Область техники
Настоящее изобретение относится к области биотехнологии, в частности, к среде для культивирования и способу культивирования для быстрого размножения первичных клеток рака кишечника, и их применению для оценки эффективности и скрининга лекарственных средств.
Уровень техники
Рак кишечника представляет собой заболевание, вызванное взаимодействием генетических факторов и факторов окружающей среды, и является одной из наиболее распространенных опухолей желудочно-кишечного тракта во всем мире. Согласно оценке (Shu Zheng et аl., Prevention of Colorectal Cancer, Chinese Journal of Oncology, 2004, vol. 13, NO. 1, pp1 - 2), рак кишечника является одной из наиболее распространенных опухолей, и общий уровень заболеваемости и уровень смертности растут, вследствие чего эта злокачественная опухоль серьезно угрожает жизни и здоровью. Хирургическое вмешательство в сочетании с послеоперационной химиотерапией в настоящее время является основным методом лечения рака кишечника. Хотя хирургические методы в последние годы улучшились, что привело к повышению уровня выживаемости пациентов с раком кишечника, метастазирование и рецидив опухоли все еще являются причиной неблагоприятного прогноза для пациентов. С точки зрения точного лечения колоректального рака, появление таргетных лекарственных средств дало надежду пациентам с колоректальным раком на поздней стадии, и будущими направлениями развития является рациональный выбор таргетных лекарственных средств и составление индивидуализированных планов лечения. Постоянное внедрение новых технологий тестирования чувствительности к лекарственным средствам обеспечивает надежную техническую поддержку для прогнозирования эффективности таргетных лекарственных средств, химиотерапевтических средств и комбинаций таргетных лекарственных средств и закладывает прочную основу для реализации индивидуализированного лечения пациентов с раком кишечника.
Известные линии клеток рака кишечника, культивируемые in vitro, в основном получают путем длительного культивирования нормальных клеток для спонтанной иммортализации или путем трансфекции онкогенами, которые способствуют иммортализации нормальных клеток. Линии клеток, созданные обычными способами, остаются основой исследований клеточной, молекулярной биологии и биологии рака. Тем не менее, эти способы изменяют генетический фон клеток, и длительно культивируемые линии клеток также склонны к нестабильности генома, что может привести к неестественным изменениям фенотипа линий опухолевых клеток и опухолевых клеток in vivo. Эти линии клеток обычно не обладают сложной гетерогенностью первичной опухоли, что ограничивает применение этих линий клеток для прогнозирования ответа опухолевых клеток и влияет на точность научных исследований и разработки лекарственных средств для лечения рака кишечника. Кроме того, в процессе культивирования клеток, полученных из тканей рака кишечника, в раковые клетки трудно получить раковые клетки обычными способами культивирования, и существуют проблемы в процессе культивирования, такие как мешающее влияние фибробластов, то, что образовавшиеся клоны не могут быть субкультивированы, что ограничивает применение первичных клеток рака кишечника человека.
В 2017 году Xuefeng Liu et ai, использовали облученные фибробласты мыши и ингибитор Rho-ассоциированной киназы (Y-27632) для размножения клеток эпителиального происхождения. Эта система обладает способностью достигать неограниченного роста клеток эпителиального происхождения без генетической манипуляции (Xuefeng Liu et ai,, Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat. Protoc. 2017, 12, 439). Тем не менее способ, разработанный Xuefeng Liu et ai., подразумевает длительный период культивирования и не может обеспечить быстрое размножение клеток, что также ограничивает применение этой технологии.
Краткое описание изобретения
Для решения вышеуказанных технических проблем в соответствии с настоящим изобретением предложены среда для культивирования (культуральная среда) и способ культивирования для быстрого размножения первичных клеток рака кишечника in vitro.
В одном аспекте настоящего изобретения предложено обеспечение среды для культивирования для первичных клеток рака кишечника, содержащей исходную среду для культивирования, ингибитор протеазы Rho, антибиотик, гастрин, А8301, заменимую аминокислоту, холерный токсин, инсулиноподобный фактор роста-1, никотинамид, инсулин, фетальную бычью сыворотку и добавку, выбранную из по меньшей мере одной из добавки В27 и добавки N2. Исходная среда выбрана из группы, состоящей из DMEM/F12, DMEM, F12 или RPMI-1640.
Предпочтительные варианты реализации настоящего изобретения включают по меньшей мере один из следующих аспектов:
(1) ингибитор протеазы Rho представляет собой один или более ингибиторов, выбранных из группы, состоящей из Y27632, фасудила и Н-1152, предпочтительно Y27632, и концентрация предпочтительно составляет 2,5-40 мкМ, более предпочтительно 2,5-5 мкМ, наиболее предпочтительно 5 мкМ;
(2) антибиотик представляет собой один или более антибиотиков, выбранных из группы, состоящей из стрептомицина/пенициллина, амфотерицина В и примоцина, в случае стрептомицина/пенициллина, концентрация стрептомицина находится в диапазоне 25-400 мкг/мл, предпочтительно 50-200 мкг/мл, более предпочтительно 200 мкг/мл, концентрация пенициллина находится в диапазоне 25-400 Ед/мл, предпочтительно 50-200 Ед/мл, более предпочтительно 200 Ед/мл; в случае амфотерицина В, его концентрация находится в диапазоне 0,25 - 4 мкг/мл, предпочтительно 0,5-2 мкг/мл; и в случае примоцина, его концентрация находится в диапазоне 25 - 400 мкг/мл, предпочтительно 50 - 200 мкг/мл;
(3) диапазон концентраций гастрина предпочтительно составляет 1,25-20 нМ, более предпочтительно 2,5-10 нМ, наиболее предпочтительно 5 нМ;
(4) диапазон концентраций А8301 предпочтительно составляет 25-200 нМ, более предпочтительно 200 нМ;
(5) заменимая аминокислота представляет собой одну или более аминокислот из группы, состоящей из глицина, аланина, аспарагина, аспарагиновой кислоты, глутаминовой кислоты, пролина и серина, и общий диапазон концентраций заменимых аминокислот составляет 25-400 мкМ, более предпочтительно 50 мкМ;
(6) диапазон концентраций холерного токсина предпочтительносоставляет 1,25-20 нг/мл, более предпочтительно 2,5-5 нг/мл, наиболее предпочтительно 5 нг/мл;
(7) диапазон концентраций инсулиноподобного фактора роста-1 предпочтительно составляет 5-405 нг/мл, более предпочтительно 45 нг/мл;
(8) диапазон концентраций никотинамида предпочтительно составляет 2-8 мМ, более предпочтительно 4-8 мМ, наиболее предпочтительно 4 мМ;
(9) диапазон концентраций инсулина предпочтительно составляет 0,5 -4 мкг/мл, более предпочтительно 1 - 2 мкг/мл, наиболее предпочтительно 2 мкг/мл;
(10) объемное отношение фетальной бычьей сыворотки к среде для культивирования предпочтительно составляет 2,5% (об./об.) - 5% (об./об.), более предпочтительно 5% (об./об.);
(11) объемное отношение добавки В27 или N2 к среде для культивирования предпочтительно составляет 1:25-1:200, более предпочтительно 1:25-1:50 и наиболее предпочтительно 1:25.
В соответствии с настоящим изобретением также предложен способ культивирования первичных клеток рака кишечника. В способе культивирования первичных клеток рака кишечника в соответствии с настоящим изобретением первичные клетки рака кишечника культивируют с применением среды для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением.
Способ культивирования первичных клеток рака кишечника в соответствии с настоящим изобретением включает следующие этапы:
1. Выделение первичных клеток рака кишечника.
(1) После промывки средой для промывки, в образцы тканей, эндоскопические образцы добавляют среду для промывки и раствор для расщепления тканей (количество добавленного раствора для расщепления тканей составляет приблизительно 5-10 мл на 1 г опухолевой ткани) в соотношении 1:3 и помещают в шейкер с постоянной температурой для расщепления при температуре расщепления от 4 до 37°С, предпочтительно 37°С; скорость вращения для расщепления составляет от 200 об/мин до 350 об/мин, предпочтительно 300 об/мин;
(2) расщепление можно остановить не ранее, чем не останется видимой тканевой массы, и время расщепления находится в диапазоне от 3 до 6 часов, предпочтительно составляет 4 часа;
(3) супернатант удаляют после центрифугирования со скоростьюцентрифугирования от 1200 до 1600 об/мин, предпочтительно 1500 об/мин, время центрифугирования составляет от 2 до 6 минут, предпочтительно 4 минуты; полученный осадок ресуспендируют с добавлением среды DMEM/F12, содержащей 10% сыворотки.
2. Культивирование с применением среды для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением.
Первичные клетки рака кишечника, полученные на этапе 1 выше, ресуспендируют в среде для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением и подсчитывают, а затем высевают на чашку для культивирования при плотности клеток 1-10×104 клеток/см2; при этом добавляют клетки трофобласта при плотности клеток 2-3×104 клеток/см2; клетки открепляют для пересевания после разрастания клеток до покрытия 90% или более площади чашки для культивирования.
В частности, состав среды для промывки, описанной на этапе 1, является следующим: среда DMEM/F12, содержащая 100 мкг/мл примоцина (приобретенного у InvivoGen, 0,2% (об./об.), концентрация коммерческого продукта составляет 50 мг/мл); состав раствора для расщепления тканей, описанного на этапе 1, является следующим: среда 1640 (Corning, 10-040-CVR), коллагеназа II (2 мг/мл), коллагеназа IV (2 мг/мл), ДНКаза (50 Ед/мл), гиалуронидаза (0,75 мг/мл), хлорид кальция (3,3 мМ), БСА (10 мг/мл); клетки трофобласта, описанные на этапе 2, представляют собой, например, облученные клетки NIH-3T3, и источник облучения представляет собой рентгеновское или γ-излучение, предпочтительно γ-излучение, при дозе облучения 20-50 Гр, предпочтительно 30 Гр.
В соответствии с настоящим изобретением также предложен способ скрининга лекарственных средств от рака кишечника, включающий следующие этапы:
(1) культивирование первичных клеток рака кишечника для скрининга лекарственных средств с применением способа культивирования первичных клеток рака кишечника в соответствии с настоящим изобретением;
(2) выбор лекарственного средства для исследования и разведение лекарственного средства на основании требуемых градиентов концентрации;
(3) добавление лекарственного средства в различных градиентах концентрации к клеткам, культивируемым и полученным на этапе (1);
(4) детектирование жизнеспособности клеток.
Техническое решение в соответствии с настоящим изобретением позволяет получить следующие технические эффекты:
(1) показатель успешности культивирования первичных клеток рака кишечника может быть улучшен, с достижением показателя успешности 80% или более;
(2) первичные клетки рака кишечника, культивированные in vitro, могут сохранять патологические характеристики, присутствующие у пациентов;
(3) культивированию первичных клеток рака кишечника не мешают мезенхимальные клетки, такие как фибробласты и адипоциты;
(4) первичные клетки рака кишечника могут быть размножены с высокой эффективностью, с успешным размножением клеток до порядка величины 106 при начальном количестве клеток на уровне 105 в течение приблизительно одной недели, и размноженные первичные клетки рака кишечника обладают способностью к непрерывному пассированию;
(5) стоимость культивирования является контролируемой, поскольку среда для культивирования не требует дорогостоящих агонистов Wnt, белков семейства R-спондинов, ингибиторов BMP, FGF10 и т.п.факторов;
(6) эта технология позволяет культивировать и обеспечить первичные клетки рака кишечника в больших количествах и с высокой однородностью, что подходит для высокопроизводительного скрининга новых соединений-кандидатов и высокопроизводительных функциональных тестов на чувствительность к лекарственным средствам in vitro для пациентов.
Краткое описание чертежей
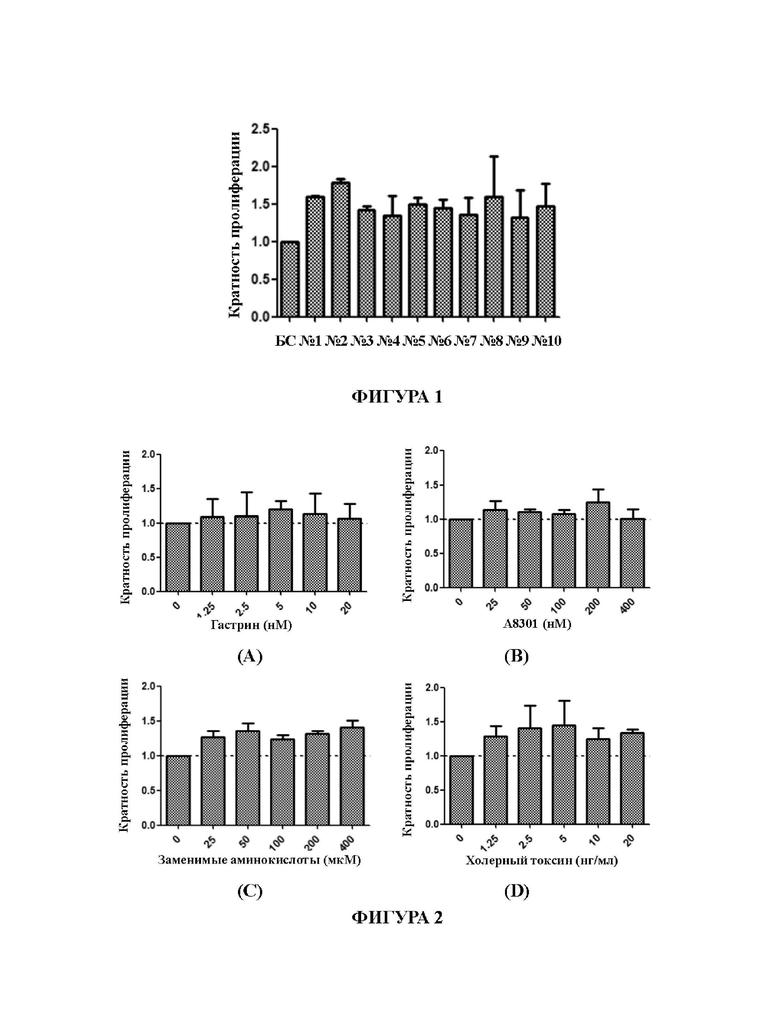
На Фиг. 1 представлен график, демонстрирующий влияние комбинаций различных факторов, добавленных в среду для культивирования для первичных клеток рака кишечника, на пролиферацию первичных клеток рака кишечника.
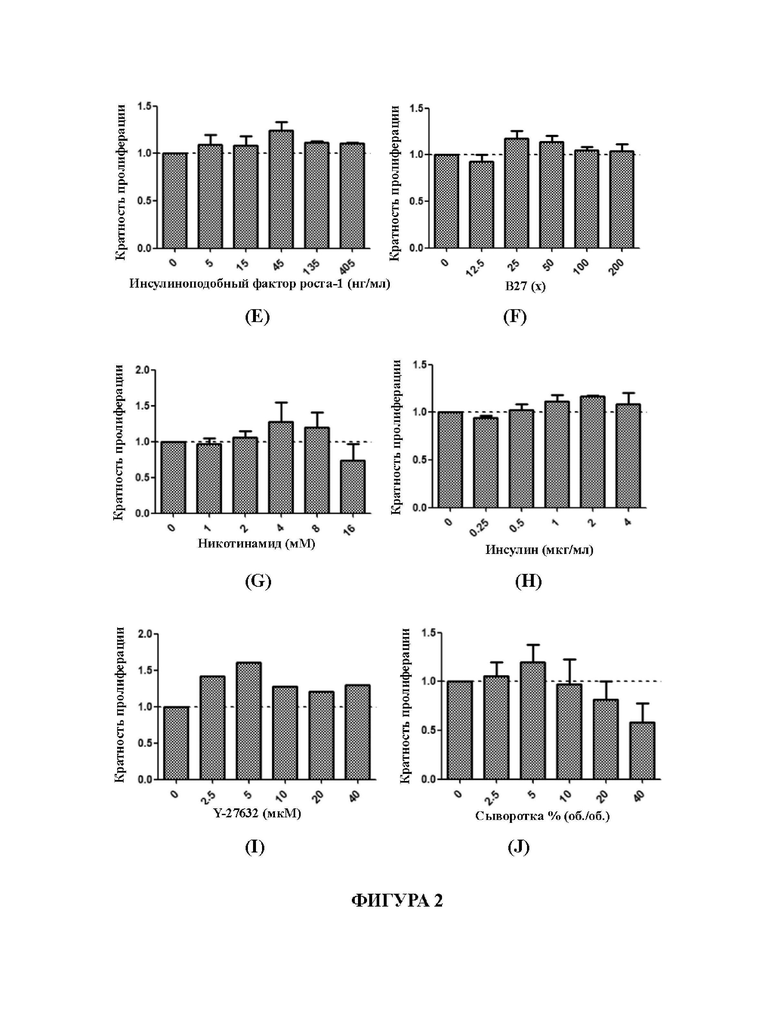
На Фиг. 2А-2J представлены графики, демонстрирующие влияние различных концентраций факторов, добавленных в среду для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением, на культуру первичных клеток рака кишечника.
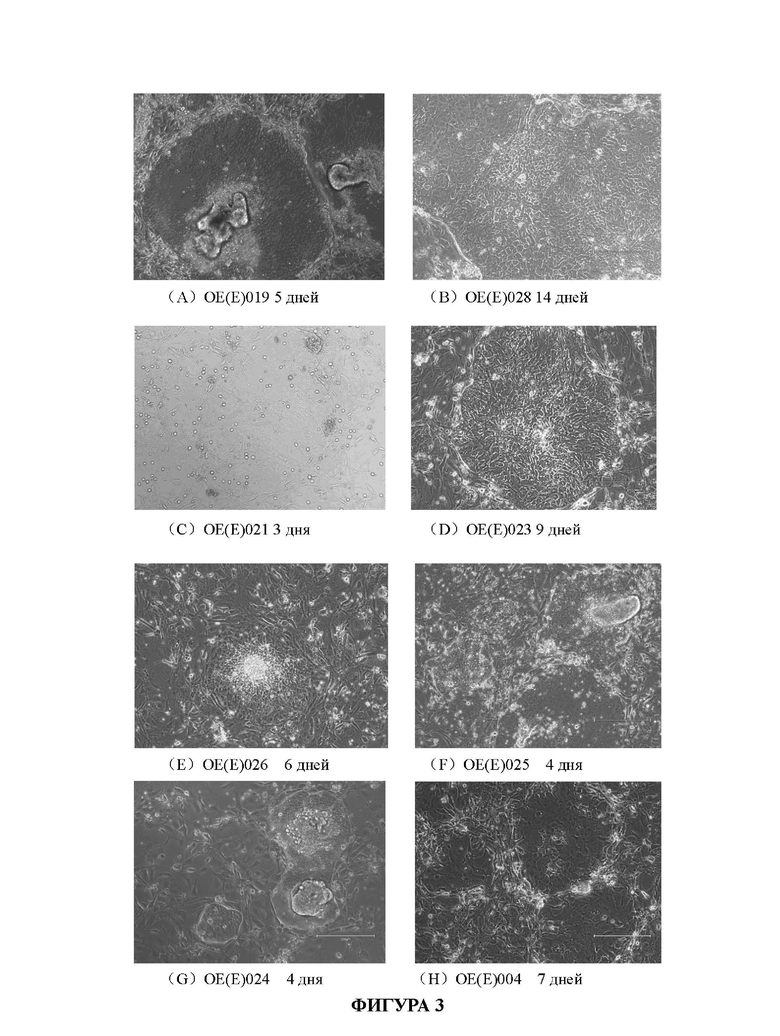
На Фиг. 3А-3Н показаны фотографии первичных клеток рака кишечника, полученные путем наблюдения с помощью микроскопа, при культивировании в среде для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением.
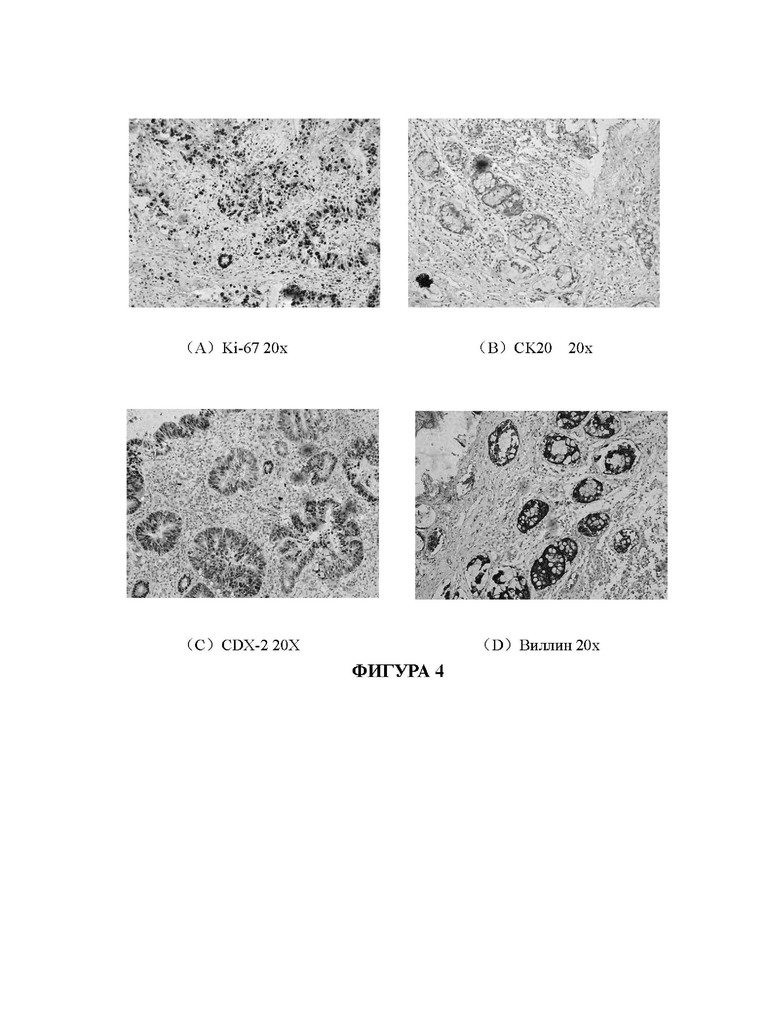
На Фиг. 4А-4D показаны результаты иммуногистохимического анализа клеток исходной ткани рака кишечника.
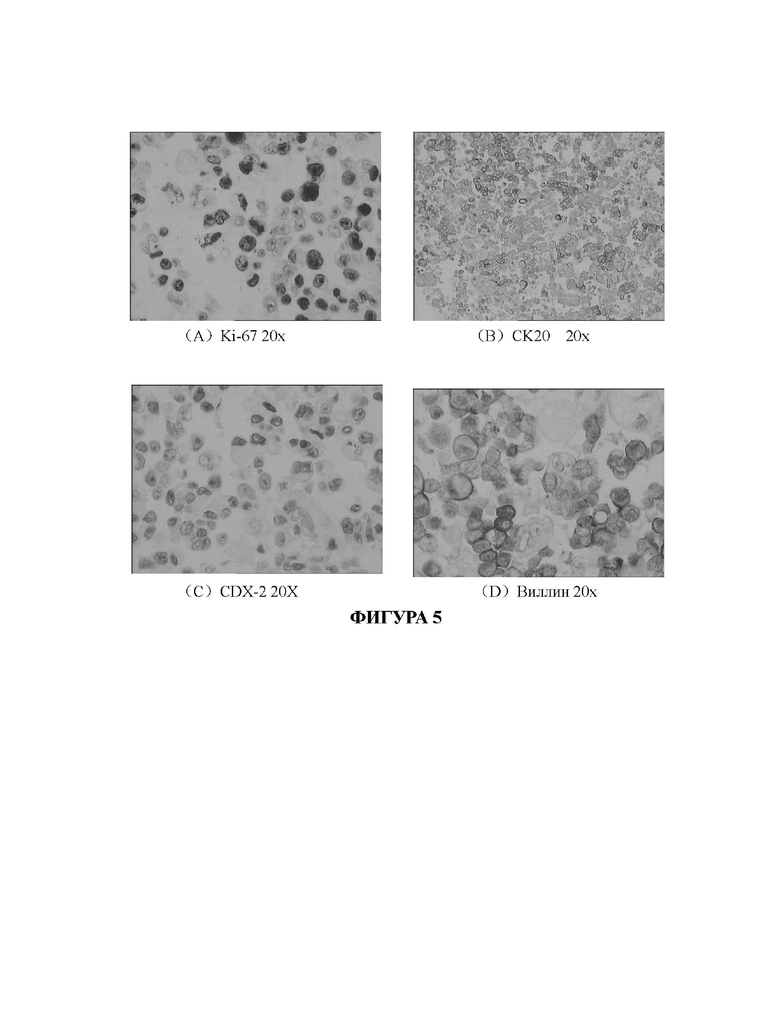
На Фиг. 5А-5D показаны результаты иммуногистохимического анализа первичных клеток рака кишечника, культивированных в среде для культивирования СА-1 в соответствии с настоящим изобретением.
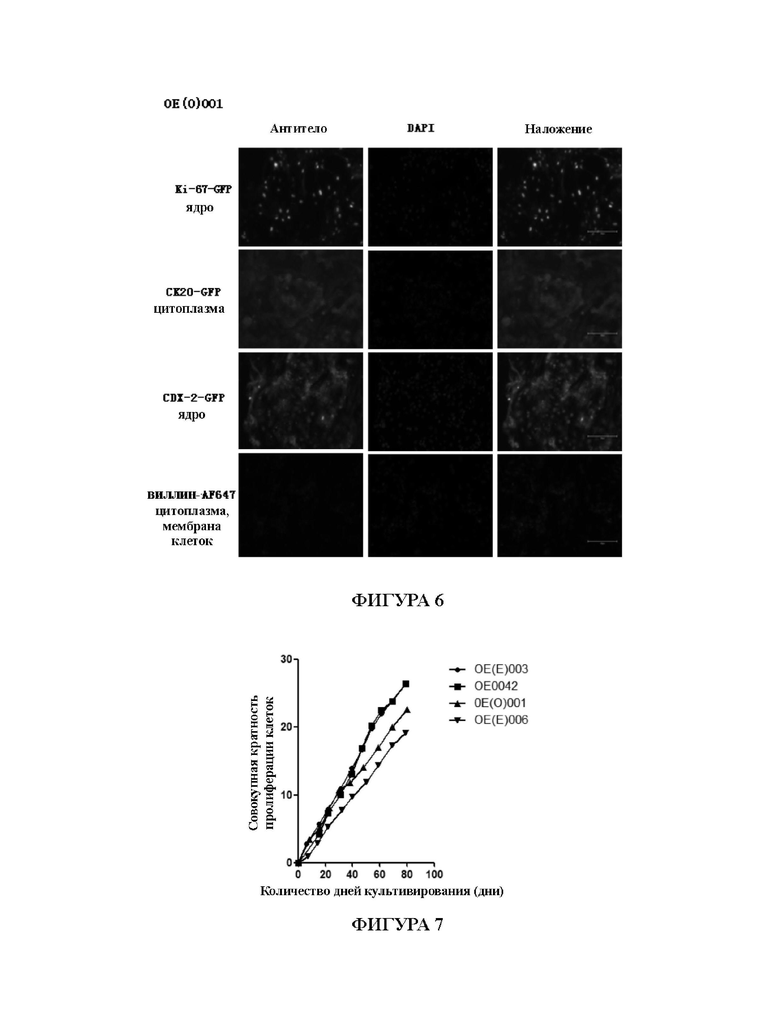
На Фиг. 6 показаны результаты иммунофлуоресцентного окрашивания первичных клеток рака кишечника, культивированных с применением среды для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением.
На Фиг. 7 показаны кривые роста первичных клеток рака кишечника, культивированных в среде для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением.
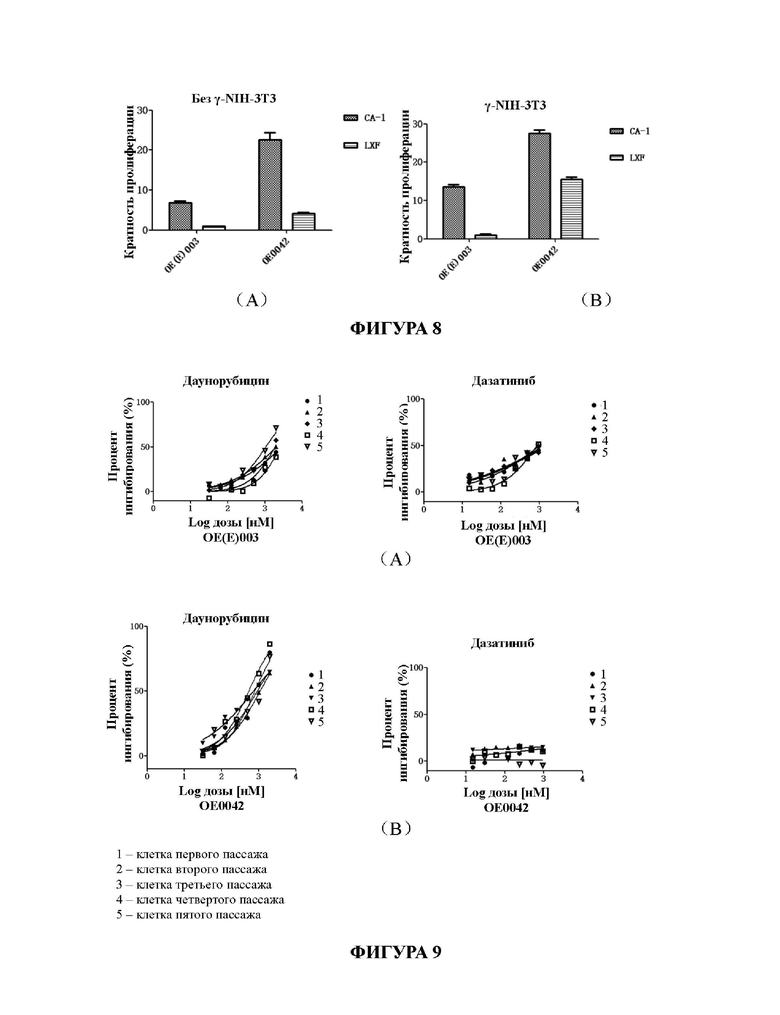
На Фиг. 8А и 8В представлены результаты сравнения культивирования первичных клеток рака кишечника с применением среды для культивирования СА-1 для первичных клеток рака кишечника в соответствии с настоящим изобретением и известной среды для культивирования LXF.
На Фиг. 9А и 9В представлены результаты скрининга лекарственных средств с применением разных поколений клеток рака кишечника, культивированных в среде для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением.
Подробное описание изобретения
Для того чтобы лучше понять настоящее изобретение, оно дополнительно описано ниже вместе с вариантами реализации и чертежами. Следующие примеры приведены исключительно с целью иллюстрирования, но не для ограничения настоящего изобретения.
Пример 1. Влияние соответствующих факторов, добавленных в среду для культивирования для первичных клеток рака кишечника, на пролиферацию первичных клеток рака кишечника.
(1) Получение среды для культивирования для первичных клеток рака кишечника.
Сначала готовили базовую среду. Состав базовой среды следующий: среда DMEM/F12 (приобретена у Corning)+5 мкМ Y27632 (приобретен у МСЕ)+5% (об./об.) фетальной бычьей сыворотки (приобретена у Excell Bio)+100 мкг/мл примоцина (приобретен у InvivoGen, 0,2% (об./об.), концентрация коммерческого продукта составляет 50 мг/мл).
Различные типы добавок (см. Таблицу 1) добавляли в базовую среду для получения сред для культивирования для первичных клеток рака кишечника, содержащих различные компоненты.
(2) Выделение и обработка первичных клеток рака кишечника
1. Выбор образца
Образцы тканей (интраоперационные/эндоскопические) солидных опухолей рака кишечника были получены от пациентов профессиональным врачом профессиональных медицинских учреждений, и все пациенты подписали информированное согласие. Размер интраоперационных образцов составлял 0,25 см3, а размер эндоскопических образцов составлял 0,025 см3; для хранения и транспортировки использовали доступный для приобретения раствор для сохранения тканей (производитель: Miltenyi Biotec).
2. Подготовка материалов.
После стерилизации поверхностей, стерильные пробирки для центрифугирования емкостью 15 мл, дозаторы, пипетки емкостью 10 мл, стерильные наконечники пипеток и т.д. помещали на ультрачистое рабочее место для облучения ультрафиолетом в течение 30 минут. Среду для промывки заранее вынимали из холодильника с температурой 4°С за 30 минут, а раствор для расщепления тканей заранее вынимали из холодильника с температурой -20°С за 30 минут.
Среда для промывки: среда DMEM/F12, содержащая 100 мкг/мл примоцина (приобретен у InvivoGen, 0,2% (об./об.), концентрация коммерческого продукта 50 мг/мл).
Раствор для расщепления тканей: среда 1640 (Corning, 10-040-CVR), коллагеназа II (2 мг/мл), коллагеназа IV (2 мг/мл), ДНКаза (50 Ед/мл), гиалуронидаза (0,75 мг/мл), хлорид кальция (3,3 мМ), БСА (10 мг/мл).
Коллагеназа II, коллагеназа IV, ДНКаза и гиалуронидаза, упомянутые выше, были приобретены у Sigma Corporation; хлорид кальция был приобретен у Sangon Biotech (Шанхай) Co., Ltd.; БСА был приобретен у Biofroxx Corporation.
3. Выделение образца.
3.1. Образцы ткани переносили с ультрачистого рабочего места в чашку для культивирования, и ткань с кровью удаляли. Образцы ткани дважды промывали средой для промывки, а затем переносили в другую чашку для культивирования и механически разрезали стерильным скальпелем на блоки ткани размером 1×1×1 мм3.
3.2. Вырезанные интраоперационные или эндоскопические ткани аспирировали в пробирку для центрифугирования емкостью 15 мл, в которую добавляли 5 мл среды для промывки, тщательно перемешивали, а затем центрифугировали при 1500 об/мин в течение 4 минут.
3.3. Супернатант удаляли, и к полученному осадку добавляли смесь 1:3 среды для промывки и раствора для расщепления тканей (количество добавленного раствора для расщепления тканей составляло приблизительно 10 мл раствора для расщепления тканей на 1 г опухолевой ткани). Образцы маркировали названиями и номерами, герметично запечатывали герметизирующей пленкой, а затем расщепляли на шейкере (Zhichu Instrument ZQLY-180N) при 37°С, 300 об/мин. Завершение расщепления определяли путем наблюдения каждые 30 минут, исходя из наличия или отсутствия видимых частиц. Время расщепления составляло 4 часа.
3.4. После того, как расщепление было завершено, нерасщепленную тканевую массу отфильтровывали через сетчатый фильтр с порами 100 мкм. Тканевую массу на фильтре промывали в пробирку для центрифугирования средой для промывки для уменьшения потери клеток. Полученный раствор центрифугировали при 25°С, 1500 об/мин в течение 4 минут.
3.5. Супернатант удаляли и визуально исследовали осадок на наличие клеток крови. Если клетки крови присутствовали, в осадок добавляли 8 мл лизирующего клетки крови реагента (Sigma) и тщательно перемешивали, лизировали при 4°С в течение 20 минут, однократно переворачивая и перемешивая во время процесса. Полученный раствор центрифугировали при 25°С, 1500 об/мин в течение 4 минут.
3.6. Супернатант удаляли и добавляли 2 мл среды DMEM/F12, содержащей 10% сыворотки (Excell Bio, FND500), для ресуспендирования клеток для сохранения.
4. Подсчет клеток и обработка.
4.1 Микроскопическое исследование: небольшое количестворесуспендированных клеток высевали в чашку для культивирования, и визуально исследовали плотность и морфологию раковых клеток под микроскопом (CNOPTEC, BDS400);
4.2 Подсчет жизнеспособных клеток: 12 мкл ресуспендированной суспензии клеток тщательно смешивали с 12 мкл окрашивающего раствора трипанового синего (Sangon Biotech (Shanghai) Co., Ltd.), а затем 20 мкл смеси добавляли в слайд-планшет для подсчета клеток (Countstar, комплектация: 50 шт. на коробку). Процент жизнеспособных крупных клеток (размер клеток>10 мкм)=количество жизнеспособных клеток/общее количество клеток ×100%, рассчитывали с помощью счетчика клеток (Countstar, IC1000).
(3) Культивирование первичных клеток рака кишечника.
Среды для культивирования с различными компонентами, представленными в Таблице 1, добавляли в 48-луночный планшет в объеме 1 мл/лунку. Первичные клетки рака кишечника, выделенные из тканей двух случаев рака кишечника (пронумерованы ОЕ0042 и ОЕ(Е)003) в соответствии с описанным выше этапом (2), высевали в 48-луночный культуральный планшет при плотности клеток 3×104 клеток/лунку и культивировали при 37°С при концентрации СО2 5%. После культивирования в течение 7-10 дней, когда клетки доросли до конфлюентности 85%, среду для культивирования удаляли и использовали 100 мкл/лунку 0,05% трипсина (приобретенного у Gibco) для однократной промывки клеток. После удаления трипсина в каждую лунку добавляли 200 мкл 0,05% трипсина. Затем планшет помещали в термостат при 37°С, 5% СО2 на 10 минут, и проводили визуальный анализ клеток под микроскопом (CNOPTEC, BDS400), который показал, что клетки полностью открепились. Для остановки открепления добавляли 300 мкл среды DMEM/F12, содержащей 10% сыворотки (Excell Bio, FND500). В планшет для подсчета клеток (Countstar, комплектация: 50 штук на коробку) добавляли 20 мкл полученной суспензии, и подсчитывали общее количество клеток с помощью счетчика клеток (Countstar, IC1000). В частности, в качестве контроля эксперимента использовали базовую среду без каких-либо добавок, и результаты эксперимента представлены в Таблице 1.
Таблица 1
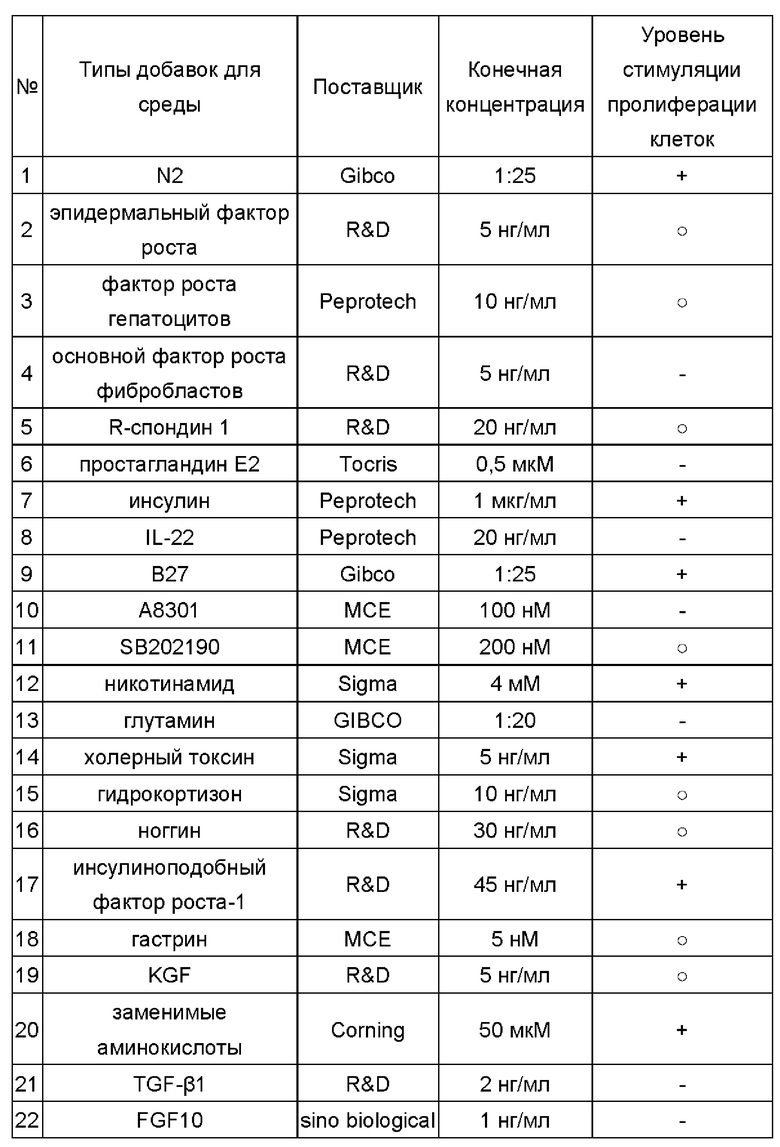
Где «+» показывает, что, по сравнению с базовой средой, среда с добавкой(-ами) оказывает эффект стимуляции пролиферации первичныхклеток рака кишечника, выделенных из ткани рака кишечника, по меньшей мере в двух случаях рака; «-» показывает, что среда с добавкой(-ами) оказывает эффект ингибирования пролиферации первичных клеток рака кишечника, выделенных из ткани рака кишечника, по меньшей мере в одном случае рака;  показывает, что среда с добавкой(-ами) не оказывает значимого эффекта на пролиферацию первичных клеток рака кишечника, выделенных из ткани рака кишечника, по меньшей мере в двух случаях рака.
показывает, что среда с добавкой(-ами) не оказывает значимого эффекта на пролиферацию первичных клеток рака кишечника, выделенных из ткани рака кишечника, по меньшей мере в двух случаях рака.
В соответствии с приведенными выше результатами для дальнейших экспериментов по культивированию были выбраны факторы, включая заменимую аминокислоту, холерный токсин, инсулиноподобный фактор роста-1, В27 или N2, никотинамид, инсулин, простагландин Е2, А8301 и гастрин.
Пример 2. Влияние комбинаций различных факторов, добавленных в среду для культивирования первичных клеток рака кишечника, на пролиферацию первичных клеток рака кишечника.
В соответствии с компонентами, представленными в Таблице 2, были получены среды для культивирования для первичных клеток рака кишечника с комбинациями различных добавленных факторов и исследованы эффекты стимуляции пролиферации первичных клеток рака кишечника комбинациями различных добавленных факторов.
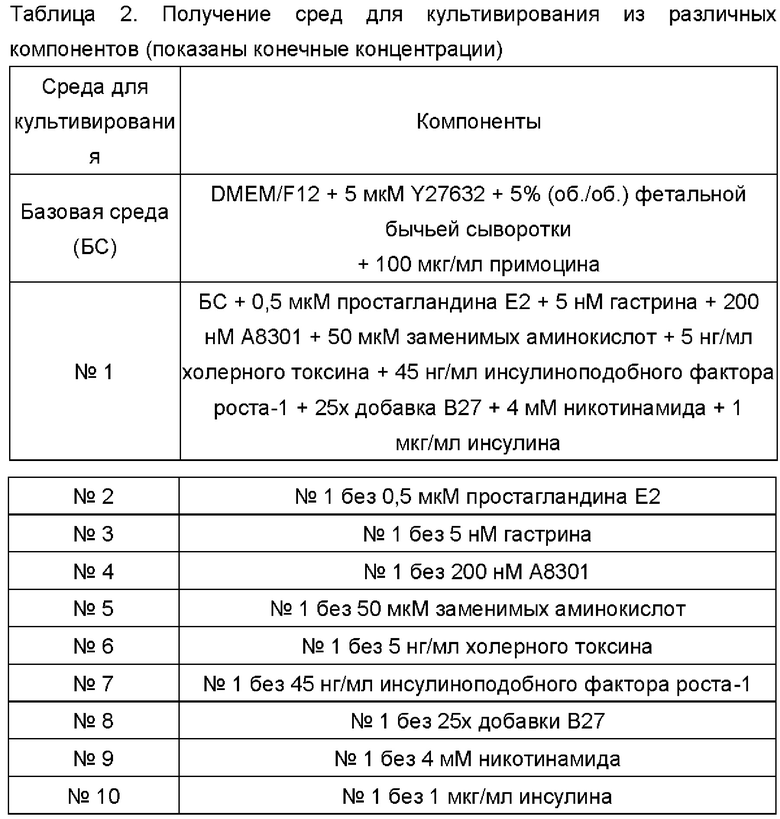
Первичные клетки рака кишечника получали из ткани рака кишечника (пронумерованной ОЕ0042, ОЕ(Е)006) в соответствии со способом, описанным на этапе (2)-3 Примера 1, и полученную суспензию клеток разделяли на 11 равных частей, которые центрифугировали при 1500 об/мин в течение 4 минут с последующим ресуспендированием в 200 мкл сред БС №1-10, соответственно. Клетки высевали в 48-луночный планшет при плотности живых клеток 2×104 клеток/см2 (20000 клеток на лунку), а затем в планшет добавляли облученные γ-излучением клетки NIH-3T3 (доза облучения 30 Гр) (приобретенные у АТСС, ресуспендированные в базовой среде (БС)) при плотности клеток 2×104 клеток/см2. Наконец, в каждую лунку 48-луночного планшета добавляли соответствующие среды для культивирования до объема 1 мл, и полученный раствор тщательно перемешивали. После дезинфекции поверхности планшет помещали в термостат при 37°С с содержанием СО25% (приобретенный у Thermo Fisher) для культивирования.
После того, как клетки наросли до покрытия 85% площади или более в 48-луночном планшете, среды удаляли. Клетки однократно промывали 100 мкл 0,05% трипсина (приобретенного у Gibco). После удаления трипсина в каждую лунку добавляли 200 мкл 0,05% трипсина. Планшет помещали в термостат при 37°С, 5% СО2 на 10 минут, и проводили визуальный анализ клеток под микроскопом (CNOPTEC, BDS400), который показал, что клетки полностью открепились. Для остановки открепления добавляли 300 мкл среды DMEM/F12, содержащей 10% сыворотки (Excell Bio, FND500). В планшет для подсчета клеток (Countstar, комплектация: 50 шт. на коробку) добавляли 20 мкл полученной суспензии и подсчитывали общее количество клеток с помощью счетчика клеток (Countstar, IC1000). Результаты, полученные для первичных клеток рака кишечника, выделенных из интраоперационного/эндоскопического образца ткани ОЕ0042, образца ткани ОЕ(Е)006 представлены на Фиг. 1.
Из результатов, показанных на Фиг. 1, можно сделать вывод, что, по сравнению с базовой средой, все указанные выше среды №1 - №10 могут на разных уровнях стимулировать пролиферацию первичных клеток рака кишечника. При применении среды для культивирования, содержащей Y27632, фетальную бычью сыворотку, гастрин, А8301, заменимые аминокислоты, холерный токсин, инсулиноподобный фактор роста-1, В27, никотинамид, инсулин (т.е. среду для культивирования №2, в дальнейшем называемую «средой СА-1»), для культивирования первичных клеток рака кишечника, был достигнут наилучший эффект пролиферации.
Пример 3. Влияние различных концентраций добавленных факторов в среде СА-1 на пролиферацию первичных клеток рака кишечника.
Первичные клетки рака кишечника получали из эндоскопических образцов ткани (пронумерованных ОЕ(Е)042, ОЕ(Е)050, ОЕ(Е)060) в соответствии со способом, описанным на этапе (2)-3 Примера 1, и культивировали клетки со средой СА-1 из Примера 2. Полученные первичные клетки рака кишечника высевали в 6-луночный планшет при плотности живых клеток 1×104 клеток/см2 (100000 клеток на лунку), а затем в планшет добавляли клетки NIH-3T3, облученные γ-излучением (доза облучения 30 Гр) при плотности клеток 2×104 клеток/см2, и хорошо перемешивали. После дезинфекции поверхности планшет помещали в термостат при 37°С, 5% СО2 (приобретенный у Thermo Fisher) для культивирования. Клетки культивировали и размножали в СА-1 до разрастания до конфлюентности 85% или более. Добавляли 500 мкл 0,05% трипсина (приобретенного у Gibco) для промывания клеток в течение 1 минуты. После удаления трипсина в каждую лунку добавляли 500 мкл 0,05% трипсина. Затем планшет помещали в термостат при 37°С, 5% СО2, на 2-10 минут до полного открепления клеток. Для остановки открепления добавляли 500 мкл среды DMEM/F12, содержащей 10% сыворотки (Excell Bio, FND500). Полученную суспензию центрифугировали при 1500 об/мин в течение 4 минут и супернатант удаляли. Осадки клеток ресуспендировали с применением DMEM/F12, и 20 мкл суспензии добавляли в планшет для подсчета клеток (производитель: Countstar, комплектация: 50 шт. накоробку). Общее количество клеток подсчитывали с помощью счетчика клеток (Countstar, IC1000). Полученные клетки использовали для следующих экспериментов по культивированию.
Затем готовили среды следующих 10 составов для экспериментов:
Состав 1: среда СА-1 без гастрина;
Состав 2: среда СА-1 без А8301;
Состав 3: среда СА-1 без заменимых аминокислот;
Состав 4: среда СА-1 без холерного токсина;
Состав 5: среда СА-1 без инсулиноподобного фактора роста-1;
Состав 6: среда СА-1 без В27;
Состав 7: среда СА-1 без никотинамида;
Состав 8: среда СА-1 без инсулина;
Состав 9: среда СА-1 без Y-27632;
Состав 10: среда СА-1 без фетальной бычьей сыворотки.
В каждую лунку добавляли 20 мкл суспензии клеток, содержащей 1×104 клеток, и использовали 1 мл указанных выше сред Состава 1-10 для разведения суспензии клеток, соответственно.
Когда использовали среду Состава 1, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного гастрина в конечных концентрациях гастрина, равных 1,25 нМ, 2,5 нМ, 5 нМ, 10 нМ, 20 нМ, соответственно; и среду Состава 1 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 2, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного А8301 в конечных концентрациях А8301, равных 25 нМ, 50 нМ, 100 нМ, 200 нМ, 400 нМ, соответственно; и среду Состава 2 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 3, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленных заменимых аминокислот в конечных концентрациях заменимых аминокислот, равных 25 мкМ, 50 мкМ, 100 мкМ, 200 мкМ, 400 мкМ, соответственно; и среду Состава 3 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 4, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного холерного токсина при конечных концентрациях холерного токсина, равных1,25 нг/мл, 2,5 нг/мл, 5 нг/мл, 10 нг/мл, 20 нг/мл, соответственно; и среду Состава 4 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 5, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного инсулиноподобного фактора роста-1 в конечных концентрациях инсулиноподобного фактора роста-1, равных 5 нг/мл, 15 нг/мл, 45 нг/мл, 135 нг/мл, 405 нг/мл, соответственно; и среду Состава 5 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 6, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного В27 в конечных концентрациях В27, равных 12,5х, 25х, 50х, 100х, 200х, соответственно; и среду Состава 6 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 7, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного никотинамида в конечных концентрациях никотинамида, равных 1 мМ, 2 мМ, 4 мМ, 8 мМ, 16 мМ, соответственно; и среду Состава 7 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 8, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного инсулина в конечных концентрациях инсулина, равных 0,25 мкг/мл, 0,5 мкг/мл, 1 мкг/мл, 2 мкг/мл, 4 мкг/мл, соответственно; и среду состава 8 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 9, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленного Y-27632 в конечных концентрациях Y-27632, равных 2,5 мкМ, 5 мкМ, 10 мкМ, 20 мкМ, 40 мкМ, соответственно; и среду Состава 9 использовали в качестве пустого контроля (ВС).
Когда использовали среду Состава 10, в 48-луночный планшет с посеянными первичными клетками добавляли 1 мл/лунку подготовленной фетальной бычьей сыворотки при соотношениях добавленной фетальной бычьей сыворотки 2,5% (об./об.), 5% (об./об.), 10% (об./об.), 20% (об./об.), 40% (об./об.), соответственно; и среду Состава 10 использовали в качестве пустого контроля (ВС).
После того, как клетки размножали до покрытия приблизительно 85% площади лунок 48-луночного планшета, их открепляли и подсчитывали.
Кратность пролиферации рассчитывали на основании количества клеток в пустом контроле (ВС), и результаты представили на Фиг. 2А-2J, соответственно. На Фиг. 2А-2J отношение представляет собой отношение количества клеток, полученных путем культивирования для первого пассажа с применением каждой среды, к количеству клеток, полученных путем культивирования для первого пассажа с применением соответствующего пустого контроля. Отношение больше 1 указывает на то, что полученная среда, содержащая различные концентрации факторов или низкомолекулярных соединений, оказывает лучшее влияние на стимуляцию пролиферации, чем среда для культивирования пустого контроля; отношение меньше 1 указывает на то, что полученная среда, содержащая различные концентрации факторов или низкомолекулярных соединений, оказывает более слабое влияние на стимуляцию пролиферации, чем среда для культивирования пустого контроля.
В соответствии с результатами, представленными на Фиг. 2А-2J, количество гастрина предпочтительно составляет 1,2-20 нМ, более предпочтительно 2,5-10 нМ, еще более предпочтительно 5 нМ; количество А8301 предпочтительно составляет 25-200 нМ, более предпочтительно 200 нМ; количество заменимых аминокислот предпочтительно составляет 25 - 400 мкМ, более предпочтительно 50 мкМ; количество холерного токсина предпочтительно составляет 1,25-20 нг/мл, более предпочтительно 2,5-5 нг/мл, еще более предпочтительно 5 нг/мл; количество инсулиноподобного фактора роста-1 предпочтительно составляет 5-405 нг/мл, более предпочтительно 45 нг/мл; объемная концентрация В27 предпочтительно составляет 25-200 х, более предпочтительно 25-50 х, еще более предпочтительно 25 х; количество никотинамида в среде для культивирования предпочтительно составляет 2-8 мМ, более предпочтительно 4-8 мМ, еще более предпочтительно 4 мМ; количество инсулина предпочтительно составляет 0,5 - 4 мкг/мл, более предпочтительно 1-2 мкг/мл, еще более предпочтительно 2 мкг/мл; количество Y-27632 предпочтительно составляет 2,5-40 мкМ, более предпочтительно 2,5-5 мкМ, еще более предпочтительно 5 мкМ; объемное содержание фетальной бычьей сыворотки предпочтительно составляет 2,5-5% (об./об.), более предпочтительно 5% (об./об.).
Пример 4. Культивирование и идентификация первичных клеток ракакишечника.
(1) Культивирование первичных клеток рака кишечника.
В соответствии со способом, описанным на стадии (2)-3 Примера 1, первичные клетки рака кишечника получали из эндоскопических образцов ткани (пронумерованных как ОЕ(Е)019, ОЕ(Е)028, ОЕ(Е)021, ОЕ(Е)023, ОЕ(Е)026, ОЕ(Е)025, ОЕ(Е)024, ОЕ(Е)004) и культивировали с применением среды СА-1 из Примера 2. Полученные первичные клетки рака кишечника высевали в 6-луночный планшет при плотности живых клеток 1×104 клеток/см2 (100000 клеток на лунку), а затем в планшет добавляли клетки NIH-3T3, облученные γ-излучением (доза облучения 30 Гр) при плотности клеток 2×104 клеток/см2, и хорошо перемешивали. После дезинфекции поверхности планшет помещали в термостат при 37°С, 5% СО2 (приобретенный у Thermo Fisher) для культивирования.
Проводили визуальный анализ культивированных первичных клеток рака кишечника с помощью микроскопа (EVOS М500, Invitrogen). На Фиг. 3А-3Н показаны фотографии, сделанные с помощью объектива с 10-кратным увеличением. Под микроскопом клетки были близко расположены и имели немного неправильную форму.
(2) Иммуногистохимическая идентификация клеток рака кишечника, субкультивированных из раковых тканей кишечника.
Из интраоперационной ткани пациента с раком кишечника (номер образца: ОЕ(О)001) брали раковую ткань размером приблизительно 0,25 см3, погружали в 1 мл 4% параформальдегида и фиксировали. Используя способ из Примера 3, образец ОЕ(О)001 непрерывно культивировали до пятого пассажа с применением среды СА-1 в соответствии с настоящим изобретением. Ткани или клетки, фиксированные 4% параформальдегидом, заливали в парафин и получали с помощью микротома срезы тканей толщиной 4 мкм. Затем проводили стандартный иммуногистохимический анализ (см. конкретные этапы в источнике Li ef ai, Nature Communication, (2018) 9: 2983). Использовали следующие первичные антитела против: ki-67, СК20, CDX-2 и виллина (все приобретены у CST).
На Фиг. 4А-4D и 5А-5D представлены сравнительные изображения результатов иммуногистохимического анализа клеток исходной ткани и опухолевых клеток рака кишечника, полученных путем культивирования клеток исходной ткани с применением среды СА-1 в соответствии с настоящим изобретением, соответственно. На Фиг. 4А и Фиг. 5Апредставлены изображения ткани рака кишечника и полученных клеток после размножения и культивирования, которые были помечены антителом против ki-67, соответственно. На Фиг. 4 В и Фиг. 5 В представлены изображения ткани рака кишечника и полученных клеток после размножения и культивирования, которые были помечены антителом против СК20, соответственно. На Фиг. 4С и Фиг. 5С представлены изображения ткани рака кишечника и клеток, полученных после размножения и культивирования, которые были помечены антителом против CDX-2, соответственно. На Фиг. 4D и Фиг. 5D представлены изображения ткани рака кишечника и клеток, полученных после размножения и культивирования, которые были помечены антителом против виллина, соответственно. Таким образом, можно подтвердить, что когда опухолевые клетки рака кишечника (номер образца: ОЕ(О)001), культивированные по технологии в соответствии с настоящим изобретением, культивируют до пятого пассажа, экспрессия связанных с раком кишечника биомаркеров на клетках по существу соответствует их экспрессии на срезах исходных тканей, из которых были получены клетки. Это указывает на то, что клетки, культивированные с применением технологии в соответствии с настоящим изобретением, сохраняют исходные патологические характеристики раковых тканей пациентов с раком кишечника.
(3) Иммунофлуоресцентная идентификация первичных клеток рака кишечника после субкультивирования.
Среду СА-1 из Примера 2 использовали для культивирования образца ОЕ(О)001 до тех пор, пока клетки не наросли до конфлюентности 85% или более, и добавляли 500 мкл 0,05% трипсина (приобретенного у Gibco) для промывки клеток в течение 1 минуты. После удаления трипсина в каждую лунку добавляли 500 мкл 0,05% трипсина. Затем планшет помещали в термостат при 37°С, 5% СО2 на 2 -10 минут до полного открепления клеток. Для остановки открепления добавляли 500 мкл среды DMEM/F12, содержащей 10% сыворотки (Excell Bio, FND500). Полученную суспензию центрифугировали при 1500 об/мин в течение 4 минут и супернатант удаляли. Полученный осадок ресуспендировали с применением 500 мкл среды СА-1. Культивированные первичные клетки рака кишечника были идентифицированы с помощью иммунофлуоресцентного окрашивания.
Культивированные первичные клетки рака кишечника высевали напредметные стекла (приобретенные у Thermo Fisher) и культивировали в термостате при 37°С, 5% СО2 до тех пор, пока клетки не прикрепились к стенке, а затем культивировали в течение еще 2 - 3 дней.
Когда клетки наросли до покрытия 80% площади дна, среду для культивирования удаляли. Клетки однократно промывали ФБР (приобретенным у Shanghai Sangon), добавляли 300 мкл 4% параформальдегида (Biosharp, BL539A), а затем выдерживали в течение 20 минут при комнатной температуре для фиксации клеток. Клетки промывали ФБР в течение 5 минут и повторяли промывку 3 раза. Затем использовали ФБР+0,3% тритон Х-100 (приобретенный у Shanghai Sangon) для приготовления 5%-ной объемной концентрации раствора БСА (приобретенного у Shanghai Sangon) для блокирования. Блокирование проводили на водяной бане при 37°С в течение 30 минут.Для разведения антител использовали буфер для разведения первичных антител (Beyotime, Р0023А), и специфические к ki-67, СК20, CDX-2 и виллину антитела (все приобретены у CST) разводили в соотношении 1:50. Удаляли блокирующий раствор и добавляли полученное первичное антитело. Предметные стекла инкубировали в течение ночи в холодильнике с температурой 4°С. В частности, СК20 экспрессировался почти во всех аденокарциномах кишечника, а аденокарциному толстой кишки идентифицировали как CDX-2(+) и виллин(+).
На следующий день предметные стекла вынимали из холодильника с температурой 4°Си уравновешивали до комнатной температуры, затем инкубировали при 37°С в течение 1 часа. Затем клетки промывали ФБР в течение 5 минут и повторяли промывку 3 раза. Готовили буфер для разведения первичных антител для разведения вторичных антител. Флуоресцентное вторичное антитело (приобретенное у Thermo Fisher, вид, из которого получено антитело, представлял собой кролика или мышь) с длиной волны возбуждения 488 нм разводили в соотношении 1:1000, инкубировали при комнатной температуре в течение 1 часа в темноте, промывали ФБР в течение 5 минут и повторяли промывку 3 раза.
DAPI (приобретенный у Sigma) разводили в ФБР в соотношении 1:1000, окрашивали при комнатной температуре в темноте в течение 5 минут, промывали ФБР в течение 5 минут и повторяли промывку 3 раза. Визуализацию, фотосъемку и запись проводили под микроскопом (EVOS М500, Invitrogen).
На Фиг. 6 показаны результаты иммунофлуоресцентного окрашивания и идентификации первичных клеток рака кишечника, культивированных из образца ОЕ(О)001 in vitro, которые представляют собой флуоресцентные фотографии, сделанные с помощью объектива с 20-кратным увеличением. На Фиг. 6 показано, что ki-67, СК20, CDX-2 и виллин экспрессировались, что указывает на то, что образец представляет собой клетки колоректальной аденокарциномы, и результаты диагностики первичных клеток, культивированных с применением среды СА-1 в соответствии с настоящим изобретением, соответствовали результатам диагностики ткани рака кишечника. Клетки, культивированные с применением технологии в соответствии с настоящим изобретением, сохраняют исходные патологические характеристики таковых у рака кишечника от пациентов.
Пример 5. Статистика начального периода культивирования и количества первичных клеток рака кишечника и расчет значения удвоения популяции (PD).
В соответствии со способом, описанным на этапе (2)-3 Примера 1, первичные клетки рака кишечника получали из образцов ткани 4 случаев рака кишечника (пронумерованных ОЕ0042, ОЕ(Е)003, ОЕ(Е)006, ОЕ(О)001). Полученные первичные клетки рака кишечника культивировали с применением среды СА-1 из Примера 2, и клетки высевали во флаконы Т25 при плотности живых клеток 2×104 клеток/см2 и культивировали. После разрастания до конфлюентности 95% клетки открепляли и подсчитывали. При этом количество дней культивирования до открепления регистрировали как один период культивирования. В этих условиях эксперимента клетки непрерывно культивировали, и полученные клетки размножали до различных пассажей. Клетки каждого пассажа подсчитывали после открепления и регистрировали соответствующий период культивирования. Значение PD вычисляли в соответствии с формулой, удвоение популяции (PD)=3,32×log10 (общее количество клеток после открепления/исходное количество высеянных клеток). Формулу см. в источнике Chapman ef а/., Stem Cell Research & Therapy 2014, 5: 60.
На Фиг. 7 показаны кривые роста первичных клеток из 4 случаев рака кишечника в условиях культивирования первичных клеток рака кишечника в соответствии с настоящим изобретением, которые были построены с помощью программного обеспечения Graphpad Prism. На оси абсцисспредставлено количество дней культивирования клеток; на оси ординат представлена совокупная кратность пролиферации клеток, представляющая собой кратность размножения клеток в течение периода культивирования, и чем больше значение, тем больше кратность размножения клеток в течение определенного периода, то есть тем больше клетки размножились; наклон показывает скорость размножения клеток. По Фиг. 7 можно убедиться, что если первичные клетки рака кишечника непрерывно культивировать и размножать в среде СА-1 в соответствии с настоящим изобретением в течение по меньшей мере 80 дней, скорость размножения клеток остается по существу неизменной, и все еще остается способность к непрерывному размножению.
Пример 6. Сравнение эффекта культивирования с таковым для известной среды для культивирования
(1) Получение среды для культивирования
Среда, известная из литературного источника (Xuefeng Liu et al, Nat. Protoc, 12(2): 439-451, 2017), имеет следующий состав: среда DMEM/F12+250 нг/мл амфотерицина В (Selleck)+10 мкг/мл гентамицина (МСЕ)+0,1 нМ холерного токсина+0,125 нг/мл EGF+25 нг/мл гидрокортизона+10 мкМ Y27632+10% ЭБС (далее называют средой “LXF”).
(2) Получение и культивирование первичных клеток рака кишечника.
Первичные клетки рака кишечника получали из интраоперационных образцов ткани (ОЕ(Е)003, ОЕ0042) в соответствии со способом, описанным на этапе (2)-3 Примера 1, а затем культивировали с клетками трофобласта и без них, соответственно.
При использовании клеток трофобласта, клетки рака кишечника высевали в указанную выше среду LXF и среду СА-1 в соответствии с Примером 2, соответственно, в 48-луночный планшет при плотности живых клеток 3×104 клеток/см2 (30000 клеток на лунку), а затем добавляли клетки NIH-3T3, облученные γ-излучением (доза облучения 30 Гр), при плотности клеток 2×104 клеток/см2. Наконец, в каждую лунку 48-луночного планшета добавляли соответствующие среды для культивирования до объема 500 мкл, и полученную суспензию тщательно перемешивали. После дезинфекции поверхности планшет помещали в термостат с содержанием 5% СО2 при 37°С (приобретенный у Thermo Fisher) для культивирования. После того, как клетки наросли до покрытия 85% площади лунок или болеев 48-луночном планшете, клетки субкультивировали.
В случае отсутствия клеток трофобласта, клетки рака кишечника высевали в указанную выше среду LXF и среду СА-1 из Примера 2, соответственно, в 48-луночный планшет при плотности живых клеток 3×104 клеток/см2 (30000 клеток на лунку), а затем в каждую лунку 48-луночного планшета добавляли соответствующие среды для культивирования до объема 500 мкл, и полученную суспензию тщательно перемешивали. После дезинфекции поверхности планшет помещали в термостат с содержанием 5% СО2 при 37°С (приобретенный у Thermo Fisher) для культивирования. После того, как клетки наросли до покрытия 85% площади лунок или более в 48-луночном планшете, клетки субкультивировали.
На 7-й день культивирования извлекали 48-луночный планшет и удаляли среду. Использовали 100 мкл 0,05% трипсина (приобретенного у Gibco) для однократной промывки клеток. После удаления трипсина в каждую лунку добавляли 200 мкл 0,05% трипсина. Планшет помещали в термостат при 37°С, 5% СО2 на 10 минут, и проводили визуальный анализ клеток под микроскопом (CNOPTEC, BDS400), который показал, что клетки полностью открепились. Затем добавляли 300 мкл среды DMEM/F12, содержащей 10% сыворотки (Excell Bio, FND500), для остановки открепления. В планшет для подсчета клеток (Countstar, комплектация: 50 шт. на коробку) добавляли 20 мкл полученной суспензии и подсчитывали общее количество клеток с помощью счетчика клеток (Countstar, IC1000). Результаты подсчета показаны на Фиг. 8А и 8 В.
По результатам, представленным на Фиг. 8А и 8 В, видно, что по сравнению со средой LXF, СА-1 может значительно стимулировать размножение первичных клеток рака кишечника, независимо от наличия или отсутствия клеток трофобласта, и ее эффект лучше, чем таковой у среды LXF, используемой в предшествующем уровне техники. Кроме того, в присутствии клеток трофобласта (Фиг. 8 В) эффект стимуляции размножения первичных клеток рака кишечника более значителен.
Пример 7. Применение первичных клеток рака кишечника, полученных путем размножения в среде для культивирования в соответствии с настоящим изобретением, для скрининга лекарственных средств.
1. Культивирование клеток и посев
В соответствии с тем же способом, который описан в Примере 1, первичные клетки рака кишечника выделяли из интраоперационных/эндоскопических образцов рака кишечника (ОЕ(Е)003, ОЕ0042) и культивировали в среде СА-1. После размножения клеток до покрытия 85% площади планшета, их открепляли в виде одного пассажа, а затем субкультивировали. Клетки открепляли и подсчитывали в соответствии с этапами, описанными в Примере 1, и клетки высевали при плотности живых клеток 5,76×104 клеток/мл и хорошо перемешивали в загрузочном слоте (приобретенном у Corning). После тщательного смешивания их высевали для культивирования в 384-луночный непрозрачный белый планшет для культивирования клеток (приобретенный у Corning), в объеме 50 мкл на лунку и при количестве клеток 3000 клеток/лунку. Планшет заполняли добавлением среды СА-1 до края планшета, и отмечали на планшете названия образцов и время тестирования CellTiter-Glo (приобретенного у Promega). Поверхность дезинфицировали 75% спиртом (приобретенным у LIRCON), и осуществляли культивирование в термостате при 37°С, 5% СО2.Для скрининга лекарственных средств отбирали первый, второй, третий, четвертый, пятый пассажи культивированных клеток, соответственно, и тестировали чувствительность к лекарственным средствам непрерывных пассажей первичных клеток, культивированных в среде в соответствии с настоящим изобретением.
2. Подготовка лекарственных средств-кандидатов.
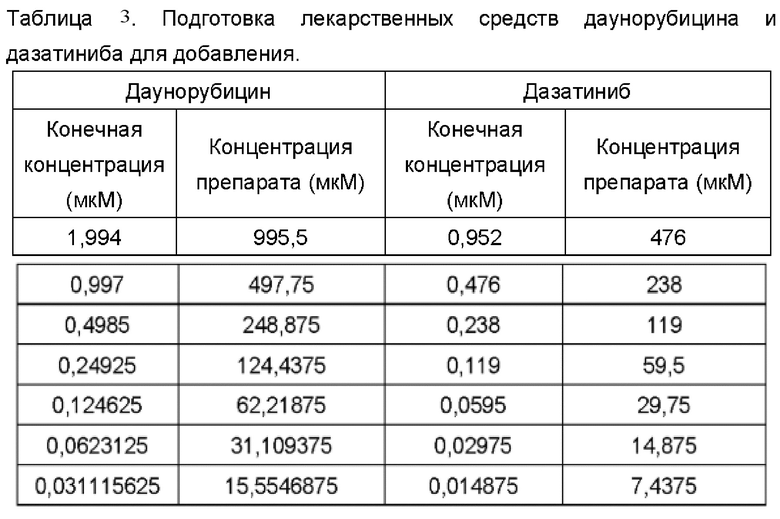
Два лекарственных средства (даунорубицин и дазатиниб; оба приобретены у МСЕ) были подготовлены в градиентах 7 концентраций в соответствии со следующей таблицей, которые были добавлены в 384-луночный планшет (приобретенный у Thermo Fisher) в объеме 30 мкл на лунку и хранились до использования.
3. Высокопроизводительная загрузка лекарственных средств Подготовленные планшеты с лекарственными средствами вынимали и помещали при комнатной температуре.
Планшеты центрифугировали в центрифуге (Beckman) при комнатной температуре, 1000 об/мин в течение 1 минуты, а затем вынимали. Для высокопроизводительной загрузки лекарственных средств использовали высокопроизводительную автоматизированную систему загрузки (JANUS, Perkin Elmer). В каждую лунку 384-луночного планшета с культивированными клетками рака кишечника добавляли 0,1 мкл лекарственных средств-кандидатов в соответствующих концентрациях. После загрузки лекарственных средств дезинфицировали поверхность 384-луночного планшета и помещали его в термостат, и через 72 часа измеряли жизнеспособность клеток.
4. Детектирование жизнеспособности клеток
Люминесцентный реагент CellTiter-Glo (приобретенный у Promega) вынимали из холодильника с температурой 4°С и добавляли 10 мл реагента в загрузочный слот; 384-луночный планшет для тестирования вынимали из термостата и добавляли 10 мкл люминесцентного реагента CellTiter-Glo в каждую лунку. После выдерживания в течение 10 минут проводили анализ с применением многофункционального устройства для прочтения микропланшетов (Envision, Perkin Elmer).
5. Обработка данных
Согласно формуле, скорость ингибирования клеток (%)=100% - значение хемилюминесценции лунки с загруженным лекарственным средством/значение хемилюминесценции контрольной лунки × 100%; рассчитывали скорость ингибирования клеток, обработанных различными лекарственными средствами, и концентрацию полумаксимального ингибирования (IC50) клеток лекарственными средствами рассчитывали сприменением программного обеспечения Graphpad Prism. Результаты представлены на Фигурах 9А и 9В.
Фигуры 9А и 9В могут подтвердить, что при использовании для скрининга лекарственных средств клеток рака кишечника, культивированных в среде для культивирования для первичных клеток рака кишечника в соответствии с настоящим изобретением, ингибирующее действие одного и того же лекарственного средства на культивируемые клетки различных пассажей остается по существу одинаковым (кривые ингибирования по существу сходны). Клетки от одного и того же пациента отличаются по чувствительности к разным лекарственным средствам при концентрации, максимальной в крови в организме человека. В соответствии с полученными результатами можно определить эффективность лекарственного средства при клиническом применении у пациентов с раком кишечника; и в то же время можно подтвердить, что чувствительность к лекарственному средству опухолевых клеток различных пассажей, полученных в соответствии со способом культивирования, предложенным в настоящей заявке, остается стабильной.
Промышленная применимость
В соответствии с настоящим изобретением предложены среда для культивирования и способ культивирования для культивирования или размножения первичных клеток рака кишечника in vitro, и культивированные клетки могут быть использованы для оценки эффективности или скрининга лекарственных средств. Таким образом, настоящее изобретение подходит для промышленного применения.
Хотя настоящее изобретение было подробно описано в настоящем документе, настоящее изобретение не ограничивается этим описанием, и специалисты в данной области техники могут вносить модификации в соответствии с принципом настоящего изобретения. Таким образом, любую модификацию, осуществленную в соответствии с принципом настоящего изобретения, следует понимать как подпадающую под объем охраны настоящего изобретения.
Изобретение относится к области биотехнологии, в частности к среде для культивирования первичных клеток рака кишечника. Среда для культивирования содержит исходную среду для культивирования, ингибитор протеазы Rho, антибиотик, гастрин, А8301, заменимую аминокислоту, холерный токсин, инсулиноподобный фактор роста-1, никотинамид, инсулин, фетальную бычью сыворотку и добавку, выбранную из по меньшей мере одной из добавки В27 и добавки N2. Также раскрыты способ культивирования первичных клеток рака кишечника и способ скрининга лекарственных средств для лечения рака кишечника. При использовании указанной среды для культивирования может быть достигнуто эффективное и быстрое размножение первичных клеток рака кишечника. Размноженные клетки сохраняют патологические характеристики, присутствующие у пациентов, и уровень успешности культивирования и уровень размножения первичных клеток рака кишечника повышаются, что обеспечивает основу для исследования персонализированного лечения пациентов. 3 н. и 3 з.п. ф-лы, 9 ил., 3 табл., 7 пр.
1. Среда для культивирования первичных клеток рака кишечника, причем указанная среда содержит:
исходную среду для культивирования, ингибитор протеазы Rho, антибиотик, гастрин, A8301, заменимую аминокислоту, холерный токсин, инсулиноподобный фактор роста-1, никотинамид, инсулин, фетальную бычью сыворотку и добавку, выбранную из по меньшей мере одной из добавки B27 и добавки N2,
причем исходная среда для культивирования выбрана из группы, состоящей из DMEM/F12, DMEM, F12 или RPMI-1640,
причем
указанный ингибитор протеазы Rho представляет собой один или более ингибиторов протеазы, выбранных из группы, состоящей из Y27632, фасудила и H-1152, и концентрация указанного ингибитора протеазы Rho составляет 2,5-40 мкМ;
указанный антибиотик представляет собой примоцин, имеющий концентрацию в диапазоне 25-400 мкг/мл;
концентрация гастрина составляет 1,25-20 нМ;
концентрация А8301 составляет 25-200 нМ;
указанная заменимая аминокислота представляет собой одну или более аминокислот, выбранных из группы, состоящей из глицина, аланина, аспарагина, аспарагиновой кислоты, глутаминовой кислоты, пролина и серина, с общей концентрацией 25-400 мкМ;
концентрация холерного токсина составляет 1,25–20 нг/мл;
концентрация инсулиноподобного фактора роста-1 составляет 5-405 нг/мл;
концентрация никотинамида составляет 2-8 мМ;
концентрация инсулина составляет 0,5-4 мкг/мл;
объемная концентрация фетальной бычьей сыворотки по отношению к среде для культивирования составляет 2,5-5% (об./об.);
объемное соотношение добавки В27 или N2 к среде для культивирования составляет 1:25-1:200.
2. Среда для культивирования для первичных клеток рака кишечника по п. 1, отличающаяся тем, что:
указанный ингибитор протеазы Rho представляет собой Y27632, и его концентрация составляет 2,5-5 мкМ;
указанный антибиотик представляет собой примоцин, имеющий концентрацию в диапазоне 50-200 мкг/мл;
концентрация гастрина составляет 2,5-10 нМ;
концентрация холерного токсина составляет 2,5-5 нг/мл;
концентрация никотинамида составляет 4-8 мМ;
концентрация инсулина составляет 1-2 мкг/мл;
объемное отношение добавки B27 или N2 к среде для культивирования составляет 1:25-1:50.
3. Способ культивирования первичных клеток рака кишечника, причем:
указанные первичные клетки рака кишечника культивируют с применением среды для культивирования первичных клеток рака кишечника по п. 1 или 2.
4. Способ культивирования по п. 3, отличающийся тем, что:
первичные клетки рака кишечника высевают в чашку для культивирования при плотности клеток 1-10×104 клетки/см2, и добавляют клетки трофобласта при плотности клеток 2-3×104 клетки/см2, и для культивирования используют среду для культивирования первичных клеток рака кишечника по п. 1 или 2.
5. Способ культивирования по п. 4, отличающийся тем, что:
клетки трофобласта представляют собой облученные клетки NIH-3T3, источником облучения является рентгеновское или γ-излучение, и доза облучения составляет 30-50 Гр.
6. Способ скрининга лекарственных средств для лечения рака кишечника, причем указанный способ включает следующие этапы:
(1) культивирование первичных клеток рака кишечника с применением способа культивирования первичных клеток рака кишечника по любому из пп. 3-5;
(2) выбор лекарственного средства для исследования и разведение указанного лекарственного средства на основании требуемых градиентов концентрации;
(3) добавление указанного лекарственного средства в различных градиентах концентрации к указанным клеткам, культивируемым и полученным на этапе (1);
(4) детектирование жизнеспособности клеток, с определением тем самым эффективности указанного лекарственного средства.
| CN 110592020 A, 20.12.2019 | |||
| WO 2020072506 A1, 09.04.2020 | |||
| CN 107988162 A, 04.05.2018 | |||
| МЕТОДИКА КУЛЬТИВИРОВАНИЯ КЛЕТОК | 2017 |
|
RU2709378C1 |
| ТРУХАН И.С | |||
| ПИТАТЕЛЬНАЯ СРЕДА КАК КЛЮЧЕВОЙ ФАКТОР КУЛЬТИВИРОВАНИЯ КЛЕТОК МЛЕКОПИТАЮЩИХ, Международный журнал прикладных и фундаментальных исследований, 2018, Номер 12 (часть 1), стр | |||
| Устройство для отыскания металлических предметов | 1920 |
|
SU165A1 |

