ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
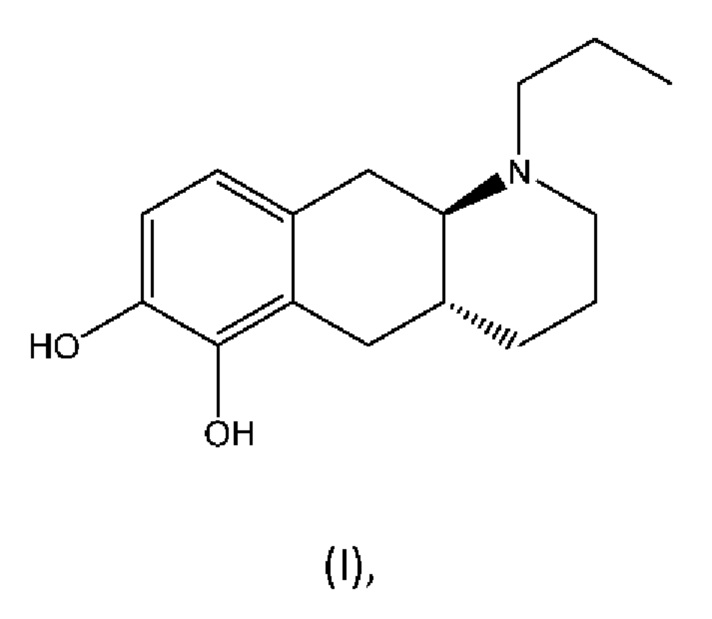
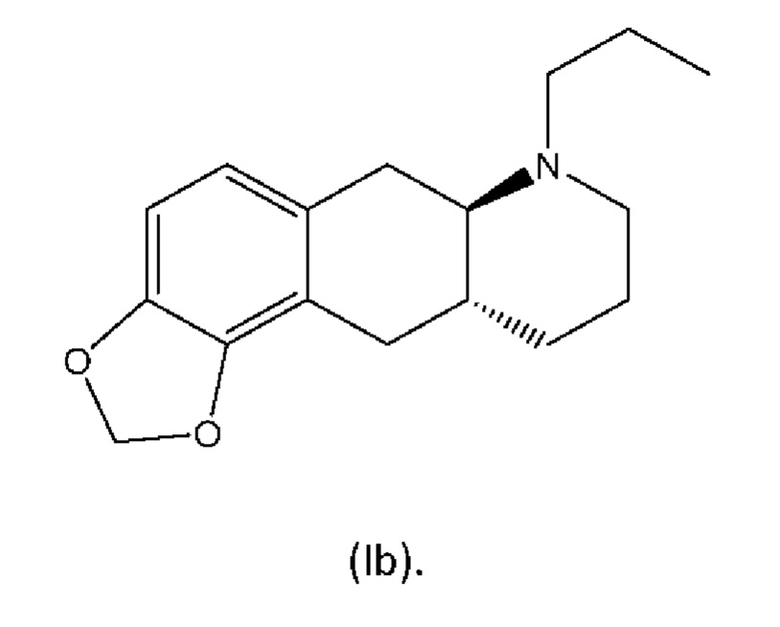
Настоящее изобретение относится к способу изготовления (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола и (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-g]хинолина и их солей, которые являются соединениями для применения в лечении нейродегенеративных заболеваний и нарушений, таких как болезнь Паркинсона. Настоящее изобретение также относится к новым промежуточным соединениям из указанного способа.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
Болезнь Паркинсона (PD) представляет собой распространенное нейродегенеративное нарушение, которое становится все более распространенным с возрастом и, по оценкам, поражает от семи до десяти миллионов человек во всем мире. Болезнь Паркинсона является многогранным заболеванием, характеризующимся как моторными, так и немоторными симптомами. Моторные симптомы включают тремор в покое (дрожание), брадикинезию/акинезию (медлительность и скудность движений), мышечную ригидность, постуральную неустойчивость и дисфункцию походки; при этом немоторные симптомы включают нейропсихиатрические нарушения (например, депрессию, психотические симптомы, тревожность, апатию, умеренное когнитивное нарушение и деменцию), а также вегетативные дисфункции и нарушения сна (Poewe et al., Nature Review, (2017) vol 3 article 17013: 1-21).
Ключевой характерной особенностью патофизиологии болезни Паркинсона является потеря пигментированных дофаминергических нейронов в компактном слое черного вещества, который обеспечивает дофаминергическую иннервацию полосатого тела и других зон головного мозга. Такая прогрессирующая нейродегенерация приводит к уменьшению уровней дофамина в полосатом теле, что в конечном итоге вызывает ряд изменений межнейронных связей в базальных ганглиях, что в конечном итоге приводит к появлению четырех основных моторных признаков болезни Паркинсона. Основной мишенью дофамина в полосатом теле являются срединные шипиковые GABA-ергические нейроны (MSN), селективно экспрессирующие рецепторы D1 или D2, продолжающиеся в виде их топографических проекций. GABA-ергический MSN, проецирующийся во внешний отдел бледного шара, также называемый стриатопаллидальным "непрямым путем", экспрессирует рецепторы D2 (MSN-2); при этом GABA-ергический MSN, проецирующийся в ретикулярную часть черного вещества и внутренний отдел бледного шара, также называемый стриатонигральным "прямым путем", экспрессирует рецепторы D1 (MSN-1). Уменьшение количества дофамина вследствие потери нейронов приводит к несбалансированной активности двух путей, что приводит к заметному уменьшению на выходе таламической и кортикальной активности и, в конечном итоге, моторным дисфункциям (Gerfen et al, Science (1990) 250: 1429-32; Delong, (1990) Trends in Neuroscience 13: 281-5; Alexander et Crutcher, (1990) Trends in Neuroscience 13: 266-71; и для обзора Poewe et al., Nature Review (2017) vol. 3 article 17013: 1-21).
Наиболее эффективными терапевтическими стратегиями, доступными для пациентов, страдающих болезнью Паркинсона, и направленными на контроль моторных симптомов, в основном являются непрямые и прямые агонисты дофамина. Классическая и общепринятая стандартная схема лечения включает постоянный пероральный прием L-3,4-дигидроксифенилаланина (L-DOPA), который декарбоксилируется в головном мозге с образованием дофамина. Другие подходы заключаются во введении агонистов дофаминовых рецепторов, таких как апоморфин, который действует как на подтипы рецепторов D1, так и на подтипы рецепторов D2, или прамипексол, ропинирол и другие, которые преимущественно направлены на подтипы рецепторов D2. Приемлемое облегчение в отношении моторных симптомов обеспечивается с помощью применения как L-DOPA, так и апоморфина за счет активации ими как подтипов рецепторов D1, так и подтипов рецепторов D2 и целостного уравновешивания непрямых и прямых путей (т.е. агонисты D2 при этом способствуют устранению только дисфункции непрямого пути).
L-DOPA и апоморфин, характеризующиеся структурами, изображенными ниже, в настоящее время представляют собой наиболее эффективные лекарственные средства для лечения PD в клинической практике.

L-DOPA представляет собой пролекарство дофамина и остается наиболее эффективным лекарственным средством при лечении болезни Паркинсона с моторными симптомами. Однако после нескольких лет лечения (т.е. периода "медового месяца") возникают осложнения вследствие присущего заболеванию прогрессирования (т.е. длительной потери дофаминергических нейронов), а также плохого фармакокинетического (РК) профиля L-DOPA. Эти осложнения включают: 1) дискинезию, которая представляет собой патологические непроизвольные движения, возникающие во время оптимального "своевременного эффекта" лекарственного средства; и 2) флуктуации в фазе "выключения", что представляет собой период, в течение которого положительный эффект L-DOPA ослабевает и симптомы снова появляются или ухудшаются (Sprenger and Poewe, CNS Drugs (2013), 27: 259-272).
Прямые агонисты дофаминовых рецепторов способны активировать ауторецепторы дофамина, а также постсинаптические дофаминовые рецепторы, расположенные на срединных шипиковых нейронах MSN-1 и MSN-2. Апоморфин относится к классу агонистов дофамина с 1,2-дигидроксибензольным (катехольным) фрагментом. В сочетании с фенетиламиновым мотивом, катехоламины часто обладают низкой пероральной би о доступностью или не обладают ей, как в случае с апоморфином. Апоморфин применяют в клинической терапии PD, хотя и с использованием непероральной доставки (как правило, посредством периодического подкожного введения или непрерывной парентеральной инфузии с помощью насоса в течение дня). В случае апоморфина исследования на животных показали, что трансдермальная доставка или доставка с помощью имплантатов могут обеспечивать возможные формы введения. Однако при исследовании доставки апоморфина из имплантатов у обезьян (Bibbiani et al., Chase Experimental Neurology (2005), 192: 73-78) было установлено, что в большинстве случаев животные должны были подвергаться лечению с помощью иммунодепрессанта дексаметазона для предупреждения местного раздражения и других осложнений после операции по имплантации. Были тщательно изучены альтернативные стратегии доставки для апоморфиновой терапии при PD, такие как ингаляционные и сублингвальные составы (см., например, Grosset et al., Acta Neurol Scand. (2013), 128:166-171 and Hauser et al., Movement Disorders (2016), Vol.32 (9): 1367-1372). Однако эти меры еще не применяются в клинической практике для лечения PD.
Альтернатива составам для неперорального применения на основе катехоламинов предусматривает применение пролекарства, обеспечивающего маскировку свободных гидроксильных групп катехола, чтобы обеспечить возможность перорального введения. Однако известной проблемой, связанной с разработкой пролекарств для клинического применения, являются трудности, связанные с прогнозированием превращения в исходное соединение в организме людей.
В литературе сообщалось о различных сложноэфирных пролекарстеах на основе катехоламинов, таких как покрытые энтеросолюбильной оболочкой N-пропилнорапорфин (NPA) и монопивалоиловый сложный эфир апоморфина для дуоденальной доставки (см., например, WO 02/100377) и агонист D1-подобного рецептора адроголид, представляющий собой пролекарство, содержащее диацетил, А-86929 (Giardina and Williams; CNS Drug Reviews (2001), Vol.7 (3): 305-316). Адроголид подвергается активному метаболизированию при первичном прохождении через печень в организме человека после перорального введения дозы и вследствие этого обладает низкой пероральной биодоступностью (прибл. 4%). У пациентов с PD адроголид, вводимый внутривенно (IV), обладает противопаркинсонической эффективностью, сравнимой с эффективностью L-DOPA (Giardina and Williams; CNS Drug Reviews (2001), Vol.7 (3): 305-316).
Помимо сложноэфирных пролекарств на основе катехоламинов, альтернативный подход к пролекарствам включает маскирование двух гидроксильных групп катехола в виде соответствующего производного, содержащего метилендиокси, или производного, содержащего диацеталил. Этот принцип получения пролекарства был описан, например, в Campbell et al., Neuropharmacology (1982); 21(10): 953-961 и в US4543256, WO 2009/026934 и WO 2009/026935.
Еще одним предлагаемым подходом для пролекарства на основе катехоламина является образование производного енона, как предлагается, например, в WO 2001/078713 и в Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444. Дополнительные примеры пролекарств на основе катехоламина см., например, в Sozio et al., Exp.Opin. Drug Disc. (2012); 7(5): 385-406.
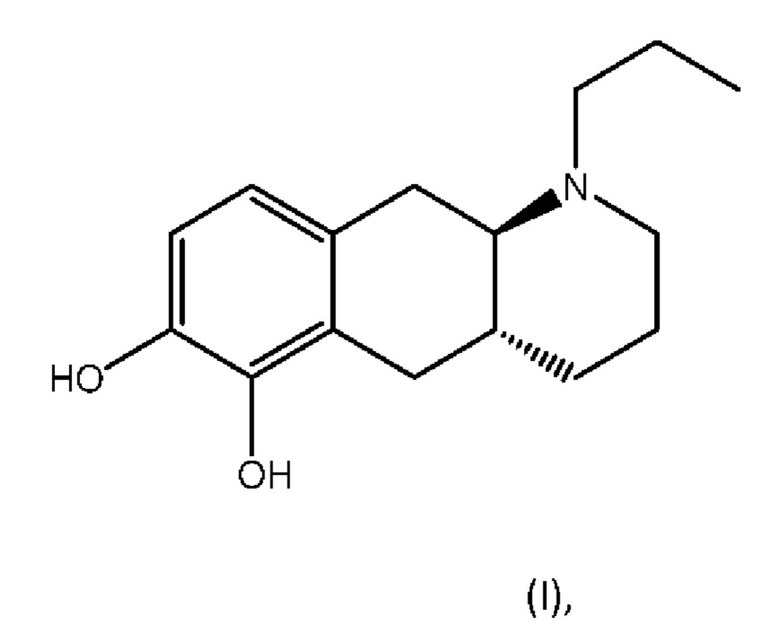
Соединение (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диол, изображенное как соединение (I) ниже, раскрыто в WO 2009/026934. Трансизомер был раскрыт ранее в Liu et al., J. Med. Chem. (2006), 49: 1494-1498 и затем в Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444, включая фармакологические данные, указывающие на то, что соединение обладает низкой пероральной биодоступностью у крыс. Рацемат впервые был раскрыт в Cannon et al., J. Heterocyclic Chem. (1980); 17:1633-1636.

Соединение (I) является агонистом дофаминовых рецепторов со смешанной активностью в отношении D1 и D2. Три пролекарственных производных соединения (I) известны из уровня техники.
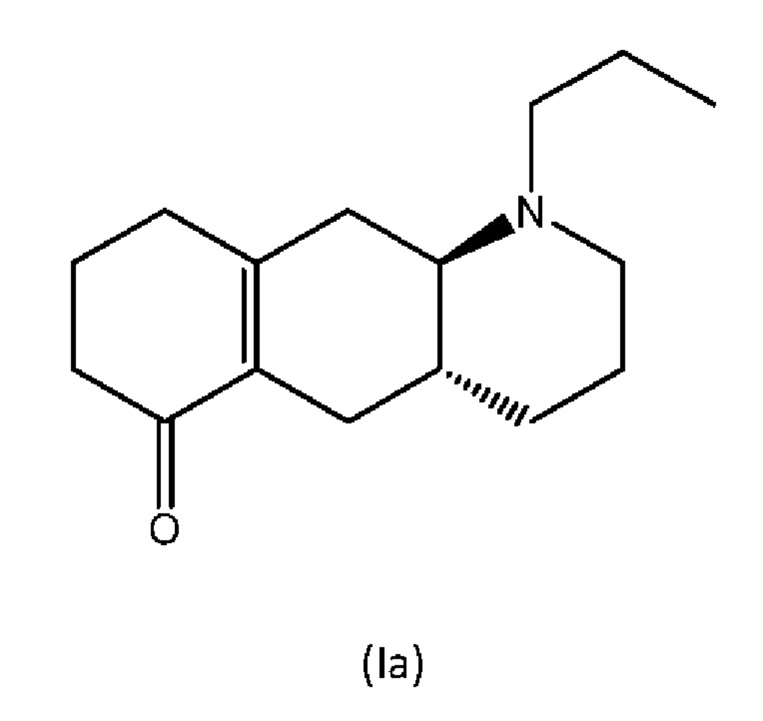
В Liu et al., J. Med. Chem. (2006), 49: 1494-1498 и Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444 раскрыто производное енона формулы (la), изображенной ниже, которое, как было показано, превращается в активное соединение (I) в организме крыс.
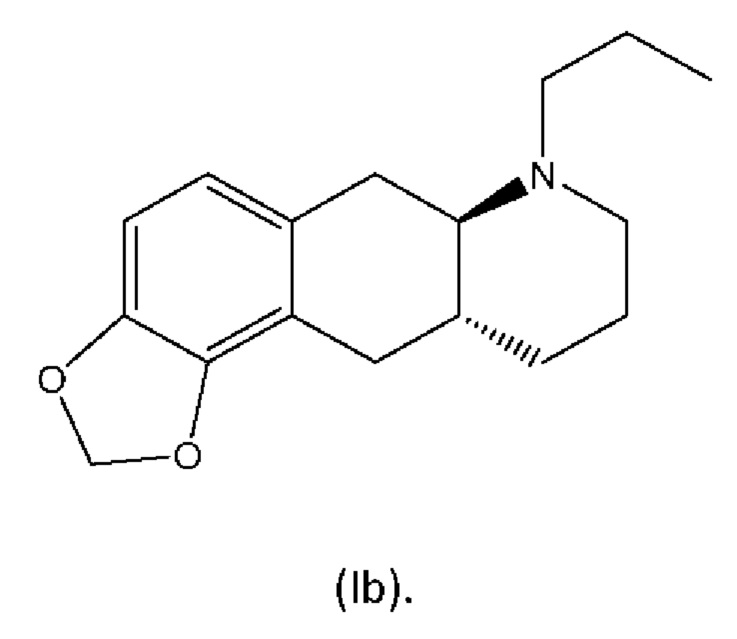
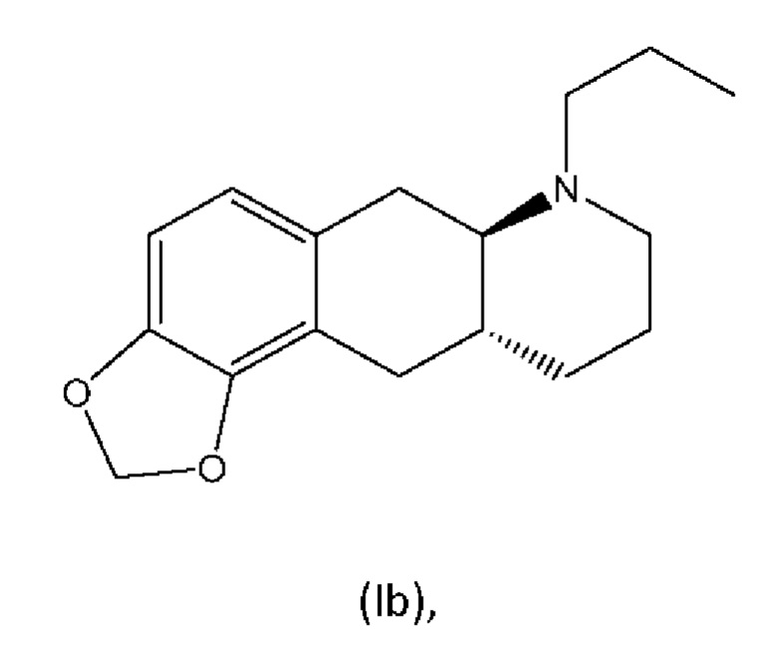
В WO 2009/026934 и WO 2009/026935 раскрыты два типа производных пролекарств на основе соединения (I), включая соединение (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-g]хинолин с нижеуказанной формулой (Ib):
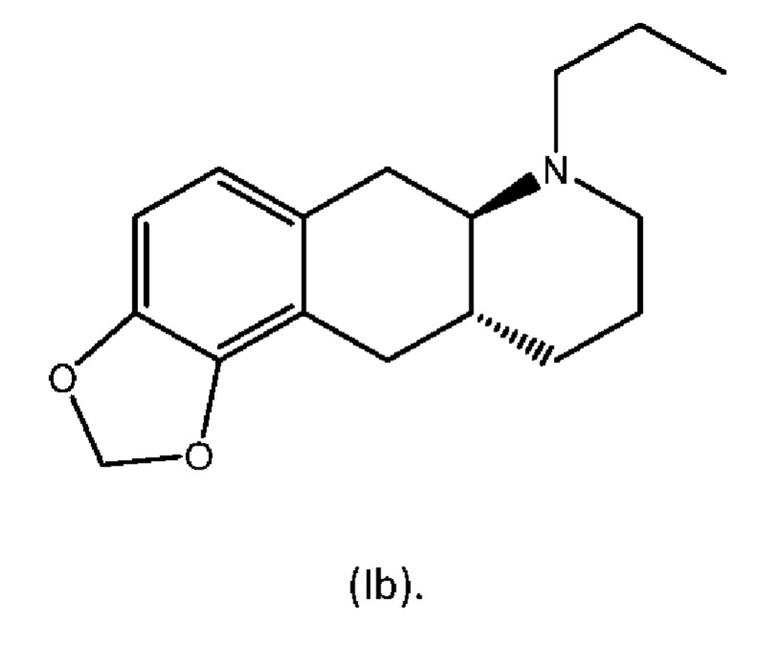
Превращение соединения (Ib) в соединение (I) в гепатоцитах крысы и человека было продемонстрировано в WO 2010/097092. Кроме того, фармакология in vivo соединений (Ia) и (Ib), а также активного "исходного соединения" (I) была исследована на различных животных моделях, соответствующих болезни Паркинсона (WO 2010/097092). Было обнаружено, что как соединение (I), так и соединения (Ia) и (Ib) являются эффективными, что указывает на то, что соединения (Iа) и (Ib) превращаются in vivo в соединение (I). Сообщалось, что все три соединения характеризовались продолжительностью действия, которая была больше, чем наблюдаемая для L-dopa и апоморфина.
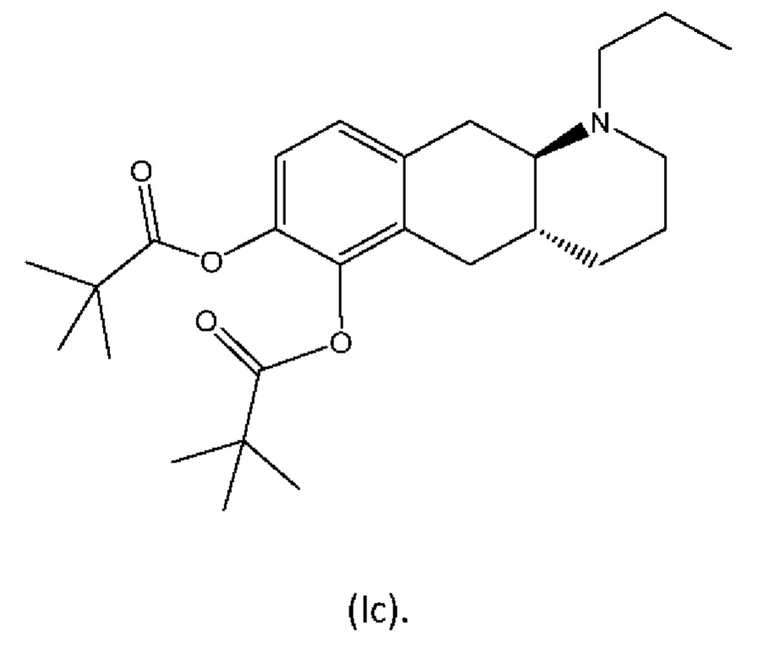
Другое пролекарство соединения (I), раскрытое в WO 2009/026934 и WO 2009/026935, является традиционным пролекарством, представляющим собой сложный эфир, согласно формуле (Ic):
Несмотря на долголетний интерес в данной области, очевидно, что по-прежнему существует неудовлетворенная потребность в разработке эффективных, хорошо переносимых и активных при применении лекарственных средств для лечения PD.
Следовательно, также существует необходимость в способе изготовления таких лекарственных средств, в частности, в способах, подходящих для крупномасштабного производства, обеспечивающего высокий выход продуктов.
В WO 2009/026934 раскрыт способ получения соединения (I) и способ получения соединения (Ib) из соединения (I). Эти способы включают множество стадий и в них используется хиральная хроматография для получения отдельных энантиомеров и, таким образом, они не являются оптимальными для крупномасштабного производства.
Таким образом, все еще существует необходимость в улучшенных способах для крупномасштабного производства соединения (I) и (Ib).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения разработали новый способ изготовления (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола (соединение (I)) и (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-g]хинолина (соединение (Ib)). Заявленный способ изготовления соединения (I) предлагает несколько преимуществ по сравнению со способом, ранее описанным в WO 2009/026934, включая 1) короткий путь синтеза, 2) улучшенный общий выход соединения (I), 3) применение разделения с помощью диастереоизомерных солей вместо разделения с помощью сверхкритической жидкостной хроматографии (SFC), последняя является неэкономичной и не подходит для крупномасштабного производства, и 4) разделение на ранней стадии пути синтеза вместо разделения на поздней стадии, как описано в WO 2009/026934, что снижает количество необходимых реагентов/растворителей и количество образующихся отходов.
Один аспект настоящего изобретения относится к новому способу изготовления (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола с нижеуказанной формулой (I) и его солей,
из соединения (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-g]хинолин с нижеуказанной формулой (Ib),
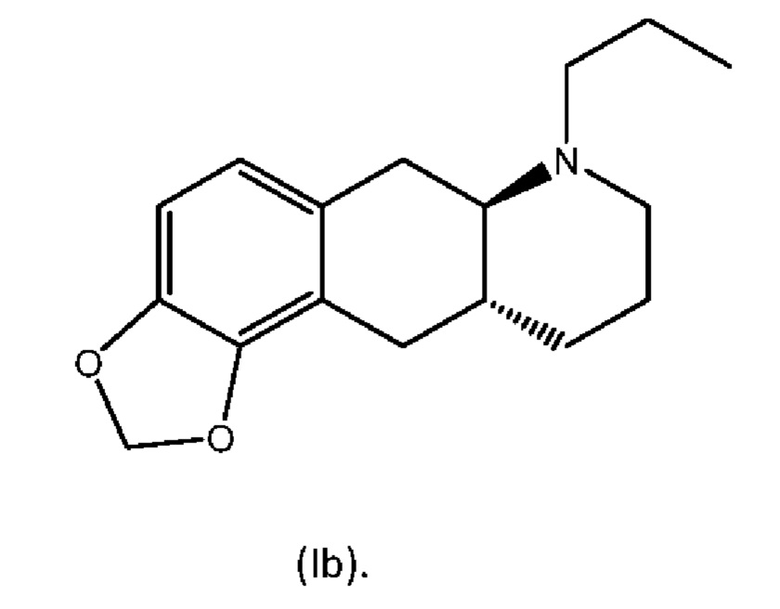
Другой аспект настоящего изобретения также обеспечивает новый способ изготовления (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[13]диоксоло[4':5':5,6]бензо[1,2-g]хинолина (соединение (Ib) и его солей.
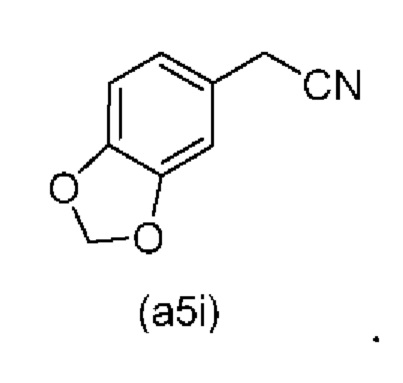
Дополнительные отдельные аспекты относятся к новым промежуточным соединениям способа. Таким образом, один аспект настоящего изобретения обеспечивает соединение формулы (А2) ниже или его соль.
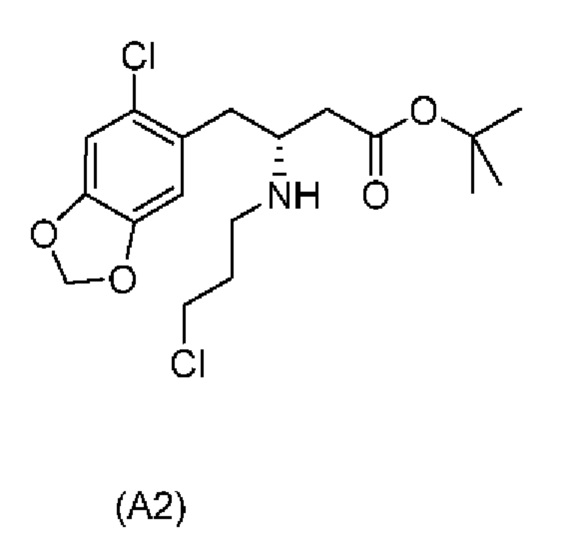
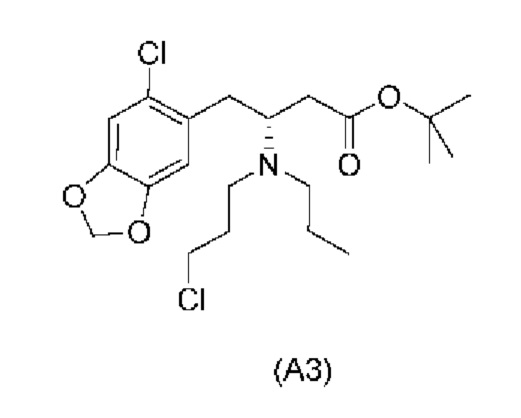
Другой аспект настоящего изобретения обеспечивает соединение формулы (A3) ниже или его соль.
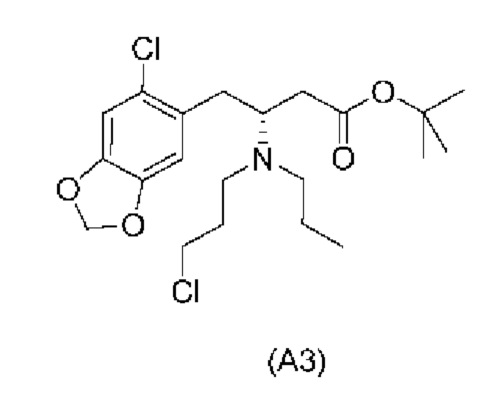
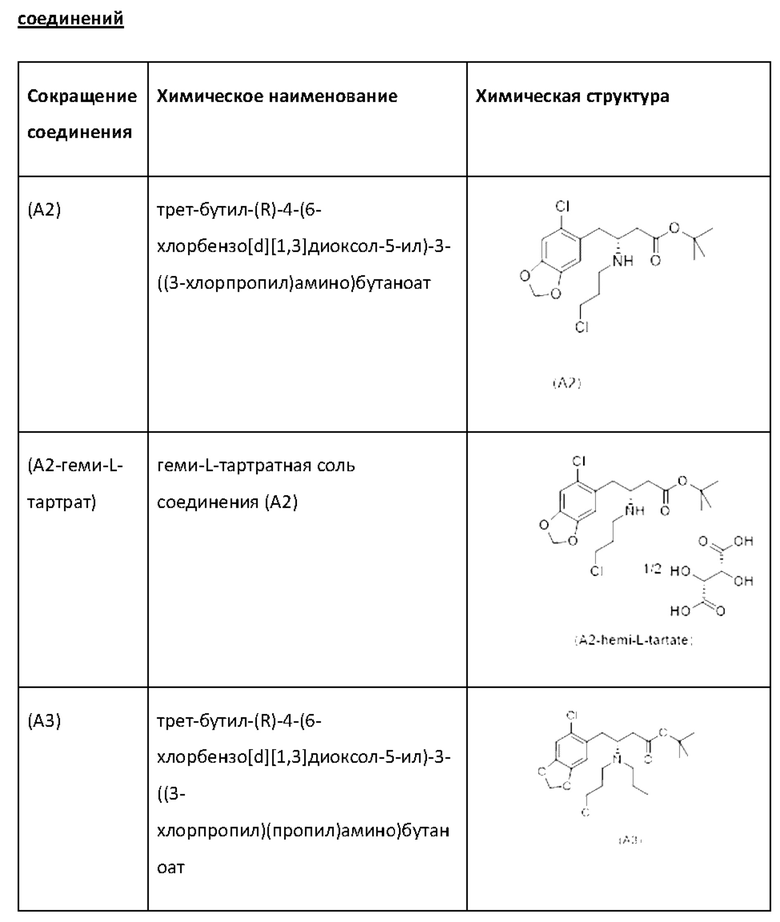
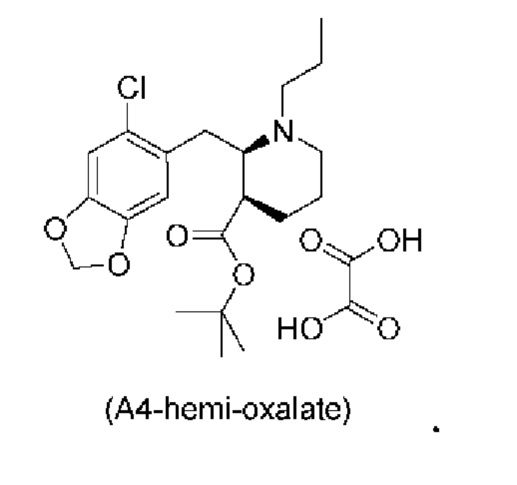
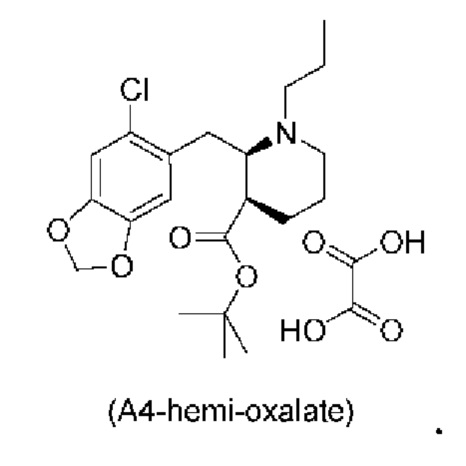
Еще один аспект настоящего изобретения обеспечивает соединение формулы (А4) ниже или его соль.
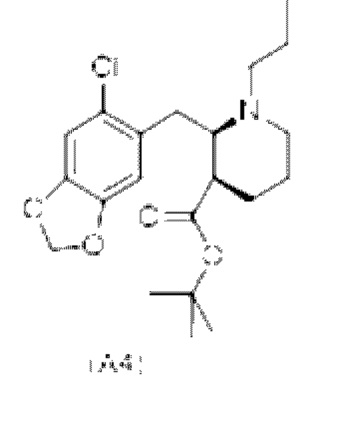
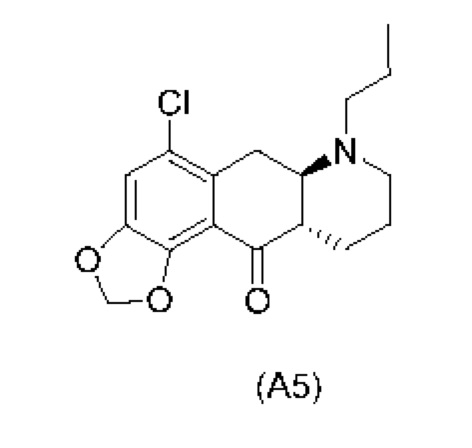
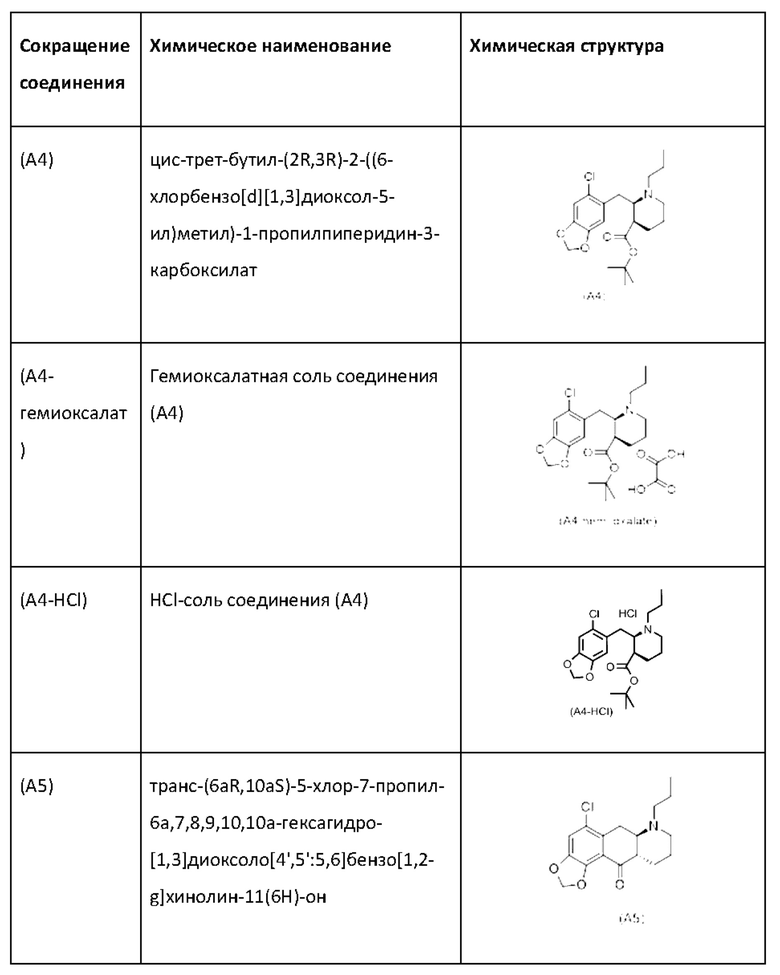
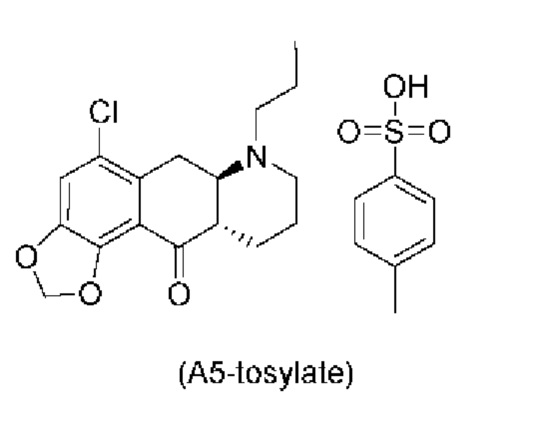
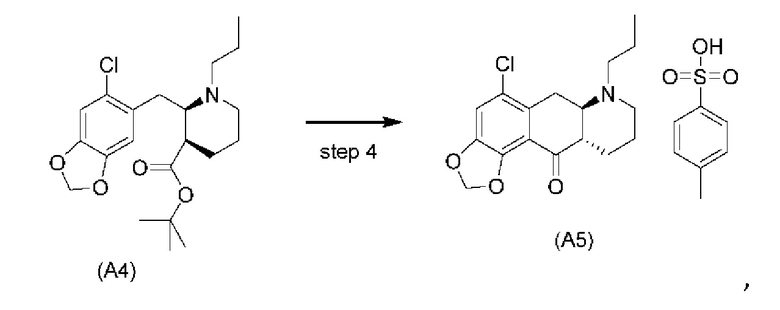
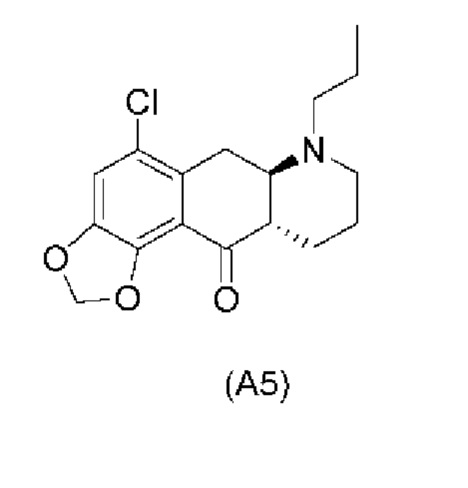
Еще один аспект настоящего изобретения обеспечивает соединение формулы (А5) ниже или его соль.
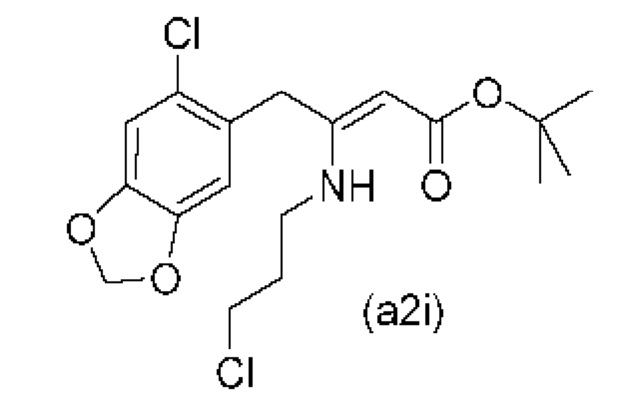
Еще один вариант осуществления настоящего изобретения обеспечивает соединение формулы (a2i) ниже или его соль.
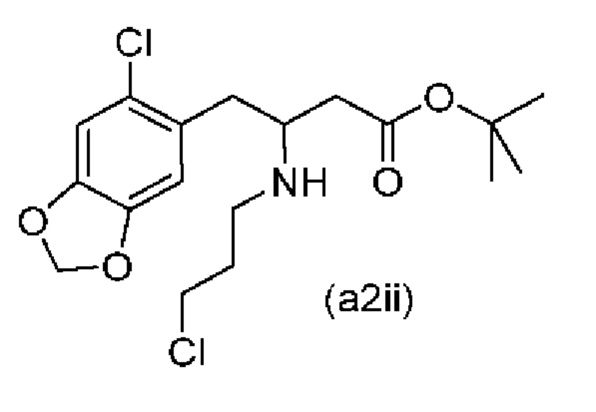
Еще один аспект настоящего изобретения обеспечивает соединение формулы (a2ii) ниже или его соль.
ОПРЕДЕЛЕНИЯ
Ссылки на соединения
Ссылки на соединение (I), соединение (Ib), соединение (А1), соединение (А2), соединение (A3), соединение (А4) или соединение (А5) включают соединения в растворе и твердые формы соединений, включая свободное вещество (например, цвиттер-ион) указанных соединений, соли указанных соединений, такие как соли присоединения кислоты или соли присоединения основания, и полиморфные и аморфные формы соединений по настоящему изобретению и его солей. Кроме того, указанные соединения и их соли могут потенциально существовать в несольватированных, а также в сольватированных формах с растворителями, такими как вода, этанол и т.п.
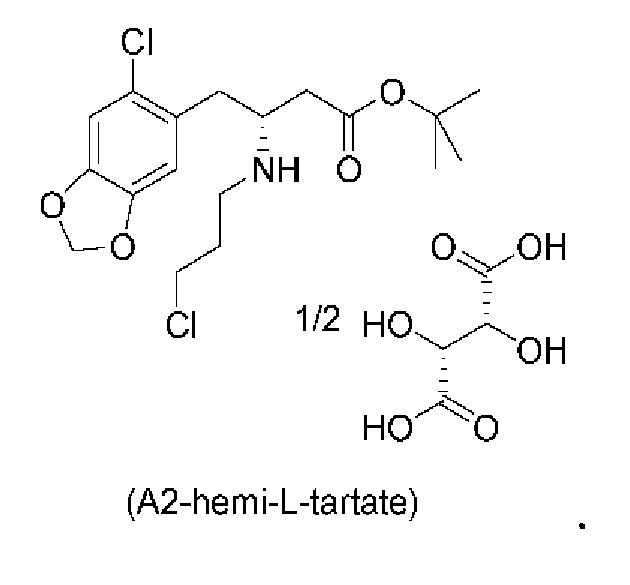
Иногда для соединения указана конкретная солевая форма, такая как, например, (А2-геми-L-тартрат), который обозначает геми-L-тартратную соль соединения (А2). Ссылка на соединение соединение (I), соединение (Ib), соединение (А1), соединение (А2), соединение (A3), соединение (А4) или соединение (А5) в виде "свободного основания" в данном контексте предназначена для обозначения того, что указанное соединение находится в несолевой форме.
Фармацевтически приемлемые соли
В контексте настоящего изобретения подразумевается, что фармацевтически приемлемые соли обозначают нетоксичные, т.е. физиологически приемлемые соли.
Термин "фармацевтически приемлемые соли" включает фармацевтически приемлемые соли присоединения кислоты, которые представляют собой соли, образованные с неорганическими и/или органическими кислотами при атоме азота в исходной молекуле. Указанные кислоты могут быть выбраны из, например, хлористоводородной кислоты, бромистоводородной кислоты, фосфорной кислоты, азотистой кислоты, серной кислоты, бензойной кислоты, лимонной кислоты, глюконовой кислоты, молочной кислоты, малеиновой кислоты, янтарной кислоты, винной кислоты, уксусной кислоты, пропионовой кислоты, щавелевой кислоты, малоновой кислоты, фумаровой кислоты, глутаминовой кислоты, пироглутаминовой кислоты, салициловой кислоты, гентизиновой кислоты, сахарина и сульфоновых кислот, таких как метансульфоновая кислота, этансульфоновая кислота, толуолсульфоновая кислота, нафталин-2-сульфоновая кислота, 2-гидроксиэтансульфоновая кислота и бензолсульфоновая кислота.
Дополнительные примеры кислот и оснований, применимых для образования фармацевтически приемлемых солей, можно найти, например, в Stahl and Wermuth (Eds) "Handbook of Pharmaceutical salts. Properties, selection, and use", Wiley-VCH, 2008.
Соединения (Ib), (A1), (A2), (A3), (A4) и (A5) можно использовать в качестве промежуточных соединений для изготовления соединения (I) или его фармацевтически приемлемой соли. Таким образом, солевая форма соединений (Ib), (A1), (А2), (A3), (А4) и (А5) не ограничена их фармацевтически приемлемыми солями. Тем не менее, фармацевтически приемлемые соли соединений (Ib), (A1), (А2), (A3), (А4) и (А5) также можно предпочтительно использовать при изготовлении соединения (I). Таким образом, в варианте осуществления настоящего изобретения соль соединения (Ib), (A1), (А2), (A3), (А4) и/или (А5) является фармацевтически приемлемой солью.
Изготовление путем химического получения
В контексте настоящего изобретения соединение, "изготовленное путем химического получения", обозначает то, что указанное соединение было получено с помощью химического способа ex vivo, такого как без ограничения один из способов, описанных в экспериментальном разделе в данном документе. Выражение "изготовление" и "изготовление путем химического получения" используют взаимозаменяемо.
Таким образом, в варианте осуществления настоящего изобретения соединение (I) изготавливают химическим способом ex vivo.
В дополнительном варианте осуществления настоящего изобретения соединение (Ib) изготавливают химическим способом ex vivo.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому способу изготовления (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола (соединение (I)), осуществляемому посредством соединения (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[1,3]диоксоло[4',5':5,6]бензо-[1,2-g]хинолин (соединение (Ib). Настоящее изобретение также относится к новому способу изготовления соединения (Ib).
Соединение (Ib) является пролекарством соединения (I), которое является двойным агонистом дофамина со смешанной активностью в отношении D1 и D2, пригодным при лечении нейродегенеративных заболеваний и нарушений, таких как болезнь Паркинсона. В WO 2009/026934 раскрыт способ изготовления соединения (I) и дополнительного способа изготовления соединения (Ib) из соединения (I).
Авторы настоящего изобретения обнаружили новый и улучшенный способ изготовления обоих соединений, причем соединение (Ib) используют в качестве промежуточного соединения при изготовлении соединения (I).
Общий способ вкратце показан на схеме 1 ниже.
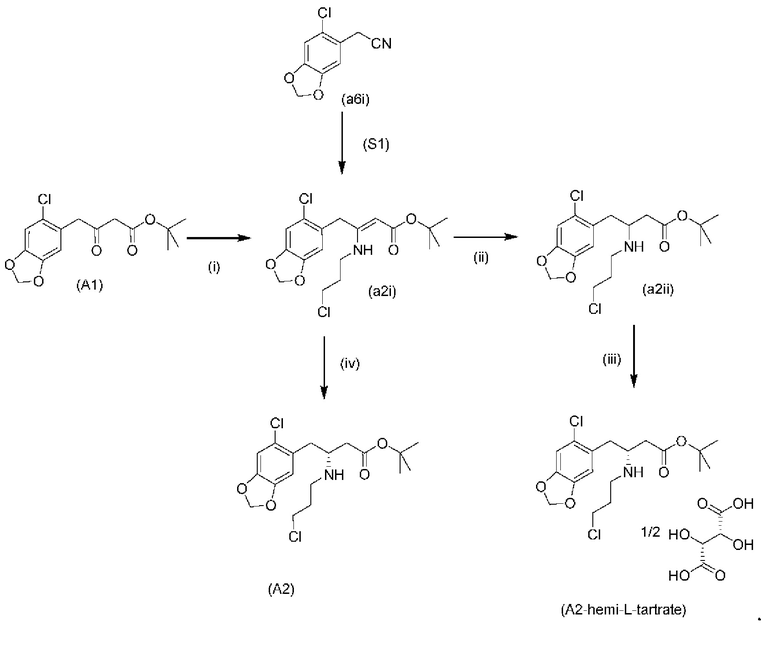
Исходный материал (А1): трет-бутил-4-(6-хлорбензо[d][1,3]диоксол-5-ил)-3-оксобутаноат можно получить с использованием известных способов, таких как описанные Brückner и соавторами в Synthesis 2008, 14: 2229-2246, или как описано ниже.
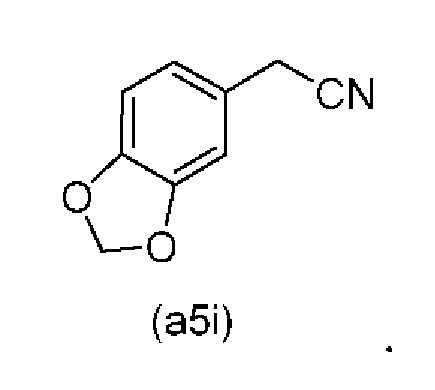
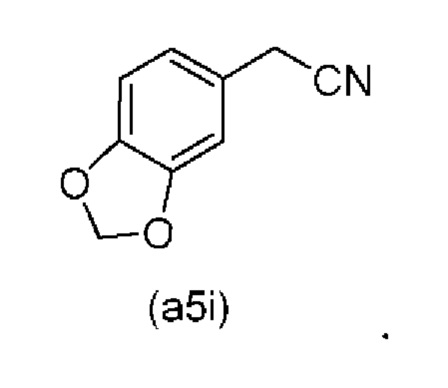
Исходный материал (a5i): 2-(бензо[d][1,3]диоксол-5-ил)ацетонитрил является коммерчески доступным.
Схема 1. Общий способ
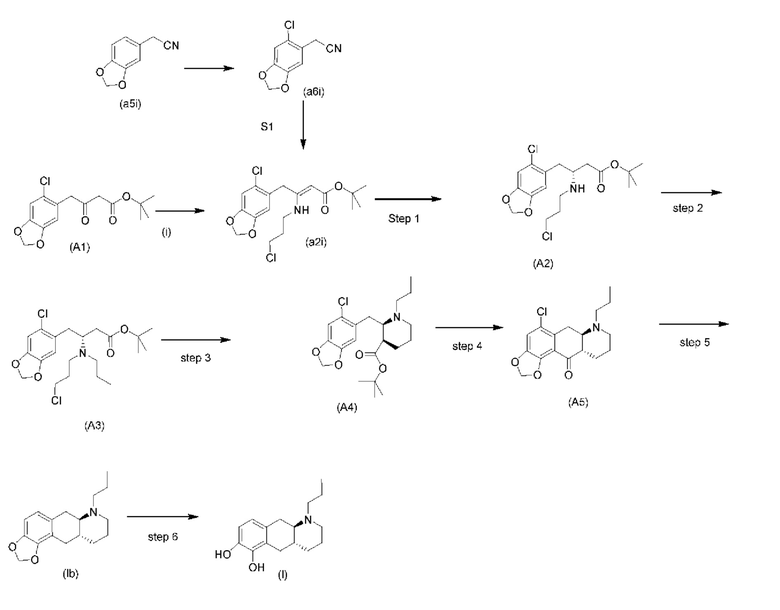
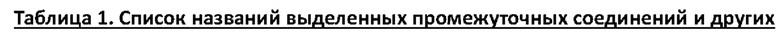
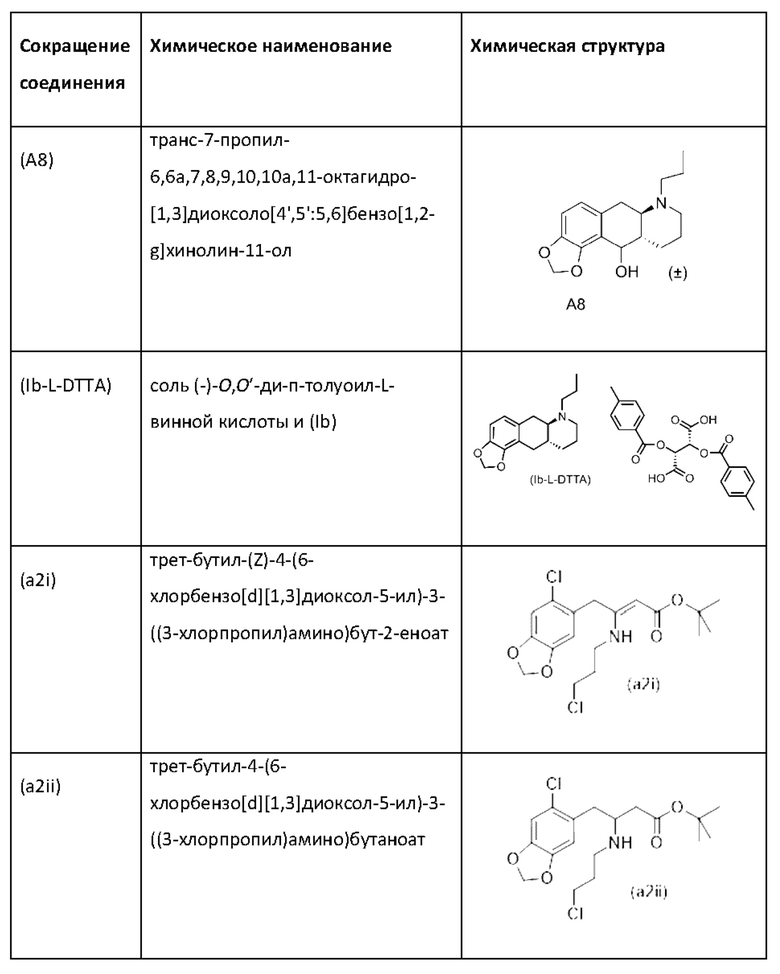

стадия 0)
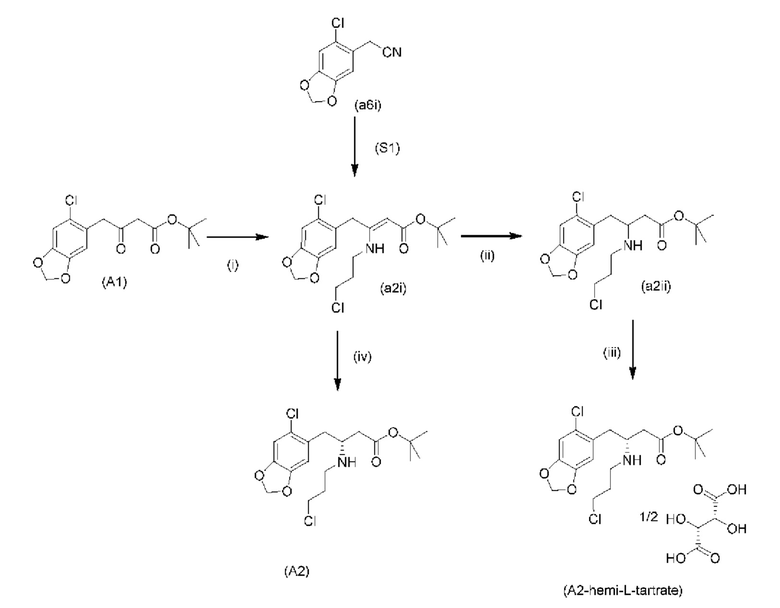
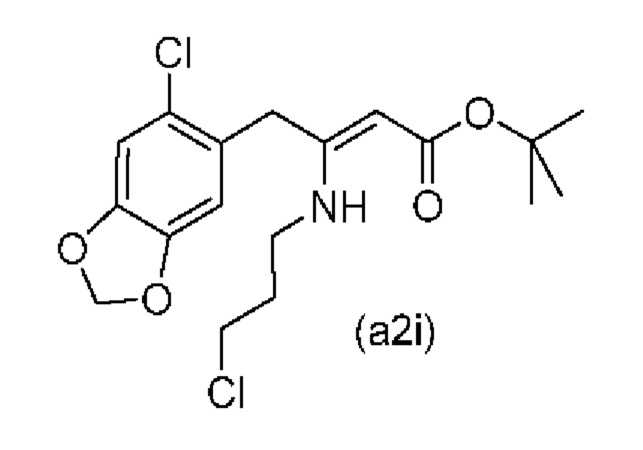
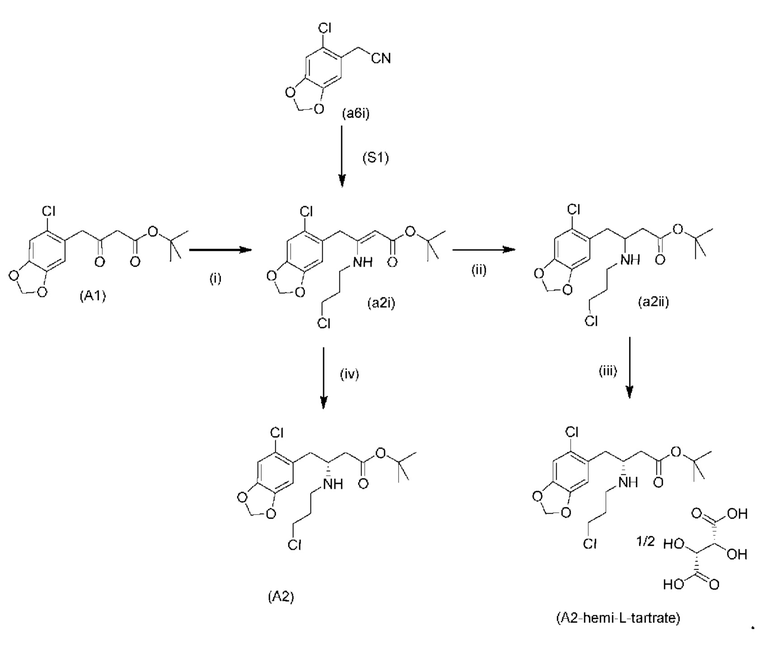
На стадии 0) образуется енаминовое промежуточное соединение (a2i). Промежуточное соединение a2i может образовываться с использованием двух альтернативных стадий с различными исходными соединениями.
Подстадия (i) стадии 0, где соединение (А1), представляющее собой сложный кетоэфир (трет-бутил-4-(6-хлорбензо[d][1,3]диоксол-5-ил)-3-оксобутаноат), превращается в енаминовое промежуточное соединение (a2i) с 3-хлорпропан-1-амином in situ с использованием каталитических количеств хлорида цинка.
Подстадия S1 стадии 0 является альтернативной для подстадии (i) выше, где соединение (a6i) подвергают реакции Блейза с трет-бутил-2-бромацетатом и цинком, а затем обрабатывают уксусной кислотой, после чего гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i). На подстадии S1 соединение (a2i) непосредственно и легко образуется из соединения (a6i).
В одном варианте осуществления настоящего изобретения подстадия S1 включает выделение соединения (a2i).
Коммерчески доступное соединение (a5i) можно хлорировать с использованием SO2Cl2 с получением соединения (a6i) с высоким выходом. Таким образом, в конкретном варианте осуществления настоящего изобретения соединение (a5i) хлорируют с использованием SO2Cl2 с получением соединения (a6i). В еще более конкретном варианте осуществления настоящего изобретения соединение (a5i) хлорируют с использованием SO2Cl2 с получением соединения (a6i) и затем используют подстадию S1 стадии 0 для получения соединения (a2i).
Стадия 0' обеспечивает альтернативный путь к получению исходного материала в виде соединения (А1) из соединения (a6i).
Коммерчески доступное соединение (a5i) можно хлорировать с использованием SO2Cl2 с получением соединения (a6i) с высоким выходом. Затем соединение (a6i) можно превращать либо в соединение (А1), либо в (a2i) в зависимости от условий реакции.
Для образования соединения (А1) соединение (a6i) подвергают реакции Блейза стрет-бутил-2-бромацетатом и цинком и после водного кислотного гидролиза получают соединение (А1).
В одном варианте осуществления настоящего изобретения используют стадию 0' для получения соединения (А1), затем используют подстадию (i) стадии 0 для образования соединения (a2i).
Стадия 11
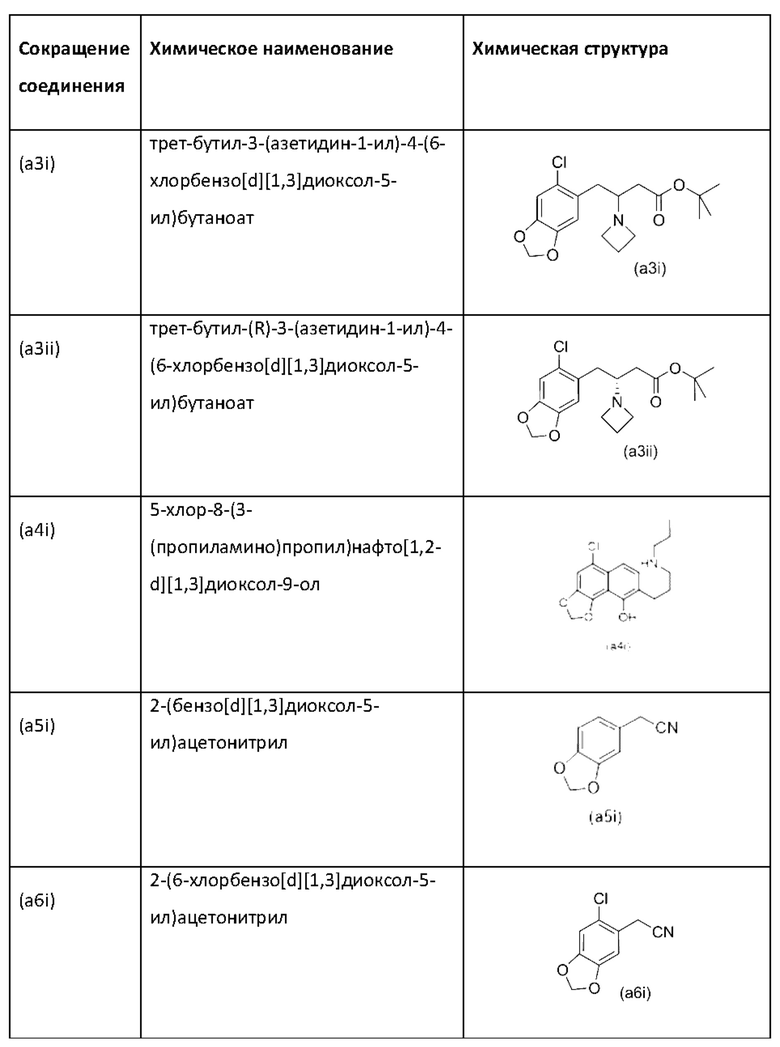
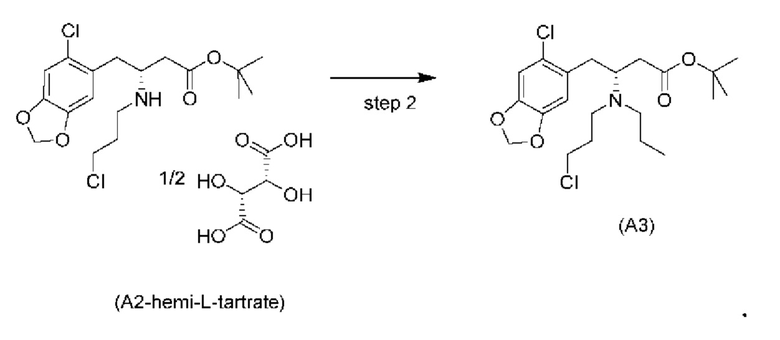
На стадии 1) соединение (a2i) превращается в необходимое энантиомерное соединение (А2) или соединение (А2-геми-L-тартрат) двумя альтернативными путями.
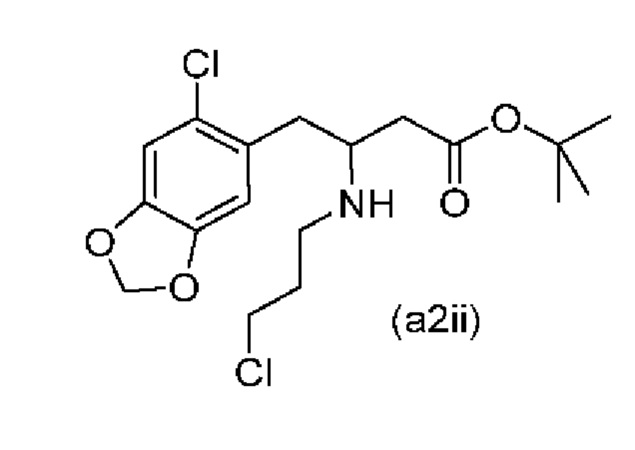
Обычно присутствие функциональных групп первичного алкилхлорида и амина в одной молекуле, как ожидается, способствует алкилированию амина, который в данном случае будет приводить к образованию азетидинов. Однако, неожиданно, соединения (a2ii) и (А2), как было обнаружено, характеризуются хорошей стабильностью несмотря на присутствие функциональных групп первичного алкилхлорида и амина в одной молекуле, поскольку они не образуют легко соответствующие азетидины (a3i) или (a3ii) соответственно (см. ниже), или самоконденсируются.
Стадия 1, подстадия (ii), затем подстадия (iii)
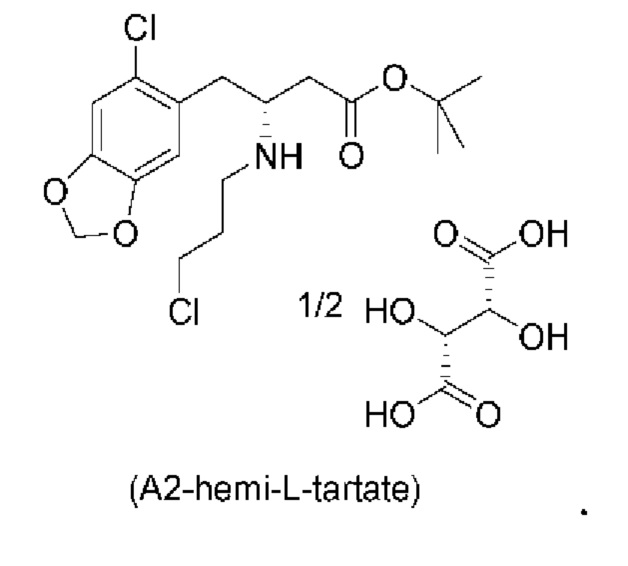
Кроме того, авторы настоящего изобретения неожиданно обнаружили, что неочищенное соединение (a2ii) может быть разделено на подстадии (iii) посредством образования диастереоизомерных солей с высоким выходом с использованием L-винной кислоты в растворителе, например, растворителе, выбранном из МеОН, EtOH и их водных смесей, с получением выделенного амина (А2) в виде геми-L-тартратной соли (А2-геми-L-тартрат) с высоким энантиомерным избытком, составляющим выше 95% (например, >99,5%), что также приводит к обеспечению высокого выхода. Таким образом, в одном варианте осуществления настоящего изобретения используют подстадию (ii) для получения соединения (a2i), и затем соединение (a2i) используют на подстадии (iii) для получения (А2-геми-L-тартрат).
На подстадии (ii) енаминовое промежуточное соединение (a2i) восстанавливают с помощью, например, цианоборгидрида натрия (NaBH3CN), триацетоксиборгидрида натрия (STAB), 5-этил-2-метилпиридинборана (РЕМВ) или NaBH4 с получением неочищенного (a2ii). В конкретном варианте осуществления настоящего изобретения подстадию (ii) осуществляют с использованием цианоборгидрида натрия.
В качестве альтернативы на подстадии (ii) стадии 1 восстановление соединения (a2i) до соединения (a2ii) можно осуществлять с использованием платинового катализатора (предпочтительно платины на углероде) в присутствии водорода в подходящем растворителе, например Me-THF.
В одном варианте осуществления настоящего изобретения подстадию (ii) осуществляют с использованием платинового катализатора при температуре от приблизительно 20°С до приблизительно 100°С, такой как от приблизительно 50°С до приблизительно 80°С, такой как от приблизительно 55°С до приблизительно 65°С, такой как приблизительно 57°С, или приблизительно 58°С, или приблизительно 59°С, или приблизительно 60°С, или приблизительно 61°С, или приблизительно 62°С, или приблизительно 63°С.
В одном варианте осуществления настоящего изобретения подстадию (ii) осуществляют с использованием платинового катализатора при давлении от приблизительно 2 до приблизительно 10 бар, таком как от приблизительно 2 бар до приблизительно 6 бар, таком как от приблизительно 3 бар до приблизительно 5 бар, таком как приблизительно 4 бар.
В одном варианте осуществления настоящего изобретения подстадию (ii) осуществляют с использованием платинового катализатора при температуре от приблизительно 50°С до приблизительно 80°С и при давлении от приблизительно 2 бар до приблизительно 6 бар.
В более конкретном варианте осуществления настоящего изобретения подстадию (ii) осуществляют с использованием платинового катализатора при температуре приблизительно 60°С и при давлении приблизительно 4 бар.
В одном варианте осуществления настоящего изобретения соединение (a2i), полученное на подстадии (i) стадии 0 выше, затем восстанавливают в том же резервуаре с использованием, например, NaBH2CN, триацетоксиборгидрида натрия (STAB), 5-этил-2-метилпиридинборана (РЕМВ) или NaBH4 с получением неочищенного (a2ii).
На подстадии (iii) соединение (a2ii) выделяют с использованием L-винной кислоты в растворителе, например, растворителе, выбранном из МеОН, EtOH и их водных смесей, с получением выделенного амина (А2) в виде геми-L-тартратной соли (А2-геми-L-тартрат).
В одном варианте осуществления настоящего изобретения соединение (a2i), полученное на подстадии (i) стадии 0 выше, затем восстанавливают в том же резервуаре с использованием, например, NaBH3CN, триацетоксиборгидрида натрия (STAB), 5-этил-2-метилпиридинборана (РЕМВ) или NaBH4 с получением неочищенного соединения (a2ii), и полученное неочищенное соединение (a2ii) затем разделяют на подстадии (iii) с получением соединения (А2-геми-L-тартрат).
В еще одном более конкретном варианте осуществления настоящего изобретения соединение (a2i), полученное на подстадии (i) стадии 0 выше, затем восстанавливают в том же резервуаре с использованием цианоборгидрида натрия с получением неочищенного соединения (a2ii), и полученное неочищенное соединение (a2ii) затем разделяют на подстадии (iii) с получением соединения (А2-геми-L-тартрат).
В качестве альтернативы стадии 1, подстадии (ii), затем подстадии (iii), как описано выше, можно использовать стадию 1, подстадию (iv) с получением соединения (А2).
Восстановление соединения (a2i) с получением соединения (А2) на подстадии (iv) стадии 1 можно осуществлять с использованием хирального катализатора, содержащего родий, иридий либо рутений, в присутствии газообразного водорода и подходящего растворителя.
В частности, асимметричную гидрогенизацию соединения (a2i) на подстадии (iv) стадии
1 можно проводить с использованием катализатора, образованного из Josiphos SL-J002-2 (cas №277306-29-3) и тетрафторбората бис(2,5-норборнадиен)родия(1) (cas №36620-11-8), в присутствии водорода и 2,2,2-трифторэтанола в качестве растворителя с получением соединения (А2) с энантиомерным избытком 96% и выходом 93% (на основе анализа с использованием LC-MS).
На схеме 2 ниже показаны более подробно подстадии стадии 0) и стадии 1).
Схема 2. Подробный обзор подстадии на стадии 0) и стадии 1)
В одном варианте осуществления настоящего изобретения используют подстадию S1 стадии 0 с получением соединения (a2i), затем используют стадию 1.
В другом более конкретном варианте осуществления настоящего изобретения за подстадией S1 стадии 0 следуют подстадия (ii) и подстадия (iii) стадии 1 с получением соединения (А2-геми-L-тартрат).
В более конкретном варианте осуществления настоящего изобретения за подстадией S1 стадии 0 следует подстадия (iv) стадии 1 с получением соединения (А2).
В одном варианте осуществления настоящего изобретения стадию 0' используют для получения соединения (А1).
Таким образом, в более конкретном варианте осуществления настоящего изобретения за стадией 0' следует подстадия (i) стадии 0 с получением соединения (a2i), а за подстадией (i) стадии 0 следует подстадия (iv) стадии 1 с получением соединения (А2).
В конкретном варианте осуществления настоящего изобретения стадию 0' используют для получения соединения (А1), и за ней следует подстадия (i) стадии 0 с получением соединения (a2i), затем за подстадией (i) стадии 0 следуют подстадия (ii) и подстадия (iii) с получением соединения (А2-геми-L-тартрат).
В другом конкретном варианте осуществления настоящего изобретения за стадией 0' следует подстадия (i) стадии 0 с получением соединения (a2i), а за подстадией (i) стадии 0 следует подстадия (iv) стадии 1 с получением соединения (А2).
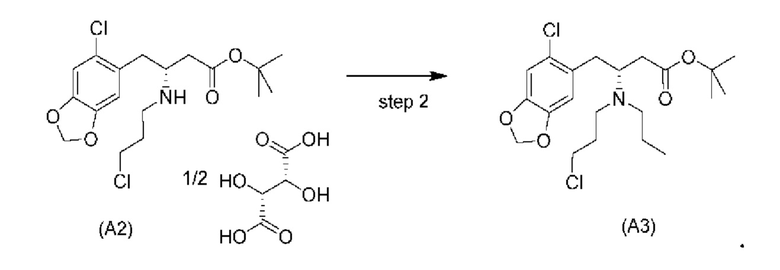
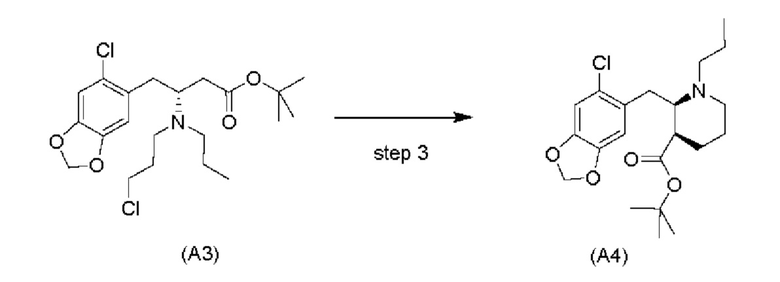
Стадия 2)
На стадии 2), как показано ниже на схеме 3, соединение (А2) или его соль подвергается дополнительному восстановительному аминированию с пропаналем с использованием восстанавливающего средства, такого как, например, NaBH3CN, триацетоксиборгидрид натрия (STAB), 5-этил-2-метилпиридинборан (РЕМВ), или платинового катализатора на носителе, таком как углерод, и водорода; в растворителе, выбранном из, например, тетрагидрофурана (THF), изопропанола (IPA) или МеОН; с получением соединения (A3).
Схема 3. Стадия 2)
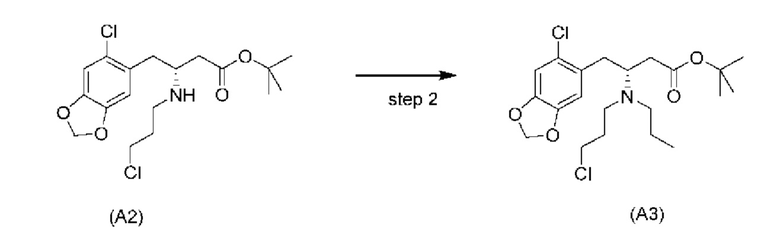
В конкретном варианте осуществления настоящего изобретения соединение (А2-геми-L-тартрат) используют на стадии 2, как описано в настоящем документе. Таким образом, в более конкретном варианте осуществления настоящего изобретения подстадию (ii) стадии 1, затем подстадию (iii) стадии 1, используют для получения соединения (А2-геми-L-тартрат), которое затем используют на стадии 2.
В еще более конкретном варианте осуществления настоящего изобретения за подстадией S1 стадии 0 следует подстадия (ii) стадии 1, за которой следует подстадия (iii) стадии 1 с получением соединения (А2-геми-L-тартрат), которое затем используют на стадии 2.
В конкретном варианте осуществления триацетоксиборгидрид натрия (STAB) используют в качестве восстанавливающего средства на стадии 2.
В одном варианте осуществления настоящего изобретения стадию 2 проводят в растворителе, выбранном из группы, состоящей из тетрагидрофурана (THF), изопропанола (IPA) и МеОН. В более конкретном варианте осуществления стадию 2 проводят в THF.
Стадия 3)
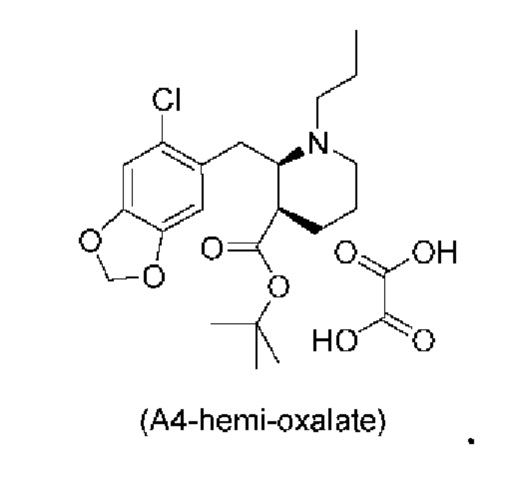
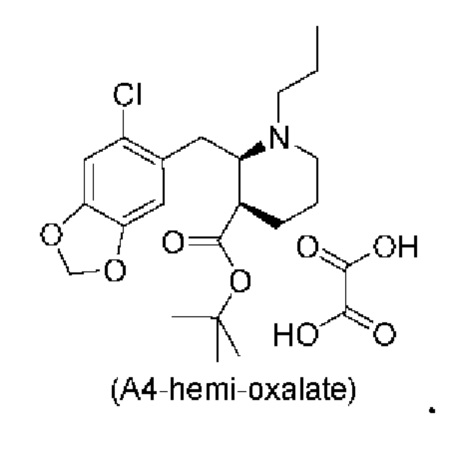
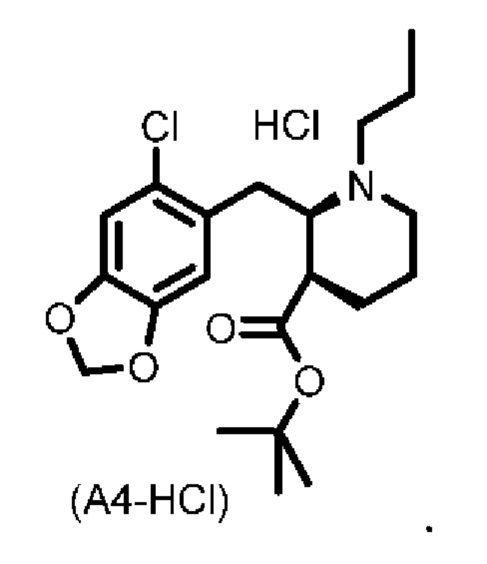
Стадия 3), как показано ниже на схеме 4, обеспечивает циклизацию соединения (A3) в основных условиях с получением соединения (А4), которое можно необязательно выделять в виде гемиоксалатной соли (А4-геми-оксалат), гидрохлоридной соли (A4-HCl) или гидробромидной соли (А4-HBr).
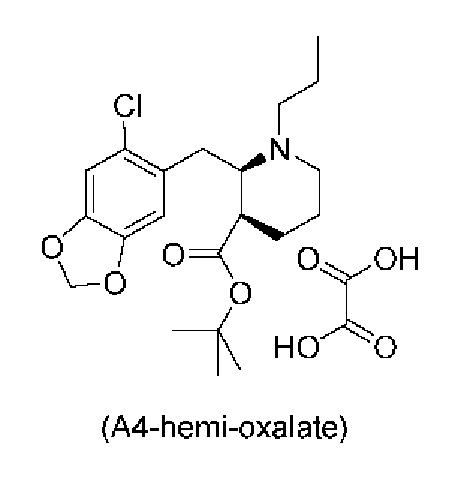
Реакцию осуществляют в присутствии сильного основания, предпочтительно бис(триметилсилил)амида натрия (NaHMDS).
В одном варианте осуществления настоящего изобретения стадию 3 осуществляют с использованием сильного основания, выбранного из группы, состоящей из бис(триметилсилил)амида натрия (NaHMDS), диизопропиламида лития (LDA), бис(триметилсилил)амида калия (KHMDS) и бис(триметилсилил)амида лития (LHMDS). В конкретном варианте осуществления настоящего изобретения стадию 3 осуществляют с использованием бис(триметилсилил)амида натрия (NaHMDS).
Подходящие растворители для стадии 3, например, представляют собой растворитель, выбранный из группы, состоящей из толуола, THF и их смеси. В конкретном варианте осуществления настоящего изобретения смесь толуола и THF используют в качестве растворителя.
Схема 4. Стадия 3)
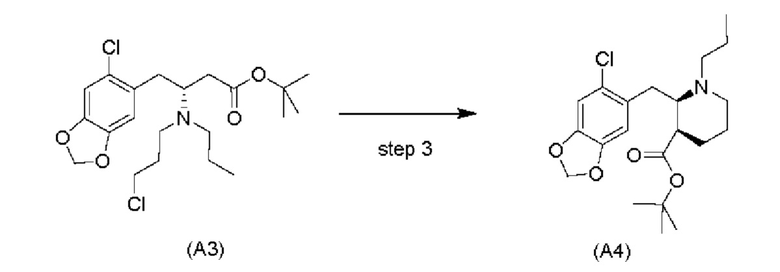
Обычно алкилирование сложных эфиров имеет недостаток, заключающийся в самоконденсации (из-за реакции Кляйзена). Уходящие группы, такие как амины, в бета-положении, как ожидается, легко отщепляются, оставляя акрилатную систему. Однако авторы настоящего изобретения обнаружили, что циклизация соединения (A3) является неожиданно чистой с получением только небольшого количества продуктов самоконденсации и/или продуктов отщепления, даже несмотря на то, что реакция проходит при приемлемой высокой температуре, составляющей -10°C. Обычно образование енолятов из сложных эфиров проходит при криогенных температурах (например, -78°С), чтобы избежать побочных реакций, например, самоконденсации (см., например, Fan et al., Bioorg. Med. Chem. Lett. 2008, 18: 6236-6239 и Kotsuki et al., J. Org. Chem. 1992, 57: 5036-5040).
В одном варианте осуществления настоящего изобретения соединение (А4) выделяют в виде гидрохлоридной соли соединения (A4-HCl).
Неожиданно авторы настоящего изобретения обнаружили, что соединение (A4-HCl) можно осаждать в виде порошка с незначительным комкообразованием или без него. Эти качества дополнительно облегчают процесс.Соединение (A4-HCl) можно получать путем обработки соединения (А4) раствором HCl.
Подходящие растворители для осаждения соли соединения (А4) могут представлять собой один или несколько растворителей, выбранных из группы, состоящей из MeTHF, EtOAc, изопропанола, iPrOAc, ацетона, толуола, гептана и их смесей.
В конкретном варианте осуществления настоящего изобретения растворитель, выбранный из группы, состоящей из MeTHF, EtOAc, iPrOAc, ацетона, толуола, смеси изопропанола (iPrOH) и гептана, смеси iPrOAc и гептана и смеси ацетона и гептана, используют для осаждения соли соединения (А4) и даже более конкретно для осаждения соединения (A4-HCl).
В конкретном варианте осуществления настоящего изобретения растворитель, выбранный из группы, состоящей из MeTHF, смеси изопропанола (iPrOH) и гептана, смеси iPrOAc и гептана и смеси ацетона и гептана, используют для осаждения соединения (A4-HCl).
В более конкретном варианте осуществления настоящего изобретения смесь изопропанола и гептана используют для осаждения соединения (A4-HCl).
В более конкретном варианте осуществления настоящего изобретения смесь ацетона и гептана используют для осаждения соединения (A4-HCl).
Если в способе по настоящему изобретению выделяют соль соединения (А4), такую как, например, соединение (A4-HCl) или соединение (А4-гемиоксалат), то можно использовать выделение основания для выделения соединения (А4) перед началом дополнительных стадий синтеза, например, стадии 5. Таким образом, в одном варианте осуществления настоящего изобретения соль соединения (А4) вступает в реакцию с подходящим основанием, таким как водный раствор Na2CO3, К2СО3 или аммиака, с получением раствора соединения (А4) в виде свободного основания.
В конкретном варианте осуществления настоящего изобретения соединение (A4-HCl) вступает в реакцию с подходящим основанием, таким как водный раствор Na2CO3, К2СО3 или аммиака, с получением соединения (А4) в виде свободного основания.
Стадия 4)
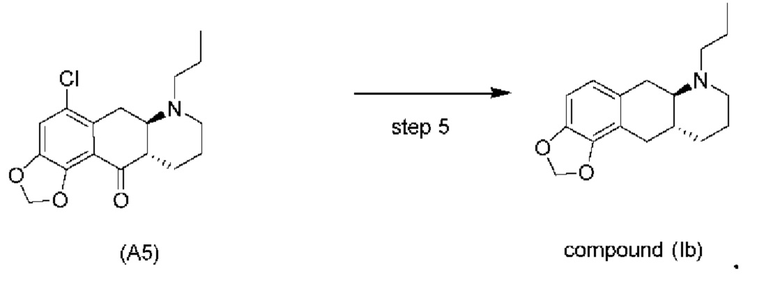
На стадии 4), как показано на схеме 5 ниже, внутримолекулярное ацилирование по Фриделю-Крафтсу соединения (А4) обеспечивает получение трициклического соединения (А5). Ацилирование по Фриделю-Крафтсу обычно осуществляют с использованием смеси Р2О5 и TFA в хлорбензоле с превращением соединения (А4) в соединение (А5).
Схема 5. Стадия 4)
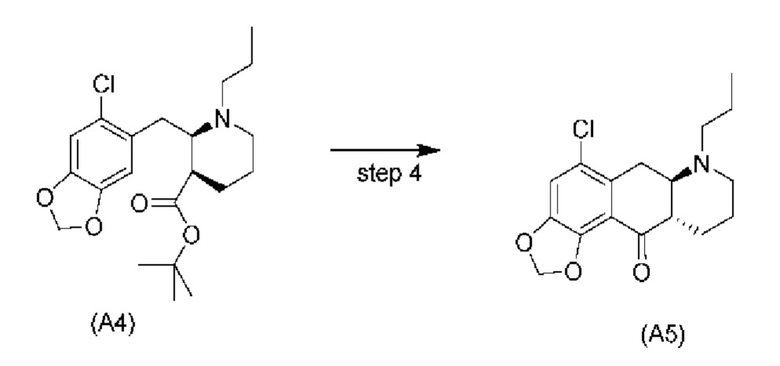
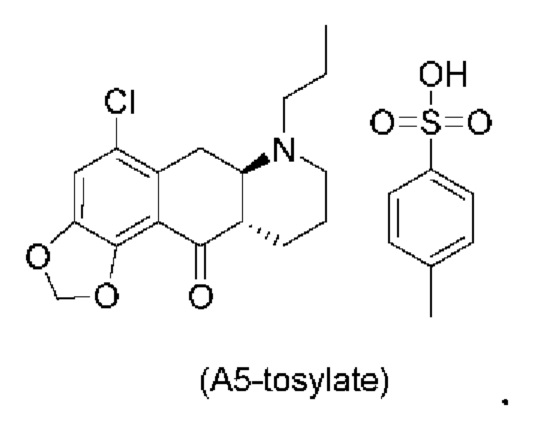
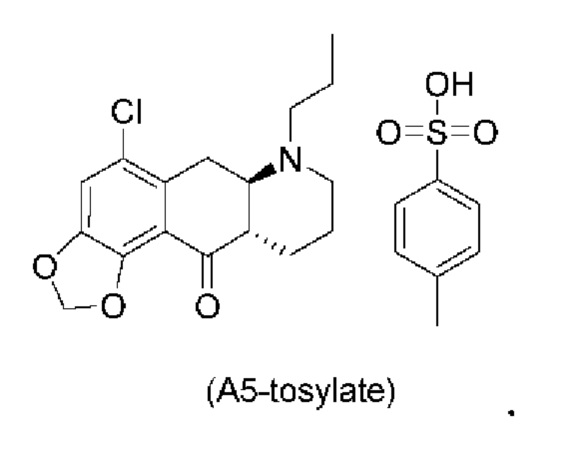
Авторы настоящего изобретения обнаружили, что соединение (А5) является неожиданно стабильным в кислотных условиях или в виде кислой соли и может быть легко выделено в виде тозилатной соли (см. формулу (А5-тозилат) ниже) с высоким выходом с последующей аккуратной нейтрализацией кислой реакционной смеси.
Напротив, если соединение (А5) выделяется в виде свободного основания, его медленно подвергают ароматизации путем отщепления аминогруппы с получением соединения (a4i) (см. формулу ниже) помимо других продуктов. Образование соединения (a4i) из соединения (А5) не наблюдается в кислотных условиях, что является неожиданным, поскольку в кислотных условиях атом азота в соединении (А5) протонируется и, как ожидается, более преимущественно является уходящей группой, чем в нейтральных или основных условиях.

Таким образом, е одном варианте осуществления настоящего изобретения стадия 4 включает выделение соединения (А5) в виде кислой соли. В более конкретном варианте осуществления настоящего изобретения стадия 4 включает выделение соединения (А5-тозилат).
Стадия 5)
Стадия 5), как показано на схеме 6 ниже, обеспечивает однореакторное гидродехлорирование и восстановление кетонов соединения (А5) или его соли в присутствии палладиевого катализатора, такого как Pd/C, и водорода с получением соединения (Ib).
Высокая эффективность реакции является неожиданной, поскольку в отличие от восстановления кетона в алкан, которому способствуют кислотные условия, реакция гидродехлорирования обычно затрудняется в кислотных условиях, и ей наоборот способствуют основные условия (Handbook of heterogeneous hydrogenation, S. Nishimura, Wiley 2001).
Схема 6. Стадия 5)
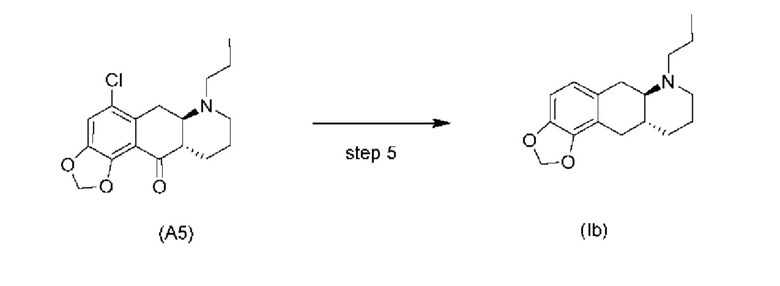
В одном варианте осуществления настоящего изобретения соединение (А5) или его соль используют на стадии 5 с получением соединения (Ib). В другом варианте осуществления настоящего изобретения соединение (А5-тозилат) используют на стадии 5 с получением соединения (Ib).
Различные растворители можно использовать для реакции на стадии 5, в частности, спиртовые растворители. В одном варианте осуществления настоящего изобретения в качестве растворителя на стадии 5 используют спирт. В более конкретном варианте осуществления настоящего изобретения стадию 5 осуществляют с использованием растворителя, который является спиртом, выбранным из группы, состоящей из МеОН, EtOH, IPA и 1-пропанола. В еще более конкретном варианте осуществления стадию 5 осуществляют с использованием EtOH в качестве растворителя.
Реакцию гидрогенизации на стадии 5 можно осуществлять с использованием палладиевого катализатора при температуре в диапазоне от приблизительно 20°С до приблизительно 100°С, например, от приблизительно 50°С до приблизительно 100°С, например, от приблизительно 60°С до приблизительно 80°С, например, приблизительно 65°C, приблизительно 67°C, или приблизительно 68°С, приблизительно 69°С, приблизительно 70°С, приблизительно 71°C, приблизительно 72°С, приблизительно 73°С, приблизительно 75°С, или приблизительно 77°С.
Реакцию гидрогенизации на стадии 5 можно осуществлять с использованием палладиевого катализатора при давлении в диапазоне от приблизительно 2 бар до приблизительно 10 бар, например, от приблизительно 3 бар до 6 бар, например, от приблизительно 3 бар до приблизительно 5 бар, например, приблизительно 3,5 бар, или, например, приблизительно 4 бар, или, например, приблизительно 4,5 бар.
В конкретном варианте осуществления настоящего изобретения стадию 5 осуществляют с использованием палладиевого катализатора, такого как Pd/C, при температуре приблизительно 70°С и при давлении приблизительно 4 бар.
Стадия 6)
Наконец, стадия 6), как показано на схеме 7 ниже, обеспечивает превращение соединения (Ib) или его соли в соединение (I) или его соль путем проведения реакции соединения (Ib) с кислотой Льюиса или кислотой Бренстеда, выбранной из группы, состоящей из BCl3, BBr3 и HBr.
Схема 7. Стадия 6)

В конкретном варианте осуществления настоящего изобретения стадия 6 включает проведение реакции соединения (Ib) с BCl3 с получением соединения (I) или его соли.
В другом конкретном варианте осуществления настоящего изобретения стадия 6 включает проведение реакции соединения (Ib) в виде свободного основания с BCl3 с получением соединения (I) или его соли.
Если соль соединения (Ib), такого как, например, соединение (Ib-L-DTTA), используют в качестве исходного материала для стадии 6, то можно использовать выделение основания для выделения соединения (Ib) перед началом дополнительных стадий синтеза, например, стадии 6. Таким образом, в одном варианте осуществления настоящего изобретения соль соединения (Ib) вступает в реакцию с основанием, таким как водный раствор Na2CO3, К2СО3 или аммиака, с получением соединения (Ib) в виде свободного основания.
В конкретном варианте осуществления настоящего изобретения соединение (Ib-L-DTTA) вступает в реакцию с подходящим основанием, таким как водный раствор Na2CO3, К2СО3 или аммиака, с поучением соединения (Ib) в виде свободного основания.
В еще более конкретном варианте осуществления настоящего изобретения стадия 6 включает проведение реакции соли (-)-O,O'-ди-п-толуоил-L-винной кислоты, соли (L-DTTA), и соединения (Ib) с водным раствором Na2CO3, К2СО3 или аммиака с получением соединения (Ib) в виде свободного основания с последующей реакцией соединения (Ib) с BCl3 с получением соединения (I) или его соли, такой как, например, HCl-соль соединения (I).
Альтернативный путь получения соединения (I)
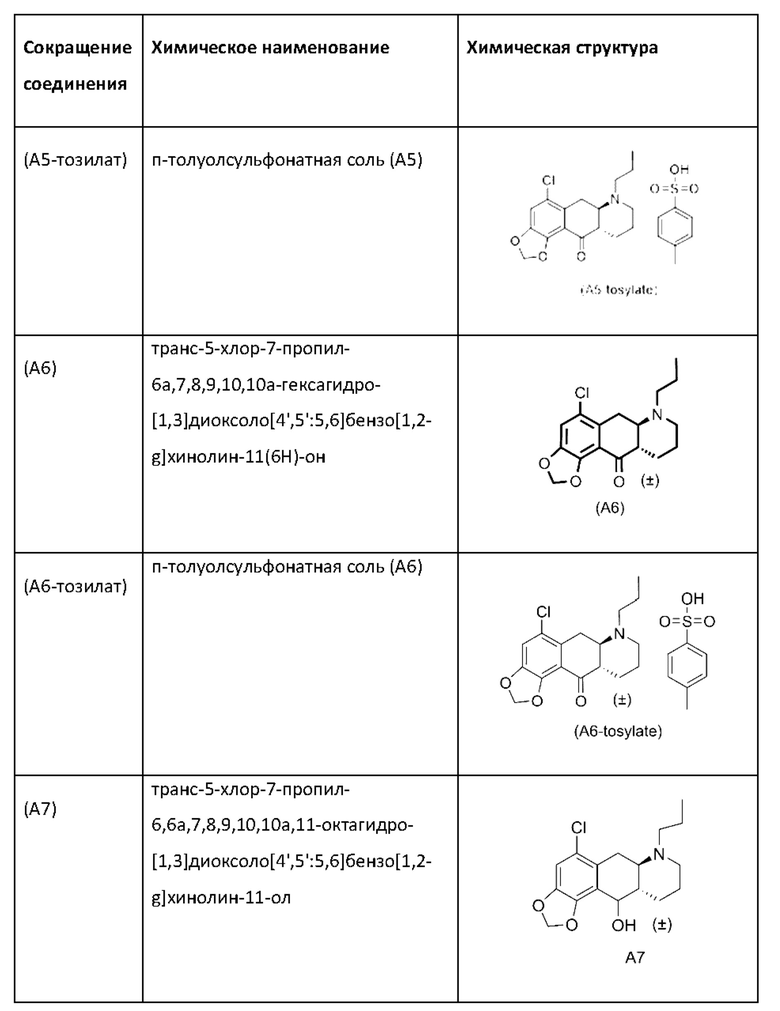
Авторы настоящего изобретения также разработали альтернативный способ получения из соединения (А6) или соединения (А6-тозилат) энантиочистого соединения (Ib) в трехстадийном процессе, причем соединение (Ib) можно получать в виде соли L-DTTA (соли (-)-O,O'-ди-п-толуоил-L-винной кислоты), как показано на схеме 8 ниже.
Схема 8. Альтернативный путь получения соединения (I)
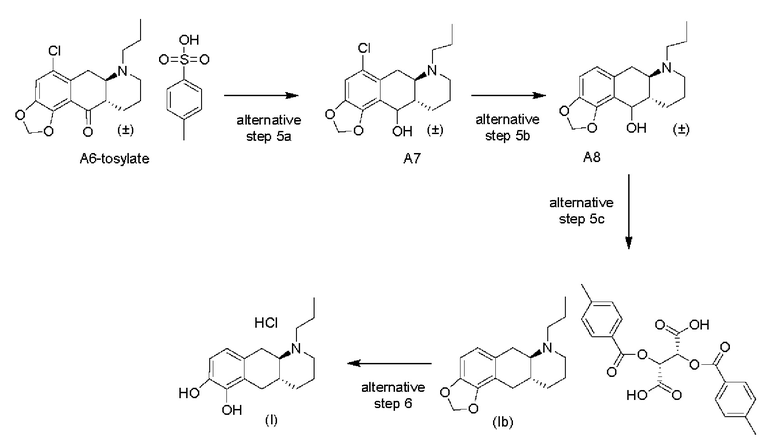
Альтернативная стадия 5а)
На альтернативной стадии 5а) на схеме 8 выше соединение (А6-тозилат) превращается в свободное основание с помощью водного раствора NaOH в Me-THF, а затем восстанавливается путем добавления водного раствора NaBH4 до получения свободного основания соединения (А6) в Me-THF с выходом 85%. Полученное соединение (А7) затем восстанавливают до соединения (А8) с выходом 90% на стадии 2 путем трансферной гидрогенизации, где восстановление облегчается катализатором Pd/C и муравьиной кислотой в смеси формиат аммония-МеОН-вода. Соединение (А8) превращается в соединение (соль Ib-L-DTTA) на стадии 3 путем подвергания его сначала гидрогенизации с использованием катализатора Pd/C и газообразного водорода с добавленным TsOH в IPA с последующим классическим разделением с помощью L-DTTA в МеОН с получением соединения (Ib-L-DTTA) с энантиомерным избытком >99%.
Альтернативная стадия 6)
Наконец, на альтернативной стадии 6) соединение (Ib-L-DTTA) превращается в свободное основание соединения (Ib), и удаляют защиту фрагмента 1,2-дигидрокси с помощью BCl3 с получением после обработки HCl-соли соединения (I).
В отличие от этого альтернативного способа (включающего альтернативную стадию 5а, альтернативную стадию 5b и альтернативную 6, как показано на схеме 8), способ, включающий стадии 5 и 6 настоящего изобретения (показанные на схеме 6 и 7), обеспечивает более удобный и эффективный способ, поскольку 1) разделение посредством образования диастереоизомерных солей осуществляют для соединения (a2ii) на более ранней стадии по сравнению с разделением соединения (Ib) на поздней стадии и 2) гидродехлорирование и восстановление кетонов осуществляют на одной стадии от соединения (А5) до соединения (Ib).
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В следующем разделе раскрыты дополнительные варианты осуществления настоящего изобретения. Первый вариант осуществления обозначен Е1, второй вариант осуществления обозначен Е2 и так далее.
Е1. Способ изготовления соединения (I) с нижеуказанной формулой,

из соединения (Ib) с нижеуказанной формулой,
Е2. Способ согласно варианту осуществления Е1, где соединение (Ib) получают с помощью способа, включающего стадии:
стадия 0)
подстадия (i) проведения реакции соединения (А1) с 3-хлорпропан-1-амином с получением соединения (a2i) или
подстадия (S1) проведения реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с получением смеси, затем
обработка смеси с подстадии (S1) уксусной кислотой, затем
проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i);
затем стадия 1)
подстадия (ii) восстановления соединения (a2i), полученного на подстадии (i) или подстадии (S1), с получением соединения (a2ii), затем
подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением соединения (А2-геми-L-тартрат); или
за подстадией (i) или подстадией (S1) следует подстадия (iv), включающая стадию подвергания полученного соединения (a2i) гидрогенизации, осуществляемой с использованием хирального катализатора в присутствии водорода и растворителя с получением соединения (А2)
согласно нижеуказанной схеме реакции:
Е3. Способ согласно варианту осуществления Е2, где соединение (a6i) или его соль получают с помощью способа, включающего стадию проведения реакции соединения (a5i) или его соли с хлорирующим средством
Е4. Способ согласно варианту осуществления Е3, где хлорирующее средство представляет собой сульфурилхлорид.
Е5. Способ согласно любому из вариантов осуществления Е2-Е4, где восстановление на подстадии (ii) стадии 1 осуществляют в присутствии восстанавливающего средства.
Е6. Способ согласно любому из вариантов осуществления Е2-Е5, где восстанавливающее средство выбрано из NaBH3CN, триацетоксиборгидрида натрия (STAB), борана, такого как 5-этил-2-метилпиридинборан (РЕМВ), и NaBH4.
Е7. Способ согласно любому из вариантов осуществления Е2-Е6, где восстановление на подстадии (ii) стадии 1 осуществляют с использованием платинового катализатора, предпочтительно платины на углероде.
Е8. Способ согласно варианту осуществления Е1-Е4, где хиральный катализатор на подстадии (iv) стадии 1 выбран из (2S)-1-[(1S)-1-[бис(1,1-диметилэтил)фосфино]этил]-2-(дифенилфосфино)ферроцена и тетрафторбората бис(2,5-норборнадиен)родия(1).
Е9. Способ согласно пунктам Е1-Е4 и Е8, где растворитель на подстадии (iv) стадии 1 представляет собой 2,2,2-трифторэтанол.
Е10. Соединение нижеуказанной формулы (А2),
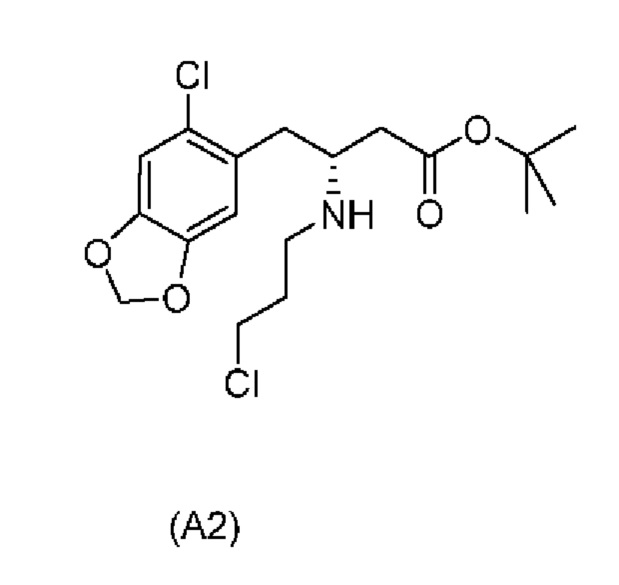
или его соль.
Е11. Соединение согласно варианту осуществления Е10, которое находится в виде геми-L-тартратной соли, как изображено ниже:
Е12. Применение соединения согласно любому из вариантов осуществления Е10 и Е11 в способе получения соединения формулы (I) или соединения формулы (Ib).
Е13. Способ согласно любому из вариантов осуществления Е1-Е9, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
2) реакция соединения (А2) или соединения (А2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства
с получением соединения (A3) согласно нижеуказанной схеме реакции а) или b):
схема а) 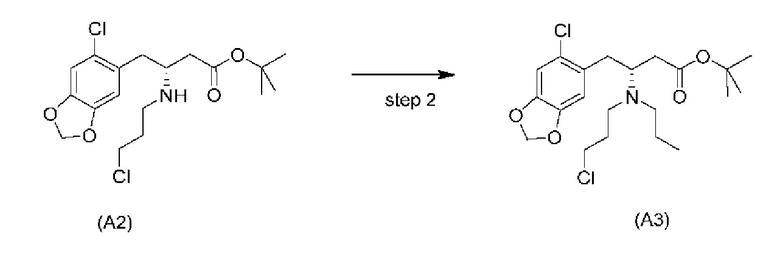 схема Ь)
схема Ь)
Е14. Способ согласно варианту осуществления Е13, где соединение (A3) получают путем проведения реакции соединения (А2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства.
Е15. Способ согласно варианту осуществления Е14, где указанное восстанавливающее средство выбрано из группы, состоящей из NaBH3CN, триацетоксиборгидрида натрия (STAB), борана, такого как 5-этил-2-метилпиридинборан (РЕМВ), и платинового катализатора, предпочтительно платины на углероде, с газообразным водородом.
Е16. Способ согласно любому из вариантов осуществления Е13-Е15, где указанную реакцию осуществляют в растворителе, выбранном из примера тетрагидрофурана (THF), изопропанола (IPA) или МеОН.
Е17. Соединение нижеуказанной формулы (A3),
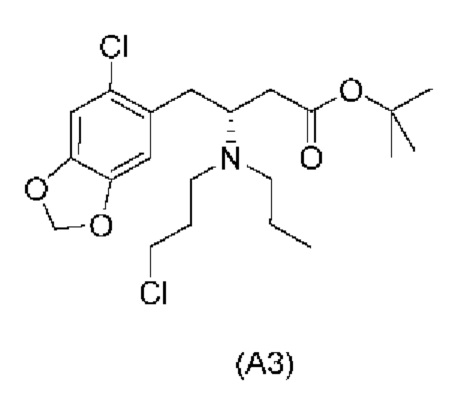
или его соль.
Е18. Применение соединения согласно варианту осуществления Е17 в способе получения соединения формулы (I) или соединения формулы (Ib).
Е19. Способ согласно варианту осуществления Е1-Е9 и Е13-Е16, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
3) проведение реакции соединения (A3) с сильным основанием с получением соединения (А4) согласно нижеуказанной схеме реакции:

за которой необязательно следует выделение соединения (А4) в виде гемиоксадатной соли, как изображено ниже:
Е20. Способ согласно варианту осуществления Е19, где указанное сильное основание представляет собой бис(триметилсилил)амид натрия (NaHMDS).
Е21. Способ согласно любому из вариантов осуществления Е19-Е20, где указанную реакцию между соединением (A3) и основанием осуществляют при температуре в диапазоне от -20 до -5°С, например, в диапазоне от -15 до -5°С или, например, при температуре приблизительно -10°С.
Е22. Способ согласно любому из вариантов осуществления Е19-Е21, где указанное соединение (А4) смешивают с щавелевой кислотой и выделяют в виде гемиоксалатной соли.
Е23. Соединение нижеуказанной формулы (А4),
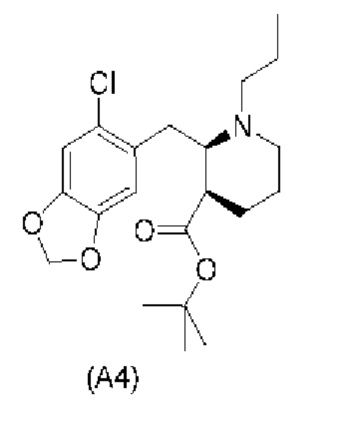
или его соль.
Е24. Соединение согласно варианту осуществления Е23, которое находится в виде гемиоксалатной соли, как изображено ниже:
Е25. Применение соединения согласно любому из вариантов осуществления Е23-Е24 в способе получения соединения формулы (I) или соединения формулы (Ib).
Е26. Способ согласно варианту осуществления Е1-Е9, Е13-Е16, Е19-Е22, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
4) проведение внутримолекулярного ацилирования по Фриделю-Крафтсу соединения (А4) с получением соединения (А5) согласно нижеуказанной схеме реакции:
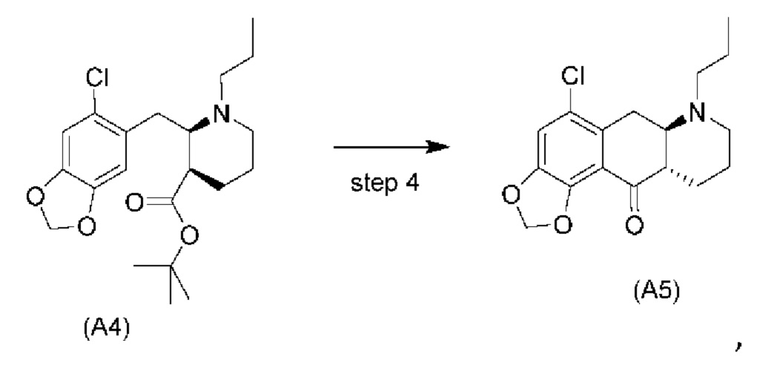
за которой необязательно следует выделение соединения (А5) в виде тозилатной соли (А5-тозилат)
Е27. Соединение нижеуказанной формулы (А5),

или его соль.
Е28. Соединение согласно варианту осуществления Е27, которое находится в виде тозилатной соли, как изображено ниже:

Е29. Применение соединения согласно любому из вариантов осуществления Е27-Е28 в способе получения соединения формулы (I) или соединения формулы (Ib).
Е30. Способ согласно варианту осуществления Е1-Е9, Е13-Е16, Е19-Е22 и Е26, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
5) восстановление соединения (А5) или его соли с получением соединения (Ib) или его соли согласно нижеуказанной схеме реакции:
Е31. Способ согласно варианту осуществления Е30, где указанное восстановление осуществляют с использованием палладиевого катализатора.
Е32. Способ согласно варианту осуществления Е1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления Е2-Е4;
стадия 1) согласно любому из вариантов осуществления Е2 и Е5-Е9; затем
стадия 2) согласно любому из вариантов осуществления Е13-Е16.
Е33. Способ согласно любому из вариантов осуществления Е1-Е7, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 2) согласно любому из вариантов осуществления Е13-Е16; затем
стадия 3) согласно любому из вариантов осуществления Е19-Е22.
Е34. Способ согласно любому из вариантов осуществления Е1-Е7 и Е11-Е13, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 3) согласно любому из вариантов осуществления Е19-Е22; затем
стадия 4) согласно варианту осуществления Е26.
Е35. Способ согласно любому из вариантов осуществления Е1-Е7, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 4) согласно любому из вариантов осуществления Е26; затем
стадия 5) согласно любому из вариантов осуществления Е30-Е31.
Е36. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления Е2-Е4;
стадия 1) согласно любому из вариантов осуществления Е2 и Е5-Е9; затем
стадия 2) согласно любому из вариантов осуществления Е13-Е16; затем
стадия 3) согласно любому из вариантов осуществления Е19-Е22.
Е37. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 2) согласно любому из вариантов осуществления Е13-Е16; затем
стадия 3) согласно любому из вариантов осуществления Е19-Е22; затем стадия 4) согласно варианту осуществления Е26.
Е38. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 3) согласно любому из вариантов осуществления Е19-Е22; затем
стадия 4) согласно варианту осуществления Е26; затем
стадия 5) согласно любому из вариантов осуществления Е30-Е31.
Е39. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления Е2-Е4; затем
стадия 1) согласно любому из вариантов осуществления Е2 и Е5-Е9; затем
стадия 2) согласно любому из вариантов осуществления Е13-Е16; затем
стадия 3) согласно любому из вариантов осуществления Е19-Е22; затем
стадия 4) согласно варианту осуществления Е26.
Е40. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 2) согласно любому из вариантов осуществления Е13-Е16; затем
стадия 3) согласно любому из вариантов осуществления Е19-Е22; затем
стадия 4) согласно варианту осуществления Е26; затем
стадия 5) согласно любому из вариантов осуществления Е30-Е31.
Е41. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления Е2-Е4; затем
стадия 1) согласно любому из вариантов осуществления Е2 и Е5-Е9; затем
стадия 2) согласно любому из вариантов осуществления Е13-Е16; затем
стадия 3) согласно любому из вариантов осуществления Е19-Е22; затем
стадия 4) согласно варианту осуществления Е26; затем
стадия 5) согласно любому из вариантов осуществления Е30-Е31.
Е42. Способ изготовления соединения (I) согласно любому из вариантов осуществления Е1-Е9, Е13-Е16, Е19-Е22, Е26 и Е30-Е31, где соединение (I) получают из соединения (Ib) с помощью следующей стадии:
6) реакция соединения (Ib) с кислотой Льюиса или кислотой Бренстеда, выбранной из группы, состоящей из BCl3, BBr3 и HBr,
с получением соединения (I) согласно нижеуказанной схеме реакции:

Е43. Способ согласно варианту осуществления Е42, где стадия 6) следует за стадией 5).
Е44. Способ изготовления соединения (I) согласно любому из вариантов осуществления Е35, Е38 и Е40-Е42, где за стадией 5) следует
стадия 6).
Е45. Способ изготовления соединения (I) согласно любому из вариантов осуществления Е1-Е9, Е13-Е16, Е19-Е22, Е26 и Е30-Е31, где способ представляет собой химический способ ex vivo.
Е46. Соединение нижеуказанной формулы (a2i),
или его соль.
Е47. Соединение нижеуказанной формулы (a2ii),
или его соль.
Е48. Способ согласно любому из вариантов осуществления Е1-Е9, Е13-Е16, Е19-Е22, Е26 и Е30-Е45, где выделяют соединение (a2i).
Е49. Способ согласно любому из вариантов осуществления Е1-Е6, Е13-Е16, Е19-Е22, Е26, и Е30-Е45, и Е48, где восстанавливающее средство, используемое на подстадии (ii) стадии 1, представляет собой цианоборгидрид натрия.
Е50. Способ согласно любому из вариантов осуществления Е1-Е5, Е7, Е13-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е49, где подстадию (ii) стадии 1 осуществляют с использованием платинового катализатора при температуре от приблизительно 20°С до приблизительно 100°С, например, от приблизительно 50°С до приблизительно 80°С, например, от приблизительно 55°С до приблизительно 65°С, например, приблизительно 57°С, или приблизительно 58°C, или приблизительно 59°С, или приблизительно 60°С, или приблизительно 61°C, или приблизительно 62°С, или приблизительно 63°С.
Е51. Способ согласно любому из вариантов осуществления Е1-Е5, Е7, Е13-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е50, где подстадию (ii) стадии 1 осуществляют с использованием платинового катализатора при давлении от приблизительно 2 до приблизительно 10 бар, например, от приблизительно 2 бар до приблизительно 6 бар, например, от приблизительно 3 бар до приблизительно 5 бар, например, приблизительно 4 бар.
Е52. Способ согласно любому из вариантов осуществления Е1-Е5, Е7, Е13-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е51, где подстадию (ii) стадии 1 осуществляют с использованием платинового катализатора при температуре от приблизительно 50°С до приблизительно 80°С и при давлении от приблизительно 2 бар до приблизительно 6 бар.
Е53. Способ согласно любому из вариантов осуществления Е1-Е5, Е7, Е13-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е52, где подстадию (ii) стадии 1 осуществляют с использованием платинового катализатора при температуре приблизительно 60°С и при давлении приблизительно 4 бар.
Е54. Способ согласно любому из вариантов осуществления Е1-Е7, Е13-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е53, где подстадию (iii) стадии 1 осуществляют с использованием спирта в качестве растворителя, такого как растворитель, выбранный из МеОН, EtOH и их водных смесей.
Е55. Способ согласно любому из вариантов осуществления Е13-Е16, Е19-Е22, Е26, Е32-Е33, Е36-Е37, Е39-Е45 и Е48-Е54, где восстанавливающее средство, используемое на стадии 2, выбрано из NaBH3CN, триацетоксиборгидрида натрия (STAB), борана, такого как 5-этил-2-метилпиридинборан (РЕМВ), и платинового катализатора, предпочтительно платины на углероде, с газообразным водородом.
Е56. Способ согласно любому из вариантов осуществления Е13-Е16, Е19-Е22, Е26, Е32-Е33, Е36-Е37, Е39-Е45 и Е48-Е55, где восстанавливающее средство, используемое на стадии 2, представляет собой триацетоксиборгидрид натрия (STAB).
Е57. Способ согласно любому из вариантов осуществления Е13-Е16, Е19-Е22, Е26, Е32-Е33, Е36-Е37, Е39-Е45 и Е48-Е56, где стадию 2 осуществляют в растворителе, выбранном из группы, состоящей из тетрагидрофурана (THF), изопропанола (IPA) и МеОН.
Е58. Способ согласно любому из вариантов осуществления Е13-Е16, Е19-Е22, Е26, Е32-Е33, Е36-Е37, Е39-Е45 и Е48-Е57, где стадию 2 проводят в THF.
Е59. Способ согласно любому из вариантов осуществления Е19-Е22, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е58, где стадию 3 осуществляют с использованием сильного основания, выбранного из группы, состоящей из бис(триметилсилил)амида натрия (NaHMDS), диизопропиламида лития (LDA), бис(триметилсилил)амида калия (KHMDS) и бис(триметилсилил)амида лития (LHMDS).
Е60. Способ согласно любому из вариантов осуществления Е19-Е22, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е59, где стадию 3 осуществляют с использованием растворителя, выбранного из группы, состоящей из толуола, THF и их смеси.
Е61. Способ согласно любому из вариантов осуществления Е19-Е21, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е60, где за стадией 3 следует выделение соединения (А4) в виде соли, выбранной из группы, состоящей из гемиоксалатной соли соединения (А4-гемиоксалат), гидрохлоридной соли соединения (A4-HCl) или гидробромидной соли соединения (А4-HBr).
Е62. Способ согласно любому из вариантов осуществления Е19-Е21, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е61, где за стадией 3 следует реакция соединения (А4) с раствором HCl и выделение гидрохлоридной соли соединения (A4-HCl).
Е63. Способ согласно любому из Е19-Е22, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е62, где за стадией 3 следует выделение соединения (А4) с использованием растворителя, выбранного из группы, состоящей из MeTHF, EtOAc, изопропанола, iPrOAc, ацетона, толуола, гептана и их смесей.
Е64. Способ согласно любому из вариантов осуществления Е19-Е21, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е63, где за стадией 3 следует выделение соединения (A4-HCl) с использованием смеси изопропанола и гептана.
Е65. Способ согласно любому из вариантов осуществления Е19-Е21, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е64, где за стадией 3 следует выделение соединения (A4-HCl) с использованием смеси ацетона и гептана.
Е66. Способ согласно любому из вариантов осуществления Е19-Е22, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е66, дополнительно включающий стадию, на которой выделенную соль соединения (А4) приводят в реакцию с основанием с получением соединения (А4) в виде свободного основания.
Е67. Способ согласно любому из вариантов осуществления Е19-Е22, Е26, Е30-Е31, Е33-Е34, Е36-Е45 и Е48-Е66, дополнительно включающий стадию, на которой соль соединения (А4) приводят в реакцию с водным раствором Na2CO3, К2СО3 или аммиаком с получением соединения (А4) в виде свободного основания.
Е68. Способ согласно любому из вариантов осуществления Е19-Е22, Е26, Е30-Е31, Е34-Е35, Е37-Е45 и Е48-Е67, дополнительно включающий стадию, на которой соединение (A4-HCl) приводят в реакцию с водным раствором Na2CO3, К2СО3 или аммиака с получением соединения (А4) в виде свободного основания.
Е69. Способ согласно любому из вариантов осуществления Е26, Е30-Е31, Е34-Е35, Е37-Е45 и Е48-Е68, где стадия 4 включает выделение соединения (А5) в виде кислой соли.
Е70. Способ согласно любому из вариантов осуществления Е26, Е30-Е31, Е34-Е35, Е37-Е45 и Е48-Е69, где ацилирование по Фриделю-Крафтсу на стадии 4 осуществляют с использованием смеси Р2О5 и TFA в подходящем растворителе, таком как хлорбензол.
Е71. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е70, где соединение (А5) или его соль используют на стадии 5 с получением соединения (Ib).
Е72. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е71, где соединение (А5-тозилат) используют на стадии 5 с получением соединения (Ib).
Е73. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е72, где спиртовой растворитель используют на стадии 5.
Е74. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е73, где растворитель, выбранный из группы, состоящей из МеОН, EtOH, IPA и 1-пропанола, используют на стадии 5.
Е75. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е74, где EtOH используют в качестве растворителя на стадии 5.
Е76. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е75, где реакцию гидрогенизации на стадии 5 осуществляют с использованием палладиевого катализатора при температуре в диапазоне от приблизительно 20°С до приблизительно 100°С, например, от приблизительно 50°С до приблизительно 100°С, например, от приблизительно 60°С до приблизительно 80°С, например, приблизительно 65°С, приблизительно 67°С, или приблизительно 68°С, приблизительно 69°С, приблизительно 70°С, приблизительно 71Т, приблизительно 72°С, приблизительно 73°С, приблизительно 75°С, или приблизительно 77°С.
Е77. Способ согласно любому из вариантов осуществления Е30-Е31, Е35, Е38, Е40-Е45 и Е48-Е76, где реакцию гидрогенизации на стадии 5 осуществляют с использованием палладиевого катализатора при давлении в диапазоне от приблизительно 2 бар до приблизительно 10 бар, например, от приблизительно 3 бар до 6 бар, например, от приблизительно 3 бар до приблизительно 5 бар, например, приблизительно 3,5 бар, или, например, приблизительно 4 бар, или, например, приблизительно 4,5 бар.
Е78. Способ согласно любому из вариантов осуществления Е42-Е45 и Е48-Е77, где стадия 6 включает проведение реакции соединения (Ib) с BCl3 с получением соединения (I) или его соли.
Е79. Способ согласно любому из вариантов осуществления Е42-Е45 и Е48-Е78, где стадия 6 включает проведение реакции соединения (Ib) в виде свободного основания с ВCl3 с получением соединения (I) или его соли.
Е80. Способ согласно любому из вариантов осуществления Е42-Е45 и Е48-Е79, где стадия 6 включает проведение реакции соли соединения (Ib) с основанием с получением соединения (Ib) в виде свободного основания, затем проведение реакции соединения (Ib) с BCl3 с получением соединения (I) или его соли.
Е81. Способ согласно любому из вариантов осуществления Е42-Е45 и Е48-Е80, где стадия 6 включает проведение реакции соли соединения (Ib) с водным раствором Na2CO3, К2СО3 или аммиака с получением соединения (Ib) в виде свободного основания, затем проведение реакции соединения (Ib) с BCl3 с получением соединения (I) или его соли.
Е82. Способ согласно любому из вариантов осуществления Е42-Е45 и Е48-Е81, где стадия 6 включает проведение реакции соли (-)-O,O'-ди-п-толуоил-L-винной кислоты, соли (L-DTTA), соединения (Ib), с основанием для получения свободного основания соединения (Ib), затем реакция соединения (Ib) с BCl3 с получением соединения (I) или его соли, такой как HCl-соль соединения (I).
Е83. Соединение согласно варианту осуществления Е23, которое находится в виде гидрохлоридной соли, как изображено ниже:
Е84. Применение соединения согласно любому из вариантов осуществления Е23 и Е83 в способе получения соединения формулы (I) или соединения формулы (Ib).
Е85. Способ изготовления соединения (I) с нижеуказанной формулой,
из соединения (Ib) с нижеуказанной формулой,
где соединение (Ib) получают с помощью способа, включающего стадии:
стадия 0)
подстадия (S1) проведения реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с получением смеси, затем
обработка смеси с подстадии (S1) уксусной кислотой, затем
проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i);
затем стадия 1)
подстадия (ii) восстановления соединения (a2i), полученного на подстадии (S1), с получением соединения (a2ii), затем
подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением соединения (А2-геми-L-тартрат)
согласно нижеуказанной схеме реакции:
Е86. Способ согласно варианту осуществления Е85, где соединение (a6i) или его соль получают с помощью способа, включающего стадию проведения реакции соединения (a5i) или его соли с хлорирующим средством
Е87. Способ согласно варианту осуществления Е86, где хлорирующее средство представляет собой сульфурилхлорид.
Е88. Способ согласно любому из еариантое осуществления Е85-Е87, где соединение (a2i) выделяют перед началом подстадии (ii) стадии 1.
Е89. Способ согласно любому из вариантов осуществления Е85-Е88, дополнительно определяемый любым из вариантов осуществления Е5-Е7, Е12-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е82.
Е90. Способ согласно любому из вариантов осуществления Е85-Е89, где соединение (A3) получают с помощью способа, включающего следующую стадию:
2) проведение реакции соединения (А2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства
с получением соединения (A3) согласно нижеуказанной схеме реакции b),
схема b)

Все ссылки, включая публикации, патентные заявки и патенты, цитируемые в данном документе, включены в данный документ посредством ссылки во всей своей полноте и в той же степени, как если бы было указано, что каждая ссылка отдельно и конкретно включена посредством ссылки и приведена во всей своей полноте (в максимальной степени, допускаемой законом).
Заголовки и подзаголовки применяются в данном документе исключительно для удобства, и их не следует рассматривать как ограничивающие настоящее изобретение каким-либо образом.
Описание в данном документе любого аспекта или аспектов настоящего изобретения с использованием таких терминов, как "включающий", "имеющий", "в том числе" или "содержащий", по отношению к элементу или элементам предназначено для подтверждения аналогичного аспекта или аспектов настоящего изобретения, который "состоит из", "состоит практически из" данного конкретного элемента или элементов или "по сути содержит" их, если не указано иное или это однозначно не противоречит контексту (например, композиция, описанная в данном документе как содержащая определенный элемент, должна также пониматься как описывающая композицию, состоящую из данного элемента, если не указано иное или это однозначно не противоречит контексту).
Использование всевозможных примеров или вводного слова перед примерами (в том числе "как например", "например", "к примеру", "такой как" и "собственно") в данном описании предназначено исключительно для лучшего освещения настоящего изобретения и не предусматривает ограничение объема настоящего изобретения, если не указано иное.
Следует понимать, что различные аспекты, варианты осуществления, реализации и признаки настоящего изобретения, упомянутые в данном документе, могут быть заявлены по отдельности или в любом сочетании.
Настоящее изобретение включает все модификации и эквиваленты объекта изобретения, изложенного в прилагаемой к данному документу формуле изобретения согласно действующему законодательству
Список пунктов
В следующем списке пунктов раскрыты некоторые дополнительные варианты осуществления настоящего изобретения. Первый вариант осуществления обозначен как ЕЕ1, второй вариант осуществления обозначен как ЕЕ2 и т.д.
ЕЕ1. Способ изготовления соединения (Ib) с нижеуказанной формулой,
включающий в себя стадии:
стадия 0)
подстадия (i) проведения реакции соединения (А1) с 3-хлорпропан-1-амином с получением соединения (a2i) или
подстадия (S1) проведения реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с получением смеси, затем
обработка смеси со стадии (S1) уксусной кислотой, затем
проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i);
затем
стадия 1)
подстадия (ii) восстановления соединения (a2i), полученного на стадии подстадии (i) или подстадии (S1), с получением соединения (a2ii), затем
подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением соединения (А2-геми-L-тартрат); или
за подстадией (i) или подстадией (S1) следует стадия подстадии (iv), включающая стадию подвергания соединения (a2i) гидрогенизации, осуществляемой с использованием хирального катализатора в присутствии водорода и растворителя, с получением соединения (А2) согласно нижеуказанной схеме реакции:
ЕЕ2. Способ согласно варианту осуществления Е1, где соединение (a6i) получают с помощью способа, включающего стадию реакции соединения (a5i) с хлорирующим средством
ЕЕ3. Способ согласно варианту осуществления ЕЕ2, где хлорирующее средство представляет собой сульфурилхлорид.
ЕЕ4. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ3, где восстановление на подстадии (ii) стадии 1 осуществляют в присутствии восстанавливающего средства.
ЕЕ5. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ4, где восстанавливающее средство выбрано из NaBH3CN, триацетоксиборгидрида натрия (STAB), борана, такого как 5-этил-2-метилпиридинборан (РЕМВ), и NaBH4.
ЕЕ6. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ5, где восстановление на подстадии (ii) стадии 1 осуществляют с использованием платинового катализатора, предпочтительно платины на углероде.
ЕЕ7. Способ согласно варианту осуществления ЕЕ1-ЕЕ3, где хиральный катализатор на подстадии (iv) стадии 1 выбран из (2S)-1-[(1S)-1-[бис(1,1-диметилэтил)фосфино]этил]-2-(дифенилфосфино)ферроцена и тетрафторбората бис(2,5-норборнадиен)родия(1).
ЕЕ8. Способ согласно пунктам ЕЕ1-ЕЕ3 и ЕЕ7, где растворитель на подстадии (iv) стадии 1 представляет собой 2,2,2-трифторэтанол.
ЕЕ9. Соединение нижеуказанной формулы (А2),

или его соль.
ЕЕ10. Соединение согласно варианту осуществления Е8, которое находится в виде геми-L-тартратной соли, как изображено ниже:
ЕЕ11. Применение соединения согласно любому из вариантов осуществления ЕЕ9 и ЕЕ10 в способе получения соединения формулы (I) или соединения формулы (Ib).
ЕЕ12. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ8, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
2) реакция соединения (А2) или соединения (А2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства
с получением соединения (A3) согласно нижеуказанной схеме реакции а) или b):
схема а)
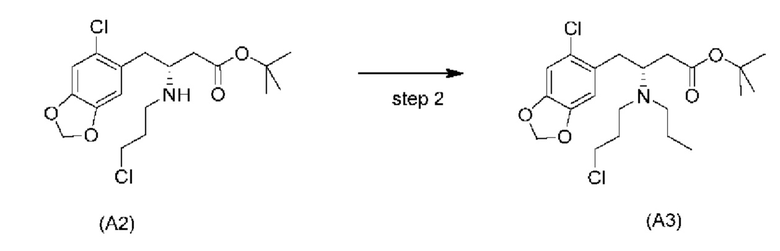
схема b)
ЕЕ13. Способ согласно варианту осуществления ЕЕ12, где соединение (A3) получают путем проведения реакции соединения (А2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства.
ЕЕ14. Способ согласно варианту осуществления ЕЕ13, где указанное восстанавливающее средство выбрано из NaBH3CN, триацетоксиборгидрида натрия (STAB), борана, предпочтительно 5-этил-2-метилпиридинборана (РЕМВ), или гидрогенизации с использованием платинового катализатора, предпочтительно платины на углероде, и газообразного водорода.
ЕЕ15. Способ согласно любому из вариантов осуществления ЕЕ12-ЕЕ14, где указанную реакцию осуществляют в растворителе, выбранном из примера тетрагидрофурана (THF), изопропанола (IPA) или МеОН.
ЕЕ16. Соединение нижеуказанной формулы (A3),
или его соль.
ЕЕ17. Применение соединения согласно варианту осуществления ЕЕ16 в способе получения соединения формулы (I) или соединения формулы (Ib).
ЕЕ18. Способ согласно варианту осуществления Е1-ЕЕ8 и ЕЕ12-ЕЕ15, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
3) проведение реакции соединения (A3) с сильным основанием с получением соединения (А4) согласно нижеуказанной схеме реакции:
за которой необязательно следует выделение соединения (А4) в виде гемиоксадатной соли, как изображено ниже:
ЕЕ19. Способ согласно варианту осуществления ЕЕ16, где указанное сильное основание представляет собой бис(триметилсилил)амид натрия (NaHMDS).
ЕЕ20. Способ согласно любому из вариантов осуществления ЕЕ16-ЕЕ17, где указанную реакцию между соединением (A3) и основанием осуществляют при температуре в диапазоне от -20 до -5°С, например, в диапазоне от -15 до -5°С или, например, при температуре приблизительно -10°С.
ЕЕ21. Способ согласно любому из вариантов осуществления ЕЕ16-ЕЕ18, где указанное соединение (А4) смешивают с щавелевой кислотой и выделяют в виде гемиоксалатной соли.
ЕЕ22. Соединение нижеуказанной формулы (А4),

или его соль.
ЕЕ23. Соединение согласно варианту осуществления ЕЕ22, которое находится в виде гемиоксалатной соли, как изображено ниже:
ЕЕ24. Применение соединения согласно любому из вариантов осуществления ЕЕ22-ЕЕ23 в способе получения соединения формулы (I) или соединения формулы (Ib).
ЕЕ25. Способ согласно варианту осуществления ЕЕ1-ЕЕ8, ЕЕ12-ЕЕ15, ЕЕ18-ЕЕ21, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
4) проведение внутримолекулярного ацилирования по Фриделю-Крафтсу соединения (А4) с получением соединения (А5) согласно нижеуказанной схеме реакции:
за которой необязательно следует стадия выделения соединения (А5) в виде тозилатной соли (А5-тозилат)

ЕЕ26. Соединение нижеуказанной формулы (А5),
или его соль.
ЕЕ27. Соединение согласно варианту осуществления ЕЕ26, которое находится в виде тозилатной соли, как изображено ниже:
ЕЕ28. Применение соединения согласно любому из вариантов осуществления ЕЕ26-ЕЕ27 в способе получения соединения формулы (I) или соединения формулы (Ib).
ЕЕ29. Способ согласно варианту осуществления ЕЕ1-ЕЕ8, ЕЕ12-ЕЕ15, ЕЕ18-ЕЕ21 и ЕЕ25, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
5) восстановление соединения (А5) или его соли с получением соединения (Ib) или его соли согласно нижеуказанной схеме реакции:

ЕЕ30. Способ согласно варианту осуществления ЕЕ29, где указанное восстановление осуществляют с использованием палладиевого катализатора.
ЕЕ31. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления ЕЕ1-ЕЕ3; затем
стадия 1) согласно любому из вариантов осуществления ЕЕ1 и ЕЕ4-ЕЕ8; затем
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15.
ЕЕ32. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ8, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15; затем
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21.
ЕЕ33. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ8 и ЕЕ12-ЕЕ15, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 3) согласно любому из вариантов осуществления ЕЕ18-21; затем
стадия 4) согласно варианту осуществления ЕЕ25.
ЕЕ34. Способ согласно любому из вариантов осуществления ЕЕ1-ЕЕ8, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 4) согласно любому из вариантов осуществления ЕЕ25; затем
стадия 5) согласно любому из вариантов осуществления ЕЕ29-ЕЕ30.
ЕЕ35. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления ЕЕ1-ЕЕ3; затем
стадия 1) согласно любому из вариантов осуществления ЕЕ1 и ЕЕ4-ЕЕ8; затем
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15; затем
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21.
ЕЕ36. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15; затем
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21; затем
стадия 4) согласно варианту осуществления ЕЕ25.
ЕЕ37. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21; затем
стадия 4) согласно варианту осуществления ЕЕ25; затем
стадия 5) согласно любому из вариантов осуществления ЕЕ29-ЕЕ30.
ЕЕ38. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 1) согласно любому из вариантов осуществления ЕЕ1 и ЕЕ4-ЕЕ8; затем
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15; затем
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21; затем
стадия 4) согласно любому из вариантов осуществления ЕЕ25.
ЕЕ39. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15; затем
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21; затем
стадия 4) согласно варианту осуществления ЕЕ25; затем
стадия 5) согласно любому из вариантов осуществления ЕЕ29-ЕЕ30.
ЕЕ40. Способ согласно варианту осуществления 1, где соединение (Ib) получают с помощью способа, включающего следующие стадии:
стадия 0) согласно любому из вариантов осуществления ЕЕ1-ЕЕ3; затем
стадия 1) согласно любому из вариантов осуществления ЕЕ1 и ЕЕ4-ЕЕ8; затем
стадия 2) согласно любому из вариантов осуществления ЕЕ12-ЕЕ15; затем
стадия 3) согласно любому из вариантов осуществления ЕЕ18-ЕЕ21; затем
стадия 4) согласно варианту осуществления ЕЕ25; затем
стадия 5) согласно любому из вариантов осуществления ЕЕ29-ЕЕ30.
ЕЕ41. Способ изготовления соединения (I) согласно любому из вариантов осуществления ЕЕ1-ЕЕ8, ЕЕ12-ЕЕ15, ЕЕ18-ЕЕ21, ЕЕ25, ЕЕ29-ЕЕ30 и ЕЕ32-ЕЕ40, где соединение (I) получают из соединения (Ib) с помощью следующей стадии.
ЕЕ42. Способ изготовления соединения (I) согласно любому из вариантов осуществления ЕЕ1-ЕЕ8, ЕЕ12-ЕЕ15, ЕЕ18-ЕЕ21, ЕЕ25, ЕЕ29-ЕЕ30 и ЕЕ32-ЕЕ40, где способ представляет химический способ exvivo.
ЕЕ43. Способ изготовления соединения (I) согласно любому из вариантов осуществления ЕЕ1-ЕЕ8, ЕЕ12-ЕЕ15, ЕЕ18-ЕЕ21, ЕЕ25, ЕЕ29-ЕЕ30 и ЕЕ31-ЕЕ42, где указанный способ дополнительно определяется любым из вариантов осуществления Е48-Е82 и Е85-Е90.
ЕЕ44. Способ изготовления соединения (Ib) с нижеуказанной формулой,

где соединение (Ib) получают с помощью способа, включающего стадии:
стадия 0)
подстадия (S1) проведения реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с получением смеси, затем
обработка смеси с подстадии (S1) уксусной кислотой, затем
проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i);
затем стадия 1)
подстадия (ii) восстановления соединения (a2i), полученного на подстадии (S1), с получением соединения (a2ii), затем
подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением соединения (А2-геми-L-тартрат)
согласно нижеуказанной схеме реакции:

ЕЕ45. Способ согласно варианту осуществления ЕЕ44, где соединение (a6i) или его соль получают с помощью способа, включающего стадию проведения реакции соединения (a5i) или его соли с хлорирующим средством
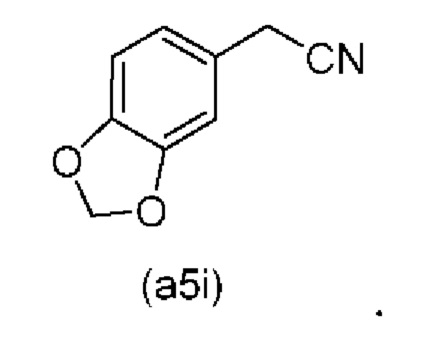
ЕЕ46. Способ согласно варианту осуществления ЕЕ45, где хлорирующее средство представляет собой сульфурилхлорид.
ЕЕ47. Способ согласно любому из вариантов осуществления ЕЕ44-Е46, где соединение (a2i) выделяют перед началом подстадии (ii) стадии 1.
ЕЕ48. Способ согласно любому из вариантов осуществления ЕЕ44-ЕЕ47, дополнительно определяемый любым из вариантов осуществления Е5-Е7, Е12-Е16, Е19-Е22, Е26, Е30-Е45 и Е48-Е82.
ЕЕ49. Способ согласно любому из вариантов осуществления ЕЕ44-ЕЕ48, где соединение (A3) получают с помощью способа, включающего следующую стадию:
2) проведение реакции соединения (А2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства с получением соединения (A3) согласно схеме реакции b) ниже:
схема b)
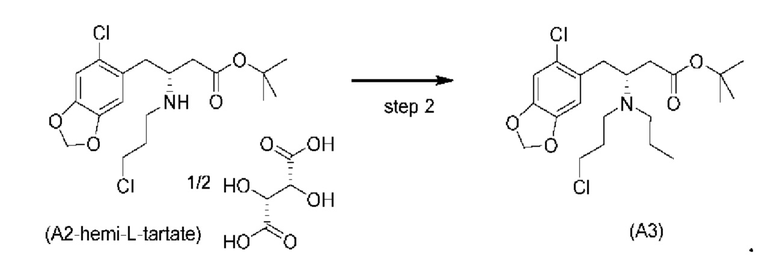
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Сокращения:
DCM: дихлорметан
DEA: Диэтиламин
ее: энантиомерный избыток
EtOAc: этилацетат
IPA: изопропанол
iPrOAc: изопропилацетат
iPrOH: изопропанол
Me-THF: 2-метилтетрагидрофуран
NaHMDS: Бис(триметилсилил)амид натрия
Pd/C: Палладий на угле
РЕМВ: 5-этил-2-метилпиридинборан
STAB: Триацетоксиборгидрид натрия
TFA: трифторуксусная кислота
THF: тетрагидрофуран
Me-THF: 2-метилтетра гидрофура н
TsOH: п-толуолсульфоновая кислота
объем/объем: Объем на объем
вес/вес: Вес на вес
ЯМР-способы
QNMR(600 МГц):
1) релаксационная задержка - 40 сек
2) время исследования - 3,76 сек
3) временной интервал - 64k
4) Размер - 32k
5) Сканы для отрицательного контроля - 4
6) Сканы - 8
7) Импульс - 30 град.
Способы LC-MS и HPLC
Аналитические данные LC-MS получали с применением способов, определенных ниже.
Способ LC-MS. LC-MS проводили на системе Waters Aquity UPLC-MS, состоящей из управляющего устройства колонки, бинарной градиентной системы управления, органайзера для образцов, детектора PDA (работающего при 254 нМ), детектора ELS и TQ-MS, оборудованного APPI-источником, работающем в режиме положительных ионов.
Условия LC. Колонка представляла собой Acquity UPLC ВЕН С18 1,7 мкм; 2,1×150 мм, работающую при 60°С с 0,6 мл/мин бинарного градиента, состоящего из воды + 0,05% трифторуксусной кислоты (А) и смеси ацетонитрил/вода (95:5)+0,05% трифторуксусной кислоты.
Градиент (линейный):
0,00 мин - 10% В
3,00 мин - 100% В
3,60 мин - 10% В
Общее время анализа: 3,6 минут
Способ хиральной HPLC. Хиральную HPLC проводили на системе Merck Hitachi HPLC серии 7000, состоящей из насоса, интерфейса, программируемого автоматического устройства для отбора образцов, колоночного термостата и УФ-детектора (работающего при 220 нм). Колонка представляла собой Chiralpak AD-H 5 мкм; 4,6×250 мм, работающая при 30°С со скоростью потока элюента, составляющей 1,0 мл/мин, состоящего из 90/10/0,1 гексана/IPA/DEA объем/объем, и общее время анализа составляло 20 минут.
Пример 1: получение соединений (a2i), (a2ii) и (А2-геми-L-тартрата) (подстадия (i) стадии 0, за которой следуют подстадии (ii) и (iii) стадии 1)
Смесь гидрохлорида 3-хлорпропан-1-амина (24,9 г, 192 ммоль), соединения (А1) (20,0 г, 63,9 ммоль), хлорида цинка в Me-THF (1,7 мл, 3,20 ммоль, 1,9 М), ацетата натрия (17,3 г, 211 ммоль) и сульфата натрия (4,54 г, 32,0 ммоль) в Me-THF (150 мл) перемешивали в течение ночи.
Затем раствор цианоборгидрида натрия (5,22 г, 83 ммоль) в Me-THF (50 мл) медленно добавляли при комнатной температуре и смесь перемешивали при 40°С в течение 4 часов. Смесь охлаждали до комнатной температуры и медленно добавляли в перемешанную смесь насыщенного водного раствора NH4Cl (100 мл) и воды (100 мл). Смесь перемешивали в течение 15 минут при комнатной температуре. Органическую фазу отделяли и промывали смесью насыщенного водного раствора NH4Cl и воды (100 мл, 1:1 объем/объем), а затем насыщенным водным раствором NaHCO3 (2х 100 мл) дважды. Органическую фазу промывали солевым раствором (100 мл), высушивали над MgSO4, фильтровали и совместно выпаривали с МеОН несколько раз до сухого состояния с получением неочищенного соединения (a2ii) (27,7 г) в виде масла.
В раствор неочищенного соединения (a2ii) в МеОН (150 мл) добавляли раствор L-винной кислоты (5,32 г, 35,5 ммоль) при перемешивании с обратным холодильником в МеОН (43 мл). Смеси обеспечивали медленное охлаждение до комнатной температуры в течение ночи при перемешивании и вводили затравку (~20 мг) при температуре несколько ниже температуры кипения с обратным холодильником. Полученную суспензию фильтровали и осадок на фильтре промывали с помощью МеОН (20 мл) и высушивали в вакуумной печи при 40°С с получением соединения (А2-геми-L-тартрата) (11,3 г, 38%) в виде твердого вещества, с соотношением S:R 1:161 (99,4% ее) соединения (А2) согласно анализу посредством хиральной HPLC.
LC-MS: RT=2,22 минуты, [М+Н]+=390,0 масса/заряд.
1Н ЯМР (600 МГц, DMSO-d6) δ 7,03 (s, 1Н), 6,95 (s, 1Н), 6,04 (dd, J=1,0, 1,0 Гц, 2Н), 4,12 (s, 2Н, L-винная кислота), 3,66 (t, J=6,5 Гц, 2Н), 3,21 (квинт, J=8,0 Гц, 1Н), 2,91 (dd, J=6,0, 13,5 Гц, 1Н), 2,75 (t, J=7,0 Гц, 2Н), 2,62 (dd, J=8,5, 13,5 Гц, 1Н), 2,31 (dd, J=7,0, 15,5 Гц, 1Н), 2,26 (dd, J=7,0, 15,5 Гц, 1Н), 1,82-1,86 (m, 2Н), 1,36 (s, 9Н).
Пример 2: получение соединения (A3) (стадия 2)
Смесь соединения (А2-геми-L-тартрат) (400 мг, 0,43 ммоль), пропаналя (80 мкл, 1,12 моль), уксусной кислоты (49 мкл, 0,86 ммоль) и THF (4,0 мл) перемешивали в течение 30 минут. Затем добавляли STAB (182 мг, 0,86 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3,5 часа. Добавляли насыщенный водный раствор NaHCO3 (4 мл) и смесь экстрагировали толуолом. Органический экстракт высушивали над MgSO4, фильтровали и выпаривали до сухого состояния с получением неочищенного соединения (A3) (167 мг, 90%) в виде твердого вещества.
LC-MS: RT=2,46 минуты, [М+Н]+=432,3 масса/заряд.
1Н ЯМР (600 МГц, CDCl3) δ 6,80 (s, 1Н), 6,62 (s, 1Н), 5,94 (s, 2Н), 3,49-3,55 (m, 2Н), 3,35-3,39 (m, 1Н), 2,90 (dd, J=3,5, 13,5 Гц, 1Н), 2,61-2,63 (m, 2Н), 2,52 (dd, J=9,0, 13,5 Гц, 1Н), 2,36-2,45 (m, 3Н), 2,16 (dd, J=5,5, 14,5 Гц, 1Н), 1,86-1,92 (m, 1Н), 1,76-1,83 (m, 1Н), 1,39-1,49 (m, 2Н), 1,41 (s, 9Н), 0,86 (t, J=7,5 Гц, 3Н).
Пример 3: получение соединения (А4) (стадия 3)
В раствор соединения (A3) (40,0 г, 92,5 ммоль) и толуола (311 мл) добавляли раствор NaHMDS в THF (93,2 мл, 185 ммоль, 2 М) при -10°C в течение периода времени, составляющего 7,5 минут. Наблюдали небольшое повышение температуры. Реакционную смесь перемешивали при -10°С в течение 50 минут. Затем температуру повышали до 5°С и выдерживали в течение ночи. Реакционной смеси затем обеспечивали достижение комнатной температуры и добавляли водный раствор NaCl (160 г, 5% вес/вес) в течение периода времени, составляющего 5 минут. Органическую фазу отделяли и выпаривали до сухого состояния под вакуумом. Остаток удаляли с ацетоном (200 мл) и остаток смешивали с ацетоном и щавелевой кислотой (8,30 г, 92,2 ммоль) и перемешивали в течение ночи при комнатной температуре. Смесь затем охлаждали на ледяной бане в течение 1 часа и фильтровали. Осадок на фильтре промывали холодным ацетоном (2х 50 мл) дважды, измельчали и высушивали под вакуумом при 50°С с получением соединения (А4-гемиоксалата) (35,0 г, 78%) в виде порошка только в виде цис-изомера, как определено посредством анализа 1Н ЯМР.
LC-MS: RT=0,61 минуты, [М+Н]+=396,3 масса/заряд.
1Н ЯМР (600 МГц, DMSO-d6) δ 7,07 (s, 1Н), 7,03 (s, 1Н), 6,06 (s, 2Н), 3,73 (br s, 1Н), 2,99-3,16 (m, 2Н), 2,73-2,90 (m, 4Н), 2,43 (br s, 1Н), 1,88-1,96 (m, 1Н), 1,60-1,80 (m, 3Н), 1,45-1,56 (m, 2Н), 1,35 (s, 9Н), 0,81 (t, J=7,5 Гц, 3Н).
Пример 4: получение соединения (А5) (стадия 4)
Получение свободного основания соединения (А4-гемиоксалата).
Смесь соединения (А4-гемиоксалата) (1000 г, 2,06 моль), гептана (8 л), воды (6 л) и водного раствора аммиака (500 мл, 6,76 моль, 25% вес/вес) перемешивали в течение 4 часов при 35-45°C. Органическую фазу отделяли и концентрировали посредством дистилляции до 1-2 литров при температуре больше 95°С. Затем добавляли хлорбензол (1,0 л) и смесь концентрировали посредством дистилляции под вакуумом до 1,5-2,0 литров с получением раствора соединения (А4) в виде свободного основания.
Образование соединения (А5).
Трифторуксусную кислоту (50,0 мл, 650 ммоль) добавляли в течение периода времени, составляющего 5-15 минут, в перемешанную смесь Р2О5 (30 г, 211 ммоль) и хлорбензола (200 мл) при 15-30°С. Полученную смесь перемешивали в течение 30 минут при комнатной температуре.
Затем добавляли раствор соединения (А4) в виде свободного основания в хлорбензоле (см. выше, соответствующего 50 г соединения (А4-гемиоксалата) (103 ммоль)) при 25-45°С в течение периода времени, составляющего 10-30 минут. Полученную смесь перемешивали при 35-40°С в течение 3 часов, а затем при комнатной температуре в течение ночи. В реакционную смесь добавляли IPA (125 мл) при комнатной температуре и смесь перемешивали при 40-45°С в течение 2 часов, а затем в течение ночи при комнатной температуре. Смесь в течение периода времени, составляющего 5-10 минут, добавляли в перемешанную смесь водного раствора аммиака (150 мл, 2,00 моль, 25% вес/вес) и воды (1,0 л) при 5-10°С. Смесь перемешивали при 35-45°С в течение 10 минут. Органическую фазу отделяли и водную фазу экстрагировали хлорбензолом (100 мл). В перемешанные объединенные органические фазы добавляли раствор TsOH-H2O (23,6 г, 124 ммоль) в IPA (150 мл). Смесь концентрировали на роторном испарителе под вакуумом при 50-70°С до объема ~50 мл. Добавляли IPA (100 мл) и смесь снова концентрировали до ~50 мл на роторном испарителе под вакуумом. Добавляли IPA (125 мл) и смесь перемешивали при комнатной температуре с обеспечением осаждения. Образованную суспензию охлаждали до 5°С и фильтровали. Осадок на фильтре дважды промывали холодным IPA (2х 20 мл, Б°С) и высушивали в вакуумной печи при 50°С с получением соединения (А5-тозилат) (35,6 г, 70%) в виде порошка.
LC-MS: RT=1,62 минуты, [М+Н]+=322,2 масса/заряд.
1Н ЯМР (600 МГц, DMSO-d6) δ 9,65 (br s, 1Н), 7,46-7,48 (m, 2H), 7,45 (s, 1H), 7,09-7,11 (m, 2H), 6,23 (d, J=5,5 Гц, 1H), 6,21 (d, J=5,5 Гц, 1H), 3,67-3,74 (m, 1H), 3,56 (dd, J=4,5, 16 Гц, 2H), 3,31-3,39 (m, 1H), 3,06-3,14 (m, 1H), 2,93-3,03 (m, 1H), 2,83-2,93 (m, 2H), 2,27 (s, 3H), 2,18-2,24 (m, 1H), 1,91-1,98 (m, 1H), 1,70-1,84 (m, 2H), 1,61-1,70 (m, 1H), 1,41-1,50 (m, 1H), 0,96 (t, J=7,5 Гц, 3Н).
Пример 5: получение соединения (Ib) (стадия 5)
Смесь соединения (А5-тозилата) (5,00 г, 10,1 ммоль), 5% Pd/C (561 мг, 0,101 ммоль, Johnson Matthey тип 424) и EtOH (50 мл) гидрировали при 4 бар водорода и 70°С в течение 48 часов. Реакционную смесь фильтровали через Arbocell ВС200 и фильтрат совместно выпаривали с толуолом несколько раз. Полученный раствор промывали разбавленным водным раствором аммиака (10% вес/вес), затем солевым раствором, высушивали над MgSO4, фильтровали и выпаривали до сухого состояния с получением соединения (Ib) (2,59 г) в виде твердого вещества.
LC-MS: RT=1,95 минуты, [М+Н]+=274,3 масса/заряд.
1H ЯМР (600 МГц, CDCl3) δ 6,63 (d, J=8,0 Гц, 1Н), 6,59 (d,J=8,0 Гц, 1Н), 5,92 (d, J=1,5 Гц, 1Н), 5,89 (d, J=1,5 Гц, 1Н), 3,15 (dd, J=5,0, 16,0 Гц, 1Н), 2,97-3,00 (m, 1Н), 2,82 (dd, J=5,0, 17,0 Гц, 1Н), 2,73 (ddd, J=5,5,10,5,13,5 Гц, 1Н), 2,56 (dd, J=11,0, 15,5 Гц, 1Н), 2,49 (ddd, J=5,5, 10,5, 13,5 Гц, 1Н), 2,21-2,30 (m, 1Н), 2,18 (dt, J=5,0, 10,5 Гц, 1Н), 1,87-1,92 (m, 1Н), 1,58-1,72 (m, 2Н), 1,42-1,58 (m, 1Н), 1,06-1,15 (m, 1Н), 0,89 (t, J=7,5 Гц, 3Н).
Пример 6: получение соединения (I) из соединения (Ib) (стадия 6)
В трехгорлую колбу объемом 2 л с механической подвесной мешалкой загружали соль (-)-O,O'-ди-п-толуоил-L-винной кислоты, соль (L-DTTA), соединения (Ib) (242 г, 367 ммоль), толуол (1250 мл), воду (375 мл) и 25% водный раствор аммиака (100 мл, 1340 ммоль). Смесь перемешивали при 20-25°С в течение 50 минут перед разделением фаз. Органическую фазу промывали смесью воды (170 мл) и 25% водного раствора аммиака (35 мл, 470 ммоль). Отделенную органическую фазу концентрировали при 50°С под вакуумом до практически сухого состояния. Остаток три раза последовательно концентрировали при 50°С под вакуумом с толуолом (3×250 мл) до практически сухого состояния. Остаток растворяли и переносили в трехгорлую колбу объемом 4 л с толуолом (1200 мл). Раствор охлаждали до 4°С и в течение 25 минут добавляли BCl3 в толуоле (800 мл, 800 ммоль, 1 М). Реакционную смесь перемешивали в течение 3 часов при 0-5°С. Еще через 1,8 часа при 0-5°С реакционную смесь гасили посредством добавления метанола (500 мл) в течение 20 минут. Полученную смесь нагревали в течение ночи при 55°С и затем нагревали с обратным холодильником, где 500 мл растворителя отгоняли. Добавляли метанол (500 мл) и смесь нагревали с обратным холодильником, где 500 мл растворителя отгоняли. Добавляли IPA (500 мл) и смесь нагревали с обратным холодильником, где 500 мл растворителя отгоняли. Добавляли IPA (1200 мл) и смесь нагревали до 75°С в течение 1,5 часа. Суспензию охлаждали до 20°С через 1,5 часа и затем продукт отфильтровывали. Осадок на фильтре два раза промывали смесью IРА/толуол (1:1, 2×200 мл). Посредством высушивания при 50°С под вакуумом получали соль HCl соединения (I) (105 г, 96%) в виде твердого вещества.
1Н ЯМР (600 МГц, DMSO-d6) δ 6,53 (d, J=8,0 Гц, 1Н), 6,37 (d, J=8,0 Гц, 1Н), 3,01 (dd, J=4,5, 15,0 Гц, 1Н), 2,89 (кажущийся d, J=11,0 Гц, 1Н), 2,79 (dd, J=5,0, 17,5 Гц, 1Н), 2,65-2,70 (m, 1Н), 2,36 (dd, J=11,0,15,5 Гц, 1Н), 2,25-2,30 (m, 1Н), 2,12 (dt, J=2,5,12,0 Гц, 1Н), 1,99-2,05 (m, 2Н), 1,79-1,84 (m, 1Н), 1,58-1,63 (m, 1Н), 1,49-1,57 (m, 1Н), 1,35-1,47 (m, 3Н), 1,05 (dq, J=4,0, 13,0 Гц, 1Н), 0,84 (t, J=7,5 Гц, 3Н).
Пример 7: получение соединения (А2-геми-L-тартрат) из соединения (a2i) (подстадия (ii) стадии 1 с применением катализатора на основе Pt и подстадия iii стадии 1)
Смесь соединения (a2i) (12,4 г, 32,0 ммоль) и платины на угле (Johnson Matthey тип 128М; 4,88% Pt; 52,6% вес/вес воды; 2,16 г, 0,256 ммоль) в Me-THF (31 мл) гидрировали при 60°С и 4 бар водорода в течение 24 часов. Реакционную смесь фильтровали и выпаривали до сухого состояния с получением неочищенного соединения (a2ii). В неочищенное соединение (a2ii) в МеОН (67 мл) при перемешивании с обратным холодильником медленно добавляли раствор L-винной кислоты (2,40 г, 16,0 ммоль) в МеОН (20 мл). Смеси обеспечивали медленное охлаждение до комнатной температуры в течение ночи при перемешивании с затравкой (~20 мг чистого соединения (А2-геми-L-тартрата)) при ~62°С. Полученную суспензию фильтровали и осадок на фильтре промывали с помощью МеОН (10 мл) и высушивали в вакуумной печи при 40°С с получением соединения (А2-геми-L-тартрата) (4,90 г, 33%) в виде белого твердого вещества с 99,2% энантиомерным избытком соединения (А2) согласно анализу посредством хиральной HPLC.
Пример 8: получение соединения (А1) из соединения (a6i) (подстадия 0')
В тщательно перемешенный порошок цинка (Umicore) (2,84 г, 43,5 ммоль) в THF (15,0 мл) добавляли метансульфоновую кислоту (83 мкл, 1,28 ммоль). Смесь нагревали с обратным холодильником в течение 30 минут. Затем добавляли раствор соединения (a6i) (5,00 г, 25,6 ммоль) в THF (15,0 мл) с последующим добавлением трет-бутил-2-бромацетата (6,48 г, 33,2 ммоль) по каплям в течение периода времени, составляющего 1 час и 40 минут, с обратным холодильником. Смесь перемешивали с обратным холодильником в течение 1 часа, нагревание прекращали и перемешанную реакционную смесь охлаждали до комнатной температуры в течение ночи. Добавляли реакционную смесь при перемешивании в водн. растворе HCl (38 мл, 76 ммоль, 2,0 М) при 0°С. Полученную смесь концентрировали под вакуумом с удалением большей части THF и затем охлаждали на бане с ледяной водой с тщательным перемешиванием и затравкой с чистым образцом соединения (А1).
Образованную суспензию фильтровали холодной (~5°С) и осадок на фильтре промывали смесью вода:ТНР с соотношением 10:1 и высушивали в вакуумной печи при 40°С в течение ночи с получением соединения (А1) (7,72 г, 97%) в виде слегка желтоватого твердого вещества.
LC-MS (способ: 555) RT=2,84 и 3,41 минуты (деа пика представляют кето- и енольный таутомер соединения (А1))
1Н ЯМР (600 МГц, DMSO-d6) δ 7,07 (s, 1Н), 6,89 (s, 1Н), 6,06 (s, 2Н), 3,90 (s, 2Н), 3,53 (s, 2Н), 1,41 (s, 9Н).
Пример 9: получение соединения (a2i) из соединения (a6i) (подстадия S1 стадии 0)
Смесь порошка цинка (Umicore) (10,0 г, 153 ммоль) и метансульфоновой кислоты (332 мкл, 5,11 ммоль) bTHF (60 мл) нагревали с обратным холодильником в течение 30 минут при перемешивании.
Добавляли раствор соединения (a6i) (20,0 г, 102 ммоль) в THF (60 мл). Затем добавляли трет-бутил-2-бромацетат (25,9 г, 133 ммоль) с обратным холодильником медленно в течение периода времени, составляющего 2 часа. Реакционную смесь перемешивали с обратным холодильником в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры.
Реакционную смесь отфильтровывали от избытка цинка и охлаждали на бане с ледяной водой. Затем медленно добавляли уксусную кислоту (7,6 мл, 133 ммоль) при перемешивании. Смесь совместно выпаривали с МеОН несколько раз с удалением THF с помощью азеотропной смеси THF-MeOH. Получали суспензию, в которую добавляли раствор гидрохлорида 3-хлорпропан-1-амина (33,2 г, 256 ммоль) в МеОН (50 мл). Смесь перемешивали в течение ночи при комнатной температуре. Смесь фильтровали и осадок на фильтре промывали небольшим количеством МеОН и высушивали в вакуумной печи при 40°С с получением соединения (a2i) (32,0 г, 81%) в виде белого твердого вещества.
LC-MS (способ: 555) RT=2,84 и 3,41 минуты (два пика представляют кето- и енольный таутомер соединения (А1), наблюдаемые вследствие гидролиза соединения (a2i) до соединения (А1) при анализе LC-MS).
1Н ЯМР (600 МГц, DMSO-d6) δ 8,57 (t, J=6,5 Гц, 1Н), 7,10 (s, 1Н), 6,89 (s, 1Н), 6,08 (s, 2Н), 3,85 (s, 1Н), 3,67 (t, J=6,5 Гц, 2Н), 3,58 (s, 2Н), 3,33 (q, J=6,5 Гц, 2Н), 1,95 (q, J=6,5 Гц, 2Н), 1,36 (s, 9Н).
Пример 10: получение соединения (a6i) из соединения (a5i)
В тщательно перемешанную смесь соединения (a5i) (30,0 г, 186 ммоль) и н-гептана (200 мл) добавляли сульфурилхлорид (17,4 мл, 214 ммоль) при 0°С. Охлаждение прекращали и смеси обеспечивали нагревание до комнатной температуры с тщательным перемешиванием. Смесь перемешивали в течение 4 часов при комнатной температуре. Образованную суспензию фильтровали и осадок промывали гептаном и высушивали под вакуумом при 40°С с получением соединения (a6i) (33,4 г, 92%) в виде белого твердого вещества.
LC-MS (способ: 555) RT=2,28 минуты, [М+Н]+=195,1 масса/заряд.
1Н ЯМР (600 МГц, CDCl3) δ 6,95 (s, 1Н), 6,88 (s, 1Н), 6,02 (s, 2Н), 3,74 (s, 2Н).
Пример 11: получение соединения A4-HCl в большом масштабе из соединения А2-геми-L-тартрата (стадии 2 и 3)
В охлажденную до 10°С суспензию, содержащую соединение (А2-геми-L-тартрат) (750 г, 805,9 ммоль, 1 экв.) и триацетоксиборгидрид натрия (597,8 г, 2820 ммоль, 3,5 экв.) в THF (3750 мл, 5 объемов), добавляли холодный (10°С) раствор пропионового альдегида (121,7 г, 150,2 мл, 2100 ммоль, 2,6 экв.) в THF (1500 мл, 2 объема) в течение периода времени, составляющего 30 минут. Затем добавляли ледяную уксусную кислоту (169,4 г, 161,5 мл, 2820 ммоль, 3,5 экв.) при 21°C в течение 5 минут и реакционную смесь перемешивали при 23°С в течение ночи. Затем 10% объема отгоняли под вакуумом при 25°С перед добавлением воды (2250 мл, 3 объема) и раствор перемешивали при 22°С в течение 1 часа. Затем добавляли толуол (2250 мл, 3 объема) с последующим добавлением 25% аммиака (880 мл, 1176 ммоль, 14,6 экв.) до рН 8 и раствор перемешивали при 22°С в течение 1 часа перед разделением двух фаз. Водную фазу экстрагировали толуолом (2250 мл, 3 объема) и объединенные органические фазы промывали 7,5% солевым раствором (1500 мл, 2 объема). Органический раствор полностью концентрировали при пониженном давлении при 65°С с получением неочищенного соединения (A3) в виде масла (696,9 г, 100%, 98,4% площади при HPLC, RT 14,96 мин).
В неочищенное соединение (A3) (696,6 г, 805,9 ммоль) добавляли толуол (6000 мл, 8 объемов) и раствор охлаждали до 0°С перед добавлением 1 М NaHMDS в THF (3380 мл, 3380 ммоль, 2,1 экв.) в течение 100 минут, при этом температура смеси не превышала 5°С. Реакционную смесь перемешивали при 2°С в течение 3,5 часов перед нагреванием раствора до 20°С в течение 13 минут. Затем реакционную смесь гасили посредством добавления 5% солевого раствора (3750 мл, 5 объемов) и смесь перемешивали в течение 10 минут перед разделением двух фаз. Органическую фазу последовательно промывали 10% уксусной кислотой (2310 мл, 4030 ммоль, 2,5 экв.) и водой (3750 мл, 5 объемов) перед полным концентрированием органической фазы при пониженном давлении при 60°С. Затем добавляли изопропанол (2250 мл, 3 объема) и органическую фазу снова полностью концентрировали при пониженном давлении при 60°С. Затем добавляли изопропанол (750 мл, 1 объем) и раствор переносили в предварительно перемешанный раствор (22°С, предварительно перемешивали в течение 1 часа) изопропанола (750 мл, 1 объем), гептана (6000 мл, 8 объемов) и ацетилхлорида (229,2 мл, 3220 ммоль, 2 экв.) и наблюдали осаждение. Смесь перемешивали при 20°С в течение 1 часа перед охлаждением суспензии до 4°С в течение 30 минут и перемешивали при этой температуре в течение 80 минут. Затем продукт отфильтровывали и осадок на фильтре промывали холодным (4°С) гептаном (750 мл, 1 объем). Влажный продукт высушивали под вакуумом при 50°C в течение ночи. Таким образом получали соединение (A4-HCl) (555,1 г, 80%) в виде белого порошка.
Чистота согласно HPLC: 99,3% площади, 99,2 вес/вес %.
1Н ЯМР (400 МГц, CDCl3) δ 12,50 (s, 1Н), 7,05 (s, 1Н), 6,82 (s, 1Н), 5,99 (m, 2Н), 3,87 (dd, J=15,8, 6,7 Гц, 1Н), 3,65 (m, 1Н), 3,45 (m, 1Н), 3,20 (ddd, J=11,0, 9,7, 4,4 Гц, 1Н), 3,08 (tdd, J=12,7, 4,9, 2,5 Гц, 1Н), 2,92 (m, 2Н), 2,77 (tdd, J=12,0, 8,5, 3,1 Гц, 1Н), 2,49 (m, 1Н), 2,21 (m, 1Н), 1,84 (m, 2Н), 1,66 (dtd, J=13,5, 11,7, 4,0 Гц, 1Н), 1,52 (m, 1Н), 1,39 (s, 9Н), 0,77 (t, J=7,3 Гц, 3Н).
СПИСОК ЛИТЕРАТУРНЫХ ИСТОЧНИКОВ
US4543256
WO 2001/078713
WO 02/100377
WO 2009/026934,
WO 2009/026935
W О2010/097092
Alexander et Crutcher, (1990) Trends in Neuroscience 13: 266-71;
Bibbiani et al., Chase Experimental Neurology (2005), 192: 73-78;
Bruckner and co-workers in Synthesis 2008,14: 2229-2246;
Campbell et al., Neuropharmacology (1982); 21(10): 953-961;
Cannon et al., J. Heterocyclic Chem. (1980); 17: 1633-1636;
Delong, (1990) Trends in Neuroscience 13: 281-5;
Fan et al., Bioorg. Med. Chem. Lett. 2008, 18: 6236-6239;
Gerfen et al, Science (1990) 250: 1429-32;
Giardina and Williams; CNS Drug Reviews (2001), Vol.7 (3): 305-316);
Grosset et al., Acta Neurol Scand. (2013), 128:166-171;
Hauser et al., Movement Disorders (2016), Vol.32 (9): 1367-1372;
Kotsuki et al., J. Org. Chem. 1992, 57: 5036-5040);
В Liu et al., J. Med. Chem. (2006), 49: 1494-1498;
Liu et al., Bioorganic Med. Chem. (2008), 16: 3438-3444;
S. Nishimura, Wiley 2001, Handbook of heterogeneous hydrogenation;
Poewe et al., Nature Review, (2017) vol 3 article 17013: 1-21;
Sprenger and Poewe, CNS Drugs (2013), 27: 259-272;
Sozio et al., Exp.Opin. Drug Disc. (2012); 7(5): 385-406;
Stahl and Wermuth (Eds) "Handbook of Pharmaceutical salts. Properties, selection, and use", Wiley-VCH, 2008;
Настоящее изобретение относится к методу получения (4aR,10aR)-1-пропил-1,2,3,4,4а,5,10,10а-октагидробензо[g]хинолин-6,7-диола (формула I) и (6aR,10aR)-7-пропил-6,6а,7,8,9,10,10а,11-октагидро-[1,3]диоксоло[4',5':5,6]бензо[1,2-g]хинолина (формула Ib) и их солей, которые применяются в лечении нейродегенеративных заболеваний и нарушений, таких как болезнь Паркинсона.
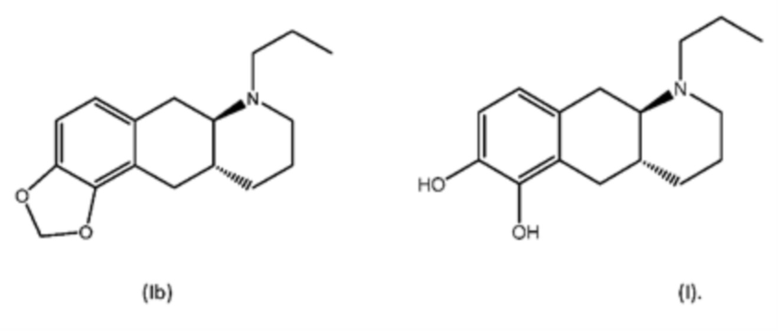

Изобретение также относится к новым промежуточным соединениям из указанного способа. Способ получения соединения с формулой (I) из соединения с формулой (Ib) включает следующие стадии. Стадия 0), подстадия (i) проведение реакции соединения (A1) с 3-хлорпропан-1-амином с получением соединения (a2i) или подстадия (S1) с проведением реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с образованием смеси, затем обработка смеси с подстадии (S1) уксусной кислотой, затем проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i). Стадия 1), подстадия (ii) восстановления соединения (a2i), полученного на подстадии (i) или подстадии (S1), с получением соединения (a2ii), затем подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением A2-геми-L-тартрата, согласно нижеуказанной схеме реакции:
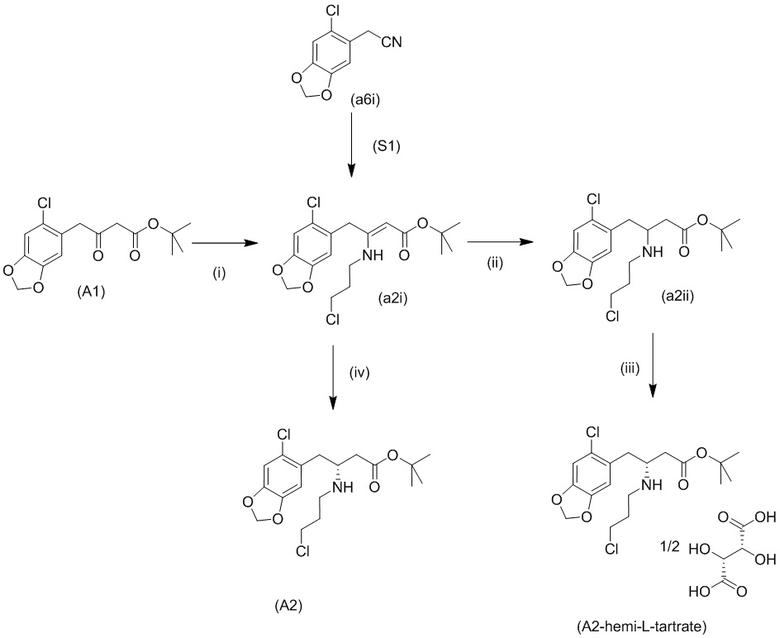
затем осуществляют стадию, на которой соединение (A2) или его соль вступает в реакцию с получением соединения (A3) или его соли согласно нижеуказанной схеме реакции:

затем проводят реакцию соединения (A3) или его соли с получением соединения A4 или его соли согласно нижеуказанной схеме реакции:
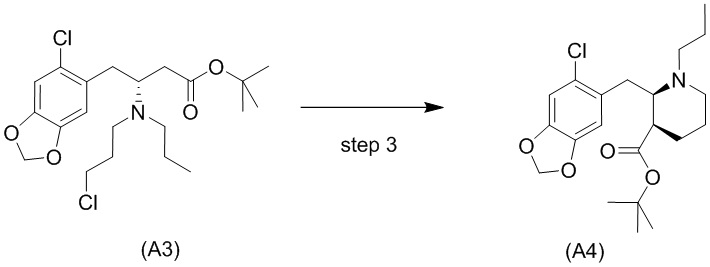
затем проводят реакцию соединения (A4) или его соли с получением соединения (A5) или его соли согласно нижеуказанной схеме реакции:

затем проводят восстановление соединения (A5) или его соли с получением соединения (Ib) или его соли согласно нижеуказанной схеме реакции:
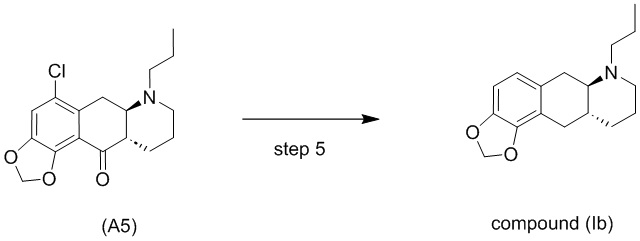 .
.
Техническим результатом изобретения являются обеспечение улучшенных способов производства целевых соединений (I) и (Ib), включающих высокий выход продуктов и исключение или уменьшение синтезов, мешающих крупномасштабному производству. 7 н. и 14 з.п. ф-лы, 11 пр.
1. Способ изготовления соединения (I) с нижеуказанной формулой
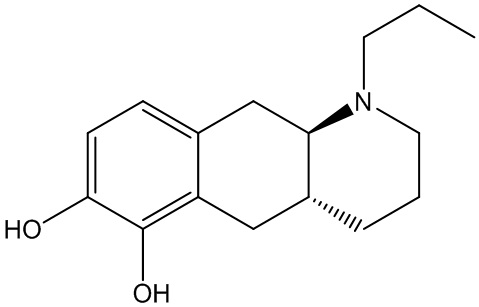
(I)
из соединения (Ib) с нижеуказанной формулой

(Ib),
где соединение (Ib) получают с помощью способа, включающего стадии:
стадия 0)
подстадия (i) проведения реакции соединения (A1) с 3-хлорпропан-1-амином с получением соединения (a2i) или
подстадия (S1) проведение реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с образованием смеси, затем обработка смеси с подстадии (S1) уксусной кислотой, затем
проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i);
затем стадия 1)
подстадия (ii) восстановления соединения (a2i), полученного на подстадии (i) или подстадии (S1), с получением соединения (a2ii), затем
подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением соединения (A2-геми-L-тартрат)
согласно нижеуказанной схеме реакции:
 ;
;
затем осуществляют стадию, на которой соединение (A2) или его соль вступает в реакцию с получением соединения (A3) или его соли согласно нижеуказанной схеме реакции:
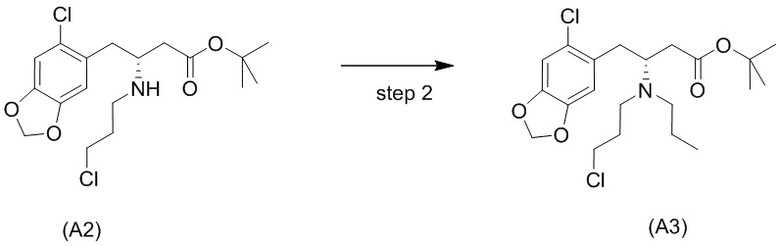 ;
;
затем проводят реакцию соединения (A3) или его соли с получением соединения A4 или его соли согласно нижеуказанной схеме реакции:
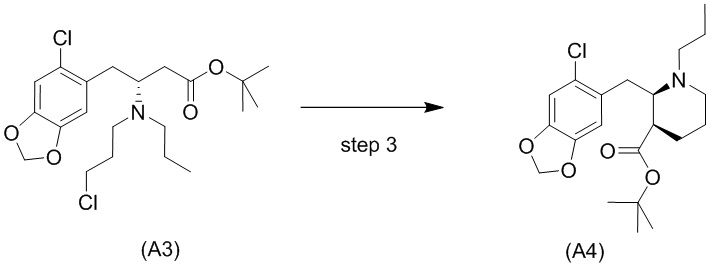 ;
;
затем проводят реакцию соединения (A4) или его соли с получением соединения (A5) или его соли согласно нижеуказанной схеме реакции:
 ;
;
затем проводят восстановление соединения (A5) или его соли с получением соединения (Ib) или его соли согласно нижеуказанной схеме реакции:
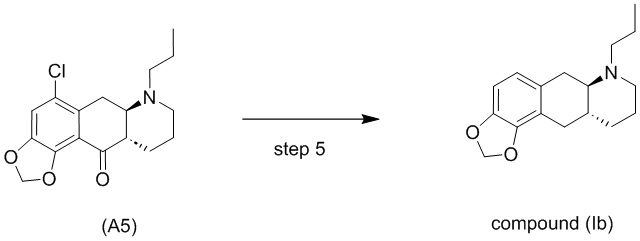 .
.
2. Способ по п. 1, где соединение (Ib) получают с помощью способа, включающего стадии:
стадия 0)
подстадия (S1)
проведение реакции соединения (a6i) с трет-бутил-2-бромацетатом и цинком с образованием смеси, затем обработка смеси с подстадии (S1) уксусной кислотой, затем проведение реакции указанной смеси с гидрохлоридом 3-хлорпропан-1-амина с получением соединения (a2i);
затем стадия 1)
подстадия (ii) восстановления соединения (a2i), полученного на подстадии (S1), с получением соединения (a2ii), затем
подстадия (iii) разделения соединения (a2ii) с использованием L-винной кислоты с получением соединения (A2-геми-L-тартрат).
3. Соединение нижеуказанной формулы (A2)
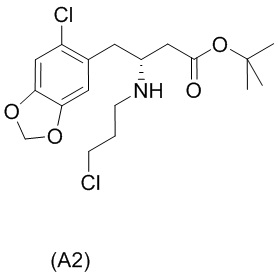
или его соль.
4. Способ по любому из пп. 1, 2, где восстановление на подстадии (ii) стадии 1) осуществляют в присутствии восстанавливающего средства, выбранного из группы, состоящей из NaBH3CN, триацетоксиборгидрида натрия (STAB), борана, 5-этил-2-метилпиридинборана (PEMB) и NaBH4.
5. Способ по любому из пп. 1, 2, где восстановление на подстадии (ii) стадии 1) осуществляют с использованием платинового катализатора, предпочтительно платины на углероде.
6. Способ по пп. 1, 2 и 4, 5, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
2) проведение реакции соединения (A2) или соединения (A2-геми-L-тартрат) с пропаналем в присутствии восстанавливающего средства с получением соединения (A3) согласно нижеуказанной схеме реакции a) или b):
схема a)
схема b)
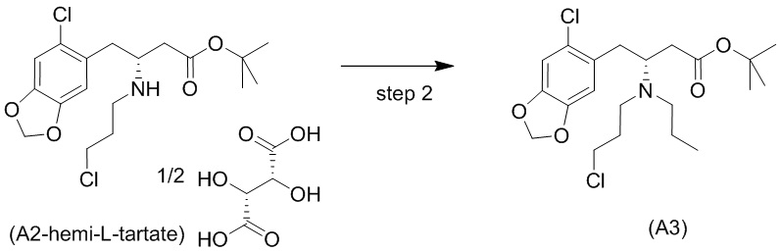 .
.
7. Соединение нижеуказанной формулы (A3)
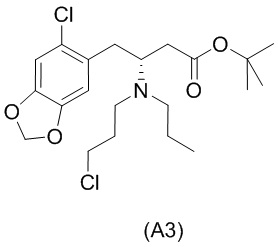
или его соль.
8. Способ по любому из пп. 1, 2, 4-6, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
3) проведение реакции соединения (A3) с сильным основанием с получением соединения A4 согласно нижеуказанной схеме реакции:
,
за которой необязательно следует выделение соединения (A4) или его соли.
9. Способ по п. 8, где A4 выделяют в виде HCl-соли, как изображено ниже:
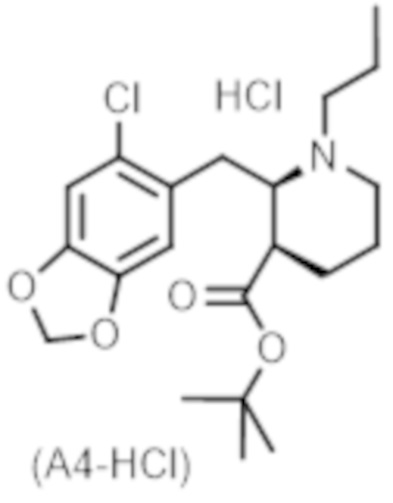 .
.
10. Соединение нижеуказанной формулы (A4)
или его соль.
11. Способ по любому из пп. 1, 2, 4-6 и 8, 9, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
4) проведение внутримолекулярного ацилирования по Фриделю-Крафтсу соединения (A4) с получением соединения (A5) согласно нижеуказанной схеме реакции:
,
за которой необязательно следует выделение соединения (A5) или его соли.
12. Соединение нижеуказанной формулы (A5)
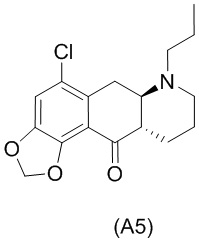
или его соль.
13. Способ по любому из пп. 1, 2, 4-6, 8, 9 и 11, где соединение (Ib) получают с помощью способа, включающего следующую стадию:
5) восстановление соединения (A5) или его соли с получением соединения (Ib) или его соли согласно нижеуказанной схеме реакции:
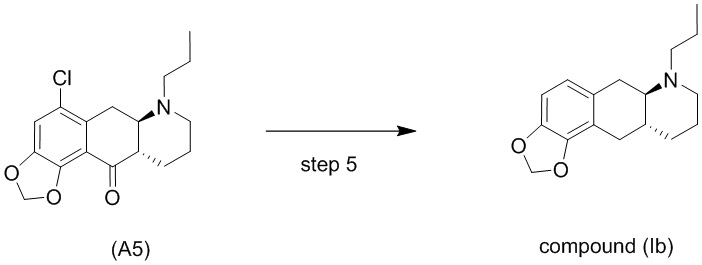 .
.
14. Способ по любому из пп. 1, 2, 4-6, 8, 9, 11 и 13, где соединение (I) получают из соединения (Ib) с помощью следующей стадии:
6) проведение реакции соединения (Ib) с кислотой Льюиса или кислотой Бренстеда, выбранной из группы, состоящей из BCl3, BBr3 и HBr, с получением соединения (I) согласно нижеуказанной схеме реакции:
 .
.
15. Способ изготовления соединения (I) с нижеуказанной формулой
(I)
из соединения (Ib) с нижеуказанной формулой
(Ib),
где соединение (Ib) получают с помощью способа, включающего стадии:
восстановления соединения (A5) или его соли с получением соединения (Ib) или его соли согласно нижеуказанной схеме реакции:
;
и проведения реакции соединения (Ib) с кислотой Льюиса или кислотой Бренстеда, выбранной из группы, состоящей из BCl3, BBr3 и HBr, с получением соединения (I) или его соли согласно нижеуказанной схеме реакции:
.
16. Способ по п. 14 или 15, где стадия 6) включает проведение реакции соединения (Ib) с BCl3 с получением соединения (I) или его соли.
17. Способ по п. 14 или 15, где стадия 6) включает проведение реакции соли соединения (Ib) с основанием с получением соединения (Ib) в виде свободного основания с последующей реакцией соединения (Ib) с BCl3 с получением соединения (I) или его соли.
18. Способ по п. 14 или 15, где стадия 6) включает проведение реакции соли соединения (Ib) с водным раствором Na2CO3, K2CO3 или аммиака с получением соединения (Ib) в виде свободного основания с последующей реакцией соединения (Ib) с BCl3 с получением соединения (I) или его соли.
19. Способ по п. 14 или 15, где стадия 6) включает проведение реакции соли (-)-O,O′-ди-п-толуоил-L-винной кислоты (соли L-DTTA) соединения (Ib) с основанием с получением соединения (Ib) в виде свободного основания с последующей реакцией соединения (Ib) с BCl3 с получением соединения (I) или его соли, такой как HCl-соль соединения (I).
20. Соединение нижеуказанной формулы (a2i)
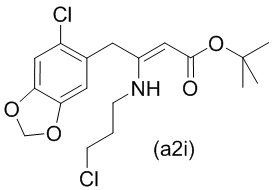
или его соль.
21. Соединение нижеуказанной формулы (a2ii)

или его соль.
| WO 2009026934 A1, 05.03.2009 | |||
| WO 2009026935 A1, 05.03.2009 | |||
| WO 2010097092 A1, 02.09.2010. |

