Настоящее изобретение относится к органической химии, более конкретно, к области растворителей, используемых в высокоэффективной жидкостной хроматографии (ВЭЖХ). А именно, предложен способ очистки ацетонитрила от аллилового спирта.
Хроматограф представляет собой весьма чувствительный комплекс механических, оптических, электрических и химических узлов, и его надежная работа во многом зависит от свойств и качества используемых растворителей. Растворители, применяемые в ВЭЖХ, должны удовлетворять следующим основным требованиям: высокая чистота, химическая инертность, совместимость с детектором, достаточная растворяющая способность по отношению к анализируемым веществам, безопастность, доступность и др. [см., например, Рудаков О.Б. и др. Тетрагидрофуран - универсальный растворитель для жидкостной хроматографии / Вестник ВГУ. Серия: Химия. Биология. Фармация, 2003, N. 2, с. 56-65].
Одним из таких растворителей, широко применяемых в ВЭЖХ, является ацетонитрил, который является практически незаменимым и одним из самых востребованных в хроматографическом анализе [см., например, Рудаков О.Б. и др. Хроматографические и экстракционные свойства ацетонитрила и его смесей с водой / Сорбционные и хроматографические процессы, 2018, Т. 18, № 4, с. 458-478]. Ацетонитрил для указанного назначения должен характеризоваться минимальным содержанием примесей, а также высокой степенью светопропускания в УФ-области. Однако коммерческий ацетонитрил может содержать различные примеси. Часть примесей может быть удалена простой ректификацией. Но некоторые примеси данным методом отделить невозможно, и для их отделения может быть использована различная предварительная химическая обработка сырья.
Одной из распространенных и трудноудаляемых примесей является аллиловый спирт. Он обладает сильным поглощением ультрафиолета, поэтому обязательно его полное удаление из ацетонитрила. При этом удаление аллилового спирта, содержащегося в качестве примеси в загрязненном ацетонитриле, путем ректификации очень затруднительно, поскольку температура кипения аллилового спирта очень близка к температуре кипения ацетонитрила [патент US 4308108 A, 29.12.1981].
Из уровня техники известны методы очистки ацетонитрила от примесей с использованием адсорбции. Так, из документа [патент RU 2149867 C1, 27.05.2000] известен способ очистки ацетонитрила, включающий селективное превращение ненасыщенных примесей с функциональными группами -C=C- или =C=O (в том числе, аллилового спирта) посредством обработки исходного ацетонитрила твердым реагентом реагентом, выбранным из сильного основания, окислителя или инициатора полимеризации, с получением продуктов превращения, имеющих физические характеристики, отличные от физических характеристик ацетонитрила, с последующим пропусканием обработанного ацетонитрила более чем через один слой адсорбента, поглощающего продукты превращения. В свою очередь, в документе [авторское свидетельство SU 1318588 A1, 23.06.1987] раскрыт способ очистки ацетонитрила, в соответствии с которым ацетонитрил, содержащий в качестве примесей аллиловый спирт и акрилонитрил, пропускают через адсорбент, в качестве которого используют окись алюминия, модифицированную перманганатом калия, при скорости пропускания ацетонитрила 1-5 мл/см2·мин, далее очищенный ацетонитрил фильтруют и при необходимости перегоняют.
Однако адсорбционный метод очистки требует периодически регенерации или замены адсорбента, что не позволяет проводить процесс разделения (очистки) непрерывно.
Из уровня техники также известны методы химической обработки примесей, содержащихся в ацетонитриле, с проведением последующей дистилляции для выделения очищенного ацетонитрила. Так, из уровня техники [авторское свидетельство
SU 1754710 A1, 15.08.1992] известен способ очистки ацетонитрила от примеси акрилонитрила и аллилового спирта, включающий обработку ацетонитрила 10%-ным водным раствором гидроксида щелочного металла при кипении и молярном соотношении воды в указанном растворе гидроксида к акрилонитрилу в исходном продукте, равном 1:1,5-2 и обработку бромом при комнатной температуре при соотношении брома к аллиловому спирту, равном 1:1,5-1,2 с последующей обработкой полученного после бромирования продукта комплексообразующим соединением (алюминий-трис(2-этоксиэтанолом), взятым в количестве 1-2 мас.% к ацетонитрилу, при кипении, с последующим выделением целевого продукта дистилляцией. В свою очередь, в документе [заявка на патент JP 2004339111 A, 02.12.2004] раскрыт способ удаления аллилового спирта из ацетонитрила, характеризующийся контактированием ангидрида кислоты (уксусного ангидрида) с ацетонитрилом (содержащим аллиловый спирт в качестве примеси), предпочтительно в присутствии катализатора (п-толуолсульфоновая кислота), с последующей перегонкой полученной смеси для отделения ацетонитрила от избытка кислотного ангидрида, катализатора и продукта реакции, при этом дополнительно к дистилляции может быть проведена адсорбционная обработка.
Однако такие методы являются многостадийными, и каждая стадия требует индивидуального аппаратурного оформления.
В документе [патент US 4308108 A, 29.12.1981] раскрыт способ очистки загрязненного ацетонитрила, содержащего цианистый водород в качестве основного примесного компонента, который включает обработку сырого ацетонитрила щелочью с последующей дистилляцией реакционной смеси при пониженном давлении. Также в этом документе раскрывается частный случай, когда ацетонитрил в качестве примеси содержит еще и аллиловый спирт, и в таком варианте способ дополнительно включает экстрактивную дистилляцию при пониженном давлении неочищенного ацетонитрила в ректификационной колонне в присутствии воды. Данное техническое решение выбрано в качестве наиболее близкого аналога (прототипа). На стадии экстрактивной дистилляции неочищенный ацетонитрил и вода подаются в промежуточную часть ректификационной колонны независимо друг от друга или в виде предварительно сформованной смеси; ацетонитрил извлекают из верхней части колонны в виде азеотропной смеси с водой (или очень близкой к азеотропной смеси), а из нижней части колонны отделяют концентрат аллилового спирта в виде смеси с водой и некоторым количеством ацетонитрила. В отличие от обычного процесса дистилляции, при котором аллиловый спирт не может быть полностью отделен от ацетонитрила, в условиях этого процесса, относительная летучесть ацетонитрила по отношению к аллиловому спирту значительно повышается, и становится возможным дистилляционное отделение аллилового спирта от ацетонитрила. При этом на эффективность отделения аллилового спирта и эффективность извлечения ацетонитрила влияет количество воды, присутствующей в системе дистилляции.
Однако в данном документе сообщается, что степень извлечения ацетонитрила, которая может быть достигнута, составляет 96-97,5%, и смесь, выходящая из нижней части колонны, содержит до 5 мас.% ацетонитрила. Таким образом, осуществление данного способа сопровождается потерями ацетонитрила. Кроме того, как уже было отмечено, данное техническое решение позволяет получить ацетонитрил только в виде азеотропной смеси с водой или близкой к азеотропной смеси (т.е. соотношение смеси ацетонитрил:вода составляет ~84 мас.%/16 мас.%).
В связи с чем, остается потребность в разработке эффективного способа удаления аллилового спирта из ацетонитрила.
Техническая задача настоящего изобретения заключается в расширении арсенала способов удаления аллилового спирта из ацетонитрила.
Технический результат настоящего изобретения заключается в получении ацетонитрила, свободного от аллилового спирта и продуктов его окисления, а также в осуществлении непрерывного способа очистки в одной единице оборудования.
Указанный технический результат достигается за счет совмещения процессов каталитического окисления аллилового спирта и одновременной ректификации получаемой смеси.
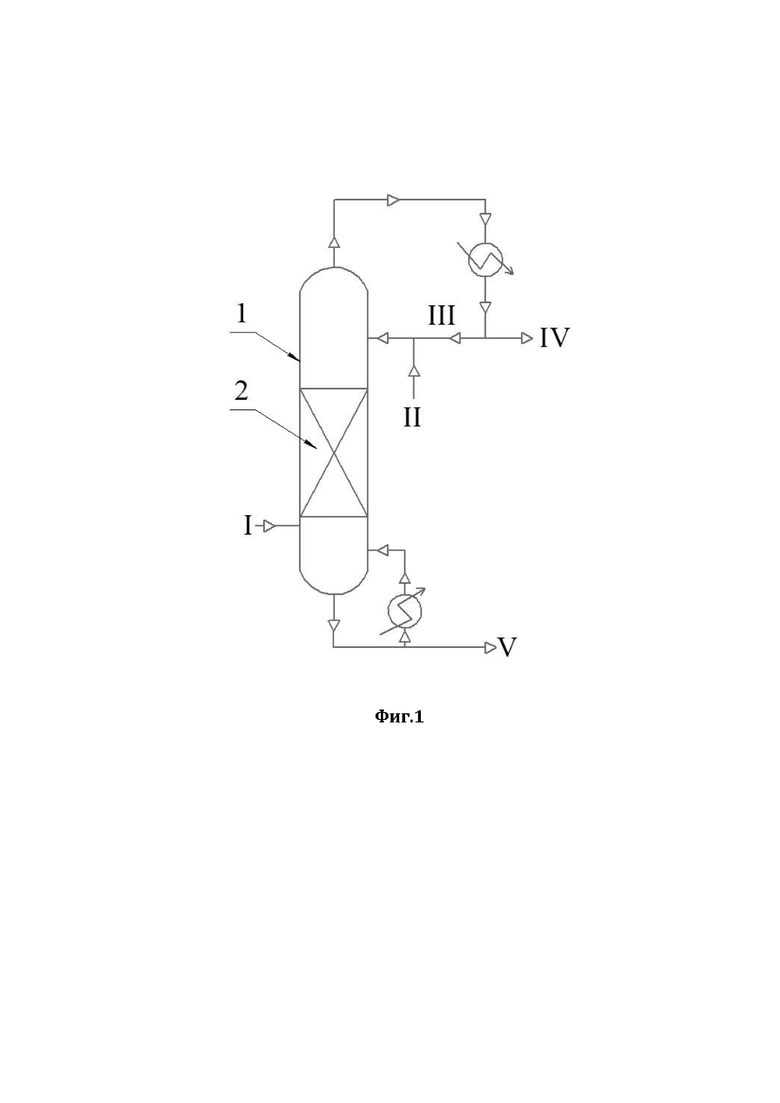
Более подробно, технический результат достигается способом очистки ацетонитрила от аллилового спирта с использованием ректификации, в соответствии с которым: в низ ректификационной колонны подают ацетонитрил, содержащий в качестве примеси аллиловый спирт, а с верха указанной ректификационной колонны противотоком подают смесь 30-35%-ного по массе водного раствора пероксида водорода и флегмы, далее очищенный от аллилового спирта ацетонитрил отделяют в виде дистиллята, а смесь глицерина, воды и непрореагировавшего пероксида водорода выводят из кубовой части указанной ректификационной колонны. В способе по настоящему изобретению применяли ректификационную колонну насадочного типа, которая была заполнена в качестве насадки керамическими шарами с нанесенным на них катализатором; при этом в качестве катализатора использовали катализатор окисления аллилового спирта - оксид вольфрама или оксид молибдена.
Краткое описание чертежей
На фиг.1 представлена схема ректификации (1 - ректификационная колона;
2 - насадки). Потоки: I - смесь ацетонитрила и аллилового спирта; II - водный раствор пероксида водорода; III - флегма; IV - ацетонитрил в виде дистиллята; V - смесь глицерина, воды и непрореагировавшего пероксида водорода.
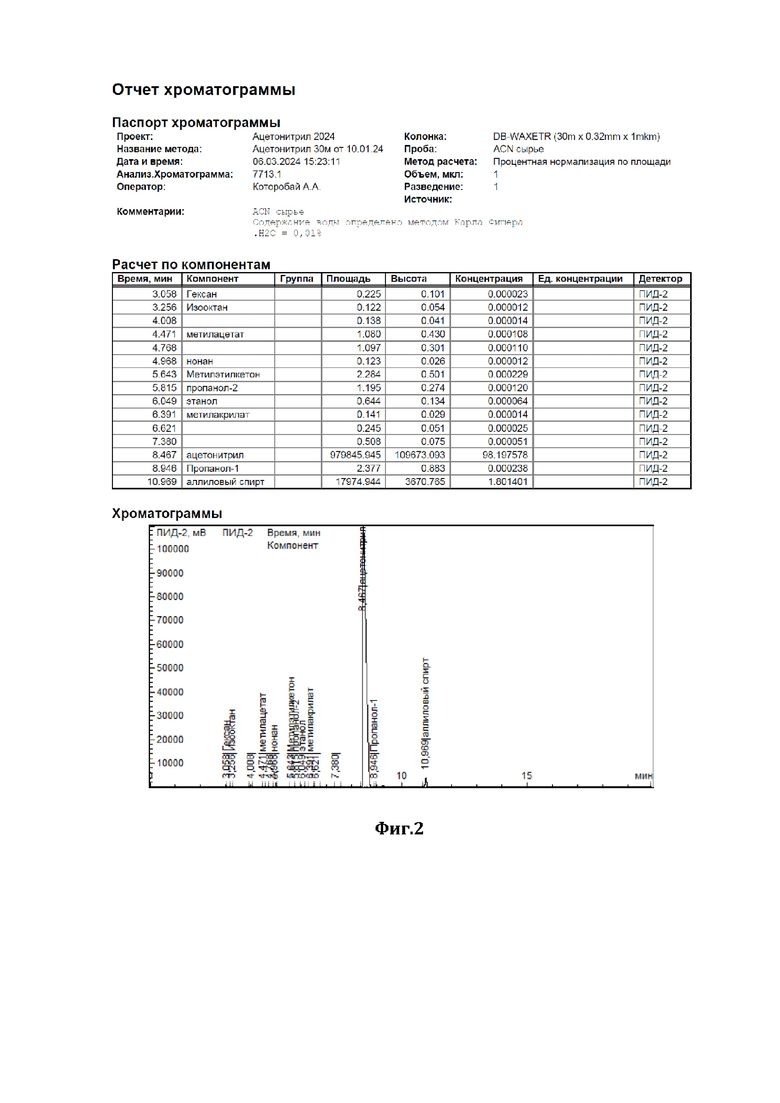
На фиг.2 показана хроматограмма исходного сырья, состоящая из 98,2 мас.% ацетонитрила и 1,8 мас.% аллилового спирта.
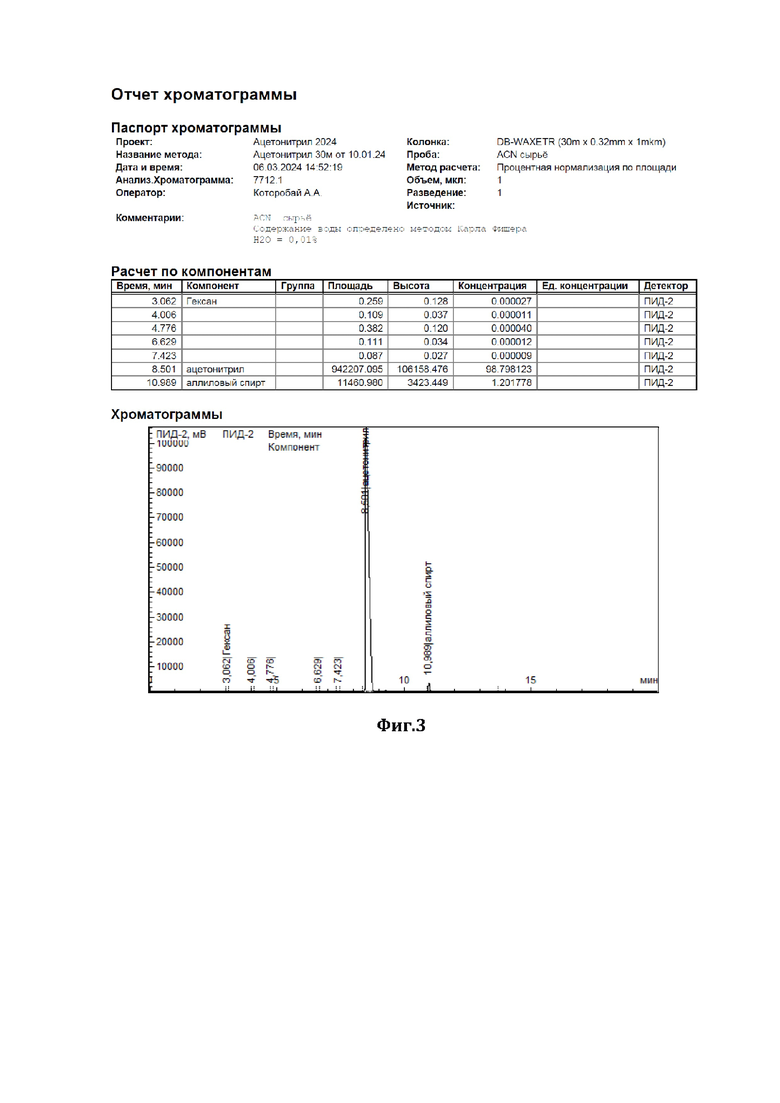
На фиг.3 показана хроматограмма исходного сырья, состоящая из 98,8 мас.% ацетонитрила и 1,2 мас.% аллилового спирта.
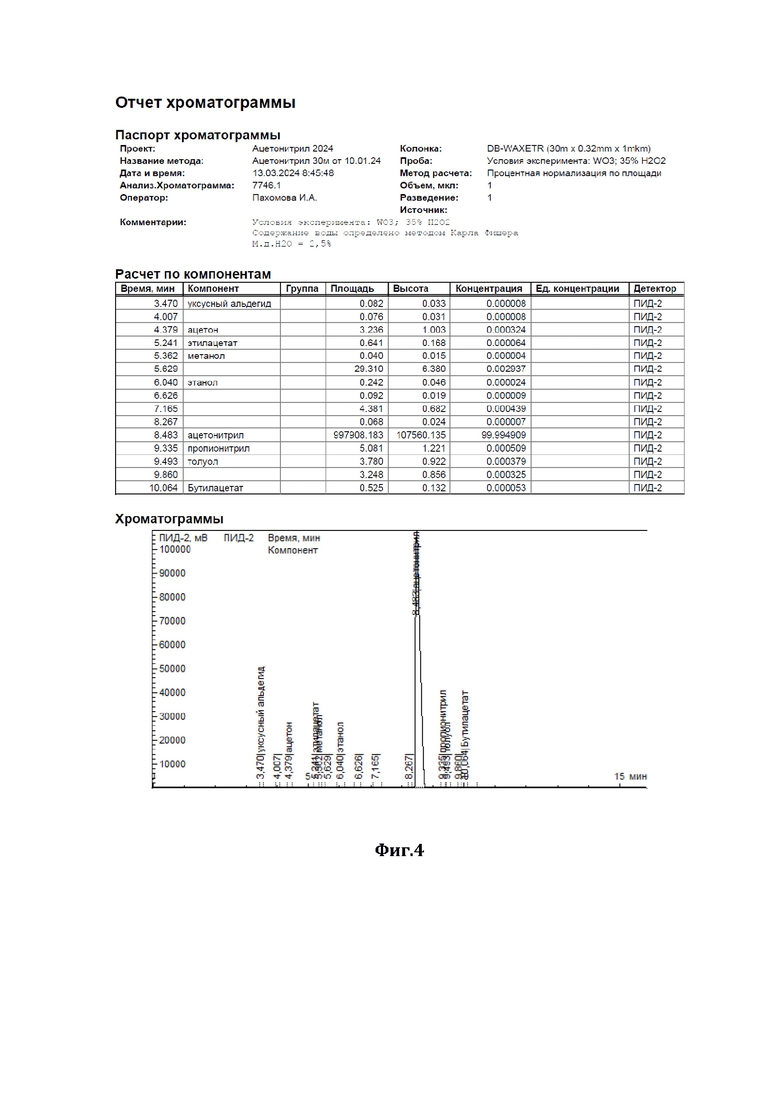
На фиг.4 показана хроматограмма результатов очистки по примеру 1.
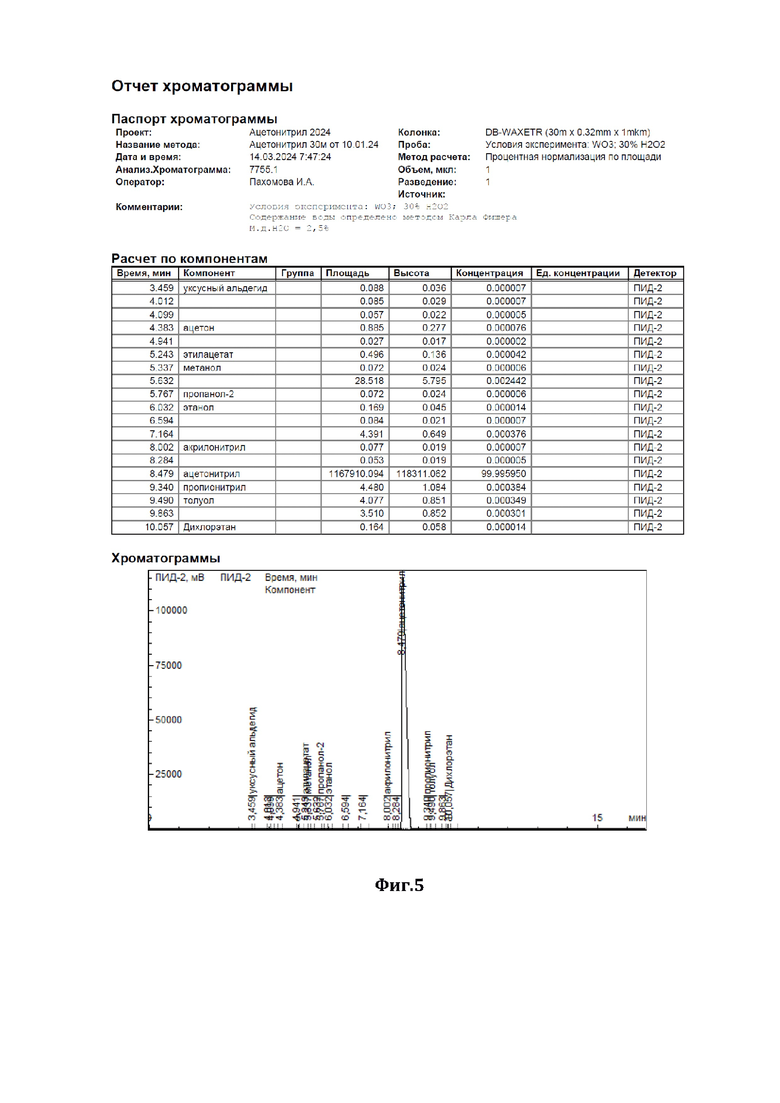
На фиг.5 показана хроматограмма результатов очистки по примеру 2.
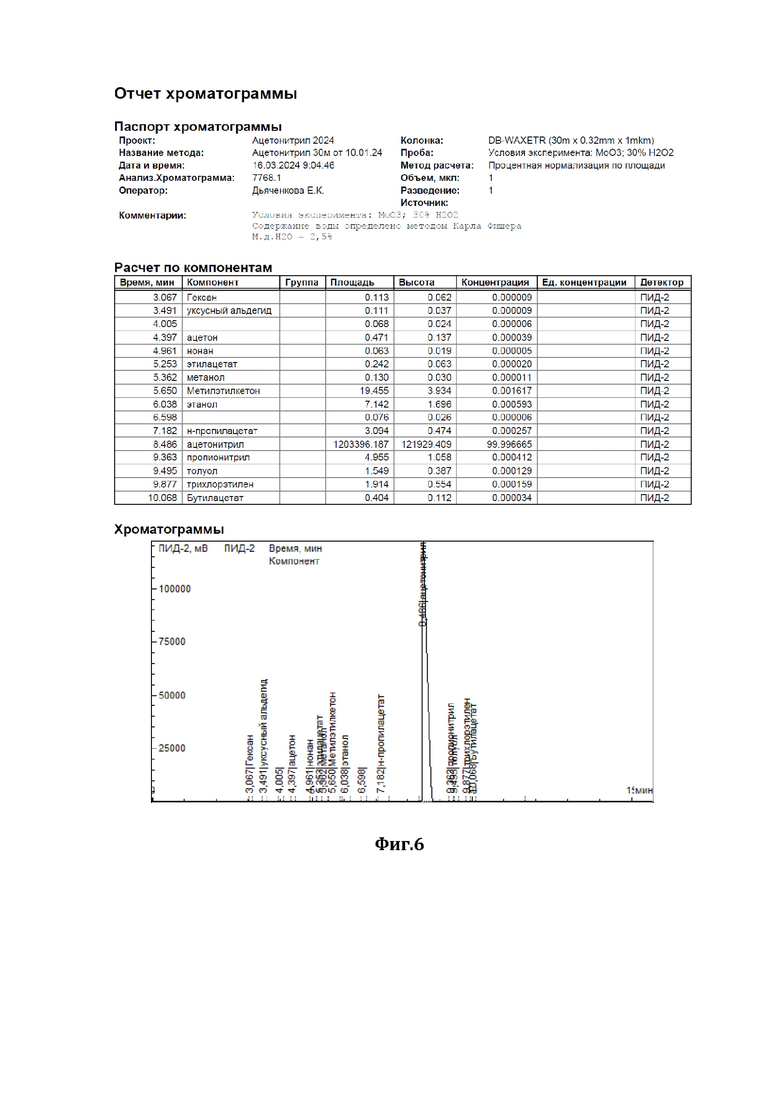
На фиг.6 показана хроматограмма результатов очистки по примеру 3.
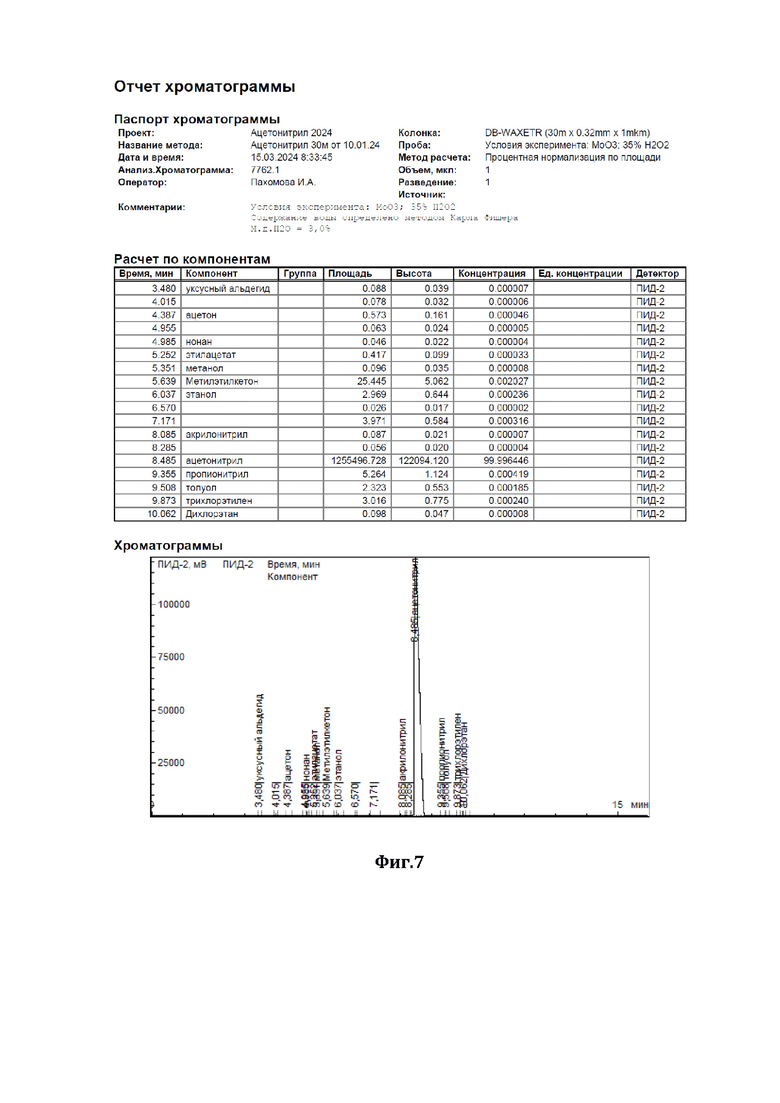
На фиг.7 показана хроматограмма результатов очистки по примеру 4.
Осуществление изобретения
В низ ректификационной колонны (1) насадочного типа, заполненной в качестве насадки (2) керамическими шарами с нанесенным на них катализатором окисления аллилового спирта (оксид вольфрама или оксид молибдена), подается в виде пара исходная смесь, состоящая из ацетонитрила и аллилового спирта (I) - ацетонитрил, содержащий в качестве примеси аллиловый спирт. С верха ректификационной колонны (I) противотоком подается смесь водного раствора пероксида водорода (II) и флегмы (III). Использовали водный раствор пероксида водорода концентрацией 30-35% по массе (данная концентрация является оптимальной для осуществления предлагаемого способа, использование более концентрированного раствора пероксида водорода взрывоопасно). В точке встречи паров исходной смеси (I) со смесью флегмы (III) и водного раствора пероксида водорода (II) на указанном катализаторе протекает реакция окисления аллилового спирта пероксидом водорода, в результате которой аллиловый спирт полностью окисляется в глицерин. При этом, за счет осуществляемой ректификации, ацетонитрил в виде дистиллята (IV) отделяется от других компонентов и выводится сверху колонны, предварительно охладившись в холодильнике. Остальные, более тяжелокипящие вещества - глицерин (продукт окисления аллилового спирта), вода, содержащаяся в водном растворе пероксида водорода, и непрореагировавший пероксид водорода (V) выводятся из кубовой части ректификационной колонны (1) и отправляются на утилизацию. Осуществление предлагаемого способа позволяет достичь полного удаления аллилового спирта из ацетонитрила. Дистиллят содержит ацетонитрил, свободный от органических примесей, с содержанием воды до 3,0 мас.%. Данный ацетонитрил, свободный от аллилового спирта, уже пригоден для дальнейшего получения из него оптически чистых квалификаций ацетонитрила. Вода, содержащаяся в небольшом количестве в дистилляте, может быть удалена дополнительной ректификацией.
Изобретение иллюстрируется следующими примерами.
Пример 1
В низ ректификационной колонны (1) насадочного типа, заполненной в качестве насадки (2) керамическими шарами (диаметром 10 мм) с нанесенным на них катализатором - оксидом вольфрама (WO3), подавали в виде пара исходную смесь (см. характеристики из Фиг.2), состоящую из 98,2 мас.% ацетонитрила и 1,8 мас.% аллилового спирта (I), расход исходной смеси составлял 1000 г/час. С верха колонны, противотоком, подавали смесь 35%-ного по массе водного раствора пероксида водорода (II), расход - 40 г/ч, и флегмы (III), расход - 30287 г/ч. Флегмовое число равно 40. Нагрев ректификационной колонны (1) осуществляли за счет пропускания кубовой жидкости через нагреватель. Температура нижней части ректификационной колонны (куба) поддерживали 87°С, а верхней части - 82°С, ректификацию и окисление аллилового спирта осуществляли при давлении 1,2 бара. Смесь ацетонитрила с водой (дистиллят) (IV) отделяли от других компонентов и выводили сверху ректификационной колонны, предварительно охладив его в холодильнике, расход дистиллята составлял 1007 г/ч.
Содержание ацетонитрила в дистилляте составляло 97,5 % мас., остальное - вода, аллиловый спирт отсутствовал (содержание компонентов дистиллята определяли методом газовой хроматографии - см. отчет хроматографии из Фиг.4). Смесь глицерина - продукта окисления аллилового спирта перекисью водорода, воды и непрореагировавшего пероксида водорода (V) выводили из кубовой части ректификационной колонны, расход смеси составлял 32,8 г/ч, и отправляли на утилизацию.
Пример 2
В низ ректификационной колонны (1) насадочного типа, заполненной в качестве насадки (2) керамическими шарами (диаметром 10 мм) с нанесенным на них катализатором - оксидом вольфрама (WO3), подавали в виде пара исходную смесь (см. характеристики из Фиг.3), состоящую из 98,8 мас.% ацетонитрила и 1,2 мас.% аллилового спирта (I), расход исходной смеси составлял 1000 г/час. С верха колонны, противотоком, подавали смесь 30%-ного по массе водного раствора пероксида водорода (II), расход - 40 г/ч, и флегмы (III), расход - 40533 г/ч. Флегмовое число равно 40. Нагрев ректификационной колонны (1) осуществляли за счет пропускания кубовой жидкости через нагреватель. Температура нижней части ректификационной колонны (куба) поддерживали 87°С, а верхней части - 82°С, ректификацию и окисление аллилового спирта осуществляли при давлении 1,2 бара. Смесь ацетонитрила с водой (дистиллят) (IV) отделяли от других компонентов и выводили сверху ректификационной колонны, предварительно охладив его в холодильнике, расход дистиллята составлял 1013 г/ч. Содержание ацетонитрила в дистилляте составляло 97,5 % мас., остальное - вода, аллиловый спирт отсутствовал (содержание компонентов дистиллята определяли методом газовой хроматографии - см. отчет хроматографии из Фиг.5). Смесь глицерина - продукта окисления аллилового спирта перекисью водорода, воды и непрореагировавшего пероксида водорода (V) выводили из кубовой части ректификационной колонны, расход смеси составлял 26,7 г/ч, и отправляли на утилизацию.
Пример 3
В низ ректификационной колонны (1) насадочного типа, заполненной в качестве насадки (2) керамическими шарами (диаметром 10 мм) с нанесенным на них катализатором - оксидом молибдена (MoO3), подавали в виде пара исходную смесь (см. характеристики из Фиг.2), состоящую из 98,2 мас.% ацетонитрила и 1,8 мас.% аллилового спирта (I), расход исходной смеси составлял 1000 г/час. С верха колонны, противотоком, подавали смесь 35%-ного по массе водного раствора пероксида водорода (II), расход - 40 г/ч, и флегмы (III), расход - 30371 г/ч. Флегмовое число равно 30. Нагрев ректификационной колонны (1) осуществляли за счет пропускания кубовой жидкости через нагреватель. Температура нижней части ректификационной колонны (куба) поддерживали 87°С, а верхней части - 82°С, ректификацию и окисление аллилового спирта осуществляли при давлении 1,2 бара. Смесь ацетонитрила с водой (дистиллят) (IV) отделяли от других компонентов и выводили сверху ректификационной колонны, предварительно охладив его в холодильнике, расход дистиллята составлял 1012 г/ч. Содержание ацетонитрила в дистилляте составляло 97,0 % мас., остальное - вода, аллиловый спирт отсутствовал (содержание компонентов дистиллята определяли методом газовой хроматографии - см. отчет хроматографии из Фиг.6). Смесь глицерина - продукта окисления аллилового спирта перекисью водорода, воды и непрореагировавшего пероксида водорода (V) выводили из кубовой части ректификационной колонны, расход смеси составлял 27,6 г/ч, и отправляли на утилизацию.
Пример 4
В низ ректификационной колонны (1) насадочного типа, заполненной в качестве насадки (2) керамическими шарами (диаметром 10 мм) с нанесенным на них катализатором - оксидом молибдена (MoO3), подавали в виде пара исходную смесь (см. характеристики из Фиг.3), состоящую из 98,8 мас.% ацетонитрила и 1,2 мас.% аллилового спирта (I), расход исходной смеси составлял 1000 г/час. С верха колонны, противотоком, подавали смесь 30%-ного по массе водного раствора пероксида водорода (II), расход - 40 г/ч, и флегмы (III), расход - 40533 г/ч. Флегмовое число равно 40. Нагрев ректификационной колонны (1) осуществляли за счет пропускания кубовой жидкости через нагреватель. Температура нижней части ректификационной колонны (куба) поддерживали 87°С, а верхней части - 82°С, ректификацию и окисление аллилового спирта осуществляли при давлении 1,2 бара. Смесь ацетонитрила с водой (дистиллят) (IV) отделяли от других компонентов и выводили сверху ректификационной колонны, предварительно охладив его в холодильнике, расход дистиллята составлял 1013 г/ч. Содержание ацетонитрила в дистилляте составляло 97,5 % мас., остальное - вода, аллиловый спирт отсутствовал (содержание компонентов дистиллята определяли методом газовой хроматографии - см. отчет хроматографии из Фиг.7). Смесь глицерина - продукта окисления аллилового спирта перекисью водорода, воды и непрореагировавшего пероксида водорода (V) выводили из кубовой части ректификационной колонны, расход смеси составлял 26,7 г/ч, и отправляли на утилизацию.
Вышеприведенные примеры и отчеты хроматограмм на Фиг.2-Фиг.7 демонстрируют, что заявленным изобретением получают ацетонитрил, свободный от аллилового спирта и продуктов его окисления, и при этом ведут непрерывно очистку в одной единице оборудования.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ ПЕРОКСИДА ВОДОРОДА | 2001 |
|
RU2205788C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВТОРИЧНОГО БУТИЛОВОГО СПИРТА | 2001 |
|
RU2206560C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛХЛОРИДА | 2009 |
|
RU2404952C1 |
| СПОСОБ ВЫДЕЛЕНИЯ КОНЦЕНТРИРОВАННОГО ЭПИХЛОРГИДРИНА ИЗ ПРОДУКТОВ ЭПОКСИДИРОВАНИЯ ХЛОРИСТОГО АЛЛИЛА ПЕРОКСИДОМ ВОДОРОДА НА ТИТАНСОДЕРЖАЩЕМ ЦЕОЛИТНОМ КАТАЛИЗАТОРЕ | 2015 |
|
RU2593205C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРОКСИДА ВОДОРОДА | 2017 |
|
RU2648887C1 |
| УДАЛЕНИЕ АЦЕТАЛЬДЕГИДА ИЗ МЕТИЛАЦЕТАТА РЕКТИФИКАЦИЕЙ ПРИ ПОВЫШЕННОМ ДАВЛЕНИИ | 2008 |
|
RU2470007C2 |
| СПОСОБ ВЫДЕЛЕНИЯ N-МЕТИЛАНИЛИНА ИЗ КАТАЛИЗАТОВ N-ГИДРОАЛКИЛИРОВАНИЯ АНИЛИНА МЕТАНОЛОМ | 2000 |
|
RU2167851C1 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ПИРОКАТЕХИНА, ГИДРОХИНОНА И ПЛАСТИФИКАТОРА БЕТОНА | 1992 |
|
RU2028288C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФОРМАЛЬДЕГИДА | 2004 |
|
RU2267479C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2003 |
|
RU2248961C1 |
Изобретение относится к области органической химии, более конкретно к области растворителей, используемых в высокоэффективной жидкостной хроматографии (ВЭЖХ). Предложен способ очистки ацетонитрила от аллилового спирта, в соответствии с которым в низ ректификационной колонны подают ацетонитрил, содержащий в качестве примеси аллиловый спирт, а с верха ректификационной колонны противотоком подают смесь 30-35%-ного по массе водного раствора пероксида водорода и флегмы. Далее очищенный от аллилового спирта ацетонитрил отделяют в виде дистиллята, а смесь глицерина, воды и непрореагировавшего пероксида водорода выводят из кубовой части ректификационной колонны. Ректификационная колонна является колонной насадочного типа, которая заполнена в качестве насадки керамическими шарами с нанесенным на них в качестве катализатора оксидом вольфрама или оксидом молибдена. Технический результат - получение ацетонитрила, свободного от аллилового спирта и продуктов его окисления, при осуществлении непрерывного способа очистки в одной единице оборудования. 7 ил., 4 пр.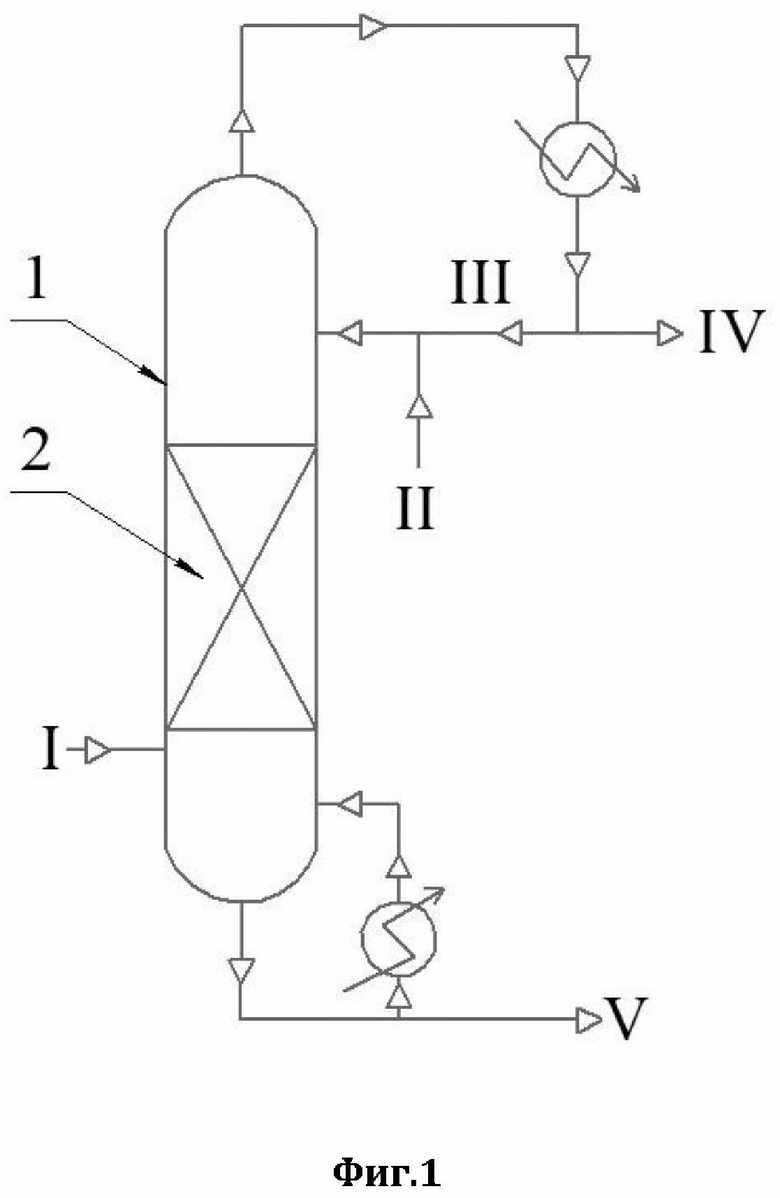
Способ очистки ацетонитрила от аллилового спирта с использованием ректификации, отличающийся тем, что в низ ректификационной колонны насадочного типа, которая заполнена в качестве насадки керамическими шарами с нанесенным на них в качестве катализатора оксидом вольфрама или оксидом молибдена, подают ацетонитрил, содержащий в качестве примеси аллиловый спирт, а с верха указанной ректификационной колонны противотоком подают смесь 30-35%-ного по массе водного раствора пероксида водорода и флегмы, далее очищенный от аллилового спирта ацетонитрил отделяют в виде дистиллята, а смесь глицерина, воды и непрореагировавшего пероксида водорода выводят из кубовой части указанной ректификационной колонны.
| 0 |
|
SU308108A1 | |
| Способ очистки ацетонитрила от примеси акрилонитрила, аллилового спирта | 1990 |
|
SU1754710A1 |
| RU 2008135049 A, 10.03.2010 | |||
| СПОСОБ ОЧИСТКИ НЕОЧИЩЕННОГО АЦЕТОНИТРИЛА | 2001 |
|
RU2267481C2 |
| Coetzee, J | |||
| F | |||
| Purification of acetonitrile and tests for impurities | |||
| Pure and Applied Chemistry, 1966, 13(3), 429-435. | |||

