Изобретение относится к химико-фармацевтической промышленности, а именно к усовершенствованию способа получения N-(2-хлорэтил)-N'-циклогексил-N-нитрозомочевины (I) (ломустина, также известного как белустина, 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины, CCNU) - противоопухолевого средства алкилирующего типа действия (код ATX L01AD02), со следующей структурной формулой
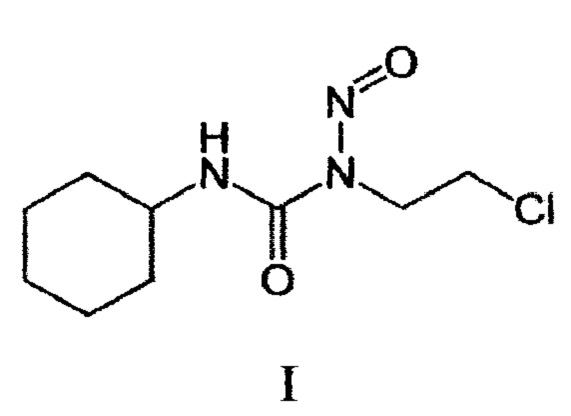
Действие ломустина основано на алкилировании ДНК и РНК, а также ингибировании отдельных этапов синтеза нуклеиновых кислот и репарации отдельных разрывов цепи ДНК, а также торможении ключевых ферментативных процессов путем изменения структуры и функции многих белков и ферментов.
T.J. Thompson, G.S. McCaleb и др. (J. Med. Chem., 1966, 9 (6): 892-911) описали два способа нитрозирования 1-(2-хлорэтил)-3-циклогексилмочевины в среде муравьиной кислоты, отличающихся друг от друга содержанием воды в реакционной смеси. При этом авторами отмечено, что при добавлении воды в реакционную смесь образуется смесь изомеров а (ломустин - X=NO; Y=H) и b (Х=Н; Y=NO) следующей формулы:

В отсутствии воды - образуется чистый изомер а, который является целевым продуктом. Содержание каждого изомеров, установлено методом ЯМР-спектроскопии на ядрах 1H составило для изомера а - 65%, для изомера b - 35%.
Также авторами предложен способ превращения смеси изомеров а и b в целевой ломустин путем обработки последней безводной муравьиной кислотой.
Известен также 3-стадийный способ получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины [J. Org. Chem., т. 46. 12, 2479-2489, 1981 г.).
Химическая схема данного процесса приведена ниже.

Согласно указанному способу, циклогексилизоцианат (II) в течение длительного времени (12 часов) обрабатывают моно этанол амином (III) в среде серного эфира; полупродукт (IV) из реакционной смеси выделяют перекристаллизацией и кипятят в среде хлористого тионила с образованием хлорэтильного производного (V). Данный процесс сопровождается осмолением и полупродукт (V) требует очистки методом колоночной хроматографии с последующей кристаллизацией, что приводит к низкому выходу по данной стадии.
Полупродукт (V) нитрозируют смесью сухого нитрита натрия в среде 98-99% муравьиной кислоты с выходом целевого продукта (ломустина) около 26,7%. Низкий выход ломустина обусловлен образованием побочных продуктов на стадии образования хлорэтильного производного (V), технологических потерь, неизбежных при перекристаллизации и очистки колоночной хроматографии, а также образованием побочного продукта реакции нитрозирования (изомер b). Целевой продукт очищают перекристаллизацией из смеси серного и петролейного эфиров.
Недостатками известного способа являются:
- использование взрывоопасных растворителей - диэтилового и петролейного эфира, что крайне нежелательно для производственных условий;
- большая продолжительность процесса - 12 часов;
- необходимость очистки продукта реакции - 1-(2-гидроксиэтил)-3-циклогексилмочевины, что усложняет технологию, и приводит к увеличению расходных норм сырья;
- кипячение в среде хлористого тионила, приводящее к осмолению, чем обусловлен низкий выход хлорэтильного производного - до 41%;
- сильно загрязненный полупродукт - 1-(2-хлорэтил)-3-циклогексилмочевина требует сложной очистки (колонка с силикагелем с последующей кристаллизацией);
- нитрозирование проводят в практически безводной (98-99%-ной) муравьиной кислоте, что также приводит к увеличению расходных норм сырья;
- образование изомера целевого продукта - 1-(2-хлорэтил)-3-циклогексил-3-нитрозомочевины, превращение которого в целевой продукт не предусмотрено.
Все вышеприведенные недостатки делают описанный процесс технологически сложным, нерентабельным и обуславливают довольно низкий выход целевого продукта - 26,7%.
В качестве прототипа использован способ получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины, защищенный RU 2192413, С07С 275/26, С07С 275/66, С07С 275/68, опубл. 10.11.2002.
Авторы проводили синтез, аналогично вышеуказанным способам; по схеме:

Согласно данному способу, для уменьшения взрывоопасности процесса синтеза на первой стадии, процесс проводят в среде ацетонитрила, использование которого позволило сократить время реакции в 3 раза. Продукт реакции (IV) выпадает из реакционной смеси в виде осадка, который отфильтровывают и без дополнительной очистки направляют на стадию взаимодействия с хлористым тионилом. Для уменьшения осмоления процесс проводят в среде толуола с использованием в качестве катализатора кислоты Льюиса (хлорида цинка). Реакционную смесь выдерживают при 80°С в течение 1 часа, отгоняют остатки хлористого тионила и толуола в вакууме. Полупродукт (V) осаждают из реакционной смеси с помощью воды.
Затем производное (V) нитрозируют смесью нитрита натрия и 85%-ной муравьиной кислоты при охлаждении реакционной смеси не выше 5°С в течение 2 часов. Кристаллический осадок желтого цвета промывают водой и сушат.
С целью трансфера нитрозогруппы из положения 7 в положение 5 и получения целевого продукта, авторами предложено обрабатывать реакционную смесь на заключительной стадии концентрированной соляной кислотой в течение 1 часа.
Технический ломустин осаждают водой. Очистку технического продукта проводят перекристаллизацией из смеси ацетон-ацетонитрил. Выход ломустина (I) составляет около 75 г, его качество соответствует требованиям Британской фармакопеи издания 2000 г. (требования аналогичны требованиям Европейской фармакопеи, издание 3): т.пл. 90,0-91,0°С, хлориды - менее 0,05%, количественное содержание 98,85%.
Из литературных данных известно, что хлористый тионил при нагревании выше температуры кипения (75,6°С) разлагается на дитиодихлорид, сернистый газ и хлор (Химическая энциклопедия в пяти томах, авторы: Научно-редакционный совет издательства "Советская энциклопедия", том 4, М, "Советская энциклопедия", 1995). В условиях реакции по способу-прототипу, данное обстоятельство вследствие протекания побочных реакций приводит к осмолению реакционной массы, уменьшению выхода полупродукта (V) и необходимости дополнительных операций по очистке, соответственно, указанные в методе-прототипе выход и чистота продукта представляются существенно завышенными.
Также качество продукта оценено только по трем показателям монографии Британской фармакопеи и, в частности, не подтверждена его подлинность.
Более того, за 20 лет, прошедших с момента опубликования способа-прототипа, появились более точные методы контроля фармацевтических субстанций, а мировые фармакопеи ужесточили требования к качеству субстанции ломустина.
В связи с этим, а также с учетом требований охраны труда на производстве активных фармацевтических субстанций, авторами была поставлена задача усовершенствования и масштабирования лабораторного способа синтеза ломустина для промышленного производства продукта фармацевтического качества, удовлетворяющего требованиям Европейской Фармакопеи издания 10.0 и Государственной Фармакопеи издания XIV (микробиологическая чистота) в килограммовом масштабе.
Настоящее изобретение направлено на достижение технического результата, заключающегося в упрощении способа получения ломустина с целью повышения безопасности синтеза и повышения выхода конечного продукта за счет устранения осмоления и замены высококипящего толуола на хлорированные углеводороды и хлорида цинка на мягкое органическое основание.
Указанный технический результат достигается тем, что способ получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины (I), заключается в том, что к охлажденному раствору моно этанол амина (III) в ацетонитриле, капельно добавляют циклогексилизоцианат (II) и перемешивают полученный состав в течение 4 час при температуре 0-5°С, выделившийся осадок отфильтровывают, промывают ацетонитрилом и после сушки в вакууме при 20°С и минимальном давлении 12 мБар, растворяют в подходящем хлорированном углеводороде с последующим охлаждением и добавлением мягкого органического основания и хлористого тионила; выдержка реакционной смеси при нагревании не более 50-60°С и последующая ее обработка дает на выходе хлорэтильное производное (V) с выходом до 75% без осмоления реакционной смеси и необходимости очистки целевого продукта, затем, для первого варианта исполнения:
- хлорэтильное производное (V) растворяют в 85%-ной муравьиной кислоте при охлаждении до (-5±2)°С, прибавляют нитрит натрия, полученную реакционную смесь при температуре до (-5±2)°С выдерживают в течение 2 час и обрабатывают концентрированной соляной кислоты в течение 1 часа с последующим осаждением конечного продукта из воды, фильтрацией и перекристаллизацией из ацетонитрила и финишной сушкой в вакууме при 28°С и минимальном давлении 12 мБар,
или, для второго варианта исполнения:
- хлорэтильное производное (V) растворяют в 98-99%-ной муравьиной кислоте при охлаждении до 0±5°С, прибавляют нитрит натрия, полученную реакционную смесь перемешивают в течение 30 мин., затем разбавляют холодной воды и перемешивают еще 30 мин, а выпавший осадок отфильтровывают и сушат в вакууме при 28°С и минимальном давлении 12 мБар.
Указанные признаки являются существенными и взаимосвязаны с образованием устойчивой совокупности существенных признаков, достаточной для получения требуемого технического результата.
Согласно настоящему изобретению, рассматривается новый способ промышленного синтеза ломустина (I), который осуществляется по следующей схеме: взаимодействием циклогексилизоцианата (II) и моноэтаноламина (III) в ацетонитриле получают 1-(2-гидрокси)-3-циклогексилмочевину (IV), которую после выделения в чистом виде, обрабатывают хлористым тионилом в присутствии органического основания в мягких условиях. Полученную 1-(2-хлорэтил)-3-циклогексилмочевину (V) после перекристаллизации нитрозируют смесью нитрита натрия и муравьиной кислоты как с добавлением воды в реакционную смесь, так и без нее. В первом случае образуется смесь изомеров: ломустин (I) и 1-циклогексил-3-(2-хлорэтил)-3-1-нитрозомочевина, последнюю изомеризуют в ломустин в сильнокислой среде; во втором случае образуется ломустин. Целевой продукт очищают перекристаллизацией из ацетонитрила.
В части получения ломустина используют один из двух альтернативных приемов, а именно:
- по первому приему получения: осадок растворяют в 85%-ной муравьиной кислоте при охлаждении до (-5±2)°С, прибавляют нитрит натрия, полученную реакционную смесь при температуре до (-5±2)°С выдерживают в течение 2 час и обрабатывают концентрированной соляной кислоты в течение 1 час с последующим осаждением конечного продукта из воды, фильтрацией и перекристаллизацией из ацетонитрила и сушкой в вакууме при 28°С и минимальном давлении 12 мБар;
- по второму приему получения: осадок растворяют в 98-99%-ной муравьиной кислоте при охлаждении до 0±5°С, прибавляют нитрит натрия, полученную реакционную смесь перемешивают в течение 30 мин., затем разбавляют холодной водой и перемешивают еще 30 мин, а выпавший осадок отфильтровывают и сушат в вакууме при 28°С и минимальном давлении 12 мБар.
Указанный способ получения ломустина позволяет получить продукт фармакопейного качества стабильный, как минимум, в течение 24 мес.
Таким образом, технический результат достигается тем, что на стадии получения 1-(2-хлорэтил)-3-циклогексилмочевины (V) высококипящий толуол заменен на хлорированные производные алканов с целевой температурой кипения 50-70°С; в качестве катализатора были использованы мягкие органические основания, такие как пиридин, диметилформамид, что позволило снизить температуру проведения реакции до 50-60°С, и провести процесс в более мягких условиях с большим выходом.
Нитрозирование хлорпроизводного (V) проводили согласно способа-прототипа (метод А) и описанному в работе (1) безводному способу (метод Б).
Перекристаллизацию технического ломустина проводили из чистого ацетонитрила. Газообразные побочные продукты реакции поглощаются раствором гидроксида натрия в воде.
Структура целевого ломустина подтверждена методом спектроскопии ядерного магнитного резонанса на ядрах 1Н и 13С и 15N. Химические сдвиги (δ, м.д.) атомов вещества молекулы ломустина в ЯМР-спектрах указаны в таблице 1, в качестве растворителя использовался d-DMSO.


где:
s - синглет, d - дублет, t - триплет, qd - квадруплет дублетов, qt - квадруплет триплетов, m - мультиплет, верхний индекс - номер атома в формуле ломустина.
Нижеследующие примеры характеризуют предлагаемое изобретение, но не ограничивают его.
Пример 1. Синтез 1-(2-гидроксиэтил)-3-циклогексилмочевины (IV)
К раствору 772 г моноэтаноламина (III) в ацетонитриле, охлажденному до температуры (-2 -0)°С, прикапывают 1540 г циклогексилизоцианата (II), перемешивают 4 часа при температуре 0-5°С. Выделившийся осадок отфильтровывают, промывают ацетонитрилом и сушат в вакууме при 20°С и минимальном давлении 12 мБар.
Получают белый кристаллический продукт с температурой плавления 96-99°С. Выход: 2150 г (93,4%).
Пример 2. Синтез 1-(2-хлорэтил)-3-циклогексилмочевины (V)
К раствору 2140 г 1-(2-гидроксиэтил)-3-циклогексилмочевины (IV) в трихлорметане прибавляют 240 мл пиридина, затем при температуре (-10±2)°С прикапывают 2640 мл тионилхлорида. Реакционную смесь выдерживают при температуре 50-60°С в течение 2 часов, затем упаривают под вакуумом на роторном испарителе до прекращения выделения погона. Остаток в колбе обрабатывают 5 л воды, выделившийся осадок фильтруют. Остаток на фильтре сушат в вакууме при 35°С и минимальном давлении 12 мБар, затем перекристаллизовывают из этилацетата.
Получают белый кристаллический продукт с температурой плавления 114-117°С. Выход: 1800 г (75,5%)
Пример 3. Синтез ломустина (I)
Метод А. К раствору 750 г 1-(2-хлорэтил)-3-циклогексилмочевины (V) в 85%-ной муравьиной кислоты при охлаждении до (-5±2)°С прибавляют 750 г нитрита натрия, реакционную смесь при той же температуре выдерживают в течение 2 часов, затем обрабатывают 1 л концентрированной соляной кислоты в течение 1 часа. Целевой продукт осаждают из воды. Осадок фильтруют, перекристаллизовывают из ацетонитрила и сушат в вакууме при 28°С и минимальном давлении 12 мБар. Выход: 754 г (88,1%).
Метод Б. К раствору 1000 г 1-(2-хлорэтил)-3-циклогексилмочевины (V) в 15 л 98-99%-ной муравьиной кислоты при охлаждении до 0±5°С прибавляют 1000 г нитрита натрия, реакционную смесь перемешивают в течение 30 мин., затем разбавляют 15 л холодной воды и перемешивают еще 30 мин. Выпавший осадок отфильтровывают и сушат в вакууме при 28°С и минимальном давлении 12 мБар.
Выход: 1023 г (89,5%)
В обоих случаях получают желтый кристаллический порошок, по физико-химическим показателям удовлетворяющий требованиям Европейской фармакопеи (издание 10). Микробиологическая чистота субстанции соответствует требованиям Фармакопеи ЕАЭС к субстанциям, использующимся в производстве нестерильных лекарственных форм.
Преимуществами предлагаемого способа являются:
- подобраны условия синтеза 1-(2-хлорэтил)-3-циклогексилмочевины (V), позволяющие избежать осмоления и увеличить выход реакции: высококипящий толуол заменен хлорированные углеводороды и хлорид цинка заменен на мягкое органическое основание;
- проведено масштабирование синтеза до 1 кг;
- с целью выполнения требований ОТ и ПБ подобраны условия нейтрализации побочных газообразных продуктов реакции;
- получен продукт, удовлетворяющий требованиям современных фармакопеи, который может быть использован в производстве нестерильных лекарственных форм; подтверждена его структура;
- разработаны и провалидированы нефармакопейные методы контроля качества субстанции; для фармакопейных методов проведена их верификация;
- субстанция, полученная предложенным методом стабильна не менее 24 мес.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1-(2-ХЛОРЭТИЛ)-3-ЦИКЛОГЕКСИЛ-1-НИТРОЗОМОЧЕВИНЫ | 2001 |
|
RU2192413C1 |
| Способ получения N-нитрозо-N-(бэта-хлорэтил)-карбамоилпептидов или их кислотно-аддитивных солей | 1982 |
|
SU1424739A3 |
| Способ получения производных N-фенил-N-(4-пиперидинил)амида или их солей | 1978 |
|
SU867304A3 |
| Способ получения N-галогеналкил-N-нитрозокарбамоилазида | 1979 |
|
SU973019A3 |
| Способ получения -галогеноалкил -нитрозокарбаматов или галогеноалкил- -нитрозоаллофанатов стероидных соединений | 1974 |
|
SU512714A3 |
| Способ получения водорастворимых производ-НыХ НиТРОзОМОчЕВиНы | 1977 |
|
SU849999A3 |
| СПОСОБ СИНТЕЗА 2-[3-(2-ХЛОРЭТИЛ)-3-НИТРОЗОУРЕИДО]-1,3-ПРОПАНДИОЛА, ОБЛАДАЮЩЕГО ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2018 |
|
RU2678846C1 |
| Способ получения 1,3-дизамещенных нитрозомочевин | 1977 |
|
SU963464A3 |
| ВСЕСОЮЗНАЯ _ nf,-;mm\.:^':'^V-ff^Mi(b-HStiiib*. •;;.^. ,».• г';!*%-!'Г-'-'А fc^••;DJ•.^•^••-'• >& '-'-'2^ | 1972 |
|
SU341234A1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИМЕТРИДАЗОЛА | 2013 |
|
RU2528025C1 |
Изобретение относится к химико-фармацевтической промышленности, а именно к способу получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины (ломустина). Способ, заключающийся в том, что к охлажденному до температуры (-2-0)°С раствору моноэтаноламина в ацетонитриле капельно добавляют циклогексилизоцианат и перемешивают полученный состав в течение 4 часов при температуре 0-5°С, выделившийся осадок отфильтровывают, промывают ацетонитрилом и после сушки в вакууме при 20°С и минимальном давлении 12 мБар, растворяют в хлорированном углеводороде с температурой кипения 50-70°C с последующим охлаждением, добавлением мягкого органического основания и хлористого тионила, выдержкой реакционной смеси при нагревании не более 50-60°C с получением хлорэтильного производного. Затем хлорэтилъное производное растворяют в 85%-ной муравьиной кислотой при охлаждении до (-5±2)°С, прибавляют нитрит натрия, полученную реакционную смесь при температуре до (-5+2)°С выдерживают в течение 2 часов и обрабатывают концентрированной соляной кислоты в течение 1 часа с последующим осаждением конечного продукта из воды, фильтрацией и перекристаллизацией из ацетонитрила и финишной сушкой в вакууме при 28°С и минимальном давлении 12 мБар, или хлорэтилъное производное растворяют в 98-99%-ной муравьиной кислотой при охлаждении до 0±5°С, прибавляют нитрит натрия, полученную реакционную смесь перемешивают в течение 30 мин, затем прибавляют холодную воду и перемешивают еще 30 мин, а выпавший осадок отфильтровывают и сушат в вакууме при 28°С и минимальном давлении 12 мБар. Технический результат заключается в упрощении способа получения ломустина с целью повышения безопасности синтеза и повышения выхода конечного продукта. 1 табл., 3 пр.
Способ получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины, заключающийся в том, что к охлажденному до температуры (-2-0)°С раствору моноэтаноламина в ацетонитриле капельно добавляют циклогексилизоцианат и перемешивают полученный состав в течение 4 часов при температуре 0-5°С, выделившийся осадок отфильтровывают, промывают ацетонитрилом и после сушки в вакууме при 20°С и минимальном давлении 12 мБар, растворяют в хлорированном углеводороде с температурой кипения 50-70°C с последующим охлаждением, добавлением мягкого органического основания и хлористого тионила, выдержкой реакционной смеси при нагревании не более 50-60°C с получением хлорэтильного производного, затем хлорэтилъное производное растворяют в 85%-ной муравьиной кислотой при охлаждении до (-5±2)°С, прибавляют нитрит натрия, полученную реакционную смесь при температуре до (-5+2)°С выдерживают в течение 2 часов и обрабатывают концентрированной соляной кислотой в течение 1 часа с последующим осаждением конечного продукта из воды, фильтрацией и перекристаллизацией из ацетонитрила и финишной сушкой в вакууме при 28°С и минимальном давлении 12 мБар, или хлорэтильное производное растворяют в 98-99%-ной муравьиной кислоте при охлаждении до 0±5°С, прибавляют нитрит натрия, полученную реакционную смесь перемешивают в течение 30 мин, затем прибавляют холодную воду и перемешивают еще 30 мин, а выпавший осадок отфильтровывают и сушат в вакууме при 28°С и минимальном давлении 12 мБар.
| СПОСОБ ПОЛУЧЕНИЯ 1-(2-ХЛОРЭТИЛ)-3-ЦИКЛОГЕКСИЛ-1-НИТРОЗОМОЧЕВИНЫ | 2001 |
|
RU2192413C1 |
| JOHNSTON T | |||
| et al., The Synthesis of Potential Anticancer Agents | |||
| XXXVI | |||
| N-Nirtosoureas | |||
| II | |||
| Haloalkyl Derivatives, Journal of Medicinal Chemistry, 1966, v | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Наборный фрезер для дерева | 1922 |
|
SU892A1 |
| LOWN J.W | |||
| et al., Synthesis and Reactions of Deuterated 2-(Alkylimino)-3-nitrosooxazolidines, 3-Alkyl-1-(2-hydroxyethyl)-1-nitrosoureas, and Related | |||
