Изобретение относится к промышленной органической химии, в частности, к получению особо чистых растворителей, конкретно, к способу очистки ацетонитрила в процессе производства ацетонитрила особой чистоты.
Ацетонитрил особой степени чистоты широко используется в аналитических процедурах, в частности, в жидкостной хроматографии, а также в лабораторной практике. Для этих целей необходима тщательная очистка ацетонитрила от органических примесей, содержащихся в исходном сырце, до уровня порядка 0,001%, а для непредельных примесей, ухудшающих пропускание в УФ-диапазоне - значительно ниже. Наиболее эффективным и широко используемым способом получения ацетонитрила является ректификационная очистка.
Известно много технических решений подобного рода (US6508917, опубл.: 2003-01-21, RU2230733, опубл.: 2004.06.20, CN116987009, опубл.: 2023-11-03, US6395142, опубл.: 2002-05-28 и иные).
Однако, примеси, имеющие температуры кипения, близкие к ацетонитрилу, отделяются только путём длительной обработки (сотни часов), что требует больших затрат труда и энергии и делает такие процессы экономически неэффективными. Основными мешающими примесями в техническом ацетонитриле, в зависимости от способа его получения, могут быть: акрилонитрил, метилакрилат, метилметакрилат, аллиловый спирт и другие соединения с близкими температурами кипения, преимущественно, непредельного характера. Некоторые примеси способны образовывать с ацетонитрилом азеотропные смеси, что дополнительно затрудняет их отделение. Для повышения эффективности процесса желательно связать указанные примеси или перевести их в соединения, кипящие при других температурах.
Существуют известные способы очистки ацетонитрила от непредельных и легко окисляющихся примесей (RU 2149867 (опубл.: 27.05.2000), SU1810332 (опубл.: 23.04.1993)) путём обработки окислителями с последующей обработкой сорбентами или без таковой.
Однако, в процессе обработки сильными окислителями теряется часть целевого продукта – ацетонитрила, который способен в присутствии окислителей как окисляться, так и подвергаться другим превращениям. Кроме того, удаление значительных количеств окисляющихся примесей требует повышенного расхода окислителей и загрязняет оборудование продуктами их распада, что ведёт к снижению тепловой эффективности установок.
Известно решение по патенту CN104744300, опул.: 2015-07-01, в котором предложен способ очистки ацетонитрила, в котором примеси в исходном ацетонитриле удаляются с помощью таких процедур, как адсорбция модифицированным активированным углем, и, таким образом, может быть получен ацетонитрил для жидкостной хроматограммы-масс-спектрометра. Применяя метод очистки ацетонитрила для жидкостного хроматограмм-масс-спектрометра, можно получить ацетонитрил, чистота которого больше или равна 99,99% (мас.), можно удовлетворить требования жидкостного хроматограмм-масс-спектрометра к высокой чистоте, очищенный ацетонитрил можно применять в области жидкостных хроматограмм-масс-спектрометров и расширить диапазон применения.
Однако, данный способ малопроизводительный и не позволяет получать большие объемы продукта.
Известен способ получения фенозановых антиоксидантов (RU2178408, опубл.: 20.01.2002), состоящий в обработке ди-(трет-бутил)-фенола или других алкилфенолов акрилатами в щелочных условиях. Этот способ весьма эффективен при температурах выше 150°С, но он неприменим для обработки ацетонитрила, поскольку он кипит при 82°С. Для практического использования требуется фенол с более реакционноспособным ядром.
Близких технических решений заявленному изобретению не выявлено.
Техническим результатом изобретения является расширение арсенала новых способов очистки ацетонитрила. Кроме того, обеспечивается возможность высокой степени очистки технического ацетонитрила от примесей непредельного характера без использования повышенного давления и при температуре кипения ацетонитрила.
Указанный технический результат достигается за счет того, что заявлен способ очистки ацетонитрила от непредельных примесей, содержащих активированную двойную связь и трудно отделяемых ректификацией (акрилонитрил, акрилаты, метакрилаты, аллиловый спирт и т.п.), характеризующийся тем, что технический ацетонитрил обрабатывают при температуре кипения ацетонитрила смесью резорцина с резорцинатом калия или резорцинатом натрия, где количество резорцина по отношению к непредельным примесям составляет не ниже 1:2 (мол.), но не более 200:1 (мол.).
Предпочтительно обработку ведут при перемешивании нагревом с обратным холодильником в течение 3-6,5 ч.
Затем обработанный ацетонитрил передают на стадию ректификации или, после выделения дистилляцией, на стадию обработки окислителем.
Осуществление изобретения
Сущность заявленного изобретения основана на реализации цели разработатки такого способа, согласно которому мешающие примеси удалялись бы без существенного расходования основного продукта. Для разработки такого способа мы использовали свойство соединений с активированной двойной связью присоединяться в орто- и пара-положение фенолов.
Известно, что наиболее реакционноспособным ядром среди доступных фенолов обладает резорцин.
Проведённые нами исследования показали, что резорцин в присутствии резорцината калия достаточно активен, чтобы поглощать из раствора в ацетонитриле ненасыщенные примеси. Особенно активно поглощаются примеси акрилатного характера. Ацетонитрил в этих реакциях не расходуется.
Основной сутью предлагаемого способа является реакция соединений с активированной двойной связью (акрилатов, акрилонитрила, метакрилатов, аллилового спирта и др.) с двухатомным фенолом – резорцином в условиях щелочного катализа.
Поскольку подобные процессы чувствительны к действию воды, целесообразно в качестве щёлочи использовать безводный резорцинат щелочного металла (калия, натрия) и подвергать обработке высушенный ацетонитрил. Использование такого высокоактивного фенола, как резорцин, позволяет проводить обработку при температуре кипения ацетонитрила без использования повышенного давления. Продолжительность обработки составляет 3-8 часов и более, и должна корректироваться в соответствии с достигнутым результатом. Количество резорцина по отношению к непредельным примесям должно быть не ниже 1:2 (мол.). При более высоком соотношении эффективность растёт. Верхний предел содержания резорцина определяется экономическими соображениями. Соотношение между резорцинатом калия или резорцина натрия с резорцином должно составлять не менее 1:200 (мол.), так как при более высоком соотношении необходимая продолжительность обработки снижается.
Способ очистки ацетонитрила от непредельных примесей, содержащих активированную двойную связь и трудно отделяемых ректификацией (акрилонитрил, акрилаты, метакрилаты, аллиловый спирт и т.п.), согласно изобретения характеризуется тем, что технический ацетонитрил обрабатывают при температуре кипения ацетонитрила смесью резорцина с резорцинатом калия или резорцином натрия, где количество резорцина по отношению к непредельным примесям составляет не ниже 1:2 (мол.), но не более 200:1 (мол.).
Обработанный технический ацетонитрил удается получать с чистотой от 99,01% до 99,99%, что позволяет его затем передавать на ректификацию без дополнительной очистки. Если же полного удаления непредельных примесей не произошло, возможно проведение последующей обработка окислителем. В этом случае, летучие продукты должны быть выделены из реакционной смеси дистилляцией, поскольку резорцин и его производные будут реагировать с окислителем, вызывая его высокий перерасход. Дистилляция позволяет полностью избавиться от производных резорцина.
Таким образом, в соответствии с настоящим изобретением, технический ацетонитрил может быть полностью, либо в высокой степени очищен от примесей непредельного характера, путём обработки резорцином в присутствии безводного резорцината щелочного металла при температуре кипения ацетонитрила.
Изобретение поясняется следующими примерами.
Пример 1.
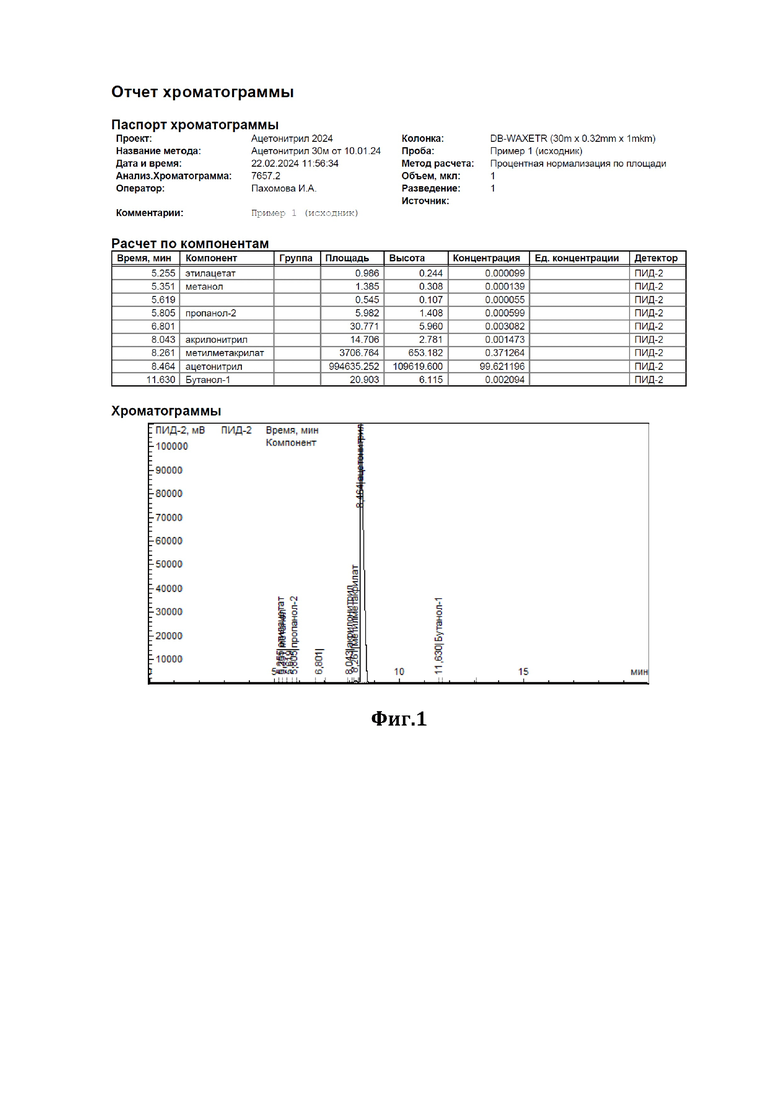
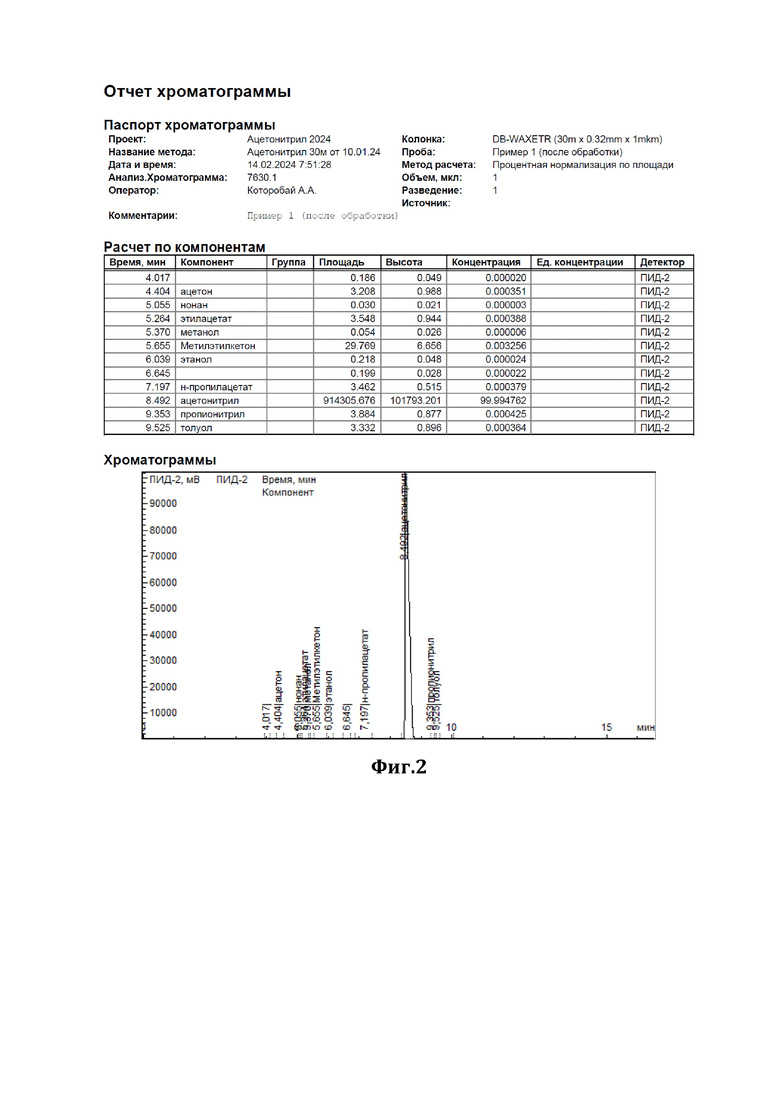
К 44 мл ацетонитрила, содержащего 0,37% метилметакрилата (см. диаграмму на Фиг.1), добавили 1,2 г сухого резорцината калия (7,5 ммоль) и 0,82 г резорцина (7,5 ммоль) и нагревали при перемешивании с обратным холодильником в течение 3,5 часов. По окончании перемешивания отогнали обработанный ацетонитрил. По результатам ГЖХ-анализа метилметакрилат в ацетонитриле отсутствует (см. диаграмму на Фиг.2).
Пример 2.
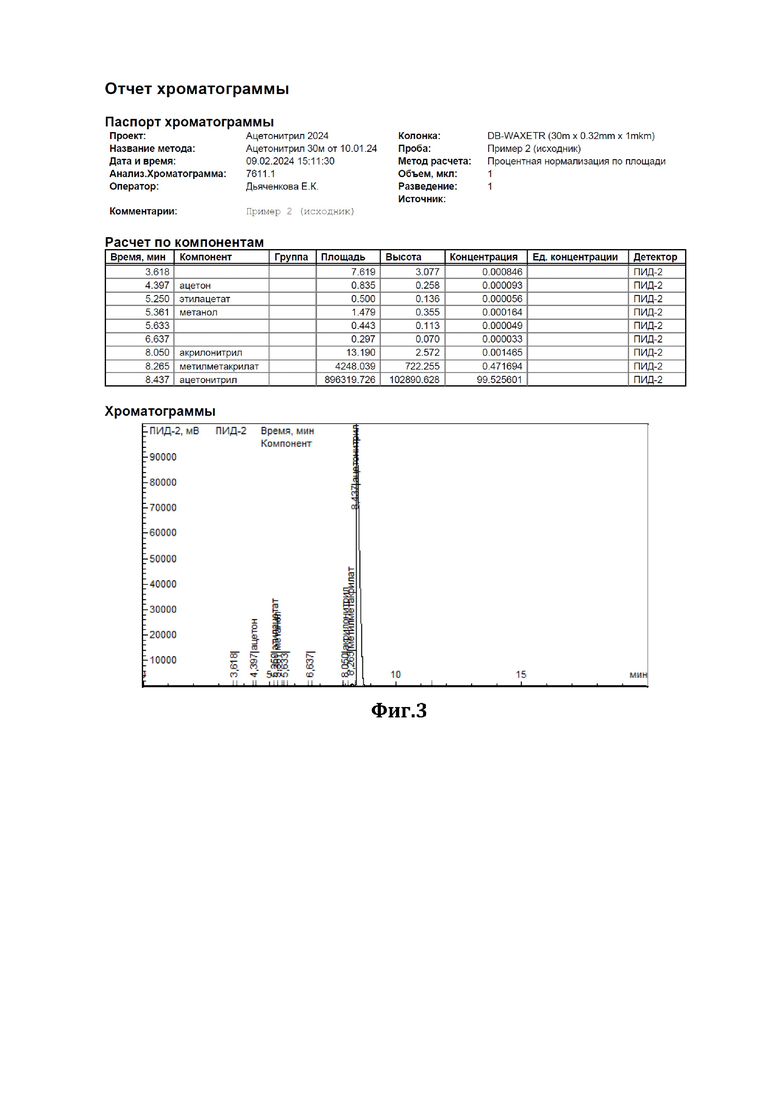
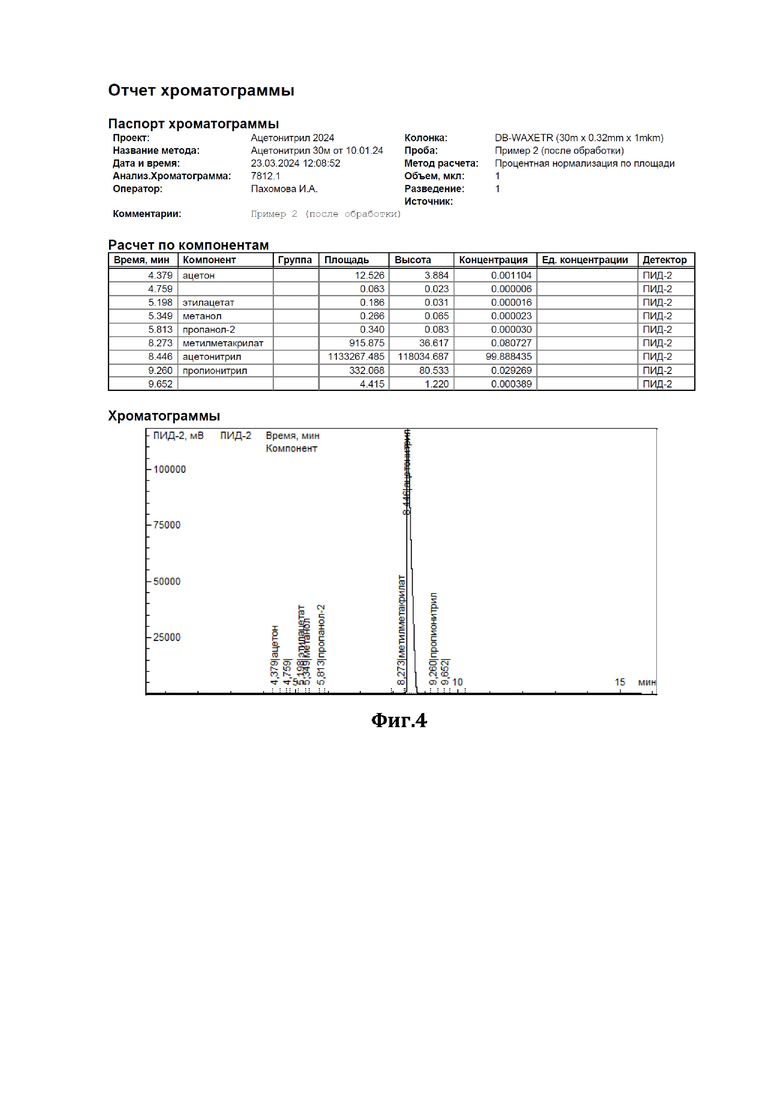
К 200 мл ацетонитрила, содержащего 0,47% метилметакрилата (см. диаграмму на Фиг.3), добавили смесь резорцина с резорцинатом калия в соотношении 1:1 (мол) в количестве 10 г. Нагревали при перемешивании с обратным холодильником в течение 3 часов. По окончании перемешивания отогнали обработанный ацетонитрил. По результатам ГЖХ-анализа содержание метилметакрилата в ацетонитриле составило 0,08% (см. диаграмму на Фиг.4).
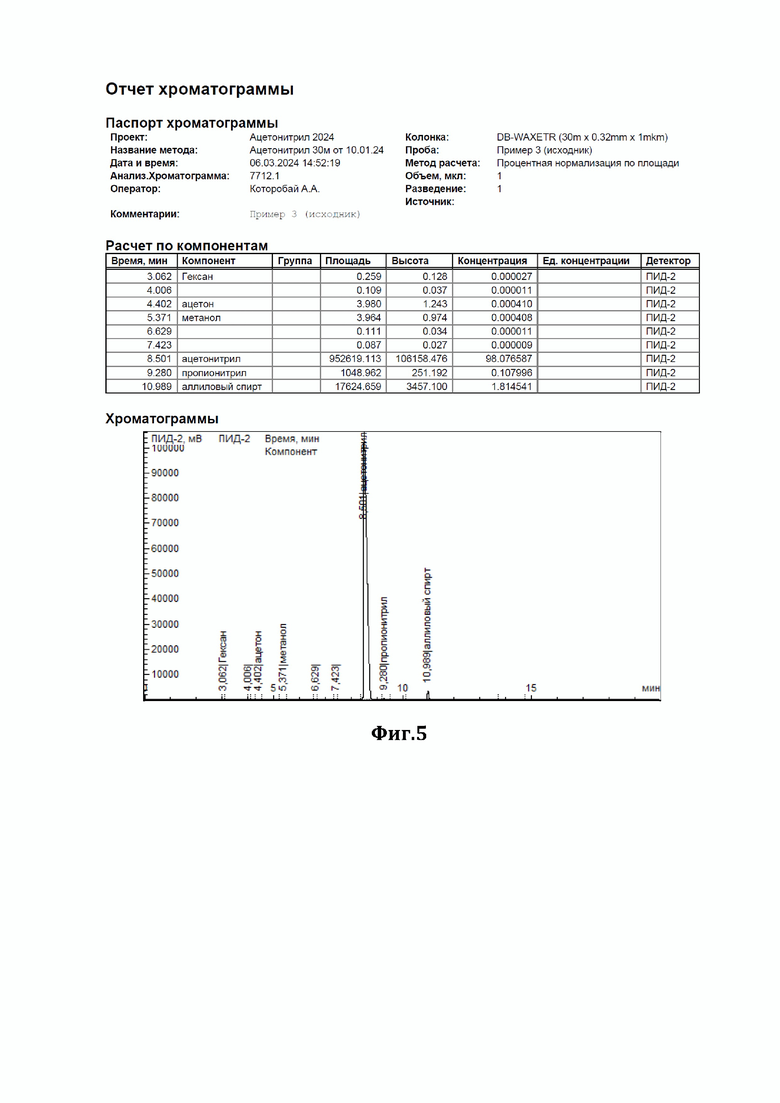
Пример 3.
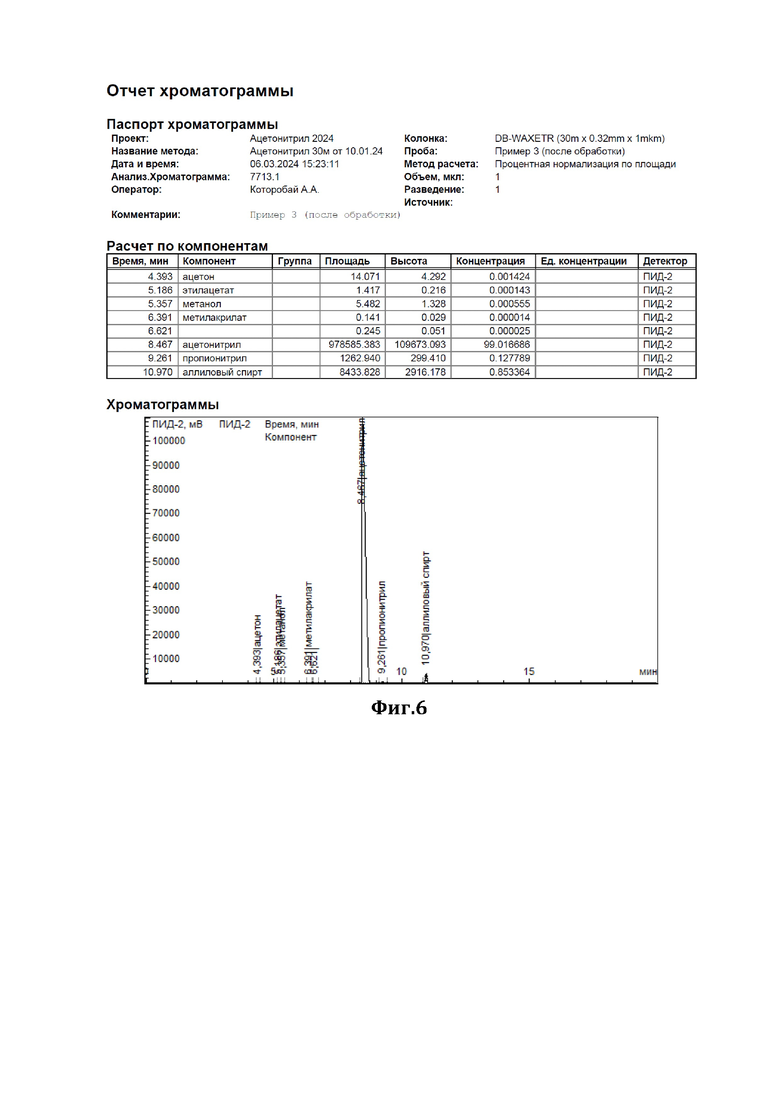
К 43 мл ацетонитрила, содержащего 1,81% аллилового спирта (см. диаграмму на Фиг.5), добавили смесь резорцина с резорцинатом калия в соотношении 1:1 (мол) в количестве 2,7 г нагревали при перемешивании с обратным холодильником в течение 4 часов. По окончании перемешивания отогнали обработанный ацетонитрил. По результатам ГЖХ-анализа содержание аллилового спирта составило 0,85% (снижение на 53%) (см. диаграмму на Фиг.6).
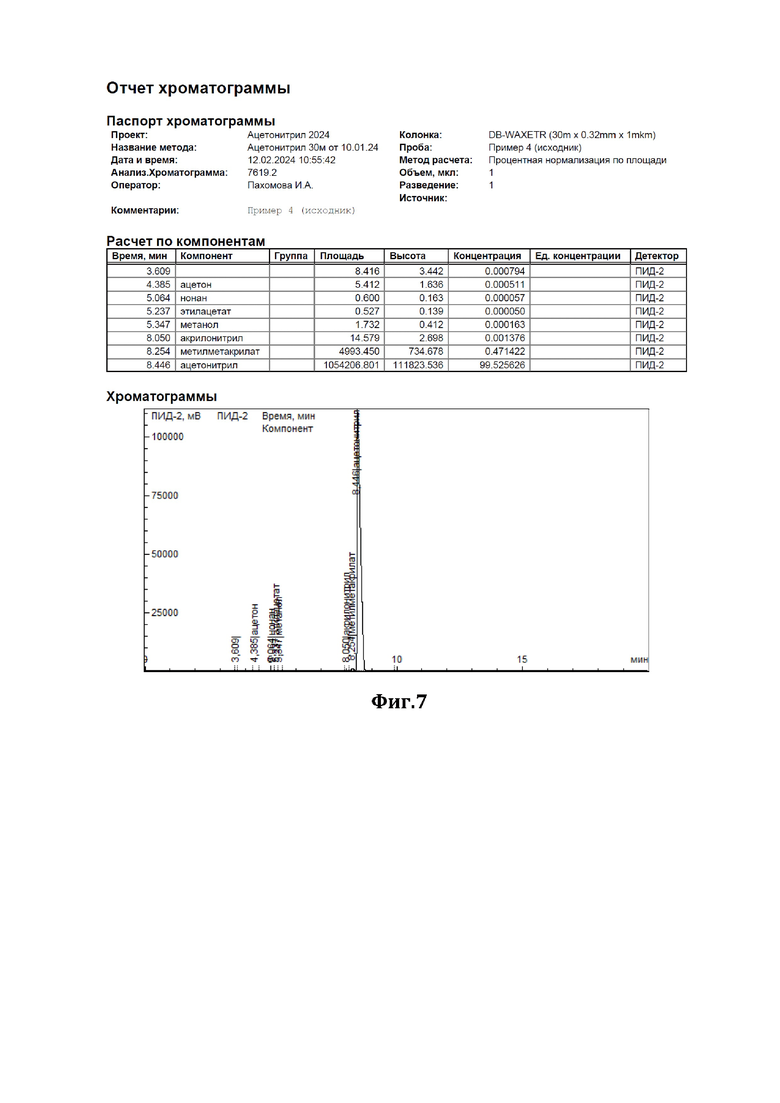
Пример 4.
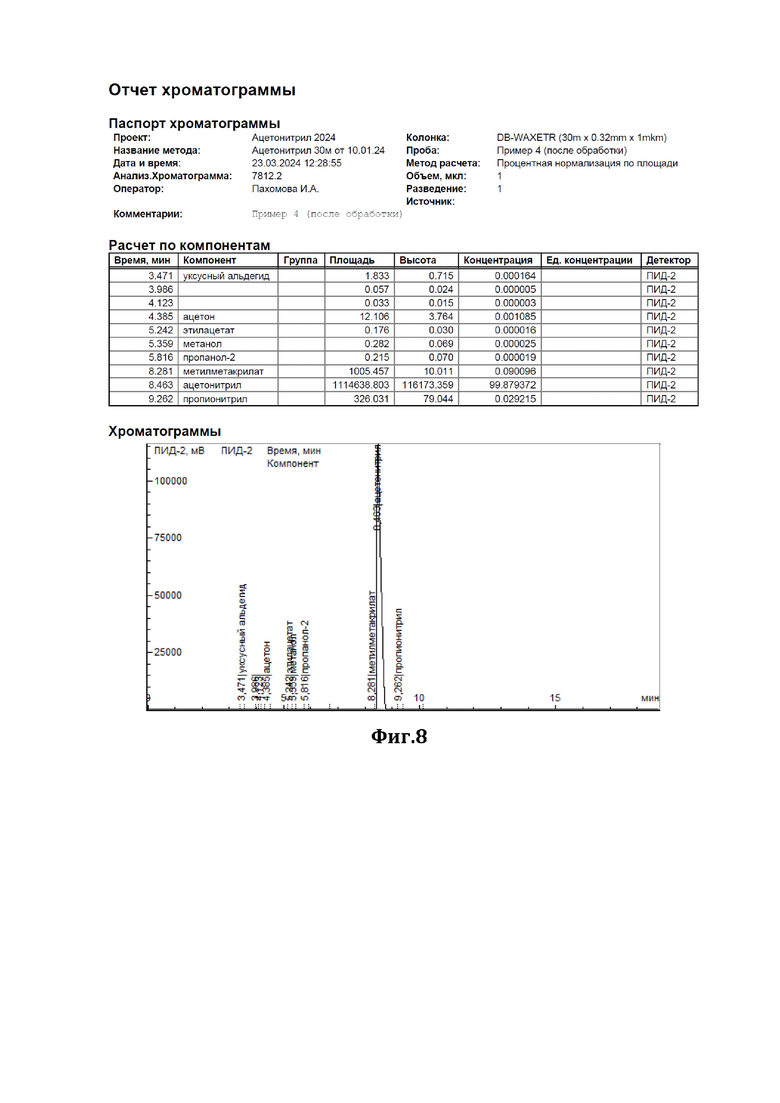
К 200 мл ацетонитрила, содержащего 0,47% метилметакрилата (см. диаграмму на Фиг.7), добавили смесь резорцина с резорцинатом натрия в соотношении 2:1 (мол) в количестве 10 г. Нагревали при перемешивании с обратным холодильником в течение 6,5 часов. По окончании перемешивания отогнали обработанный ацетонитрил. По результатам ГЖХ-анализа содержание метилметакрилата в ацетонитриле составило 0,09% (см. диаграмму на Фиг.8).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ АЦЕТОНИТРИЛА ОТ АЛЛИЛОВОГО СПИРТА | 2024 |
|
RU2825957C1 |
| Способ получения м-феноксифенола | 1990 |
|
SU1740365A1 |
| СПОСОБ ОЧИСТКИ АЦЕТОНИТРИЛА | 1995 |
|
RU2149867C1 |
| Способ очистки ацетонитрила от примеси акрилонитрила, аллилового спирта | 1990 |
|
SU1754710A1 |
| Способ очистки нафталинсодержащих продуктов | 1981 |
|
SU1047897A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(ФТОРМЕТОКСИ)-1,1,1,3,3,3-ГЕКСАФТОРИЗОПРОПАНА (СЕВОФЛУРАНА) | 2024 |
|
RU2830201C1 |
| Способ получения перфтор-4-(фторсульфонил)бутилвинилового эфира | 2022 |
|
RU2800857C1 |
| РЕАГЕНТ ДЛЯ ВВЕДЕНИЯ ПЕРФТОР-ТРЕТ-БУТИЛЬНОЙ ГРУППЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПРИМЕНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ПЕРФТОР-ТРЕТ-БУТИЛ-ЗАМЕЩЕННЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2602240C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-ЗАМЕЩЕННЫХ-3-(3-ФЕНОКСИФЕНИЛ)-2-ПРОПЕНЕНИТРИЛОВ | 2008 |
|
RU2366647C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТА-ФЕНОКСИФЕНОЛА | 2005 |
|
RU2287516C1 |
Изобретение относится к технологии удаления примесей из органических соединений, конкретно к способу очистки ацетонитрила от непредельных примесей, содержащих активированную двойную связь и трудно отделяемых ректификацией. Метод характеризуется тем, что технический ацетонитрил обрабатывают при температуре кипения ацетонитрила смесью резорцина с резорцинатом калия или резорцинатом натрия, где мольное количество резорцина по отношению к непредельным примесям составляет 1:2-200:1. Технический результат изобретения заключается в предоставлении способа высокой степени очистки целевого соединения от примесей непредельного характера без использования повышенного давления. 2 з.п. ф-лы, 8 ил., 4 пр.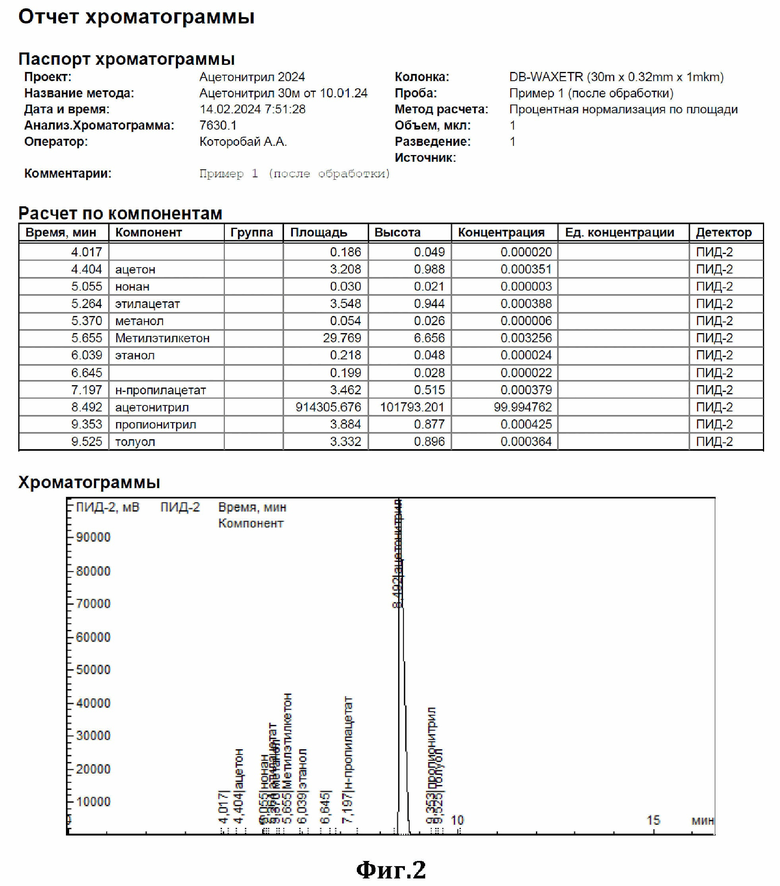
1. Способ очистки ацетонитрила от непредельных примесей, содержащих активированную двойную связь и трудно отделяемых ректификацией, характеризующийся тем, что технический ацетонитрил обрабатывают при температуре кипения ацетонитрила смесью резорцина с резорцинатом калия или резорцинатом натрия, где мольное количество резорцина по отношению к непредельным примесям составляет не ниже 1:2, но не более 200:1.
2. Способ по п.1, отличающийся тем, что обработку ведут нагревом при перемешивании с обратным холодильником в течение 3-6,5 ч.
3. Способ по п.1 или 2, отличающийся тем, что обработанный ацетонитрил передают на стадию ректификации или, после выделения дистилляцией, на стадию обработки окислителем.
| СПОСОБ ОЧИСТКИ АЦЕТОНИТРИЛА | 1995 |
|
RU2149867C1 |
| Способ очистки ацетонитрила от примеси акрилонитрила, аллилового спирта | 1990 |
|
SU1754710A1 |
| Способ очистки ацетонитрила | 1985 |
|
SU1318588A1 |
| Способ очистки ацетонитрила | 1991 |
|
SU1810332A1 |
| СПОСОБ ОЧИСТКИ НЕОЧИЩЕННОГО АЦЕТОНИТРИЛА | 2001 |
|
RU2267481C2 |
| US 10294197 B2, 21.05.2019 | |||
| US 4228297 A, 14.10.1980. | |||

