ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области биомедицины, в частности к 2-аминопиримидиновым соединениям, фармацевтическим композициям и их применению.
УРОВЕНЬ ТЕХНИКИ
Семейство толл-подобных рецепторов (TLR) играет важную роль в распознавании патогенов и активации врожденного иммунитета. Толл-подобный рецептор 8 (TLR8) преимущественно экспрессируется иммунными клетками костного мозга, и активация этого рецептора запускает широкий спектр иммунных реакций. Сигнальный путь TLR8 может быть активирован бактериальными одноцепочечными РНК, синтетическими агонистами и микроРНК. Активация TLR8 приводит к выработке полярных цитокинов Thl, таких как ИЛ-12, ИЛ-18, ФИО-а и ИФН-γ, а также различных костимулирующих факторов, таких как CD80 и CD86. Эти цитокины могут активировать и усиливать реакции врожденного и приобретенного иммунитета и оказывать терапевтическое воздействие при различных заболеваниях, связанных с аутоиммунитетом, воспалением, аллергией, астмой, отторжением трансплантата, реакцией «трансплантат против хозяина», инфекциями, опухолями и иммунодефицитом. Например, при гепатите В активация TLR8 на специализированных антигенпрезентирующих клетках (рАРС) и других внутрипеченочных иммунных клетках сопровождается индукцией ИЛ-12 и провоспалительных цитокинов, что, как ожидается, усилит HBV-специфический Т-клеточный ответ, активирует внутрипеченочные NK-клетки и будет способствовать формированию противовирусного иммунитета.
В настоящее время на рынке отсутствует регулятор толл-подобных рецепторов, таких как TLR8. Учитывая потенциал для лечения широкого спектра заболеваний, имеется высокая потребность в разработке регуляторов TLR8 с высокой активностью, высокой селективностью, низкой токсичностью и превосходными фармакокинетическими свойствами.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является предоставление соединения формулы I и его применение в области противовирусных, противоинфекционных средств, а также для лечения аутоиммунных, опухолевых и других заболеваний.
В первом аспекте настоящего изобретения предлагается соединение, представленное формулой I, или стереоизомер, или фармацевтически приемлемая соль этого соединения:
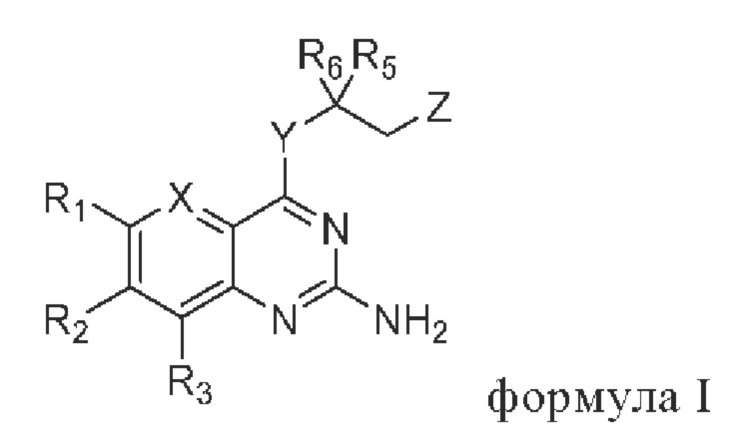
где
X выбирают из группы, состоящей из N и CR7;
Y выбирают из группы, состоящей из NH и О;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы состоящей из водорода, галогена, C1-C6 алкила, гидроксила и амино; алкил необязательно замещен 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, гидроксила, C1-С6 алкиламино, C1-С6 алкокси, циано, C1-C6 алкил-С(O)NH-, C1-C6 алкил-NH-С(О)-, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, галогензамещенный C1-С6 алкила, С3-С10 гетероарила и С6-С12 арила;
R2 выбирают из группы состоящей из С3-С10 циклоалкила, С3-С10 гетероциклоалкила, -NR8R9, -OR8, -SR8, -S(O)2R8, -N(R10)COR11, -N(R10)S(O)2R11, -CONR10R11, -COR10 и -S(O)2NR10R11; циклоалкил и гетероциклоалкил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, гидроксила, C1-C6 алкиламино, C1-C6 алкокси, циано, C1-С6 алкил-С(O)NH-,=O, C1-C6 алкил-NH-С(О)-, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, галогензамещенный C1-C6 алкила, С3-С10 гетероарила и С6-С12 арила;
R4, R5 и R6 независимо выбирают из группы состоящей из галогена, C1-C6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С12 арила; алкил, циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, гидроксила, амино, C1-C6 алкиламино, C1-C6 алкокси, циано, C1-C6 алкил-С(O)NH-, C1-C6 алкил-NH-C(O)-, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, галогензамещенный C1-С6 алкила, С3-С10 гетероарила и С6-С12 арила;
R7 выбирают из группы, состоящей из водорода, галогена и C1-С6 алкила; алкил необязательно замещен 13 заместителями, выбранными из группы, состоящей из водорода, галогена, гидроксила и амино;
R8 выбирают из группы состоящей из С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С10 арила; циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, гидроксила, амино, C1-C6 алкиламино, C1-C6 алкокси, циано, C1-C6 алкил-С(O)NH-, C1-C6 алкил-NH-С(О)-, C1-C6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, галогензамещенный C1-C6 алкила, С3-С10 гетероарила и С6-С12 арила;
R9, R10 и R11 независимо выбирают из группы состоящей из водорода, гидроксила, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С10 арила; алкил, циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, гидроксила, амино, (C1-C6 алкил)2N-, C1-C6 алкокси, циано, C1-C6 алкил-C(O)NH-, C1-C6 алкил-NH-С(O)-, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, галогензамещенный C1-C6 алкила, С3-С10 гетероарила и C6-C12 арила;
или R10 и R11 и соответствующие N, С или S, с которыми они связаны, образуют С3-С10 гетероциклоалкил, 6-12-членный конденсированный гетероциклил или С3-С10 гетероарил; гетероциклоалкил может быть замещен 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, гидроксила, амино, (C1-С6 алкил)2N-, C1-С6 алкокси, циано, C1-C6 алкил-С(O)NH-, C1-C6 алкил-NH-С(О)-, C1-C6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, галогензамещенный C1-C6 алкила, C1-C6 алкокси, С3-С10 гетероарила и С6-С12 арила;
и гетероциклоалкил включает 1, 2 или 3 гетероатома, выбранных из N, О или S;
гетероарил включает 1, 2 или 3 гетероатома, выбранных из N, О или S;
6-12-членный конденсированный гетероциклил включает 1, 2, 3 или 4 гетероатома, выбранных из N, О или S;
В другом предпочтительном варианте осуществления изобретения X выбирают из группы, состоящей из N и CR7;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы, состоящей из водорода, галогена и C1-C6 алкила, и алкил может быть замещен 1-3 заместителями, выбранными из группы, состоящей из водорода и галогена;
R2 выбирают из группы состоящей из С3-С10 циклоалкила, С3-С10 гетероциклоалкила, -NR8R9, -OR8, -SR8, -S(O)2R8, -N(R10)COR11, -N(R10)S(O)2R11, -CONR10R11, -COR10 и -S(O)2NR10R11; циклоалкил и гетероциклоалкил необязательно замещены 13 заместителями, выбранными из группы состоящей из водорода, =O и галогена;
R4 выбирают из группы, состоящей из C1-С6 алкила, С3-С10 циклоалкила и С3-С10 гетероциклоалкила; алкил, циклоалкил и гетероциклоалкил могут быть замещены 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена и C1-C6 алкиламино;
R5 и R6 независимо представляют собой C1-C6 алкил соответственно; алкил может быть замещен 1-3 заместителями, выбранными из группы, состоящей из водорода и галогена;
R7 выбирают из группы, состоящей из водорода, галогена и C1-С6 алкила; алкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода и галогена;
R8 выбирают из группы, состоящей из С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С12 арила; циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена и гидроксила;
R9, R10 и R11 независимо выбирают из группы состоящей из водорода, гидроксила, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероариал и С6-С12 арила; алкил, циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 13 заместителями, выбранными из группы состоящей из водорода, галогена, (C1-C6 алкил)2N- и C1-C6 алкила;
или R10 и R11 и соответствующие N, С или S, с которыми они связаны, образуют С3-С10 гетероциклоалкил, 6-12-членный конденсированный гетероциклил или С3-С10 гетероарил; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, гидроксила, (C1-С6 алкил)2N-, C1-С6 алкокси и С3-С10 гетероциклоалкила.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляент собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы, состоящей из водорода, галогена и C1-C6 алкила, и алкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода и галогена;
R2 выбирают из группы, состоящей из -NR8R9, -CONR10R11, -COR10 и -S(O)2NR10R11;
R4 выбирают из группы, состоящей из C1-C6 алкила и С3-С10 циклоалкила; алкил и циклоалкил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена и C1-C6 алкиламино;
R5 и R6 независимо представляют собой C1-C6 алкил соответственно; алкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода и галогена;
R8 выбирают из группы состоящей из С3-С10 циклоалкила; циклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена и гидроксила;
R9 представляет собой Н;
R10 и R11 независимо выбирают из группы, состоящей из водорода, гидроксила, C1-C6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С12 арила; алкил, циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, (C1-C6 алкил)2N- и C1-C6 алкила;
или R10 и R11 и N, с которыми они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил, который необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, гидроксила, (C1-С6 алкил)2N-, C1-C6 алкокси и С3-С10 гетероциклоалкила.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы, состоящей из водорода и галогена;
R2 представляет собой-CONR10R11;
R4 представляет собой СН3- или 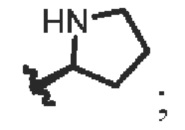 R5 и R6 независимо являются C1-C6 алкилами соответственно;
R5 и R6 независимо являются C1-C6 алкилами соответственно;
R10 и R11 независимо выбирают из группы, состоящей из водорода, C1-С6 алкила и С3-С10 циклоалкила; и алкил и циклоалкил необязательно замещены 13 заместителями, выбранными из группы, состоящей из водорода, галогена, (C1-C6 алкил)2N- и C1-C6 алкила;
или R10 и R11 и N, с которыми они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы состоящей изводорода, галогена, гидроксила, (C1-C6 алкил)2N- и C1-C6 алкокси;
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы, состоящей из водорода и галогена;
R2 представляет собой -CONR10R11;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 и R11 независимо выбирают из группы, состоящей из водорода, C1-С6 алкила и С3-С10 циклоалкила; алкил и циклоалкил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, (C1-C6 алкил)2N- и C1-C6 алкила;
или R10 и R11 и N, с которыми они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, гидроксила, (C1-C6 алкил)2N- и C1-C6 алкокси.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы, состоящей из водорода и галогена;
R2 представляет собой -CONR10R11;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 и R11 независимо выбирают из группы состоящей из водорода, C1-С6 алкила и С3-С10 циклоалкила; алкил и циклоалкил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, (C1-C6 алкил)2N - и C1-C6 алкила.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящеий из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы представляет собой водорода и галогена;
R2 представляет собой -COR10;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 выбирают из группы состоящей из гидроксила, C1-C6 алкила, С3-С10 циклоалкила и С3-С10 гетероарила; алкил, циклоалкил и гетероарил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена и C1-C6 алкила.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы, состоящей из водорода и галогена;
R2 представляет собой -COR10;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 выбирают из группы состоящей из C1-C6 алкила и С3-С10 циклоалкила В другом предпочтительном варианте осуществления изобретения X представляет собой N;
Y представляет собой NH;
Z выбирают из группы состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы состоящей из водорода и галогена;
R2 представляет собой -NR8R9;
R4 выбирают из группы состоящей из СН3- и 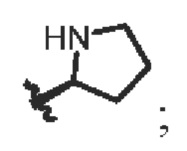
R5 представляет собой метил;
R6 представляет собой н-бутил;
R8 представляет собой С3-С10 циклоалкил; циклоалкил необязательно замещен 1-3 заместителями, выбранными из группы состоящей из водорода, галогена и гидроксила;
R9 представляет собой водород.
В другом предпочтительном варианте осуществления изобретения X выбирают из группы состоящей из N и CR7;
Y представляет собой NH;
Z выбирают из группы, состоящей из-N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы состоящей из водорода и галогена;
R4 выбирают из группы, состоящей из СН3-, 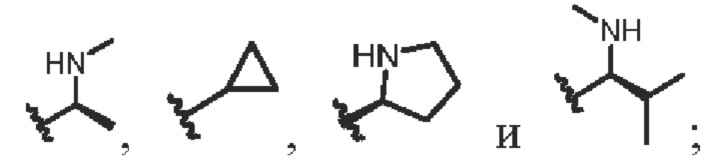
R5 и R6 независимо являются C1-C6 алкилами соответственно;
R7 выбирают из группы, состоящей из водорода и галогена;
В другом предпочтительном варианте осуществления изобретения X выбирают из группы, состоящей из N и CR7; Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 независимо выбирают из группы состоящей из водорода и галогена;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R7 выбирают из группы, состоящей из водорода и галогена.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
R2 выбирают из группы состоящей из -CONR10R11, -COR10 и -S(O)2NR10R11;
R10 и R11 независимо выбирают из группы состоящей из водорода, гидроксила, C1-С6 алкила, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С12 арила; алкил, циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, (C1-C6 алкил)2N- и C1-C6 алкила;
или R10 и R11 и N, с которыми они связаны, образуют С3-С10 гетероциклоалкил, 6-12-членный конденсированный гетероциклил или С3-С10 гетероарил; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из водорода, галогена, гидроксила, (C1-C6 алкил)2N-, C1-C6 алкокси и С3-С10 гетероциклоалкила.
В другом предпочтительном варианте осуществления изобретения X представляет собой N;
R2 выбирают из группы состоящей из -NR8R9, -OR8, -SR8 и -S(O)2R8.
R8 выбирают из группы, состоящей из С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С12 арила; циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена и гидроксила;
R9 выбирают из группы состоящей из водорода, гидроксила, C1-С6 алкил, С3-С10 циклоалкила, С3-С10 гетероциклоалкила, С3-С10 гетероарила и С6-С12 арила; алкил, циклоалкил, гетероциклоалкил, гетероарил и арил необязательно замещены 1-3 заместителями, выбранными из группы состоящей из водорода, галогена, (C1-C6 алкил)2N- и C1-С6 алкила.
В другом предпочтительном варианте осуществления изобретения
R2 выбирают из группы состоящей из С3-С10 циклоалкила и С3-С10 гетероциклоалкила; циклоалкил и гетероциклоалкил могут быть замещены 1-3 заместителями, выбранными из группы состоящей из водорода, =O и галогена.
В другом предпочтительном варианте осуществления изобретения соединение выбирают из группы, состоящей из:
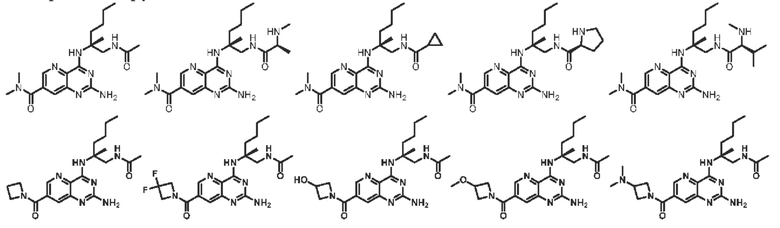

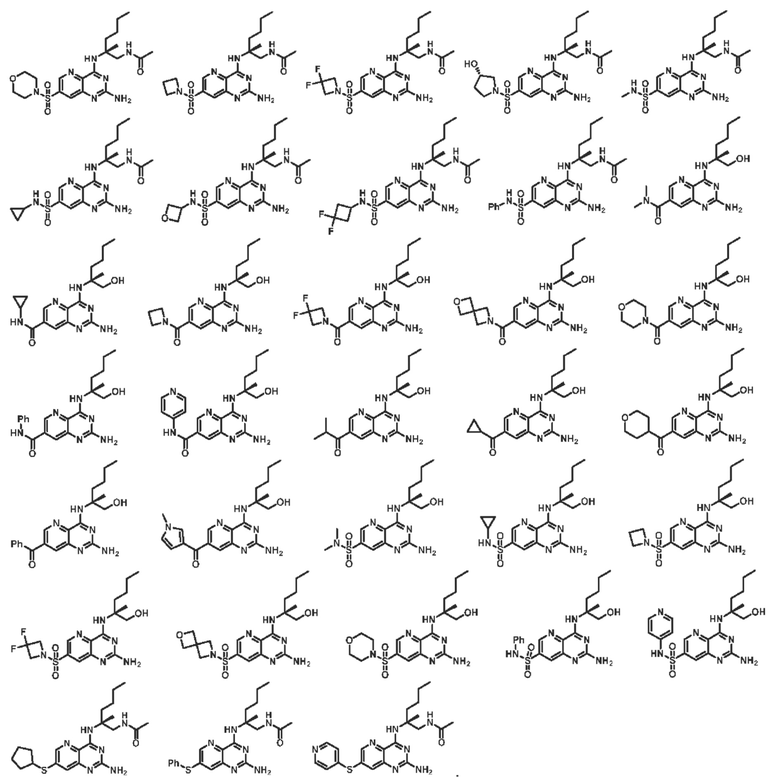
В другом предпочтительном варианте осуществления изобретения соединение выбирают из группы, состоящей из:
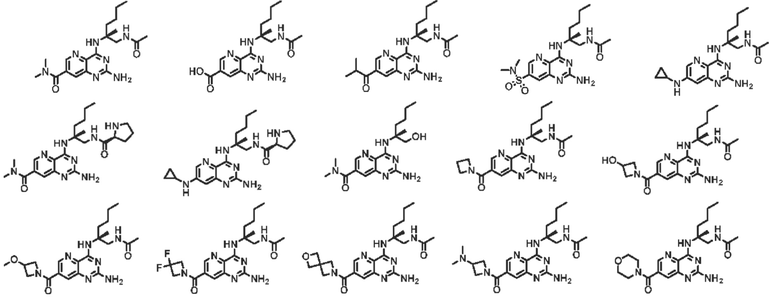
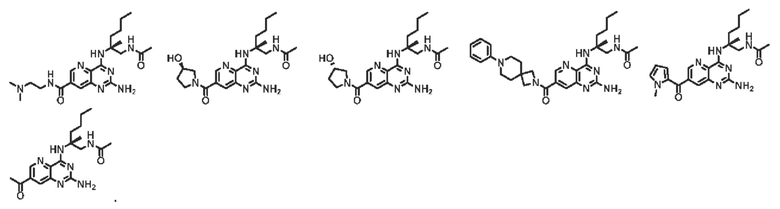
Во втором аспекте настоящего изобретения представлена фармацевтическая композиция, включающая:
i) одно или более соединений или стереоизомеров или их фармацевтически приемлемых солей в соответствии с первым аспектом настоящего изобретения;
ii) фармацевтически приемлемые носители.
В третьем аспекте настоящего изобретения обеспечивается применение соединений или стереоизомеров или их фармацевтически приемлемых солей в соответствии с первым аспектом настоящего изобретения, где они предлагаются для использования для применения, выбранного из группы, состоящей из:
1) приготовления противоопухолевых препаратов;
2) приготовления противовирусных препаратов;
3) приготовления противоинфекционных препаратов;
4) приготовления препаратов для профилактики и/или лечения аутоиммунных заболеваний.
В другом предпочтительном варианте осуществления изобретения опухоль выбирают из группы, включающей рак легкого, рак печени, рак пищевода, рак молочной железы, злокачественные образования головного мозга, рак простаты, меланому и лейкоз.
В другом предпочтительном варианте осуществления изобретения вирус выбирают из группы, включающей вирус гриппа, вирус Коксаки, вирус гепатита В (HBV), вирус гепатита С (HCV), вирус иммунодефицита человека (ВИЧ), вирус денге и полиомавирус.
В другом предпочтительном варианте осуществления изобретения инфекцию выбирают из группы, включающей из вирусные, хламидийные, микоплазменные, бактериальные, грибковые инфекции и паразитарные инвазии.
В другом предпочтительном варианте осуществления изобретения аутоиммунные заболевания выбирают из группы, включающей ревматоидный артрит, системную красную волчанку, рассеянный склероз, инсулинзависимый сахарный диабет, артериосклероз и воспалительные заболеваний кишечника.
В четвертом аспекте настоящего изобретения описан модулятор TLR8, представляющий собой одно или более соединений или стереоизомеров или их фармацевтически приемлемых солей в соответствии с первым аспектом настоящего изобретения.
В пятом аспекте настоящего изобретения описан метод модуляции TLR8 путем введения нуждающемуся в этом испытуемому представляющий собой одно или более соединений или стереоизомеров или их фармацевтически приемлемых солей в достаточном для эффективной модуляции количестве в соответствии с первым аспектом настоящего изобретения.
Следует понимать, что в настоящем изобретении любой из технических признаков, конкретно описанный выше или ниже (в частности, в приведенных Примерах) может быть объединен с другим признаком, которые не будут избыточно подробно описаны в данном документе по отдельности.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
После длительного и интенсивного исследования представленные изобретатели неожиданно получили класс соединений формулы I, обладающих значительно более высокой активностью и селективностью в отношении TLR8 за счет структурного улучшения в определенных положениях (особенно в положениях R2). По сравнению с аналогичными соединениями, известными в данной области, соединения по настоящему изобретению обладают более высокой активностью, лучшей селективностью, более высокой эффективностью in vivo, лучшей безопасностью, лучшей фармакокинетикой и лучшей возможностью разработки лекарственных препаратов. Исходя из этого изобретатели осуществили настоящее изобретение.
ТЕРМИНЫ
В настоящем изобретении, если не указано иное, используемые термины имеют общее значение, хорошо известное специалистам в данной области.
В настоящем изобретении термин «галоген» обозначает F, Cl, Br или I.
В настоящем изобретении термин «C1-С6 алкил» обозначает линейный или разветвленный алкил, включающий от 1 до 6 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, неопентил, третпентил или т.п.
В настоящем изобретении термин «С3-С10 циклоалкил» обозначает циклический алкил, имеющий от 3 до 10 атомов углерода в кольце, включая, помимо прочих, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и другие моноциклоалкилы, спироциклоалкилы и мостиковые циклоалкилы.
В настоящем изобретении термин «C1-С6 алкокси» обозначает линейный или разветвленный алкокси, имеющий от 1 до 6 атомов углерода, включая, помимо прочих, метокси, этокси, пропокси, изопропокси, бутокси и т.д. Предпочтительно С1-С4 алкокси.
В настоящем изобретении термин «гетероциклоалкил» обозначает 4-10-членный гетероциклил, содержащий 1, 2 или 3 гетероатома, выбранных из N, О или S, включая, помимо прочих, моно-гетероциклилы, такие как 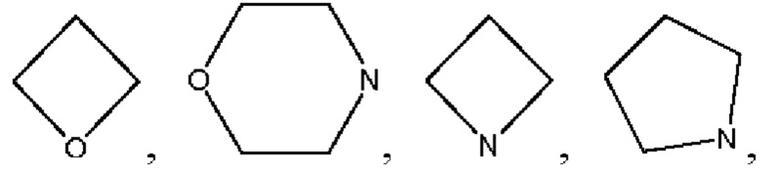
 и т.п., а также спирогетероциклил, такой как
и т.п., а также спирогетероциклил, такой как  В настоящем изобретении термин «конденсированный гетероциклил» обозначает конденсированное кольцо, образованное путем совместного использования двух соседних атомов углерода двумя структурами, выбранными из группы, включающей С3-С10 циклоалкил, С3-С10 гетероциклоалкил, С3-С10 гетероарил и С6-С12 арил, и конденсированный гетероциклил содержит 1, 2, 3 или 4 гетероатома, выбранных из N, О и S, например,
В настоящем изобретении термин «конденсированный гетероциклил» обозначает конденсированное кольцо, образованное путем совместного использования двух соседних атомов углерода двумя структурами, выбранными из группы, включающей С3-С10 циклоалкил, С3-С10 гетероциклоалкил, С3-С10 гетероарил и С6-С12 арил, и конденсированный гетероциклил содержит 1, 2, 3 или 4 гетероатома, выбранных из N, О и S, например, 
В настоящем изобретении термин «ароматическое кольцо» или «арил» имеет одинаковое значение, предпочтительно «С6-С10 арил». Термин «С6-С10 арил» обозначает ароматическое кольцо, имеющее от 6 до 10 атомов углерода и не имеющему гетероатомов в кольце, например, фенил, нафтил и т.п.
В настоящем изобретении термин «аминогруппа» обозначает следующую структуру: -NH2.
В настоящем изобретении термин «C1-С6 алкиламино» обозначает следующую структуру: C1-С6 алкил-NH-.
В настоящем изобретении термин «ароматический гетероцикл» или «гетероарил» имеет одно и то же значение и обозначает гетер о ароматическую группу, содержащую от одного до нескольких гетероатомов. Например, «С3-С10 гетероарил» обозначает ароматический гетероциклил, содержащий от 1 до 4 гетероатомов, выбранных из кислорода, серы или азота, и от 3 до 10 атомов углерода. Примеры, помимо прочих, включают следующие: фурил, тиенил, пиридил, пиразолил, пирролил, N-алкилпирролил, пиримидинил, пиразинил, имидазолил, тетразолил и т.п. Гетероарильное кольцо может быть соединено с арильным, гетероциклическим или циклоалкильным кольцом, при этом кольцо, соединенное с исходной структурой, представляет собой гетероарильное кольцо. Гетероарильные группы могут быть как замещенными, так и незамещенными.
В настоящем изобретении термин «галогенированный» означает замещенный галогеном.
В настоящем изобретении термин «замещенный» означает, что один или несколько атомов водорода в определенной группе замещены определенным заместителем. Конкретные заместители представляют собой заместители, ранее описанные соответствующим образом, или заместители, указанные в соответствующих Примерах. Если не указано иное, замещенная группа может иметь заместитель, выбранный из определенной группы, в любом замещаемом положении группы, причем заместители могут быть одинаковыми или разными в каждом положении. Специалисты в данной области поймут, что комбинации заместителей, предусмотренные настоящим изобретением, представляют собой комбинации, которые являются стабильными или химически достижимыми. Заместителями являются, помимо прочих, галоген, гидроксил, карбоксил (-СООН), C1-C6 алкил, С3-C8 циклоалкил, 3-12-членный гетероциклил, арил, гетероарил, аминогруппа, C1-C6 алкокси и т.п.
Соединение
В изобретении описано соединение, представленное формулой I, или стереоизомер, или фармацевтически приемлемая соль этого соединения:
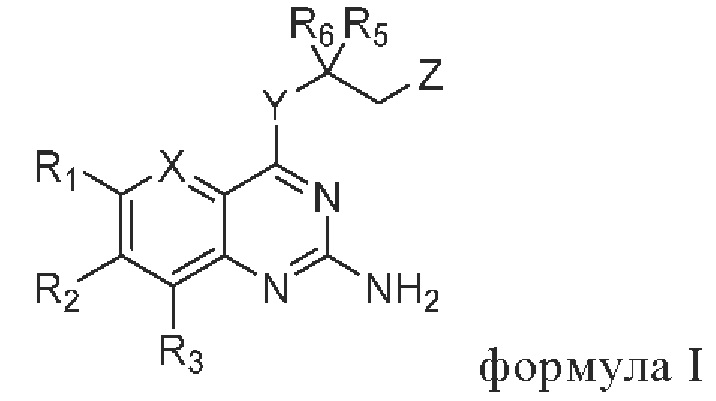
где каждая группа определена согласно приведенной выше информации.
В другом предпочтительном варианте осуществления изобретения любой из X, Y, Z, R1, R2, R3, R4, R5, R6, R7, R6, R9, R10 и R11 в соединении является соответствующей группой в конкретном соединении настоящего изобретения соответственно.
Термин «стереоизомер», или «оптический изомер», означает, что хиральный атом углерода, входящий в соединение по настоящему изобретению, может быть в конфигурации R или S или в их сочетании.
В настоящем изобретении термин «фармацевтически приемлемая соль» обозначает соль, образованная соединением по настоящему изобретению и кислотой или основанием, подходящую для использования в качестве лекарственного средства.
Фармацевтически приемлемые соли включают неорганические и органические соли. Предпочтительным классом солей являются соли соединений по настоящему изобретению, образованные с кислотами. Подходящие кислоты для образования солей включают, помимо прочих, неорганические кислоты, такие как хлористоводородная кислота, бромная кислота, фтористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота; органические кислоты, такие как муравьиная, уксусная, трифторуксусная, пропионовая, щавелевая, малоновая, янтарная, фумаровая, малеиновая, молочная, яблочная, винная, лимонная, пикриновая, бензойная, метилсульфоновая, этансульфоновая, п-толуолсульфоновая, бензолсульфоновая, нафталинсульфоновая; и аминокислоты, такие как пролин, фенилаланин, аспарагиновая и глутаминовая кислоты.
Другим предпочтительным классом солей являются соли соединений по настоящему изобретению, образованные основаниями, такими как соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли магния или кальция), соли аммония (например, низкомолекулярные алканол-аммониевые соли и другие фармацевтически приемлемые соли аминов), такие как соль метиламина, соль этиламина, соль пропиламина, соль диметиламина, соль триметиламина, соль диэтиламина, соль триэтиламина, соль трет-бутиламина, соль этилендиамина, соль гидроксиэтиламина, соль дигидроксиэтиламина, соль тригидроксиэтиламина и соль амина, образованная из морфолина, пиперазина и лизина соответственно.
Фармацевтическая композиция и способ применения
Настоящее изобретение также представляет фармацевтическую композицию, включающую:
i) одно или более указанных выше соединений, стереоизомеров или их фармацевтически приемлемых солей;
ii) фармацевтически приемлемые носители.
Фармацевтическая композиция по настоящему изобретению включает безопасное и эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, а также фармацевтически приемлемое вспомогательное вещество или носитель.
Термин «Безопасное и эффективное количество» относится к такому количеству соединения, которого достаточно для существенного улучшения состояния без появления серьезных побочных эффектов. Обычно фармацевтическая композиция содержит от 0,1 до 2000 мг соединения по настоящему изобретению/доза, более предпочтительно от 0,1 до 200 мг соединения по настоящему изобретению/доза. Предпочтительно, «доза» представляет собой капсулу или таблетку.
«Фармацевтически приемлемый носитель» означает один или несколько наполнителей в твердой или жидкой форме или желеобразные материалы, которые пригодны для использования для человека и должны иметь достаточную чистоту и достаточно низкую токсичность. «Совместимый» означает, что каждый компонент такой композиции может быть смешан с соединениями по настоящему изобретению, а также друг с другом без существенного снижения эффективности этих соединений. Некоторые примеры фармацевтически приемлемых носителей, включают в себя целлюлозу и ее производные (такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза натрия, ацетат целлюлозы и т.д.), желатин, тальк, твердые смазки (такие как стеариновая кислота, стеарат магния), сульфат кальция, растительные масла (такие как соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (такие как Tween®), смачивающий агент (такой как додецилсульфат натрия), красители, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и пр.
Фармацевтическая композиция представляет собой раствор для инъекций, капсулу, таблетку, пилюлю, порошок или гранулы.
Пути введения соединений или фармацевтических композиций по настоящему изобретению особым образом не ограничены, а типичные способы введения включают, помимо прочих, прием внутрь, ректальное, парентеральное (внутривенное, внутримышечное или подкожное) и местное введение.
Твердые лекарственные формы для приема внутрь включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых лекарственных формах активная фармацевтическая субстанция смешивается по меньшей мере с одним обычным инертным вспомогательным веществом (или носителем), таким как натрия цитрат или дикальция фосфат, или с любым из следующих компонентов: (а) наполнители или вещества для улучшения совместимости, например, крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующие вещества, например, гидроксиметилцеллюлоза, альгинат, желатин, поливинилпирролидон, сахароза и аравийская камедь; (с) увлажнители, такие как глицерин; (d) разрыхлители, такие как агар, кальция карбонат, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые сложные силикаты и натрия карбонат; (е) агенты, замедляющие растворение, такие как парафин; (f) ускорители абсорбции, например, четвертичные аммониевые соединения; (g) смачивающие агенты, такие как цетиловый спирт и глицерилмоностеарат; (h) адсорбенты, например, каолин; и (i) смазывающие вещества, такие как тальк, кальция стеарат, магния стеарат, твердый полиэтиленгликоль, натрия додецилсульфат или их смесь. В капсулах, таблетках и пилюлях лекарственные формы могут также содержать буферные вещества.
Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, могут быть приготовлены с покрытиями и оболочками, такими как кишечнорастворимые оболочки и другие материалы, известные в этой области. Они могут содержать замутнители, и высвобождение действующего вещества или соединения в таких композициях может происходить в определенной части пищеварительного тракта с задержкой. Примерами таких компонентов, которые могут быть использованы с этой целью, являются полимерные и восковые материалы. При необходимости действующее вещество может также находиться в микрокапсулированной форме с одним или несколькими вышеупомянутыми вспомогательными веществами.
Жидкие лекарственные формы для приема внутрь включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. В дополнение к активному соединению жидкая лекарственная форма может содержать инертные разбавители, обычно используемые в этой области, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид и масла, в частности хлопковое масло, арахисовое масло, масло зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло или смеси этих веществ.
Кроме этих инертных разбавителей, композиции могут содержать вспомогательные вещества, такие как смачиватели, эмульгаторы и суспендирующие вещества, подсластители, ароматизаторы и вкусовые добавки.
Кроме действующего вещества, суспензия может содержать суспендирующее средство, например, этоксилированный изооктадеканол, полиоксиэтилен сорбитол и дегидрированный эфир сорбитана, микрокристаллическую целлюлозу, алюминия метоксид и агар, их смесь или иные соединения.
Композиции для парентерального введения могут включать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, которые могут быть повторно растворены в стерильных инъекционных растворах или дисперсиях. Подходящие водные и неводные носители, разбавители, растворители или наполнители включают в себя воду, этанол, многоатомные спирты и любые их подходящие смеси.
Лекарственные формы соединений по настоящему изобретению для местного применения включают мази, порошки, пластыри, спреи и ингаляторы. Активную фармацевтическую субстанцию смешивают в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферными веществами или пропеллентами, которые могут потребоваться при необходимости.
Соединения по настоящему изобретению можно применять самостоятельно или в сочетании с другими фармацевтически приемлемыми соединениями.
Лечение с использованием настоящего изобретения может назначаться отдельно или в комбинации с другими способами лечения или лекарственными препаратами.
При использовании фармацевтической композиции безопасное и эффективное количество соединения по настоящему изобретению применяют для млекопитающего, нуждающегося в лечении (например, для человека). При этом дозировка во время приема представляет собой фармацевтически эффективную дозу, для людей с массой тела 60 кг суточная доза обычно составляет от 0,1 до 2000 мг, предпочтительно от 0,1 до 500 мг. Конечно, при выборе конкретных доз следует учитывать и такие факторы, как способ введения, здоровье пациента и т.д., находящиеся в компетенции квалифицированного врача.
Показания
Соединения по настоящему изобретению обладает очень высокой активностью и селективностью в отношении TLR8, поэтому соединения по настоящему изобретению и фармацевтические композиции, содержащие соединения по настоящему изобретению в качестве основного действующего вещества, могут использоваться для лечения, профилактики и симптоматической терапии родственных заболеваний. Согласно уровню техники, соединения по настоящему изобретению могут быть использованы для лечения в том числе аутоиммунных заболеваний, воспаления, аллергии, астмы, отторжения трансплантата, реакции «трансплантат против хозяина», инфекций, опухолей, иммунодефицитов и т.д.
По сравнению с предшествующим уровнем техники основными преимуществами настоящего изобретения являются:
1) Соединение обладает значительно более высокой активностью и селективностью по отношению к TLR8.
2) Соединение обладает более высокой активностью, лучшей селективностью, более высокой эффективностью in vivo, лучшей безопасностью, лучшей фармакокинетикой и лучшей возможностью разработки лекарственных препаратов.
3) Настоящее изобретение также обеспечивает фармацевтическую композицию, содержащую соединение, и применение соединения или композиции для разработки противоопухолевых, антивирусных, антиинфекционных, аутоиммунных и других лекарственных препаратов.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что эти примеры используются только для иллюстрации настоящего изобретения, а не для ограничения области настоящего изобретения. Экспериментальные методы, в которых не указаны конкретные условия в следующих примерах, обычно используют стандартные условия (например, условия, описанные в Sambrook et al., Molecular Cloning: Laboratory Manual (New York: Cold SpRing Harbor Laboratory PRess, 1989)) или условия, рекомендованные производителем. Если не указано иное, доли и проценты рассчитаны по массе.
Если не указано иное, вся профессиональная и научная терминология, используемая в тексте, имеет те же значения, которые известны специалистам в данной области. Кроме того, к методам изобретения могут быть применены любые методы и материалы, аналогичные или равные по заявленному содержанию. Описанный здесь способ предпочтительного варианта осуществления и материал предназначены только для демонстрационных целей.
Пример 1. Получение соединения А
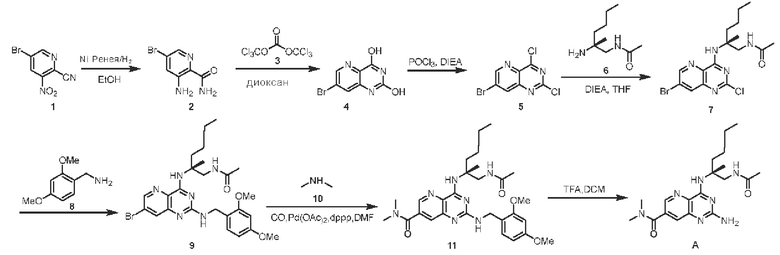
Этап 1. Соединение 2
К безводному этанолу (50 мл) добавляли 1,5 г никеля Ренея, затем добавляли соединение 1 (3,0 г, 13 ммоль), воздух в системе заменяли водородом и перемешивали для протекания реакции при комнатной температуре в течение 30 часов в атмосфере водорода (водородный баллон). После замены водорода в системе на воздух реакционный раствор фильтровали, фильтрат концентрировали и очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: петролейный эфир/этилацетат=10/1) с получением соединения 2 (твердое вещество желтого цвета, 1,3 г, выход 46%).
ЖХ/МС: [М+Н]+=216,0, [М+Н]+=218,0.
Этап 2. Соединение 4
Соединение 2 (800 мг, 3,7 ммоль) и трифосген (1,1 г, 3,7 ммоль) растворяли в 1,4-диоксане (10 мл), реакционный раствор нагревали до 100°С и перемешивали в течение 2 часов. После охлаждения реакционного раствора до комнатной температуры получали твердое вещество желтого цвета путем фильтрации и промывали 1,4-диоксаном с получением соединения 4 (твердое вещество желтого цвета, 870 мг, выход 97%).
ЖХ/МС: [М+Н]+=242,0, [М+Н]+=243,9.
Этап 3. Соединение 5
Соединение 4 (500 мг, 2,08 ммоль) растворяли в фосфора оксихлориде (10 мл), добавляли N,N-диизопропилэтиламин (ДИЭА, 538 мг, 4,16 ммоль), реакционный раствор нагревали до 130°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, концентрировали, добавляли ледяную воду, экстрагировали этилацетатом (3 × 100 мл), органическую фазу промывали насыщенным раствором натрия хлорида (100 мл), высушивали над безводным натрия сульфатом, концентрировали органическую фазу и очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: петролейный эфир/этилацетат=3/1) с получением соединения 5 (твердое вещество желтого цвета, 522 мг, выход 91%).
ЖХ/МС: [М+Н]+=277,9, [М+Н]+=279,9, [М+Н]+=281,9, [М+Н]+=283,9.
Этап 4. Соединение 7
Соединение 5 (100 мг, 0,36 ммоль), соединение 6 (123 мг, 0,72 ммоль, ссылка на патент WO 2018233648) и ДИЭА (231 мг, 1,79 ммоль) растворяли в тетрагидрофуране (5 мл), реакционный раствор нагревали до 40°С и перемешивали в течение 16 часов. После охлаждения реакционного раствора до комнатной температуры добавляли этилацетат (30 мл), полученный раствор промывали водой (30 мл) и насыщенным раствором натрия хлорида (2 × 30 мл) соответственно, органическую фазу высушивали над безводным натрия сульфатом, концентрировали и очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: дихлорметан/метанол=20/1) с получением соединения 7 (маслянистое вещество желтого цвета, 100 мг, выход 67%).
ЖХ/МС: [М+Н]+=414,1, [М+Н]+=416,0.
Этап 5. Соединение 9
Соединение 7 (100 мг, 0,24 ммоль) растворяли в 2,4-диметоксибензамине (1 мл), нагревали до 100°С и перемешивали в течение 2 часов. Реакционный раствор очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: дихлорметан/метанол=10/1) с получением соединения 9 (маслянистое вещество желтого цвета, 60 мг, выход 46%).
ЖХ/МС: [М+Н]+=545,2, [М+Н]+=547,2.
Этап 6. Соединение 11
Соединение 9 (600 мг, 1,10 ммоль) растворяли в диметилформамиде (ДМФА) (20 мл), затем последовательно добавляли 1,3-бис(дифенилфосфино)пропан (ДФФП) (227 мг, 0,55 ммоль), палладия ацетат (123 мг, 0,55 ммоль) и раствор соединения 10 в ТГФ (2 моль/л, 2,5 мл, 5 ммоль). Воздух в реакционном растворе трижды заменяли углерода монооксидом, реакционный раствор нагревали до 80°С в атмосфере углерода монооксида и перемешивали в течение 16 часов, затем температуру повышали до 120°С и перемешивали в течение 8 часов. После охлаждения до комнатной температуры реакционный раствор разбавляли этилацетатом (300 мл), органическую фазу последовательно промывали водой (100 мл) и насыщенным раствором натрия хлорида (100 мл), высушивали над безводным натрия сульфатом, концентрировали органическую фазу и очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: дихлорметан/метанол=100/3) с получением соединения 11 (200 мг, выход 34%).
ЖХ/МС: [М+Н]+=538,4.
Этап 7. Соединение А
Соединение 11 (390 мг, 0,72 ммоль) растворяли в дихлорметане (20 мл), затем добавляли трифторуксусную кислоту (4 мл) и перемешивали реакционный раствор при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали для удаления трифторуксусной кислоты, остаток растворяли с помощью этилацетата (200 мл), а органическую фазу последовательно промывали насыщенным водным раствором натрия бикарбоната (100 мл) и насыщенным раствором натрия хлорида (100 мл), высушивали над безводным натрия сульфатом. Органическую фазу концентрировали и очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: дихлорметан/этилацетат/метанол=5/5/1) с получением соединения А (твердое вещество желтого цвета, 133,8 мг, выход 49%). ЖХ/МС: [М+Н]+=388,3
1H ЯМР (400 МГц, CDCl3) δ 8,41 (дублет, J=2,0 Гц, 1H), 7,66 (дублет, J=2,0 Гц, 1Н), 7,19 (широкий синглет, 1H), 6,61 (широкий синглет, 1H), 5,19 (широкий синглет, 2Н), 3,84-3,69 (мультиплет, 2Н), 3,16 (синглет, 3Н), 3,02 (синглет, 3Н), 2,10-2,06 (мультиплет, 1Н), 2,00 (синглет, 3Н), 1,75-1,65 (мультиплет, 1Н), 1,46 (синглет, 3Н), 1,42-1,24 (мультиплет, 4Н), 0,90 (триплет, J=7,2 Гц, 3Н).
Пример 2. Получение соединения В
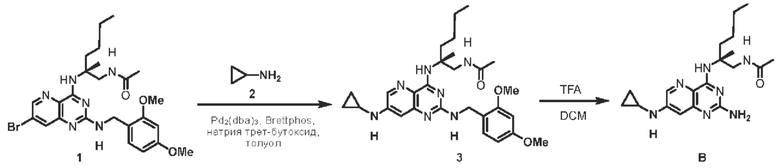
Этап 1. Соединение 3
Соединение 1 (50 мг, 0,092 ммоль), соединение 2 (52 мг, 0,092 ммоль), Pd2(dba)3 (17 мг, 0,018 ммоль), BRettPhos (10 мг, 0,018 ммоль) и натрия трет-бутоксид (26 мг, 0,276 ммоль) добавляли к 5 мл толуола при комнатной температуре. После замещения атмосферы азотом температуру повышали до 80°С и выдерживали для проведения реакции в течение 18 часов. После концентрирования реакционный раствор очищали с помощью хроматографии на колонке с силикагелем (подвижная фаза: дихлорметан/метанол=100/5) для получения соединения 3 (25 мг, 52%) в виде маслянистого вещества бледно-желтого цвета.
ЖХ/МС: [М+Н]+=522,3.
Этап 2. Соединение В
Соединение 3 (25 мг, 0,048 ммоль) растворяли в дихлорметане (2 мл), добавляли трифторуксусную кислоту (1 мл) и перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали и очищали методом обращенно-фазовой хроматографии (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения В (11,2 мг, выход 63%) в виде твердого вещества белого цвета. ЖХ/МС: [М+Н]+=372,3
1H ЯМР (400 МГц, MeOD) δ 8,54 (синглет, 1H), 8,03 (дублет, J=2,2 Гц, 1Н), 6,84 (дублет, J=2,3 Гц, 1H), 3,90 (дублет, J=13,8 Гц, 1H), 3,55 (дублет, J=13,8 Гц, 1Н), 2,49 (триплет дублетов, J=6,5, 3,4 Гц, 1H), 2,19-2,10 (мультиплет, 1H), 1,96-1,79 (мультиплет, 4Н), 1,50-1,27 (мультиплет, 7Н), 0,88 (двойной дублет дублетов, J=11,4, 10,0, 5,8 Гц, 5Н), 0,57-0,49 (мультиплет, 2Н).
Пример 3. Получение соединения С
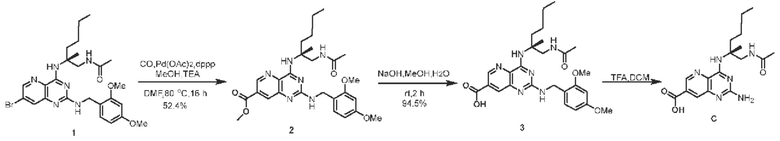
Этап 1. Соединение 2 Соединение 1 (50 мг, 0,092 ммоль, 1,0 экв.) растворяли в смеси ДМФА/метанол (4 мл/1 мл), затем последовательно добавляли палладия ацетат (5 мг, 0,018 ммоль, 0,2 экв.), 1,3-бис(дифенилфосфин)пропан (10 мг, 0,018 ммоль, 0,2 экв.) и триэтиламин (40 мг, 0,368 ммоль, 4 экв.). Реакционный раствор нагревали до 80°С в атмосфере углерода монооксида под давлением 1 атм. и выдерживали для проведения реакции в течение 16 часов. После охлаждения до комнатной температуры реакционный раствор переливали в воду (40 мл) и затем экстрагировали этилацетатом (дважды по 20 мл). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а сырой продукт, полученный после концентрирования, очищали методом препаративной тонкослойной хроматографии для получения соединения 2 (25 мг, 52,4%). ЖХ/МС: [М+Н]+=525,2
Этап 2. Соединение 3
Соединение 2 (25 мг, 0,048 ммоль, 1 экв.) растворяли в метаноле (2 мл), затем добавляли водный раствор натрия гидроксида (2 моль/л, 1 мл). Реакционный раствор перемешивали при комнатной температуре в течение 2 часов, затем корректировали значение рН до 5-6 с использованием 2 М водного раствора хлористоводородной кислоты и экстрагировали этилацетатом (дважды по 10 мл). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а сырой продукт, полученный после концентрирования, очищали методом препаративной тонкослойной хроматографии для получения соединения 3 (23 мг, 94,5%).
ЖХ/МС: [М+Н]+=511,3
Этап 3. Соединение С
Соединение 3 (30 мг, 0,059 ммоль) растворяли в ДХМ (2 мл), затем добавляли трифторуксусную кислоту (1 мл) и проводили реакцию при комнатной температуре в течение 2 часов. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения С (4,3 мг, 20%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=361,2
1H ЯМР (400 МГц, CD3OD) δ 8,86 (синглет, 1Н), 8,70-8,24 (мультиплет, 2Н), 4,50-3,71 (мультиплет, 2Н), 2,67-2,11 (мультиплет, 1H), 1,99 (синглет, 3Н), 1,89-1,80 (мультиплет, 1Н), 1,71-1,35 (мультиплет, 7Н), 1,05-0,89 (мультиплет, 3Н).
Пример 4. Получение соединения D
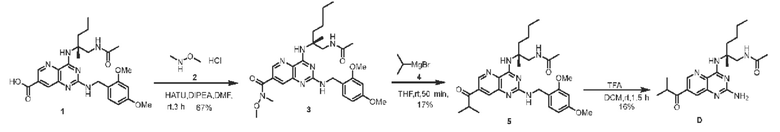
Этап 1. Соединение 3
Соединение 1 (260 мг, 0,509 ммоль, 1,0 экв.) растворяли в ДМФА (7 мл), затем последовательно добавляли соединение 2 (124 мг, 1,273 ммоль, 2,5 экв.), HATU (290 мг, 0,763 ммоль, 1,5 экв.) и ДИЭА (394 мг, 3,055 ммоль, 6,0 экв.) и перемешивали реакционную смесь при комнатной температуре в течение 3 часов. Реакционный раствор разводили этилацетатом (50 мл), затем последовательно промывали водой и насыщенным солевым раствором, высушивали над безводным натрия сульфатом и концентрировали, а остаток очищали с помощью хроматографии на колонке с силикагелем (дихлорметан/метанол=10/1) для получения соединения 3 (190 мг, 67%) в виде твердого вещества коричневого цвета.
ЖХ/МС: [М+Н]+=554,3.
Этап 2. Соединение 5 Соединение 3 (118 мг, 0,213 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (8 мл), добавляли соединение 4 (1 моль/л раствор, 2,13 мл, 2,13 ммоль, 10,0 экв.) при комнатной температуре и перемешивали реакционный раствор при комнатной температуре в течение 50 минут. Реакционный раствор переливали в насыщенный водный раствор аммония хлорида, а затем экстрагировали этилацетатом (трижды по 20 мл). Органические фазы объединяли и промывали насыщенным солевым раствором, высушивали над безводным натрия сульфатом и концентрировали, а остаток очищали методом препаративной тонкослойной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 5 (20 мг, 17%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=537,3.
Этап 3. Соединение D
Соединение 5 (20 мг, 0,0372 ммоль, 1,0 экв.) растворяли в дихлорметане (0,5 мл), затем добавляли трифторуксусную кислоту (1,5 мл) и перемешивали реакционный раствор при комнатной температуре в течение 1,5 часов. Реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) для получения соединения D (2,3 мг, 16%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=387,3
1Н ЯМР (400 МГц, CDCl3) δ 8,89 (дублет, J=2,0 Гц, 1H), 8,17 (дублет, J=2,0 Гц, 1H), 7,52 (синглет, 1H), 6,35 (синглет, 1H), 3,87 (двойной дублет, J=14,0, 5,6 Гц, 1Н), 3,68 (двойной дублет, J=14,0, 6,0 Гц, 1H), 3,55-3,50 (мультиплет, 1H), 2,11-2,05 (мультиплет, 1Н), 2,01 (синглет, 3Н), 1,77 (синглет, 1H), 1,49 (синглет, 3Н), 1,40-1,29 (мультиплет, 4Н), 1,25 (дублет, J=6,8 Гц, 6Н), 0,90 (триплет, J=7,0 Гц, 3Н).
Пример 5. Получение соединения Е
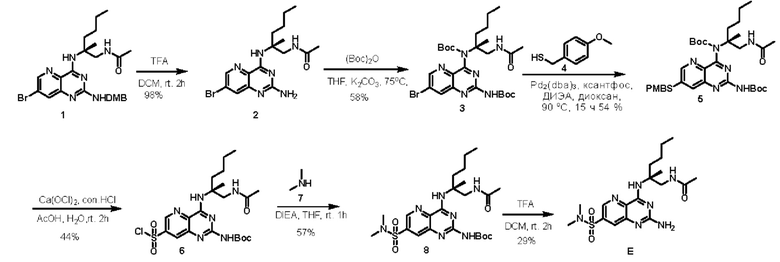
Этап 1. Соединение 2
Соединение 1 (400 мг, 0,78 ммоль, 1,0 экв.) растворяли в ДХМ (10 мл), затем добавляли трифторуксусную кислоту (5 мл) и перемешивали при комнатной температуре в течение 2 ч, после завершения реакции концентрировали, корректировали уровень рН до >7 раствором натрия бикарбоната, экстрагировали этилацетатом, органическую фазу сушили над безводным натрия сульфатом и концентрировали для получения соединения 2 (300 мг, 98%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=395,1.
Этап 2. Соединение 3
Соединение 2 (300 мг, 0,76 ммоль, 1,0 экв.), (Вос)2O (497 мг, 2,28 ммоль, 3,0 экв.) и калия карбонат (315 мг, 2,28 ммоль, 3,0 экв.) растворяли в тетрагидрофуране (30 мл), затем нагревали до температуры 75°С для проведения реакции в течение 18 ч. После завершения реакции реакционный раствор концентрировали, полученный остаток очищали методом колоночной хроматографии с силикагелем (этилацетат/петролейный эфир=70/30) для получения соединения 3 (260 мг, 58%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=595,1.
Этап 3. Соединение 5
Соединение 3 (260 мг, 0,44 ммоль, 1,0 экв.), Pd2(dba)3 (40 мг, 0,044 ммоль, 0,1 экв.), ксантФос (25 мг, 0,044 ммоль, 0,1 экв.), ДИПЭА (170 мг, 1,32 ммоль, 3,0 экв.) и соединение 4 (101 мг, 0,66 ммоль, 1,5 экв.) растворяли в диоксане (15 мл), замещали азотом, затем повышали температуру до 90°С и подвергали реакции в течение 15 ч. После завершения реакции реакционный раствор концентрировали, полученный остаток очищали методом колоночной хроматографии с силикагелем (этилацетат/петролейный эфир=2/1) для получения соединения 5 (160 мг, 54%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=669,1
Этап 4. Соединение 6
Соединение 5 (150 мг, 0,22 ммоль, 1,0 экв.) растворяли в уксусной кислоте (5 мл) и воде (1,5 мл), замещали азотом, затем вводили достаточное количество газообразного хлора (20 мл концентрированной хлористоводородной кислоты добавляли по каплям к 20 г кальция гипохлорита для образования газообразного хлора), перемешивали при комнатной температуре в течение 2 ч, быстро охлаждали ледяной водой после завершения реакции, уровень рН корректировали до >7 насыщенным раствором натрия бикарбоната, экстрагировали этилацетатом, органическую фазу сушили над безводным натрия сульфатом, концентрировали для получения сырого соединения 6 (50 мг, 44%) в виде твердого вещества желтого цвета, которое непосредственно использовали на следующем этапе.
ЖХ/МС: [М+Н]+=515,0
Этап 5. Соединение 8
Соединение 6 (50 мг, 0,1 ммоль, 1,0 экв.) и ДИЭА (39 мг, 0,3 ммоль, 3,0 экв.) растворяли в тетрагидрофуране (5 мл), затем добавляли соединение 7 (2 М в растворе тетрагидрофурана, 0,5 мл, 1 ммоль, 10,0 экв.) и перемешивали при комнатной температуре в течение 1 ч.
Завершали реакцию и быстро охлаждали водой, экстрагировали этилацетатом, органическую фазу сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 8 (30 мг, 54%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=524,1
Этап 6. Соединение Е
Соединение 8 (30 мг, 0,06 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (3 мл) и дихлорметане (3 мл) и подвергали реакции при комнатной температуре в течение 2 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения Е (7,3 мг, 29%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=424,2
1Н ЯМР (400 МГц, CD3OD) δ 8,66 (дублет, J=2,0 Гц, 1H), 7,93 (дублет, J=2,0 Гц, 1Н), 3,95-3,92 (мультиплет, 1Н), 3,61-3,58 (мультиплет, 1H), 2,78 (синглет, 6Н), 2,23-2,17 (мультиплет, 1Н), 1,95 (синглет, 3Н), 1,87-1,80 (мультиплет, 1Н), 1,49 (синглет, 3Н), 1,37-1,31 (мультиплет, 4Н), 0,92-0,89 (мультиплет, 3Н).
Пример 6. Получение соединения F
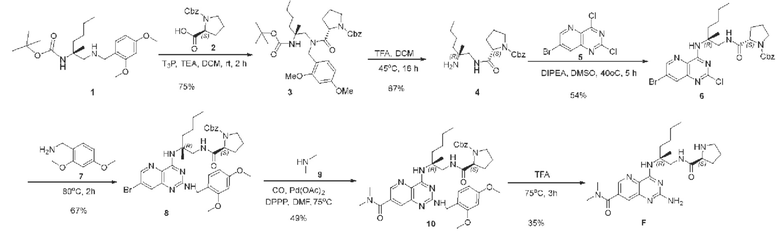
Этап 1. Соединение 3
Соединение 1 (2 г, 5,3 ммоль, 1,0 экв.) растворяли в ДХМ (30 мл) и затем добавляли триэтиламин (2,7 г, 26,3 ммоль, 5,0 экв.), соединение 2 (1,44 г, 5,8 ммоль, 1,1 экв.) и 1-пропилфосфатный ангидрид (5 г, 15,8 ммоль, 3,0 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=3/1) для получения соединения 3 (2,4 мг, 75%) в виде масла коричневого цвета.
ЖХ/МС: [M+Na]+=634,3
Этап 2. Соединение 4
Соединение 3 (0,5 г, 0,82 ммоль, 1,0 экв.) растворяли в смеси дихлорметана (2,0 мл) и трифторуксусной кислоты (2,0 мл) и перемешивали реакционную смесь при температуре 45°С в течение 16 ч. В реакционный раствор для корректировки уровня рН до 7-8 добавляли охлажденный до комнатной температуры насыщенный раствор натрия бикарбоната и экстрагировали водную фазу дихлорметаном (20 × 3 мл). Органические фазы объединяли и сушили над безводным натрия сульфатом, а остаток, полученный после концентрирования, очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=50:1-10:1) для получения соединения 4 (200 мг, 67%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=362,2.
Этап 3. Соединение 6
Соединение 4 (200 мг, 0,55 ммоль, 1,0 экв.) растворяли в диметилсульфоксиде (5 мл) и затем последовательно добавляли соединение 5 (462 мг, 1,66 ммоль, 3 экв.) и ДИПЭА (214 мг, 1,66 ммоль, 3 экв.). Реакционный раствор подвергали реакции при температуре 40°С в течение 5 ч. Охлаждали до комнатной температуры, добавляли 50 мл воды и экстрагировали реакционный раствор этилацетатом (3 × 15 мл). Органические фазы объединяли и сушили над безводным натрия сульфатом, а остаток, полученный после концентрирования, очищали методом колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=20/1-2/1) для получения соединения 6 (180 мг, 54%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=603,1
Этап 4. Соединение 8
Соединение 6 (180 мг, 0,29 ммоль, 1,0 экв.) растворяли в соединении 7 (0,5 мл), нагревали реакционный раствор до 80°С в защитной азотной атмосфере и перемешивали в течение 2 ч. Охлажденную до комнатной температуры реакционную смесь очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=100/1) для получения сырого продукта, сырой продукт снова очищали методом колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=100/1-1/1) для получения соединения 8 (140 мг, 67%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=734,2
Этап 5. Соединение 10
Соединение 8 (34 мг, 0,1 ммоль, 1,0 экв.) растворяли в ДМФА (10 мл), затем последовательно добавляли ДФФП (42 мг, 0,1 ммоль, 1,0 экв.), палладия ацетат (23 мг, 0,1 ммоль, 1,0 экв.) и раствор соединения 9 в ТГФ (2 М, 2 мл, 4 ммоль, 40 экв.). После замещения углерода монооксидом три раза смесь нагревали до температуры 75°С в атмосфере монооксида углерода и перемешивали в течение 16 ч. Реакционный раствор охлаждали до комнатной температуры, разводили этилацетатом (100 мл), промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали. Остаток разделяли и очищали методом колоночной хроматографии с силикагелем (этилацетат:метанол=30:1) для получения соединения 10 (36 мг, 39%).
ЖХ/МС: [М+Н]+=727,4
Этап 6. Соединение F
Соединение 10 (34 мг, 0,046 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (3 мл) при комнатной температуре. Реакционный раствор перемешивали при температуре 75°С в течение 3 ч. Реакционный раствор концентрировали для удаления трифторуксусной кислоты, остаток растворяли с помощью этилацетата (200 мл), органическую фазу последовательно промывали насыщенным водным раствором натрия бикарбоната и насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали. Сырой продукт очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения F (7,1 мг, 35%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=443,3
1H ЯМР (400 МГц, MeOD) δ 8,40 (синглет, 1H), 7,65 (синглет, 1Н), 4,24-4,15 (мультиплет, 1Н), 4,00 (дублет, J=13,7 Гц, 1Н), 3,84 (дублет, J=13,7 Гц, 1H), 3,30-3.21 (мультиплет, 2Н), 3,15 (синглет, 3Н), 3,04 (синглет, 3Н), 2,40-2,20 (мультиплет, 2Н), 1,96 (широкий синглет, 3Н), 1,80-1,65 (мультиплет, 1H), 1,48 (синглет, 3Н), 1,43-1.22 (мультиплет, 4Н), 0,91 (триплет, J=7,2 Гц, 3Н).
Пример 7. Получение соединения G
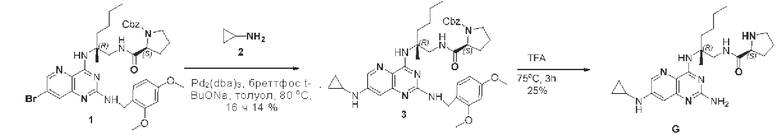
Этап 1. Соединение 3 Соединение 1 (75 мг, 0,1 ммоль, 1 экв.), соединение 2 (30 мг, 0,5 ммоль, 5 экв.), Pd2(dba)3 (27 мг, 0,05 ммоль, 0,5 экв.), БреттФос (46 мг, 0,05 ммоль, 0,5 экв.) и натрия трет-бутоксид (30 мг, 0,3 ммоль, 3 экв.) добавляли в толуол (10 мл) при комнатной температуре, после замещения азотом температуру повышали до 80°С для проведения реакции в течение 16 ч. После концентрирования реакционный раствор очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=100/5) для получения соединения 3 (10 мг, 14%) в виде твердого вещества бледно-желтого цвета.
ЖХ/МС: [М+Н]+=711,1
Этап 2. Соединение G
Соединение 3 (10 мг, 0,014 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (2 мл), нагревали реакционный раствор до температуры 75°С и подвергали реакции в течение 3 ч. После завершения реакции реакционный раствор сначала концентрировали, затем добавляли насыщенный водный раствор натрия бикарбоната для корректировки рН до >7 и затем для экстрагирования использовали этилацетат (50 мл * 3). Органические фазы объединяли, сушили над безводным натрия сульфатом и концентрировали. Остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) для получения соединения G (1,5 мг, 25%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=427,3
1H ЯМР (400 МГц, CD3OD) δ 8,54 (широкий синглет, 1H), 8,01 (синглет, 1H), 6,86 (синглет, 1H), 4,61 (синглет, 1Н), 4,09-3,90 (мультиплет, 2Н), 3,73 (дублет, J=13,6 Гц, 1H). 3,18-3,12 (мультиплет, 2Н), 2,50 (синглет, 1H), 2,26-2,17 (мультиплет, 2Н), 1,88-1,73 (мультиплет.4Н), 1,47-1,33 (мультиплет, 7Н), 0,95-0,81 (мультиплет, 4Н), 0,54 (синглет, 2Н).
Пример 8. Получение соединения Н
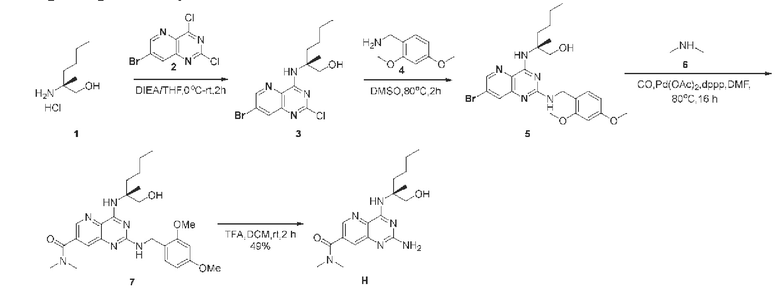
Этап 1. Соединение 3
Соединение 1 (5,5 г, чистота 55%, 18,37 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (100 мл), добавляли ДИЭА (11,87 г, 91,84 ммоль, 5,0 экв.) и перемешивали реакционный раствор при комнатной температуре в течение 0,5 ч. В реакционный раствор добавляли раствор соединения 2 (5,12 г, 18,37 ммоль, 1,1 экв.) в тетрагидрофуране (100 мл) и затем перемешивали реакционный раствор при комнатной температуре в течение ночи. Реакционный раствор концентрировали, а полученный остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=20/1) для получения соединения 3 (4,2 г, 61%) в виде твердого вещества желтого цвета. ЖХ/МС: [М+Н]+=375,0
Этап 2. Соединение 5
Соединение 3 (4,2 г, 11,24 ммоль, 1,0 экв.) растворяли в ДМСО (20 мл), затем добавляли соединение 4 (15 г, 89,92 ммоль, 8,0 экв.) и перемешивали реакционный раствор при температуре 80°С в течение 2 ч. После охлаждения до комнатной температуры в реакционный раствор добавляли воду (80 мл), рН корректировали до уровня между 7 и 8 с помощью водного раствора лимонной кислоты (1 М) и затем экстрагировали с помощью ЭА (3 × 100 мл). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали. Полученный остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 5 (3,52 г, 62%) в виде твердого вещества желтого цвета. ЖХ/МС: [М+Н]+=504,2.
Этап 3. Соединение 7
Соединение 5 (3,02 г, 5,99 ммоль, 1,0 экв.) растворяли в ДМФА (50 мл), затем быстро добавляли ДФФП (1,23 г, 3,0 ммоль, 0,5 экв.), палладия ацетат (671 мг, 3,0 ммоль, 0,5 экв.) и раствор соединения 6 в ТГФ (2 М, 48 мл, 96 ммоль, 16 экв.). Систему замещали углерода моноокисдом три раза и затем нагревали до температуры 80°С в атмосфере монооксида углерода, и перемешивали в течение 16 ч. Реакционный раствор охлаждали до комнатной температуры, разводили этилацетатом (300 мл), последовательно промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали. Полученный остаток разделяли и очищали методом колоночной хроматографии с силикагелем (ДХМ-ДХМ: МеОН=100:3) для получения сырого продукта 7 (2,75 г, 92%).
ЖХ/МС: [М+Н]+=497,2
Этап 4. Соединение Н
Соединение 7 (2,66 г, 5,36 ммоль, 1,0 экв.) растворяли в дихлорметане (20 мл) и затем добавляли трифторуксусную кислоту (20 мл). Реакционный раствор перемешивали при температуре 25°С в течение 2 ч. Реакционный раствор концентрировали, добавляли метанол и затем концентрировали, и эту операцию проводили три раза. Полученный остаток растворяли в этилацетате (300 мл), рН полученного этилацетатного раствора корректировали до 8 водным раствором Na2CO3, экстрагированную органическую фазу промывали насыщенным солевым раствором, сушили над безводным натрия сульфата и концентрировали. Полученный сырой продукт очищали методом колоночной хроматографии с силикагелем (отношение ДХМ к DCM:MeOH=10:1) для получения соединения Н (1,07 г, 58%) в виде твердого вещества бледно-желтого цвета.
ЖХ/МС: [М+Н]+=347,1
1H ЯМР (400 МГц, CDCl3) δ 8,44 (синглет, 1H), 7,75 (синглет, 1H), 7,62 (синглет, 1H), 6,72 (широкий, 2Н), 3,89-3,76 (мультиплет, 2Н), 3,14 (синглет, 3Н), 3,00 (синглет, 3Н), 2,01-1,94 (мультиплет, 1H), 1,79-1,73 (мультиплет, 1Н), 1,45 (синглет, 3Н), 1,42-1,25 (мультиплет, 4Н), 0,91 (триплет, J=7,2 Гц, 3Н).
Пример 9. Получение соединения I
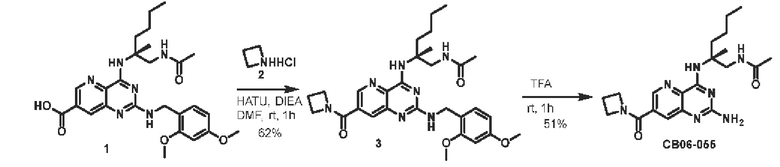
Этап 1. Соединение 3
Соединение 1 (30 мг, 0,059 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), затем добавляли HATU (34 мг, 0,09 ммоль, 1,5 экв.), ДИЭА (23 мг, 0,18 ммоль, 3,0 экв.) и соединение 2 (7,2 мг, 0,076 ммоль, 1,3 экв.) для проведения реакции при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой и затем экстрагировали этилацетатом (50 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=100/8) для получения соединения 3 (20 мг, 62%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=550,3
Этап 2. Соединение I
Соединение 3 (20 мг, 0,036 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (3 мл) и подвергали реакции при комнатной температуре в течение 1 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения I (7,3 мг, 51%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=400,3
1H ЯМР (400 МГц, CD3OD) δ 8,59 (дублет, J=2,0 Гц, 1Н), 7,80 (дублет, J=2,0 Гц, 1H), 4,42 (триплет, J=7,6 Гц, 2Н), 4,24 (триплет, J=7,6 Гц, 2Н), 3,93 (дублет, J=13,6 Гц, 1H), 3,63 (дублет, J=13,6 Гц, 1H), 2,45-2,27 (мультиплет, 2Н), 2,24-2,17 (мультиплет, 1Н), 1,95 (синглет, 3Н), 1,83-1,76 (мультиплет, 1H), 1,47 (синглет, 3Н), 1,41-1,26 (мультиплет, 4Н), 0,90 (триплет, J=6,8 Гц, 3Н).
Пример 10. Получение соединения J
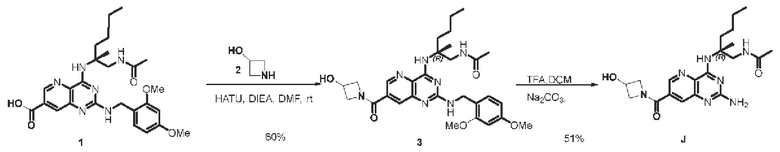
Этап 1. Соединение 3
Соединение 1 (30 мг, 0,059 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), затем добавляли HATU (67 мг, 0,176 ммоль, 3,0 экв.), ДИЭА (90 мг, 0,696 ммоль, 11,8 экв.) и соединение 2 (38,6 мг, 0,352 ммоль, 6,0 экв.) для проведения реакции в течение 3 ч. Реакционный раствор разводили водой и затем экстрагировали этилацетатом (50 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 3 (20 мг, 60%).
ЖХ/МС: [М+Н]+=566,4
Этап 2. Соединение J
Соединение 3 (20 мг, 0,035 ммоль) растворяли в ДХМ (3 мл), затем по каплям добавляли трифторуксусную кислоту (1 мл) и перемешивали реакционный раствор при комнатной температуре в течение 2 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения J (7,3 мг, 51%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=416,3
1Н ЯМР (400 МГц, CD3OD) δ 8,63 (синглет, 1Н), 8,43 (синглет, 1Н), 7,84 (синглет, 1Н), 4,70-4,62 (мультиплет, 2Н), 4,48-4,39 (мультиплет, 1H), 4,23-4,15 (мультиплет, 1Н), 4,00-3,96 (мультиплет, 1Н), 3,93 (дублет, J=14,0 Гц, 1H), 3,60 (дублет, J=14,0 Гц, 1Н), 2,26-2,14 (мультиплет, 1Н), 1,95 (синглет, 3Н), 1,88-1,76 (мультиплет, 1Н), 1,49 (синглет, 3Н), 1,36-1,26 (мультиплет, 4Н), 0,90 (триплет, J=6,8 Гц, 3Н).
Пример 11. Получение соединения K
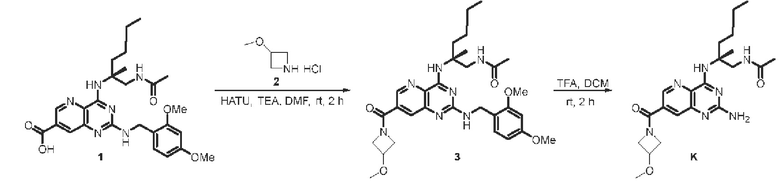
Этап 1. Соединение 3
Соединение 1 (33 мг, 0,065 ммоль, 1,0 экв.) растворяли в безводном ДМФА (5 мл), затем добавляли HATU (32 мг, 0,084 ммоль, 1,3 экв.), триэтиламин (0,3 мл, 0,194 ммоль, 3,0 экв.) и соединение 2 (12 мг, 0,084 ммоль, 1,5 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 2 ч. В реакционный раствор добавляли 20 мл воды и экстрагировали этилацетатом (3 × 20 мл). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали. Полученный остаток очищали методом колоночной хроматографии с силикагелем (петролейный эфир/этилацетат=2/1) для получения соединения 3 (30 мг, 80%) в виде масла желтого цвета.
ЖХ/МС: [М+Н]+=580,3
Этап 2. Соединение K
Соединение 3 (30 мг, 0,052 ммоль, 1,0 экв.) растворяли в дихлорметане (1 мл), затем добавляли трифторуксусную кислоту (3 мл). Реакционный раствор перемешивали при комнатной температуре в течение 2 ч. Остаток, полученный концентрированием реакционного раствора, очищали методом препаративной ВЭЖХ (0,1% аммиак/ацетонитрил/вода) для получения соединения K (5,8 мг, 21%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=430,2
1H ЯМР (400 МГц, ДМСО) δ 8,45 (синглет, 1H), 7,87 (широкий синглет, 1H), 7,67 (синглет, 1Н), 7,14 (широкий синглет, 1Н), 6,53 (широкий синглет, 2Н), 4,49-4,45 (мультиплет, 1Н), 4,30-4,26 (мультиплет, 2Н), 4,18-4,16 (мультиплет, 1H), 3,88 (дублет, 7=7,3 Гц, 1H), 3,74 (дублет дублетов, J=13,6, 5,5 Гц, 1H), 3,49-3,44 (мультиплет, 1Н), 3,22 (синглет, 3Н), 2,10-2,04 (мультиплет, 1H), 1,82 (синглет, 3Н), 1,76-1,71 (мультиплет, 1Н), 1,37 (синглет, 3Н), 1,27-1,23 (мультиплет, 4Н), 0,84 (триплет, J=6,5 Гц, 3Н).
Пример 12. Получение соединения L
Этап 1. Соединение 3
Соединение 2 (7 мг, 0,08 ммоль, 1,3 экв.) растворяли в ДМФА (3 мл) и добавляли HATU (30 мг, 0,08 ммоль, 1,3 экв.) и соединение 1 (30 мг, 0,06 ммоль, 1,0 экв.), затем в реакционный раствор добавляли ДИЭА (23 мг, 0,17 ммоль, 3,0 экв.) и перемешивали ее при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой, экстрагировали этилацетатом (3 × 30 мл), промывали насыщенным солевым раствором (30 мл), сушили на безводный натрия сульфатом, фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=20/1) для получения соединения 3 (30 мг, 87%) в виде твердого вещества белого цвета
ЖХ/МС: [М+Н]+=586,3
Этап 2. Соединение L
Соединение 3 (30 мг, 0,05 ммоль, 1,0 экв.) растворяли в дихлорметане (1 мл) и затем добавляли трифторуксусную кислоту (5 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали, а остаток очищали методом обращенно-фазовой хроматографии (0,05% муравьиная кислота/CH3CN/Н2О) для получения соединения L (14,3 мг, 66%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=436,2. 1H ЯМР (400 МГц, CD3OD) δ 8,61 (дублет, J=2,0 Гц, 1H), 7,83 (дублет, J=2,0 Гц, 1H), 4,83-4,74 (мультиплет, 2Н), 4,58 (синглет, 2Н), 3,92 (дублет, J=14,0 Гц, 1Н), 3,62 (дублет, J=14,0 Гц, 1H), 2,23-2,17 (мультиплет, 1H), 1,95 (синглет, 3Н), 1,84-1,77 (мультиплет, 1Н), 1,47 (синглет, 3Н), 1,40-1,27 (мультиплет, 4Н), 0,90 (триплет, J=6,8 Гц, 3Н).
Пример 13. Получение соединения М
Этап 1. Соединение 3
Соединение 1 (30 мг, 0,059 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), затем добавляли HATU (67 мг, 0,176 ммоль, 3,0 экв.), ДИЭА (76 мг, 0,18 ммоль, 3,0 экв.) и соединение 2 (35 мг, 0,352 ммоль, 6,0 экв.) для проведения реакции при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой и затем экстрагировали этилацетатом (50 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 3 (30 мг, 85%).
ЖХ/МС: [М+Н]+=592,4
Этап 2. Соединение М
Соединение 3 (20 мг, 0,036 ммоль, 1,0 экв.) растворяли в ДХМ (3 мл), затем по каплям добавляли трифторуксусную кислоту (1 мл) и перемешивали реакционный раствор при комнатной температуре в течение 2 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения М (6,6 мг, 41%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=442,3
1H ЯМР (400 МГц, CD3OD) δ 8,57 (синглет, 1H), 7,79 (синглет, 1H), 4,83-4,81 (мультиплет, 4Н), 4,57 (синглет, 2Н), 4,37 (синглет, 2Н), 3,93 (дублет, J=13,6 Гц, 1Н), 3,62 (дублет, 7=13,6 Гц, 1H), 2,26-2,15 (мультиплет, 1Н), 1,95 (синглет, 3Н), 1,86-1,74 (мультиплет, 1Н), 1,47 (синглет, 3Н), 1,43-1,20 (мультиплет, 4Н), 0,90 (триплет, J=6,8 Гц, 3Н).
Пример 14. Получение соединения N
Этап 1. Соединение 3 Соединение 2 (10 мг, 0,1 ммоль, 1,3 экв.) растворяли в ДМФА (3 мл) и добавляли HATU (38 мг, 0,1 ммоль, 1,3 экв.) и соединение 1 (40 мг, 0,08 ммоль, 1,0 экв.), затем в реакционный раствор добавляли ДИЭА (30 мг, 0,23 ммоль, 3,0 экв.) и перемешивали ее при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой, экстрагировали этилацетатом (3 × 30 мл), промывали насыщенным солевым раствором (30 мл), сушили на безводный натрия сульфатом, фильтровали и концентрировали. Остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=20/1) для получения соединения 3 (20 мг, 43%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=593,3
Этап 2. Соединение N
Соединение 3 (20 мг, 0,03 ммоль, 1,0 экв.) растворяли в дихлорметане (1 мл) и затем добавляли трифторуксусную кислоту (5 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали, а остаток очищали методом обращенно-фазовой хроматографии (0,05% муравьиная кислота/CH3CN/H2O) для получения соединения N (6,0 мг, 40%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=443,3.
1H ЯМР (400 МГц, CD3OD) δ 8,64 (синглет, 1Н), 7,85 (синглет, 1Н), 4,61 (синглет, 1Н), 4,45 (синглет, 1H), 4,32-4,18 (мультиплет, 2Н), 4,05-4,03 (мультиплет, 1H), 3,95-3,92 (мультиплет, 1Н), 3,62-3,59 (мультиплет, 1H), 2,24 (синглет, 6Н), 2,21-2,17 (мультиплет, 1Н), 1,95 (синглет, 3Н), 1,86-1,81 (мультиплет, 1Н), 1,49 (синглет, 3Н), 1,37-1,29 (мультиплет, 4Н), 0,92-0,89 (мультиплет, 3Н).
Пример 15. Получение соединения Р
Этап 1. Соединение 3
Соединение 1 (30 мг, 0,0587 ммоль, 1,0 экв.) растворяли в ДМФА (1,5 мл), затем последовательно добавляли соединение 2 (8 мг, 0,0881 ммоль, 1,5 экв.), HATU (29 мг, 0,0763 ммоль, 1,3 экв.) и ДИЭА (23 мг, 0,176 ммоль, 3,0 экв.) и перемешивали реакционную смесь при комнатной температуре в течение 2 ч. Реакционный раствор разводили этилацетатом (30 мл), затем последовательно промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 3 (22 мг, 64%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=580,3
Этап 2. Соединение Р
Соединение 3 (22 мг, 0,0379 ммоль, 1,0 экв.) растворяли в дихлорметане (0,5 мл), затем добавляли трифторуксусную кислоту (1,5 мл) и перемешивали реакционный раствор при комнатной температуре в течение 2 ч. Реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) для получения соединения Р (6,4 мг, 39%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=430,3
1Н ЯМР (400 МГц, CD3OD) δ 8,49 (дублет, J=1,8 Гц, 1Н), 8,36 (синглет, 1H), 7,70 (дублет, J=1,8 Гц, 1H), 3,97-3,87 (мультиплет, 1Н), 3,79 (синглет, 4Н), 3,66 (синглет, 2Н), 3,61-3,55 (мультиплет, 1Н), 3,48 (синглет, 2Н), 2,22-2,17 (мультиплет, 1H), 1,95 (синглет, 3Н), 1,89-1,83 (мультиплет, 1H), 1,50 (синглет, 3Н), 1,36-1,29 (мультиплет, 4Н), 0,91 (триплет, J=7,2 Гц, 3Н).
Пример 16. Получение соединения Q
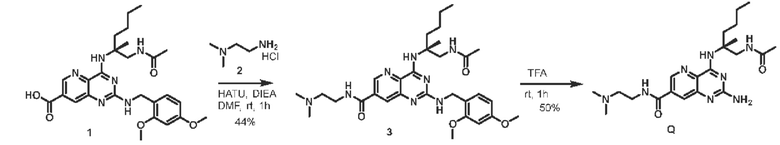
Этап 1. Соединение 3 Соединение 1 (30 мг, 0,059 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), затем добавляли HATU (34 мг, 0,09 ммоль, 1,5 экв.), ДИЭА (23 мг, 0,18 ммоль, 3,0 экв.) и соединение 2 (10,9 мг, 0,088 ммоль, 1,5 экв.) для проведения реакции при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой и затем экстрагировали этилацетатом (50 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=100/8) для получения соединения 3 (15 мг, 44%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=581,3
Этап 2. Соединение Q
Соединение 3 (15 мг, 0,026 ммоль, 1,0 экв.) растворяли в трифторуксусной кислоте (3 мл) и подвергали реакции при комнатной температуре в течение 1 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения Q (5,7 мг, 50%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=431,2
1Н ЯМР (400 МГц, CD3OD) δ 8,82 (синглет, 1Н), 8,06 (синглет, 1Н), 3,95-3,92 (мультиплет, 1Н), 3,76 (широкий синглет, 2Н), 3,62-3,58 (мультиплет, 1H), 3,24 (широкий синглет, 2Н), 2,86 (синглет, 6Н), 2,24-2,17 (мультиплет, 1Н), 1,95 (синглет, 3Н), 1,88-1,81 (мультиплет, 1Н), 1,50 (синглет, 3Н), 1,37-1,32 (мультиплет, 4Н), 0,90 (триплет, J=6,8 Гц, 3Н)
Пример 17. Получение соединения R
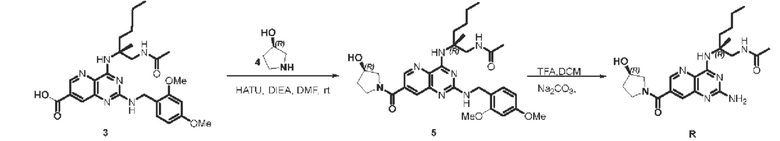
Этап 1. Соединение 5
Соединение 1 (30 мг, 0,059 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), затем добавляли HATU (67 мг, 0,176 ммоль, 3,0 экв.), ДИЭА (76 мг, 0,18 ммоль, 3,0 экв.) и соединение 4 (35 мг, 0,352 ммоль, 6,0 экв.) для проведения реакции при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой и затем экстрагировали этилацетатом (50 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 5 (30 мг, 88%).
ЖХ/МС: [М+Н]+=580,3
Этап 2. Соединение R
Соединение 5 (30 мг, 0,051 ммоль, 1,0 экв.) растворяли в ДХМ (3 мл), затем по каплям добавляли трифторуксусную кислоту (1 мл) для проведения реакции при комнатной температуре в течение 2 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения R (9,1 мг, 41%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=430,3
1Н ЯМР (400 МГц, CD3OD) δ 8,54 (дублет, J=4,4 ГЦ, 1Н), 7,76 (дублет, J=7,2 Гц, 1H), 4,46 (дублет, J=44 Гц, 1H), 3,94 (дублет, J=13,6 Гц, 1H), 3,80-3,53 (мультиплет, 4Н), 3,36-3,37 (мультиплет, 1Н), 2,23-1,90 (мультиплет, 6Н), 1,85-1,79 (мультиплет, 1Н), 1,49 (синглет, 3Н), 1,43-1,21 (мультиплет, 4Н), 0,91 (триплет, J=6,8 Гц, 3Н)
Пример 18. Получение соединения S
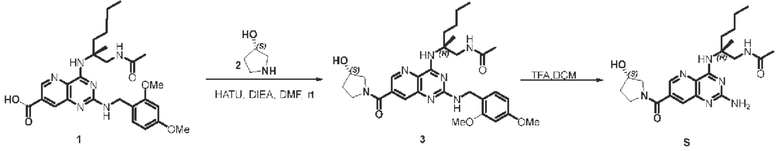
Этап 1. Соединение 3
Соединение 1 (30 мг, 0,059 ммоль, 1,0 экв.) растворяли в ДМФА (3 мл), затем добавляли HATU (67 мг, 0,176 ммоль, 3,0 экв.), ДИЭА (76 мг, 0,18 ммоль, 3,0 экв.) и соединение 2 (35 мг, 0,352 ммоль, 6,0 экв.) для проведения реакции при комнатной температуре в течение 1 ч. Реакционный раствор разводили водой и затем экстрагировали этилацетатом (50 мл * 3). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом колоночной хроматографии с силикагелем (дихлорметан/метанол=10/1) для получения соединения 3 (30 мг, 88%).
ЖХ/МС: [М+Н]+=580,3
Этап 2. Соединение S
Соединение 3 (20 мг, 0,036 ммоль, 1,0 экв.) растворяли в ДХМ (3 мл), затем по каплям добавляли трифторуксусную кислоту (1 мл) для проведения реакции при комнатной температуре в течение 2 ч. После завершения реакции реакционный раствор концентрировали, остаток очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения S (19 мг, 86%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=430,3
1H ЯМР (400 МГц, CD3OD) δ 8,47-8,45 (мультиплет, 1H), 7,70-7,68 (мультиплет, 1H), 4,45 (дублет, J=44 Гц, 1H), 3,94 (дублет, J=14,0 Гц, 1H), 3,82-3,47 (мультиплет, 4Н), 3,31-3,30 (мультиплет, 1Н), 2,25-1,92 (мультиплет, 6Н), 1,82-1,74 (мультиплет, 1Н), 1,47 (синглет, 3Н), 1,40-1,24 (мультиплет, 4Н), 0,90 (триплет, J=6,8 Гц, 3Н).
Пример 19. Получение соединения Т
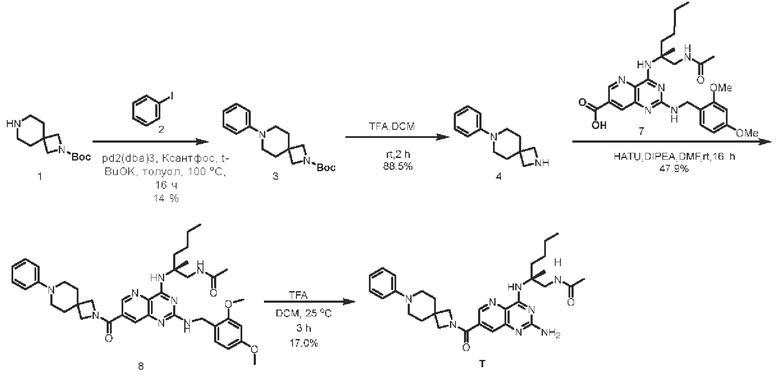
Этап 1. Соединение 3
Соединение 1 (1,2 г, 5,4 ммоль, 1,1 экв.) растворяли в толуоле (20 мл), затем последовательно добавляли соединение 2 (1 г, 4,9 ммоль, 1, 0 экв.), Pd2(dba)3 (450 мг, 0,5 ммоль, 0,1 экв.), КсантоФос (290 мг, 0,5 ммоль, 0,1 экв.) и калия трет-бутоксид (1,2 г, 10 ммоль, 2 экв.). Реакционный раствор перемешивали при температуре 100°С в течение 16 ч. Сырой продукт, полученный концентрированием реакционного раствора, очищали методом колоночной флэш-хроматографии для получения соединения 3 (760 мг, 47%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]+=303,2
Этап 2. Соединение 4
Соединение 3 (76 мг, 0,25 ммоль, 1 экв.) растворяли в дихлорметане (5 мл) и затем добавляли трифторуксусную кислоту (1 мл). Реакционный раствор перемешивали при комнатной температуре в течение 2 ч. Для корректировки уровня рН до 7-8 добавляли насыщенный раствор натрия бикарбоната и затем экстрагировали дихлорметаном (10 мл × 2). Затем органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали для получения соединения 4 (45 мг, 88,5%).
ЖХ/МС: [М+Н]+=203,2
Этап 3. Соединение 8
Соединение 7 (23 мг, 0,045 ммоль, 1,0 экв.) растворяли в ДМФА (2 мл), затем последовательно добавляли соединение 4 (18 мг, 0,090 ммоль, 2,0 экв.), HATU (21 мг, 0,054 ммоль, 1,2 экв.) и ДИПЭА (12 мг, 0,090 ммоль, 2 экв.). Реакционный раствор перемешивали при комнатной температуре в течение 16 ч и затем вливали в воду (20 мл). Смесь экстрагировали этилацетатом (10 мл × 2), органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а сырой продукт, полученный после концентрирования, очищали методом препаративной тонкослойной хроматографии (дихлорметан/метанол=10:1) для получения соединения 8(15 мг, 47,9%).
ЖХ/МС: [М+Н]+=695,4
Этап 4. Соединение Т
Соединение 8(15 мг, 0,022 ммоль, 1 экв.) растворяли в дихлорметане (4 мл) и затем добавляли трифторуксусную кислоту (1 мл). Реакционный раствор перемешивали при комнатной температуре в течение 3 ч и затем добавляли насыщенный раствор натрия бикарбоната для корректировки уровня рН до 7-8. Смесь экстрагировали этилацетатом (10 мл × 2), органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а сырой продукт, полученный после концентрирования, очищали методом ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) для получения соединения Т (2,0 мг, 17,0%) в виде твердого вещества белого цвета.
ЖХ/МС: [М+Н]=545,3
1Н ЯМР (400 МГц, CDCl3) δ 8,66 (синглет, 1H), 7,84 (синглет, 1H), 7,25-7,21 (мультиплет, 2Н), 6,93 (дублет, J=8,0 Гц, 2Н), 6,87 (триплет, J=8,0 Гц, 1H), 6,59 (синглет, 1H), 5,43-5,26 (мультиплет, 2Н), 4,08 (синглет, 2Н), 3,99 (синглет, 2Н), 3,89-3,82 (мультиплет, 1Н), 3,75-3,69 (мультиплет, 1H), 3,20-3,00 (мультиплет, 4Н), 2,09 (триплет, J=8,0 Гц, 1Н), 2,00 (синглет, 3Н), 1,96-1,91 (мультиплет, 3Н), 1,74-1,67 (мультиплет, 1Н), 1,46 (синглет, 3Н), 1,40-1,21 (мультиплет, 6Н), 0,90 (триплет, J=4,0 Гц, 3Н).
Пример 20. Получение соединения U
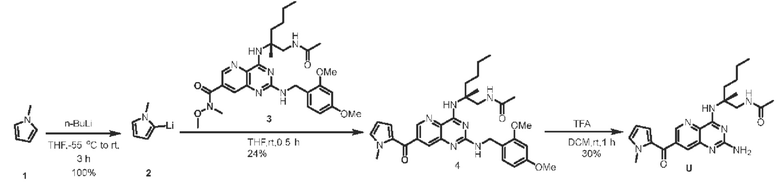
Этап 1. Соединение 2 Соединение 1 (500 мг, 6,164 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (7 мл), охлаждали до температуры -55°С и затем по каплям добавляли раствор п-бутиллития (2,4 М, 2,56 мл, 1,0 экв.). Реакционный раствор перемешивали при температуре -55°С в течение 5 мин, затем температуру повышали до комнатной температуры и перемешивали раствор при комнатной температуре в течение 3 ч для получения раствора промежуточного продукта 2 (0,65 М, 9,5 мл, 6,164 ммоль, 100%) в тетрагидрофуране, который непосредственно использовали для следующей реакции. Этап 2. Соединение 4
Соединение 3 (100 мг, 0,180 ммоль, 1,0 экв.) растворяли в тетрагидрофуране (5 мл), добавляли раствор соединения 2 в тетрагидрофуране (0,65 М, 2,77 мл, 1,806 ммоль, 10,0 экв.) при комнатной температуре и перемешивали реакционный раствор при комнатной температуре в течение 0,5 ч. Реакционный раствор вливания в ледяную воду и затем экстрагировали этилацетатом (3 × 10 мл). Органические фазы объединяли и промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и концентрировали, а остаток очищали методом препаративной тонкослойной хроматографии с силикагелем (дихлорметан/метанол=12/1) для получения соединения 4 (25 мг, 24%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=574,4
Этап 3. Соединение U
Соединение 4 (25 мг, 0,0436 ммоль, 1,0 экв.) растворяли в дихлорметане (0,7 мл), затем добавляли трифторуксусную кислоту (2 мл) и перемешивали реакционный раствор при комнатной температуре в течение 1 ч. Реакционный раствор концентрировали, остаток растворяли в дихлорметане (20 мл), затем последовательно промывали водным раствором натрия карбоната (0,5 М, 20 мл) и насыщенным солевым раствором, сушили надо безводным натрия сульфатом и концентрировали. Полученный сырой продукт очищали методом препаративной ВЭЖХ (0,1% муравьиная кислота/ацетонитрил/вода) для получения соединения U (5,6 мг, 30%) в виде твердого вещества желтого цвета.
ЖХ/МС: [М+Н]+=424,1
1Н ЯМР (400 МГц, CDCl3) δ 8,76 (дублет, J=1,6 Гц, 1H), 8,06 (дублет, J=2,0 Гц, 1H), 7,72 (синглет, 1H), 7,00 (синглет, 1H), 6,73 (дублет, J=4,0 Гц, 1H), 6,48 (синглет, 1Н), 6,20-6,18 (мультиплет, 1Н), 5,99 (широкий синглет, 2Н), 4,06 (синглет, 3Н), 3,90-3,87 (мультиплет, 1Н), 3,71-3,68 (мультиплет, 1H), 2,11-2,05 (мультиплет, 1Н), 2,02 (синглет, 3Н), 1,81-1,75 (мультиплет, 1H), 1,50 (синглет, 3Н), 1,42-1,27 (мультиплет, 4Н), 0,91 (триплет, J=6,8 Гц, 3Н).
Пример 21. Получение соединения V
Этап 1. Соединение 3
Соединение 1 (100 мг, 0,183 ммоль, 1,0 экв.), соединение 2 (200 мг, 0,550 ммоль, 3,0 экв.), Pd(dppf)Cl2 (134 мг, 0,183 ммоль, 1,0 экв.) растворяли в диоксане (20 мл), смесь трижды замещали азотом, затем смесь нагревали до температуры 85°С в азотной атмосфере и перемешивали в течение 16 ч. После охлаждения до комнатной температуры реакционный раствор концентрировали, а остаток разделяли и очищали методом колоночной хроматографии с силикагелем (ПЭ:ЭА=10:1 к ЭА) для получения сырого продукта 3 (200 мг).
ЖХ/МС: [М+Н]+=537,3
Этап 2. Соединение V
Соединение 3 (200 мг, 0,183 ммоль, сырой продукт) растворяли в ДХМ (10 мл), затем по каплям добавляли трифторуксусную кислоту (5 мл) и перемешивали реакционный раствор при комнатной температуре в течение ночи. После завершения реакции реакционный раствор концентрировали, остаток разделяли и очищали методом колоночной хроматографии с силикагелем (ДХМ/метанол=10/1) для получения сырого продукта (100 мг), а сырой продукт очищали методом препаративной ВЭЖХ (0,05% муравьиная кислота/ацетонитрил/вода) для получения соединения V (22,6 мг, общий выход двух этапах составил 34%).
ЖХ/МС: [М+Н]+=359,2
1Н ЯМР (400 МГц, CD3OD) δ 8,93 (синглет, 1H), 8,14 (синглет, 1H), 3,94 (дублет, J=14,0 Гц, 1H), 3,60 (дублет, J=14,0 Гц, 1H), 2,69 (синглет, 3Н), 2,26-2,13 (мультиплет, 1Н), 1,94 (синглет, 3Н), 1,88-1,75 (мультиплет, 1H), 1,49 (синглет, 3Н), 1,42-1,23 (мультиплет, 4Н), 0,90 (триплет, J=6,4 Гц, 3Н).
Пример 22. Скрининговый эксперимент по активности связывания с рецептором in vitro толл-подобного рецептора человека 7 (TLR7) и толл-подобного рецептора человека 8 (TLR8)
1. Цель эксперимента
Способность соединения настоящего изобретения активировать TLR7 и TLR8 соответственно оценивали с помощью экспериментов по связыванию с рецептором in vitro.
2. Принцип эксперимента
Клетки НЕК-Blue™hTLR7 (номер артикула: hkb-hTLR7) и HEK-Blue™hTLR8 (номер артикула: hkb-hTLR8) покупали у компании InvivoGen. Клетки HEK-Blue™hTLR7/8 могут показывать активность TLR7/8 путем определения уровня активации сигнального пути NF-кВ. Клетки HEK-Blue™hTLR7/8 получены из 293 клеток почки эмбриона человека, стабильно экспрессируют TLR7 человека или TLR8 человека соответственно и могут индуцировать экспрессию секреторной щелочной фосфатазы (SEAP) в системе репортерного гена. Экспрессия репортерного гена SEAP регулируется упрощенным промотором IFN-β, который состоит из 5 последовательностей связывания NF-κВ и последовательностей связывания АР-1. Поэтому, когда в этих двух клетках активируется TLR7/8, активируются и NF-κВ и АР-1, и индуцируется экспрессия и внеклеточная секреция SEAP. Уровень активности TLR7/8 может быть показан путем определения активности SEAP вне клетки.
3. Этапы эксперимента
(1) Соединение добавляли на планшет для клеточной культуры после трехкратного градиентного разведения, всего с 10 концентрациями, с двумя лунками для каждой концентрации. В лунки с отрицательным контролем добавляли 0,5 мкл ДМСО на лунку, в лунки с положительным контролем добавляли 0,5 мкл, 2,0 мг/мл (5,7 мМ) R848 (структурная формула:  на лунку;
на лунку;
(2) Клетки HEK-Blue™hTLR7/8 раздельно ферментировали, подсчитывали и в 96-луночный планшет для клеточной культуры, содержащий соединение, добавляли по 5,0 X 104 клеток на лунку;
(3) Клетки, в которые было добавлено соединение, помещали в инкубатор с 5% СО2 при температуре 37°С на 24 ч;
(4) Обнаружение активности соединения: брали 20 мкл/лунка надосадочной жидкости клеточной культуры и добавляли ее в 96-луночный планшет, содержащий 180 мкл реактива QUANTI-BLUE™. Инкубировали при температуре 37°С в течение 1 ч и определяли значение оптической плотности (ОП650) при длине волны 650 нм с помощью многофункционального микропланшетного ридера;
(5) Обнаружение активности клеток: использовали в соответствии с инструкциями к Celltiter-Glo и детектировали хемилюминесцентный сигнал (RLU) с помощью многофункционального микропланшетного ридера; (6) Анализ данных:
Активность соединения: значение ОП650 анализировали с помощью программного обеспечения GraphPad PRism и аппроксимировали кривую доза-эффект соединения для вычисления значения ЕС50 соединения.
Обнаружение активности клеток: активность клеток анализировали с помощью программного обеспечения GraphPad PRism и аппроксимировали кривую доза-эффект соединения для вычисления значения СС50 соединения.
4. Результаты эксперимента:
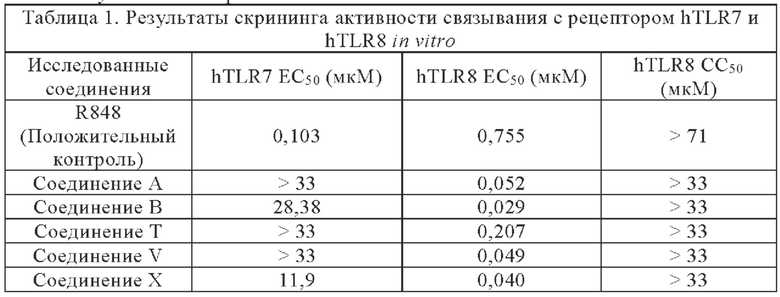
По вышеуказанным результатам эксперимента видно, что соединения настоящего изобретения обладают специфической способностью активации TLR8 в экспериментах in vitro по активности связывания с рецептором TLR7 и TLR8, и цитотоксичность отсутствует. Соединение X известно в литературе, и его структура: 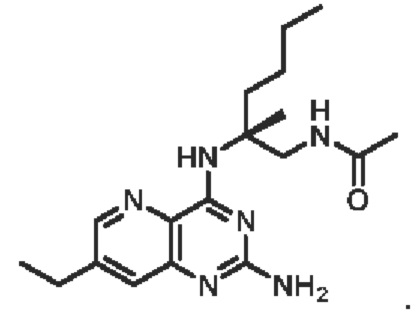 По вышеуказанным результатам теста видно, что соединения А, В и V настоящего изобретения обалдеют большей селективностью активации TLR8, чем соединение X.
По вышеуказанным результатам теста видно, что соединения А, В и V настоящего изобретения обалдеют большей селективностью активации TLR8, чем соединение X.
Пример 23. Эксперимент с мононуклеарными клетками периферической крови человека (РВМС)
1. Цель эксперимента
Для определения индуцирующей активности IL-12p40 соединения согласно настоящему изобретению в мононуклеарных клетках периферической крови человека (РВМСВ) используется эксперимент с РВМС человека и метод ИФА IL-12p40.
2. Принцип эксперимента
Толл-подобные рецепторы (TLR) представляют собой ряд переключателей, регулирующих иммунную систему человека, и играют важную роль как во врожденном (неспецифическом), так и в специфическом иммунитете. TLR8 преимущественно экспрессируют в моноциты, макрофаги и миелоидные дендритные клетки. TLR8 экспрессирует на внутренней оболочке эндосомы клетки и распознает одноцепочечную РНК с высоким содержанием GU (гуанилат/уроновая кислота). После активации сигнального пути TLR8 он активирует секрецию ряда последующих цитокинов (IL-12, TNF-альфа, IFN-гамма и т.д.), вызывая противовирусный иммунный ответ.В этом эксперименте соединение использовали для стимуляции РВМС человека, и определяли уровень белка IL-12p40 в надосадочной жидкости культуры для оценки уровня активации соединения в сигнальном пути TLR8.
3. Этапы эксперимента
(1) Соединение добавляли на планшет для клеточной культуры после трехкратного градиентного разведения, всего с 8 концентрациями, с двумя лунками для каждой концентрации. В лунки с отрицательным контролем добавляли 1 мкл ДМСО на лунку, а в лунки с положительным контролем добавляли 1 мкл, 5,0 мг/мл (14,25 мМ) R848 на лунку;
(2) Замороженные мононуклеарные клетки периферической крови человека помещали на водяную баню при температуре 37°С, после их размораживания переносили в центрифужную пробирку вместимостью 15 мл, постепенно добавляли 10 мл подогретой среды RPMI1640, хорошо перемешивали, центрифугировали при 200 х g в течение 15 мин и удаляли надосадочную жидкость. После ресуспендирования подсчитывали количество клеток и доводили их количество до 2,5 × 106 клеток/мл с помощью питательной среды. Затем в каждую лунку 96-луночного планшета, содержащего соединение, добавляли 200 мкл разведенных клеток (5,0 × 105 клеток/лунка);
(3) Соединения и клетки инкубировали при температуре 37°С в инкубаторе с 5% СО2 в течение 24 ч;
(4) Обнаружение активности соединения: брали 70 мкл/лунка надосадочной жидкости клеточной культуры, использовали в соответствии с инструкцией к набору для ИФА для определения содержания IL-12p40 в надосадочной жидкости и определяли значение оптической плотности для каждой лунки при длине волны 450 нм (ОП450) с помощью многофункционального микропланшетного ридера;
(5) Обнаружение активности клеток: использовали в соответствии с инструкциями к Celltiter-Glo и детектировали хемилюминесцентный сигнал (RLU) с помощью многофункционального микропланшетного ридера;
(6) Анализ данных:
Активность соединения: значение ОП450 анализировали с помощью программного обеспечения GraphPad PRism и аппроксимировали кривую доза-эффект соединения для вычисления значения ЕС50 соединения.
Обнаружение активности клеток: активность клеток анализировали с помощью программного обеспечения GraphPad PRism и аппроксимировали кривую доза-эффект соединения для вычисления значения СС50 соединения.
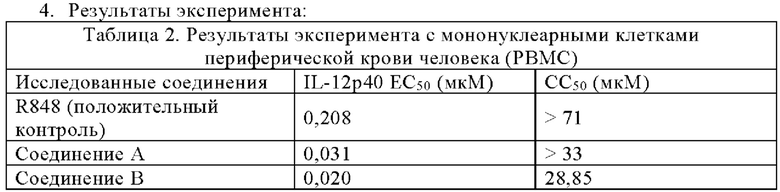
По вышеуказанным результатам эксперимента видно, что соединение настоящего изобретения обладает высокой активностью и безопасностью в тесте на активность индукции IL-12p40 в мононуклеарных клетках периферической крови человека. Значения ЕС50 индуцированной активности соединений А и В на IL-12p40 составили 0,031 мкМ и 0,020 мкМ соответственно. Значения СС50 цитотоксичности соединений А и В для мононуклеарных клеток периферической крови человека были больше 10 мкМ.
Пример 24. Исследование фармакокинетики
1) Цель исследования: получить фармакокинетические характеристики и распределение исследуемого соединения в печени у самцов крыс линии SD.
2) Суть эксперимента
Фармакокинетическое исследование: 6 здоровых самцов крыс линии SD (квалификация SPF) разделили на 2 группы по 3 крысы в группе. После голодания в течение ночи (только для группы перорального введения) соединение А вводили внутривенно с дозировкой 5 мг/кг и перорально с дозировкой 50 мг/кг, брали кровь путем пункции яремной вены в моменты времени 0,083 (только ВВ), 0,25, 0,5, 1, 2, 4, 6 (только ПО), 8 и 24 ч и не менее 0,3 мл цельной крови брали в пробирку с антикоагулянтом ЭДТА-K2. В течение получаса отбирали плазму крови после центрифугирования (6000 об/мин, 8 мин, 4°С) и замораживали при температуре -20°С для дальнейшего использования.
Исследование отношения печень-кровь: 4 здоровых самца крыс линии SD (квалификация SPF) разделили на 2 группы по 2 крысы в группе. После голодания в течение ночи перорально вводили соединение А и соединение X соответственно. Кровь брали путем пункции сердца в моменты времени 1 и 4 ч, не менее 0,5 мл цельной крови брали в пробирку с антикоагулянтом ЭДТА-K2 и в течение получаса отбирали плазму крови после центрифугирования (6000 об/мин, 8 мин, 4°С), и замораживали при температуре -20°С для дальнейшего использования. Одновременно брали ткань печени, промывали физиологическим раствором, затем промокали досуха абсорбирующей бумагой, взвешивали и замораживали при температуре -20°С для дальнейшего использования.
Результаты эксперимента: для вычисления фармакокинетических параметров после введения использовали некомпартментную модель программного обеспечения WinNonlin® 8.1 (Certara, США) согласно полученным данным концентрации лекарственного вещества в крови.
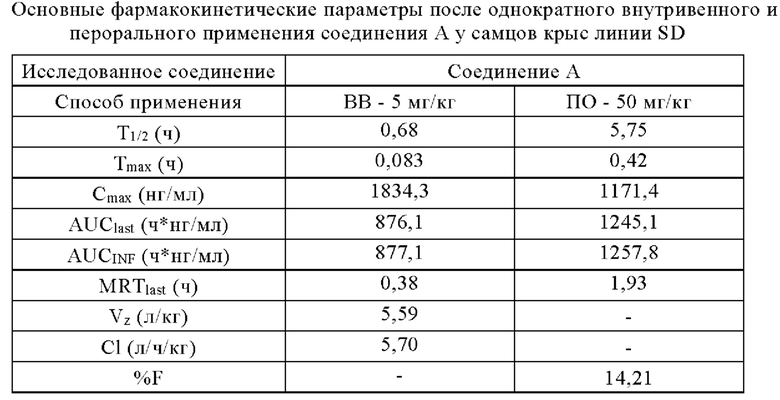

Все литературные источники, упомянутые в настоящем изобретении, включены в настоящий документ в виде ссылок, как если бы они были включены при помощи ссылки по отдельности. Кроме того, следует понимать, что после изучения вышеприведенного описания специалистами в данной области техники могут быть внесены многочисленные изменения и модификации, и такие эквиваленты также попадают в объем, определяемый прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ НЕЙРОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2802457C1 |
| СОЕДИНЕНИЕ, НАЦЕЛЕННОЕ НА БЕЛОК И ЕГО ДЕГРАДАЦИЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2829459C1 |
| СОЕДИНЕНИЕ, ИМЕЮЩЕЕ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ КИНАЗ | 2021 |
|
RU2831403C1 |
| ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ СОЕДИНЕНИЯ НА ОСНОВЕ КЕТОАМИДА | 2021 |
|
RU2819346C1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800153C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ АМИДНОЕ СОЕДИНЕНИЕ, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2823875C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА | 2020 |
|
RU2839897C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ | 2020 |
|
RU2830113C2 |
| ЗАМЕЩЕННЫЕ 2-МЕТИЛИДЕН-5-(ФЕНИЛАМИНО)-2,3-ДИГИДРОТИОФЕН-3-ОНЫ ДЛЯ ЛЕЧЕНИЯ ЛЕЙКОЗОВ С ТРАНСЛОКАЦИЯМИ MLL-ГЕНА И ДРУГИХ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2656603C1 |
Группа изобретений относится к фармацевтической химии и включает 2-аминопиримидиновое производное формулы I, его стереоизомер или фармацевтически приемлемую соль, фармацевтическую композицию и применение. В формуле I X представляет собой N; Y представляет собой NH; Z выбирают из группы, состоящей из -N(H)C(О)R4 и -ОН; R1 и R3 представляют собой водород; R2 выбирают из группы, состоящей из -NR8R9, -CONR10R11, -COR10 и -S(O)2NR10R11; R4, R5 и R6 независимо выбирают из группы, состоящей из C1-С6 алкила, С3-С10 гетероциклоалкила; гетероциклоалкил включает 1, 2 или 3 гетероатома, выбранных из N, О или S; R8 представляет собой С3-С10 циклоалкил; R9, R10 и R11 независимо выбирают из группы, состоящей из водорода, гидроксила, C1-С6 алкила, С3-С10 гетероарила; гетероарил включает 1, 2 или 3 гетероатома, выбранных из N, О или S; алкил, гетероарил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из (C1-С6 алкил)2N-, C1-С6 алкила; или R10 и R11 и N, с которым они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из N или О; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из галогена, гидроксила, (C1-С6 алкил)2N-, C1-С6 алкокси и C6-C12 арила. Технический результат: 2-аминопиримидиновое производное формулы I, обладающее свойствами селективного активатора TLR8. 3 н. и 7 з.п. ф-лы, 2 табл., 22 пр.
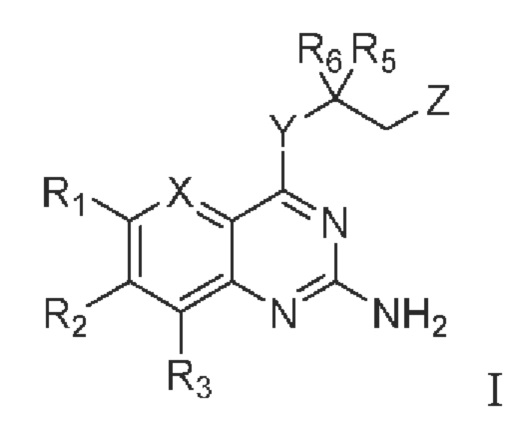
1. Соединение, представленное формулой I, или стереоизомер, или фармацевтически приемлемая соль этого соединения:
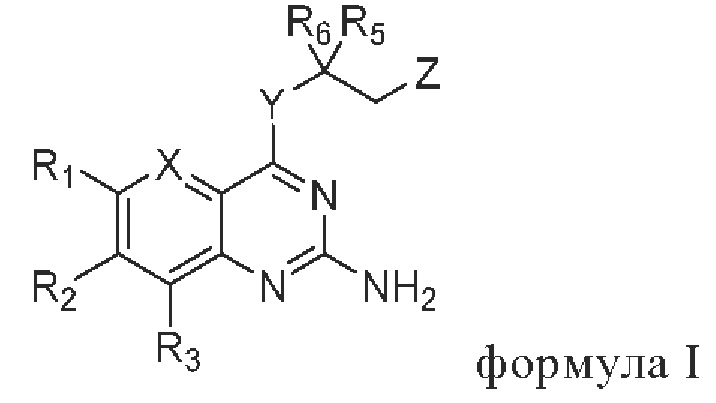 ,
,
где
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(О)R4 и -ОН;
R1 и R3 независимо представляют собой водород;
R2 выбирают из группы, состоящей из -NR8R9, -CONR10R11, -COR10 и -S(O)2NR10R11;
R4, R5 и R6 независимо выбирают из группы, состоящей из C1-С6 алкила, С3-С10 гетероциклоалкила;
R8 представляет собой С3-С10 циклоалкил;
R9, R10 и R11 независимо выбирают из группы, состоящей из водорода, гидроксила, C1-С6 алкила, С3-С10 гетероарила; алкил, гетероарил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из (C1-С6 алкил)2N-, C1-С6 алкила;
или R10 и R11 и N, с которым они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из N или О; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из галогена, гидроксила, (C1-С6 алкил)2N-, C1-С6 алкокси и C6-C12 арила;
и, гетероциклоалкил включает 1, 2 или 3 гетероатома, выбранных из N, О или S;
гетероарил включает 1, 2 или 3 гетероатома, выбранных из N, О или S.
2. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 1, которое характеризуется следующим:
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 представляют собой водород;
R2 выбирают из группы, состоящей из -NR8R9, -CONR10R11, -COR10 и -S(O)2NR10R11;
R4 представляет собой C1-С6 алкил;
R5 и R6 независимо представляют собой C1-С6 алкил соответственно;
R8 представляет собой С3-С10 циклоалкил;
R9 представляет собой Н;
R10 и R11 независимо выбирают из группы, состоящей из водорода, гидроксила, C1-С6 алкила, С3-С10 гетероарила; и алкил, гетероарил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из (C1-С6 алкил)2N- и C1-С6 алкила;
или R10 и R11 и N, с которыми они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из N или О; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из галогена, гидроксила, (C1-С6 алкил)2N-, C1-С6 алкокси.
3. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 2, которое характеризуется следующим:
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 представляют собой водород;
R2 представляет собой -CONR10R11;
R4 представляет собой СН3-;
R5 и R6 независимо представляют собой C1-С6 алкил соответственно;
R10 и R11 независимо выбирают из группы, состоящей из водорода и C1-С6 алкила, и алкил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из (C1-С6 алкил)2N- и C1-С6 алкила;
или R10 и R11 и N, с которыми они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из N или О; и гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из галогена, гидроксила, (C1-С6 алкил)2N- и C1-С6 алкокси.
4. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 3, которое характеризуется следующим:
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 представляют собой водород;
R2 представляет собой -CONR10R11;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 и R11 независимо выбирают из группы, состоящей из водорода и C1-С6 алкила, и алкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из (C1-С6 алкил)2N- и C1-С6 алкила;
или R10 и R11 и N, с которыми они соответствующим образом связаны, образуют С3-С10 гетероциклоалкил, содержащий 1 или 2 гетероатома, выбранных из N или О; гетероциклоалкил необязательно замещен 1-3 заместителями, выбранными из группы, состоящей из галогена, гидроксила, (C1-С6 алкил)2N- и C1-С6 алкокси.
5. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 2, которое характеризуется следующим:
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 представляют собой водород;
R2 представляет собой -COR10;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 выбирают из группы, состоящей из гидроксила, C1-С6 алкила и С3-С10 гетероарила; алкил и гетероарил необязательно замещены 1-3 заместителями, выбранными из группы, состоящей из C1-С6 алкила.
6. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 2, которое характеризуется следующим:
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 представляет собой водород;
R2 представляет собой -COR10;
R4 представляет собой метил;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R10 выбирают из группы, состоящей из C1-С6 алкила.
7. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 2, которое характеризуется следующим:
X представляет собой N;
Y представляет собой NH;
Z выбирают из группы, состоящей из -N(H)C(O)R4 и -ОН;
R1 и R3 представляют собой водород;
R2 представляет собой -NR8R9;
R4 представляет собой СН3-;
R5 представляет собой метил;
R6 представляет собой н-бутил;
R8 представляет собой С3-С10 циклоалкил;
R9 представляет собой водород.
8. Соединение, стереоизомер или фармацевтически приемлемая соль по п. 1, где соединение выбирают из группы, состоящей из следующих соединений:
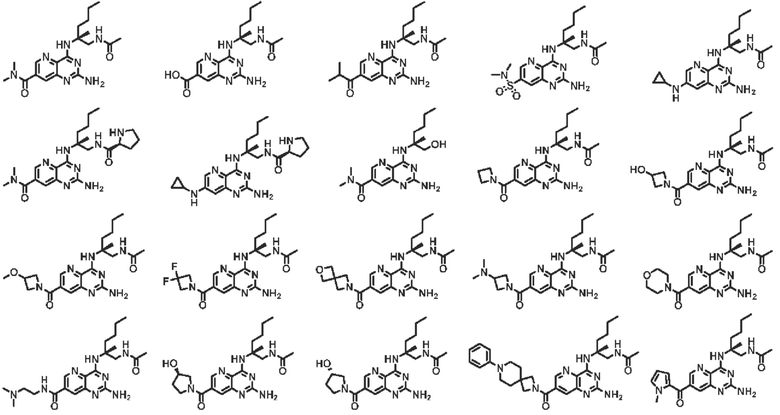
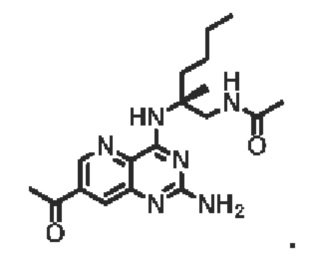
9. Фармацевтическая композиция для активирования TLR8, включающая:
i) одно или более соединений или стереоизомеров, или их фармацевтически приемлемых солей по п. 1; и
ii) фармацевтически приемлемые носители.
10. Применение соединения или стереоизомера, или фармацевтически приемлемой соли по п. 1 как активатора TLR8, отличающееся тем, что их используют для применения, выбранного из группы, состоящей из:
1) приготовления противоопухолевых препаратов, где опухоль выбрана из группы, состоящей из рака легкого, рака печени, рака пищевода, рака молочной железы, рака головного мозга, рака предстательной железы, меланомы и лейкемии;
2) приготовления противовирусных препаратов;
3) приготовления противоинфекционных препаратов;
4) приготовления препаратов для лечения аутоиммунных заболеваний.
| CN 107108615 A, 29.08.2017 | |||
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| CN 103748081 A, 23.04.2014 | |||
| Приспособление для проверки и запиловки передних углов лерки | 1931 |
|
SU27792A1 |
| Ping Yin et al., Synthesis of 2,4-Diaminoquinazolines and Tricyclic Quinazolines by Cascade Reductive Cyclization of Methyl N-Cyano-2-nitrobenzimidates | |||
| J | |||
| Org | |||
| Chem | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |

