ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтики и относится к дендримеру, нагруженному лекарственным средством и фармакокинетическим модификатором, и, в частности, к связыванию лекарственного средства с дендримером посредством определенного линкера.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Хотя исследование и разработка лекарственных средств в настоящее время значительно продвинулись вперед, все еще трудно получить подходящие композиции для многих лекарственных средств для введения в клинических испытаниях из-за их физических свойств (таких как растворимость), или композиции являются непригодными из-за токсических эффектов, которые возникают на стадии с высокой концентрацией лекарственного средства, или плохих терапевтических показателей после введения. Кроме того, недостатки композиций также включают, например, плохую абсорбцию, низкую биодоступность, плохую стабильность in vivo, системные побочные эффекты из-за плохого нацеливания и неспособность контролировать их биораспределение, метаболизм и почечный или печеночный клиренс в организме после введения. По мере быстрого развития исследований в области лекарственных средств были открыты некоторые новые области исследований и разработаны некоторые технические подходы с большим потенциалом для облегчения разработки лекарственных средств. Эти подходы включают, например, составление лекарственных средств в виде липосомных, мицелловых или полимерных мицелловых композиций, ковалентное присоединение лекарственных средств к каркасам гидрофильных полимеров и т.д. Хотя эти подходы позволяют солюбилизировать фармацевтически активные агенты и в некоторых случаях улучшить биодоступность и нацеливание, существует трудности с точки зрения высвобождения фармацевтически активных агентов. В некоторых случаях носитель быстро разлагается для высвобождения фармацевтически активного агента до того, как молекула лекарственного средства достигает органа-мишени. Во многих случаях скорость высвобождения фармацевтически активного агента из носителя является переменной, что делает лекарственное средство неспособным достигать терапевтически эффективной дозы in vivo или в органе-мишени.
В последние годы были достигнуты значительные успехи в отношении дендримеров в области биотехнологии и применения лекарственных средств (Xiangyang Shi et al., Sci China Mater, 2018, 61(11), 1387-1403). Дендримеры представляют собой особый класс полимеров, которые имеют плотные разветвленные структуры. Они представляют собой древовидные макромолекулы, возникающие в результате повторяемого разветвления молекулы ядра наружу. Другими словами, ветвь ядра вырастает до определенной длины, а затем разделяется на две ветви, и это повторяется до тех пор, пока молекула не станет настолько плотной, что образует сферический куст (V Gajbhiye et al., Journal of Pharmacy and Pharmacology, 2009, 61, 989-1003). Дендримеры характеризуются более высокой концентрацией функциональных групп/единичного молекулярного объема, чем обычные полимеры. В частности, уникальные свойства дендримеров, такие как их высокая степень разветвления, мультивалентность, сферические структуры и хорошо определенные молекулярные массы, делают их перспективными для использования в новых каркасах для доставки лекарственных средств. За последнее десятилетие возрос интерес к исследованиям по разработке и синтезу биосовместимых дендримеров и их применению во многих областях биологических наук, включая доставку лекарственных средств.
Starpahrma Inc. из Австралии использует собственно разработанный дендритный полилизин для осуществления нагрузки и транспортировки противораковых лекарственных средств для усиления фармакологических свойств лекарственных средств, гарантируя, что лекарственные средства будут доставлены в соответствующие части организма в надлежащее время. Этот подход называется «доставка лекарственных средств» («drug delivery»), и эта методика выпускается под торговой маркой DEP®. DЕР®-доцетаксел, DЕР®-кабазитаксел и DЕР®-иринотекан являются 3 основными противораковыми лекарственными средствами, разработанными компанией с использованием методики DEP®, которые в настоящее время находятся на стадии клинических исследований и имеют большие перспективы.
CN103796684A раскрывает макромолекулы, полученные путем связывания лекарственных средств с дендримерами посредством дикислотных линкеров, в частности насыщенных разветвленных или линейных С1-С10 дикислотных линкеров, прерванных атомами кислорода, азота или серы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к макромолекуле, которая содержит:
i) дендример D, имеющий поверхностные аминогруппы, где по меньшей мере две разные концевые группы ковалентно связаны с поверхностными аминогруппами дендримера;
ii) первую концевую группу, которая представляет собой фармацевтически активный агент, содержащий гидрокси, амино или сульфгидрильную группу, или представляет собой его остаток А; и
iii) вторую концевую группу, которая является фармакокинетическим модификатором;
где первая концевая группа ковалентно связана с поверхностной аминогруппой дендримера посредством линкера -X1-L-X2-; где X1 представляет собой группу связывания линкера с фармацевтически активным агентом или его остатком А, Х2 представляет собой группу связывания линкера с дендримером D, и X1 и Х2 оба представляют собой -С(О)-; L представляет собой линейный или разветвленный С1-10 алкилен, где линейный или разветвленный С1-10 алкилен замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, гидрокси, С3-7 циклоалкила, С3-7 циклоалкилена, С1-6 алкокси, галогеналкила, галогеналкокси, галогена, нитро, циано, ацила, сульфгидрила, тиоэфирной группы, сульфинила, сульфонила, -NR1R2, арила, гетероарила и гетероциклила;
каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, гидрокси, C1-6 алкила, циклоалкила и C1-6 алкокси.
В некоторых вариантах осуществления L представляет собой линейный или разветвленный С1-10 алкилен, где линейный или разветвленный С1-10 алкилен замещен одним или более заместителями, выбранными из группы, состоящей из дейтерия, галогена, -OR1, -SR1, -NR1R2 и -C(O)R3;
где каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, гидрокси, C1-6 алкила, С3-7 циклоалкила, C1-6 алкокси и C(O)R4, и указанные C1-6 алкил, С3-7 циклоалкил и C1-6 алкокси необязательно замещены одним или более заместителями, выбранными из группы, состоящей из гидрокси, галогена, C1-6 алкила, C1-6 алкокси, С2-6 алкенила, С2-6 алкинила, С1-6 галогеналкила, С1-6 галогеналкокси, нитро, циано, амино и C1-6 алкиламино;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и C1-6 алкокси.
В некоторых вариантах осуществления R1 не представляет собой водород.
В некоторых вариантах осуществления линейный или разветвленный С1-10 алкилен замещен одним или более заместителями, выбранными из группы, состоящей из -OR1, -SR1 и -NR1R2, где каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, C1-6 алкила, С3-7 циклоалкила, C1-6 алкокси и C(O)R4, и указанные C1-6 алкил, С3-7 циклоалкил и C1-6 алкокси необязательно замещены одним или более заместителями, выбранными из группы, состоящей из гидрокси, галогена, С1-6 алкила, С1-6 алкокси, С1-6 галогеналкила, C1-6 галогеналкокси, амино и C1-6 алкиламино; R4 выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и C1-6 алкокси.
В некоторых вариантах осуществления линейный или разветвленный C1-10 алкилен замещен одним или более -NR1R2, где каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, С1-6 алкила, С3-7 циклоалкила, С1-6 алкокси и C(O)R4, и указанные C1-6 алкил, С3-7 циклоалкил и C1-6 алкокси необязательно замещены одним или более заместителями, выбранными из группы, состоящей из гидрокси, галогена, С1-6 алкила, С1-6 алкокси, C1-6 галогеналкила, C1-6 галогеналкокси, амино и C1-6 алкиламино; R4 выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и C1-6 алкокси.
В некоторых вариантах осуществления линейный или разветвленный С1-10 алкилен замещен одним или более -NR1R2, где каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, C1-6 алкила и C(O)R4, и R4 выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 галогеналкила и C1-6 алкокси; предпочтительно каждый из R1 и R2 независимо выбран из группы, состоящей из водорода и C1-6 алкила, и R1 и R2 одновременно не представляют собой водород.
В некоторых вариантах осуществления линейный или разветвленный C1-10 алкилен представляет собой линейный или разветвленный C1-6 алкилен.
В некоторых вариантах осуществления, в линкере -X1-L-X2-, X1 представляет собой -С(О)- и связан с фармацевтически активным агентом или его остатком А; Х2 представляет собой -С(О)- и связан с поверхностной аминогруппой дендримера D с образованием амидной связи.
В некоторых вариантах осуществления фармацевтически активный агент или его остаток А содержит гидроксигруппу, которая образует сложноэфирную связь с Х1.
В некоторых конкретных вариантах осуществления макромолекула имеет структуру, выбранную из группы, состоящей из:
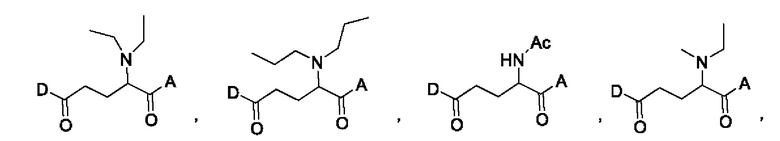
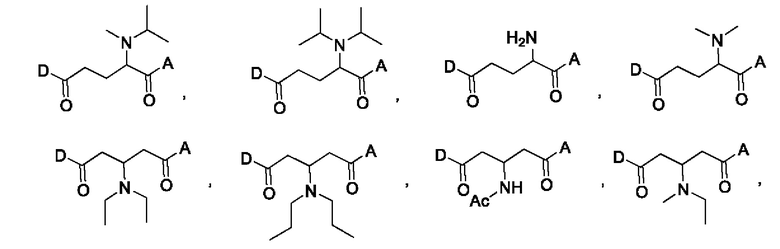
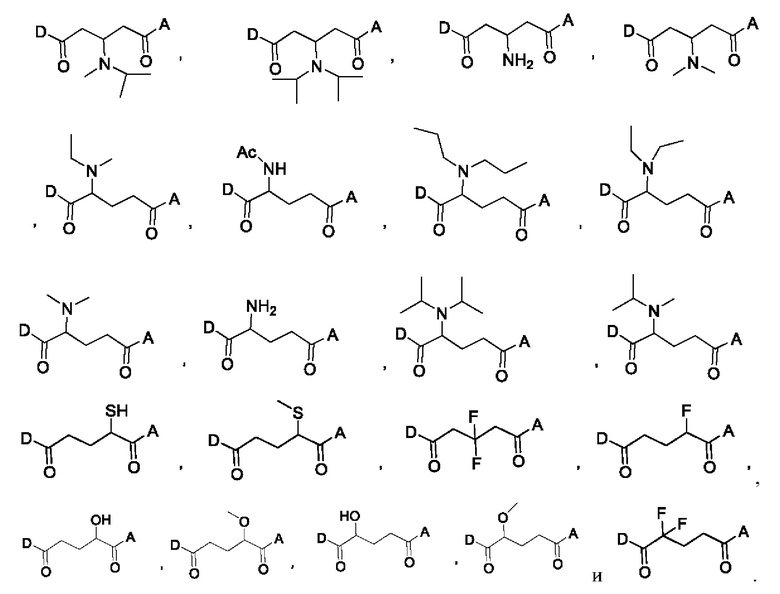
В некоторых вариантах осуществления линкер по настоящему изобретению может быть выбран для обеспечения желаемой скорости высвобождения лекарственного средства, например, быстрого высвобождения или медленного высвобождения.
В некоторых вариантах осуществления фармацевтически активный агент макромолекулы высвобождается быстрее, возможно по меньшей мере в 2 раза быстрее, чем при доставке независимо от макромолекулы. В некоторых вариантах осуществления фармацевтически активный агент макромолекулы высвобождается медленнее, возможно в два, три, четыре, пять, шесть, семь, восемь, девять, десять или более, пятнадцать или более, двадцать или более, тридцать или более раз медленнее, чем при доставке независимо от макромолекулы. Макромолекула с низкой скоростью высвобождения подходит для составления в форме лекарственного средства, которое обеспечивает медленное высвобождение в течение длительного периода времени, например, от 1 недели до 3 месяцев, от 1 месяца до 6 месяцев или более 6 месяцев. Быстрое высвобождение предпочтительно представляет собой высвобождение более 50% фармацевтически активного агента в течение от 0 до 8 ч, предпочтительно от 0 до 4 ч, в частности от 0 до 2 ч, и более конкретно от 5 до 60 мин. Промежуточное высвобождение предпочтительно представляет собой высвобождение более 50% фармацевтически активного агента в течение от 1 до 72 ч, предпочтительно в течение от 2 до 48 ч. Скорость высвобождения фармацевтически активного агента может контролироваться путем выбора подходящего линкера. Скорость высвобождения также зависит от свойств фармацевтически активного агента. В некоторых вариантах осуществления фармацевтически активный агент связан с дендримером посредством идентичных линкеров. В некоторых других вариантах осуществления фармацевтически активный агент связан с дендримером двумя или более типами линкера, так что фармацевтически активный агент может высвобождаться из макромолекулы с различными скоростями.
В некоторых вариантах осуществления первая концевая группа и вторая концевая группа присутствуют в соотношении от 1:2 до 2:1, предпочтительно 1:2, 1:1 или 2:1. В некоторых вариантах осуществления макромолекула содержит третью концевую группу, которая представляет собой блокирующую группу, лекарственное средство или нацеливающую группу. Блокирующая группа может представлять собой ацильную группу. В некоторых вариантах осуществления первая концевая группа, вторая концевая группа и третья концевая группа находятся в соотношении от 1:1:1 до 1:2:2, предпочтительно 1:2:1. В некоторых вариантах осуществления по меньшей мере 50% концевых групп включают одну из первой концевой группы и второй концевой группы. В конкретных вариантах осуществления фармацевтически активный агент связывается с более чем 14%, 25%, 27%, 30%, 39%, 44% или 48% поверхностных аминогрупп. В некоторых вариантах осуществления фармакокинетический модификатор связывается с более чем 15%, 25%, 30%, 33% или 46% поверхностных аминогрупп.
Фармацевтически активный агент по настоящему изобретению может быть выбран из группы, состоящей из анестетиков, антацидов, антител, противоинфекционных препаратов, биологических препаратов, сердечно-сосудистых препаратов, контрастных агентов, диуретических средств, агентов, повышающих количество гемоглобина в крови, иммунодепрессантов, гормонов и аналогов, нутрицевтиков, офтальмологических лекарственных средств, обезболивающих средств, респираторных лекарственных средств, адъювантов, анаболиков, противоартритных средств, противосудорожных средств, антигистаминных агентов, противовоспалительных лекарственных средств, противоязвенных лекарственных средств, модифицирующих поведение лекарственных средств, онкологических лекарственных средств, лекарственных средств для лечения центральной нервной системы, противозачаточных средств, лекарственных средств для лечения диабета, лекарственных средств для лечения бесплодия, стимуляторов роста, кровоостанавливающих средств, иммуностимуляторов, миорелаксантов, лекарственных средств для лечения ожирения, лекарственных средств для лечения остеопороза, пептидов, седативных средств и транквилизаторов, подкислителей для мочевыводящих путей и витаминов.
В некоторых вариантах осуществления фармацевтически активный агент представляет собой онкологическое лекарственное средство, стероид, опиоидное анальгетическое средство, респираторное лекарственное средство, лекарственное средство для лечения центральной нервной системы (ЦНС), лекарственное средство для лечения гиперхолестеринемии, антигипертензивное лекарственное средство, антибактериальное средство, иммунодепрессивное лекарственное средство, антибиотик, агонист лютеинизирующего гормона-рилизинга (ЛГРГ), антагонист ЛГРГ, противовирусное лекарственное средство, антиретровирусное лекарственное средство, модулятор рецепторов эстрогена, аналог соматостатина, противовоспалительное лекарственное средство, аналог витамина D2, синтетический тироксин, антигистаминный агент, противогрибковый агент или нестероидное противовоспалительное лекарственное средство (НПВС), предпочтительно онкологическое лекарственное средство.
В некоторых вариантах осуществления онкологическое лекарственное средство включает таксаны (такие как паклитаксел, кабазитаксел и доцетаксел), камптотецины и их аналоги (такие как иринотекан и топотекан), нуклеозиды (такие как гемцитабин, кладрибин, флударабин, капецитабин, децитабин, азацитидин, клофарабин и неларабин), ингибиторы киназы (такие как дазатиниб (спирцел), темизиролимус, AZD6244, AZD1152, PI-103, R-росковитин, оломоуцин и пурваланол А) и аналоги эпотилона В (такие как иксабепилон), антроциклины (такие как амрубицин, доксорубицин, эпирубицин и валрубицин), производные эктеинасцидина (такие как трабектецин и лурбинектедин), ингибиторы протеасом (такие как бортезомиб) и другие ингибиторы топоизомеразы, интеркаляторы и алкилирующие агенты, ингибиторы микротрубочек (такие как эрибулин и винфлюнин) и т.д., или структурные модификации этих молекул лекарственных средств.
В некоторых вариантах осуществления фармацевтически активный агент выбран из группы, состоящей из таксанов, производных камптотецина, нуклеозидов, антрациклинов, производных эктеинасцидина, ингибиторов протеасом, ингибиторов микротрубочек, ингибиторов BCL-2, ингибиторов BCL-XL, селективного ингибитора ядерного экспорта, антиметаболитов, ингибиторов тирозинкиназы, ингибиторов PLK1, ингибиторов CDK4/6, ингибиторов ВТK, антагонистов рецепторов нестероидных гормонов и стероидов, предпочтительно из группы, состоящей из таксанов, производных камптотецина, ингибиторов BCL-2 и ингибиторов BCL-XL.
В некоторых вариантах осуществления фармацевтически активный агент выбран из группы, состоящей из доцетаксела, иринотекана, гемцитабина, капецитабина, децитабина, азацитидина, доксорубицина, эпирубицина, трабектедина, лурбинектедина, бортезомиба, эрибулина, селинексора, венетоклакса, тезетаксела, пеметрекседа, кабазитаксела, кабозантиниба, онвансертиба, соединения 2 и соединения 3, показанных ниже, и структурных модификаций этих молекул лекарственного средства,
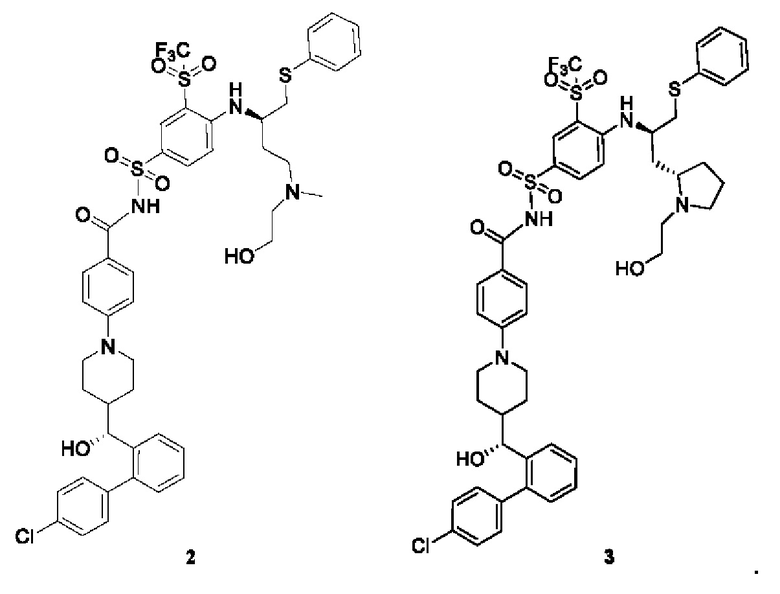
В некоторых вариантах осуществления стероиды включают синтетические стероиды (такие как тестостерон, дигидротестостерон и этинилэстрадиол) и кортикостероиды (такие как кортизон, преднизилон, будесонид, триамцинолон, флутиказон, мометазон, амцинонид, флуцинолон, флуоцинанид, дезонид, гальцинонид, предникарбат, флуокортолон, дексаметазон, бетаметазон и флупреднидин).
В некоторых вариантах осуществления опиоидное анальгетическое средство включает морфин, оксиморфон, налоксон, кодеин, оксикодон, метилналтрексон, гидроморфон, бупренорфин и эторфин. В некоторых вариантах осуществления респираторное лекарственное средство включает бронхолитические средства, ингаляционные стероиды и противоотечные средства, в частности, сальбутамол, ипратропия бромид, монтелукаст и формотерол. В некоторых вариантах осуществления лекарственное средство для лечения ЦНС включает нейролептические препараты (такие как кветиапин) и антидепрессанты (такие как венлафаксин). В некоторых вариантах осуществления лекарственное средство для лечения гиперхолестеринемии включает эзетимиб и статины, такие как симвастатин, ловастатин, аторвастатин, флувастатин, питавастатин, правастатин и розувастатин. В некоторых вариантах осуществления антигипертензивное лекарственное средство включает лозартан, олмесартан, медоксомил, метролол, травопрост и бозентан. В некоторых вариантах осуществления иммунодепрессивное лекарственное средство включает глюкокортикоиды, цитостатические средства, фрагменты антител, антииммунофилины, интерфероны, белки, связывающие TNF, и особенно ингибиторы кациневрина, такие как такролимус, микофеноловая кислота и ее производные, такие как микофенолат мофетил и циклоспорин. В некоторых вариантах осуществления антибактериальный агент включает антибиотики, такие как амоксициллин, меропенем и клавулановая кислота. В некоторых вариантах осуществления агонист ЛГРГ включает гозерелина ацетат, деслорелин и лейпрорелин. В некоторых вариантах осуществления антагонист ЛГРГ включает цетрореликс, ганиреликс, абареликс и дегареликс.В некоторых вариантах осуществления противовирусный агент включает нуклеозидные аналоги, такие как ламивудин, зидовудин, абакавир и энтекавир, и антиретровирусное лекарственное средство включает ингибиторы протеазы, такие как атазанавир, лапинавир и ритонавир. В некоторых вариантах осуществления модулятор рецептора эстрогена включает ралоксифен и фулвестрант.В некоторых вариантах осуществления аналог соматостатина включает октреотид. В некоторых вариантах осуществления противовоспалительное лекарственное средство включает месалазин, и подходящие НПВС включают парацетамол (ацетаминофен). В некоторых вариантах осуществления аналог витамина D2 включает парикальцитол. В некоторых вариантах осуществления синтетический тироксин включает левотироксин. В некоторых вариантах осуществления антигистаминный агент включает фексофенадин. В некоторых вариантах осуществления противогрибковый агент включает азолы, такие как вириконазол.
В некоторых вариантах осуществления фармацевтически активный агент представляет собой эрибулин. В некоторых вариантах осуществления фармацевтически активный агент представляет собой доцетаксел. В некоторых вариантах осуществления фармацевтически активный агент представляет собой трабектедин. В некоторых вариантах осуществления фармацевтически активный агент представляет собой лурбинектедин.
В некоторых вариантах осуществления фармацевтически активный агент является умеренно растворимым или нерастворимым в водном растворе.
Вторая концевая группа представляет собой фармакокинетический модификатор, который может модифицировать или модулировать фармакокинетические свойства фармацевтически активного агента или макромолекулы, включая абсорбцию, распределение, метаболизм и/или экскрецию. В конкретных вариантах осуществления фармакокинетический модификатор продлевает период полувыведения из плазмы фармацевтически активного агента, так что фармацевтически активный агент, связанный с макромолекулой, имеет более длительный период полувыведения, чем один фармацевтически активный агент или фармацевтически активный агент на недендримерном носителе. Предпочтительно, макромолекула или композиция имеет период полувыведения, который по меньшей мере в 3 раза дольше, более предпочтительно по меньшей мере в 11 раз дольше, чем у одного фармацевтически активного агента или фармацевтически активного агента на недендримерном носителе.
Фармакокинетический модификатор может быть выбран из группы, состоящей из полиэтиленгликоля, полиэтилоксазолина, поливинилпирролидона, полипропиленгликоля, фолата и производного фолата лиганда рецептора клеточной поверхности. В некоторых вариантах осуществления фармакокинетический модификатор представляет собой полиэтиленгликоль. В некоторых вариантах осуществления полиэтиленгликоль имеет молекулярную массу в диапазоне от 220 до 5500 Да; например, молекулярная масса может составлять от 220 до 2500 Да, от 570 до 2500 Да, от 220 до 1100 Да, от 570 до 1100 Да, от 1000 до 5500 Да, от 1000 до 2500 Да или от 1000 до 2300 Да. В некоторых вариантах осуществления фармакокинетический модификатор образует амидную связь с поверхностной аминогруппой дендримера.
Нацеливающая группа представляет собой агент, который связывается с биологической целевой клеткой, органом или тканью с некоторой селективностью, тем самым помогая направлять макромолекулу к конкретной мишени в организме и позволяя ей накапливаться в этой целевой клетке, органе или ткани. Кроме того, нацеливающая группа может обеспечить механизм для активного введения макромолекулы в клетку или ткань путем опосредованного рецепторами эндоцитоза. Конкретные примеры включают лектины и антитела и другие лиганды (включая малые молекулы) рецепторов клеточной поверхности. Взаимодействие может происходить через любой тип связи или ассоциации (включая ковалентную, ионную и водородную связь и силы Ван-дер-Ваальса). Подходящие нацеливающие группы включают группы, которые связываются с рецепторами клеточной поверхности, например, фолатный рецептор, адренергический рецептор, рецептор гормона роста, рецептор лютеинизирующего гормона, рецептор эстрогена, рецептор эпидермального фактора роста, рецептор фактора роста фибробластов (например, FGFR2), рецептор IL-2, CFTR и рецептор фактора роста эпителия сосудов (VEGF).
В некоторых вариантах осуществления нацеливающая группа представляет собой лютеинизирующий гормон-рилизинг (ЛГРГ) или его производное, которое связывается с рецептором лютеинизирующего гормон-рилизинга. В некоторых вариантах осуществления нацеливающая группа представляет собой LYP-1, пептид, который нацелен на лимфатическую систему опухолей, но не на лимфатическую систему нормальной ткани. В некоторых вариантах осуществления нацеливающая группа может представлять собой RGD (аргинилглициласпарагиновая кислота)-пептид. RGD-пептиды представляют собой пептиды, содержащие последовательность -Arg-Gly-Asp-, которая является первичным сайтом распознавания интегрина во внеклеточных матричных белках. В некоторых вариантах осуществления нацеливающая группа может представлять собой фолиевую кислоту. Эстрогены также могут быть использованы для клеток-мишеней, экспрессирующих рецепторы эстрогена.
В некоторых вариантах осуществления нацеливающая группа может связываться с ядром дендримера непосредственно или, предпочтительно, через группу связывания. Группа связывания может представлять собой любую двухвалентную группу, способную связываться с функциональной группой ядра и функциональной группой нацеливающей группы.
Макромолекула по настоящему изобретению содержит дендример, в котором самая внешняя генерация структурного звена имеет поверхностные аминогруппы. Свойства дендримера макромолекулы не являются особенно существенными при условии, что он имеет поверхностные аминогруппы. Например, дендример может представлять собой дендример полилизина, аналога полилизина, полиамидоамина (РАМАМ), полиэтиленимина (PEI) или полиэфира гидроксиламина (РЕНАМ). В некоторых вариантах осуществления дендример представляет собой полилизин или аналог полилизина. Полилизин или аналог полилизина содержит ядро и 2-7 генераций лизина или аналога лизина, например, 2, 3, 4, 5, 6 или 7 генераций лизина или аналога лизина.
В некоторых вариантах осуществления лизин имеет структуру, показанную в 1:
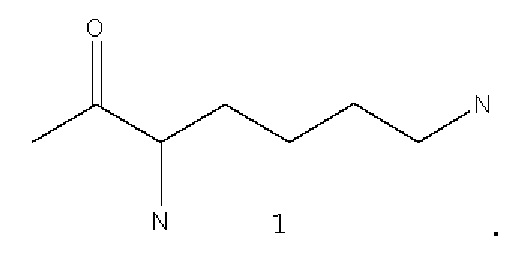
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 2: 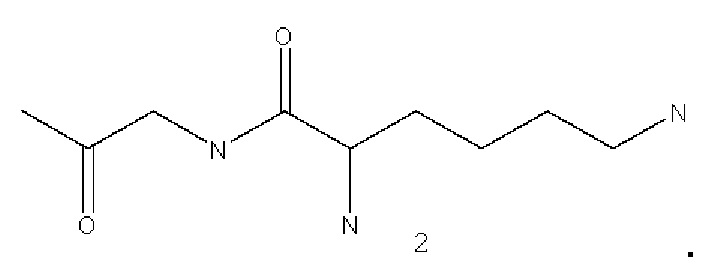
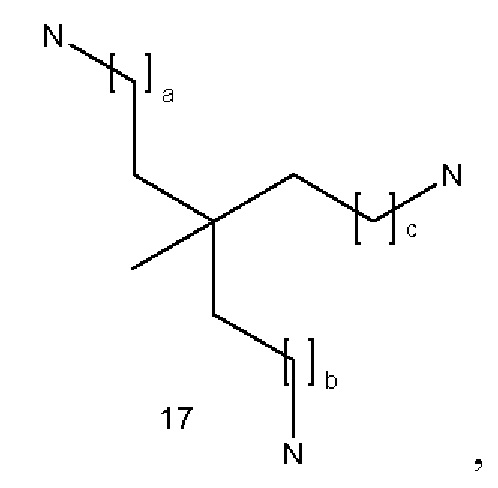
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 3: 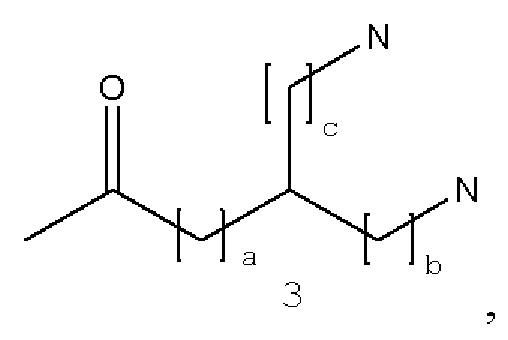 где а равно 1 или 2; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 4.
где а равно 1 или 2; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 4.
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 4: 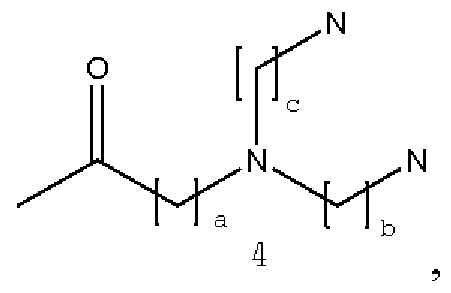 где а представляет собой целое число от 0 до 2; b и с являются одинаковыми или различными и представляют собой целые числа от 2 до 6.
где а представляет собой целое число от 0 до 2; b и с являются одинаковыми или различными и представляют собой целые числа от 2 до 6.
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 5: 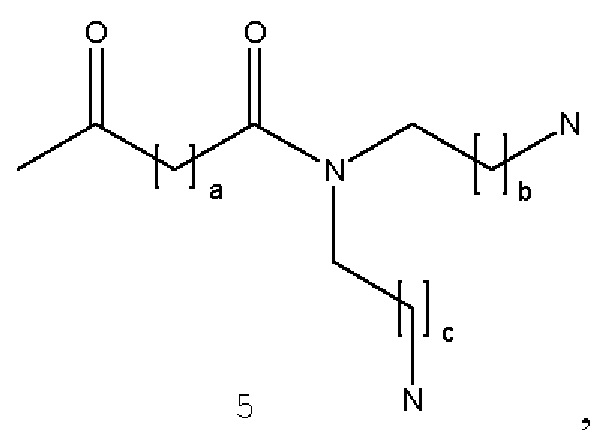 где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 6: 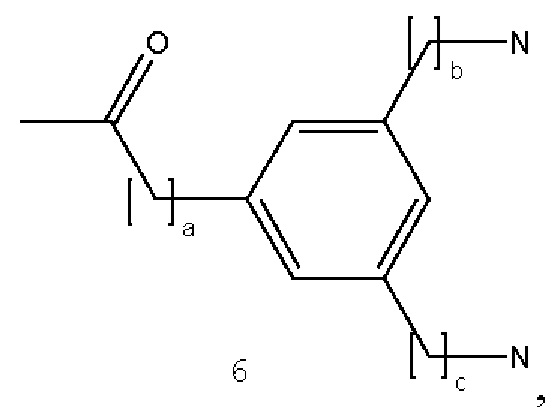 где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 0 до 5.
где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 0 до 5.
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 7: 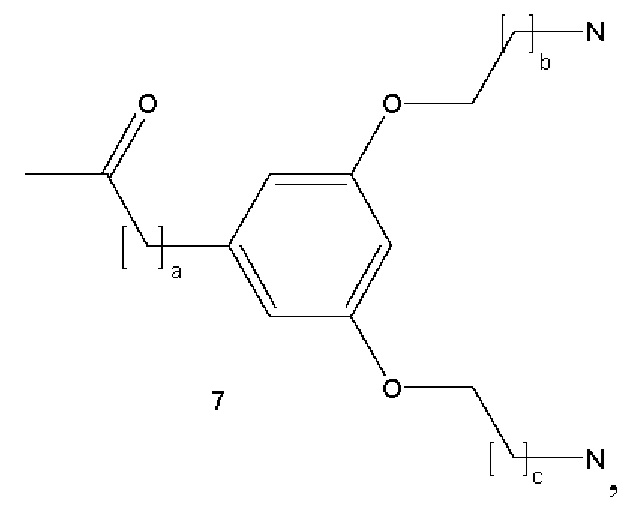 где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
В некоторых вариантах осуществления аналог лизина имеет структуру показанную в 8: 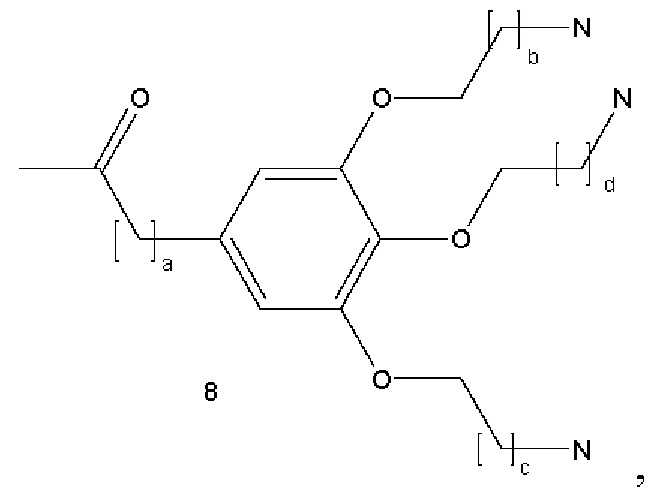 где а представляет собой целое число от 0 до 5; b, с и d являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
где а представляет собой целое число от 0 до 5; b, с и d являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
В некоторых вариантах осуществления аналог лизина имеет структуру, показанную в 9: 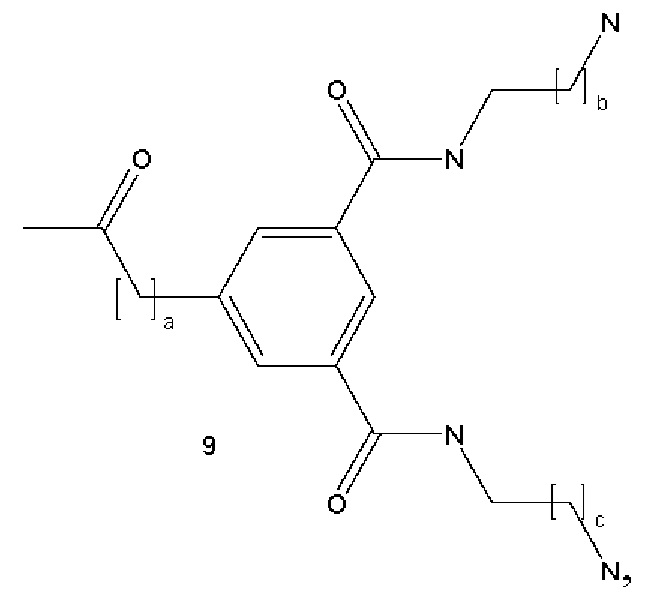 где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
где а представляет собой целое число от 0 до 5; b и с являются одинаковыми или различными и представляют собой целые числа от 1 до 5.
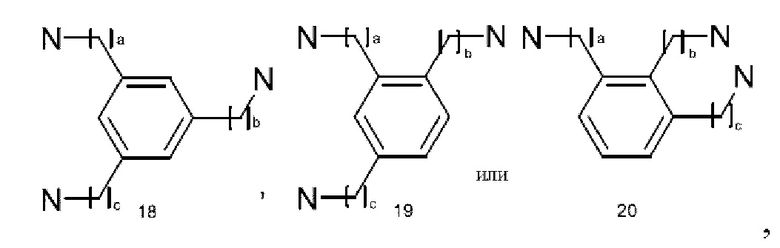
Ядро дендримера, в частности, полилизин или аналог полилизина, описанный в настоящем описании, может быть выбран из группы, состоящей из бензгидриламина (ВНА), бензгидриламида лизина (BHALys) или аналога лизина, или:
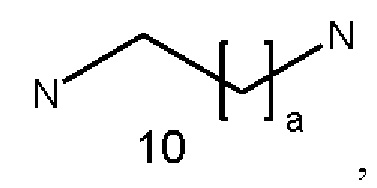 где а представляет собой целое число от 1 до 9, предпочтительно от 1 до 5;
где а представляет собой целое число от 1 до 9, предпочтительно от 1 до 5;
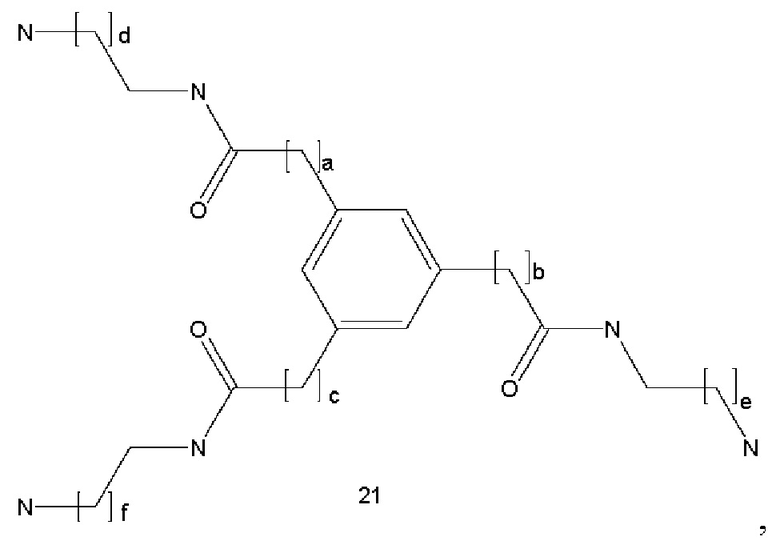
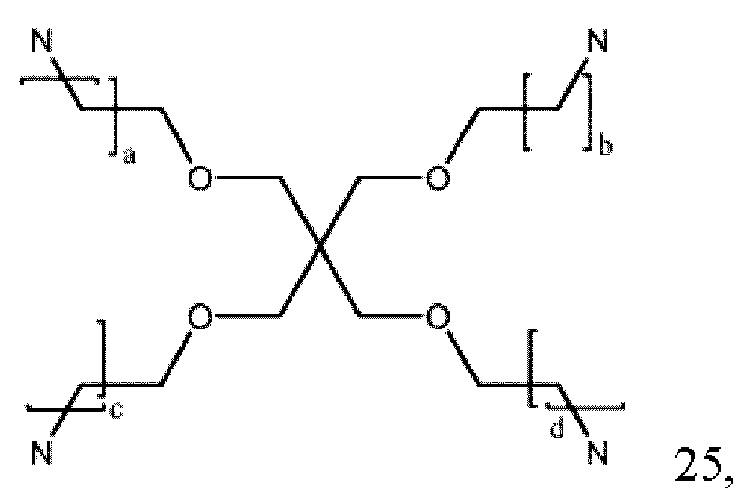
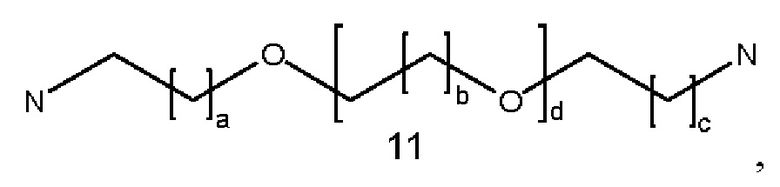 где a, b и с могут быть одинаковыми или различными и представляют собой целые числа от 1 до 5, и d представляет собой целое число от 0 до 100, предпочтительно от 1 до 30;
где a, b и с могут быть одинаковыми или различными и представляют собой целые числа от 1 до 5, и d представляет собой целое число от 0 до 100, предпочтительно от 1 до 30;
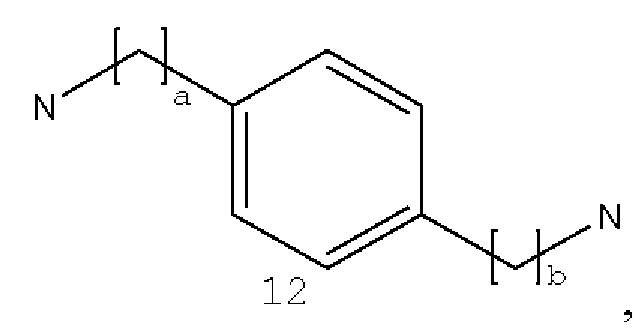 где а и b могут быть одинаковыми или различными и представляют собой целые числа от 0 до 5;
где а и b могут быть одинаковыми или различными и представляют собой целые числа от 0 до 5;
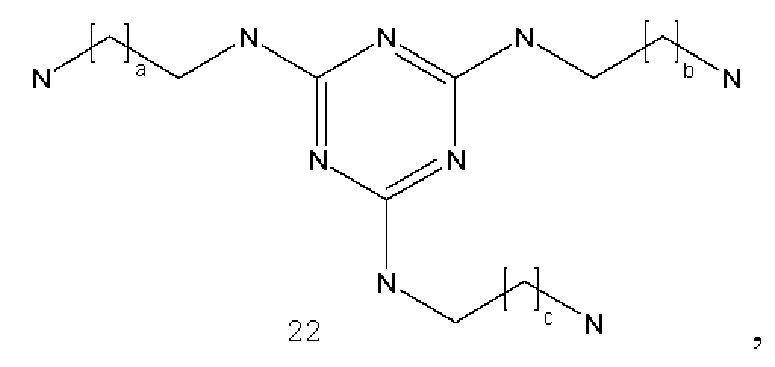
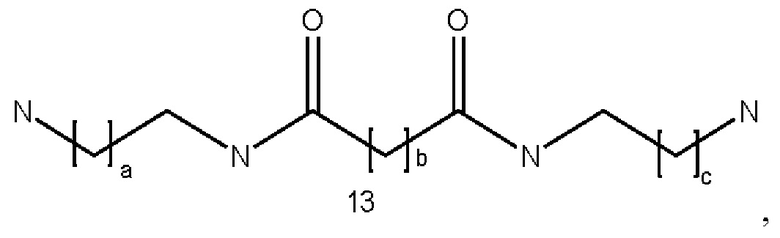 где а и с могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6, и b представляет собой целое число от 0 до 6;
где а и с могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6, и b представляет собой целое число от 0 до 6;
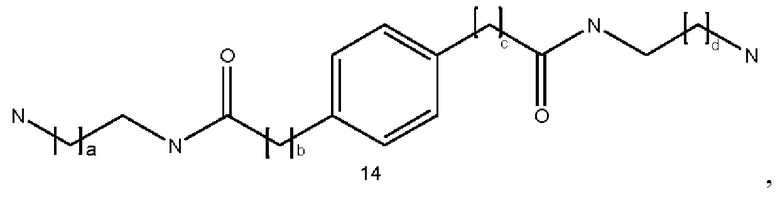 где a и d могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6, и b и с могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6;
где a и d могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6, и b и с могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6;
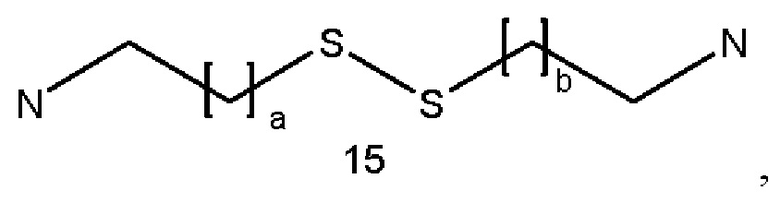 где а и b являются одинаковыми или различными и представляют собой целые числа от 1 до 5, в частности от 1 до 3, и особенно 1;
где а и b являются одинаковыми или различными и представляют собой целые числа от 1 до 5, в частности от 1 до 3, и особенно 1;
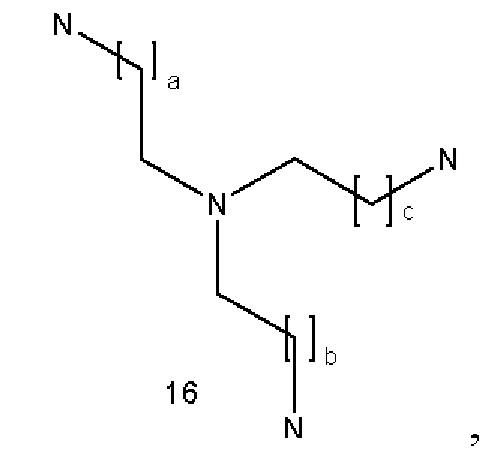 где a, b и с являются одинаковыми или различными и представляют собой целые числа, выбранные из группы, состоящей из 1-6;
где a, b и с являются одинаковыми или различными и представляют собой целые числа, выбранные из группы, состоящей из 1-6;
 где a, b и с являются одинаковыми или различными и представляют собой целые числа, выбранные из группы, состоящей из 0-6;
где a, b и с являются одинаковыми или различными и представляют собой целые числа, выбранные из группы, состоящей из 0-6;
 где a, b и с являются одинаковыми или различными и представляют собой целые числа, выбранные из группы, состоящей из 0-6;
где a, b и с являются одинаковыми или различными и представляют собой целые числа, выбранные из группы, состоящей из 0-6;
 где a, b и с могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6, и d, е и f могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6;
где a, b и с могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6, и d, е и f могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6;
 где a, b и с могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6;
где a, b и с могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6;
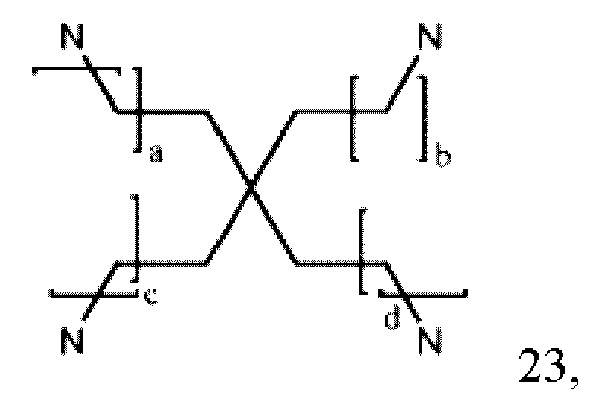
 где а, b, с и d могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6;
где а, b, с и d могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6;
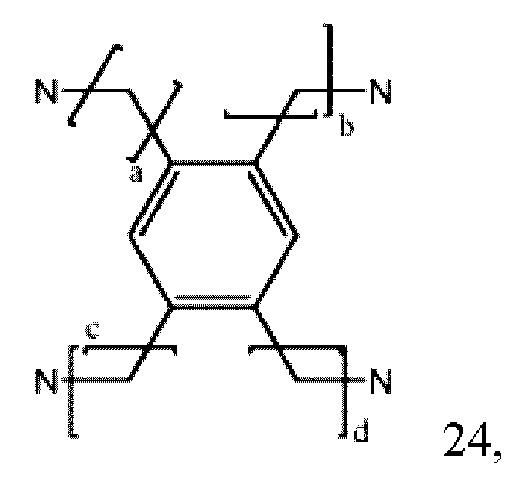
 где а, b, с и d могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6; или
где а, b, с и d могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6; или
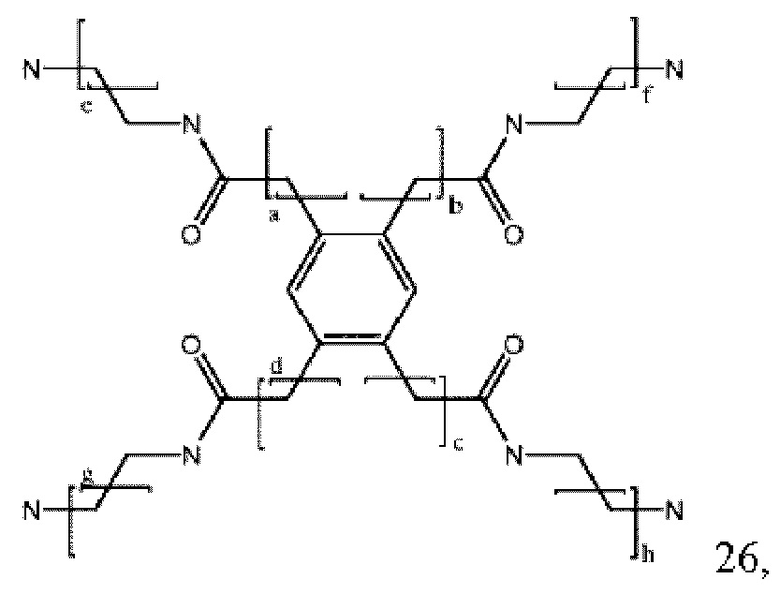 где a, b, с и d могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6, и е, f, g и h могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6. В некоторых вариантах осуществления макромолекула содержит:
где a, b, с и d могут быть одинаковыми или различными и представляют собой целые числа от 0 до 6, и е, f, g и h могут быть одинаковыми или различными и представляют собой целые числа от 1 до 6. В некоторых вариантах осуществления макромолекула содержит:
i) дендример D, имеющий поверхностные аминогруппы, где по меньшей мере две разные концевые группы ковалентно связаны с поверхностными аминогруппами дендримера;
ii) первую концевую группу, которая представляет собой фармацевтически активный агент, содержащий гидрокси, амино или сульфгидрильную группу или представляет собой его остаток А; и
iii) вторую концевую группу, которая является фармакокинетически модифицированным полиэтиленгликолем;
где первая концевая группа ковалентно связана с поверхностной аминогруппой дендримера посредством линкера, выбранного из группы, состоящей из
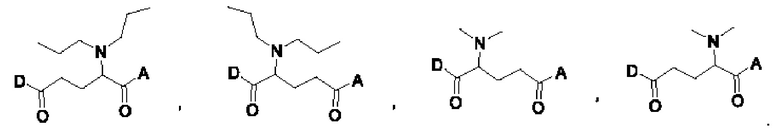
Дендример D выбран из группы, состоящей из BHALys[Lys]16, BHALys[Lys]32 и BHALys[Lys]64.
Полиэтиленгликоль имеет молекулярную массу в диапазоне от 1000 до 2500 Да.
Настоящее изобретение также относится к фармацевтической композиции, которая содержит макромолекулу по настоящему изобретению и фармацевтически приемлемый носитель. В некоторых вариантах осуществления фармацевтическая композиция не содержит солюбилизирующих эксципиентов, таких как полиэтоксилированные касторовые масла и полисорбаты. В некоторых вариантах осуществления фармацевтическую композицию вводят трансдермально, перорально, путем инъекции и т.д.
Макромолекула согласно настоящему изобретению составлена в виде композиции, включая композиции, подходящие для перорального, ректального, местного, назального, ингаляционного, аэрозольного, офтальмологического или парентерального (включая внутрибрюшинную, внутривенную, подкожную или внутримышечную инъекцию) введения. Композиция может быть для удобства представлена в стандартной лекарственной форме и может быть получена с использованием любого из способов, хорошо известных в области фармации. Все способы включают стадию приведения макромолекулы в сочетание с носителем, который представляет собой один или более вспомогательных ингредиентов. Как правило, композицию получают путем приведения макромолекулы в сочетание с жидким носителем с образованием раствора или суспензии или приведения макромолекулы в сочетание с компонентами композиции, подходящими для образования твердого продукта, необязательно, в виде частиц, а затем, при необходимости, формования продукта в желаемую форму доставки. Твердая композиция по настоящему изобретению, в случае частиц, как правило, будет содержать частицы с размерами от около 1 нанометра до около 500 микрон. Как правило, для твердых композиций, предназначенных для внутривенного введения, частицы обычно находятся в диапазоне от около 1 нм до около 10 микрон в диаметре. Композиция может содержать макромолекулу по настоящему изобретению, которая представляет собой наночастицу имеющую диаметр частиц менее 1000 нм, например, от 5 до 1000 нм, предпочтительно от 5 до 500 нм, в частности от 5 до 400 нм (например, от 5 до 50 нм и предпочтительно от 5 до 20 нм). В конкретных вариантах осуществления композиция содержит макромолекулы со средним размером от 5 до 20 нм. В некоторых вариантах осуществления макромолекула является полидисперсной в композиции с PDI (коэффициент полидисперсности) от 1,01 до 1,8, в частности от 1,01 до 1,5, предпочтительно от 1,01 до 1,2. В конкретных вариантах осуществления макромолекула является монодисперсной в композиции. Особенно предпочтительными являются стерильные лиофилизированные композиции, которые перед инъекцией восстанавливают в водном носителе.
В конкретных вариантах осуществления композиция содержит макромолекулы со средним размером от 5 до 20 нм. В некоторых вариантах осуществления макромолекулы имеют размер частиц D90 или D50 менее 1000 нм, например, от 5 до 1000 нм, в частности от 5 до 500 нм, предпочтительно от 5 до 400 нм (например, от 5 до 50 нм, предпочтительно от 5 до 20 нм). В конкретных вариантах осуществления композиция содержит макромолекулы, имеющие D50 от 5 до 20 нм.
Макромолекула по настоящему изобретению также может быть использована для получения композиций с контролируемым высвобождением и/или замедленным высвобождением фармацевтически активного агента. В композиции с замедленным высвобождением ингредиенты композиции выбраны так, чтобы происходило высвобождение макромолекулы из композиции в течение длительного периода времени (например, дней, недель или месяцев). Такие композиции включают трансдермальные пластыри или находятся в имплантируемых устройствах, которые могут быть нанесены подкожно или введены внутривенно, подкожно, внутримышечно, интраэпидурально или внутричерепно. В композициях с контролируемым высвобождением дикислотный линкер выбран для высвобождения большей части своего фармацевтически активного агента в заданное временное окно. Например, когда известно время, необходимое для накопления большей части макромолекулы в целевом органе, ткани или опухоли, линкер может быть выбран для высвобождения большей части своего фармацевтически активного агента после истечения времени накопления. Это может позволить обеспечить высокую нагрузку лекарственным средством в заданный момент времени в месте, где его действие желательно. Альтернативно, линкер выбран для высвобождения фармацевтически активного агента на терапевтическом уровне в течение длительного периода времени. В некоторых вариантах осуществления композиция может иметь множество характеристик с точки зрения контролируемого высвобождения. Например, композиция содержит макромолекулы, в которых лекарственное средство связано разными линкерами; это позволяет осуществлять «взрывное» высвобождение лекарственного средства с последующим более медленным высвобождением при низких, но постоянных терапевтических уровнях в течение длительного периода времени. В некоторых вариантах осуществления композиция может иметь характеристики замедленного высвобождения и контролируемого высвобождения. Например, ингредиенты композиции могут быть выбраны для высвобождения макромолекулы в течение длительного периода времени, и линкер выбран для доставки постоянного низкого терапевтического уровня фармацевтически активного агента. В некоторых вариантах осуществления фармацевтически активный агент связан с одной и той же молекулой разными линкерами. В некоторых вариантах осуществления каждая комбинация лекарственное средство-линкер связана с разными макромолекулами в одной и той же композиции.
В некоторых вариантах осуществления в фармацевтической композиции макромолекула составлена таким образом, чтобы высвобождать более 50% фармацевтически активного агента в течение от 5 до 60 минут. В некоторых вариантах осуществления в фармацевтической композиции макромолекула составлена таким образом, чтобы высвобождать более 50% фармацевтически активного агента в течение от 2 до 48 часов. В некоторых вариантах осуществления в фармацевтической композиции макромолекула составлена таким образом, чтобы высвобождать более 50% фармацевтически активного агента в течение от 5 до 30 дней.
Другой аспект настоящего изобретения относится к способу лечения или ингибирования роста опухоли, который включает введение эффективного количества макромолекулы или фармацевтической композиции по настоящему изобретению, где фармацевтически активный агент первой концевой группы представляет собой онкологическое лекарственное средство. Опухоль, описанная в настоящем изобретении, выбрана из группы, состоящей из меланомы, опухоли головного мозга, рака пищевода, рака желудка, рака печени, рака поджелудочной железы, колоректального рака, рака легких, рака почки, рака молочной железы, рака яичников, рака предстательной железы, рака кожи, нейробластомы, саркомы, остеохондромы, остеомы, остеосаркомы, семиномы, опухоли яичек, рака матки, опухоли головы и шеи, множественной миеломы, злокачественной лимфомы, истинной полицитемии, лейкоза, опухоли щитовидной железы, опухоли мочеточника, рака мочевого пузыря, рака желчного пузыря, рака желчных протоков, хорионической эпителиомы и опухолей детского возраста (семейная саркома Юинга, опухоль Вильмса, рабдомиосаркома, ангиосаркома, эмбриональный рак яичка, нейробластома, ретинобластома, гепатобластома, нефробластома и др.).
В другом аспекте настоящего изобретения предложен способ снижения гиперчувствительности при лечении онкологическим лекарственным средством, который включает введение фармацевтической композиции по настоящему изобретению, где композиция по существу не содержит солюбилизирующих эксципиентов, таких как кремофор EL и полисорбат 80.
В другом аспекте настоящего изобретения предложен способ снижения токсичности онкологического лекарственного средства или композиции онкологического лекарственного средства, который включает введение макромолекулы по настоящему изобретению, причем онкологическое лекарственное средство представляет собой первую концевую группу макромолекулы. В некоторых вариантах осуществления токсичность, которая снижается, представляет собой гематологическую токсичность, неврологическую токсичность, желудочно-кишечную токсичность, кардиотоксичность, гепатотоксичность, нефротоксичность, ототоксичность или энцефалотоксичность.
В другом аспекте настоящего изобретения предложен способ уменьшения побочных эффектов, связанных с онкологическим лекарственным средством или композицией онкологического лекарственного средства, который включает введение макромолекулы по настоящему изобретению, причем онкологическое лекарственное средство представляет собой первую концевую группу макромолекулы. В некоторых вариантах осуществления уменьшенные побочные эффекты выбраны из группы, состоящей из нейтропении, лейкопении, тромбоцитопении, миелотоксичности, миелосупрессии, нейропатии, усталости, неспецифических нейрокогнитивных проблем, головокружения, энцефалопатии, анемии, дисгевзии, одышки, запора, анорексии, заболевания ногтей, задержки жидкости, астении, боли, тошноты, рвоты, мукозита, алопеции, кожных реакций, миалгии и гиперчувствительности.
В некоторых вариантах осуществления макромолекула или фармацевтическая композиция, содержащая макромолекулу согласно настоящему изобретению, может снижать или устранять необходимость в предхирургическом лекарственном средстве с такими агентами, как кортикостероиды и антигистаминные агенты.
Способы получения дендримеров известны в данной области техники. Например, дендример макромолекулы может быть получен с использованием расходящегося метода или сходящегося метода или их комбинации.
В расходящемся методе каждая генерация структурных единиц последовательно добавляется к ядру или предыдущей генерации. Поверхностная генерация, имеющая одну или две поверхностные аминогруппы, защищена. Если одна из аминогрупп защищена, свободную аминогруппу подвергают взаимодействию с одним из: линкера, линкера-фармацевтически активного агента и фармакокинетического модификатора. Если обе аминогруппы защищены, они защищены разными защитными группами, одна из которых может быть удалена без удаления другой. Одну из аминозащитных групп удаляют и проводят реакцию с одним из: линкера, линкера-фармацевтически активного агента и фармакокинетического модификатора. После того, как исходная концевая группа была связана с дендримером, удаляют другую аминозащитную группу и добавляют другие первую и вторую концевые группы. Эти группы связаны с поверхностными аминогруппами посредством образования амида, известного в данной области техники.
В сходящемся методе каждая генерация структурных единиц строится на предыдущей генерации с образованием дендрона. Первая и вторая концевые группы могут быть связаны с поверхностными аминогруппами, как описано выше, до или после связывания дендрона с ядром.
При комбинированном подходе каждая генерация структурных единиц добавляется к ядру или предыдущей генерации структурных единиц. Однако перед добавлением последней генерации к дендримеру поверхностные аминогруппы функционализируют концевыми группами (например, первой и второй концевыми группами, первой и третьей концевыми группами или второй и третьей концевыми группами). Затем функционализированную конечную генерацию добавляют к подповерхностному слою структурных единиц, и дендрон связывается с ядром.
Фармацевтически активный агент взаимодействует с одной из карбоновых кислот линкера путем образования сложного эфира, известного в данной области техники. Например, образуется активированная карбоновая кислота. Например, хлорангидрид кислоты или ангидрид используют и повергают взаимодействию с гидроксильной группой фармацевтически активного агента. Если фармацевтически активный агент имеет более чем одну гидроксигруппу, другие гидроксигруппы могут быть защищены.
Если нацеливающий агент связан с ядром, функциональная группа на ядре может быть защищена во время образования дендримера, затем подвергнута снятию защиты и взаимодействию с нацеливающим агентом, группой связывания или нацеливающим агентом-группой связывания. Альтернативно, ядро может вступать в реакцию с группой связывания или нацеливающим агентом-группой связывания до образования дендримера.
Подходящие защитные группы и способы их введения и удаления описаны в Greene&Wuts, Protecting Groups in Organic Synthesis, Third Edition, 1999.
Настоящее изобретение также включает различные дейтерированные формы макромолекулы или ее фармацевтически приемлемых солей, где каждый доступный атом водорода в макромолекуле может быть независимо заменен атомом дейтерия. Специалисты в данной области техники будут знать, как синтезировать дейтерированные формы макромолекулы по настоящему изобретению или ее фармацевтически приемлемых солей.
Настоящее изобретение также включает макромолекулы, которые мечены изотопами. Один или более атомов в макромолекулах заменяют атомами с атомной массой или массовым числом, которое отличается от наиболее распространенной атомной массы или массового числа, встречающихся в природе. Примеры изотопов для макромолекулы согласно настоящему изобретению включают изотопы водорода, углерода, азота, кислорода, фтора, йода и хлора, такие как 3Н, 11C, 14С, 18F, 123I или 125I.
Макромолекула по настоящему изобретению может присутствовать в форме фармацевтически приемлемых солей. Однако следует понимать, что не фармацевтически приемлемые соли также подпадают в объем настоящего изобретения, поскольку они могут быть пригодны в качестве промежуточных соединений при получении фармацевтически приемлемых солей или могут быть пригодны для хранения или транспортировки. Подходящие фармацевтически приемлемые соли включают, но не ограничиваются, соли фармацевтически приемлемых неорганических кислот (таких как соляная кислота, серная кислота, фосфорная кислота, азотная кислота, карбоновая кислота, борная кислота, сульфаминовая кислота и бромистоводородная кислота) или соли фармацевтически приемлемых органических кислот (таких как уксусная кислота, пропионовая кислота, масляная кислота, винная кислота, малеиновая кислота, гидроксималеиновая кислота, фумаровая кислота, лимонная кислота, молочная кислота, муциновая кислота, глюконовая кислота, бензойная кислота, янтарная кислота, щавелевая кислота, фенилуксусная кислота, метансульфоновая кислота, толуолсульфоновая кислота, бенезолсульфоновая кислота, салициловая кислота, сульфаниловая кислота, аспарагиновая кислота, глутаминовая кислота, этилендиаминтетрауксусная кислота, стеариновая кислота, пальмитиновая кислота, олеиновая кислота, лауриновая кислота, пантотеновая кислота, дубильная кислота, аскорбиновая кислота и валериановая кислота). Основные соли включают, но не ограничиваются, соли, образованные с фармацевтически приемлемыми катионами (такими как натрий, калий, литий, кальций, магний, аммоний и алкиламмоний).
Если не указано иное, следующие термины, используемые в описании и формуле изобретения, имеют приведенные ниже значения.
«Галоген» относится к фтору, хлору, брому или йоду.
«Галоген» означает замещение одним или более атомами, выбранными из группы, состоящей из фтора, хлора, брома и йода.
«Алкил» относится к линейной или разветвленной насыщенной углеводородной группе, имеющей от 1 до 10 атомов углерода. В соответствующих случаях алкил может иметь указанное количество атомов углерода, например, С1-4 алкил, который включает алкильные группы, имеющие 1, 2, 3 или 4 атома углерода в линейном или разветвленном расположении. Примеры подходящих алкильных групп включают, но не ограничиваются, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, 2-метилбутил, 3-метилбутил, 4-метилбутил, н-гексил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 5-метилпентил, 2-этилбутил, 3-этилбутил, гептил, октил, нонил и децил.
«Алкилен» относится к линейной или разветвленной двухвалентной алкильной группе, имеющей от 1 до 10 атомов углерода.
Термин «алкенил» включает разветвленные и линейные алкены, имеющие от 2 до 12 атомов углерода, или алкены, содержащие алифатические углеводородные группы; если указано количество атомов углерода, имеется в виду указанное количество. Например, «С2-6 алкенил» относится к алкенильной группе, имеющей 2, 3, 4, 5 или 6 атомов углерода. Примеры алкенильных групп включают, но не ограничиваются, этенил, аллил, 1-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-метилбут-2-енил, 3-метилбут-1-енил, 1-пентенил, 3-пентенил и 4-гексенил.
Термин «алкинил» включает разветвленный и линейный алкинил, имеющий от 2 до 12 атомов углерода, или алкинил, содержащий алифатические углеводородные группы, или алкинил, содержащий определенное количество атомов углерода (если определено конкретное количество), например, этинил, пропинил (например, 1-пропинил, 2-пропинил), 3-бутинил, пентинил, гексинил и 1-метилпент-2-инил.
«Алкенилен» и «алкинилен» относятся к частично ненасыщенной разветвленной или линейной двухвалентной углеводородной группе, полученной из алкенила или алкинила. В некоторых вариантах осуществления такие алкениленовые группы необязательно замещены. Неограничивающие примеры алкениленовых групп включают этенилен, пропенилен, бутенилен, пентенилен, гексенилен, гептенилен, октенилен, ноненилен, деценилен и тому подобное; неограничивающие примеры алкиниленовых групп включают этинилен, пропинилен, бутинилен, пентинилен, гексинилен и тому подобное.
«Циклоалкил» относится к насыщенному или ненасыщенному циклическому углеводороду. Циклоалкильное кольцо может содержать указанное количество атомов углерода. Например, 3-8-членная циклоалкильная группа включает 3, 4, 5, 6, 7 или 8 атомов углерода. Примеры подходящих циклоалкильных групп включают, но не ограничиваются, циклопропил, циклобутил, циклопентанил, циклопентенил, циклогексанил, циклогексенил, 1,4-циклогексадиенил, циклогептанил и циклооктанил.
«Циклоалкилен» относится к двухвалентной циклической углеводородной группе, полученной из циклоалкила, например, 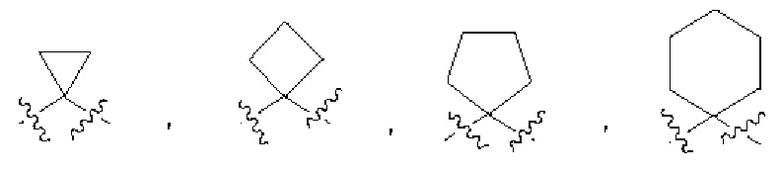 и т.д.
и т.д.
Термин «алкокси» относится к группе -О-(алкил) и -O-(незамещенный циклоалкил), где алкил является таким, как определено выше. Неограничивающие примеры алкоксигруппы включают: метокси, этокси, пропокси, бутокси, циклопропокси, циклобутокси, циклопентилокси и циклогексилокси. Алкокси может быть замещенным или незамещенным. При замещении алкокси может быть замещен одним или более заместителями, предпочтительно независимо выбранными из группы, состоящей из Н, D, галогена, алкила, алкокси, галогеналкила, гидрокси, гидроксиалкила, циано, амино, нитро, циклоалкила, гетероциклила, арила и гетероарила.
«Арил» означает любое стабильное, моноциклическое или бициклическое углеродное кольцо, содержащее вплоть до 7 атомов в каждом кольце, где по меньшей мере одно кольцо является ароматическим. Примеры таких арильных групп включают, но не ограничиваются, фенил, нафтил, тетрагидронафтил, инданил, бифенил и бинафтил.
Термин «гетероциклоалкил» или «гетероциклил» относится к циклическому углеводороду, в котором от одного до четырех атомов углерода были заменены гетероатомами, независимо выбранными из группы, состоящей из N, N(R), S, S(O), S(O)2 и О. Гетероциклильное кольцо может быть насыщенным или ненасыщенным. Примеры подходящих гетероциклильных групп включают тетрагидрофуранил, тетрагидротиенил, пирролидинил, пирролинил, пиразолинил, пиранил, пиперидинил, пиразолинил, дитиолил, оксатиолил, диоксанил, диоксинил, морфолино и оксазинил.
«Гетероарил» представляет собой стабильное моноциклическое или бициклическое кольцо до 7 атомов в каждом кольце, где по меньшей мере одно кольцо представляет собой ароматическое и по меньшей мере одно кольцо содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, N и S. Гетероарильные группы в рамках этого определения включают, но не ограничиваются, акридинил, карбазолил, циннолинил, хиноксалинил, хиназолинил, пиразолил, индолил, бензотриазолил, фуранил, тиенил, тиофенил, 3,4-пропилендиокситиофенил, бензотиенил, бензофуранил, бензодиоксан, бензодиоксин, хинолинил, изохинолинил, оксазолил, изоксазолил, имидазолил, пиразинил, пиридазинил, пиридинил, пиримидинил, пирролил, тетрагидрохинолин, тиазолил, изотиазолил, 1,2,4-триазолил, 1,2,3-триазолил, 1,2,4-оксадиазолил, 1,2,4-тиадиазолил, 1,3,5-триазинил, 1,2,4-триазинил, 1,2,4,5-тетразинил и тетразолил.
«Дендример» относится к молекуле, содержащей ядро и по меньшей мере один дендрон, связанный с ядром. Каждый дендрон состоит из по меньшей мере одного слоя или генерации разветвленных структурных единиц, в результате чего с каждой генерацией структурных единиц образуется разветвленная структура с растущим числом ветвей. Максимальное количество дендронов, связанных с ядром, ограничено количеством функциональных групп на ядре.
«Структурная единица» относится к разветвленной молекуле, имеющей по меньшей мере три функциональные группы, одну для связывания с ядром или предыдущей генерацией структурных единиц и по меньшей мере две функциональные группы для связывания со следующей генерацией структурных единиц или формирования поверхности дендримера.
«Генерация» относится к количеству слоев структурных единиц, которые составляют дендрон или дендример. Например, дендример из одной генерации будет иметь один слой разветвленных структурных единиц, связанных с ядром, например, ядро-[[структурная единица]]u, где и представляет собой количество дендронов, связанных с ядром. Дендример из двух генераций имеет два слоя структурных единиц в каждом дендроне, связанном с ядром, и когда структурная единица имеет одну точку разветвления, дендример может представлять собой: ядро [[структурная единица][структурная единица]2]u. Дендример из трех генераций имеет три слоя структурных единиц в каждом дендроне, связанном с ядром, например, ядро-[[структурная единица][структурная единица]2[структурная единица]4]u. Дендример из 6 генераций имеет шесть слоев структурных единиц, связанных с ядром, например, ядро-[[структурная единица] [структурная единица]2[структурная единица]4[структурная единица]8[структурная единица]16[структурная единица]32]u и тому подобное. Последняя генерация структурных единиц (самое внешняя генерация) обеспечивает функционализацию поверхности дендримера и количество функциональных групп, доступных для связывания с концевыми группами. Например, в дендримере, имеющем два дендрона, связанных с ядром (u=2), если каждая структурная единица имеет одну точку разветвления и существует 6 генераций, самая внешняя генерация имеет 64 структурных единицы и 128 функциональных групп, доступных для связывания с концевыми группами.
«Умеренно растворимый» относится к лекарственному средству или фармацевтически активному агенту, который имеет растворимость в воде от 1 до 10 мг/мл. Лекарственные средства с растворимостью в воде менее 1 мг/мл считаются нерастворимыми.
«Солюбилизирующий эксципиент» относится к добавке композиции, которая используется для солюбилизации нерастворимого или умеренно растворимого фармацевтически активного агента в водной композиции. Примеры включают поверхностно-активные вещества, такие как полиэтоксилированные касторовые масла, включая Кремофор EL, Кремофор RH40 и Кремофор RH60, D-α-токоферол-полиэтиленгликоль 1000 сукцинат, полисорбат 20, полисорбат 80, раствор HS15, сорбитан моноолеат, полоксамер 407, лабразол и т.д.
Термин «необязательный» или «необязательно» означает, что событие или обстоятельство, описанное далее, может, но необязательно, иметь место, и что такое описание включает случаи, когда событие или обстоятельство происходит или не происходит. Например, «L представляет собой линейный C1-10 алкилен, необязательно прерванный одним или более атомами кислорода, серы или азота», означает, что линейный C1-10 алкилен может быть, но не обязательно, прерван атомами кислорода, серы или азота, и это описание включает случай, когда линейный C1-10 алкилен прерывается атомами кислорода, серы или азота, и случай, когда линейный C1-10 алкилен не прерывается атомами кислорода, серы или азота.
Термин «замещен» означает, что один или более, предпочтительно вплоть до 5, более предпочтительно от 1 до 3 атомов водорода в группе независимо замещены соответствующим количеством заместителей. Само собой разумеется, что заместитель находится только в своем возможном химическом положении, и специалисты в данной области техники смогут определить (экспериментально или теоретически) возможное или невозможное замещение без особых усилий.
В химической структуре соединения, описанного в настоящем документе, связь  не указана с конфигурацией, то есть связь
не указана с конфигурацией, то есть связь  может быть
может быть  или
или  или включает обе конфигурации
или включает обе конфигурации  и
и  В химической структуре соединения, описанного в настоящем документе,
В химической структуре соединения, описанного в настоящем документе,  связь не указана с конфигурацией, то есть она может иметь Z-конфигурацию или Е-конфигурацию или включает обе конфигурации.
связь не указана с конфигурацией, то есть она может иметь Z-конфигурацию или Е-конфигурацию или включает обе конфигурации.
Любое изотопно меченное производное соединения, или его фармацевтически приемлемой соли, или изомера, описанного в настоящем документе, охватывается настоящим изобретением. Атомы, которые могут быть изотопно мечены, включают, но не ограничиваются, водород, углерод, азот, кислород, фосфор, фтор, хлор, йод и т.д. Они могут быть заменены изотопами 2H(D), 3Н, 11C, 13С, 14С, 15N, 18F, 31Р, 32Р, 35S, 36Cl, 125I и т.д. Если не указано иное, когда положение конкретно обозначено как дейтерий (D), это положение следует понимать как дейтерий, имеющий содержание, которое по меньшей мере в 3000 раз больше, чем природное содержание дейтерия (которое составляет 0,015%) (то есть, включение по меньшей мере 45% дейтерия).
Фармацевтически активный агент или его остаток, фармацевтически активный агент и фармацевтически активный агент или его остаток А используют взаимозаменяемо в настоящем описании, и все они относятся к молекуле или группе с фармацевтической активностью.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг.1 показаны ФК (фармакокинетические) профили лекарственных средств в плазме крыс в тестовом примере 1.
На фиг.2 показаны ФК профили в плазме собак породы бигль в тестовом примере 2 (0-168 часов).
На фиг.3 показаны ФК профили в плазме собак породы бигль в тестовом примере 2 (0-24 часа).
На фиг.4 показаны кривые роста объема опухоли для групп мышей в модели подкожной трансплантатной опухоли клетками рака легкого человека А549 в тестовом примере 3.
На фиг.5 показаны ФК профили в плазме собак породы бигль в тестовом примере 4.
На фиг.6 показан терапевтический эффект соединения тестового примера 6 в отношении подкожных трансплантатных опухолей у мышей с острым лимфобластным лейкозом человека RS4; 11.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение дополнительно описано и объяснено ниже со ссылкой на примеры, но эти примеры не предназначены для ограничения объема настоящего изобретения.
Экспериментальные процедуры без условий, указанные в примерах настоящего изобретения, как правило, проводили в соответствии со стандартными условиями или в соответствии с условиями, рекомендованными производителем исходных материалов или коммерческих продуктов. Реагенты без указания конкретного происхождения являются коммерчески доступными общепринятыми реагентами.
Структуры соединений идентифицировали с помощью ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (МС). Спектры ЯМР измеряли с использованием прибора ядерного магнитного резонанса Bruker AVANCE-400 с дейтерированным диметилсульфоксидом (ДМСО-d6), дейтерированным хлороформом (CDCl3) и дейтерированным метанолом (CD3OD) в качестве растворителей определения и тетраметилсиланом (TMS) в качестве внутреннего стандарта. Химические сдвиги приведены в единицах измерения 10-6 (млн-1).
МС-анализ проводили с использованием масс-спектрометра FINNIGAN LCQAd (ESI) (производитель: Thermo, модель: Finnigan LCQ advantage MAX).
Анализ высокоэффективной жидкостной хроматографии (ВЭЖХ) проводили с использованием жидкостного хроматографа высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и жидкостного хроматографа высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Пластины силикагеля Yantai Huanghai HSGF254 или Qingdao GF254 с толщиной слоя от 0,15 до 0,2 мм использовали для анализа тонкослойной хроматографии (ТСХ) и толщиной слоя от 0,4 до 0,5 мм для разделения и очистки ТСХ.
Силикагель Yantai Huanghai 200-300 меш обычно использовали в качестве носителя в колоночной хроматографии.
Известные исходные материалы по настоящему изобретению могут быть синтезированы с использованием или в соответствии со способами, известными в данной области техники, или могут быть приобретены у ABCR GmbH & Co.KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc, Chembee Chemicals и других компаний.
В примерах реакции проводили в атмосфере аргона или атмосфере азота, если не указано иное.
Атмосфера аргона или атмосфера азота означает, что реакционная колба соединена с баллоном, содержащим около 1 л аргона или азота.
Атмосфера водорода означает, что реакционная колба соединена с баллоном, содержащим около 1 л водорода.
Гидрогенизатор Parr 3916ЕKХ, гидрогенизатор Qinglan QL-500 или гидрогенизатор HC2-SS использовали в реакциях гидрирования под давлением.
Реакция гидрирования обычно включает 3 цикла вакуумирования и продувки водородом.
В реакциях, проводимых под воздействием микроволнового излучения, использовали микроволновый реактор СЕМ Discover-S 908860.
В примерах раствор в реакции относится к водному раствору, если не указано иное.
В примерах температура реакции представляет собой комнатную температуру, если не указано иное.
Комнатная температура является оптимальной температурой реакции, находящейся в диапазоне от 20 до 30°С.
Получение буфера PBS (фосфатно-солевой буферный раствор), имеющего рН 6,5, в примерах: 8,5 г KН2РO4, 8,56 г K2НРO4×3Н2O, 5,85 г NaCl и 1,5 г EDTA (этилендиаминтетрауксусная кислота) добавляли в колбу, доводили раствор до объема 2 л, все компоненты растворяли с помощью ультразвука и раствор хорошо встряхивали.
Элюирующая система для колоночной хроматографии и система проявляющего растворителя для тонкослойной хроматографии, используемая для очистки соединения, включают: А: дихлорметановую и изопропанольную систему, В: дихлорметановую и метанольную систему и С: петролейноэфирную и этилацетатную систему. Объемное соотношение растворителей корректировали в соответствии с полярностью соединения или добавляли небольшое количество триэтиламина и кислотного или основного реагента.
Некоторые из соединений по настоящему изобретению характеризовали с помощью анализа Q-TOF LC/MS. Анализ Q-TOF LC/MS выполняли с помощью квадрупольного времяпролетного масс-спектрометра с точной массой Agilent 6530 и ультравысокоэффективного жидкостного хроматографа Agilent 1290-Infinity (хроматографическая колонка Agilent Poroshell 300SB-C8 5 мкм, 2,1×75 мм).
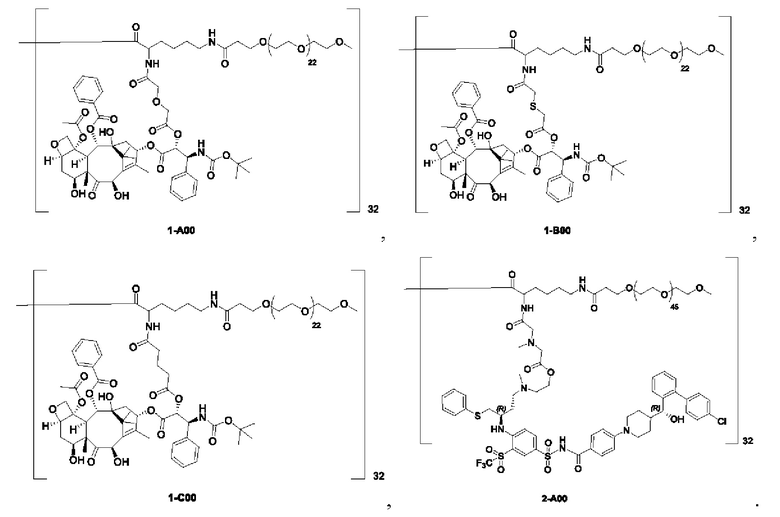
Дендример, как показано ниже, синтезировали со ссылкой на способ синтеза патента CN 110312531 А. Его первая концевая аминогруппа используется для связывания с фармацевтически активным агентом, и его вторая концевая группа используется для связывания с фармакокинетический модификатором PEG (полиэтиленгликоль).
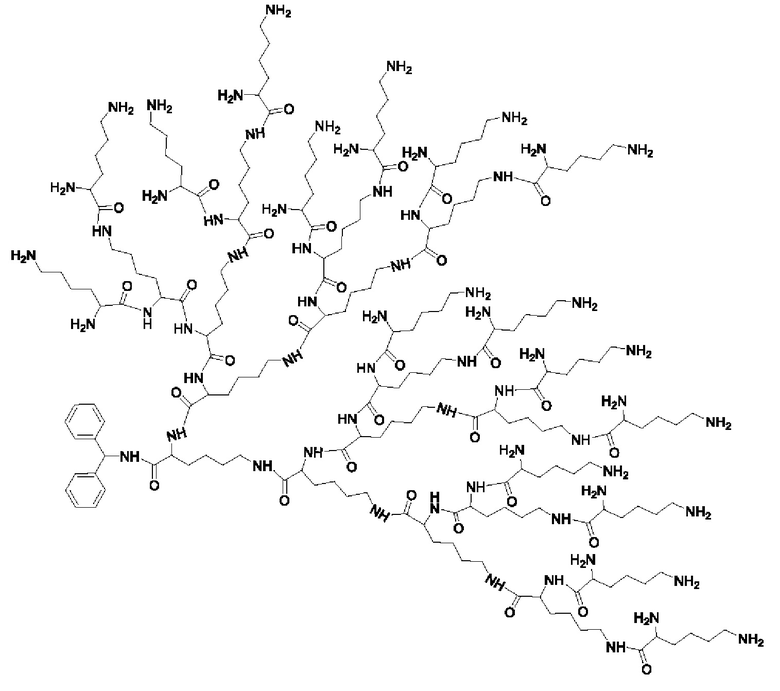
Дендримеры, представленные в приведенных ниже примерах, включают ссылки на ядро и структурные единицы в самой внешней генерации дендримера. Генерации от 1 до подповерхностной не показаны. Дендример BHALys[Lys]32 представляет собой дендример из 5 генераций, имеющий формулу BHALys[Lys]2[Lys]4[Lys]8[Lys]16[Lys]32; 64 поверхностные аминогруппы доступны для связывания с концевыми группами.
Получение дендримерных каркасов BHALys[Lys]32[α-NH2 TFA]32[ε-PEG570]32, ВНALys[Lys]32[α-NH2 TFA]32[ε-PEG1100]32, BHALys[Lys]32[α-NH2 TFA]32[ε-t-PEG2300]32, BHALys[Lys]32[α-4-HSBA]32[ε-PEG1100]32,
BHALys[Lys]32[α-GILGVP-NH2.TFA]32[ε-PEG1100]32 и BHALys[Lys]32[α-GILGVP-NH2 TFA]32[ε-t-PEG2300]32 можно найти в Kaminskas et al., J Control. Release (2011) (doi: 10.1016/j.jconrel.2011.02.005). Получение дендримерного каркаса 4-азидобензамид-PEG12-NEOEOEN[Su(NPN)2][Lys]16[NH2 TFA]32 можно найти в WO08/017122.
1-А00, 1-В00 и 1-С00 синтезировали в соответствии с патентом WO2012167309, и фармацевтически активный агент, с которым они были связаны, представлял собой доцетаксел. 2-А00 синтезировали в соответствии с патентом WO2018154004A, и фармацевтически активный агент, с которым он был связан, представлял собой соединение 2 (синтезированное в соответствии с патентом WO2012017251). Дендримерный каркас BHALys[Lys]32[α-NH2 TFA]32[ε-PEG1100]32 обозначен в примерах как дендример 1, и BHALys[Lys]32[α-NH2 TFA]32[ε-PEG2100]32 обозначен в примерах как дендример 1-PEG2K. Их синтезировали согласно способу WO2018154004A:
Общая процедура Общая процедура А
Установка линкеров к лекарственным средствам
К перемешиваемому с магнитом раствору линкера карбоновой кислоты (0,2-0,5 ммоль) в растворителе DMF (диметилформамид) или ацетонитриле (1-5 мл) при 0°С добавляли связывающий агент EDC или DCC (1,2 эквивалентов). Смесь оставляли перемешиваться в течение 5 мин, а затем по каплям добавляли раствор растворителя (1 мл), содержащий смесь лекарственного средства (0,4-1 эквивалента) и DMAP (4-диметиламинопиридин, 0,4-1 эквивалента). Смесь выдерживали при 0°С в течение 1 ч, а затем нагревали до температуры окружающей среды. Затем летучие вещества удаляли в вакууме (in vacuo) и остаток очищали с помощью препаративной ВЭЖХ (ВЕН300 Waters XBridge С18, 5 мкМ, 30×150 мм, 40-80% ACN (ацетонитрил)/вода (5-40 мин), без буфера) с получением целевого продукта.
Общая процедура В
Установка линкеров к лекарственным средствам
К перемешиваемому с магнитом раствору лекарственного средства (0,3-1,0 ммоль) и ангидрида (2 эквивалента) в DMF (3-5 мл) добавляли DIPEA (диизопропилэтиламин, 3 эквивалента). Смесь перемешивали при температуре окружающей среды в течение ночи. Затем летучие вещества удаляли в вакууме и остаток очищали с помощью препаративной ВЭЖХ (ВЕН300 Waters XBridge C18, 5 мкМ, 30 × 150 мм, 40-70% ACN/вода (5-40 мин), без буфера, RT=34 мин). Соответствующие фракции концентрировали в вакууме с получением желаемого продукта.
Общая процедура С
Нагрузка дендримера лекарственным средством-линкером
К перемешиваемой с магнитом смеси BHALys[Lys]32[α-NH2 TFA]32[ε-PEG1100]32 (0,5-1,0 мкмоль) и DIPEA (1,2 эквивалента/амин) в DMF при комнатной температуре добавляли линкер-лекарственное средство (1,2 эквивалента/амин) и РуВОР (гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония, 1,2 эквивалента/амин). После 1,5 ч перемешивания при комнатной температуре летучие вещества удаляли и остаток очищали с помощью SEC (гель-проникающая хроматография, сефадекс, LH20, МеОН). Соответствующие фракции (как определяли по ВЭЖХ) объединяли и концентрировали с получением желаемого продукта.
Общая процедура D
Клик-реакция
К перемешиваемому с магнитом раствору дендримера (0,5-1,0 ммоль) в 1:1 Н2О/t-BuOH (около 0,5 мл) добавляли алкиновый реагент (2 эквивалента), раствор аскорбата натрия (2 эквивалента) и раствор CuSO4 (20 мол.%). Раствор нагревали до 80°С и контролировали с помощью ВЭЖХ. Дополнительные количества аскорбата натрия и CuSO4 добавляли по мере необходимости, чтобы привести реакцию к завершению. После того, как реакция была признана завершенной, реакционную смесь концентрировали в вакууме и затем очищали.
Макромолекула по настоящему изобретению может быть синтезирована согласно схеме, выбранной из группы, состоящей из следующего, с PG в качестве защитной группы карбоновой кислоты:
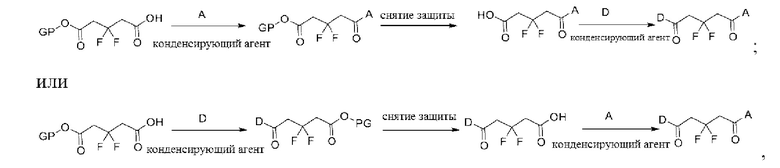
или
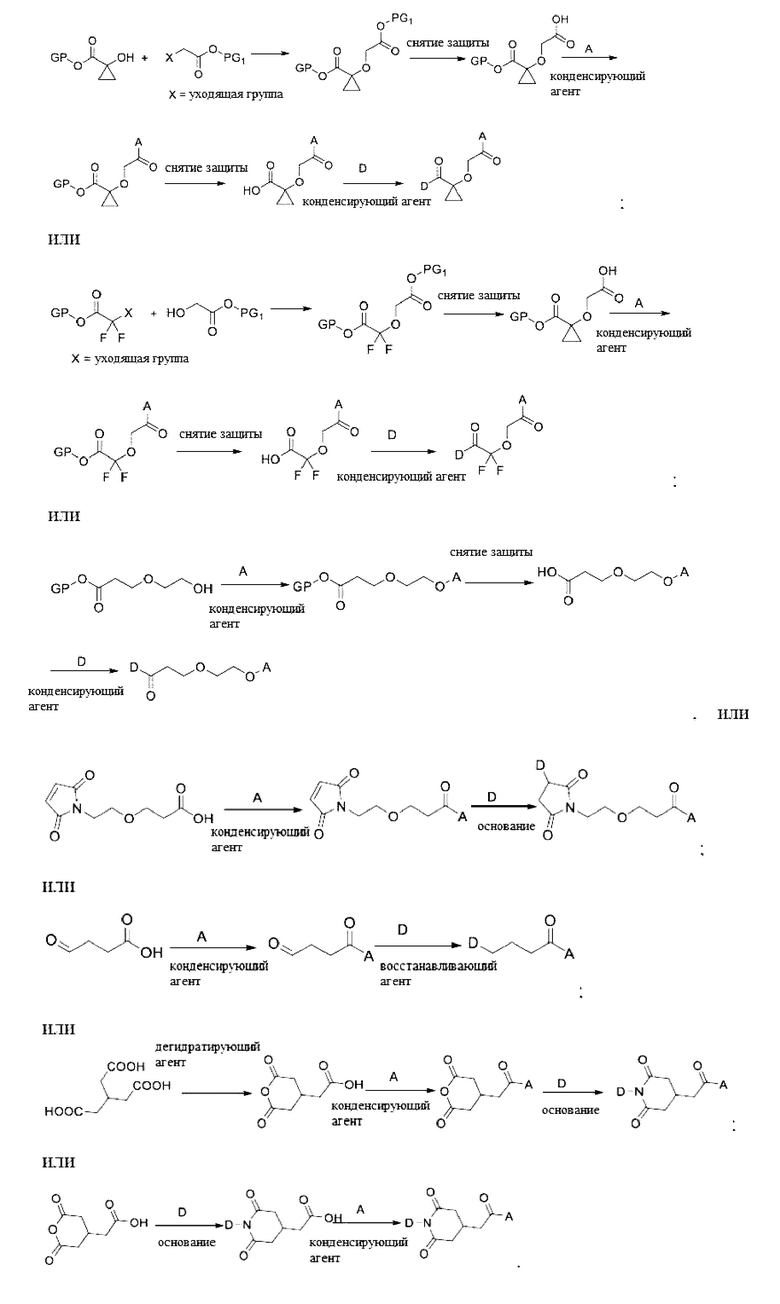
Пример 1: Получение Соединения 3
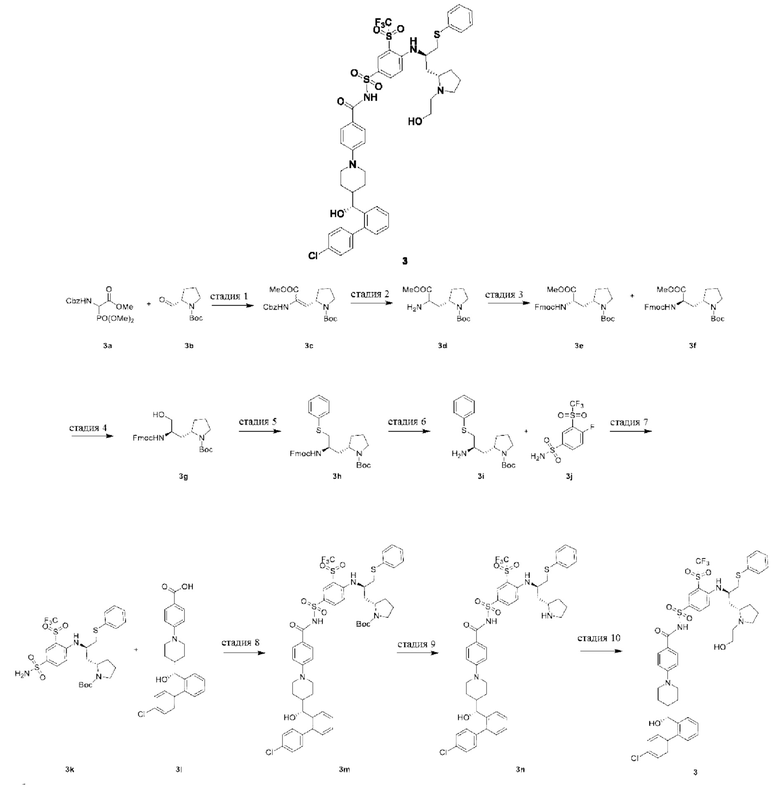
Стадия 1
Трет-бутил-(S)-2-(2-(((бензилокси)карбонил)амино)-3-метокси-3-оксопроп-1-ен-1-ил)пирролидин-1-карбоксилат 3с
3а (2,49 г, 7,53 ммоль, Accela ChemBio) растворяли в тетрагидрофуране (20 мл). Раствор охлаждали до 0°С в условиях ледяной бани. По каплям добавляли N,N-диизопропиламид лития (661 мг, 6,17 ммоль). Смесь перемешивали при 0°С в течение 1 ч, реакционную систему охлаждали до минус 78°С и по каплям добавляли раствор трет-бутил-(S)-2-формилпирролидин-1-карбоксилата 3b (1,00 г, 5,02 ммоль, PharmaBlock) в тетрагидрофуране (20 мл). После добавления по каплям, ванну с сухим льдом и ацетоном удаляли, и смесь нагревали до комнатной температуры и оставляли реагировать в течение 12 ч. Смесь гасили насыщенным раствором хлорида аммония (20 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фракцию сушили над безводным сульфатом натрия и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой В с получением указанного в заголовке продукта 3с (1,90 г, выход: 94,1%).
МС m/z (ESI): 305,0 [М+Н-100].
Стадия 2
Трет-бутил-(2S)-2-(2-амино-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 3d
3с (1,90 г, 4,7 ммоль) растворяли в изопропаноле (50 мл). Добавляли палладий на угле (380 мг, 10% нагрузка, на сухой основе). Систему продували 3 раза водородом. Смесь перемешивали при комнатной температуре в течение 8 ч. Реакционную смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом (20 мл) и метанолом (20 мл). Фильтрат концентрировали с получением указанного в заголовке продукта 3d (неочищенный, 1,28 г). Продукт непосредственно использовали на следующей стадии без очистки.
Стадия 3
Трет-бутил-(S)-2-((S)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 3е
Трет-бутил-(S)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-метокси-3-оксопропил)пирролидин-1-карбоксилат 3f
Неочищенный 3d (1,28 г, 4,7 ммоль) растворяли в 1,4-диоксане (40 мл). Добавляли воду (10 мл), бикарбонат натрия (1,17 г, 14,1 ммоль) и флуоренилметоксикарбонил хлорид (1,21 г, 4,7 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Смесь гасили водой (30 мл) и экстрагировали этилацетатом (20 мл × 3). Органическую фракцию сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке продукта 3е (1,03 г, 44,2%, пик 1) и 3f (513 мг, 22,1%, пик 2).
МС m/z (ESI): 495,2 [М+1].
Пик 1: анализ УВЭЖХ: время удерживания: 2,77 мин (колонка: ACQUITY UPLC ВЕНС18 50×2,1 мм, 1,7 мкм; подвижная фаза: ацетонитрил/вода/муравьиная кислота=10/90/0,1 (об./об./об.) до 95/5/0,1 (об./об./об.) градиентное элюирование).
Пик 2: анализ УВЭЖХ: время удерживания: 2,69 мин (колонка: ACQUITY UPLC ВЕНС18 50×2,1 мм, 1,7 мкм; подвижная фаза: ацетонитрил/вода/муравьиная кислота=50/50/0,1 (об./об./об.) до 95/5/0,1 (об./об./об.) градиентное элюирование).
Стадия 4
Трет-бутил-(S)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-гидроксипропил)пирролидин-1-карбоксилат 3g
3f (169 мг, 0,34 ммоль) растворяли в тетрагидрофуране (8 мл). Добавляли этанол (8 мл), боргидрид натрия (77 мг, 2,05 ммоль) и хлорид лития (94 мг, 2,22 ммоль). Смесь перемешивали при 28°С в течение 2,5 ч и добавляли по каплям насыщенный хлорид аммония (5 мл) для гашения реакции. Раствор концентрировали при пониженном давлении. Остаток растворяли в этилацетате (10 мл) и последовательно промывали водой (10 мл) и насыщенным хлоридом натрия (10 мл). Органическую фракцию сушили над безводным сульфатом натрия, фильтровали и концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке продукта 3g (125 мг, 78,4%).
МС m/z (ESI): 467,2 [М+1].
Стадия 5
Трет-бутил-(S)-2-((R)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 3h
3g (120 мг, 0,26 ммоль) растворяли в толуоле (5 мл). Добавляли дифенилдисульфид (168 мг, 0,77 ммоль) и трибутилфосфин (156 мг, 0,77 ммоль). Реакционную систему нагревали до 80°С и перемешивали в течение 12 ч в атмосфере азота. Раствор концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке продукта 3h (135 мг, 93,9%).
МС m/z (ESI): 559,2 [М+1].
Стадия 6
Трет-бутил-(S)-2-((R)-2-амино-3-(фенилтио)пропил)пирролидин-1-карбоксилат 3i
3h (135 мг, 0,24 ммоль) растворяли в дихлорметане (4 мл). Добавляли диэтиламин (4 мл). Смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении для удаления органического растворителя с получением указанного в заголовке продукта 3i (81,6 мг). Продукт непосредственно использовали на следующей стадии без очистки.
Стадия 7
Трет-бутил-(S)-2-((R)-4-(фенилтио)-3-((4-сульфамоил-2-((трифторметил)сульфонил)фенил)амино)бутил)пирролидин-1-карбоксилат 3k
Неочищенный 3i (81,6 мг, 0,24 ммоль) растворяли в N,N-диметилформамиде (5 мл). Добавляли 4-фтор-3-(трифторметилсульфонил)бензолсульфонамид 3j (82 мг, 0,27 ммоль) и N,N-диизопропилэтиламин (314 мг, 2,4 ммоль). Реакционную систему нагревали до 50°С и перемешивали в течение 12 ч в атмосфере азота. Смесь гасили водой (10 мл) и экстрагировали этилацетатом (5 мл × 3). Органическую фракцию сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с элюирующей системой А с получением указанного в заголовке продукта 3k (105 мг, 69,3%).
MC m/z (ESI): 622,1 [М-1].
Стадия 8
Трет-бутил-(S)-2-((R)-2-((4-(N-(4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)бензоил)сульфамоил)-2-((трифторметил)сульфонил)фенил)амино)-3-(фенилтио)пропил)пирролидин-1-карбоксилат 3m
3k (30 мг, 0,048 ммоль) растворяли в дихлорметане (3 мл). Добавляли 31 (24 мг, 0,058 ммоль, синтезированное с использованием способа, представленного для промежуточного соединения 40 на стр. 79 описания в патенте «CN103153954 В»), а затем последовательно добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (19 мг, 0,096 ммоль) и 4-диметиламинопиридин (12 мг, 0,096 ммоль). После добавления смесь перемешивали при комнатной температуре в течение 4 ч в атмосфере азота и добавляли 31 (12 мг, 0,038 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (19 мг, 0,096 ммоль) и 4-диметиламинопиридин (12 мг, 0,096 ммоль). После добавления смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор последовательно промывали водой (5 мл) и насыщенным солевым раствором (5 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали тонкослойной хроматографией с системой проявляющего растворителя А с получением указанного в заголовке продукта 3m (30 мг, выход: 60,7%).
MC m/z (ESI): 1027,2 [М+1].
Стадия 9
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-(фенилтио)-3-((S)-пирролидин-2-ил)пропан-2-ил)амино)-3-((трифторметил)сульфонил)фенил)сульфонил)бензамид 3n
3m (30 мг, 0,029 ммоль) растворяли в дихлорметане (2 мл). Добавляли 4 М раствор хлористого водорода в 1,4-диоксане (2 мл). Смесь перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь концентрировали при пониженном давлении для удаления органического растворителя. Остаток растворяли в дихлорметане и затем концентрировали при пониженном давлении до сухости; процесс повторяли три раза. Полученный остаток очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep С18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил). Соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 3n (25 мг, выход: 92,3%).
MC m/z (ESI): 927,1 [М+1].
1H ЯМР (500 МГц, CD3OD) δ 8,24 (d, 1H), 8,05-8,01 (m, 1H), 7,82 (d, 2Н), 7,60 (d, 1Н), 7,45-7,39 (m, 3Н), 7,37 (d, 2Н), 7,34-7,28 (m, 3Н), 7,23 (t, 2Н), 7,20-7,14 (m, 2Н), 6,80 (d, 2Н), 6,72 (d, 1Н), 4,42 (d, 1H), 3,97 (br, 1H), 3,84-3,74 (m, 1Н), 3,27-3,21 (m, 2Н), 3,19-3,07 (m, 3Н), 3,06-2,97 (m, 1Н), 2,69-2,61 (m, 1Н), 2,55-2,48 (m, 1H), 2,16-2,00 (m, 5Н), 1,98-1,89 (m, 2Н), 1,88-1,80 (m, 1H), 1,78-1,69 (m, 1H), 1,63-1,57 (m, 2Н), 1,54-1,49 (m, 1H), 1,26-1,20 (m, 1H), 1,15-1,04 (m, 1Н), 1,01-0,94 (m, 1Н).
Стадия 10
4-(4-((R)-(4'-хлор-[1,1'-бифенил]-2-ил)(гидрокси)метил)пиперидин-1-ил)-N-((4-(((R)-1-((S)-1-(2-гидроксиэтил)пирролидин-2-ил)-3-(фенилтио)пропан-2-ил)амино)-3-((трифтор метил)сульфонил)фенил)сульфонил)бензамид 3
3n (15 мг, 0,016 ммоль) растворяли в ацетонитриле (2 мл). Последовательно добавляли триэтиламин (16 мг, 0,16 ммоль) и бромэтанол (20 мг, 0,16 ммоль). Реакционную систему нагревали до 70°С и подвергали реакции в течение 12 ч в атмосфере азота. Реакционную смесь очищали с помощью высокоэффективной жидкостной хроматографии (условия разделения: колонка: Sharpsil-T Prep C18; подвижная фаза: бикарбонат аммония, вода, ацетонитрил). Соответствующую фракцию собирали и концентрировали при пониженном давлении с получением указанного в заголовке продукта 3 (10 мг, выход: 63,4%).
МС m/z (ESI): 971,2 [М+1].
1H ЯМР (500 МГц, CD3OD) δ 8,25 (d, 1H), 8,06 (dd, 1H), 7,80 (d, 2Н), 7,61 (dd, 1Н), 7,49-7,35 (m, 5Н), 7,35-7,28 (m, 3Н), 7,24 (dd, 2Н), 7,20-7,14 (m, 2Н), 6,79 (t, 3Н), 4,42 (d, 1H), 4,02 (t, 1H), 3,84-3,70 (m, 3Н), 3,61 (d, 2Н), 3,28-3,14 (m, 2Н), 3,06 (br, 1H), 2,96 (br, 1Н), 2,71-2,59 (m, 1H), 2,56-2,45 (m, 1H), 2,45-2,35 (m, 1Н), 2,32-2,22 (m, 1Н), 2,10-1,97 (m, 4Н), 1,96-1,86 (m, 1H), 1,85-1,69 (m, 2Н), 1,60 (t, 2Н), 1,26-1,18 (m, 1H), 1,06-1,08 (m, 1Н), 1,01-0,92 (m, 1H), 0,92-1,86 (m, 1Н).
Пример 2: Получение Соединения 3-TMS
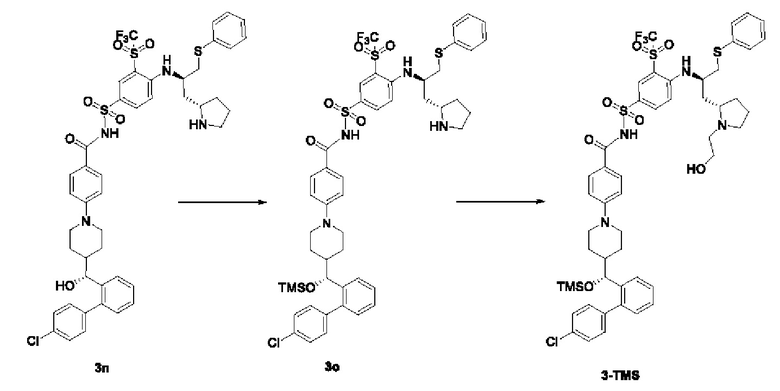
Стадия 1
3n (6 г, 6,47 ммоль) растворяли в DCM (дихлорметан, 120 мл). Добавляли триэтиламин (3,22 г, 32,35 ммоль). Смесь охлаждали до 0°С на водяной бане со льдом. Триметилсилил трифторметансульфонат (7,2 г, 32,35 ммоль) медленно добавляли по каплям к реакционной смеси. Смесь перемешивали на водяной бане со льдом в течение 2 ч. Смесь гасили добавлением по каплям метанола (12 мл) в условиях водяной бани со льдом, перемешивали на водяной бане со льдом в течение 10 мин и концентрировали при пониженном давлении для удаления растворителя с получением 3о (неочищенный, 16,5 г), который использовали на следующей стадии без очистки.
MC-ESI: m/z 999,1 [М+1]+.
Стадия 2
3о (16,5 г, рассчитанное как 6,47 ммоль) растворяли в ацетонитриле (90 мл). Добавляли триэтиламин (7,86 г, 77,64 ммоль) и бромэтанол (9,7 г, 77,64 ммоль). Систему трижды продували азотом. Смесь нагревали до 70°С и оставляли реагировать в течение ночи (около 16 ч). Растворитель удаляли при пониженном давлении, а затем добавляли ЕА (120 мл) и воду (120 мл). Смесь перемешивали. Водную фазу отделяли и экстрагировали ЕА (60 мл × 2). Органические фазы объединяли, промывали водой (120 мл), промывали насыщенным солевым раствором (120 мл), сушили и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (DСМ:СН3ОН = от 20:1 до 10:1) с получением соединения 3-TMS (5,03 г, выход на двух стадиях: 74,5%, чистота: 95,9%).
MC-ESI: m/z 1043,05 [M+1]+.
1Н ЯМР (400 МГц, CDCl3) δ 8,29 (s, 1H), 8,05 (s, 1H), 7,74 (d, 2Н), 7,55 (t, 1H), 7,43-7,33 (m, 3Н), 7,29 (dd, 3Н), 7,24-7,06 (m, 6Н), 6,97 (d, 1H), 6,72 (d, 2Н), 6,63 (d, 1H), 4,54 (d, 1Н), 3,98 (s, 1H), 3,82-3,46 (m, 5Н), 3,02 (d, 5Н), 2,82-2,44 (m, 4Н), 2,26 (dd, 1H), 2,07 (d, 1H), 1,90 (dd, 3Н), 1,74 (d, 2Н), 1,58-1,41 (m, 1H), 1,40-1,15 (m, 4Н), -0,06 (s, 9Н).
Пример 3: Получение Соединения 1-Z00
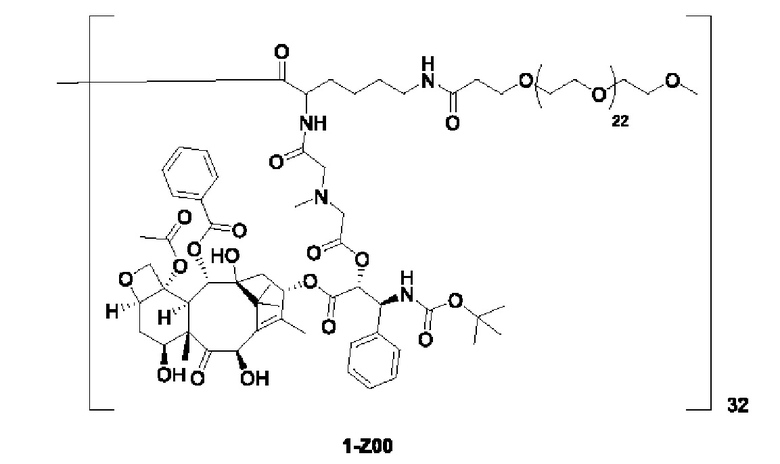
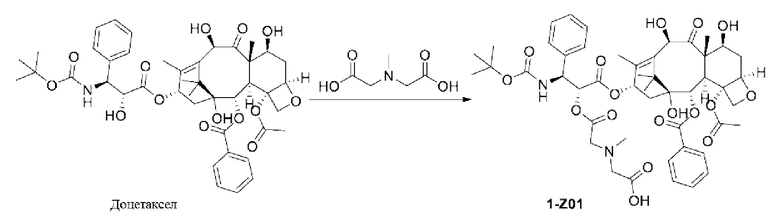
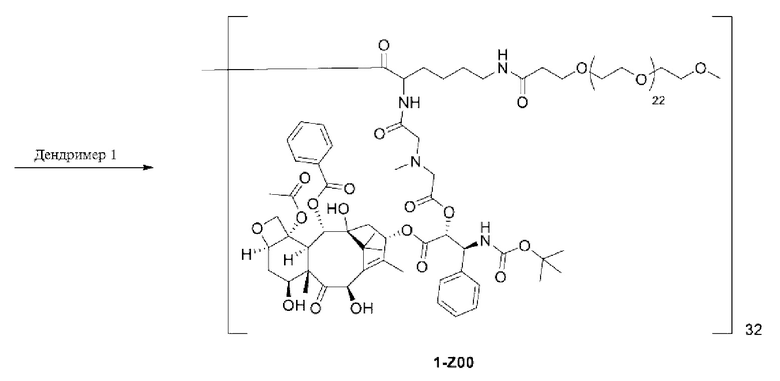
Стадия 1
В реакционную колбу последовательно добавляли DMF (4 мл), метилиминодиуксусную кислоту (218 мг, 1,48 ммоль) и DCC (245 мг, 1,18 ммоль). Смесь охлаждали на водяной бане со льдом. Добавляли доцетаксел (400 мг, 0,495 ммоль) и DMAP (60 мг, 0,495 ммоль). Смесь перемешивали при комнатной температуре в течение 1,5 ч. Добавляли дополнительное количество DCC (102 мг, 0,495 ммоль). Смесь перемешивали при комнатной температуре в течение еще часа. Реакцию останавливали после того, как она была признана завершенной. Смесь фильтровали. Осадок на фильтре промывали этилацетатом (15 мл). Фильтрат концентрировали при пониженном давлении для удаления этилацетата. Оставшуюся маточную жидкость очищали с помощью препаративной ВЭЖХ с получением соединения 1-Z01 (86 мг, выход: 19%).
MC-ESI: m/z 937,4 [M+H]+.
Стадия 2
1-Z01 (1,59 г, 1,70 ммоль) и РуВОР (1,70 г, 3,27 ммоль) растворяли в безводном DMF (28,0 мл) в атмосфере азота. Раствор хорошо перемешивали. Затем дендример 1 (2,00 г, 4,25×10-2 ммоль) и раствор DIPEA (0,42 г, 3,27 ммоль) в безводном DMF (28,0 мл) по каплям добавляли к вышеуказанному реакционному раствору. Смесь оставляли реагировать в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом (56,0 мл) и затем очищали ультрафильтрацией с ацетонитрилом в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (2,70 г). Продукт растворяли в чистой воде (150 мл) путем перемешивания с помощью вортекса. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 1-Z00 (2,40 г, выход: 85%).
1Н ЯМР показала 25 DTX/дендример. Фактическая молекулярная масса составляла около 66,4 кДа (30,39 мас. % DTX).
1H ЯМР (400 МГц, CD3OD) δ (млн-1) 8,05-8,24 (m, 64 Н), 7,11-7,78 (m, 272 Н), 5,98-6,25 (m, 25 Н), 5,55-5,71 (m, 29 Н), 5,19-5,49 (m, 84 Н), 4,93-5,07 (s, 47 Н), 4,10-4,64 (m, 162 Н), 3,40-4,08 (m, 3200 Н), 3,33-3,37 (S, 96 Н), 3,02-3,23 (m, 126 Н), 2,14-2,63 (m, 271 Н), 0,99-2,10 (m, 1109 Н).
Пример 4: Получение Соединения 1-Z00-J
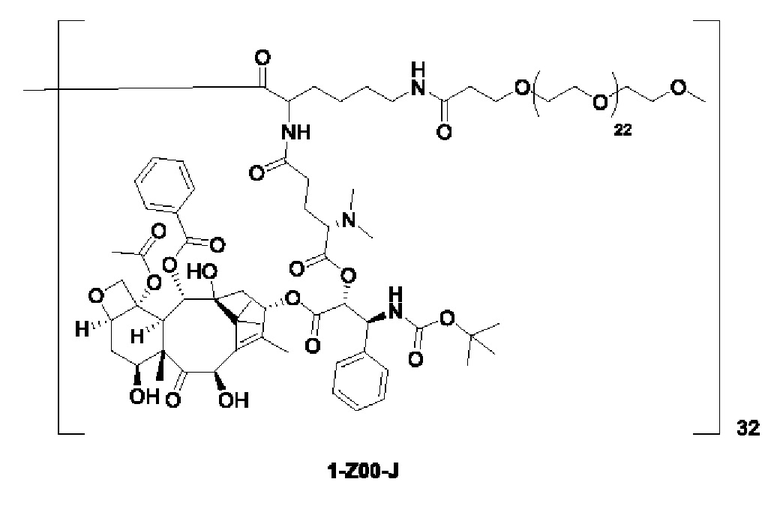
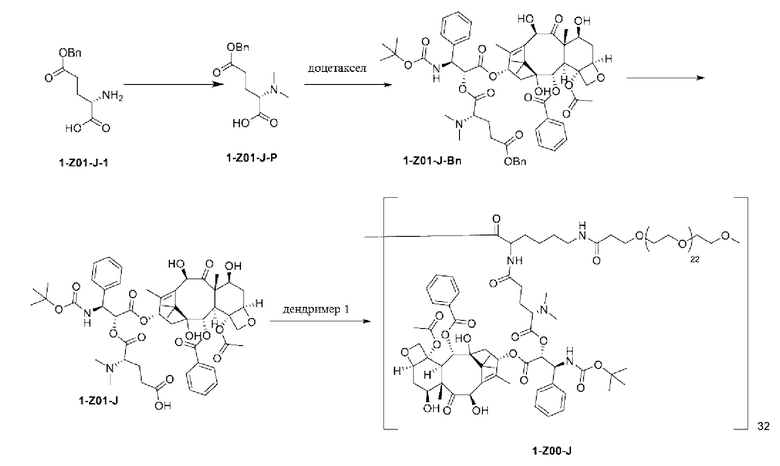
Стадия 1
Соединение 1-Z01-J-1 (7 г, 29,5 ммоль) растворяли в метаноле (150 мл). Добавляли раствор формальдегида (15 мл) и NaBH(AcO)3 (63 г, 295 ммоль). Смесь оставляли реагировать при комнатной температуре в течение 1 ч. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-J-P (5,9 г, выход: 75%).
1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,36-7,32 (m, 5Н), 5,09 (s, 2Н), 3,12 (t, J=7,2 Гц, 1H), 2,5-2,44 (m, 2Н), 2,38 (s, 6Н), 1,87-1,84 (,, 2Н). MC-ESI: m/z 266,2 [М+Н]+.
Стадия 2
Соединение 1-Z01-J-P (1,44 г, 5,45 ммоль) и доцетаксел (4 г, 4,96 ммоль) растворяли в дихлорметане (50 мл). Добавляли EDCI (1-этил-3-3-диметиламинопропил-карбодиимид, 1,04 г, 5,45 ммоль) и DMAP (665 мг, 5,45 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили водой и экстрагировали дихлорметаном. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-J-Bn (5 г, выход: 95%).
MC-ESI: m/z 1055,5 [М+Н]+.
Стадия 3
Соединение 1-Z01-J-Bn (5,75 г, 5,46 ммоль) растворяли в тетрагидрофуране (60 мл). Добавляли Pd/C (600 мг, 10 мас. %). Систему продували три раза водородом. Смесь оставляли реагировать при комнатной температуре в течение ночи. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-J (3,2 г, выход: 57%).
MC-ESI: m/z 965,3 [М+Н]+.
Стадия 4
Соединение 1-Z01-J (819 мг, 849,5 мкмоль) и РуВОР (853 мг, 1640 мкмоль) растворяли в безводном N,N-диметилформамиде (14 мл) в атмосфере азота. По каплям добавляли раствор соединения дендримера 1 (1000 мг, 21,24 мкмоль) и DIPEA (212 мг, 1640 мкмоль) в безводном N,N-диметилформамиде (14 мл). Смесь оставляли реагировать в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом (28 мл) и затем очищали ультрафильтрацией с ацетонитрилом в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (1,42 г). Продукт растворяли в чистой воде (100 мл). Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 1-Z00-J (1,42 г, выход: 95%).
1Н ЯМР показала 25 DTX/дендример. Фактическая молекулярная масса составляла около 67,1 кДа (30,0 мас. % DTX).
1Н ЯМР (400 МГц, CD3OD) δ 7,15-8,31 (m, 405Н), 6,00-6,31 (m, 25Н), 5,57-5,73 (m, 25Н), 5,20-5,54 (m, 83Н), 4,96-5,08 (m, 35Н), 4,10-4,68 (m, 141Н), 3,40-4,08 (m, 3200Н), 3,33-3,37 (s, 103Н), 3,07-3,24 (m, 92Н), 2,14-2,69 (m, 260Н), 1,00-2,10 (m, 1183Н).
Пример 5: Получение Соединения 1-Z00-K
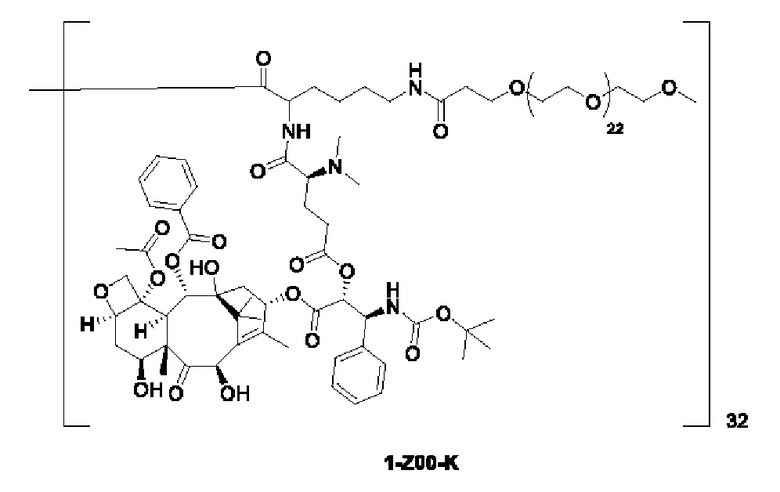
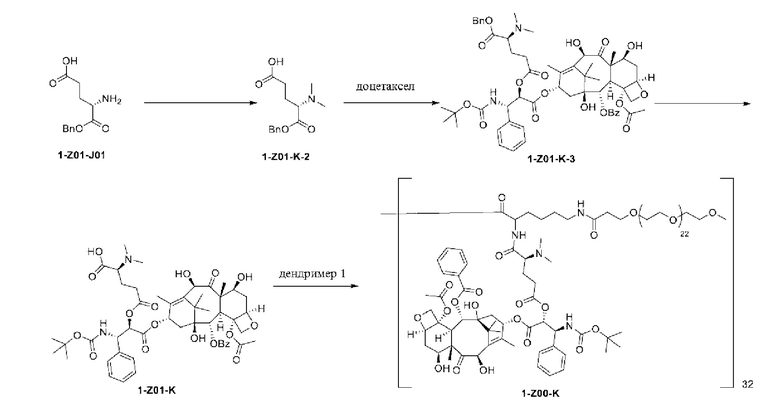
Стадия 1
Соединение 1-Z01-J01 (5,0 г, 21,09 ммоль, GL Biochem (Shanghai) Ltd.) растворяли в МеОН (100 мл) при перемешивании, а затем добавляли водный раствор формальдегида (37 мас. %, 10 мл). Добавляли NaBH(AcO)3 (22,3 г, 105,4 ммоль) партиями в условиях ледяной бани. После добавления смесь подвергали взаимодействию при комнатной температуре в течение 18 ч. После того, как реакция была признана завершенной, реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с получением соединения 1-Z01-K-2 (3,2 г, выход: 57%).
MC-ESI: m/z 266,1 [М+Н]+.
1Н-ЯМР: (CDCl3, 400 МГц) δ 7,36-7,27 (m, 5Н), 5,18 (dd, J1=20,0 Гц, J2=12,0 Гц, 2Н), 3,42-3,36 (m, 2Н), 2,48-2,46 (m, 2Н), 2,45 (s, 6Н), 2,05-2,00 (m, 2Н).
Стадия 2
В реакционную колбу в атмосфере азота добавляли соединение 1-Z01-K-2 (1,0 г, 1,23 ммоль), доцетаксел (1,0 г, 1,23 ммоль), EDCI (261 мг, 1,36 ммоль) и DMAP (166 мг, 1,36 ммоль), а затем безводный DMF (10 мл). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный раствор бикарбоната натрия и этилацетат. Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с получением соединения 1-Z01-K-3 (1,021 г, выход: 71%).
MC-ESI: m/z 1055,4 [М+Н]+.
Стадия 3
В реакционную колбу добавляли исходный материал 1-Z01-K-3 (1,0 г, 0,947 ммоль) и влажный палладий на угле (10 мас. %, 200 мг), а затем тетрагидрофуран (20 мл). Систему продували 3 раза водородом. Смесь подвергали взаимодействию при комнатной температуре в течение 18 ч. Реакционную смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом. Фильтрат концентрировали и сушили в вакууме с получением соединения 1-Z01-K (736 мг, выход: 80%).
MC-ESI: m/z 965,3 [М+Н]+.
Стадия 4
Соединение 1-Z01-K (162 мг, 0,168 ммоль) и РуВОР (168 мг, 0,323 ммоль) растворяли в безводном DMF (6,0 мл) в атмосфере азота и раствор хорошо перемешивали. Затем к вышеуказанному реакционному раствору по каплям добавляли раствор дендримера 1 (200 мг, 4,2×10-3 ммоль) и DIPEA (42 мг, 0,323 ммоль) в безводном DMF (6,0 мл). Смесь оставляли реагировать в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом и затем очищали ультрафильтрацией с ацетонитрилом в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,215 г). Продукт растворяли в чистой воде (50 мл). Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 1-Z00-K (0,21 г, выход: 76%).
1Н ЯМР показала 24 DTX/дендример. Фактическая молекулярная масса составляла около 66,1 кДа (29,30 мас. % DTX).
1Н ЯМР (400 МГц, CD3OD) δ (млн-1) 8,00-8,23 (m, 55Н), 7,11-7,78 (m, 239Н), 5,95-6,30 (m, 24Н), 5,51-5,75 (m, 24Н), 5,15-5,45 (m, 72Н), 4,93-5,07 (s, 19Н), 4,10-4,64 (m, 157Н), 3,40-4,08 (m, 3200Н), 3,33-3,37 (s, 102Н), 2,93-3,23 (m, 167Н), 2,12-2,63 (m, 212Н), 0,99-2,10 (m, 1145Н).
Пример 6: Получение Соединения 1-Z00-L
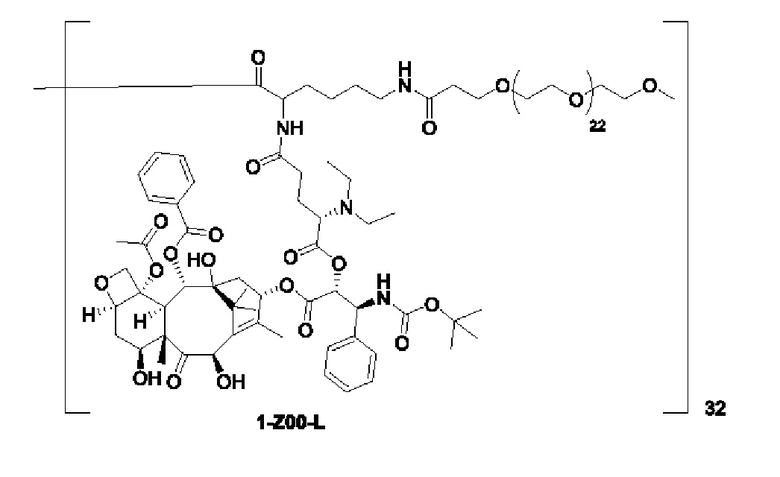
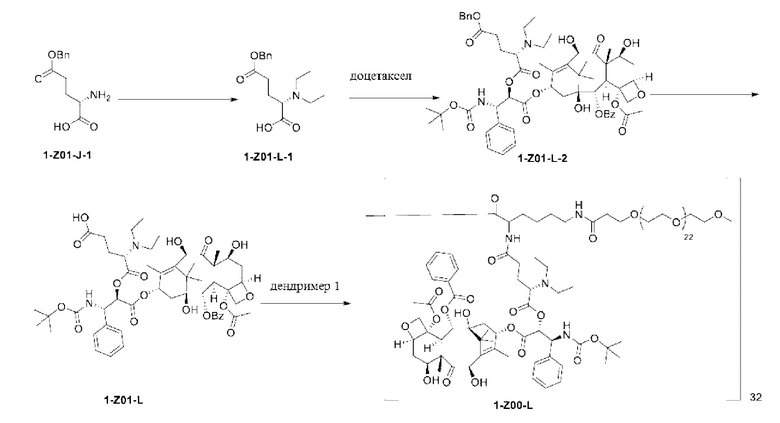
Стадия 1
Соединение 1-Z01-J-1 (1,0 г, 4,2 ммоль, Shanghai Bide Pharmatech) растворяли в МеОН (20 мл) при перемешивании, а затем добавляли водный раствор ацетальдегида (40 мас. %, 2 мл). Добавляли NaВН(АсО)3 (4,472 г, 21,0 ммоль) в условиях ледяной бани. Смесь подвергали взаимодействию при комнатной температуре в течение 20 ч. После концентрирования при пониженном давлении остаток очищали колоночной хроматографией с получением соединения 1-Z01-L-1 (733 мг, выход: 59,6%).
MC-ESI: m/z 294,1 [М+Н]+.
1H-ЯМР: (CDCl3, 400 МГц) δ 7,35-7,28 (m, 5Н), 5,10 (dd, J1=18 Гц, J2=12,4 Гц, 2Н), 4,15 (brs 1Н), 3,56 (t, J=6,8 Гц 1H), 3,27-3,22 (m, 2Н), 3,07-3,02 (m, 2Н), 2,86-2,69 (m, 2Н), 2,05-2,00 (m, 2Н), 1,30 (t, J=7,6 Гц, 6Н).
Стадия 2
В реакционную колбу добавляли соединение 1-Z01-L-1 (399 мг, 1,36 ммоль), доцетаксел (1,0 г, 1,23 ммоль), EDCI (261 мг, 1,36 ммоль) и DMAP (166 мг, 1,36 ммоль), а затем безводный DMF (10 мл). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный раствор бикарбоната натрия и этилацетат. Водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией с получением соединения 1-Z01-L-2 (798 мг, выход: 54%).
MC-ESI: m/z 1083,4 |М+Н]+.
Стадия 3
В реакционную колбу добавляли соединение 1-Z01-L-2 (790 мг, 0,729 ммоль) и влажный палладий на угле (10 мас. %, 158 мг), а затем тетрагидрофуран (16 мл). Систему продували 3 раза водородом. Смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом. Фильтрат концентрировали и сушили с получением соединения 1-Z01-L (616 мг, выход: 85%).
MC-ESI: m/z 993,4 [М+Н]+.
Стадия 4
Соединение 1-Z01-L(530 мг, 0,53 ммоль) и РуВОР (537 мг, 1,03 ммоль) растворяли в безводном DMF (9,0 мл) в атмосфере азота и раствор хорошо перемешивали. Затем к вышеуказанному реакционному раствору по каплям добавляли раствор дендримера 1 (629 мг, 13,4×10-3 ммоль) и DIPEA (133 мг, 1,03 ммоль) в безводном DMF (9,0 мл). Смесь оставляли реагировать в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом и затем очищали ультрафильтрацией с ацетонитрилом в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,66 г). Продукт растворяли в чистой воде (100 мл). Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 1-Z00-L(0,65 г, выход: 71%).
1Н ЯМР показала 26 DTX/дендример. Фактическая молекулярная масса составляла около 68,8 кДа (30,51 мас. % DTX).
1H ЯМР (400 МГц, CD3OD) δ (млн-1) 7,20-8,25 (m, 288Н), 5,98-6,30 (m, 26Н), 5,52-5,78 (m, 28Н), 5,15-5,45 (m, 84Н), 4,92-5,08 (s, 31Н), 4,10-4,64 (m, 212Н), 3,40-4,08 (m, 3200Н), 3,33-3,37 (S, 102Н), 2,93-3,23 (m, 172Н), 2,12-2,63 (m, 262Н), 0,99-2,10 (m, 1186Н).
Пример 7: Получение Соединения 1-Z00-M
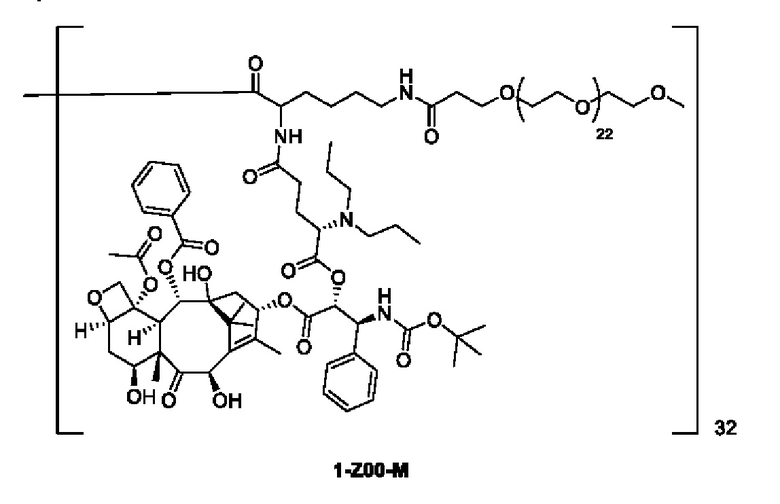
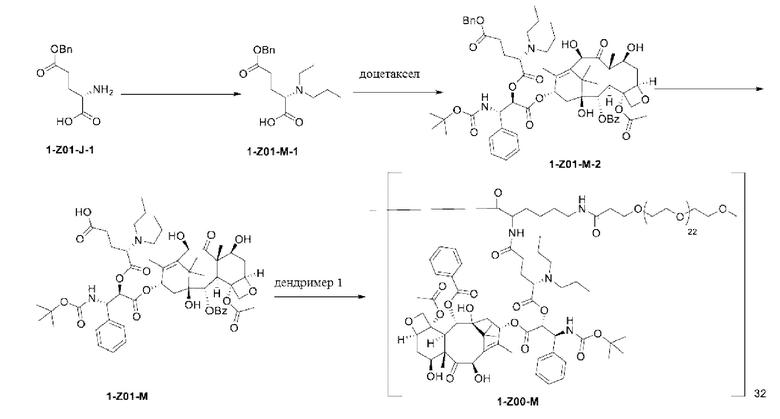
Стадия 1
Соединение 1-Z01-J-1 (5,1 г, 21,496 ммоль, Shanghai Bide Pharmatech) растворяли в МеОН (100 мл) при перемешивании, а затем добавляли пропиональдегид (6,3 г, 108,47 ммоль). Добавляли NaBH(AcO)3 (22,9 г, 108,02 ммоль) с охлаждением на ледяной бане. После добавления смесь подвергали взаимодействию при комнатной температуре в течение 20 ч. Реакционную смесь концентрировали при пониженном давлении для удаления метанола. Остаток перемешивали с водой и отфильтровывали нерастворимое вещество. Фильтрат очищали колоночной хроматографией с получением соединения 1-Z01-M-1 (4,85 г, выход: 68,7%).
МС (ESI) m/z=322,2 [М+Н]+.
1H-ЯМР (CDCl3, 400 МГц) δ 7,37-7,26 (m, 5Н), 5,12 (s, 2Н), 3,53 (brs 1Н), 3,04-2,72 (m, 7Н), 2,09-1,98 (m, 2Н), 1,79-1,67 (m, 4Н), 0,97-0,93 (m, 6Н).
Стадия 2
В реакционную колбу в атмосфере азота добавляли соединение 1-Z01-M-1 (367 мг, 1,1 ммоль), доцетаксел (801 г, 0,991 ммоль), EDCI (212 мг, 1,1 ммоль) и DMAP (136 мг, 1,1 ммоль), а затем безводный DMF (10 мл). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли насыщенный раствор бикарбоната натрия и этилацетат.Водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией с получением соединения 1-Z01-M-2 (732 мг, выход: 66%).
MC-ESI: m/z 1111,4 [М+Н]+.
Стадия 3
В реакционную колбу добавляли соединение 1-Z01-M-2 (732 мг, 0,658 ммоль) и влажный палладий на угле (10 мас. %, 133 мг), а затем тетрагидрофуран (18 мл). Систему продували 3 раза водородом. Смесь перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом. Фильтрат концентрировали при пониженном давлении с получением соединения 1-Z01-M (669 мг, выход: 99%).
MC-ESI: m/z 1021,4 [М+Н]+.
Стадия 4
Соединение 1-Z01-M (500 мг, 0,49 ммоль) и РуВОР (490 мг, 0,943 ммоль) растворяли в безводном DMF (7,5 мл) в атмосфере азота и раствор хорошо перемешивали. Затем к вышеуказанному реакционному раствору по каплям добавляли раствор дендримера 1 (543 мг, 12,25×10-3 ммоль) и DIPEA(122 мг, 0,943 ммоль) в безводном DMF (7,5 мл). Смесь оставляли реагировать в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом и затем очищали ультрафильтрацией с ацетонитрилом в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,64 г). Продукт растворяли в чистой воде (100 мл). Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 1-Z00-M (0,63 г, выход: 83%).
1Н ЯМР показала 21 DTX/дендример. Фактическая молекулярная масса составляла около 61,7 кДа (27,4 мас. % DTX).
1Н ЯМР (400 МГц, CD3OD) δ (млн-1) 8,00-8,22 (m, 45Н), 7,09-7,81 (m, 198Н), 5,95-6,30 (m, 21Н), 5,51-5,75 (m, 20Н), 5,13-5,45 (m, 62Н), 4,93-5,07 (s, 17Н), 4,10-4,64 (m, 175Н), 3,40-4,08 (m, 3000Н), 3,33-3,37 (S, 91Н), 2,93-3,23 (m, 127Н), 2,12-2,63 (m, 217Н), 0,79-2,10 (m, 1396Н).
Пример 8: Получение Соединения 1-Z00-Q
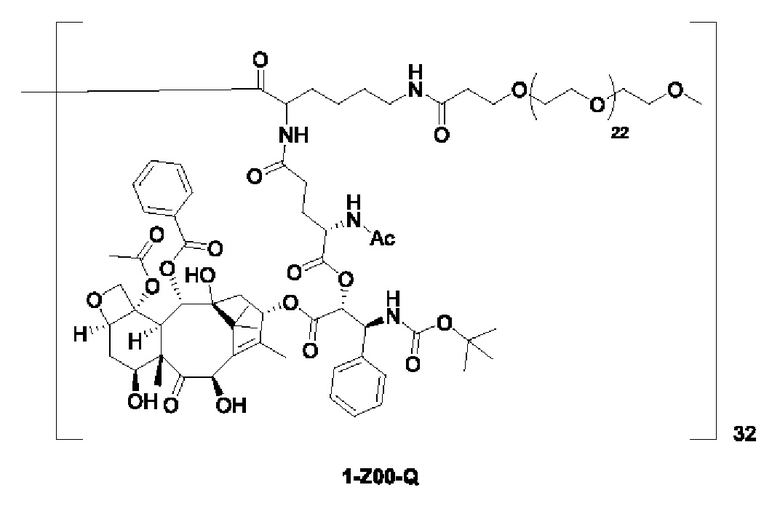
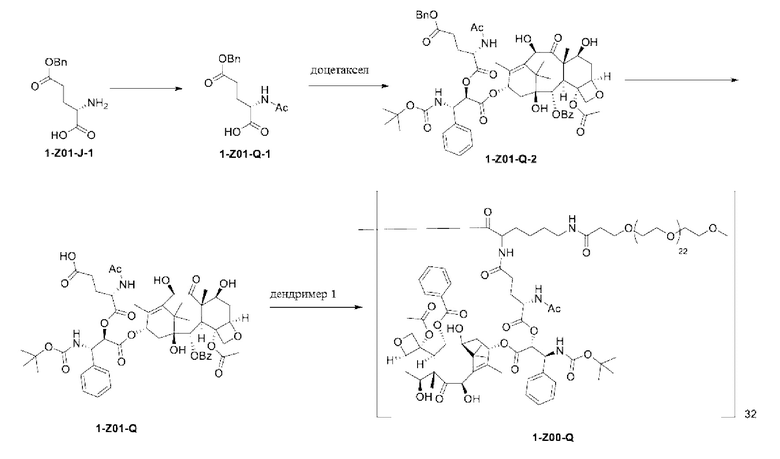
Стадия 1
Соединение 1-Z01-J-1 (5 г, 21,1 ммоль, Shanghai Bide Pharmatech) растворяли в DCM (50 мл). Добавляли триэтиламин (6,41 г, 63,3 ммоль) в атмосфере азота. Смесь охлаждали на водяной бане со льдом. По каплям добавляли уксусный ангидрид (2,37 г, 23,2 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. Реакционную смесь промывали водой. Водные фазы объединяли, доводили до рН 1 концентрированной соляной кислотой в условиях водяной бани со льдом и трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением белого твердого вещества (5,8 г). Белое твердое вещество растирали в порошок со смешанным раствором этилацетата и петролейного эфира в течение ночи. Смесь фильтровали с получением соединения 1-Z01-Q-1 (4,9 г, выход: 83%). Продукт непосредственно использовали на следующей стадии.
MC-ESI: m/z 280,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 12,62 (br, 1Н), 8,14 (d, J=8 Гц, 1Н), 7,40-7,31 (m, 5Н), 5,09 (s, 2Н), 4,24-4,18 (m, 1Н), 2,48-2,38 (m, 2Н), 2,05-2,00 (m, 1Н), 1,98-1,77 (m, 1Н), 1,97 (s, 3Н).
Стадия 2
Соединение 1-Z01-Q-1 (377 мг, 1,350 ммоль), доцетаксел (1 г, 1,239 ммоль) и DMAP (166 мг, 1,363 ммоль) взвешивали в реакционной колбе. Сухой DMF (10 мл) и EDCI (261 мг, 1,363 ммоль) добавляли в атмосфере азота и условиях водяной бани со льдом. После добавления смесь оставляли реагировать при комнатной температуре в течение ночи. К реакционной смеси добавляли этилацетат. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия и насыщенным раствором NaCl, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией с получением соединения 1-Z01-Q-2 (805 мг, выход: 60,8%).
MC-ESI: m/z 1069,5 [М+Н]+.
Стадия 3
Соединение 1-Z01-Q-2 (1,1 г, 1,029 ммоль) растворяли в THF (20 мл). Добавляли влажный палладий на углероде (10 мас. %, 220 мг). Систему три раза продували водородом, а затем смесь перемешивали в течение ночи при комнатной температуре в атмосфере водорода. Реакционную смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении и сушили в вакууме с получением соединения 1-Z01-Q (962 мг, выход: 95,5%).
MC-ESI: m/z 979,4 [М+Н]+.
Стадия 4
Соединение 1-Z01-Q (500 мг, 0,511 ммоль) и РуВОР (513 мг, 0,986 ммоль) растворяли в безводном DMF (7,9 мл) в атмосфере азота и раствор хорошо перемешивали. Затем к вышеуказанному реакционному раствору по каплям добавляли раствор дендримера 1 (567 мг, 12,8×10-3 ммоль) и DIPEA(127 мг, 0,986 ммоль) в безводном DMF (7,9 мл). Смесь оставляли реагировать в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом и затем очищали ультрафильтрацией с ацетонитрилом в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,625 г). Продукт растворяли в чистой воде (100 мл). Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 1-Z00-Q (0,62 г, выход: 74%).
1Н ЯМР показала 26 DTX/дендример. Фактическая молекулярная масса составляла около 65,7 кДа (31,96 мас. % DTX).
1Н ЯМР (400 МГц, CD3OD) δ 8,00-8,22 (m, 55Н), 7,09-7,81 (m, 253Н), 5,95-6,31 (m, 26Н), 5,51-5,77 (m, 28Н), 5,13-5,45 (m, 91Н), 4,92-5,08 (s, 41Н), 4,10-4,64 (m, 225Н), 3,40-4,08 (m, 3000Н), 3,33-3,37 (s, 91Н), 2,93-3,23 (m, 102Н), 2,12-2,63 (m, 310Н), 0,96-2,10 (m, 1119Н).
Пример 9: Получение Соединения 3-L00
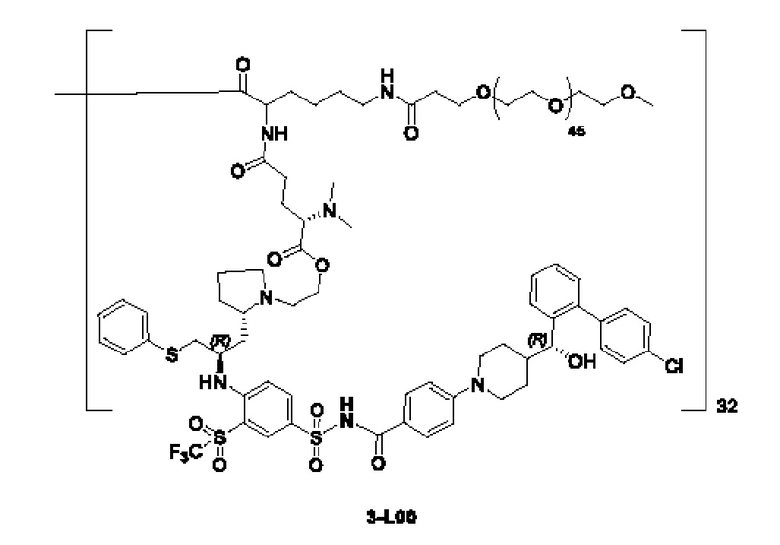
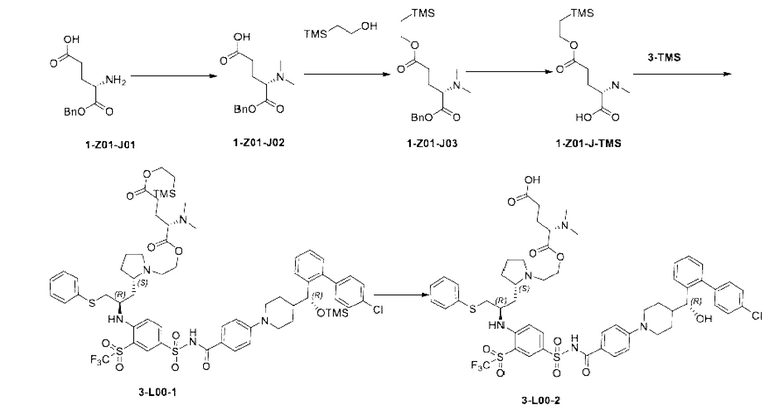
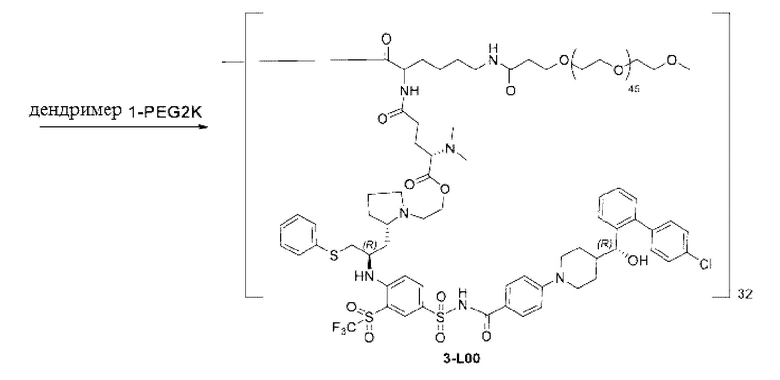
Стадия 1
Соединение 1-Z01-J01 (5,0 г, 18,3 ммоль) растворяли в метаноле (100 мл). Раствор охлаждали до 0°С. Добавляли водный раствор формальдегида (10 мл, 40 мас. %) и NaВН(ОАс)3 (20,0 г, 91,6 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-J02 (3,4 г, выход: 61%).
MC-ESI: m/z 266,1 [M+H]+.
Стадия 2
Соединение 1-Z01-J02 (3,4 г, 12,8 ммоль), триметилсилилэтанол (3,0 г, 26,0 ммоль), DMAP (780 мг, 6,4 ммоль) и N,N-диизопропилэтиламин (6,6 г, 51,2 ммоль) растворяли в безводном тетрагидрофуране (30 мл). Раствор охлаждали до 0°С. Добавляли HATU (1-бис(диметиламино)метилен-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид-гексафторфос фат, 6,3 г, 16,6 ммоль). Смесь оставляли реагировать при комнатной температуре в течение 16 ч. Смесь гасили водой (50 мл) и экстрагировали этилацетатом (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-J03 (4,0 г, выход: 85%).
MC-ESI: m/z 366,2 [М+Н]+.
Стадия 3
Соединение 1-Z01-J03 (4,0 г, 10,9 ммоль) растворяли в безводном тетрагидрофуране (40 мл). Добавляли Pd/C (200 мг, 10 мас. %). Систему продували три раза водородом. Смесь оставляли реагировать при комнатной температуре в течение 16 ч. После фильтрации и концентрирования получали неочищенный продукт (3,6 г). Продукт растирали в порошок с безводным тетрагидрофураном (30 мл) в течение 30 мин. Смесь фильтровали с получением соединения 1-Z01-J-TMS (1,6 г, выход: 45%).
MC-ESI: m/z 276,1 [М+Н]+.
1H ЯМР (400 МГц, CDCl3) δ 4,14-4,18 (m, 2Н), 3,44-3,47 (m, 1Н), 2,87 (s, 6Н), 2,66-2,70 (m, 2Н), 2,03-2,12 (m, 2Н), 0,96-0,99 (m, 2Н), 0,04 (s, 9Н).
Стадия 4
Соединение 1-Z01-J-TMS (118 мг, 0,431 ммоль), 3-TMS (300 мг, 0,287 ммоль) и DMAP (17,5 мг, 0,216 ммоль) растворяли в безводном дихлорметане (10 мл). Добавляли DCC (88,8 мг, 0,431 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 3-L00-1 (373 мг, выход: 98%).
MC-ESI: m/z 650,8 [1/2М+Н]+.
Стадия 5
Соединение 3-L00-1 (373 мг, 0,288 ммоль) растворяли в безводном тетрагидрофуране (3 мл). Добавляли TBAF (фторид тетрабутиламмония, 2,9 мл, 2,88 ммоль, 1,0 моль/л в THF). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Смесь гасили водой (10 мл) и экстрагировали этилацетатом (10 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-L00-2 (238 мг, выход: 73%).
MC-ESI: m/z 564,5 [1/2M+H]+.
Стадия 6
Соединение 3-L00-2 (0,390 г, 0,346 ммоль) и РуВОР (0,216 г, 0,415 ммоль) растворяли в безводном N,N-диметилформамиде (5,4 мл) в атмосфере азота. По каплям добавляли раствор дендримера 1-PEG2K (0,535 г, 6,92 мкмоль, синтезированный в соответствии с Journal of Controlled Release, 2011, 152, 241-248) и NMM (N-метилморфолин, 0,139 г, 1,38 ммоль) в безводном N,N-диметилформамиде (5,4 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом (11 мл) и затем очищали ультрафильтрацией с ацетонитрилом (0,55 л, 25 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart) с получением неочищенного продукта (0,625 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-L00 (0,62 г, выход: 89%).
1Н ЯМР показала 24 молекулы лекарственного средства/дендример. Фактическая молекулярная масса составляла около 100,2 кДа (23,22 мас. % соединения 3).
1Н ЯМР (400 МГц, CD3OD) δ 6,40-8,51 (m, 498Н), 4,19-4,68 (m, 111Н), 3,40-4,10 (m, 5900Н), 3,33-3,36 (s, 106H), 2,79-3,20 (m, 112H), 2,22-2,74 (m, 246H), 0,61-1,92 (m, 643H).
Пример 10: Получение Соединения 3-M00
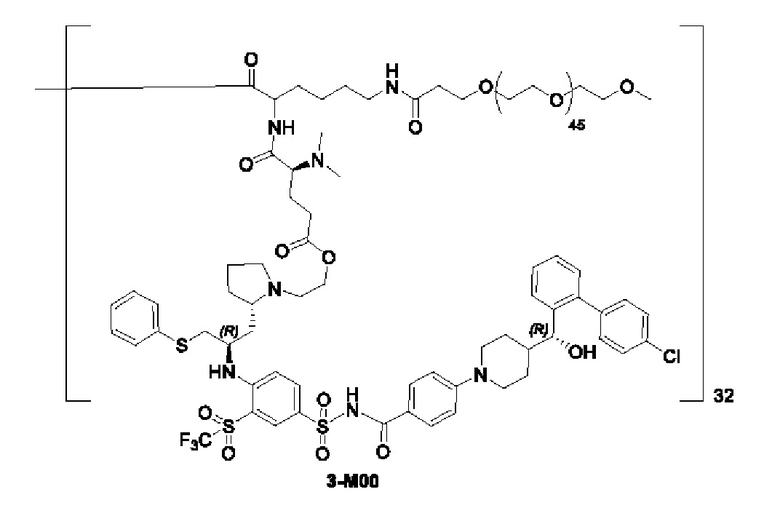
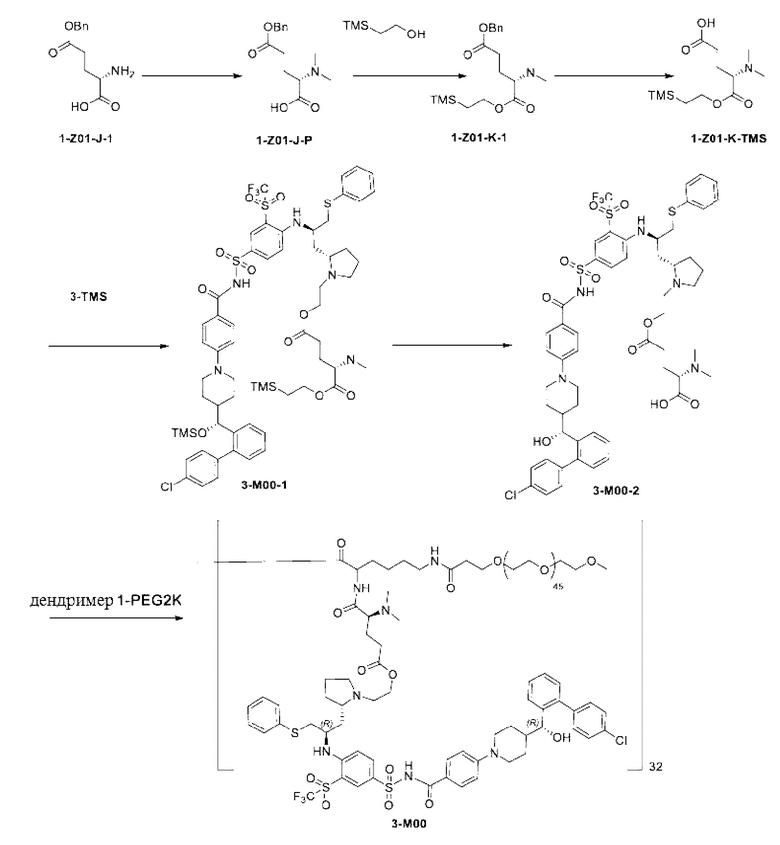
Стадия 1
Соединение 1-Z01-J-1 (7 г, 29,5 ммоль) растворяли в метаноле (150 мл). Добавляли водный раствор формальдегида (15 мл, 40 мас. %) и NaBH(OAc)3 (63 г, 295 ммоль). Смесь оставляли реагировать при комнатной температуре в течение 1 ч. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-J-P (5,9 г, выход: 75%).
MC-ESI: m/z 266,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСО-d6) δ 7,36-7,32 (m, 5Н), 5,09 (s, 2Н), 3,12 (t, J=7,2 Гц, 1Н), 2,5-2,44 (m, 2Н), 2,38 (s, 6Н), 1,87-1,84 (m, 2Н).
Стадия 2
Соединение 1-Z01-J-P (4,2 г, 15,8 ммоль), триметилсилилэтанол (2,24 г, 19 ммоль), DMАР (0,193 г, 1,58 ммоль) и N,N-диизопропилэтиламин (6,1 г, 47,5 ммоль) растворяли в дихлорметане (42 мл). Раствор охлаждали до 0°С. Добавляли HATU (7,2 г, 19 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. Смесь гасили насыщенным раствором бикарбоната натрия (150 мл) и экстрагировали дихлорметаном (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-K-1 (4,9 г, выход: 86%).
MC-ESI: m/z 366,2 [М+Н]+.
Стадия 3
Соединение 1-Z01-K-1 (900 мг, 2,46 ммоль) растворяли в тетрагидрофуране (18 мл) в атмосфере азота. Добавляли Pd/C (180 мг, 10 мас. %). Систему продували три раза водородом. Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. Смесь фильтровали и концентрировали с получением соединения 1-Z01-K-TMS (670 мг, выход: 99%).
MC-ESI: m/z 276,1 [М+Н]+.
1Н ЯМР (400 МГц, СDCl3) δ 4,22 (dd, J=10,0 Гц, J=8,4 Гц, 2Н), 3,39 (t, J=7,2 Гц, 1Н), 2,65-2,58 (m, 1Н), 2,49 (s, 6Н), 2,47-2,39 (m, 1H), 2,02 (dd, J=12,8 Гц, J=6,8 Гц, 2Н), 1,04-1,00 (m, 2Н), 0,04 (s, 9Н).
Стадия 4
Соединение 3-TMS (800 мг, 0,767 ммоль), 1-Z01-K-TMS (316 мг, 0,767 ммоль) и DMAP (46 мг, 0,383 ммоль) растворяли в дихлорметане (16 мл) в атмосфере азота. Раствор охлаждали до 0°С. Добавляли DCC (237 мг, 1,15 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 18 ч. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 3-М00-1 (1,036 г, выход: 100%).
MC-ESI: m/z 650,8 [1/2М+Н]+.
Стадия 5
Соединение 3-М00-1 (1,0 г, 0,768 ммоль) растворяли в тетрагидрофуране (10 мл) в атмосфере азота. Раствор охлаждали до 0°С. Добавляли TBAF (7,7 мл, 7,68 ммоль, 1,0 моль/л в THF). Смесь оставляли реагировать при комнатной температуре в течение 1 ч. Смесь гасили водой (30 мл) и экстрагировали этилацетатом (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-М00-2 (670 мг, выход: 77%).
MC-ESI: m/z 1128,3 [М+Н]+.
Стадия 6
Соединение 3-М00-2 (0,400 г, 0,355 ммоль) и РуВОР (0,222 г, 0,426 ммоль) растворяли в безводном N,N-диметилформамиде (5,6 мл) в атмосфере азота. По каплям добавляли раствор дендримера 1-PEG2K (0,548 г, 7,10 мкмоль) и NMM (0,144 г, 1,42 ммоль) в безводном N,N-диметилформамиде (5,6 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом (11 мл) и затем очищали ультрафильтрацией с ацетонитрилом (0,55 л, 25 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,614 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-М00 (0,61 г, выход: 88%).
1Н ЯМР показала 22 молекулы лекарственного средства/дендример. Фактическая молекулярная масса составляла около 98,0 кДа (21,77 мас. % соединения 3).
1H ЯМР (400 МГц, CD3OD) δ 6,40-8,52 (m, 440Н), 4,13-4,70 (m, 137Н), 3,40-4,10 (m, 5900H), 3,33-3,36 (s, 109H), 2,79-3,25 (m, 99H), 2,22-2,74 (m, 246H), 0,61-1,92 (m, 622H).
Пример 11: Получение Соединения 3-N00
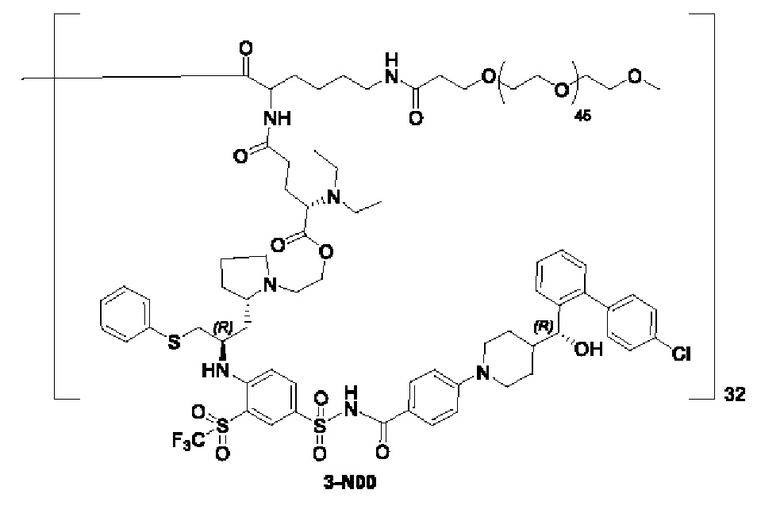
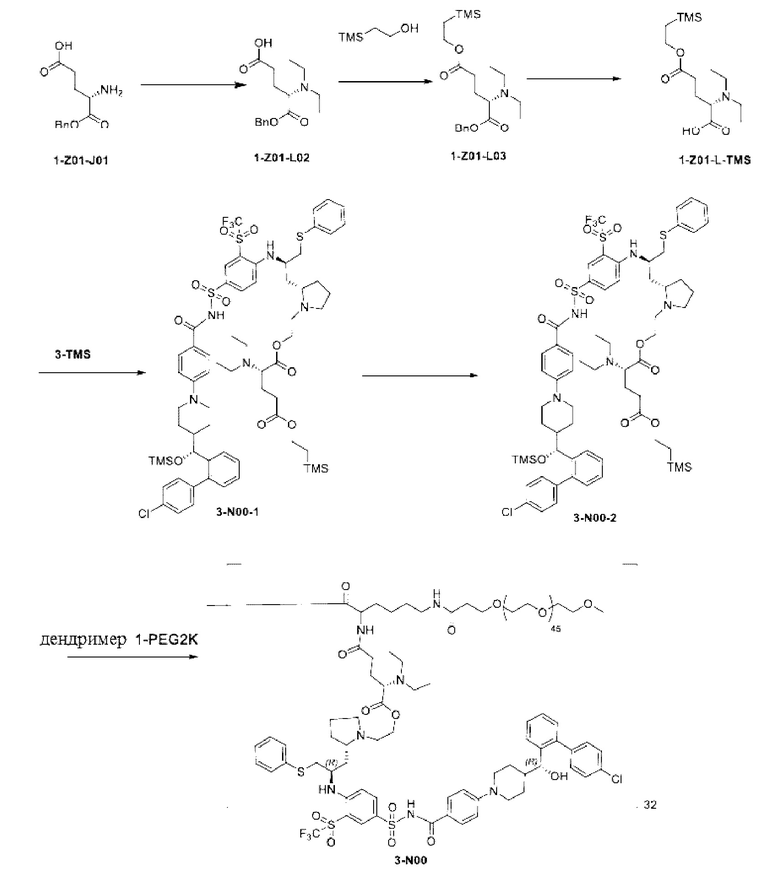
Стадия 1
Соединение 1-Z01-J01 (10,0 г, 36,6 ммоль) растворяли в метаноле (200 мл). Раствор охлаждали до 0°С. Добавляли водный раствор ацетальдегида (20 мл, 40 мас. %), а затем NaBH(OAc)3 (40,0 г, 183,2 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-L02 (5,4 г, выход: 50,4%).
MC-ESI: m/z 294,1 [М+Н]+.
Стадия 2
Соединение 1-Z01-L02 (500 мг, 1,7 ммоль), триметилсилилэтанол (404 мг, 3,4 ммоль), DMАР (104 мг, 0,85 ммоль) и N,N-диизопропилэтиламин (658 мг, 5,1 ммоль) растворяли в безводном тетрагидрофуране (10 мл). Раствор охлаждали до 0°С. Добавляли HATU (840 мг, 2,2 ммоль). Смесь оставляли реагировать при комнатной температуре в течение 16 ч. Смесь гасили водой (20 мл) и экстрагировали этилацетатом (20 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-L03 (476 мг, выход: 71%).
MC-ESI: m/z 394,2 [М+Н]+.
Стадия 3
Соединение 1-Z01-L03 (476 мг, 1,2 ммоль) растворяли в безводном тетрагидрофуране (10 мл). Добавляли Pd/C (50 мг, 10 мас. %). Систему продували три раза водородом. Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. Смесь фильтровали и концентрировали с получением соединения 1-Z01-L-TMS (366 мг, выход: 100%).
MC-ESI: m/z 304,2 [М+Н]+.
1Н ЯМР (400 МГц, CD3OD) δ 4,14-4,18 (m, 2Н), 3,58-3,60 (m, 1Н), 3,26-3,30 (m, 2Н), 3,06-3,13 (m, 2Н), 2,66-2,77 (m, 2Н), 2,00-2,06 (m, 2Н), 1,33-1,38 (m, 6Н), 0,94-0,99 (m, 2Н), 0,04 (s, 9Н).
Стадия 4
Соединение 1-Z01-L-TMS (366 мг, 1,21 ммоль), 3-TMS (800 мг, 0,767 ммоль) и DMAP (47,0 мг, 0,38 ммоль) растворяли в безводном дихлорметане (25 мл). Добавляли DCC (246,0 мг, 1,2 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 3-N00-1 (1,0 г, выход: 98%).
MC-ESI: m/z 664,8 [1/2М+Н]+.
Стадия 5
Соединение 3-N00-1 (1,0 г, 0,75 ммоль) растворяли в безводном тетрагидрофуране (10 мл). Добавляли TBAF (7,5 мл, 7,5 ммоль, 1,0 моль/л в THF). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Смесь гасили водой (20 мл) и экстрагировали этилацетатом (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-N00-2 (740 мг, выход: 85%).
MC-ESI: m/z 578,7 [1/2М+Н]+.
Стадия 6
Соединение 3-N00-2 (0,400 г, 0,346 ммоль) и РуВОР (0,215 г, 0,414 ммоль) растворяли в безводном N,N-диметилформамиде (5,6 мл) в атмосфере азота. По каплям добавляли раствор соединения дендримера 1-PEG2K (0,535 г, 6,90 мкмоль) и NMM (0,139 г, 1,38 ммоль) в безводном N,N-диметилформамиде (5,6 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом (11 мл) и затем очищали ультрафильтрацией с ацетонитрилом (0,56 л, 25 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart) с получением неочищенного продукта (0,631 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-N00 (0,625 г, выход: 91%).
1Н ЯМР показала 23 молекулы лекарственного средства/дендример. Фактическая молекулярная масса составляла около 99,8 кДа (22,36 мас. % соединения 3).
1H ЯМР (400 МГц, CD3OD) δ 6,40-8,52 (m, 477Н), 4,19-4,68 (m, 184Н), 3,40-4,10 (m, 5900H), 3,33-3,36 (s, 108H), 2,79-3,20 (m, 101H), 2,22-2,74 (m, 158H), 0,61-1,92 (m, 726H).
Пример 12: Получение Соединения 3-000
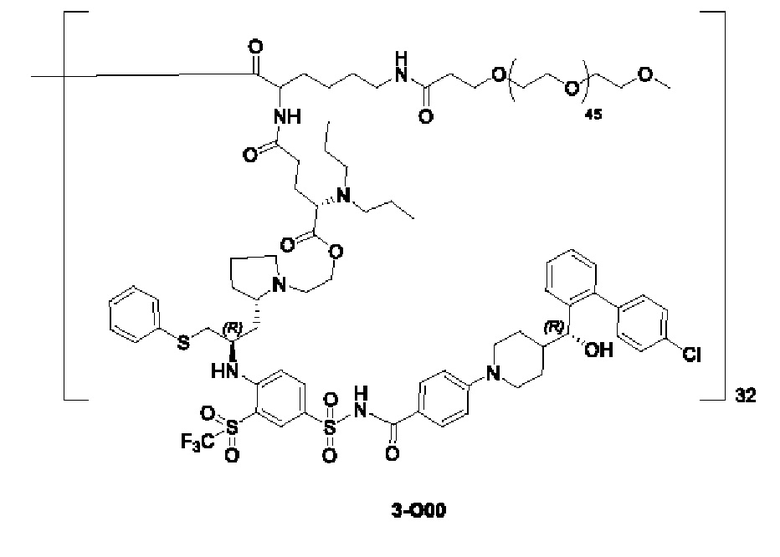
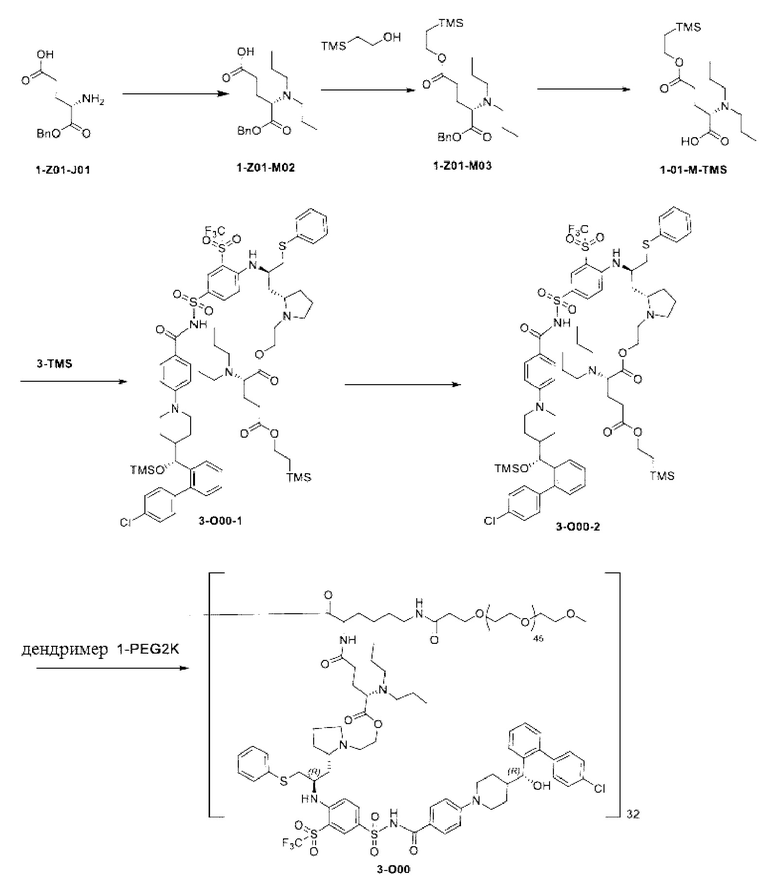
Стадия 1
Соединение 1-Z01-J01 (5,0 г, 21,1 ммоль) растворяли в метаноле (100 мл). Раствор охлаждали до 0°С. Добавляли пропиональдегид (6,1 г, 105,0 ммоль) и NаВН(ОАс)3 (31,1 г, 105,0 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-M02 (5,0 г, выход: 74%).
MC-ESI: m/z 322,1 [М+Н]+.
Стадия 2
Соединение 1-Z01-M02 (890 мг, 2,8 ммоль), триметилсилилэтанол (393,3 мг, 3,3 ммоль), DMAP (33,8 мг, 0,3 ммоль) и N,N-диизопропилэтиламин (1,1 г, 9,3 ммоль) растворяли в безводном дихлорметане (20 мл). Раствор охлаждали до 0°С. Добавляли HATU (1,3 г, 3,3 ммоль). Смесь оставляли реагировать при комнатной температуре в течение 16 ч. Смесь гасили водой (40 мл) и экстрагировали этилацетатом (20 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-M03 (1 г, выход: 86%).
MC-ESI: m/z 422,2 [М+Н]+.
Стадия 3
Соединение 1-Z01-M03 (1 г, 2,4 ммоль) растворяли в безводном тетрагидрофуране (30 мл). Добавляли Pd/C (200 мг, 10 мас. %). Систему продували три раза водородом. Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. Смесь фильтровали и концентрировали с получением соединения 1-Z01-M-TMS (766 мг, выход: 100%).
MC-ESI: m/z 332,2 [М+Н]+.
1H ЯМР (400 МГц,СDCl3) δ 4,10-4,14 (m, 2Н), 3,50-3,53 (m, 1Н), 2,84-3,04 (m, 4Н), 2,63-2,69 (m, 2Н), 1,95-2,01 (m, 2Н), 1,69-1,77 (m, 4Н), 0,90-0,96 (m, 8Н), 0,04 (s, 9Н).
Стадия 4
Соединение 1-Z01-M-TMS (381,7 мг, 1,2 ммоль), 3-TMS (800 мг, 0,767 ммоль) и DMAP (46,08 мг, 0,38 ммоль) растворяли в безводном дихлорметане (20 мл). Добавляли DCC (237,0 мг, 1,2 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 3-О00-1 (890 мг, выход: 86%).
MC-ESI: m/z 678,8 [1/2М+Н]+.
Стадия 5
Соединение 3-О00-1 (890 мг, 0,66 ммоль) растворяли в безводном тетрагидрофуране (10 мл). Добавляли TBAF (6,6 мл, 6,6 ммоль, 1,0 моль/л в THF). Смесь оставляли реагировать при комнатной температуре в течение 3 ч. Смесь гасили водой (20 мл) и экстрагировали этилацетатом (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-О00-2 (810 мг, выход: 100%).
MC-ESI: m/z 592,8 [1/2М+Н]+.
Стадия 6
Соединение 3-О00-2 (0,409 г, 0,346 ммоль) и РуВОР (0,216 г, 0,415 ммоль) растворяли в безводном N,N-диметилформамиде (5 мл) в атмосфере азота. По каплям добавляли раствор дендримера 1-PEG2K (0,52 г, 6,8 мкмоль) и NMM (0,137 г, 1,36 ммоль) в безводном N,N-диметилформамиде (5 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом (10 мл) и затем очищали ультрафильтрацией с ацетонитрилом (0,5 л, 25 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,605 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-О00 (0,60 г, выход: 87%).
1Н ЯМР показала 24 молекулы лекарственного средства/дендример. Фактическая молекулярная масса составляла около 101,5 кДа (22,92 мас. % соединения 3).
1Н ЯМР (400 МГц, CD3OD) δ 6,40-8,51 (m, 496Н), 4,19-4,68 (m, 126Н), 3,40-4,10 (m, 5900H), 3,33-3,36 (s, 100H), 2,79-3,20 (m, 136H), 2,22-2,74 (m, 199H), 0,61-1,92 (m, 683H).
Пример 13: Получение Соединения 3-R00
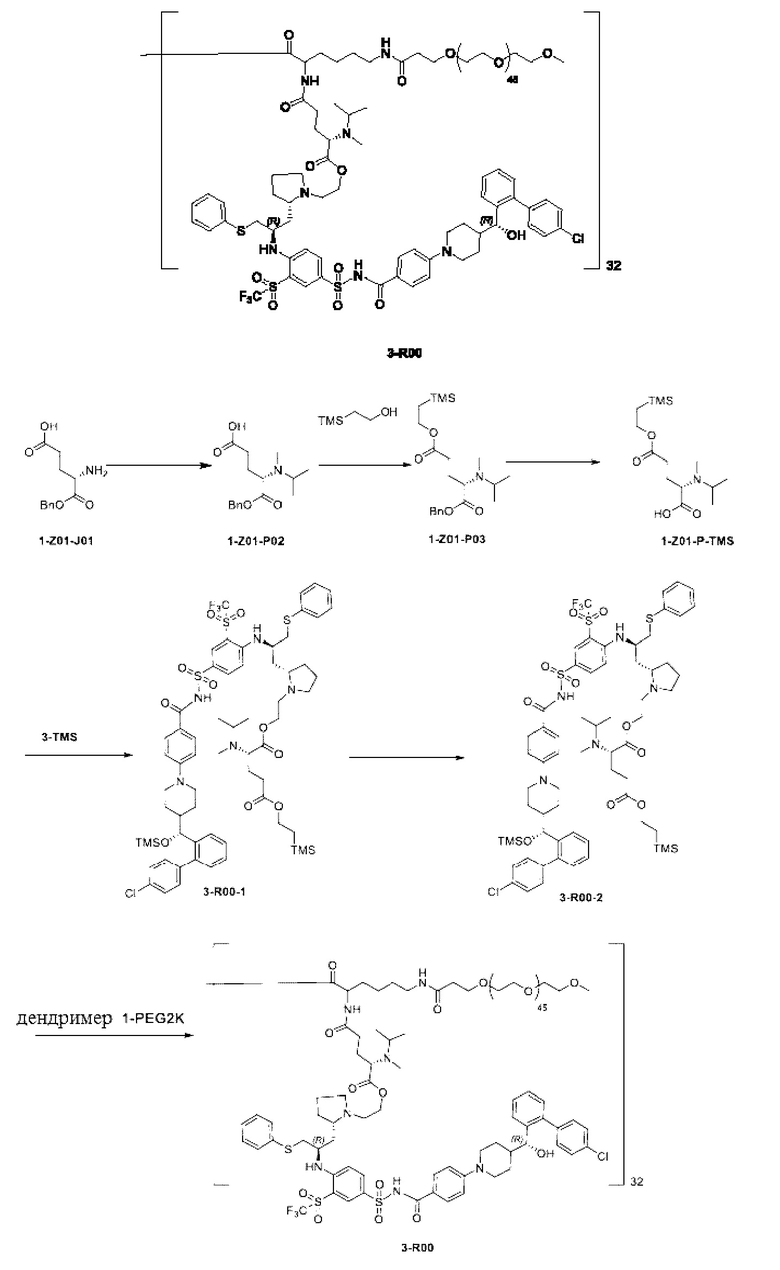
Стадия 1
Соединение 1-Z01-J01 (5,01 г, 18,3 ммоль) растворяли в метаноле (100 мл). Раствор охлаждали до 0°С. Добавляли ацетон (5,5 г, 94,7 ммоль) и NaBH(OAc)3 (11,9 г, 56,13 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 2 ч. Добавляли водный раствор формальдегида (14 мл, 40 мас. %) и NaBH(OAc)3 (8,26 г, 38,96 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-P02 (5 г, выход: 93%).
MC-ESI: m/z 294,1 [М+Н]+.
Стадия 2
Соединение 1-Z01-P02 (2,58 г, 8,8 ммоль), триметилсилилэтанол (2 г, 16,9 ммоль), DMAP (540 мг, 4,43 ммоль) и N,N-диизопропилэтиламин (2,98 г, 23,1 ммоль) растворяли в безводном тетрагидрофуране (20 мл). Добавляли HATU (3,38 г, 8,89 ммоль). Смесь оставляли реагировать при комнатной температуре в течение 17 ч. Смесь гасили водой (20 мл) и экстрагировали этилацетатом (20 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-P03 (3,09 г, выход: 89%).
MC-ESI: m/z 394,2 [М+Н]+.
Стадия 3
Соединение 1-Z01-P03 (3 г, 7,62 ммоль) растворяли в безводном тетрагидрофуране (30 мл). Добавляли Pd/C (152 мг, 10 мас. %). Систему продували три раза водородом. Смесь подвергали взаимодействию при комнатной температуре в течение 15 ч. После фильтрации и концентрирования получали 1-Z01-P-TMS (неочищенный, 2,32 г). Неочищенный продукт растирали в порошок с метил-трет-бутиловым эфиром (20 мл) в течение 30 мин. Смесь фильтровали с получением соединения 1-Z01-P-TMS (0,92 г, выход: 39%).
MC-ESI: m/z 304,2 [М+Н]+.
1Н ЯМР (400 МГц, ДМСO-d6) δ 4,05-4,10 (m, 2Н), 3,26-3,30 (m, 1H), 3,12-3,15 (m, 1H), 3,35-3,43 (m, 2Н), 2,31 (s, 3Н), 1,75-1,85 (m, 2Н), 1,03-1,07 (m, 6Н), 0,89-0,94 (m, 2Н), 0,01 (s, 9Н).
Стадия 4
Соединение 1-Z01-P-TMS (379 мг, 1,25 ммоль), 3-TMS (805 мг, 0,772 ммоль) и DMАР (54,0 мг, 0,44 ммоль) растворяли в безводном дихлорметане (16 мл). Добавляли DCC (239,0 мг, 1,16 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 16 ч. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 3-R00-1 (863 мг, выход: 84%).
MC-ESI: m/z 664,8 [1/2M+H]+.
Стадия 5
Соединение 3-R00-1 (847 мг, 0,64 ммоль) растворяли в безводном тетрагидрофуране (6 мл). Добавляли TBAF (6 мл, 6 ммоль, 1,0 моль/л в THF). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Смесь гасили водой (20 мл) и экстрагировали этилацетатом (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-R00-2 (702 мг, выход: 95%).
MC-ESI: m/z 578,8 [1/2М+Н]+.
Стадия 6
Соединение 3-R00-2 (0,299 г, 0,259 ммоль) и РуВОР (0,162 г, 0,311 ммоль) растворяли в безводном N,N-диметилформамиде (4 мл) в атмосфере азота. По каплям добавляли раствор соединения дендримера 1-PEG2K (0,400 г, 5,177 мкмоль) и NMM (0,209 г, 2,071 ммоль) в безводном N,N-диметилформамиде (4 мл). Смесь нагревали до 38°С и подвергали взаимодействию в течение 3 ч. Реакционную смесь разбавляли ацетонитрилом и затем очищали ультрафильтрацией с ацетонитрилом (0,4 л, 13 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart®) с получением неочищенного продукта (0,470 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-R00 (0,465 г, выход: 88%).
1Н ЯМР показала 25 молекул лекарственного средства/дендример. Фактическая молекулярная масса составляла около 102,0 кДа (23,76 мас. % соединения 3).
1H ЯМР (400 МГц, CD3OD) δ 6,40-8,51 (m, 514Н), 4,13-4,72 (m, 123Н), 3,40-4,10 (m, 5900Н), 3,33-3,36 (s, 93Н), 2,79-3,25 (m, 73Н), 2,22-2,74 (m, 206Н), 0,61-1,92 (m, 810Н).
Пример 14: Получение Соединения 3-S00
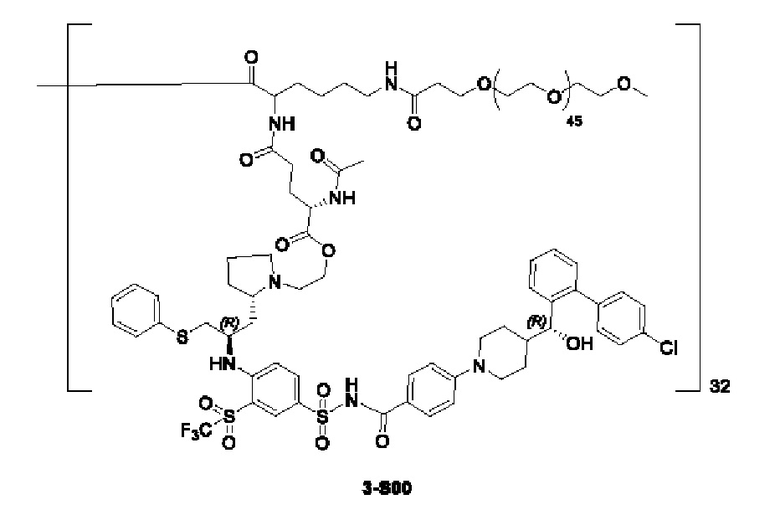
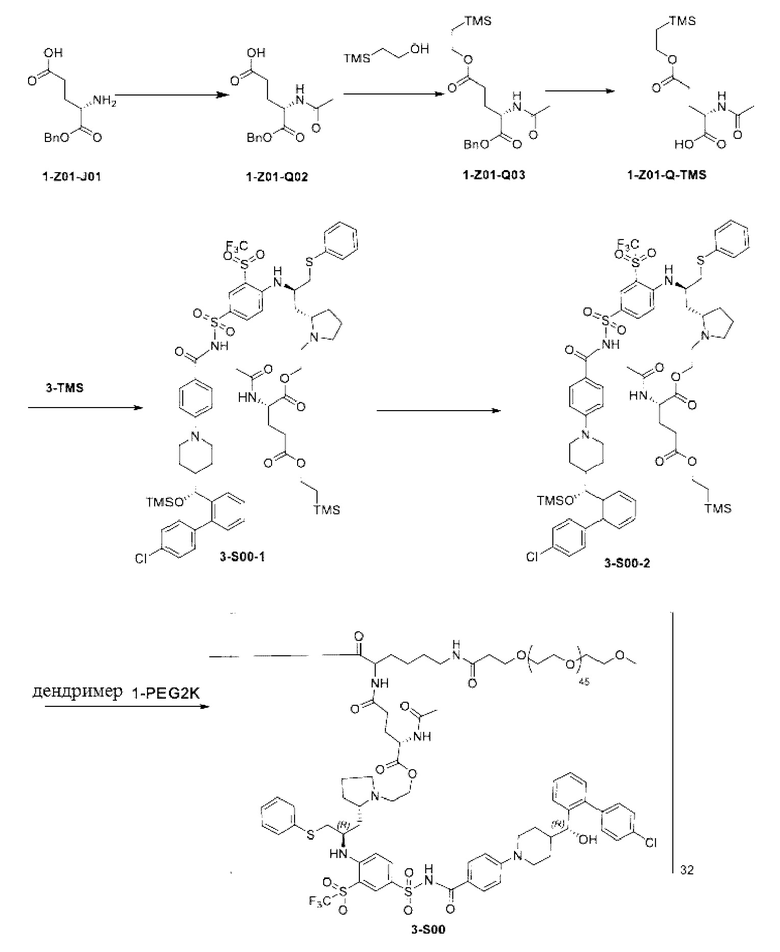
Стадия 1
Соединение 1-Z01-J01 (5 г, 18,3 ммоль) растворяли в дихлорметане (50 мл) в атмосфере азота. Добавляли триэтиламин (5,5 г, 54,9 ммоль). Смесь охлаждали до 0°С. По каплям добавляли уксусный ангидрид (2 г, 18,7 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. Смесь промывали водой (20 мл × 2). Водные фазы объединяли, доводили до рН 1 концентрированной соляной кислотой и экстрагировали этилацетатом (50 мл × 3). После концентрирования получали соединение 1-Z01-Q-02 (4,7 г, выход: 91%).
MC-ESI: m/z 280,2 [М+Н]+.
Стадия 2
Соединение 1-Z01-Q-02 (4,5 г, 16,1 ммоль) и триметилсилилэтанол (5,7 г, 48,3 ммоль) растворяли в дихлорметане (5 мл) в атмосфере азота. Раствор охлаждали до 0°С. Добавляли тионилхлорид (1,9 г, 16,1 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. После концентрирования остаток очищали колоночной хроматографией с получением соединения 1-Z01-Q-03 (1,6 г, выход: 26%).
MC-ESI: m/z 380,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 7,39-7,28 (m, 5Н), 6,20 (d, J=7,6 Гц, 1H), 5,14 (s, 2Н), 4,61-4,63 (m, 1Н), 4,19-4,04 (m, 2Н), 2,21-2,26 (m, 3Н), 2,02-1,94 (m, 4Н), 1,02-0,90 (m, 2Н), 0,05-0,02 (m, 9Н).
Стадия 3
Соединение 1-Z01-Q-03 (0,9 г, 2,3 ммоль) растворяли в тетрагидрофуране (10 мл). Добавляли влажный Pd/C (180 мг, 10 мас. %). Систему продували три раза водородом. Смесь оставляли реагировать при комнатной температуре в течение ночи. Смесь фильтровали и концентрировали с получением соединения 1-Z01-Q-TMS (621 мг, выход: 93%).
MC-ESI: m/z 290,3 [М+Н]+.
1Н ЯМР (400 МГц, CDCl3) δ 6,75 (d, J=6,9 Гц, 1H), 4,54-4,42 (m, 1H), 4,22-4,07 (m, 2Н), 2,56-2,32 (m, 2Н), 2,24-2,12 (m, 1H), 2,09-1,90 (m, 4Н), 1,03-0,90 (m, 2Н), 0,00 (s, 9Н).
Стадия 4
Соединение 1-Z01-Q-TMS (400 мг, 1,3 ммоль) растворяли в дихлорметане (20 мл) в атмосфере азота. Добавляли DMAP (56 мг, 0,46 ммоль), 3-TMS (962 мг, 0,92 ммоль) и DCC (285 мг, 1,3 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. Добавляли дополнительное количество 1-Z01-Q-TMS (150 мг, 0,5 ммоль) и DCC (143 мг, 0,6 ммоль). Смесь оставляли реагировать при комнатной температуре в течение ночи. После фильтрации и концентрирования остаток очищали колоночной хроматографией с получением соединения 3-S00-1 (560 мг, выход: 33%).
MC-ESI: m/z 1314,3 [М+Н]+.
Стадия 5
Соединение 3-S00-1 (560 мг, 0,426 ммоль) растворяли в тетрагидрофуране (8 мл) в атмосфере азота. Добавляли TBAF (4,3 мл, 4,3 ммоль, 1,0 моль/л в THF). Смесь оставляли реагировать в течение 2 ч. Смесь гасили водой (10 мл) и экстрагировали этилацетатом (50 мл × 3). После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-S00-2 (420 мг, выход: 86%).
MC-ESI: m/z 1142,3 [М+Н]+.
Стадия 6
Соединение 3-S00-2 (0,074 г, 0,065 ммоль) и РуВОР (0,041 г, 0,078 ммоль) растворяли в безводном N,N-диметилформамиде (1 мл) в атмосфере азота. По каплям добавляли раствор дендримера 1-PEG2K (0,100 г, 1,30 мкмоль) и NMM (0,026 г, 0,258 ммоль) в безводном N,N-диметилформамиде (1 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом, затем очищали ультрафильтрацией с ацетонитрилом (0,3 л, 25 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart®) и концентрировали с получением неочищенного продукта (0,124 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-S00 (0,12 г, выход: 87%).
1Н ЯМР показала 30 молекул лекарственного средства/дендример. Фактическая молекулярная масса составлила около 107,3 кДа (27,12 мас. % соединения 3).
1Н ЯМР (400 МГц, CD3OD) δ 6,40-8,52 (m, 609Н), 4,19-4,68 (m, 184Н), 3,40-4,10 (m, 5900Н), 3,33-3,36 (s, 102Н), 2,79-3,20 (m, 102Н), 2,22-2,74 (m, 146Н), 0,61-1,92 (m, 767Н).
Пример 15: Получение Соединения 3-Т00
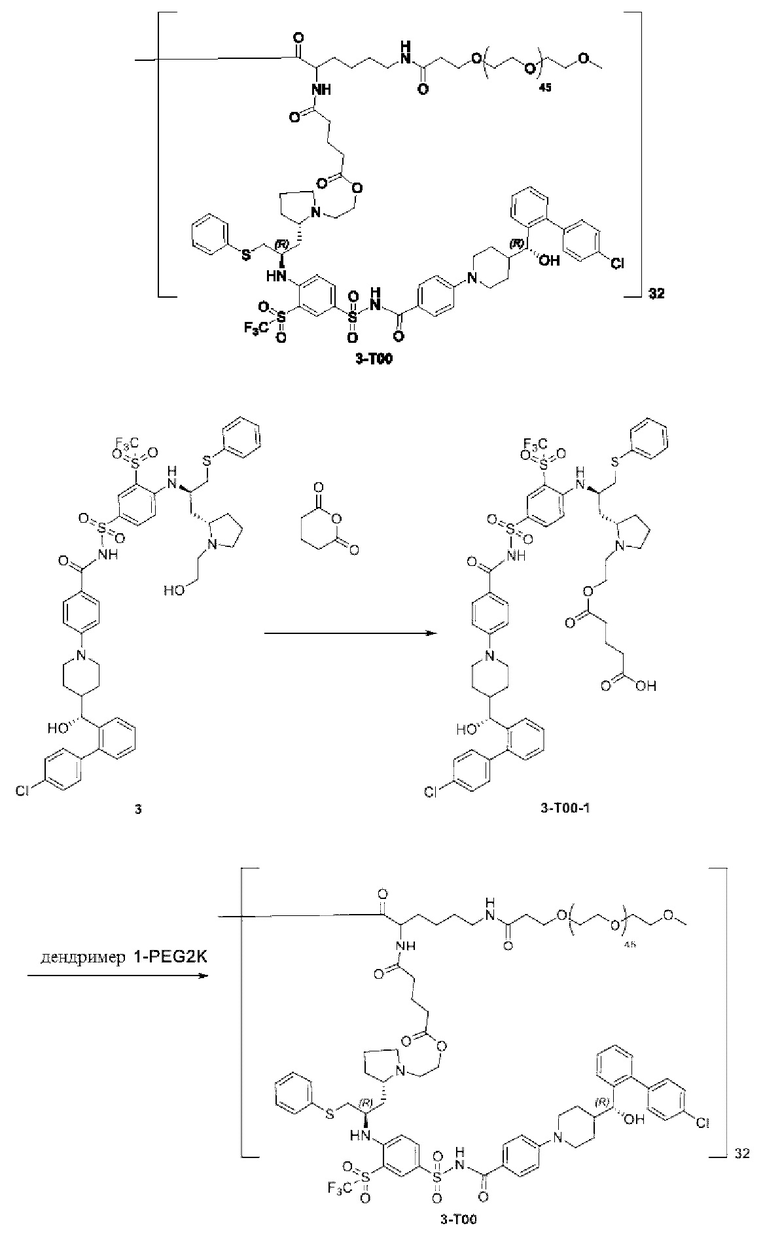
Стадия 1
Соединение 3 (1,0 г, 1,02 ммоль) растворяли в дихлорметане (20 мл) в атмосфере азота. По каплям добавляли раствор NMM (322 мг, 3,18 ммоль) и N,N-диизопропилэтиламина (89 мг, 0,69 ммоль) в дихлорметане (5 мл). Добавляли глутаровый ангидрид (141 мг, 1,23 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 20 ч. Добавляли глутаровый ангидрид (94 мг, 0,82 ммоль). Смесь подвергали взаимодействию при комнатной температуре в течение 3 ч. После концентрирования остаток очищали колоночной хроматографией с получением соединения 3-Т00-1 (1,030 г, выход: 92%).
MC-ESI: m/z 1085,2 [М+Н]+.
Стадия 2
Соединение 3-Т00-1 (0,352 г, 0,325 ммоль) и РуВОР (0,203 г, 0,390 ммоль) растворяли в безводном N,N-диметилформамиде (5 мл) в атмосфере азота. По каплям добавляли раствор дендримера 1-PEG2K (0,500 г, 6,50 мкмоль) и NMM (0,131 г, 1,300 ммоль) в безводном N,N-диметилформамиде (5 мл). Смесь оставляли реагировать при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли ацетонитрилом, затем очищали ультрафильтрацией с ацетонитрилом (0,5 л, 25 об.) в устройстве для ультрафильтрации (10 кДа, Hydrosart) и концентрировали с получением неочищенного продукта (0,585 г). Продукт растворяли в чистой воде. Раствор фильтровали через фильтрующую мембрану (0,22 мкм) и лиофилизировали с получением соединения 3-Т00 (0,580 г, выход: 84%).
1Н ЯМР показала 31 молекулу лекарственного средства/дендример. Фактическая молекулярная масса составляла около 106,6 кДа (28,19 мас. % соединения 3).
1Н ЯМР (400 МГц, CD3OD) δ 6,40-8,52 (m, 623Н), 4,19-4,67 (m, 173Н), 3,40-4,10 (m, 5900Н), 3,33-3,36 (s, 95Н), 2,75-3,10 (m, 73Н), 2,22-2,74 (m, 217Н), 0,61-1,92 (m, 613Н).
Биологическая оценка
Тестовый пример 1: Фармакокинетическое исследование введения крысам различных соединений путем однократных внутривенных инъекций 1.
Тестируемые образцы
Доцетаксел (G1) и соединения по настоящему изобретению 1-Z00 (G2), 1-Z00-J (G3), 1-Z00-K (G4), 1-Z00-M (G5) и 1-С00 (G6).
Получение образцов:
(1) Получение доцетаксела: коммерчески доступную инъекцию доцетаксела (0,5 мл:20 мг) разбавляли до 4 мг/мл физиологическим раствором;
(2) Получение других тестируемых соединений: соединения растворяли в ДМСО с конечной концентрацией 5%, и растворы разбавляли до 4 мг/мл физиологическим раствором.
2. Тестируемые животные
Самки крыс SD, весом 160-180 г, возрастом 6 недель, SPF (свободный от патогенной флоры)
3. Способ
Кровь собирали в разные моменты времени после внутривенного введения (доза: 10 мг/кг) (группа G1: 0 ч, 0,0833 ч, 0,5 ч, 1 ч, 4 ч, 8 ч и 24 ч; группы G2-6: 0 ч, 0,0833 ч, 1 ч, 4 ч, 8 ч, 24 ч, 48 ч, 96 ч и 168 ч) и антикоагулировали EDTAK2. Образцы плазмы отделяли и добавляли ингибитор эстеразы DDVP (диметилдихлорвинилфосфат). Образцы хранили при минус 80°С и тестировали на уровень доцетаксела (N=3).
4. Результаты
Как показано в таблице 2 ниже и на фиг.1, после внутривенного введения 10 мг/кг группы 1-Z00-J, группы 1-Z00-K и группы 1-Z00-M имели значительно лучшее значение AUCo-t, чем другие группы, и группа 1-Z00-J и группа 1-Z00-M имели более низкий плазменный клиренс, что указывает на лучший эффект с точки зрения замедленного высвобождения.
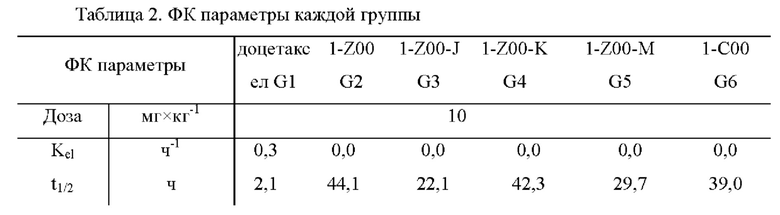
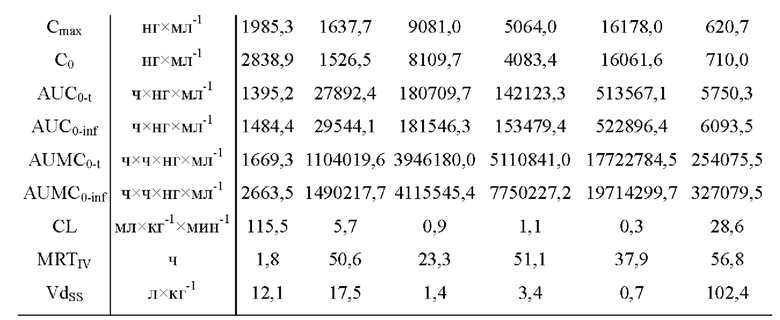
5. Заключение
По сравнению с обычной инъекцией доцетаксела, которая продемонстрировала быстрый плазменный клиренс и низкую экспозицию, 1-Z00-J, 1-Z00-K и 1-Z00-M все продемонстрировали значительный эффект с точки зрения замедленного высвобождения и более высокую экспозицию. По сравнению с 1-С00 и 1-Z00, соединения по настоящему изобретению имеют значительно улучшенную экспозицию лекарственного средства и более низкий клиренс лекарственного средства.
Тестовый пример 2: Фармакокинетическое исследование введения собакам породы бигль различных соединений путем однократных внутривенных инъекций
1. Тестируемые образцы
Соединения по настоящему изобретению 1-В00, 1-Z00 и 1-Z00-J и доцетаксел
Получение образцов:
1-В00, 1-Z00 и 1-Z00-J: к тестируемым соединениям (84 мг/флакон) добавляли соответствующий объем физиологического раствора для получения 0,6 мг/мл растворов для внутривенного введения.
Доцетаксел (84 мг/флакон, коммерчески доступный) растворяли в 5% (конечный объем) ДМСО+30% PEG300+5% твин 80+60% деионизированной воды с получением 0,6 мг/мл композиций для внутривенного введения.
2. Тестируемые животные
Собаки породы бигль, подвергавшиеся воздействию, полученные от Medicilon: 999М-004.
3. Способ
Способ введения: измеряли массу и рассчитывали дозировку на основе массы. Лекарственные средства медленно вводили путем внутривенной инъекции. Кровь собирали из яремной вены или с помощью других подходящих средств, около 1 мл на образец. Образцы плазмы собирали до введения и в разные моменты времени после введения и тестировали на уровень свободного доцетаксела в плазме. Для группы доцетаксела сбор проводили перед введением и через 0,083 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения. Для других групп тестируемых соединений сбор проводили перед введением и через 0,083 ч, 1 ч, 4 ч, 8 ч, 24 ч, 48 ч, 72 ч, 96 ч, 120 ч, 144 ч и 168 ч после введения.
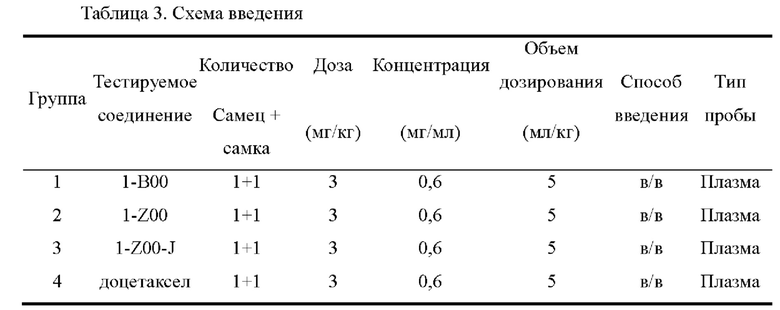
4. Результаты
Фармакокинетика для каждой группы показана в таблице 4 ниже и на фиг.2 и 3.
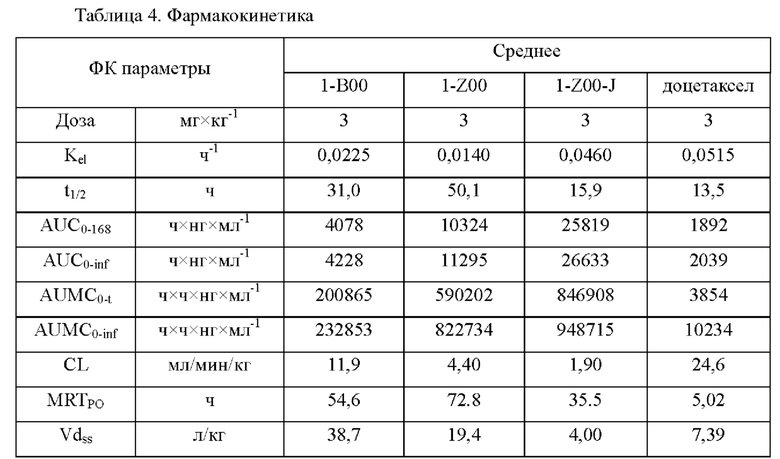
Как видно из результатов, 1-Z00-J имеет более высокую экспозицию in vivo и более низкий клиренс лекарственного средства, чем доцетаксел и 1-Z00.
Тестовый пример 3: Фармакодинамика и эффективность лекарственного средства в сочетании с гематологическими оценками модели подкожной ксенотрансплантатной опухоли клетками рака легкого человека А549 у голых мышей BALB/C
1. Тестируемые образцы
Доцетаксел, 1-Z00 и 1-Z00-J
Образцы готовят в соответствии с таблицей 5 ниже.



2. Экспериментальные животные
Голые мыши BALB/C, самки, возраст 6-7 недель (возраст, когда была выполнена инокуляция опухолевых клеток), масса 16,5-22,6 г, приобретенные у GemPharmatech Co., Ltd., сертификат №320727210100243463. Условия содержания: SPF.
3. Способ
3.1. Культура клеток
Клетки А549 (National Collection of Authenticated Cell Cultures/Cell Bank of Shanghai Institutes for Biological Sciences (cellbank.org.cn, SIBS)) культивировали в среде F12K (Gibco), содержащей 10% фетальную бычью сыворотку. Клетки собирали в экспоненциальной фазе роста и ресуспендировали в PBS до подходящей концентрации для применения в подкожной инокуляции опухоли у мышей.
3.2. Моделирование и случайное группирование животных
5×106 клеток А549 подкожно инокулировали в правый бок самок мышей (объем инокуляции: 0,1 мл/мышь). День инокуляции был определен как день 0. Когда средний объем опухоли составил 142,96 мм3 (на день 13 после инокуляции клеток), мышей случайным образом группировали в соответствии с размером опухоли, как показано в таблице 6 ниже (объем дозирования: 10 мкл/г).

3.3. Наблюдение и сбор данных
После инокуляции опухоли параметры рутинного мониторинга включали влияние роста опухоли и лечения на нормальное поведение животных, включая активность, потребление пищи и воды, увеличение или потерю массы (массу тела измеряли два раза в неделю), аномалии в глазах, волосах и т.д. Клинические симптомы, наблюдаемые во время эксперимента, были зарегистрированы в необработанных данных. Формула расчета объема опухоли: объем опухоли (мм3)=1/2×(a×b2) (где а представляет собой длинный диаметр, и b представляет собой короткий диаметр). В эксперименте сбор данных, включая измерение длинного и короткого диаметров опухолей и массы тела животных, проводили с использованием программного обеспечения StudyDirectorTM (версия: 3.1.399.19; поставщик: Studylog System, Inc., Сан-Франциско, Калифорния, США). Необработанные данные, измеренные с помощью весов и штангенциркулей, были непосредственно импортированы в программное обеспечение. Любые изменения данных фиксировались в программном обеспечении.
3.4. Лечение потери массы у животных в ходе эксперимента
Когда масса тела отдельного животного уменьшалась более чем на 15% (BWL больше или равно 15%), прием лекарственного средства для отдельного животного/группы животных прекращали до тех пор, пока его/их потеря массы тела не возвращалась в пределы 15% (BWL меньше 15%).
Когда масса тела отдельного животного уменьшалась на более 20%, его подвергали эвтаназии согласно критериям гумманного обращения с животным.
3.5. Критерии оценки эффективности лекарственного средства Относительная степень пролиферации опухоли, Т/С (%), относится к процентному
значению относительного объема опухоли или массы опухоли для группы лечения и контрольной группы в определенный момент времени. Формула для расчета: T/C%=TRTV/CRTV×100% (TRTV: средний RTV для группы лечения; CRTV: средний RTV для контрольной группы; RTV=Vt/V0, V0 представляет собой объем опухоли животного на момент группирования, и Vt представляет собой объем опухоли животного после лечения).
Расчетная формула для относительного ингибирования опухоли, TGI (%), представляет собой TGI%=(1-Т/С)×100% (Т и С представляют собой относительный объем опухоли (RTV) или массу опухоли (TW) для группы лечения и контрольной группы, соответственно, в определенный момент времени).
3.6. Конечная точка
После последнего введения плазму собирали в моменты времени, указанные в таблице 7 (использовали пробирки с антикоагулянтом EDTA (на момент забора крови добавляли ингибитор эстеразы DDVP)). После того, как сбор плазмы в последний момент времени был завершен, мышей подвергали эвтаназии, и опухоли собирали, замораживали и хранили.

3.7 Статистический анализ
Все результаты выражены как средний объем опухоли ± SEM (стандартная ошибка среднего). Лучшую точку лечения лекарственным средством (обычно после последнего введения) выбирали для статистического анализа между различными группами. Двухфакторный ANOVA (дисперсионный анализ) использовали для определения того, существует ли значительная разница в объеме опухоли между группой лечения и контрольной группой, р менее 0,05 указывает на статистически значимую разницу.
Статистический анализ и построение графиков проводили в языковой среде R (версия 3.3.1). Все тестирования были двусторонними, если не указано иное. Значение р менее 0,05 считалось статистически значимым.
4. Результаты
4.1. Результаты тестирования противоопухолевого эффекта лекарственных средств в модели подкожной трансплантатной опухоли клетками рака легкого человека А549
У мышей в контрольной группе носителя средний объем опухоли составлял 1620,93 мм на 21 день после начала введения.
В группе доцетаксела в дозе 20 мг/кг средний объем опухоли составлял 564,93 мм3 на 21 день после начала введения, что статистически достоверно отличалось (р менее 0,001) от такого занчения в контрольной группе; относительное ингибирование опухоли TGI (%) составляло 66,24%.
Средний объем опухоли в группе 1-Z00 в дозе 20 мг/кг составлял 576,02 мм3 на 21 день после начала введения, что статистически достоверно отличалось (р менее 0,001) от такого значения в контрольной группе; относительное ингибирование опухоли TGI (%) составляло 64,85%.
В группе 1-Z00-J в дозе 10 мг/кг средний объем опухоли составлял 285,12 мм3 на 21 день после начала введения, что статистически достоверно отличалось (р менее 0,001) от такого значения в контрольной группе; относительное ингибирование опухоли TGI (%) составляло 83,02%.
Рост опухоли для каждой группы лечения и контрольной группы показан в Таблице 8 и Таблице 9 ниже и на фиг.4.


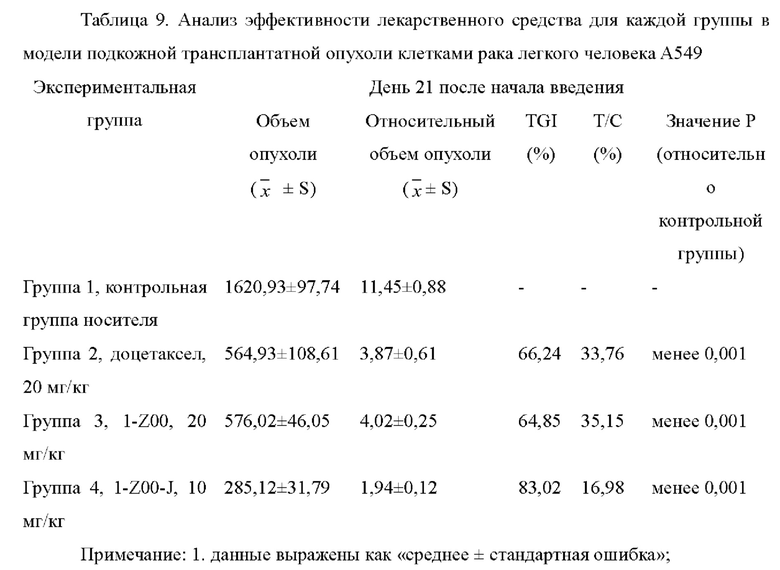
2. Т/С%=TRTV/CRTV×100%; TGI%=(1-Т/С)×100%.
3. Значение Р: полученное с помощью двухфакторного ANOVA для сравнения объема опухоли группы лечения с объемом опухоли контрольной группы.
4.2. Эффективность лекарственного средства в сочетании с результатами гематологического тестирования лекарственных средств в отношении модели подкожной трансплантатной опухоли клетками рака легкого человека А549
Как показано в таблице 10 ниже, лейкоциты, лимфоциты и мононуклеарные клетки в крови у группы мышей, которым внутривенно вводили 20 мг/кг доцетаксела, были снижены в 2-3 раза по сравнению с контрольной группой носителя, что указывает на значительную лимфатическую токсичность доцетаксела. Внутривенное введение 10 мг/кг 1-Z00-J было значительно более безопасным для лимфатической системы, чем внутривенное введение 20 мг/кг доцетаксела.

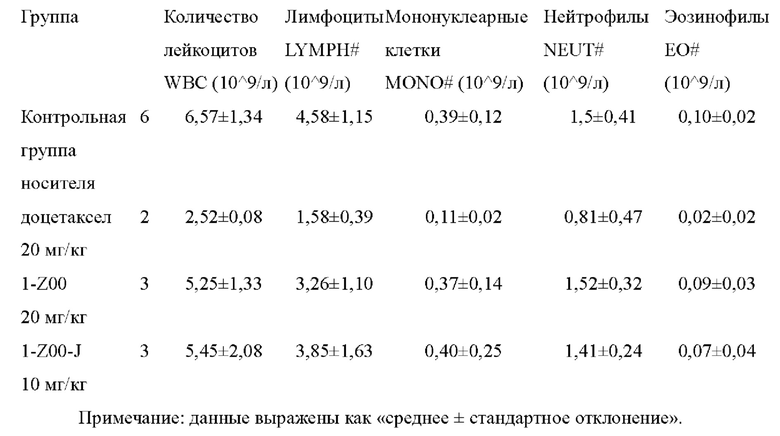
5. Заключение
1-Z00-J имел значительно лучший эффект, чем обычная инъекция доцетаксела и 1-Z00, когда он вводился в половине дозы. Между тем, молекула 1-Z00-J продемонстрировала значительно сниженную гематологическую токсичность, такую как снижение лимфоцитов и нейтрофилов, по сравнению с инъекцией доцетаксела.
Тестовый пример 4: Фармакокинетическое исследование введения собакам породы бигль различных соединений путем однократных внутривенных инъекций
1. Тестируемые образцы
Соединения по настоящему изобретению: 3-L00, 3-М00, 3-N00, 3-О00, 3-R00 и 3-Т00.
Получение образцов:
К тестируемым соединениям 3-L00, 3-М00, 3-N00, 3-000 и 3-R00 (80 мг/флакон) добавляли 5% (конечный объем) ДМСО и 95% физиологического раствора, и тестируемые соединения растворяли на вортексе с получением 1,5 мг/мл композиций для введения.
3-Т00: к тестируемому соединению (120 мг/флакон) добавляли 5% (конечный объем) ДМСО и 95% физиологического раствора, и тестируемое соединение растворяли на вортексе с получением 1,5 мг/мл композиций для введения.
2. Тестируемые животные
Собаки породы бигль, подвергавшиеся воздействию, полученные от Medicilon: 999М-004.
3. Способ
Способ введения: измеряли массу и рассчитывали дозу на основе массы; схема введения показана в таблице 11 ниже. Моменты времени отбора образцов крови представляли собой 0,083 ч, 1 ч, 4 ч, 8 ч, 24 ч, 48 ч, 72 ч и 96 ч после введения. Кровь собирали из яремной вены или другими подходящими средствами в определенные моменты времени после введения, около 1 мл на образец. Собранные образцы крови помещали в пробирки для сбора крови с антикоагулянтом EDTA-K2, и каждая группа образцов крови должна была быть специально обработана ингибитором эстеразы. В пробирки для сбора крови с антикоагулянтом EDTA-K2 добавляли 200 мМ DDVP в количестве 1/40 по объему собранной крови (если количество собранной крови составляло 1 мл, добавляли 25 мкл 200 мМ DDVP). Собранные образцы крови помещали на лед, и плазму отделяли центрифугированием в течение 1 ч (условия центрифугирования: 2200 g, 10 мин, 2-8°С). Затем определяли концентрацию лекарственного средства в плазме и производили фармакокинетический расчет. Образцы плазмы перед тестированием хранили в морозильной камере при температуре минус 70°С.
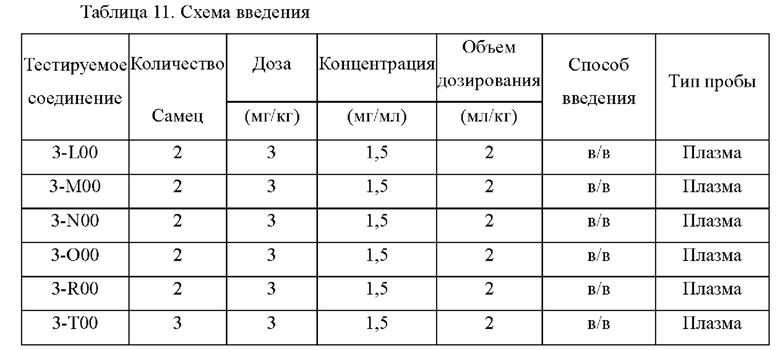
4. Результаты
Фармакокинетические (ФК) результаты для 3-L00, 3-М00, 3-N00, 3-О00, 3-R00 и 3-Т00 у собак породы бигль показаны на фиг.5 и в таблице 12.
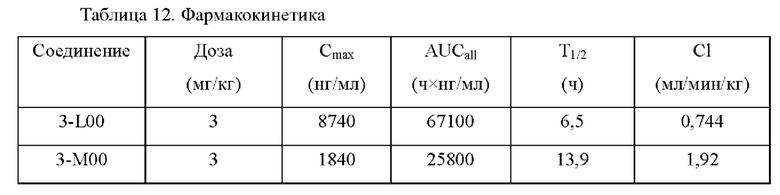
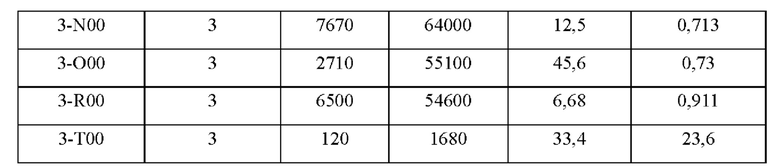
Результаты показывают, что дендримеры 3-L00, 3-М00, 3-N00, 3-О00 и 3-R00 по настоящему изобретению имели значительно более высокую экспозицию (AUC), чем 3-Т00, демонстрируя значительный эффект с точки зрения замедленного высвобождения. Дендримеры по настоящему изобретению объединяют алкильные линкеры, которые имеют азотные связи на боковых цепях через дендримеры. По сравнению с линкером с цельноуглеродной цепью указанные алкильные линкеры могут способствовать высвобождению малых молекул.
Тестовый пример 5: Влияние однократной внутривенной инъекции Соединения 3, 3-М00 и 3-О00 собакам породы бигль на количество тромбоцитов
1. Тестируемые образцы
Соединения по настоящему изобретению: соединение 3, 3-М00 и 3-О00. Получение образцов:
К тестируемому соединению, соединение 3 (120 мг/флакон), добавляли 5% (конечный объем) ДМСО, 5% твин 80 и 90% физиологического раствора, и тестируемое соединение растворяли на вортексе с получением 1,5 мг/мл композиций для введения.
К тестируемым соединениям 3-М00 и 3-О00 (80 мг/флакон) добавляли 5% (конечный объем) ДМСО и 95% физиологического раствора, и тестируемые соединения растворяли на вортексе с получением 1,5 мг/мл композиций для введения.
2. Тестируемые животные
Собаки породы бигль, подвергавшиеся воздействию, полученные от Medicilon: 999М-004.
3. Способ
Способ введения: измеряли массу тела и рассчитывали дозу на основании массы; схема введения показана в таблице 13 ниже. Лекарственные средства медленно вводили путем внутривенной инъекции. Кровь собирали из яремной вены или с помощью других подходящих средств, около 1 мл на образец. Цельную кровь собирали до введения и через 24 часа после введения и проводили гематологические тесты.
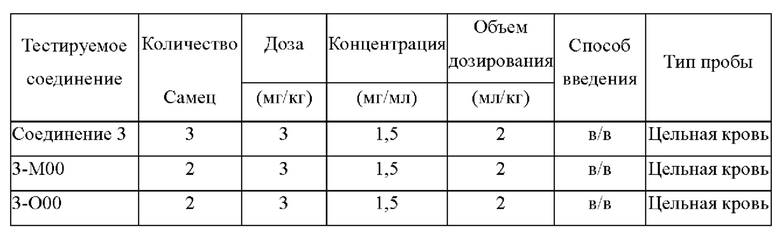
4. Результаты
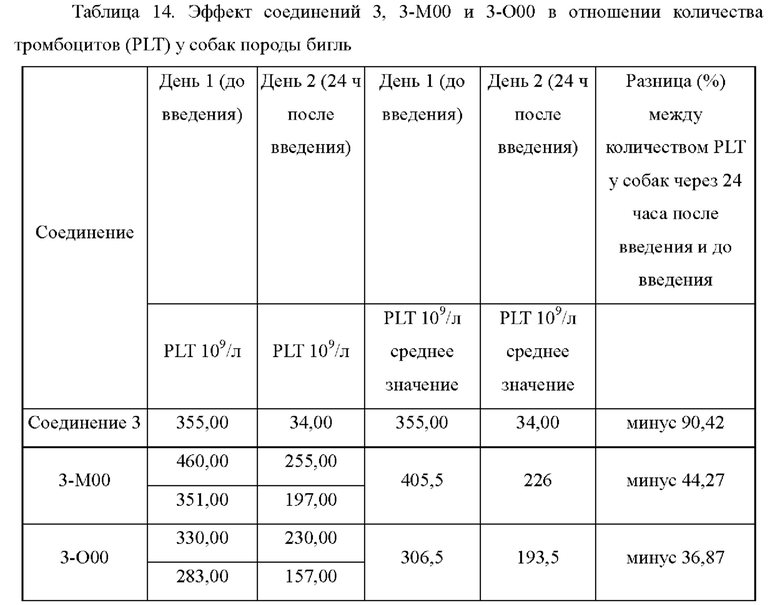
Результаты показывают, что дендримеры 3-М00 и 3-O00 по настоящему изобретению могут значительно уменьшать снижение количества тромбоцитов, на которые нацелено низкомолекулярное соединение 3. В этом эксперименте 3 собаки породы бигль в группе лечения низкомолекулярным соединением 3 характеризовались непереносимостью однократной внутривенной инъекции 3 мг/кг соединения 3, и 2 животных умерли. Однако собаки породы бигль в группах, получавших дендримеры 3-М00 и 3-О00, не погибли, что дополнительно указывает на то, что дендримеры по настоящему изобретению значительно улучшили безопасность низкомолекулярного соединения 3.
Тестовый пример 6: Терапевтический эффект 3-L00, 3-М00, 3-N00, 3-О00, 3-R00 и 3-Т00 на подкожно трансплантатные опухоли у мышей с острым лимфобластным лейкозом человека RS4; 11
1. Тестируемые образцы
3-L00, 3-М00, 3-N00, 3-О00, 3-R00 и 3-Т00.
Получение образцов:
3-L00, 3-М00, 3-N00, 3-О00, 3-R00 и 3-Т00: в каждый флакон добавляли 0,25 мл ДМСО для растворения соединений с получением 20 мг/мл рабочих маточных растворов, а затем добавляли 4,75 мл физиологического раствора; соединения растворяли на вортексе с получением 1 мг/мл композиций для введения.
2. Тестируемые клетки и животные
Клетки острого лимфобластного лейкоза человека RS4;11 были приобретены в American Type Culture Collection. Клетки RS4;11 культивировали в чашке Петри 10 см с культуральной средой RPMI 1640 (Gibco), содержащей 10% фетальную бычью сыворотку (Gibco) и пенициллин и стрептомицин, в инкубаторе при 37°С, содержащем 5% СО2. Клетки пересеевали два раза в неделю, а затем собирали, подсчитывали и инокулировали, когда они находились в экспоненциальной фазе роста.
Мыши NOD-Scid, возраст 5 недель, самки, приобретенные у Beijing Huafukang Biotechnology Co., Ltd. с лицензией на производство №: SCXK (Пекин) 2019-0008, и сертификатами №110322211100992176 и 110322211101069023. Условия содержания: SPF.
Использование и благополучие лабораторных животных осуществляли в соответствии с положениями Международной ассоциации по оценке и аккредитации содержания лабораторных животных (AAALAC). Здоровье и смерть животных контролировали ежедневно. Рутинные обследования включали наблюдение за влиянием тестируемых соединений и лекарственных средств на ежедневное поведение животных, такое как поведенческая активность, изменения массы и внешний вид.
3. Способ
Каждой мыши подкожно инокулировали 1х107 клеток RS4;11. Когда опухоли вырастали до 150-200 мм, мышей группировали в соответствии с объемом опухоли и внутривенно (IV) вводили лекарственные средства один раз в неделю (1 р/нед, QW), и в общей сложности вводили 4 дозы. Объем инъекции составлял 0,1 мл/10 г массы тела; дозы и схема введения приведены в таблице 15 ниже. Экспериментальный индекс позволял исследовать влияние лекарственного средства на рост опухоли, и специальный индекс представлял собой Т/С% или ингибирование роста опухоли (TGI%).
Диаметры опухоли измеряли два раза в неделю с помощью штангенциркуля, и объем опухоли (V) рассчитывали по следующей формуле:
V=1/2×а×b2, где а и b обозначают соответственно длину и ширину.
Т/С (%)=(Т-Т0)/(С-С0)×100,
где Т и С представляют собой объемы опухолей в конце эксперимента; Т0 и С0 представляют собой объемы опухолей в начале эксперимента.
% ингибирования роста опухоли (TGI%)=100 Т/С (%).

4. Результаты

Результаты показывают, что в модели подкожной трансплантатной опухоли мышей, несущих острый лимфобластный лейкоз человека RS4;11, 3-Т00 не удалось эффективно высвобождать низкомолекулярное соединение 3, не демонстрируя противоопухолевую активность, в то время как 3-L00, 3-М00, 3-N00, 3-О00 и 3-R00 имели линкеры, которые позволили эффективно способствовать высвобождению низкомолекулярного соединения 3, демонстрируя значительную противоопухолевую активность.
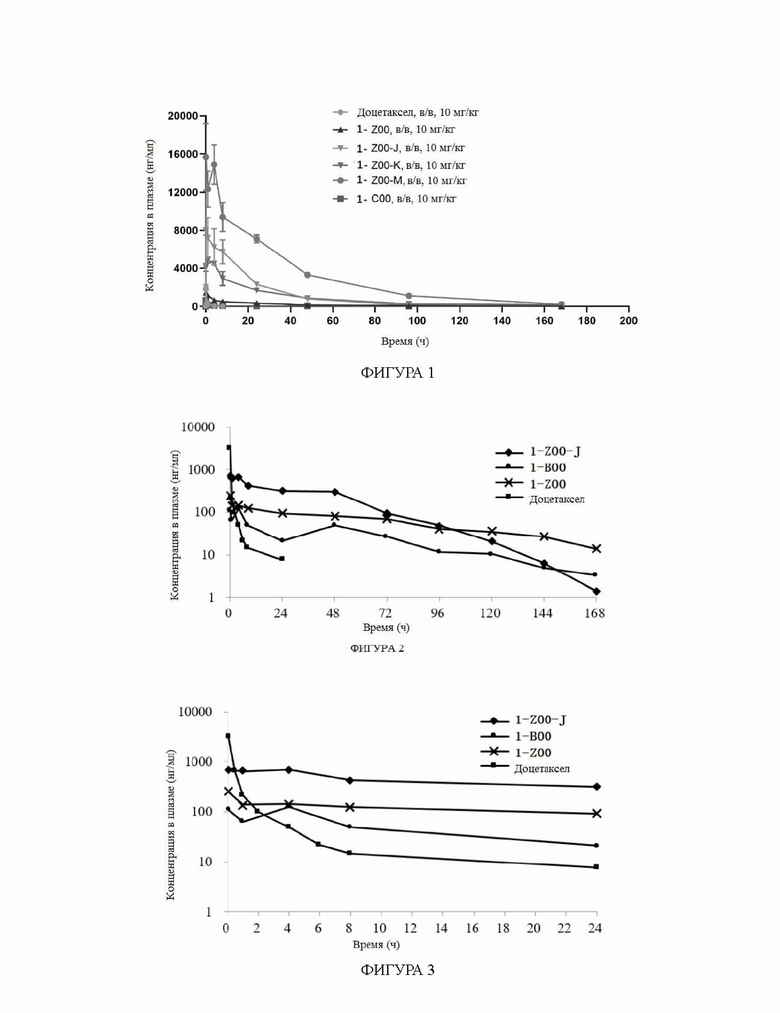
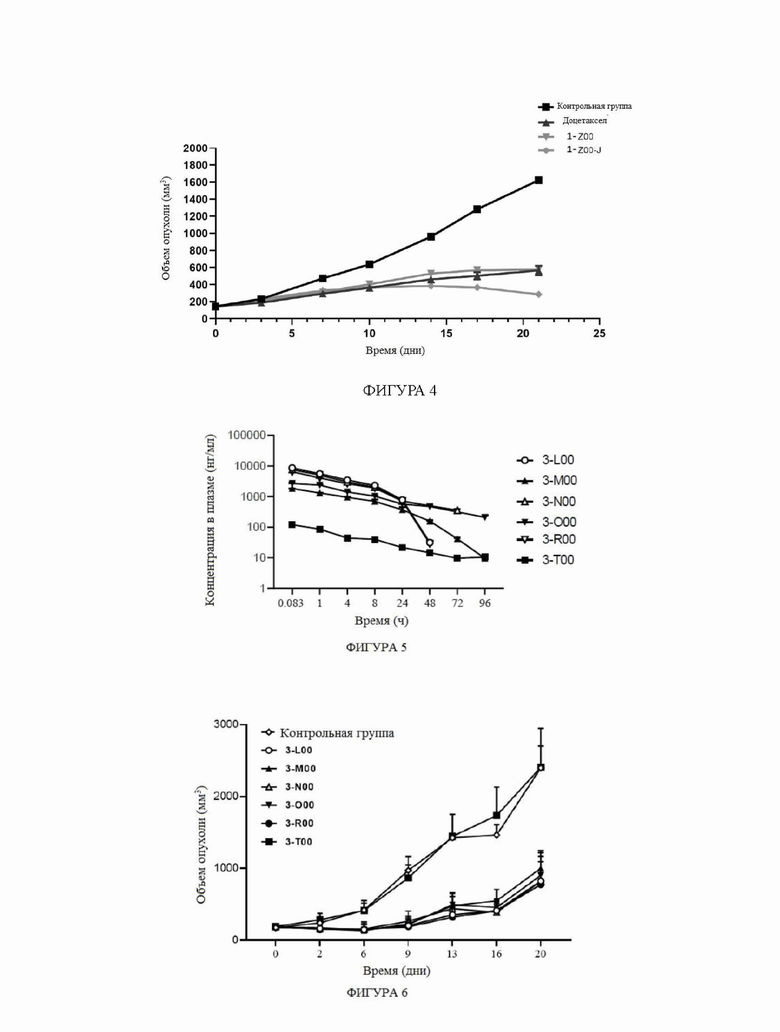
Группа изобретений относится к регулированию скорости высвобождения лекарственного средства. Макромолекула содержит дендример D, имеющий поверхностные аминогруппы, где по меньшей мере две разные концевые группы ковалентно связаны с поверхностными аминогруппами дендримера; первая концевая группа - фармацевтически активный агент; вторая концевая группа - фармакокинетический модификатор; первая концевая группа ковалентно связана с поверхностной аминогруппой дендримера посредством линкера -X1-L-X2-, где X1 представляет собой группу связывания линкера с фармацевтически активным агентом, X2 представляет собой группу связывания линкера с дендримером, X1 и X2 - -C(O)-, L - C3 алкилен, замещенный -NR1R2, где R1 и R2 независимо выбран из водорода, C1-6 алкила и C(O)R4, R4 - C1-6 алкил; причем дендример D представляет собой BHALys[Lys]32, фармакокинетический модификатор - полиэтиленгликоль, который имеет молекулярную массу в диапазоне от 1000 до 2300 Да, фармацевтически активный агент - доцетаксел или другие соединения. Также раскрыты фармацевтическая композиция и применение макромолекулы для получения лекарственного средства для лечения опухолей. Группа изобретений обеспечивает эффективное высвобождение, в частности замедленное высвобождение лекарственного средства. 3 н. и 1 з.п. ф-лы, 6 ил., 16 табл., 6 пр.
1. Макромолекула, содержащая:
i) дендример D, имеющий поверхностные аминогруппы, где по меньшей мере две разные концевые группы ковалентно связаны с поверхностными аминогруппами дендримера;
ii) первую концевую группу, которая представляет собой фармацевтически активный агент, содержащий гидрокси, амино или сульфгидрильную группу; и
iii) вторую концевую группу, которая представляет собой фармакокинетический модификатор;
где первая концевая группа ковалентно связана с поверхностной аминогруппой дендримера посредством линкера -X1-L-X2-, где X1 представляет собой группу связывания линкера с фармацевтически активным агентом, X2 представляет собой группу связывания линкера с дендримером и X1 и X2 оба представляют собой -C(O)-, L представляет собой C3 алкилен, замещенный -NR1R2, где каждый из R1 и R2 независимо выбран из группы, состоящей из водорода, C1-6 алкила и C(O)R4, и R4 представляет собой C1-6 алкил;
причем дендример D представляет собой BHALys[Lys]32, фармакокинетический модификатор представляет собой полиэтиленгликоль, который имеет молекулярную массу в диапазоне от 1000 до 2300 Да, фармацевтически активный агент представляет собой доцетаксел, соединение 2 или соединение 3:
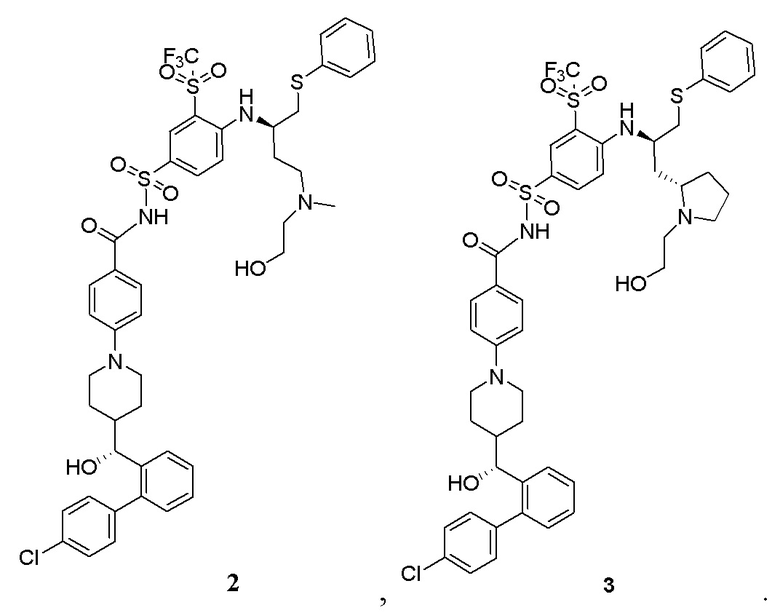
2. Макромолекула по п. 1, где в линкере -X1-L-X2- X1 представляет собой -C(O)- и связан с фармацевтически активным агентом, X2 представляет собой -C(O)- и связан с поверхностной аминогруппой дендримера D с образованием амидной связи; и макромолекула имеет структуру, как показано ниже:
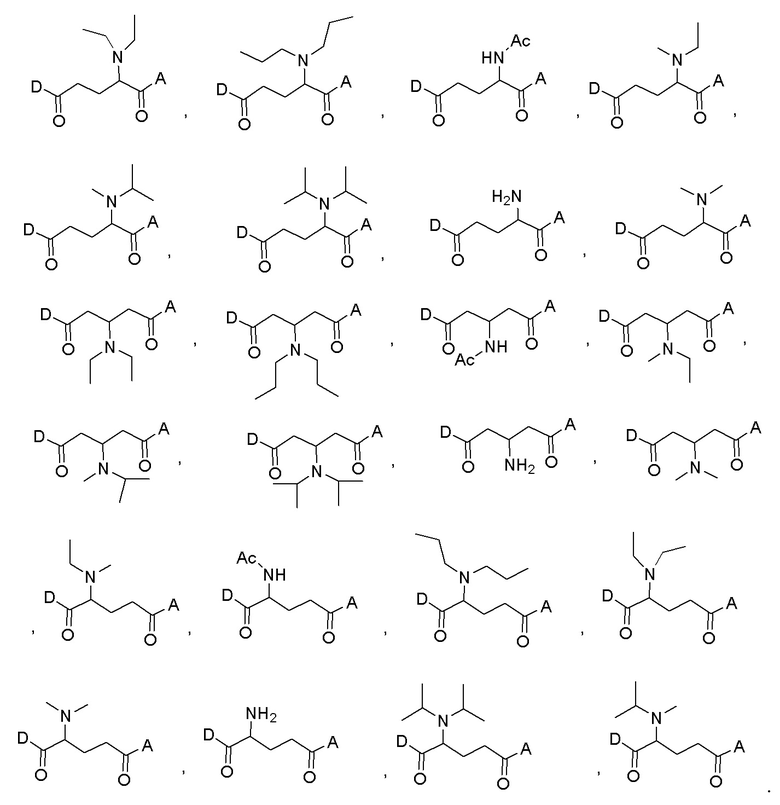
3. Фармацевтическая композиция для лечения опухолей, содержащая макромолекулу по п. 1 или 2 и фармацевтически приемлемый носитель.
4. Применение макромолекулы по п. 1 или 2 для получения лекарственного средства для лечения опухолей.
| WO 2012167309 A1, 13.12.2012 | |||
| WO 2020102852 A1, 28.05.2020 | |||
| RU 2017114360 A, 26.10.2018 | |||
| CN 102917699 A, 06.02.2013 | |||
| БЕЛИКОВ В.Г | |||
| Фармацевтическая химия // Высшая школа, М., 1993, Т.1, с.43-47 | |||
| СУТЯГИН В.М | |||
| и др | |||
| Химия и физика полимеров: Учебное пособие | |||
| - Томск: Изд-во ТПУ, 2003 | |||
| Гидравлическая или пневматическая передача | 0 |
|
SU208A1 |

