phe Asp Tyr
1 5 10
<210> 20
<211> 11
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечание="Описание Искусственной Последовательности:
Синтетический Пептид"
<400> 20
Lys Ala Ser Gln Asp Val Ser Ile Gly Val Ala
1 5 10
<210> 21
<211> 7
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечание="Описание Искусственной Последовательности:
Синтетический Пептид"
<220>
<221> ВАРИАНТ
<222> (5)..(5)
<223> /замена="Leu"
<220>
<221> ВАРИАНТ
<222> (6)..(6)
<223> /замена="Glu"
<220>
<221> ВАРИАНТ
<222> (7)..(7)
<223> /замена="Ser"
<220>
<221> MISC_FEATURE
<222> (5)..(7)
<223> /примечание=" Остаток, указанный в последовательности, не имеет
предпочтений по сравнению с остатком, указанным в аннотации
для указанной позиции"
<400> 21
Ser Ala Ser Tyr Arg Tyr Thr
1 5
<210> 22
<211> 9
<212> БЕЛОК
<213> Искусственная Последовательность
<220>
<221> источник
<223> /примечание="Описание Искусственной Последовательности:
Синтетический Пептид"
<400> 22
Gln Gln Tyr Tyr Ile Tyr Pro Tyr Thr
1 5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ АНТИ-HER2 БИПАРАТОПНЫХ АНТИТЕЛ-ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2806049C2 |
| ВАРИАНТЫ ПЕРТУЗУМАБА И ИХ АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА | 2014 |
|
RU2737727C2 |
| БИСПЕЦИФИЧЕСКИЕ АНТИГЕНСВЯЗЫВАЮЩИЕ КОНСТРУКЦИИ ПРОТИВ HER2 | 2014 |
|
RU2737882C2 |
| БИСПЕЦИФИЧЕСКИЕ АНТИГЕНСВЯЗЫВАЮЩИЕ ПОЛИПЕПТИДЫ | 2015 |
|
RU2723940C2 |
| САЙТ-СПЕЦИФИЧЕСКИЕ КОНЪЮГАТЫ АНТИТЕЛА К HER2 И ЛЕКАРСТВЕННОГО СРЕДСТВА | 2016 |
|
RU2745565C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ВАРИАНТ РН20 ГИАЛУРОНИДАЗЫ ЧЕЛОВЕКА, И ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПОДКОЖНОГО ВВЕДЕНИЯ | 2020 |
|
RU2810952C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ БИСПЕЦИФИЧЕСКОЙ АНТИГЕНСВЯЗЫВАЮЩЕЙ КОНСТРУКЦИИ, НАЦЕЛЕННОЙ НА HER2, ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКОГО ЗАБОЛЕВАНИЯ ЖЕЛЧНЫХ ПРОТОКОВ | 2019 |
|
RU2819802C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА ПУТЕМ ВНУТРИВЕННОГО ВВЕДЕНИЯ РЕКОМБИНАНТНОГО MVA И АНТИТЕЛА | 2018 |
|
RU2795103C2 |
| Биспецифическое антитело анти-HER2/анти-4-1BB и его применение | 2020 |
|
RU2831838C2 |
| HER-2-НАПРАВЛЕННЫЕ АНТИГЕНСВЯЗЫВАЮЩИЕ МОЛЕКУЛЫ, СОДЕРЖАЩИЕ 4-1BBL | 2019 |
|
RU2815451C2 |






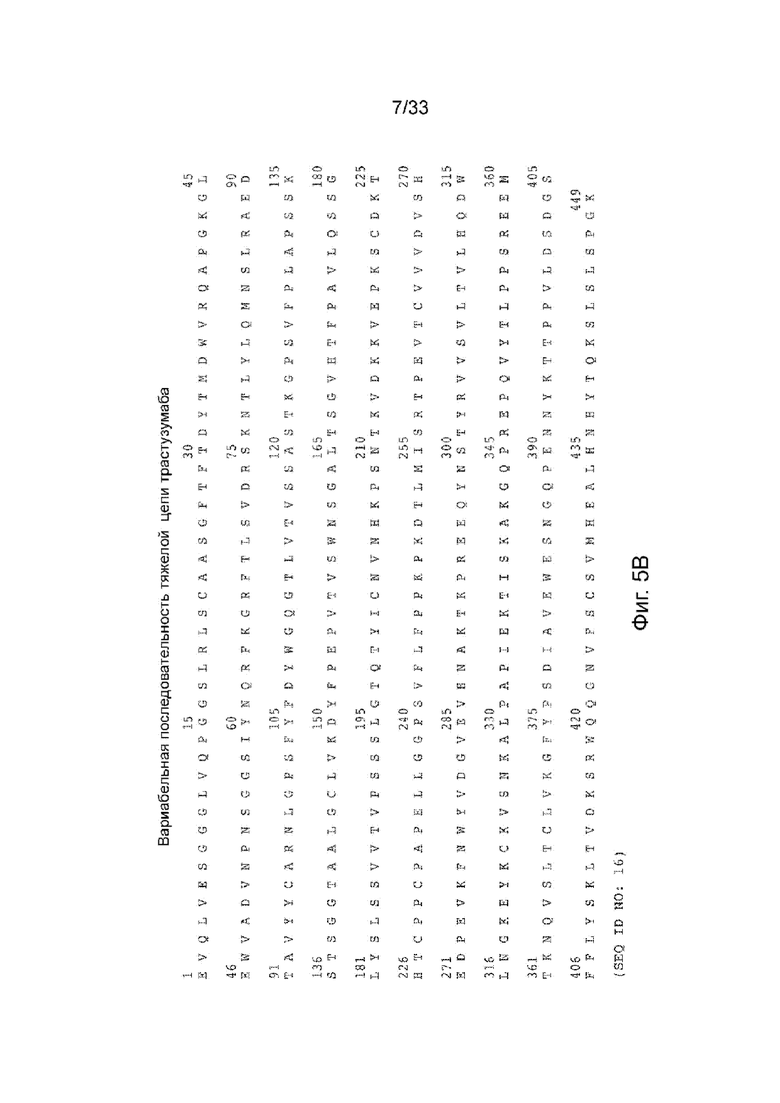
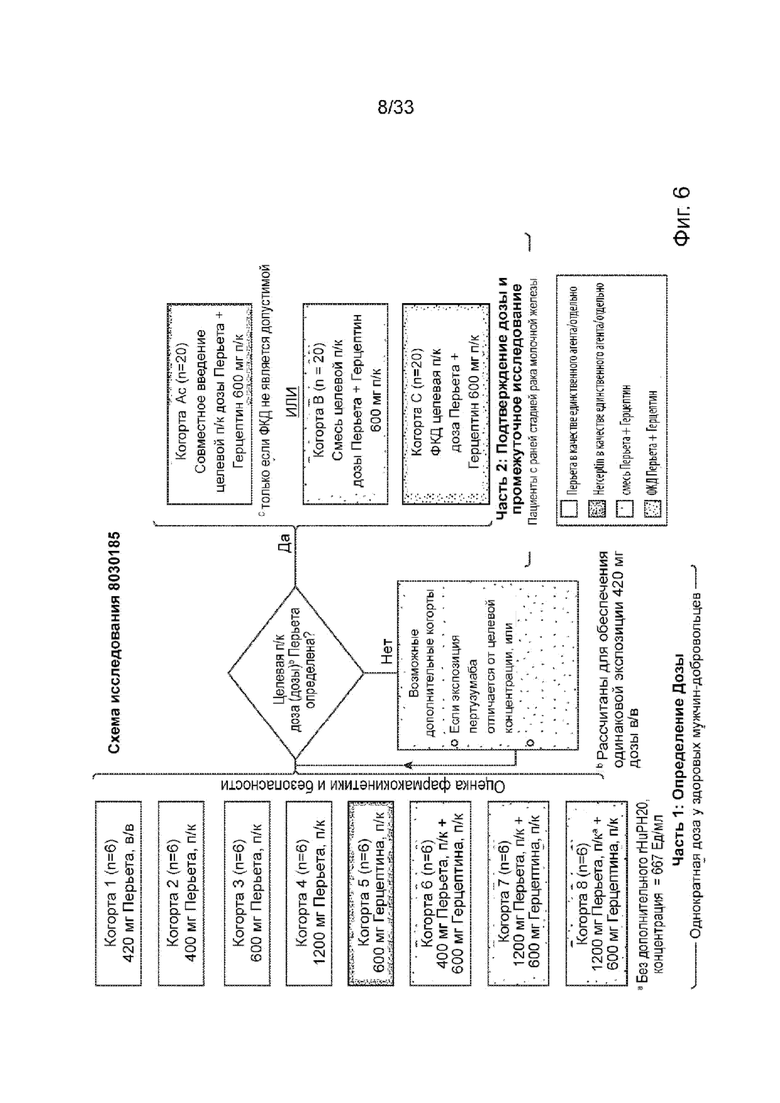
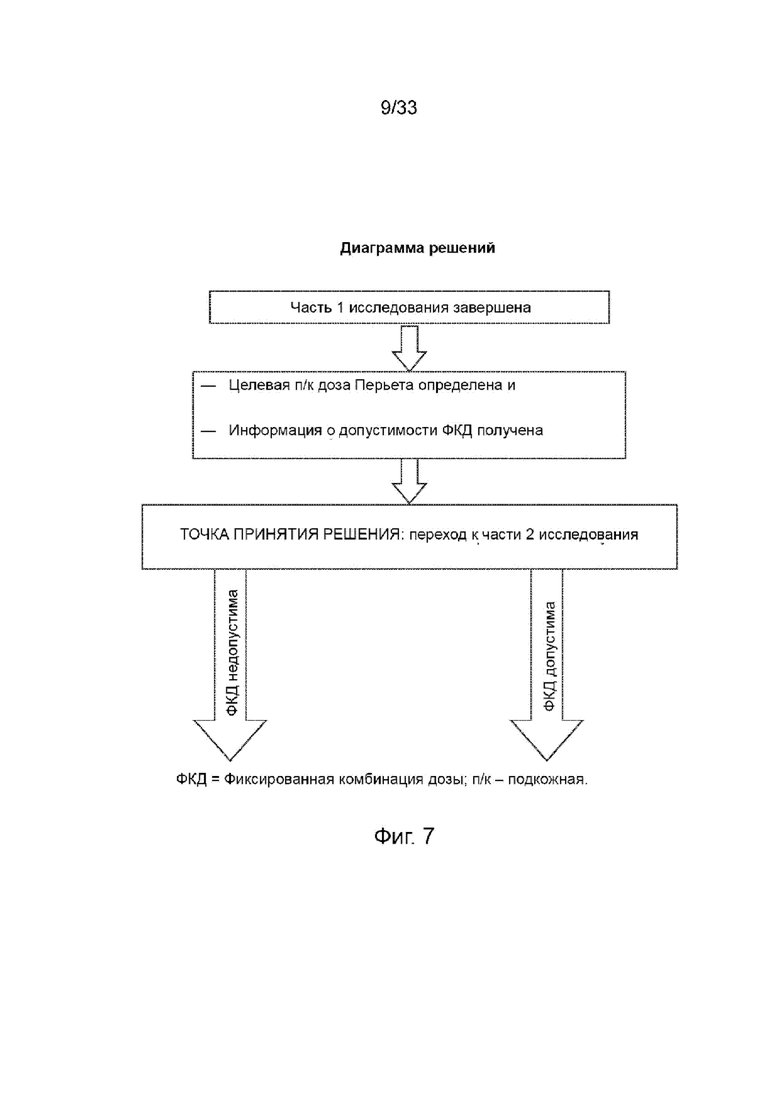
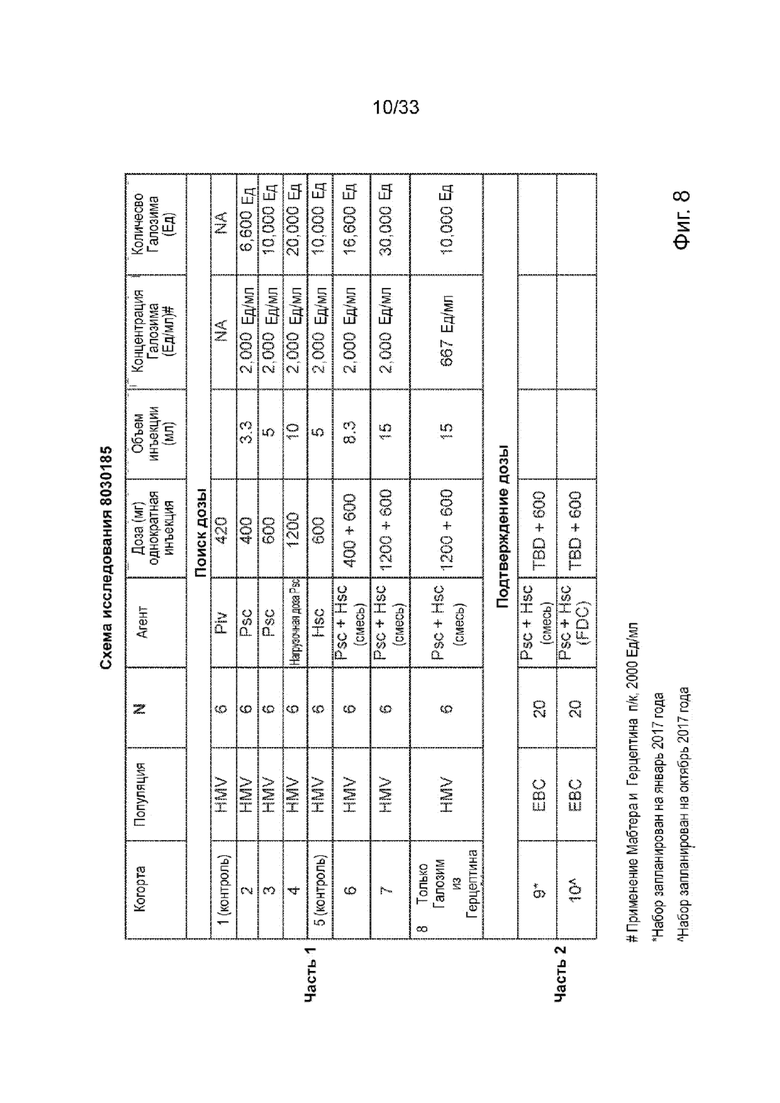
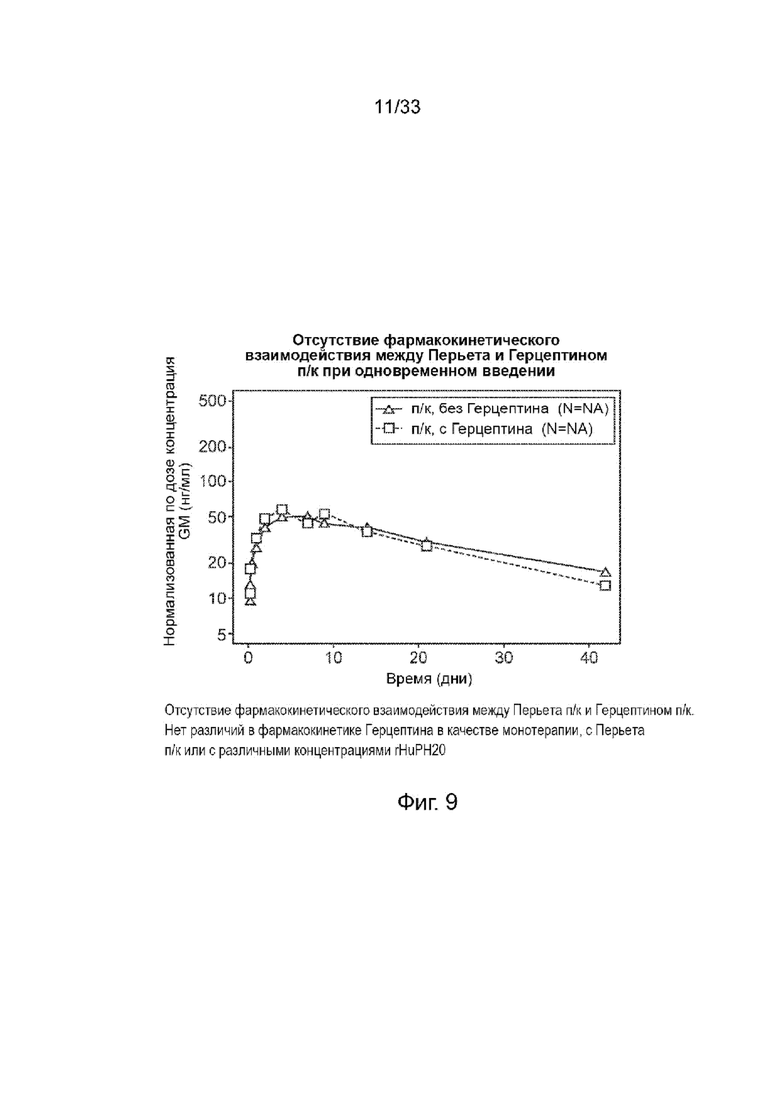

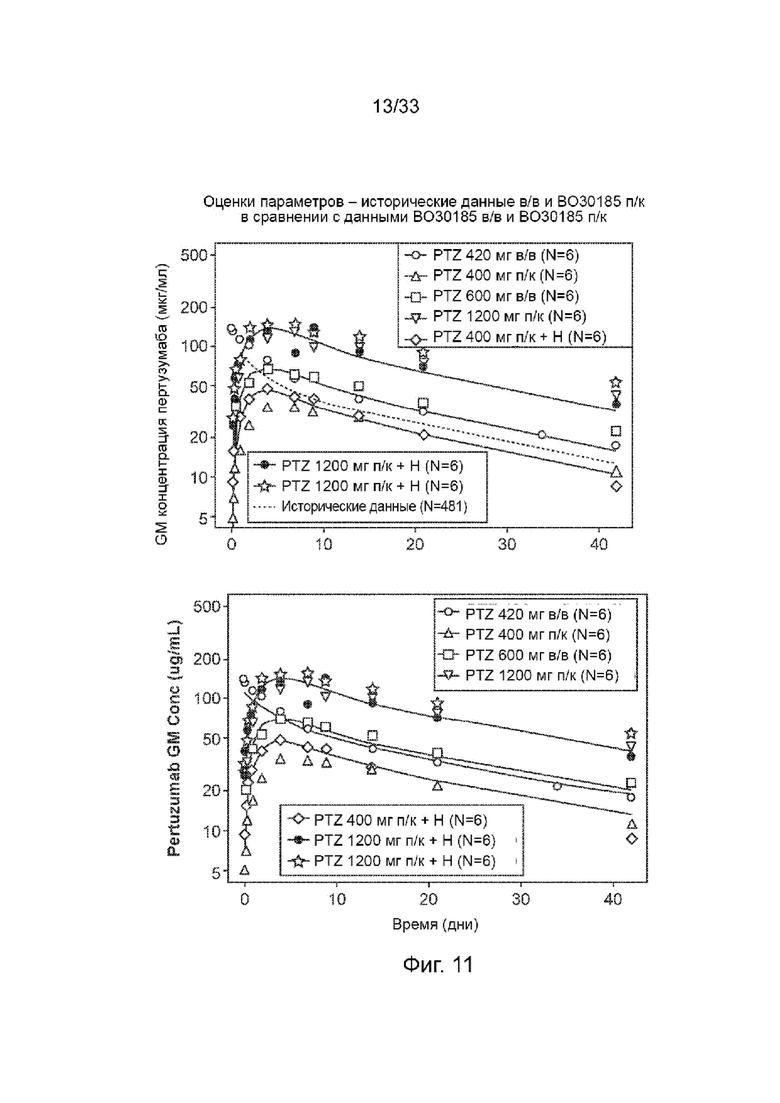
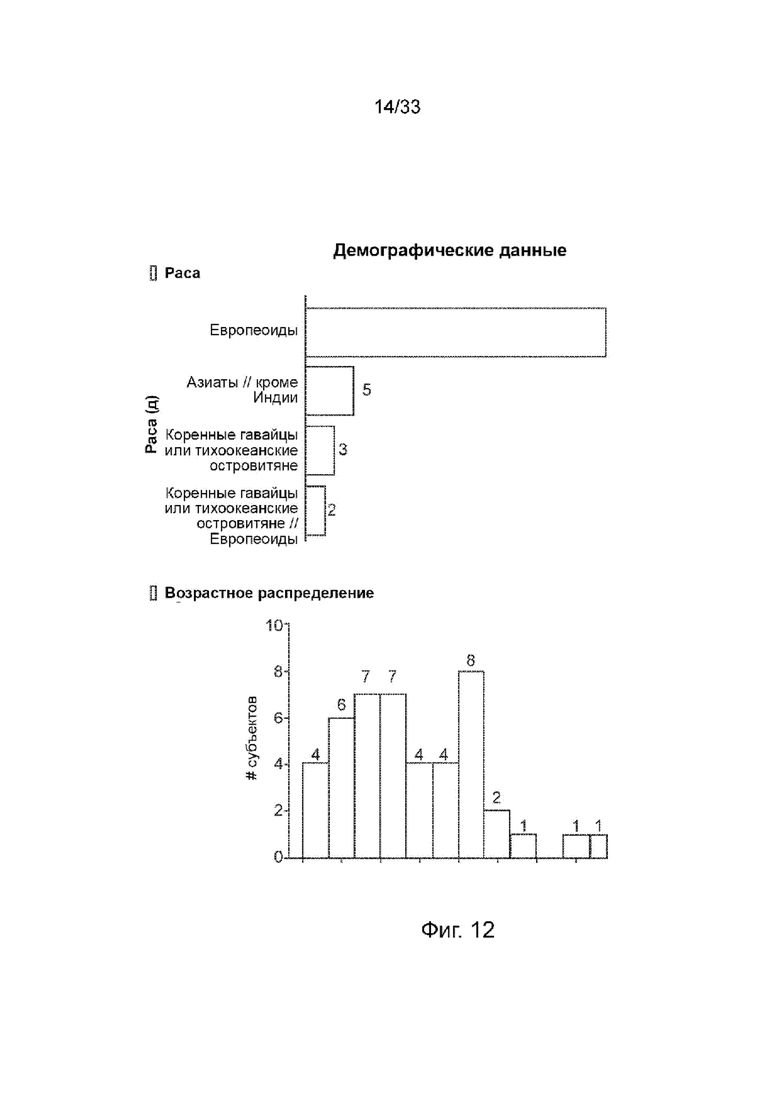
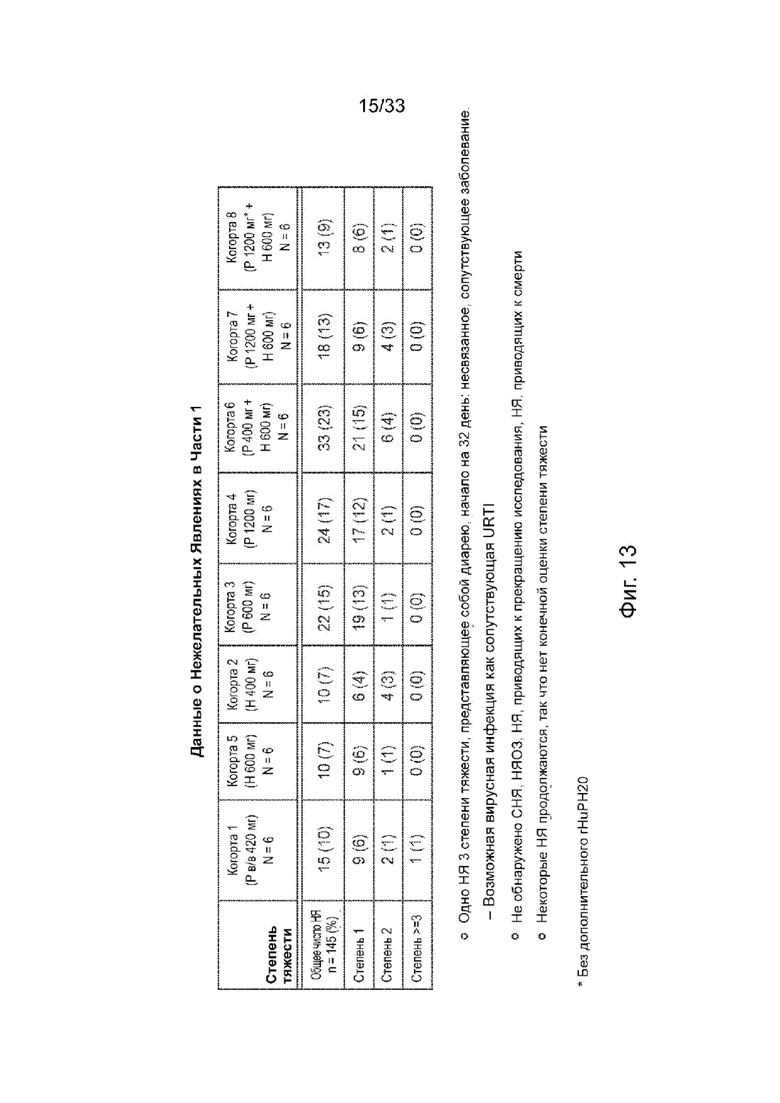
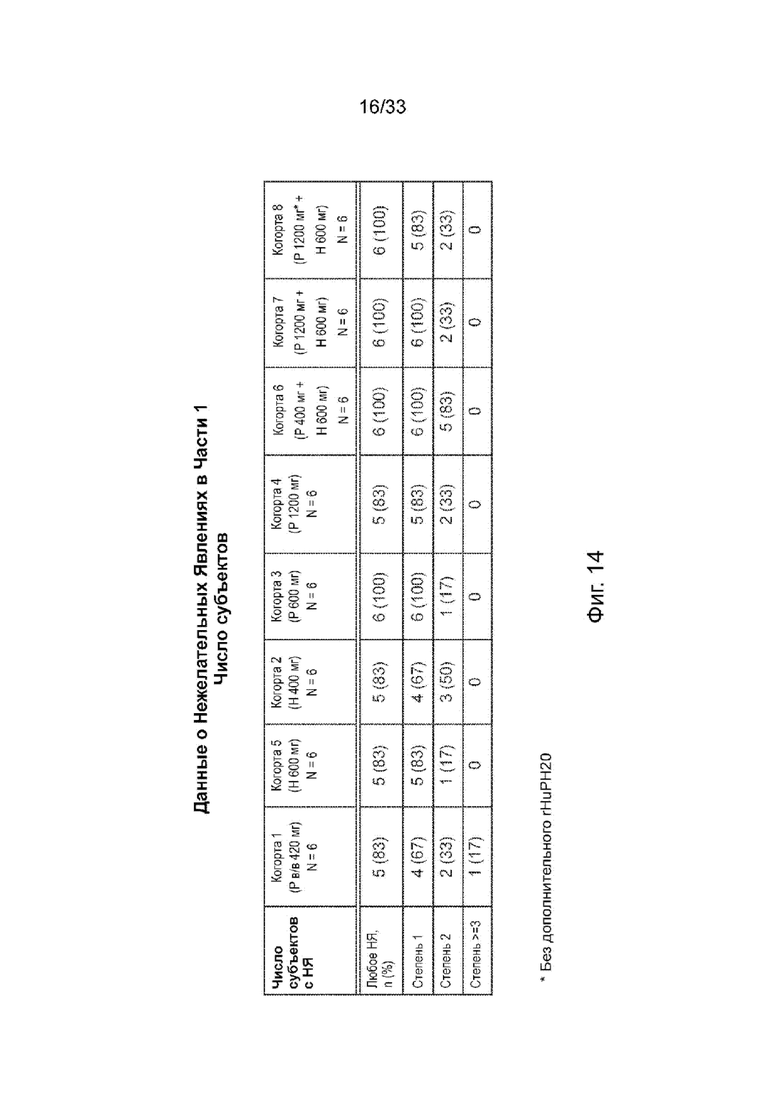
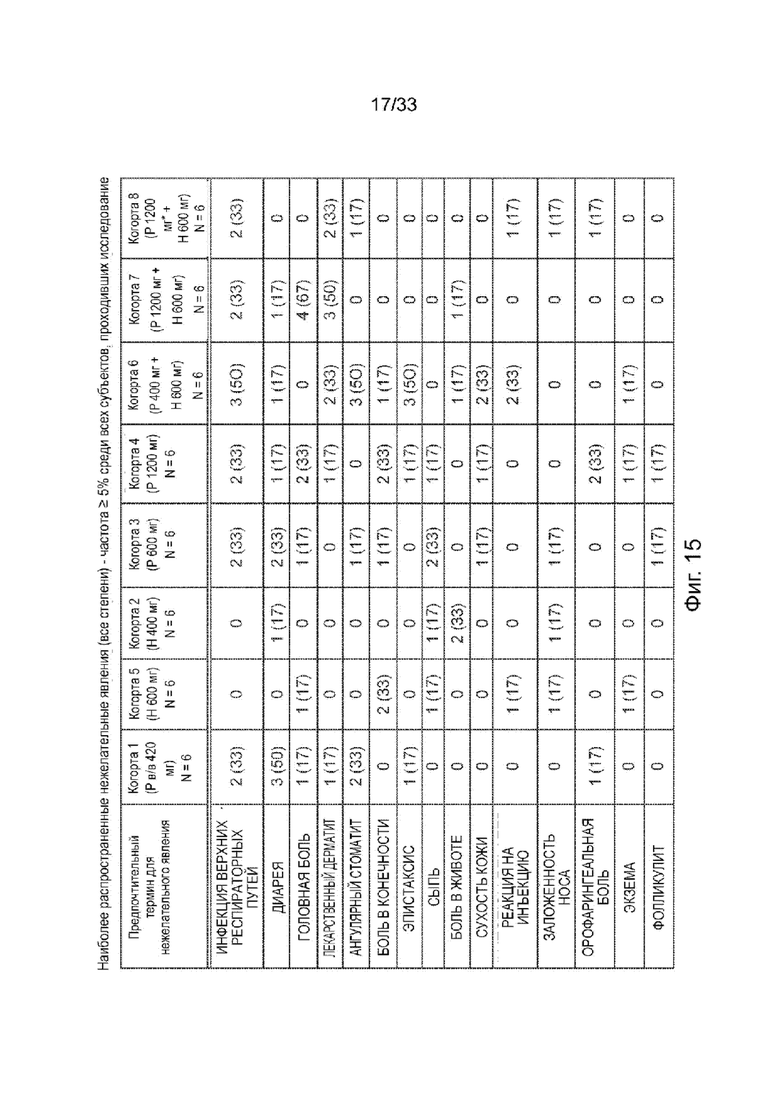
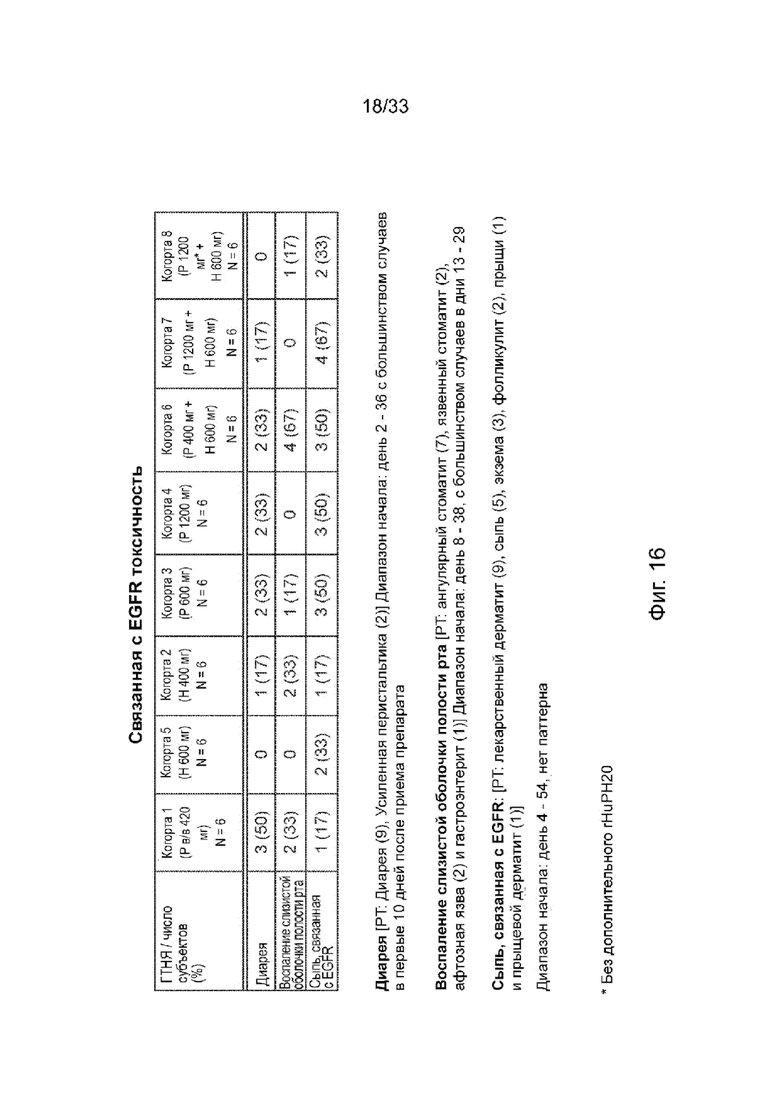



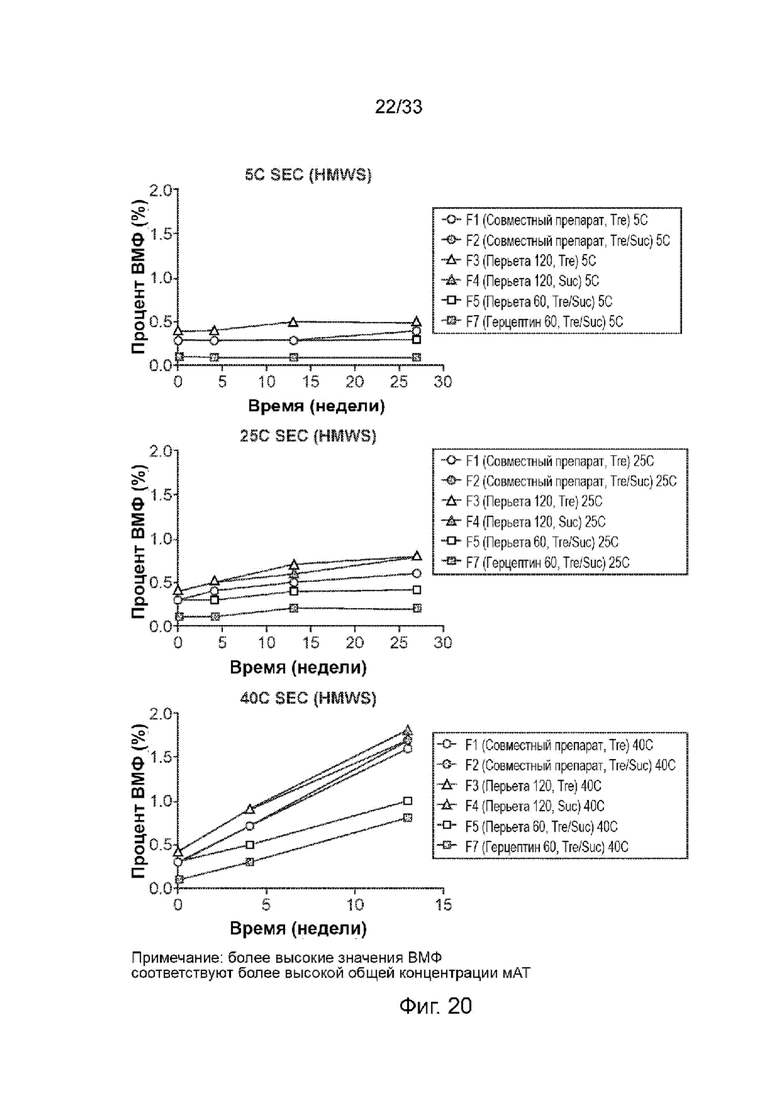



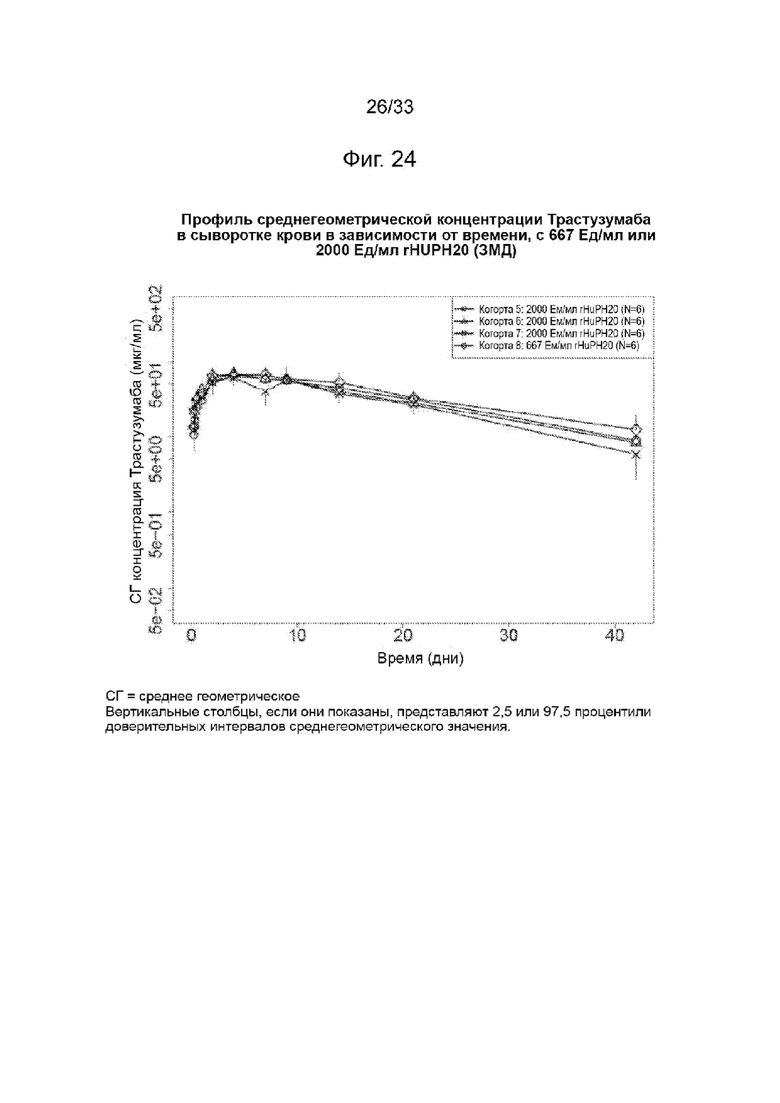
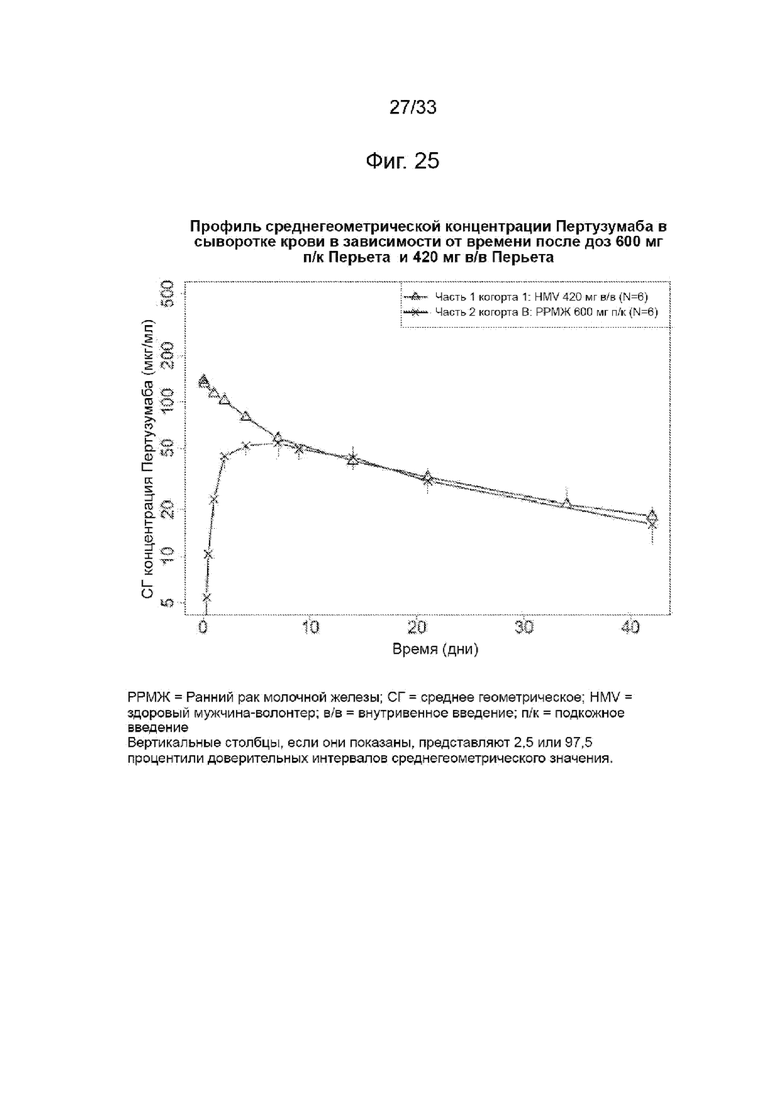

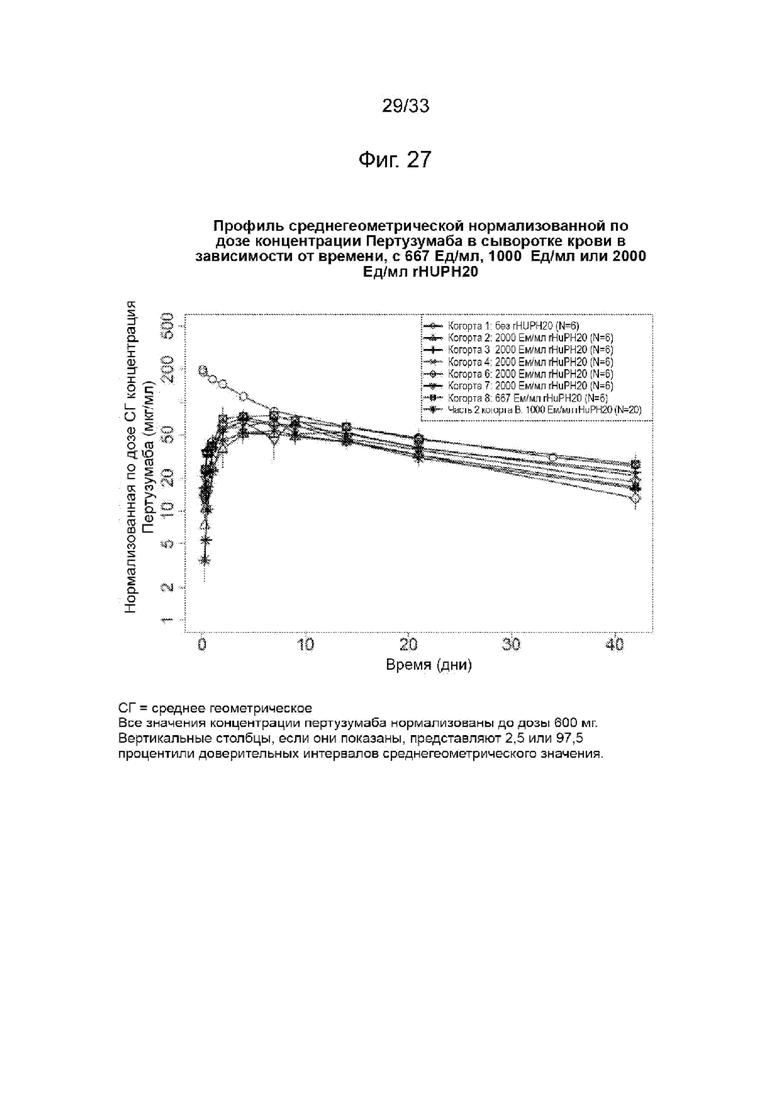
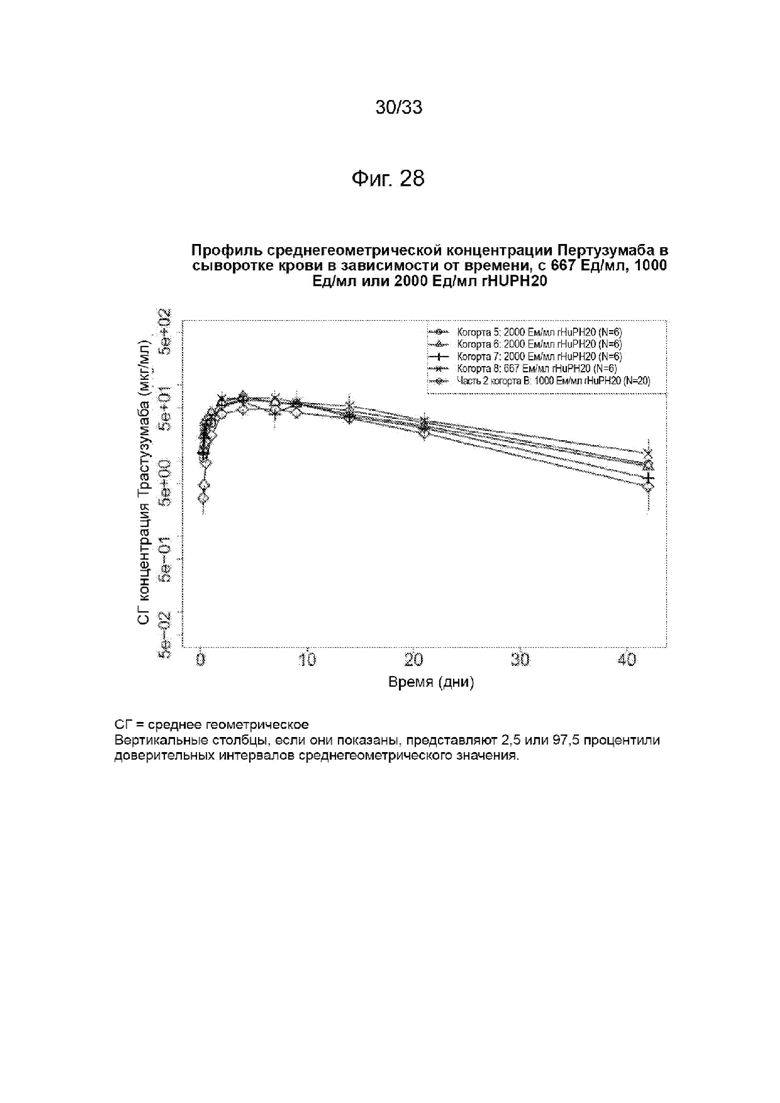


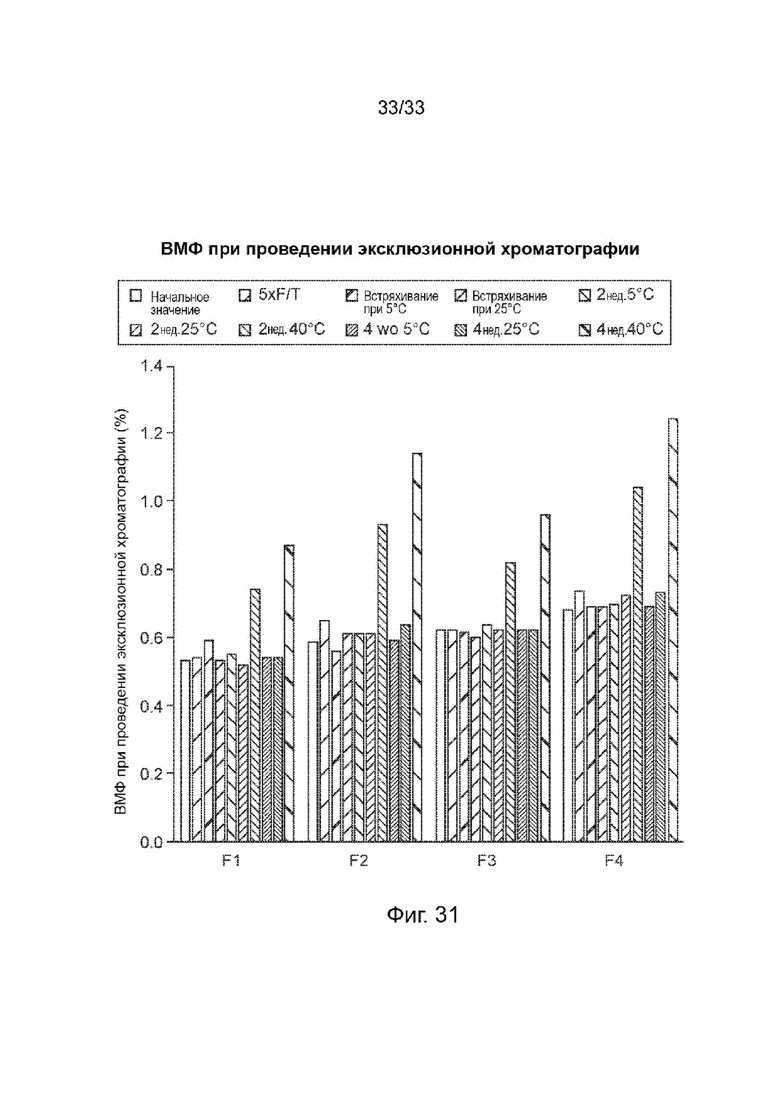
Изобретение относится к области биотехнологии, в частности к применению пертузумаба для лечения HER2-положительного онкологического заболевания. При этом пертузумаб вводят подкожно в комбинации с подкожно вводимым трастузумабом. Изобретение позволяет эффективно осуществлять лечение HER2-положительного онкологического заболевания. 15 з.п. ф-лы, 31 ил., 6 табл., 2 пр.
1. Применение пертузумаба для лечения HER2-положительного онкологического заболевания, при этом пертузумаб вводят подкожно в комбинации с подкожно вводимым трастузумабом, и причем пертузумаб вводят в фиксированной нагрузочной дозе 1200 мг в комбинации с 600 мг трастузумаба с последующей, по меньшей мере, одной поддерживающей дозой 600 мг пертузумаба в комбинации с 600 мг трастузумаба.
2. Применение по п. 1, в котором после введения фиксированной нагрузочной дозы следует введение нескольких поддерживающих доз пертузумаба.
3. Применение по п. 2, в котором первую поддерживающую дозу пертузумаба вводят спустя приблизительно две недели или приблизительно три недели после введения фиксированной нагрузочной дозы пертузумаба.
4. Применение по любому из пп. 2 или 3, в котором поддерживающие дозы пертузумаба вводят приблизительно каждые 2 недели или приблизительно каждые 3 недели.
5. Применение по п. 4, в котором поддерживающие дозы пертузумаба вводят приблизительно каждые 3 недели.
6. Применение по п. 1, в котором HER2-положительное онкологическое заболевание выбрано из группы, состоящей из рака молочной железы, перитонеального рака, рака маточной трубы, рака легких, колоректального рака, рака желчных путей и рака мочевого пузыря.
7. Применение по п. 6, в котором HER2-положительное онкологическое заболевание представляет собой рак молочной железы.
8. Применение по п. 7, в котором рак молочной железы представляет собой ранний рак молочной железы (РРМЖ) или метастатический рак молочной железы (МРМЖ).
9. Применение по п. 1, в котором пертузумаб смешивают с трастузумабом и вводят в виде единой подкожной инъекции.
10. Применение по п. 1, в котором пертузумаб и трастузумаб вводят в виде одного совместного препарата для подкожного введения.
11. Применение по п. 10, в котором совместный препарат дополнительно включает рекомбинантную гиалуронидазу человека (rHuPH20), необязательно, где концентрация rHuPH20 в совместном препарате составляет от 600 Ед/мл до 2000 Ед/мл.
12. Применение по п. 11, в котором совместный препарат представляет собой жидкую фармацевтическую композицию.
13. Применение по п. 1, дополнительно включающее введение химиотерапевтического агента пациенту.
14. Применение по п. 13, в котором химиотерапевтический агент представляет собой таксан или антрациклин.
15. Применение по п. 13, в котором химиотерапевтический агент представляет собой таксан и таксан содержит паклитаксел или доцетаксел.
16. Применение по п. 13, в котором химиотерапевтический агент представляет собой антрациклин и антрациклин содержит даунорубицин, доксорубицин или эпирубицин.
| US2013095172 A1, 18.04.2013 | |||
| US2011044977 A1, 24.02.2011 | |||
| HOFFMANN-LA ROCHE, A Dose-Finding Study of Pertuzumab (Perjeta) in Combination With Trastuzumab (Herceptin) in Healthy Male Participants and Women With Early Breast Cancer (EBC), 14.04.2016 | |||
| HAMIZI S | |||
| et al., Subcutaneous trastuzumab: development of a new formulation for treatment of |

