Изобретение относится к области медицины и может быть использовано в молекулярно-генетической диагностике болезни Вильсона-Коновалова, вызывающей избыток меди в организме, приводящий к отложению меди в различных органах человека.
Болезнь Вильсона-Коновалова (БВК, WD) (OMIM 277900) или гепатолентикулярная дегенерация (ГЛД) - это аутосомно-рецессивное наследственное заболевание, обусловленное нарушением метаболизма меди, которое возникает в результате различных мутаций в гене ATP7B. Несмотря на давнюю историю, хорошую изученность и успешное лечение, БВК до сих пор остается тяжело протекающим заболеванием, которое приводит к утрате трудоспособности, деструктивно влияя на качество жизни пациентов [Еремина Е.Ю. Болезнь Вильсона-Коновалова. Вестник современной клинической медицины. 2011;4(1):38-46.]. Согласно официальной статистике выявляемость БВК составляет 1:35 000-45 000 населения. Вместе с тем, периодически появляются указания на регионы с высокими показателями заболеваемости (превышающие традиционные в 3 - 4 раза) [Shimizu N, Fujiwara J, Ohnishi S, Sato M, Kodama H, Kohsaka T, Inui A, Fujisawa T, Tamai H, Ida S, Itoh S, Ito M, Horiike N, Harada M, Yoshino M, Aoki T. Effects of long-term zinc treatment in Japanese patients with Wilson disease: efficacy, stability, and copper metabolism. Transl Res. 2010 Dec;156(6):350-7. doi: 10.1016/j.trsl.2010.08.007. Epub 2010 Sep 23. PMID: 21078496.; Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, Klaffke S, Joyce CJ, Dhawan A, Hadzic N, Mieli-Vergani G, Kirk R, Elizabeth Allen K, Nicholl D, Wong S, Griffiths W, Smithson S, Giffin N, Taha A, Connolly S, Gillett GT, Tanner S, Bonham J, Sharrack B, Palotie A, Rattray M, Dalton A, Bandmann O. A genetic study of Wilson's disease in the United Kingdom. Brain. 2013 May;136(Pt 5):1476-87. doi: 10.1093/brain/awt035. Epub 2013 Mar 21. PMID: 23518715; PMCID: PMC3634195.]. В России ежегодная выявляемость БВК составляет 1:167000, при гетерозиготном носительстве 1:100 и расчетной распространенности - 1:10000, что указывает на проблемы диагностики данного заболевания [Баязутдинова Г.М., Щагина О.А., Поляков А.В. Мутация с.3207C>A гена АТР7В - наиболее частая причина гепатолентикулярной дегенерации в России: частота и причина распространения. Медицинская генетика. 2018;17(4):25-30. DOI: 10.25557/2073-7998.2018.04.25-30.].
БВК принадлежит к группе заболеваний, которые требуют диагностики на ранних этапах развития болезни. Диагноз БВК определяется сочетанием клинических проявлений и лабораторных показателей, которые указывают на нарушение обмена меди с ее накоплением в печени и мозговой ткани - снижением содержания церулоплазмина в сыворотке крови и повышения суточной экскреции меди с мочой. Однако эти стандартные тесты на раннем этапе заболевания могут давать как ложноположительные, так и ложноотрицательные результаты. Невозможность диагностировать пациента с БВК на раннем этапе заболевания может привести или к потере возможностей патогенетической терапии, или к несоответствующему введению потенциально токсичных препаратов таким пациентам при ложноположительной диагностике [European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease. J Hepatol 2012;56(03):671-685.; Ferenci P, Stremmel W, Członkowska A, Szalay F, Viveiros A, Stättermayer AF, Bruha R, Houwen R, Pop TL, Stauber R, Gschwantler M, Pfeiffenberger J, Yurdaydin C, Aigner E, Steindl-Munda P, Dienes HP, Zoller H, Weiss KH. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease. Hepatology. 2019 Apr;69(4):1464-1476. doi: 10.1002/hep.30280. Epub 2019 Mar 1. PMID: 30232804.].
Клиническая диагностика БВК затруднена в связи с широким спектром клинических проявлений и с отсутствием типичных симптомов у пациентов с этим заболеванием. Подозрение на БВК должно возникать в каждом случае необъяснимой печеночной, неврологической или психической дисфункции. При этом методы и тесты для диагностики БВК значительно отличаются у пациентов с печеночной дисфункцией и психоневрологической симптоматикой [European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease. J Hepatol 2012;56(03):671-685.; Ferenci P, Stremmel W, Członkowska A, Szalay F, Viveiros A, Stättermayer AF, Bruha R, Houwen R, Pop TL, Stauber R, Gschwantler M, Pfeiffenberger J, Yurdaydin C, Aigner E, Steindl-Munda P, Dienes HP, Zoller H, Weiss KH. Age and Sex but Not ATP7B Genotype Effectively Influence the Clinical Phenotype of Wilson Disease. Hepatology. 2019 Apr;69(4):1464-1476. doi: 10.1002/hep.30280. Epub 2019 Mar 1. PMID: 30232804.].
В настоящее время ни один из доступных лабораторных тестов нельзя признать универсальным и специфичным для ранней диагностики БВК [European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease. J Hepatol 2012;56(03):671-685.]. Для точной постановки диагноза используется соответствие критериям Лейпцига, которые были приняты на 8-м Международном совещании по БВК и болезни Менкеса и приняты в Европейской ассоциации по изучению печени [European Association for Study of Liver. EASL clinical practice guidelines: Wilson’s disease. J Hepatol 2012;56(03):671-685.]. Ключевыми клиническими диагностическими признаками, которые легли в основу формирования критериев Лейпцига, стали признаки печеночной дисфункции, острый гемолиз с почечной недостаточностью, расстройства двигательных функций, нервно-психические расстройства и кольца Кайзера-Флейшера на роговице [Ferenci P, Merle U, FolhoVer A, Evstatiev R, Yurdaydin C, Bruha R et al (2005) Phenotype-genotype correlations in Wilson Disease (WD)-results of a multinational study. Hepatology 42(Suppl 1):258A]. Данная система оценки БВК обеспечивает хорошую диагностическую точность у пациентов с ярко выраженными симптомами болезни, но не способна продемонстрировать высокую эффективность у пациентов на ранних этапах заболевания или у их родственников [Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010 Dec;52(6):1948-56. doi: 10.1002/hep.23910. Epub 2010 Oct 21. PMID: 20967755.].
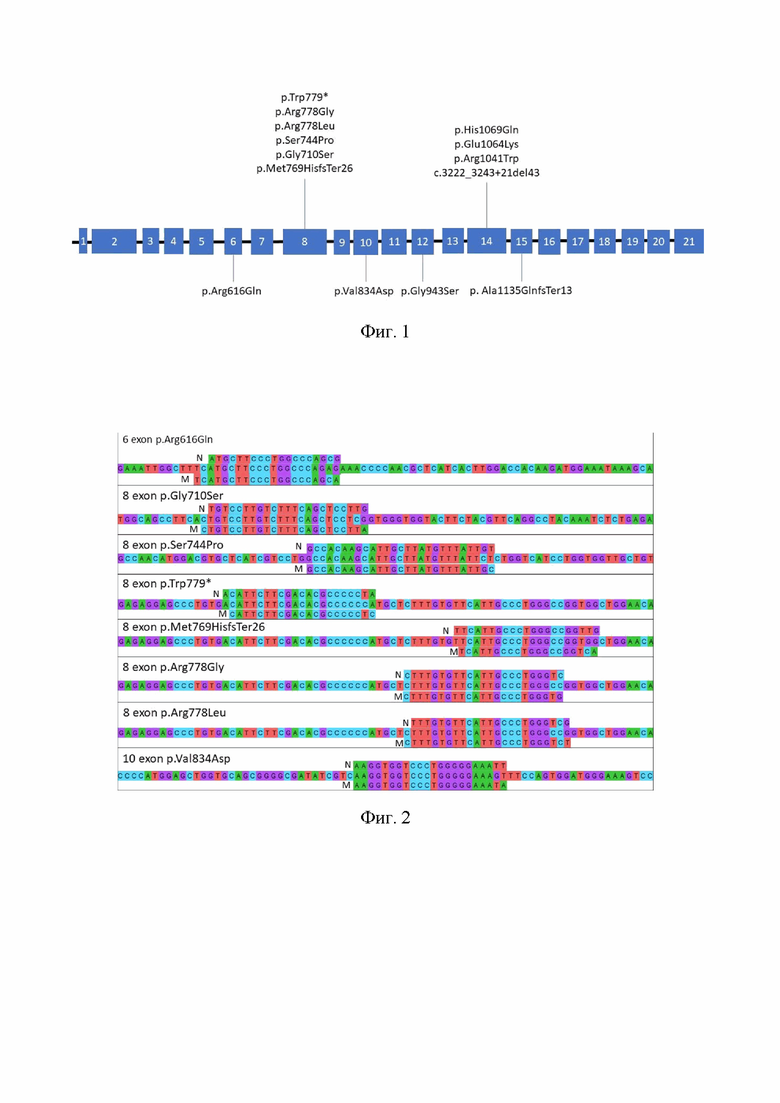
Прямая молекулярно-генетическая диагностика затруднена из-за наличия 800 возможных мутаций в гене ATP7B (http://www.wilsondisease.med.ualberta.ca/ найдено 10.04.2023). Однако тип мутации играет роль в прогнозировании клинических проявлений. Так же, несколько частых мутаций демонстрируют своеобразное глобальное распределение [Ferenci P. 2006. Regional distribution of mutations of the ATP7B gene in patients with Wilson Disease: impact on genetic testing. Hum Genet 120:151-159.]. Эта особенность используется для установления характерного спектра мутаций для региона и дальнейшего облегчения молекулярно-генетической диагностики и разработки тест-систем.
На сегодняшний день известно несколько технических решений для выявления мутаций гена ATP7B.
Известен способ поиска мутантных аллелей гена ATP7B с помощью метода парно-концевого чтения на основе платформы массового параллельного секвенирования (NGS, next generation sequencing), позволяющее выявить весь спектр мутаций [Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, Klaffke S, Joyce CJ, Dhawan A, Hadzic N, Mieli-Vergani G, Kirk R, Elizabeth Allen K, Nicholl D, Wong S, Griffiths W, Smithson S, Giffin N, Taha A, Connolly S, Gillett GT, Tanner S, Bonham J, Sharrack B, Palotie A, Rattray M, Dalton A, Bandmann O. A genetic study of Wilson's disease in the United Kingdom. Brain. 2013 May;136(Pt 5):1476-87. doi: 10.1093/brain/awt035. Epub 2013 Mar 21. PMID: 23518715; PMCID: PMC3634195.]. Осуществление данного изобретения предполагает выделение ДНК из образцов биоматериалов пациентов с последующим полным прочтением всего гена ATP7B, представляющего собой очень протяженный фрагмент ДНК длиной не менее 6,5 тыс. пар нуклеотидов. Несмотря на то, что такой подход является достаточно надежным, он имеет несколько существенных недостатков, препятствующих его широкому использованию в клинической практике, таких как высокая цена проведения исследования и необходимого для него оборудования, высокая трудоемкость и высокие требования к квалификации лабораторного работника [Chang IJ, Hahn SH. The genetics of Wilson disease. Handb Clin Neurol. 2017;142:19-34. doi: 10.1016/B978-0-444-63625-6.00003-3. PMID: 28433102; PMCID: PMC5648646.].
Таким образом, предложенный способ не может быть применен массово в повседневной практике медицинских учреждений, а с учетом достаточной распространенности мутаций в данном гене среди населения, для оказания своевременной медицинской помощи необходимо проведение скрининга на выявление молекулярно-генетической причины заболевания фактически в каждом крупном населенном пункте или районом центре. Что не может быть осуществлено посредством заявленного выше изобретения.
Другим техническим решением для выявления генетических дефектов гена ATP7B является использование секвенирования по Сэнгеру [Lepori MB, Zappu A, Incollu S, Dessì V, Mameli E, Demelia L, Nurchi AM, Gheorghe L, Maggiore G, Sciveres M, Leuzzi V, Indolfi G, Bonafé L, Casali C, Angeli P, Barone P, Cao A, Loudianos G. Mutation analysis of the ATP7B gene in a new group of Wilson's disease patients: contribution to diagnosis. Mol Cell Probes. 2012 Aug;26(4):147-50. doi: 10.1016/j.mcp.2012.03.007. Epub 2012 Mar 29. PMID: 22484412.]. Благодаря высокой надежности и высокоинформативным результатам этот метод заслужил широкое распространение в большинстве стран мира.
Минусами данного метода является то, что для проведения секвенирования необходимо пройти множество этапов: выделить ДНК из целевого органа или его части, провести ПЦР, очистить смесь для дальнейшего использования, провести пробоподготовку для секвенирования и само секвенирование. Такая многоэтапность увеличивает вероятность ошибки, является достаточно дорогой из-за использования большого количества лабораторного оборудования и увеличивает время исследования. Поэтому, в настоящее время, секвенирование по Сенгеру не используется широко в целях диагностики заболевания, но благодаря, высокой точности и информативности он остается «золотым стандартом» и применяется для подтверждения результатов других методов диагностики [Chang IJ, Hahn SH. The genetics of Wilson disease. Handb Clin Neurol. 2017;142:19-34. doi: 10.1016/B978-0-444-63625-6.00003-3. PMID: 28433102; PMCID: PMC5648646.].
Среди аналогов так же выделяется MLPA-анализ (Multiplex ligation probe amplification или мультиплексная амплификация лигированных зондов). Эта разновидность мультиплексной ПЦР позволяет амплифицировать несколько (до 60 зондов) образцов только с одной парой праймеров. После денатурации ДНК образца к нему добавляют смесь MLPA-зондов, которые состоят из двух олигонуклеотидов. Если в образце присутствует последовательность комплементарная зонду, то олигонуклеотиды гибридизируются друг за другом так, чтобы быть сшитыми лигазой в единый зонд после чего зонды разной длины амплифицируются. В результате происходит амплификация не ДНК образца, а смеси зондов уникальной длины (от 130 до 500 п.о.). Так же один из праймеров несет флуорисцентную метку для визуализации продуктов амплификации в ходе капиллярного электрофореза. При наличии мутаций в исследуемом образце высота его пиков при анализе будет ниже, чем высота пиков референсного образца. MLPA является высокочувствительным методом и позволяет одновременно определять относительно большое количество копий всех экзонов.
Однако, в конечном этапе, результат часто зависит от опыта лабораторного работника и является субьективным. При использовании MLPA сложнее обнаружить однонуклеотидные замены [Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002 Jun 15;30(12):e57. doi: 10.1093/nar/gnf056. PMID: 12060695; PMCID: PMC117299.].
Наиболее близким техническим решением с заявляемому изобретению, принятому за прототип, является способ выявления мутаций гена ATP7B, принцип действия которого основан на сочетании методов ПЦР в реальном времени и мультиплексной аллель-специфичной ПЦР. Данный метод заключается в том, что после выделения ДНК из образца крови пациента по одному из стандартных протоколов проводится аллель-специфическая ПЦР с последующим электрофорезом продуктов амплификации по стандартному протоколу. Отмечено что данный способ с высокой точностью детектирует мутации Arg778Leu, Ala874Val, Leu1083Phe и Asn1270Ser расположенные в 8, 11, 15 и 18 экзонах (Заявка KR № KR20100083487A, МПК C12N15/11, опубл. 22.07.2010).
Однако он также не лишен недостатков, в частности, выражающиеся в малом количестве выявляемых мутаций и различиях в спектре мутаций среди азиатских и европейских популяций. Несмотря на глобальное распределение мутаций гена ATP7B большое количество пациентов является носителями редких мутаций. Данный недостаток при диагностике заболевания может привести к ложноотрицательному результату, что повлечет за собой ошибку в диагнозе и угрозу для здоровья пациента. Другим недостатком прототипа является достаточно высокая стоимость метода ПЦР в реальном времени, заключающаяся как в стоимости наборов реагентов, так и в необходимости использования дорогостоящего ПЦР-амплификатора с оптическим модулем для детекции продуктов ПЦР непосредственно в ходе процесса амплификации нуклеиновых кислот. Что с учетом распространенности мутаций в гене ATP7B и необходимости проведения массового скрининга молекулярных причин болезни Вильсона фактически в каждом населенном пункте или районном центре, является существенным затруднением, препятствующим эффективной диагностике болезни и своевременной помощи больным.
Технической проблемой, поставленной перед данным изобретением, является:
- повышение количества выявляемых мутантных аллелей для более корректной и всеобъемлющей диагностики.
- удешевление и упрощение метода для его более широкого применения в клинической лабораторной диагностике.
Заявленный технический результат достигается тем, что в известном способе выявления мутаций гена ATP7B, в котором предусматривается выделение ДНК из образцов крови пациентов по одному из стандартных протоколов с последующим анализом при помощи аллель-специфической ПЦР с последующим электрофорезом продуктов амплификации по стандартному протоколу, согласно изобретению, устанавливают мутации в экзонах 6, 8, 10, 12, 14 и 15, при этом используют следующий набор олигонуклеотидных праймеров:
- для мутации 1847G>A в экзоне 6 используют прямые праймеры: SEQ ID NO: 1 (мутантный); SEQ ID NO: 2 (дикий тип) и обратный SEQ ID NO: 3;
- для мутации 2304insC в экзоне 8 используют прямые праймеры: SEQ ID NO: 4 (мутантный); SEQ ID NO: 5 (дикий тип) и обратный SEQ ID NO: 10;
- для мутации 2128G>A в экзоне 8 применяют прямые праймеры: SEQ ID NO: 6 (мутантный); SEQ ID NO: 7 (дикий тип) и обратный SEQ ID NO: 10.;
- для мутации 2230T>C в экзоне 8 используют прямые праймеры: SEQ ID NO: 8 (мутантный); SEQ ID NO: 9 (дикий тип) и обратный обратный SEQ ID NO: 10;
- для мутации 2333G>T в экзоне 8 применяют прямые праймеры: SEQ ID NO: 11 (мутантный); SEQ ID NO: 12 (дикий тип) и обратный SEQ ID NO: 17;
- для мутации 2332C>G в экзоне 8 используют прямые праймеры: SEQ ID NO: 13 (мутантный); SEQ ID NO: 14 (дикий тип) и обратный SEQ ID NO: 17;
- для мутации 2336G>A в экзоне 8 применяют прямые праймеры: SEQ ID NO: 15 (мутантный); SEQ ID NO: 16 (дикий тип) и обратный SEQ ID NO: 17;
- для мутации 2501T>A в экзоне 10 используют прямые праймеры: SEQ ID NO: 18 (мутантный); SEQ ID NO: 19 (дикий тип) и обратный SEQ ID NO: 20;
- для мутации 2827G>A в экзоне 12 необходимо используют праймеры: SEQ ID NO: 21 (мутантный); SEQ ID NO: 22 (дикий тип) и обратный SEQ ID NO: 23;
- для мутации 3207C>A в экзоне 14 используют прямые праймеры: SEQ ID NO: 24 (мутантный); SEQ ID NO: 25 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3190G>A в экзоне 14 используют прямые праймеры: SEQ ID NO: 26 (мутантный); SEQ ID NO: 27 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3121C>T в экзоне 14 используют прямые праймеры: SEQ ID NO: 28 (мутантный); SEQ ID NO: 29 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3222_3243+21del43 в экзоне 14 используют прямые праймеры: SEQ ID NO: 30 (мутантный); SEQ ID NO: 31 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3402delC в экзоне 15 применяют прямые праймеры: SEQ ID NO: 33 (мутантный); SEQ ID NO: 34 (дикий тип) и обратный SEQ ID NO: 35.
Другим важным техническим результатом заявляемого изобретения является использования метода аллель-специфической ПЦР вместо метода ПЦР в реальном времени, используемом в прототипе. Эта особенность позволяет снизить требования к лабораторному оборудованию и используемым реактивам, позволяя существенно удешевить и сделать более доступной ПЦР-диагностику болезни Вильсона. Так же гибкость данной системы позволяет многократно расширить список выявляемых мутаций, что повышает точность диагностики, проводимой данным методом.
Заявленный способ решает поставленные проблемы и достигает заявленного технического результата за счет:
- повышения количества выявляемых мутаций до 14 вариантов, встречающихся в экзонах 6, 8, 10, 12, 14 и 15, что соответствует полученным современным данным о спектре наиболее часто встречающихся мутаций гена ATP7B, часть из которых наиболее характерна для Европейской популяции.
- удешевление и упрощение метода за счет взятого за основу простого ПЦР вместо ПЦР в реальном времени.
Таким образом, предложенный способ идентификации генных мутаций и полиморфизмов осуществляют посредством проведения аллель-специфической ПЦР в присутствии специализированных олигонуклеотидных праймеров, соответствующих мутантным и нормальным аллелям гена ATP7B. Использование данных праймеров позволяет упростить диагностику болезни Вильсона и обеспечивают высокую эффективность диагностики за счет выявления как самых распространенных патогенных мутаций гена ATP7B, так и мутаций с умеренным уровнем распространения, но значительно представленными среди Европейской популяции.
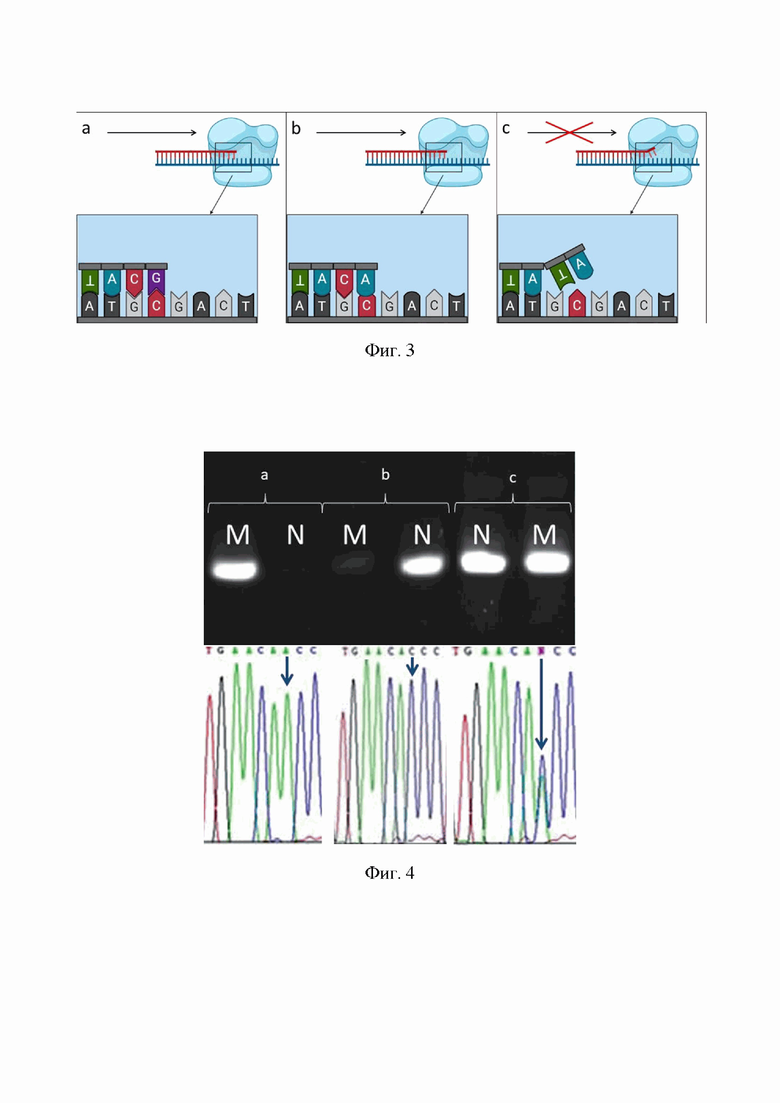
Аллель-специфическая ПЦР строится на использовании модифицированных праймеров, подбираемых строго на места мутаций. Зачастую для реализации технологии могут быть использованы два варианта универсальных праймеров: первый вариант - праймер должен быть строго комплементарен по своему 3'-концевому нуклеотиду, соответствующему нуклеотиду матричной ДНК. Второй вариант - универсальный праймер содержит 3'-концевой нуклеотид, всегда некомплементарный матрице, а мутантный нуклеотид матрицы попадает в его внутреннюю часть. В этом случае продукты ПЦР отсутствуют, если в гибриде во внутреннюю часть праймера попадает любой некомплементарный мутантный нуклеотид матричной ДНК вне зависимости от его точной локализации. Такие праймеры позволяют обнаруживать любые точечные мутации (SNP) в гомозиготном состоянии. Для выявления гетерозиготного состояния анализируемого гена могут быть использованы «мутантный» и «нормальный» праймеры в двух разных пробирках. Для проведения данного теста используют SNPdetect полимеразу. Наша модификация заключается в разработке уникальных праймеров, модифицированных одним нуклеотидным несоответствием в 2-3 положении 3'-конца в дополнении к несоответствию в мутантном праймере. Модификация происходит по правилу сильных и слабых несоответсвий нуклеотидов: сильные несоответствия - это C-C, G-A и A-A; слабые несовпадения - это T-T, T-C, T-G, G-G и A-C [Old, J. M. (n.d.). The Amplification Refractory Mutation System. Nucleic Acid Protocols Handbook, The, 723-727. doi:10.1385/1-59259-038-1:723]. Данная модификация позволяет использовать Taq ДНК-полимеразу, что удешевляет анализ. Визуализация результатов проводится с помощью электрофореза.
Изобретение иллюстрируется следующими фигурами:
На фиг.1. приведена карта-схема экзонов гена ATP7B и расположение обнаруженных на них мутаций, которые были использованы для создания праймеров.
На фиг.2. приведена карта-схема расположения праймеров для поиска мутаций в разных экзонах гена ATP7B, где N - праймер с нормальным аллелем, М - праймер с мутантным аллелем.
На фиг.3. приведена схема присоединения модифицированного праймера и возникновения «плавающего хвоста» на примере:
(а) несоответствия нет;
(b) наличие одного несоответствия;
(в) наличие двух несоответствий, препятствующих получению фрагмента.
На фиг.4. приведены примеры результатов работы аллель-специфической ПЦР у пациентов с различными мутациями: а) мутация в изучаемом аллеле, находится в гомозиготном состоянии; (б) дикий тип; в) мутация в изучаемом аллеле, но находится в гетерозиготном состоянии.
На фиг.5. приведены электрофореграммы результатов аллель-специфической ПЦР. WD - условное обозначение, присваеваемое пациентам для обезличивания; М - мутантный аллель; Н - нормальный аллель, при совместном проявлении мутантного и нормального аллеля пациент является гетерозиготным носителем.
Способ осуществляют следующим образом.
Перед реализацией заявленного метода предварительно выделяется ДНК из образца венозной крови пациента. После чего проводится аллель-специфическая ПЦР по стандартному протоколу, вариант которого приведен ниже.
Реакционная смесь для аллель-специфической ПЦР готовили из расчета на итоговый объем 20 мкл, для чего смешивали следующие компоненты: деионизированная вода - 5,5 мкл; DreamTaq Green PCR Master Mix (Thermo Scientific™) - 10 мкл; смесь праймеров (включающая мутантный прямой праймер, нормальный прямой и обратный праймеры в соответствии с прилагаемым списком олигонуклеотидных последовательностей для каждого аллельного варианта, взятые в концентрации 10 пикомоль/мкл) - по 1 мкл; образец ДНК пациента (10 нг/мкл) - 2,5 мкл. Процедуру аллель-специфической ПЦР проводили при следующих условиях: предварительная денатурация 95°С - 5 мин.; далее для 35 циклов: 95°С денатурация - 30 сек., 65°С отжиг - 30 сек., 72°С синтез - 40 сек.; заключительный цикл 72°С - 3 мин. Визуализацию результатов осуществляли с помощью электрофореза в 1% агарозном геле с добавлением раствора бромистого этидия (10 мг/мл, Evrogen) в TAE буфере (40 мМ Трис-ацетатный буферный раствор, рН 7,6, 1 мМ ЭДТА) и фотографировали на ChemiDoc MP Imaging System (BioRad) в проходящем ультрафиолетовом свете. Для определения длины фрагментов использовали ДНК маркер 1000 пар нуклеотидов (пн) (Evrogen). Олигонуклеотидные праймеры для проведения аллель-специфической ПЦР (ARMS PCR) для выявления 14 мутаций в гене ATP7B приведены в списке последовательностей в конце описания изобретения. Аллель-специфическую ПЦР проводили с праймерами, помещенными в стандартную ПЦР смесь, распределенными по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные последовательности, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. Полученные фрагменты ДНК анализируют методом гель-электрофореза и на основании наличия или отсутствия полос, соответствующих продукту нормального и мутантного варианта, делают заключение о гомо- или гетерозиготном носительстве мутаций в гене ATP7B, или об отсутствии мутации в случае наличии только продукта амплификации аллеля дикого типа.
Пример 1
Для реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяли ДНК из образцов крови пациента WD23, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерам SEQ ID NO: 1 (мутантный) и SEQ ID NO: 2 (дикий тип) на мутацию 1847G>A в экзоне 6 происходила наработка фрагмента, соответствующего мутантному аллелю, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого мутантного и обратного праймеров, что свидетельствует о наличии мутации 1847G>A в гомозиготном варианте (Фиг. 5 WD23). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4a).
Пример 2
Другим примером демонстрации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, является кровь пациента WD63. Из образца выделяли ДНК затем проводили амплификацию при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 4 (мутантный) и SEQ ID NO: 5 (дикий тип) на мутацию 2304insC в экзоне 8 происходила наработка фрагмента, соответствующего мутантному и аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующих полос. Таким образом, полосы на электрофорезе появилась в лунках, содержащих смесь из прямого мутантного и прямого праймера дикого типа, что свидетельствует о наличии мутации 2304insC в гетерозиготном варианте (Фиг. 5 WD63). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4c).
Пример 3
Еще одним примером реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, является кровь пациента WD2. Из образца выделяли ДНК затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 6 (мутантный) и SEQ ID NO: 7 (дикий тип) на мутацию 2128G>A в экзоне 8 происходила наработка фрагмента, соответствующего аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого праймера дикого типа и обратного праймеров, что свидетельствует об отсутствии мутации 2128G>A (Фиг. 4b). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа. Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4b).
Пример 4
Так же возможна реализация способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяя ДНК из образцов крови пациента WD41, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами 8 (мутантный) и SEQ ID NO: 9 (дикий тип) на мутацию 2230T>C в экзоне 8 происходила наработка фрагмента, соответствующего мутантному аллелю, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого мутантного и обратного праймеров, что свидетельствует о наличии мутации 2230T>C в гомозиготном варианте (Фиг. 5 WD41). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4a).
Пример 5
Так же возможна реализация способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяя ДНК из образцов крови пациента WD64, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 11 (мутантный) и SEQ ID NO: 12 (дикий тип) на мутацию 2333G>T в экзоне 8 происходила наработка фрагмента, соответствующего мутантному и аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующих полос. Таким образом, полосы на электрофорезе появилась в лунках, содержащих смесь из прямого мутантного и прямого праймера дикого типа, что свидетельствует о наличии мутации 2333G>T в гетерозиготном варианте (Фиг. 5 WD64). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4a).
Пример 6
Другим примером демонстрации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, является кровь пациента WD4. Из выделяли ДНК затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 13 (мутантный) и SEQ ID NO: 14 (дикий тип) на мутацию 2332C>G в экзоне 8 происходила наработка фрагмента, соответствующего аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого праймера дикого типа и обратного праймеров, что свидетельствует об отсутствии мутации 2332C>G (Фиг. 4b). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа. Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4b).
Пример 7
Помимо вышеперечисленного, для реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяли ДНК из образцов крови пациента WD85, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 15 (мутантный) и SEQ ID NO: 16 (дикий тип) на мутацию 2336G>A в экзоне 8 происходила наработка фрагмента, соответствующего мутантному аллелю, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого мутантного и обратного праймеров, что свидетельствует о наличии мутации 2336G>A в гомозиготном Фиг. 5 WD85). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4a).
Пример 8
Еще одним примером реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, является кровь пациента WD75. Из образца выделяли ДНК затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 18 (мутантный) и SEQ ID NO: 19 (дикий тип) на мутацию 2501T>A в экзоне 10 происходила наработка фрагмента, соответствующего мутантному и аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующих полос. Таким образом, полосы на электрофорезе появилась в лунках, содержащих смесь из прямого мутантного и прямого праймера дикого типа, что свидетельствует о наличии мутации 2501T>A в гетерозиготном варианте (Фиг. 5 WD75). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4c).
Пример 9
Для реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяли ДНК из образцов крови пациента WD5, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 21 (мутантный) и SEQ ID NO: 22 (дикий тип) на мутацию 2827G>A в экзоне 12 происходила наработка фрагмента, соответствующего аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого праймера дикого типа и обратного праймеров, что свидетельствует об отсутствии мутации 2827G>A. Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4b).
Пример 10
Помимо вышеперечисленного, для реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяли ДНК из образцов крови пациента WD25, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 24 (мутантный) и SEQ ID NO: 25 (дикий тип) на мутацию 3207C>A в экзоне 14 происходила наработка фрагмента, соответствующего мутантному аллелю, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого мутантного и обратного праймеров, что свидетельствует о наличии мутации 3207C>A в гомозиготном варианте (Фиг. 5 WD25). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4a).
Пример 11
Так же возможна реализация способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяя ДНК из образцов крови пациента WD21, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 26 (мутантный) и SEQ ID NO: 27 (дикий тип) на мутацию 3190G>A в экзоне 14 происходила наработка фрагмента, соответствующего мутантному и аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующих полос. Таким образом, полосы на электрофорезе появилась в лунках, содержащих смесь из прямого мутантного и прямого праймера дикого типа, что свидетельствует о наличии мутации 3190G>A в гетерозиготном варианте (Фиг. 5 WD21). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4c).
Пример 12
Другим примером демонстрации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, является кровь пациента WD9. Из образца выделяли ДНК затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 28 (мутантный) и SEQ ID NO: 29 (дикий тип) на мутацию 3121C>T в экзоне 14 происходила наработка фрагмента, соответствующего аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого праймера дикого типа и обратного праймеров, что свидетельствует об отсутствии мутации 3121C>T. Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4b).
Пример 13
Помимо вышеперечисленного, для реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяли ДНК из образцов крови пациента WD23, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 30 (мутантный); SEQ ID NO: 31 (дикий тип) на мутацию 3222_3243+21del43 в экзоне 14 происходила наработка фрагмента, соответствующего мутантному аллелю, что было видно по результатам электрофореза в виде соответствующей полосы. Таким образом, полоса на электрофорезе появилась только в лунке, содержащей смесь из прямого мутантного и обратного праймеров, что свидетельствует о наличии мутации 3222_3243+21del43 в гомозиготном варианте (Фиг. 5 WD23). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4a).
Пример 14
Для реализации способа диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, выделяли ДНК из образцов крови пациента WD76, затем проводили амплификацию ДНК при помощи аллель-специфической ПЦР с последующим анализом продуктов реакции гель-электрофорезом в 1% агарозе в соответствии со стандартным протоколом, описанным выше. Аллель-специфическая ПЦР включала праймеры, описанные в формуле изобретения и помещенные в стандартную ПЦР смесь, распределенную по 28 пробиркам, соответствующим 14 парам праймеров, выявляющим мутантные аллели, и 14 парам праймеров, выявляющим соответствующие аллели дикого типа. В лунке с праймерами SEQ ID NO: 33 (мутантный) и SEQ ID NO: 34 (дикий тип) на 3402delC в экзоне 15 происходила наработка фрагмента, соответствующего мутантному и аллелю дикого типа, что было видно по результатам электрофореза в виде соответствующих полос. Таким образом, полосы на электрофорезе появилась в лунках, содержащих смесь из прямого мутантного и прямого праймера дикого типа, что свидетельствует о наличии мутации 3402delC в гетерозиготном варианте (Фиг. 5 WD76). Остальные лунки продемонстрировали отсутствие мутаций и показали наработку исключительно фрагментов гена дикого типа (Фиг. 4b). Данные результаты были подтверждены данными секвенирования фрагментов ДНК по методу Сенгеру (Фиг. 4c).
Заявляемый способ поиска мутаций гена ATP7B, позволяет исключить выявленные недостатки известных способов и достичь заявляемого технического результата. В отличии от указанного прототипа, заявляемый способ использует обычную амплификацию фрагментов ДНК по протоколу стандартной ПЦР с последующим анализом продуктов стандартным методом электрофореза в агарозном геле, что сильно удешевляет диагностику. Так же заявляемый способ обеспечивает большую эффективность диагностики поскольку выявляет 14 мутаций, расположенных в 6 экзонах, в отличие от прототипа, выявляющего исключительно 4 мутации в 4 экзонах, что еще больше увеличивает точность диагностики, особенно применительно к Европейской популяции, для которой выявляемые мутации по заявляемому способу представляют собой наиболее часто встречающиеся патологические варианты.
--->
Список олигонуклеотидных последовательностей:
<110> Гарбуз Михаил Максимович
<120> Способ поиска мутаций гена ATP7B
<160> 35
<210> 1
<211> 21
<212> ДНК
<213>Homosapiens
<400> 1
TCATGCTTCCCTGGCCCAGCA
<210> 2
<211> 19
<212> ДНК
<213>Homosapiens
<400> 2
ATGCTTCCCTGGCCCAGCG
<210> 3
<211> 24
<212> ДНК
<213>Homosapiens
<400> 3
TGCTCACTAGCTACATGTGACTAG
<210> 4
<211> 21
<212>ДНК
<213> Homo sapiens
<400> 4
CATTCTTCGACACGCCCCCTC
<210> 5
<211> 22
<212>ДНК
<213> Homo sapiens
<400> 5
ACATTCTTCGACACGCCCCCTA
<210> 6
<211> 24
<212>ДНК
<213> Homo sapiens
<400> 6
CTGTCCTTGTCTTTCAGCTCCTTA
<210> 7
<211> 24
<212>ДНК
<213> Homo sapiens
<400> 7
CTGTCCTTGTCTTTCAGCTCCTTG
<210> 8
<211> 27
<212>ДНК
<213> Homo sapiens
<400> 8
GCCACAAGCATTGCTTATGTTTATTGC
<210> 9
<211> 27
<212>ДНК
<213> Homo sapiens
<400> 9
GCCACAAGCATTGCTTATGTTTATTGT
<210> 10
<211> 26
<212>ДНК
<213> Homo sapiens
<400> 10
TTTGGAGATTAGTGACTAGAGCACCT
<210> 11
<211> 24
<212>ДНК
<213> Homo sapiens
<400> 11
CTTTGTGTTCATTGCCCTGGGTCT
<210> 12
<211> 23
<212>ДНК
<213> Homo sapiens
<400> 12
TTTGTGTTCATTGCCCTGGGTCG
<210> 13
<211> 23
<212>ДНК
<213> Homo sapiens
<400> 13
CTTTGTGTTCATTGCCCTGGGTG
<210> 14
<211> 23
<212>ДНК
<213> Homo sapiens
<400> 14
CTTTGTGTTCATTGCCCTGGGTC
<210> 15
<211> 20
<212>ДНК
<213> Homo sapiens
<400> 15
TCATTGCCCTGGGCCGGTCA
<210> 16
<211> 21
<212>ДНК
<213> Homo sapiens
<400> 16
TTCATTGCCCTGGGCCGGTTG
<210> 17
<211> 27
<212>ДНК
<213> Homo sapiens
<400> 17
GACCAACTACATATTCAGTTTTGCACC
<210> 18
<211> 22
<212>ДНК
<213> Homo sapiens
<400> 18
AAGGTGGTCCCTGGGGGAAATT
<210> 19
<211> 22
<212>ДНК
<213> Homo sapiens
<400> 19
AAGGTGGTCCCTGGGGGAAATA
<210> 20
<211> 27
<212>ДНК
<213> Homo sapiens
<400> 20
GTCTGATTTCCCAGAACTCTTCACATA
<210> 21
<211> 28
<212>ДНК
<213> Homo sapiens
<400> 21
TTGACGTTGGTGGTATGGATTGTAATTA
<210> 22
<211> 26
<212>ДНК
<213> Homo sapiens
<400> 22
GACGTTGGTGGTATGGATTGTAATTG
<210> 23
<211> 24
<212>ДНК
<213> Homo sapiens
<400> 23
CAACTGAGCACCAATTGGTGTCTG
<210> 24
<211> 22
<212>ДНК
<213> Homo sapiens
<400> 24
TGCGGAGGCCAGCAGTGAATAA
<210> 25
<211> 21
<212>ДНК
<213> Homo sapiens
<400> 25
GCGGAGGCCAGCAGTGAATAC
<210> 26
<211> 20
<212>ДНК
<213> Homo sapiens
<400> 26
GGCTGTGGTGGGGACTGCTA
<210> 27
<211> 19
<212>ДНК
<213> Homo sapiens
<400> 27
GCTGTGGTGGGGACTGCTG
<210> 28
<211> 19
<212>ДНК
<213> Homo sapiens
<400> 28
GGCGTCCCCAGGGTCACGT
<210> 29
<211> 18
<212>ДНК
<213> Homo sapiens
<400> 29
GCGTCCCCAGGGTCACGC
<210> 30
<211> 21
<212>ДНК
<213> Homo sapiens
<400> 30
CAGGGTCATGCTGGCCACACT
<210> 31
<211> 18
<212>ДНК
<213> Homo sapiens
<400> 31
GCGGGTGCTCCTGCTGGG
<210> 32
<211> 21
<212>ДНК
<213> Homo sapiens
<400> 32
AGACTGCCCGTACTCCCCAAG
<210> 33
<211> 24
<212>ДНК
<213> Homo sapiens
<400> 33
CTTGGGATACTGCACGGACTTTAG
<210> 34
<211> 24
<212>ДНК
<213> Homo sapiens
<400> 34
TTGGGATACTGCACGGACTTCTAG
<210> 35
<211> 27
<212>ДНК
<213> Homo sapiens
<400> 35
CTCTATGAATTTAAGGCAGCCATAAGC
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ идентификации полиморфизмов Cys1079Gly и Cys1079Phe медь-транспортной АТФ-азы Вильсона | 2020 |
|
RU2756112C1 |
| Способ генотипирования полиморфного локуса rs36210422 (G>A) гена KCNH2 | 2025 |
|
RU2837877C1 |
| Способ генотипирования полиморфного локуса rs139794067 (G>T) гена MYL3 | 2025 |
|
RU2837876C1 |
| МОДУЛЯТОРЫ ЭКСПРЕССИИ ГЕНА ОТКРЫТОЙ РАМКИ СЧИТЫВАНИЯ 72 ХРОМОСОМЫ 9 И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2828216C2 |
| Тест-система и способ выявления делеций гена SESN1 | 2021 |
|
RU2772504C1 |
| Тест-система и способ выявления A, B, D мутаций гена NPM1 для количественного определения минимальной остаточной болезни | 2023 |
|
RU2830545C1 |
| Способ выявления мутации p.L265P в гене MYD88 | 2020 |
|
RU2756909C1 |
| СПОСОБ АНАЛИЗА СОМАТИЧЕСКИХ МУТАЦИЙ В ГЕНЕ PI3K С ИСПОЛЬЗОВАНИЕМ LNA-БЛОКИРУЮЩЕЙ МУЛЬТИПЛЕКСНОЙ ПЦР И ПОСЛЕДУЮЩЕЙ ГИБРИДИЗАЦИЕЙ С ОЛИГОНУКЛЕОТИДНЫМ БИОЛОГИЧЕСКИМ МИКРОЧИПОМ (БИОЧИПОМ) | 2013 |
|
RU2549682C1 |
| Набор для определения CCR5delta32 мутации в геноме человека | 2020 |
|
RU2748998C1 |
| ПАНЕЛЬ БИОМАРКЕРОВ И СПОСОБЫ ВЫЯВЛЕНИЯ МИКРОСАТЕЛЛИТНОЙ НЕСТАБИЛЬНОСТИ ПРИ РАЗНЫХ ВИДАХ РАКА | 2019 |
|
RU2795410C2 |

Изобретение относится к биотехнологии. Описан способ идентификации генных мутаций и полиморфизмов. Проводят аллель-специфическую ПЦР в присутствии 35 видов праймеров, соответствующих мутантным и нормальным аллелям гена ATP7B. При этом 14 пар праймеров направлены на выявление мутантных последовательностей и 14 пар праймеров на выявление последовательностей дикого типа, что обеспечивает поиск самых частых мутаций гена ATP7B, встречающихся в экзонах 6, 8, 10, 12, 14 и 15. Использование данных праймеров позволяет упростить диагностику болезни Вильсона-Коновалова и увеличивает достоверность получаемых результатов, поскольку выявляет 14 наиболее распространенных мутаций данного гена. Использование способа позволяет выполнить быстрый скрининг мутаций гена ATP7B для оперативной постановки диагноза болезни Вильсона-Коновалова. 5 ил., 14 пр.
Способ диагностики болезни Вильсона посредством выявления мутаций в гене ATP7B, предусматривающий выделение ДНК из образцов крови пациентов по одному из стандартных протоколов с последующим анализом при помощи аллель-специфической ПЦР и электрофореза продуктов амплификации по стандартному протоколу, отличающийся тем, что устанавливают мутации в экзонах 6, 8, 10, 12, 14 и 15, применяя следующий набор из 35 олигонуклеотидных праймеров:
- для мутации 1847G>A в экзоне 6 используют прямые праймеры: SEQ ID NO: 1 (мутантный); SEQ ID NO: 2 (дикий тип) и обратный SEQ ID NO: 3;
- для мутации 2304insC в экзоне 8 используют прямые праймеры: SEQ ID NO: 4 (мутантный); SEQ ID NO: 5 (дикий тип) и обратный SEQ ID NO: 10;
- для мутации 2128G>A в экзоне 8 применяют прямые праймеры: SEQ ID NO: 6 (мутантный); SEQ ID NO: 7 (дикий тип) и обратный SEQ ID NO: 10.;
- для мутации 2230T>C в экзоне 8 используют прямые праймеры: SEQ ID NO: 8 (мутантный); SEQ ID NO: 9 (дикий тип) и обратный обратный SEQ ID NO: 10;
- для мутации 2333G>T в экзоне 8 применяют прямые праймеры: SEQ ID NO: 11 (мутантный); SEQ ID NO: 12 (дикий тип) и обратный SEQ ID NO: 17;
- для мутации 2332C>G в экзоне 8 используют прямые праймеры: SEQ ID NO: 13 (мутантный); SEQ ID NO: 14 (дикий тип) и обратный SEQ ID NO: 17;
- для мутации 2336G>A в экзоне 8 применяют прямые праймеры: SEQ ID NO: 15 (мутантный); SEQ ID NO: 16 (дикий тип) и обратный SEQ ID NO: 17;
- для мутации 2501T>A в экзоне 10 используют прямые праймеры: SEQ ID NO: 18 (мутантный); SEQ ID NO: 19 (дикий тип) и обратный SEQ ID NO: 20;
- для мутации 2827G>A в экзоне 12 используют праймеры: SEQ ID NO: 21 (мутантный); SEQ ID NO: 22 (дикий тип) и обратный SEQ ID NO: 23;
- для мутации 3207C>A в экзоне 14 используют прямые праймеры: SEQ ID NO: 24 (мутантный); SEQ ID NO: 25 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3190G>A в экзоне 14 используют прямые праймеры: SEQ ID NO: 26 (мутантный); SEQ ID NO: 27 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3121C>T в экзоне 14 используют прямые праймеры: SEQ ID NO: 28 (мутантный); SEQ ID NO: 29 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3222_3243+21del43 в экзоне 14 используют прямые праймеры: SEQ ID NO: 30 (мутантный); SEQ ID NO: 31 (дикий тип) и обратный SEQ ID NO: 32;
- для мутации 3402delC в экзоне 15 применяют прямые праймеры: SEQ ID NO: 33 (мутантный); SEQ ID NO: 34 (дикий тип) и обратный SEQ ID NO: 35.
| Способ получения библиотеки генов для диагностики патологий печени | 2020 |
|
RU2743858C1 |
| US 20200393473 A1, 17.12.2020 | |||
| ГЛЮКУРОНИДИРОВАННЫЙ АЦЕТАМИНОФЕН КАК МАРКЕР РАССТРОЙСТВ ПЕЧЕНИ | 2009 |
|
RU2555331C2 |

