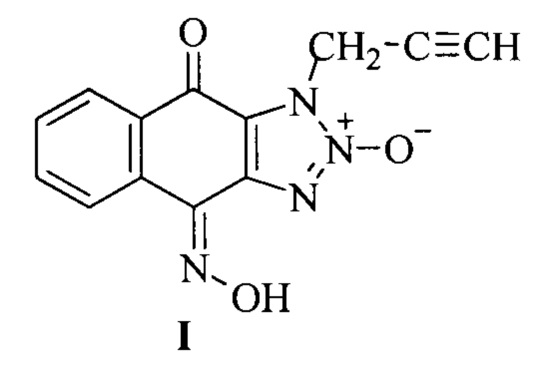
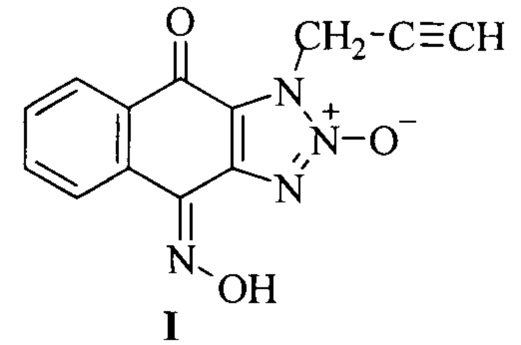
Изобретение относится к области органической химии и медицины и касается способа получения биологически активных химических соединений, обладающих цитотоксической активностью, а именно к способу получения 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I), содержащего несколько фармакофорных групп - терминальный остаток ацетилена, триазолоксидный фрагмент и оксимную группу.
Известно, что производные хинонов, содержащих терминальные ацетиленовые остатки и их производные, обладают различными видами биологической активности [Shimbashi A., Nishiyama S. Synthesis of chloroquinocin, a pyranonaphthoquinone antibiotic against Gram-positive bacteria // Tetrahedron Lett., 48, 1545 (2007), https://doi.org/10.1016/j.tetlet.2007.01.020].
На основе 2,3-дибром-1,4-нафтохинона получена серия 2,3-диалкинил-1,4-нафтохинонов, которые проявили себя как цитотоксические агенты на трех линиях опухолевых клеток: аденокарценома яичника (OVCAR-8), метастатический рак предстательной железы (РС-3М) и бронхоальвеолярный рак легкого (NCI-H358M) [Романов B.C., Иванчикова И.Д., Мороз А.А., Шварцберг М.С. Замещение галогена на ацетиленовые группы в хиноидном цикле // Известия Академии Наук. Серия химическая, 2005, С. 1636. Silva M.G., Camara С.А., Silva T.M.S., Feitosa A.C.S., Meira A.S., Pessoa C. Synthesis of 2,3-diyne-1,4-naphthoquinone derivatives and evaluation of cytotoxic activity against tumor cell lines //J. Braz. Chem. Soc, 24, 1420 (2013)].
С другой стороны, производные полициклических хиноидных триазол-N-оксидов проявляют антипролиферативную активность, причем некоторые из них не менее активны, чем широко используемый антрациклиновый противоопухолевый препарат доксорубицин [Gornostaev L.M., Tsvetkov V.B., Markova А.А., Lavrikova Т.I., Khalyavina Yu.G., Kuznetsova A.S., Kaluzhny D.N., Shunayev A.V., Tsvetkova M.V., Glazunova V.A., Chernyshev V.V., Shtil A.A. The oxime derivatives of 1-R-1H-Naphtho[2,3-d][1,2,3]triazole-4,9-dione 2-oxides: Synthesis and properties // Anti-Cancer Agents in Medicinal Chemistry, 2017, V. 17, №13, P. 1814-1823.].
Кроме того, известно, что замена карбонильной группы в хиноидных соединениях на имино- или оксимную группу может приводить к уменьшению кардиотоксичности при сохранении цитотоксической активности [Tseng С.-Н., Chen Y.-L., Yang S.-H., Peng S.-L, Cheng C.-M., Han C.-H., Lin S.-R., Tzeng C.-C. Synthesis and antiproliferative evaluation of certain iminonaphtho[2,3-b]furan derivatives // Bioorg. Med. Chem., 2010, Vol. 18, №. 14, P. 5172-5182.].
Наиболее близким к заявляемому изобретению, является способ получения 1-R-4,9-диоксо-1H-нафто[2,3-d][1,2,3]триазол-4-оксим-2-оксидов, обладающих цитотоксической активностью, описанный в известном прототипе (патент 2545091, МПК C07D 249/22 (2006.01), A61K 31/4192 (2006.01), оп. 27.03.2015):
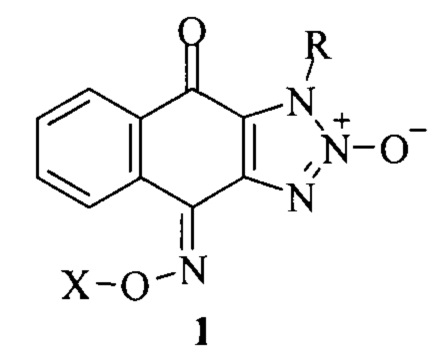
где: R=Alk, бензил; X=H,-C(=O)R', где R'=метил, фенил. Биологические активные химические соединения 1-R-4,9-диоксо-1H-нафто[2,3-d][1,2,3]триазол-2-оксиды получали путем циклизации 2-азидо-3-N-нитрозо-алкиламино-1,4-нафтохинонов в 1-алкил-4,9-диоксо-1Н-нафто[2,3][1,2,3]триазол-2-оксиды; нагревание этих продуктов с гидрохлоридом гидроксиламина в пиридине приводило к 1-R-4,9-диоксо-1H-нафто[2,3-d][1,2,3]триазол-4-оксим-2-оксидам. Полученные оксимы далее ацилировали уксусным ангидридом или бензоилхлоридом в пиридине, что приводило к соответствующим 1-R-1нафто[2,3-d][1,2,3]триазол-4,9-дион-4-(O-ацилоксим)-2-оксидам.
К недостаткам известного технического решения можно отнести отсутствие в известных веществах фармакофорного пропаргильного остатка в положении 1.
Задачей настоящего изобретения является разработка экономичного и простого способа получения 1-(2-пропаргил)-1H-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I).
Техническим результатом от реализации изобретения является получение продукта, обладающего антипролиферативной активностью и сохраняющего возможности дополнительной функционализации, при этом реакции проводятся без очистки выделенных промежуточных продуктов, что позволяет сократить число стадий и добиться высокого суммарного выхода целевого продукта - получения 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида, содержащего фармакофорные группы - терминальный остаток ацетилена, триазолоксидный фрагмент и оксимную группу.
Это достигается за счет того, что способ получения 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида, содержащего фармакофорные группы - терминальный остаток ацетилена, триазолоксидный фрагмент и оксимную группу, формулы
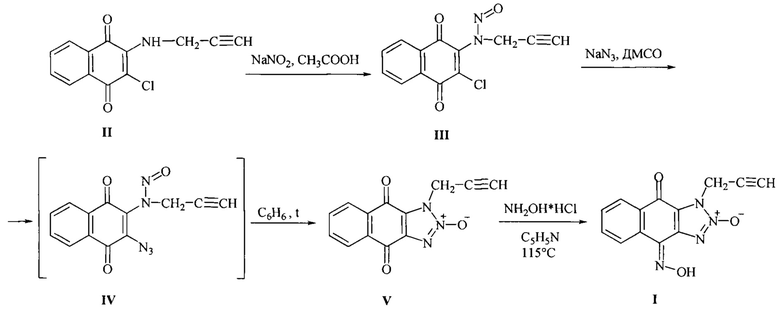
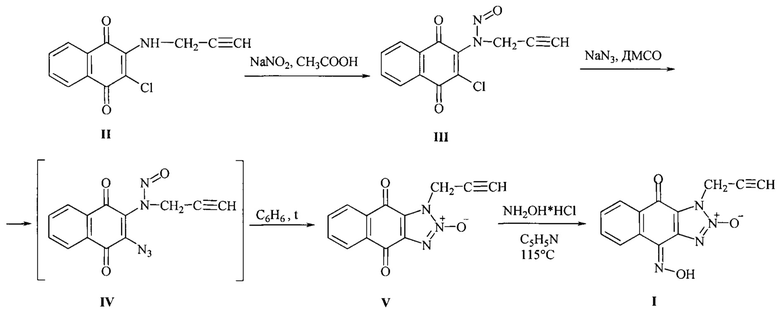
согласно изобретению, осуществляют по схеме, без очистки выделяемых промежуточных продуктов, в качестве исходного вещества используют - 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто [2,3-d][1,2,3]триазол-2-оксид (V), при этом к 2-пропаргиламино-3-хлор-1,4-нафтохинону (II) в уксусной кислоте прибавляют пятикратный молярный избыток нитрита натрия небольшими порциями в течение 1 часа, реакционную массу перемешивают 30 мин при 20-22°С, затем разбавляют водой со льдом, желтый осадок 2-(N-нитрозопропаргиламино)-3-хлор-1,4-нафтохинона (III) отделяют фильтрованием, промывают водой, высушивают в темноте без дополнительной очистки растворяют в диметилсульфоксиде (ДМСО) и добавляют раствор азида натрия в воде, реакционную массу перемешивают 30 мин при 30°С. Полученный азид (IV) выделяют вливанием в десятикратный объем ледяной воды, осадок азида (IV) отфильтровывают, растворяют в бензоле и кипят в течение 30 минут, затем раствор концентрируют, охлаждают до 20°С, выпавший в осадок 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксид (V) отделяют фильтрованием, промывают двумя порциями воды, затем этанолом, высушивают. Затем полученный триазолоксид (V) оксимируют гидрохлоридом гидроксиламина при кратковременном кипячении в пиридине:

Принципиальным отличием вещества I от прототипа является присутствие в нем нескольких фармакофорных фрагментов - триазолоксидного и терминального ацетиленового фрагмента, а также оксимной группы.
Способ осуществляют следующим образом. Исходным продуктом в предлагаемом способе получения 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I) является 2-пропаргилгамино-3-хлор-1,4-нафтохинон (II), полученный по известной методике [Mezeiova Е., Janockova J., Andrys R., Soukup О., Kobrlova Т., Muckova L., Pejchal J., Simunkova M., Handl J., Micankova P., Capek J., Rousar Т., Hrabinova M., Nepovimova E., Marco-Contelles J.L., Valko M., Korabecny J., 2-Propargylamino-naphthoquinone derivatives as multipotent agents for the treatment of alzheimer's disease // European Journal of Medicinal Chemistry, 2021, V. 211; Art.No: 113112 https://doi.org/10.1016/j.ejmech.2020.113112].
К 4.90 г (20 ммоль) 2-пропаргиламино-3-хлор-1,4-нафтохинона (II) в 40 мл уксусной кислоты прибавляли пятикратный избыток 6.90 г (100 ммоль) нитрита натрия небольшими порциями в течение 1 часа. Реакционную массу перемешивали 30 мин при 20-22°С, затем разбавили водой со льдом до объема 300 мл. Желтый осадок 2-(N-нитрозопропаргиламино)-3-хлор-1,4-нафтохинона (III) отделяли фильтрованием, промывали водой, высушивали в темноте. Полученный N-нитрозамин (III) без дополнительной очистки растворяли в 40 мл ДМСО и добавляли в течение 5 мин раствор 2.0 г (30 ммоль) азида натрия в 15 мл воды. Реакционную массу перемешивали 30 мин при 30°С. Полученный азид (IV) выделяли вливанием в десятикратный объем ледяной воды. Осадок азида (IV) отфильтровывали, растворяли в 200 мл бензола и кипятили в течение 30 минут. Затем раствор концентрировали отгонкой бензола до 40 мл, охлаждали до 20°С. Выпавший в осадок 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксида (V) отделяли фильтрованием, промывали 2 мл этанола, высушивали. Выход триазолоксида (V) составил 2.0 г (40%) в пересчете на исходный амин II), т.пл. 230-233°С. УФ спектр, λмакс., нм (lg ε): 230 (4.48), 281(3.69), 340 (2.71). ИК спектр, см-1: 3290 (С≡C-H). 2946 (СН2), 2138 (С≡С), 1684 (С=O), 1662 (С=O), 1257 (NO). Спектр ЯМР13С (DMSO-d6, δ, м.д., J, Гц): 36.33 (СН2), 74.29 (С≡С), 76.90 (С≡С), 125.51 (Саром.), 126.51 (Саром.), 126.80 (Саром.), 132.01 (Саром.), 132.03 (Саром.), 134.60 (Саром.), 134.97 (Саром.), 135.30 (Саром.), 172.19 (С=O), 174.82 (С=O). Спектр ЯМР1Н (DMSO-d6, δ, м.д., J, Гц): 3.62 (1H, С≡С-Н), 5.41 (2Н,СН2), 7.94 (2Н, Саром.), 8.13 (2Н, Саром.). Масс-спектр, m/z, %: 254 ((М+1)+, 8), 253 (М+, 50), 223 (29), 167 (10), 159 (10), 158 (100), 157 (13), 140 (11), 105 (19), 104 (16), 102 (51), 76 (29), 75 (13), 50 (12), 39 (27). Найдено, %: С 61.58; Н 2.69; N 16.61. C13H17N3O3. Вычислено, %: С 61.66; Н 2.79; N 16.59. Mr 253,05.
Способ синтеза целевого продукта - 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I).
К 0.50 г (2 ммоль) 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксида (V) в 7 мл пиридина добавили 0.40 г (6 ммоль) гидрохлорида гидроксиламина. Реакционную массу кипятили в течение 15 мин, охлаждали до 20-22°С, осадок фильтровали, промывали двумя порциями воды по 10 мл, затем 2 мл этанола. Выход: 0.27 г (50%), т.пл. >300°С. УФ спектр, λмакс., нм (lg ε): 230 (4.50), 259 (3.74), 290 (3.62), 365 (3.45). ИК спектр, см-1: 3244 (ОС-C-H), 2950 (СН2), 2128 (C≡С), 1652 (С=O), 1268 (NO). Спектр ЯМР13С (DMSO-d6, δ, м.д., J, Гц): 36.04 (СН2), 74.83 (C≡С), 76.58 (C≡С), 120.11 (Саром.), 123.73 (Саром.), 125.91 (Саром.), 129.58 (Саром.), 129.71 (Саром.), 131.72 (Саром.), 132.62 (Саром.), 133.67(Саром.), 136.21 (Саром.), 171.10 (С=O). Спектр ЯМР1Н (DMSO-d6, δ, м.д., J, Гц): 3.56 (1H, C≡С-Н), 5.45 (2Н, СН2), 7.70 (1H, Саром.), 8.80 (1Н, Саром.), 8.16 (1Н, Саром.), 8.33 (1H, Саром.), 13.62 (1H, N-OH). Масс-спектр, m/z, %: 268 (М+, 68), 220 (34), 173 (56), 146 (55), 130 (100), 115 (40), 114 (48), 102 (76), 90 (35), 88 (37), 76 (34), 75 (23), 39 (67), 30 (29). Найдено, %: С 58.11; Н 3.08; N 20.49. C13H8N4O3. Вычислено, %: С 58.21; Н 3.01; N 20.89. Mr 268.
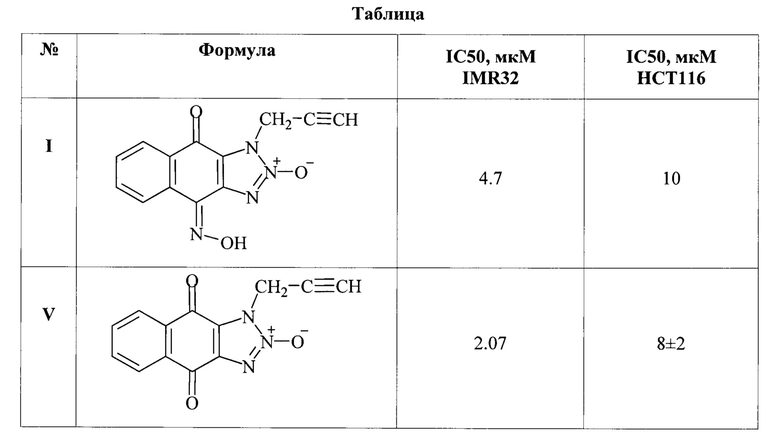
Исследование цитотоксической активности 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I) и его предшественника - 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол 2-оксида (V).
Клетки линий нейробластомы человека IMR32 и рака толстой кишки НСТ116 культивировали в среде DMEM (Dulbecco's modified Eagles medium) с добавлением следующих компонентов до конечных концентраций: 10% эмбриональной телячьей сыворотки, 2 mM L-глутамина, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина (ПанЭко, Россия), инкубация проводилась при 37°С, 5% CO2 в увлажненной атмосфере. В экспериментах использованы клетки в логарифмической фазе роста. Для профилактики микоплазменного заражения использовался препарат Mycokill (ПанЭко, Россия). Перед началом экспериментов проводилось не менее трех пассажей на свободной от антимикоплазменного препарата среде.
Исследуемые соединения I, V растворяли в ДМСО до концентрации стокового раствора 10 мМ.
Цитотоксическое действие соединений исследовали в МТТ-тесте (по восстановлению бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия в темно-синий кристаллический формазан митохондриями живых клеток). По результатам исследования цитотоксичности определены значения IC50. Клетки рассеивали в лунки 96-луночного планшета (NUNC, США) (5000 клеток в 150 мкл культуральной среды), инкубировали 24 часа при 37°С, 5% CO2, в увлажненной атмосфере. Вносили по 50 мкл раствора исследуемых веществ в культуральной среде, приготовленных серийными разведениями из исходного раствора, до 9 конечных концентраций от 0,195 до 50 мкМ. Контролем в эксперименте служили клетки без препарата (интактные). Клетки инкубировали 72 ч при 37°С, 5% СО2, в увлажненной атмосфере. За 1 час до окончания инкубации в лунки вносили по 10 мкл водного раствора МТТ (5 мг/мл, ПанЭко, Россия). После окончания инкубации культуральную среду отбирали, клетки ресуспендировали в 200 мкл ДМСО и измеряли оптическую плотность раствора на планшетном спектрофотометре Multiscan FC (Thermo Scientific, США) при длине волны 570 нм. Процент клеток, выживших при действии каждой дозы соединения, подсчитывали как частное от деления средней оптической плотности в лунках после инкубации с данной дозой к средней оптической плотности контрольных лунок (значения последних приняты за 100%).
Изобретение иллюстрируется таблицей.
В таблице приведены результаты исследования цитотоксической активности биологически активных химических соединений 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I) и его предшественника - 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол 2-оксида (V) для культуры клеток нейробластомы человека IMR32 и клеток рака толстой кишки человека НСТ116.
Для соединения I средняя IC50 составила 10 мкМ, для соединения V средняя IC50 равна 8 мкМ на линии рака толстой кишки НСТ116. Для соединения I средняя IC50 равна 4,7 мкМ, для соединения V средняя IC50 равна 2.07 мкМ на линии клеток нейробластомы IMR32.
Результаты цитотоксической активности соединений I, V свидетельствуют об активности исследуемых соединений в микромолярных концентрациях.
Изобретение относится к области органической химии и медицины и касается способа получения биологически активных химических соединений, обладающих цитотоксической активностью. Способ получения 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида (I) осуществляют по схеме, без очистки выделяемых промежуточных продуктов, в качестве исходного вещества используют 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксид (V), при этом к 2-пропаргиламино-3-хлор-1,4-нафтохинону (II) в уксусной кислоте прибавляют пятикратный молярный избыток нитрита натрия небольшими порциями в течение 1 ч, реакционную массу перемешивают 30 мин при 20-22°С, затем разбавляют водой со льдом, желтый осадок 2-(N-нитрозопропаргиламино)-3-хлор-1,4-нафтохинона (III) отделяют фильтрованием, промывают водой, высушивают в темноте без дополнительной очистки, растворяют в диметилсульфоксиде (ДМСО) и добавляют раствор азида натрия в воде, реакционную массу перемешивают 30 мин при 30°С. Полученный азид (IV) выделяют вливанием в десятикратный объем ледяной воды, осадок азида (IV) отфильтровывают, растворяют в бензоле и кипятят в течение 30 мин, затем раствор концентрируют, охлаждают до 20°С, выпавший в осадок 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксид (V) отделяют фильтрованием, промывают двумя порциями воды, затем этанолом, высушивают. Затем полученный триазолоксид (V) оксимируют гидрохлоридом гидроксиламина при кратковременном кипячении в пиридине:
 .
.
Техническим результатом изобретения является получение продукта, обладающего антипролиферативной активностью и сохраняющего возможности дополнительной функционализации, при этом реакции проводятся без очистки выделяемых промежуточных продуктов, что позволяет сократить число стадий и добиться высокого суммарного выхода целевого продукта. 1 табл., 2 пр.
Способ получения 1-(2-пропаргил)-1Н-нафто[2,3-d][1,2,3]триазол-4,9-дион-4-оксим-2-оксида, содержащего фармакофорные группы - терминальный остаток ацетилена, триазолоксидный фрагмент и оксимную группу,
отличающийся тем, что способ осуществляют по схеме, без очистки выделяемых промежуточных продуктов, в качестве исходного вещества используют 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксид (V), при этом к 2-пропаргиламино-3-хлор-1,4-нафтохинону (II) в уксусной кислоте прибавляют пятикратный молярный избыток нитрита натрия небольшими порциями в течение 1 ч, реакционную массу перемешивают 30 мин при 20-22°С, затем разбавляют водой со льдом, желтый осадок 2-(N-нитрозопропаргиламино)-3-хлор-1,4-нафтохинона (III) отделяют фильтрованием, промывают водой, высушивают в темноте, без дополнительной очистки растворяют в диметилсульфоксиде (ДМСО) и добавляют раствор азида натрия в воде, реакционную массу перемешивают 30 мин при 30°С, полученный азид (IV) выделяют вливанием в десятикратный объем ледяной воды, осадок азида (IV) отфильтровывают, растворяют в бензоле и кипят в течение 30 мин, затем раствор концентрируют, охлаждают до 20°С, выпавший в осадок 4,9-диоксо-1-(2-пропаргил)-4,9-дигидро-1Н-нафто[2,3-d][1,2,3]триазол-2-оксид (V) отделяют фильтрованием, промывают двумя порциями воды, затем этанолом, высушивают, затем полученный триазолоксид (V) оксимируют гидрохлоридом гидроксиламином при кратковременном кипячении в пиридине:
| 1-R-4,9-ДИОКСО-1H-НАФТО[2,3-d][1,2,3]ТРИАЗОЛ-4-ОКСИМ-2-ОКСИДЫ И ИХ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ ЦИТОТОКСИЧЕСКОЙ АКТИВНОСТЬЮ | 2014 |
|
RU2545091C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2007 |
|
RU2349317C1 |
| СПОСОБ СИНТЕЗА МОЛЕКУЛ, СОДЕРЖАЩИХ ФУНКЦИОНАЛЬНУЮ ГРУППУ НИТРИЛОКСИДА | 2016 |
|
RU2724105C2 |
| Silva M.G | |||
| et | |||
| al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Способ окисления алкоголей | 1915 |
|
SU1420A1 |

