Область техники
Настоящее изобретение относится к области медицинских технологий, в частности,лёгкого к среде для культивирования и способу культивирования или размножения in vitro первичных эпителиальных клеток рака лёгкого, а также к способу и применению культивируемых клеток при оценке эффективности и скрининге лекарственных средств.
Уровень техники
Рак лёгкого в настоящее время является самой распространенной опухолью дыхательных путей в мире. Ежегодно во всем мире насчитывается более 1,8 миллиона новых случаев рака лёгкого. Как наиболее распространенная злокачественная опухоль в клинической практике, рак лёгкого в основном лечится с помощью хирургического вмешательства, химиотерапии, лучевой терапии, молекулярной таргетной терапии и иммунотерапии, среди которых наиболее часто используемыми вариантами лечения являются хирургическое вмешательство, химиотерапия и лучевая терапия. Тем не менее, существуют ограничения в отношении подходящей популяции пациентов для этих клинических способов лечения. В последние годы с ростом и развитием молекулярной биологии фармакотерапия опухолей показала разнообразие направлений, среди которой молекулярно-нацеленные лекарственные средства стали «горячей» темой исследований в области клинического лечения рака лёгкого благодаря их эффективному таргетированию и высокой безопасности. Однако среди множества вариантов клинических способов терапии особенно важно выбрать тот, который подходит конкретному пациенту. Хотя генетическое тестирование и используют в качестве индикатора, для некоторых пациентов, у которых нет мутации гена, или для некоторых пациентов, у которых есть определенная мутация, но существует несколько таргетных препаратов, нацеленных на эту мутацию, в клинической практике может быть трудно определить план лечения. В дополнение к секвенированию генов, первичные культуры клеток образцов от пациентов с раком лёгкого in vitro стали важным средством для прогнозирования эффективности in vitro и определения направления клинической медицины в будущем. Однако быстрое получение первичных клеток рака лёгкого in vitro всегда было неотложной технической проблемой, которую необходимо решать.
Функциональное тестирование относится к способу определения чувствительности к противоопухолевым лекарственным средствам на клетках имеющего рак пациента in vitro. Ключевым для применения этого способа является разработка клеточных моделей опухолей, которые имеют короткий цикл роста и могут отражать конкретные биологические характеристики пациентов с раком лёгкого. Кроме того, клеточная модель должна быть простой в обращении, чтобы быстро и результативно предсказывать эффективность клинической терапии, в целях предоставления своевременных точных рекомендаций по лечению пациентов с раком. Однако низкие в среднем показатели успешности и длительные циклы роста при создании in vitro клеточных моделей из первичных опухолевых клеток от пациентов с раком и такие проблемы, как избыточная пролиферация мезенхимальных клеток (таких как фибробласты и т.п.), ограничивают развитие в этой области. В настоящее время существуют две методики культивирования первичных эпителиальных/стволовых клеток, которые являются относительно зрелыми, в области функционального тестирования с использованием опухолевых клеток. Одной из них является методика, в которой используют облученные фидерные клетки и ROCK-ингибитор Y27632 для стимуляции роста первичных эпителиальных клеток с целью исследования чувствительности к лекарственным средствам отдельных пациентов, то есть методика кондиционного перепрограммирования клеток (Liu et al., Am J Pathol, 180: 599-607, 2012). Другая методика представляет собой 3D-культуру in vitro стволовых клеток взрослого организма для получения органоидов, подобных тканям и органам (Hans Clevers et al., Cell, 11; 172 (1-2): 373-386, 2018).
Однако обе методики имеют определенные ограничения. Перепрограммирование клеток - это методика, в которой первичные аутологичные эпителиальные клетки пациента культивируют с фидерными клетками, происходящими от мыши. Когда первичные клетки пациента тестируют на чувствительность к лекарственным средствам, присутствие этих клеток, происходящих от мыши, может влиять на результаты тестирования на чувствительность к лекарственным средствам первичных аутологичных клеток этого пациента; однако, если удалить указанные фидерные клетки, происходящие от мыши, указанные первичные аутологичные клетки пациента будут оторваны от перепрограммирующего окружения, а скорость пролиферации клеток и внутриклеточные сигнальные пути претерпят значительные изменения (Liu et al., Am J Pathol, 183 (6): 1862-1870, 2013; Liu et al., Cell Death Dis., 9 (7): 750, 2018), тем самым значительно влияя на ответ указанных первичных аутологичных клеток пациента на лекарственное средство. Получение органоида - это методика, в которой аутологичные первичные эпителиальные клетки пациента внедряют во внеклеточный матрикс для трехмерного культивирования in vitro, и влияние фидерных клеток, происходящих от мыши, отсутствует, поскольку фидерные клетки в этой методике не требуются. Однако среда для культивирования в органоидной методике должна быть дополнена различными специфическими факторами роста (такими как белки Wnt и белки семейства R-спондинов), что дорого и не подходит для широкого клинического применения. Кроме того, такой органоид должен быть заключен в гелеобразный внеклеточный матрикс в течение всего процесса культивирования, а этапы инокуляции клеток, пересева и тестирования на чувствительность к лекарственным средствам являются трудоемкими и затратными по времени в сравнении с операциями 20-культивирования. Также размер органоида, сформированного с помощью этой методики, трудно контролировать, и некоторые органоиды могут вырасти до слишком большого размера, что вызывает внутренний некроз. Таким образом, органоидная методика имеет худшую реализуемость и применимость, чем методика 2D-культуры. Для ее реализации необходим квалифицированный технический персонал, и поэтому она не подходит для масштабного и широкого клинического применения для тестирования на чувствительность к лекарственным средствам in vitro (Nick Barker, Nat Cell Biol, 18 (3): 246-54, 2016).
Ввиду ограничений вышеуказанных методик необходимо разработать методику культивирования первичных эпителиальных клеток рака лёгкого в клинических условиях, которая может обеспечить короткий период культивирования, контролируемую стоимость и удобство работы без влияния экзогенных клеток. Когда такая методика применяется для создания клеточной модели опухоли из первичных клеток рака лёгкого, культивированные клетки рака лёгкого могут представлять биологические характеристики самих пациентов с раком лёгкого. Путем оценки in vitro чувствительности к противоопухолевым лекарственным средствам в указанных клеточных моделях, полученных из клеток от отдельных пациентов с раком, можно улучшить в клинике степень ответа на противоопухолевое лекарственное средство, а проблемы, вызванные неподходящими для указанных пациентов лекарственными средствами, и расход медицинских ресурсов могут быть сокращены.
Краткое описание изобретения
С учетом указанных недостатков в уровне техники, настоящее изобретение направлено на обеспчение среды для культивирования для культивирования первичных эпителиальных клеток рака лёгкого и способа культивирования первичных эпителиальных клеток рака лёгкого с использованием среды для культивирования. Указанные среда для культивирования и способ культивирования первичных эпителиальных клеток рака лёгкого по настоящему изобретению могут обеспечить указанные короткий период культивирования in vitro, контролируемую стоимость и удобство работы без влияния экзогенных клеток. При применении этой методики для создания клеточной модели опухоли из первичных клеток рака лёгкого можно получить указанные первичные клетки рака лёгкого с биологическими характеристиками указанных пациентов с раком лёгкого, которые можно применять при скрининге новых лекарственных средств и в тестировании на чувствительность к лекарственным средствам in vitro.
Одним из аспектов настоящего изобретения является обеспечение среды для культивирования первичных клеток для культивирования первичных эпителиальных клеток рака лёгкого, содержащей ингибитор киназы MST1/2, по меньшей мере один ROCK-ингибитор, выбранный из группы, состоящей из Y27632, фасудила и Н-1152; фактор роста фибробластов 7 (FGF7, fibroblast growth factor 7); по меньшей мере одну добавку, выбранную из группы, состоящей из добавки В27 и добавки N2; фактор роста гепатоцитов (HGF, hepatocyte growth factor); инсулиноподобный фактор роста 1 (IGF-1, insulin-like growth factor 1); интерлейкин 6 (IL-6); по меньшей мере один ингибитор рецептора TGFβ (transforming growth factor β) I типа, выбранный из группы, состоящей из А83-01, SB431542, Repsox, SB505124, SB525334, SD208, LY36494 и SJN2511, где ингибитор киназы MST1/2 включает Соединение по Формуле (I), или его фармацевтически приемлемую соль, или сольват,
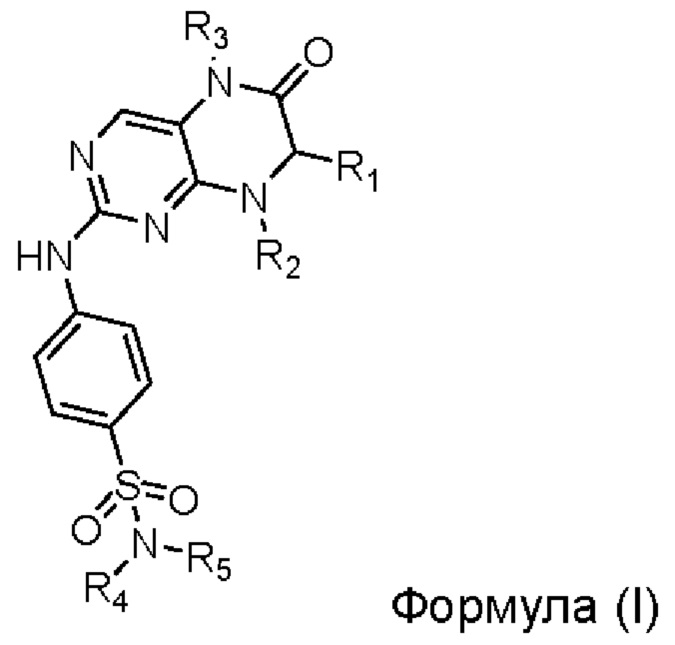
где
радикал R1 выбран из С1-С6-алкила, С3-С6-циклоалкила, С4-С8-циклоалкилалкила, С2-С6-спироциклоалкила и арила (например, фенила и нафтила, и т.д.), необязательно замещенного 1-2 независимыми радикалами R6, арил-С1-С6-алкила (например, фенилметила и т.д.), необязательно замещенного 1-2 независимыми радикалами R6, и гетероарила (например, тиенила и т.д.), необязательно замещенного 1-2 независимыми радикалами R6;
каждый из радикалов R2 и R3 независимо выбран из С1 -С6-алкила, предпочтительно С1-С3-алкила, более предпочтительно метила;
каждый из радикалов R4 и R5 независимо выбран из водорода, С1-С6-алкила, С3-С6-циклоалкила, С4-С8-циклоалкилалкила, гидроксил-С1-С6-алкила, С1 -Сб-галогеналкила,
С1-С6-алкиламино-С1-С6-алкила, С1-С6-алкокси-С1-С6-алкила и С3-С6-гетероциклил-С1-С6-алкила (гетероциклил выбран, например, из пиперидила, тетрагидропиранила и т.д.);
радикал R6 выбран из галогена (предпочтительно фтора и хлора, более предпочтительно фтора), С1-С6-алкила (предпочтительно метила), С1-С6-алкокси (предпочтительно метокси) и С1-С6-галогеналкила (предпочтительно трифторметила).
В предпочтительном варианте реализации изобретения, ингибитор киназы MST1/2 включает Соединение Формулы (Ia), или его фармацевтически приемлемую соль, или сольват,
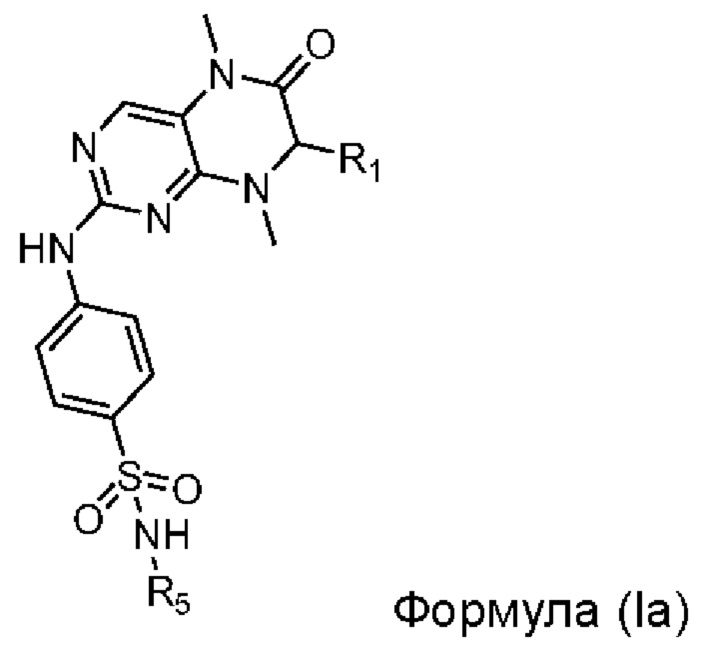
где
радикал R1 выбран из С1-С6-алкила, фенила, необязательно замещенного 1-2 независимыми радикалами R6, тиенила, необязательно замещенного 1-2 независимыми радикалами R6, и фенилметила, необязательно замещенного 1-2 независимыми радикалами R6; более предпочтительно, радикал R1 представляет собой фенил, необязательно замещенный 1-2 независимыми радикалами R6;
радикал R5 выбран из водорода, С1-С6-алкила и С3-С6-циклоалкила; более предпочтительно, радикал R5 представляет собой водород;
радикал R6 независимо выбран из галогена, С1-С6-алкила и С1-С6-галогеналкила; более предпочтительно, радикал R6 представляет собой фтор, метил или трифторметил.
Предпочтительно ингибитор киназы MST1/2 представляет собой по меньшей мере одно соединение, выбранное из следующих соединений, или его фармацевтически приемлемую соль, или сольват.
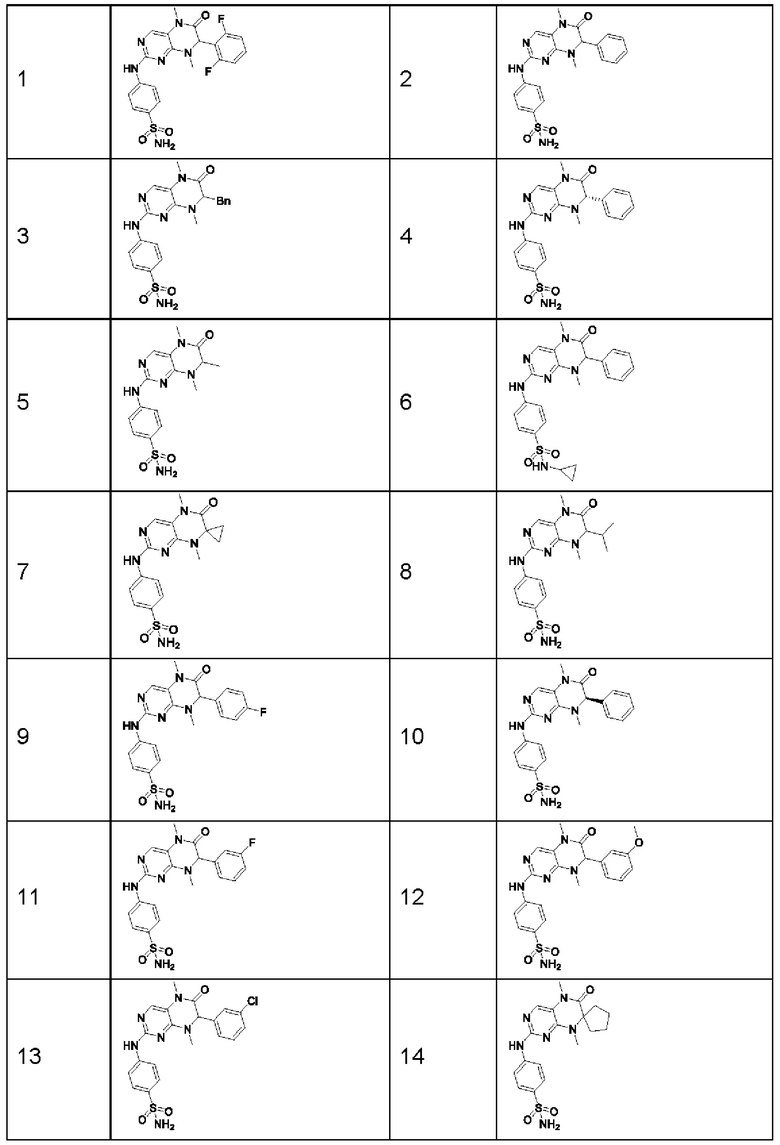
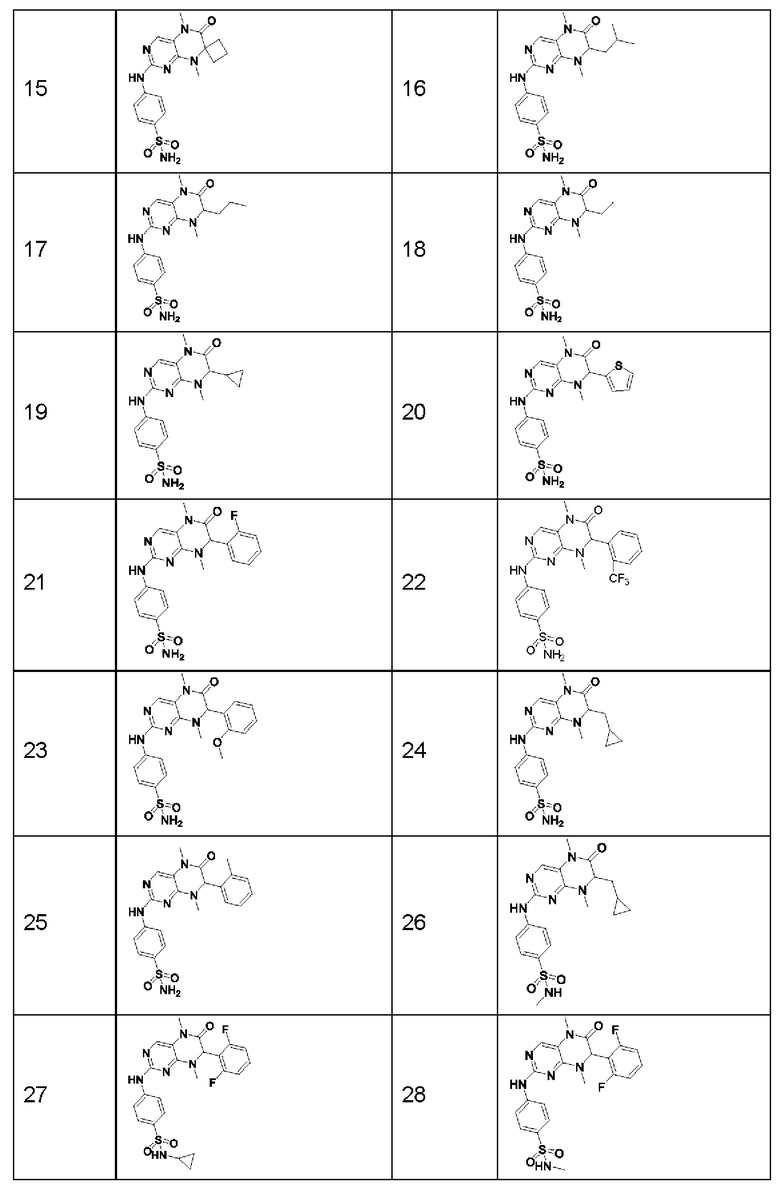
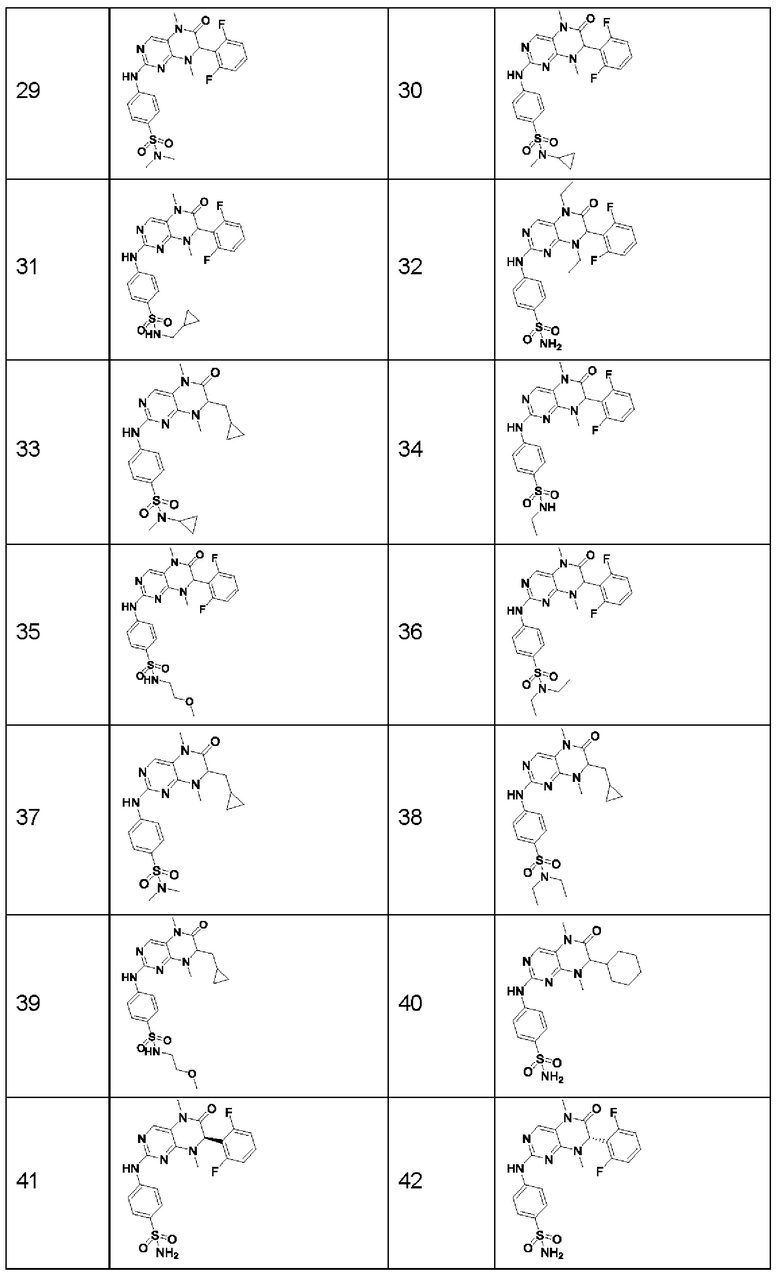
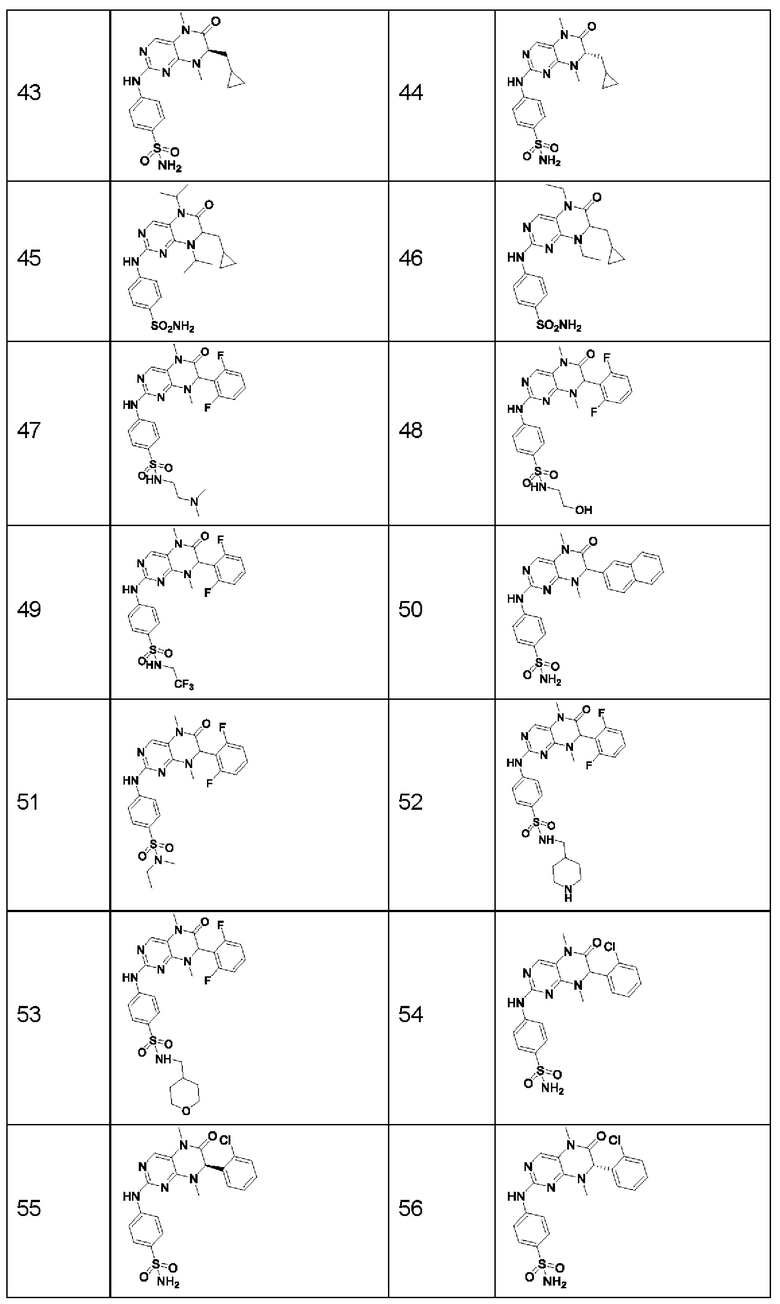
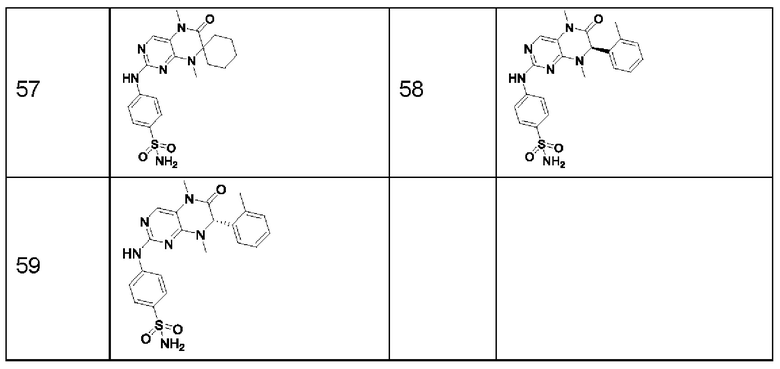
Наиболее предпочтительно, ингибитор киназы MST1/2 по изобретению представляет собой Соединение Формулы (I).
В варианте реализации изобретения количество ингибитора киназы MST1/2 в среде для культивирования обычно составляет 2,5-10 мкМ, предпочтительно 5-7,5 мкМ.
В еще одном варианте реализации ROCK-ингибитор предпочтительно представляет собой Y27632. Кроме того, предпочтительно количество ROCK-ингибитора в среде для культивирования обычно составляет 2,5-15 мкМ, предпочтительно 5-15 мкМ, и более предпочтительно 10-12,5 мкМ.
В предпочтительном варианте реализации количество фактора роста фибробластов 7 составляет 5-160 нг/мл, более предпочтительно 10-80 нг/мл, более предпочтительно 20-40 нг/мл; объемная концентрация добавки В27 или добавки N2 в среде для культивирования составляет 1:25-1:800, более предпочтительно 1:25-1:200, более предпочтительно 1:50-1:100; количество фактора роста гепатоцитов составляет 5-160 нг/мл, более предпочтительно 20-80 нг/мл; количество инсулиноподобного фактора роста 1 составляет 5-160 нг/мл, более предпочтительно 20-80 нг/мл; количество интерлейкина 6 составляет 2,5-40 нг/мл, более предпочтительно 5-40 нг/мл; ингибитор рецептора TGFβ I типа предпочтительно представляет собой А83-01, а количество ингибитора рецептора TGFβ I типа составляет 62,5-500 нМ, предпочтительно 250-500 нМ.
Состав среды по настоящему изобретению также включает исходную среду, выбранную из группы, состоящей из DMEM/F12, DMEM, F12 или RPMI-1640, и один или более одного антибиотиков, выбранных из группы, состоящей из стрептомицина/пенициллина, амфотерицина В и примоцина. Согласно некоторым вариантам реализации настоящего изобретения исходная среда предпочтительно представляет собой DMEM/F12, и антибиотик предпочтительно представляет собой примоцин. В еще одном предпочтительном варианте реализации количество примоцина в среде для культивирования составляет 25-400 мкг/мл, предпочтительно 50-200 м кг/мл.
По сравнению с композицией среды для кондиционного перепрограммирования клеток и композицией среды для органоидов эпителиальных клеток рака лёгкого, композиция среды по настоящему изобретению дополнена ингибитором киназы MST1/2, но не содержит вариабельных компонентов с неуточненными параметрами, таких как сыворотка, экстракт бычьего гипофиза или подобных, узкоспециализированных факторов, которые необходимы для культивирования органоидов, таких как агонисты Wnt, белки семейства R-спондинов, ингибиторы BMP или подобные, а также не содержит никотинамид или N-ацетилцистеин, что значительно снижает стоимость среды, упрощает рабочий процесс приготовления среды и обеспечивает осуществление культивирования in vitro первичных эпителиальных клеток рака лёгкого с контролируемой стоимостью и удобством работы.
Согласно настоящему изобретению, первичные эпителиальные клетки рака лёгкого могут представлять собой эпителиальные клетки рака лёгкого, нормальные эпителиальные клетки рака лёгкого или эпителиальные стволовые клетки рака лёгкого.
Одним из аспектов настоящего изобретения является обеспечение способа культивирования первичиных эпителиальных клеток рака лёгкого, включающего следующие этапы:
(1) Приготовление первичной среды для культивирования клеток по изобретению в соответствии с описанным выше составом.
(2) Покрытие сосуда для культивирования раствором гелеобразного внеклеточного матрикса.
В частности, в качестве гелеобразного внеклеточного матрикса можно использовать гелеобразный внеклеточный матрикс с низким содержанием фактора роста, например, можно использовать коммерчески доступный матрикс Матригель (приобретено у Corning) или ВМЕ (приобретено у Trevigen). Более конкретно, гелеобразный внеклеточный матрикс разбавляют бессывороточной средой для культивирования, которая может представлять собой среду DMEM/F12 (приобретено у Corning). Степень разбавления гелеобразного внеклеточного матрикса составляет 1:50-1:400, предпочтительно 1:100-1:200. Способ нанесения покрытия включает внесение разбавленного гелеобразного внеклеточного матрикса в сосуд для культивирования до полного покрытия поверхности дна сосуда для культивирования и выдерживание в течение 30 минут или более, предпочтительно выдерживание и покрытие при 37°С, предпочтительно в течение времени покрытия 30-60 минут. После завершения нанесения покрытия излишки разбавленного гелеобразного внеклеточного матрикса удаляют, и сосуд для культивирования готов к дальнейшему использованию.
(3) Выделение первичных эпителиальных клеток рака лёгкого из ткани рака лёгкого.
Такие первичные эпителиальные клетки рака лёгкого могут быть получены, например, из хирургических образцов рака лёгкого и образцов-биоптатов. Например, указанные образцы раковой ткани лёгкого получают из образцов раковой ткани путем хирургической резекции от пациентов с раком лёгкого, которые дали информированное согласие на эту процедуру, и образцы биопсии отбирают из внутрилёгочного поражения под ультразвуковым контролем. Отбор вышеуказанных образцов ткани осуществляют в течение получаса после хирургического иссечения или взятия биопсии у пациента. Более конкретно, в стерильных условиях вырезают образец ткани из ненекротических участков объемом 5 мм3 или более, а затем образец ткани помещают в 10-15 мл предварительно охлажденной среды DMEM/F12, помещенной в пластиковую стерильную центрифужную пробирку с крышкой, и транспортируют в лабораторию на льду. При этом среда DMEM/F12 содержит ингибитор киназы MST1/2 (например, Соединение Формулы (1)) по изобретению и 0,2-0,4 об. % примоцина (здесь и далее такая среда именуется транспортировочной средой для ткани). В случае использования ингибитора киназы MST1/2 по изобретению диапазон концентраций составляет 0,3-10 мкМ, предпочтительно 2-5 мкМ, и более предпочтительно 3 мкМ; и в случае использования примоцина диапазон концентраций составляет 25-400 мкг/мл, предпочтительно 50-200 мкг/мл, более предпочтительно 100 мкг/мл.
В боксе биологической безопасности образец ткани переносят в чашку для культивирования, а затем промывают транспортировочной средой и смывают клетки крови с поверхности образца ткани. Промытый образец ткани переносят в другую, новую, культуральную чашку с добавлением 1-3 мл транспортировочной среды и с помощью стерильного лезвия скальпеля и пинцета разделяют образец ткани на фрагменты ткани объемом менее 3 мм3.
Фрагменты образца ткани переносят в центрифужную пробирку, которую центрифугируют при 1000-3000 об/мин в течение 3-5 минут в настольной центрифуге (Sigma, 3-18K); после удаления супернатанта добавляют транспортировочную среду и раствор для ферментативной диссоциации тканей в соотношении 1:1 (доза составляет приблизительно 5 мл раствора для ферментативной диссоциации тканей на 10 мг ткани; способ приготовления раствора для ферментативной диссоциации тканей включает: растворение 1-2 мг/мл коллагеназы II, 1-2 мг/мл коллагеназы IV, 50-100 Ед/мл дезоксирибонуклеиновой кислоты I, 0,5-1 мг/мл гиалуронидазы, 0,1-0,5 мг/мл хлорида кальция, 5-10 мг/мл бычьего сывороточного альбумина в солевом растворе Хэнкса HBSS и среде для культивирования RPMI-1640 с объемным соотношением 1:1); затем образец пронумеровывают и плотно закрывают герметизирующей лабораторной пленкой, а затем диссоциируют в шейкере с постоянной температурой (Zhichu Instrument ZQLY-180N) при 37°С, при 200-300 оборотах; завершенность диссоциации определяется путем наблюдения каждый час; если не обнаружено видимого фрагмента ткани, диссоциация может быть прекращена; в противном случае диссоциация продолжается до тех пор, пока не станет достаточной, при этом время диссоциации составляет от 4 до 8 часов. После завершения ферментативной диссоциации недиссоциированные фрагменты ткани отфильтровывают через сетчатый клеточный фильтр (размер ячеек сетчатого клеточного фильтра составляет, например, 70 мкм); фрагменты ткани на сетке фильтра промывают транспортировочной средой; оставшиеся клетки смывают в центрифужную пробирку и центрифугируют в настольной центрифуге при 1000-3000 об/мин в течение 3-5 минут. После удаления супернатанта проверяют, содержит ли полученная клеточная масса клетки крови; при наличии клеток крови добавляют 3-5 мл раствора для лизиса клеток крови (приобретено у Sigma), затем хорошо перемешивают, лизируют при 4°С в течение 10-20 минут при встряхивании и тщательном перемешивании каждые 5 минут; после лизиса полученный продукт извлекают и центрифугируют при 1000-3000 об/мин в течение 3-5 минут. После удаления супернатанта добавляют среду для культивирования клеток по изобретению для ресуспендирования. Общее количество клеток находят путем подсчета с помощью счетчика для визуализации клеток в потоке (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.).
(4) Инокуляция первичных эпителиальных клеток рака лёгкого, выделенных на этапе (3), в сосуд для культивирования с покрытием и культивирование с использованием среды для культивирования первичных клеток, полученной на этапе (1).
Более конкретно, первичные клетки рака лёгкого сначала инокулируют в лунку многолуночного планшета с плотностью от 2×104 до 8×104 клеток/см2 (например, 4×104 клеток/см2); добавляют 2-3 мл среды для культивирования первичных эпителиальных клеток, а затем планшет культивируют в инкубаторе для клеток в течение 8-16 дней, например, в условиях 37°С, 5% СО2; свежую среду для культивирования первичных клеток заменяют каждые 4 дня во время культивирования; диссоциацию и пассирование выполняют, когда первичные эпителиальные клетки рака лёгкого вырастают до плотности клеток, которая составляет от 80% до 90% площади поверхности дна многолуночного планшета.
Этот этап инокуляции не требует использования фидерных клеток и, таким образом, устраняет этапы культивирования и облучения фидерных клеток по сравнению с методикой кондиционного перепрограммирования клеток. По сравнению с органоидной методикой, на этом этапе не требуется равномерное смешивание первичных клеток с гелеобразным матриксом на льду для образования капель геля и ожидания затвердевания геля перед добавлением среды для культивирования, и, таким образом, предварительно покрытый матриксом сосуд для культивирования может быть непосредственно использован для инокуляции первичных клеток. Кроме того, по сравнению с органоидной методикой, для покрытия сосуда для культивирования требуется лишь небольшое количество разбавленного гелеобразного внеклеточного матрикса, что экономит количество указанного дорогостоящего гелеобразного внеклеточного матрикса, а также упрощает операции.
Необязательно, после культивирования инокулированных первичных эпителиальных клеток рака лёгкого в течение 8-16 дней, когда образовавшиеся в культуральном планшете клеточные клоны сливаются вместе и покрывают 80% площади дна, супернатант удаляют и добавляют 0,5-2 мл 0,05%-ного раствора трипсина (приобретено у Thermo Fisher) для диссоциации клеток, а затем инкубируют при комнатной температуре в течение 5-20 минут. Разделенные клетки ресуспендируют в 1-4 мл среды для культивирования DMEM/F12, содержащей, например, 5% (об./об.) эмбриональной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина, и центрифугируют при 1000-3000 об/мин в течение 3-5 минут. Диссоциированные отдельные клетки ресуспендируют с использованием среды для культивирования для культивирования первичных клеток по изобретению, и полученную суспензию клеток помещают в культуральный флакон Т25, покрытый гелеобразным внеклеточным матриксом, для длительного культивирования. Этап нанесения покрытия на культуральный флакон Т25 аналогичен этапу (2).
Размноженные эпителиальные клетки рака лёгкого растут в 2D-формате, без формирования неоднородных по размеру органоидов и внутреннего некроза в чрезмерно выросших органоидах, которые могут возникнуть при размножении с использованием органоидной методики.
Согласно другому аспекту изобретения эпителиальные клетки рака лёгкого, особенно клетки рака лёгкого, выращиваемые способом культивирования первичных эпителиальных клеток рака лёгкого по настоящему изобретению, можно применять для оценки эффективности и скрининга лекарственных средств, что включает следующие этапы:
(1) получение первичных эпителиальных клеток рака лёгкого, более предпочтительно, получение образцов раковой ткани или образцов биопсии раковой ткани, полученных от пациентов с раком лёгкого; выделение первичных эпителиальных клеток рака лёгкого и культивирование и размножение первичных эпителиальных клеток рака лёгкого (в частности, первичных клеток рака лёгкого) в соответствии со способом культивирования первичных эпителиальных клеток рака лёгкого, описанным выше, до получения клеток в количестве по меньшей мере 105, предпочтительно по меньшей мере 106.
(2) Выбор испытываемого лекарственного средства.
(3) В зависимости от максимальной концентрации в плазме Cmax указанного лекарственного средства, взятой в качестве референсной, использование 2-5-кратной концентрации Cmax в качестве начальной и серийное разведение указанного лекарственного средства с получением градиента различных концентраций указанного лекарственного средства, например, градиента 5-10 концентраций указанного лекарственного средства, предпочтительно градиента 6-8 концентраций указанного лекарственного средства.
(4) Диссоциация эпителиальных клеток рака лёгкого, культивированных на этапе (1), в суспензию отдельных клеток, подсчет количества клеток с помощью счетчика для визуализации клеток в потоке, разведение суспензии отдельных клеток средой для культивирования первичных клеток по изобретению, равномерное внесение в многолуночный планшет разведенной суспензии клеток с плотностью 2000-4000 клеток на лунку, например, 50 мкл разведенной суспензии клеток на лунку, и прикрепление в течение ночи.
Этот этап позволяет избежать проблемы способа перепрограммирования клеток, заключающейся в том, что присутствие фидерных клеток может мешать подсчету первичных клеток и последующему анализу жизнеспособности первичных клеток, а также устраняет необходимость в трудоемком этапе смешивания и заключения суспензии клеток в гелеобразный матрикс на льду с последующим посевом, как в органоидной методике, что значительно упрощает процесс работы и повышает удобство использования и практичность способа. Поскольку инокулированные клетки представляют собой суспензии одиночных клеток, а не трехмерные структуры, такие как органоиды, этот способ может позволить получить более равномерное распределение клеток при посеве и меньшие различия в количестве клеток между лунками по сравнению с органоидной методикой, что делает этот способ более подходящим для последующих манипуляций по высокопроизводительному скринингу лекарственных средств.
(5) Добавление выбранных лекарственных средств-кандидатов, таких как традиционные химиотерапевтические лекарственные средства, препараты направленного действия, препараты антител, или их комбинации, в серийных разведениях с градиентом концентраций, к прикрепленным клеткам, полученным на этапе (4), с использованием высокопроизводительной автоматизированной рабочей станции.
(6) Через несколько часов после добавления лекарственного средства, например, через 72 часа после добавления лекарственного средства, жизнеспособность эпителиальных клеток рака лёгких определяли с помощью набора для люминесцентного анализа жизнеспособности клеток Cell-Titer Glo (приобретено у Promega) для скрининга активности лекарственного средства.
В частности, в каждую лунку добавляют, например, 10 мкл реагента Cell Titer-Glo (приобретено у Promega), и после равномерного встряхивания для каждой лунки измеряют интенсивность хемилюминесценции с помощью флуоресцентного микропланшетного ридера. Принимая концентрацию лекарственного средства за абсциссу, а интенсивность флуоресценции за ординату, используют программное обеспечение GraphPad Prism для построения кривой «доза-эффект» для лекарственного средства на основе измеренных значений, и рассчитывают ингибирующую активность лекарственного средства в отношении пролиферации тестируемых клеток.
При применении первичных клеток рака лёгкого по настоящему изобретению для скрининга лекарственных средств и обнаружения чувствительности к лекарственным средствам in vitro фидерные клетки не будут мешать результатам детекции, как в методике перепрограммирования клеток, поскольку не используют систему совместного культивирования клеток. Кроме того, из-за роста клеток в 20-плоскости время взаимодействия с препаратом короче, чем время детекции для препарата в органоидной методике (среднее время воздействия в органоидной методике составляет 6 дней).
Полезные эффекты изобретения также включают следующее:
(1) показатель успешности культивирования первичных эпителиальных клеток рака лёгкого улучшается, с достижением показателя успешности 80% или более;
(2) обеспечение воспроизведения первичными эпителиальными клетками рака лёгкого, культивированными in vitro, патологического фенотипа и гетерогенности первичных клеток пациента-хозяина;
(3) культивированным первичным эпителиальным клеткам рака лёгкого не мешают фибробласты, и могут быть получены очищенные эпителиальные клетки рака лёгкого;
(4) состав среды не содержит сыворотки, а значит, на него не влияет качество и количество сыворотки из разных партий;
(5) эпителиальные клетки рака лёгкого размножаются с высокой эффективностью, при этом величина 106 эпителиальных клеток рака лёгкого успешно достигается в течение приблизительно двух недель с момента начала подсчета клеток на уровне в 104 клеток, и размноженные эпителиальные клетки рака лёгкого обладают способностью к длительному пассированию;
(6) нет необходимости работать на льду и отделять гелеобразный матрикс на этапе пересева, а отделение и пересев клеток может быть выполнен в течение 10-15 минут;
(7) стоимость культивирования является контролируемой, поскольку среда для культивирования первичных клеток рака лёгкого не требует применения дорогостоящих агонистов Wnt, белков семейства R-спондинов, ингибиторов BMP и подобных факторов, что является упрощением и улучшением состава существующей среды для культивирования для органоида из первичных эпителиальных клеток рака лёгкого; для инокуляции клеток не требуется использование более высокой концентрации внеклеточного матрикса, смешанного с первичными клетками для образования капель геля, а также необходимо использовать только небольшое количество разбавленного гелеобразного внеклеточного матрикса, что позволяет экономить количество более дорогостоящего внеклеточного матрикса;
(8) операции удобны: по сравнению с методикой кондиционного перепрограммирования, эта методика не требует культивирования и облучения фидерных клеток и, таким образом, позволяет избежать проблемы, заключающейся в том, что качество и количество фидерных клеток из разных партий могут влиять на эффективность первичной клеточной культуры; объектами посева и тестирования при скрининге лекарственных средств являются только первичные эпителиальные клетки рака лёгкого, без влияния со стороны фидерных клеток, как в системе совместного культивирования, требуемой в методике кондиционного перепрограммирования клеток; по сравнению с органоиднаой методикой, в способе нанесения покрытия из гелеобразного внеклеточного матрикса на сосуды для культивирования, используемом в изобретении, сосуды для культивирования могут быть подготовлены заранее, и нет необходимости заключать клетки в гелеобразный матрикс, как в органоидной методике, и, таким образом, этапы операции просты и удобны;
(9) эта методика позволяет культивировать и обеспечивать эпителиальные клетки рака лёгкого в большом количестве и с высокой однородностью, что подходит для высокопроизводительного скрининга новых веществ-кандидатов и высокопроизводительных функциональных тестов на чувствительность in vitro к лекарственным средствам для пациентов.
Применение среды для культивирования клеток по этому варианту реализации позволяет культивировать происходящие из организма человека или других млекопитающих эпителиальные клетки рака лёгкого, включая клетки рака лёгкого, нормальные эпителиальные клетки лёгкого, эпителиальные стволовые клетки рака лёгкого, или ткани, содержащие по меньшей мере какие-либо из этих клеток. Наряду с этим среду для культивирования по изобретению также можно использовать для разработки набора для размножения и культивирования in vitro первичных клеток рака лёгкого.
Кроме того, клетки, полученные способом культивирования по этому варианту реализации, можно использовать в регенеративной медицине, в фундаментальных медицинских исследованиях эпителиальных клеток рака лёгкого, при скрининге ответа на лекарственные средства и при разработке новых лекарственных средств для лечения рака лёгкого, и т.п.
Описание чертежей
На Фиг. 1A-1D показано влияние различных факторов в среде для культивирования на пролиферацию первичных клеток рака лёгкого.
На Фиг. 2 показано влияние возрастающих количеств факторов в среде для культивирования на пролиферацию первичных клеток рака лёгкого.
На Фиг. 3А-3Н показано влияние концентрации каждого из факторов на пролиферацию первичных клеток рака лёгкого.
Фиг. 4А и 4В представляют собой сделанные под инвертированным микроскопом фотографии клеток рака лёгкого, которые культивировали из клеток, выделенных из одного клинического образца ткани рака лёгкого, с использованием среды для культивирования FLM по изобретению, на 4-й и 12-й день, соответственно.
Фиг. 5A-5D представляют собой фотографии, сделанные под инвертированным микроскопом, после 12 дней культивирования в условиях 4 различных сред для культивирования клеток, выделенных из одного хирургически резецированного образца рака лёгкого.
Фиг. 6 представляет собой сравнительную таблицу эффектов клеточной пролиферации, полученных при культивировании клеток, выделенных из девяти хирургически резецированных образцов рака лёгкого, в течение 16 дней культивирования в условиях 4 различных сред для культивирования.
Фиг. 7 представляет собой сравнительный график кривых роста клеток, полученных при культивировании клеток, выделенных из одного клинического образца ткани рака лёгкого, при культивировании в условиях 4 различных сред для культивирования.
На Фиг. 8 показано сравнение результатов исследования методом иммуногистохимии клеток рака лёгкого, полученных при культивировании клеток, выделенных из одного хирургически резецированного образца рака лёгкого, с использованием среды для культивирования FLM по изобретению.
Фиг. 9 представляет собой сравнительный график кривых роста клеток, полученный при культивировании клеток, выделенных из одного клинического образца ткани рака лёгкого, в условиях среды для культивирования FLM по изобретению и сред для культивирования, полученных путем удаления различных компонентов из FLM, соответственно.
Фиг. 10 представляет собой сравнительную диаграмму подсчета клеток после того, как клетки, выделенные из одного клинического образца ткани рака лёгкого, культивировали в течение 8 дней в среде для культивирования FLM по изобретению и в средах для культивирования, полученных путем удаления различных компонентов из FLM, соответственно.
На Фиг. 11А и 11В показаны кривые «доза-эффект» для первичных клеток рака лёгкого в ответ на различные химиотерапевтические лекарственные средства и лекарственные средства направленного действия, где первичные клетки рака лёгкого получали путем культивирования хирургически резецированных образцов опухолевой ткани от двух разных пациентов с раком лёгкого в условиях среды для культивирования FLM по изобретению, соответственно.
Подробное описание изобретения
В настоящем описании изобретения эпителиальные клетки включают дифференцированные эпителиальные клетки и эпителиальные стволовые клетки, полученные из эпителиальных тканей. Термин «Эпителиальные стволовые клетки» относится к клеткам, обладающим способностью к долговременному самообновлению и дифференцировке в эпителиальные клетки, и к стволовым клеткам, происходящим из эпителиальных тканей. Примеры эпителиальных тканей включают роговицу, слизистую оболочку полости рта, кожу, конъюнктиву, мочевой пузырь, почечные канальцы, почки, органы пищеварения (пищевод, желудок, двенадцатиперстную кишку, тонкую кишку (включая тощую и подвздошную кишку), толстую кишку (включая толстую кишку)), печень, поджелудочную железу, молочную железу, слюнную железу, слезную железу, предстательную железу, корень волоса, трахею, лёгкое и др. Среда для культивирования клеток по этому варианту реализации настоящего изобретения предпочтительно представляет собой среду для культивирования эпителиальных клеток, происходящих из лёгких.
Кроме того, в данном описании термин «эпителиальные опухолевые клетки» относится к клеткам, получаемым в результате опухолевой трансформации клеток, происходящих из вышеупомянутых эпителиальных тканей.
В настоящем описании изобретения термин «органоид» относится к трехмерной органоподобной ткани из клеток, образованной путем спонтанной организации и агрегации клеток с высокой плотностью в контролируемом пространстве.
[Примеры получения ингибиторов киназы MST1/2]
В настоящем описании изобретения ингибитор киназы MST1/2 относится к любому ингибитору, который прямо или косвенно отрицательно регулирует передачу сигналов MST1/2. Как правило, ингибиторы киназы MST1/2 снижают активность киназы MST1/2, например, связываясь с ней. Поскольку MST1 и MST2 имеют сходную структуру, ингибиторами киназы MST1/2 могут быть, например, соединения, которые связываются с MST1 или MST2 и снижают их активность.
1. Получение ингибитора киназы MST1/2, Соединения по Формуле (I). 4-((7-(2,6-дифторфенил)-5,8-диметил-6-оксо-5,6,7,8-тетрагидроптеридин-2-ил)амино)бензсульфамид (Соединение по Формуле (I)):
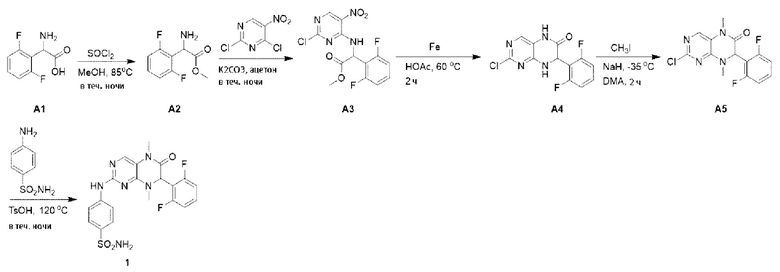
Метил-2-амино-2-(2,6-дифторфенил)ацетат (соединение А2): 2-амино-2-(2,6-дифторфенил)уксусную кислоту (2,0 г) и затем метанол (30 мл) добавляли в круглодонную лабораторную колбу с последующим добавлением по каплям тионилхлорида (1,2 мл) на ледяной бане. Реакцию в реакционной смеси осуществляли в течение ночи при 85°С. После завершения реакции смесь выпаривали при пониженном давлении для удаления растворителя, и полученное белое твердое вещество использовали непосредственно на следующей стадии.
Метил-2-((2-хлор-5-нитропиримидин-4-ил)амино)-2-(2,6-дифторфенил) ацетат (соединение A3): метил-2-амино-2-(2,6-дифторфенил)ацетат (2 г), а затем ацетон (30 мл) и карбонат калия (2,2 г) добавляли в круглодонную лабораторную колбу, после чего смесь охлаждали до -10°С на ледяной солевой бане, а затем медленно добавляли раствор 2,4-дихлор-5-нитропиримидина (3,1 г) в ацетоне. Реакционную смесь перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную смесь фильтровали, растворитель удаляли из фильтрата при пониженном давлении и осадок очищали с помощью колоночной хроматографии на силикагеле под давлением с получением соединения A3. Жидкостная хромато-масс-спектрометрия, ЖХ/МС (Liquid chromatography-mass spectrometry, LC/MS): M+H 359,0.
2-хлор-7-(2,6-дифторфенил)-7,8-дигидроптеридин-6(5Н)-он (соединение А4): в круглодонную лабораторную колбу добавляли метил-2-((2-хлор-5-нитропиримидин-4-ил)амино)-2-(2,6-дифторфенил)ацетат (2,5 г), затем уксусную кислоту (50 мл) и порошок железа (3,9 г). Реакционную смесь перемешивали при 60°С в течение двух часов. После завершения реакции реакционную смесь выпаривали при пониженном давлении, чтобы удалить растворитель, и полученную смесь нейтрализовали до щелочной реакции насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным раствором соли и сушили безводным сульфатом натрия. Органическую фазу фильтровали и упаривали досуха при пониженном давлении для получения неочищенного продукта. Неочищенный продукт промывали диэтиловым эфиром с получением соединения А4. Жидкостная хромато-масс-спектрометрия, ЖХ/МС: М+Н 297,0.
2-хлор-7-(2,6-дифторфенил)-5,8-диметил-7,8-дигидроптеридин-6(5Н)-он (соединение А5): 2-хлор-7-(2,6-дифторфенил)-7,8-дигидроптеридин-6(5Н)-он (2 г) и N,N-диметилацетамид (10 мл) добавляли в круглодонную лабораторную колбу и охлаждали до -35°С с последующим добавлением йодметана (0,9 мл), а затем гидрида натрия (615 мг), и реакционную смесь перемешивали в течение двух часов. После завершения реакции реакционную смесь гасили добавлением воды и экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным раствором соли, соответственно, и сушили безводным сульфатом натрия. Органическую фазу фильтровали и упаривали досуха при пониженном давлении для получения неочищенного продукта. Неочищенный продукт промывали диэтиловым эфиром с получением соединения А5. Жидкостная хромато-масс-спектрометрия, ЖХ/МС: М+Н 325,0.
4-((7-(2,6-дифторфенил)-5,8-диметил-6-оксо-5,6,7,8-тетрагидроптеридин-2-ил)амино)бензсульфамид (Соединение по Формуле (I)): 2-хлор-7-(2,6-дифторфенил)-5,8-диметил-7,8-дигидроптеридин-6(5Н)-он (100 мг), сульфаниламид (53 мг), п-толуолсульфокислоту (53 мг) и вторичный бутиловый спирт (5 мл) добавляли в круглодонную лабораторную колбу. Реакционную смесь перемешивали при 120°С в течение ночи. После завершения реакции реакционную смесь фильтровали и промывали метанолом и диэтиловым эфиром с получением Соединения по Формуле (I). Жидкостная хромато-масс-спектрометрия, ЖХ/МС: М+Н 461,1.
2. Получение других соединений-ингибиторов MST1/2 по изобретению.
Другие соединения-ингибиторы MST1/2 по изобретению синтезировали способом, аналогичным способу синтеза Соединения по Формуле (I), и их структуры и данные масс-спектра показаны в следующей таблице.
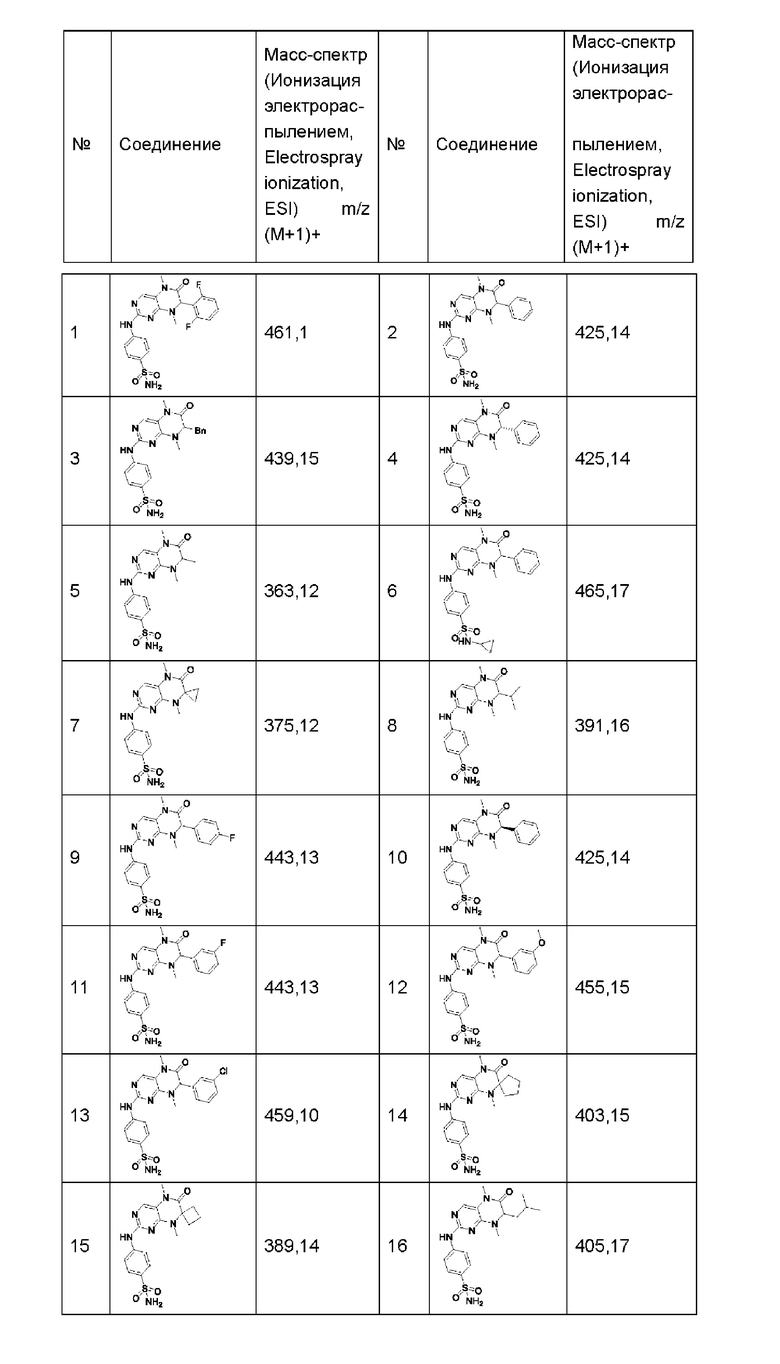

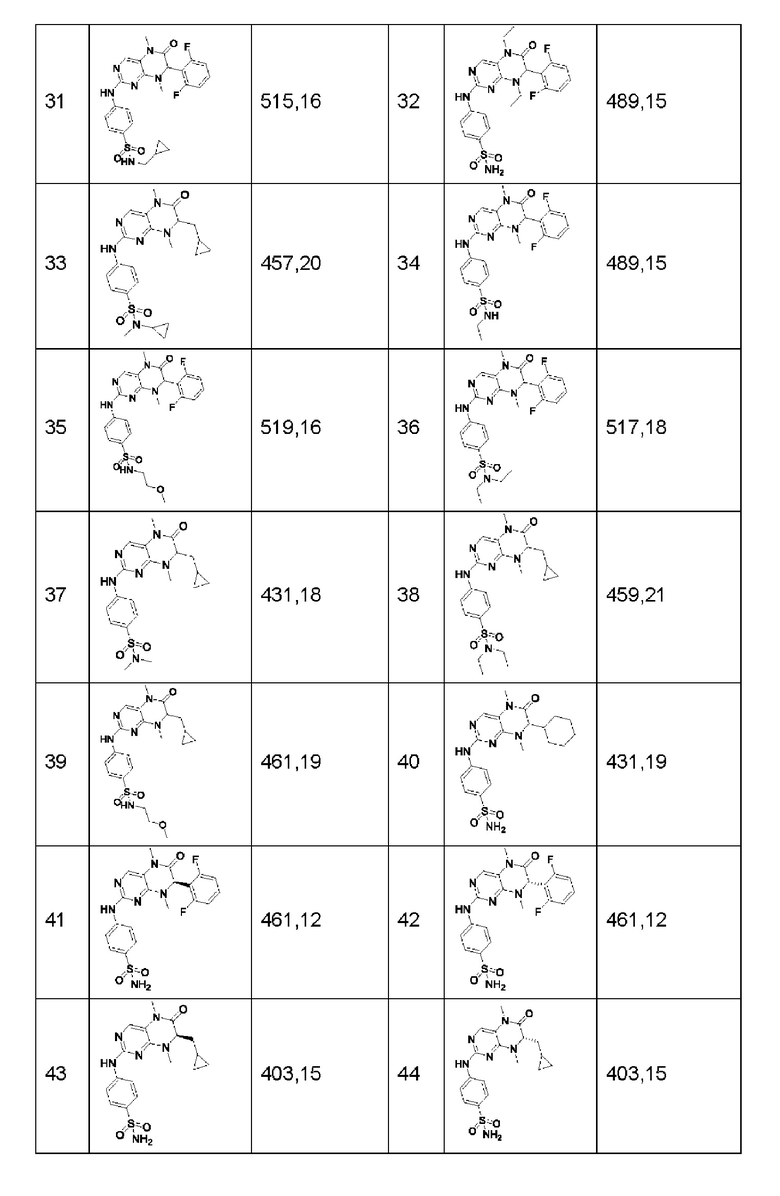
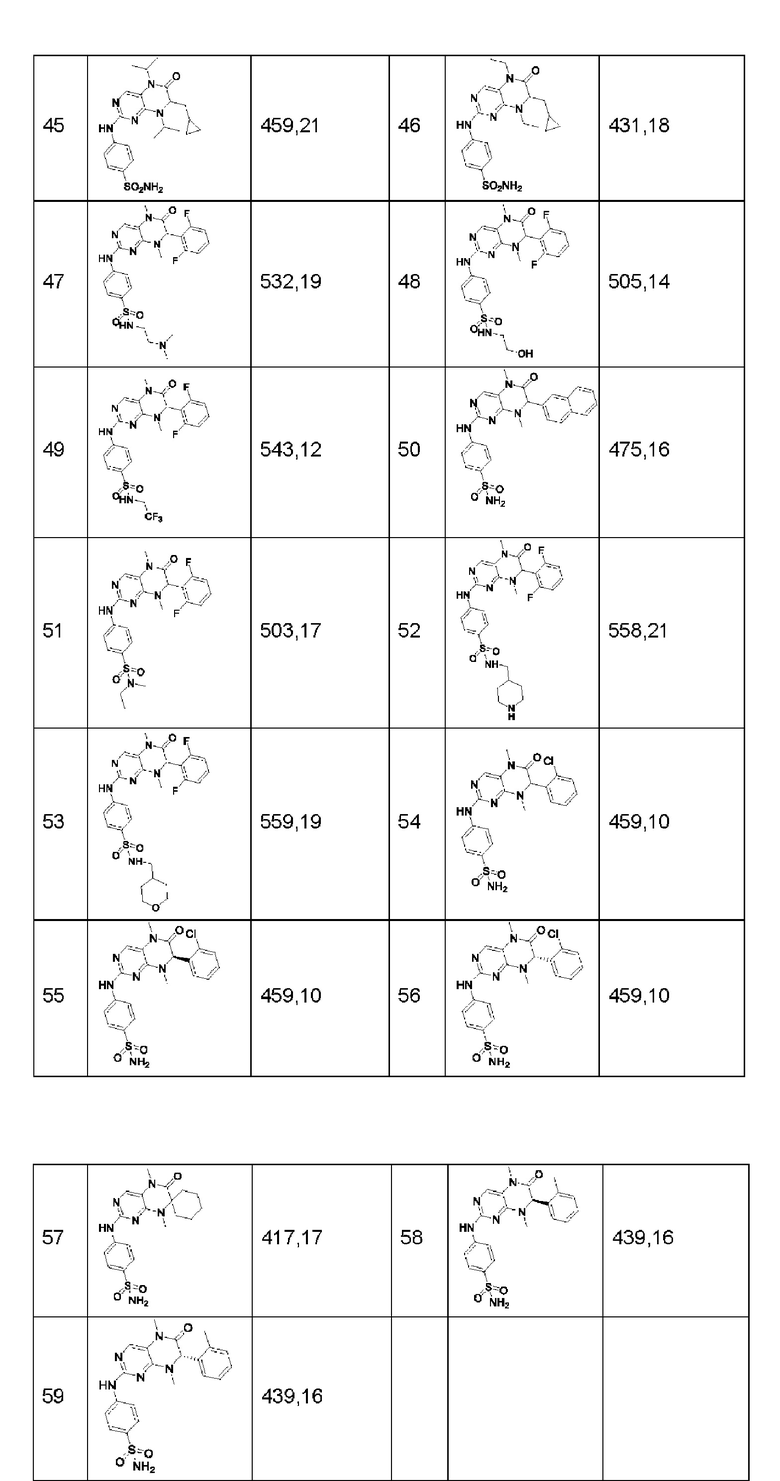
[Пример 1]
Выделение первичных эпителиальных клеток рака лёгкого человека
Образцы ткани рака лёгкого получали из образцов раковой ткани путем хирургической резекции от пациентов с раком лёгкого, которые дали информированное согласие. Один из образцов (№ В4) описан ниже.
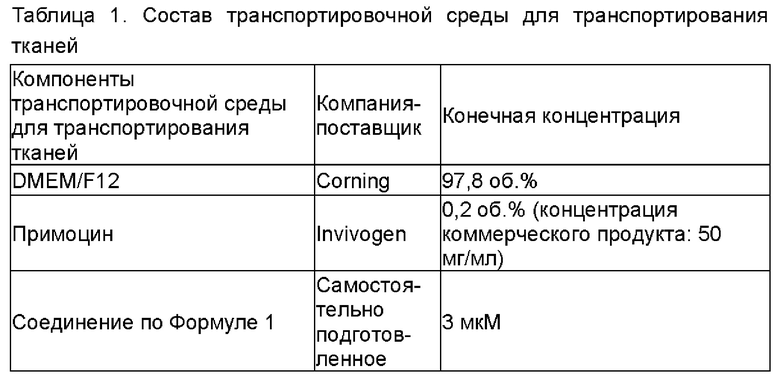
Вышеуказанные образцы тканей отбирали в течение получаса после хирургического иссечения или взятия биопсии у пациента. Более конкретно, в стерильных условиях образцы ткани вырезали из ненекротических участков объемом 0,5 см3 или более и помещали в 4 мл предварительно охлажденной транспортировочной среды для тканей (конкретный состав показан в Таблице 1). Транспортировочную среду с образцом ткани помещали в стерильную пластиковую пробирку для криоконсервации объемом 5 мл с крышкой (приобретено у Guangzhou Jet Bio-Filtration Co., Ltd.) и транспортировали в лабораторию с использованием холодовой цепи (0-10°С).

В боксе биологической безопасности образец ткани (№ В4) переносили в культуральную чашку диаметром 100 мм (приобретено у NEST). Образец ткани промывали транспортировочной средой для тканей. Остатки крови на поверхности образца ткани смывали. Излишки тканей, такие как жир на поверхности образца ткани, удаляли. Промытый образец ткани переносили в другую новую культуральную чашку диаметром 100 мм; добавляли 2 мл транспортировочной среды и стерильным лезвием скальпеля, и пинцетом разделяли образец ткани на фрагменты ткани объемом менее 3 мм3.
Фрагменты образца ткани переносили в центрифужную пробирку объемом 15 мл и центрифугировали при 1500 об/мин в течение 4 минут в настольной центрифуге (Sigma, 3-18K); после удаления супернатанта добавляли транспортировочную среду и раствор для ферментативной диссоциации тканей в соотношении 1:1 (дозировка составляет приблизительно 5 мл раствора для ферментативной диссоциации тканей на 10 мг ткани; точный состав показан в Таблице 2); далее образец пронумеровывали и плотно закрывали герметизирующей лабораторной пленкой, а затем проводили обработку ферментами на шейкере с постоянной температурой (Zhichu Instrument ZQLY-180N) при 37°С, при 300 оборотах; завершенность ферментативной диссоциации определяли путем наблюдения через каждый час.
После завершения ферментативной диссоциации недиссоциированные фрагменты ткани отфильтровывали с помощью клеточного фильтра с размером ячеек 70 мкм; фрагменты ткани на сетке фильтра промывали транспортировочной средой; оставшиеся клетки смывали в центрифужную пробирку и центрифугировали при 1500 об/мин в течение 4 минут.
После удаления супернатанта проверяли, содержит ли полученная клеточная масса клетки крови; при наличии клеток крови добавляли 3 мл раствора для лизиса клеток крови (приобретено у Sigma), затем хорошо перемешивали, лизировали при 4°С в течение 15 минут, при встряхивании и тщательном перемешивании каждые 5 минут; после лизиса полученный продукт извлекали и центрифугировали при 1500 об/мин в течение 4 минут. Супернатант удаляли для получения диссоциированных и выделенных первичныхклеток рака лёгкого, к которым добавляли основную среду (ВМ, basic medium) для ресуспендирования. Основную среду готовили путем добавления 0,2 об. % примоцина (приобретено у Invivogen, концентрация 50 мг/мл) к коммерческой среде DMEM/F-12 до получения конечной концентрации 100 мкг/мл. Общее количество клеток составило 1210000, что получали путем подсчета с помощью счетчика для визуализации клеток в потоке (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.).
[Пример 2]
Оптимизация среды для культивирования для первичных эпителиальных клеток рака лёгкого
(1) Влияние различных факторов
Гелеобразный внеклеточный матрикс (Матригель, Matrigel®, производство BD Biosciences) разбавляли бессывороточной средой DMEM/F12 в соотношении 1:100 для получения разбавленного внеклеточного матрикса. Разбавленный внеклеточный матрикс добавляли в 48-луночный культуральный планшет по 500 мкл/лунку до полного покрытия дна лунок культурального планшета. Планшет для культивирования оставляли на 1 час в инкубаторе при 37°С. Через 1 час разбавитель внеклеточного матрикса удаляли, с получением планшета, покрытого матриксом Матригель.
Приготовление основной среды (сокращенно ВМ): ВМ готовили путем добавления 0,2 об. % примоцина (приобретено у Invivogen, концентрация 50 мг/мл) к коммерческой среде DMEM/F-12 до получения конечной концентрации 100 мкг/мл.
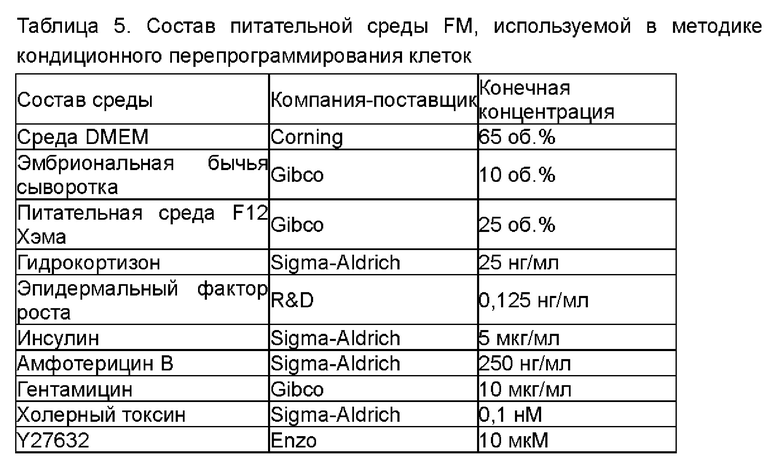
Затем к основной среде (ВМ) добавляли различные виды и концентрации дополнительных факторов (Таблица 3) для приготовления сред для культивирования для эпителиальных клеток рака лёгкого, содержащих различные дополнительные компоненты.
Таблица 3. Приготовление сред для культивирования, содержащих различные компоненты (показаны конечные концентрации)

Среды для культивирования с различными компонентами добавляли в объеме 500 мкл/лунку в 48-луночные планшеты, покрытые гелеобразным внеклеточным матриксом (Матригель). Клетки рака лёгкого (образец № В5), выделенные из тканей рака лёгкого в соответствии с тем же способом, который описан в Примере 1, инокулировали с плотностью клеток 2×104 клеток/см2 в вышеуказанные 48-луночные культуральные планшеты, покрытые матриксом Матригель. После дезинфекции поверхности планшеты помещали в инкубатор в условиях 37°С, 5% СО2 (приобретено у Thermo Fisher), и такое же количество свежевыделенных опухолевых клеток образца рака лёгкого (№ В5) культивировали в различных по составу средах. После начала культивирования среды для культивирования заменяли каждые 4 дня. Через 12 дней культивирования проводили подсчет клеток. Основную среду (ВМ) без добавления каких-либо добавок использовали в качестве контроля. Результаты представлены на Фиг. 1A-D. Ордината на Фигурах представляет собой отношение количества клеток, полученных после культивирования в различных средах, к количеству клеток, полученных после культивирования в основной среде ВМ. Как показано на рисунках, добавление различных концентраций различных факторов к среде ВМ в соответствии с Таблицей 3 приводило к различным воздействиям на пролиферацию клеток. В том числе добавка В27, добавка N2, фактор роста фибробластов 7, интерлейкин 6, фактор роста гепатоцитов, инсулиноподобный фактор роста 1, Соединение по Формуле 1, Y27632 и А83-01 оказывали определенные стимулирующие эффекты на пролиферацию клеток в определенных диапазонах концентраций.
(3) Влияние различных возрастающих концентраций факторов в средах для культивирования на пролиферацию первичных клеток рака лёгкого, полученных способом по изобретению
Гелеобразный внеклеточный матрикс Матригель (Matrigel®, приобретено у BD Biosciences) разбавляли бессывороточной средой DMEM/F12 в соотношении 1:100 для получения разбавленного внеклеточного матрикса. Разбавленный внеклеточный матрикс добавляли в 48-луночный культуральный планшет по 500 мкл/лунку, так чтобы дно лунок культурального планшета было полностью покрыто. Планшет для культивирования оставляли на 1 час в инкубаторе при 37°С. Через 1 час разбавитель внеклеточного матрикса удаляли, с получением планшета, покрытого матриксом Матригель.
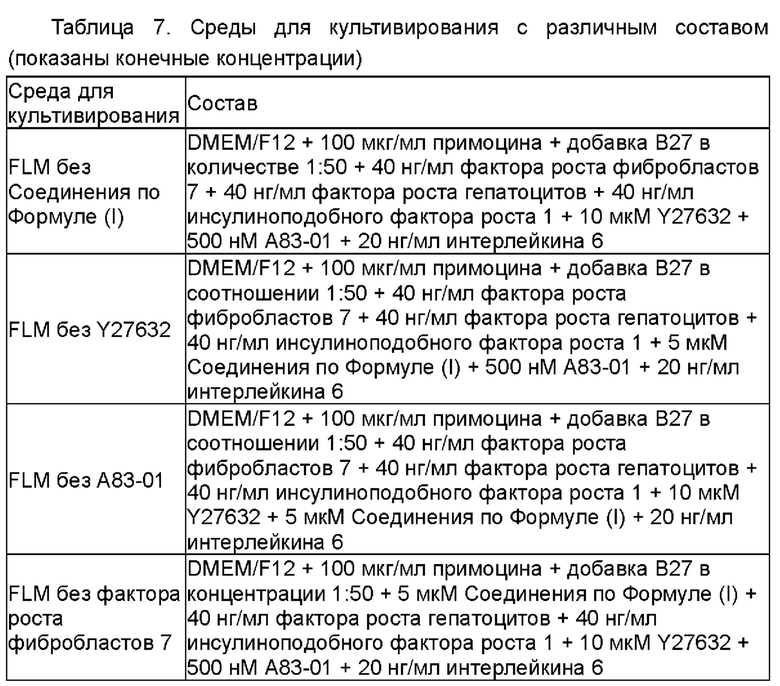
Различные малые молекулы, добавки и факторы роста (Таблица 4) последовательно добавляли к основной среде ВМ, соответственно, с получением сред для культивирования для эпителиальных клеток рака лёгкого, содержащих различные дополнительные компоненты.

Среды для культивирования с различными компонентами добавляли в объеме 500 мкл/лунку в 48-луночные планшеты, покрытые гелеобразным внеклеточным матриксом (Матригель), при этом в качестве контроля использовали среду ВМ. Клетки рака лёгкого (№ В5), выделенные из тканей рака лёгкого в соответствии со способом, описанным в Примере 1, инокулировали с плотностью клеток 2×104 клеток/см2 в 48-луночные культуральные планшеты, покрытые матриксом Матригель. После дезинфекции поверхности планшеты помещали в инкубатор в условиях 37°С, 5% СО2 (приобретено у Thermo Fisher), и такое же количество свежевыделенных опухолевых клеток из образца рака лёгкого (№ В5) культивировали в различных по составу средах. Через 10 дней культивирования проводили подсчет клеток. Результаты представлены на Фиг. 2. Как показано на этой Фигуре, установили, что среда №8 (далее сокращенно FLM) является наиболее предпочтительной средой для культивирования по настоящей заявке для культивирования и размножения первичных клеток рака лёгкого.
(4) Влияние различных концентраций дополнительных факторов на пролиферацию первичных клеток рака лёгкого, полученных согласно настоящему изобретению
Гелеобразный внеклеточный матрикс (Матригель, Matrigel®, производства BD Biosciences) разбавляли бессывороточной средой DMEM/F12 в соотношении 1:100 для получения разбавителя внеклеточного матрикса. Разбавитель внеклеточного матрикса добавляли в 48-луночный культуральный планшет по 200 мкл/лунку, чтобы полностью покрыть дно лунок культурального планшета. Планшет для культивирования оставляли на 1 час в инкубаторе при 37°С. Через 1 час растворитель внеклеточного матрикса удаляли, с получением планшета, покрытого матриксом Матригель.
Приготовление среды для культивирования для первичных эпителиальных клеток рака лёгкого по изобретению (сокращенно FLM): к основной среде (ВМ) добавляли фактор роста фибробластов 7 (FGF7) до конечной концентрации 40 нг/мл, фактор роста гепатоцитов (HGF) до конечной концентрации 40 нг/мл, инсулиноподобный фактор роста 1 (IGF-1) до конечной концентрации 40 нг/мл, добавку В27 до конечного объемного соотношения 1:50, Соединение по Формуле (I) до конечной концентрации 5 мкМ, Y27632 до конечной концентрации 10 мкМ, ингибитор TGFβ1 А83-01 до конечной концентрации 500 нМ и интерлейкин 6 (IL-6) до конечной концентрации 20 нг/мл для приготовления среды для культивирования для первичных эпителиальных клеток рака лёгкого.
Эпителиальные клетки рака лёгкого, полученные из опухолевых тканей, выделяли из опухолевой ткани пациента с раком лёгкого (образец № В6) с использованием того же способа, что и в Примере 1. Затем эпителиальные клетки рака лёгкого, полученные из раковых тканей, подсчитывали с помощью счетчика для визуализации в потоке(JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) для вычисления общего количества клеток. Затем клетки инокулировали с плотностью 4×104 клеток/см2 в 48-луночный планшет, покрытый матриксом Матригель (Matrigel®, приобретено у BD Biosciences). 2 мл приготовленной среды для культивирования FLM для первичных эпителиальных клеток рака лёгкого добавляли в 48-луночный планшет, который затем помещали в инкубатор в условиях 37°С, 5% СО2 (приобретено у Thermo Fisher) для культивирования. Когда клетки в культуральном планшете вырастали и покрывали приблизительно 80% площади поверхности дна, среду удаляли и в 48-луночный планшет добавляли 500 мкл 0,05%-ного раствора трипсина (приобретено у Gibco) для диссоциации клеток, а затем инкубировали при 37°С в течение 10 минут до полной диссоциации клеток, за которой наблюдали под микроскопом (EVOS М500, Invitrogen); затем ферментативную диссоциацию останавливали, используя 1 мл среды для культивирования DMEM/F12, содержащей 5% (об./об.) эмбриональной бычьей сыворотки (приобретено у ExCell Bio), 100 Ед/мл пенициллина (приобретено у Corning) и 100 мкг/мл стрептомицина (приобретено у Corning); полученную смесь собирали в центрифужную пробирку объемом 15 мл и центрифугировали при 1500 об/мин в течение 4 минут, а затем удаляли надосадочную жидкость. Отцентрифугированный клеточный осадок ресуспендировали в основной среде ВМ и клетки подсчитывали с помощью счетчика для визуализации клеток в потоке (JIMBIOFIL, Jiangsu Jimbio Technology Co., Ltd.) для вычисления общего количества клеток. Полученные клетки использовали в следующих экспериментах по культивированию.
Далее для экспериментов готовили следующие 8 составов питательной среды:
Состав 1: композиция среды FLM без добавки В27;
Состав 2: композиция среды FLM без фактора роста фибробластов 7;
Состав 3: композиция среды FLM без инсулин-подобного фактора роста 1;
Состав 4: композиция среды FLM без фактора роста гепатоцитов;
Состав 5: композиция среды FLM без Y27632;
Состав 6: композиция среды FLM без Соединения по Формуле (I);
Состав 7: композиция среды FLM без А83-01;
Состав 8: композиция среды FLM без интерлейкина 6.
Суспензию диссоциированных клеток разбавляли вышеуказанными составами 1-8, соответственно, и затем инокулировали в 48-луночный планшет в объеме 250 мкл в количестве 10000 клеток на лунку.
При использовании среды с Составом 1 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленную добавку В27 в объеме 250 мкл на лунку, при этом конечные концентрации добавки В27 составляли 1:800, 1:400, 1:200, 1:100, 1:50, 1:25, соответственно; лунку пустого контроля (Blank Control, ВС) заполняли средой с Составом 1.
При использовании среды с Составом 2 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленный фактор роста фибробластов 7 в объеме 250 мкл на лунку, при этом конечные концентрации фактора роста фибробластов 7 составляли 160 нг/мл, 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл, 5 нг/мл; лунку пустого контроля (ВС) заполняли средой с Составом 2.
При использовании среды с Составом 3 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленный инсулиноподобный фактор роста 1 в объеме 250 мкл на лунку, при этом конечные концентрации инсулиноподобного фактора роста 1 составляли 160 нг/мл, 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл, 5 нг/мл, соответственно; лунку пустого контроля (ВС) заполняли средой с Составом 3.
При использовании среды с Составом 4 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленный фактор роста гепатоцитов в объеме 250 мкл на лунку, при этом конечные концентрации фактора роста гепатоцитов составляли 160 нг/мл, 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл и 5 нг/мл, соответственно; лунку пустого контроля (ВС) заполняли средой с Составом 4.
При использовании среды с Составом 5 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленный фактор Y27632 в объеме 250 мкл на лунку, при этом конечные концентрации Y27632 составляли 20 мкМ, 15 мкМ, 12,5 мкМ, 10 мкМ, 5 мкМ и 2,5 мкМ, соответственно; лунку пустого контроля (ВС) заполняли средой с Составом 5.
При использовании среды с Составом 6 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленное Соединение по Формуле (I) в объеме 250 мкл на лунку, при этом конечные концентрации Соединения по Формуле (I) составляли 20 мкМ, 10 мкМ, 7,5 мкМ, 5 мкМ, 2,5 мкМ и 1,25 мкМ, соответственно; лунку пустого контроля (ВС) заполняли средой с Составом 6.
При использовании среды с Составом 7 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленный фактор А83-01 в объеме 250 мкл на лунку, при этом конечные концентрации А83-01 составляли 2000 нМ, 1000 нМ, 500 нМ, 250 нМ, 125 нМ и 62,5 нМ, соответственно; лунку пустого контроля (ВС) заполняли средой с Составом 7.
При использовании среды с Составом 8 в 48-луночный планшет с инокулированными первичными клетками добавляли подготовленный интерлейкин 6 в объеме 250 мкл на лунку, при этом конечные концентрации интерлейкина 6 составляли 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл, 5 нг/мл и 2,5 нг/мл, соответственно; лунку пустого контроля (ВС) заполняли средой с Составом 8.
После того, как клетки разрастались приблизительно до 85% площади лунок в 48-луночном планшете, клетки подвергали ферментативной диссоциации и подсчитывали их количество относительно количества клеток в лунке пустого контроля (ВС), и результаты показаны на Фиг. 3А-3Н. На каждой из диаграмм на Фиг. 3А-3Н указанное соотношение представляет собой отношение числа клеток первого пассажа, культивированных с использованием каждой из сред для культивирования, к числу клеток первого пассажа, полученных при культивировании в соответствующей контрольной лунке. Соотношение, превышающее 1, указывает на то, что стимулирующее пролиферацию действие приготовленной среды, содержащей различные факторы или низкомолекулярные соединения в различных концентрациях, более сильное, чем действие среды в лунке контрольного образца; соотношение меньше 1 указывает на то, что стимулирующее пролиферацию действие приготовленной среды, содержащей различные факторы или низкомолекулярные соединения в различных концентрациях, более слабое, чем действие среды в лунке контрольного образца.
Согласно результатам на Фиг. 3А-3Н, объемная концентрация добавки В27 в среде для культивирования предпочтительно составляет 1:25-1:800, более предпочтительно 1:25-1:200 и еще более предпочтительно 1:50-1:100; количество фактора роста фибробластов 7 предпочтительно составляет 5-160 нг/мл, более предпочтительно 10-80 нг/мл и еще более предпочтительно 20-40 нг/мл; количество инсулиноподобного фактора роста 1 предпочтительно составляет 5-160 нг/мл и более предпочтительно 20-80 нг/мл; количество фактора роста гепатоцитов предпочтительно составляет 5-160 нг/мл и более предпочтительно 20-80 нг/мл; количество Y27632 предпочтительно составляет 2,5-15 мкМ, более предпочтительно 5-15 мкМ и еще более предпочтительно 10-12,5 мкМ; количество Соединения по Формуле (I) предпочтительно составляет 2,5-10 мкМ и более предпочтительно 5-7,5 мкМ; количество А83-01 предпочтительно составляет 62,5-500 нМ и более предпочтительно 250-500 нМ; количество интерлейкина 6 предпочтительно составляет 2,5-40 нг/мл и более предпочтительно 5-40 нг/мл.
[Пример 3]
Культивирование первичных клеток рака лёгкого, полученных из тканей рака лёгкого
Эпителиальные клетки рака лёгкого, полученные из опухолевых тканей, выделяли из опухолевых тканей пациентов с раком лёгкого (образец № В7) с использованием того же способа, что и в Примере 1. Затем эпителиальные клетки рака лёгкого, полученные из раковой ткани, подсчитывали с помощью счетчика для визуализациив потоке (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) для вычисления общего количества клеток. Затем клетки инокулировали в 12-луночный планшет, покрытый матриксом Матригель (Matrigel®, приобретено у BD Biosciences), с плотностью 4×104 клеток/см2. 2 мл приготовленной среды для культивирования FLM для первичных эпителиальных клеток рака лёгкого добавляли в 12-луночный планшет, который затем помещали в инкубатор в условиях 37°С, 5% СО2 (приобретено у Thermo Fisher) для культивирования.
Фиг. 4А представляет собой микроскопическое изображение (полученное с помощью фотосъемки через инвертированный фазово-контрастный микроскоп 40х) клеток, которые культивировали до 4-го дня после инокуляции с плотностью 4×104 клеток/см2 в 12-луночном планшете, покрытом матриксом Матригель в соответствии с настоящим Примером. Микроскопическое исследование показывает, что культивированные первичные клетки рака лёгкого, полученные из опухолевых тканей, имели высокую чистоту, фибробласты отсутствовали. Фиг. 4 В представляет собой микроскопическое изображение (полученное с помощью фотосъемки через инвертированный фазово-контрастный микроскоп 40х) клеток, которые культивировали до 12-го дня после инокуляции, в соответствии с настоящим Примером. Из Фиг. 4А и 4В видно, что образование заметного клона уже можно наблюдать под микроскопом, когда выделенные первичные клетки рака лёгкого культивировали in vitro в течение 4 дней, и количество клеток значительно увеличивалось после 12 дней культивирования, что позволяет предположить, что методика данного изобретения является эффективной методикой для выращивания эпителиальных клеток рака лёгкого in vitro.
[Пример 4]
Влияние различных сред для культивирования на стимулирование пролиферации первичных клеток рака лёгкого, полученных из тканей рака лёгкого
(1) Сравнение влияния различных сред для культивирования на образование клонов первичных клеток первого поколения и сравнение их влияния на пролиферацию.
Среду FLM для культивирования первичных эпителиальных клеток рака лёгкого готовили тем же способом, что и в Примере 2, а основную среду ВМ готовили в качестве контроля. В качестве другого контроля дополнительно готовили среду для культивирования FM, используемую в методике кондиционного перепрограммирования клеток. Об этапах подготовки см. у Liu et al., Nat Protoc, 12(2): 439-451, 2017. Состав среды для культивирования показан в Таблице 5. Кроме того, в качестве дополнительного контроля у компании STEMCELL Technologies приобретали коммерческую среду для культивирования EpiCult™ Plus Medium (далее именуемая как «среда EpiM»), и состав среды для культивирования показан в Таблице 6.

С использованием того же способа, что и в Примере 1, из тканей рака лёгкого получали первичные клетки рака лёгкого (образец № В8). Затем клетки культивировали с той же плотностью (4×104 клеток/см2) в следующих пяти условиях культивирования:
A. Способ по настоящему изобретению: первичные клетки рака лёгкого инокулировали в 24-луночный планшет, покрытый матриксом Матригель (Matrigel®, приобретено у BD Biosciences), с плотностью 4×104 клеток/см2, культивировали с использованием 2 мл среды для культивирования FLM для первичных эпителиальных клеток рака лёгкого по настоящему изобретению;
B. Методика кондиционного перепрограммирования клеток: первичные клетки рака лёгкого инокулировали в 24-луночный планшет, в который предварительно помещали клетки клеточной линии J2 фибробластов мыши, облученных Y-лучами (приобретено у Kerafast), с плотностью 4×104 клеток/см2, культивировали в 24-луночном планшете с использованием среды FM для кондиционного перепрограммирования клеток (подробное описание этапов см. Liu et al., Am J Pathol, 183(6): 1862-1870,2013);
C. Первичные клетки рака лёгкого инокулировали в 24-луночный планшет, покрытый матриксом Матригель (Matrigel®, производства BD Biosciences), с плотностью 4×104 клеток/см2, и культивировали в 24-луночном планшете с использованием 2 мл коммерческой среды для культивирования EpiM;
D. Первичные клетки рака лёгкого инокулировали в 24-луночный планшет, покрытый матриксом Матригель (Matrigel®, производства BD Biosciences), с плотностью 4×104 клеток/см2, и культивировали в 24-луночном планшете с использованием 2 мл основной среды ВМ.
В указанных выше четырех культурах клетки культивировали в перечисленных различных условиях культивирования (4 случая), при этом среду обновляли каждые 4 дня. В то же время наблюдали за образованием клеточных клонов и статусом пролиферации клеток при культивировании в каждой из сред в 24-луночном планшете, а состояние роста клеток регистрировали путем фотографирования с помощью микроскопа (EVOS М500, Invitrogen).
В отношении первичных клеток рака лёгкого (No. В8), культивируемых по методике изобретения, когда клетки в культуральном планшете вырастали и покрывали приблизительно 80% площади поверхности дна, среду удаляли и в 24-луночный планшет добавляли 500 мкл 0,05%-ного раствора трипсина (приобретено у GIBCO) для диссоциации клеток, а затем инкубировали при 37°С в течение 10 минут до полной диссоциации клеток, за которой наблюдали под микроскопом (EVOS М500, Invitrogen); затем ферментативную диссоциацию останавливали, используя 1 мл среды для культивирования DMEM/F12, содержащей 5% (об./об.) эмбриональной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина; полученную смесь собирали в центрифужную пробирку объемом 15 мл и центрифугировали при 1500 об/мин в течение 4 минут, а затем удаляли надосадочную жидкость. Центрифугированный клеточный осадок ресуспендировали в среде для культивирования по изобретению и клетки подсчитывали с помощью счетчика для визуализации клеток в потоке (JIMBIOFIL, Jiangsu Jimbio Technology Co., Ltd.) для вычисления общего количества клеток, составившего 464000. Клетки, выращенные в других трех условиях культивирования, подвергали ферментативной диссоциации и подсчитывали с использованием описанного выше рабочего способа. Общее количество клеток, культивируемых с использованием сред FM, EpiM и ВМ, составило 350000, 110000 и 68000, соответственно.
Фотографии клеток на Фиг. 5A-5D представляют собой микроскопические фотографии (полученные под инвертированным фазово-контрастным микроскопом 40х) клеток образца под номером В8, культивируемых в четырех различных условиях культивирования до 12 дня: Фиг. 5А представляет собой микроскопическую фотографию клеток образца В8, культивируемых до 12-го дня с использованием базовой среды ВМ; Фиг. 5 В представляет собой микроскопическую фотографию клеток образца В8, культивируемых до 12-го дня с использованием среды для культивирования FLM по изобретению; Фиг. 5С представляет собой микроскопическую фотографию клеток образца В8, культивируемых до 12-го дня с использованием коммерческой среды для культивирования EpiM; Фиг. 5D представляет собой микроскопическую фотографию клеток образца В8, культивируемых до 12-го дня с использованием среды FM, используемой в методике кондиционного перепрограммирования клеток. Как видно из представленных фигур, образец В8 не может образовывать клоны клеток при культивировании в основной среде ВМ (фиг. 5А) в течение 12 дней; только небольшое количество клонов клеток образуется при культивировании в среде EpiM (фиг. 5С) в течение 12 дней, и клетки находятся в плохом состоянии; клетки демонстрируют определенную степень размножения при культивировании в среде FM, используемой в методике кондиционного перепрограммирования клеток (фиг. 5D) в течение 12 дней, но плотность клеток и количество клеток не сопоставимы с таковыми в среде FLM по изобретению.
Фиг. 6 представляет собой сравнительную таблицу эффектов клеточной пролиферации, полученных при культивировании первичных клеток рака лёгкого, выделенных из девяти образцов, взятых от пациентов с раком лёгкого, согласно Примеру 1, в течение 16 дней в условиях четырех различных сред для культивирования, где знак √ означает умеренную способность к образованию клонов и к эффекту стимулирования пролиферации, знак √√ означает значительную способность к образованию клонов и к эффекту стимулирования пролиферации, знак √√√ означает чрезвычайно выраженную способность к образованию клонов и к эффекту стимулирования пролиферации, и знак × означает отсутствие образования клонов. Из Фиг. 6 следует, что среда для культивирования по изобретению имеет значительное превосходство над другими тремя средами с другими условиями культивирования с точки зрения способности к образованию клонов, стимулирующего действия на пролиферацию клеток и показателя успешности в получении культуры при культивировании первичных клеток, полученных из тканей рака лёгкого.
(2) Длительное культивирование и кривая роста первичных клеток рака лёгкого в различных средах для культивирования
Среду для культивирования FLM для первичных эпителиальных клеток рака лёгкого го и среды для культивирования ВМ, FM и EpiM в качестве контролей получали с использованием того же способа, что и в пункте (1) данного Примера.
Первичные клетки рака лёгкого, полученные из раковой ткани лёгкого (образец № В9) культивировали в четырех различных условиях культивирования с использованием того же способа, что и в пункте (1) данного Примера, а затем подвергали ферментативной диссоциации, пересевали и подсчитывали.
Когда после пересева клетки вырастали в культуральном планшете и снова покрывали приблизительно 80% площади поверхности дна планшета, культивируемые клетки подвергали ферментативной диссоциации, собирали и подсчитывали с использованием описанного выше рабочего способа. Клетки снова инокулировали с плотностью 4×104 клеток/лунку и длительно культивировали.
Ниже приведена формула для расчета показателя удвоения клеточной популяции первичных эпителиальных клеток лёгкого в различных условиях культивирования:
Удвоение популяции (PD, Population Doubling)=3,32*log10 (общее число собранных при ферментативной диссоциации клеток/число клеток при начальной инокуляции), см. Chapman et al., Stem Cell Research & Therapy 2014, 5: 60.
На Фиг. 7 показаны кривые роста клеток, полученных из образца В9, в четырех различных условиях культивирования, построенные с использованием программного обеспечения Graphpad Prism. По оси абсцисс представлена продолжительность культивирования клеток в днях, а по оси ординат - кратность пролиферации, представляет собой кратность размножения клеток в течение цикла культивирования. Чем больше значение, тем больше раз клетки размножались в течение определенного цикла, то есть тем больше клеток амплифицируется. Значение наклона кривой представляет собой скорость размножения клеток. Из рисунка следует, что скорость пролиферации эпителиальных клеток рака лёгкого, культивируемых в среде для культивирования FLM по изобретению, была выше, чем в других четырех условиях культивирования, и также следует, что среда для культивирования по настоящему изобретению позволяет длительно культивировать первичные эпителиальные клетки рака лёгкого, и скорость размножения остается неизменной до 30 дней.
[Пример 5]
Иммуногистохимическая идентификация первичных тканей рака лёгкого и клеток рака лёгкого после пассирования культуры
Опухолевую ткань (Образец № В12) размером с фасолину брали из хирургически резецированного образца от пациента с раком лёгкого и погружали в 1 мл 4%-ного раствора параформальдегида. Эпителиальные клетки рака лёгкого (Образец № В12) получали из оставшейся опухолевой ткани тем же способом, что и в Примере 1. Клетки образца В12 длительно культивировали до четвертого пассажа с использованием среды для культивирования FLM по изобретению, в соответствии со способом Примера 3.
Иммуногистохимический анализ использовали для выявления экспрессии значимых биомаркеров, связанных с раком лёгкого, в исходной ткани образца В12 и первичных клетках, полученных путем непрерывного культивирования до четвертого пассажа. Ткань фиксировали 4%-ным раствором параформальдегида, заливали в парафин и нарезали на срезы ткани толщиной 4 мкм с помощью микротома. Затем проводили обычное иммуногистохимическое исследование (подробно об этапах см. у Li et al., Nature Communication, (2018) 9: 2983). В качестве первичных антител использовали антитела к TTF-1 (приобретено у CST) и антитела к Ki-67 (приобретено у R&D).
Фиг. 8 подтверждает, что экспрессия связанных с раком биомаркеров на клетках, культивированных из клеток рака лёгкого (Образец № В12) в среде для культивирования по настоящему изобретению до четвертого пассажа, по существу соответствовала экспрессии маркеров на исходном срезе ткани, из которой получали клетки. Это свидетельствует о том, что клетки, культивированные в среде для культивирования по настоящему изобретению, сохраняют исходные патологические характеристики опухолевых тканей пациентов с раком лёгкого.
[Пример 6]
Влияние удаления одного фактора из среды для культивирования на длительное размножение и культивирование первичных клеток рака лёгкого
Среду для культивирования FLM для первичных эпителиальных клеток рака лёгкого получали с использованием того же способа, что и в Примере 2. В качестве контроля использовали тот же способ, что и в Примере 2, для получения основной среды ВМ. Кроме того, другие 8 сред для культивирования готовили в соответствии с Таблицей 7.

В одном случае первичные клетки рака лёгкого (образец № В13), полученные из тканей рака лёгкого, получали с использованием того же способа, что и в Примере 1. Первичные клетки рака лёгкого инокулировали в 48-луночный планшет, покрытый матриксом Матригель (Matrigel®, производства BD Biosciences), с плотностью 4×104 клеток/см2, и культивировали с 2 мл среды для культивирования (FLM) для первичных эпителиальных клеток рака лёгкого по изобретению в инкубаторе в условиях 37°С, 5% СО2 (приобретено у Thermo Fisher).
Когда клетки в культуральном планшете вырастали и покрывали приблизительно 80% площади поверхности дна, среду в 48-луночном планшете удаляли и добавляли 200 мкл 0,05%-ного раствора трипсина (приобретено у Gibco) для диссоциации клеток, а затем инкубировали при 37°С в течение 10 минут до полной диссоциации клеток, за которой наблюдали под микроскопом (EVOS М500, Invitrogen); затем ферментативную диссоциацию останавливали, используя 800 мкл среды для культивирования DMEM/F12, содержащей 5% (об./об.) эмбриональной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина; полученную смесь собирали в центрифужную пробирку объемом 15 мл и центрифугировали при 1500 об/мин в течение 4 минут, а затем удаляли надосадочную жидкость. Отцентрифугированный клеточный осадок ресуспендировали в среде для культивирования по изобретению и подсчитывали клетки с помощью счетчика для визуализации клеток в потоке (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) для вычисления общего количества клеток. Клетки инокулировали в другой 48-луночный планшет, покрытый гелеобразным внеклеточным матриксом, с плотностью 2×104 клеток/см2 для дальнейшего культивирования.
Клетки, культивированные в других 8 средах для культивирования и в среде БМ, диссоциировали, пересевали и подсчитывали с использованием тех же способов, что и выше, и культивировали с использованием различных сред.
Когда после пересева клетки вырастали в культуральном планшете и снова покрывали приблизительно 80% площади поверхности дна планшета, культивируемые клетки подвергали ферментативной диссоциации, собирали и подсчитывали с использованием описанного выше рабочего способа. Клетки снова инокулировали с плотностью 2×104 клеток/см2 и длительно культивировали.
Ниже приведена формула для расчета показателя удвоения клеточной популяции первичных эпителиальных клеток рака лёгкого в условиях культивирования в различных средах для культивирования:
Удвоение популяции (PD, Population Doubling)=3,32*log10 (общее число собранных при ферментативной диссоциации клеток/число клеток при начальной инокуляции), см. Chapman et al., Stem Cell Research & Therapy 2014, 5: 60.
Фигура 9 представляет собой график кривых роста клеток в условиях десяти различных по составу сред для культивирования, построенный с использованием программного обеспечения Graphpad Prism. По оси абсцисс представлена продолжительность культивирования клеток в днях, а по оси ординат - кратность пролиферации, представляет собой кратность размножения клеток в течение цикла культивирования. Чем больше значение, тем больше раз клетки размножались в течение определенного цикла, то есть тем больше клеток амплифицируется. Значение наклона кривой представляет собой скорость размножения клеток.
На Фиг. 10 показаны результаты сбора и подсчета клеток после культивирования в течение 8 дней в условиях десяти различных по составу сред для культивирования.
Из результатов на Фиг. 9 и 10 видно, что после удаления по отдельности различных малых молекул и добавочных факторов из среды для культивирования по изобретению эффект пролиферации клеток в определенной степени ослабляется. Это позволяет предположить, что указанные компоненты в среде для культивирования по изобретению необходимы для пролиферации клеток и длительного культивирования.
[Пример 7]
Эксперименты по туморогенезу с помощью ксенотрансплантации первичных клеток рака лёгкого, полученных из раковых тканей, на мышах
Клетки рака лёгкого (образец № В15) выделяли и получали из опухолевых тканей пациента с диагнозом рака лёгкого с использованием того же способа, что и в Примере 1. Клетки образца В15 культивировали с использованием среды для культивирования FLM по изобретению по способу Примера 3, и когда количество клеток рака лёгкого достигало числа 1×107, клетки рака лёгкого диссоциировали и собирали с использованием того же способа, что и в Примере 4. Среду для культивирования FLM для клеток рака лёгкого по изобретению и матрикс Матригель (Matrigel®, приобретено у BD Biosciences) смешивали в соотношении 1:1 и 100 мкл среды для культивирования, смешанной с матриксом, использовали для ресуспендирования клеток рака лёгкого в количестве 5×106, и полученную в результате суспензию инъецировали в жировую клетчатку подмышечной впадины правой передней конечности 6-недельных самок мышей с тяжелым иммунодефицитом (NCG-мыши) (приобретено в Нанкинском центре исследования модельных животных, Nanjing Model Animal Research Center), соответственно. Объемы и скорость роста у мышей опухолей, полученных из клеток рака лёгкого, обследовали и делали фотографии каждые три дня.
Образование опухоли можно наблюдать в обоих из двух мест инокуляции опухолевых клеток у мышей на 15-й день после инокуляции опухолевых клеток. С 15-го по 30-й день пролиферация опухоли у мышей становилась заметной. Это указывает на то, что происходящие из раковой ткани клетки рака лёгкого, культивированные с применением способа культивирования по настоящему изобретению, обладают туморогенностью у мышей.
[Пример 8]
Функциональный тест на чувствительность к лекарственным средствам клеток рака лёгкого, полученных из тканей рака
С использованием извлеченного путем хирургической резекции образца от пациента с раком лёгкого в качестве примера подтверждено, что клетки рака лёгкого, культивированные из происходящих от пациента образцов рака лёгкого, могут применяться для тестирования чувствительности опухолевых клеток пациента к различным лекарственным средствам.
1. Посев первичных клеток рака лёгкого: Суспензии выделенных клеток рака лёгкого (образцы № В13 и № В15), полученные в соответствии со способом Примера 1, инокулировали в 12-луночный планшет, покрытый матриксом Матригель (Matrigel®, приобретено у BD Biosciences), с плотностью 4×104 клеток/см2. 2 мл приготовленной среды для культивирования FLM для первичных эпителиальных клеток рака лёгкого добавляли в 12-луночный планшет, который затем помещали в инкубатор в условиях 37°С, 5% СО2 (приобретено у Thermo Fisher) для культивирования. Когда клетки в культуральном планшете вырастали и покрывали приблизительно 80% площади поверхности дна, среду удаляли и в 12-луночный планшет добавляли 500 мкл 0,05%-ного раствора трипсина (приобретено у Gibco) для диссоциации клеток, а затем инкубировали при 37°С в течение 10 минут до полной диссоциации клеток, за которой наблюдали под микроскопом (EVOS М500, Invitrogen); затем ферментативную диссоциацию останавливали, используя 1 мл среды для культивирования DMEM/F12, содержащей 5% (об./об.) эмбриональной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина; полученную смесь собирали в центрифужную пробирку объемом 15 мл и центрифугировали при 1500 об/мин в течение 4 минут, а затем удаляли надосадочную жидкость. Отцентрифугированный клеточный осадок ресуспендировали в среде для культивирования FLM и подсчитывали клетки с помощью счетчика для визуализации в потоке (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) для вычисления общего количества клеток, которое составило 830000 и 768000, соответственно. Клетки инкубировали в 384-луночном планшете с плотностью 2000-4000 клеток/лунку, и клетки прикреплялись к поверхности в течение ночи.
2. Эксперименты с градиентом (серийными разведениями) лекарственных средств:
(1) Планшеты для хранения лекарственных средств готовили способом серийных разведений: 10 мкл исходных растворов лекарственного средства (исходные растворы лекарственного средства готовили с концентрацией, в 2 раза превышающей максимальную концентрацию Cmax в плазме в организме человека), подлежащего тестированию, соответствующим образом пипетировали и добавляли в пробирки ЕР (типа эппендорф) объемом 0,5 мл, содержащие 20 мкл ДМСО; 10 мкл растворов из указанных выше пробирок ЕР снова переносили пипетированием во вторые пробирки ЕР объемом 0,5 мл, в которые добавлено 20 мкл ДМСО, таким образом, разбавляя препараты в соотношении 1:2 (т.е. в 3 раза). Вышеописанный способ повторяли для поэтапного серийного разведения и получали 8 концентраций, необходимых для дозирования. Различные концентрации лекарственных средств добавляли в 384-луночный планшет для хранения лекарственных средств. В каждую лунку контрольной группы растворителя добавляли равный объем ДМСО в качестве контроля. В этом примере тестируемыми препаратами были паклитаксел (приобретено у МСЕ), гемцитабин (приобретено у МСЕ), афатиниб (приобретено у МСЕ) и эрлотиниб (приобретено у МСЕ).
(2) Используя высокопроизводительную автоматизированную рабочую станцию (JANUS, Perkin Elmer), различные концентрации лекарственных средств и контролен растворителя в 384-луночных планшетах для хранения лекарственных средств добавляли в 384-луночные планшеты для культивирования клеток с клетками рака лёгкого. Группы лекарственных средств и контрольные группы с растворителем готовились в 3 дублирующих лунках. Объем лекарственных средств, добавляемых в каждую лунку, составлял 100 мкл.
(3) Тест на жизнеспособность клеток: через 72 часа после добавления лекарственного средства использовали набор Cell Titer-Glo (приобретено у Promega) для определения значения хемилюминесценции культивируемых клеток после добавления лекарственного средства. Величина значения хемилюминесценции отражает жизнеспособность клеток и влияние лекарственного средства на жизнеспособность клеток. В каждую лунку добавляли 10 мкл приготовленного раствора для детекции Cell Titer-Glo и после перемешивания использовали устройство для считывания микропланшетов (Envision, Perkin Elmer) для определения значения хемилюминесценции.
(4) Тест на жизнеспособность клеток: В соответствии с формулой, выживаемость клеток (%)=(значение хемилюминесценции в лунке с лекарственным средством/значение хемилюминесценции в контрольной лунке)*100%, рассчитывали показатели выживаемости клеток, обработанных различными лекарственными средствами. Графики строили с использованием программного обеспечения Graphpad Prism и рассчитывали значения показателя концентрации полуингибирования IC50.
(5) Результаты тестирования на чувствительность к лекарственным средствам представлены на Фиг. 11.
На Фиг. 11А и 11В, соответственно, представлена чувствительность клеток рака лёгкого, полученных из извлеченных путем хирургической резекции образцов раковой ткани от двух разных пациентов с раком лёгкого (образец № В13 и образец № В15), к двум химиотерапевтическим препаратам, паклитакселу и гемцитабину, а также к двум препаратам направленного действия, афатинибу и эрлотинибу. В частности, на Фиг. 11А показаны результаты исследования чувствительности клеток рака лёгкого, полученных из образца № В13, к четырем лекарственным средствам; на Фиг. 11 В показаны результаты исследования чувствительности клеток рака лёгкого, полученных из образца № В15, к четырем лекарственным средствам. Результаты показывают, что клетки одного и того же пациента имеют разную чувствительность к разным препаратам, и клетки разных пациентов также имеют разную чувствительность к одному и тому же препарату.
Несмотря на то, что изобретение было подробно описано в общем и в конкретных вариантах реализации выше, для специалистов в данной области техники понятно, что на основе этого описания могут быть произведены некоторые модификации или усовершенствования. Таким образом, указанные модификации или усовершенствования, которые не отступают от существа изобретения, входят в объем охраны, заявленный настоящим изобретением.
Промышленная применимость
Согласно изобретению, предложены среда для культивирования и способ культивирования, для культивирования или размножения первичных эпителиальных клеток рака лёгкого in vitro. Культивируемые клетки можно использовать для оценки эффективности и скрининга лекарственных средств. Следовательно, изобретение применимо в промышленности.
Хотя изобретение подробно описано в настоящем документе, изобретение не ограничивается этим, и специалисты в данной области могут внести изменения в соответствии с принципом изобретения. Поэтому все модификации, выполненные в соответствии с принципом изобретения, должны рассматриваться как входящие в объем охраны изобретения.
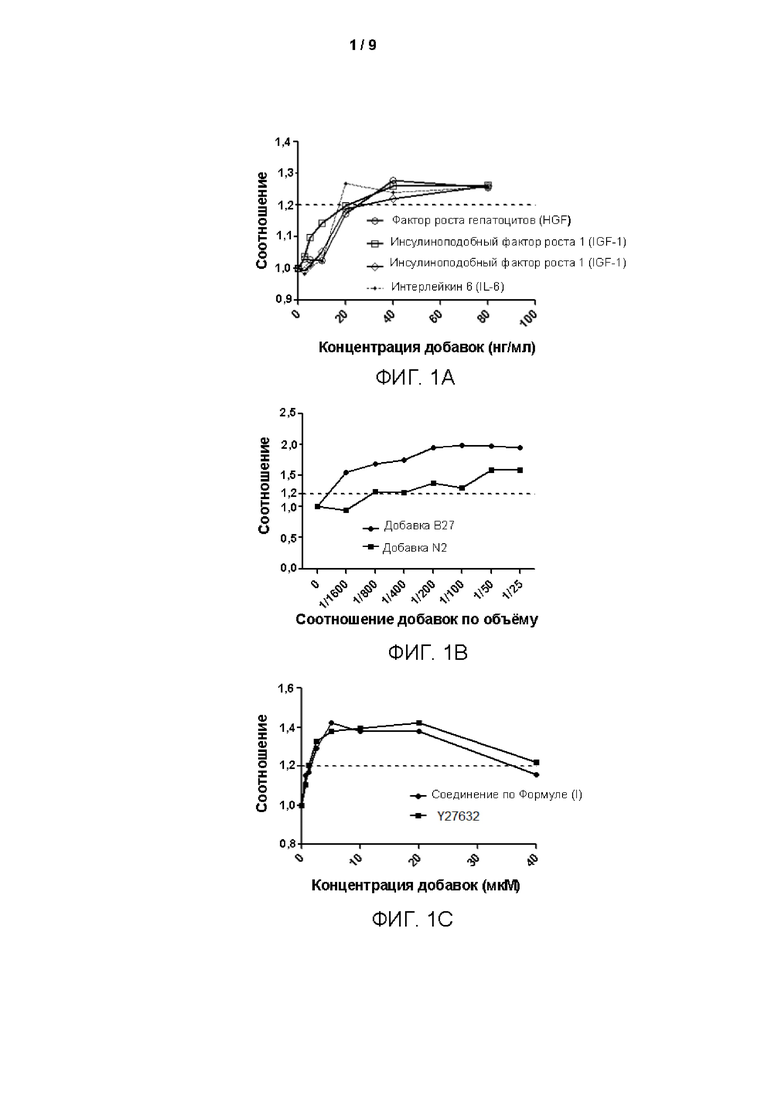
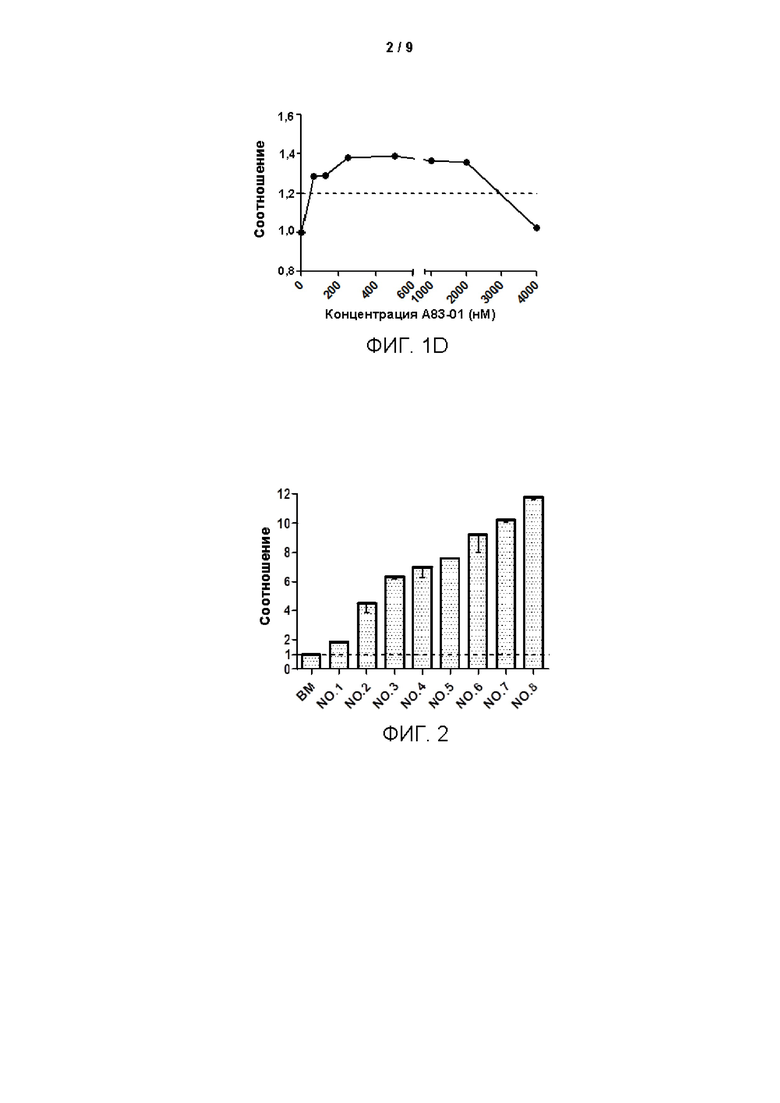


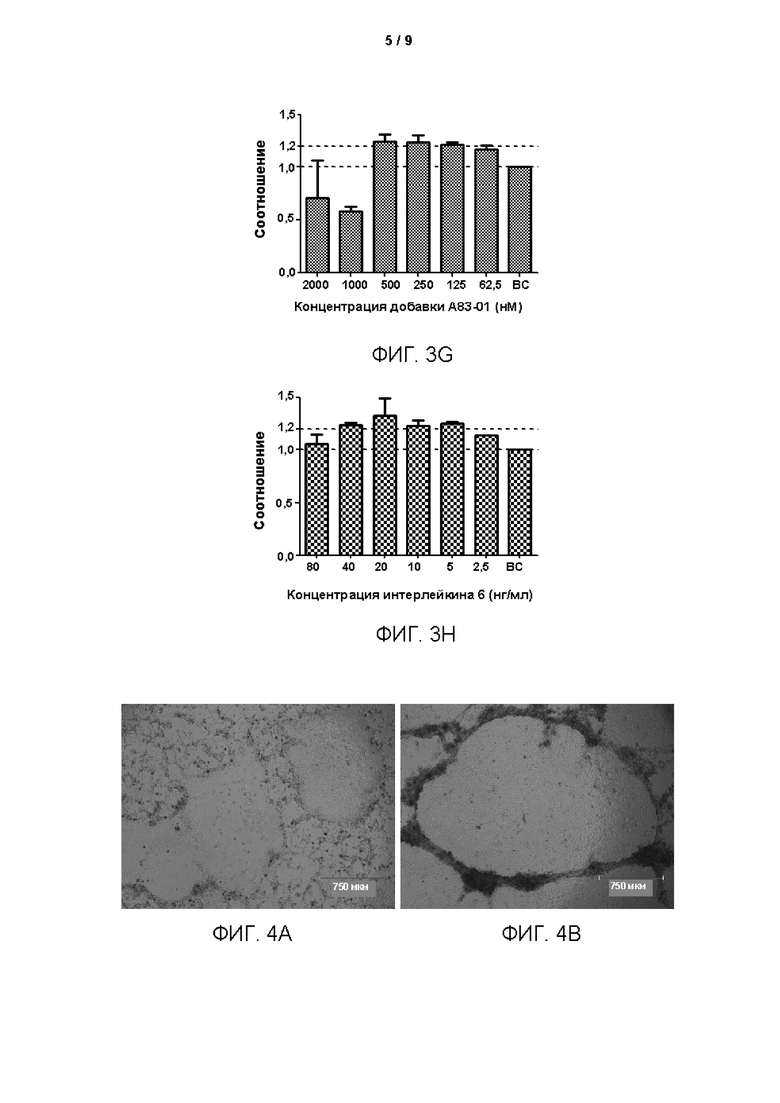

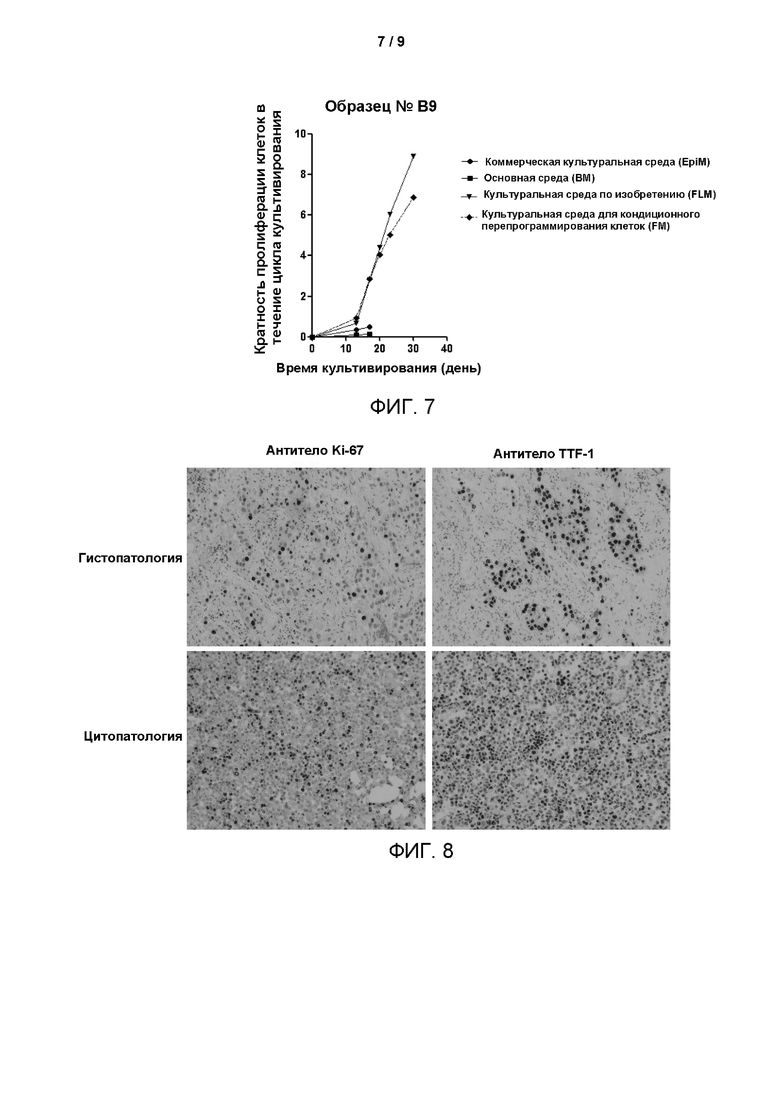
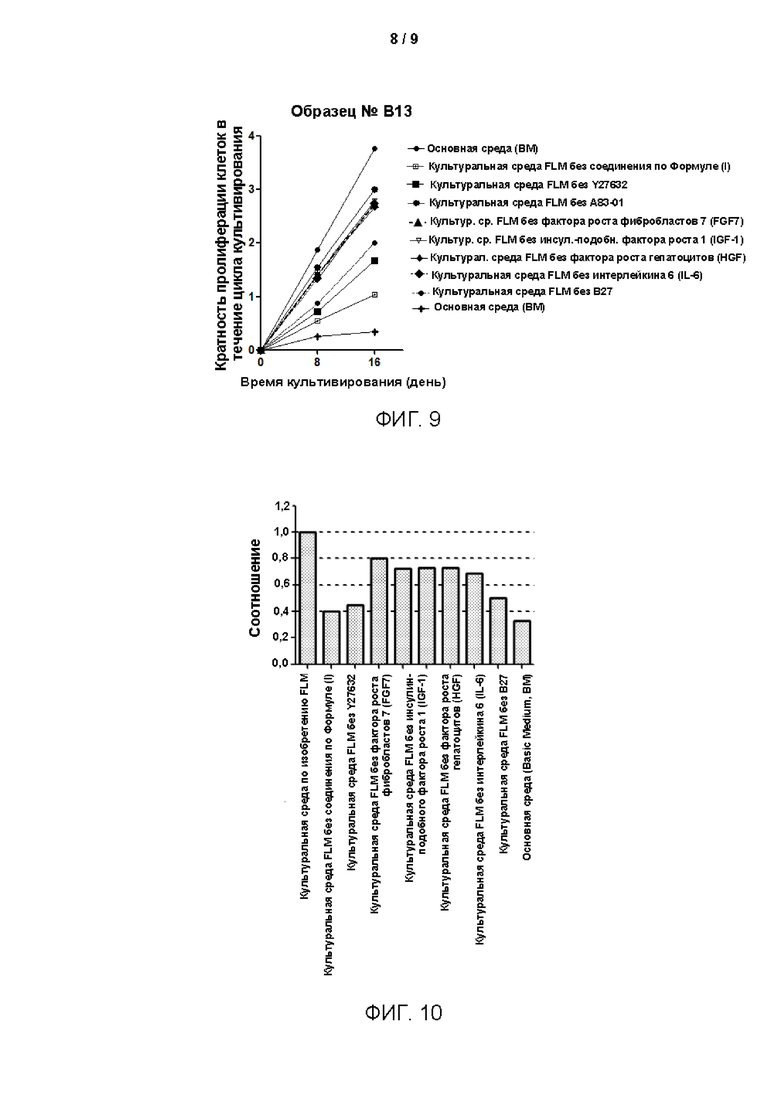
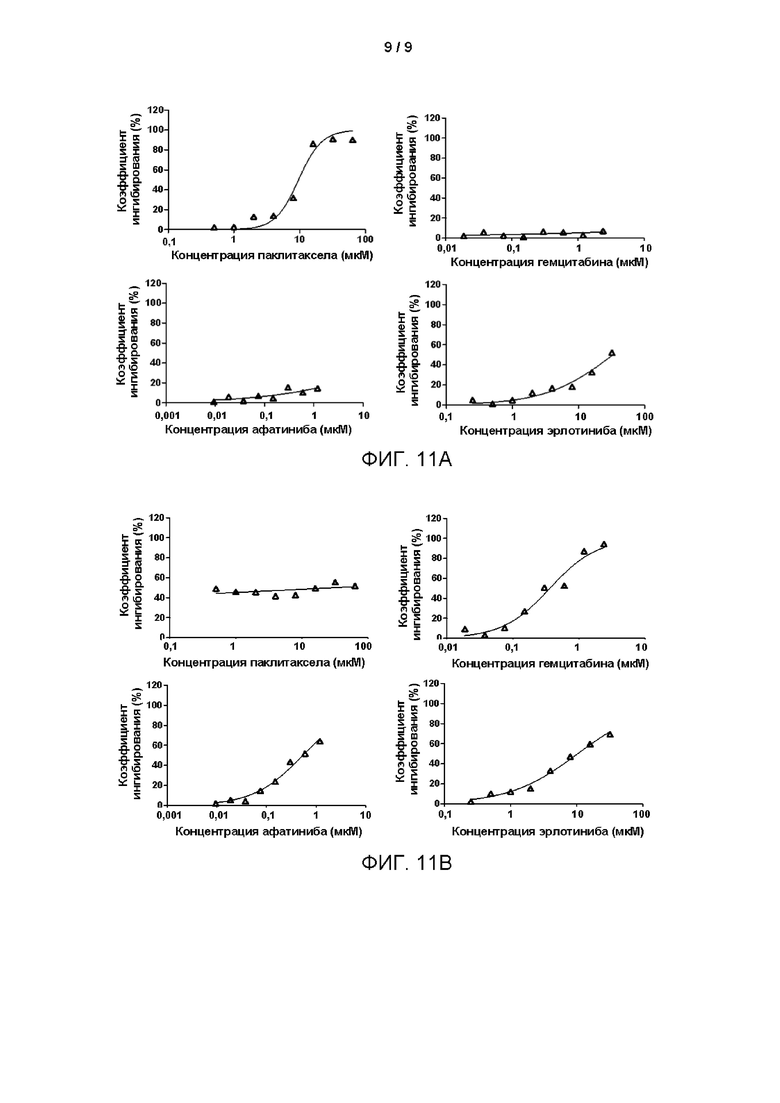
Группа изобретений относится к биотехнологии. Предложены среда для культивирования первичных эпителиальных клеток рака лёгкого, способ их культивирования и способ определения чувствительности к лекарственному средству. Среда для культивирования содержит ингибитор киназы MST1/2, по меньшей мере один ингибитор ROCK, фактор роста фибробластов 7, по меньшей мере одну добавку, выбранную из добавки B27 и добавки N2, фактор роста гепатоцитов, инсулиноподобный фактор роста 1, интерлейкин 6, по меньшей мере один ингибитор рецептора TGFβ I типа, выбранный из A83-01, SB431542, Repsox, SB505124, SB525334, SD208, LY36494 и SJN2511, исходную среду, выбранную из DMEM/F12, DMEM, F12 или RPMI-1640, и один или более антибиотиков, выбранных из стрептомицина/пенициллина, амфотерицина B и примоцина. Указанную среду используют для культивирования первичных эпителиальных клеток рака лёгкого в сосуде, покрытом гелеобразным внеклеточным матриксом. Полученную клеточную модель применяют для скрининга лекарственных средств. Изобретения обеспечивают короткий период культивирования in vitro без влияния экзогенных клеток. 3 н. и 8 з.п. ф-лы, 11 ил., 7 табл., 8 пр.
1. Среда для культивирования первичных эпителиальных клеток рака лёгкого, причем указанная среда содержит:
ингибитор киназы MST1/2 в количестве 2,5-10 мкМ; по меньшей мере один ингибитор ROCK, выбранный из группы, состоящей из Y27632, фасудила и H-1152, в количестве 2,5-15 мкМ; фактор роста фибробластов 7 в количестве 5-160 нг/мл; по меньшей мере одну добавку, выбранную из группы, состоящей из добавки B27 и добавки N2, объёмная концентрация которой в среде для культивирования составляет 1:25-1:800; фактор роста гепатоцитов в количестве 5-160 нг/мл; инсулиноподобный фактор роста 1 в количестве 5-160 нг/мл; интерлейкин 6 в количестве 2,5-40 нг/мл; по меньшей мере один ингибитор рецептора TGFβ I типа, выбранный из группы, состоящей из A83-01, SB431542, Repsox, SB505124, SB525334, SD208, LY36494 и SJN2511, в количестве 62,5-500 нМ; исходную среду, выбранную из группы, состоящей из DMEM/F12, DMEM, F12 или RPMI-1640; и один или более антибиотиков, выбранных из группы, состоящей из стрептомицина/пенициллина, амфотерицина B и примоцина;
причем указанный ингибитор киназы MST1/2 включает соединение Формулы (I), или его фармацевтически приемлемую соль, или сольват:
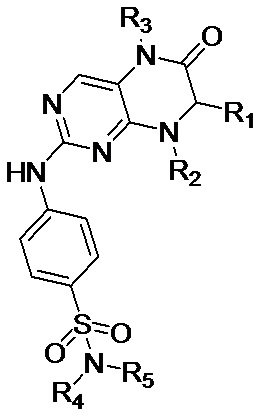 Формула (I),
Формула (I),
где:
R1 выбран из C1-C6-алкила, C3-C6-циклоалкила, C2-C6-спироциклоалкила, фенила, необязательно замещённого 1-2 независимыми R6, и тиенила, необязательно замещённого 1-2 независимыми R6;
каждый из R2 и R3 независимо выбраны из C1-C3-алкила;
каждый из R4 и R5 независимо выбраны из водорода, C1-C6-алкила, C3-C6-циклоалкила и C4-C8-циклоалкилалкила;
радикал R6 выбран из галогена, C1-C6-алкила и C1-C6-галогеналкила.
2. Среда для культивирования клеток по п. 1, отличающаяся тем, что указанный ингибитор киназы MST1/2 содержит соединение Формулы (Ia), или его фармацевтически приемлемую соль, или сольват:
 Формула (Ia),
Формула (Ia),
где:
R1 выбран из C1-C6 алкила, фенила, необязательно замещённого 1-2 независимыми R6, и тиенила, необязательно замещённого 1-2 независимыми R6;
R5 выбран из водорода, C1-C6-алкила и C3-C6-циклоалкила;
R6 независимо выбран из галогена, C1-C6-алкила и C1-C6-галогеналкила.
3. Среда для культивирования клеток по п. 2, отличающаяся тем, что:
R1 представляет собой фенил, необязательно замещённый 1-2 независимыми R6;
R5 представляет собой водород;
R6 представляет собой фтор, метил или трифторметил.
4. Среда для культивирования клеток по п. 1, отличающаяся тем, что указанный ингибитор киназы MST1/2 представляет собой по меньшей мере одно соединение, выбранное из следующих соединений или их фармацевтически приемлемых солей:

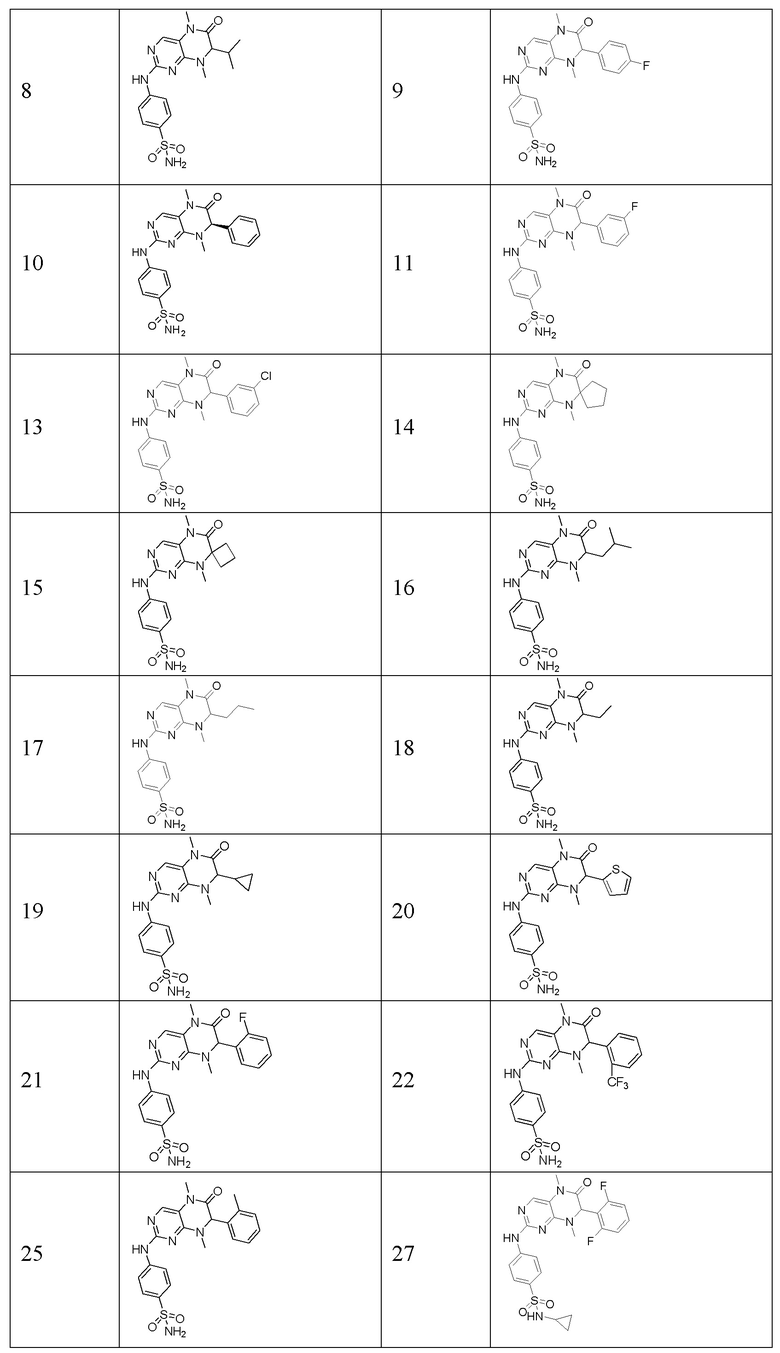
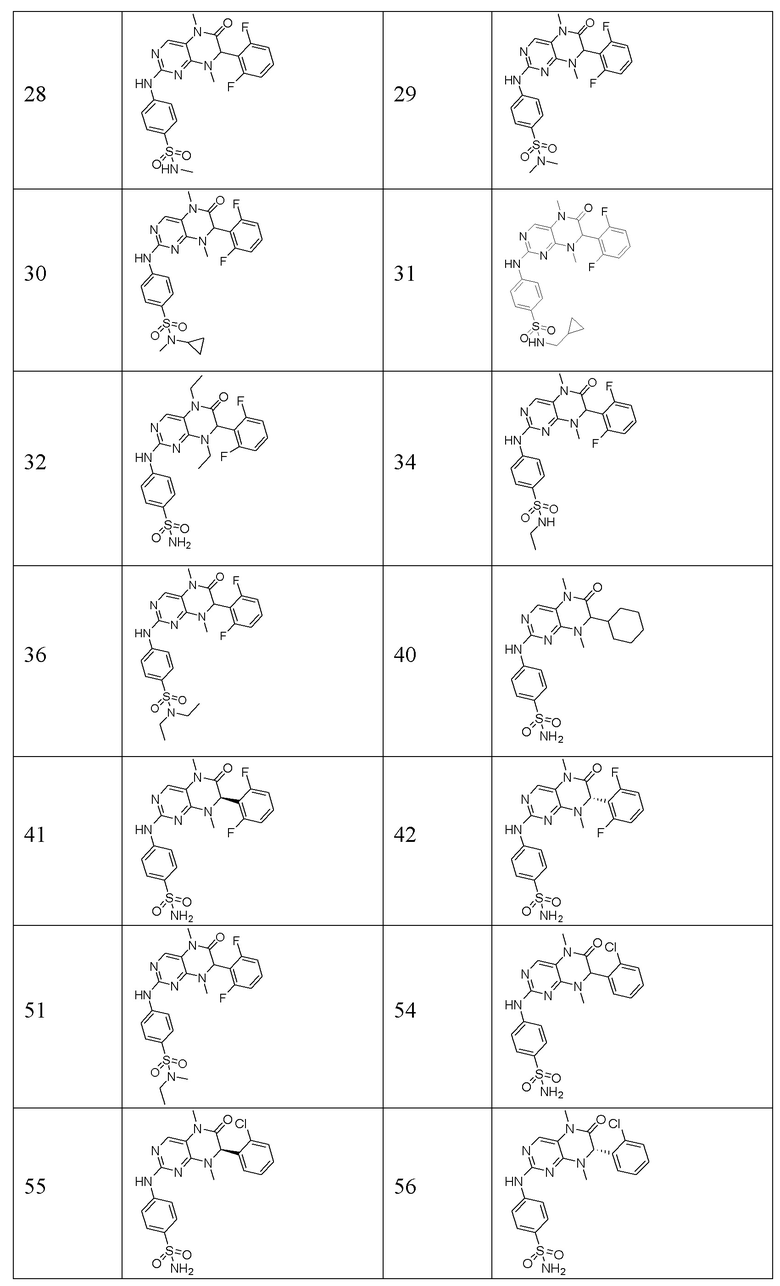
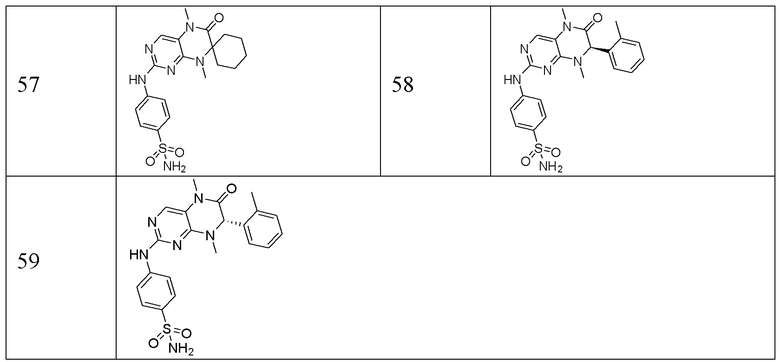
5. Среда для культивирования клеток по любому из пп. 1-4, отличающаяся тем, что количество указанного ингибитора киназы MST1/2 в указанной среде для культивирования составляет 5-7,5 мкМ.
6. Среда для культивирования клеток по любому из пп. 1-4, отличающаяся тем, что указанная среда для культивирования удовлетворяет любому одному или более или всем из следующих условий:
количество ингибитора ROCK в среде для культивирования составляет 5-15 мкМ, предпочтительно 10-12,5 мкМ;
количество фактора роста фибробластов 7 составляет 10-80 нг/мл, предпочтительно 20-40 нг/мл;
объёмная концентрация добавки B27 или добавки N2 в среде для культивирования составляет 1:25-1:200, предпочтительно 1:50-1:100;
количество фактора роста гепатоцитов составляет 20-80 нг/мл;
количество инсулиноподобного фактора роста 1 составляет 20-80 нг/мл;
количество интерлейкина 6 составляет 5-40 нг/мл;
количество ингибитора рецептора TGFβ I типа составляет 250-500 нМ.
7. Среда для культивирования клеток по любому из пп. 1-4, отличающаяся тем, что указанная среда для культивирования удовлетворяет любому одному или более или всем из следующих условий:
ингибитор киназы MST1/2 представляет собой соединение Формулы (I);
ингибитор ROCK представляет собой Y27632;
ингибитор рецептора TGFβ I типа представляет собой A83-01.
8. Среда для культивирования клеток по любому из пп. 1-4, отличающаяся тем, что указанная среда не содержит сыворотки, экстракта бычьего гипофиза, агонистов Wnt, белков семейства R-спондинов, ингибиторов BMP, никотинамида или N-ацетилцистеина.
9. Среда для культивирования клеток по любому из пп. 1-4, отличающаяся тем, что первичные эпителиальные клетки рака лёгкого выбраны из группы, состоящей из:
клеток рака лёгкого, нормальных эпителиальных клеток рака лёгкого и эпителиальных стволовых клеток рака лёгкого.
10. Способ культивирования первичных эпителиальных клеток рака лёгкого, причем указанный способ включает следующие этапы:
(1) приготовление среды для культивирования клеток по любому из пп. 1-9;
(2) покрытие сосуда для культивирования разбавленным раствором гелеобразного внеклеточного матрикса, выбранного из Матригеля и BME;
(3) инокуляция первичных эпителиальных клеток рака лёгкого, выделенных из тканей рака лёгкого, в сосуд для культивирования, который покрыт гелеобразным внеклеточным матриксом, и культивирование с использованием среды для культивирования клеток, приготовленной на этапе (1).
11. Способ определения чувствительности первичных эпителиальных клеток рака лёгкого к лекарственному средству, причем указанный способ включает следующие этапы:
(1) культивирование эпителиальных клеток рака лёгкого способом культивирования по п. 10;
(2) выбор испытываемого лекарственного средства и серийное разведение с получением различных концентраций указанного лекарственного средства;
(3) добавление указанного лекарственного средства, которое было серийно разведено, к эпителиальным клеткам рака лёгкого, полученным на этапе (1), и определение жизнеспособности клеток.
| CN 108624561 A, 09.10.2018 | |||
| CN 111039944 A, 21.04.2020 | |||
| ПИРИДИНОВЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ СЕМЬ АТОМОВ В КОЛЬЦЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ УКАЗАННЫЕ СОЕДИНЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2748696C2 |

