ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится, главным образом, к области лечения рассеянного склероза.
УРОВЕНЬ ТЕХНИКИ
Рассеянный склероз (РС) представляет собой хроническое иммуноопосредуемое демиелинизирующее заболевание центральной нервной системы (ЦНС). Иммунная система атакует миелиновую оболочку нервов в ЦНС и сами нервные волокна. РС является наиболее распространенным аутоиммунным нарушением, поражающим ЦНС, и основной причиной нетрудоспособности у молодых взрослых людей. Заболевание обычно начинается в возрасте от 20 до 50 лет. В 2015 году количество страдающих им людей по всему миру составило приблизительно 2,3 миллиона.
Существует несколько форм РС, либо с новыми симптомами, возникающими во время отдельных атак (рецидивирующие формы), либо с постепенным прогрессированием заболевания с течением времени без типичных рецидивов (прогрессирующие формы). Прогрессирующие формы включают первично-прогрессирующий РС и вторично-прогрессирующий РС.
Несмотря на интенсивные исследования механизмы патогенеза заболевания остаются неясными и, хотя существует множество лекарственных средств против РС, одобренных FDA, все еще отсутствует излечивающее средство. Среди этих лекарственных средств, большинство одобрено для лечения ремиттирующего РС, в то время как только одно лекарственное средство одобрено FDA для лечения первично-прогрессирующего РС. Современные лекарственные средства, используемые для лечения как ремиттирующих, так и прогрессирующих форм РС, хотя и являются умеренно эффективными, могут иметь серьезные побочные эффекты или могут плохо переноситься. Кроме того, многие из этих лекарственных средств должны вводиться парентеральным путем, что является неудобством для пациентов в контексте хронического заболевания, такого как РС.
Таким образом, существует потребность в новом лекарственном средстве для лечения РС, в частности, в лекарственных средствах, которые также могут быть эффективными против прогрессирующих форм заболевания и/или которые демонстрируют меньше побочных эффектов, чем современные способы терапии, и которые можно вводить пероральным путем. Настоящее изобретение удовлетворяет эти и другие потребности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к новым способам лечения рассеянного склероза с использованием (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата.
Таким образом, настоящее изобретение относится к (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амину или его фармацевтически приемлемой соли или сольвату для применения для лечения рассеянного склероза.
Кроме того, настоящее изобретение относится к способу лечения рассеянного склероза у пациента (предпочтительно человека), включающему введение пациенту терапевтически эффективного количества (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата.
Кроме того, настоящее изобретение относится к применению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для изготовления лекарственного средства для лечения рассеянного склероза.
Кроме того, настоящее изобретение относится к применению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для лечения рассеянного склероза.
В некоторых вариантах осуществления рассеянный склероз представляет собой хронический прогрессирующий рассеянный склероз, в частности, первично-прогрессирующий рассеянный склероз или вторично-прогрессирующий рассеянный склероз.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
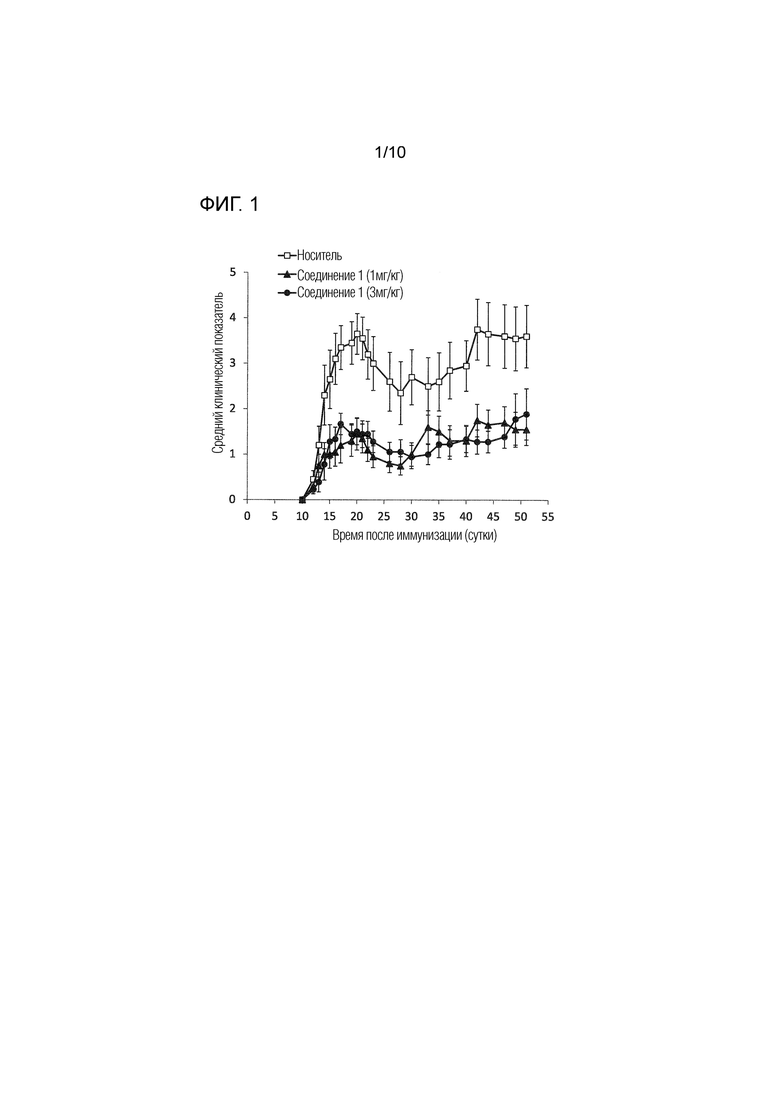
На фиг.1 представлены результаты, полученные для соединения 1 в дозе 1 и 3 мг/кг п/о в модели экспериментального аутоиммунного энцефаломиелита (EAE) у мышей, как описано в примере 3.1 и 3.2. Данные отражают прогрессирование заболевания для каждой группы, определенное в качестве среднего клинического показателя (±SEM).
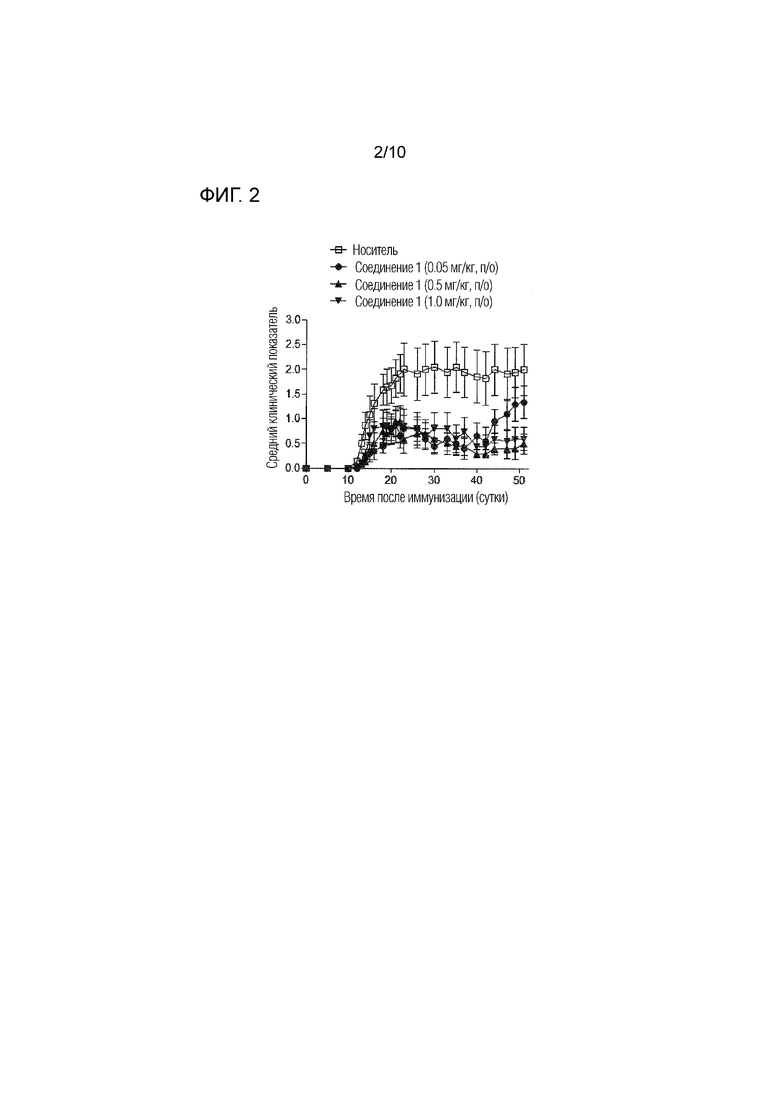
На фиг.2 представлены эффекты соединения 1 в дозе 1, 0,5 и 0,05 мг/кг п/о в модели EAE, как описано в примере 3.3. Данные отражают прогрессирование заболевания для каждой группы, определенное в качестве среднего клинического показателя (±SEM).
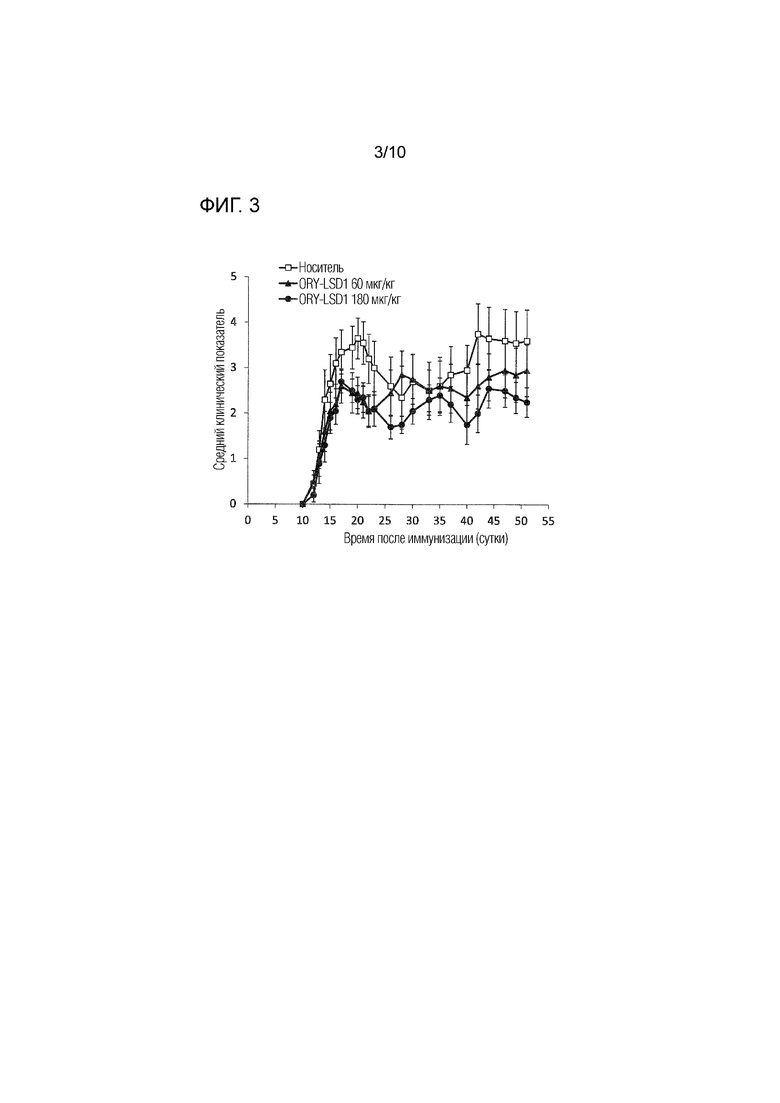
На фиг.3 представлены эффекты ингибитора LSD1, обозначаемого как «ORY-LSD1» (как дополнительно определено в примере 1) в дозе 0,06 и 0,180 мг/кг п/о в модели EAE, как описано в примере 3,4. Данные отражают прогрессирование заболевания для каждой группы, определенное в качестве среднего клинического показателя (±SEM).
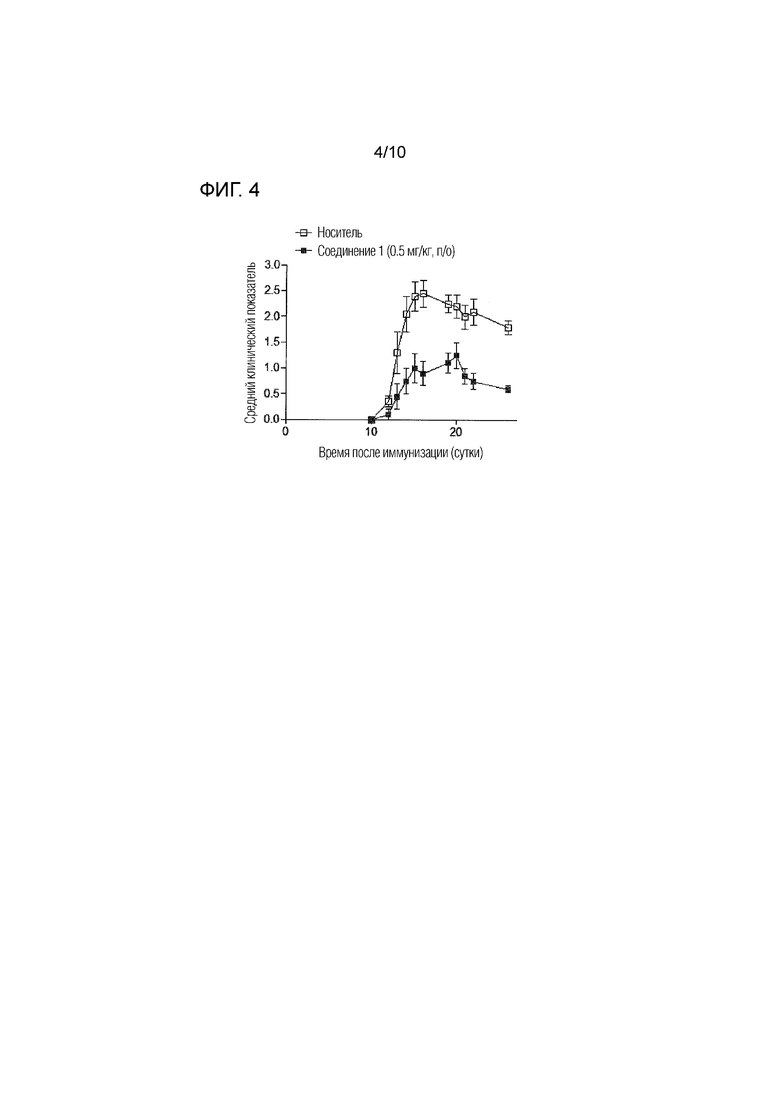
На фиг.4 представлены эффекты соединения 1 в дозе 0,5 мг/кг п/о в анализе с EAE, как описано в примере 4. Данные отражают прогрессирование заболевания для каждой группы, определенное в качестве среднего клинического показателя (±SEM).
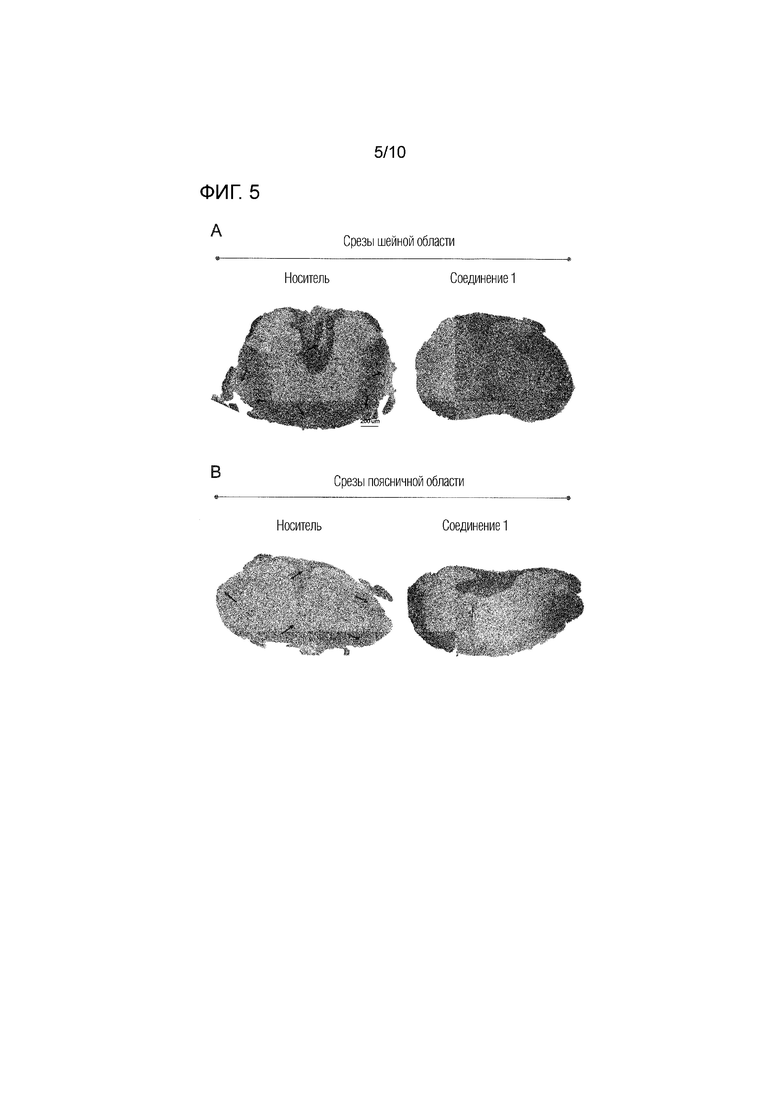
На фиг.5 представлены результаты гистопатологического анализа спинного мозга, извлеченного в конце лечения (через 26 суток после иммунизации) из животных, которым вводили соединение 1 в дозе 0,5 мг/кг п/о или носитель в анализе с EAE, как описано в примере 4. Представленные изображения соответствуют поперечным срезам шейного (A) и поясничного (B) отдела спинного мозга на пике клинического заболевания, окрашенным по Клювер-Баррера. Стрелки указывают на области демиелинизации и инфильтрации воспалительных клеток. Горизонтальной планкой указан масштаб 200 мкм.
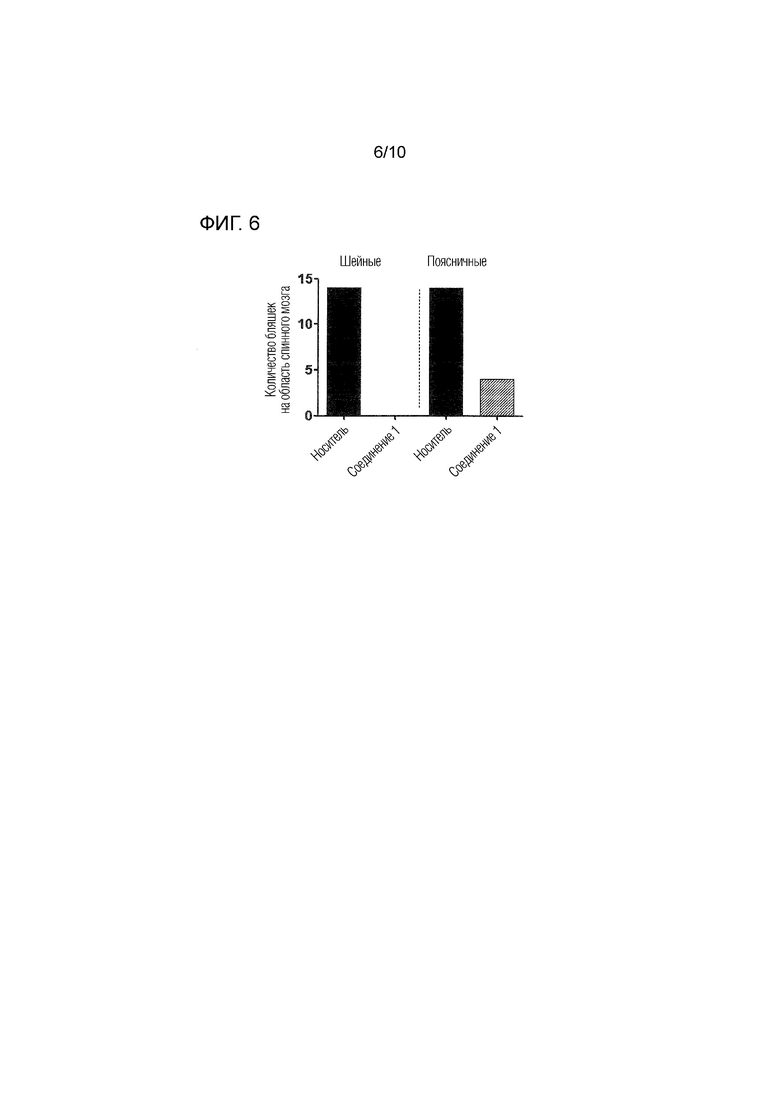
На фиг.6 представлено среднее количество бляшек демиелинизации в поясничной и шейной областях, соответствующих спинному мозгу, извлеченному согласно примеру 4, демонстрирующее отсутствие или значительное снижение демиелинизации в срезах шейной и поясничной области спинного мозга, соответственно, животных, которым вводили соединение 1.
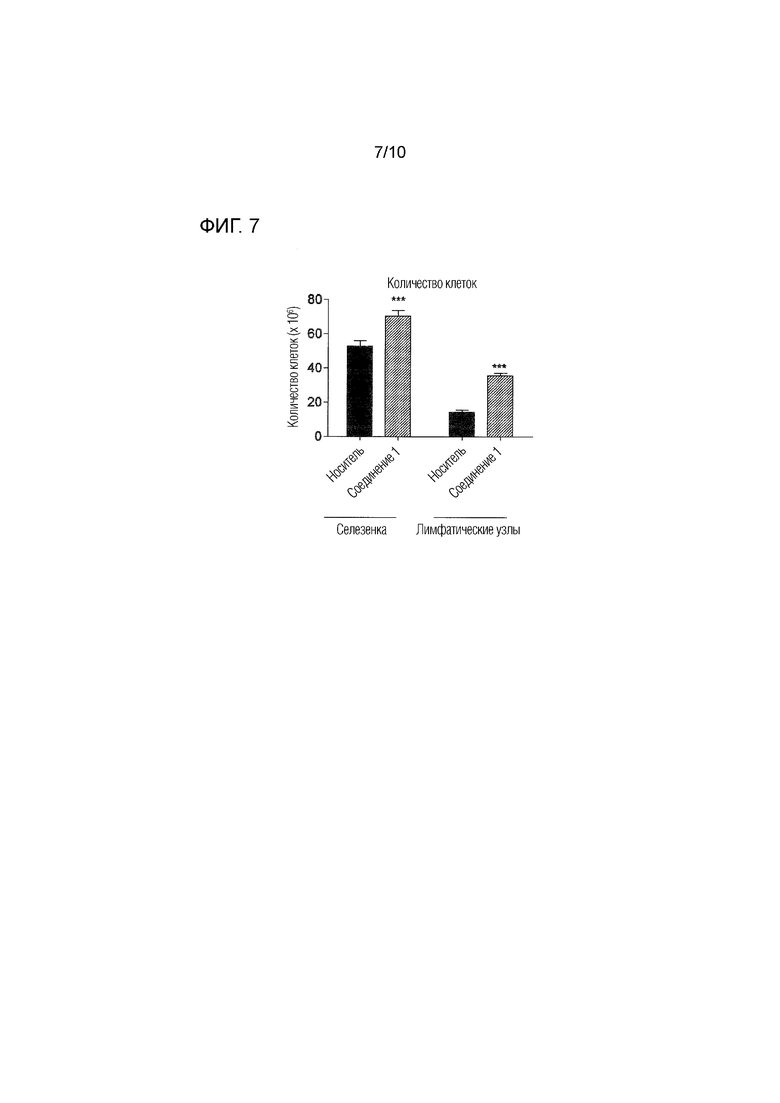
На фиг.7 представлено количество иммунных клеток, выделенных из селезенки и лимфатических узлов животных, которым вводили соединение 1 в дозе 0,5 мг/кг п/о или носитель согласно примеру 4, демонстрирующее значительное увеличение количества T-клеток, остающихся в селезенке и лимфатических узлах животных, которым вводили соединение 1, что указывает на снижение выхода лимфоцитов из иммунных тканей.
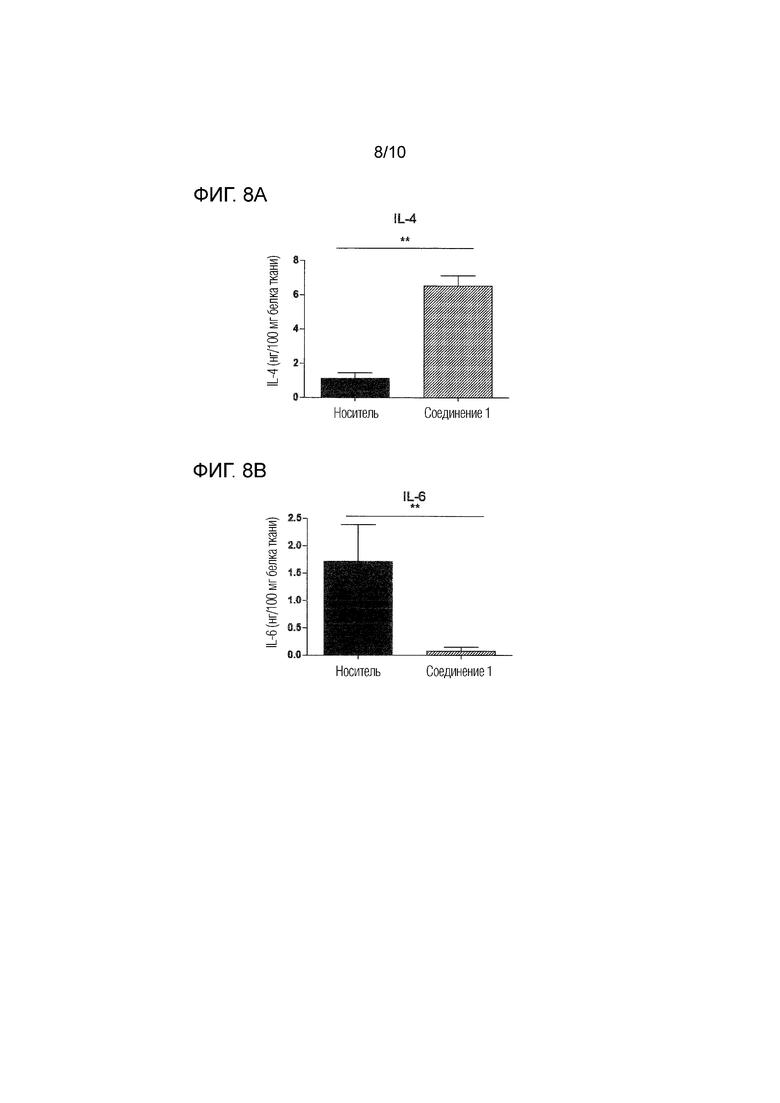
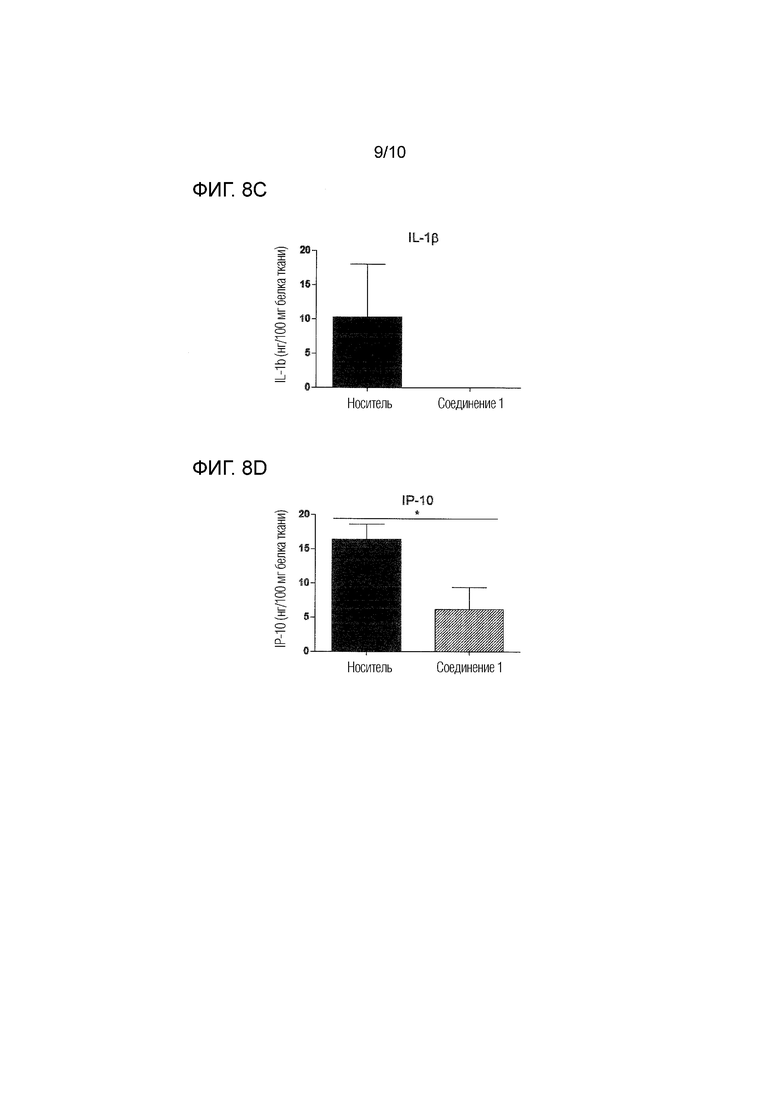
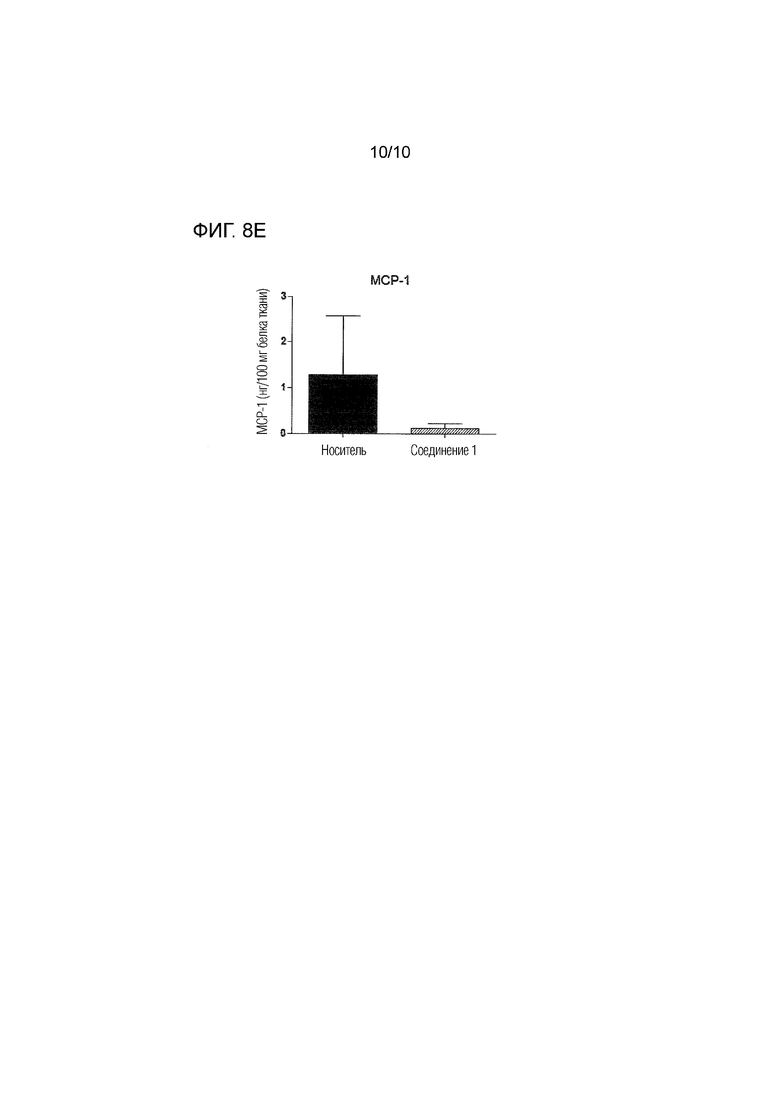
На фиг.8 представлены уровни нескольких цитокинов и хемокинов, определенные посредством ELISA в спинном мозге, извлеченном через 26 суток после иммунизации животных, которым вводили соединение 1 в дозе 0,5 мг/кг п/о или носитель согласно примеру 4. Фиг.8A: IL-4; фиг.8B: IL-6; фиг.8C: IL1-бета; фиг.8D: IP-10; фиг.8E: MCP-1. Уровни выражены в качестве нг/100 мг белка ткани.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В основе настоящего изобретения лежит идентификация соединения (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина в качестве высокоэффективного терапевтического средства для лечения рассеянного склероза, как более подробно пояснено в настоящем описании ниже и проиллюстрировано в разделе «Примеры». Это соединение, (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин, обозначается в примерах и на чертежах как соединение 1 (или Comp. 1). Наименования «(-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин», «Соединение 1» или «Comp. 1» используются в настоящем описании взаимозаменяемо.
Таким образом, настоящее изобретение относится к (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амину или его фармацевтически приемлемой соли или сольвату для применения для лечения рассеянного склероза.
Кроме того, настоящее изобретение относится к способу лечения рассеянного склероза у пациента (предпочтительно человека), включающему введение пациенту терапевтически эффективного количества (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата.
Кроме того, настоящее изобретение относится к применению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для изготовления лекарственного средства для лечения рассеянного склероза.
Кроме того, настоящее изобретение относится к применению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для лечения рассеянного склероза.
В некоторых вариантах осуществления рассеянный склероз представляет собой хронический прогрессирующий рассеянный склероз (например, первично-прогрессирующий рассеянный склероз или вторично-прогрессирующий рассеянный склероз).
Таким образом, настоящее изобретение, кроме того, относится к (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амину или его фармацевтически приемлемой соли или сольвату для применения для лечения хронического прогрессирующего рассеянного склероза.
Кроме того, настоящее изобретение относится к способу лечения хронического прогрессирующего рассеянного склероза у пациента (предпочтительно человека), включающему введение пациенту терапевтически эффективного количества (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата.
Кроме того, настоящее изобретение относится к применению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для изготовления лекарственного средства для лечения хронического прогрессирующего рассеянного склероза.
Кроме того, настоящее изобретение относится к применению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для лечения хронического прогрессирующего рассеянного склероза.
Предпочтительно, соединение (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин (или его фармацевтически приемлемую соль или сольват) вводят перорально. Иллюстративные составы, которые можно вводить путем перорального применения (или проглатывания), более подробно описаны в настоящем описании ниже.
Как объяснено выше, настоящее изобретение относится к соединению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амину или фармацевтически приемлемой соли или сольвату указанного соединения для применения для лечения рассеянного склероза. Таким образом, изобретение относится к соединению (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амину в виде свободного основания (в несолевой форме) для применения для лечения рассеянного склероза (например, хронического прогрессирующего рассеянного склероза) и, более того, изобретение также относится к фармацевтически приемлемой соли или сольвату (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина для применения для лечения рассеянного склероза (например, хронического прогрессирующего рассеянного склероза).
Как проиллюстрировано в разделе «Примеры», соединение (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин обеспечивает заметные терапевтические эффекты в моделях рассеянного склероза на животных. В частности, соединение 1 было протестировано с использованием модели экспериментального аутоиммунного энцефаломиелита (EAE). EAE демонстрирует патологическое и клиническое сходство с РС человека и широко используется в качестве модельной системы для тестирования потенциальных терапевтических средств против РС. В частности, модель EAE на мышах, как описано в разделе «Примеры», с использованием MOG35-55 и линии мышей C57BL/6 считается проверенной доклинической моделью хронической прогрессирующей формы РС.
Эффекты соединения 1 на хронический активный EAE были оценены в терапевтическом режиме, т.е. при введении соединения после появления симптомов заболевания. Как более подробно проиллюстрировано в примере 3 и на фиг.1, 2 и 4, введение соединения 1 в значительной степени ингибировало развитие EAE и снижало заболеваемость и тяжесть, определяемые с использованием ежедневного среднего клинического показателя. Например, в анализе с EAE, где соединение 1 вводили в дозе 1 или 3 мг/кг п/о, в то время как у мышей, которым вводили носитель, развивались признаки EAE от умеренных до тяжелых, и они продемонстрировали смертность вследствие тяжелого паралича, в группах, в которых вводили соединение 1, 40-70% мышей имели мягкие симптомы и 30% из них практически полностью выздоровели через 40 суток после начала заболевания. Было обнаружено, что соединение 1 является эффективным в этой модели РС в дозах уже 0,05 мг/кг п/о, как показано в примере 3.3 и на фиг.2. Важно, что защитный эффект соединения 1 сохранялся в течение длительного периода времени после прекращения введения.
Примечательно, что соединение 1 демонстрирует быстрое начало действия против прогрессирования заболевания, проявляя благоприятные эффекты на ежедневный клинический показатель уже вскоре после начала введения, как показано, например, на фиг.1. Соединение 1, таким образом, может быть полезным для обеспечения раннего смягчения острых атак РС или быстро-прогрессирующего рассеянного склероза, и может обеспечить альтернативу стандартному лечению высокой дозой в/в кортикостероидами, особенно в случаях гиперчувствительности или аллергии на кортикостероиды.
Как проиллюстрировано в примере 4 и на фиг.5 и 6, соединение 1 является пригодным для уменьшения инфильтрации иммунных клеток в спинной мозг, а также для уменьшения демиелинизации в спинном мозге, как показано у мышей с EAE. Лечение соединением 1 снижает выход лимфоцитов из иммунных тканей, на что указывает значительное увеличение количества иммунных клеток, остающихся в селезенке и лимфатических узлах, как более подробно описано в примере 4 и на фиг.7. Соединение 1 также снижает уровень провоспалительных цитокинов, таких как IL-6 и IL-1-бета, и хемокинов, таких как IP-10 и MCP-1, в спинном мозге (см. фиг.8). Уровень цитокина IL-4 был значительно увеличен в спинном мозге животных, которых лечили соединением 1, что указывает на противовоспалительный ответ Th2 (фиг.8A).
Важно, что терапевтические эффекты соединения 1 при РС могут быть достигнуты в дозах, которые не вызывают клинически значимых эффектов на гематологию или количество циркулирующих лимфоцитов, что является общим побочным эффектом лекарственных средств против РС, и/или без признаков желудочно-кишечной токсичности. Таким образом, соединение 1 можно использовать для лечения РС, включая прогрессирующий РС без возникновения клинически значимых эффектов на гематологию и количество циркулирующих лимфоцитов.
Было обнаружено, что терапевтические эффекты соединения 1 при лечении РС неожиданно являются выдающимися также по сравнению с эффектами других ингибиторов LSD1. Соединение 1 представляет собой необратимый ингибитор LSD1 на основе циклопропиламино. С использованием EAE, являющегося моделью для РС, согласно примеру 3.1, эффекты соединения 1 сравнивали с другим необратимым ингибитором LSD1 на основе циклопропиламино - соединением, обозначаемым как ORY-LSD1, более подробно описанным в примере 1. Соединение 1 демонстрирует IC50 в отношении LSD1 90 нМ, в то время как ORY-LSD1 имеет IC50 в отношении LSD1 10 нМ, как более подробно описано в примере 2. Поскольку эти два соединения имеют различную эффективность in vitro против LSD1, ORY-LSD1 было протестировано в модели EAE в примере 3 в дозах, эквивалентных дозам, используемых для соединения 1 для ингибирования LSD1 in vivo. В то время как ORY-LSD1 продемонстрировало явную тенденцию к улучшению (фиг.3), Соединение 1 было значительно более эффективным, чем ORY-LSD1. Таким образом, соединение 1 является особенно пригодным ингибитором LSD1 для применения для лечения рассеянного склероза.
Фармацевтические составы
Хотя является возможным введение соединения 1 для применения в терапии непосредственно в чистом виде, его обычно вводят в форме фармацевтической композиции, которая содержит соединение 1 в качестве активного фармацевтического ингредиента вместе с одним или несколькими фармацевтически приемлемыми эксципиентами или носителями. Любое упоминание соединения 1 в настоящем описании включает соединение в виде свободного основания и любой его фармацевтически приемлемой соли или сольвата.
Соединение 1 можно вводить любым способом, который обеспечивает достижение цели. Примеры включают введение пероральным, парентеральным, внутривенным, подкожным или местным путем.
Для пероральной доставки соединение 1 можно включать в состав, который включает фармацевтически приемлемые носители, такие как связующие вещества (например, желатин, целлюлоза, трагакантовая камедь), эксципиенты (например, крахмал, лактоза), смазывающие вещества (например, стеарат магния, диоксид кремния), дезинтегрирующие вещества (например, альгинат, примогель и кукурузный крахмал), и подсластители или вкусовые добавки (например, глюкоза, сахароза, сахарин, метилсалицилат и мята перечная). Состав можно доставлять перорально в форме закрытых желатиновых капсул или прессованных таблеток. Капсулы и таблетки можно получать любым общепринятым способом. Капсулы и таблетки также можно покрывать различными покрытиями, известными в данной области, для модификации запаха, вкуса, цвета и формы капсул и таблеток. Кроме того, также в капсулы могут быть включены жидкие носители, такие как жирное масло.
Подходящие пероральные составы также могут иметь форму суспензии, сиропа, жевательной резинки, пастилки, эликсира и т.п. Если желательно, также могут быть включены общепринятые средства для модификации запаха, вкуса, цвета и формы конкретных форм. Кроме того, для удобного введения посредством трубки для энтерального питания у пациентов, неспособных глотать, активные можно растворять в приемлемом липофильном носителе на основе растительного масла, такого как оливковое масло, кукурузное масло и сафлоровое масло.
Соединение 1 также можно вводить парентеральным путем в форме раствора или суспензии, или лиофилизированной форме, которая может быть преобразована в форму раствора или суспензии перед применением. В таких составах можно использовать разбавители или фармацевтически приемлемые носители, такие как стерильная вода и физиологический солевой буфер. Могут быть включены другие общепринятые растворители, pH-буферы, стабилизаторы, антибактериальные средства, поверхностно-активные вещества и антиоксиданты. Например, пригодные компоненты включают хлорид натрия, ацетатные, цитратные или фосфатные буферы, глицерин, декстрозу, жирные масла, метилпарабены, полиэтиленгликоль, пропиленгликоль, бисульфат натрия, бензиловый спирт, аскорбиновую кислоту и т.п. Парентеральные составы можно хранить в любых общепринятых контейнерах, таких как флаконы и ампулы.
Для местного введения соединение 1 можно составлять в форме лосьонов, кремов, мазей, гелей, порошков, паст, спреев, суспензий, капель и аэрозолей. Таким образом, в составы может быть включен один или несколько загустителей, увлажнителей и стабилизаторов. Примеры таких средств включают, но не ограничиваются ими, полиэтиленгликоль, сорбит, ксантановую смолу, вазелин, пчелиный воск или минеральное масло, ланолин, сквален и т.п. Конкретной формой местного введения является доставка через трансдермальный пластырь. Способы получения трансдермальных пластырей описаны, например, в Brown, et al. (1988) Ann. Rev. Med. 39:221-229, которая включена в настоящее описание в качестве ссылки.
Также подходящим путем введения может быть подкожная имплантация состава соединения 1 с замедленным высвобождением. Это влечет за собой хирургические процедуры по имплантации активного соединения в любом подходящем составе в подкожное пространство, например, передней стенки живота. См., например, Wilson et al. (1984) J. Clin. Psych. 45:242-247. В качестве носителя для замедленного высвобождения активных соединений можно использовать гидрогели. Гидрогели в основном известны в данной области. Их, как правило, получают путем сшивания высокомолекулярных биосовместимых полимеров в сеть, которая набухает в воде с образованием гелеобразного материала. Предпочтительно, гидрогели являются биодеградируемыми и биорассасывающимися. Для целей настоящего изобретения могут быть использованы гидрогели, изготовленные из полиэтиленгликолей, коллагена или сополимера гликолевой и L-молочной кислот. См., например, Phillips et al. (1984) J. Pharmaceut. Sci., 73: 1718-1720.
Соединение 1 также можно конъюгировать с растворимым в воде неиммуногенным непептидным высокомолекулярным полимером с образованием конъюгата полимера. Например, соединение 1 можно ковалентно связывать с полиэтиленгликолем с получением конъюгата. Как правило, такой конъюгат демонстрирует улучшенную растворимость, стабильность и сниженную токсичность и иммуногенность. Таким образом, при введении пациенту соединение 1 в конъюгате может иметь большее время полужизни в организме и может демонстрировать лучшую эффективность. См., главным образом, Burnham (1994) Am. J. Hosp. Pharm. 15:210-218. Пегилированные белки в настоящее время используют в белок-заместительной терапии и для других терапевтических применений. Например, пегилированный интерферон (PEG-INTRON A®) клинически используют для лечения гепатита B. Пегилированную аденозиндезаминазу (ADAGEN®) используют для лечения тяжелого комбинированного иммунодефицита (SCID). Пегилированную L-аспарагиназу (ONCAPSPAR®) используют для лечения острого лимфобластного лейкоза (ALL). Является предпочтительным, чтобы ковалентная связь между полимером и активным соединением и/или в самом полимере была гидролитически деградируемой в физиологических условиях. Такие конъюгаты, известные как «пролекарства», могут без труда высвобождать активное соединение внутрь организма. Контролируемого высвобождения активного соединения также можно достигать путем включения активного ингредиента в микрокапсулы, нанокапсулы или гидрогели, в основном известные в данной области. Другие фармацевтически приемлемые пролекарства соединения 1 включают, но не ограничиваются ими, сложные эфиры, карбонаты, тиокарбонаты, N-ацильные производные, N-ацилоксиалкильные производные, четвертичные производные третичных аминов, N-основания Манниха, основания Шиффа, конъюгаты аминокислот, фосфатные сложные эфиры, соли металлов и сульфонатные сложные эфиры.
Также в качестве носителей для активного соединения можно использовать липосомы. Липосомы представляют собой мицеллы, изготовленные из различных липидов, таких как холестерин, фосфолипиды, жирные кислоты и их производные. Также можно использовать различные модифицированные липиды. Липосомы могут снижать токсичность активных соединений и повышать их стабильность. Способы получения липосомальных суспензий, содержащих активные ингредиенты, в основном известны в данной области. См., например, патент США № 4522811; Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N. Y. (1976).
Фармацевтические композиции, такие как пероральные и парентеральные композиции, можно составлять в виде единичных дозированных форм для простоты введения и единообразия дозировки. Как используют в рамках изобретения, «единичные дозированные формы» относятся к физически дискретным единицам, пригодным в качестве единичных дозировок для введения индивидуумам, причем каждая единица содержит заданное количество активного ингредиента, вычисленное для обеспечения желаемого терапевтического эффекта, совместно с одним или несколькими подходящими фармацевтическими носителями.
В терапевтических применениях фармацевтические композиции вводят способом, пригодным для заболевания, подлежащего лечению, как может определить специалист в области медицины. Подходящая доза и подходящая длительность и частота введения определяются такими факторами, как состояние пациента, тип и тяжесть заболевания, конкретная форма активного ингредиента, способ введения, среди прочих. Как правило, посредством надлежащей дозы и режима введения предоставляют фармацевтическую композицию в количестве, достаточном для обеспечения терапевтической пользы, например, улучшенного клинического исхода, такого как более частые полные или частичные ремиссии или более длительная свободная от заболевания и/или общая выживаемость, или уменьшение тяжести симптомов, или любое другое поддающееся объективному определению улучшение, которое может клинический специалист. Эффективные дозы, как правило, можно оценивать или экстраполировать с использованием экспериментальных моделей, таких как кривые доза-эффект на основе модельных тест-систем in vitro или на животных, таких как тест-системы, проиллюстрированные в разделе «Примеры».
Фармацевтические композиции по изобретению могут быть включены в контейнер, упаковку или дозатор вместе с инструкциями по введению.
Соединение 1 является перорально активным и эффективным для лечения РС при пероральном введении, как проиллюстрировано в примерах 3 и 4. Таким образом, предпочтительно, чтобы соединение 1 вводили для лечения РС пероральным путем.
Определения
Если не определено иначе, все технические и научные термины, используемые в настоящем описании, обладают тем же значением, которое обычно подразумевает специалист в области, к которой настоящее изобретение относится.
В настоящем описании и формуле изобретения применимы следующие определения, если нет иных конкретных указаний.
«Пациент» или «индивидуум» для целей настоящего изобретения включает как человека, так и других животных, в частности, млекопитающих, и другие организмы. Таким образом, способы применимы как к терапии человека, так и к ветеринарным применениям. В предпочтительном аспекте индивидуумом или пациентом является млекопитающее и в наиболее предпочтительном аспекте индивидуум или пациент является человеком.
Термины «лечение», «осуществление лечения» и т.п. используют в настоящем описании для обозначения в общем способа достижения желаемого фармакологического и/или физиологического эффекта. Эффект может быть профилактическим с точки зрения полного или частичного предупреждения заболевания или его симптома и/или может быть терапевтическим с точки зрения частичного или полного излечения заболевания и/или неблагоприятного эффекта, связанного с заболеванием. Термин «лечение», как используют в рамках изобретения, охватывает любое лечение заболевания у пациента и включает: (a) предупреждение заболевания у пациента, который может быть предрасположенным/иметь риск развития заболевания; (b) ингибирование заболевания, т.е. остановку его развития; или (c) смягчение заболевания, т.е. обеспечение регрессии заболевания. Как используют в рамках изобретения, термин «осуществление лечения заболевания» или «лечение заболевания» относится, в частности, к замедлению или обращению вспять прогрессирования заболевания. Осуществление лечения заболевания включает лечение симптома и/или уменьшение симптомов заболевания.
Как используют в рамках изобретения, термин «терапевтически эффективное количество» относится к количеству, достаточному для достижения желаемого биологического эффекта (например, терапевтического эффекта) у индивидуума. Таким образом, терапевтически эффективное количество соединения может представлять собой количество, достаточное для лечения заболевания и/или отсрочивания возникновения или прогрессирования заболевания, и/или для смягчения одного или нескольких симптомов заболевания при введении индивидууму, страдающему заболеванием или предрасположенному к нему.
Как используют в рамках изобретения, «фармацевтически приемлемая соль» означает соль, которая сохраняет биологическую эффективность свободных кислот и оснований определенного соединения и которая не является с биологической или иной точки зрения нежелательной. Соединение может обладать достаточно кислотными, достаточно основными группами, или и теми, и другими, и, таким образом, может реагировать с любым из ряда неорганических или органических оснований, и неорганических и органических кислот с образованием фармацевтически приемлемой соли. Иллюстративные фармацевтически приемлемые соли включают соли, полученные реакцией соединения 1 с минеральной или органической кислотой, такие как гидрохлориды, гидробромиды, сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, нитраты, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиоляты, оксалаты, малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бутин-1,4 диоаты, гексин-l,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, гамма-гидроксибутираты, гликоляты, тартраты, метан-сульфонаты, этан-сульфонаты, пропансульфонаты, бензолсульфонаты, толуолсульфонаты, трифторметансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, миндаляты, пируваты, стеараты, аскорбаты или салицилаты. Когда соединение содержит кислотную часть, его подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, например, соли натрия или калия; соли щелочноземельных металлов, например, соли кальция или магния; и соли, образованные с подходящими органическими лигандами, такими как аммиак, алкиламины, гидроксиалкиламины, лизин, аргинин, N-метилглюкамин, прокаин и т.п. Фармацевтически приемлемые соли хорошо известны в данной области.
Как используют в рамках изобретения, «фармацевтически приемлемый сольват» относится к комплексу с переменной стехиометрией, образованному растворенным веществом и фармацевтически приемлемым растворителем, таким как вода, этанол и т.п. Комплекс с водой известен как гидрат.
Как используют в рамках изобретения, «фармацевтически приемлемый носитель» или «фармацевтически приемлемый эксципиент» относится к веществам, не являющимся API (API означает активный фармацевтический ингредиент), таким как разрыхлители, связующие вещества, наполнители и смазывающие вещества, используемые для составления фармацевтических продуктов. Их введение обычно является безопасным для человека в соответствии с принятыми государственными стандартами, включая стандарты, объявленные Food and Drug Administration США и European Medical Agency. Фармацевтически приемлемые носители или эксципиенты хорошо известны специалистам в данной области.
ПРИМЕРЫ
Следующие примеры иллюстрируют различные аспекты изобретения. Безусловно, следует понимать, что примеры только иллюстрируют определенные варианты осуществления изобретения и не ограничивают объем изобретения. Результаты также представлены и описаны на чертежах и на легендах к чертежам.
Пример 1: Материалы
Соединение 1 представляет собой соединение (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин, которое может быть получено, как описано в WO 2012/013728.
ORY-LSD1 представляет собой соединение N-((1R,2S)-2-(2-фторфенил)циклопропил)пиперидин-4-амин, которое может быть получено, как описано в WO 2013/057320.
Пример 2: Биохимические анализы in vitro
2.1 LSD1
Ингибиторную активность представляющего интерес соединения в отношении LSD1 можно исследовать с использованием способа, описанного ниже:
Использовали рекомбинантный белок LSD1 человека от BPS Bioscience Inc (каталожный справочный номер 50100: рекомбинантный LSD1 человека, номер доступа GenBank № NM_015013, аминокислоты с 158 до конца с N-концевой GST-меткой, ММ: 103 кДа). Для мониторинга ферментативной активности LSD1 и/или скорости его ингибирования исследуемым соединением в качестве субстрата был выбран диметилированный пептид H3-K4 (Anaspec). Активность деметилазы оценивали в аэробных условиях путем количественного определения высвобождения продуцированного H2O2 в ходе каталитического процесса с использованием набора для анализа пероксидводорода/пероксидазы Amplex® Red (Invitrogen).
В кратком изложении, фиксированное количество LSD1 инкубировали на льду в течение 15 минут в отсутствие и/или в присутствии по меньшей мере восьми 3-кратных серийных разведений соответствующего ингибитора (например, от 0 до 75 мкМ, в зависимости от концентрации ингибитора). Для контроля ингибирования использовали транилципромин (Biomol International). В каждом эксперименте каждую концентрацию ингибитора тестировали в двух экземплярах. После обеспечения возможности ферменту взаимодействовать с ингибитором, в каждую реакционную смесь добавляли KM диметилированного пептида H3-K4 и экспериментальную смесь оставляли на 30 минут при 37°C в темноте. Ферментативные реакции проводили в 50 мМ натрий-фосфатном буфере, pH 7,4. В конце инкубации к реакционной смеси добавляли реагент Amplex® Red и раствор пероксидазы хрена (HPR) в соответствии с рекомбинациями поставщика (Invitrogen) и оставляли инкубироваться в течение дополнительных 5 минут при комнатной температуре в темноте. В качестве контроля эффективности набора использовали 1 мкМ раствор H2O2. Мониторинг конвертирования реагента Amplex® Red в резоруфин вследствие присутствия H2O2 в анализируемой смеси проводили по флуоресценции (возбуждение при 540 нм, испускание при 590 нм) с использованием устройства для считывания микропланшетов (Infinite 200, Tecan). Для измерения уровня H2O2, продуцированного в отсутствие и/или в присутствии ингибитора, использовали произвольные единицы.
Максимальную активность деметилазы LSD1 достигали в отсутствие ингибитора и в нее вносили поправку на фоновую флуоресценцию в отсутствие LSD1. Величину IC50 каждого ингибитора вычисляли с использованием программного обеспечения GraphPad Prism.
2.2 МОНОАМИНОКСИДАЗА A (MAO-A) И B (MAO-B)
LSD1 имеет высокую степень структурного сходства и идентичности/гомологии аминокислот с флавин-зависимыми аминоксидазами: моноаминоксидазами A (MAO-A) и B (MAO-B). Для определения уровня селективности ингибитора LSD1 против MAO-A и MAO-B, ингибиторную активность представляющего интерес соединения против MAO-A и MAO-B можно тестировать с использованием способа, описанного ниже:
Рекомбинантные белки моноаминоксидазы MAO-A и MAO-B человека приобретали от Sigma Aldrich. MAO катализируют окислительное дезаминирование первичных, вторичных и третичных аминов. Для мониторинга ферментативной активности MAO и/или скорости их ингибирования посредством представляющего интерес ингибитора(ов) проводили скрининговый анализ (ингибитора) на основе флуоресценции. В качестве субстрата использовали 3-(2-аминофенил)-3-оксопропанамин (кинурамина дигидробромид, Sigma Aldrich), нефлуоресцентное соединение. Кинурамин является неспецифическим субстратом активности как MAO-A, так и MAO-B. Подвергаясь окислительному дезаминированию посредством активности MAO, кинурамин конвертируется в 4-гидроксихинолин (4-HQ), конечный флуоресцентный продукт.
Активность моноаминоксидазы оценивали путем количественного определения конвертирования кинурамина в 4-гидроксихинолин. Анализ проводили в 96-луночных черных планшетах с прозрачным дном (Corning) в конечном объеме 100 мкл. Буфер для анализа представлял собой 100 мМ HEPES, pH 7,5. Каждый эксперимент проводили в двух экземплярах в каждом эксперименте.
В кратком изложении, фиксированное количество MAO инкубировали на льду в течение 15 минут в реакционном буфере в отсутствие и/или в присутствии по меньшей мере восьми 3-кратных серийных разведений каждой из них. В качестве контроля специфического ингибирования MAO-A и MAO-B, соответственно, использовали клоргилин и депренил (Sigma Aldrich).
После обеспечения возможности ферменту(ам) взаимодействовать с ингибитором, в каждую реакционную смесь добавляли KM кинурамина для анализа MAO-B и MAO-A, соответственно, и реакционную смесь оставляли стоять в течение 1 часа при 37°C в темноте. Окислительное дезаминирование субстрата останавливали добавлением 50 мкл 2 Н NaOH. Мониторинг конвертирования кинурамина в 4-гидроксихинолин проводили по флуоресценции (возбуждение при 320 нм, испускание при 360 нм) с использованием устройства для считывания микропланшетов (Infinite 200, Tecan). Для количественного определения уровней флуоресценции в отсутствие и/или в присутствии ингибитора использовали произвольные единицы.
Максимум активности окислительного дезаминирования был достигнут путем определения количества 4-гидроксихинолина, образовавшегося в результате дезаминирования кинурамина в отсутствие ингибитора, и внесения поправки на фоновую флуоресценцию в отсутствие ферментов MAO. Величины IC50 для каждого ингибитора вычисляли с использованием программного обеспечения GraphPad Prism.
2.3 РЕЗУЛЬТАТЫ
Иллюстративные величины IC50 в отношении LSD1, MAO-A и MAO-B, полученные с использованием описанных выше способов для соединения 1 и ORY-LSD1, представлены в таблице ниже:
IC50 (мкМ)
Как можно видеть из приведенных выше данных, соединение 1 является мощным двойным ингибитором LSD1/MAO-B. ORY-LSD1 является мощным ингибитором LSD1 с селективностью в отношении LSD1 относительно MAO-A и MAO-B.
Пример 3: Оценка эффективности соединения 1 в отношении экспериментального аутоиммунного энцефаломиелита у мышей
Модель экспериментального аутоиммунного энцефаломиелита (EAE) демонстрирует патологическое и клиническое сходство с рассеянным склерозом (РС) у человека и широко используется в качестве модели РС. В частности, модель EAE на мышах, как описано в настоящем описании, с использованием MOG35-55 и линии мышей C57BL/6, считается подтвержденной доклинической моделью хронической прогрессирующей формы РС.
3.1 СПОСОБ
Для индукции хронического EAE посредством активной иммунизации мышей C57BL/6 имменизировали п/к посредством 100 мкг гликопротеина миелина олигодендроцитов MOG35-55, эмульгированного в полном адъюванте Фрейнда (CFA), содержащем 4 мг/мл H37 RA Mycobacterium tuberculosis. Мышам также проводили в/б инъекции 200 нг коклюшного токсина на 0 и 2 сутки.
Лечение состояло в пероральном введении соединения 1 (в дозе 1 мг/кг или 3 мг/кг) после возникновения заболевания (12 сутки после иммунизации), один раз в сутки, в течение пяти последовательных суток с 12 суток по 16 сутки после иммунизации и с 19 суток по 23 сутки после иммунизации. Контрольным мышам перорально вводили носитель [2% об./об. Tween-80+98% HPβCD (13% масс./об.)] по тому же режиму введения, что и для соединения 1. n=10 мышей/группа за исключением группы, в которой вводили соединение 1 в дозе 3 мг/кг, где n=9.
Мышей оценивали каждые сутки в отношении признаков EAE по следующей клинической системе оценки: 0, нет клинических признаков; 0,5, частичная утрата тонуса хвоста; 1, полная утрата тонуса хвоста; 2, дряблый хвост и нарушение походки; 3, паралич задних конечностей; 4, паралич задних конечностей с парезом нижней части тела; 5, паралич задних и передних конечностей; и 6, сметь.
3.2 РЕЗУЛЬТАТЫ
У контрольных мышей развивались признаки EAE от умеренных (30% животных достигали максимального клинического показателя 1,5-3) до тяжелых (70% животных достигали максимального клинического показателя 3,5-6), и продемонстрировали показатель смертности 40% вследствие тяжелого паралича. Лечение соединением 1 значительно ингибировало развитие EAE и снижало встречаемость и тяжесть заболевания при измерении с помощью ежедневного клинического показателя, как показано на фиг.1. В группе, в которой проводили лечение соединением 1, 40-70% мышей имели мягкие симптомы и 30% практически полностью выздоровели через 40 суток после начала заболевания. Защитный эффект соединения 1 сохранялся в течение длительного периода времени после прекращения лечения.
Исходя из результатов, полученных в этом анализе, ожидается, что соединение 1 будет пригодным для лечения рассеянного склероза, в том числе хронической прогрессирующей формы рассеянного склероза.
3.3 СОЕДИНЕНИЕ 1 ЯВЛЯЕТСЯ ЭФФЕКТИВНЫМ УЖЕ В ДОЗАХ 0,05 МГ/КГ
С использованием того же протокола анализа EAE, который описан в примере 3,1 выше, Соединение 1 далее тестировали в дозе 1, 0,5 и 0,05 мг/кг п/о, начиная на 12 сутки после иммунизации, один раз в сутки, в течение пяти последовательных дней с 12 суток до 16 суток после иммунизации с 19 суток до 23 суток после иммунизации. Контрольным мышам перорально вводили носитель [2% масс./масс. Tween-80+98% HPβCD (13% масс./об.)] в соответствии с тем же режимом введения. Мышей оценивали каждые сутки в отношении признаков EAE в соответствии с клинической оценочной системой, описанной в примере 3.1. n=10 мышей/группа.
Как показано на фиг.2, Соединение 1 продемонстрировало явный эффект на EAE, снижая клинический показатель уже в дозах 0,05 мг/кг п/о.
3.4 СРАВНЕНИЕ ЭФФЕКТОВ СОЕДИНЕНИЯ 1 С ДРУГИМ ИНГИБИТОРОМ LSD1
С использованием модели EAE согласно примеру 3.1, авторы изобретения протестировали другой необратимый ингибитор LSD1 на основе циклопропиламино, ORY-LSD1, более подробно описанный в примере 1. ORY-LSD1 является мощным и селективным ингибитором LSD1.
Для возможности сравнения результатов, полученных для соединения 1 в примере 3.1, с ORY-LSD1 и поскольку два соединения имеют различную эффективность in vitro против LSD1 (см. пример 2 для их величин IC50), ORY-LSD1 вводили в анализе с EAE в дозах, выбранных так, чтобы они были эквивалентными дозам, использованным для соединения 1 в примере 3.1 в отношении ингибирования LSD1 in vivo. ORY-LSD1 вводили в дозе 0,06 и 0,180 мг/кг п/о. ORY-LSD1 и носитель (тот же, что и в примере 3.1) вводили согласно схеме введения, описанной в примере 3.1 (n=10 мышей/группа).
Результаты, полученные для ORY-LSD1, представлены на фиг.3. В то время как ORY-LSD1 демонстрировало явную тенденцию к улучшению, ORY-LSD1 было значительно менее эффективным, чем соединение 1. Таким образом, соединение 1 оказалось особенно пригодным соединением для лечения рассеянного склероза.
Пример 4: Дальнейшая характеризация терапевтических эффектов соединения 1 в модели EAE у мышей
Для дальнейшей характеризации терапевтических эффектов соединения 1 в модели EAE примера 3, соединение 1 далее тестировали в дозе 0,5 мг/кг п/о и проводили анализ белков гистопатологический анализ.
Введение соединения 1 проводили по той же схеме, которая описана в примере 3.1, т.е. начиная на 12 сутки после иммунизации, один раз в сутки в течение пяти последовательных дней с 12 суток по 16 сутки и с 19 суток по 23 сутки после иммунизации. Контрольным мышам перорально вводили носитель [2% об./об. Tween-80+98% HPβCD (13% масс./об.)] в соответствии с тем же режимом введения, что и для соединения 1. Мышей оценивали каждые сутки в отношении признаков EAE с использованием способа оценки, описанного в примере 3.1. Животных умерщвляли на 26 сутки после иммунизации и проводили взятие образцов и обработку, как описано ниже. n=10 мышей/группа.
4.1 СПОСОБЫ
Сбор тканей и выделение клеток. На 26 сутки после иммунизации извлекали селезенку, дренирующие лимфатические узлы (DLN: шейные, паховые и подмышечные) и спинной мозг. Сегменты спинного мозга шейной и поясничной областей подготавливали по отдельности и обрабатывали для экстракции белка и гистопатологического анализа. Суспензии отдельных клеток получали из селезенки и объединенных лимфатических узлов, образцы гомогенизировали и общее количество клеток количественно определяли с использованием камеры Нойбауэра.
Обработка образцов для гистопатологического анализа. Шейные и поясничные сегменты спинного мозга отделяли и обрабатывали для заключения в парафин и получения срезов. Сегменты спинного мозга сразу фиксировали забуференным 10% формалином в течение 48 ч, дегидратировали и заливали парафином с использованием стандартных способов. Поперечные срезы (толщиной 4 мкм) окрашивали быстрым голубым Luxol, крезиловым фиолетовым и гематоксилином по методу Клювер-Баррера и анализировали на присутствие областей демиелинизации и инфильтрации клеток с использованием светового микроскопа (Leica, DM2000).
Экстракция белка и анализ цитокинов/хемокинов. Белки экстрагировали из шейного и поясничного сегментов спинного мозга посредством гомогенизации 50 мг ткани/мл) в лизирующем буфере (50 мМ Tris-HCl, pH 7,4, 0,5 мМ DTT и 10 мкг/мл ингибиторов протеаз PMSF, пепстатина и леупептина). Образцы центрифугировали (20000×g, 15 мин, 4°C) и супернатанты анализировали в отношении концентрации белка (с использованием способа Брэдфорда) и в отношении содержания цитокинов/хемокинов с использованием специфического сэндвич-ELISA для IL-4, IL-6, IL-1-бета, IP-10 и MCP-1, в соответствии с рекомендациями изготовителя с использованием следующих антител и рекомбинантных белков:
Рекомбинантный IL-4 мыши. BD Pharmingen. 0,2 мг/мл. Ссылочный номер: 550067.
Биотинилированное антитело крысы против IL-4 мыши. BD Pharmingen. 0,5 мг/мл. Ссылочный номер: 554390
Рекомбинантный IL-6 мыши. BD Pharmingen. 0,1 мг/мл. Ссылочный номер: 554582.
Биотинилированное антитело крысы против IL-6 мыши. BD Pharmingen. 0,5 мг/мл. Ссылочный номер: 554402.
Рекомбинантный IL-1-бета мыши. Peprotech. 0,1 мг/мл. Ссылочный номер: 211-11B.
Биотинилированное антитело кролика против IL-1-бета мыши. Peprotech. 0,4 мг/мл.Ссылочный номер: 500-P51Bt.
Ссылочный номер: 500-P129.
Рекомбинантный IP-10 мыши (CXCL10). Peprotech. 0,1 мг/мл. Ссылочный номер: 250-16.
Биотинилированное подвергнутое аффинной очистке с антигеном антитело против IP-10 мыши. Peprotech. Ссылочный номер: 500-P129Bt. 0,5 мг/мл
Рекомбинантный JE/MCP-1 мыши (CCL2). Peprotech. 0,1 мг/мл. Ссылочный номер: 250-10.
Биотинилированное подвергнутое аффинной очистке с антигеном поликлональное антитело против JE мыши. Peprotech. 0,5 мг/мл. Ссылочный номер: 500-P113Bt.
Статистический анализ: анализ количества клеток в лимфатических узлах и селезенке: статистические отличия указаны как ***p<0,001 против носителя с использованием теста ANOVA. Анализ уровней цитокинов/хемокинов: статистические отличия указаны как *p<0,05, **p<0,005, с использованием критерий Манна-Уитни; для анализа уровня IP-10 использовали непарный t-критерий.
4.2 РЕЗУЛЬТАТЫ
Введение соединения 1 в дозе 0,5 мг/кг п/о хорошо переносилось мышами на протяжении длительного лечения, значительно ингибировало развитие EAE и заболеваемость и тяжесть заболевания, как определяли с использованием ежедневного клинически показателя, как также показано на фиг.4.
Соединение 1 значительно снижало инфильтрацию воспалительных клеток и демиелинизацию в спинном мозге мышей с EAE, как показано на фиг.5. Стрелками на данной фигуре указаны области демиелинизации и инфильтрации воспалительных клеток. В контрольных (животные, которым вводили носитель) образцах, как шейной, так и поясничной областей, наблюдали множество областей демиелинизации и инфильтрации воспалительных клеток, в то время как в образцах, обработанных соединением 1, не наблюдали ни инфильтрации воспалительных клеток, ни демиелинизации. На фиг.6 показано среднее количество бляшек демиелинизации в поясничной и шейной областях спинного мозга животных, которым вводили соединение 1 или носитель, демонстрирующее отсутствие или значительное снижение демиелинизации в срезах шейной и поясничной областей у животных, которым вводили соединение 1. Эти результаты, как также проиллюстрировано на фиг.5 и 6, демонстрируют, что соединение 1 снижает инфильтрацию иммунных клеток в спинной мозг и защищает спинной мозг от демиелинизации в модели рассеянного склероза - EAE.
Как показано на фиг.7, введение соединения 1 приводило к значительному увеличению количества иммунных клеток, оставшихся в селезенке и лимфатических узлах подвергнутых введению животных, что указывает на сниженный выход лимфоцитов из иммунных тканей. Кроме того, лечение соединением 1 модулирует воспалительные и аутоиммунные ответы, как проиллюстрировано на фиг.8A-8E. Уровень противовоспалительного цитокина IL-4 был значительно увеличен в спинном мозге животных, которым вводили соединение 1, что указывает на противовоспалительный ответ Th2 (фиг.8A). Уровни провоспалительных цитокинов IL-6 и IL-1-бета в спинном мозге были снижены после введения соединения 1 (фиг.8B и 8C). Кроме того, соединение 1 значительно снижало уровни различных хемокинов в органе-мишени, включая IP-10 (фиг.8D) и MCP-1 (фиг.8E), которые вовлечены в привлечение воспалительных и энцефалитогенных Th1-клеток в спинной мозг. Эти результаты дополнительно подтверждают, что соединение 1 является особенно пригодным в качестве терапевтического средства для лечения рассеянного склероза.
Все цитированные в настоящем описании публикации, патенты и патентные заявки включены в настоящее описание в качестве ссылок в полном объеме.
Публикации, патенты и патентные заявки, упомянутые в описании, предоставлены только для их раскрытия до даты подачи настоящей заявки. Ничто в настоящем описании не следует истолковывать как допущение того, что они являются уровнем техники для настоящей заявки.
Хотя изобретение описано применительно к его конкретным вариантам осуществления, будет понятно, что оно может быть подвергнуто дальнейшим модификациям и предполагается, что его применение охватывает любые варианты, применения или адаптации изобретения в соответствии с общими принципами изобретения и включает такие отклонения от настоящего изобретения в соответствии с известной или привычной практикой в области, к которой относится изобретения, и как может быть применено к основным признакам, указанным выше и вытекающим из прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ LSD1 НА ОСНОВЕ АРИЛЦИКЛОПРОПИЛАМИНА И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2011 |
|
RU2611437C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА С ИСПОЛЬЗОВАНИЕМ СОЕДИНЕНИЙ ПИРИМИДИНА И ПИРИДИНА С BTK ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2016 |
|
RU2779287C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЗМЕНЕНИЙ ПОВЕДЕНИЯ | 2018 |
|
RU2799049C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПОГРАНИЧНОГО РАССТРОЙСТВА ЛИЧНОСТИ | 2020 |
|
RU2833298C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2681211C2 |
| ОСНОВНЫЕ ЭФИРЫ ЖИРНЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ ИЛИ ИММУНОМОДУЛИРУЮЩИХ СРЕДСТВ | 2003 |
|
RU2337093C2 |
| ЛИЗИНСПЕЦИФИЧЕСКИЕ ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ-1 И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2599248C2 |
| ЛИЗИНСПЕЦИФИЧЕСКИЕ ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ-1 И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2602814C2 |
| Амидные соединения, содержащие их фармацевтические композиции и способы их применения | 2017 |
|
RU2759913C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АПОПТОЗА НЕЙРОНОВ ИЛИ НЕЙРОДЕГЕНЕРАЦИИ | 2011 |
|
RU2586772C2 |
Изобретение относится к способу лечения рассеянного склероза у пациента, включающему введение пациенту терапевтически эффективного количества (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата. Технический результат – разработан новый способ лечения склероза на основе (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина, который демонстрирует меньше побочных эффектов и который можно вводить пероральным путем. 3 н. и 12 з.п. ф-лы, 8 ил., 4 пр.
1. Способ лечения рассеянного склероза у пациента, включающий введение пациенту терапевтически эффективного количества (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата.
2. Способ по п.1, где рассеянный склероз представляет собой хронический прогрессирующий рассеянный склероз.
3. Способ по п.1 или 2, где способ включает введение пациенту терапевтически эффективного количества (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина.
4. Способ по любому из пп.1-3, где указанный (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин или его фармацевтически приемлемую соль или сольват вводят перорально.
5. Способ по любому из пп.1-4, где пациентом является человек.
6. Применение (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для изготовления лекарственного средства для лечения рассеянного склероза.
7. Применение по п.6, где рассеянный склероз представляет собой хронический прогрессирующий рассеянный склероз.
8. Применение по п.6 или 7, где (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин используют для производства указанного лекарственного средства.
9. Применение по любому из пп.6-8, где указанное лекарственное средство предназначено для перорального введения.
10. Применение по любому из пп.6-9, где указанное лекарственное средство предназначено для лечения человека.
11. Применение (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амина или его фармацевтически приемлемой соли или сольвата для лечения рассеянного склероза.
12. Применение по п.11, где рассеянный склероз представляет собой хронический прогрессирующий рассеянный склероз.
13. Применение по п.11 или 12, где (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин используют для лечения рассеянного склероза.
14. Применение по любому из пп.11-13, где указанный (-)5-((((транс)-2-(4-(бензилокси)фенил)циклопропил)амино)метил)-1,3,4-оксадиазол-2-амин или его фармацевтически приемлемую соль или сольват вводят перорально.
15. Применение по любому из пп.11-14, где пациентом, подвергаемым лечению, является человек.
| WO 2012013728 A1, 02.02.2012 | |||
| LAUREN G FRIEDMAN et al., ALZHEIMERS RES THER, vol | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| A | |||
| SCOUMANNE et al., Journal of Biological Chemistry, vol | |||
| ПОРШНЕВОЙ ДВИГАТЕЛЬ | 1916 |
|
SU282A1 |
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕСТОНАХОЖДЕНИЯ УТЕЧЕК В МАГИСТРАЛЬНЫХ ТРУБОПРОВОДАХ | 2004 |
|
RU2258865C1 |
| Цепной нож для жатвенных машин | 1922 |
|
SU7179A1 |

