Область техники
[0001] Настоящее изобретение относится к новым соединениям, способным ингибировать определенные ферменты аминоксидазы. Эти соединения полезны для лечения множества показаний, например, фиброза, рака и/или ангиогенеза у людей, а также у домашних животных и скота. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим эти соединения, а также к их различным применениям.
Уровень техники
[0002] Ферменты представляют собой лизилоксидазу (LOX), первый описанный член семейства и LOX-like1 (LOXL1), LOXL2, LOXL3 и LOXL4 (J Cell Biochem 2003; 88: 660 - 672). Изоферменты лизилоксидазы представляют собой медьзависимые аминоксидазы, которые инициируют ковалентное поперечное сшивание коллагена и эластина. Основная функция изоферментов лизилоксидазы заключается в облегчении поперечного сшивания коллагена и эластина путем окислительного дезаминирования аминокислотных боковых цепей лизина и гидроксилизина до альдегидов, которые спонтанно реагируют с соседними остатками. Полученные поперечно сшитые нити способствуют стабильности внеклеточного матрикса (ВКМ) и делают его менее восприимчивым к протеолитической деградации ферментами, такими как матриксные металлопротеиназы (ММП). Активность ферментов лизилоксидазы имеет решающее значение для поддержания нормальных свойств растяжения и эластичности соединительной ткани многих систем органов тела.
[0003] Изоферменты лизилоксидазы принадлежат к более широкой группе аминоксидаз, которая включает флавин-зависимые и медь-зависимые оксидазы, которые описываются природой каталитического кофактора. Флавин-зависимые ферменты включают моноаминоксидазу-A (MAO-A), моноаминоксидазу-B (MAO-B), полиаминоксидазу и лизиндеметилазу (LSD1) и медь-зависимые ферменты включают семикарбазид-чувствительную аминоксидазу (адгезивный сосудистый белок-1, SSAO/VAP-1), ретинальную аминоксидазу, диаминоксидазу и изоферменты лизилоксидазы. Медь-зависимые аминоксидазы имеют второй кофактор, который слегка варьируется от фермента к ферменту. В SSAO/VAP-1 это окисленный остаток тирозина (TPQ, окисленный до хинона), тогда как в изоферментах лизилоксидазы TPQ дополнительно подвергается процессингу путем добавления соседнего остатка лизина (с образованием LTQ) (J Cell Biochem 2003; 88: 660 - 672).
[0004] Изоферменты лизилоксидазы проявляют различные паттерны экспрессии in vivo, что предполагает, что определенные изоферменты будут выполнять определенные биологические роли. Каталитически активные формы LOX были идентифицированы в цитозольном и ядерном компартментах, и в настоящее время проводятся исследования для определения их роли в этих компартментах. Сама LOX, например, играет важную роль в эпителиально-мезенхимальном переходе (ЭМП), миграции клеток, адгезии, трансформации и регуляции генов. Различные паттерны экспрессии/активности LOX были связаны с различными патологическими процессами, включая фиброзные заболевания, болезнь Альцгеймера и другие нейродегенеративные процессы, а также с прогрессированием опухоли и метастазированием (Am J Surg 2005; 189: 297 - 301).
[0005] Прямая замена мертвых или поврежденных клеток соединительной тканью после травмы представляет собой механизм выживания, который сохраняется на протяжении всей эволюции и, по-видимому, наиболее выражен у людей, играя ценную роль после травматического повреждения, инфекции или заболеваний. Прогрессирующее рубцевание может возникнуть после более хронических и/или повторяющихся травм, которые вызывают нарушение функции частей или всего пораженного органа. Различные причины, такие как хронические инфекции, хроническое воздействие алкоголя и других токсинов, аутоиммунные и аллергические реакции или хирургическое вмешательство, радио- и химиотерапия, могут привести к фиброзу. Таким образом, этот патологический процесс может возникать практически в любом органе или ткани тела и, как правило, является результатом ситуаций, длящихся в течение нескольких недель или месяцев, в которых одновременно происходят воспаление, разрушение и восстановление тканей. В этом случае фиброз чаще всего поражает легкие, печень, кожу, почки и сердечнососудистую систему.
[0006] Фиброз печени, например, может возникать как осложнение гемохроматоза, болезни Вильсона, алкоголизма, шистосомоза, вирусного гепатита, непроходимости желчных протоков, воздействия токсинов и метаболических нарушений. Фиброз печени характеризуется накоплением внеклеточного матрикса, который можно качественно отличить от такового в нормальной печени. Этот фиброз может прогрессировать до цирроза, печеночной недостаточности, рака и, в конечном итоге, смерти (Pathology – Research and Practice 1994; 190: 910 - 919).
[0007] Фиброзные ткани могут накапливаться в сердце и кровеносных сосудах в результате гипертонии, гипертонической болезни сердца, атеросклероза и инфаркта миокарда, когда накопление внеклеточного матрикса или фиброзные отложения приводят к жесткости сосудистой сети и жесткости самой сердечной ткани (Am J Physiol Heart Circ Physiol 2010; 299: H1 - H9).
[0008] Легочная артериальная гипертензия (ЛАГ) - редкое и быстро приводящее к летальному состоянию заболевание, характеризующееся повышенным легочным артериальным давлением и вызванное повышенным сопротивлением легочных сосудов. Несмотря на то, что это гетерогенное состояние с широким спектром причин, все чаще признается, что ЛАГ связана с другими заболеваниями, такими как заболевание соединительной ткани и склеродермия. Патологические признаки ЛАГ включают ремоделирование сосудистой стенки с избыточным отложением внеклеточного матрикса (ВКМ) и перекрестное сшивание. Лизилоксидазы нарушают регуляцию легочной сосудистой сети у пациентов с идиопатической легочной артериальной гипертензией (ИЛАГ) и способствуют сохранению компонентов ВКМ и неправильному ремоделированию коллагена и эластина посредством перекрестного сшивания (Arterioscler. Thromb Vasc. Biol. 2014; 34: 1446 - 1458). Прогноз для пациентов с ЛАГ плохой. Фармакологическое воздействие на лизилоксидазы может обеспечить терапевтическое вмешательство там, где в настоящее время его мало или совсем нет.
[0009] Была продемонстрирована сильная связь между фиброзом и повышенной активностью лизилоксидазы. Например, в экспериментальном фиброзе печени у крыс (Proc. Natl. Acad. Sci. USA 1978; 75: 2945 - 2949), в моделях фиброза легких (J Pharmacol Exp Ther 1981; 219: 675 - 678), при артериальном фиброзе. (Arteriosclerosis 1981; 1: 287 - 291), при фиброзе кожи (Br J Dermatol 1995; 133: 710 - 715) и при индуцированном адриамицином фиброзе почек у крыс (Nephron 1997; 76: 192 - 200). Из этих экспериментальных моделей заболеваний человека наиболее поразительное увеличение активности ферментов было замечено на крысиной модели фиброза печени, индуцированного CCl4. В этих исследованиях низкий уровень активности ферментов в здоровой печени увеличивался в 15-30 раз в фиброзной печени.
[0010] У людей также существует значительная связь между активностью лизилоксидазы, измеренной в плазме, и прогрессированием фиброза печени. Уровень активности лизилоксидазы обычно низкий в сыворотке здоровых людей, но значительно повышается при хроническом активном гепатите и даже в большей степени при циррозе. Следовательно, лизилоксидаза может служить маркером внутреннего фиброза.
[0011] Изоферменты лизилоксидазы в высокой степени регулируются индуцируемым гипоксией фактором 1α (ГИФ-1α) и TGF-β, двумя наиболее известными факторами роста, вызывающими фиброз (Cell Biol 2009; 29: 4467 - 4483). Поперечное сшивание коллагена происходит при любом типе фиброза, поэтому ингибитор изофермента лизилоксидазы можно использовать при идиопатическом фиброзе легких, склеродермии, фиброзе почек или печени.
[0012] При нормальном заживлении ран образование грануляционной ткани - это кратковременный процесс, обеспечивающий основу для реэпителизации и восстановления. Впоследствии ткань ремоделируется и образуется нормотрофический рубец. Однако после травмы человек не может восстановить нормальную кожу. Вместо этого процесс восстановления (или заживления) приводит к образованию рубцов (рубцеванию). Шрамы эстетически и функционально уступают коже. Шрамы являются хронической проблемой, и чрезмерное или гипертрофическое рубцевание и сопровождающие его эстетические, функциональные и психологические последствия остаются ключевыми проблемами при лечении глубоких повреждений кожи и ожогов. Ключевым фактором плохого внешнего вида и гибкости рубцов, в особенности гипертрофических, являются изменения коллагена в дермальном слое. В рубцовой ткани коллаген (преимущественно Коллаген I) более плотно упакован и выровнен в параллельные пучки. В нормальной коже коллаген не упакован плотно и имеет структуру типа «плетение корзины». Эти изменения, как в структуре, так и в количестве коллагена, в значительной степени лежат в основе плохого внешнего вида рубца и приводят к потере гибкости, дискомфорту и функциональным проблемам.
[0013] Дермальный фиброз или чрезмерное рубцевание кожи является следствием чрезмерной реакции заживления и характеризуется непропорциональной пролиферацией фибробластов и образованием внеклеточного матрикса (ВКМ) в дерме. Клинически фиброз кожи проявляется в виде утолщенных, стянутых и затвердевших участков кожи. Спектр фиброзных кожных заболеваний широк, включая, помимо прочего: гипертрофические рубцы, келоиды, склеродермию (диффузные и ограниченные подтипы), склередему (болезнь Бушке), системный амилоидоз, липодерматосклероз, прогероидные расстройства, синдром жесткой кожи, контрактуру Дюпюитрена, нефрогенную фиброзирующую дермопатию (НФД), смешанное заболевание соединительной ткани, склеромикседема, болезнь трансплантат против хозяина (БТПХ) и эозинофильный фасциит. Хотя каждое из этих расстройств имеет свою этиологию и клинические характеристики, все они связаны с чрезмерной выработкой коллагена и измененным ремоделированием коллагена. Одним из возможных механизмов измененного ремоделирования ВКМ является ковалентное перекрестное сшивание. На патогенез кожного фиброза напрямую влияет семейство ферментов LOX (Laboratory investigation 2019; 99: 514 - 527). Экспрессия LOX и LOXL1-4 повышена в фибробластах рубца по сравнению с фибробластами нормальной кожи, при этом LOX и LOXL1 являются доминирующими изоформами, обнаруживаемыми в ткани кожи.
[0014] Исследования включали две комплементарные кожоподобные модели in vitro - эквивалент кожи человека (ЭКч) и самоорганизующиеся стромальные ткани, идентифицировали LOXL4 как ключевую изоформу, опосредующую фиброзные фенотипы, индуцированные TGF-бета (Lab. Invest. 2019; 99: 514 - 527).
[0015] Процессы рубцевания представляют собой серьезную проблему для глаза и окружающих структур. Глазное рубцевание играет важную роль либо в первичном заболевании (например, рубцевание роговицы и конъюнктивы), либо в неэффективности лечения (например, послеоперационная трабекулэктомия) (Ocular Surgery News U. S. Edition, 1 октября 2002 г.).
[0016] Глаукома - это заболевание, при котором повреждается зрительный нерв, что приводит к прогрессирующей и необратимой потере зрения. Повышенное внутриглазное давление (ВГД) - один из основных факторов риска развития и прогрессирования глаукомы. Большинство методов лечения глаукомы нацелены на снижение внутриглазного давления либо за счет уменьшения образования водянистой жидкости в глазу, либо, как в случае фильтрующей хирургии глаукомы, за счет увеличения оттока жидкости из глаза. Трабекулэктомия - текущий золотой стандарт лечения ВГД - представляет собой фильтрующую хирургию, при которой создается отверстие в передней камере из-под частичного склерального лоскута, чтобы обеспечить отток воды из глаза. Послеоперационные рубцы являются основной причиной неудач лечения. Антиметаболиты митомицин-C (MMC) и 5-фторурацил (5-FU) используются в современной клинической практике для ограничения образования послеоперационных рубцов на глазах. Хотя было показано, что эти агенты улучшают результат ВГД при фильтрующей хирургии, они делают это неселективным образом и связаны со значительными побочными эффектами (Arch. Ophthalmol. 2002; 120: 297 - 300). Необходимы более безопасные, более направленные противофиброзные средства.
[0017] Фиброматоз десен - это редкая и разнородная группа заболеваний, которые развиваются как медленно прогрессирующие, локальные или диффузные, фиброзные увеличения ороговевшей десны (разрастание или увеличение десны). В тяжелых случаях избыток ткани может покрывать коронки зубов, вызывая жевательные, эстетические, фонетические, функциональные и пародонтальные проблемы. Разрастание десен может быть унаследованным, иметь идиопатическое происхождение, связанное с воспалительными заболеваниями полости рта или связанное с другими системными заболеваниями. Однако в большинстве случаев это связано с побочными эффектами системных лекарств, таких как противосудорожный препарат фенитоин, иммунодепрессант циклоспорин А и некоторые антигипертензивные дигидропиридиновые блокаторы кальциевых каналов, в частности нифедипин (crit rev oral biol 2004; 15: 165 - 175). Патологическое проявление чрезмерного роста десны включает чрезмерное накопление белков внеклеточного матрикса, из которых Коллаген I является наиболее преобладающим. Одной из признанных концепций механизма избыточного роста десен, вызванного лекарственными средствами, является ЭМП, процесс, в котором взаимодействие десневых клеток и внеклеточного матрикса ослабляется, поскольку эпителиальные клетки трансдифференцируются в фиброгенные фибробластоподобные клетки (AJP 2010; 177: 208 - 218). Поврежденный эпителий, базальная мембрана и подлежащая строма приводят к образованию TGF-β стимуляции активности фермента лизилоксидазы и способствуют фиброзу соединительной ткани (Lab Invest 1999; 79: 1655 - 1667).
[0018] Обоснование последовательного и сильного ингибирования фиброза блокаторами изоферментов лизилоксидазы заключается в том, что недостаток активности поперечного сшивания делает коллаген восприимчивым к деградации протеолитическими ферментами, такими как ММП. Следовательно, любой тип фиброза должен быть обращен лечением ингибиторами изофермента лизилоксидазы. Учитывая различное участие всех изоферментов лизилоксидазы в фиброзе, ингибитор, который демонстрирует устойчивое сильное ингибирование всех изоферментов лизилоксидазы, т.е. пан-ингибитор LOX должен быть наиболее эффективным.
[0019] Ревматоидный артрит (РА) - это системное аутоиммунное заболевание, характеризующееся хроническим болезненным воспалением слизистой оболочки суставов. Однако у некоторых людей состояние может прогрессировать, включая болезненный отек и воспаление окружающей ткани и других систем организма, включая кожу, глаза, легкие, сердце и кровеносные сосуды. Таким образом, ревматоидный артрит является болезненным и изнурительным заболеванием, которое может привести к значительной потере функции и подвижности рук, запястий и стоп. Активный ревматоидный артрит возникает в нескольких суставах, но впоследствии может прогрессировать и поражать несколько суставов. Синовиальная гиперплазия с вовлечением инфильтрированных иммунных клеток и резидентных синовиальных фибробластов (СФ) является типичным признаком РА. Синовиальные фибробласты при ревматоидном артрите (РАСФ) являются наиболее распространенным типом клеток в местах инвазии и являются основным виновником разрушения суставов. Активированные РАСФ способны мигрировать и, как таковые, участвуют в распространении артрита между суставами. Цитокины инфильтрированных иммунных клеток индуцируют активацию и пролиферацию синовиальных фибробластов. Эти активированные СФ, в свою очередь, генерируют патогенную строму для сохранения хронического воспаления, что в конечном итоге приводит к разрушению хрящей и костей. Путем имплантации РАСФ вместе с человеческим хрящом мышам с тяжелым комбинированным иммунодефицитом было продемонстрировано, что активированные РАСФ мигрируют in vivo, распространяя болезнь на участки имплантированного человеческого хряща. Более того, в то время как РАСФ активно разрушают хрящ, контроли, с имплантированными синовиальными фибробластами от пациентов с остеоартритом (ОА), и кожными фибробластами от здоровых доноров этого не сделали (Nat. Med. 2009; 15: 1414 - 1420). РАСФ отличаются от неактивированных здоровых фибробластов своей морфологией и экспрессией генов. РАСФ характеризуются экспрессией антиапоптотических, протоонкогенов и недостатком экспрессии генов-супрессоров опухолей. Производство провоспалительных цитокинов и хемокинов с помощью РАСФ дополнительно способствует привлечению иммунных клеток к синовиальной оболочке. Кроме того, производство ферментов матриксной металлопротеиназы (ММП) способствует инвазии и разрушению хряща.
[0020] Модель коллаген-индуцированного артрита (КИА) типа II является широко используемой животной моделью для РА, поскольку она хорошо воспроизводит характерные иммунологические, патологические и артритные проявления, наблюдаемые при РА у людей. Крысы КИА демонстрировали высокие уровни экспрессии LOX в синовиальных оболочках, синовиальной жидкости и сыворотке. Было обнаружено, что ингибирование LOX бета-аминопропионитрилом (BAPN; пан ингибитор LOX) ослабляет воспаление, синовиальную гиперплазию, ангиогенез и экспрессию MMП-2 и MMП-9, что указывает на то, что LOX способствует синовиальной гиперплазии и ангиогенезу у крыс КИА. Кроме того, нокдаун LOXL2 и антител против LOXL2 ослабляет отложение коллагена, пролиферацию и инвазию РАСФ (Mol. Med. Rep. 2017: 6736 - 6742).
[0021] Хотя лечения РА не существует, существует ряд способов лечения, облегчающих симптомы и изменяющих прогрессирование заболевания. Однако такое лечение сопровождается значительными побочными эффектами, отчасти связанными с супрессией иммунной системы. Селективные препараты, нацеленные на РАСФ, представляют собой более полезную терапию для РА.
[0022] Остеоартрит (ОА) - это заболевание, характеризующееся дегенерацией суставного хряща и подлежащей кости. В основном в результате «износа», ОА вызывает боль и жесткость сустава. Наиболее часто поражаются суставы пальцев, коленей, спины и бедер. В отличие от других форм артрита (например, РА), остеоартрит поражает только суставы. Часто суставы на одной стороне тела поражаются сильнее, чем на другой. ОА - это прогрессирующее и изнурительное заболевание, которое может существенно повлиять на работу и повседневную деятельность.
[0023] Синовиальный фиброз является ключевым фактором ОА и является проявлением пролиферации фибробластов и дисбаланса синтеза коллагена и деградации коллагена. Этот дисбаланс приводит к чрезмерному отложению коллагена во внеклеточном матриксе (ВКМ) и приводит к утолщению и жесткости синовиальной мембраны.
[0024] Было показано, что гены, кодирующие ряд ферментов семейства лизилоксидаз, включая LOX, LOXL2, LOXL3 и LOXL4, высоко экспрессируются у мышей с экспериментальным ОА и у людей с конечной стадией ОА (Arthritis and Rheumatology 2014; 66: 647 - 656).
[0025] Учитывая различный вклад многих членов семейства ферментов лизилоксидазы в развитие как ревматоидного артрита, так и остеоартрита, пан-ингибитор LOX может обеспечить потенциально более эффективную терапию.
[0026] BAPN является широко используемым необратимым ингибитором лизилоксидазы на основе неселективного механизма. С 1960-х годов BAPN использовался в исследованиях на животных (в основном на крысах, мышах и хомяках) и был эффективным в снижении содержания коллагена в различных моделях (например, CCl4, блеомицин, кварц, рак) и тканях (например, печень, легкие и дерма). (J Cell Biochem 2003; 88: 660 - 672). Однако исследования на людях, больных склеродермией, показали, что BAPN плохо переносится и подчеркивает необходимость более безопасных альтернатив (Clin. Pharmacol. Ther. 1967: 593 - 602).
[0027] Поперечное сшивание коллагена катализируемое лизилоксидазой может протекать двумя путями: ализиновый путь и гидроксиализиновый путь. В гидроксиализиновом пути, сначала образуются незрелые двухвалентные поперечные сшивки, включая дегидродигидроксилизинонорлейцин (deH-DHLNL) и дегидрогидроксилизинонорлейцин (deH-HLNL), и затем они дополнительно прогрессируют (через независимые от лизилоксидазы реакции) до зрелых трехвалентных поперечных сшивок между тремя молекулами коллагена с образованием дезоксипиридинолина (DPD) и пиридинолина (PYD). Эти зрелые и незрелые поперечные сшивки можно измерить при помощи ЖХ-МС/МС (PLoS One 2014; 9 (11), e112391).
[0028] Изоферменты лизилоксидазы не только участвуют в поперечном сшивании эластина и коллагена во время заживления ран и фиброза, но также регулируют движение клеток и передачу сигналов. Их внутриклеточная и внутриядерная функция, связанная с генной регуляцией и может привести к онкогенезу и прогрессии опухоли (Inflammapharmacol 2011; 19: 117 - 129). Описаны как понижающая, так и повышающая регуляция изоферментов лизилоксидазы в опухолевых тканях и линиях раковых клеток, что предполагает двойную роль изоферментов лизилоксидазы и пропептида LOX как гена промотора метастазирования, а также гена-супрессора опухоли.
[0029] Помимо своей роли в ремоделировании тканей, изоферменты LOX также играют важную роль в первичном раке и метастазировании. Рост опухоли связан с реактивной стромой, которая преимущественно состоит из фибробластов; называемые фибробласты ассоциированны с раком (РАФ). Известно, что мыши, подкожно инокулированные равной смесью опухолевых и РАФ-клеток, обладают более высокой скоростью роста и более высокой частотой метастазов (Trends Mol Med. 2013; 19(8): 447 - 453). Модели с нокаутом РАФ показали свою проонкогенность, однако это довольно абстрактный сценарий по сравнению с микросредой опухоли пациента. Было показано, что РАФ обладают повышенной экспрессией LOX по сравнению с нормальными фибробластами (Dis Model Mech. 2018; 11 (4)). Использование ингибитора LOX в условиях рака потенциально повлияет как на опухоль, так и на стромальный компартмент, что поможет уменьшить рост опухоли и метастазирование.
[0030] Новые данные свидетельствуют о связи между идиопатическим фиброзом легких и раком легких, однако необходимы дополнительные исследования. Химический или радиационно-индуцированный фиброз на моделях мышей как в легких, так и в печени вызывает увеличение альфа-актина гладких мышц (маркер фибробластов), экспрессию LOX и рост метастатической опухоли, который нейтрализуется антителом LOX (Cancer Res. 2013; 73 (6): 1721 - 1732).
[0031] На сегодняшний день увеличение мРНК и/или белка в изоферментах лизилоксидазы наблюдается в линиях клеток рака груди, ЦНС, сквамозная клеточная карцинома головы и шеи, пищеводе, почках, легких, простате, прозрачной клетке и карциномах легких, яичниках, матке, образцах пациентов с меланомой и остеосаркомой из The Cancer Genome Atlas (TCGA). В Таблице 1 представлены данные об экспрессии гена пациента TCGA для семейства LOX. Знак плюс указывает на то, что экспрессия гена выше среднего в этом наборе данных.
Таблица 1
Данные об экспрессии гена пациента TCGA для семейства LOX
Рак
LOX
LOXL1
LOXL2
LOXL3
LOXL4
[0032] Статистически значимые клинические корреляции между экспрессией изоферментов лизилоксидазы и прогрессированием опухоли наблюдались в сквамозных опухолях груди, головы и шеи, миелофиброзе, простатической, панкреатической, яичниковой и светлоклеточной почечно-клеточной карциномах. Роль изоферментов лизилоксидазы в опухолевой прогрессии была наиболее широко изучена при раке груди с использованием моделей миграции/инвазии in vitro и моделей онкогенеза и метастазирования мышей in vivo (Nature. 2006; 440 (7088): 1222 - 1226). Повышенная экспрессия изоферментов лизилоксидазы была обнаружена у пациентов с гипоксией и была связана с отрицательным статусом рецепторов эстрогена (РЭ-), снижением общей выживаемости у РЭ- пациентов и пациентов без поражений лимфатических узлов, которые не получали адъювантное системное лечение, а также с более короткой свободной от метастазирования в кости выживаемостью у РЭ-пациентов и пациентов без поражений лимфатических узлов; модели in vivo продемонстрировали, что ингибиторы LOX обладают потенциалом у пациентов с раком груди с метастазами в кости путем модуляции гомеостаза кости независимо от RANKL (Nature. 2015; 522 (7554): 106 - 110). Было продемонстрировано, что мРНК изоферментов лизилоксидазы активируется в инвазивных и метастатических клеточных линиях (MDA-MB-231 и Hs578T), а также в более агрессивных клеточных линиях рака груди и отдаленных метастатических тканях по сравнению с тканями первичного рака (Cancer Res. 2002; 62 (15): 4478 - 4483).
[0033] Патогенные процессы при первичном миелофиброзе включают первичную взвешенную на мегакариоциты клональную миелопролиферацию и паранеопластическую стромальную реакцию, которая включает фиброз костного мозга, остеосклероз, ангиогенез и экстрамедуллярный гематопоэз. Реакция костного мозга включает избыточное отложение белков внеклеточного матрикса, таких как фибриллярный коллаген, гипоцеллюлярность, активацию и рекрутирование фибробластов костного мозга, избыточное производство цитокинов и факторов роста и другие изменения, которые приводят к снижению гематопоэтической емкости. Вторичный миелофиброз может быть результатом истинной красной полицитемии или эссенциального тромбоцитоза. При миелофиброзе прогрессирование заболевания коррелирует с увеличением количества мегакариоцитов, которые сверхэкспрессируют LOX. В модели миелофиброза на мышах GATA 1low прогрессирование заболевания (включая увеличение числа мегакариоцитов, фиброза и размера селезенки) было значительно ослаблено пан-ингибитором LOX (J Biol Chem. 2011; 286 (31): 27630 - 27638).
[0034] При большинстве типов опухолей первой линией лечения является хирургическая резекция. Реакция на заживление ран инициируется хирургическим вмешательством и может коррелировать с увеличением метастатического распространения. Модели рака груди показали, что абдоминальная хирургия увеличивает метастазирование в легких. Кроме того, было показано, что это вызвано системной LOX. Инъекция плазмы, собранной у мышей абдоминальной хирургии (которая содержала LOX), мышам с опухолью приводила к увеличению метастазов в легких. Системная LOX, вызванная хирургическим вмешательством, блокировалась BAPN, уменьшая метастазирование и увеличивая выживаемость (Cell Rep. 2017; 19 (4): 774 - 784).
[0035] На моделях рака толстой кишки, рака груди и меланомы было показано, что эндотелиальные клетки, ассоциированные с опухолью, имеют повышенную экспрессию LOX, которая стимулирует ангиогенез и рост опухоли (Cancer Res. 2015; 73 (2): 583 - 594).
[0036] У пациентов с раком поджелудочной железы, груди, легких, яичников и толстой кишки высокое содержание коллагена коррелирует с высокой экспрессией гена LOX, устойчивостью к химиотерапии и значительным снижением выживаемости (Oncogene. 2018; 37(36) 4921 - 4940, EMBO Mol Med. 2015; 7(8) 1063 - 1076, Oncotarget. 2016; 7(22) 32100 - 32112). Ингибиторы LOX (как BAPN, так и антитело LOX) и стандартные химиотерапевтические препараты были объединены в моделях десмопластической опухоли мышей, чтобы снизить интерстициальное давление опухоли, вызывающее расширение сосудов (Oncotarget. 2016; 7 (22) 32100 - 32112). Повышенный сосудистый кровоток увеличивает концентрацию химиотерапевтического агента в месте первичной опухоли, что приводит к снижению метастатической нагрузки и увеличению выживаемости (Oncotarget. 2016 May 31; 7(22) 32100 - 32112).
[0037] При сквамозной клеточной карциноме головы и шеи была обнаружена повышенная экспрессия изофермента лизилоксидазы связанная с CA-IX, маркером гипоксии, и была связана со снижением специфической выживаемости при раке, снижением общей выживаемости и более низкой выживаемостью без метастазов (Oncotarget. 2016; 7 (31): 50781 - 50804). При сквамозной клеточной карциноме полости рта экспрессия мРНК изофермента лизилоксидазы повышала регуляцию по сравнению с нормальной слизистой оболочкой.
[0038] Профили экспрессии генов глиом выявили сверхэкспрессию изофермента лизилоксидазы как часть молекулярной сигнатуры, указывающей на инвазию, и ассоциированную с опухолями более высокого уровня, которые сильно коррелируют с плохой выживаемостью пациентов (PloS ONE. 2015 Mar 19; 10(3) e0119781). Экспрессия белка изофермента лизилоксидазы была увеличена в тканях глиобластомы и астроцитомы, а также в культивируемых клетках астроцитомы инвазивных U343 и U251.
[0039] В тканях мРНК изофермента лизилоксидазы повышала регуляцию при раке простаты по сравнению с доброкачественной гипертрофией простаты, коррелировала с оценкой Глисона и ассоциировалась как с высокой степенью, так и с коротким временем до рецидива (Oncol Rep 2008; 20: 1561 - 1567).
[0040] В светлоклеточной ПКР, курение ассоциировалось с аллельным дисбалансом в хромосоме 5q23.1, где локализован ген LOX, и может включать дупликацию гена (Cancer Genet Cytogenet. 2005; 163(1)7: 7 - 11).
[0041] Клетки рака шейки матки SiHa продемонстрировали повышенную инвазию in vitro в условиях гипоксии/аноксии; это подавлялось ингибированием внеклеточной каталитически активной активности лизилоксидазы лечением BAPN, а также антисмысловыми олигонуклеотидами LOX, антителом LOX, кшРНК LOX или внеклеточным хелатором меди (Oncol Rep. 2013; 29 (2), 541 - 548).
[0042] В моделях генно-инженерных мышей с раком яичников (нокаутный ApoE) образуется десмопластическая опухоль с повышенной экспрессией гена LOX. Лечение BAPN значительно увеличивало выживаемость и уменьшало метастазирование в легких (J Exp Clin Cancer Res. 2018; 37: 32). Некоторые опухоли от пациентов с раком яичников имеют однонуклеотидный полиморфизм G473A гена LOX. Два независимых исследования показали, что люди с выраженным полиморфизмом G473A имеют повышенные шансы на развитие рака яичников (J Int Med Res. 2012; 40(3): 917 - 923; Genet Test Mol Biomarkers. 2012; 16 (8): 915 - 919).
[0043] При первичной сквамозной клеточной карциноме ротовой полости человека (СККР) уровни фермента лизилоксидазы (в частности, LOX и LOXL2) и экспрессия лизилгидроксилазы значительно повышены и заметно повышены на поздних стадиях опухолей, (RLNM)-позитивных по метастазам в регионарных лимфатических узлах. Как восстанавливаемые, или незрелые поперечные сшивки (deH-DHLNL и deH-HLNL), так и невосстанавливаемые или зрелые поперечные сшивики (DPD и PYD) значительно выше в СКРР по сравнению с нормальными тканями (J Dent Res 2019; 98(5): 517 – 525).
[0044] Результаты, описанные в данном документе, дают веское основание для комбинированной терапии с участием ингибиторов изофермента LOX и противоопухолевой терапии у пациентов.
[0045] Совсем недавно CCT365623, обратимый пан-ингибитор LOX, был использован в модели рака груди (MMTV-PyMT) для уменьшения метастазирования и увеличения выживаемости (Nat Commun. 2017; 18 (8): 14909).
[0046] В научной и патентной литературе описаны низкомолекулярные ингибиторы изоферментов лизилоксидазы и антитела LOX и LOXL2 с терапевтическими эффектами на животных моделях фиброза и метастазировании рака. Сообщается также, что некоторые известные ингибиторы МАО ингибируют изофермент лизилоксидазы (например Мофегилин - ингибитор МАО-B, показанный ниже). Этот ингибитор является членом семейства галогеналлиламина - ингибиторов МАО; галоген в Мофегилине представляет собой фтор. Ингибиторы фтораллиламина описаны в патенте US № 4,454,158. Выданы патенты на фтораллиламины и хлораллиламины, например MDL72274 (проиллюстрировано ниже) в качестве ингибиторов лизилоксидазы (патенты US 4,943,593; 4,965,288; 5,021,456; 5,059,714; 5,182,297; 5,252,608). Сообщается также, что многие из соединений, заявленных в этих патентах, являются сильнодействующими ингибиторами MAO-B и SSAO/VAP-1.
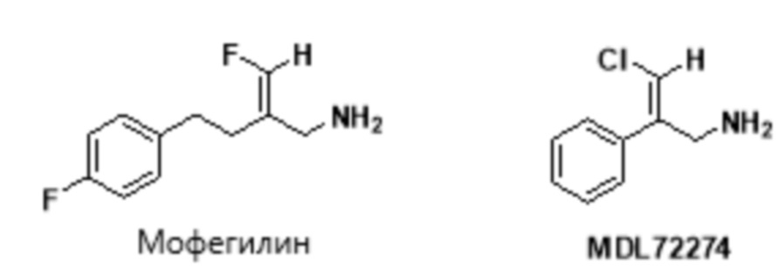
[0047] Дополнительные ингибиторы фтораллиламина описаны в патенте US 4,699,928. Другие примеры, структурно связанные с Мофегилином, можно найти в WO 2007/120528.
[0048] WO 2009/066152 описывает семейство 3-замещенных 3-галогеналлиламинов, которые являются ингибиторами SSAO/VAP-1 полезными для лечения множества показаний, включая воспалительное заболевание. Ни в одном из этих документов конкретно не описаны фтораллиламинные соединения формулы (I) согласно настоящему изобретению.
[0049] Антитела к LOX и LOXL2 описаны в US 2009/0053224 со способами диагностического и терапевтического применения. Антитела анти-LOX и анти-LOXL2 можно использовать, чтобы выявлять и лечить такие состояния, как фиброзное состояние, ангиогенез, или предотвращать переход от состояния эпителиальных клеток к состоянию мезенхимальных клеток: US 2011/0044907.
[0050] WO 2017/136871 и WO 2017/136870 описывают галогеналлиламин-индол и производные азаиндола в качестве ингибиторов лизилоксидаз и их применение.
[0051] WO 2018/157190 описывает производные галогеналлиламинпиразола в качестве ингибиторов лизилоксидаз и их применение.
[0052] WO 2017/141049 и WO 2019/073251 описывают семейства производных метиламина и мостикового гомопиперазина, соответственно, в качестве ингибиторов лизилоксидазы и их применение для лечения рака и заболеваний, связанных с фиброзом.
[0053] WO 2003/097612, WO 2006/053555 и US 2008/0293936 описывают другой класс ингибиторов лизилоксидазы.
[0054] WO 2018/048930, WO 2017/015221, WO 2017/003862, WO 2016/144702 и WO 2016/144703 описывают другие ингибиторы LOXL2.
СУТЬ ИЗОБРЕТЕНИЯ
[0055] Настоящее изобретение относится к замещенным фтораллиламиновым соединениям, которые ингибируют лизилоксидазу (LOX), лизилоксидазоподобный 2 (LOXL2) и другие изоферменты лизилоксидазы. Неожиданно модификация структур 3-замещенного-3-фтораллиламина, описанная ранее, привела к открытию новых соединений, которые являются мощными ингибиторами изоферментов LOX и LOXL человека. Кроме того, некоторые из этих новых соединений также селективно ингибируют определенные изоферменты LOX и LOXL по сравнению с другими ферментами семейства аминоксидаз.
[0056] Первый аспект изобретения обеспечивает соединение Формулы I:
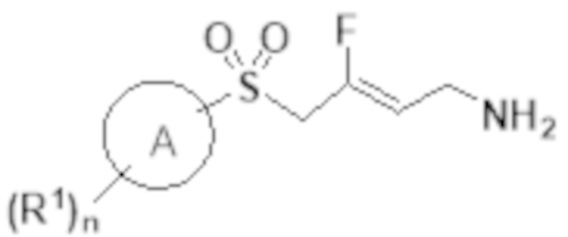
Формула I
или его фармацевтически приемлемую соль, полиморфную форму, сольват, гидрат или таутомерную форму; в которой:
А представляет собой арил или гетероарил;
каждый R1 независимо выбирают из группы, состоящей из X-R2, галогена, дейтерия, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -OC1-4алкила, -CF3, -CH2CF3 и –O-CF3;
X выбирают из группы, состоящей из O, CH2, OCH2, CH2O, CH2S(O)2, CONH и NHCO;
R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила; где каждый R2 необязательно замещен одним или несколькими R7;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -OC1-4алкила, -CF3, -CH2CF3 и –O-CF3;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3;
R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1, 2, 3, 4, 5 или 6.
[0057] Второй аспект изобретения обеспечивает фармацевтическую композицию, содержащую соединение согласно первому аспекту изобретения или его фармацевтически приемлемую соль или сольват, и по меньшей мере один фармацевтически приемлемый эксципиент, носитель или разбавитель.
[0058] Третий аспект изобретения обеспечивает способ ингибирования активности аминоксидазы любого из LOX, LOXL1, LOXL2, LOXL3 или LOXL4 у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения согласно первому аспекту изобретения, или его фармацевтически приемлемую соль или сольват, или фармацевтическую композицию согласно второму аспекту изобретения.
[0059] Четвертый аспект изобретения обеспечивает способ лечения состояния, связанного с любым одним из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения согласно первому аспекту изобретения, или его фармацевтически приемлемой соли или сольвата, или фармацевтической композиции согласно второму аспекту изобретения.
[0060] Пятый аспект изобретения обеспечивает применение соединения согласно первому аспекту изобретения или его фармацевтически приемлемой соли или сольвата для производства лекарственного средства для лечения состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
[0061] Шестой аспект изобретения обеспечивает соединение согласно первому аспекту изобретения или его фармацевтически приемлемую соль или сольват для применения при лечении состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
[0062] В одном варианте реализации способов и применений настоящего изобретения состояние выбрано из фиброза, рака и ангиогенеза.
[0063] В данном документе рассматривается комбинированная терапия, в которой способы дополнительно включают совместное введение дополнительных терапевтических агентов, которые используются для лечения рака, фиброза, ангиогенеза, воспаления, гипертензии, иммуносупрессии и метаболических состояний.
Определения
[0064] Ниже приведены некоторые определения, которые могут быть полезны для понимания описания настоящего изобретения. Они предназначены как общие определения и никоим образом не должны ограничивать объем настоящего изобретения только этими терминами, но приведены для лучшего понимания следующего описания.
[0065] Если контекст не требует иного или специально не указано иное, целые числа, стадии или элементы изобретения, изложенные в данном документе как единственные целые числа, стадии или элементы, явно охватывают формы единственного и множественного числа перечисленных целых чисел, стадий или элементов.
[0066] Во всем этом описании, если контекст не требует иного, слово «содержать» или варианты, такие как «содержит» или «содержащий», будет пониматься как подразумевающее включение указанной стадии или элемента, или целого числа, или группы стадий или элементов, или целых чисел, но не исключение любой другой стадии, элемента, целого числа или группы элементов или целых чисел. Таким образом, в контексте данного описания термин «содержащий» означает «включая в основном, но не обязательно исключительно».
[0067] Специалисты в данной области техники поймут, что описанное в данном документе изобретение допускает изменения и модификации, отличные от конкретно описанных. Следует понимать, что изобретение включает все такие варианты и модификации. Изобретение также включает все стадии, признаки, композиции и соединения, упомянутые или указанные в данном описании, по отдельности или вместе, и любые и все комбинации или любые две или более из указанных стадий или признаков.
[0068] Используемый в данном документе термин «алкил» включает в своем значении одновалентные («алкил») и двухвалентные («алкилен») насыщенные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода, например, 1, 2, 3, 4, 5 или 6 атомов углерода. Алкильная группа с прямой или разветвленной цепью присоединяется в любой доступной точке с образованием стабильного соединения. Например, термин "алкил" включает, но не ограничивается ими, метил, этил, 1-пропил, изопропил, 1-бутил, 2-бутил, изобутил, трет-бутил, амил, 1,2-диметилпропил, 1,1-диметилпропил, пентил, изопентил, гексил, 4-метилпентил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 2,2-диметилбутил, 3,3-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 1,2,2-триметилпропил, 1,1,2-триметилпропил и тому подобное.
[0069] Термин «алкокси» или «алкилокси», используемый в данном документе, относится к алкилокси-группам с прямой или разветвленной цепью (т.е. O-алкил), где алкил определен как указанно выше. Примеры алкоксигрупп включают метокси, этокси, н-пропокси и изопропокси.
[0070] Используемый в данном документе термин «циклоалкил» включает в своем значении одновалентные («циклоалкил») и двухвалентные («циклоалкилен») насыщенные, моноциклические, бициклические, полициклические или конденсированные аналоги. В контексте настоящего описания циклоалкильная группа может иметь от 3 до 10 атомов углерода. Конденсированный аналог циклоалкила означает моноциклическое кольцо, конденсированное с арильной или гетероарильной группой, в которой точка присоединения находится на неароматической части. Примеры циклоалкила и его конденсированных аналогов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, тетрагидронафтил, декагидронафтил, инданил, адамантил и тому подобное.
[0071] Используемый в данном документе термин «арил» или варианты, такие как «арилен», относятся к одновалентным («арил») и двухвалентным («арилен») одиночным, многоядерным, сопряженным и конденсированным аналогам ароматических углеводородов, содержащих от 6 до 10 атомов углерода. Конденсированный аналог арила означает арильную группу, конденсированную с моноциклической циклоалкильной или моноциклической гетероциклильной группой, в которой точка присоединения находится на ароматической части. Примеры арила и его конденсированных аналогов включают фенил, нафтил, инданил, инденил, тетрагидронафтил, 2,3-дигидробензофуранил, тетрагидробензопиранил, 1,4-бензодиоксанил и тому подобное. «Замещенный арил» представляет собой арил, который независимо замещен одним или несколькими, предпочтительно 1, 2 или 3 заместителями, присоединенными к любому доступному атому с образованием стабильного соединения.
[0072] Термин «алкиларил», используемый в данном документе, включает в своем значении одновалентные («арил») и двухвалентные («арилен»), одиночные, полиядерные, сопряженные и конденсированные ароматические углеводородные радикалы, присоединенные к двухвалентным, насыщенным, линейным или разветвленным алкиленовым радикалам. Примеры алкиларильных групп включают бензил.
[0073] Термин «гетероарил» и варианты, такие как «гетероароматическая группа» или «гетероарилен», используемые в данном документе, включают в своем значении одновалентные («гетероарил») и двухвалентные («гетероарилен»), одиночные, полиядерные, сопряженные и конденсированные гетероароматические радикалы, имеющие от 5 до 10 атомов, где от 1 до 4 атомов в кольце или от 1 до 2 атомов в кольце представляют собой гетероатомы, независимо выбранные из O, N, NH и S. Гетероарил также включает окисленные S или N, такие как сульфинил, сульфонил и N-оксид азота третичного кольца. Атом углерода или азота является местом присоединения гетероарильной кольцевой структуры, так что образуется стабильное соединение. Гетероароматическая группа может быть C1-9гетероароматической. Конденсированный аналог гетероарила означает гетероарильную группу, конденсированную с моноциклической циклоалкильной или моноциклической гетероциклильной группой, в которой точка присоединения находится на ароматической части. Примеры гетероарильных групп и их конденсированных аналогов включают пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тетразолил, триазинил, тиенил, бензоксазолил, бензотиазолил, бензимидазолил, бензофуранил, бензотиофенил, фуро(2,3-b)пиридил, индолил, изохинолинил, имидазопиридил, пиримидинил, пиридазинил, пиразинил, пиридонил, фенантролинил, хинолил, изохинолинил, имидазолинил, тиазолинил, пирролил, фуранил, тиофенил, оксазолил, изоксазолил, изотиазолил, триазолил и т.п. «Азотсодержащий гетероарил» относится к гетероарилу, в котором любые гетероатомы представляют собой N. «Замещенный гетероарил» представляет собой гетероарил, который независимо замещен одним или несколькими, предпочтительно 1, 2 или 3 заместителями, присоединенными к любому доступному атому с образованием стабильного соединения.
[0074] Термин «гетероциклил» и варианты, такие как «гетероциклоалкил», используемые в данном документе, включают в себя одновалентные («гетероциклил») и двухвалентные («гетероциклилен»), насыщенные или частично насыщенные (неароматический), моноциклические, бициклические, полициклические или конденсированные углеводородные радикалы, содержащие от 3 до 10 атомов в кольце, где от 1 до 4 или от 1 до 2 атомов в кольце представляют собой гетероатомы, независимо выбранные из O, N, NH или S, SO или SO2, в которых точка присоединения может быть углеродом или азотом. Конденсированный аналог гетероциклила означает моноциклический гетероцикл, конденсированный с арильной или гетероарильной группой, в которой точка присоединения находится на неароматической части. Гетероциклильная группа может представлять собой C3-8гетероциклил. Гетероциклоалкильная группа может представлять собой C3-6 гетероциклил. Гетероциклильная группа может представлять собой C3-5 гетероциклил. Примеры гетероциклильных групп и их конденсированных аналогов включают пирролидинил, тиазолидинил, пиперидинил, пиперазинил, имидазолидинил, 2,3-дигидрофуро(2,3-b)пиридил, бензоксазинил, тетрагидрохинолинил, тетрагидроизохинолинил, дигидроиндолил, хинуклидинил, азетидинил, морфолинил, тетрагидротиофенил, тетрагидрофуранил, тетрагидропиранил и тому подобное. Термин также включает частично ненасыщенные моноциклические кольца, которые не являются ароматическими, такие как 2- или 4-пиридоны, присоединенные через азот или N-замещенные урацилы.
[0075] Термин «галоген» или его варианты, такие как «галид» или «галоген», используемые в данном документе, относятся к фтору, хлору, брому и йоду.
[0076] Термин «гетероатом» или его варианты, такие как «гетеро-» или «гетерогруппа», используемые в данном документе, относятся к O, N, NH и S.
[0077] В общем, «замещенный» относится к органической группе, как определено в данном документе (например, алкильной группе), в которой одна или несколько связей с содержащимся в ней атомом водорода заменены связью с неводородными или неуглеродными атомами. Замещенные группы также включают группы, в которых одна или несколько связей с атомом углерода(ов) или водорода(ов) заменены одной или несколькими связями, включая двойные или тройные связи, с гетероатомом. Таким образом, замещенная группа будет замещена одним или несколькими заместителями, если не указано иное. В некоторых вариантах реализации замещенная группа замещена 1, 2, 3, 4, 5 или 6 заместителями.
[0078] Термин «необязательно замещенный», используемый в данном документе, означает группу, к которой этот термин относится, которая может быть незамещенной или может быть замещенной одной или несколькими группами, независимо выбранными из алкила, алкенила, алкинила, циклоалкила, циклоалкенила, гетероциклоалкила, галогена, галоалкила, гидроксила, гидроксиалкил, алкокси, тиоалкокси, алкенилокси, галоалкокси, NO2, NH(алкил), N(алкил)2, алкиламино, диалкиламино, ацил, алкеноил, алкиноил, ациламино, диациламино, ацилокси, алкилсульфонил, алкилсульфонилокси, сульфонамидо, гетероциклокси, гетероциклоамино, гетероциклоалкил, аликлсульфенил, алкилкарбонилокси, фосфорсодержащие группы, такие как фосфоно и фосфинил, арил, гетероарил, алкиларил, аралкил, алкилгетероарил, циано, CO2H, CO2алкил, C(O)NH2, -C(O)NH(алкил) и -C(O)N(алкил)2. Предпочтительные заместители включают галоген, C1-C6алкил, C1-C6галогеналкил, C1-C6алкокси, гидрокси(C1-6)алкил, C3-C6циклоалкил, C(O)OH, NHC(O)C1-C4алкил, C(O)C1-C4алкил, NH2, NHC1-C4алкил, N(C1-C4алкил)2, SO2(C1-C4алкил), OH и CN. Особенно предпочтительные заместители включают C1-4алкил, C1-4алкокси, SO2(C1-C4алкил), галоген, OH, гидрокси(C1-3)алкил (например, C(CH3)2OH) и C1-3галогеналкил (например, CF3, CH2CF3).
[0079] Данное изобретение включает в себя соединения в одной или нескольких таутомерных формах, включая как одиночные таутомеры, так и смеси таутомеров. В объем настоящего изобретения также включены все полиморфы и кристаллические формы описанных в данном документе соединений.
[0080] Настоящее изобретение включает в себя изотопы различных атомов. Любой атом, специально не обозначенный как конкретный изотоп, означает любой стабильный изотоп этого атома. Таким образом, следует понимать, что настоящее описание включает изотопы водорода - дейтерий и тритий.
[0081] Все ссылки, цитируемые в этой заявке, специально включены в качестве перекрестных ссылок во всей их полноте. Ссылка на любые такие документы не должна толковаться как признание того, что документ является частью общеизвестных знаний или уровня техники.
[0082] В контексте данного описания термин «введение» и варианты этого термина, включая «ввести» и «введение», включают приведение в контакт, нанесение, доставку или обеспечение соединения или композиции изобретения в организме или на поверхности любым подходящим средства. В контексте данного описания термин «лечение» относится к любому и всем применениям, которые лечат состояние или симптомы заболевания, предотвращают возникновение заболевания или иным образом предотвращают, препятствуют, замедляют или обращают развитие заболевания или других нежелательных симптомов каким-либо образом.
[0083] В контексте данного описания термин «местное введение» или варианты этого термина, включая «местное нанесение», включает в своем значении нанесение, приведение в контакт, доставку или обеспечение соединения или композиции изобретения на коже или локализованных областях тела.
[0084] В контексте данного описания термин «локальное введение» или варианты этого термина, включая «локальное нанесение», включает в своем значении нанесение, приведение в контакт, доставку или обеспечение соединения или композиции настоящего изобретения на коже или локализованных областях тела.
[0085] В контексте данного описания термин «эффективное количество» включает в своем значении достаточное, но нетоксичное количество соединения или композиции настоящего изобретения для обеспечения желаемого эффекта. Таким образом, термин «терапевтически эффективное количество» включает в своем значении достаточное, но нетоксичное количество соединения или композиции изобретения, чтобы обеспечить желаемый терапевтический эффект. Точное необходимое количество будет варьироваться от субъекта к субъекту в зависимости от таких факторов, как вид, которого лечат, пол, возраст и общее состояние субъекта, тяжесть состояния, которое лечат, конкретный вводимый агент, способ введения, и так далее. Таким образом, невозможно указать точное «эффективное количество». Однако для любого конкретного случая подходящее «эффективное количество» может определить специалист средней квалификации в данной области, используя только рутинные эксперименты.
Краткое описание графических материалов
[0086] На Фиг. 1(a) и 1(b) показаны кривые выживаемости, сравнивающие высокую и низкую экспрессию генов у пациентов с аденокарциномой поджелудочной железы на основе набора данных TCGA. А. Экспрессия гена LOX. B. Экспрессия гена LOXL2.
[0087] Фиг. 2(а-с) изображают дозозависимое блокирование ферментативной активности лизилоксидазы Соединениями 1 и 33. Измерение активности лизилоксидазы по сравнению с необработанным контролем в тканях крысы (а) ухо (через 24 часа после однократной пероральной дозы 10 и 30 мг/кг, Соединение 1); (b) ухо (4, 24, 48 и 120 часов после однократной пероральной дозы 30 мг/кг Соединения 33) и; (c) аорта (однократная пероральная доза 5, 10 или 30 мг/кг, Соединение 33)
[0088] Фиг. 3(а) показывает уменьшение количества коллагена в рубцовой ткани мыши после травмы при лечении Соединением 1 путем местного применения.
[0089] Фиг. 3(b) гистология показывает толстые параллельные пучки коллагена в контрольной рубцовой ткани.
[0090] Фиг. 3(c) ткань, обработанная Соединением 1, показывает уменьшенную плотность пучков и более «нормальную» структуру коллагена.
[0091] Фиг. 4(a-b) показывает снижение активности LOX и общего коллагена после ежедневного местного лечения в течение 4 недель 3% раствором Соединения 1 по сравнению с контролем.
[0092] Фиг. 4(c-f) общая морфология показывает аналогичные раны во время травмы (c, контроль; d, обработанные), и во время эвтаназии раны кажутся более зажившими (e, контроль, f обработанные).
[0093] Фиг. 4 (g-h) гистология показывает толстые полосы коллагена в необработанной рубцовой ткани (выделены стрелками, g). Похоже, что в обработанной ткани он уменьшается (h).
[0094] Фиг. 5 (a-e) данные о росте опухоли на модели ксенотрансплантированнго ортотопического рака поджелудочной железы человека: Данные об эффективности. A. Диаграмма стратегии роста и лечения. B. Мониторинг роста опухоли in vivo при помощи биолюминесцентного изображения. C. Биолюминесцентный сигнал ex vivo об общей опухолевой нагрузке. D. Биолюминесцентный сигнал ex vivo первичной опухоли. E. Биолюминесцентный сигнал ex vivo метастатической нагрузки.
[0095] Фиг. 6(a-c) гистологический анализ модели кожи мышей со склерозом при местном лечении Соединением 1. A. Оценка композитной кожи. B. Средняя оценка по коллагену. C. Средняя оценка LOX.
[0096] Фиг. 7(a-e) анализ селезенки на модели первичного миелофиброза (GATA-1low), обработанной Соединением 19. A. Окрашивание селезенки серебром по Гомори. B. Масса селезенки. C. Количественная оценка ретикулинового фиброза селезенки. D. Изображение селезенки, окрашенное H&E. E. Количественное определение мегакариоцитов в селезенке.
[0097] Фиг. 8(a-d) анализ костного мозга из модели первичного миелофиброза (GATA-1low), обработанной Соединением 19. A. Окрашивание костного мозга серебром по Гомори. B. Количественная оценка ретикулинового фиброза костного мозга. C. Изображение костного мозга, окрашенное H&E. D. Количественная оценка мегакариоцитов в костном мозге.
[0098] Фиг. 9 изображает изменения в области фиброза в модели ООМ мыши.
[0099] Фиг. 10 изображает способность Соединения 33 уменьшать индуцированный блеомицином фиброз легких (оценка Эшкрофта) и увеличение веса.
[0100] Фиг. 11(a-d) изображает способность Соединения 33 уменьшать ассоциированные с фиброзом метастазы на модели фиброза печени мыши, индуцированного CCl4, с ортотопически инъецированной клеточной линией рака груди (4t). (а) Схема дизайна исследования; (b) клиническая оценка фиброза печени; (c) концентрация поперечных сшивок в печени; (d) измерение метастазов в печени.
Подробное описание
[0101] Настоящее изобретение относится к замещенным фтораллиламиновым соединениям, которые ингибируют лизилоксидазу (LOX), лизилоксидазоподобную 2 (LOXL2) и другие изоферменты лизилоксидазы. В частности, настоящее изобретение относится к замещенным производным фтораллиламина с сульфоновым линкером.
[0102] В частности, настоящее изобретение относится к соединениям Формулы I:
Формула I
или его фармацевтически приемлемой солиполиморфной форме, сольвату, гидрату или таутомерной форме; в которой:
А представляет собой арил или гетероарил;
каждый R1 независимо выбирают из группы, состоящей из X-R2, галогена, дейтерия, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
X выбирают из группы, состоящей из O, CH2, OCH2, CH2O, CH2S(O)2, CONH и NHCO;
R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила; где каждый R2 необязательно замещен одним или несколькими R7;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -OC1-4алкила, -CF3, -CH2CF3 и –O-CF3;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3;
R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1, 2, 3, 4, 5 или 6.
[0103] В одном варианте реализации соединений настоящего изобретения, A выбран из арила и гетероарила. В другом варианте реализации соединений настоящего изобретения, A выбирают из группы, состоящей из фенила, нафтила, пиридинила, хинолинила, бензотиазолила и индолила. В дополнительном варианте реализации соединений настоящего изобретения A выбирают из группы, состоящей из
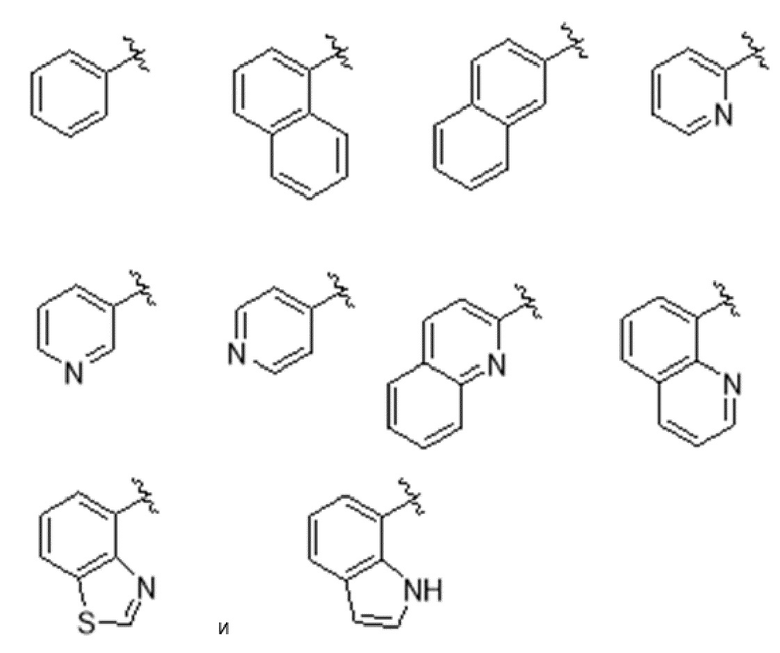 .
.
В еще одном дополнительном варианте реализации соединений настоящего изобретения A выбирают из группы, состоящей из  В дополнительном варианте реализации A выбирают из группы, состоящей из
В дополнительном варианте реализации A выбирают из группы, состоящей из 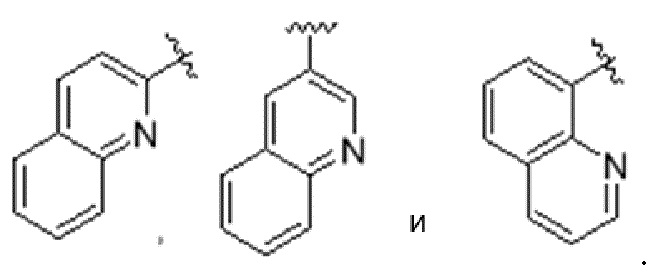 В еще одном дополнительном варианте реализации A представляет собой
В еще одном дополнительном варианте реализации A представляет собой 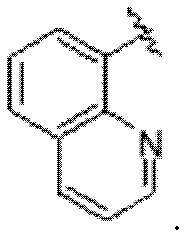 В другом варианте реализации A представляет собой фенил. В дополнительном варианте реализации A представляет собой гетероарил.
В другом варианте реализации A представляет собой фенил. В дополнительном варианте реализации A представляет собой гетероарил.
[0104] В одном варианте реализации соединений настоящего изобретения R1 независимо выбирают из группы, состоящей из X-R2, галогена, дейтерия, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3. В другом варианте реализации соединений настоящего изобретения каждый R1 независимо выбирают из группы, состоящей из -X-R2, C1-6алкила, C1-6галогеналкила, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6. В дополнительном варианте реализации соединений настоящего изобретения каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, C 1-6 галогеналкила и -S(O)2R6. В одном варианте реализации соединений настоящего изобретения по меньшей мере один из R1 представляет собой X-R2. В другом варианте реализации соединений настоящего изобретения один из R1 представляет собой X-R2.
[0105] В одном варианте реализации соединений настоящего изобретения X выбирают из группы, состоящей из O, CH2, OCH2, CH2O, CH2S(O)2, CONH и NHCO. В другом варианте реализации соединений настоящего изобретения X выбирают из группы, состоящей из O, CH2, OCH2, CONH и NHCO. В другом варианте реализации соединений настоящего изобретения X выбирают из группы, состоящей из O, OCH2, и CONH. В дополнительном варианте реализации соединений настоящего изобретения X выбирают из группы, состоящей из O, CH2 и OCH2. В другом варианте реализации соединений настоящего изобретения X выбирают из группы, состоящей из CONH и NHCO. В дополнительном варианте реализации соединений настоящего изобретения X представляет собой О. В другом варианте реализации соединений настоящего изобретения X представляет собой ОСН2. В дополнительном варианте реализации соединений настоящего изобретения X представляет собой CONH.
[0106] В одном варианте реализации соединений настоящего изобретения R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила, где каждый R2 необязательно замещен одним или несколькими R7. В другом варианте реализации соединений настоящего изобретения R2 выбирают из группы, состоящей из арила и циклоалкила, где каждый R2 необязательно замещен одним или несколькими R7. В дополнительном варианте реализации соединений настоящего изобретения R2 представляет собой циклоалкил, где каждый R2 необязательно замещен одним или несколькими R7. В другом варианте реализации соединений настоящего изобретения R2 представляет собой арил, необязательно замещенный одним или несколькими R7. В другом варианте реализации соединений настоящего изобретения R2 представляет собой фенил, замещенный одним R7. В другом варианте реализации соединений настоящего изобретения R2 представляет собой адамантил или фенил, где каждый R2 необязательно замещен одним или несколькими R7. В другом варианте реализации R 2 представляет собой адамантил или фенил, необязательно замещенный –S(O)2NR4R5. В дополнительном варианте реализации R2 представляет собой адамантил. В другом варианте реализации R2 представляет собой фенил, необязательно замещенный –S(O)2NR4R5.
[0107] В одном варианте реализации соединений настоящего изобретения R2 замещен одним R7. В другом варианте реализации соединений настоящего изобретения R2 замещен двумя R7. В другом варианте реализации соединений настоящего изобретения R2 замещен тремя R7. В другом варианте реализации соединений настоящего изобретения R2 замещен четырьмя или пятью R7.
[0108] В одном варианте реализации соединений настоящего изобретения R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -OC1-4алкила, -CF3, -CH2CF3 и –O-CF3. В другом варианте реализации соединений настоящего изобретения R3 представляет собой водород. В дополнительном варианте реализации соединений настоящего изобретения R3 представляет собой C1-6алкил или C3-7циклоалкил. В еще одном дополнительном варианте реализации соединений настоящего изобретения R3 представляет собой водород или C1-6алкил. В другом варианте реализации соединений настоящего изобретения, R3 представляет собой C1-6алкил. В дополнительном варианте реализации соединений настоящего изобретения R3 представляет собой метил или этил. В другом варианте реализации соединений настоящего изобретения R3 выбирают из группы, состоящей из водорода, метила и этила.
[0109] В одном варианте реализации соединений настоящего изобретения R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3. В другом варианте реализации соединений настоящего изобретения R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила. В другом варианте реализации соединений настоящего изобретения R4 и R5 представляют собой водород. В дополнительном варианте реализации соединений настоящего изобретения R4 и R5 представляют собой C1-6алкил. В другом варианте реализации соединений настоящего изобретения R4 и R5 оба представляют собой метил. В дополнительном варианте реализации соединений настоящего изобретения R4 и R5 оба представляют собой изопропил. В одном варианте реализации соединений настоящего изобретения R4 представляет собой водород, и R5 представляет собой изопропил. В дополнительном варианте реализации соединений настоящего изобретения R4 и R5 независимо выбирают из группы, состоящей из водорода и C3-7циклоалкила. В другом варианте реализации соединений настоящего изобретения R4 представляет собой водород и R5 представляет собой C1-6алкил. В одном варианте реализации соединений настоящего изобретения R4 представляет собой водород, и R5 представляет собой метил. В дополнительном варианте реализации соединений настоящего изобретения R4 представляет собой водород, и R5 представляет собой C3-7циклоалкил.
[0110] В одном варианте реализации соединений настоящего изобретения R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца. В дополнительном варианте реализации соединений настоящего изобретения R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего 1 дополнительный гетероатом в качестве членов кольца. В другом варианте реализации R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего 0 дополнительных гетероатомов в качестве членов кольца.
[0111] В одном варианте реализации соединений настоящего изобретения R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3. В другом варианте реализации R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила. В другом варианте реализации R6 представляет собой C1-6алкил. В дополнительном варианте реализации R6 представляет собой C3-7циклоалкил.
[0112] В одном варианте реализации соединений настоящего изобретения R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH. В другом варианте реализации соединений настоящего изобретения, R7 выбирают из группы, состоящей из галогена, C1-6алкила, -C(O)NR4R5, -S(O)2NR4R5 и -S(O)2R6. В дополнительном варианте реализации соединений настоящего изобретения R7 выбирают из группы, состоящей из -C(O)NR4R5, -S(O)2NR4R5 и -S(O)2R6. В другом варианте реализации соединений настоящего изобретения, R7 представляет собой -S(O)2NR4R5. В дополнительном варианте реализации соединений настоящего изобретения, R7 представляет собой -S(O)2N(CH3)2.
[0113] В одном варианте реализации соединений настоящего изобретения R8 представляет собой водород или C1-6алкил. В другом варианте реализации соединений настоящего изобретения R8 представляет собой водород. В дополнительном варианте реализации соединений настоящего изобретения R8 выбирают из группы, состоящей из водорода, метила и этила. В другом варианте реализации соединений настоящего изобретения R8 представляет собой водород или метил.
[0114] В одном варианте реализации соединений настоящего изобретения R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3. В другом варианте реализации R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила. В другом варианте реализации R9 представляет собой C1-6алкил. В дополнительном варианте реализации R9 представляет собой C3-7циклоалкил.
[0115] В одном варианте реализации соединений настоящего изобретения R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца. В другом варианте реализации R8 и R9 объединены с образованием 5-7-членного кольца, имеющего 1 дополнительным гетероатомом в качестве члена кольца. В другом варианте реализации R8 и R9 объединены с образованием 5-7-членного кольца, имеющего 0 дополнительных гетероатомов в качестве членов кольца.
[0116] В одном из вариантов реализации соединений настоящего изобретения n равно 0, 1, 2, 3, 4 или 5. В другом варианте реализации соединений настоящего изобретения n равно 0. В дополнительном варианте реализации соединений настоящего изобретения n равно 0, 1 или 2. В другом варианте реализации соединений настоящего изобретения n равно 1, 2 или 3. В другом варианте реализации соединений настоящего изобретения n равно 1 или 2. В дополнительном варианте реализации соединений настоящего изобретения n равно 1. В другом варианте реализации соединений настоящего изобретения n равно 2. В дополнительном варианте реализации соединений настоящего изобретения n равно 3. В другом варианте реализации соединений настоящего изобретения n равно 4. В дополнительном варианте реализации соединений настоящего изобретения n равно 5. В другом варианте реализации соединений настоящего изобретения n равно 6.
[0117] В одном варианте реализации настоящее изобретение также относится к соединениям Формулы Ia:
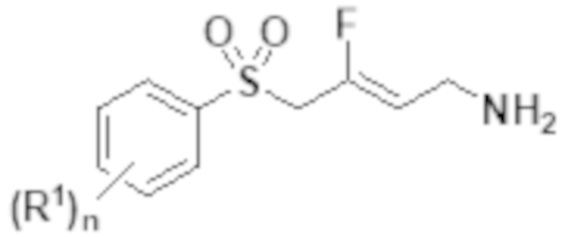
Формула Ia
или их фармацевтически приемлемой соли, полиморфной форме, сольвату, гидрату или таутомерной форме; в которой:
каждый R1 независимо выбирают из группы, состоящей из X-R2, галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
X выбирают из группы, состоящей из O, CH2, OCH2, CH2O, CH2S(O)2, CONH и NHCO;
R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила; где каждый R2 необязательно замещен одним или несколькими R7;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1, 2 или 3.
[0118] В одном варианте реализации настоящее изобретение также относится к соединениям Формулы Ib:
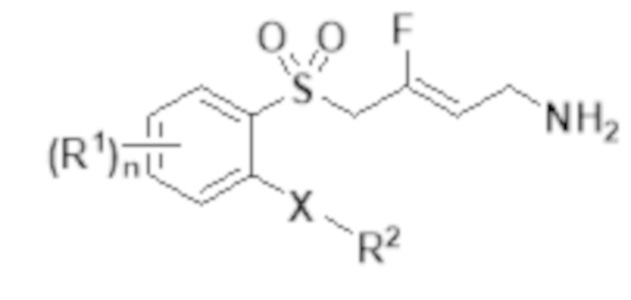
Формула Ib
или их фармацевтически приемлемым солям или сольватам; в которой:
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
X выбирают из группы, состоящей из O, CH2, OCH2, CH2O, CH2S(O)2, CONH и NHCO;
R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила; где каждый R2 необязательно замещен одним или несколькими R7;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1 или 2.
[0119] В одном варианте реализации соединений Формулы Ib изобретения, X выбирают из группы, состоящей из O, OCH2 и CONH; R2 выбирают из группы, состоящей из адамантила и фенила; где каждый R2 необязательно замещен одним или несколькими R7; R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила; R7 представляет собой -S(O)2NR4R5; и n равно 0.
[0120] В другом варианте реализации настоящее изобретение также относится к соединениям Формулы Ic:
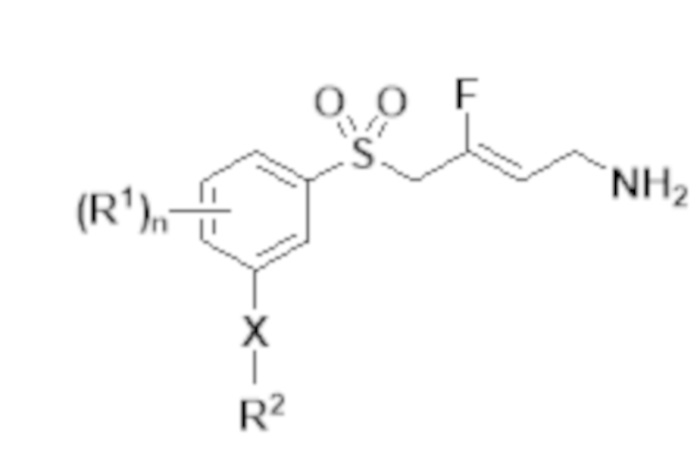
Формула Ic
или их фармацевтически приемлемым солям или сольватам; в которой:
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
X выбирают из группы, состоящей из O, CH2, OCH2, CH2O, CH2S(O)2, CONH и NHCO;
R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила; где каждый R2 необязательно замещен одним или несколькими R7;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1 или 2.
[0121] В одном варианте реализации соединений Формулы Ic изобретения, X выбирают из группы, состоящей из OCH2 и CONH; R2 выбирают из группы, состоящей из адамантила и фенила; где каждый R2 необязательно замещен одним или несколькими R7; R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила; R7 представляет собой -S(O)2NR4R5; и n равно 0.
[0122] В другом варианте реализации настоящее изобретение также относится к соединениям Формулы Id:
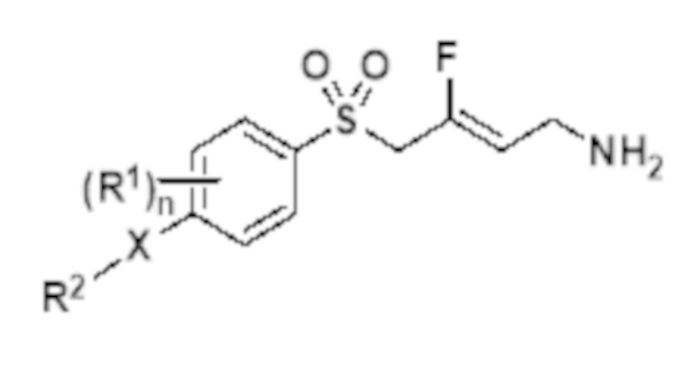
Формула Id
или их фармацевтически приемлемым солям или сольватам; в которой:
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
X выбирают из группы, состоящей из O, CH2, OCH2, CONH и NHCO;
R2 выбирают из группы, состоящей из циклоалкила, гетероциклоалкила, арила и гетероарила; где каждый R2 необязательно замещен одним или несколькими R7;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R7 выбирают из группы, состоящей из галогена, OH, C1-6алкила, O-C1-6алкила, C3-7циклоалкила, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; где каждый C1-6алкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена и –OH;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1 или 2.
[0123] В одном варианте реализации соединений Формулы Id изобретения, X выбирают из группы, состоящей из OCH2 и CONH; R2 выбирают из группы, состоящей из адамантила и фенила; где каждый R2 необязательно замещен одним или несколькими R7; R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила; R7 представляет собой -S(O)2NR4R5; и n равно 0.
[0124] В другом варианте реализации настоящее изобретение также относится к соединениям Формулы Ie:
Формула Ie
или их фармацевтически приемлемым солям или сольватам; в которой:
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1 или 2.
[0125] В другом варианте реализации настоящее изобретение также относится к соединениям Формулы If:
Формула If
или их фармацевтически приемлемым солям или сольватам; в которой:
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1 или 2.
[0126] В другом варианте реализации настоящее изобретение также относится к соединениям Формулы Ig:
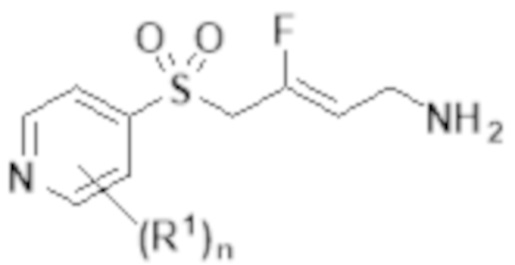
Формула Ig
или их фармацевтически приемлемым солям или сольватам; в которой:
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, арила, гетероарила, циклоалкила, гетероциклоалкила, -CN, -C(O)OR3, -C(O)NR4R5, -S(O)2NR4R5, -S(O)2R6, -NR8C(O)R9 и -NR8S(O)2R9; где каждый C1-6алкил, арил, гетероарил, циклоалкил и гетероциклоалкил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -SO2CH3, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и –O-CF3;
R3 выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила;
R4 и R5 независимо выбирают из группы, состоящей из водорода, C1-6алкила и C3-7циклоалкила; или
R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
R6 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из группы, состоящей из C1-6алкила и C3-7циклоалкила; где каждый C1-6алкил и C3-7циклоалкил необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, -OH, -C1-4алкила, -O-C1-4алкила, -CF3, -CH2CF3 и -O-CF3; или
R8 и R9 объединены с образованием 5-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца;
и
n равно 0, 1 или 2.
[0127] В другом варианте реализации соединений Формул Ie, If и Ig изобретения, каждый R1 представляет собой C1-6алкил, и n равно 0 или 1.
[0128] В другом варианте реализации соединений Формулы I изобретения, каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, C1-6галогеналкила, C1-6алкилокси, C1-6галогеналкокси, -CN, -C(O)OR3, -C(O)NR4R5, -NR4C(O)R6, -S(O)2NR4R5, -NR4S(O)2R6 и -S(O)2R6; R3 выбирают из группы, состоящей из водорода, необязательно замещенного C1-6алкила и необязательно замещенного C3-7циклоалкила; R4 и R5 независимо выбирают из группы, состоящей из водорода, необязательно замещенного C1-6алкила и необязательно замещенного C3-7циклоалкила; или R4 и R5, когда они присоединены к одному и тому же атому азота, объединены с образованием 4-7-членного кольца, имеющего от 0 до 1 дополнительных гетероатомов в качестве членов кольца; R6 выбирают из группы, состоящей из необязательно замещенного C1-6алкила, необязательно замещенного C3-7циклоалкила и необязательно замещенного C1-6галогеналкила; и n равно 0, 1, 2 или 3.
[0129] В другом варианте реализации соединений Формулы I изобретения каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, C1-6галогеналкила и -S(O)2R6; R6 представляет собой C1-6алкил; и n равно 0, 1 или 2.
[0130] В другом варианте реализации соединений Формулы I изобретения каждый R1 независимо выбирают из группы, состоящей из хлора, фтора, метила, изопропила, ОСН3, фенила и SO2 CH3; и n равно 1 или 2.
[0131] В дополнительном варианте реализации соединений Формулы I изобретения, A представляет собой  и n равно 0.
и n равно 0.
[0132] В контексте настоящего описания любой один или несколько аспектов или вариантов реализации могут быть объединены с любыми другими аспектами или вариантами реализации.
[0133] Примеры соединений согласно настоящему изобретению включают соединения, указанные в Таблице 2:
Таблица 2
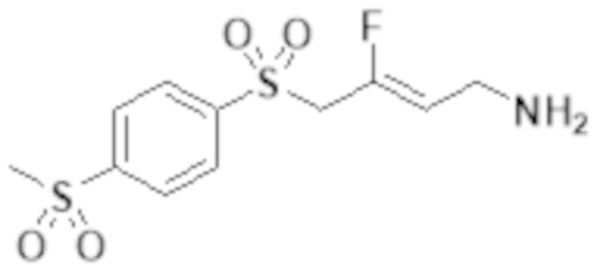
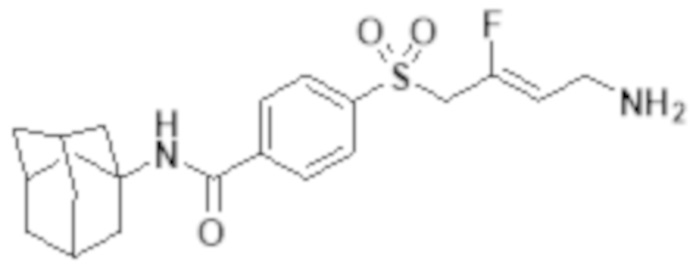
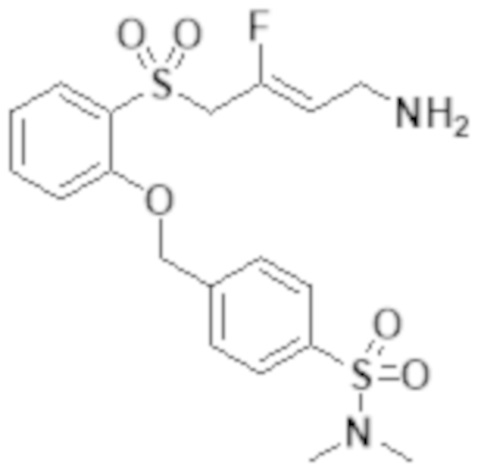
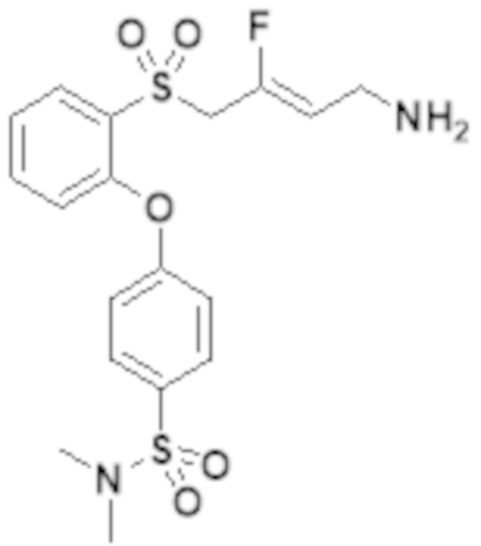
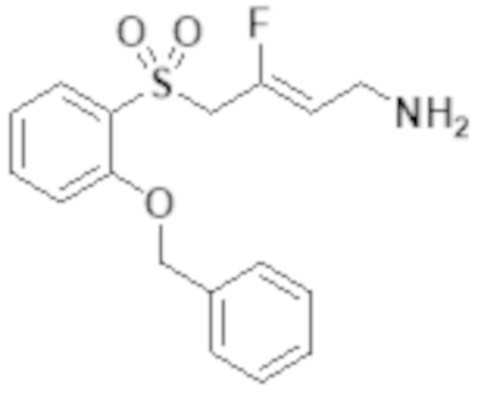
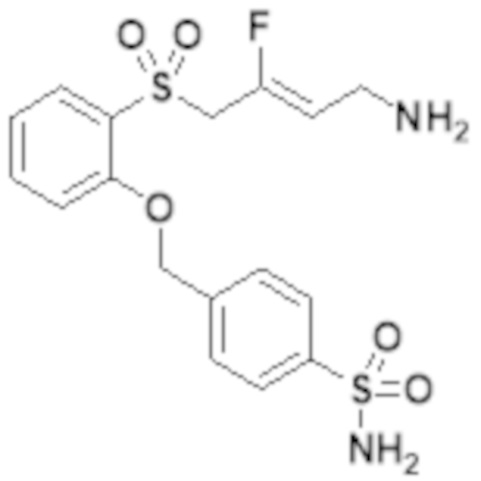
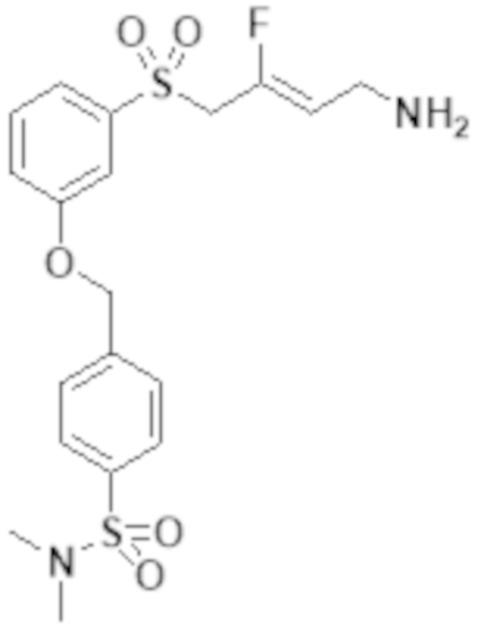
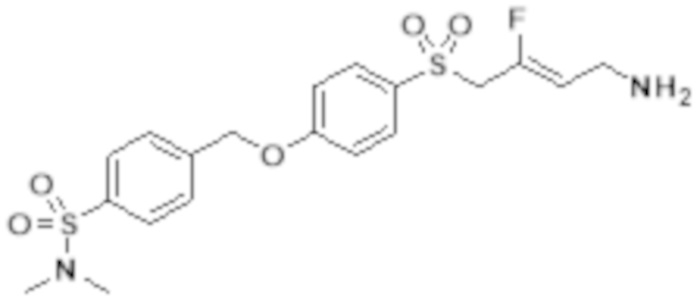
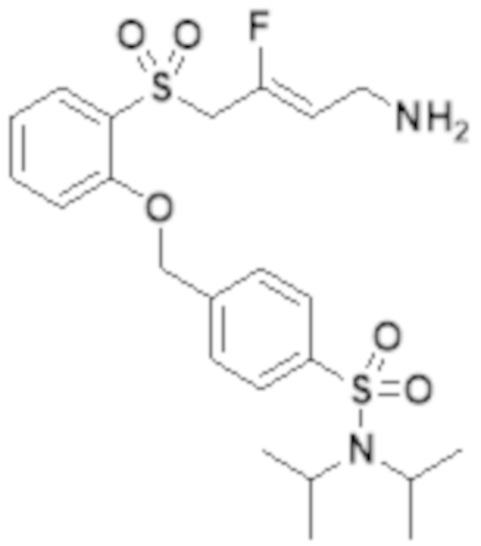
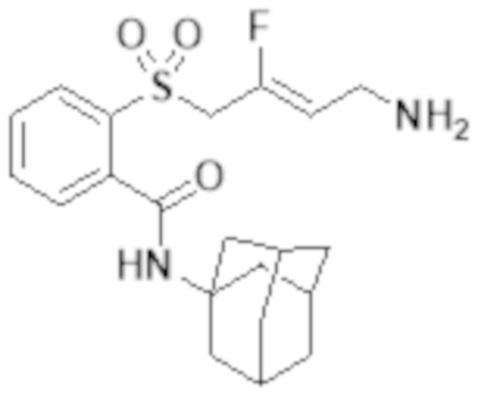
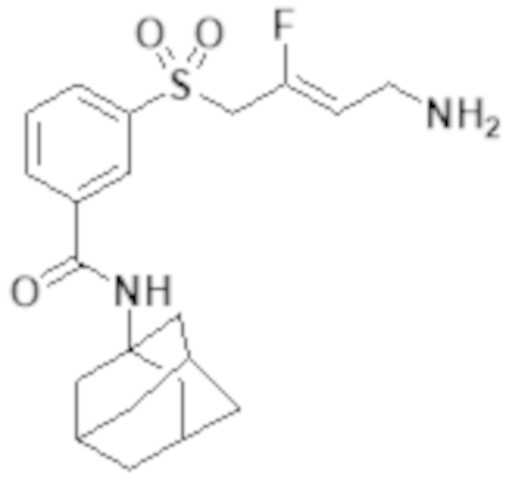
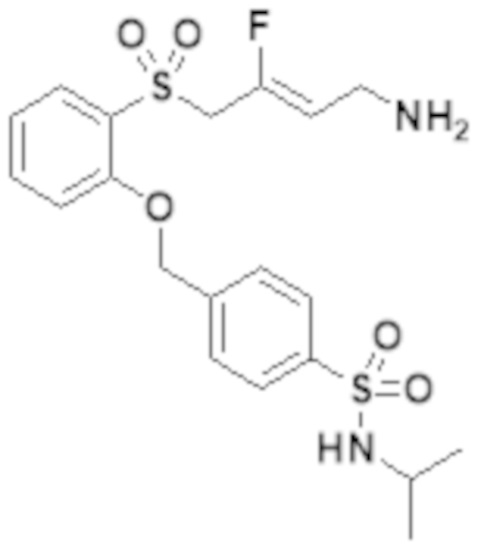
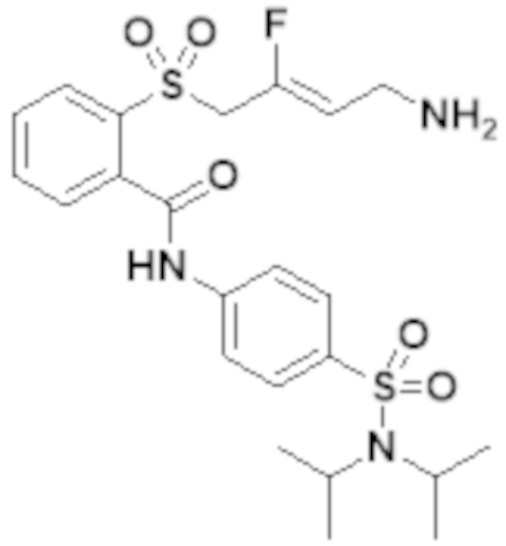
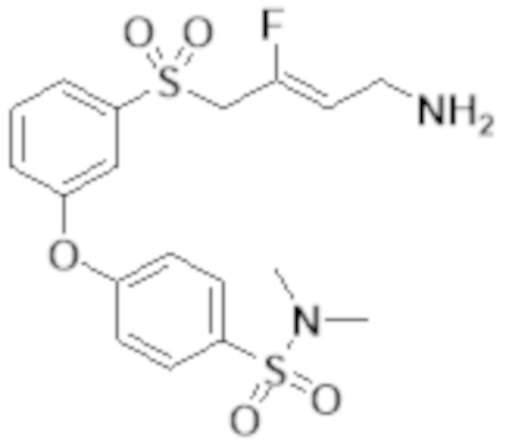
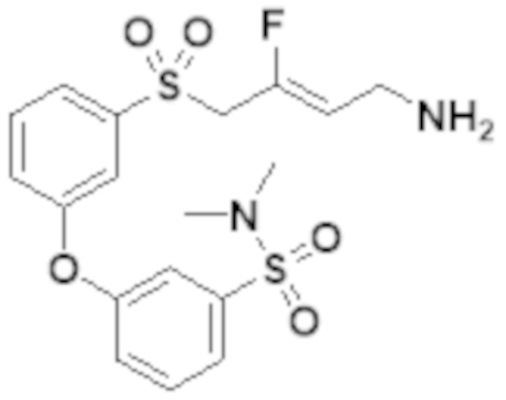
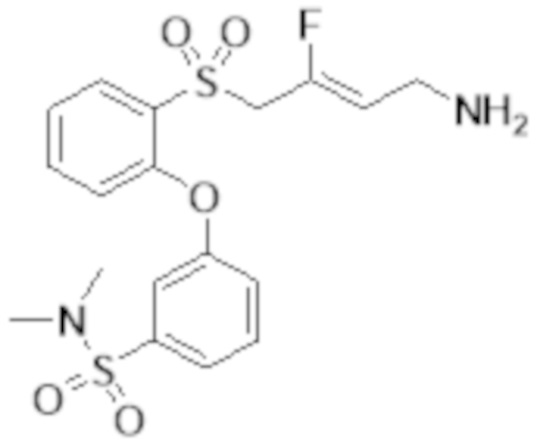
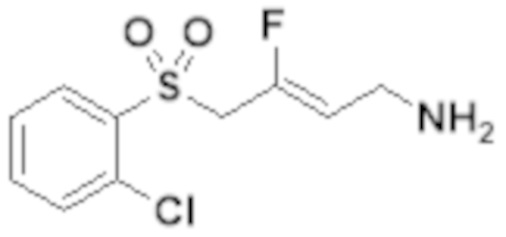
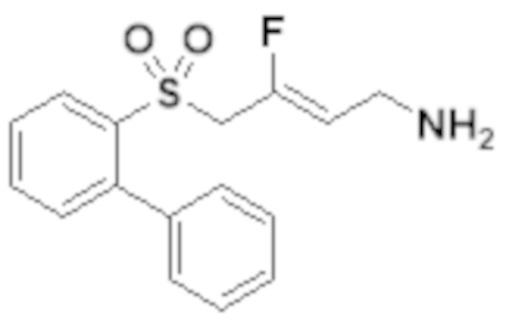
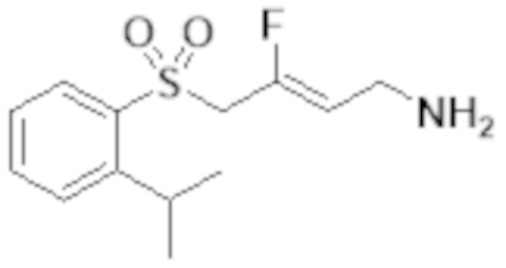
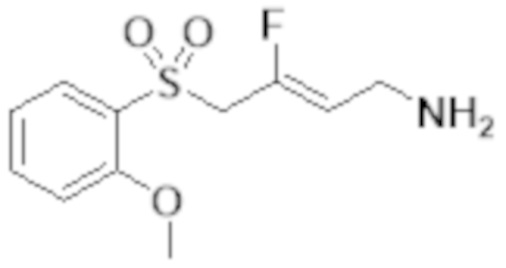
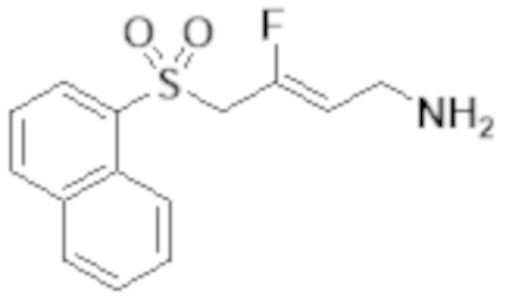
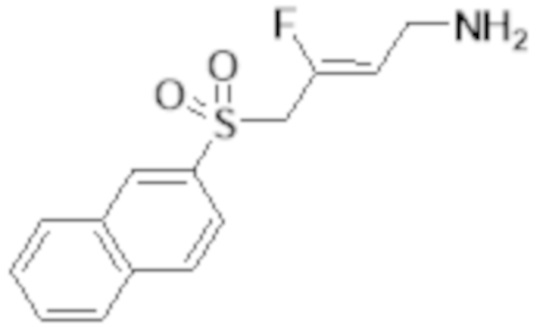
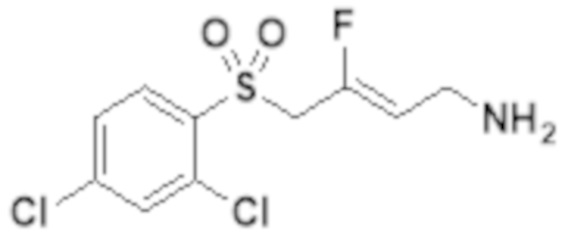
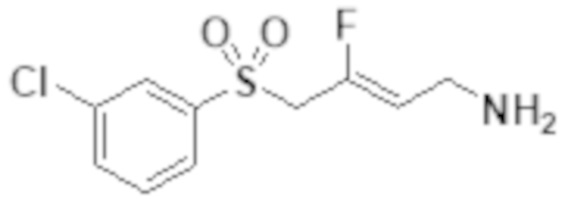
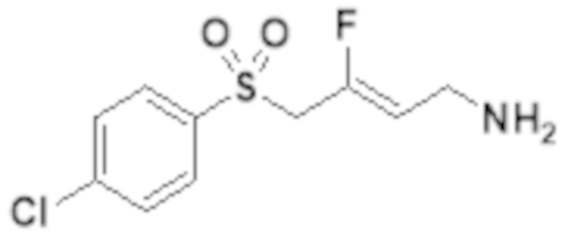
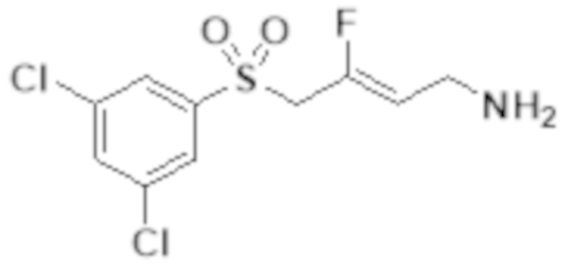
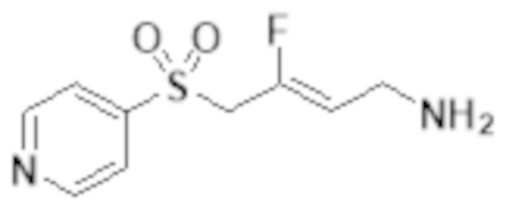
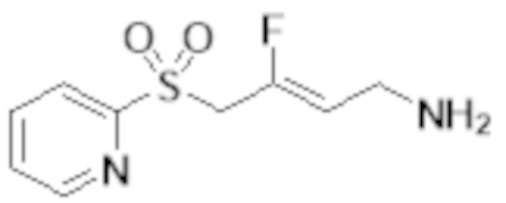
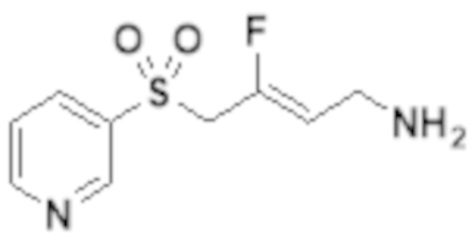
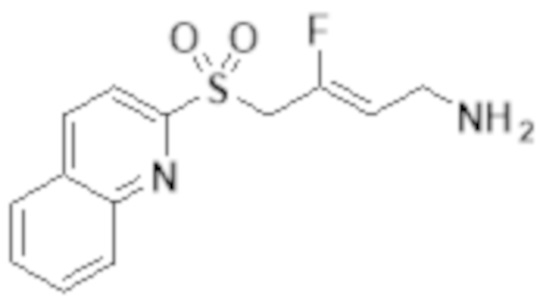
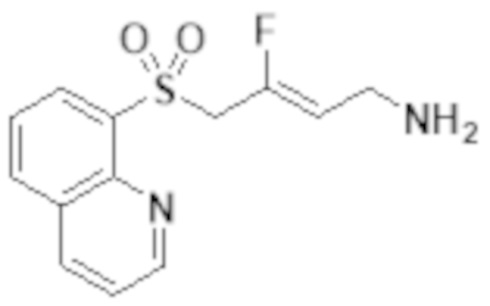
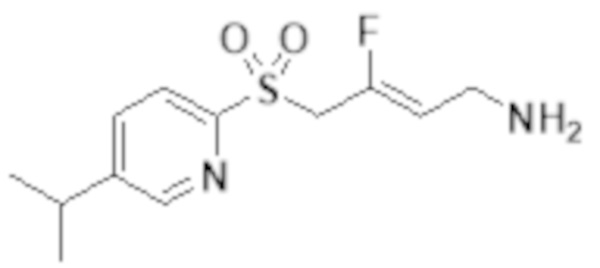
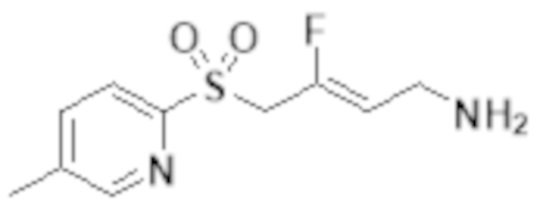
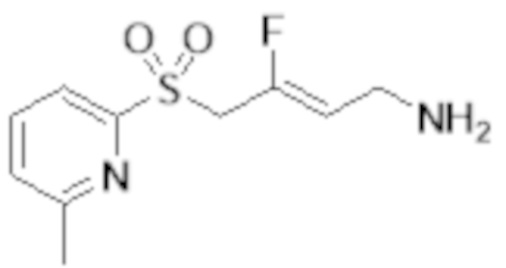
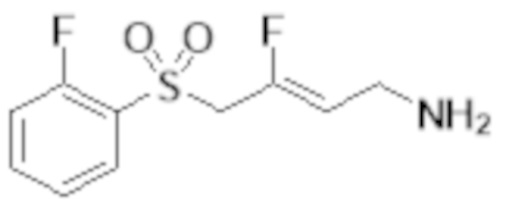
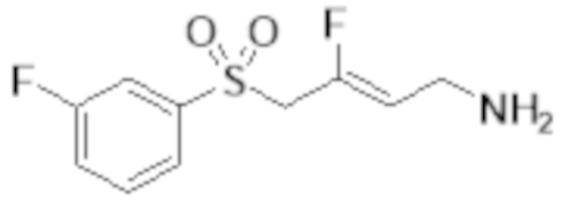
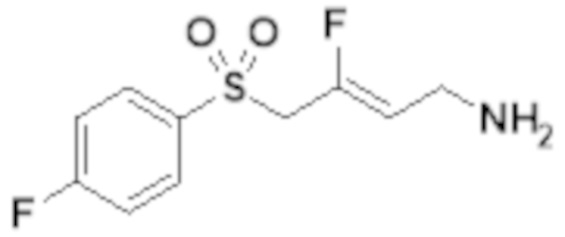
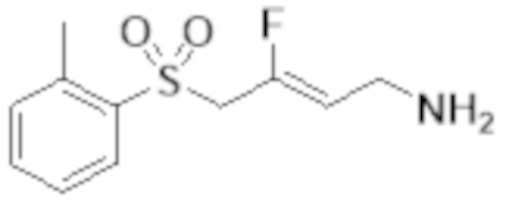
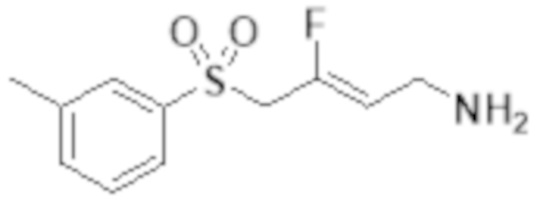
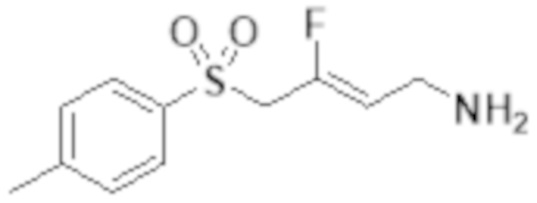
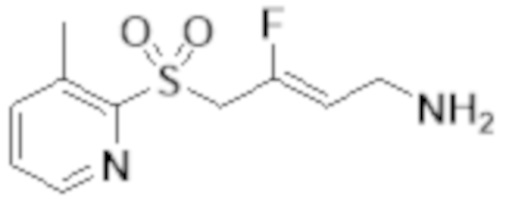
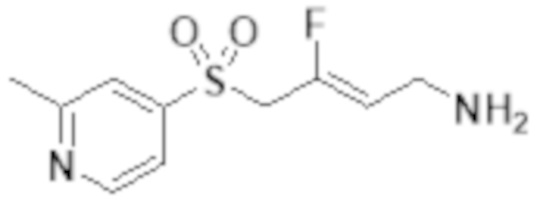
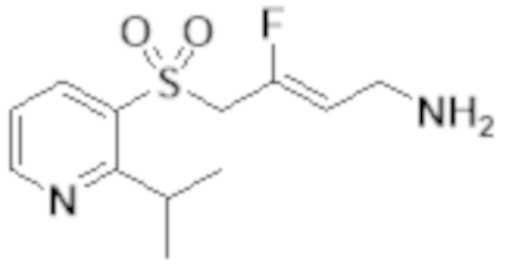
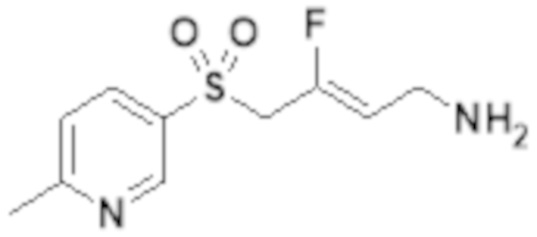
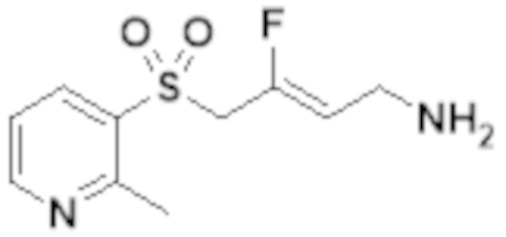
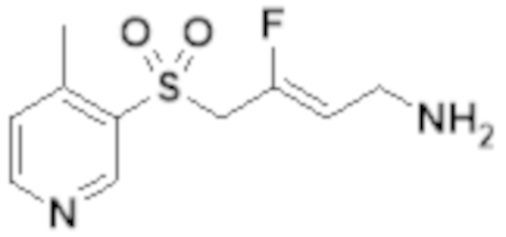
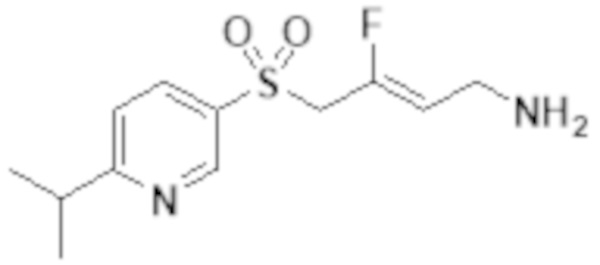
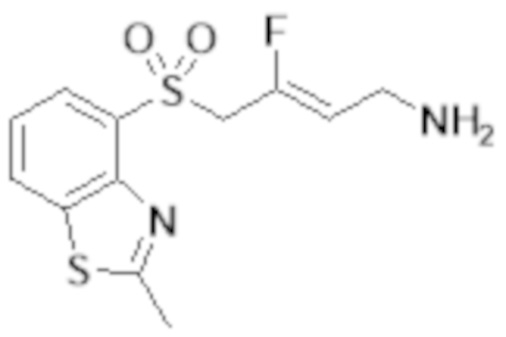
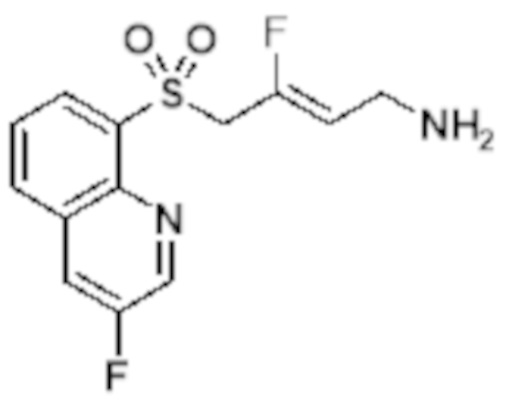
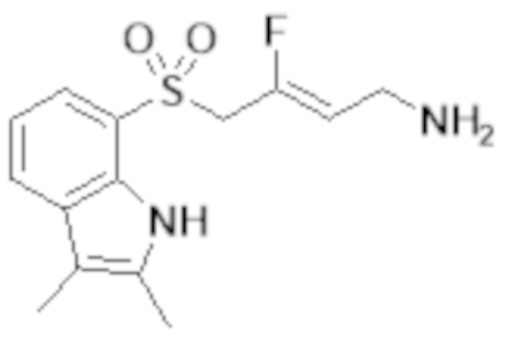
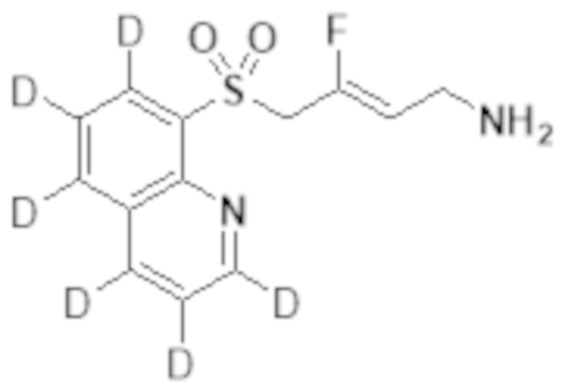
[0134] В одном варианте реализации соединение настоящего изобретения выбрано из группы, состоящей из и , или их фармацевтически приемлемых солей или сольватов. В другом варианте реализации соединение настоящего изобретения представляет собой или его фармацевтически приемлемую соль или сольват. В дополнительном варианте реализации соединение настоящего изобретения представляет собой или его фармацевтически приемлемую соль или сольват.
[0135] В одном варианте реализации соединение настоящего изобретения выбрано из группы, состоящей из и , или их фармацевтически приемлемых солей или сольватов. В другом варианте реализации соединение настоящего изобретения представляет собой или его фармацевтически приемлемую соль или сольват. В дополнительном варианте реализации соединение настоящего изобретения представляет собой или его фармацевтически приемлемую соль или сольват.
Получение Соединений Формулы I
[0136] Соединения Формулы I могут быть легко получены специалистами в данной области с использованием способов и материалов, известных в данной области техники и со ссылкой на стандартные учебники, такие как “Advanced Organic Chemistry” by Jerry March (third edition, 1985, John Wiley and Sons) или “Comprehensive Organic Transformations” by Richard C. Larock (1989, VCH Publishers).
[0137] Соединения Формулы I можно синтезировать, как описано ниже. На следующих схемах представлен обзор типичных неограничивающих вариантов реализации изобретения. Специалисты в данной области поймут, что аналоги Формулы I, включая различные изомерные формы, также могут быть получены из аналогичных исходных материалов.
Схема 1:
[0138] Получение соединений, описываемых Формулой Ib, где X представляет собой –OCH2-, описано на Схеме 1 ниже. Специалист в данной области техники поймет, что соединения, описанные Формулами Ic, Id, Ie, If и Ig, могут быть получены с использованием аналогичных синтетических методик с подходящими исходными материалами.
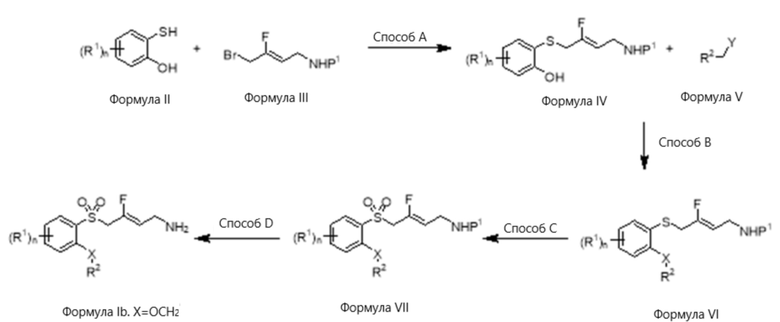
Схема 1
[0139] P1 представляет собой функциональную группу, используемую для защиты азотной группы. Примерами P1 являются группы, образующие карбамат, такие как трет-бутилоксикарбонильная (BOC), 9-флуоренилметилоксикарбонильная (FMOC) и бензилоксикарбонильная (CBZ) группы.
[0140] В общей Схеме 1 исходный материал, R1-замещенный гидрокситиофенол, описываемый Формулой II, может быть получен из коммерческих источников или может быть получен многими способами, хорошо известными в данной области техники. Хотя существует множество способов достижения реакции, описанной Способом A, один удобный способ включает взаимодействие соединений, описанных Формулами II и III, с основанием, таким как карбонат калия, в растворителе, таком как N,N-диметилформамид, при температуре окружающей среды в течение нескольких часов. Следуя стандартным методам экстракции и очистки, продукт, описываемый Формулой IV, может быть получен с хорошим выходом и чистотой.
[0141] Хотя существует множество способов достижения реакции, описанной Способом B, один удобный способ включает взаимодействие соединений, описанных Формулами IV и V (в которых Y представляет собой подходящую уходящую группу, такую как Br, I, OT и OMs) с основанием, таким как карбонат калия в растворителе, таком как N, N-диметилформамид, при температуре окружающей среды в течение нескольких часов. Продукт, описываемый Формулой VI, может быть восстановлен стандартными способами обработки.
[0142] Одним из удобных способов превращения соединений, описанных Формулой VI, в соединения, описанные Формулой VII, является способ C, который включает обработку раствора соединения, описанного Формулой VI, и основания, такого как гидрокарбонат натрия, в растворителе, таком как дихлорметан, окислителем, таким как mCPBA (3-хлорпероксибензойная кислота), при температуре от 0 °C до температуры окружающей среды в течение нескольких часов. Продукт, описываемый Формулой VII, может быть восстановлен стандартными способами обработки.
[0143] Существует множество хорошо установленных химических способов снятия защиты с соединений, описанных Формулой VII, до соединений, описанных Формулой Ib (Способ D). Например, если P1 представляет собой BOC-защитную группу, соединения, описанные Формулой VII, можно обрабатывать кислым веществом, таким как сухой хлористый водород, в растворителе, таком как диэтиловый эфир, с получением соединений, описываемых Формулой Ib, в виде гидрохлоридных солей. Как правило, свободные аминосоединения превращаются в кислотно-аддитивные соли для простоты обращения и для улучшения химической стабильности. Примеры кислотно-аддитивных солей включают, но не ограничиваются ими, гидрохлоридные, гидробромидные, 2,2,2-трифторацетатные и метансульфонатные соли.
Схема 2:
[0144] Получение соединений, описываемых Формулой Ib, где X представляет собой -CONH-, описано на Схеме 2 ниже. Специалист в данной области техники поймет, что соединения, описанные Формулами Ic, Id, Ie, If и Ig, могут быть получены с использованием аналогичных синтетических методик с подходящими исходными материалами.
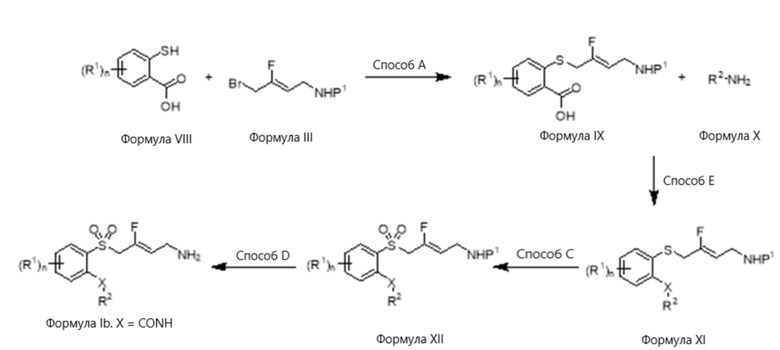
Схема 2
[0145] В общей Схеме 2 исходный материал, R1-замещенная меркаптобензойная кислота, может быть получена из коммерческих источников или может быть получена многими способами, хорошо известными в данной области техники.
[0146] Соединения, описанные Формулой XI, могут быть получены реакцией соответственно замещенного фрагмента бензойной кислоты (описанного формулой IX) с фрагментом амина (формула X) в присутствии подходящего связывающего реагента (такого как HATU) и основания (такого как триэтиламин) в растворителе, таком как N,N-диметилформамид, при температуре окружающей среды в течение нескольких часов (Способ E). Продукт, описываемый Формулой XI, может быть восстановлен с помощью стандартных способов обработки.
Схема 3:
[0147] Получение соединений, описываемых Формулой Ib, где X представляет собой –O-, описано на Схеме 3 ниже. Специалист в данной области техники поймет, что соединения, описанные Формулами Ic, Id, Ie, If и Ig, могут быть получены с использованием аналогичных синтетических методик с подходящими исходными материалами.
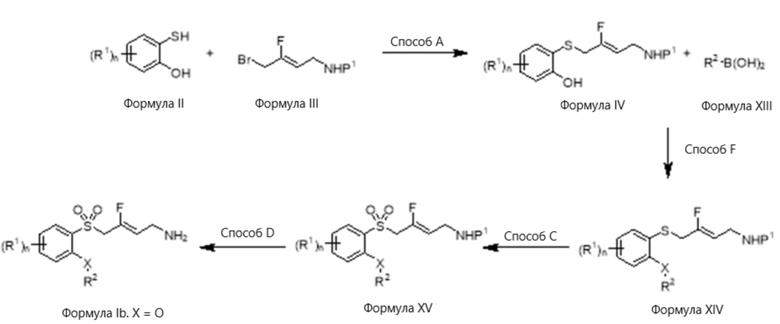
Схема 3
[0148] В общей Схеме 3 исходный материал, R1-замещенный гидрокситиофенол, может быть получен из коммерческих источников или может быть получен многими способами, хорошо известными в данной области техники.
[0149] Модификация катализируемой медью реакции Ульмана может быть использована для связывания соединений, описанных Формулами IV и XIII (Способ F). В литературе описаны многочисленные варианты этого типа реакции, одним из которых является модификация Чана-Эванса-Лама. Соединения, описанные Формулами IV и XIII, в присутствии пиридина можно растворить в растворителе, таком как дихлорметан, а затем обработать ацетатом меди (II) при температуре окружающей среды в течение нескольких часов. Следуя стандартным способам экстракции и очистки, связанный продукт, описанный Формулой XIV, может быть получен с хорошим выходом и чистотой.
Схема 4:
[0150] Получение соединений, описываемых Формулой Ia, описано на Схеме 4 ниже.
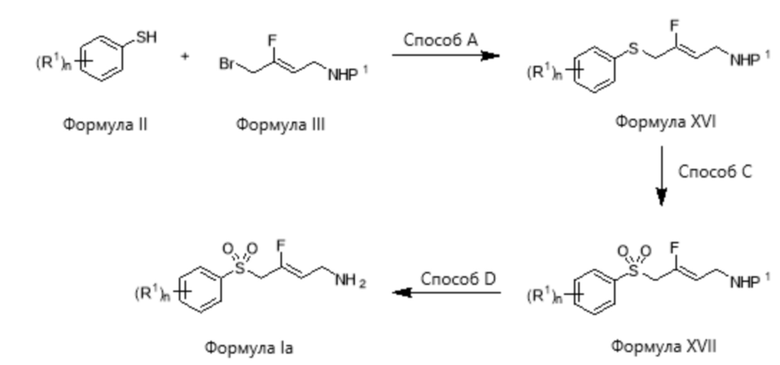
Схема 4
[0151] В общей Схеме 4 исходный материал, R1-замещенный тиол, может быть получен из коммерческих источников или может быть получен многими способами, хорошо известными специалистам в данной области техники.
Схема 5:
[0152] Получение соединений, описываемых Формулой Ia, описано на Схеме 5 ниже.
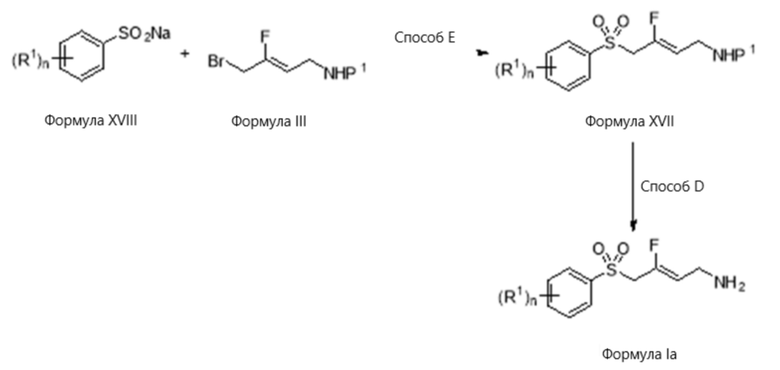
Схема 5
[0153] В общей Схеме 5 исходный материал, R1-замещенный арилсульфинат, описываемый Формулой XVIII, может быть получен из коммерческих источников или может быть получен многими способами, хорошо известными специалистам в данной области техники. Один удобный способ для достижения превращения, описанного Способом E, включает реакцию соединений, описанных Формулами XVIII и III, с основанием, таким как карбонат калия, в растворителе, таком как N,N-диметилформамид, при температуре окружающей среды в течение нескольких часов. Следуя стандартным способам экстракции и очистки, продукт, описанный Формулой XVII, может быть получен с хорошим выходом и чистотой.
[0154] Специалист в данной области техники поймет, что соединения Формулы I, где A представляет собой гетероарил, могут быть получены способами, аналогичными описанным выше.
[0155] Цис/транс (E/Z) изомеры можно разделить обычными способами, хорошо известными специалистам в данной области техники, например, хроматографией и фракционной кристаллизацией.
Терапевтическое использование и составы
[0156] Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение Формулы I или его фармацевтически приемлемую соль или стереоизомер вместе с фармацевтически приемлемым разбавителем, эксципиентом или адъювантом.
[0157] Настоящее изобретение также относится к применению соединений Формулы I в терапии, в частности, для ингибирования членов семейства лизилоксидазы, LOX, LOXL1, LOXL2, LOXL3 и LOXL4. В одном варианте реализации изобретение обеспечивает селективное ингибирование определенных изоферментов лизилоксидазы. В другом варианте реализации изобретение обеспечивает одновременное ингибирование 2, 3 или 4 изоферментов LOX. Относительные ингибирующие способности соединений можно определить по количеству, необходимому для ингибирования аминоксидазной активности LOX, LOXL1, LOXL2, LOXL3 и LOXL4 различными способами, например, в анализе in vitro с рекомбинантным или очищенным человеческим белком или с рекомбинантным или очищенным нечеловеческим ферментом, в клеточных анализах, экспрессирующих нормальный фермент грызунов, в клеточных анализах, которые были трансфицированы человеческим белком, в тестах in vivo на грызунах и других видах млекопитающих и т.п.
[0158] В одном варианте реализации соединения настоящего изобретения являются ингибиторами длительного действия членов семейства лизилоксидаз LOX, LOXL1, LOXL2, LOXL3 и LOXL4. В одном варианте реализации соединений настоящего изобретения являются ингибиторами длительного действия ферментов LOX или LOXL1-4, если ингибирование продолжает превышать 50% активности ферментов LOX или LOXL1-4 после того, как концентрация соединения была снижена ниже IC50. В одном варианте реализации соединения настоящего изобретения демонстрируют устойчивое ингибирование ферментов LOX или LOXL1-4 в течение 24 часов. В одном варианте реализации соединения настоящего изобретения являются необратимыми ингибиторами членов семейства лизилоксидаз LOX, LOXL1, LOXL2, LOXL3 и LOXL4.
[0159] Соответственно, дополнительный аспект изобретения направлен на способ ингибирования активности аминоксидазы любой из LOX, LOXL1, LOXL2, LOXL3 или LOXL4 у субъекта, нуждающегося в этом, включающий введение субъекту эффективного количества соединения Формулы I, или его фармацевтически приемлемой соли или сольвата, или его фармацевтической композиции.
[0160] В одном варианте реализации настоящее изобретение направлено на способ ингибирования активности аминоксидазы LOXL2. В другом варианте реализации настоящее изобретение направлено на ингибирование активности аминоксидазы LOX и LOXL2. В дополнительном варианте реализации настоящее изобретение направлено на способ ингибирования активности аминоксидазы LOX.
[0161] Как обсуждалось ранее, ферменты LOX и LOXL1-4 являются членами большого семейства флавин-зависимых и медь-зависимых аминоксидаз, которое включает SSAO/VAP-1, моноаминоксидазу-B (MAO-B) и диаминоксидазу (DAO). В одном варианте реализации соединения настоящего изобретения селективно ингибируют членов семейства изоферментов лизилоксидазы в отношении SSAO/VAP-1, MAO-B, DAO и других членов семейства аминоксидаз.
[0162] Настоящее изобретение также описывает способы использования соединений, описанных Формулой I, для ингибирования одного или нескольких изоферментов лизилоксидазы (LOX, LOXL1, LOXL2, LOXL3 и LOXL4) у пациентов, страдающих фиброзным заболеванием, и способы лечения фиброзных заболеваний. Кроме того, настоящее изобретение описывает способы использования соединений, описанных Формулой I, для ингибирования одного или нескольких изоферментов лизилоксидазы (LOX, LOXL1, LOXL2, LOXL3 и LOXL4) у пациентов, страдающих раком, включая метастатический рак, и способы лечения рака и метастатического рака.
[0163] В дополнительном аспекте изобретения обеспечивается способ лечения состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения Формулы I, или его фармацевтически приемлемой соли или сольвата, или его фармацевтической композиции.
[0164] В другом аспекте обеспечивается способ лечения состояния, модулируемого любой из LOX, LOXL1, LOXL2, LOXL3 или LOXL4, включающий введение субъекту, который в этом нуждается, терапевтически эффективного количества соединения Формулы I или его фармацевтически приемлемой соли или сольвата, или его фармацевтической композиции.
[0165] В одном варианте реализации способов настоящего изобретения состояние выбрано из группы, состоящей из фиброза, рака и ангиогенеза.
[0166] В другом аспекте настоящее изобретение обеспечивает способ уменьшения образования внеклеточного матрикса путем лечения субъектов-людей, домашних животных и скота фтораллиламиновыми ингибиторами семейства изоферментов лизилоксидазы Формулы I, как описано в данном документе.
[0167] Вышеописанные способы применимы при фиброзе. Используемый в данном документе термин «фиброз» включает такие заболевания, как муковисцидоз, идиопатический фиброз легких, фиброз печени, фиброз почек, склеродермия, радиационно-индуцированный фиброз, болезнь Пейрони, рубцевание и другие заболевания, при которых чрезмерный фиброз способствует патологии заболевания.
[0168] В одном варианте реализации фиброз выбирают из группы, состоящей из медиастинального фиброза, миелофиброза, забрюшинного фиброза, прогрессирующего массивного фиброза, нефрогенного системного фиброза, болезни Крона, келоида, системного склероза, артрофиброза, контрактуры Дюпюитрена, адгезивного капсулита, фиброза поджелудочной железы, фиброза кишечника, фиброза печени, фиброза легких, фиброза почек, фиброза сердца, фибростеноза, муковисцидоза, идиопатического фиброза легких, фиброза, вызванного радиацией, болезни Пейрони и склеродермии или связанного с респираторным заболеванием, аномальным заживлением и восстановлением ран, рубцеванием, гипертрофическим рубцеванием/келоидом, рубцы после операции, остановки сердца и все состояния, при которых избыточное или аберрантное отложение волокнистого материала связано с заболеванием, травмой, имплантатами или операцией. В другом варианте реализации фиброз выбирают из группы, состоящей из фиброза печени, фиброза легких, фиброза почек, фиброза сердца, рубцевания и склеродермии. В дополнительном варианте реализации фиброз выбирают из группы, состоящей из миелофиброза, системного склероза, фиброза печени, фиброза легких, фиброза почек, фиброза сердца и фиброза, вызванного радиацией.
[0169] В одном варианте реализации фиброз почек включает, но не ограничивается ими, диабетическую нефропатию, пузырно-мочеточниковый рефлюкс, тубулоинтерстициальный почечный фиброз, гломерулонефрит или гломерулярный нефрит, включая фокальный сегментарный гломерулосклероз и мембранозный гломерулонефрит, нефропатию IgA и мезангиокапиллярный гломерулонефрит. В одном из вариантов реализации фиброз печени приводит к циррозу и включает связанные состояния, такие как хронический вирусный гепатит, неалкогольное жировое заболевание печени (НАЖЗП), алкогольный стеатогепатит (АСГ), неалкогольный стеатогепатит (НАСГ), первичный билиарный цирроз (ПБЦ), билиарный цирроз и аутоиммунный гепатит.
[0170] В одном варианте реализации фиброз выбран из келоидов, рубцов, рубцов на глазах, гипертрофических рубцов, склеродермии, контрактуры Дюпюитрена и болезни Пейрони. В одном варианте реализации гипертрофическое рубцевание возникает в результате ожога. В одном варианте реализации гипертрофическое рубцевание вызвано внешними повреждениями. В другом варианте реализации гипертрофическое рубцевание вызвано хирургическими процедурами. В одном варианте реализации келоид вызван внешними повреждениями. В другом варианте реализации келоид вызван хирургическими процедурами. В дополнительном варианте реализации келоид является результатом повреждения кожи, вызванного акне, ожогами, ветряной оспой, проколом уха, царапинами, хирургическими порезами или участками вакцинации.
[0171] Вышеописанные способы также применимы, когда состояние представляет собой пролиферативное заболевание, например, рак. В одном варианте реализации рак выбирают из группы, состоящей из рака легких; рака молочной железы; колоректального рака; анального рак; рака поджелудочной железы; рака простаты; рака яичников; карциномы печени и желчных протоков; карциномы пищевода; мезотелиомы, неходжкинская лимфома; карциномы мочевого пузыря; карциномы матки; глиомы, глиобластомы, медуллабластомы и других опухолей головного мозга; миелофиброза, рака почки; рака головы и шеи; рака желудка; множественной миеломы; рака яичек; опухоли половых клеток; нейроэндокринной опухоли; рака шейки матки; рака ротовой полости, карциноиды желудочно-кишечного тракта, груди и других органов; карциномы из перстневидных клеток; мезенхимальных опухолей, включая саркомы, фибросаркомы, гемангиому, ангиоматоз, гемангиоперицитому, псевдоангиоматозную стромальную гиперплазию, миофибробластому, фиброматоз, воспалительную миофибробластическую опухоль, липому, ангиолипому, гранулярно-клеточную опухоль, нейрофиброма, шванному, ангиосаркому, липосаркому, рабдомиосаркому, остеосаркому, лейомиому или лейомиосаркому.
[0172] В одном варианте реализации рак выбирают из группы, состоящей из рака груди, сквамозного рака головы и шеи, рака мозга, рака простаты, почечно-клеточной карциномы, рака печени, рака легких, рака полости рта, рака шейки матки и метастазов опухоли.
[0173] В одном варианте реализации рак легкого включает аденокарциному легкого, сквамозную клеточную карциному, крупноклеточную карциному, бронхоальвеолярную карциному, немелкоклеточную карциному, мелкоклеточную карциному и мезотелиому. В одном варианте реализации рак груди включает протоковую карциному, лобулярную карциному, воспалительный рак груди, светлоклеточную карциному и муцинозную карциному. В одном варианте реализации колоректальный рак включает рак толстой кишки и рак прямой кишки. В одном варианте реализации рак поджелудочной железы включает аденокарциному поджелудочной железы, карциному островковых клеток и нейроэндокринные опухоли.
[0174] В одном варианте реализации карцинома яичников включает эпителиальную карциному яичников или поверхностную эпителиально-стромальную опухоль, включая серозную опухоль, эндометриоидную опухоль и муцинозную цистаденокарциному, и опухоль стромы полового тяжа яичников. В одном варианте реализации карцинома печени и желчных протоков включает гепатоцеллюлярную карциному, холангиокарциному и гемангиому. В одном варианте реализации карцинома пищевода включает аденокарциному пищевода и сквамозную клеточную карциному. В одном варианте реализации карцинома матки включает аденокарциному эндометрия, папиллярную серозную карциному, светлоклеточную карциному матки, саркомы матки и лейомиосаркомы и смешанные опухоли Мюллера. В одном варианте реализации рак почки включает почечно-клеточную карциному, светлоклеточную карциному и опухоль Вильма. В одном варианте реализации рак головы и шеи включает сквамозные клеточные карциномы. В одном варианте реализации рак желудка включает аденокарциному желудка и опухоль стромы желудочно-кишечного тракта.
[0175] В одном варианте реализации рак выбирают из группы, состоящей из рака толстой кишки, рака яичников, рака легких, карциномы пищевода, рака груди и рака простаты. В одном варианте реализации рак выбирают из группы, состоящей из рака поджелудочной железы, рака печени, рака груди, миелофиброза и мезотелиомы.
[0176] В одном варианте реализации соединения настоящего изобретения могут использоваться для лечения неметастатического рака. В другом варианте реализации, соединения настоящего изобретения могут использоваться для лечения метастатического рака. В дополнительном варианте реализации соединения настоящего изобретения могут использоваться для предотвращения или лечения метастазов опухоли.
[0177] Вышеописанные способы применимы, когда состоянием является ангиогенез.
[0178] В одном варианте реализации способов настоящего изобретения субъект выбирают из группы, состоящей из людей, домашних животных и домашнего скота. В другом варианте реализации способов настоящего изобретения субъектом является человек.
[0179] Дальнейший аспект изобретения обеспечивает использование соединения Формулы I или его фармацевтически приемлемой соли или сольвата для производства лекарственного средства для лечения состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
[0180] Другой аспект изобретения предусматривает применение соединения Формулы I или его фармацевтически приемлемой соли или сольвата для производства лекарственного средства для лечения состояния, модулируемого любой из LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
Фармацевтические и/или терапевтические составы
[0181] В другом варианте реализации настоящего изобретения обеспечены композиции, содержащие соединение Формулы I и по меньшей мере один его фармацевтически приемлемый эксципиент, носитель или разбавитель. Соединение(я) Формулы I также может присутствовать в виде подходящих солей, включая фармацевтически приемлемые соли.
[0182] Фраза «фармацевтически приемлемый носитель» относится к любому носителю, известному специалистам в данной области техники как подходящий для конкретного способа введения. Кроме того, соединения могут быть составлены в виде единственного фармацевтически активного ингредиента в композиции или могут быть объединены с другими активными ингредиентами.
[0183] Фраза «фармацевтически приемлемая соль» относится к любому солевому препарату, который подходит для использования в фармацевтике. Под фармацевтически приемлемой солью подразумеваются те соли, которые в рамках здравого медицинского суждения подходят для использования в контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и т. п. и соизмеримы с разумным соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области и включают соли присоединения кислоты и основания. Также могут образовываться полусоли кислот и оснований. Фармацевтически приемлемые соли включают соли аминов минеральных кислот (например, гидрохлориды, гидробромиды, сульфаты и тому подобное); и аминные соли органических кислот (например, формиаты, ацетаты, лактаты, малаты, тартраты, цитраты, аскорбаты, сукцинаты, малеаты, бутираты, валераты, фумараты и тому подобное).
[0184] Для соединений Формулы (I), имеющих основный участок, подходящими фармацевтически приемлемыми солями могут быть соли присоединения кислоты. Например, подходящие фармацевтически приемлемые соли таких соединений могут быть получены путем смешивания фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, метансульфоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, бензойная кислота, фосфорная кислота, уксусная кислота, щавелевая кислота, угольная кислота, винная кислота или лимонная кислота с соединениями изобретения.
[0185] S. M. Berge et al. подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 1977, 66:1-19. Соли могут быть получены in situ во время окончательного выделения и очистки соединений изобретения или отдельно путем взаимодействия функциональной группы свободного основания с подходящей органической кислотой. Типичные соли присоединения кислоты включают ацетат, адипат, альгинат, аскорбат, аспарат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, диглюконат, циклопентанепропионат, додецилсульфат, этансульфонат, фумарат, глюкогептонат, глицерофосфат, гемисульфат, гептонат, гексаноат, гидробромид, гидрохлорид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, толуолсульфонат, ундеканоат, валератные соли и т.п. Подходящие основные соли образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Типичные соли щелочных или щелочноземельных металлов включают натрий, литий, калий, кальций, магний и т.п., а также нетоксичные катионы аммония, четвертичного аммония и аминов, включая, но не ограничиваясь ими, аммоний, тетраметиламмоний, тетраэтиламмоний, диметиламин, триметиламин, триэтиламин, этиламин, триэтаноламин и тому подобное.
[0186] Фармацевтически приемлемые соли соединений Формулы I могут быть получены способами, известными специалистам в данной области техники, включая, например:
(i) путем взаимодействия соединения формулы I с желаемой кислотой или основанием;
(ii) путем удаления неустойчивой к кислотам или основаниям защитной группы из подходящего предшественника соединения формулы I или путем раскрытия цикла подходящего циклического предшественника, например, лактона или лактама, с использованием желаемой кислоты или основания; или
(iii) путем превращением одной соли соединения формулы I в другую реакцией с подходящей кислотой или основанием или при помощи подходящей ионообменной колонки.
[0187] Вышеупомянутые реакции (i)-(iii) обычно проводят в растворе. Полученная соль может выпадать в осадок и собираться фильтрованием или может быть выделена путем выпаривания растворителя. Степень ионизации полученной соли может варьироваться от полностью ионизированной до почти неионизированной.
[0188] Таким образом, например, подходящие фармацевтически приемлемые соли соединений настоящего изобретения могут быть получены путем смешивания фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, метансульфоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, бензойная кислота, фосфорная кислота, уксусная кислота, угольная кислота, винная кислота или лимонная кислота с соединениями изобретения. Подходящие фармацевтически приемлемые соли соединений настоящего изобретения, следовательно, включают соли присоединения кислот.
[0189] Соединения изобретения могут существовать как в несольватированной, так и в сольватированной формах. Термин «сольват» используется в данном документе для описания молекулярного комплекса, содержащего соединение изобретения и стехиометрическое количество одной или нескольких фармацевтически приемлемых молекул растворителя, например этанола. Термин «гидрат» используется, когда растворителем является вода.
[0190] В одном из вариантов реализации соединения Формулы I можно вводить в форме «пролекарства». Фраза «пролекарство» относится к соединению, которое при введении in vivo метаболизируется за одну или несколько стадий или процессов или иным образом превращается в биологически, фармацевтически или терапевтически активную форму соединения. Пролекарства могут быть получены модификацией функциональных групп, присутствующих в соединении, таким образом, что модификации расщепляются либо обычными способами, либо in vivo до соединения, описанного в данном документе. Например, пролекарства включают соединения настоящего изобретения, в которых гидрокси, амино или углеводная группа связана с любой группой, которая при введении субъекту-млекопитающему может расщепляться с образованием свободной гидроксильной, свободной амино или свободной группы карбоновой кислоты, соответственно. Типичные пролекарства включают, например, амиды, сложные эфиры, простые эфиры енола, сложные эфиры енола, ацетаты, формиаты, бензоатные производные и т.п. спиртовых и аминовых функциональных групп в соединениях настоящего изобретения. Форму пролекарства можно выбрать из таких функциональных групп, как -C(O)алкил, -C(O)циклоалкил, -C(O)арил, -C(O)-арилалкил, C(O)гетероарил, -C(O)-гетероарилалкил или тому подобное. На основании знаний о фармакодинамических процессах и метаболизме лекарственных средств in vivo специалисты в данной области техники, как только станет известно фармацевтически активное соединение, могут разработать пролекарства соединения (см., например , Nogrady (1985) Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, pages 388–392).
[0191] Композиции в данном документе содержат одно или несколько соединений, представленных в настоящем документе. В одном варианте реализации соединения входят в состав подходящих фармацевтических препаратов, таких как растворы, суспензии, таблетки, кремы, гели, диспергируемые таблетки, пилюли, капсулы, порошки, составы с замедленным высвобождением или эликсиры, для перорального введения или в стерильных растворах или суспензиях для парентерального введения, а также составы трансдермальных пластырей и ингаляторы сухого порошка. В одном варианте реализации описанные выше соединения составляют в фармацевтические композиции с использованием способов и процедур, хорошо известных в данной области (см., например, Ansel Introduction to Pharmaceutical Dosage Forms, Fourth Edition 1985, 126).
[0192] В композициях эффективные концентрации одного или нескольких соединений или их фармацевтически приемлемых производных смешиваются с подходящим фармацевтическим носителем. Соединения могут быть дериватизированы в виде соответствующих солей, сложных эфиров, простых или сложных эфиров енолов, ацеталей, кеталей, ортоэфиров, гемиацеталей, гемикеталей, кислот, оснований, сольватов, гидратов или пролекарств перед составлением, как описано выше. Концентрации соединений в композициях эффективны для доставки количества при введении, которое лечит, предотвращает или облегчает один или несколько симптомов заболеваний или расстройств, подлежащих лечению.
[0193] В одном варианте реализации композиции составлены для введения разовой дозы. Для составления композиции массовая доля соединения растворяется, суспендируется, диспергируется или иным образом смешивается в выбранном носителе в эффективной концентрации, так что излеченное состояние облегчается, предотвращается или облегчается один или несколько симптомов.
[0194] Активное соединение включается в фармацевтически приемлемый носитель в количестве, достаточном для оказания терапевтически полезного эффекта при отсутствии нежелательных побочных эффектов у пациента, которого лечат. Терапевтически эффективная концентрация может быть определена эмпирически путем тестирования соединений в системах in vitro и in vivo, описанных в настоящем документе, и затем экстраполирована оттуда для доз для людей.
[0195] Концентрация активного соединения в фармацевтической композиции будет зависеть от скорости всасывания, распределения, инактивации и элиминации активного соединения, физико-химических характеристик соединения, схемы дозирования и вводимого количества, а также других факторов, известных специалистам в данной области техники.
[0196] В одном варианте реализации терапевтически эффективная доза должна обеспечивать концентрацию сыворотки активного ингредиента от около 0,1 нг/мл до около 50 - 100 мкг/мл. В другом варианте реализации фармацевтические композиции должны обеспечивать дозу от около 0,001 мг до около 2000 мг соединения на килограмм веса тела в день. Фармацевтические стандартные лекарственные формы получают для обеспечения от около 0,01 мг, 0,1 мг или от 1 мг до около 500 мг, 1000 мг или 2000 мг и в одном варианте реализации от около 10 мг до около 500 мг активного ингредиента или комбинации основных ингредиентов на единичную стандартную лекарственную форму.
[0197] Дозирование может происходить с интервалами в минуты, часы, дни, недели, месяцы или годы или непрерывно в течение любого из этих периодов. Подходящие дозировки лежат в диапазоне от около 0,1 нг на кг массы тела до 0,1 г на кг массы тела на дозу. Дозировка предпочтительно находится в диапазоне от 10 мкг до 0,1 г на кг массы тела на дозу, например, в диапазоне от 0,1 мг до 0,01 г на кг массы тела на дозу. Соответственно, доза находится в диапазоне от 10 мкг до 50 мг на кг массы тела на дозу, например, от 10 мкг до 20 мг на кг массы тела на дозу, или от 10 мкг до 10 мг на кг массы тела на дозу. Другие подходящие дозировки могут находиться в диапазоне от 0,1 мг до 25 мг на кг массы тела, включая от 0,1 мг до 10, 20, 50 или 100 мг на кг массы тела на дозу.
[0198] Альтернативно, эффективная доза может составлять до около 10 мг/см2 или может составлять до около 1 мг/см2, около 0,5 мг/см2, около 0,2 мг/см2, около 0,1 мг/см2, около 0,05 мг/см2, около 0,02 мг/см2 или около 0,01 мг/см2. Эффективная доза может быть, например, в диапазоне от около 10 мкг/см2 до около 1 мг/см2, от около 10 мкг/см2 до около 0,1 мг/см2, от около 10 мкг/см2 до около 0,01 мг/см2, от около 10 мкг/см2 до около 500 мкг/см2, от около 10 мкг/см2 до около 200 мкг/см2, от около 10 мкг/см2 до около 100 мкг/см2, от около 10 мкг/см2 до около 50 мкг/см2, от около 20 мкг/см2 до около 1 мг/см2, от около 50 мкг/см2 до около 1 мг/см2, от около 100 мкг/см2 до около 1 мг/см2, от около 200 мкг/см2 до около 1 мг/см2, от около 500 мкг/см2 до около 1 мг/см2, от около 50 мкг/см2 до около 500 мкг/см2, от около 50 мкг/см2 до около 200 мкг/см2, от около 100 мкг/см2 до около 500 мкг/см2 или от около 200 мкг/см2 до около 500 мкг/см2.
[0199] Подходящие количества доз и режимы дозирования могут быть определены лечащим врачом и могут зависеть от конкретного состояния, которое лечат, серьезности состояния, а также общего состояния здоровья, возраста и массы субъекта.
[0200] В случаях, когда соединения проявляют недостаточную растворимость, можно использовать способы солюбилизации соединений. Такие способы известны специалистам в данной области техники, и включают в себя, но не ограничиваются ими, использование сорастворителей, таких как диметилсульфоксид (DMSO), использование поверхностно-активных веществ, таких как TWEEN®, растворение в водном растворе бикарбоната натрия, составление соединений, представляющих интерес в виде наночастиц и тому подобное. Производные соединений, такие как пролекарства соединений, также можно использовать при составлении эффективных фармацевтических композиций.
[0201] При смешивании или добавлении соединения(й) полученная смесь может представлять собой раствор, суспензию, эмульсию и т.п. Форма полученной смеси зависит от ряда факторов, включая предполагаемый способ введения и растворимость соединения в выбранном носителе или растворителе. Эффективная концентрация достаточна для облегчения симптомов заболевания, расстройства или состояния, из которого проводится лечение, и может быть определена эмпирически.
[0202] Фармацевтические композиции предназначены для введения людям и животным в стандартных лекарственных формах, таких как таблетки, капсулы, пилюли, порошки, гранулы, стерильные парентеральные растворы или суспензии, и пероральные растворы или суспензии, и эмульсии масло-вода, содержащие подходящие количества соединения или их фармацевтически приемлемые производные. Фармацевтически терапевтически активные соединения и их производные в одном варианте реализации составляют и вводят в виде однократных дозированных форм или многократных дозированных форм. Активный ингредиент можно вводить сразу или можно разделить на несколько меньших доз для введения через определенные промежутки времени. Используемые в данном документе однократные дозированные формы относятся к физически дискретным единицам, подходящим для людей и животных и упакованным индивидуально, как известно в данной области. Каждая однократная доза содержит заранее определенное количество терапевтически активного соединения, достаточное для получения желаемого терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем, растворителем или разбавителем. Примеры однократных дозированных форм включают ампулы и шприцы и индивидуально упакованные таблетки или капсулы. Однократные дозированные формы можно вводить дробно или кратно. Многократная дозированная форма представляет собой множество идентичных форм однократной дозы, упакованных в один контейнер, для введения в отдельной форме однократной дозы. Примеры форм с многократными дозами включают флаконы, сосуды с таблетками или капсулами или сосуды с пинтами или галлонами. Следовательно, форма многократной дозы представляет собой множество однократных доз, которые не разделены в упаковке.
[0203] Фактические способы получения таких дозированных форм известны или будут очевидны специалистам в данной области техники; например, см. Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 15th Edition, 1975.
[0204] Могут быть получены дозированные формы или композиции содержащие активный ингредиент в диапазоне от 0,005% до 100% (мас.%), при этом остальное составляет нетоксичный носитель. Способы получения этих композиций известны специалистам в данной области. Предполагаемые композиции могут содержать 0,001% -100% (мас.%) активного ингредиента, в одном варианте реализации 0,1-95% (мас.%), в другом варианте 75-85% (мас.%).
Режимы введения
[0205] Удобные способы введения включают инъекцию (подкожную, внутривенную и т.д.), пероральное введение, ингаляцию, трансдермальное применение, кремы или гели или порошки для местного применения, вагинальное или ректальное введение. В зависимости от пути введения, состав и/или соединение могут быть покрыты материалом для защиты соединения от действия ферментов, кислот и других природных условий, которые могут инактивировать терапевтическую активность соединения. Соединение также можно вводить парентерально или внутрибрюшинно.
Композиции для перорального введения
[0206] Фармацевтические дозированные формы для перорального применения могут быть твердыми, гелевыми или жидкими. Твердые дозированные формы представляют собой таблетки, капсулы, гранулы и сыпучие порошки. Типы пероральных таблеток включают прессованные жевательные пастилки и таблетки, которые могут иметь энтеросолюбильное покрытие, сахарное покрытие или пленочное покрытие. Капсулы могут быть твердыми или мягкими желатиновыми капсулами, тогда как гранулы и порошки могут быть предоставлены в не шипучей или шипучей форме с комбинацией других ингредиентов, известных специалистам в данной области техники.
Твердые композиции для перорального применения
[0207] В некоторых вариантах реализации составы представляют собой твердые дозированные формы, в одном варианте реализации представляют собой капсулы или таблетки. Таблетки, пилюли, капсулы, троше и т.п. могут содержать один или несколько из следующих ингредиентов или соединений аналогичной природы: связующее; смазывающее; разбавитель; скользящее; разрыхлитель; краситель; подсластитель; ароматизатор; смачивающий агент; рвотное покрытие; и пленочное покрытие. Примеры связующих включают микрокристаллическую целлюлозу, трагакантовую камедь, раствор глюкозы, слизь акации, раствор желатина, мелассу, полвинилпирролидин, повидон, кросповидоны, сахарозу и крахмальную пасту. Смазывающие включают тальк, крахмал, стеарат магния или кальция, ликоподий и стеариновую кислоту. Разбавители включают, например, лактозу, сахарозу, крахмал, каолин, соль, маннит и дикальцийфосфат. Глиданты включают, но не ограничиваются ими, коллоидный диоксид кремния. Разрыхлители включают кросскармеллозу натрия, крахмалгликолят натрия, альгиновую кислоту, кукурузный крахмал, картофельный крахмал, бентонит, метилцеллюлозу, агар и карбоксиметилцеллюлозу. Красители включают, например, любые утвержденные сертифицированные водорастворимые красители FD и C, их смеси; и водонерастворимые красители FD и C, суспендированные на гидрате оксида алюминия. Подсластители включают сахарозу, лактозу, маннит и искусственные подсластители, такие как сахарин, и любое количество ароматизаторов, высушенных распылением. Ароматизаторы включают натуральные ароматизаторы, экстрагированные из растений, таких как фрукты, и синтетические смеси соединений, которые вызывают приятное ощущение, такие как, помимо прочего, перечная мята и метилсалицилат. Смачивающие агенты включают моностеарат пропиленгликоля, моноолеат сорбитана, монолаурат диэтиленгликоля и полиоксиэтиленлауральный эфир. Рвотные покрытия включают жирные кислоты, жиры, воски, шеллак, аммонизированный шеллак и фталаты ацетата целлюлозы. Пленочные покрытия включают гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу натрия, полиэтиленгликоль 4000 и фталат ацетата целлюлозы.
[0208] Соединение или его фармацевтически приемлемое производное может быть представлено в композиции, которая защищает его от кислой среды желудка. Например, композиция может быть получена в виде энтеросолюбильного покрытия, которое сохраняет свою целостность в желудке и высвобождает активное соединение в кишечнике. Композиция также может быть получена в комбинации с антацидом или другим подобным ингредиентом.
[0209] Когда стандартная дозированная форма представляет собой капсулу, она может содержать, помимо материала вышеуказанного типа, жидкий носитель, такой как жирное масло. Кроме того, однократные дозированные формы могут содержать различные другие материалы, которые модифицируют физическую форму дозированной единицы, например, покрытия из сахара и других энтеросолюбильных агентов. Соединения также можно вводить в виде компонента эликсира, суспензии, сиропа, капсулы-импланта, посыпки, жевательной резинки и т.п. Сироп может содержать, помимо активных соединений, сахарозу в качестве подслащивающего агента, и некоторые консерванты, краски, красители и ароматизаторы.
[0210] Активные материалы также могут быть смешаны с другими активными материалами, которые не ухудшают желаемое действие, или с материалами, которые дополняют желаемое действие, такими как антациды, блокаторы H2 и диуретики. Активный ингредиент представляет собой соединение или его фармацевтически приемлемое производное, как описано в данном документе. Могут быть включены более высокие концентрации, примерно до 98% по массе активного ингредиента.
[0211] Во всех вариантах реализации на составы таблеток и капсул может быть нанесено покрытие, известное специалистам в данной области, чтобы модифицировать или поддерживать растворение активного ингредиента. Так, например, они могут быть покрыты обычным энтеросолюбильным покрытием, таким как фенилсалицилат, воски и фталат ацетата целлюлозы.
Жидкие композиции для перорального введения
[0212] Жидкие пероральные лекарственные формы включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, восстановленные из не шипучих гранул, и шипучие препараты, восстановленные из шипучих гранул. К водным растворам относятся, например, эликсиры и сиропы. Эмульсии либо масло в воде, либо вода в масле.
[0213] Жидкие фармацевтически вводимые композиции можно, например, получить путем растворения, диспергирования или иного смешивания активного соединения, как определено выше, и необязательных фармацевтических адъювантов в носителе, таком как, например, вода, физиологический раствор, водная декстроза, глицерин, гликоли, этанол и т. п. с образованием раствора или суспензии. Если желательно, фармацевтическая композиция для введения может также содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие агенты, эмульгирующие агенты, солюбилизирующие агенты, буферные агенты pH и тому подобное, например, ацетат, цитрат натрия, производные циклодекстрина, монолаурат сорбитана, триэтаноламин ацетата натрия, триэтаноламина олеат и другие подобные агенты.
[0214] Эликсиры - это прозрачные подслащенные водно-спиртовые препараты. Фармацевтически приемлемые носители, используемые в эликсирах, включают растворители. Сиропы представляют собой концентрированные водные растворы сахара, например, сахарозы, и могут содержать консервант. Эмульсия представляет собой двухфазную систему, в которой одна жидкость диспергирована в виде небольших шариков в другой жидкости. Фармацевтически приемлемые носители, используемые в эмульсиях, представляют собой неводные жидкости, эмульгирующие агенты и консерванты. В суспензиях используются фармацевтически приемлемые суспендирующие агенты и консерванты. Фармацевтически приемлемые вещества, используемые в нешипучих гранулах, которые должны быть восстановлены в жидкую пероральную дозированную форму, включают разбавители, подсластители и смачивающие вещества. Фармацевтически приемлемые вещества, используемые в шипучих гранулах, подлежащих восстановлению в жидкую пероральную дозированную форму, включают органические кислоты и источник диоксида углерода. Красители и ароматизаторы используются во всех вышеуказанных лекарственных формах.
[0215] Растворители включают глицерин, сорбит, этиловый спирт и сироп. Примеры консервантов включают глицерин, метил и пропилпарабен, бензойную кислоту, бензоат натрия и этанол. Примеры неводных жидкостей, используемых в эмульсиях, включают минеральное масло и хлопковое масло. Примеры эмульгаторов включают желатин, гуммиарабик, трагакант, бентонит и поверхностно-активные вещества, такие как моноолеат полиоксиэтиленсорбитана. Суспендирующие агенты включают карбоксиметилцеллюлозу натрия, пектин, трагакант, Veegum и гуммиарабик. Подслащивающие вещества включают сахарозу, сиропы, глицерин и искусственные подсластители, такие как сахарин. Смачивающие агенты включают моностеарат пропиленгликоля, моноолеат сорбитана, монолаурат диэтиленгликоля и лауриловый эфир полиоксиэтилена. Органические кислоты включают лимонную и винную кислоты. Источники диоксида углерода включают бикарбонат натрия и карбонат натрия. Красители включают любые утвержденные сертифицированные водорастворимые красители FD и C, и их смеси. Ароматизаторы включают натуральные ароматизаторы, экстрагированные из растений, таких как фрукты, и синтетические смеси соединений, которые вызывают приятное вкусовое ощущение.
[0216] Для твердой дозированной формы раствор или суспензия, например, в пропиленкарбонате, растительных маслах или триглицеридах, в одном варианте реализации инкапсулируют в желатиновую капсулу. Для жидкой дозированной формы раствор, например, например, в полиэтиленгликоле, может быть разбавлен достаточным количеством фармацевтически приемлемого жидкого носителя, например, воды, чтобы его можно было легко измерить для введения.
[0217] Альтернативно, жидкие или полутвердые пероральные составы могут быть получены путем растворения или диспергирования активного соединения или соли в растительных маслах, гликолях, триглицеридах, сложных эфирах пропиленгликоля (например, пропиленкарбонате) и других подобных носителях и инкапсуляции этих растворов или суспензий в твердые или мягкие желатиновые оболочки капсул. Другие полезные составы включают составы, изложенные в патентах US №№ RE28,819 и 4,358,603. Вкратце, такие составы включают, но не ограничиваются ими, те, которые содержат соединение, представленное в настоящем документе, диалкилированный моно- или полиалкиленгликоль, включая, но не ограничиваясь ими, 1,2-диметоксиметан, диглим, триглим, тетраглим, полиэтиленгликоль-350-диметиловый эфир, полиэтиленгликоль-550-диметиловый эфир, полиэтиленгликоль-750-диметиловый эфир, где 350, 550 и 750 относятся к приблизительной средней молекулярной массе полиэтиленгликоля, и одного или нескольких антиоксидантов, таких как бутилированный гидрокситолуол (BHT), бутилированный гидроксианизол (BHA), пропилгаллат, витамин E, гидрохинон, гидроксикумарины, этаноламин, лецитин, цефалин, аскорбиновая кислота, яблочная кислота, сорбитол, фосфорная кислота, тиодипропионовая кислота и ее эфиры, а также дитиокарбаматы.
[0218] Другие составы включают, но не ограничиваются ими, водно-спиртовые растворы, включая фармацевтически приемлемый ацеталь. Спирты, используемые в этих составах, представляют собой любые фармацевтически приемлемые смешиваемые с водой растворители, имеющие одну или несколько гидроксильных групп, включая, но не ограничиваясь ими, пропиленгликоль и этанол. Ацетали включают, но не ограничиваются ими, ди(низший алкил)ацетали низших алкилальдегидов, такие как диэтилацеталь ацетальдегида.
Инъекции, растворы и эмульсии
[0219] В данном документе также рассматривается парентеральное введение в одном варианте реализации, характеризующееся инъекцией подкожно, внутримышечно или внутривенно. Инъекционные препараты могут быть получены в обычных формах, либо в виде жидких растворов или суспензий, твердых форм, подходящих для растворения или суспендирования в жидкости перед инъекцией, либо в виде эмульсий. Инъекции, растворы и эмульсии также содержат один или несколько эксципиентов. Подходящими эксципиентами являются, например, вода, физиологический раствор, декстроза, глицерин или этанол. Кроме того, при желании вводимые фармацевтические композиции могут также содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие или эмульгирующие агенты, буферные агенты pH, стабилизаторы, усилители растворимости и другие подобные агенты, такие как, например, натрий ацетат, монолаурат сорбитана, олеат триэтаноламина и циклодекстрины.
[0220] В данном документе также рассматривается имплантация системы с медленным или замедленным высвобождением, так что поддерживается постоянный уровень дозировки. Вкратце, соединение, представленное в данном документе, диспергировано в твердой внутренней матрице, например, полиметилметакрилат, полибутилметакрилат, пластифицированный или непластифицированный поливинилхлорид, пластифицированный нейлон, пластифицированный полиэтилентерефталат, натуральный каучук, полиизопрен, полиизобутилен, полибутадиен, полиэтилен, сополимеры этилен-винилацетатата, силиконовый каучук, полидиметилсилоксаны, сополимеры карбоната силикона, гидрофильные полимеры, такие как гидрогели сложных эфиров акриловой и метакриловой кислоты, коллаген, поперечно сшитый поливиниловый спирт и поперечно сшитый частично гидролизованный поливинилацетат, который окружен внешней полимерной мембраной, например полиэтиленом, полипропиленом, сополимеры этилена/пропилена, сополимеры этилена/этилакрилата, сополимеры этилена/винилацетата, силиконовые каучуки, полидиметилсилоксаны, неопреновый каучук, хлорированный полиэтилен, поливинилхлорид, сополимеры винилхлорида с винилацетатом, винилиденхлорид, этилен и пропилен, иономерный полиэтилентерефталат, бутилкаучуковые эпихлоргидриновые каучуки, сополимер этилена/винилового спирта, терполимер этилена/винилацетата/винилового спирта и сополимер этилена/винилоксиэтанола, который нерастворим в жидкостях организма. Соединение диффундирует через внешнюю полимерную мембрану на стадии регулирования скорости высвобождения. Процент активного соединения, содержащегося в таких парентеральных композициях, сильно зависит от его конкретной природы, а также от активности соединения и потребностей пациента.
[0221] Парентеральное введение композиций включает внутривенное, подкожное и внутримышечное введение. Препараты для парентерального введения включают стерильные растворы, готовые к инъекции, стерильные сухие растворимые продукты, такие как лиофилизированные порошки, готовые к смешиванию с растворителем непосредственно перед использованием, включая таблетки для подкожных инъекций, стерильные суспензии, готовые к инъекции, стерильные сухие нерастворимые продукты, готовые к употреблению в комбинации с растворителем непосредственно перед использованием и стерильными эмульсиями. Растворы могут быть водными или неводными.
[0222] При внутривенном введении подходящие носители включают физиологический раствор или фосфатно-солевой буфер (PBS), и растворы, содержащие загущающие и солюбилизирующие агенты, такие как глюкоза, полиэтиленгликоль и полипропиленгликоль, и их смеси.
[0223] Фармацевтически приемлемые носители, используемые в парентеральных препаратах, включают водные растворители, неводные растворители, противомикробные агенты, изотонические агенты, буферы, антиоксиданты, местные анестетики, суспендирующие и диспергирующие агенты, эмульгирующие агенты, секвестрирующие или хелатирующие агенты и другие фармацевтически приемлемые вещества.
[0224] Примеры водных растворителей включают инъекцию хлорида натрия, инъекцию Рингера, инъекцию изотонической декстрозы, инъекцию стерильной воды, инъекцию декстрозы и инъекцию лактата Рингера. Неводные растворители для парентерального введения включают жирные масла растительного происхождения, оливковое масло, хлопковое масло, кукурузное масло, кунжутное масло и арахисовое масло. Противомикробные агенты в бактериостатических или фунгистатических концентрациях должны добавляться к парентеральным препаратам, упакованным в многодозовые контейнеры, которые включают фенолы или крезолы, ртути, бензиловый спирт, хлорбутанол, сложные эфиры метил и пропил-пара-гидроксибензойной кислоты, тимеросал, хлорид бензалкония и хлорид бензетония. Изотонические агенты включают хлорид натрия и декстрозу. Буферы включают фосфат и цитрат. Антиоксиданты включают бисульфат натрия. Местные анестетики включают гидрохлорид прокаина. Суспендирующие и диспергирующие агенты включают карбоксиметилцеллюлозу натрия, гидроксипропилметилцеллюлозу и поливинилпирролидон. Эмульгирующие агенты включают Полисорбат 80 (TWEEN® 80). Агенты, секвестрирующие или хелатирующие ионы металлов включают EDTA. Фармацевтические носители также включают этиловый спирт, полиэтиленгликоль и пропиленгликоль для смешивающихся с водой растворителей; и гидроксид натрия, соляная кислота, лимонная кислота или молочная кислота для регулирования pH.
[0225] Концентрация фармацевтически активного соединения регулируется таким образом, чтобы инъекция обеспечивала эффективное количество для получения желаемого фармакологического эффекта. Точная доза зависит от возраста, массы и состояния пациента или животного, как известно в данной области.
[0226] Однократные дозы парентеральных препаратов расфасованы в ампулы, флаконы или шприцы с иглой. Все препараты для парентерального введения должны быть стерильными, как известно и практикуется в данной области.
[0227] Например, внутривенная или внутриартериальная инфузия стерильного водного раствора, содержащего активное соединение, является эффективным способом введения. Другой вариант реализации представляет собой стерильный водный или масляный раствор или суспензию, содержащую активное вещество, вводимое по мере необходимости для получения желаемого фармакологического эффекта.
[0228] Инъекционные препараты предназначены для местного и системного применения. В одном варианте реализации терапевтически эффективная дозировка составляется таким образом, чтобы она содержала концентрацию от по меньшей мере около 0,1% мас./мас. до около 90% мас./мас. или более, в некоторых вариантах реализации более 1% мас./мас. активного соединения к обработанной ткани (ям).
[0229] Соединение может быть суспендировано в микронизированной или другой подходящей форме или может быть дериватизировано для получения более растворимого активного продукта или для получения пролекарства. Форма полученной смеси зависит от ряда факторов, включая предполагаемый способ введения и растворимость соединения в выбранном носителе или растворителе. Эффективная концентрация достаточна для облегчения симптомов состояния и может быть определена эмпирически.
Лиофилизированные порошки
[0230] В данном документе интерес представляют также лиофилизированные порошки, которые можно восстановить для введения в виде растворов, эмульсий и других смесей. Они также могут быть восстановлены и получены в виде твердых веществ или гелей.
[0231] Стерильный лиофилизированный порошок получают растворением соединения, представленного в данном документе, или его фармацевтически приемлемого производного в подходящем растворителе. Растворитель может содержать эксципиент, улучшающий стабильность, или другой фармакологический компонент порошка или восстановленного раствора, полученного из порошка. Эксципиенты, которые можно использовать, включают, но не ограничиваются ими, декстрозу, сорбитал, фруктозу, кукурузный сироп, ксилит, глицерин, глюкозу, сахарозу или другой подходящий агент. Растворитель также может содержать буфер, такой как цитрат, фосфат натрия или калия, или другой такой буфер, известный специалистам в данной области техники, в одном варианте реализации при около нейтральном pH. Последующая стерильная фильтрация раствора с последующей лиофилизацией в стандартных условиях, известных специалистам в данной области, обеспечивает желаемый состав. В одном варианте реализации полученный раствор будет распределяться по флаконам для лиофилизации. Каждый флакон будет содержать одну или несколько доз соединения. Лиофилизированный порошок можно хранить в соответствующих условиях, например при температуре от около 4 °C до комнатной.
[0232] Восстановление этого лиофилизированного порошка водой для инъекций обеспечивает состав для парентерального введения. Для восстановления лиофилизированный порошок добавляют в стерильную воду или другой подходящий носитель. Точное количество зависит от выбранного соединения. Такое количество можно определить эмпирически.
Местное применение
[0233] Смеси для местного нанесения готовят, как описано для местного и системного применения. Полученная смесь может быть раствором, суспензией, эмульсиями и т. п. и составлены в виде кремов, гелей, мазей, эмульсий, растворов, эликсиров, лосьонов, суспензий, настоек, паст, пен, аэрозолей, орошений, спреев, суппозиториев, повязок, кожных пластырей или любых других составов, подходящих для местного применения.
[0234] Соединения или их фармацевтически приемлемые производные могут быть получены в виде аэрозолей для местного применения, например, для ингаляции. Эти составы для введения в дыхательные пути могут быть в форме аэрозоля или раствора для распылителя или в виде мелкодисперсного порошка для инсуффляции, отдельно или в комбинации с инертным носителем, таким как лактоза. В таком случае частицы состава в одном варианте реализации будут иметь диаметр менее 50 микрон, в одном варианте менее 10 микрон.
[0235] Соединения могут быть составлены для локального или местного применения, например, для местного нанесения на кожу и слизистые оболочки, например, в глаза, в форме гелей, кремов и лосьонов, а также для нанесения на глаза или для интрацистернального или интраспинального нанесения. Предусматривается местное применение для трансдермальной доставки, и для применения в глаза или слизистую оболочку или для ингаляционной терапии. Также можно вводить назальные растворы активного соединения отдельно или в комбинации с другими фармацевтически приемлемыми эксципиентами.
[0236] Эти растворы, особенно те, которые предназначены для офтальмологического применения, могут быть составлены в виде 0,01-10% (об.%) изотонических растворов, pH около 5-7, с соответствующими солями.
Композиции для других путей введения
[0237] В данном документе также предусмотрены другие пути введения, такие как трансдермальные пластыри, включая устройства для ионтофоретики и электрофоретики, вагинальное и ректальное введение.
[0238] Трансдермальные пластыри, включая устройства для ионтофоретики и электрофоретики, хорошо известны специалистам в данной области техники. Например, фармацевтические дозированные формы для ректального введения представляют собой ректальные суппозитории, капсулы и таблетки для системного действия. Используемые в данном документе ректальные суппозитории означают твердые тела для введения в прямую кишку, которые плавятся или размягчаются при температуре тела, высвобождая один или несколько фармакологически или терапевтически активных ингредиентов. Фармацевтически приемлемые вещества, используемые в ректальных суппозиториях, представляют собой основы или растворители и агенты для повышения температуры плавления. Примеры основ включают масло какао (масло теобромы), глицерин-желатин, карбовакс (полиоксиэтиленгликоль) и соответствующие смеси моно-, ди- и триглицеридов жирных кислот. Могут использоваться комбинации различных оснований. К агентам, повышающим точку плавления суппозиториев, относятся спермацет и воск. Ректальные суппозитории могут быть получены прессованным способом или формованием. В одном варианте реализации масса ректального суппозитория составляет от около 2 до 3 граммов.
[0239] Таблетки и капсулы для ректального введения производятся с использованием того же фармацевтически приемлемого вещества и теми же способами, что и составы для перорального введения.
Целевые составы
[0240] Предлагаемые в данном документе соединения или их фармацевтически приемлемые производные также могут быть составлены для нацеливания на конкретную ткань, рецептор или другую область тела субъекта, подлежащего лечению. Многие такие способы нацеливания хорошо известны специалистам в данной области. Все такие способы нацеливания рассматриваются в настоящем документе для использования в настоящих композициях.
[0241] В одном варианте реализации липосомные суспензии, включая липосомы, нацеленные на ткань, такие как липосомы, нацеленные на опухоль, также могут быть подходящими в качестве фармацевтически приемлемых носителей. Их можно получить способами, известными специалистам в данной области. Например, липосомные композиции могут быть получены, как описано в патенте US № 4,522,811. Вкратце, липосомы, такие как многослойные везикулы (MLV), могут быть образованы путем высушивания яичного фосфатидилхолина и фосфатидилсерина мозга (молярное соотношение 7:3) внутри колбы. Добавляют раствор соединения, обеспеченного в настоящем документе, в забуференном фосфатом физиологическом растворе, не содержащем двухвалентных катионов (PBS), и встряхивают колбу до диспергирования липидной пленки. Полученные везикулы промывают для удаления неинкапсулированного соединения, осаждают центрифугированием и затем ресуспендируют в PBS.
Совместное применение с другими препаратами
[0242] В соответствии с другим аспектом настоящего изобретения предполагается, что соединения Формулы I, как описано в данном документе, можно вводить субъекту, нуждающемуся в этом, в комбинации с лекарством, которое специалисты в данной области считают текущим стандартом лечения интересующего состояния. Такие комбинации обеспечивают одно или несколько преимуществ для субъекта, например, требуя уменьшенных доз для достижения аналогичного эффекта, получения желаемого паллиативного эффекта за меньшее время и т. п.
[0243] Соединения в соответствии с настоящим изобретением можно вводить как часть терапевтического режима с другими лекарственными средствами. Может быть желательно ввести комбинацию активных соединений, например, с целью лечения конкретного заболевания или состояния. Соответственно, в рамках настоящего изобретения две или несколько фармацевтических композиций, по меньшей мере одна из которых содержит соединение Формулы (I) настоящего изобретения, могут быть скомбинированы в форме набора, подходящего для совместного использования композиций.
[0244] В одном варианте реализации способов настоящего изобретения соединение Формулы I можно вводить со вторым терапевтическим агентом. В одном варианте реализации второй терапевтический агент может быть выбран из одной или нескольких из следующих категорий:
(i) Противораковые агенты, такие как цисплатин, оксалиплатин, карбоплатин, циклофосфамид, азотистый иприт, урациловый иприт, бендамустин, мелфалан, хлорамбуцил, хлорметин, бусульфан, темозоломид, нитрозомочевины, ифозамид, пипобромин, триэтиленмеламин, триэтилентиофосфорамин, кармустин, ломустин, строптозоцин и дакарбазин, гемцитабин, фосгемцитабин, палабенамид, 5-фторурацил, тегафур, ралтитрексед, метотрексат, пеметрексед, лейковорин, цитозинарабинозид, флоксуридин, цитарабин, 6-меркаптопурин, 6-тиогуанин, флударабин фосфат, пентостатин, гидроксимочевина, трифлуридин, трифурацил, адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-C, дактиномицин, митрамицин, винкристин, винбластин, виндезин и винорелбин, таксол, таксотер, эрибулин, карфилзомиб, бортезомиб, этопозид, тенипозид, амсакрин, топотекан, иринотекан, митоксантрон, камптотецин, дактиномицин, даунорубицин, альдоксорубицин, эпирубицин, идарубицин, ара-С, паклитаксел (Taxol™), набпаклитаксел, доцетаксел, дезоксикоформицин, L-аспарагиназа, IFN-альфа, азацитидин, децитабин, вориностат, MS-275, панобиностат, ромидепсин, вальпроевая кислота, моцетиностат, прациностат, белиностат, ирабектедин, тамоксифен, фульвестрант, торемифен, ралоксифен, дролоксифен, йодоксифен, бикалутамид, флутамид, нилутамид, ципротерона ацетат, гозерелин, лейпрорелин, бусерелин, прогестоген, мегестрола ацетат, анастрозол, летрозол, воразол, экземестан, финастерид, навелбен, капецитабин и дролоксафин; и абиратерон, энзалутамид, ланреотид, дазатиниб, босутиниб, трастузумаб, пертузумаб, панитумумаб, цетуксимаб, гефитиниб, эрлотиниб, афатиниб, вандетаниб, осимертиниб, рокилетиниб, лапатиниб, иматиниб, нилотиниб, сорафениб, типифарниб, и лонафарниб, вемирафениб, дубрафениб, траметиниб, кобиметиниб, понатиниб, палдоциклиб, эверолимус, руксолитиниб, пакритиниб, яктиниб, иметельстат, плитидепсин, певонедистат, ибрутиниб, церитиниб, кризотиниб, эктиниб, кабозантирсиб, висмодегибан, сонидегиб, регофарениб, вандетаниб, ваталаниб, сунитиниб, акситиниб, пазопаниб, ленватиниб, талимоген, лахерпарепвек, деносумаб, обинулузумаб, блинатомумаб, динутуксимаб, идаруцизумаб, даратумумаб, нецитумумаб, элотузумаб, оларатумаб, алемтузанаб, ритуксимаб, ибритумомаб тиуксетан, офатумумаб, пегинтерферон альфа-2b, Гардасил, Церварикс, Онкофаг, Сипулеуцел-Т, ниволумаб, пембролизумаб, атезолизумаб, индоксимод, ниволумаб, ипилумумаб, брентуксимаб ведотин, ирастузумаб, эмтанзин, флударибин, кладрибин, пентостатин, иделалисиб, перифосин, биринапант, бортезомиб, иксазомиб, карфилзомиб, маризомиб, олапариб, рукапариб, венетоклакс, навитоклакс, обатоклакс, гласдегиб, пакриностат, бупаралисиб, мемолетиниб, итакитинуб, умбралисиб, гусацитиниб, таграксофусп, рибоциклиб, абемациклиб, нирапариб, трабадектин, пофимер, винфлунин, напабукасин, лурбинектин, таземетостат, акалабрутиниб, леватиниб, нератиниб, памипариб, эпакадостат, энзастаурин, селинексор, маситиниб, эвафосфамид, глуфосфамид, роксадустат, стрептозоцин, девимистат, галунисертиб, биниметиниб, велипариб, энтиностат, пексидартиниб, талазопариб, энтректиниб.
(ii) Противовоспалительные агенты, такие как мелоксикам, феопрофен, оксапрозин, сальсалат, эторикоксиб, теноксикам, аспирин, набуметон, флурбипрофен, мефенамовая кислота, фенилбутазон, лорноксикам, индометацин, этодолак, дифлунизал, кетопрофен, вальдекоксиб, толфенамовая кислота, пироксикам, сулиндак, толметин, кеторолак, локсопрофен, ацетаминофен, бромфенак, диклофенак, ибупрофен, меклофенамат, набуметон, напроксен, непафенак, целекоксиб, триамцинолона ацетонид, гидрокортизон, ацетат гидрокортизона, метилпреднизолон, дипропионат аклометазона, эмриказан, BI 1467335, намоденозон, GLPG-1690, тергурид.
(iii) Антигипертензивное средство, такое как гидрохлоротиазид, хлорталидон, фуросемид, спиронолактон, триамтерен, амилорид, беназеприл, каптоприл, лизиноприл, эналаприл, рамиприл, фозиноприл, моэксиприл, периндоприл, квинаприл, трандолаприл, лозартан, кандесартан, валсартан, телмисартан, клонидин, метилдопа, пропранолол, надолол, тимолол, пиндолол, лабетолол, метопролол, атенолол, эсмолол, бетаксолол, карведилол, празозин, теразозин, доксазозин, феноксибензамин, фентоламин, верапамил, дилтиазем, нифедипин, фелодипин, амлодипин, нимодипин, диазоксид, миноксидил, пинацидил, никорандил, гидралазин, нитропруссид натрия, бозентан, эпопростенол, илопрост, берапрост, эсуберапрост, ралинепаг, мацитентан, ситаксентан, амбрисентан, риоцигуат, трепростинил, убенимекс, селексипаг, левосимендан, уденафил, тадалафил и силденафил.
(iv) Антифиброзный агент, такой как пирфенидон, нинтеданиб, ценикривирок, селонсертиб, ланифибранор, нимацимаб, нитразоксанид, NGM282, апараренон, типелукаст, Actimmune, понатиниб, ленватиниб, довитиниб, люцитаниб, данусертиниб, бриватиниб, эрдафитиниб, белапектин, PD173074, PD166866, AZD4547, BGJ398, LY2874455, TAS-120, ARQ087, BLU9931, FGF401, BAY-1163877, ENMD-2076, IMCA1, FGF401, DEBIO1347, FIIN-2, GP-369, PRO-001, H3B-6527, BAY1187982, MFGR1877S, FP-1039, BLU554, PRN1371, S49076, SU6668, SU5416, PBI-4050, KD-025.
(v) Средство против ангиогенеза, такое как акситиниб, бевацизумаб, кабозантиниб, леналидомид, ленватиниб, пазопаниб, рамуцирумаб, вандетаниб, ваталаниб, сунитиниб, зив-афлиберцепт, талидомид, помалидомид, леналидомид.
(vi) Иммуносупресант, такой как преднизон, будесонид, преднизолон, тофацитиниб, циклоспорин, такролимус, сиролимус, эверолимус, азатиоприн, лефлуномид, микофенолат, абатацепт, адалимумаб, анакинра, цертолизумаб, этанерцепт, голимумаб, инфликсимаб, иксекизумаб, натализумаб, ритуксимаб, секукинумаб, тоцилизумаб, устекинумаб, ведолизумаб, базиликсимаб, даклизумаб, диметилфумарат, микофенолят мофетиллат.
(vii) Метаболический агент, такой как обетихолевая кислота, элафибранор, арамхол, селадельпар, MGL-3196, тропифексор, MSDC-0602K, BMS-986036, семаглутид, EDP-305, гемкабен, PF-05221304, PF-06865571, PF-06835919, LIK066, LMB763, витамин E, акарбоза, миглитол, прамлинтид, алоглиптан, линаглиптан, саксаглиптин, ситаглиптин, альбиглутид, дулаглутид, экзенатид, лираглутид, ликсисенатид, инсулин, натеглинид, репаглинид, метформин, канаглифлозин, дапаглифлозин, эмпаглифлозин, хлорпропамид, глимепирид, глипизид, глибурид, толазамид, толбутамид, розиглитазон, пиоглитазон, аторвастатин, амлодипин, симвастатин, эзетимиб, ловастатин, ситаглиптин, холестирамин, колесевелам, колестипол, фенофибрат, гемфиброзил, фенофибриновая кислота, ниацин, икозапент, мипомерсен, ломитапид, эволокумаб, алирокумаб, флувастатин, правастатин, розувастатин, питавастатин, симвастатин, церивастатин, аллопуринол, лесинурад, пеглотиказа, фебуксостат, расбуриказа, ивакафтор, велаглюцераза альфа, имиглюцераза, альглюкозидаза альфа, ларонидаза, церлипоназа альфа, альглюцераза, идурсульфаза, талиглюцераза альфа, агалсидаза бета, себелипаза альфа, вестронидаза альфа, галсульфаза, элосульфаза альфа, элиглюстат, буросумаб, мигаластат, сапроптерин, метрелептин, нитисинон, пегвалиаза, асфотаза альфа, инотерсен, миглустат, орлистат, фенилбутират натрия, глицерин фенилбутират.
[0245] В одном варианте реализации соединения настоящего изобретения можно вводить в комбинации с другими терапевтическими способами лечения. Например, соединения настоящего изобретения можно вводить в комбинации с лучевой терапией или химиотерапией. В одном варианте реализации соединения настоящего изобретения можно вводить в комбинации с одним или несколькими дополнительными противоопухолевыми средствами и/или радиотерапией для лечения рака.
[0246] Когда два или более активных ингредиента вводятся совместно, активные ингредиенты можно вводить одновременно, последовательно или раздельно. В одном варианте реализации соединение Формулы I вводят одновременно со вторым терапевтическим агентом. В другом варианте реализации соединение Формулы I и второй терапевтический агент вводят последовательно. В следующем варианте реализации соединение Формулы I и второй терапевтический агент вводят отдельно.
[0247] Теперь изобретение будет описано более подробно только в качестве иллюстрации со ссылкой на следующие неограничивающие примеры. Примеры предназначены для иллюстрации изобретения и не должны рассматриваться как ограничение общности раскрытия описания в данной спецификации.
Эксперимент: Общие способы
[0248] Коммерчески доступные растворители и реагенты использовались в полученном виде. При необходимости реакции проводят в атмосфере аргона. За реакциями следили либо аналитической тонкослойной хроматографией (ТСХ), либо аналитической жидкостной хроматографией-масс-спектрометрией (ЖХМС), записанной либо на приборе Shimadzu LCMS 2020, либо на приборе Agilent LC/MSD 1200 с использованием условий обращения фазы. При необходимости очистку промежуточных и конечных соединений проводили с помощью колоночной хроматографии или препаративной ВЭЖХ. Колоночную хроматографию с нормальной фазой проводили при среднем давлении либо на силикагеле, либо на предварительно набитых картриджах с силикагелем, используя систему флэш-хроматографии (CombiFlash Rf200, Teledyne Isco systems, США). Обращенно-фазовую колоночную хроматографию проводили при низком давлении на предварительно набитых картриджах C18 с использованием системы флэш-хроматографии (Reveleris® X2). Элюенты контролировали УФ-светом (λ = 254/280 нм). Спектры 1H-ЯМР и 19F-ЯМР регистрировали с использованием ЯМР-спектрометра Bruker 300 МГц, ЯМР-спектрометра Bruker Avance III plus 400 МГц или спектрометра Varian III plus 300 МГц. Химические сдвиги (δ) выражены в миллионных долях (м.д.) относительно тетраметилсилана (ТМС; внутренний стандарт). Для кратностей используются следующие сокращения: с = синглет; шс = широкий синглет; д = дублет; и = триплет; к = квартет; м = мультиплет; и шм = широкий мультиплет. Масс-спектры низкого разрешения (МС) получали как масс-спектры электроспрей - ионизация при атмосферном давлении (ЭС-АДИ), которые записывали либо на приборе Shimadzu LCMS 2020, либо на приборе Agilent LC/MSD 1200 с использованием условий обращенно-фазовой обработки. Все эксперименты на животных проводились в соответствии с установленными правилами и с одобрения местных комитетов по этике.
ПРИМЕР 1
[0249] Получение (Z)-трет-бутил (4-бром-3-фторбут-2-ен-1-ил)карбамата
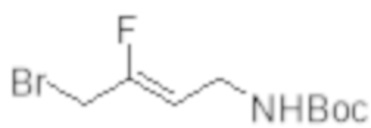
Методика А: Получение трет- бутил-2-оксоэтилкарбамата

[0250] К перемешиваемому раствору 3-амино-1,2-пропандиола (20,0 г, 0,22 моль) в воде (200 мл) при 0-5 °С добавляли ди-трет-бутилдикарбонат (55,5 мл, 0,24 моль). После доведения щелочности раствора до pH~9 добавлением водн. NaOH (6 н.), смесь оставляли перемешиваться при комнатной температуре (к.т.) на 18 часов. Реакционную смесь охлаждали до 0-5 °С, и затем подкисляли до pH~6 перед добавлением метапериодата натрия (56,3 г, 0,26 моль). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Смесь фильтровали для удаления всех твердых веществ, и фильтрат переносят в делительную воронку и экстрагируют этилацетатом (200 мл). К водному слою добавляли хлорид натрия до получения насыщенного раствора. Затем водный слой дополнительно экстрагировали этилацетатом (100 мл). Объединенные органические слои сушили над Na2SO4, и затем концентрировали в вакууме с получением неочищенного трет-бутил 2-оксоэтилкарбамата (45,7 г) в виде желтой смолы. Неочищенный материал использовали на следующей стадии без очистки.
Методика B: Получение ( E )-этил 4-( трет- бутоксикарбониламино)-2-фторбут-2-еноата и ( Z )-этил 4-( трет- бутоксикарбониламино)-2-фторбут-2-еноата

[0251] К перемешиваемой суспензии неочищенного трет-бутил 2-оксоэтилкарбамата (43,7 г, 0,22 моль) и сульфата магния (32,0 г) в ацетонитриле (200 мл) при 0 °C в атмосфере N2 добавляли последовательно этил 2-фторфосфоноацетат (55,7 мл, 0,27 моль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (32,8 мл, 0,22 моль). Реакционной смеси давали нагреться до комнатной температуры и перемешивание продолжали в течение 3 часов. После удаления растворителя при пониженном давлении остаток помещали в этилацетат (200 мл) и затем переносили в делительную воронку. Органические слои последовательно промывали водн. HCl (2 M; 100 мл x 2), водн. NaOH (2 M; 100 мл x 2) и рассолом (100 мл). После сушки над MgSO4 органические слои концентрировали в вакууме с получением неочищенного желаемого продукта в виде смеси изомеров E/Z (2:3; 57,0 г). Этот неочищенный материал использовали на следующей стадии без очистки.
Методика С: Получение (Е) - трет -бутил-3-фтор-4-гидроксибут-2-енилкарбамата и (Z) - трет -бутил-3-фтор-4-гидроксибут-2-енилкарбамата
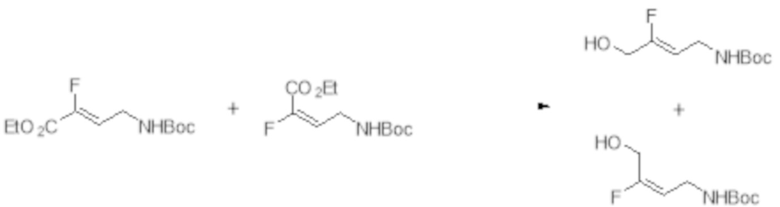
[0252] К перемешиваемому раствору неочищенного E/Z-этил 4-(трет-бутоксикарбониламино)-2-фторбут-2-еноата (18,0 г, 72,8 ммоль) в THF (150 мл) при 0 °С в атмосфере N2 добавляли диизо-бутилалюминий гидрид (1 М в толуоле, 182 мл, 182 ммоль) по каплям в течение 45 мин. После завершения добавления смесь оставляли перемешиваться при 0 °C в течение 3 ч. Реакционную смесь переносили в делительную воронку и по каплям добавляли к перемешиваемой смеси льда (100 г) и водн. NaOH (2 М; 200 мл). После добавления смесь перемешивали в течение 2 часов. Гашенную реакционную смесь экстрагировали диэтиловым эфиром (100 мл × 2) и объединенные органические слои промывали рассолом (100 мл). После сушки над MgSO4 органические слои концентрировали в вакууме, с получением неочищенного спирта в виде смеси изомеров E/Z. Эту смесь очищают на силикагеле (135 г), элюируют 25% этилацетатом в н-гексане, с получением (Z)-трет-бутил 3-фтор-4-гидроксибут-2-енилкарбамата (6,20 г, 30% после трех стадий) и (E)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамат (1,85 г, 8,9% после трех стадий). (E)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамат: 1H-ЯМР (200 МГц; CDCl3) δ м.д.: 1,43 (9H, с), 3,72 (2H, дд, J 7,5, 5,4 Гц), 4,25 (2H, д, J 21,5 Гц), 4,85 (1H, шс), 5,18 (1H, шт, J 19,2, 8,5 Гц). (Z)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамат: 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,46 (9H, с), 3,84 (2H, дд, J 6,2, 6,2 Гц), 4,13 (2H, д, J 13,9 Гц), 4,68 (1H, шс), 5,03 (1H, дт, J 36,0, 7,1 Гц).
Методика D: Получение ( Z )- трет- бутил-4-бром-3-фторбут-2-енилкарбамата

[0253] К перемешиваемому раствору (Z)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамата (6,20 г, 30,2 ммоль) и триэтиламина (6,32 мл, 45,3 ммоль) в ацетоне (100 мл) при 0 °C по каплям добавляли метансульфонилхлорид (2,81 мл, 36,3 ммоль). После завершения добавления смесь оставляли перемешиваться при 0 °C в течение 30 мин. По истечении этого времени порциями добавляли бромид лития (13,1 г, 0,15 моль) и полученную суспензию перемешивали еще 2 часа. Реакционную смесь фильтровали для удаления всех твердых веществ, и фильтрат концентрировали при пониженном давлении. Остаток разделяли между водой (50 мл) и CH2Cl2 (50 мл) и водный слой дополнительно экстрагировали CH2Сl2 (50 мл x 2). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Неочищенный остаток очищали на силикагеле (100 г), элюируя н-гексаном, а затем 25% этилацетатом в н-гексане, с получением (Z)-трет-бутил 4-бром-3-фторбут-2-енилкарбамата (7,00 г, 86%) в виде бесцветного твердого вещества. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,46 (9H, с), 3,85 (2H, дд, J 6,2, 6,2 Гц), 3,93 (2H, д, J 19,5 Гц), 4,66 (1H, шс), 5,16 (1H, дт, J 34,0, 6,5 Гц).
ПРИМЕР 2
[0254] Следующее соединение получали согласно методикам E, F, G, H и I.
[0255] Получение гидрохлорида (Z)-4-((2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил) фенокси)метил)- N,N-диизопропилбензолсульфонамида (Соединение 11)
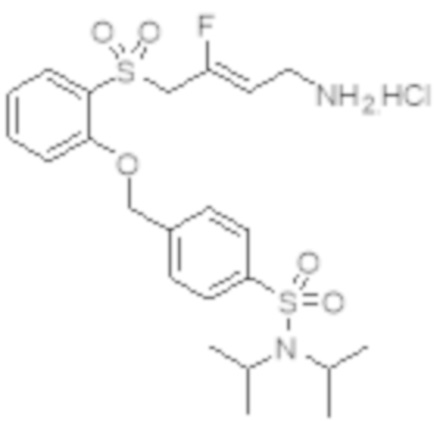
Методика E: Получение 4-(бромметил)- N,N -ди изо пропилбензолсульфонамида
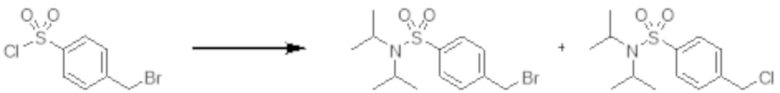
[0256] К перемешиваемому раствору 4-(бромметил)бензолсульфонилхлорида (500 мг, 1,86 ммоль) в CH2Cl2 (10 мл) при 0 °С добавляли ди-изо-пропиламин (0,65 мл, 4,63 ммоль) по каплям. После добавления полученную смесь оставляли перемешиваться при этой температуре в течение 30 минут, после чего давали нагреться до комнатной температуры и перемешивали еще 48 часов. Реакционную смесь разделяли между водн. HCl (1 M, 20 мл) и CH2Cl2 (20 мл). Органический слой промывали водн. HCl (1 M; 20 мл) и водой (20 мл), сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединения (желтое масло, 190 мг) в виде смеси с 4-(хлорметил)-N,N-диизопропилбензолсульфонамидом, который использовали как таковое на следующей стадии.
Методика F: Получение ( Z )- трет- бутил-(3-фтор-4-((2-гидроксифенил)тио)бут-2-ен-1-ил)карбамата
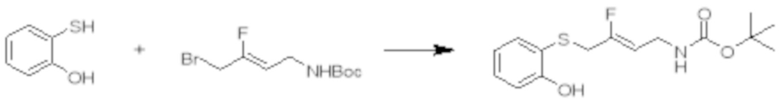
[0257] К раствору 2-меркаптофенола (235 мг, 1,86 ммоль) и (Z)-трет-бутил(4-бром-3-фторбут-2-ен-1-ил)карбамата (500 мг, 1,86 ммоль) в ацетоне (3 мл) при комнатной температуре добавляли карбонат калия (387 мг, 2,70 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 16 часов. Затем реакционную смесь разделяли между EtOAc (20 мл) и водой (20 мл) и фазы разделяли. Водную фазу экстрагировали EtOAc (20 мл x 2), и затем органические фазы объединяли и промывали (рассол; 20 мл), сушили (Na2SO4) и концентрировали в вакууме, с получением (Z)-трет-бутил-(3-фтор-4-((2-гидроксифенил)тио)бут-2-ен-1-ил)карбамата (580 мг, 99%) в виде светло-желтого твердого вещества. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,45 (9H, с), 3,31 (2H, д, J = 19,7 Гц), 3,69 (2H, около т, J = 6,7 Гц), 4,47 (1H , дт, J = 34,6, 7,2 Гц), 4,49 (1H, шс), 6,67 (1H, с), 6,90 (1H, ддд, J = 7,6, 7,6 1,3 Гц), 7,02 (1H, дд, J = 8,2, 1,2 Гц), 7,31 (1H, ддд, J = 8,2, 7,3, 1,6 Гц), 7,45 (1H, дд, J = 7,7, 1,7 Гц).
Методика G: Получение (Z) - трет -бутил-(4-((2-((4- (N,N -диизопропилсульфамоил)бензил)окси)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата
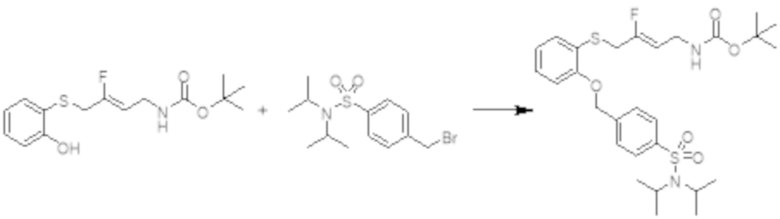
[0258] К перемешиваемому раствору (Z)-трет-бутил(3-фтор-4-((2-гидроксифенил)тио)бут-2-ен-1-ил)карбамата (100 мг, 0,32 ммоль) и 4-(бромметил)-N,N-диизопропилбензолсульфонамида (107 мг, 0,32 ммоль) в DMF (1 мл) при комнатной температуре добавляли карбонат калия (66 мг, 0,48 ммоль). Полученную суспензию перемешивали при этой температуре в течение 16 часов. Затем реакционную смесь разделяли между EtOAc (10 мл) и водой (10 мл) и фазы разделяли. Водную фазу экстрагировали EtOAc (10 мл × 2), и затем органические фазы объединяли и промывали (насыщ. водн. NH4Cl, затем рассолом), сушили (Na2SO4) и концентрировали в вакууме, с получением (Z)-трет-бутил (4-((2-((4-(N,N-диизопропилсульфамоил)бензил)окси)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата (180 мг, 99%) в виде желтой смолы, которую использовали на следующей стадии без очистки.
Методика Н: Получение (Z) - трет -бутил-(4-((2-((4- (N,N- ди изо пропилсульфамоил)бензил)окси)фенил)сульфонил)-3-фторбут-2-ен-1-ил)карбамата
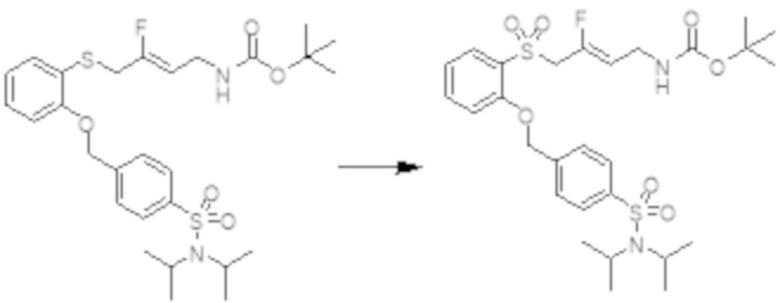
[0259] К перемешиваемой суспензии (4-((2-((4-(N,N-диизопропилсульфамоил)бензил)окси)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата (180 мг , 0,32 ммоль) и гидрокарбоната натрия (133 мг, 1,59 ммоль) в CH2Cl2 (2 мл) и воде (2 мл) при 0 °C добавляли 3-хлорпероксибензойную кислоту (178 мг, 0,79 ммоль) тремя порциями в течение 5 минут. Полученную суспензию перемешивали при 0 °C в течение 2 часов, прежде чем разбавить насыщ. водн. NaHCO3 (15 мл) и экстрагировали CH2Cl2 (10 мл). Водную фазу дополнительно экстрагировали CH2Cl2 (10 мл x 2), и органические фазы объединяли, сушили (Na2SO4) и концентрировали в вакууме. Неочищенный материал очищали флэш-колонкой, элюируя смесью 40% EtOAc/гексан, затем 2% MeOH в 50% EtOAc/гексане с получением (Z)-трет-бутил-(4-((2-((4-(N,N-диизопропилсульфамоил)бензил)окси)фенил)сульфонил)-3-фторбут-2-ен-1-ил)карбамата (160 мг, 84%) в виде твердого вещества белого цвета. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,29 (12 H, д, J = 6,8 Гц), 1,43 (9H, с), 3,67-3,80 (3H, м), 4,15 (2H, д, J = 18,9 Гц), 4,52 (1H, шс), 4,93 (1H, дт, J = 34,4, 6,9 Гц), 5,33 (2H, с), 7,09 (1H, д, J = 8,0 Гц), 7,18 (1H , ддд, J = 8,3, 7,8, 0,8 Гц), 7,63 (1H, ддд, J = 8,4, 7,6, 1,7 Гц), 7,66 (2H, д, J = 8,4), 7,93 (2H, д, J = 8,5 Гц ), 7,99 (1H, дд, J = 7,9, 1,7 Гц). В качестве модификации этой методики для получения дополнительных соединений окисление осуществляли путем медленного добавления водного раствора OXONE® (4 экв. в 1,2 мл H2O на ммоль OXONE®) к раствору исходного сульфанилового эфира в MeOH:THF (1 : 1, около 3 мл каждого на ммоль сульфанилового эфира) при комнатной температуре, и давали возможность прореагировать до тех пор, пока контроль ЖХ-МС не показал высокую степень превращения в желаемый сульфоновый продукт. Затем смесь разделяли между избытком насыщенного водного раствора метабисульфита натрия и EtOAc, промывали рассолом, сушили (Na2SO4), концентрировали в вакууме и очищали колоночной хроматографией.
Методика I: Получение гидрохлорида (Z) -4-((2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил)- N,N -ди изо пропилбензолсульфонамида (Соединение 11)
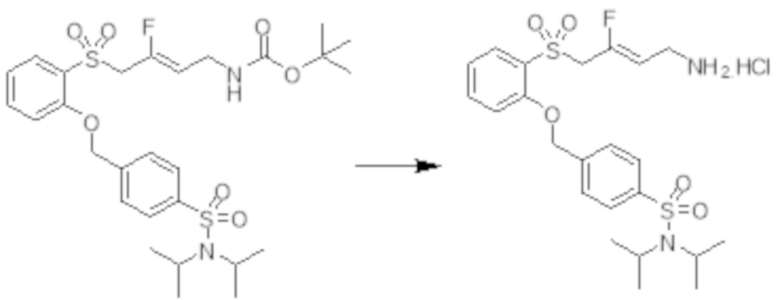
[0260] К перемешиваемому раствору (Z)-трет-бутил-(4-((2-((4-(N,N-диизопропилсульфамоил)бензил)окси)фенил)сульфонил)-3-фторбут-2-ен-1-ил)карбамата (160 мг, 0,27 ммоль) в MeOH (1 мл) при комнатной температуре добавляли эфирный HCl (2 M; 4,00 мл, 8,00 ммоль), и полученную смесь оставляли перемешиваться в течение 1 часа. По истечении этого времени осаждалось белое твердое вещество, которое собирали фильтрованием и сушили в высоком вакууме с получением гидрохлорида (Z)-4-((2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил)-N,N-диизопропилбензол-сульфонамида (79 мг, 55%). Белое твердое вещество; т.п. 222-224 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 1,27 (12H, д, J = 6,8 Гц), 3,59 (2H, дд, J = 7,4, 1,8 Гц), 3,79 (2H, гепт, J = 6,8 Гц), 4,45 (2H, д, J = 19,2 Гц), 5,16 (1H, дт, J = 32,8, 7,4 Гц), 5,46 (2H, с), 7,23 (1H, ддд, J = 7,4, 7,4, 0,9 Гц ), 7,38 (1H, д J = 7,9 Гц), 7,74 (1H, ддд, J = 8,5, 7,5, 1,7 Гц), 7,79 (2H, д, J = 8,6 Гц), 7,91-7,95 (3H, м).
ПРИМЕР 3
[0261] Следующие ниже соединения получали в соответствии с методиками E-I с использованием соответственно функционализированного исходного тиолового материала.
Гидрохлорид (Z)-4-((2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил)-N,N-диметилбензолсульфонамида (Соединение 5)
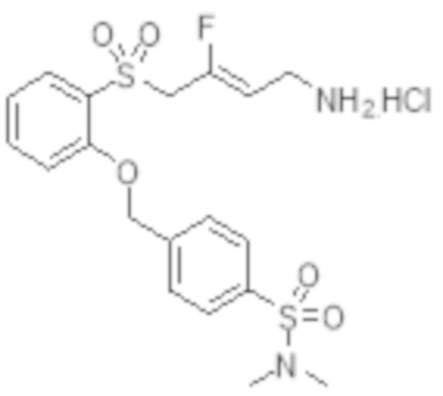
[0262] Белое твердое вещество; т.п. 235–236 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 2,72 (6H, с), 3,60 (2H, дд, J = 7,4, 1,7 Гц), 4,47 (2H, д, J = 19,2 Гц), 5,18 ( 1H, дт, J = 32,9, 7,4 Гц), 5,49 (2H, с), 7,24 (1H, ддд, J = 7,9, 7,9, 0,9 Гц), 7,39 (1H, дд, J = 8,5, 0,7 Гц), 7,76 (1H, ддд, J = 8,4, 7,4, 1,7 Гц), 7,86 (4H, шс), 7,95 (1H, дд, J = 7,9, 1,8 Гц).
Гидрохлорид (Z)-4-((2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил) бензолсульфонамида (Соединение 8)
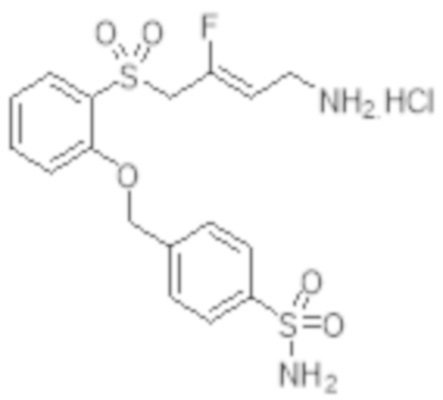
[0263] Не совсем белое твердое вещество; т.п. 233-235 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 3,59 (2H, дд, J = 7,4, 1,6 Гц), 4,44 (2H, д, J = 19,2 Гц), 5,14 (1H, дт, J = 32,8 , 7,4 Гц), 5,45 (2H, с), 7,23 (1H, дд, J = 7,3, 7,3 Гц), 7,38 (1H, д, J = 8,3 Гц), 7,74 (1H, ддд, J = 8,6, 8,6, 1,7 Гц), 7,78 (2H, д, J = 8,2 Гц), 7,94 (1H, дд, J = 8,1, 1,6 Гц), 7,97 (2H, д, J = 8,5 Гц).
Гидрохлорид (Z)-4-((3-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил)-N,N-диметилбензолсульфонамида (Соединение 9)
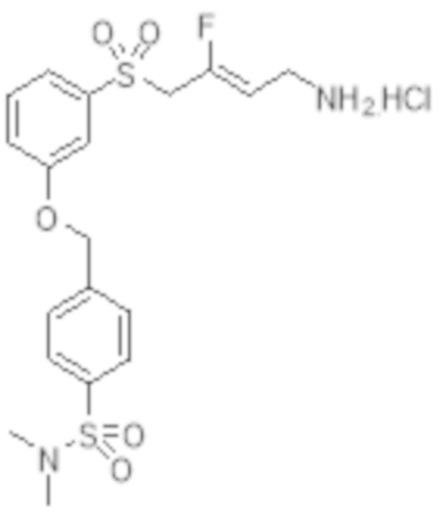
[0264] Белое твердое вещество; т.п. 211-213 °C; 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 2,63 (6H, с), 3,48 (2H, шс), 4,65 (2H, д, J = 19,6 Гц), 5,17 (1H, дт, J = 34,6, 7,2 Гц), 5,36 (2H, с), 7,45 (1H, ддд, J = 8,1, 2,5, 1,0 Гц), 7,52-7,57 (2H, м), 7,63 (1H, дд, J = 8,1, 8,1 Гц), 7,75 (2H, д, J = 8,5 Гц), 7,81 (2H, д, J = 8,6 Гц), 8,11 (3H, шс).
Гидрохлорид (Z)-4-((4-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил)-N,N-диметилбензолсульфонамида (Соединение 10)
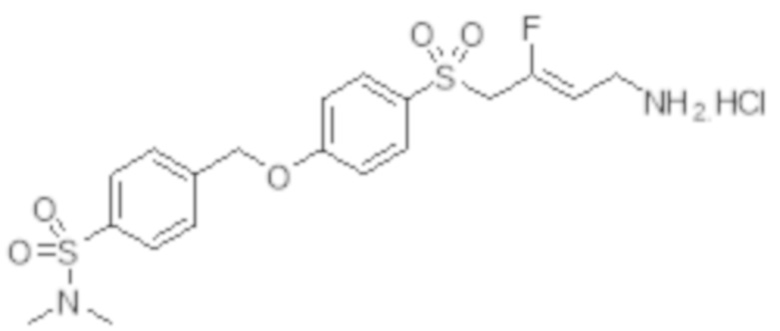
[0265] Белое твердое вещество; т.п. 216-218 °C; 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 2,63 (6H, с), 3,48 (2H, шс), 4,55 (2H, д, J = 19,7 Гц), 5,12 (1H, дт, J = 34,8, 7,1 Гц), 5,38 (2H, с), 7,29 (2H, дд, J = 9,0, 1,9 Гц), 7,74 (2H, дд, J = 8,5, 1,8 Гц), 7,81 (2H, дд, J = 8,5, 1,9 Гц), 7,88 (2H, дд, J = 8,9, 2,0 Гц), 8,03 (3H, шс).
Гидрохлорид (Z)-4-((2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)метил)- N-изопропилбензолсульфонамида (Соединение 14)
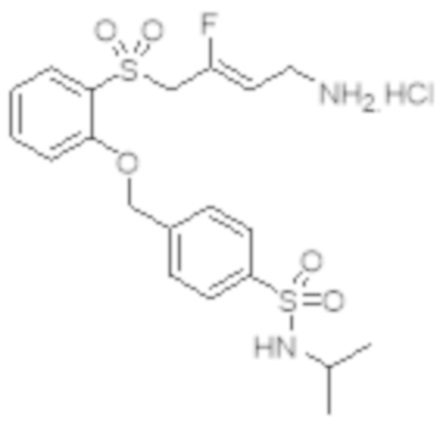
[0266] Белое твердое вещество; т.п. 248–250 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 1,05 (6H, д, J = 6,6 Гц), 3,40 (1H, гепт, J = 6,6 Гц), 3,59 (2H, приб. д, J = 7,3 Гц), 4,45 (2H, д, J = 19,1 Гц), 5,16 (1H, дт, J = 33,0, 7,4 Гц), 5,46 (2H, с), 7,23 (1H, ддд, J = 8,0, 8,0, 1,0 Гц. ), 7,38 (1H, д, J = 8,1 Гц), 7,74 (1H, ддд, J = 8,4, 7,4, 1,8 Гц), 7,80 (2H, д, J = 8,7 Гц), 7,91-7,95 (3H, м).
ПРИМЕР 4
[0267] Следующее соединение получали в соответствии с методиками F-I, используя соответствующий исходный тиоловый материал.
Гидрохлорид (Z)-4-((2-(бензилокси)фенил)сульфонил)-3-фторбут-2-ен-1-амина (Соединение 7)
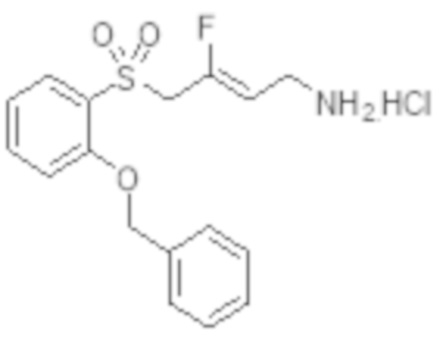
[0268] Белое твердое вещество; т.п. 205-207 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 3,57 (2H, приб. д, J = 7,0 Гц), 4,44 (2H, д, J = 19,1 Гц), 5,14 (1H, дт, J = 32,8, 7,3 Гц), 5,36 (2H, с), 7,20 (1H, дд, J = 7,4, 0,9 Гц), 7,35-7,46 (4H, м), 7,55-7,60 (2H, м), 7,73 (1H, ддд, J = 8,5, 7,4, 1,7 Гц), 7,92 (1H, дд, J = 7,9, 1,7 Гц).
ПРИМЕР 5
[0269] Следующие ниже соединения получали в соответствии с методиками F, H и I с использованием соответственно функционализированного исходного тиолового материала.
Гидрохлорид (Z)-4-((3,5-бис(трифторметил)фенил)сульфонил)-3-фторбут-2-ен-1-амина (Соединение 2)
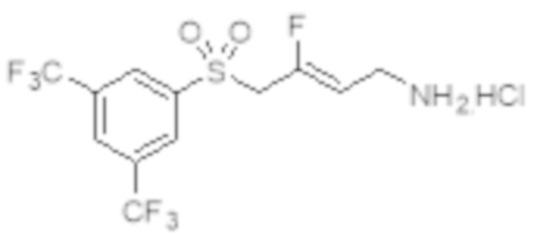
[0270] Белое твердое вещество; т.п. 217–220 °C; 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 3,48 (2H, прибл. д, J = 7,1 Гц), 4,96 (2H, д, J = 19,6 Гц), 5,19 (1H, дт, J = 34,8, 7,2 Гц), 8,10 (3H, шс), 8,55 (2H, с), 8,67 (1H, с).
Гидрохлорид (Z)-4-(бифенил-2-илсульфонил)-3-фторбут-2-ен-1-амина (Соединение 20)
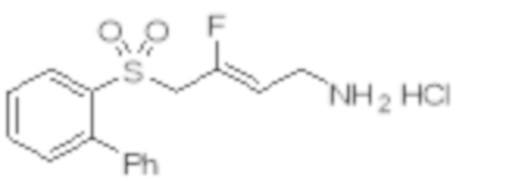
[0271] Белое твердое вещество; т.п. 170 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,18 (дд, J = 8,0, 1,4 Гц, 1H), 7,81 (тд, J = 7,5, 1,4 Гц, 1H), 7,68 (тд, J = 7,6, 1,6 Гц, 1H), 7,52 - 7,44 (м, 6H), 5,03 (дт, J = 32,9, 7,4 Гц, 1H), 3,83 (д, J = 18,9 Гц, 2H), 3,56 (дд, J = 7,4, 1,3 Гц, 2H).
Гидрохлорид (Z)-3-фтор-4-(2-изопропилфенилсульфонил)бут-2-ен-1-амина (Соединение 21)
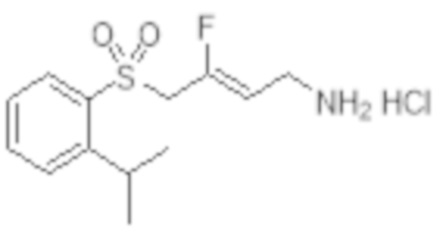
[0272] Белое твердое вещество; т.п. 205-215 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,00 - 7,95 (м, 1H), 7,77 - 7,67 (м, 2H), 7,48 - 7,41 (м, 1H), 5,25 (дт, J = 32,8, 7,4 Гц, 1H), 4,34 (д, J = 19,2 Гц, 2H), 3,88 (гепт, J = 6,9 Гц, 1H), 3,63 (дд, J = 7,5, 1,9 Гц, 2H), 1,36 (д, J = 6,8 Гц, 6H).
Гидрохлорид (Z)-3-фтор-4-(2-метоксифенилсульфонил)бут-2-ен-1-амина (Соединение 22)
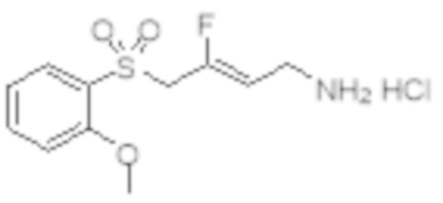
[0273] Белое твердое вещество; т.п. 228-221 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,89 (дд, J = 7,9, 1,7 Гц, 1H), 7,75 (ддд, J = 8,4, 7,4, 1,8 Гц, 1H), 7,32 (дд, J = 8,5, 0,9 Гц, 1H), 7,19 (тд, J = 7,6, 1,0 Гц, 1H), 5,23 (дт, J = 32,8, 7,4 Гц, 1H), 5,17 (т, J = 7,4 Гц, 0H), 4,48 (д, J = 19,3 Гц, 2H), 4,05 (с, 3H), 3,61 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z)-3-фтор-4-(нафталин-1-илсульфонил)бут-2-ен-1-амина (Соединение 23)
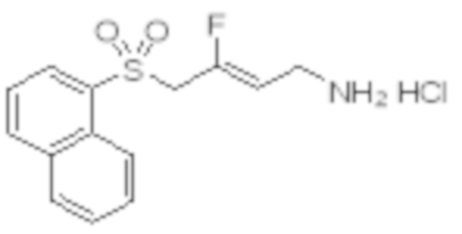
[0274] Белое твердое вещество; т.п. 230–240 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,76 (ддд, J = 8,7, 1,2, 0,6 Гц, 1H), 8,33 (дд, J = 7,8 Гц, 2H), 8,13 (ддд, J = 8,2, 1,5, 0,8 Гц, 1H), 7,81 (ддд, J = 8,6, 6,9, 1,5 Гц, 1H), 7,72 (тд, J = 8,0, 1,9 Гц, 2H), 5,13 (дт, J = 32,7, 7,4 Гц, 1H ), 4,49 (д, J = 19,2 Гц, 2H), 3,57 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z)-3-фтор-4-(нафталин-2-илсульфонил)бут-2-ен-1-амина (Соединение 24)
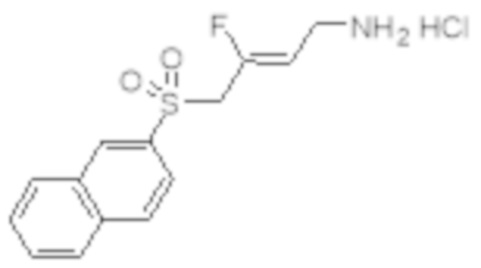
[0275] Белое твердое вещество; т.п. 215–220 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,60 (д, J = 1,9 Гц, 1H), 8,18 - 8,11 (м, 2H), 8,06 (дд, J = 8,2, 1,4 Гц, 1H), 7,95 (дд, J = 8,7, 1,9 Гц, 1H), 7,81 - 7,68 (м, 2H), 5,19 (дт, J = 32,8, 7,4 Гц, 1H), 4,46 (д, J = 19,2 Гц, 2H), 3,63 (дт, J = 7,4, 1,3 Гц, 2H).
Гидрохлорид (Z)-4-(2,4-дихлорфенилсульфонил)-3-фторбут-2-ен-1-амина (Соединение 25)
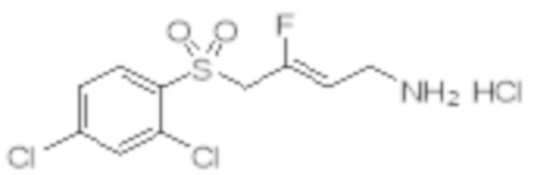
[0276] Белое твердое вещество; т.п. 220 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,10 (д, J = 8,6 Гц, 1H), 7,85 (д, J = 2,0 Гц, 1H), 7,66 (дд, J = 8,6, 2,0 Гц, 1H ), 5,28 (дт, J = 32,9, 7,4 Гц, 1H), 4,60 (д, J = 19,1 Гц, 2H), 3,63 (ддд, J = 7,4, 2,0, 0,6 Гц, 2H).
Гидрохлорид (Z)-4-(3-хлорфенилсульфонил)-3-фторбут-2-ен-1-амина (Соединение 26)
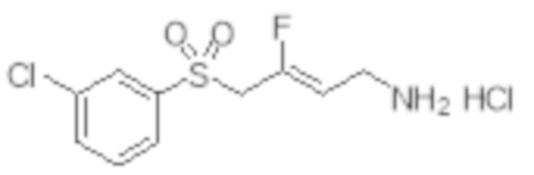
[0277] Белое твердое вещество; т.п. 225-235 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,00 (т, J = 1,9 Гц, 1H), 7,92 (ддд, J = 7,8, 1,8, 1,1 Гц, 1H), 7,82 (ддд, J = 8,1, 2,1, 1,1 Гц, 1H), 7,68 (д, J = 8,3 Гц, 1H), 5,22 (дт, J = 32,9, 7,4 Гц, 1H), 4,44 (д, J = 19,1 Гц, 2H), 3,65 (дд, J = 7,4, 1,9 Гц, 2H)
Гидрохлорид (Z)-4-(4-хлорфенилсульфонил)-3-фторбут-2-ен-1-амина (Соединение 27)
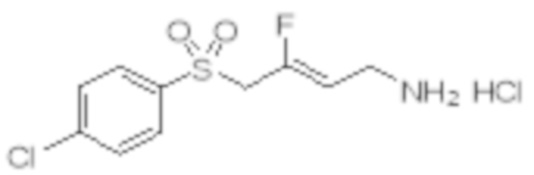
[0278] Белое твердое вещество; т.п. 240 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,96 (дт, J = 8,8, 2,3 Гц, 2H), 7,71 (дт, J = 8,3, 1,9 Гц, 2H), 5,20 (дт, J = 32,9, 7,4 Гц, 1H), 4,40 (д, J = 19,1 Гц, 2H), 3,65 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z)-4-(3,5-дихлорфенилсульфонил)-3-фторбут-2-ен-1-амина (Соединение 28)
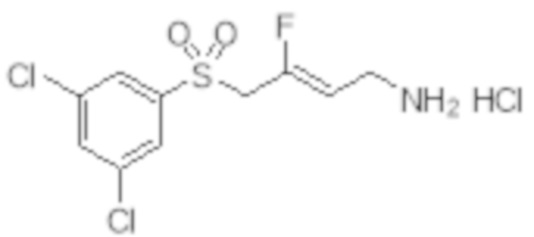
[0279] Белое твердое вещество; т.п. 250 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,97 (д, J = 1,8 Гц, 2H), 7,93 (т, J = 1,9 Гц, 1H), 5,27 (дт, J = 33,0, 7,4 Гц, 1H ), 4,50 (д, J = 19,0 Гц, 2H), 3,67 (дд, J = 7,4, 2,0 Гц, 2H).
Дигидрохлорид (Z)-3-фтор-4-(пиридин-4-илсульфонил)бут-2-ен-1-амина (Соединение 29)
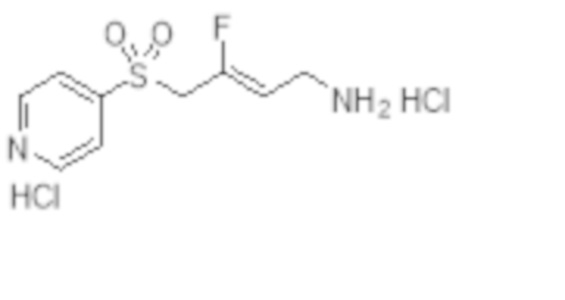
[0280] Белое твердое вещество; т.п. 162–164 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,07 (дд, J = 4,9, 1,7 Гц, 2H), 8,22 (дд, J = 4,6, 1,6 Гц, 2H), 5,31 (дт, J = 33,1, 7,4 Гц, 1H), 4,63 (д, J = 19,0 Гц, 2H), 3,67 (д, J = 7,4 Гц, 2H)
Дигидрохлорид (Z)-3-фтор-4-(хинолин-2-илсульфонил)бут-2-ен-1-амина (Соединение 32)
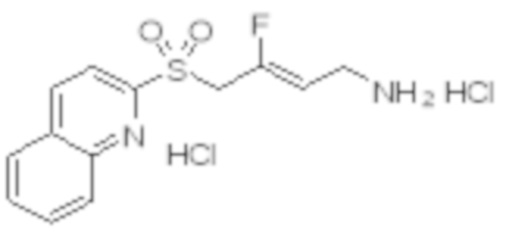
[0281] Белое твердое вещество; т.п. 203-205 °C; 1H ЯМР (300 МГц, d6-DMSO) δ м.д.: 8,82 (д, J = 8,6 Гц, 1H), 8,29 - 8,20 (м, 2H), 8,16 (д, J = 8,5 Гц, 1H), 8,12 - 7,93 (м, 3H), 7,92 - 7,83 (м, 1H), 5,26 (дт, J = 34,8, 7,2 Гц, 1H), 4,92 (д, J = 19,6 Гц, 2H), 3,48 (с, 2H).
Дигидрохлорид (Z)-3-фтор-4-(хинолин-8-илсульфонил)бут-2-ен-1-амина (Соединение 33)
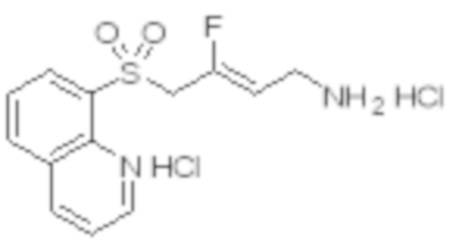
[0282] Белое твердое вещество; т.п. 150–153 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,18 (д, J = 4,7 Гц, 1H), 8,70 (дд, J = 8,4, 2,6 Гц, 1H), 8,57 (д, J = 7,4 Гц, 1H ), 8,45 (д, J = 8,5 Гц, 1H), 7,99 - 7,68 (м, 2H), 5,22 (дт, J = 32,9, 7,4 Гц, 1H), 5,00 (д, J = 19,4 Гц, 2H), 3,60 (д, J = 7,7 Гц, 2H); ЖХМС: для C13H13FN2O2S вычислено 280,1, найдено 281,1 [M+1]+.
Гидрохлорид (Z)-3-фтор-4-(2-фторфенилсульфонил)бут-2-ен-1-амина (Соединение 37)
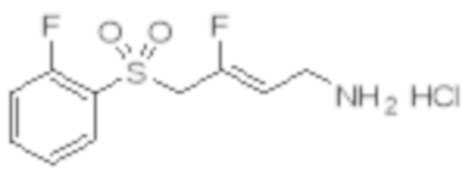
[0283] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,01 - 7,90 (м, 1H), 7,89 - 7,77 (м, 1H), 7,53 - 7,39 (м, 2H), 5,29 (дт, J = 32,8, 7,4 Гц, 1H), 4,49 (д, J = 19,1 Гц, 2H), 3,63 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z)-3-фтор-4-(3-фторфенилсульфонил)бут-2-ен-1-амина (Соединение 38)
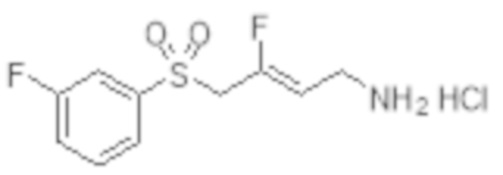
[0284] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,83 (ддд, J = 7,8, 1,7, 1,1 Гц, 1H), 7,81-7,66 (м, 2H), 7,56 (тдд, J = 8,4, 2,6, 1,1 Гц, 1H), 5,23 (дт, J = 32,9, 7,4 Гц, 1H), 4,44 (д, J = 19,1 Гц, 2H), 3,65 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z) -3-фтор-4-(4-фторфенилсульфонил)бут-2-ен-1-амина (Соединение 39)
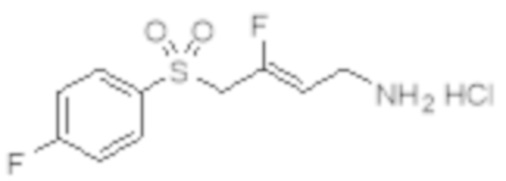
[0285] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,13 - 7,99 (м, 2H), 7,51 - 7,34 (м, 2H), 5,19 (дт, J = 32,8, 7,5 Гц, 1H), 4,39 (д, J = 19,1 Гц, 2H), 3,62 (ддт, J = 7,4, 1,9, 0,6 Гц, 2H).
Гидрохлорид (Z)-3-фтор-4-(о-толилсульфонил)бут-2-ен-1-амина (Соединение 40)
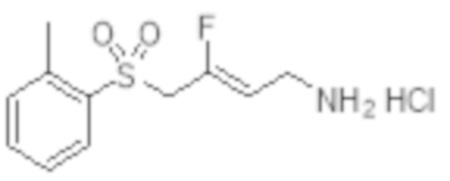
[0286] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,99 (дд, J = 7,6, 1,1 Гц, 1H), 7,71-7,59 (м, 1H), 7,53-7,39 (м, 2H), 5,22 (дт, J = 32,8, 7,4 Гц, 1H), 4,35 (дд, J = 19,3, 0,5 Гц, 2H), 3,63 (ддт, J = 7,4, 2,0, 0,6 Гц, 2H), 2,73 (с, 3H).
Гидрохлорид (Z)-3-фтор-4-(м-толилсульфонил)бут-2-ен-1-амина (Соединение 41)
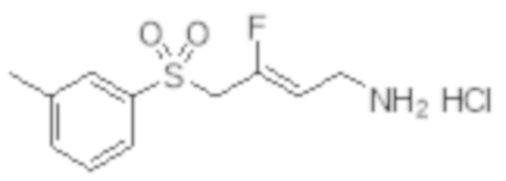
[0287] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,84 - 7,72 (м, 2H), 7,67 - 7,49 (м, 2H), 5,44 - 5,08 (м, 1H), 4,35 (дк, J = 19,1, 0,5 Гц, 2H), 3,64 (ддт, J = 7,4, 2,0, 0,6 Гц, 2H), 2,56 - 2,38 (м, 3H).
Гидрохлорид (Z)-3-фтор-4-(п-толилсульфонил)бут-2-ен-1-амина (Соединение 42)
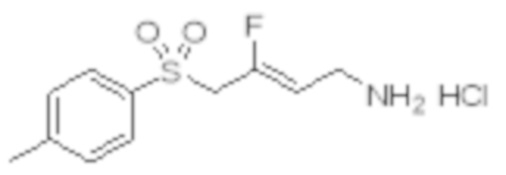
[0288] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,88 - 7,82 (м, 2H), 7,54 - 7,45 (м, 2H), 5,17 (дт, J = 32,8, 7,4 Гц, 1H), 4,32 (дд, J = 19,2, 0,5 Гц, 2H), 3,64 (ддт, J = 7,4, 2,0, 0,6 Гц, 2H), 2,49 (с, 3H)
Дигидрохлорид (Z)-3-фтор-4-((3-фторхинолин-8-ил)сульфонил)бут-2-ен-1-амина (Соединение 51)
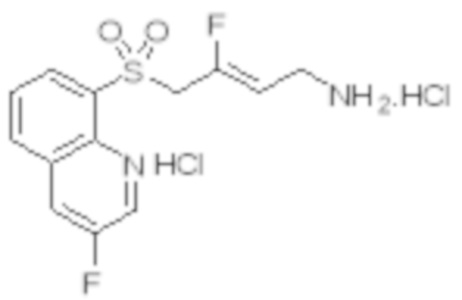
[0289] 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,09 (дд, J = 2,9, 0,6 Гц, 1H), 8,49 (ддд, J = 7,4, 1,4, 0,4 Гц, 1H), 8,38 (ддд, J = 8,3, 1,4, 0,4 Гц, 1H), 8,31 (дд, J = 8,8, 2,9 Гц, 1H), 7,87 (ддд, J = 8,3, 7,3, 0,8 Гц, 1H), 5,20 (дд, J = 32,9, 7,4 Гц , 1H), 4,99 (д, J = 19,3 Гц, 2H), 3,59 (д, J = 7,4 Гц, 2H).
ПРИМЕР 6
[0290] Следующие ниже соединения получали в соответствии с методиками F, J, H и I с использованием соответственно функционализированного исходного тиолового материала.
Методика J: Получение ( Z )- трет- бутил-(3-фтор-4-((4-(метилсульфонил)фенил)сульфонил)бут-2-ен-1-ил)карбамата
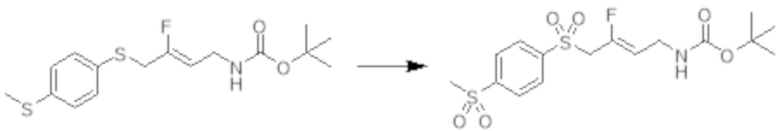
[0291] К перемешиваемой суспензии (Z)-трет-бутил(3-фтор-4-((4-(метилтио)фенил)тио)бут-2-ен-1-ил)карбамата (120 мг, 0,35 ммоль) и натрия гидрокарбоната (150 мг, 1,79 ммоль) в CH2Cl2 (4 мл) и воде (2 мл) при 0 °C добавляли 3-хлорпероксибензойную кислоту (378 мг, 2,19 ммоль) тремя порциями в течение 5 минут. Полученную суспензию перемешивали при 0 °C в течение 1,5 часов перед разбавлением водн. NaOH (2 M; 1 мл), водой (10 мл) и CH2Cl2. Затем фазы разделяли и водную фазу экстрагировали CH2Cl2 (10 мл × 2). Органические фазы объединяли, сушили (Na2SO4) и концентрировали в вакууме. Неочищенный материал очищали флэш-колонкой, элюируя 30% EtOAc/гексаном, с получением (Z)-трет-бутил-(3-фтор-4-((4-(метилсульфонил)фенил)сульфонил)бут-2-ен-1-ил)карбамата (17 мг, 12%) в виде белого твердого вещества. 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 1,37 (9H, с), 3,33 (3H, с), 3,54 (2H, прибл. т, J = 5,6 Гц), 4,62 (2H, д, J = 19,4 Гц), 4,93 (1H, дт, J = 36,4, 6,8 Гц), 7,05 (1H, т, J = 5,8 Гц), 8,15 (2H, дд, J = 8,7, 2,1 Гц), 8,21 (2H, дд, J = 8,7, 2,1 Гц).
(Z)-4-((3,5-бис(трифторметил)фенил)сульфонил)-3-фторбут-2-ен-1-аминотрифторацетат (Соединение 3)
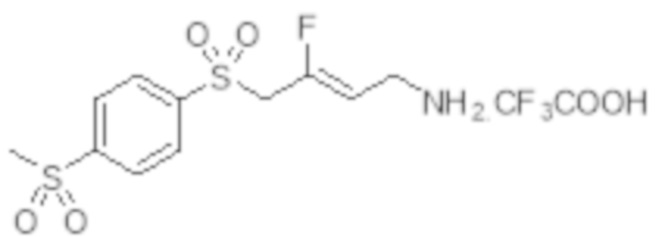
[0292] Белое твердое вещество; т.п. 155-157 °C; 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 3,35 (3H, с), 3,50 (2H, прибл. д, J = 6,7 Гц), 4,80 (2H, д, J = 19,7 Гц), 5,12 (1H, дт, J = 34,7, 7,3 Гц), 7,88 (3H, шс), 8,19 (2H, дд, J = 8,8, 2,5 Гц), 8,24 (2H, дд, J = 8,9, 2,4 Гц).
ПРИМЕР 7
[0293] Следующее соединение получали в соответствии с методиками K, L и M, затем F, N, H и I.
Получение гидрохлорида (Z)-4-(2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)-N,N-диметилбензолсульфонамида (Соединение 6)
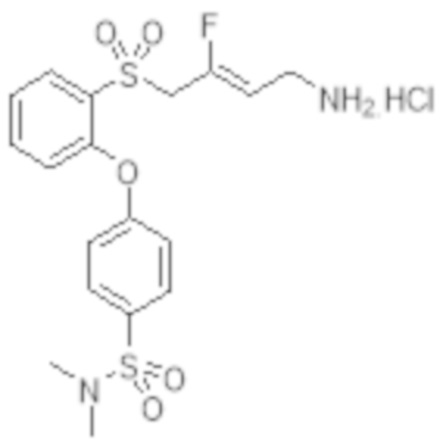
Методика К: Получение 4-бром- N,N ,-диметилбензолсульфонамида
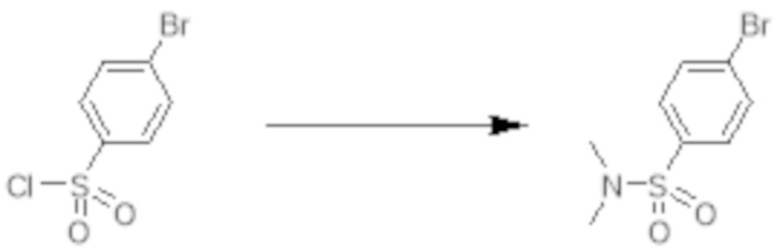
[0294] К перемешиваемому раствору диметиламина (12 мл, 40% мас./мас. водный раствор) в THF (20 мл) при 5 °C добавляли раствор 4-бромбензолсульфонилхлорида (5,00 г, 19,6 ммоль) в THF (10 мл) в течение 5 минут. После добавления смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали в вакууме и полученный остаток разделяли между водой (25 мл) и CH2Cl2 (20 мл), и водный слой экстрагировали дополнительным количеством CH2Cl2 (20 мл x 2). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме, с получением 4-бром-N,N,-диметилбензолсульфонамида (4,83 г, 93%) в виде белого твердого вещества. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 2,74 (6H, с), 7,64-7,73 (4H, м).
Методика L: Получение N,N- диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензолсульфонамида
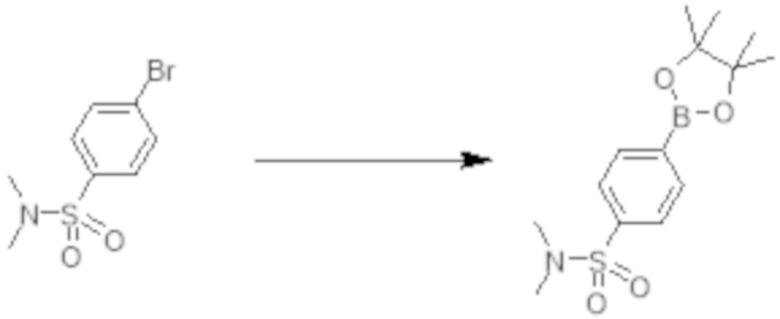
[0295] Перемешиваемую суспензия 4-бром-N,N-диметилбензолсульфонамида (1,00 г, 3,79 ммоль), 4,4,5,5-тетраметил-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолана (1,15 г, 4,54 ммоль) и ацетата калия (1,11 г, 11,4 ммоль) в 1,4-диоксане (25 мл) продували азотом в течение 15 минут перед добавление комплекса дихлорида 1,1'-бис(дифенилфосфино)ферроцен-палладия (II) и дихлорметана (155 мг, 0,19 ммоль). Полученную суспензию нагревали при 80 °C в атмосфере азота в течение 16 часов. Смесь охлаждали до комнатной температуры, разделяли между EtOAc (40 мл) и водой (30 мл) и фильтровали через целит. Органический слой отделяли, а водный слой дополнительно экстрагировали EtOAc (20 мл × 2). Затем объединенные органические слои промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме с получением N,N-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензолсульфонамида (1,60 г, 68%) в виде серого твердого вещества. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,38 (12H, с), 2,71 (6H, с), 7,77 (2H, дд, J = 8,4, 1,0 Гц), 7,98 (2H, дд, J = 8.4, 0.9 Гц).
Методика М: Получение (4-( N , N- диметилсульфамоил)фенил)бороновой кислоты
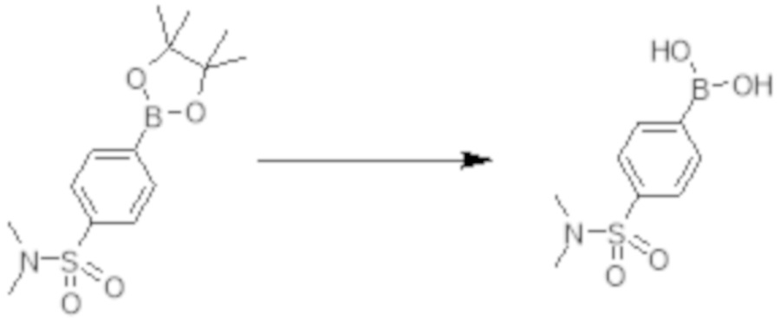
[0296] К перемешиваемому раствору N,N-диметил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензолсульфонамида (1,00 г, 2,25 ммоль) в THF (20 мл) и воде (5 мл) при 0 °C добавляли перйодат натрия (2,06 г, 9,64 ммоль). Смесь перемешивали в течение 5 минут при этой температуре, а затем давали нагреться до комнатной температуры и перемешивали еще 30 минут. Добавляли водный HCl (1 M; 1,57 мл, 1,57 ммоль), и полученную смесь перемешивали при комнатной температуре в течение еще 1 часа. Реакционную смесь разбавляли водой (30 мл) и экстрагировали EtOAc (20 мл × 3). Затем органические слои объединяли и промывали (рассолом), сушили (Na2SO4) и концентрировали в вакууме. Неочищенный материал очищали флэш-колонкой, элюируя смесью 50% EtOAc/гексан, а затем 10% MeOH в 50% EtOAc/гексане с получением (4-(N,N-диметилсульфамоил)фенил)бороновой кислоты (470 мг, 91%) в виде коричневого твердого вещества. 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 2,69 (6H, с), 7,75 (2H, д, J = 8,2 Гц), 7,88-7,98 (2H, м).
Методика N: Получение ( Z )- трет- бутил(4-((2-(4-( N , N- диметилсульфамоил)фенокси)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата
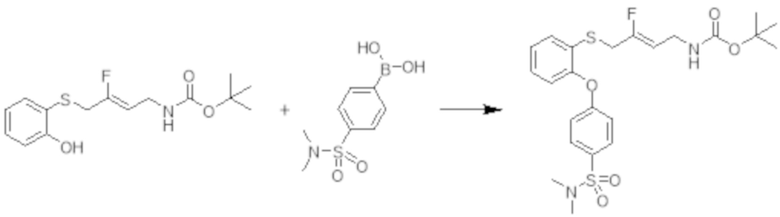
[0297] К перемешиваемому раствору (Z)-трет-бутил(3-фтор-4-((2-гидроксифенил)тио)бут-2-ен-1-ил)карбамата (150 мг, 0,48 ммоль), (4-(N,N-диметилсульфамоил)фенил)бороновой кислоты (219 мг, 0,96 ммоль) и пиридина (0,19 мл, 2,39 ммоль) в CH2Cl2 (6 мл) при комнатной температуре добавляли ацетат меди (II) (87 мг, 0,48 ммоль) одним порцией. Полученную смесь перемешивали при этой температуре в течение 16 часов. По истечении этого времени реакционную смесь разбавляли добавлением CH2Cl2 (30 мл), фильтровали через целит и промывали водн. HCl (1 M; 20 мл) с последующим насыщ. водн. NaHCO3 (20 мл) и рассолом (20 мл). Затем органическую фазу сушили (Na2SO4) и концентрировали в вакууме. Неочищенное вещество очищали флэш-колонкой, элюируя 25% EtOAc/гексаном, с получением (Z)-трет-бутил-(4-((2-(4-(N,N-диметилсульфамоил)фенокси)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата (80 мг, 34%) в виде желтого масла. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,45 (9H, с), 2,73 (6H, с), 3,55 (2H, д, J = 17,1 Гц), 3,73 (2H, около т, J = 5,6 Гц), 4,46 (1H, шс), 4,80 (1H, дт, J = 34,8, 6,8 Гц), 7,02 (2H, д, J = 8,7 Гц), 7,06 (1H, дд, J = 8,2, 1,0 Гц), 7,24 (1H, ддд, J = 7,5, 7,5, 1,1 Гц), 7,35 (1H, ддд, J = 7,6, 7,6, 1,6 Гц), 7,52 (1H, дд, J = 7,7, 1,5 Гц), 7,75 (2H, д, J = 8,6 Гц).
Гидрохлорид (Z)-4-(2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)фенокси)-N,N-диметилбензолсульфонамида (Соединение 6)
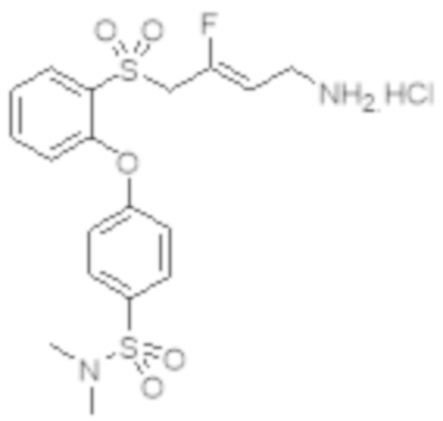
[0298] Белое твердое вещество; т.п. 153-156 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 2,73 (6H, с), 3,64 (2H, прибл. д, J = 6,9 Гц), 4,57 (2H, д, J = 19,1 Гц), 5,30 ( 1H, дт, J = 32,8, 7,3 Гц), 7,25 (1H, дд, J = 8,3, 0,7 Гц), 7,30 (2H, дд, J = 8,9, 2,0 Гц), 7,49 (1H, ддд, J = 7,9, 7,9, 1,0 Гц), 7,81 (1H, ддд, J = 7,8, 8,3, 1,7 Гц), 7,86 (2H, дд, J = 8,6, 2,0 Гц), 8,07 (1H, дд, J = 7,9, 1,6 Гц).
ПРИМЕР 8
[0299] Следующее соединение получают согласно методикам K, L, M, N, H и I.
Гидрохлорид (Z)-4-(3-(4-амино-2-фторбут-2-енилсульфонил)фенокси)-N,N-диметилбензолсульфонамида (Соединение 16)
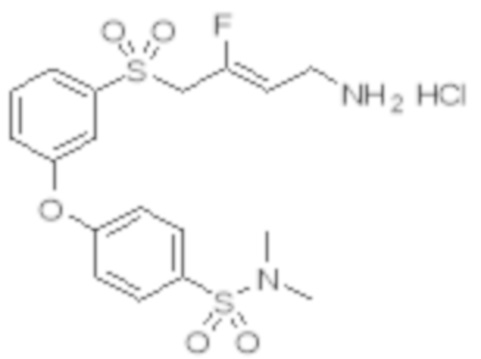
[0300] Белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,89 - 7,82 (м, 3H), 7,76 (тд, J = 8,0, 0,5 Гц, 1H), 7,68 (т, J = 2,4, 1,7 Гц, 1H), 7,53 (ддд, J = 8,1, 2,5, 1,1 Гц, 1H), 7,29 - 7,22 (м, 2H), 5,28 (дт, J = 33,2, 7,4 Гц, 1H), 4,90 (с, 6H), 4,43 (д, J = 19,1 Гц, 2H), 3,65 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z)-3-(3-(4-амино-2-фторбут-2-енилсульфонил)фенокси)-N,N-диметилбензолсульфонамида (Соединение 17)
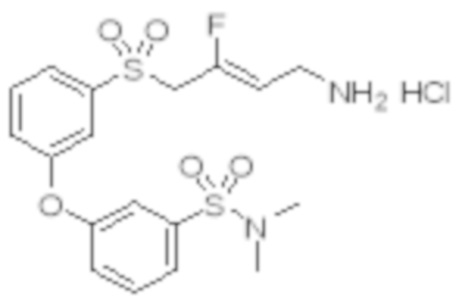
[0301] Белое твердое вещество; т.п. 220 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 7,81 (ддд, J = 7,8, 1,7, 1,2 Гц, 1H), 7,77-7,69 (м, 2H), 7,66-7,59 (м, 2H), 7,50 (ддд, J = 8,0, 2,5, 1,2 Гц, 1H), 7,46-7,38 (м, 2H), 5,24 (дт, J = 32,9, 7,4 Гц, 1H), 4,90 (с, 6H), 4,41 (д, J = 19,1 Гц, 2H), 3,65 (дд, J = 7,4, 1,9 Гц, 2H).
Гидрохлорид (Z)-3-(2-(4-амино-2-фторбут-2-енилсульфонил)фенокси)-N,N-диметилбензолсульфонамида (Соединение 18)
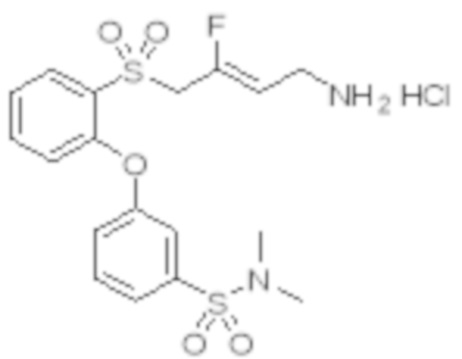
[0302] Белое твердое вещество; т.п. 205 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,06 (ддд, J = 7,9, 1,7, 0,3 Гц, 1H), 7,83 - 7,73 (м, 1H), 7,70 (дд, J = 7,7, 0,7 Гц, 1H), 7,65 (дт, J = 7,8, 1,4 Гц, 1H), 7,51 - 7,42 (м, 3H), 7,20 (дд, J = 8,3, 1,0 Гц, 1H), 5,31 (дт, J = 33,0, 7,4 Гц , 1H), 4,90 (с, 6H), 4,60 (д, J = 19,2 Гц, 2H), 3,64 (дд, J = 7,4, 1,9 Гц, 2H).
ПРИМЕР 9
[0303] Следующее соединение получают в соответствии с методиками O и P, затем F, Q, H и I.
Получение (Z)-2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)-N-(4-(N,N-диизопропилсульфамоил)фенил)бензамида (Соединение 15)
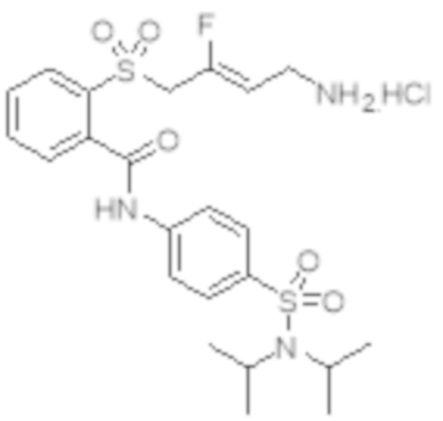
Методика О: Получение N,N - диизо -пропил-4 нитробензолсульфонамида
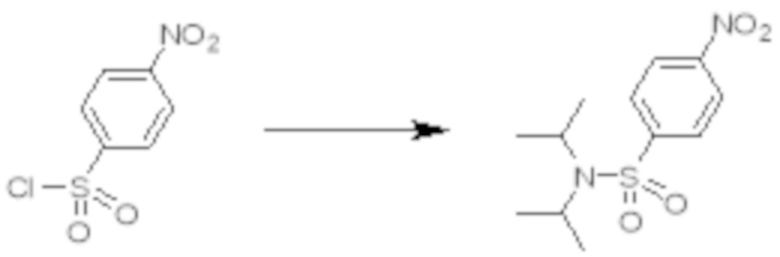
[0304] К перемешиваемому раствору диизопропиламина (3,16 мл, 22,6 ммоль) в THF (10 мл) при 0-5 °C добавляли раствор 4-нитробензолсульфонилхлорида (2,00 г, 9,02 ммоль) в THF (5 мл) в течение 5 минут. После добавления смесь оставляли перемешиваться при комнатной температуре в течение 16 часов. Затем реакционную смесь концентрировали в вакууме и полученный остаток разделяли между водой (25 мл) и CH2Cl2 (20 мл). Фазы разделяли и водный слой дополнительно экстрагировали CH2Cl2 (20 мл × 2). Затем объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Неочищенный материал очищали флэш-колонкой, элюируя 20% EtOAc/гексан, с получением N,N-диизопропил-4-нитробензолсульфонамида (285 мг, 11% выход) в виде желтого твердого вещества. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,30 (12H, д, J = 6,7 Гц), 3,79 (1H, гепт, J = 6,9 Гц), 8,07 (2H, дд, J = 9,2, 2,2 Гц), 8,35 (2H, дд, J = 8,9, 1,9 Гц).
Методика Р: Получение 4-амино- N,N- ди изо пропилбензолсульфонамида

[0305] К перемешиваемому раствору N,N-диизопропил-4-нитробензолсульфонамида (260 мг, 0,91 ммоль) в метаноле (10 мл) при комнатной температуре добавляли суспензию палладия на угле (10% мас./мас.; 50 мг) в воду (50 μл) в атмосфере азота. Полученную смесь перемешивали в атмосфере водорода в течение 3 часов. Смесь фильтровали через целит и концентрировали в вакууме с получением 4-амино-N,N-диизопропилбензолсульфонамида (200 мг, 86%) в виде коричневого твердого вещества. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,27 (12H, д, J = 6,8 Гц), 3,66 (2H, гепт, J = 6,8 Гц), 4,04 (2H, шс), 6,67 (2H, дд, J = 8,7, 2,1 Гц), 7,65 (2H, дд, J = 8,6, 1,9 Гц).
Методика Q: Получение (Z)-трет-бутил-(4-((2-((4-(N,N-диизопропилсульфамоил)фенил)карбамоил)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата
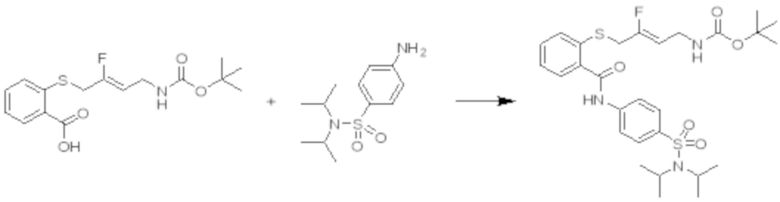
[0306] К перемешиваемому раствору (Z)-2-((4-((трет-бутоксикарбонил)амино)-2-фторбут-2-ен-1-ил)тио)бензойной кислоты (180 мг, 0,53 ммоль), 4-амино-N,N-диизопропилбензолсульфонамида (203 мг, 0,79 ммоль) и триэтиламина (0,26 мл, 1,85 ммоль) в DMF (0,8 мл) при комнатной температуре добавляли HATU (301 мг, 0,79 ммоль) и полученный раствор перемешивали при этой температуре в течение 16 ч. Затем реакционную смесь разделяли между водой (10 мл) и EtOAc (10 мл), и фазы разделяли. Водную фазу снова экстрагировали EtOAc, и органические фазы объединяли и промывали HCl (1 M; 20 мл), затем водой (20 мл x 3) и рассолом (20 мл). Органическую фазу сушили (Na2SO4) и концентрировали в вакууме. Сырой продукт очищали флэш-хроматографией, элюируя смесью 50-100% EtOAc/гексан с получением (Z)-трет-бутил-(4-((2-((4-(N,N-диизопропилсульфамоил)фенил)карбамоил)фенил)тио)-3-фторбут-2-ен-1-ил)карбамата (93 мг, 30%) в виде масла бледно-сиреневого цвета. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,31 (12H, д, J = 6,7 Гц), 1,44 (9H, с), 3,57 (2H, д, J = 18,5 Гц), 3,63 (2H, около т, J = 5,6 Гц), 3,74 (2H, гепт, J = 6,7 Гц), 4,40 (1H, дт, J = 34,8, 6,9 Гц), 4,75 (1H, шс), 7,41 (1H, дд, J = 7,1, 7,1 Гц), 7,47 (1H, ддд, J = 7,8, 7,8, 1,8 Гц), 7,62 (1H, дд, J = 7,8, 1,2 Гц), 7,75 (1H, дд, J = 7,3 Гц), 7,88 (4H, шс).
Гидрохлорид (Z)-2-((4-амино-2-фторбут-2-ен-1-ил)сульфонил)-N-(4-(N,N-диизопропилсульфамоил)фенил)бензамида (Соединение 15)
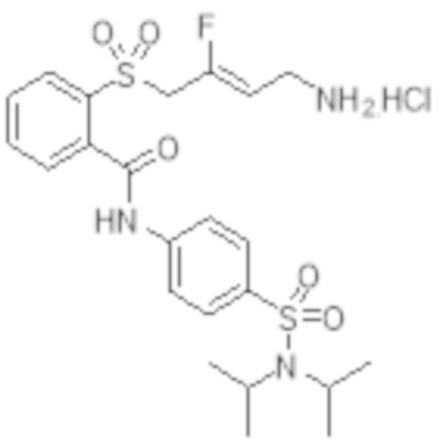
[0307] Белое твердое вещество; т.п. 248–250 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 1,29 (12H, д, J = 6,9 Гц), 3,64 (2H, дд, J = 7,4, 1,5 Гц), 3,78 (2H, гепт, J = 6,7 Гц), 4,69 (2H, д, J = 19,2 Гц), 5,23 (1H, дт, J = 32,4, 7,4 Гц), 7,76-7,82 (2H, м), 7,85-7,93 (5H, м), 8,10-8,13 (1H, м).
ПРИМЕР 10
[0308] Следующие соединения получали в соответствии с методиками Q, H и I с использованием соответствующих исходных тиоловых и амино веществ.
Гидрохлорид N-(адамантан-1-ил)-4-(((Z)-4-амино-2-фторбут-2-ен-1-ил)сульфонил)бензамида (Соединение 4)
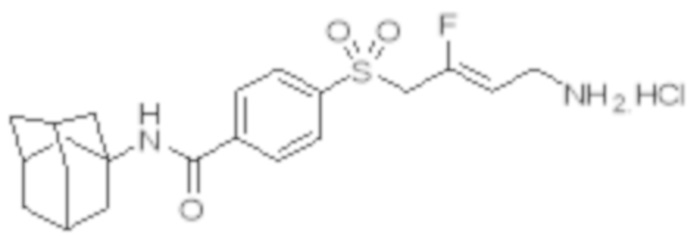
[0309] Не совсем белое твердое вещество; т.п. 231-233 °C; 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 1,67 (6H, шс), 2,08 (9H, шс), 3,46 (2H, шс), 4,67 (2H, д, J = 19,7 Гц), 5,10 (1H, дт, J = 34,6, 7,1 Гц), 7,97 (2H, дд, J = 8,6, 1,5 Гц), 8,00 (2H, дд, J = 8,8, 1,4 Гц), 8,06 (3H , шс).
Гидрохлорид N-(адамантан-1-ил)-2-(((Z)-4-амино-2-фторбут-2-ен-1-ил)сульфонил)бензамида (Соединение 12)
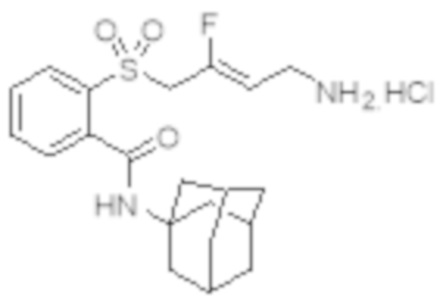
[0310] Белое твердое вещество; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 1,77-1,80 (6H, м), 2,09-2,15 (3H, м), 2,16-2,21 (6H, м), 3,61 (2H, около д, J = 7,2 Гц), 4,62 (2H, д, J = 19,5 Гц), 5,16 (1H, дт, J = 32,6, 7,4 Гц), 7,56 (1H, дд, J = 7,5, 1,0 Гц), 7,67 (1H, ддд, J = 7,7, 7,7, 1,2 Гц), 7,78 (1H, ддд, J = 7,5, 7,5, 1,1 Гц), 8,01 (1H, дд, J = 7,8, 1,0 Гц).
Гидрохлорид N-(адамантан-1-ил)-3-(((Z)-4-амино-2-фторбут-2-ен-1-ил)сульфонил)бензамида (Соединение 13)
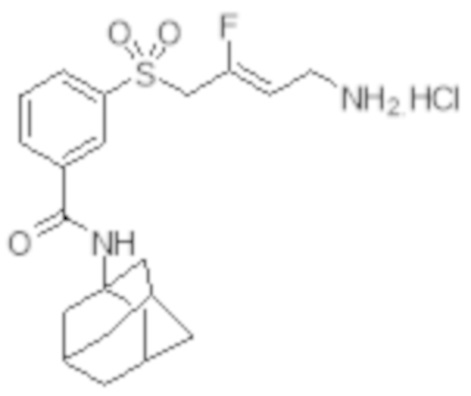
[0311] Белое твердое вещество; т.п. 230-232 °C; 1H-ЯМР (300 МГц; d6-DMSO) δ м.д.: 1,68 (6H, шс), 2,05-2,12 (9H, м), 3,49 (2H, шс), 4,68 (2H, д, J 19,8 Гц), 5,12 (1H, дт, J 34,7, 7,0 Гц), 7,73 (1H, дд, J 7,7, 7,7 Гц), 7,98 (3H, шс), 8,03 (1H, ддд, J 7,8, 1,7, 0,9 Гц), 8,16 (1H, ддд, J 7,8, 1,4, 1,0 Гц), 8,28 (1H, дд, J 1,6, 1,1 Гц).
ПРИМЕР 11
[0312] Следующие соединения получали в соответствии с методиками R и I.
Гидрохлорид (Z)-3-фтор-4-(фенилсульфонил)бут-2-ен-1-амина (Соединение 1)
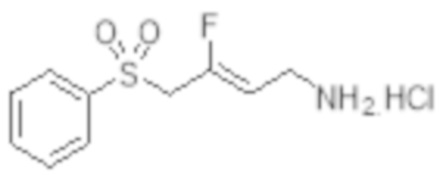
Методика R: Получение трет- бутил ( Z )-(3-фтор-4-(фенилсульфонил)бут-2-ен-1-ил)карбамата
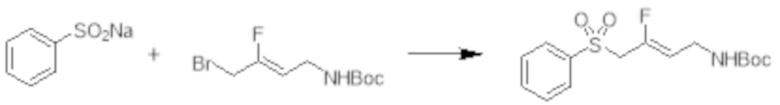
[0313] К перемешиваемому раствору трет-бутил (Z)-(4-бром-3-фторбут-2-ен-1-ил)карбамата (60,0 г, 224 ммоль) в DMF (300 мл) добавляли бензолсульфинат натрия (44,1 г, 269 ммоль) порциями при комнатной температуре в течение 15 минут. Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разбавляли водой (2,7 л) и продолжали перемешивание при комнатной температуре в течение еще 15 минут. Полученное осажденное твердое вещество фильтровали, промывали водой (50 мл x 2) и затем сушили в печи при 60°С с получением трет-бутил (Z)-(3-фтор-4-(фенилсульфонил)бут-2-ен-1-ил)карбамата (74,0 г, 100%) в виде белого твердого вещества, которое использовали как таковое на следующей стадии. 1H ЯМР (300 МГц, CDCl3) δ м.д.: 7,98 - 7,91 (м, 2H), 7,76 - 7,67 (м, 1H), 7,61 (ддт, J = 8,3, 6,6, 1,3 Гц, 2H), 4,94 (дт, J = 34,3, 7,0 Гц, 1H), 4,59 (с, 1H), 3,94 (д, J = 18,4 Гц, 2H), 3,79 (т, J = 6,5 Гц, 2H), 1,46 (с, 9H).
Гидрохлорид (Z)-3-фтор-4-(фенилсульфонил)бут-2-ен-1-амина (Соединение 1)
[0314] Не совсем белое твердое вещество; т.п. 209-211 °C; 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 3,64 (2H, дд, J = 7,3, 1,2 Гц), 4,36 (2H, д, J = 19,1 Гц), 5,18 (1H, дт, J = 32,7 , 7,4 Гц), 7,65-7,71 (2H, м), 7,79 (1H, тт, J = 7,4, 1,2 Гц), 7,96-8,00 (2H, м); ЖХМС: для C10H12FNO2S вычислено 229,1, найдено 230,1 [M+1]+.
ПРИМЕР 12
[0315] Следующие соединения получали в соответствии с методиками R и I.
Гидрохлорид (Z)-4-(2-хлорфенилсульфонил)-3-фторбут-2-ен-1-амина (Соединение 19)
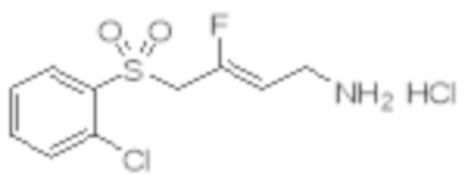
[0316] Белое твердое вещество; т.п. 205-207 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,13 (ддд, J = 7,9, 1,5, 0,6 Гц, 1H), 7,81-7,69 (м, 2H), 7,62 (ддд, J = 7,9, 6,4, 2,2 Гц, 1H), 5,27 (дт, J = 32,8, 7,4 Гц, 1H), 4,59 (д, J = 19,1 Гц, 2H), 3,62 (дд, J = 7,4, 1,9 Гц, 2H); ЖХМС: для C10H11ClFNO2S вычислено 263,0, найдено 264,0 [M+1]+.
Дигидрохлорид (Z)-3-фтор-4-(пиридин-2-илсульфонил)бут-2-ен-1-амина (Соединение 30)
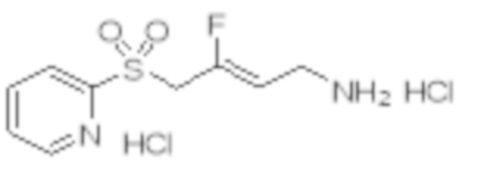
[0317] Белое твердое вещество; т.п. 153-155 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,88 - 8,73 (м, 1H), 8,25 - 8,10 (м, 2H), 7,77 (ддд, J = 6,8, 4,7, 1,9 Гц, 1H), 5,26 (дт, J = 32,9, 7,4 Гц, 1H), 4,59 (д, J = 19,1 Гц, 2H), 3,63 (дд, J = 7,3, 1,8 Гц, 2H).
Дигидрохлорид (Z)-3-фтор-4-(пиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 31)
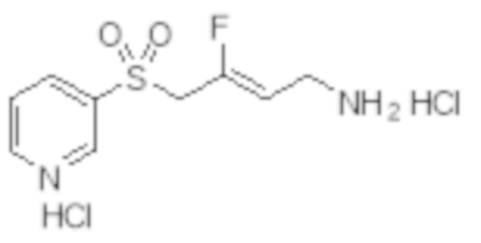
[0318] Белое твердое вещество; т.п. 168–170 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,23 (дд, J = 2,3, 0,8 Гц, 1H), 9,04 (дд, J = 5,2, 1,5 Гц, 1H), 8,64 (дт, J = 8,2, 1,9 Гц, 1H), 7,94 (ддд, J = 8,2, 5,2, 0,8 Гц, 1H), 5,30 (дт, J = 33,1, 7,4 Гц, 1H), 4,59 (д, J = 19,1 Гц, 2H), 3,68 (д, J = 7,4 Гц, 2H).
ПРИМЕР 13
[0319] Следующее соединение получали в соответствии с методиками S, F, H и I.
Дигидрохлорид (Z)-3-фтор-4-(3-метилпиридин-2-илсульфонил)бут-2-ен-1-амина (Соединение 43)
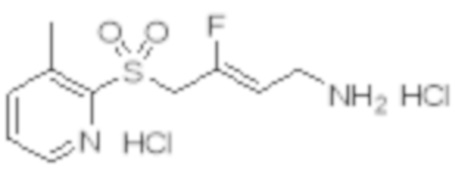
Методика S: Получение 3-метилпиридин-2-тиола
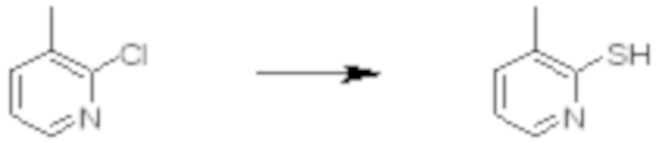
[0320] 2-Хлор-3-метилпиридин (500 мг, 3,93 ммоль) и гидросульфид натрия (2,21 г, 39,36 ммоль) помещали в DMF (2,0 мл). Полученную смесь нагревали при 120 °C в течение 12 ч. После завершения реакции (ТСХ) реакционную смесь охлаждали до комнатной температуры, разбавляли водой (20 мл), подкисляли (pH = 5) добавлением уксусной кислоты и экстрагировали EtOAc (20 мл x 3). Объединенный органический экстракт сушили над Na2SO4 и концентрировали при пониженном давлении с получением 3-метилпиридин-2-тиола (1,50 г, 50,5%). 1H ЯМР (600 МГц, d6-DMSO) δ м.д.: 13,5 - 13,3 (м, 1H), 7,6-7,58 (м, 1H), 7,51 (д, J = 6 Гц, 1H), 2,21 (с, 3H).
Дигидрохлорид (Z)-3-фтор-4-(3-метилпиридин-2-илсульфонил)бут-2-ен-1-амина (Соединение 43)
[0321] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,53 (дд, J = 4,7, 1,3 Гц, 1H), 7,92 (д, J = 7,6 Гц, 1H), 7,60 (дд, J = 7,8, 4,5 Гц, 1H), 5,37 (дт, J = 32,9, 7,2 Гц, 1H), 4,75 (д, J = 19,0 Гц, 2H), 3,67 (д, J = 7,1 Гц, 2H), 2,68 (с, 3H).
ПРИМЕР 14
[0322] Следующие соединения получали в соответствии с методиками S, F, H и I.
Дигидрохлорид (Z)-3-фтор-4-(2-метилпиридин-4-илсульфонил)бут-2-ен-1-амина (Соединение 44)
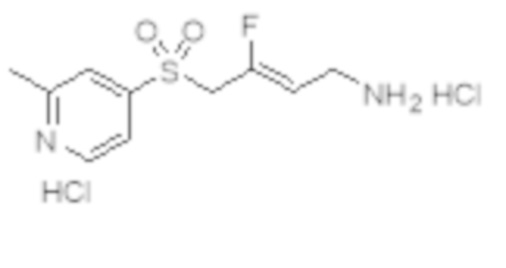
[0323] Бледно-желтое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ 9,00 (д, J = 5,6 Гц, 1H), 8,35 (с, 1H), 8,23 (д, J = 5,4 Гц, 1H), 5,36 (дт, J = 33,0, 7,1 Гц, 1H), 4,68 (д, J = 18,9 Гц, 2H), 3,69 (д, J = 6,7 Гц, 2H), 2,90 (с, 3H).
Дигидрохлорид (Z)-3-фтор-4-(5-изопропилпиридин-2-илсульфонил)бут-2-ен-1-амина (Соединение 34)
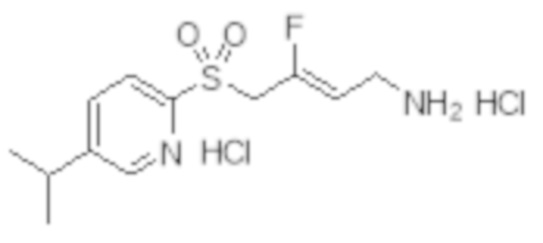
[0324] Белое стеклообразное твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ 8,70 (т, J = 1,3 Гц, 1H), 8,12-7,94 (м, 2H), 5,28 (дт, J = 32,9, 7,4 Гц, 1H), 4,55 (д, J = 19,1 Гц, 2H), 3,64 (дд, J = 7,6, 1,7 Гц, 2H), 3,15 (гепт, J = 6,9 Гц, 1H), 1,36 (д, J = 6,9 Гц, 6H).
Дигидрохлорид (Z)-3-фтор-4-(5-метилпиридин-2-илсульфонил)бут-2-ен-1-амина (Соединение 35)
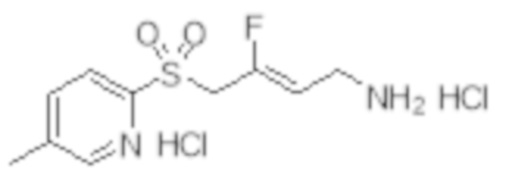
[0325] Белое твердое вещество; т.п. 155-157 °C; 1H ЯМР (300 МГц, d6-DMSO) δ 8,67 (д, J = 0,9 Гц, 1H), 8,09 (с, 3H), 7,98 (т, J = 1,4 Гц, 2H), 5,20 (дт, J = 34,8, 7,2 Гц, 1H), 4,70 (д, J = 19,6 Гц, 2H), 3,46 (т, J = 5,9 Гц, 2H), 2,45 (с, 3H).
Дигидрохлорид (Z)-3-фтор-4-(6-метилпиридин-2-илсульфонил)бут-2-ен-1-амина (Соединение 36)
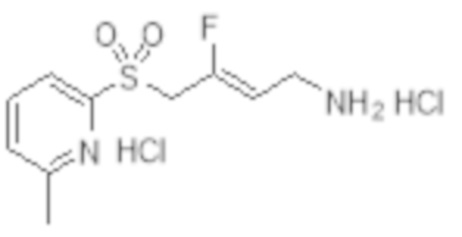
[0326] Бледно-желтое твердое вещество; т.п. 174–176 °C; 1H ЯМР (300 МГц, d6-DMSO) δ 8,18 (с, 3H), 8,06 (т, J = 7,8 Гц, 1H), 7,88 (д, J = 7,7 Гц, 1H), 7,66 (д, J = 7,5 Гц, 1H), 5,23 (дт, J = 34,8, 7,1 Гц, 1H), 4,70 (д, J = 19,6 Гц, 2H), 3,46 (т, J = 5,9 Гц, 2H), 2,61 (с, 3H).
ПРИМЕР 15
[0327] Следующее соединение получали в соответствии с методиками T, U и V, затем F, H и I.
Дигидрохлорид (Z)-3-фтор-4-(6-изопропилпиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 49)
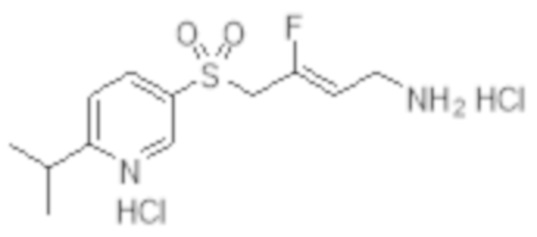
Методика T: Получение метил-3-((6-хлорпиридин-3-ил)тио)пропаноата

[0328] В повторно закрываемой реакционной пробирке объемом 50 мл растворяли 2-хлор-5-йодпиридин (2,50 г, 10,5 ммоль) в дегазированном 1,4-диоксане (25 мл) при комнатной температуре в атмосфере азота. Последовательно добавляли Pd2(dba)3 (100 мг, 0,11 ммоль), ксантфос (125 мг, 0,22 ммоль), метил 3-меркаптопропаноат (1,25 г, 10,5 ммоль) и DIPEA (2,50 мл, 14,4 ммоль) в атмосфере азота. Раствор дегазировали продувкой газообразным азотом в течение 15 мин, и затем постепенно нагревали до 70 °С. Полученную реакционную смесь перемешивали при этой температуре в течение 6 часов. По завершении реакции (ТСХ) реакционную смесь охлаждали до комнатной температуры, разбавляли холодной водой и экстрагировали этилацетатом (3 × 30 мл). Объединенные органические слои промывали рассолом и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (100-200 меш), элюируя 10% EtOAc-гексаны, с получением метил-3-((6-хлорпиридин-3-ил)тио)пропаноата (2,40 г, 99%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,36 (д, J = 2,4 Гц, 1H), 7,67 (дд, J = 8,4, 2,4 Гц, 1H), 7,28 (д, J = 9,2 Гц, 1H), 3,69 (с, 3H), 3,17 (т, J = 7,2 Гц, 2H), 2,64 (т, J = 7,2 Гц, 2H).
Методика U: Получение метил-3-((6-изопропилпиридин-3-ил)тио)пропаноата

[0329] В круглодонной колбе на 1000 мл растворяли метил-3-((6-хлорпиридин-3-ил)тио)пропаноат (5,00 г, 21,6 ммоль) в безводном THF (200 мл) и 1-метил-2-пирролидиноне (25 мл) в атмосфере азота. Раствор охлаждали до -55 °С, и добавляли раствор ацетилацетоната железа (III) (1,70 г, 4,80 ммоль) в THF (50 мл), поддерживая атмосферу азота. Полученную смесь перемешивали при -55 °C в течение 15 мин, за это время по каплям добавляли раствор изопропилмагнийхлорида в THF (2 M, 50 мл) при -55 °C в атмосфере азота. Полученную реакционную смесь перемешивали при -40 °C еще 1 час. После завершения реакции (ТСХ) реакционную смесь охлаждали до 0 °C, гасили насыщенным раствором NH4Cl (50 мл) и продукт экстрагировали этилацетатом (100 мл x 3). Объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (100-200 меш), элюируя 10% EtOAc-гексанами, с получением метил-3-((6-изопропилпиридин-3-ил)тио)пропаноата (2,60 г, 50%) в виде бледно-желтой жидкости.
Методика V: Получение 6-изопропилпиридин-3-тиола
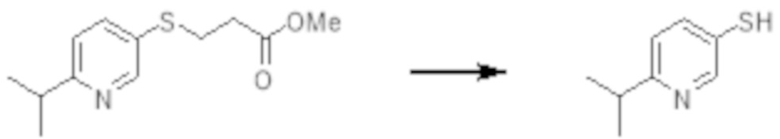
[0330] В круглодонной колбе на 100 мл растворяли метил-3-((6-изопропилпиридин-3-ил)тио)пропаноат (860 мг, 3,59 ммоль) в безводном THF (20 мл) и охлаждали раствор до -78 °С в атмосфере азота. К указанной выше смеси добавляли раствор трет-бутоксида калия в THF (1,0 М, 3,5 мл, 3,59 ммоль) в атмосфере азота. Полученную смесь перемешивали при -78 °C в течение 1 ч. По завершении реакции (ТСХ) реакционную смесь нагревали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток промывали н-гексаном, с получением 6-изопропилпиридин-3-тиола (655 мг) в виде светло-коричневого твердого вещества. Соединение использовали на следующей стадии без дополнительной очистки.
Дигидрохлорид (Z)-3-фтор-4-(6-изопропилпиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 49)
[0331] 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,29 (дд, J = 2,1, 0,7 Гц, 1H), 9,02 (ддд, J = 8,6, 3,5, 2,1 Гц, 1H), 8,32 (дд, J = 8,6, 3,2 Гц, 1H), 5,48 (т, J = 7,3 Гц, 1H), 4,74 (д, J = 19,0 Гц, 2H), 3,81 - 3,65 (м, 2H), 3,55 (дк, J = 7,0, 2,2 Гц, 1H), 1,53 (дд, J = 7,0, 0,8 Гц, 6H).
ПРИМЕР 16
[0332] Следующее соединение получают в соответствии с методиками T, U и V, затем F, H и I.
Дигидрохлорид (Z)-3-фтор-4-(2-изопропилпиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 45)
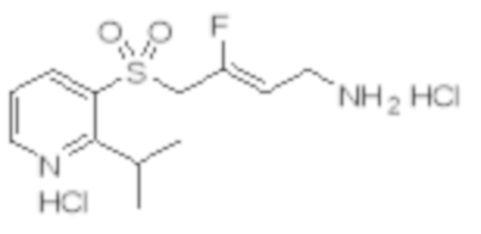
[0333] 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,95 (дд, J = 5,2, 1,7 Гц, 1H), 8,66 (дд, J = 8,2, 1,8 Гц, 1H), 7,78 (дд, J = 8,0, 5,0 Гц, 1H), 5,39 (дт, J = 33,2, 7,3 Гц, 1H), 4,58 (д, J = 19,0 Гц, 2H), 4,08 (p, J = 6,8 Гц, 1H), 3,72 - 3,62 (м, 2H), 1,45 (д, J = 6,7 Гц, 6H).
ПРИМЕР 17
[0334] Следующее соединение получают в соответствии с методиками T и V, затем F, H и I.
Дигидрохлорид (Z)-3-фтор-4-(6-метилпиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 46)
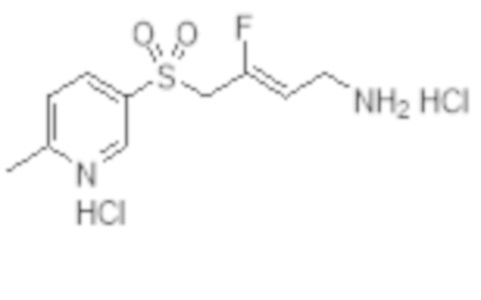
[0335] 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,17 (д, J = 2,2 Гц, 1H), 8,67 (д, J = 8,5 Гц, 1H), 7,96 (д, J = 8,4 Гц, 1H), 5,33 (дт, J = 33,0, 7,3 Гц, 1H), 4,62 (д, J = 19,1 Гц, 2H), 3,75 - 3,63 (м, 2H), 2,85 (с, 3H).
Дигидрохлорид (Z)-3-фтор-4-(2-метилпиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 47)
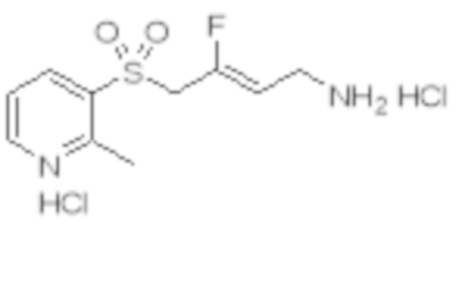
[0336] 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,00 (дд, J = 5,6, 1,5 Гц, 1H), 8,91 (дд, J = 8,1, 1,5 Гц, 1H), 8,05 (дд, J = 8,1, 5,6 Гц, 1H), 5,42 (дт, J = 33,2, 7,3 Гц, 1H), 4,67 (д, J = 19,0 Гц, 2H), 3,79 - 3,64 (м, 2H), 3,12 (с, 3H).
Дигидрохлорид (Z)-3-фтор-4-(4-метилпиридин-3-илсульфонил)бут-2-ен-1-амина (Соединение 48)
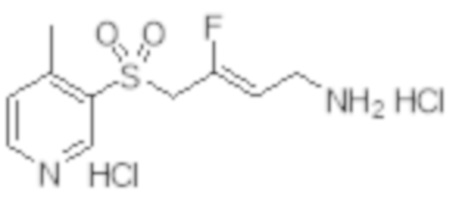
[0337] Не совсем белое твердое вещество; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,25 (с, 1H), 8,97 (д, J = 5,8 Гц, 1H), 8,07 (д, J = 5,9 Гц, 1H), 5,42 (дт, J = 33,2, 7,3 Гц, 1H), 4,68 (д, J = 19,1 Гц, 2H), 3,70 (дд, J = 7,4, 1,9 Гц, 2H), 2,99 (д, J = 0,6 Гц, 3H).
Гидрохлорид (Z)-3-фтор-4-((2-метилбензо[d]тиазол-4-ил)сульфонил)бут-2-ен-1-амина (Соединение 50)
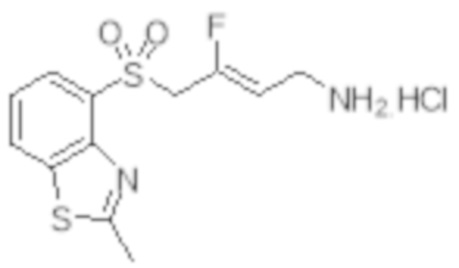
[0338] Бледно-желтое твердое вещество; т.п. 243–245 °C; 1H ЯМР (300 МГц, CD3OD) δ м.д.: 8,37 (дд, J = 8,1, 1,2 Гц, 1H), 8,10 (дд, J = 7,7, 1,2 Гц, 1H), 7,63 (т, J = 7,9 Гц, 1H), 5,20 (дт, J = 32,8, 7,4 Гц, 1H), 4,82 (д, J = 19,0 Гц, 2H), 3,60 (д, J = 7,4 Гц, 2H), 2,97 (с, 3H).
ПРИМЕР 18
[0339] Следующее соединение получают согласно методиками W, X, Y, Z, затем H и I.
Гидрохлорид (Z)-4-((2,3-диметил-1H-индол-7-ил)сульфонил)-3-фторбут-2-ен-1-амина (Соединение 52)
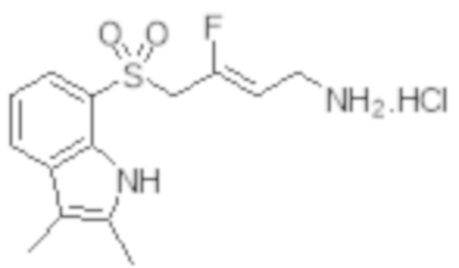
Методика W: Получение (2-йодфенил)гидразина
[0340] В круглодонной колбе на 1 л, раствор 2-йоданилина (40,0 г, 0,182 ммоль) в конц. HCl (250 мл) обрабатывали раствором NaNO2 (15,1 г, 0,22 ммоль) в воде (40 мл) при 0°С. Реакционную смесь перемешивали при 0°C в течение 2 ч и медленно обрабатывали SnCl2 (86,6 г, 0,46 ммоль) при 0°С. Температуру реакции постепенно повышают до комнатной температуры и перемешивали еще 6 часов. По завершении реакции (ТСХ) реакционную смесь фильтровали и промывали н-пентаном (50 мл) и диэтиловым эфиром (50 мл) с получением указанного в заголовке соединения (42 г, 98,26%). 1H ЯМР (300 МГц, d6-DMSO) δ м.д.: 10,2 (шс, 2H), 7,79-7,76 (м, 1H), 7,7-7,45 (ш, 1H), 7,36 (т, J = 8,1 Гц. , 1H), 7,02 (д, J = 7,8 Гц, 1H), 6,75 (т, J = 7,8 Гц, 1H).
Методика X: Получение 7-йод-2,3-диметил-1 Н -индола

[0341] В круглодонной колбе на 250 мл, раствор (2-йодофенил)гидразина (10 г, 42,73 ммоль) в уксусной кислоте (80 мл) постепенно нагревали до 60°C и перемешивали в течение 1 часа. 2-Бутанон (6,18 г, 85,36 ммоль) медленно добавляли при 60°C. Полученную реакционную смесь затем нагревали при 80°C в течение 5 ч. По завершении реакции (ТСХ) реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Полученный остаток разбавляли холодной водой и экстрагировали этилацетатом (100 мл × 2). Объединенный органический экстракт промывали рассолом и концентрировали при пониженном давлении. Полученный остаток очищали на силикагеле, элюируя 10% EtOAc-гексаны с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (2,00 г, 18%). 1H ЯМР (400 МГц, d6-DMSO) δ м.д.: 10,54 (с, 1H), 7,37-7,24 (м, 2H), 6,73 (т, J = 8 Гц, 1H), 2,32 (с, 3H), 2,12 (с, 3H).
Методика Y: Получение метил-3-((2,3-диметил-1 H- индол-7-ил)тио)пропаноата
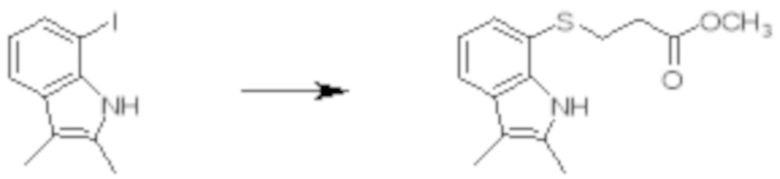
[0342] В повторно закрываемой реакционной пробирке объемом 50 мл дегазировали раствор 7-йод-2,3-диметил-1H-индола (1,60 г, 10,5 ммоль) в 1,4-диоксане (10 мл) в атмосфере азота. Последовательно добавляли Pd2(dba)3 (50,0 мг, 0,06 ммоль), ксантфос (100 мг, 0,18 ммоль), метил 3-меркаптопропаноат (0,70 г, 5,90 ммоль) и DIPEA (2,00 мл, 11,80 ммоль) в атмосфере азота. Раствор дегазировали продувкой газообразным аргоном в течение 15 мин, постепенно нагревали до 110°C и перемешивали при этой температуре в течение 12 часов. После завершения реакции (ТСХ) реакционную смесь охлаждали до комнатной температуры, разбавляли холодной водой и экстрагировали EtOAc (2 × 20 мл). Объединенный органический экстракт промывали рассолом, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный остаток очищали на силикагеле, элюируя смесью 10% EtOAc-гексаны, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (1,50 г, 96%). 1H ЯМР (400 МГц, CDCl3) δ м.д.: 8,6 (ш, 1H), 7,44 (д, J = 8 Гц, 1H), 7,22 (д, J = 7,6 Гц, 1H), 7,03 (т, J = 7,6 Гц, 1H), 3,7 (с, 3H), 3,08 (т, J = 6,8 Гц, 2H), 2,57 (т, J = 7,2 Гц, 2H), 2,41 (с, 3H), 2,22 (с, 3H).
Методика Z: Получение трет- бутил ( Z )-(4-((2,3-диметил-1 H- индол-7-ил)тио)-3-фторбут-2-ен-1-ил)карбамата
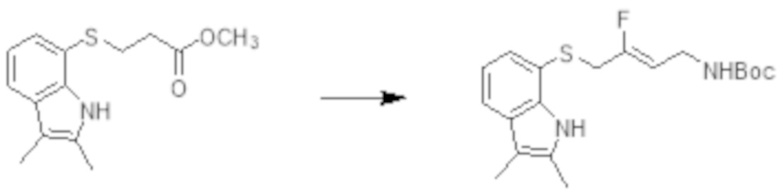
[0343] В круглодонной колбе на 100 мл карбонат цезия (1,49 г, 4,56 ммоль) добавляли к перемешиваемому раствору метил 3-((2,3-диметил-1H-индол-7-ил)тио)пропаноата (0,40 г 1,52 ммоль) и трет-бутил (Z)-(4-бром-3-фторбут-2-ен-1-ил)карбамата (0,41 г, 1,53 ммоль) в DMF (15 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. По завершении реакции (ТСХ) реакционную смесь гасили добавлением ледяной воды (10 мл) и экстрагировали этилацетатом (10 мл x 3). Объединенный органический экстракт сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Неочищенное вещество очищали на силикагеле, элюируя 20% этилацетатом в гексанах с получением указанного в заголовке соединения в виде бледно-желтой жидкости (350 мг, 63%). 1H ЯМР (300 МГц, d6-DMSO) δ м.д.: 10,65 (с, 1H), 7,31 (д, J = 7,5 Гц, 1H), 7,07 (д, J = 7,8 Гц, 1H), 6,93-6,83 (м, 2H), 4,66 (дт, J = 36,3, 7,2 Гц, 1H), 3,67 (д, J = 19,5 Гц, 2H), 3,47 (т, J = 6 Гц, 2H), 2,32 (с, 3H), 2,13 (с, 3H), 1,34 (с, 9H).
Гидрохлорид (Z)-4-((2,3-диметил-1H-индол-7-ил)сульфонил)-3-фторбут-2-ен-1-амина (Соединение 52)
[0344] 1H ЯМР (300 МГц, CD3OD) δ м.д.: 10,25 (с, 1H), 7,80 (дд, J = 7,8, 1,0 Гц, 1H), 7,55 (дд, J = 7,7, 1,1 Гц, 1H), 7,20 (т, J = 7,7 Гц, 1H), 5,05 (дт, J = 32,7, 7,4 Гц, 1H), 4,34 (д, J = 19,2 Гц, 2H), 3,55 (дд, J = 7,4, 1,9 Гц, 2H), 2,49 - 2,39 (м, 3H), 2,26 (д, J = 0,8 Гц, 3H).
ПРИМЕР 19
Получение (Z)-трет-бутил (4-бром-3-фторбут-2-ен-1-ил)карбамата
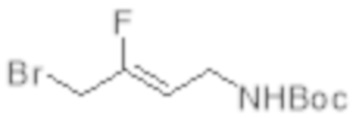
Методика AA: получение трет- бутил-2-оксоэтилкарбамата

[0345] Сосуд, загруженный 3-амино-1,2-пропандиолом (50,0 кг, 549 моль) и этилацетатом (150 л), охлаждали до 0-5 °C. В сосуд добавляли порциями ди-трет-бутилдикарбонат (120 кг, 550 моль). Полученную смесь перемешивали при 20-25 °C в течение 12 часов. После охлаждения реакционной смеси до 0-5 ° C порциями добавляли периодат натрия (120 кг, 561 моль). Суспензию перемешивали при этой температуре 1 час. Затем реакционную смесь фильтровали, и «лепешку» на фильтре промывали этилацетатом (180 л). Объединенный фильтрат промывали водным NaCl (10% мас./мас., 300 л), сушили над Na2SO4 и затем концентрировали в вакууме с получением неочищенного трет-бутил 2-оксоэтилкарбамата (70,0 кг, 80%). Неочищенный материал использовали на следующей стадии без очистки.
Методика AB: Получение ( E )-этил-4-( трет- бутоксикарбониламино)-2-фторбут-2-еноата

[0346] Сосуд, загруженный этил 2-фторфосфоноацетатом (46,0 кг, 190 моль), ацетонитрилом (250 л) и 1,8-диазабицикло[5.4.0]ундец-7-еном (DBU) (38,0 кг, 250 моль), охлаждали до 0 - 10 °С. В сосуд добавляли по каплям трет-бутил 2-оксоэтилкарбамат (68,0 кг, 427 моль) и полученную смесь перемешивали при 0 - 10 °C в течение 4 часов. Реакционную смесь разбавляли трет-бутилметиловым эфиром (500 л) и водой (500 л) и продолжали перемешивать в течение 30 мин. После еще 30 мин отстаивания водный слой удаляли. Органический слой промывали водой (250 л), и затем концентрировали в вакууме. Остаток растворяли в этилацетате (56 л) и петролейном эфире (240 л) и очищали на силикагеле (40 кг, размер меш: 100 - 200), элюируя этилацетатом в петролейном эфире (1:10), с получением неочищенного продукта (E)-этил 4-(трет-бутоксикарбониламино)-2-фторбут-2-еноата (90 кг). Неочищенный материал использовали на следующей стадии без дополнительной очистки.
Методика AC: Получение ( Z )-т рет- бутил 3-фтор-4-гидроксибут-2-енилкарбамата
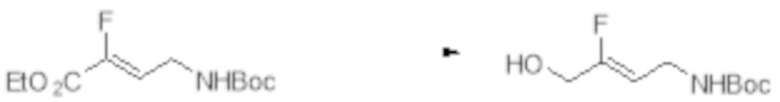
[0347] В реакционный сосуд загружали неочищенный (E)-этил 4-(трет-бутоксикарбониламино)-2-фторбут-2-еноат (90,0 кг, 364 моль), THF (314 л) и метанол (36 л) при 0 - 10 °C, добавляли боргидрид натрия (16,6 кг, 439 моль) порциями в течение 4 часов. После завершения добавления полученную смесь перемешивали при 0 - 10 °C в течение 3 часов. Реакцию гасили добавлением водной HCl (0,5 н, 900 л). Затем продукт экстрагировали этилацетатом (720 л × 2). Объединенные органические слои промывали водой (450 л) и концентрировали в вакууме. Оставшуюся воду удаляли совместным упариванием с THF (200 л × 3) с получением неочищенного (Z)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамата (85 кг). Этот материал использовали на следующей стадии без дополнительной очистки.
Методика AD: Получение ( Z )-4-((трет-бутоксикарбонил)амино)-2-фторбут-2-ен-1-ил 3,5-динитробензоата
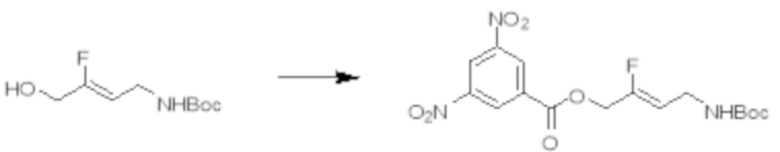
[0348] В реакционный сосуд содержащий неочищенный (Z)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамат (170 кг, 828 моль), THF (765 л) и триэтиламин (174 л, 1245 моль) добавляли порциями 3,5-динитробензоилхлорид (190 кг, 824 моль) при 0 - 20 °C. Полученную смесь перемешивали при 0 - 20 °C в течение 2 ч, и затем разбавляли водой (850 л) и этилацетатом (1700 л). Водный слой удаляли, и органические слои промывали водным Na2CO3 (10% мас., 850 л x 2), и затем водой (850 л). После концентрирования в вакууме (примерно до 190 л) добавляли этилацетат (245 л). Смесь перемешивали при 55 °C с получением прозрачного раствора. Смесь охлаждали до 10 - 20 °C, добавляли н-гептан (730 л) и перемешивали смесь в течение 6 часов. Образовавшееся при этом твердое вещество выделяли фильтрованием и промывали смесью н-гептан/этилацетат (4:1, 170 л). Анализ ВЭЖХ показал содержание E-изомера 30%. Содержание E- изомера снижают до 4,3% последующим осаждением. Твердое вещество растворяли в этилацетате (570 л). К раствору добавляли н-гептан и полученную суспензию перемешивали при 20-30 °C в течение 4-6 часов. Твердое вещество отделяли фильтрованием и «лепешку» на фильтре промывали смесью н-гептан/этилацетат (4:1). Этот процесс повторяют еще раз, с получением (Z)-4-((трет-бутоксикарбонил)амино)-2-фторбут-2-ен-1-ил-3,5-динитробензоата (84 кг в виде влажной «лепешки»). Этот материал использовали на следующей стадии.
Методика AE: Получение ( Z )-т рет- бутил 3-фтор-4-гидроксибут-2-енилкарбамата
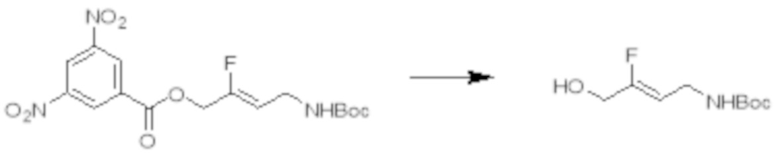
[0349] Реакционный сосуд загруженный (Z)-4-((трет-бутоксикарбонил)амино)-2-фторбут-2-ен-1-ил-3,5-динитробензоатом (84 кг, 210 моль), THF (187 л) и водным NaOH (1,00 М, 420 л) перемешивали при 15-25 °C в течение 2-5 часов. За ходом реакции следили при помощи ТСХ. Реакционную смесь разбавляли изопропилацетатом (966 л) и продолжали перемешивание в течение 10 - 30 мин. После отстаивания в течение 10 - 30 мин и последующего разделения слоев водный слой дополнительно экстрагировали изопропилацетатом (483 л). Объединенные органические слои промывали водным Na2CO3 (10% мас./мас., 420 л x 2), водным NaCl (10% мас./мас., 420 л), и затем концентрировали до около 84 л. Оставшуюся воду удаляли путем совместного упаривания с THF (468 л) с получением (Z)-трет-бутил-3-фтор-4-гидроксибут-2-енилкарбамата (29,0 кг, 67%). Анализ ВЭЖХ показала, что содержание Е-изомера составляло 3,8%.
Методика AF: Получение ( Z )- трет- бутил-4-бром-3-фторбут-2-енилкарбамата

[0350] Реакционный сосуд загруженный (Z)-трет-бутил 3-фтор-4-гидроксибут-2-енилкарбаматом (29,0 кг, 141 моль) в THF (290 л) и диизопропилэтиламином (DIPEA) (74,0 л, 425 моль) охлаждали до 0 - 10 °С. К раствору добавляли по каплям раствор метансульфонового ангидрида (50,0 кг, 287 моль) в THF (145 л). После завершения добавления, добавляли порциями бромид лития (74,0 кг, 852 моль), поддерживая температуру между 0 - 10 °C. Полученную смесь перемешивали при 0 - 10 °C в течение 4 часов. ТСХ по истечении этого времени указывает на полное израсходование исходного материала. Реакционную смесь разбавляли водой (290 л) и продукт экстрагировали этилацетатом (290 л + 145 л). Объединенные органические слои промывали водой (150 л), и затем концентрировали досуха. Неочищенный остаток растворяли в этилацетате (67 л) и н-гептане (440 л), и проводят очистку на силикагеле (40 кг; размер меш 100 - 200), элюируя смесью этилацетат/н-гептан (1:5). Все фракции, содержащие желаемый продукт, концентрировали досуха. Добавляли этилацетат (56 л) и продолжали перемешивание при 40-50 °C до получения прозрачного раствора. К раствору добавляли по каплям н-гептан (280 л). Смесь охлаждали до 10-15 °C и перемешивали 8 ч. Полученное твердое вещество собирали фильтрованием. Анализ ВЭЖХ показал, что содержание Е- изомера составляет 0,9%. Для дополнительного снижения содержания E- изомера процесс осаждения повторяют следующим образом. Добавляли этилацетат (33 л) и продолжали перемешивание при 40-50 °C до получения прозрачного раствора. К раствору добавляли по каплям н-гептан (164 л). Смесь охлаждали до 10-15 °C и перемешивали 8 ч. Полученное твердое вещество собирали фильтрованием и сушили с получением (Z)-трет-бутил 4-бром-3-фторбут-2-енилкарбамата (29,5 кг, 68%). Анализ ВЭЖХ показал, что содержание Е-изомера составляет 0,1%. 1H-ЯМР (300 МГц; CDCl3) δ м.д.: 1,46 (9H, с), 3,85 (2H, дд, J 6,2, 6,2 Гц), 3,93 (2H, д, J 19,5 Гц), 4,66 (1H, шс), 5,16 (1H, дт, J = 34,0, 6,5 Гц).
ПРИМЕР 20
[0351] Следующее соединение получают в соответствии с методиками AG и AH.
Получение гидрохлорида (Z)-3-фтор-4-(фенилсульфонил)бут-2-ен-1-амина (Соединение 1)

Методика AG: Получение трет- бутил ( Z )-(3-фтор-4-(фенилсульфонил)бут-2-ен-1-ил)карбамата
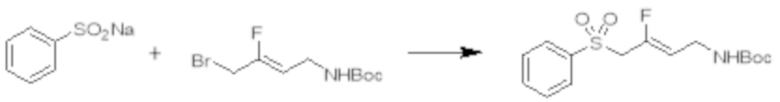
[0352] Сосуд загруженный трет-бутил (Z)-(4-бром-3-фторбут-2-ен-1-ил)карбаматом (2,40 кг, 8,95 моль) и DMF (12,0 л), охлаждали до 15 - 20 °С. К раствору добавляли бензолсульфинат натрия (2,20 кг, 13,4 моль), и полученную смесь перемешивали при 15 - 20 °C в течение 4 часов. Реакционную смесь разбавляли водой (12,0 л) и продолжали перемешивание при комнатной температуре в течение еще 1 часа. Образовавшееся при этом твердое вещество отделяли фильтрованием, и «лепешку» на фильтре дополнительно промывали водой (6,0 л). Затем твердое вещество сушили в вакууме при 50 - 55 °C в течение 20 часов, с получением трет-бутил(Z)-(3-фтор-4-(фенилсульфонил)бут-2-ен-1-ил)карбамата (2,70 кг, 92%). Время удерживания (ВУ) = 13,75 мин; Способ - Agilent LC/MSD 1200 Series; колонка: ZORBAX SB-C18, ODS 2000 (50 × 4,6 мм, 5 мкм), работающая в режиме ионизации ES (+) или (-); скорость потока 1,5 мл/мин; Температура (T) = 30 °C; длина волны обнаружения: 214 нм; подвижная фаза: от 5% ацетонитрила (содержащего 0,05% трифторуксусной кислоты (TFA)) и 95% воды (содержащей 0,05% TFA) до 90% ацетонитрила и 10% воды в течение 24 мин.
Методика AH: получение гидрохлорида ( Z )-3-фтор-4-(фенилсульфонил)бут-2-ен-1-амина (Соединение 1)

[0353] В сосуд, загруженный HCl (4,00 M в этилацетате; 13,5 л, 54,0 моль) и охлажденный до 10-20 °C, добавляли отфильтрованный раствор (гифло) трет-бутил (Z)-(3-фтор-4-(фенилсульфонил)бут-2-ен-1-ил)карбамата (2,70 кг, 8,20 моль) в этилацетате (27,0 л). Реакционную смесь перемешивали при 10 – 20 °C в течение 6 часов. Образовавшееся твердое вещество отделяли фильтрованием, и «лепешку» на фильтре промывали этилацетатом (8,0 л). Затем твердое вещество сушили в вакууме при 50-55 °C в течение 40 часов с получением гидрохлорида (Z)-3-фтор-4-(фенилсульфонил)бут-2-ен-1-амина (Соединение 1) (2,10 кг; 96 %). 1H-ЯМР (300 МГц; CD3OD) δ м.д.: 3,64 (2H, дд, J = 7,3, 1,2 Гц), 4,36 (2H, д, J = 19,1 Гц), 5,18 (1H, дт, J = 32,7, 7,4 Гц), 7,65-7,71 (2H, м), 7,79 (1H, тт, J = 7,4, 1,2 Гц), 7,96-8,00 (2H, м); ЖХМС: для C10H12FNO2S вычислено 229,1, найдено 230,1 [M+1]+.
ПРИМЕР 21
[0354] Следующее соединение получают согласно методикам AI, AJ и AK.
Получение моногидрата дигидрохлорида (Z)-3-фтор-4-(хинолин-8-илсульфонил)бут-2-ен-1-амина (Соединение 33)
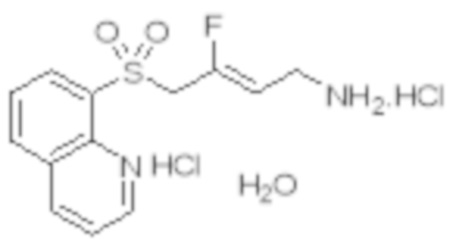
Методика AI: Получение хинолин-8-сульфината натрия

[0355] Сосуд, загруженный Na2SO3 (6,70 кг, 53,2 моль) и водой (21,0 л), перемешивали при комнатной температуре в течение 20 минут. В сосуд добавляли Na2CO3 (5,50 кг, 51,9 моль) и продолжали перемешивание при комнатной температуре в течение 20 минут. Затем порциями добавляли хинолин-8-сульфонилхлорид (6,00 кг, 26,4 моль), поддерживая температуру ниже 40 °C. Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь фильтровали, и «лепешку» на фильтре промывали метанолом (7,0 л). Фильтрат концентрировали досуха в вакууме и к полученному остатку добавляли метанол (7,0 л). После перемешивания при комнатной температуре в течение 1 часа смесь фильтровали и фильтрат концентрировали досуха. Во втором и последнем цикле промывания, остаток растворяли в метаноле (10,0 л) и продолжали перемешиванием при комнатной температуре в течение 1 часа. Смесь фильтровали и фильтрат концентрировали в вакууме, с получением хинолин-8-сульфината натрия (4,10 кг, 72%).
Методика AJ: Получение трет- бутил ( Z )-(3-фтор-4-(хинолин-8-илсульфонил) бут-2-ен-1-ил)карбамата
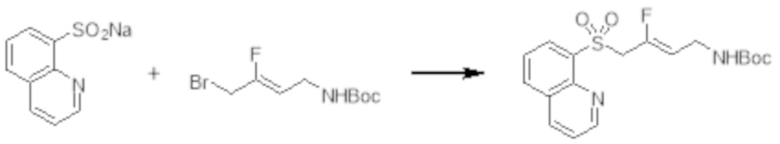
[0356] Сосуд загруженный трет-бутил (Z)-(4-бром-3-фторбут-2-ен-1-ил)карбаматом (3,50 кг, 13,1 моль), хинолин-8-сульфинатом натрия (4,20 кг, 19,5 моль) и DMF (17,5 л) охлаждали до 15 - 20 °C. Полученную смесь перемешивали при этой температуре в течение 20 часов. Затем смесь разбавляли этилацетатом (35,0 л) и водой (35,0 л), и продолжали перемешивание еще 10 мин. Затем органический слой отделяли и промывали водой (20,0 л × 2). После концентрирования органического слоя до около 20 л по каплям добавляли н-гептан (42,0 л). Полученную суспензию перемешивали при 20 - 30 °C в течение 20 часов. Твердое вещество выделяли фильтрованием, промывали н-гептаном, и затем сушили в вакууме при 50 - 55 °C в течение 20 часов с получением трет-бутил (Z)-(3-фтор-4-(хинолин-8-илсульфонил)бут-2-ен-1-ил)карбамата (3,80 кг, 77%). ВУ = 12,97 мин; Способ - Agilent LC/MSD 1200 Series; колонка: ZORBAX SB-C18, ODS 2000 (50 × 4,6 мм, 5 мкм), работающая в режиме ионизации ES (+) или (-); скорость потока 1,5 мл/мин; Температура (T) = 30 °C; длина волны обнаружения: 214 нм; подвижная фаза: от 5% ацетонитрила (содержащего 0,05% трифторуксусной кислоты (TFA)) и 95% воды (содержащей 0,05% TFA) до 90% ацетонитрила и 10% воды в течение 24 мин.
Методика AK: Получение моногидрата дигидрохлорида ( Z )-3-фтор-4-(хинолин-8-илсульфонил)бут-2-ен-1-амина (Соединение 33)

[0357] В сосуд, загруженный HCl (1,5 М в этилацетате; 53 л) при 10 - 20 °C, добавляли трет-бутил (Z)-(3-фтор-4-(хинолин-8-илсульфонил)бут-2-ен-1-ил)карбамата (5,3 кг, 14 моль). Смесь перемешивали при 15 °C в течение 4 часов. Полученное твердое вещество выделяли фильтрованием и промывали этилацетатом (20 л). В колбу, содержащую твердое вещество, добавляли этилацетат (53 л). Затем суспензию перемешивали при 10 – 20 °C в течение 2 часов. Твердое вещество выделяли фильтрованием и промывали этилацетатом (20 л). Твердое вещество растворяли в метаноле (53 л) и раствор фильтровали. Затем к веществу по каплям добавляли воду (500 мл) и трет-бутилметиловый эфир (53 л) и продолжали перемешивание при 15 °C в течение еще 20 часов. Твердое вещество собирали фильтрованием и сушили в вакууме при 55 - 60 °C с получением моногидрата дигидрохлорида (Z)-3-фтор-4-(хинолин-8-илсульфонил)бут-2-ен-1-амина (Соединение 33) (4,4 кг, 90%). 1H ЯМР (300 МГц, CD3OD) δ м.д.: 9,18 (д, J = 4,7 Гц, 1H), 8,70 (дд, J = 8,4, 2,6 Гц, 1H), 8,57 (д, J = 7,4 Гц, 1H ), 8,45 (д, J = 8,5 Гц, 1H), 7,99 - 7,68 (м, 2H), 5,22 (дт, J = 32,9, 7,4 Гц, 1H), 5,00 (д, J = 19,4 Гц, 2H), 3,60 (д, J = 7,7 Гц, 2H); ЖХМС: для C13H13FN2O2S вычислено 280,1, найдено 281,1 [M+1]+.
ПРИМЕР 22
[0358] Следующее соединение получают в соответствии с методиками AL и AM.
Получение дигидрохлорида (Z)-3-фтор-4-((хинолин-8-ил-d6)сульфонил)бут-2-ен-1-амина (Соединение 53)
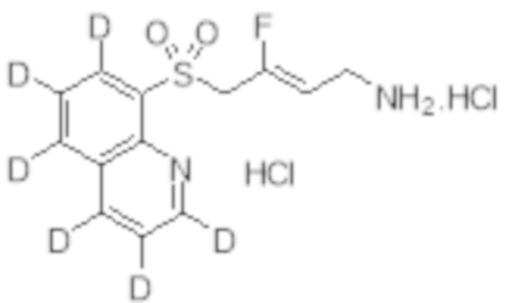
Методика AL: Получение трет- бутил ( Z )-(3-фтор-4-((хинолин-8-ил- d 6)сульфонил)бут-2-ен-1-ил)карбамата
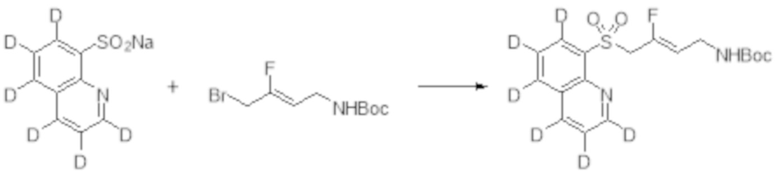
[0359] К перемешиваемому раствору трет-бутил (Z)-(4-бром-3-фторбут-2-ен-1-ил)карбамата (606 мг, 2,26 ммоль) в DMF (4,0 мл) добавляли 2,3,4,5,6,7-гексадейтериохинолин-8-сульфинат натрия (500 мг, 2,26 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь разбавляли водой (40 мл) и продукт экстрагировали этилацетатом (20 мл x 3). Объединенный органический слой промывали насыщ. водн. NH4Cl (20 мл × 3) и рассолом (20 мл), сушили над Na2SO4 и затем концентрировали в вакууме. Неочищенный продукт очищали нормально-фазовой хроматографией (Reveleris), элюируя 20 - 50% этилацетатом в гексане, с получением трет-бутил (Z)-(3-фтор-4-((хинолин-8-ил-d6)сульфонил)бут-2-ен-1-ил)карбамата (470 мг, 53%) в виде не совсем белого твердого вещества.
Методика AM: Получение дигидрохлорида ( Z )-3-фтор-4-((хинолин-8-ил- d 6)сульфонил)бут-2-ен-1-амина (Соединение 53)
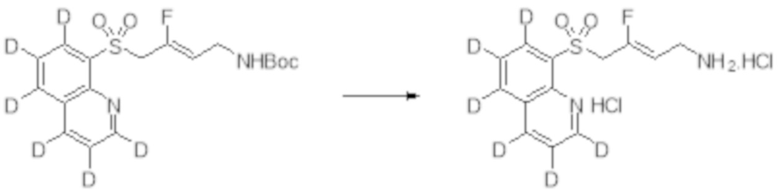
[0360] К перемешиваемому раствору трет-бутил (Z)-(3-фтор-4-((хинолин-8-ил-d6)сульфонил)бут-2-ен-1-ил)карбамата (450 мг, 1,22 ммоль) в метаноле (1,0 мл) при комнатной температуре добавляли HCl (2,0 М в диэтиловом эфире; 4,0 мл, 8,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, в течение которого выпадал осадок. Реакционную смесь переносили в сосуд и центрифугируют (4000 об/мин, 4 мин). Супернатант осторожно декантировали и оставшуюся твердую «лепешку» сушили в высоком вакууме, с получением дигидрохлорида (Z)-3-фтор-4-((хинолин-8-ил-d6)сульфонил)бут-2-ен-1-амина (355 мг, 81%) в виде белого твердого вещества. 1H ЯМР (300 МГц, CD3OD) δ м.д.: 5,20 (дт, J = 32,8, 7,4 Гц, 1H), 5,08 - 4,96 (м, 2H), 3,59 (д, J = 7,4 Гц, 2H).
ПРИМЕР 23
Способ определения способности соединений изобретения ингибировать LOX и LOXL1-4 из разных источников
[0361] Лизилоксидаза (LOX) представляет собой внеклеточный медьзависимый фермент, который окисляет остатки пептидиллизина и гидроксилизина в коллагене и остатки лизина в эластине с образованием пептидил-альфа-аминоадипин-дельта-полуальдегидов. Эта каталитическая реакция может быть необратимо ингибирована β-аминопропионитрилом (BAPN), который связывается с активным центром LOX (Tang S.S., Trackman P.C. and Kagan H.M., Reaction of aortic lysyl oxidase with beta-aminoproprionitrile. J Biol Chem 1983; 258: 4331-4338). Существует пять членов семьи LOX; это LOX, LOXL1, LOXL2, LOXL3 и LOXL4. Члены семейства LOX и LOXL могут быть приобретены в виде рекомбинантных активных белков из коммерческих источников или извлечены из тканей животных, таких как аорта, сухожилия, кожа свиньи; или получены из клеточных культур. Ингибирующие эффекты соединений настоящего изобретения были протестированы против данного препарата LOX-LOXL с использованием способа, основанного на обнаружении перекиси водорода при помощи анализа окисления Amplex Red (Zhou et al. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 1997; 253, 162-168). Анализ был разработан с использованием формата на 384 или 96 лунок. Вкратце, в стандартном анализе в 384-луночный планшет с черным прозрачным дном с 25 мкл разведения любого из изоферментов и ортологов в 1,2 М мочевине, 50 мМ натрий-боратном буфере (pH 8,2) добавляли в каждую лунку в присутствии 1 мкМ мофегилин и 0,5 мМ паргилин (для ингибирования SSAO и MAO-B и MAO-A, соответственно; не требуется, если фермент из рекомбинантной или очищенной формы). Тестируемые соединения растворяли в DMSO и тестировали на кривой зависимости от концентрации (КЗК) с 11 точками данных, обычно в микромолярном или наномолярном диапазоне после инкубации с ферментом в течение 30 минут при 37 °C. Двадцать пять мкл реакционной смеси, содержащей двойную концентрацию КМ путресцина (Sigma Aldrich, например, 20 мМ для LOX, или 10 мМ для LOXL2 и LOXL3), 120 мкМ Amplex Red (Sigma Aldrich) и 1,5 ед/мл пероксидазы хрена (Sigma Aldrich), приготовленной в 1,2 М мочевины, 50 мМ натрий-боратном буфере (pH 8,2) затем добавляли в соответствующие лунки. Вышеуказанные объемы удваивали в случае 96-луночного планшета. Флуоресценцию (RFU) считывают каждые 2,5 мин в течение 30 мин в диапазоне температур от 37 °C, возбуждения 565 нм и эмиссии 590 (Optima; BMG labtech). Наклон кинетики для каждой лунки рассчитывают с использованием программного обеспечения анализа данных MARS (BMG labtech), и это значение использовали для определения значения IC50 (Dotmatics). Способность соединений изобретения ингибировать активность аминоксидазы LOX и других членов семейства показана в Таблице 3.
Таблица 3
[0362] Ингибирующие активности LOX и LOXL2 примерами соединений изобретения
ПРИМЕР 24
Соединения настоящего изобретения демонстрируют устойчивое ингибирование LOXL1 и LOXL2
[0363] Для значимого фармакологического эффекта в присутствии высокой концентрации субстрата, соединения, которые проявляют устойчивое, продолжительное ингибирование LOX и LOXL1-4, имеют сильное преимущество перед конкурентными ингибиторами, поскольку фармакологический эффект длится дольше, чем присутствие несвязанного ингибитора. В предпочтительном варианте реализации соединения настоящего изобретения демонстрируют устойчивое ингибирование LOX и LOXL1-4.
Способ определения устойчивого ингибирования LOX и LOXL1-4 соединениями изобретения
[0364] Эксперимент с скачкообразным разбавлением: Анализ был разработан с использованием 96-луночного формата, и исходная концентрация фермента была установлена в 100 раз выше, чем для исследований ингибирования. Фермент инкубируют в течение 40 минут при 37 °C в присутствии 10-кратной или (при необходимости, чтобы убедиться, что концентрация ингибитора превышает концентрацию фермента) 30-кратной IC50 концентрации тестируемого ингибитора. После инкубации смесь разводят в 50 раз в буфере для анализа с последующим дополнительным 2-кратным разведением в реакционной смеси Amplex Red-пероксидаза хрена-путресцин (как в исследованиях ингибирования) перед измерением флуоресценции. Результаты выражали в % восстановления сигнала по истечении заданного времени по сравнению с неингибированными контролями. Результаты представлены в Таблице 4.
Таблица 4
[0365] Измерение длительного ингибирования LOXL1 и LOXL2 соединениями изобретения
ПРИМЕР 25
Фармакокинетика in vivo
[0366] Тестируемые соединения (10 мг/кг и 30 мг/кг в PBS, перорально n = 3) вводили самцам крыс линии Wistar в возрасте 7-9 недель. Образцы крови тестируемого соединения собирали в моменты времени от 0,25 ч до 8,00 ч (Соединение 1); 0,5 ч - 8,00 ч после введения (Соединение 33) из хвостовой вены.
[0367] Получение образца: 20 мкл калибровочных образцов (в одном экземпляре), образцы для контроля качества (в двух экземплярах) и образцы плазмы крыс смешивали с 60 мкл ацетонитрила, содержащего внутренний стандарт (IS; 200 нг/мл толбутамида и 50 нг/мл пропанолола) в пробирках Эппендорфа. Смесь встряхивали в течение 1 мин, затем центрифугировали в течение 10 мин, 50 мкл супернатанта переносили в 96-луночный планшет со 100 мкл воды. После встряхивания в течение 10 минут 10 мкл вводили в систему жидкостной хроматографии и масс-спектрометрии (ЖХ-МС/МС) для анализа.
Метод ЖХ (Соединение 1)
ВЭЖХ: Agilent 1100; Масс-спектрометр: API 4000
Колонка: Phenomenex Gemini C18 5 мкм 50 x 4,6 мм
Подвижная фаза: 0,1 % муравьиная кислота в воде, 0,1 % муравьиная кислота в ацетонитриле
Метод ЖХ (Соединение 33)
ВЭЖХ: Shimadzu LC30AD; Масс-спектрометр: API 4000
Колонка: Agilent SB C18 1,8 мкм 50 x 2,1 мм
Подвижная фаза: 0,1 % муравьиная кислота в воде, 0,1 % муравьиная кислота в ацетонитриле
[0368] Для образцов разведения: 10 мкл образца добавляли к 90 мкл холостой плазмы крысы. Смесь осаждали 300 мкл ацетонитрила, содержащего IS, в пробирках Эппендорфа. Смесь встряхивали в течение 1 мин, затем центрифугировали в течение 10 мин, 50 мкл супернатанта переносили в 96-луночный планшет со 100 мкл воды. После встряхивания в течение 10 минут, 10 мкл вводили в систему ЖХ-МС/МС для анализа. Определенные средние концентрации в плазме Соединений 1 и 33 показаны в Таблице 5 и Таблице 6 соответственно.
Таблица 5
Средняя концентрация Соединения 1 в плазме после перорального введения 10 и 30 мг/кг
(часы)
Таблица 6
Средняя концентрация Соединения 33 в плазме после перорального введения 10 и 30 мг/кг
(часы)
ПРИМЕР 26
Целевое взаимодействие
[0369] Одной дозы ингибитора на основе механизма может быть достаточно для блокирования ферментативной активности in vivo на длительный период.
Измерение активности лизилоксидазы - анализ среза уха
[0370] Уши, собранные сразу после умерщвления крыс (обработанных или необработанных ингибитором LOX), быстро замораживают в сухом льду для хранения при -80 °C. Для анализа вырезают образцы размером 5x5 мм (все еще наполовину размороженные), заливают в агарозный гель и разрезают поперечные срезы толщиной 200 мкм (вибратом). Тонкие срезы собирали в охлажденном льдом PBS, содержащем ингибиторы протеаз, и оставляли в бане на 2-3 часа, чтобы растворимые загрязнители могли диффундировать до анализа. Для формата 96-луночного планшета: собирали три тонких среза, осторожно промокали насухо на Kimwipe и переносили в каждую лунку, содержащую 100 мкл буфера для анализа (1,2 М мочевина, 50 мМ буфер бората натрия, pH 8,2), на 30-минутную предварительную инкубацию при 37 °C в присутствии 1 мкМ мофегилина и 500 мкМ паргилина. BAPN (500 мкМ) добавляли во все лунки с «низким контролем». Для каждого условия было выполнено минимум три повторения.
[0371] После предварительной инкубации во все лунки добавляли 100 мкл смеси Amplex Red и пероксидазы хрена (HRP) (120 мкМ Amplex Red, 1,5 ед/мл HRP, 20 мМ путресцин), и данные с планшета считывали при 37 °C каждые 2,5 минуты, 13 раз (возбуждение 544 нм и эмиссия 590 нм, на BMG Clariostar в орбитальном режиме с верхним считыванием). Наклон кинетических кривых, вычтенный из значений, полученных в низком контроле (в присутствии BAPN), считали мерой специфической активности лизилоксидазы в образце.
ПРИМЕР 27
Измерение активности лизилоксидазы в аорте
[0372] Подготовка образцов: все действия по приготовлению проводили на льду с буферами в присутствии ингибиторов протеаз (PMSF: Sigma P7626, 0,25 мМ, апротинин: Sigma A6279, 1 мкл на мл) (Methods in Cell Biology, 2018; 143: 147 - 156). Окружающие адвентицию и мышцы/жир из замороженного образца аорты удаляли в ледяном буфере для промывания (0,15 М NaCl, 50 мМ борат натрия, pH = 8,0, с ингибиторами протеазы) с помощью тонких щипцов под препарирующим микроскопом. Аорту промокали на Kimwipe, взвешивали в пробирке Эппендорфа на 1,5 мл и оставляли на льду. После мгновенного замораживания аорты в жидком азоте использовали ступку и пестик (хранились при -80 °C) для измельчения ткани. Измельченную ткань переносили в специальную пробирку, содержащую металлические шарики, 10 x об./мас. промывочный буфер + ингибиторы протеаз. Ткани гомогенизировали в течение 5 секунд при помощи бисерной мельницы. Гомогенат центрифугировали при 10000 g в течение 10 минут при 4 °C, и супернатант выбрасывали. Стадии гомогенизации и промывания повторяли еще дважды. Полученный осадок ресуспендировали в 3 x об./мас. 6 М буфера мочевины (50 мМ бората натрия, 6 М мочевины, pH = 8,2) + ингибиторы протеазы, и затем встряхивают. Затем образец оставляли при 4 °C на валике на 3 часа экстракции. После экстракции образец центрифугировали (10000 g при 4 °C в течение 20 минут), и супернатант сохраняли. Равный объем всех образцов переносили в свежую пробирку для разведения (минимум 33 мкл). Образцы разбавляли до 2,4 М мочевины (образец на этой стадии составляет ~ 4,5 М мочевины) натрий-боратным буфером (50 мМ борат натрия, pH 8,2) + ингибиторы протеазы и использовали в последующем анализе.
[0373] Анализ: к образцам добавляли паргилин (конечная концентрация 0,5 мМ) и мофегилин (конечная концентрация 1 мкМ). Дублирующие лунки помещали в черный планшет 384-луночный, 25 мкл на лунку. К одному дубликату добавляли 0,5 мкл 30 мМ BAPN (ингибитор pan-lox); обеспечение фонового значения (низкий контроль). Затем образцы инкубируют в течение 30 минут при 37 °C. Реакционную смесь (25 мкл) добавляли в каждую лунку (концентрации были показаны как удвоенные конечные в анализе: 120 мкМ Amplex red, 1,5 ед/мл HRP, 20 мМ путресцин). Затем флуоресценцию планшета измеряли каждые 2,5 мин (Ex: 544 нм/ Em: 590 нм/ усиление: 1260; 37 °C) на Fluostar™.
[0374] Молодым самцам крыс Wistar давали однократную пероральную дозу (10 или 30 мг/кг) Соединения 1 или 33 (Таблицы 5 и 6 соответственно), и ферментативную активность измеряли в тканях с высокой базальной активностью в этом возрасте, в ухе (Фиг. 2a и b; Соединение 1 и 33 соответственно) и аорте (Фиг. 3c; только Соединение 33). Ответы были нормализованы к активности, измеренной у животных, получавших физиологический раствор (контроль).
[0375] Описанное в данном документе Соединение 1 оказывает длительное ингибирование LOX. В то время как концентрации Соединения 1 в плазме крови значительно ниже IC 50 через 8 часов (Таблица 5) в плазме крыс, устойчивое снижение активности LOX в зависимости от дозы можно измерить через 24 часа после однократной пероральной дозы (Фиг. 2a); > 20 часов после того, как концентрация Соединения 1 в плазме упала ниже IC50.
[0376] Было обнаружено, что однократная высокая (30 мг/кг) доза Соединения 33 полностью устраняет активность лизилоксидазы. Хотя концентрации Соединения 33 в плазме намного ниже IC50 через 8 часов (Таблица 6), период полувыведения составляет от 2-3 дней (ухо) до 24 часов (аорта) (Фиг. 2b и 2c). Таким образом, Соединение 33 вызывает длительное ингибирование, которое сохраняется дольше присутствия активного соединения в плазме.
ПРИМЕР 28
Метод определения способности соединений Формулы I ингибировать рекомбинантный человеческий SSAO/VAP-1
[0377] Активность рекомбинантной аминоксидазы SSAO/VAP-1 человека определяют с использованием комбинированного колориметрического способа, как описано для моноаминоксидазы, медьсодержащих аминоксидаз и родственных ферментов (Holt A. and Palcic M., A peroxidase-coupled continuous absorbance plate-reader assay for flavin monoamine oxidases, copper-containing amine oxidases and related enzymes. Nat Protoc 2006; 1: 2498-2505). Вкратце, собирали клонированную матрицу кДНК, соответствующую остаткам 34-763 человеческого SSAO/VAP-1 и включающую сигнальную последовательность мышиного Ig каппа (κ), флажок N-терминальной метки эпитопа и сайт расщепления вируса гравировки табака (ВГТ) в векторе экспрессии млекопитающих (pLO-CMV) от Geneart AG. Этот вектор, содержащий остатки SSAO/VAP-1 человека, трансфицируют в линию клеток мутантного гликозилирования CHO-K1, Lec 8. Клон, стабильно экспрессирующий человеческий SSAO/VAP-1, выделяли и культивировали в больших масштабах. Активный человеческий SSAO/VAP-1 очищали и выделяли при помощи иммуноаффинной хроматографии. Это использовали в качестве источника для активности SSAO/VAP-1. Был разработан высокопроизводительный колориметрический анализ с использованием формата 96 или 384 лунок. Вкратце, в стандартном 96-луночном планшете для анализа в каждую лунку добавляли 50 мкл очищенного человеческого SSAO/VAP-1 (0,25 мкг / мл) в 0,1 М натрий-фосфатном буфере (pH 7,4). Тестируемые соединения растворяли в DMSO и тестировали на кривой зависимости от концентрации (КЗК) с 4-11 точками данных, обычно в микромолярном или наномолярном диапазоне после инкубации с человеческим SSAO/VAP-1 в течение 30 минут при 37 °C. После 30 мин инкубации 50 мкл реакционной смеси, содержащей 600 мкМ бензиламина (Sigma Aldrich), 120 мкМ Amplex Red (Sigma Aldrich) и 1,5 ед/мл пероксидазы хрена (Sigma Aldrich), полученной в 0,1 М натрий-фосфатном буфере (pH 7,4) добавляли в соответствующую лунку. Единицу флуоресценции (RFU) считывали каждые 2,5 мин в течение 30 мин при 37 °C, возбуждение 565 нм и эмиссия 590 (Optima; BMG labtech). Наклон кинетики для каждой лунки рассчитывали с использованием программного обеспечения анализа данных MARS (BMG labtech), и это значение использовали для определения значения IC50 (Dotmatics). Способность соединений Формулы I ингибировать SSAO/VAP-1 показана в Таблице 7.
ПРИМЕР 29
Способ определения способности соединений Формулы I ингибировать рекомбинантный MAO - B человека
[0378] Специфичность соединений настоящего изобретению проверяют путем определения их способности ингибировать активность MAO-B in vitro. Рекомбинантный человеческий МАО-В (0,06 мг/мл; Sigma Aldrich) использовали в качестве источника активности фермента МАО-В. Анализ проводят таким же образом, как и для SSAO/VAP-1 человека (Пример 28), за исключением того, что использовали субстрат бензиламин в концентрации 100 мкМ. Способность соединений Формулы I ингибировать MAO-B показана в Таблице 7.
Таблица 7
[0379] Селективность Соединений Формулы I в отношении LOX и LOXL2 по сравнению с SSAO/VAP-1 и MAO-B
[0380] Ферменты LOX и LOXL1-4 являются членами большого семейства флавин-зависимых и медьзависимых аминоксидаз, в которое входят SSAO/VAP-1 и моноаминоксидаза-B (MAO-B). Соединения настоящего изобретения селективно ингибируют членов семейства ферментов LOX по отношению к SSAO/VAP-1, MAO-B и другим членам семейства аминоксидаз. Примеры величины селективности можно увидеть в Таблице 7.
ПРИМЕР 30
Модель травмы грызунов
[0381] Мыши получали травму путем сечения кожной ткани. В группе лечения, 1 % раствор Соединения 1 применяли местно с 24 часов после травмы до 1 недели после травмы. Затем раны оставляли для заживления еще на неделю. Мышей умерщвляют через 14 дней после травмы, и ткань анализировали на содержание коллагена I, и изменения общей морфологии и гистологии.
[0382] Количество коллагена I (измеренное при помощи ЖХМС и нормализованное по содержанию белка) было снижено в обработанной ткани по сравнению с контролем (Фиг. 3а). Гистология показала толстые параллельные пучки коллагена в контрольной рубцовой ткани. Ткань, обработанная Соединением 1, показала уменьшенную плотность пучков и более «нормальную» структуру коллагена (Фиг. 3b и 3c).
ПРИМЕР 31
Модель ожога свиньи
[0383] Свиньи получали 4 глубоких ожога кожи по 25 см2, по 2 на каждом боку. С момента повторной эпителизации, на каждой свинье 2 раны обрабатывали 3% кремом Соединения 1 ежедневно в течение 4 недель и 2 раны получали контрольный крем. Свиней умерщвляли и ткани обрабатывали для анализа.
[0384] Активность LOX была значительно снижена в обработанной ткани (Фиг. 4a), как и общий белок коллагена I (Фиг. 4b). Общая морфология (Фиг. 4c-f) и гистология (Фиг. 4g-h) подтверждают снижение плотности толстых коллагеновых волокон в обработанных ранах.
ПРИМЕР 32
Мышиная модель рака поджелудочной железы
[0385] Самки бестимусных мышей (возраст 4-5 недель) получали инокуляцию MiaPaca-2luc (линия клеток поджелудочной железы человека) непосредственно в поджелудочную железу. Измеренные интенсивность биолюминесцентного сигнала и массу тела использовали для стратификации опухолей по группам лечения на 14 день после первоначальной инокуляции. Группы лечения включали 1. растворитель, в/б. 3 раза в неделю. 2. Абраксан (10 мг/кг, в/б. 2 раза в неделю) и гемцитабин (начальная доза 100 мг/кг в/б., для второй дозы она была снижена до 60 мг/кг в/б. из-за острой токсичности и принималась дважды в неделю). 3. Абраксан (дозированный, как описано выше) и гемцитабин (дозированный, как описано выше) и Соединение 33 (10 мг/кг в/б. 3 раза в неделю) (см. Фиг. 5а). Мышей умерщвляли на 43 день после инокуляции опухоли. Дважды в неделю у мышей наблюдали состояние и изменение веса. Рост опухоли контролируют in vivo при помощи биолюминесцентной визуализации на протяжении исследований (Фиг. 5b), и органы - ex vivo во время сбора ткани (Фиг. 5c-e).
ПРИМЕР 33
Мышиная модель склероза
[0386] Подкожный блеомицин вводили через день (в общей сложности 20 дней) самке C57BL/6 для индуцирования фиброза кожи в качестве модели склероза. Поражения обрабатывали либо растворителем, либо 0,5%, 1% или 3% Соединением 1. Гистология завершали через 21 день. Гистологический анализ показан на Фиг. 6 (а-с).
ПРИМЕР 34
Мышиная модель первичного миелофиброза
[0387] Самцам и самкам мышей GATA1low в возрасте от пятнадцати до шестнадцати недель внутрибрюшинно вводили либо растворитель (оливковое масло), либо Соединение 19 в дозе 15 мг/кг четыре раза в неделю в течение 10 недель. Затем мышей умерщвляли и собирали селезенки и бедренные кости для гистологии и анализа (Фиг. 7(a-e) и Фиг. 8 (a-d)).
[0388] Мыши, получавшие Соединение 19, имели значительно более низкий вес селезенки, скорректированный с учетом массы тела, по сравнению с группой растворителя (242,25 ± 18 мг против 305,11 ± 22,4 мг, p < 0,05). Не было различий в гематологических параметрах до лечения, однако у мышей в группе лечения было значительно меньшее количество тромбоцитов по сравнению с мышами, получавшими растворитель, после лечения (77,5 ± 4,4 К/мкл против 106 ± 12, p < 0,05). Костный мозг (КМ) и фиброз селезенки значительно снизились у мышей в группе лечения по сравнению с растворителем. Были подсчитаны морфологические мегакариоциты (МК) BM на окрашивании H&E, и они были уменьшены у мышей в группе лечения по сравнению с растворителем (24,65 ± 0,6 на 20-кратном увеличении против 32,91 ± 0,71, p < 0,001).
ПРИМЕР 35
Модель односторонней обструкции мочеточника (ООМ)
[0389] 14-дневная односторонняя обструкция мочеточника (ООМ) острого фиброза почек у мышей была проведена для имитации различных стадий обструктивной нефропатии, приводящей к тубулоинтерстициальному фиброзу в ускоренном режиме. Операцию ООМ выполняли путем перевязки левого мочеточника и индуцированного уменьшения толщины левой почки и атрофии (на что указывало значительное уменьшение веса почки и соотношения веса почки/тела по сравнению с противоположной стороной), а также значительного увеличения фиброза почки. Группы лечения получали Соединение 33 (10 мг/кг в день) перорально на протяжении всего исследования. Соединение 33 увеличивало вес и толщину почек и уменьшало площадь фиброза, как измерено с помощью Picrosirius Red (Фиг. 9). Каптоприл (~32 мг/кг/день в питьевой воде) использовали в качестве положительного контроля.
ПРИМЕР 36
Модель фиброза легких, индуцированного блеомицином
[0390] Мышам C57Bl/6 вводят 1,5 ед./кг общего клинического блеомицина (Бленоксан) через ротоглотку. Соединение 33 вводили ежедневно через желудочный зонд в течение 21 дня, после чего ткани собирали и анализировали. Как показано на Фиг. 10, Соединение 33 значительно уменьшает оценку Эшкрофта и вес легких. Как и ожидалось для ингибитора лизилоксидазы, количество незрелых (DHLNL) и зрелых (PYD) поперечных сшивок на легкое также уменьшилось на 37 и 45 процентов, соответственно.
ПРИМЕР 37
Соединение 33 уменьшает метастазы, связанные с фиброзом
[0391] Все больше данных свидетельствует о том, что для возникновения метастазов рака необходимо создать дометастатическую нишу, чтобы способствовать экстравазации и метастатической колонизации. Считается, что фиброзное микроокружение усиливает инвазию опухоли и метастазирование.
[0392] Фиброз печени индуцировали обработкой мышей BALB/c два раза в неделю тетрахлоридом углерода (CCl4) в дозе 1 мг/кг в течение 8 недель. Параллельно проводили обработку Соединением 33 (20 мг/кг ежедневно внутрибрюшинно), которое значительно уменьшало фиброз печени (Фиг. 11a и b). В конце недели 4 ортотопически вводили линию клеток рака груди мыши (4t1) (как показано на Фиг. 11а). Лечение Соединением 33 значительно уменьшало фиброз печени (Фиг. 11b), поперечные сшивки коллагена и метастатическую нагрузку в печени (Фиг. 11c и 11d). Никаких различий в росте первичной опухоли или поперечных сшивках коллагена не наблюдалось.
ПРИМЕР 38
Соединения настоящего изобретения обладают пониженной субстратной активностью в SSAO по сравнению с Е- алкеновыми, изомерными аналогами
[0393] Сведение к минимуму нецелевой активности является ключевым моментом при планировании и разработке соединений, предназначенных для терапевтического применения. Соединения, включающие E-1, были представлены в качестве ингибиторов семикарбазид-чувствительной аминоксидазы (SSAO) [WO2009/066152]. Сообщалось, что молекулы этого типа также могут превращаться в субстраты для SSAO, генерируя потенциально токсичные метаболиты (Foot et. Al., 2012). В предпочтительном варианте реализации соединения настоящего изобретения не являются ни ингибиторами, ни субстратами SSAO.
Измерение оборота субстрата по SSAO
[0394] Вкратце, используемый анализ определяет склонность соединения к субстрату относительно фона (только диметилсульфоксид). Окисление соединений rhSSAO измеряют флуорометрическим анализом (Holt and Palcic, 2006). Вкратце, rhSSAO инкубируют в течение 2 часов при 37 °C в буфере HEPES перед добавлением равного объема Amplex Red (20 мМ), пероксидазы хрена (4 ед./мл) и Соединения (2,5 мМ) в том же буфере. Кинетику образования резоруфина немедленно измеряют при помощи ридера Optima (BMG Labtech GmbH, Ortenburg, Germany) при 37 °C. Репрезентативные результаты показаны в Таблице 8.
Таблица 8
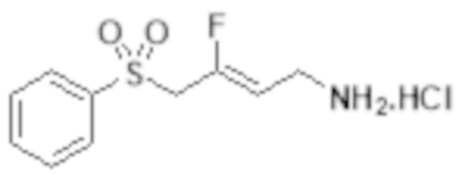
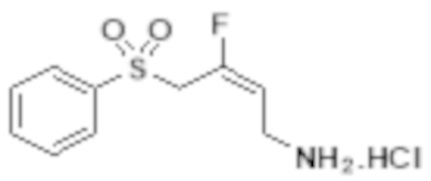
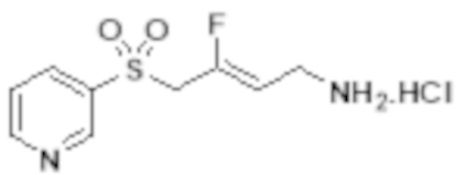
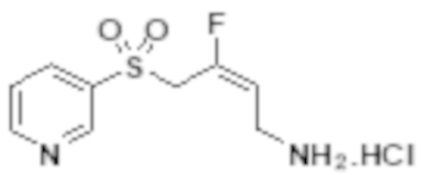
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТАГОНИСТЫ АРИЛСУЛЬФОНАМИДА CCR3 | 2010 |
|
RU2539591C2 |
| 2,5-ДИЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДНЫЕ АНТАГОНИСТЫ CCR3 | 2010 |
|
RU2532515C2 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| ИНГИБИТОРЫ БЕЛКОВЫХ ТИРОЗИНФОСФАТАЗ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2833220C2 |
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2016 |
|
RU2720678C2 |
| НОВОЕ ИМИДАЗООКСАЗИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2012 |
|
RU2578608C2 |
| ИНГИБИТОРЫ ПРОТЕИН-ТИРОЗИНФОСФАТАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2799446C2 |
| ЗАМЕЩЕННЫЕ 4-(АРИЛАМИНО) СЕЛЕНОФЕНОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2011 |
|
RU2566293C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ ATR | 2009 |
|
RU2604066C2 |
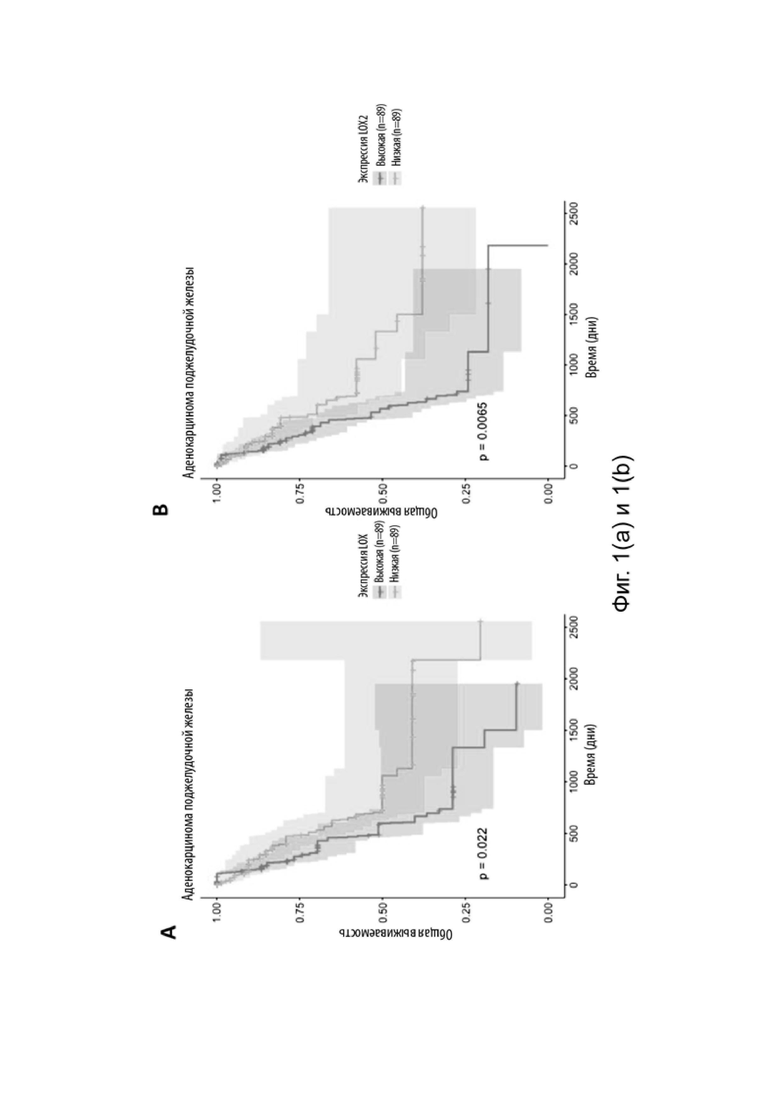
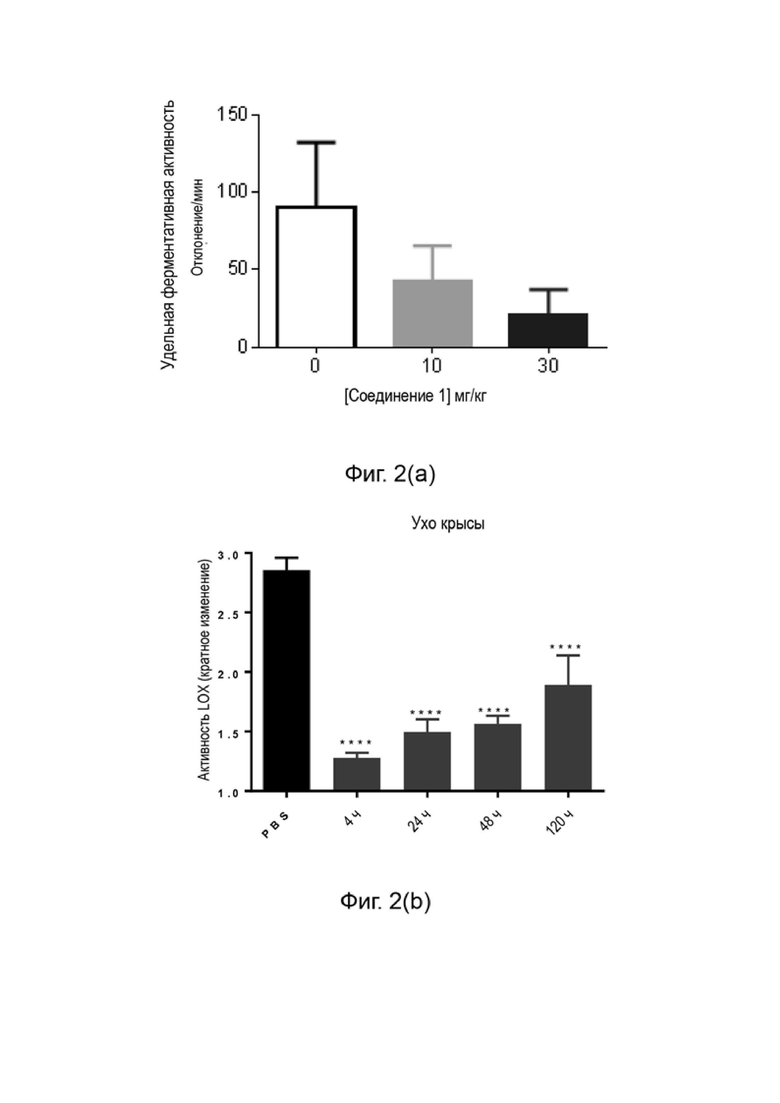
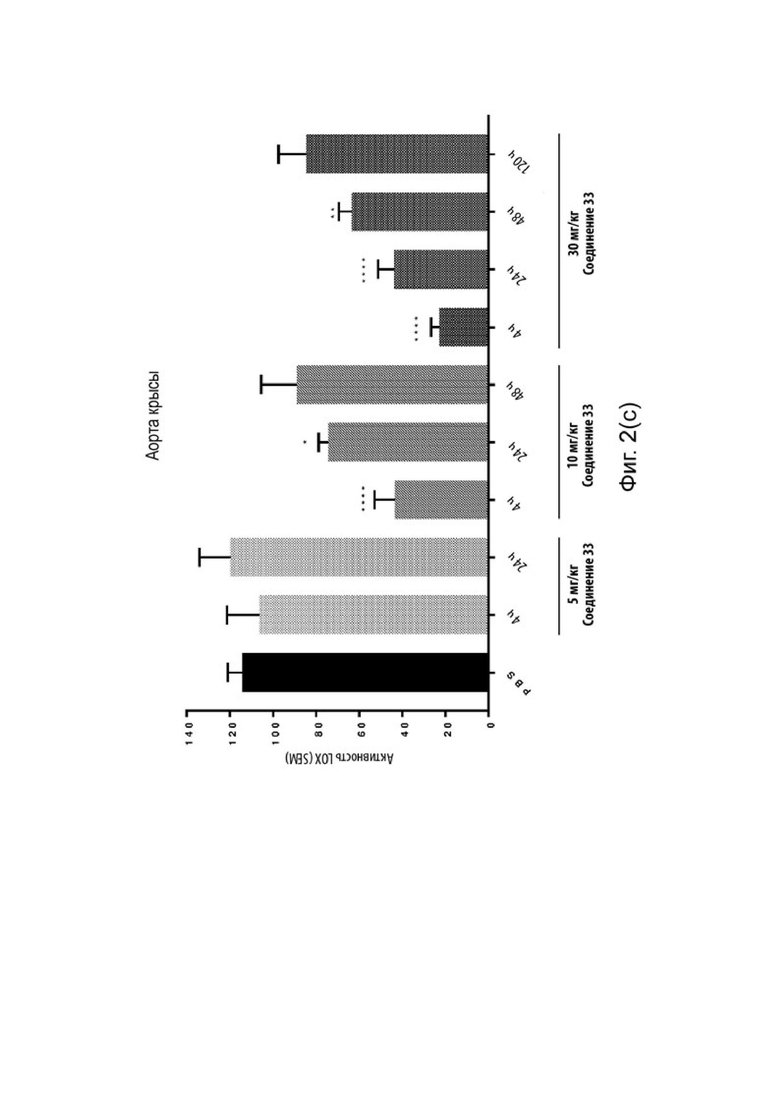
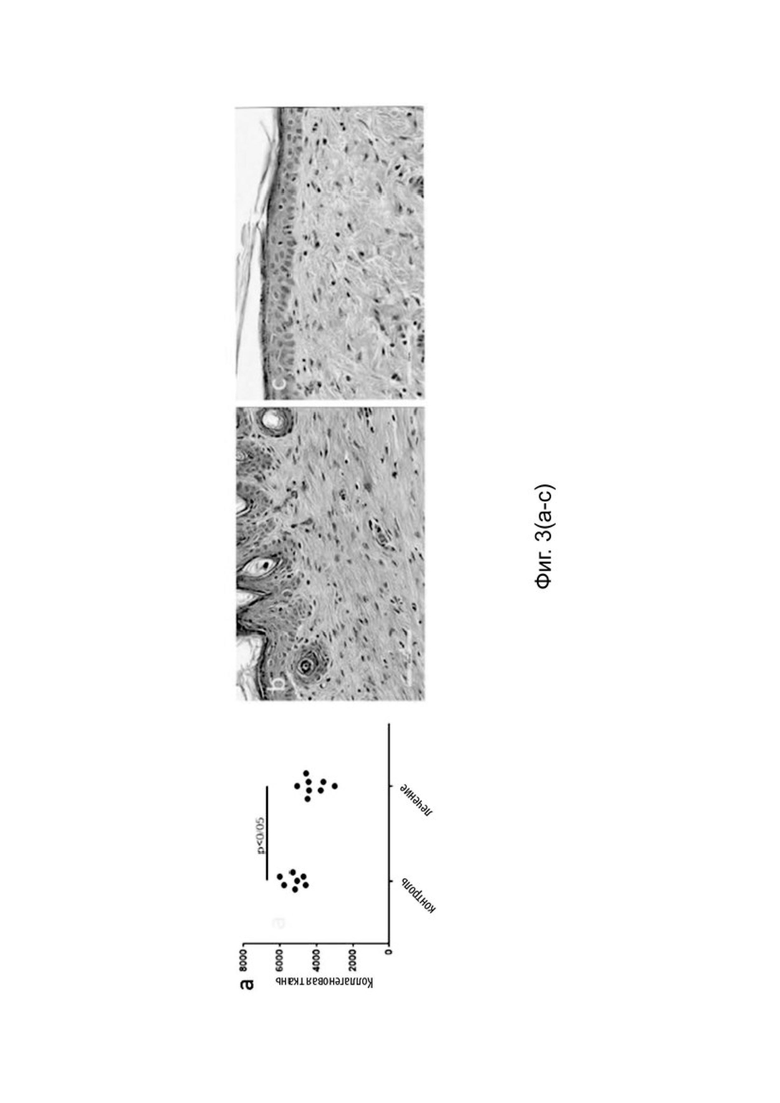
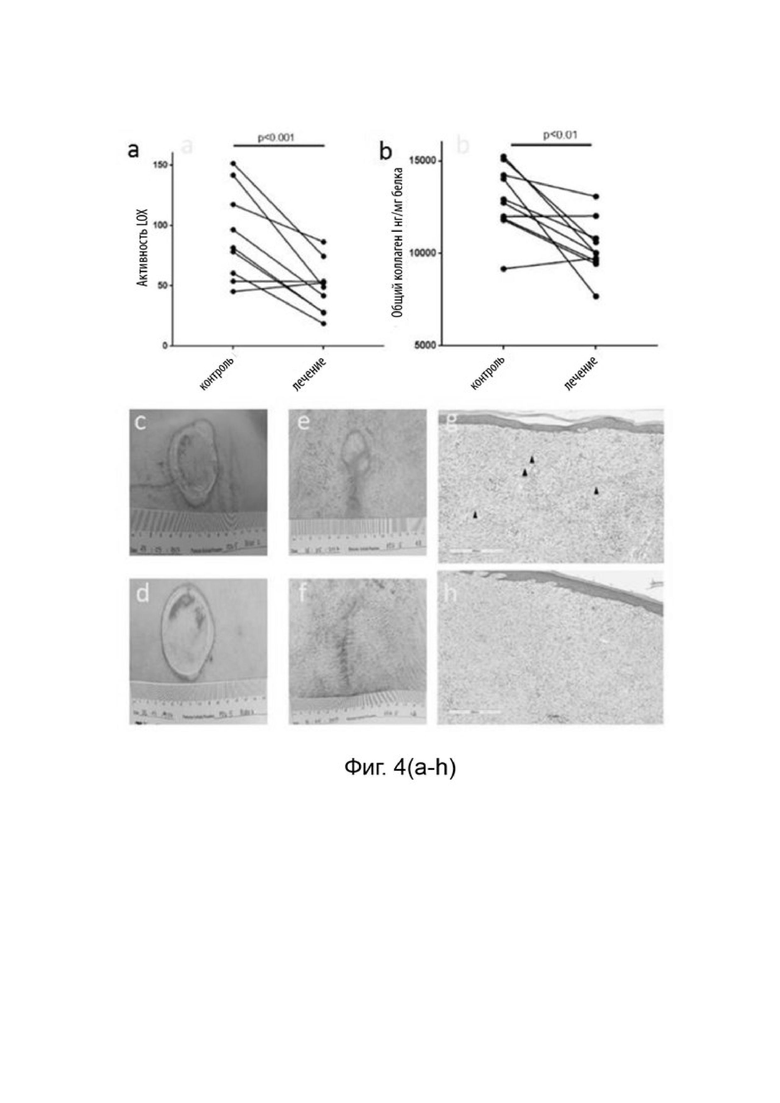
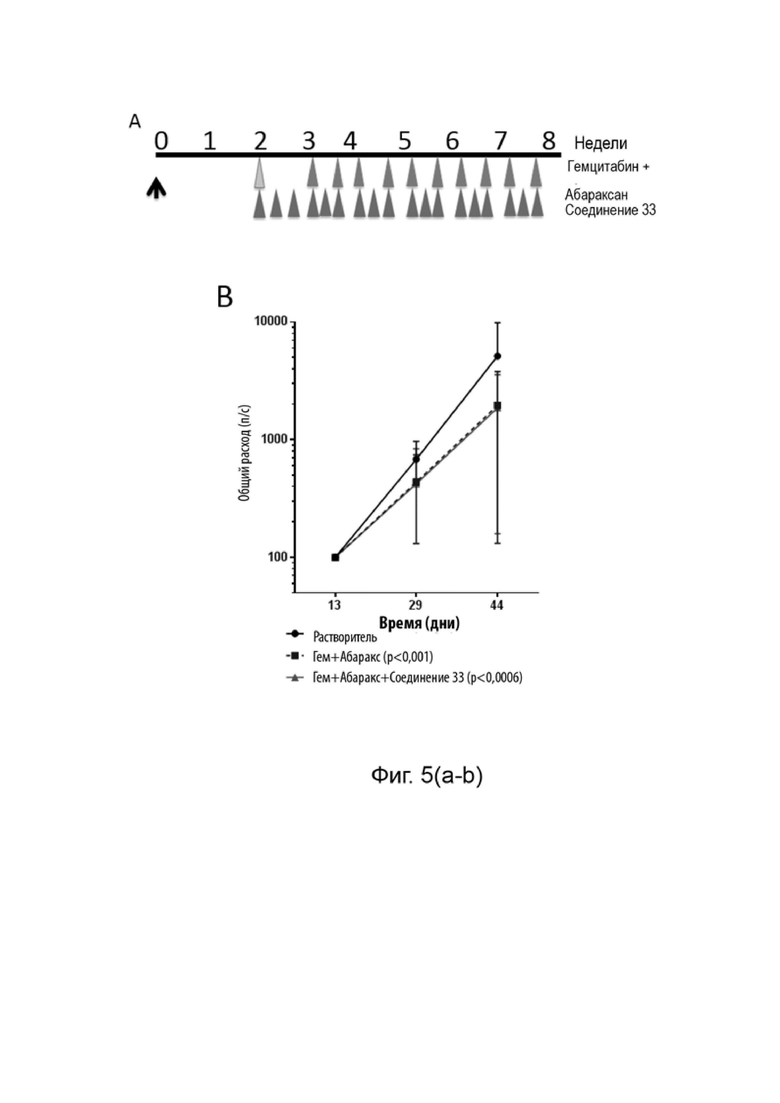
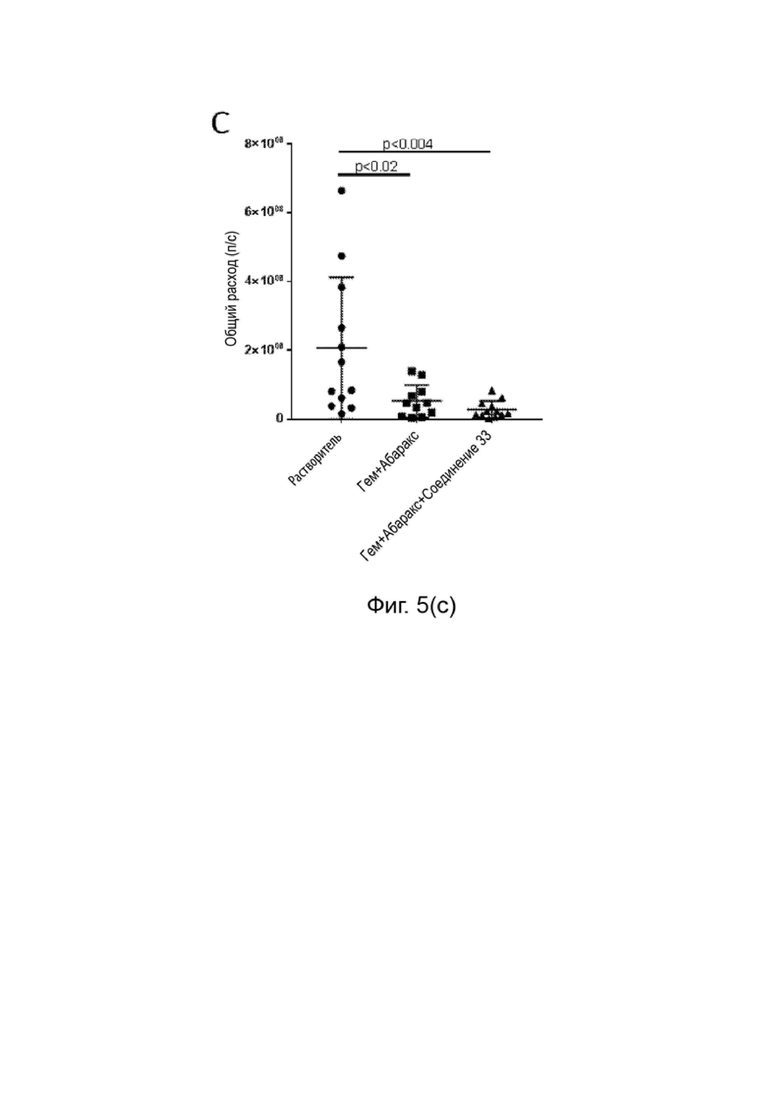
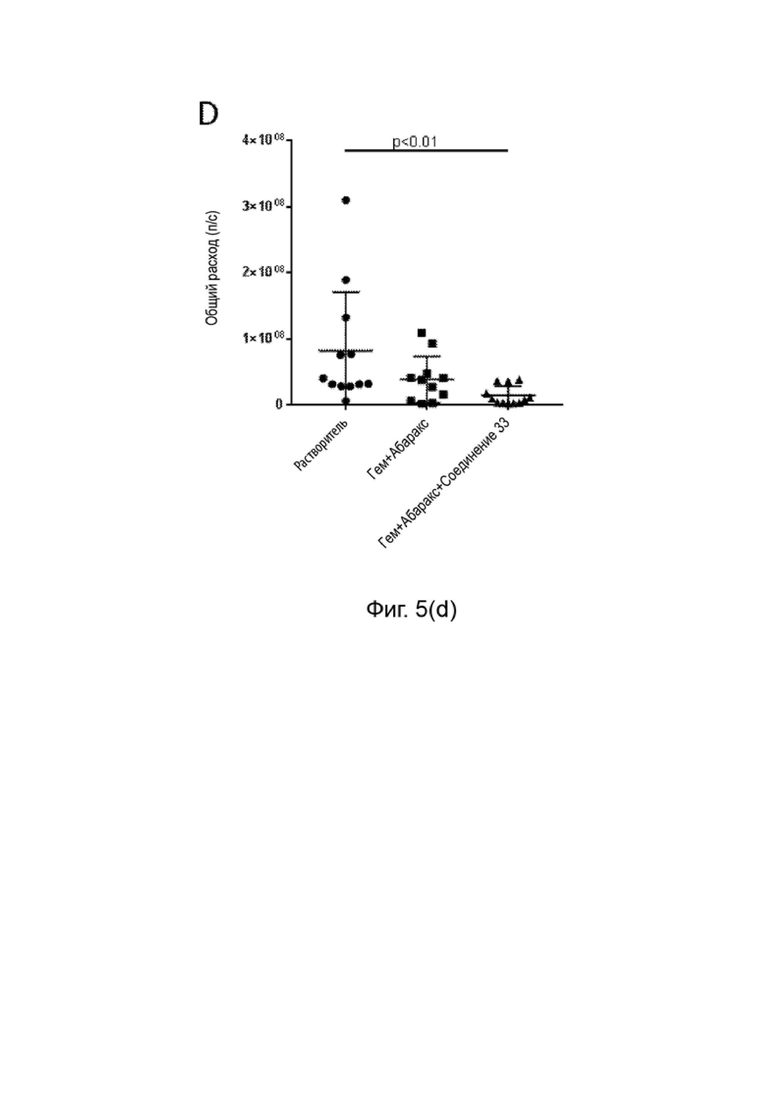
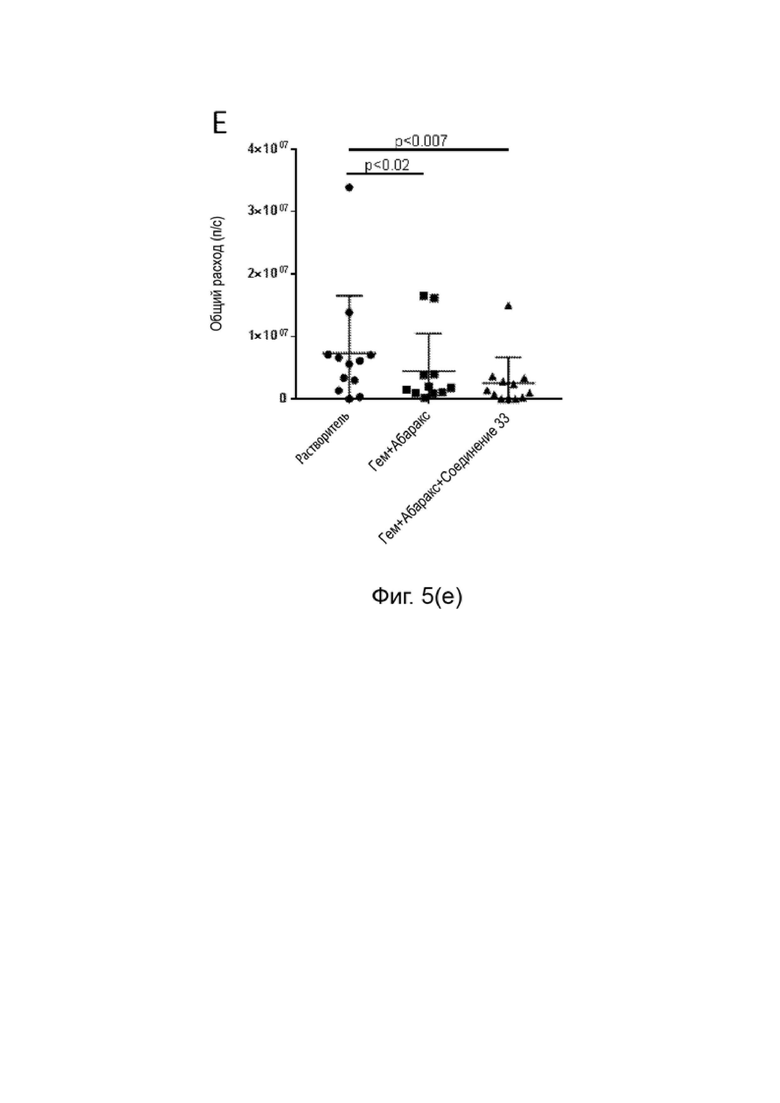
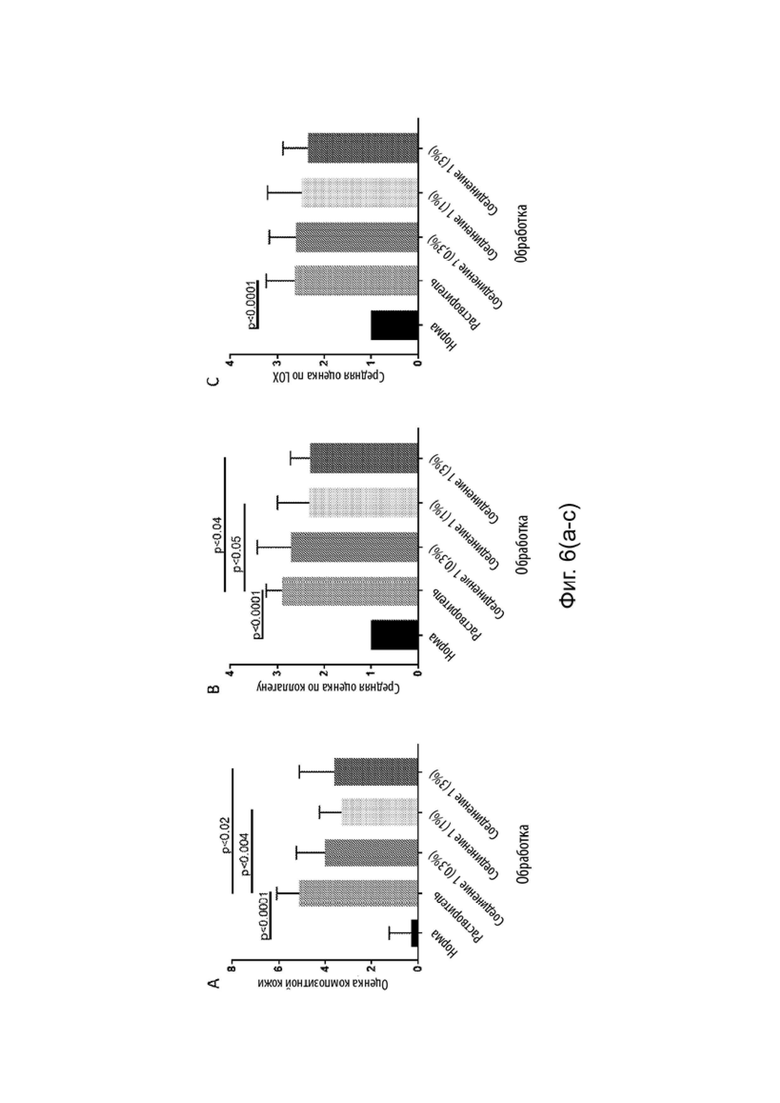
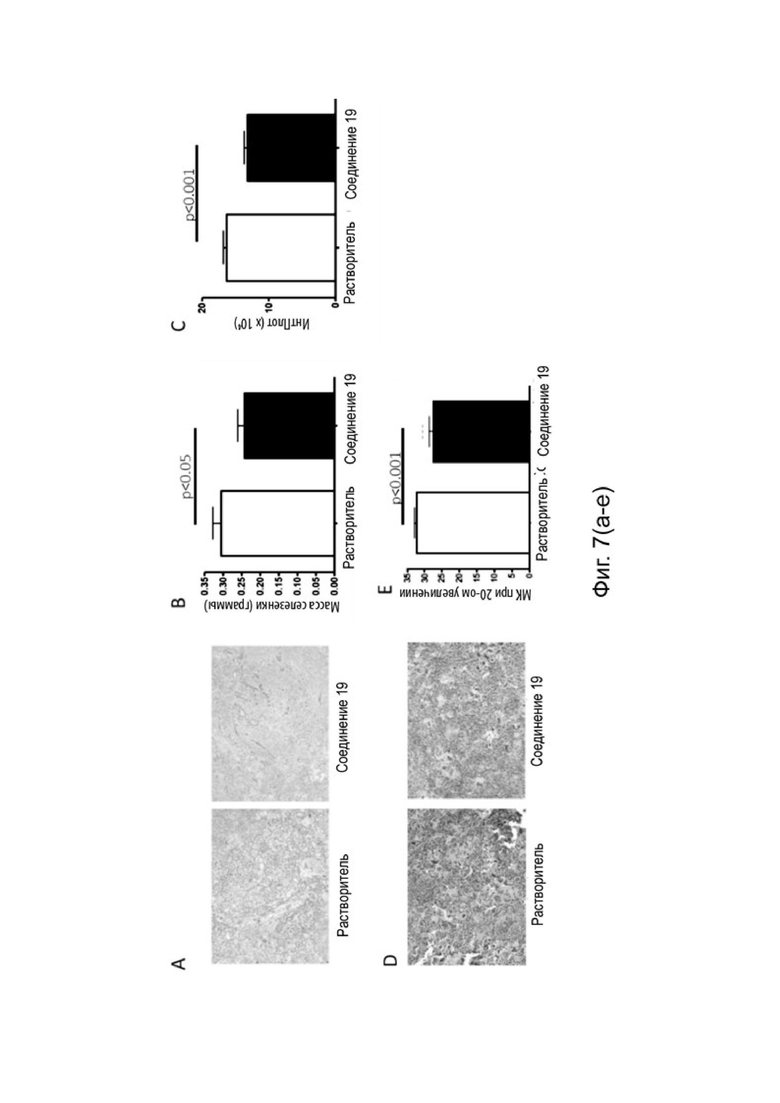
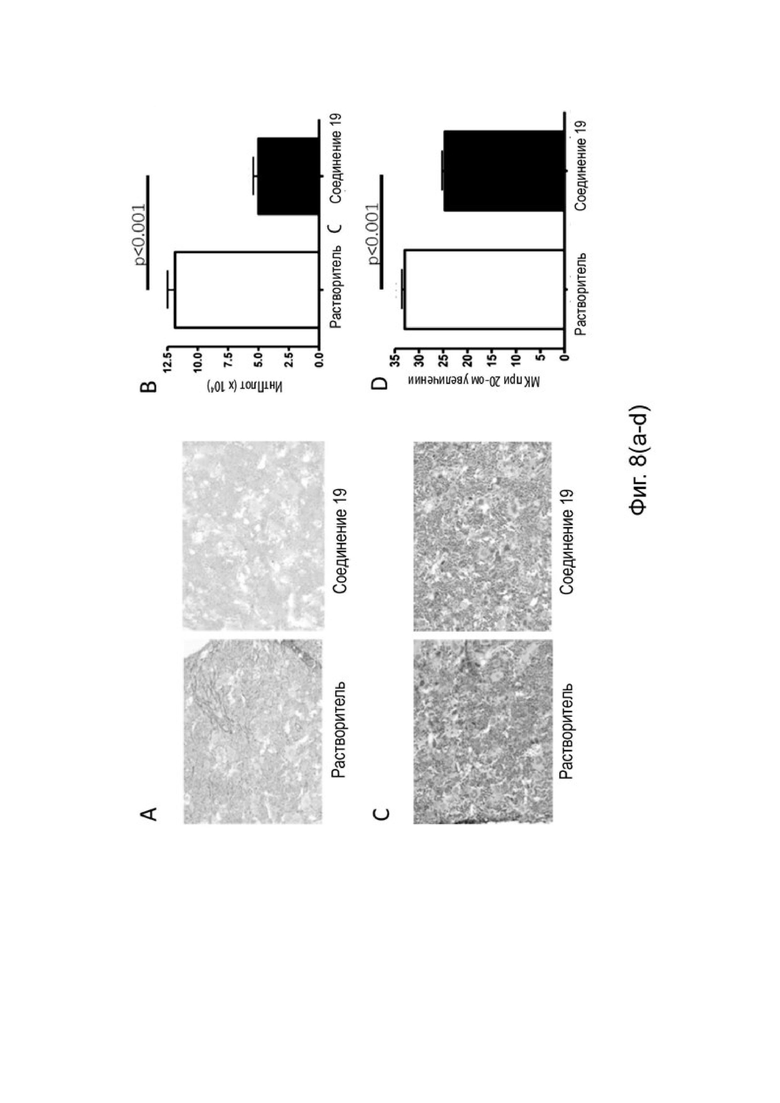
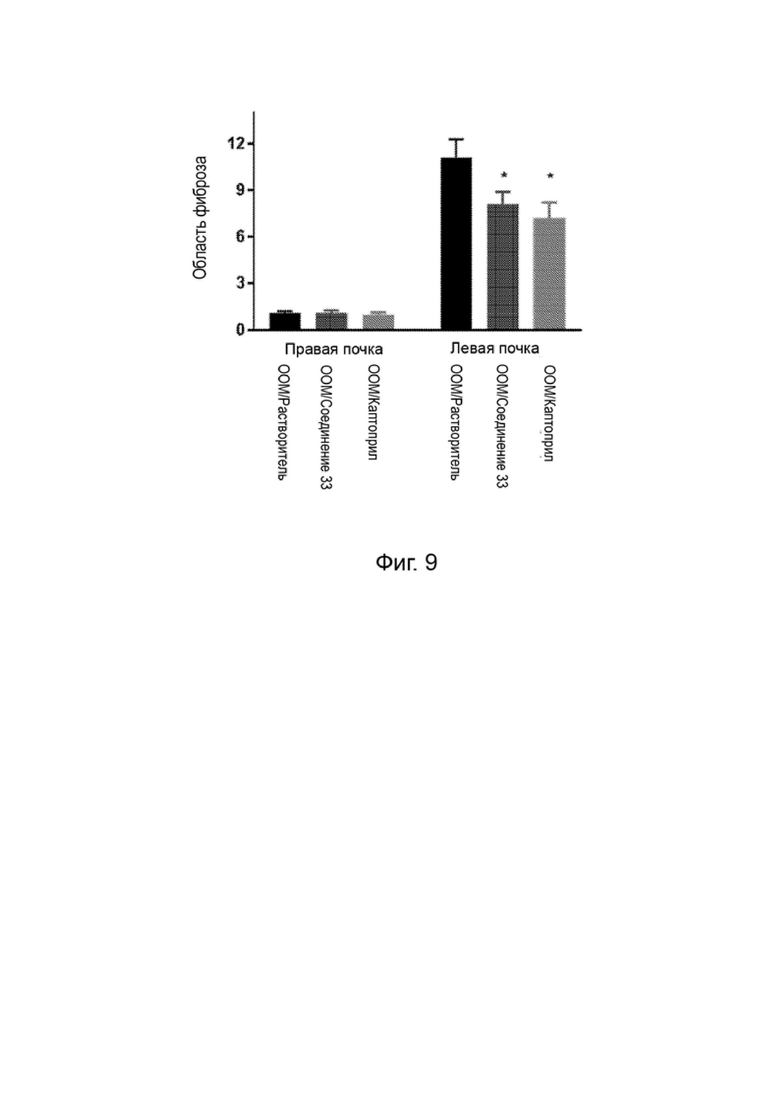
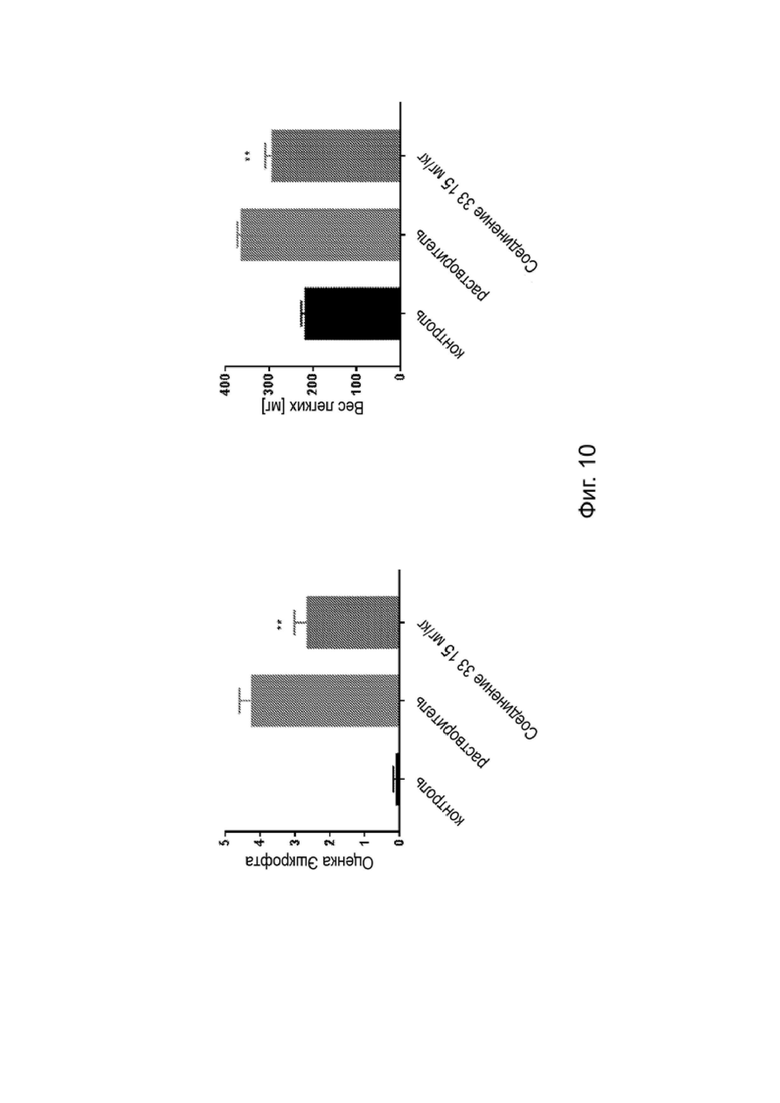
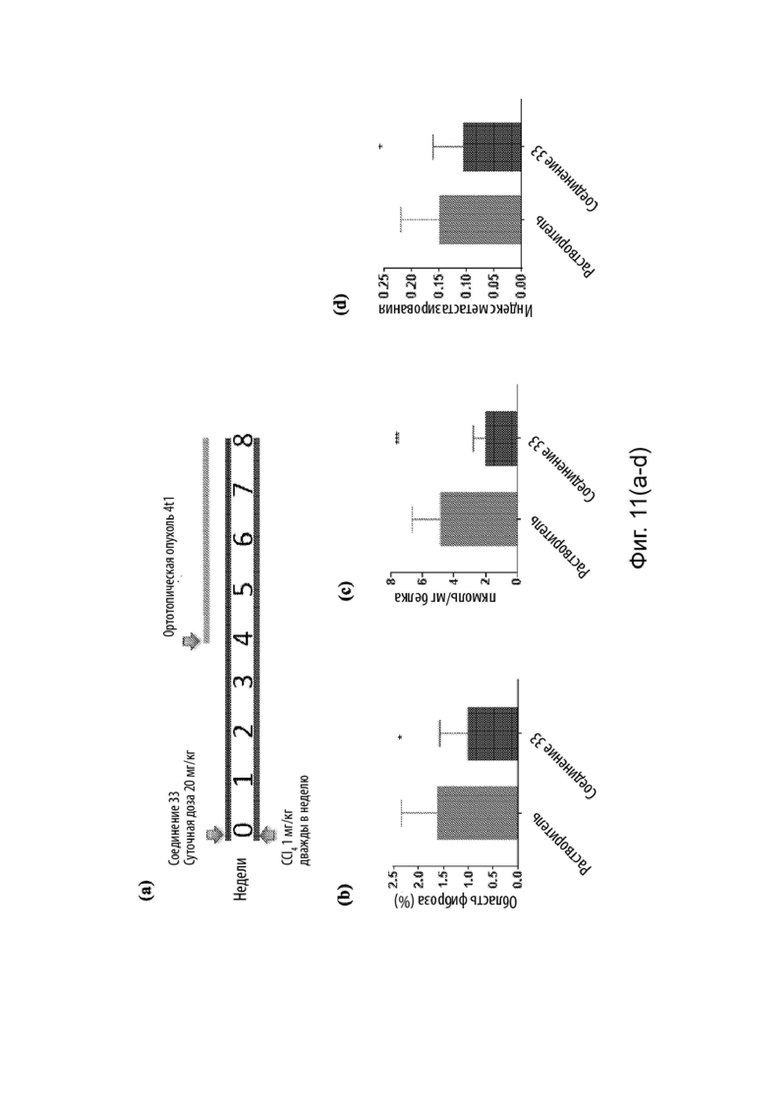
Изобретение относится к соединению - Z-изомеру формулы I, его фармацевтически приемлемой соли или гидрату, которые обладают ингибирующей активностью по отношению к лизилоксидазам LOX, LOXL1, LOXL2, LOXL3 и LOXL4. В указанной формуле А представляет собой C6-10арил или 5-10-членный гетероарил, в котором 1-2 кольцевых атома представляют собой гетероатомы, независимо выбранные из N и S; каждый R1 независимо выбирают из группы, состоящей из X-R2, галогена, дейтерия, C1-6алкила, O-C1-6алкила, C6-10арила, -C(O)NR4R5 и -S(O)2R6, где каждый C1-6алкил и C6-10арил необязательно замещен одним или несколькими галогенами; X выбирают из группы, состоящей из O, OCH2 и CONH; R2 выбирают из группы, состоящей из C3-10циклоалкила и C6-10арила, где каждый R2 необязательно замещен одним или несколькими R7; R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила; R6 представляет собой C1-6алкил; R7 представляет собой -S(O)2NR4R5; и n равно 0, 1, 2, 3, 4, 5 или 6. Изобретение также относится к фармацевтической композиции для ингибирования аминоксидазной активности любой из LOX, LOXL1, LOXL2, LOXL3 и LOXL4, содержащей соединение общей формулы I, его применению для ингибирования вышеуказанной аминоксидазной активности, лечения состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 и LOXL4, и для производства лекарственного средства для лечения этого состояния. 5 н. и 15 з.п. ф-лы, 11 ил., 8 табл., 38 пр.
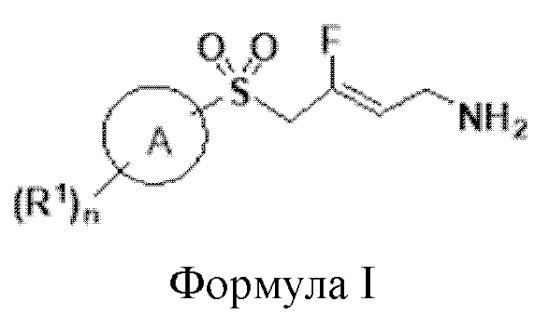
1. Соединение-Z-изомер Формулы I
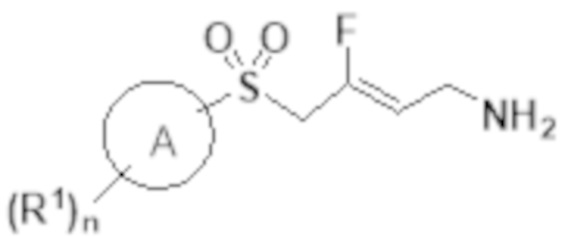
Формула I
или его фармацевтически приемлемая соль или гидрат, в которой:
А представляет собой C6-10арил или 5-10-членный гетероарил, в котором 1-2 кольцевых атома представляют собой гетероатомы, независимо выбранные из N и S;
каждый R1 независимо выбирают из группы, состоящей из X-R2, галогена, дейтерия, C1-6алкила, O-C1-6алкила, C6-10арила, -C(O)NR4R5 и -S(O)2R6, где каждый C1-6алкил и C6-10арил необязательно замещен одним или несколькими галогенами;
X выбирают из группы, состоящей из O, OCH2 и CONH;
R2 выбирают из группы, состоящей из C3-10циклоалкила и C6-10арила, где каждый R2 необязательно замещен одним или несколькими R7;
R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила;
R6 представляет собой C1-6алкил;
R7 представляет собой -S(O)2NR4R5; и
n равно 0, 1, 2, 3, 4, 5 или 6.
2. Соединение по п. 1, отличающееся тем, что A выбирают из группы, состоящей из фенила, нафтила, пиридинила, хинолинила, бензотиазолила и индолила.
3. Соединение по п. 1 или 2, отличающееся тем, что A выбирают из группы, состоящей из:
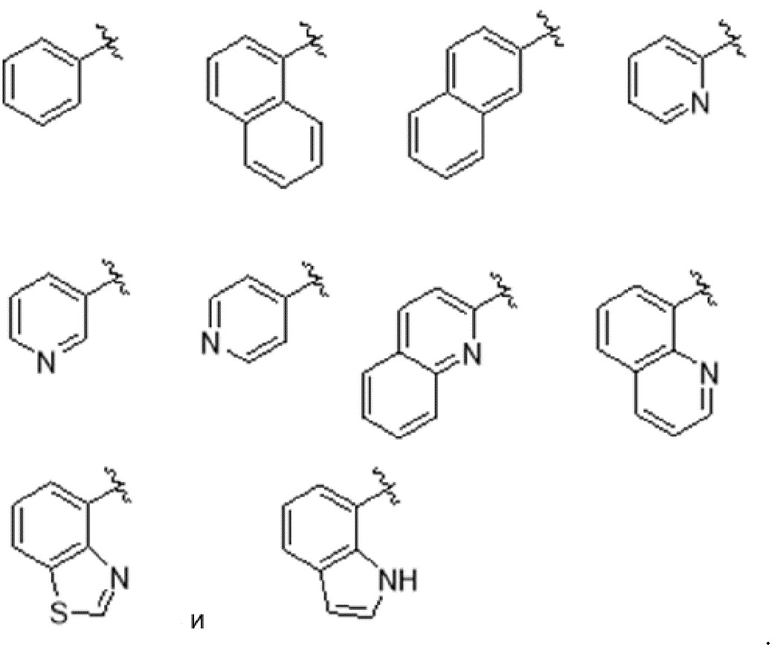
4. Соединение по п. 1, отличающееся тем, что A представляет собой 5-10-членный гетероарил, в котором 1-2 кольцевых атома представляют собой гетероатомы, независимо выбранные из N и S.
5. Соединение по п. 1, отличающееся тем, что
A выбирают из группы, состоящей из  и
и  ;
;
R1 представляет собой метил или изопропил; и
n равно 0 или 1.
6. Соединение по п. 1, отличающееся тем, что A представляет собой 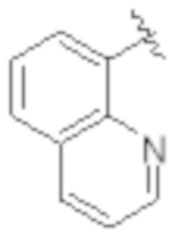 и n равно 0.
и n равно 0.
7. Соединение по п. 1 Формулы Ia
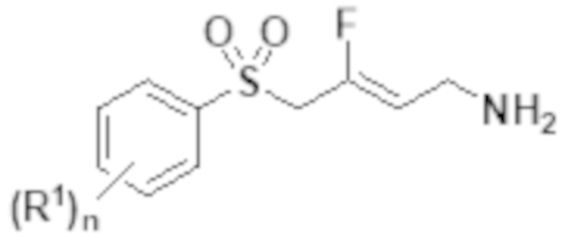
Формула Ia
или его фармацевтически приемлемая соль, в которой:
каждый R1 независимо выбирают из группы, состоящей из X-R2, галогена, C1-6алкила, O-C1-6алкила, C6-10арила, -C(O)NR4R5 и -S(O)2R6, где каждый C1-6алкил и C6-10арил необязательно замещен одним или несколькими галогенами;
X выбирают из группы, состоящей из O, OCH2 и CONH;
R2 выбирают из группы, состоящей из C3-10циклоалкила и C6-10арила, где каждый R2 необязательно замещен одним или несколькими R7;
R4 и R5 независимо выбирают из группы, состоящей из водорода и C1-6алкила;
R6 представляет собой C1-6алкил;
R7 представляет собой -S(O)2NR4R5; и
n равно 0, 1, 2 или 3.
8. Соединение по п. 7, отличающееся тем, что n равно 0.
9. Соединение по п. 7, отличающееся тем, что
каждый R1 независимо выбирают из группы, состоящей из галогена, C1-6алкила, O-C1-6алкила, C6-10арила и -S(O)2R6, где каждый C1-6алкил необязательно замещен одним или несколькими галогенами;
R6 представляет собой C1-6алкил; и
n равно 1 или 2.
10. Соединение по п. 1, выбранное из группы, состоящей из
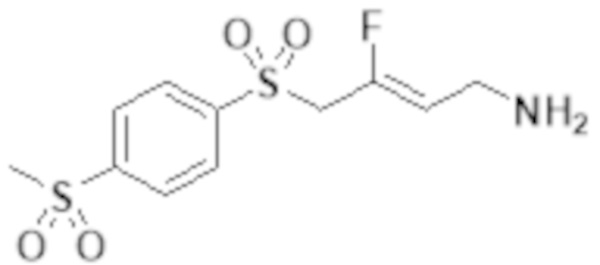
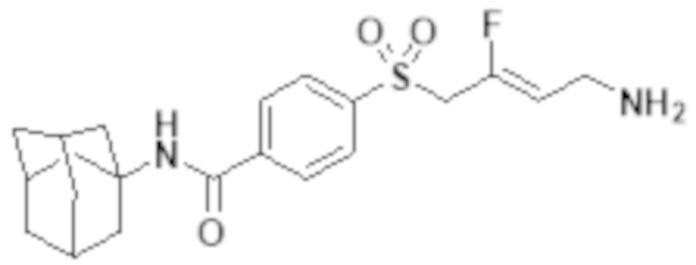
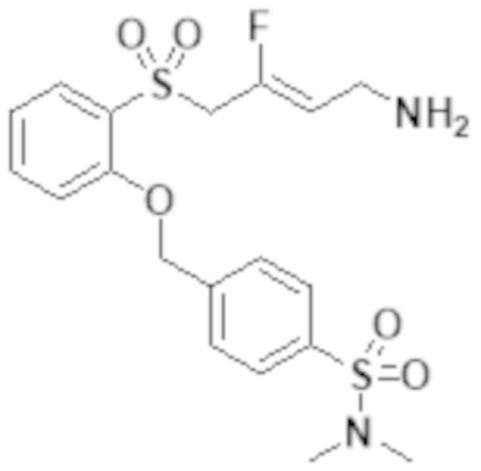
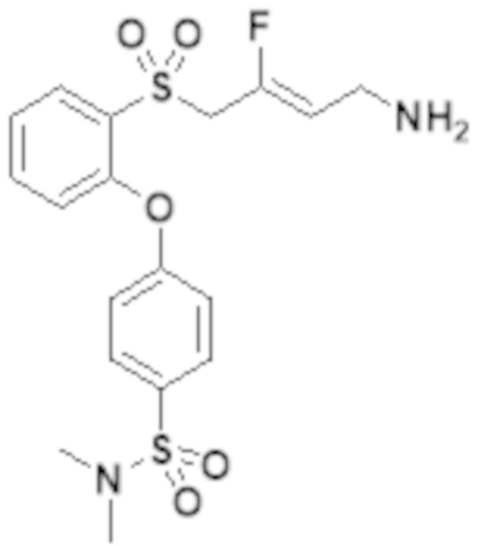
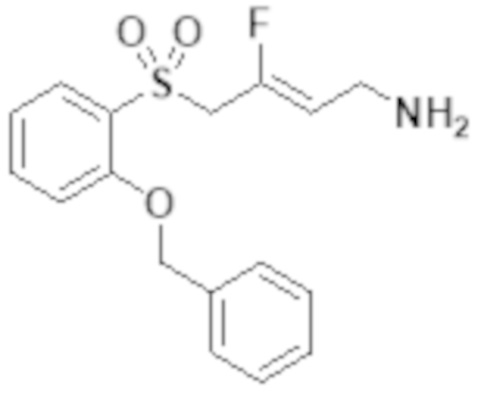
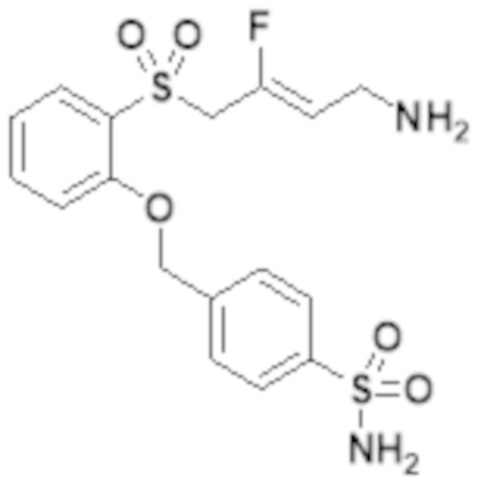
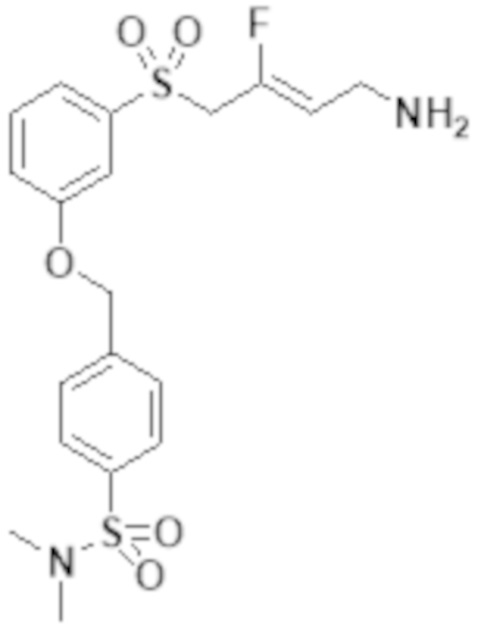
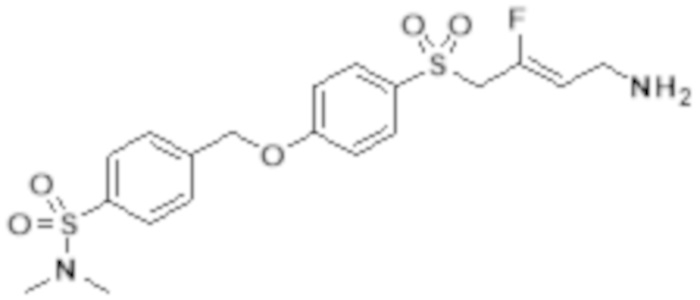
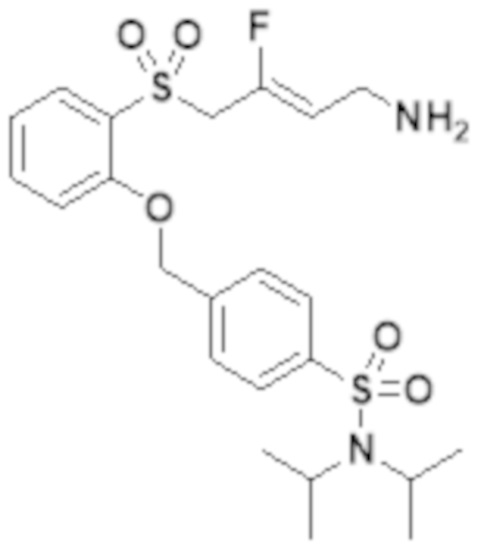
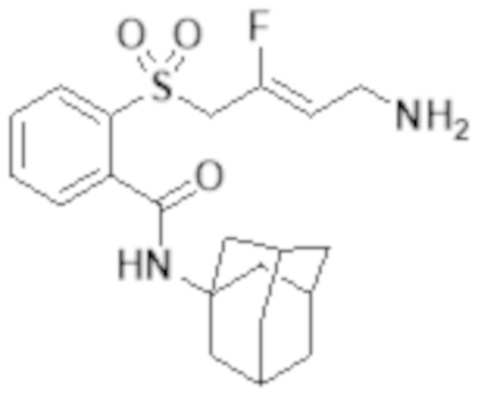
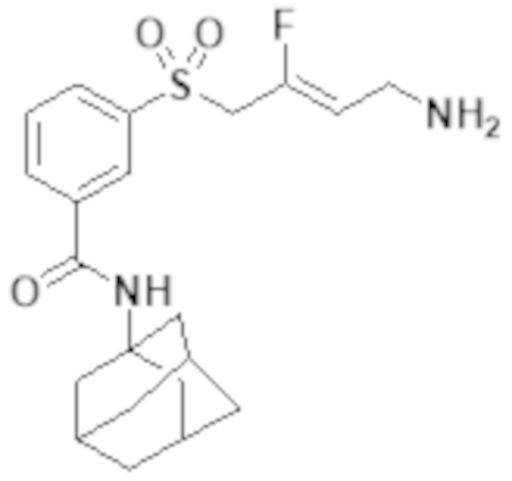
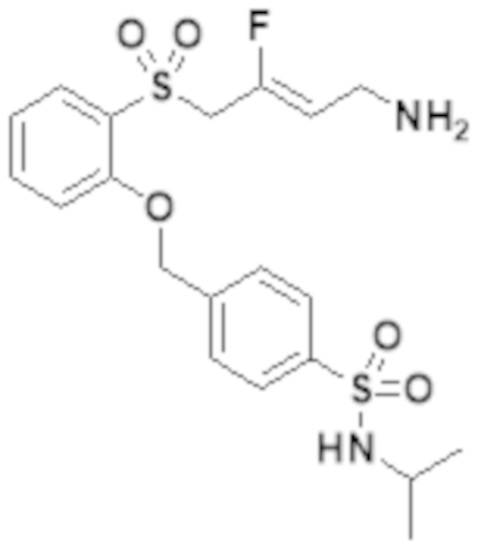
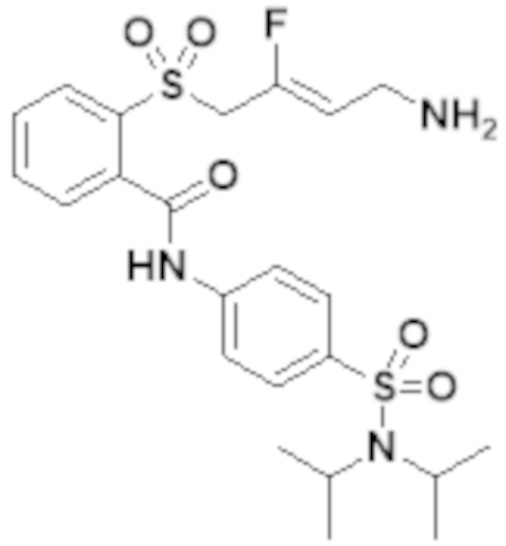
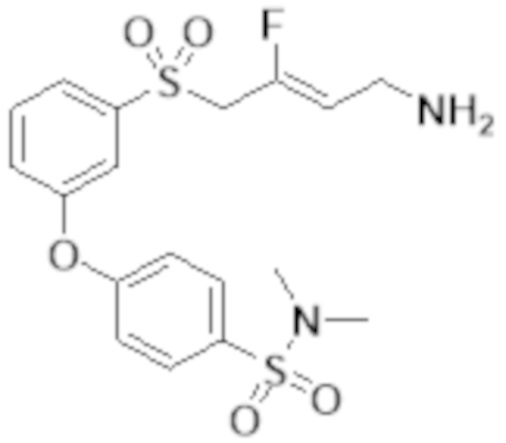
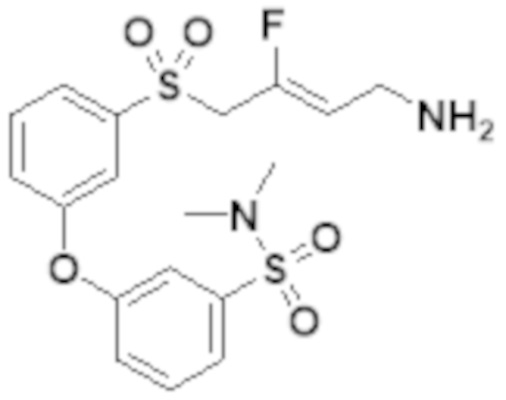
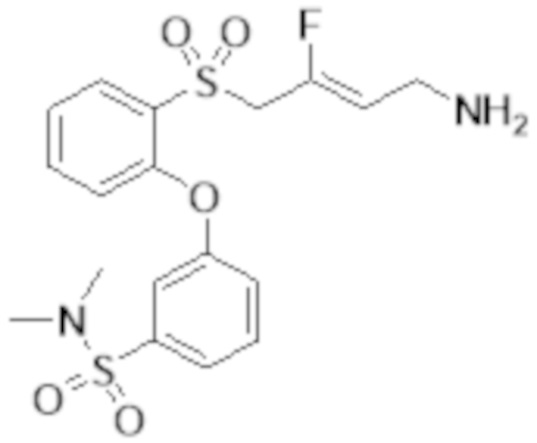
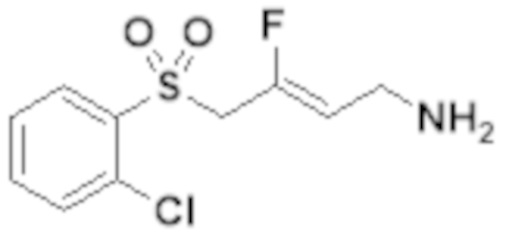
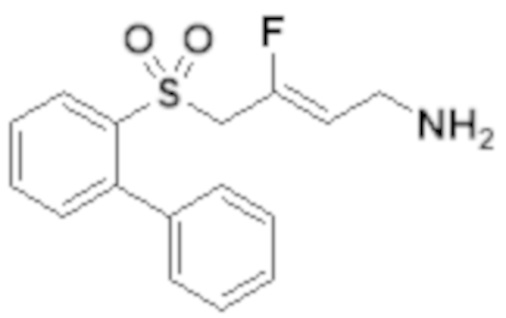
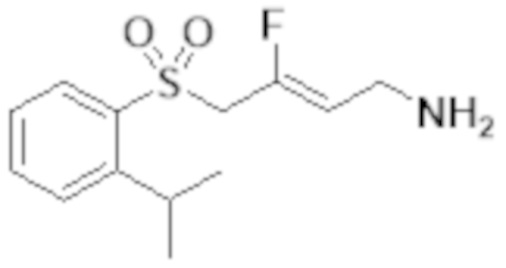
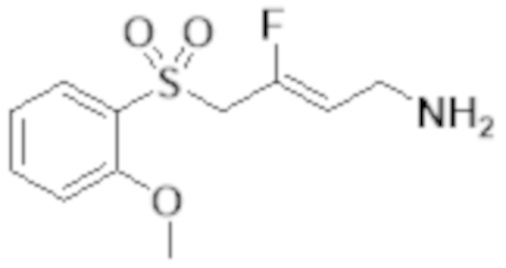
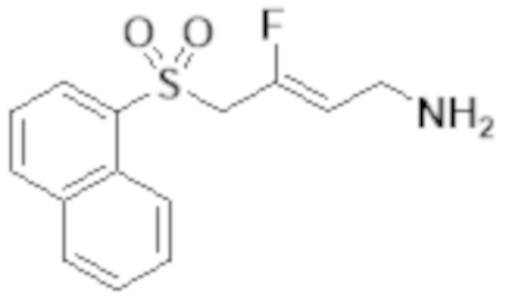
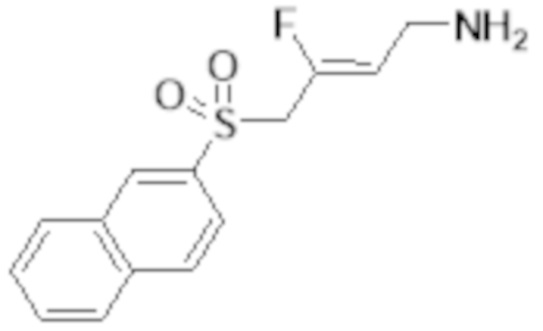
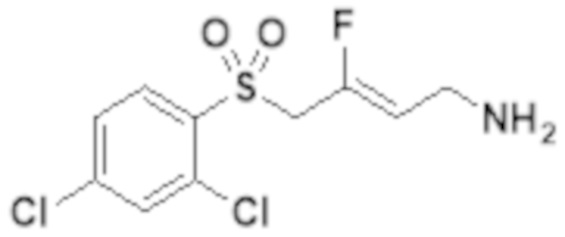
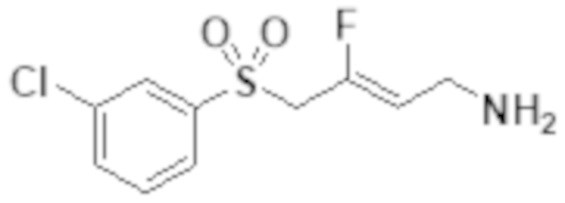
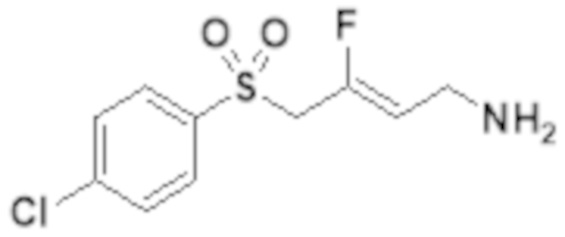
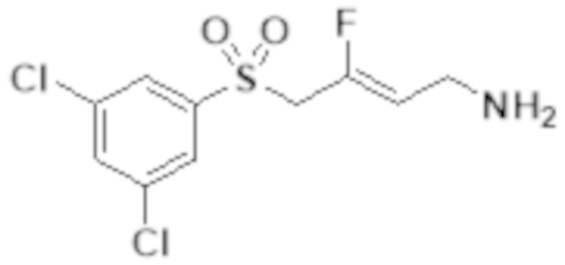
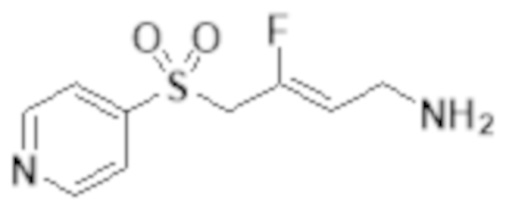
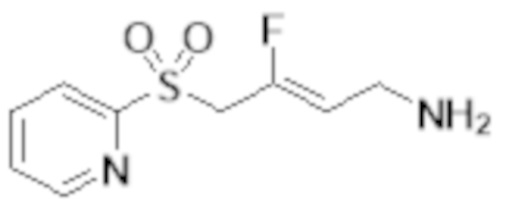
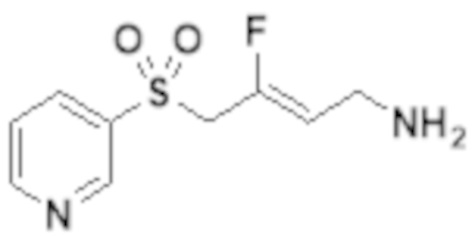
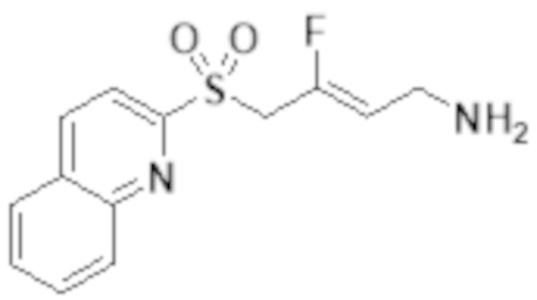
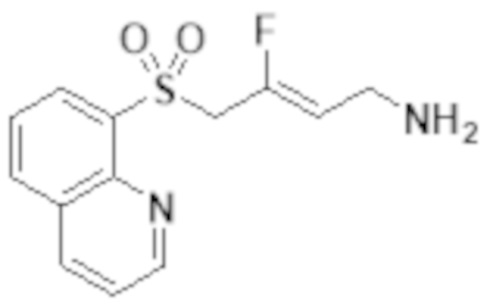
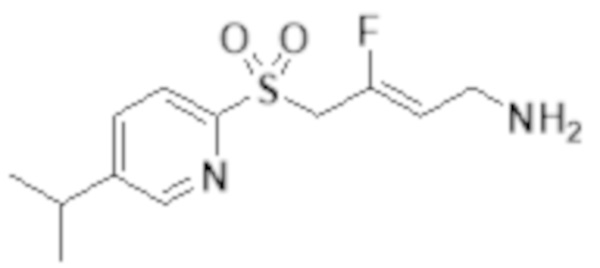
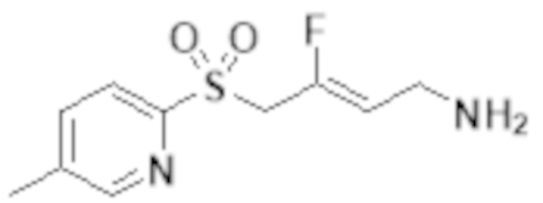
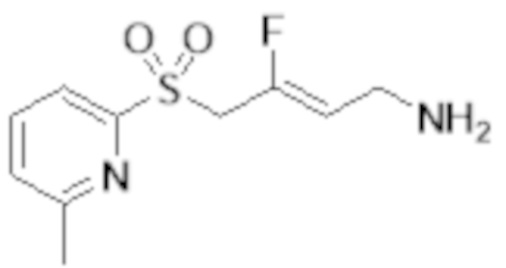
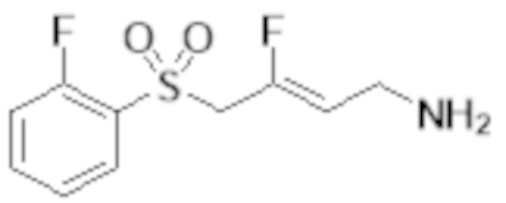
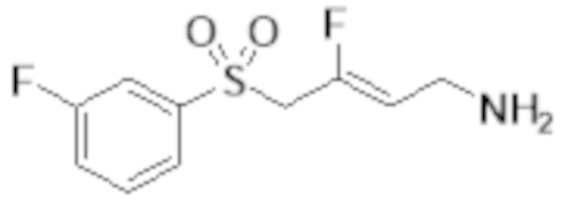
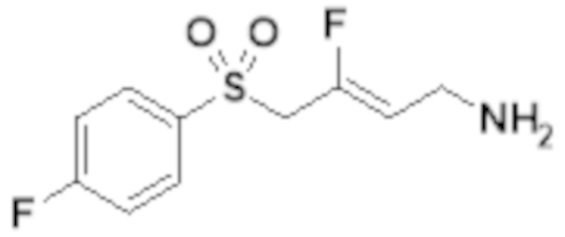
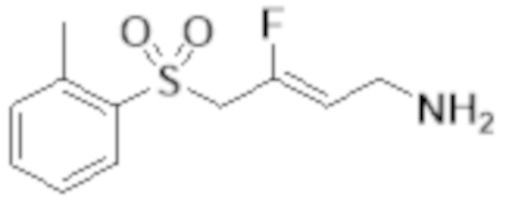
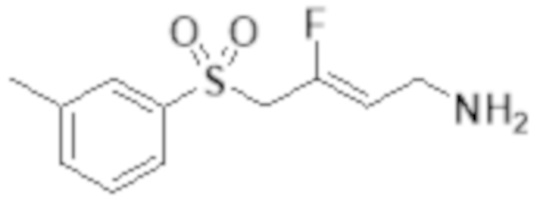
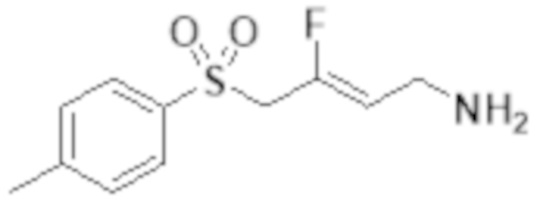
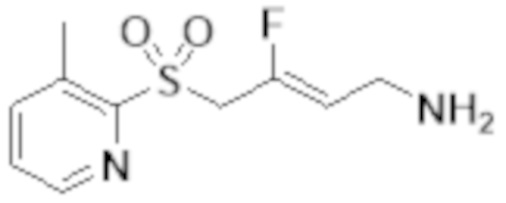
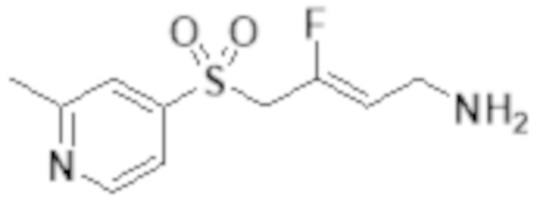
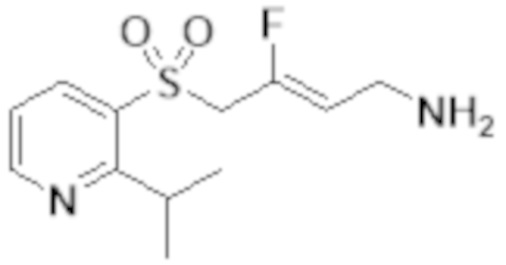
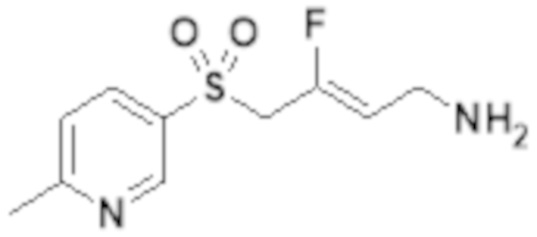
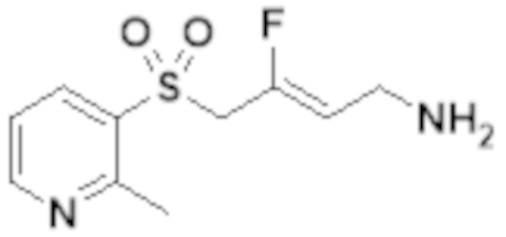
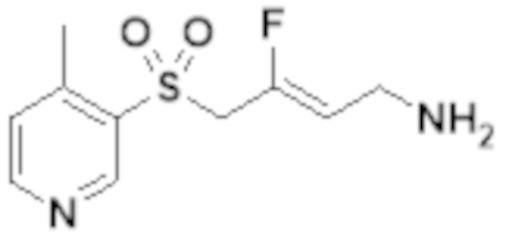
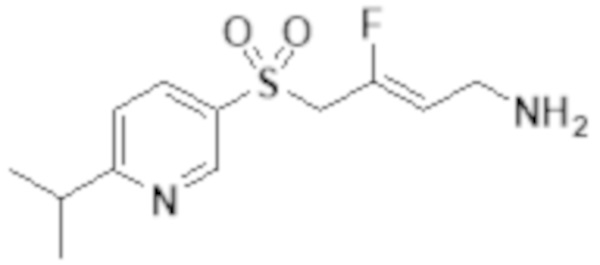
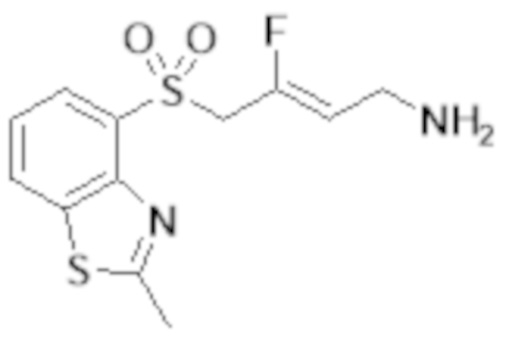
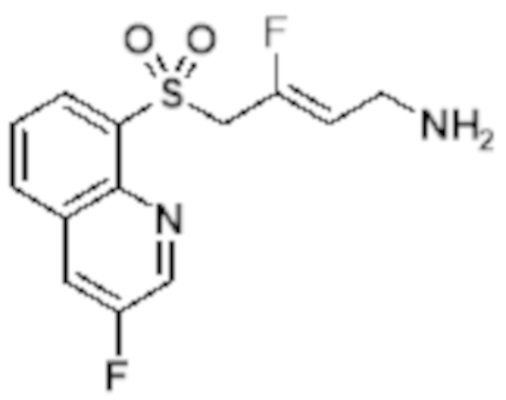
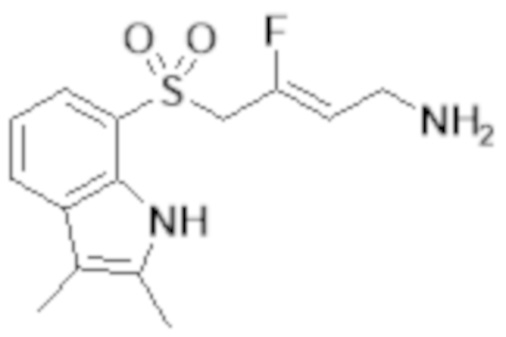
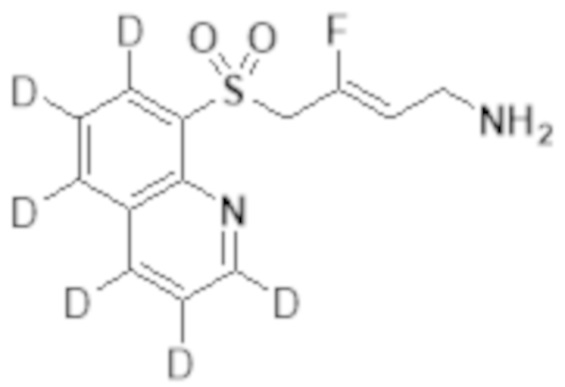
или его фармацевтически приемлемая соль или гидрат.
11. Соединение по п. 1, выбранное из группы, состоящей из и , или их фармацевтически приемлемых солей.
12. Соединение по п. 1, выбранное из группы, состоящей из и , или их фармацевтически приемлемых солей или гидратов.
13. Фармацевтическая композиция для ингибирования аминооксидазной активности любой из LOX, LOXL1, LOXL2, LOXL3 или LOXL4, содержащая эффективное количество соединения по любому из пп. 1-12 или его фармацевтически приемлемой соли или гидрата и по меньшей мере один фармацевтически приемлемый экципиент, носитель или разбавитель.
14. Применение эффективного количества соединения по любому из пп. 1-12 или его фармацевтически приемлемой соли или гидрата или фармацевтически приемлемой композиции по п. 13 для ингибирования активности аминоксидазы любой из LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
15. Применение эффективного количества соединения по любому из пп. 1-12 или его фармацевтически приемлемой соли или гидрата или фармацевтически приемлемой композиции по п. 13 для лечения состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
16. Применение по п. 15, отличающееся тем, что состояние выбрано из группы, состоящей из фиброза, рака и ангиогенеза.
17. Применение по п. 16, отличающееся тем, что в случае фиброза, фиброз выбирают из группы, состоящей из медиастинального фиброза, миелофиброза, забрюшинного фиброза, прогрессирующего массивного фиброза, нефрогенного системного фиброза, болезни Крона, келоида, системного склероза, артрофиброза, контрактуры Дюпюитрена, адгезивного капсулита, фиброза поджелудочной железы, фиброза кишечника, фиброза печени, фиброза легких, фиброза почек, фиброза сердца, фибростеноза, муковисцидоза, идиопатического фиброза легких, фиброза, вызванного радиацией, болезни Пейрони и склеродермии или связанного с респираторным заболеванием, аномальным заживлением и восстановлением ран, рубцевания, гипертрофического рубцевания/келоида, рубцов после операции, остановки сердца и всех состояний, при которых избыточное или аберрантное отложение волокнистого материала связано с заболеванием, травмой, имплантатами или операцией; предпочтительно фиброз выбирают из группы, состоящей из миелофиброза, системного склероза, фиброза печени, фиброза легких, фиброза почек, фиброза сердца и фиброза, вызванного радиацией; и
в случае, если заболевание представляет собой рак, рак выбирают из группы, состоящей из рака легких; рака молочной железы; колоректального рака; анального рака; рака поджелудочной железы; рака простаты; рака яичников; карциномы печени и желчных протоков; карциномы пищевода; мезотелиомы; неходжкинской лимфомы; карциномы мочевого пузыря; карциномы матки; глиомы, глиобластомы, медуллабластомы и других опухолей головного мозга; миелофиброза, рака почек; рака головы и шеи; рака желудка; множественной миеломы; рака яичек; опухоли половых клеток; нейроэндокринной опухоли; рака шейки матки; рака ротовой полости, карциноидов желудочно-кишечного тракта, груди и других органов; карциномы из перстневидных клеток; мезенхимальных опухолей, включая саркомы, фибросаркомы, гемангиому, ангиоматоз, гемангиоперицитому, псевдоангиоматозную стромальную гиперплазию, миофибробластомы, фиброматоз, воспалительную миофибробластическую опухоль, липому, ангиолипому, гранулярно-клеточную опухоль, нейрофиброму, шванному, ангиосаркому, липосаркому, рабдомиосаркому, остеосаркому, лейомиому или лейомиосаркому.
18. Применение по любому из пп. 15-17, дополнительно включающее введение второго терапевтического агента.
19. Применение по п. 18, отличающееся тем, что второй терапевтический агент выбирают из группы, состоящей из противоракового агента, противовоспалительного агента, антигипертензивного агента, антифиброзного агента, антиангиогенного агента, иммуносупресаната и метаболического агента.
20. Применение эффективного количества соединения по любому из пп. 1-12 или его фармацевтически приемлемой соли или гидрата для производства лекарственного средства для лечения состояния, связанного с любым из белков LOX, LOXL1, LOXL2, LOXL3 или LOXL4.
| WO 2009066152 A2, 28.05.2009 | |||
| WO 2017136871 A1, 17.08.2017 | |||
| WO 2017136870 A1, 17.08.2017 | |||
| WO 2013163675 A1, 07.11.2013 | |||
| ИНГИБИТОР ФИБРОЗА | 2009 |
|
RU2497525C2 |

