ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способу получения и промежуточным продуктам для получения кристаллической или некристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида. Настоящее изобретение также относится к фармацевтическим композициям, содержащим кристаллическую форму, и к способам применения кристаллической формы для лечения разных заболеваний.
УРОВЕНЬ ТЕХНИКИ
N-((1S,3S)-3-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид обладает химической формулой C14H21N5O2S и следующей структурной формулой:
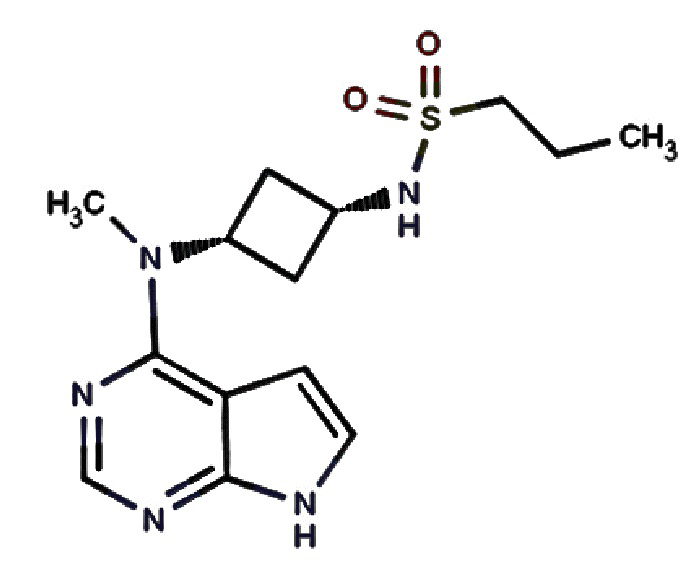
Препаративный синтез N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида описан в принадлежащей тому же правообладателю заявке US9035074, содержание которой во всей его полноте включено в настоящее изобретение в качестве ссылки. Кристаллическая форма свободного основания N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида применима в качестве ингибитора протеинкиназ, таких как фермент киназа Janus (JAK), и сама по себе применима в терапии в качестве иммуносупрессорного средства при трансплантации органа, ксенотрансплантации, волчанке, рассеянном склерозе, ревматоидном артрите, псориазе, диабете типа I и осложнениях при диабете, раке, астме, атопическом дерматите, аутоиммунных нарушениях щитовидной железы, язвенном колите, болезни Крона, болезни Альцгеймера, лейкозе и других показаниях, когда желательна иммуносупрессия. Настоящее изобретение относится к способу получения и промежуточным продуктам для получения кристаллической или некристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, где кристаллическая форма обеспечивает некоторые улучшенные характеристики для применения при получении фармацевтической дозированной формы, в особенности пероральных и местных дозированных форм.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения кристаллической или некристаллической формы свободного основания N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида. Настоящее изобретение также относится к композициям, включая фармацевтические композиции, содержащие кристаллическое свободное основание 3-((3R,4R)-4-метил-3-[метил-(7H-пирроло[2,3-d]пиримидин-4-ил)-амино]-пиперидин-1-ил)-3-оксопропионитрила. Настоящее изобретение также относится к способу лечения заболевания у млекопитающего, включающему введение нуждающемуся в нем млекопитающему терапевтически эффективного количества кристаллического N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида или его фармацевтически приемлемой соли или фармацевтической композиции, указанное заболевание выбрано из группы, включающей ревматоидный артрит, волчанку, псориаз, атопический дерматит, витилиго и воспалительную болезнь кишечника.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
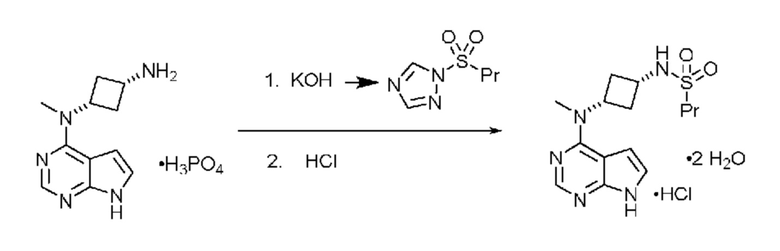
На фиг. 1 приведена порошковая рентгенограмма кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
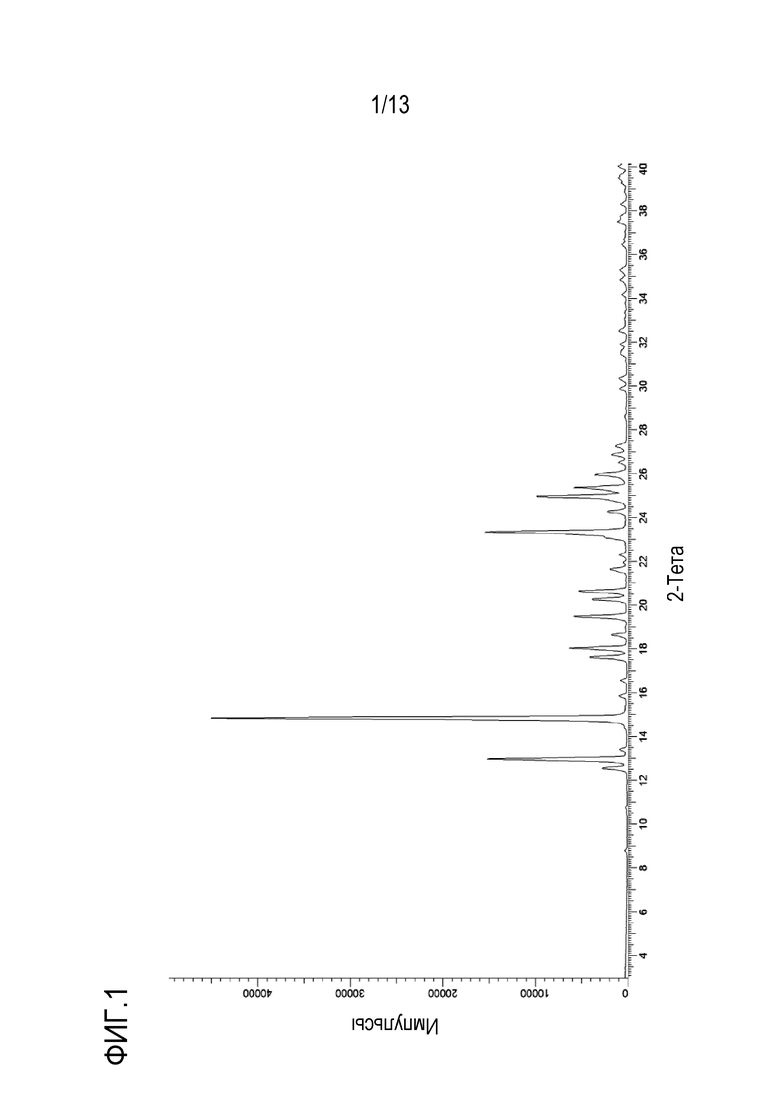
На фиг. 2 приведен спектр комбинационного рассеяния кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
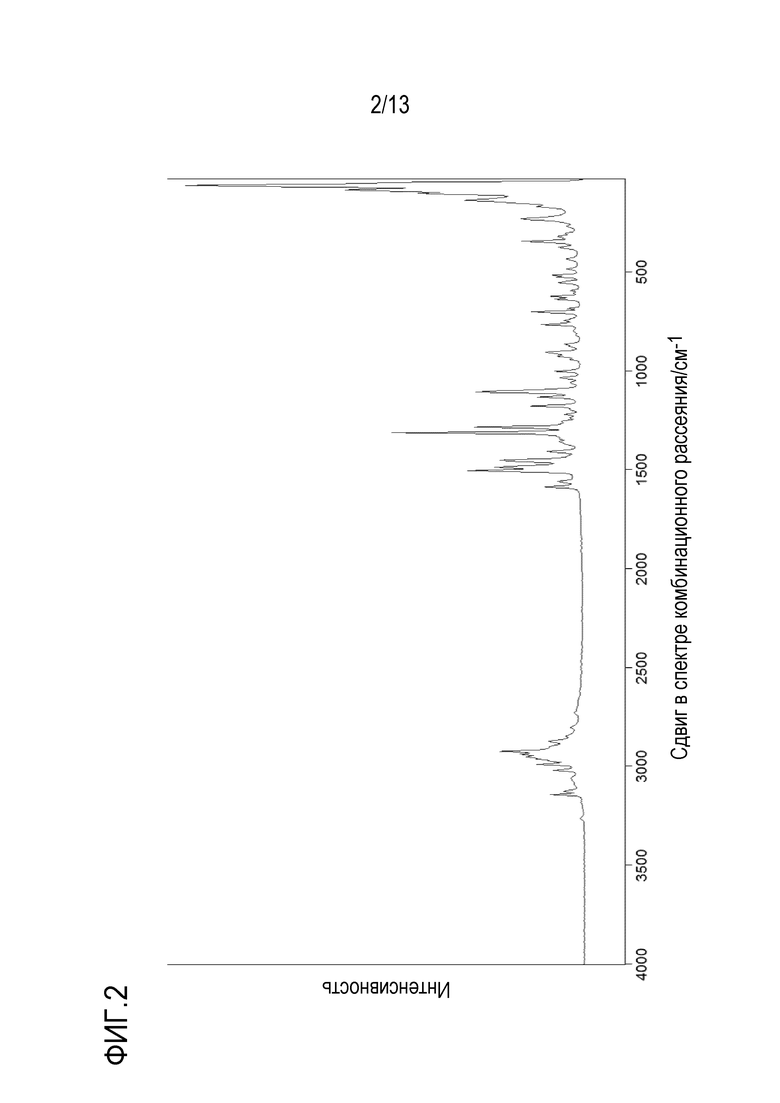
На фиг. 3 приведен FT-IR спектр кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
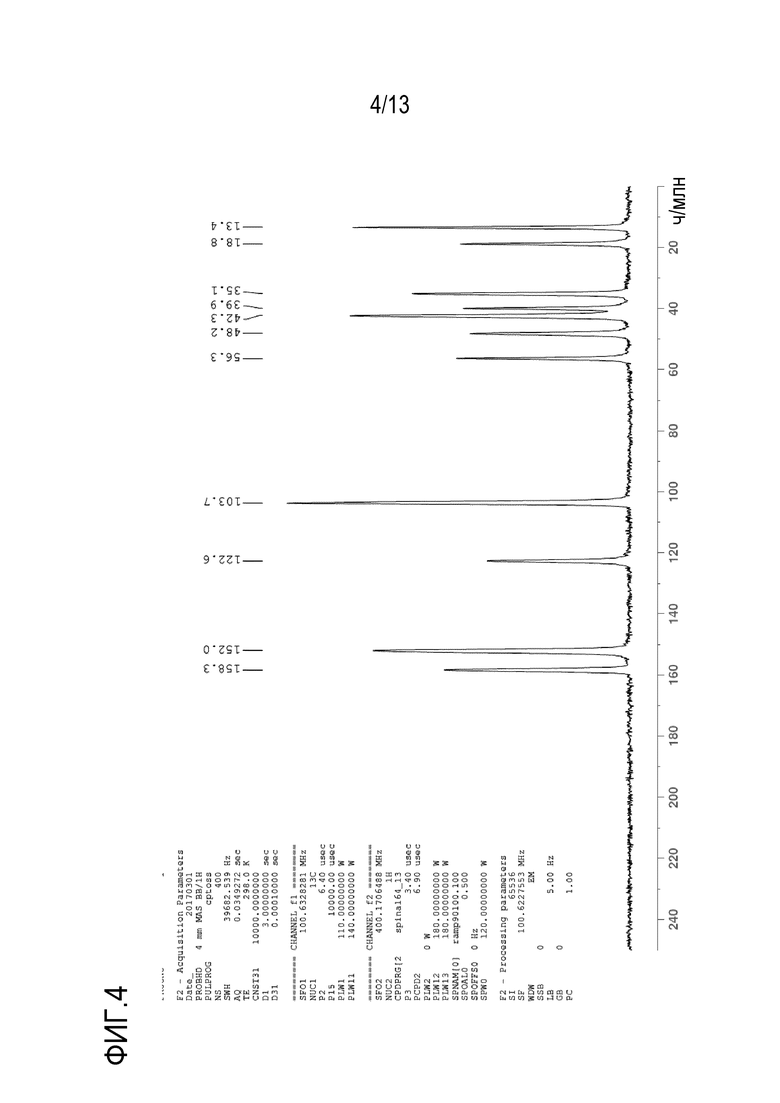
На фиг. 4 приведен твердофазный спектр 13C ядерного магнитного резонанса кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида. Боковые полосы вращения отмечены звездочкой.
На фиг. 5 приведена порошковая рентгенограмма безводной моно-соли с HCl кристаллической формы A N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
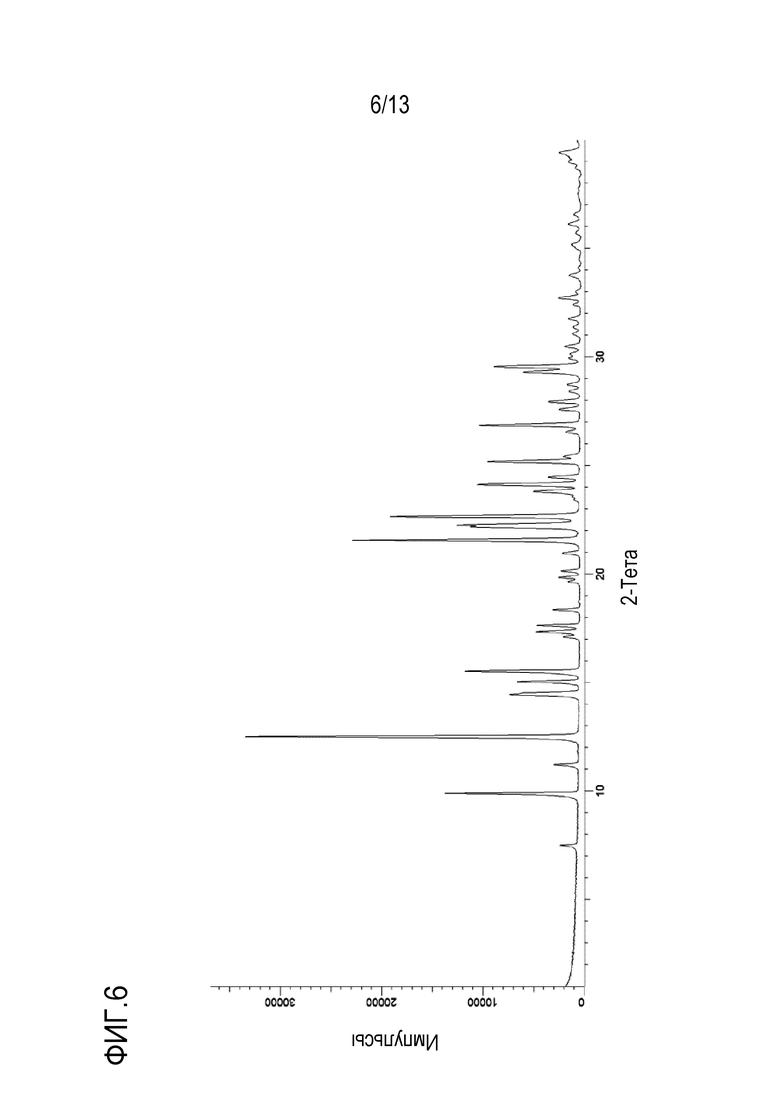
На фиг. 6 приведена порошковая рентгенограмма моногидрата моно-соли с HCl формы B кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
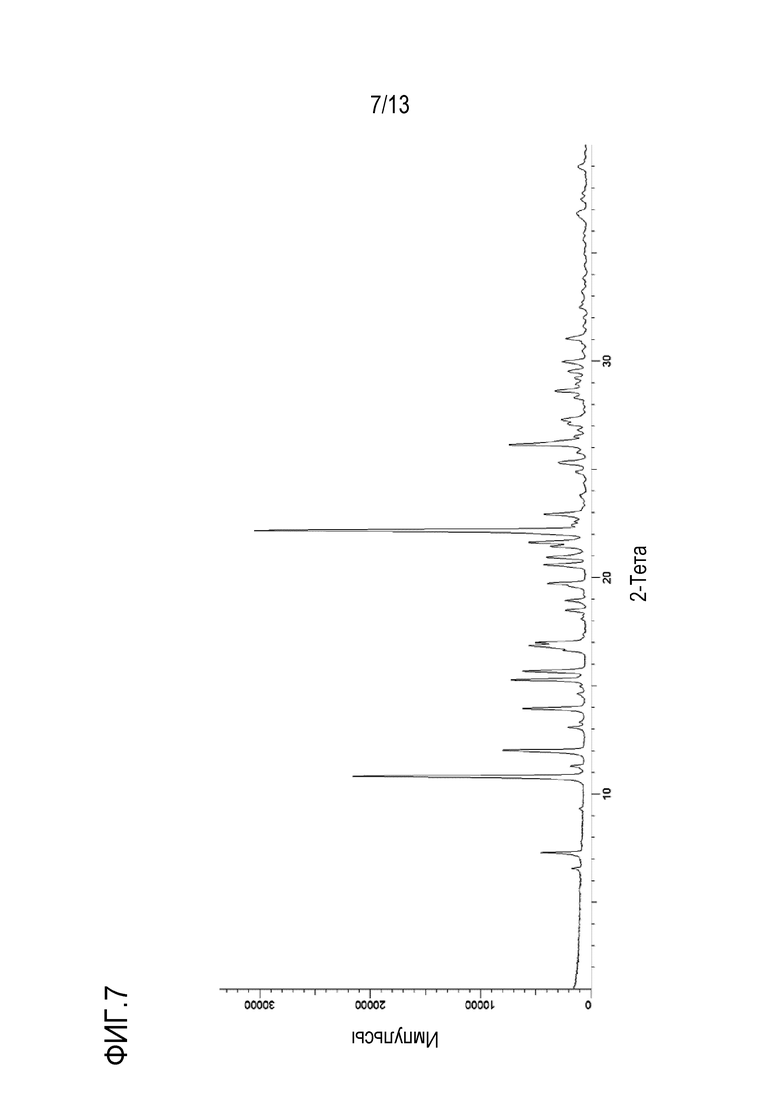
На фиг. 7 приведена порошковая рентгенограмма безводной моно-соли с HCl формы C кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
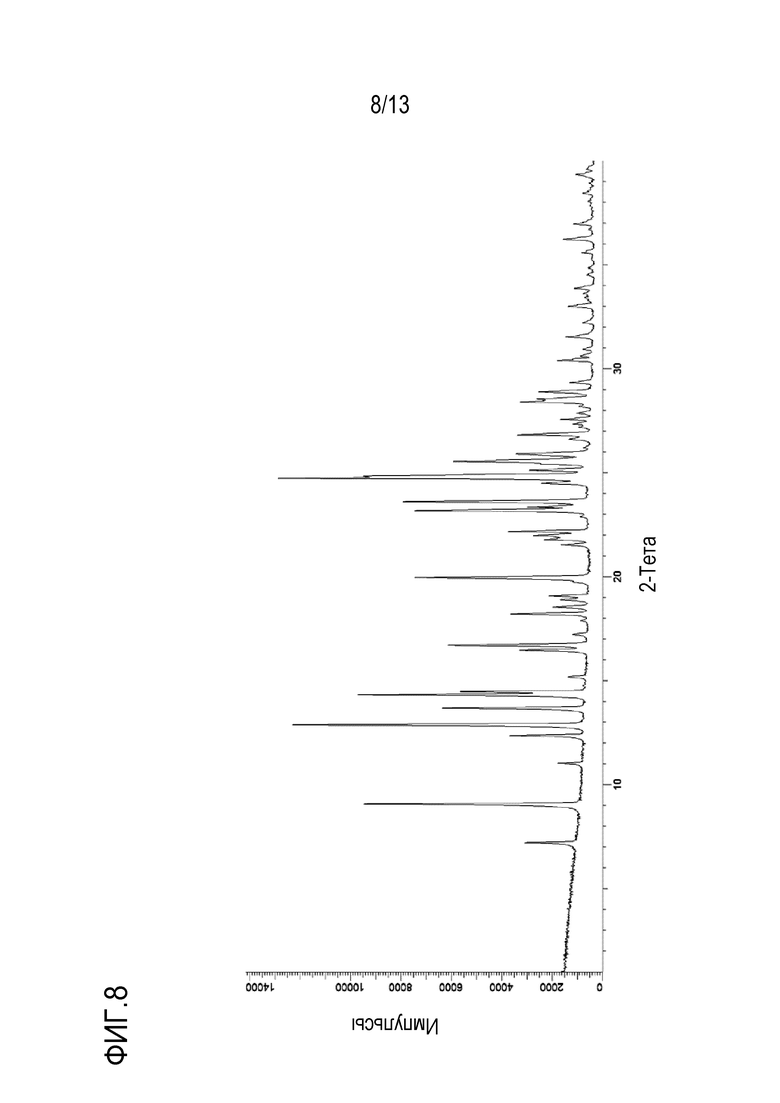
На фиг. 8 приведена порошковая рентгенограмма дигидрата моно-соли с HCl формы E кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
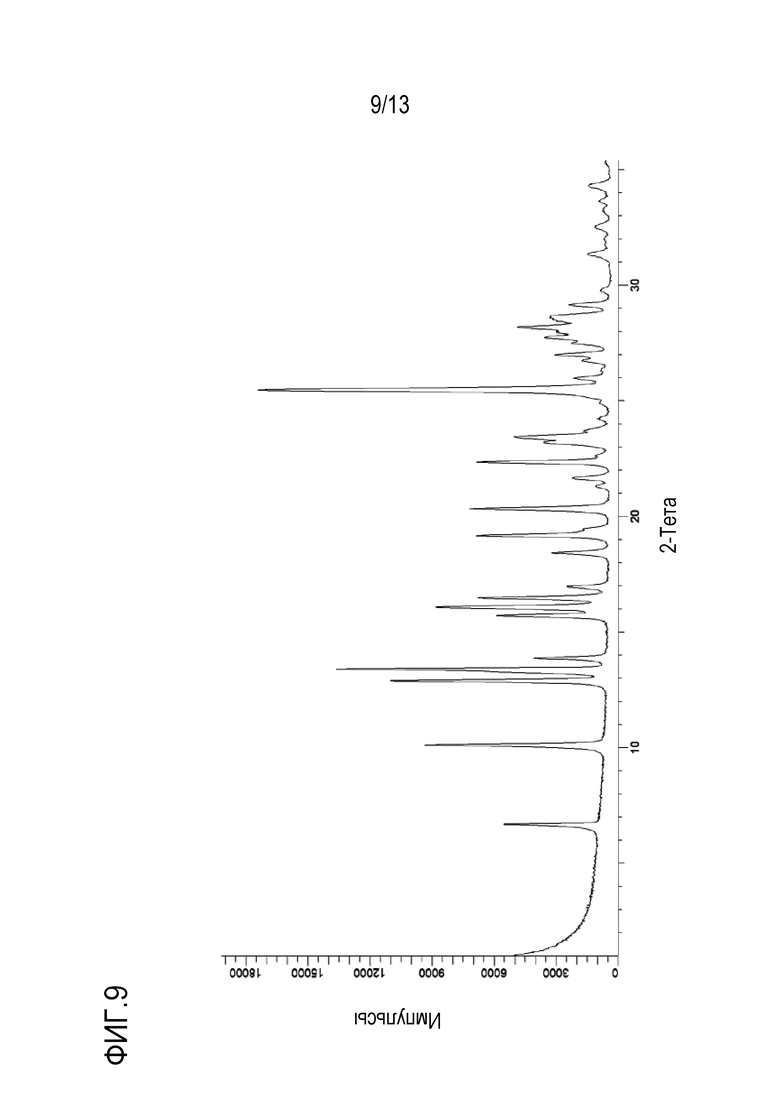
На фиг. 9 приведена порошковая рентгенограмма безводной моно-соли с HCl формы G кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
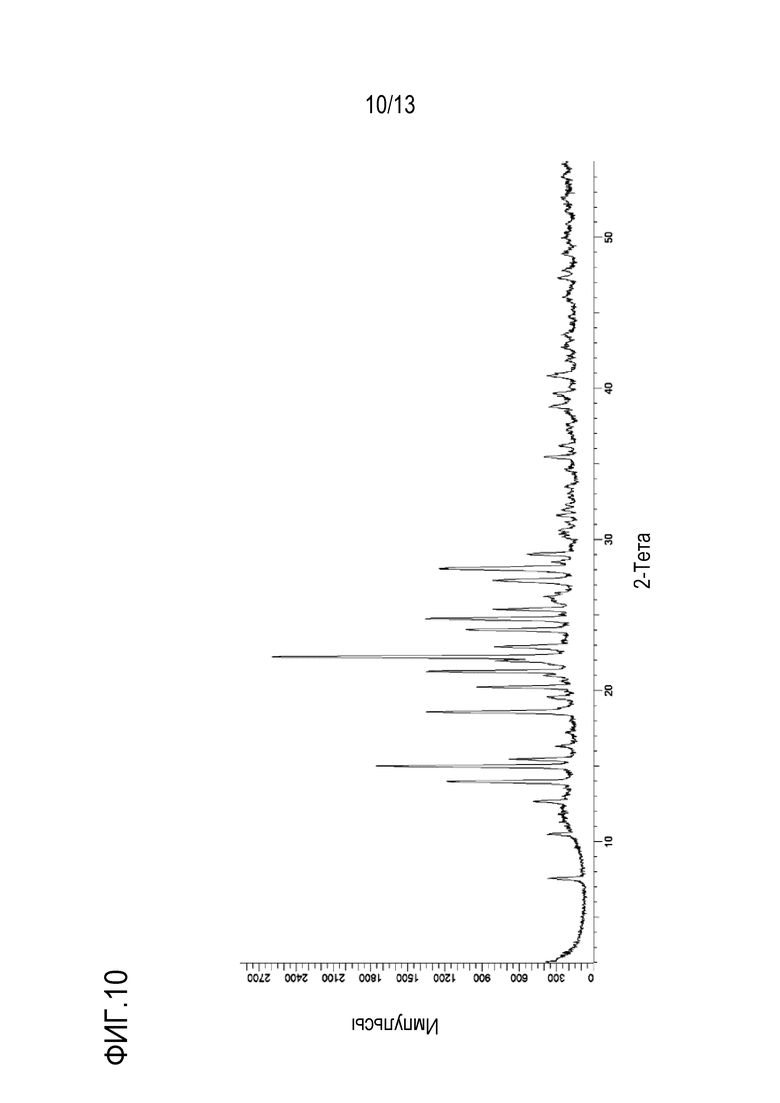
На фиг. 10 приведена порошковая рентгенограмма кристаллической формы гемисульфата N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
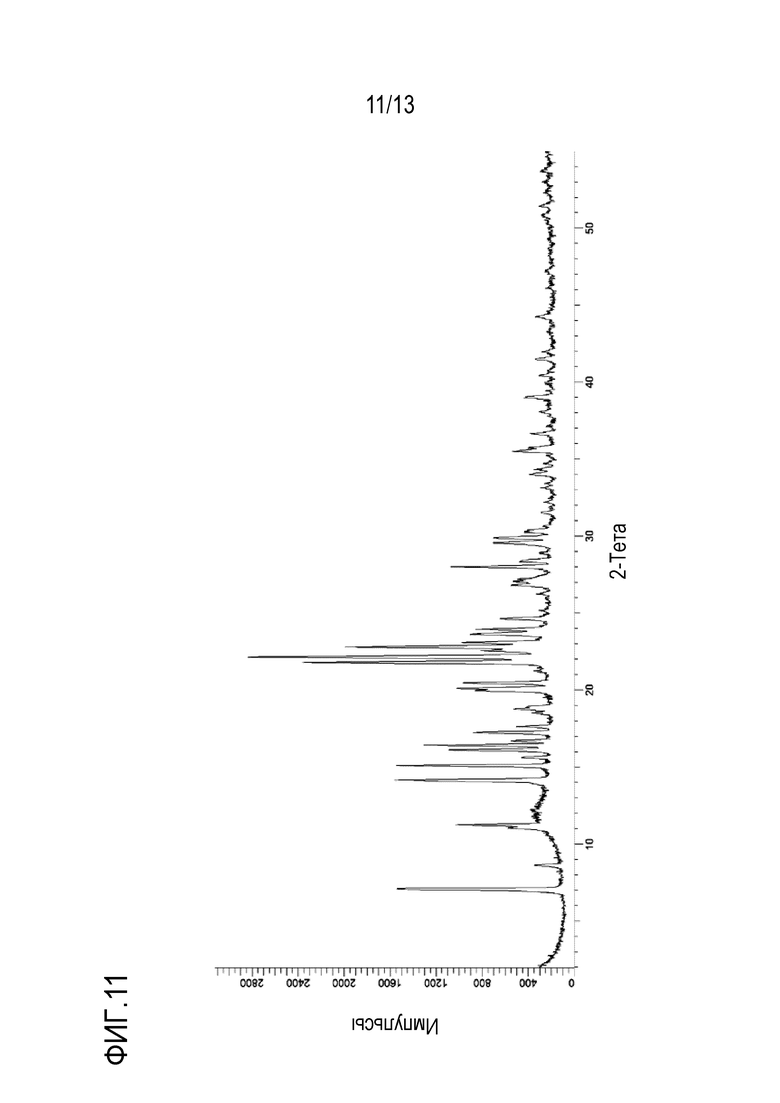
На фиг. 11 приведена порошковая рентгенограмма кристаллической формы мезилата N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
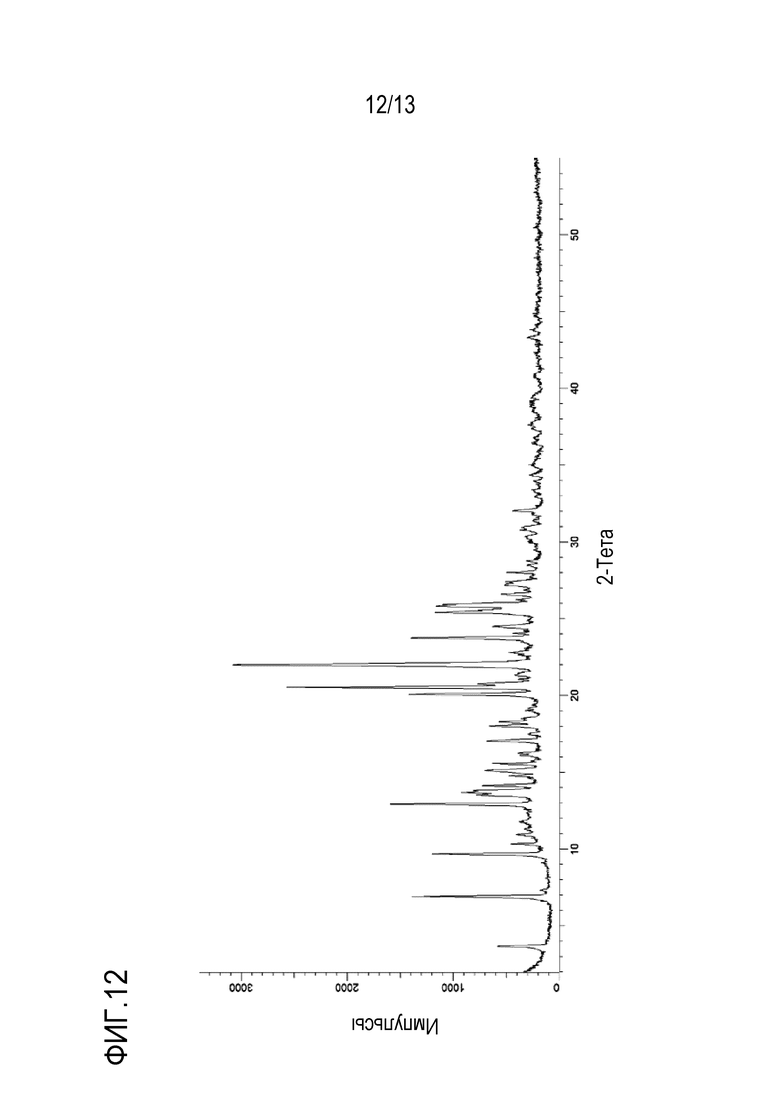
На фиг. 12 приведена порошковая рентгенограмма кристаллической формы п-тозилата N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
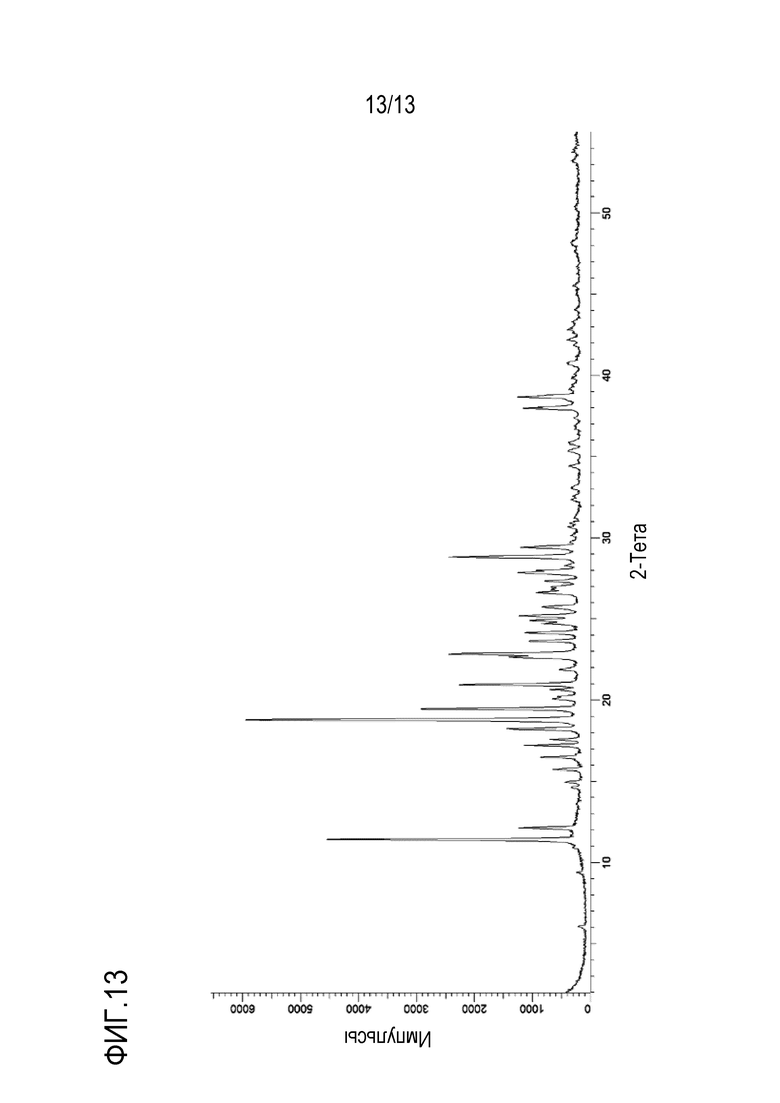
На фиг. 13 приведена порошковая рентгенограмма кристаллической формы совместного кристалла фумаровой кислоты и N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
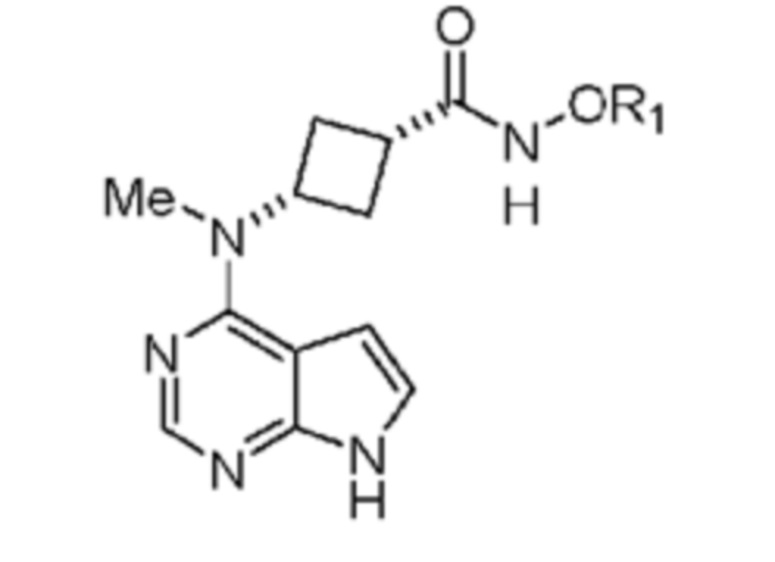
Настоящее изобретение относится к соединению, применимому для получения N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, обладающего структурой:
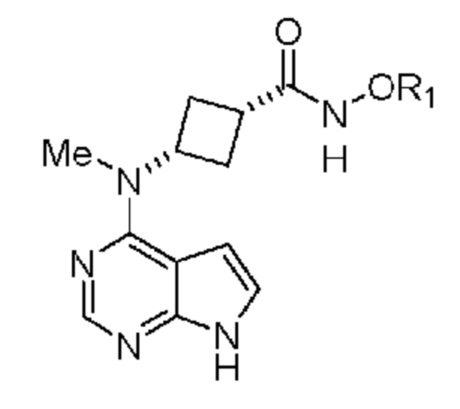
в которой R1 выбран из группы, включающей: водород, замещенный или незамещенный фенил, замещенный или незамещенный пиридил, замещенный или незамещенный имидазолил, (C1-C6)алкил, (C3-C10)циклоалкил, который необязательно может быть замещен 1, 2 или 3 группами, независимо выбранными из группы, включающей галоген, (C1-C3)алкил и (C1-C3)алкилоксигруппу, или его соли, выбранной из группы, включающей соль натрия, калия, лития, магния и кальция. В предпочтительном объекте настоящее изобретение относится к соединению, в котором R1 означает водород, или его соли.
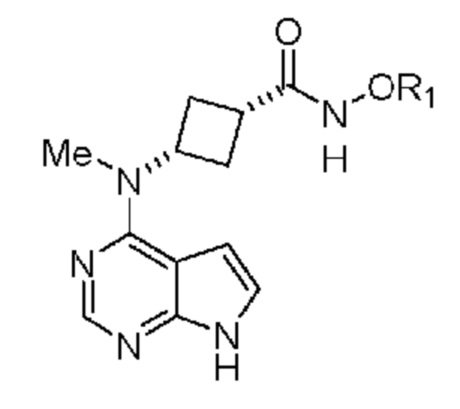
Настоящее изобретение также относится к (1s,3s)-N-гидрокси-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутан-1-карбоксамиду, обладающему структурой:
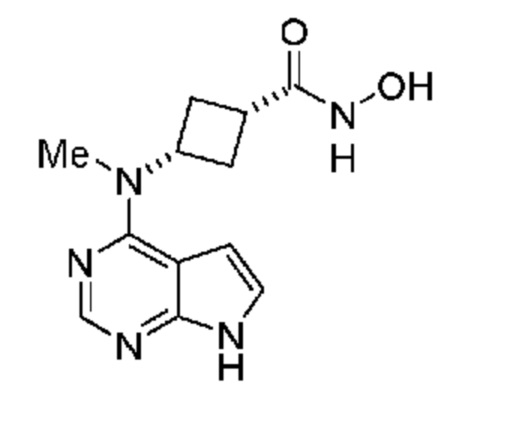
или его соли.
Настоящее изобретение также относится к соединению, обладающему структурой:
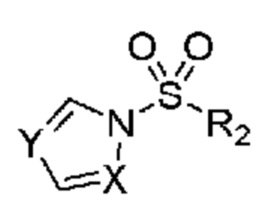
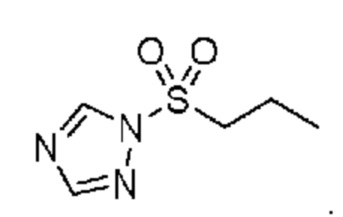
в которой R2 выбран из группы, включающей (C3-C5)алкил и (C3-C4)циклоалкил; и X и Y независимо выбраны из группы, включающей CR3 и N, где R3 выбран из группы, включающей: водород и (C1-C6)алкил. В некоторых объектах настоящее изобретение относится к соединению, в котором X и Y оба означают N; и, R2 означает (C3-C5)алкил. В некоторых других объектах настоящее изобретение относится к соединению, в котором R2 означает линейную или разветвленную пропильную группу. В некоторых других объектах настоящее изобретение относится к соединению, в котором R2 означает линейную пропильную группу. В некоторых других объектах настоящее изобретение относится к соединению, в котором X означает CR3, где R3 означает водород; и Y означает N и, кроме того, где R2 означает линейную или разветвленную пропильную группу. В предпочтительном объекте настоящее изобретение относится к соединению, в котором R2 означает линейную пропильную группу. Соответственно, настоящее изобретение относится к 1-(пропилсульфонил)-1H-1,2,4-триазолу, обладающему структурой:
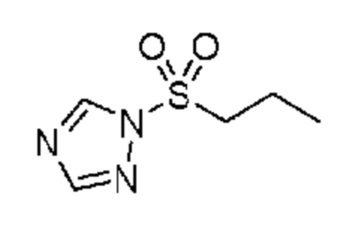
Настоящее изобретение также относится к соли соединения, обладающего структурой:
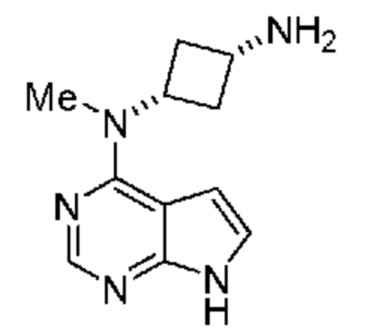
указанная соль выбрана из группы, включающей соль хлористоводородной кислоты, соль фосфорной (моно-, бис-, трис-) кислоты, соль (1S)-(+)-10-камфорсульфоновой кислоты, соль 1,2-этандисульфоновой кислоты, соль дибензоил-L-винной кислоты, соль дибензоил-D-винной кислоты, соль лимонной кислоты, соль янтарной кислоты, соль фумаровой кислоты, соль малеиновой кислоты, соль щавелевой кислоты, соль п-толуолсульфоновой кислоты, соль L-(+)-винной кислоты, соль D-(-)-винной кислоты, соль бромистоводородной кислоты, соль кислоты, мезилат и соль малоновой кислоты. В предпочтительном объекте настоящее изобретение относится к соли фосфорной кислоты соединения.
Настоящее изобретение также относится к соли с кислотой соединения, обладающего структурой:
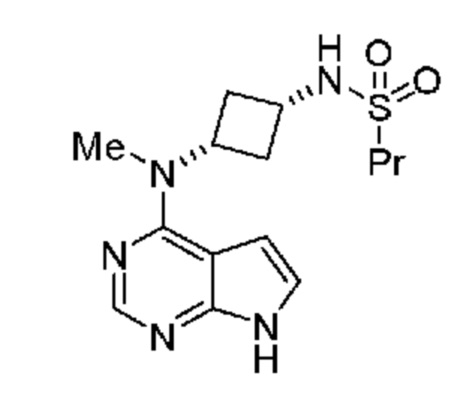
где указанная соль с кислотой выбрана из группы, включающей гидрохлорид, фосфат, соль янтарной кислоты, цитрат, соль п-толуолсульфоновой кислоты, мезилат, гемисульфат, гемифумарат и соль малоновой кислоты. В одном объекте настоящее изобретение относится к соли с кислотой соединения, обладающего структурой:
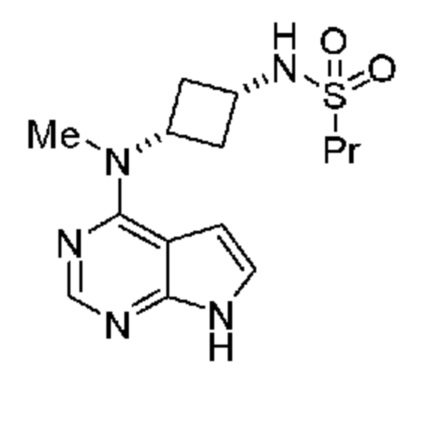
где солью с кислотой является гидрохлорид.
Настоящее изобретение также относится к соединению, обладающему структурой:
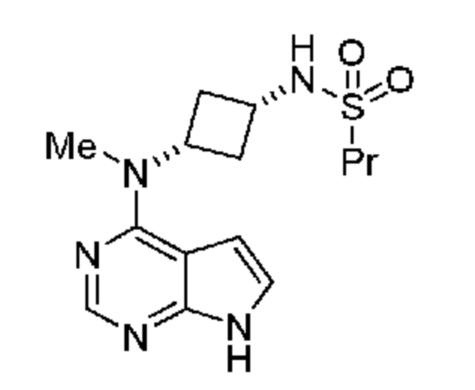
полученному из соли с кислотой, выбранной из группы, включающей гидрохлорид, фосфат, соль янтарной кислоты, соль п-толуолсульфоновой кислоты, мезилат, гемисульфат, цитрат, гемифумарат и соль малоновой кислоты. В предпочтительном объекте настоящее изобретение относится к соединению, полученному из гидрохлорида указанного соединения в подходящей щелочной среде. В особенности настоящее изобретение относится к соединению, обладающему порошковой рентгенограммой, включающей пики, выраженные в углах 2и, при 13,0°, 14,8° и 23,3° 2и ± 0,2° 2и.
Настоящее изобретение также относится к соединению, обладающему структурой:
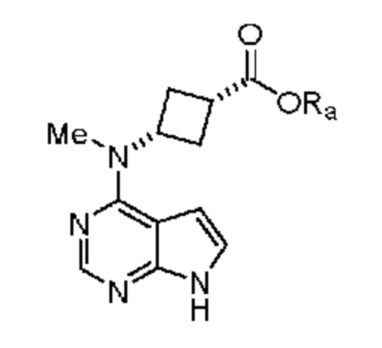
в которой Ra выбран из группы, включающей водород, (C1-C6)алкил, (C3-C10)циклоалкил, и замещенный или незамещенный фенил, где указанный алкил и циклоалкил необязательно может быть замещен 1, 2 или 3 группами, независимо выбранными из группы, включающей галоген, (C1-C3)алкил и (C1-C3)алкилоксигруппу, или его соли. В предпочтительном объекте настоящее изобретение относится к указанному выше соединению, в котором Ra означает водород. Другим предпочтительным объектом настоящего изобретения является указанное выше соединение, в котором Ra означает метил или изопропил.
Кроме того, настоящее изобретение относится к способу получения соединения, обладающего структурой:
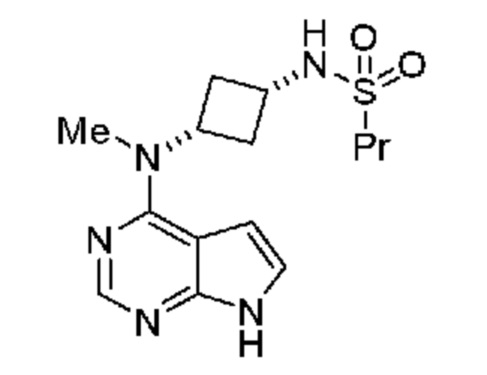
который включает (a) получение гидрохлорида указанного соединения при подходящих условиях и затем (b) взаимодействие указанной соли с подходящим основанием при подходящих условиях с получением соединения, где указанное подходящее основание выбрано из группы, включающей карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия, гидроксид натрия, гидроксид калия или триэтиламин. В одном объекте настоящее изобретение относится к способу, в котором указанным подходящим основанием является бикарбонат натрия или калия.
Настоящее изобретение также относится к способу получения соединения, обладающего структурой:
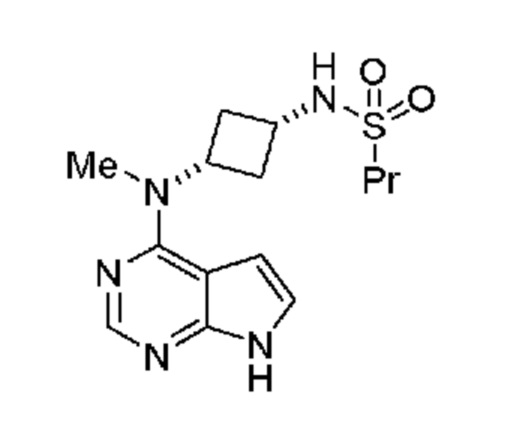
который включает (a) получение гидроксиламина, обладающего структурой:
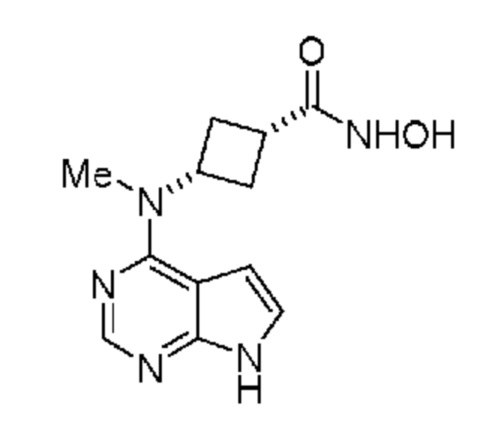
(b) введение в реакцию указанного гидроксиламина при подходящих условиях с получением амина, обладающего структурой:
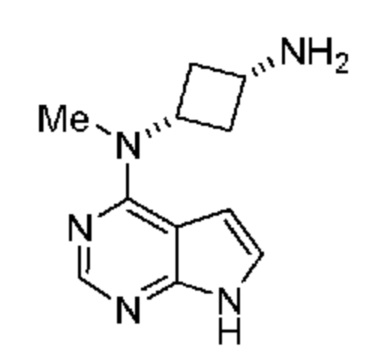
и затем (c) обработку указанного амина подходящим н-пропилсульфонирующим реагентом при подходящих условиях с получением соединения. В некоторых объектах настоящее изобретение относится к способу, в котором н-пропилсульфонирующим реагентом является соединение, обладающее структурой:
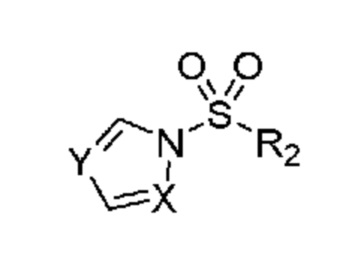
в которой R2 означает н-пропил; и X и Y независимо выбраны из группы, включающей CR3 и N, в которой R3 выбран из группы, включающей: водород и (C1-C6)алкил. В предпочтительных объектах настоящее изобретение относится к способу, в котором X и Y оба означают N. В других объектах настоящее изобретение также относится к способу, в котором н-пропилсульфонирующим реагентом является 1-(пропилсульфонил)-1H-1,2,4-триазол, обладающий структурой:
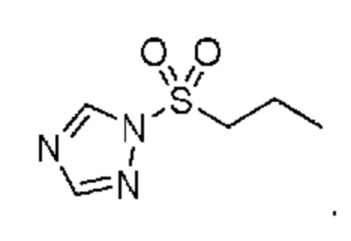 .
.
Еще одним объектом настоящего изобретения является способ, в котором соединение, обладающее структурой:
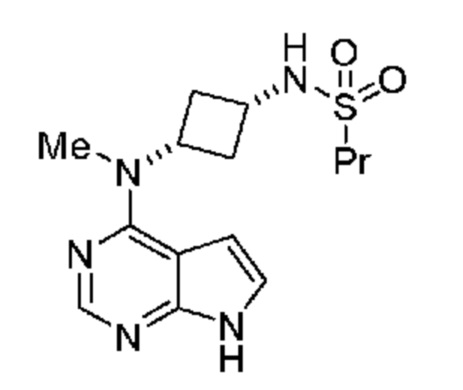
является кристаллической формой N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, обладающей порошковой рентгенограммой, включающей пики, выраженные в углах 2и, при 13,0°, 14,8° и 23,3° 2и ± 0,2° 2и.
Настоящее изобретение также относится к способу получения соединения, обладающего структурой:
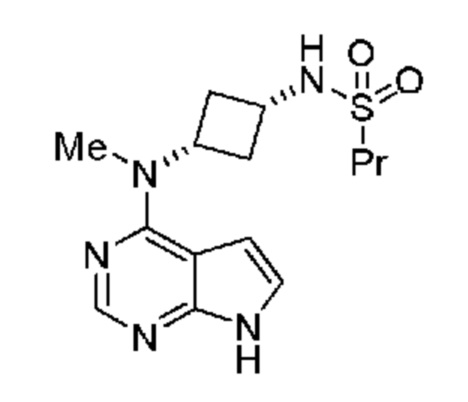
который включает (a) получение амина, обладающего структурой:
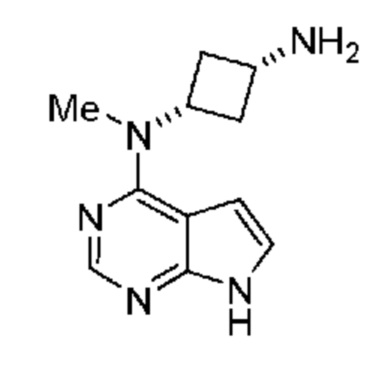
и затем (b) обработку указанного амина подходящим н-пропилсульфонирующим реагентом при подходящих условиях с получением соединения. В некоторых объектах настоящее изобретение относится к способу, в котором н-пропилсульфонирующим реагентом является соединение, обладающее структурой:
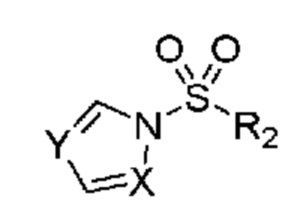
в которой R2 означает н-пропил; и X и Y независимо выбраны из группы, включающей CR3 и N, где R3 выбран из группы, включающей водород и (C1-C6)алкил. В некоторых объектах настоящее изобретение относится к способу, в котором X и Y оба означают N. В некоторых других объектах настоящее изобретение относится к способу, в котором н-пропилсульфонирующим реагентом является 1-(пропилсульфонил)-1H-1,2,4-триазол, обладающий структурой:
 .
.
Настоящее изобретение также относится к способу, в котором соединение, обладающее структурой:
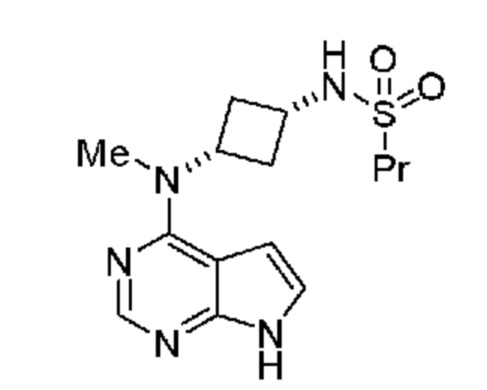
является кристаллической формой N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, обладающей порошковой рентгенограммой, включающей пики, выраженные в углах 2и, при 13,0°, 14,8° и 23,3° 2и ± 0,2° 2и.
Кроме того, настоящее изобретение относится к фармацевтической композиции соединения, обладающего структурой:
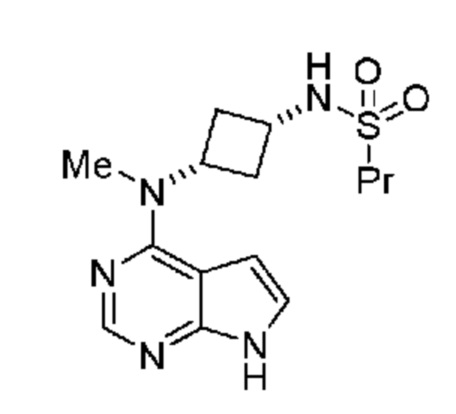
где указанную композицию получают из указанного соединения, обладающего кристаллической формой, обладающей порошковой рентгенограммой, включающей пики, выраженные в углах 2и, при 13,0°, 14,8° и 23,3° 2и ± 0,2° 2и; и она дополнительно содержит фармацевтически приемлемый носитель. В некоторых объектах настоящее изобретение относится к фармацевтической композиции, включающей препарат для местного применения, выбранный из группы, включающей крем, чрескожный пластырь, мазь, глазные капли, примочку и гель. В частности, настоящее изобретение относится к фармацевтической композиции, где препарат для местного применения содержит от примерно 0,1% до примерно 5,0% (мас./об.) N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид.
Настоящее изобретение также относится к способу лечения заболевания у млекопитающего, включающему введение нуждающемуся в нем млекопитающему терапевтически эффективного количества фармацевтической композиции, раскрытой выше в настоящем изобретении, в котором заболевание выбрано из группы, включающей волчанку, ревматоидный артрит, IBD (воспалительная болезнь кишечника), язвенный колит, болезнь Крона, витилиго, алопецию, псориаз и атопический дерматит.
Настоящее изобретение также относится к способу местного лечения заболевания у млекопитающего, включающему введение местным путем введения нуждающемуся в нем млекопитающему терапевтически эффективного количества фармацевтической композиции, раскрытой выше в настоящем изобретении, в котором заболевание выбрано из группы, включающей витилиго, алопецию, псориаз и атопический дерматит.
Настоящее изобретение также относится к фармацевтической композиции, раскрытой выше в настоящем изобретении, для применения в качестве лекарственного средства.
Настоящее изобретение также относится к фармацевтической композиции, раскрытой выше в настоящем изобретении, для применения для лечения нарушения, выбранного из группы, включающей волчанку, ревматоидный артрит, IBD, язвенный колит, болезнь Крона, витилиго, алопецию, псориаз и атопический дерматит.
Настоящее изобретение также относится к применению фармацевтической композиции, раскрытой выше в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения нарушения, выбранного из группы, включающей волчанку, ревматоидный артрит, IBD, язвенный колит, болезнь Крона, витилиго, алопецию, псориаз и атопический дерматит.
Прибор и методики анализа:
Рассчитанные порошковые рентгенограммы: Порошковые рентгенограммы рассчитывали по рентгенографическим данным для монокристалла с использованием программного обеспечения SHELXTL, включая XFOG (SHELXTL, Bruker AXS, XFOG, Version 5.100, 1997) и XPOW (SHELXTL, Bruker AXS, XPOW, Version 5.102, 1997-2000). Соответствующую длину волны, необходимую для наложения графики, добавляли с использованием программы обмена файлов XCH (SHELXTL, Bruker AXS, XCH, Version 5.0.4, 1995-2001).
Порошковая рентгенография:
Анализ с помощью порошковой рентгенографии проводили с помощью дифрактометра Bruker AXS D8 Advance, снабженного источником излучения Cu, снабженного зеркалом Гебеля. Дифрагированное излучение детектировали детектором LYNXEYE_EX со щелями с электроприводом. Для первичного и вторичного пучка использовали щели Соллера 2.5. Напряжение и силу тока на рентгеновской трубке устанавливали равными 40 кВ и 40 мА соответственно. Данные накапливали с помощью тета-тета гониометра при сканировании по схеме "тета-два тета" при длине волны Cu K-альфа от 3,0 до 40,0 градусов 2-тета с использованием 1204 шагов и скорости сканирования 0,50 с на шаг. Образцы готовили путем их помещения в кремниевый держатель с низким фоном и при сборе данных вращали. Данные собирали с использованием программного обеспечения Bruker DIFFRAC Plus. Анализ проводили с использованием программного обеспечения EVA diffract plus. Файл данных PXRD не обрабатывало до поиска пиков. С использованием алгоритма поиска пиков в программном обеспечении EVA для предварительного отнесения пиков использовали пики с пороговым значением, равным 1. Для обеспечения правильности отнесения проводили вручную; результаты автоматического отнесения проверяли визуально и положения пиков устанавливали по максимумам пиков. Обычно выбирали пики с относительной интенсивностью ≥ 2%. Неразрешенные пики и пики, согласующиеся с шумом, не выбирали. Типичная погрешность положения пика по данным PXRD в USP установлена равной +/- 0,2° 2-Тета (USP-941).
Отнесения отражений PXRD: Для визуализации и анализа спектров PXRD использовали программное обеспечение Eva Application 9.0. Положения пиков относили к максимуму интенсивности данного отражения. Все отражения, обладающие относительной интенсивностью, превышающей 10%, включены в последующие таблицы.
Настоящее изобретение относится к фармацевтической композиции получали из кристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)-циклобутил)пропан-1-сульфонамида, которую можно идентифицировано с помощью одной или большего количества твердофазных аналитических методик.
Перечень пиков PXRD для кристаллической формы при 23°C приведен в таблице 1.
Таблица 1: Перечень пиков PXRD для формы 1. Положения пиков представляют характеристические пики формы 1 безводного свободного основания N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида.
b) Определены, как высоты пиков. Интенсивности могут меняться в зависимости от фактических установленных экспериментальных параметров CPMAS и термической предыстории образца. Интенсивности CPMAS необязательно являются количественными. *Плечо пика
Соответственно, настоящее изобретение относится к фармацевтическим композициям, полученным из кристаллической формы, и к способам получения таких форм, а также фармацевтические композиции для применения в медицине и для применения для лечения таких заболеваний, как волчанка, ревматоидный артрит, IBD, язвенный колит, болезнь Крона, витилиго, алопеция, псориаз и атопический дерматит. Настоящее изобретение также относится к применению таких фармацевтических композиций для получения лекарственного средства для лечения таких заболеваний, как волчанка, ревматоидный артрит, IBD, язвенный колит, болезнь Крона, витилиго, алопеция, псориаз и атопический дерматит.
Способы лечения заболеваний и синдромов, указанные в настоящем изобретении, следует понимать, как включающие введение индивидууму, нуждающемуся в таком лечении, терапевтически эффективного количества фармацевтической композиции полиморфа, полученной способом, предлагаемым в настоящем изобретении. При использовании в настоящем изобретении термин "лечение" применительно к заболеванию означает предупреждение, подавление и/или улучшение протекания заболевания.
При использовании в настоящем изобретении термин "индивидуум" или "пациент", использующиеся взаимозаменяемым образом, означают любое животное, включая млекопитающих, предпочтительно мышей, крыс, других грызунов, кроликов, собак, кошек, свиней, крупный рогатый скот, овец, коз, лошадей или приматов и наиболее предпочтительно людей. При использовании в настоящем изобретении выражение "терапевтически эффективное количество" означает количество активного соединения или фармацевтического средства, которое приводит к биологическому или медицинскому ответу в ткани, системе, у животного, индивидуума или человека, который необходим исследователю, ветеринару, врачу или другому клиницисту или оказывающему помощь, или индивидууму, который включает одно или большее количество из следующих:
(1) предупреждение заболевания; например, предупреждение заболевания, патологического состояния или нарушения у индивидуума, который может быть предрасположен к заболеванию, патологическому состоянию или нарушению но который не ощущает или у которого не наблюдаются патология или симптоматика заболевания;
(2) подавление заболевания; например, подавление заболевания, патологического состояния или нарушения у индивидуума, который ощущает или у которого наблюдаются патология или симптоматика заболевания, патологического состояния или нарушения (т. е. остановка или замедление дальнейшего развития патологии и/или симптоматики); и
(3) улучшение протекания заболевания; например, улучшение протекания заболевания, патологического состояния или нарушения у индивидуума, который ощущает или у которого наблюдаются патология или симптоматика заболевания, патологического состояния или нарушения (т. е. обращение патологии и/или симптоматики).
Дозы и препарат
Настоящее изобретение также включает фармацевтические композиции, описанные в настоящем изобретении, включая одно или большее количество следующих: фармацевтически приемлемые носители, инертные наполнители, разбавители и т. п. Фармацевтическую композицию, предлагаемую в настоящем изобретении, вводят в количестве, эффективном для лечения патологического состояния, описанного в настоящем изобретении, и ее можно получить из самого кристаллического соединения, или, альтернативно, в виде его фармацевтически приемлемой соли.
Фармацевтическую композицию, предлагаемую в настоящем изобретении, вводят любым подходящим путем в виде фармацевтической композиции, адаптированной для такого пути, в дозе эффективной для назначенного лечения. Соединения, предлагаемые в настоящем изобретении, можно вводить перорально, ректально, вагинально, парентерально или местно.
Фармацевтическую композицию, предлагаемую в настоящем изобретении, можно вводить перорально. Пероральное введение может включать проглатывание, так что соединение попадает в желудочно-кишечный тракт, или можно использовать буккальное или сублингвальное введение, с помощью которого соединение попадает в кровоток непосредственно изо рта.
В другом варианте осуществления фармацевтическую композицию, предлагаемую в настоящем изобретении, также можно вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие пути для парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, внутрижелудочковый, внутриуретральный, надчревный, внутричерепной, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) шприцы, безыгольные шприцы и устройства для вливания.
В другом варианте осуществления фармацевтическую композицию, предлагаемую в настоящем изобретении, также можно вводить местно на кожу или слизистую оболочку, т. е. накожно или чрескожно. В другом варианте осуществления фармацевтическую композицию, предлагаемую в настоящем изобретении, также можно вводить интраназально или путем ингаляции. В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, можно вводить ректально или вагинально. В другом варианте осуществления фармацевтическую композицию, предлагаемую в настоящем изобретении, также можно вводить непосредственно в глаз или ухо.
Режим дозирования фармацевтических композиций, предлагаемых в настоящем изобретении, основан на множестве факторов, включая тип, возраст, массу, пол и состояние здоровья пациента; тяжесть патологического состояния; путь введения; и активность конкретного использующегося соединения. Таким образом, режим дозирования может значительно меняться. В одном варианте осуществления полная суточная доза соединения обычно равна от примерно 0,01 до примерно 100 мг/кг (т. е. количество миллиграммов соединения, предлагаемого в настоящем изобретении, на 1 кг массы тела) для лечения указанных патологических состояний, рассмотренных в настоящем изобретении. В другом варианте осуществления полная суточная доза соединения равна от примерно 0,1 до примерно 50 мг/кг и в еще одном варианте осуществления от примерно 0,5 до примерно 30 мг/кг.
Для перорального введения композиции можно использовать в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 75,0, 100, 125, 150, 175, 200, 250 и 500 мг активного ингредиента для симптоматического подбора дозы для пациента. Лекарственное средство обычно содержит от примерно 0,01 мг до примерно 500 мг активного ингредиента или в другом варианте осуществления от примерно 1 мг до примерно 100 мг активного ингредиента. Внутривенные дозы могут находиться в диапазоне от примерно 0,01 до примерно 10 мг/кг/мин при постоянной скорости вливания.
Подходящие субъекты в контексте настоящего изобретения включают млекопитающих. Млекопитающие, подходящие для настоящего изобретения, включают собачьих, кошачьих, бычьих, козлиных, лошадиных, овечьих, свиных, грызунов, зайцеобразных, приматов и т. п., и включают млекопитающих в утробе. В одном варианте осуществления подходящими субъектами являются люди. Люди могут быть любого пола и на любой стадии развития.
В другом варианте осуществления настоящее изобретение относится к фармацевтическим композициям, содержащим соединение вместе с фармацевтически приемлемым носителем. Также могут содержаться другие фармакологически активные вещества. При использовании в настоящем изобретении "фармацевтически приемлемый носитель" включает любые и все растворители, диспергирующие среды, покрытия, антибактериальные и фунгицидные агенты, изотонические агенты и агенты, задерживающие всасывание и т. п., которые являются физиологически совместимыми. Примеры фармацевтически приемлемых носителей включают одно или большее количество следующих: вода, физиологический раствор, забуференный фосфатом физиологический раствор, декстроза, глицерин, этанол и т. п., а также их комбинации, и могут содержать в композиции изотонические агенты, например, сахара, хлорид натрия или многоатомные спирты, такие как маннит, или сорбит. Фармацевтически приемлемые вещества, такие как смачивающие агенты или небольшие количества вспомогательных веществ, таких как смачивающие или эмульгирующие агенты, консерванты или буферы, которые увеличивают длительность действия или эффективность антител или фрагментов антител.
Композиции, предлагаемые в настоящем изобретении, могут находиться в разных формах. Они включают, например, жидкие, полужидкие и твердые дозированные формы, такие как жидкие растворы (например, растворы для инъекции и вливания), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Форма зависит от назначенного пути введения и терапевтического применения.
Типичные композиции находятся в форме растворов для инъекции или вливания, такие как композиции, аналогичные используемым для пассивной общей иммунизации людей антителами. Одним путем введения является парентеральный (например, внутривенный, подкожный, внутрибрюшинный, внутримышечный). В другом варианте осуществления антитела вводят путем внутривенного вливания или инъекции. В еще одном варианте осуществления антитела вводят путем внутримышечной или подкожной инъекции.
Дозированная форма для перорального введения твердого вещества может, например, быть представлена в отдельных порциях, таких как твердые или мягкие капсулы, пилюли, облатки, пастилки или таблетки, каждая из которых содержат заданное количество по меньшей мере одного соединения, предлагаемого в настоящем изобретении. В другом варианте осуществления пероральное введение может проводиться в форме порошка или гранулы. В другом варианте осуществления пероральная дозированная форма является сублингвальной, такой как, например, пастилка. В таких твердых дозированных формах кристаллическое соединение обычно объединяют с одним или большим количеством вспомогательных веществ. Такие капсулы или таблетки могут представлять собой препарат регулируемого высвобождения. В случае капсул, таблеток и пилюль дозированные формы также могут содержать буферные реагенты или их можно приготовить с энтеросолюбильными покрытиями.
В другом варианте осуществления пероральное введение может проводиться с помощью жидкой дозированной формы. Жидкие дозированные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно использующиеся в данной области техники (например, воду). Такие композиции также могут содержать вспомогательные вещества, такие как смачивающие, эмульгирующие, суспендирующие, придающие вкус (например, подсластители) и/или отдушки.
В другом варианте осуществления настоящее изобретение относится к парентеральной дозированной форме. "Парентеральное введение" включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинные, внутримышечные инъекции, надчревные инъекции и вливание. Препараты для инъекции (т. е. стерильные водные или масляные суспензии для инъекции) можно приготовить в соответствии с известным уровнем техники с использованием подходящих диспергирующих, смачивающих агентов и/или суспендирующих агентов.
В другом варианте осуществления настоящее изобретение относится к дозированной форме для местного применения. "Местное введение" включает, например, чрескожное введение, такое как с помощью чрескожных пластырей или устройств для ионтофореза, внутриглазное введение или интраназальное или ингаляционное введение. Композиции для местного введения также включают, например, гели, спреи, мази и кремы для местного применения. Препарат для местного применения может включать кристаллическое соединение, которое усиливает поглощение или проникновение активного ингредиента через кожу или другие пораженные участки. Если кристаллическое соединение, предлагаемое в настоящем изобретении, вводят с помощью чрескожного устройства, введение проводят с помощью пластыря резервуарного и пористого мембранного типа, или разных твердых матриц. Типичные препараты для этой цели включают гели, гидрогели, примочки, растворы, кремы, мази, присыпки, повязки, пенки, пленки, кожные пластыри, пластинки, имплантаты, губки, волокна, перевязочные материалы и микроэмульсии. Также можно использовать липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкое вазелиновое масло, белое вазелиновое масло, глицерин, полиэтиленгликоль и пропиленгликоль. Можно включать средства, улучшающие проницаемость - см., например, B. C. Finnin and T. M. Morgan, J. Pharm. Sci., vol. 88, pp. 955-958, 1999.
Соответственно, препараты для местного применения, полученные из кристаллической или некристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида способом, предлагаемым в настоящем изобретении, можно вводить в виде таких препаратов, включая все обычные способы введения через поверхность тела и внутренние выстилки пассажей организма, включая эпителиальные ткани и слизистые оболочки, включая чрескожный, эпидермальный, буккальный, легочный, глазной, интраназальный, вагинальный и ректальный пути введения. Типичные носители включают спирт, воду, минеральное масло, жидкое вазелиновое масло, белое вазелиновое масло, глицерин, полиэтиленгликоль и пропиленгликоль. Такие препараты для местного применения можно получить в комбинации с дополнительными фармацевтически приемлемыми инертными наполнителями. Инертный наполнитель, который может быть важен для обеспечения клинической эффективности, представляет собой один или большее количество средств, улучшающих проницаемость, таких как один или большее количество насыщенных или цис-ненасыщенных C10-C18 жирных спиртов. Такие жирные спирты включают C16-C18 жирные спирты и наиболее предпочтительным является C18 жирный спирт. Примеры цис-ненасыщенных C16-C18 жирных спиртов включают олеиловый спирт, линолеиловый спирт, г-линолениловый спирт и линолениловый спирт. Насыщенные C10-C18 жирные спирты, применимые, как средства, улучшающие проницаемость включают дециловый спирт, лауриловый спирт, миристиловый спирт, цетиловый спирт и стеариловый спирт. Альтернативно, другие средства, улучшающие проницаемость, которые можно использовать для получения препаратов для местного применения включают C10-C18 жирные кислоты, которые, если они являются насыщенными, могут включать каприновую кислоту, лауриновую кислоту, миристиновую кислоту, пальмитиновую кислоту, стеариновую кислоту и арахидиновую кислоту. Альтернативно, средство, улучшающее проницаемость предпочтительно может представлять собой цис-ненасыщенную жирную кислоту, такую как пальмитолеиновая кислота (цис-9-гексадеценовая кислота), олеиновая кислота (цис-9-октадеценовая кислота), цис-вакценовая кислота (цис-11-октадеценовая кислота), линолевая кислота (цис-9,12-октадекадиеновая кислота), г-линоленовая кислота (цис-6,9,12-октадекатриеновая кислота), линоленовая кислота (цис-9,12,15-октадекатриеновая кислота) и арахидоновая кислота (цис-5,8,11,14-эйкозатетраеновая кислота). Средства, улучшающие проницаемость, например, выбранные из числа C10-C18 жирных спиртов, используют в количествах в диапазоне от примерно 0,1 до примерно 5% (мас./об.), более предпочтительно, от 1 примерно до 4%, еще более предпочтительно от 1 примерно до 3% (мас./об.).
Препараты для местного применения, содержащие N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид в терапевтически эффективных количествах, можно вводить нуждающимся в них пациентам один или два раза в сутки. Эти количества находятся в диапазоне от примерно 0,1% до примерно 5,0% (мас./об.), более предпочтительно, от примерно 0,1% до примерно 3,0% (мас./об.). В число других инертных наполнителей, которые увеличивают стабильность этих препаратов, входят поглотители альдегидов, такие как глицерин и пропиленгликоль, и антиоксиданты, такие как бутилгидроксианизол (BHA), бутилгидрокситолуол (BHT), пропилгаллат, аскорбиновая кислота (витамин C), полифенолы, токоферолы (витамин E) и их производные.
Препараты, подходящие для местного введения в глаза, включают, например, глазные капли, в которых соединение, предлагаемое в настоящем изобретении, растворено или суспендировано в подходящем носителе. Типичный препарат, подходящий для введения в глаза или уши, может находиться в форме капель микронизированной суспензии или раствора в изотоническом с установленным pH стерильном физиологическом растворе. Другие препараты, подходящие для введения в глаза или уши, включают мази, биологически разрушающиеся (т. е. впитывающие гель губки, коллаген) и биологически неразрушающиеся (т. е. силиконовые) имплантаты, пластинки, линзы и измельченные или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновую кислоту, полимер целлюлозы, например, гидроксипропилметилцеллюлозу, гидроксиэтилцеллюлозу или метилцеллюлозу, или полимерный гетерополисахарид, например, геллановую камедь, можно включить вместе с консервантом, таким как бензалконийхлорид. Такие препараты также можно доставить с помощью ионтофореза.
Для интраназального введения или введения путем ингаляции кристаллическое соединение, предлагаемое в настоящем изобретении, обычно доставляют в форме раствора или суспензии из контейнера с насосом для распыления, который сжимает или использует для накачивания пациент, или системы распыления аэрозоля из контейнера под давлением или распылителя с использованием подходящего пропеллента. Препараты, подходящие для интраназального введения обычно вводят в форме сухого порошка (чистого, в виде смеси, например, сухой смеси с лактозой, или смешанных частиц компонента, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора для сухого порошка или с распылением аэрозоля из контейнера под давлением, с помощью насоса, распылителя, атомизатора (предпочтительно атомизатора с использованием электродинамического устройства для получения тонкого тумана), или распылителя с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан. Для интраназального применения, порошок может содержать биоадгезивный агент, например, хитозан или циклодекстрин.
В другом варианте осуществления настоящее изобретение относится к ректальной дозированной форме, полученной из кристаллической или некристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида способом, предлагаемым в настоящем изобретении. Такая ректальная дозированная форма может находиться в форме, например, суппозитория. Масло какао является традиционной основой для суппозитория, но можно использовать разные альтернативы, если они являются подходящими.
Также можно использовать другие материалы носителей и пути введения, известные в фармацевтике. Фармацевтические композиции, предлагаемые в настоящем изобретении, можно получить по любой из хорошо известных методик фармацевтики, таким как эффективные процедуры получения и введения. Приведенные выше соображения в отношении эффективных препаратов и методик введения хорошо известны в данной области техники и описаны в стандартных учебниках. Составление лекарственных средств рассмотрено, например, в Hoover, John E., Remington’s Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Kibbe et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999.
Кристаллическую или некристаллическую форму N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, полученную способом, предлагаемым в настоящем изобретении, можно использовать по отдельности или в комбинации с другими терапевтическими средствами. Настоящее изобретение относится к любым из применений, способов или композиций, определенных в настоящем изобретении, где кристаллическую или некристаллическую форму, указанную в настоящем изобретении, или фармацевтически приемлемый сольват указанного соединения, используют в комбинации с одним или большим количеством других терапевтических средств, рассмотренных в настоящем изобретении.
Введение двух или большего количества соединений "в комбинации" означает, что все соединения вводят в близкие моменты времени, так что наличие одного средства может изменить биологические воздействия другого соединения (соединений). Два или большее количество соединений можно вводить одновременно, совместно или последовательно. Кроме того, одновременное введение можно провести путем смешивания соединений до введения или путем введения соединений в один и тот же момент времени, но в виде отдельных дозированных форм на один или разные участки введения.
Выражения "одновременное введение", "совместное введение", "синхронное введение" и "введение одновременно" означает, что соединения вводят в комбинации.
В другом варианте осуществления настоящее изобретение относится к способам лечения, которые включают введение кристаллической или некристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, полученной способом, предлагаемым в настоящем изобретении, в комбинации с одним или большим количеством других фармацевтических средств, где одно или большее количество других фармацевтических средств можно выбрать из числа средств, рассмотренных в настоящем изобретении.
Эти средства и кристаллическую или некристаллическую форму N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, полученную способом, предлагаемым в настоящем изобретении, можно объединить с фармацевтически приемлемыми разбавителями, такими как физиологический раствор, раствор Рингера, раствор декстрозы и т. п. Конкретный режим введения, т. е. доза, время и количество повторов, зависит от конкретного индивидуума и истории болезни индивидуума.
Приемлемые носители, инертные наполнители или стабилизаторы нетоксичны для реципиента в использующихся дозах и концентрациях и могут содержать буферы, такие как фосфатный, цитратный и включающий другие органические кислоты; соли, такие как хлорид натрия; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как октадецилдиметилбензилхлорид аммония; гексаметонийхлорид; бензалконийхлорид, бензэтонийхлорид; фенол, бутиловый или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; пирокатехин; резорцин; циклогексанол; 3-пентанол; и м-крезол); обладающие низкой молекулярной массой (менее примерно 10 остатков) полипептиды; балки, такие как сывороточный альбумин, желатин или Igs; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатные агенты, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как ионы натрия; комплексы металлов (например, комплексы Zn-белок); и/или неионогенные поверхностно-активные вещества, такие как TWEENTM, PLURONICSTM или полиэтиленгликоль (PEG).
Липосомы, содержащие эти средства и/или соединения, предлагаемые в настоящем изобретении, получают по методикам, известным в данной области техники, таким как описанные в патентах U.S. №№ 4485045 и 4544545. Липосомы с увеличенным временем обращения раскрыты в патенте U.S. № 5013556. Особенно полезные липосомы можно получить по методике обращенно-фазового выпаривания с использованием липидной композиции, содержащей фосфатидилхолин, холестерин и дериватизированный с помощью PEG фосфатидилэтаноламин (PEG-PE). Липосомы экструдируют через фильтры с определенным размером отверстий и получают липосомы желательного диаметра.
Эти средства и/или кристаллическую или некристаллическую форму N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, полученную способом, предлагаемым в настоящем изобретении, также можно поместить в микрокапсулы, получаемые, например, по методикам коацервации или с помощью межфазной полимеризации, например, гидроксиметилцеллюлозы, или желатиновые микрокапсулы и поли(метилметакрилатные) микрокапсулы соответственно для коллоидных систем доставки лекарственного средства (например, липосомы, микросферы альбумина, микроэмульсии, наночастицы и нанокапсулы) или включить в макроэмульсии. Такие методики раскрыты в Remington, The Science and Practice of Pharmacy, 20th Ed., Mack Publishing (2000).
Можно использовать препараты пролонгированного высвобождения. Подходящие примеры препаратов пролонгированного высвобождения включают полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие антитела/соединение, предлагаемое в настоящем изобретении, и эти матрицы находятся в виде формованных изделий, например, пленок или микрокапсул. Примеры матриц пролонгированного высвобождения включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды (патент U.S. № 3773919), сополимеры L-глутаминовой кислоты и 7-этил-L-глутамата, неразлагающиеся сополимеры этилен-винилацетат, разлагающиеся сополимеры молочная кислота-гликолевая кислота, такие как использующиеся в LUPRON DEPOTTM (микросферы для инъекции, состоящие из сополимера молочная кислота-гликолевая кислота и лейпролидацетата), ацетат-изобутират сахарозы и поли-D-(-)-3-гидроксимасляную кислоту.
Препараты для использования для внутривенного введения должны быть стерильными. Это легко обеспечить, например, путем стерильного фильтрования через мембраны. Кристаллическую или некристаллическую форму N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, полученную способом, предлагаемым в настоящем изобретении, обычно помещают в контейнер, содержащий стерильное впускное отверстие, например, мешок с раствором для внутривенного введения или флакон с пробкой, прокалываемой иглой шприца для подкожной инъекции.
Подходящие эмульсии можно получить с использованием имеющихся в продаже эмульсии жиров, таких как IntralipidTM, LiposynTM, InfonutrolTM, LipofundinTM и LipiphysanTM. Активный ингредиент можно растворить в предварительно смешанной композиции эмульсии или, альтернативно, его можно растворить в масле (таком как, соевое масло, сафлоровое масло, хлопковое масло, кунжутное масло, кукурузное масло или миндальное масло) и эмульсии, образовавшейся после перемешивания с фосфолипидом (таким как яичные фосфолипиды, соевые фосфолипиды или соевый лецитин) и водой. Следует понимать, что можно добавить другие ингредиенты, например, глицерин или глюкозу, для регулирования тоничности эмульсии. Подходящие эмульсии обычно содержат до 20% масла, например, от 5 до 20%. Эмульсия жира может содержать капельки жира размером от 0,1 до 1,0 мкм, предпочтительно от 0,1 до 0,5 мкм, и обладают pH в диапазоне от 5,5 до 8,0.
Реагенты, использующиеся для получения кристаллической или некристаллической формы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида способом, предлагаемым в настоящем изобретении, могут находиться в продаже или их можно получить по стандартным методикам, описанным в литературе. Например, кристаллическую или некристаллическую форму, предлагаемую в настоящем изобретении, можно получить по методикам, приведенным в последующих- примерах.
В описании настоящего изобретения используются разные аббревиатуры, хорошо известные специалистам в данной области техники, включая следующие:
CH3CN: Ацетонитрил
DCM: Дихлорметан
DMF: N,N-Диметилформамид
DMSO: Диметилсульфоксид
EtOAc: Этилацетат
EtOH: Этанол
FT-IR: Инфракрасная Фурье-спектроскопия
HOAc: Уксусная кислота
MeOH: Метанол
PXRD: порошковая рентгенография
ss 13C ЯМР: твердофазный 13C ядерный магнитный резонанс
ТГФ: Тетрагидрофуран
ТСХ: Тонкослойная хроматография
ПРИМЕРЫ
Последующие неограничивающие примеры приведены только для иллюстрации настоящего изобретения. Специалист в данной области техники должен понимать, что имеются многочисленные эквиваленты и варианты, не приведенные в качестве примеров, которые все же являются частью настоящего изобретения.
Пример 1
Получение формы I N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)-циклобутил)пропан-1-сульфонамида
Искомое соединение получали в соответствии с примером 2 патента US № 9035074. Неочищенное вещество нагревали в 10 объемах (100 мг/мл) смеси состава 2:1 EtOH/вода при 80°C (до полного растворения) и затем подвергали фильтрованию тонкой очистки и медленно охлаждали до кристаллизации продукта. После фильтрования, вещество сушили в вакууме при 45-55°C.
Пример 2
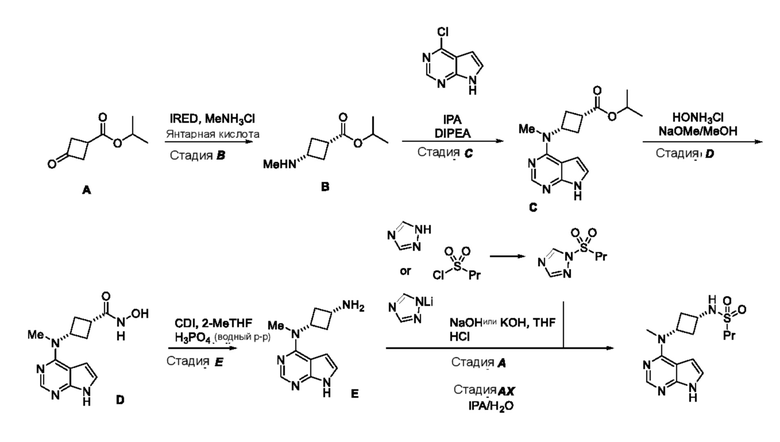
Альтернативное получение N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида
Изопропил-3-оксоциклобутан-1-карбоксилат (A)
Имеющуюся в продаже 3-циклобутанонкарбоновую кислоту (175 г) растворяли в 2-пропаноле (1050 мл) и добавляли моногидрат п-толуолсульфоновой кислоты (11,85 г, 4 мол.%). Раствор нагревали при 80°C и перемешивали в течение 19 ч. По данным UPLCMS полагали, что реакция завершилась и реакционную смесь охлаждали. Реакционную смесь концентрировали и получали светло-желтое масло и разбавляли с помощью 1000 мл MTBE. Раствор промывали с помощью 300 мл насыщенного раствора бикарбоната натрия и разделяли. Водный слой отбрасывали и промывали с помощью еще 200 мл насыщенного раствора бикарбоната натрия. Слои разделяли и водный слой отбрасывали. Содержащий MTBE слой сушили с помощью 200 мл рассола и затем сульфата магния. Затем раствор в MTBE концентрировали и получали бледно-желтое масло. 1H ЯМР (400 МГц, DMSO-d6) д 4,95 (гептет, J=6,3 Гц, 1H), 3,38-3,18 (m, 5H), 1,22 (d, J=6,3 Гц, 6H).
Изопропил-(1s,3s)-3-(метиламино)циклобутан-1-карбоксилат (свободное основание) (B)
В реактор помещали водный раствор метиламина (75 мл, 40 мас.%), затем фосфатный буфер (700 мл, pH 7,2, 100 мМ), состоящий из моно- и дифосфата калия. Значение pH раствора устанавливали равным 8,8 путем медленного добавления концентрированной хлористоводородной кислоты. Затем добавляли NADP+ (700 мг), GDH (350 мг) и глюкозу (150 г). В реактор помещали фермент IRED (50 мл лизата). Субстрат изопропил-3-оксоциклобутан-1-карбоксилат (100 г) разбавляли с помощью DMSO (25 мл) и помещали м реактор. Реакционную смесь нагревали при 30°C и pH поддерживали равным 7,5 с помощью pH-метра и дозирующего устройства (2 н. гидроксид натрия). За протеканием реакции следили путем анализа с помощью GC и UPLC. Когда полагали, что реакция завершена, смесь фильтровали через целит™ (50 г) и осадок на целите™ промывали водой (100 мл). В делительную воронку помещали водный раствор и MTBE (500 мл) и встряхивали. Слой MTBE отделяли и отбрасывали. Затем водный слой подщелачивали гидроксидом натрия (50% водный раствор) до pH=12. Затем добавляли MTBE (1 л) и делительную воронку встряхивали. Фазы разделялись и MTBE собирали и отставляли. Водный слой один раз экстрагировали дополнительным количеством MTBE (700 мл) и слои разделяли. Водный слой отбрасывали и все растворы в MTBE фильтровали через дополнительное количество целита™ (36,5 г). Затем растворы в MTBE объединяли и сушили над безводным сульфатом натрия. 1H ЯМР (400 МГц, DMSO-d6) д 4,86 (гептет, J=6,3 Гц, 1H), 2,96 (tt, J=8,7, 7,0 Гц, 1H), 2,67 (tt, J=9,7, 8,1 Гц, 1H), 2,41-2,26 (m, 2H), 2,15 (s, 3H), 1,78 (dtd, J=9,8, 8,8, 2,6 Гц, 2H), 1,17 (d, J=6,2 Гц, 6H).
Изопропил-(1s,3s)-3-(метиламино)циклобутан-1-карбоксилат (сукцинат) (сукцинат B)
Неочищенную биокаталитическую реакционную смесь (1125 мл) концентрировали примерно до половины объема (530 мл). В отдельный реактор помещали янтарную кислоту (75,6 г) и 2MeТГФ (1100 мл) и нагревали при 60°C для растворения кислоты. Раствор охлаждали до 50°C и добавляли примерно половину раствора амина. Полученный мутный раствор выдерживали в течение 30 мин и получали жидкую суспензию. Затем добавляли остаток раствора амина. Раствор охлаждали до 20°C и затем нагревали до 50°C и получали суспензию. Суспензию выдерживали в течение 30 мин при 50°C и затем охлаждали до 20°C и перемешивали в течение ночи. Суспензию фильтровали и промывали двумя порциями 2-MeТГФ (по 100 мл) и вещество сушили в сушильном шкафу. Выделяли 95,7 г (выход 52%).
1H ЯМР (400 МГц, DMSO-d6) д 4,88 (гептет, J=6,2 Гц, 1H), 3,30 (tt, J=8,8, 7,3 Гц, 1H), 2,82 (tt, J=9,8, 8,2 Гц, 1H), 2,43-2,34 (m, 2H), 2,31 (d, J=2,5 Гц, 7H), 2,13-2,00 (m, 2H), 1,18 (d, J=6,3 Гц, 6H).
Изопропил-(1s,3s)-3-(метиламино)циклобутан-1-карбоксилат (B соль с HCl)
Неочищенный амин (8 г) растворяли в MTBE (80 мл) и нагревали при 50 C. Затем добавляли хлористоводородную кислоту в диоксане (11 мл, 4M). Наблюдали перемешиваемую суспензию и реакционную смесь выдерживали при 50°C в течение 1 ч. Суспензию охлаждали до 20°C и затем фильтровали и промывали двумя порциями MTBE (по 20 мл). Затем вещество сушили в вакууме. Выделяли 8,7 г (выход 96%). 1H ЯМР (400 МГц, DMSO-d6) д 9,26 (s, 2H), 4,89 (гептет, J=6,3 Гц, 1H), 3,52 (tt, J=9,0, 7,5 Гц, 1H), 2,92 (tt, J=9,8, 8,2 Гц, 1H), 2,45-2,35 (m, 5H), 2,34-2,24 (m, 2H), 1,18 (d, J=6,3 Гц, 6H).
Изопропил-(1s,3s)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)цикло-бутан-1-карбоксилат (C; CSNAr)
Сукцинат амина (сукцинат B, 75,4 г) и хлорпирролопиримидин (40,0 г) объединяли в реакторе. Добавляли 2-пропанол (200 мл) и получали суспензию. Добавляли диизопропилэтиламин (114 мл) и получали жидкую суспензию. Реакционную смесь нагревали при 80°C, что давало раствор. Реакционную смесь выдерживали при 80°C, пока по данным UPLCMS не полагали, что реакция завершена (примерно через 48 ч). Реакционную смесь охлаждали до 20°C и получали суспензию. Твердые вещества отфильтровывали и промывали двумя порциями 2-пропанола (по 80 мл). Выделяли 61 г вещества (81%). 1H ЯМР (400 МГц, DMSO-d6) д 11,64 (s, 1H), 8,11 (s, 1H), 7,16 (dd, J=3,6, 2,4 Гц, 1H), 6,62 (dd, J=3,6, 1,9 Гц, 1H), 5,24 (tt, J=9,4, 8,0 Гц, 1H), 4,91 (гептет, J=6,3 Гц, 1H), 3,25 (s, 3H), 2,87 (tt, J=9,2, 8,0 Гц, 1H), 2,48-2,35 (m, 4H), 1,20 (d, J=6,3 Гц, 6H).
(1s,3s)-N-Гидрокси-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)цикло-бутан-1-карбоксамид (D; амид гидроксамовой кислоты)
В реактор помещали метанол (500 мл) и метоксид натрия в метаноле (93,7 мл, 25 мас.%) в атмосфере азота. К реакционной смеси при комнатной температуре добавляли гидроксиламингидрохлорид (15,1 г), что приводило к небольшому поглощению тепла. Затем добавляли изопропил-(1s,3s)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутан-1-карбоксилат (C; 50 г) и получали белую суспензию. Реакционную смесь нагревали при 40°C и перемешивали в течение ночи и по данным UPLCMS полагали, что реакция завершена. Затем в густую суспензию добавляли хлористоводородную кислоту (1M) до установления pH=7,0. Затем суспензию фильтровали и промывали метанолом (100 мл). Вещество сушили в вакуумном сушильном шкафу в течение ночи и получали 42,7 г белого твердого вещества (выход 94%). 1H ЯМР (400 МГц, DMSO-d6) д 11,63 (s, 1H), 10,46 (s, 1H), 8,75 (s, 1H), 8,10 (s, 1H), 7,15 (dd, J=3,6, 1,9 Гц, 1H), 6,61 (dd, J=3,7, 1,4 Гц, 1H), 5,21 (p, J=9,6 Гц, 1H), 3,28 (s, 3H), 2,60 (p, J=8,3 Гц, 1H), 2,49-2,37 (m, 2H), 2,35-2,26 (m, 2H).
(1s,3s)-N1-Метил-N1-(7H-пирроло[2,3-d]пиримидин-4-ил)циклобутан-1,3-диамин (E; фосфат)
В реактор помещали амид гидроксамовой кислоты D (19,4 г), затем 2-MeТГФ (388 мл) и получали белую суспензию. Суспензию нагревали при 30°C и добавляли карбонилдиимидазол (16,1 г). Реакционную смесь перемешивали в течение ночи. По данным UPLCMS полагали, что реакция завершена. Готовили раствор фосфорной кислоты (14,7 M в воде, 25,5 мл), разбавленный водой (78 мл), и медленно добавляли к суспензии. Суспензия растворялась и реакционную смесь нагревали при 60°C и выдерживали в течение нескольких часов. Полагали, что реакция завершена и гидроксид натрия (20 мас.% в воде, 16,4 m) добавляли к для установления pH, равного примерно 6. Затем реакционную смесь нагревали при 80°C и затем охлаждали до 25°C. Медленно добавляли 2-пропанол (58 мл) и твердые вещества отфильтровывали. Осадок на фильтре промывали смесью 2-пропанол/вода (1:1, 40 мл) и сушили в вакуумном сушильном шкафу и получали 19,1 г (выход 81%). 1H ЯМР (600 МГц, оксид дейтерия, 35°C) д 8,08 (s, 1H), 7,20 (d, J=3,6 Гц, 1H), 6,66 (d, J=3,6 Гц, 1H), 4,81 (tt, J=9,6, 7,4 Гц, 1H), 3,67 (tt, J=8,9, 7,3 Гц, 1H), 3,28 (s, 3H), 2,96-2,73 (m, 2H), 2,47 (qd, J=9,4, 2,9 Гц, 2H). 31P ЯМР (243 МГц, оксид дейтерия, 35°C) д 0,31.
(1s,3s)-N1-Метил-N1-(7H-пирроло[2,3-d]пиримидин-4-ил)циклобутан-1,3-диамин (свободное основание амина)
Аминофосфат E (10 г) растворяли в H2O (30 мл). Добавляли хлористоводородную кислоту (6 н.) до установления pH=2 и получали прозрачный раствор. Значение pH повышали до 12 гидроксидом натрия (50 мас.%) и получали густую суспензию. Суспензию очищали с помощью хроматографии с обращенной фазой (10:90 CH3CN:H2O 2 объема колонки, градиентный режим до 40:60 CH3CN:H2O, 4 объема колонки). Соответствующие фракции собирали, объединяли и концентрировали в вакууме, и получали свободное основание амина E в виде белого твердого вещества (6,05 г, выход 88%). 1H ЯМР (400 МГц, оксид дейтерия) д 7,78 (s, 1H), 6,91 (d, J=3,6 Гц, 1H), 6,24 (d, J=3,6 Гц, 1H), 4,25 (ddd, J=9,7, 7,4, 2,3 Гц, 1H), 3,08 (td, J=7,9, 7,1, 1,8 Гц, 1H), 2,95 (s, 3H), 2,58-2,39 (m, 2H), 1,86 (dd, J=9,4, 2,8 Гц, 2H).
1-(Пропилсульфонил)-1H-1,2,4-триазол (триазольный реагент)
1,2,4-Триазол (11,98 г) и ТГФ (40 мл) помещали в реактор, снабженный верхним перемешивающим устройством. Суспензию перемешивали в течение 10 мин, затем добавляли 1-пропансульфонилхлорид (7,89 мл, 68,0 ммоля) при 20°C. Полученную суспензию перемешивали при 20°C, пока по данным 1H ЯМР не израсходовались исходные вещества. После завершения реакционную смесь фильтровали и фильтрат переносили в делительную воронку, где его разбавляли водой (20 мл) и экстрагировали дихлорметаном (50 мл). Слои разделяли и слой с DCM промывали водой (2Ч20 мл) и рассолом (1Ч20 мл). Органический слой сушили над MgSO4, фильтровали, и концентрировали в вакууме, и получали сульфонилтриазол в виде вязкого прозрачного бесцветного масла (10,66 г, выход 89%). 1H ЯМР (400 МГц, хлороформ-d) д 8,67 (s, 1H), 8,13 (s, 1H), 3,55-3,46 (m, 2H), 1,76 (h, J=7,5 Гц, 2H), 1,03 (t, J=7,5 Гц, 3H).
Пример 3
Альтернативное получение N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)-амино)циклобутил)пропан-1-сульфонамида
Сульфонилирование с использованием триазольного реагента, 1-(пропилсульфонил)-1H-1,2,4-триазола
В реактор помещали воду (18 мл) и фосфат амина E (3 г), затем гидроксид натрия в воде (19M, 1,5 мл). Реакция протекала с небольшим выделением тепла и реакционную смесь охлаждали до 25°C. 1-(Пропилсульфонил)-1H-1,2,4-триазол (4,3 г) растворяли в ТГФ (12 мл) и добавляли в реакционную смесь с гидроксидом. Когда полагали, что реакция завершена, добавляли воду (18 мл) и вещество отфильтровывали и сушили. Получали 2,57 г (выход 84%).
In-situ получение реагента
Литиевую соль 1,2,4-триазола (1,0 г) и ТГФ (8 мл) помещали в реактор при 20°C. Затем добавляли 1-пропансульфонилхлорид (1,47 мл). Суспензию перемешивали при 20°C, пока по данным 1H ЯМР не израсходовался сульфонилхлорид. В отдельной колбе аминофосфат (2,0 г) растворяли в H2O (12 мл) при 20°C и затем добавляли гидроксид натрия (1,0 мл, 50 мас.%), поддерживая температуру ниже 30°C. Водный раствор охлаждали до 10°C, затем добавляли раствор в ТГФ реагента 1-пропансульфонилтриазола, поддерживая температуру ниже 20°C. Полученную суспензию перемешивали, пока по данным UPLCMS не оставалось <5% амина, затем добавляли гидроксид натрия (0,67 мл, 50 мас.%) и реакционную смесь нагревали при 50°C. Когда по данным UPLC израсходовался сульфонилтриазольный реагент, реакционную смесь охлаждали до 20°C и значение pH устанавливали равным 5-6 с помощью хлористоводородной кислоты (6 н.). Полученную суспензию охлаждали до 10°C и выдерживали в течение 30 мин и фильтровали. Осадок на фильтре промывали смесью 75:25 H2O:ТГФ (10 мл) и твердые вещества сушили при 50°C в вакуумном сушильном шкафу, и получали искомый продукт в виде почти белого твердого вещества.
Сульфонилирование с использованием 1-пропансульфонилхлорида
В реактор помещали фосфат амина E (3,07 г), затем воду (18 мл). Затем к суспензии добавляли гидроксид натрия (2,9 мл, 10M) и смесь перемешивали при комнатной температуре. Добавляли 2-MeТГФ (12 мл) и смесь охлаждали до 10°C. Добавляли 1-пропансульфонилхлорид (1,6 мл) добавляли, что приводило к выделению тепла. За протеканием реакции следили и полагали, что реакция завершена. Затем добавляли воду (18 мл) и суспензию гранулировали при 10°C. Суспензию фильтровали, промывали водой (5 мл) и сушили в вакууме. Выделяли 3,0 г бледно-желтого твердого вещества (выход 96%).
Пример 4
Альтернативное получение (1s,3s)-N1-метил-N1-(7H-пирроло[2,3-d]пиримидин-4-ил)циклобутан-1,3-диамина
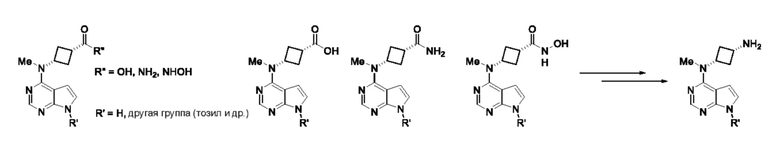
Получение (1s,3s)-3-(метил-(7-тозил-7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутан-1-карбоксамида
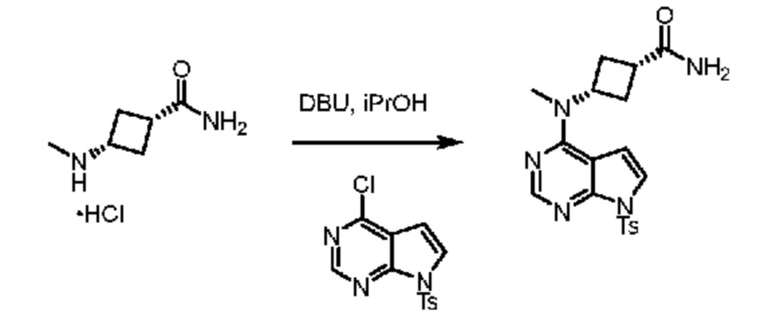
В пробирку Шленка объемом 20 мл помещали гидрохлорид амино-амида (500 мг, 3,04 ммоля 1,0 экв.) и изопропанол (4 мл), затем хлорпирролопиримидин (1,03 г, 3,34 ммоля, 1,1 экв.) и DBU (0,97 г, 6,38 ммоля, 2,1 экв.). Полученную смесь нагревали до 85°C и перемешивали, пока по данным UPLC реакция не завершалась. После завершения смесь охлаждали до 40°C, затем добавляли воду (20 мл) и получали прозрачный раствор. Продолжали охлаждение до 20°C, что приводило к осаждению твердых веществ из реакционной смеси. Твердые вещества отфильтровывали и осадок на фильтре промывали с помощью H2O (30 мл). Неочищенные твердые вещества (1,18 г) очищали с помощью хроматографии с обращенной фазой (градиентный режим от 4:6 MeOH:H2O до 100% MeOH, 20 объемов колонки). Искомые фракции объединяли и метанол удаляли в вакууме, что приводило к осаждению твердых веществ. Твердые вещества отфильтровывали, промывали с помощью H2O (10 мл) и сушили в вакуумном сушильном шкафу при 50°C, и получали искомый продукт реакции SNAr в виде белого твердого вещества (686 мг, выход 57%). 1H ЯМР (400 МГц, DMSO-d6) д 8,24 (s, 1H), 7,97 (d, J=8,4 Гц, 2H), 7,63 (d, J=4,1 Гц, 1H), 7,43 (d, J=8,6 Гц, 2H), 7,31 (s, 1H), 6,96 (d, J=4,1 Гц, 1H), 6,82 (s, 1H), 5,07 (p, J=8,6 Гц, 1H), 3,22 (s, 3H), 2,70 (p, J=8,6 Гц, 1H), 2,40-2,26 (m, 7H). 13C ЯМР (101 МГц, DMSO-d6) д 175,6, 157,1, 152,8, 151,7, 146,2, 134,9, 130,4, 128,2, 122,0, 106,9, 104,8, 47,3, 32,2, 31,7, 31,0, 21,6.
Получение (1s,3s)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутан-1-карбоновой кислоты
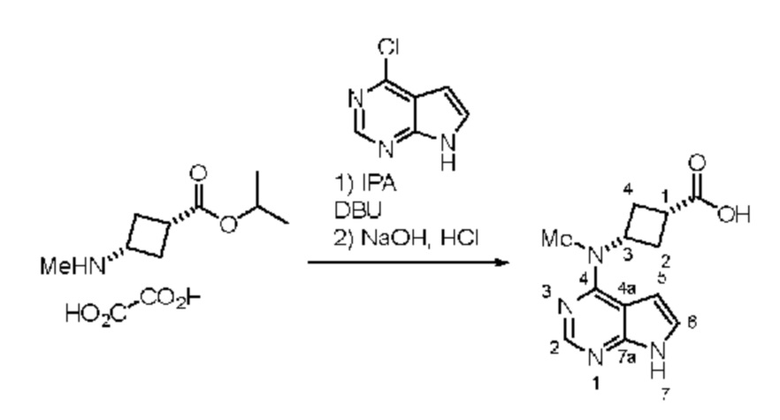
В реактор помещали хлорпирролопиримидин (10,0 г) и добавляли оксалат амино-амида (18,7 г). Добавляли IPA (100 мл) и суспензию перемешивали. Добавляли DBU (39 мл) и смесь нагревали при 80°C. Когда полагали, что реакция завершена, добавляли воду и гидроксид натрия, и реакционную смесь перемешивали при 80°C до образования густой суспензии. Добавляли IPA (60 мл) добавляли и суспензию перемешивали и фильтровали. Выделяли 26,39 г аддукта кислота-DBU. 10,0 г аддукта с DBU растворяли в воде (125 мл) при 45°C. Добавляли хлористоводородную кислоту (4 мл, 6M) и полученный осадок перемешивали и отфильтровывали. Твердые вещества промывали водой (10 мл). Твердые вещества сушили (2,47g). 1H ЯМР (400 МГц, DMSO-d6) д 2,44 (d, J=8,8 Гц, 4H), 2,44 (d, J=8,8 Гц, 4H), 2,77-2,91 (m, 1H), 3,26 (s, 3H), 3,31-3,41 (m, 1H), 5,23 (t, J=8,7 Гц, 1H), 6,54-6,72 (m, 1H), 7,08-7,26 (m, 1H), 8,12 (s, 1H), 11,65 (br s, 1H), 12,13-12,43 (m, 1H).
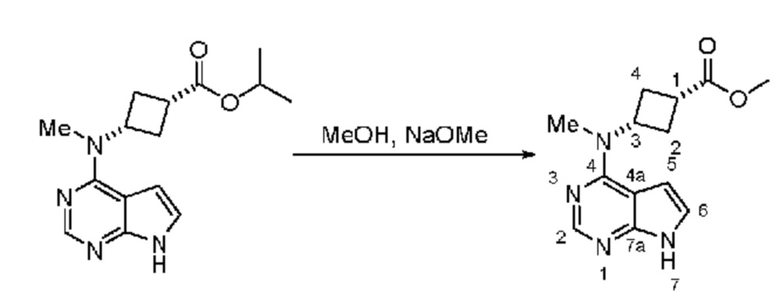
Получение метил-(1s,3s)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутан-1-карбоксилата
Изопропиловый эфир изопропил-(1s,3s)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутан-1-карбоксилат, 15,03 г) помещали в реактор с метанолом (75 мл) и перемешивали при 25°C. По каплям добавляли метоксид натрия (25 мас.% в метаноле, 15 мл) и реакционную смесь перемешивали, пока по данным UPLCMS не полагали, что реакция завершена. Твердые вещества отфильтровывали, промывали метанолом (20 мл) и сушили в вакуумном сушильном шкафу и получали 11,27 г белого твердого вещества. 1H ЯМР (400 МГц, хлороформ-d) д 11,48 (s, 1H), 8,33 (s, 1H), 7,09 (d, J=3,6 Гц, 1H), 6,58 (d, J=3,6 Гц, 1H), 5,58-5,23 (m, 1H), 3,72 (s, 3H), 3,39 (s, 3H), 2,88 (dq, J=10,0, 8,1 Гц, 1H), 2,69-2,52 (m, 4H). 13C ЯМР (101 МГц, CDCl3) д 175,36, 157,64, 152,11, 150,87, 120,46, 103,41, 102,02, 52,06, 47,38, 32,03, 31,80, 31,20.
Пример 5
Общая методика получения соли соединения E:
Соединение E растворяли в разных растворителях, указанных в таблице 1, затем добавляли до 4 экв. противоионов/веществ для совместного образования, указанных в таблице 2. Все образцы обрабатывали путем циклического изменения температуры и твердые вещества выделяли для характеризации.
Таблица 1
Таблица 2
Пример 6
Альтернативное получение N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида из аминоциклобутана
Получение сульфонилтриазола:
В колбу объемом 250 мл помещали воду (75 мл) и 1,2,4-триазол (37,24 г, 1,7 экв.), и перемешивали до растворения. После образования гомогенного раствора его переносили в сосуд объемом 2 л, содержащий ТГФ (400 мл) и снабженный верхним перемешивающим устройством. Раствор нагревали до 35°C, затем добавляли безводный порошкообразный LiOH (13,18 г, 1,7 экв.). Полученную суспензию перемешивали в течение 30 мин или до полного растворения всех твердых веществ. После обеспечения гомогенности медленно добавляли 1-пропансульфонилхлорид (58,84 мл, 1,6 экв.), поддерживая внутреннюю температуру ниже 40°C. После завершения добавления перемешивание раствора продолжали при 35°C в течение 30 мин, затем охлаждали до 0°C и выдерживали до завершения образования фосфата свободного основания амина E.
Образование свободного основания E из фосфата E:
В сосуд объемом 1 л, снабженный верхним перемешивающим устройством, помещали воду (300 мл) и ТГФ (200 мл), затем 11,5M водный раствор KOH (82,75 мл, 3,0 экв.) и нагревали до 25°C. Пятью порциями добавляли фосфат амина E (100 г, 1,0 экв.) и перемешивание продолжали до растворения всех твердых веществ. Перемешивание прекращали фазам давали разделиться. Водную (нижнюю) фазу отбрасывали. Перемешивание органической фазы продолжали при охлаждении до 8°C.
Первичная процедура:
Охлажденный раствор амина (свободного основания) переносили в раствор сульфонилтриазола, поддерживая внутреннюю температуру ниже 10°C. При необходимости сосуд, содержавший амин, промывали минимальным количеством ТГФ. После завершения переноса полученный раствор нагревали до 20°C со скоростью 1°C/мин. Перемешивали в течение 90 мин, затем добавляли 11,5 M водный раствор KOH (13,8 мл, 0,5 экв.). Перемешивание продолжали, пока не оставалось <5% амина E. После завершения добавляли воду (1,14 л), затем 11,5 M водный раствор KOH (110,3 мл, 4 экв.). Смесь нагревали до 40°C и выдерживали в течение 4 ч, затем охлаждали до 10°C. После охлаждения начинали перегонку в вакууме (100 мбар), медленно нагревая до внутренней температуры, равной 45-50°C, затем конечный объем раствора составлял 1,65-1,70 л, это показывало, что перегонка завершена. Смесь охлаждали до 40°C, затем добавляли концентрированную HCl (149 мл, 5,7 экв.), устанавливая pH равным 2,0-3,0, поддерживая внутреннюю температуру равной 40-45°C. Смесь нагревали до 65°C, выдерживали в течение 15 мин, затем охлаждали со скоростью 0,5°/мин до 53°C. По окончании добавляли затравочные кристаллы N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)-циклобутил)пропан-1-сульфонамида.HCl (500 мг, 0,005 г/г). Охлаждали до 50°C и перемешивали в течение 30 мин, затем охлаждали до 10°C со скоростью 0,1°C/мин. Перемешивали при 10°C в течение 1 ч, затем фильтровали через воронку Бюхнера объемом 600 мл с крупнозернистой фриттой. Осадок на фильтре промывали с помощью H2O, охлажденной до 10°C (200 мл). Твердые вещества сушили в воронке, доводя конечное содержание воды до 9,5% по данным титрования по Карлу Фишеру. 1H ЯМР (400 МГц, DMSO-d6) д 12,81 (s, 1H), 8,34 (s, 1H), 7,64 (d, J=9,3 Гц, 1H), 7,52-7,43 (m, 1H), 7,00 (dd, J=3,7, 1,8 Гц, 1H), 4,75 (t, J=8,3 Гц, 1H), 3,63 (h, J=8,4 Гц, 1H), 3,00-2,90 (m, 2H), 2,73 (dtd, J=10,0, 7,4, 2,9 Гц, 2H), 2,35 (qd, J=9,1, 2,7 Гц, 2H), 1,68 (h, J=7,5 Гц, 2H), 0,98 (t, J=7,4 Гц, 3H). 13C ЯМР (101 МГц, DMSO-d6) д 152,2, 146,3, 143,2, 124,5, 105,0, 102,2, 54,2, 47,2, 41,3, 36,6, 34,0, 17,3, 13,2.
N-((1S,3S)-3-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид
В реактор помещали N-((1S,3S)-3-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид.HCl (8 г), затем помещали изопропанол (56 мл) и воду (20 мл). Смесь нагревали при 60°C. Раствор бикарбонат калия (2,4 г) в воде (8 мл) готовили и добавляли к нагретой смеси. Смесь нагревали при 80°C, затем охлаждали до 65°C. Добавляли затравочные кристаллы искомого соединения (70 мг). Затем реактор охлаждали до 20°C и затем содержимое размалывали. Вещество фильтровали с помощью вакуума и промывали двумя порциями смеси изопропанол/вода состава 2/1 (по 14 мл). Осадок на фильтре сушили в вакууме и получали 6,09 г (выход 87%) искомого соединения. 1H ЯМР (400 МГц, DMSO-d6) д 11,65 (s, 1H), 8,13 (s, 1H), 7,50 (d, J=9,2 Гц, 1H), 7,15 (dd, J=3,6, 1,6 Гц, 1H), 6,63 (d, J=3,5 Гц, 1H), 4,91 (tt, J=9,6, 7,4 Гц, 1H), 3,70-3,47 (m, 1H), 3,26 (s, 3H), 3,04-2,87 (m, 2H), 2,61 (ddd, J=11,7, 8,9, 5,2 Гц, 2H), 2,24 (dt, J=11,8, 9,1 Гц, 2H), 1,79-1,55 (m, 2H), 0,98 (t, J=7,4 Гц, 3H).
Пример 7
Общее получение солей N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида
N-((1S,3S)-3-(Метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамид растворяли в ацетоне, этаноле, изопропаноле, воде или их смесях. Добавляли некоторое количество кислоты (хлористоводородной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, фумаровой кислоты или серной кислоты) и образцы обрабатывали путем циклического изменения температуры. Твердые вещества выделяли для характеризации.
Для специалистов в данной области техники очевидны изменения, модификации и другие реализации того, что описано в настоящем изобретении, вводимые без отклонения от сущности и основных характеристик настоящего изобретения. Соответственно, объем настоящего изобретения следует определять не по предшествующему иллюстративному описанию, а по прилагаемой формуле изобретения, и предполагается, что все изменения в соответствии со смыслом и диапазоном эквивалентности формулы изобретения входят в объем настоящего изобретения.
Каждая из печатных публикаций, включая, но не ограничиваясь только ими, патенты, заявки на патенты, книги, технические документы, отраслевые издания и статьи в журналах, которые описаны или на которые даны ссылки в настоящем описании, во всей своей полноте включены в настоящее изобретение в качестве ссылки для всех объектов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕЧЕНИЕ ГИДРАДЕНИТА ИНГИБИТОРАМИ JAK | 2020 |
|
RU2805595C1 |
| ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ И 7-ДЕАЗАПУРИНОВЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2606514C2 |
| ПРОИЗВОДНЫЕ 5-(7Н-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)-5-АЗАСПИРО[2.5]ОКТАН-8-КАРБОНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2761626C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ НА ОСНОВЕ ИНГИБИТОРОВ PRMT5 | 2020 |
|
RU2830439C1 |
| ХИМИЧЕСКОЕ СОЕДИНЕНИЕ | 2019 |
|
RU2803741C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ PAH | 2020 |
|
RU2786588C1 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 1, ИХ КОМПОЗИЦИИ, СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2824349C2 |
| ИНГИБИТОРЫ RMT5 | 2019 |
|
RU2814198C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-d]ПИРИМИДИНА | 2021 |
|
RU2819004C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |

Изобретение относится к промежуточному продукту указанной ниже формулы или его соли, где R1 представляет собой водород, используемый в способе получения N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида. Технический результат: разработан эффективный способ получения N-((1S,3S)-3-(метил(7H-пирроло[2,3-d]пиримидин-4-ил)амино)циклобутил)пропан-1-сульфонамида, полезного в качестве ингибитора протеинкиназ, таких как фермент киназа Janus (JAK), с использованием нового промежуточного соединения. 2 н.п. ф-лы, 13 ил.
1. Соединение, обладающее структурой:
 ,
,
в которой R1 представляет собой водород или его соль.
2. Способ получения соединения, обладающего структурой:
 ,
,
который включает (a) получение гидроксиламина, обладающего структурой:
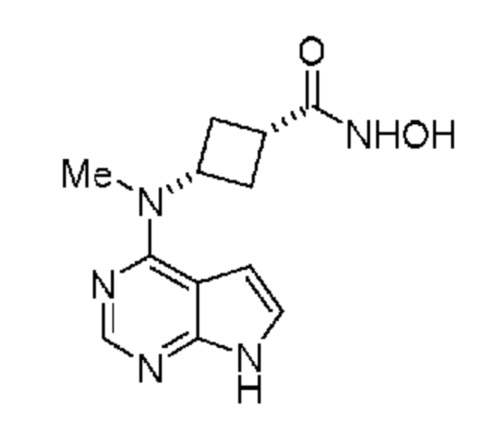 ,
,
(b) введение в реакцию указанного гидроксиламина при подходящих условиях с получением амина, обладающего структурой:
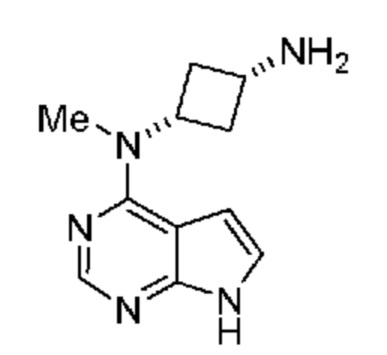 ,
,
и затем (c) обработку указанного амина н-пропилсульфонирующим реагентом при подходящих условиях с получением указанного соединения;
в котором н-пропилсульфонирующим реагентом является соединение, обладающее структурой:
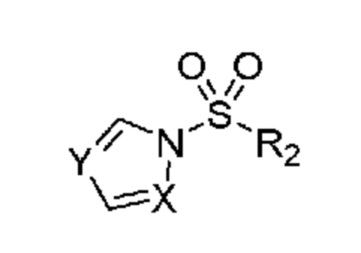 ,
,
в которой R2 означает н-пропил и
X и Y оба представляют собой N.
| David M | |||
| Hodgson и др | |||
| Organolithium-induced enantioselective alkylative double ring-opening of epoxides: synthesis of enantioenriched unsaturated amino alcohols, Tetrahedron, 60(16), 2004, с.3611-3624 | |||
| Vazquez, Michael L | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

