ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицинской технологии, в частности к культуральной среде и способу культивирования или размножения in vitro эпителиальных клеток первичного рака гортани, а также к способу и применению культивированных клеток при оценке эффективности и скрининге лекарственных средств.
УРОВЕНЬ ТЕХНИКИ
Уровень заболеваемости раком гортани составляет от 5,7 до 7,6% от всех злокачественных опухолей в организме. В области отоларингологии он занимает третье место только после носоглоточной карциномы и рака носовой полости и придаточных пазух носа. Преобладающий возраст составляет от 50 до 70 лет. В Китае уровень заболеваемости самый высокий на северо-востоке, при этом большинство пациентов являются мужчинами. Этиология не очень ясна, но в городах с сильным загрязнением воздуха уровень заболеваемости выше, чем в городах с небольшим загрязнением. Поскольку молекулярный патогенез рака гортани все еще неясен, а способы лечения пациентов с раком гортани, особенно пациентов на средней и поздней стадиях, все еще ограничены, это приводит к низкому уровню пятилетней выживаемости в клинической практике и отсутствию персонифицированных и точных рекомендаций по применению лекарственных препаратов.
Функциональное тестирование относится к способу определения in vitro чувствительности раковых клеток пациентов к противоопухолевым средствам. Ключевым фактором для применения такого способа является разработка моделей опухолевых клеток, которые имеют короткий цикл роста и могут представлять собой биологические характеристики пациентов с раком гортани. Кроме того, клеточная модель должна быть простой в обращении, чтобы быстро и эффективно предсказывать эффективность клинических препаратов, и, таким образом, своевременно давать точные рекомендации по лечению онкологических больных. Однако обычно низкие уровни успеха при создании in vitro клеточных моделей из первичных опухолевых клеток больных раком, длительные циклы роста и такие проблемы, как чрезмерная пролиферация мезенхимальных клеток (таких как фибробласты и т.п.), все это ограничивает разработку в данной области. В настоящее время в области функционального тестирования опухолевых клеток существует две технологии культивирования относительно зрелых первичных эпителиальных/стволовых клеток. Одна из них представляет собой технологию, в которой для исследования чувствительности к лекарственным средствам отдельных пациентов используют облученные трофобластные клетки и ингибитор киназы ROCK для стимуляции роста первичных эпителиальных клеток, то есть технологию условного перепрограммирования клеток. (Liu et al., Am J Pathol, 180: 599-607, 2012). Другой способ представляет собой культивирование in vitro в трехмерном пространстве взрослых стволовых клеток с получением органоидов, аналогичных тканям и органам. (Hans Clevers et al., Cell, 11; 172(1-2): 373-386, 2018).
Технология выращивания органоидов представляет собой технологию, которая позволяет встраивать аутологичные первичные эпителиальные клетки пациента во внеклеточный матрикс для трехмерного культивирования in vitro; однако культуральная среда при применении такой технологии требует добавления ряда специфических факторов роста (таких как белки Wnt и белки семейства R-спондин), что является дорогостоящим и не подходит для широкого клинического применения. Кроме того, органоид необходимо встраивать в гель внеклеточного матрикса в течение всего процесса культивирования, при этом стадии посева, включающие инокуляцию клеток, пассирование и тестирование на чувствительность к лекарственным средствам, являются громоздкими и трудоемкими по сравнению с процессами двухмерного культивирования. Кроме того, размер органоида, формируемого с применением такой технологии, трудно регулировать, и некоторые органоиды могут достигать слишком больших размеров и вызывать внутренний некроз. Соответственно, технология выращивания органоидов имеет меньшую функциональность и применимость, чем технология двухмерного культивирования. Для работы такая технология требует квалифицированный технический персонал и поэтому она не подходит для масштабного и широкого клинического применения для тестирования in vitro чувствительности к лекарственным средствам (Nick Barker, Nat Cell Biol, 18(3): 246-54, 2016).
Перепрограммирование клеток представляет собой способ, в котором аутологичные первичные эпителиальные клетки пациента культивируют совместно с питающими клетками, полученными от мышей. Однако в литературе не сообщалось об исследовании образцов рака гортани, поэтому неизвестно, могут ли компоненты среды, указанные в литературе, способствовать быстрому размножению клеток первичного рака гортани.
С учетом ограничений описанных выше технологий, необходимо разработать технологию культивирования эпителиальных клеток первичного рака гортани в клинических условиях, которая может обеспечить короткий период культивирования, регулируемую стоимость и удобную технологическую операцию. При применении такой технологии для создания модели рака гортани на основе первичных опухолевых клеток культивированные клетки рака гортани могут представлять собой биологические характеристики пациентов с раком гортани. Путем оценки in vitro чувствительности к противоопухолевым средствам на клеточных моделях, полученных от отдельных пациентов с раком, можно улучшить частоту ответов на противоопухолевое средство в клинических условиях, а также можно уменьшить боли, вызванные введением пациентам неподходящих лекарственных средств, и ненужные траты медицинских ресурсов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Принимая во внимание недостатки предшествующего уровня техники, настоящее изобретение предназначено для обеспечения культуральной среды для культивирования эпителиальных клеток первичного рака гортани и способа культивирования эпителиальных клеток первичного рака гортани с применением указанной культуральной среды. Культуральная среда и способ культивирования эпителиальных клеток первичного рака гортани согласно настоящему изобретению позволяют обеспечить достижение цели, состоящей в коротком периоде культивирования in vitro, регулируемой стоимости и удобной технологической операции. При применении такой технологии для создания модели первичных опухолевых клеток рака гортани можно получать клетки первичного рака гортани с биологическими характеристиками пациентов с раком гортани, которые можно использовать при скрининге новых лекарственных средств и в тесте in vitro на чувствительность к лекарственным средствам.
В одном из аспектов настоящего изобретения предложена культуральная среда для первичных клеток для культивирования эпителиальных клеток первичного рака гортани, содержащая ингибитор киназы MST1/2, при этом указанный ингибитор киназы MST1/2 включает соединение формулы (I) или его фармацевтически приемлемую соль или сольват.

где
R1 выбран из С1-С6 алкила, С3-С6 циклоалкила, С4-С8 циклоалкилалкила, С2-С6 спироциклоалкила и арила (например, фенила и нафтила и т.д.), необязательно независимо содержащего в качестве заместителя от 1 до 2 R6, арил-С1-С6 алкила (например, фенилметила и т.д.), необязательно независимо содержащего в качестве заместителя от 1 до 2 R6, и гетероарила (например, тиенила и т.д.), необязательно независимо содержащего в качестве заместителя от 1 до 2 R6;
R2 и R3 каждый независимо выбраны из С1-С6 алкила, предпочтительно С1-С3 алкила, более предпочтительно метила;
R4 и R5 каждый независимо выбраны из водорода, С1-С6 алкила, С3-С6 циклоалкила, С4-С8 циклоалкилалкила, гидроксил-С1-С6 алкила, С1-С6 галогеналкила, С1-С6 алкиламино-С1-С6 алкила, С1-С6 алкокси-С1-С6 алкила и С3-С6 гетероциклил-С1-С6 алкила (при этом гетероциклил выбран, например, из пиперидила, тетрагидропиранила и т.д.);
R6 выбран из галогена (предпочтительно фтора и хлора, более предпочтительно фтора), С1-С6 алкила (предпочтительно метила), С1-С6 алкокси (предпочтительно метокси) и С1-С6 галогеналкила (предпочтительно трифторметила).
В предпочтительном варианте реализации ингибитор киназы MST1/2 содержит соединение формулы (Ia) или его фармацевтически приемлемую соль или сольват,

где
R1 выбран из С1-С6 алкила, фенила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6, тиенила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6, и фенилметила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6; более предпочтительно R1 представляет собой фенил, необязательно независимо содержащий в качестве заместителя от 1 до 2 R6;
R5 выбран из водорода, С1-С6 алкила и С3-С6 циклоалкила; более предпочтительно R5 представляет собой водород;
R6 независимо выбран из галогена, С1-С6 алкила и С1-С6 галогеналкила; более предпочтительно R6 представляет собой фтор, метил или трифторметил.
Ингибитор киназы MST1/2 предпочтительно представляет собой по меньшей мере соединение, выбранное из следующих соединений или их фармацевтически приемлемой соли или сольвата.
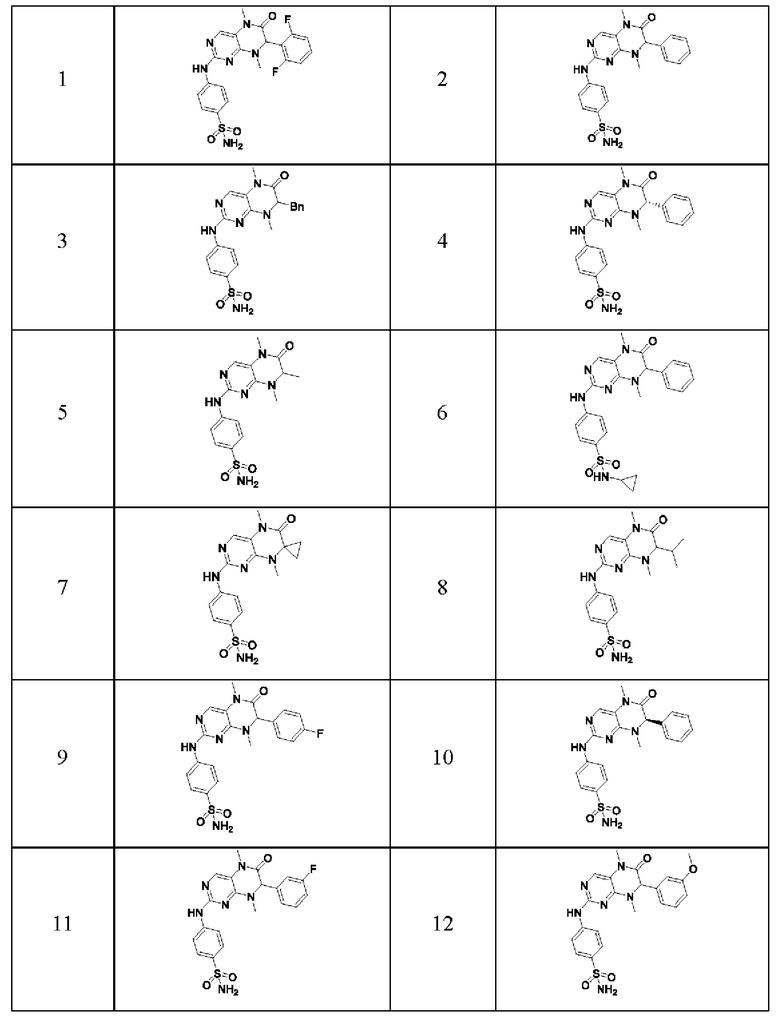


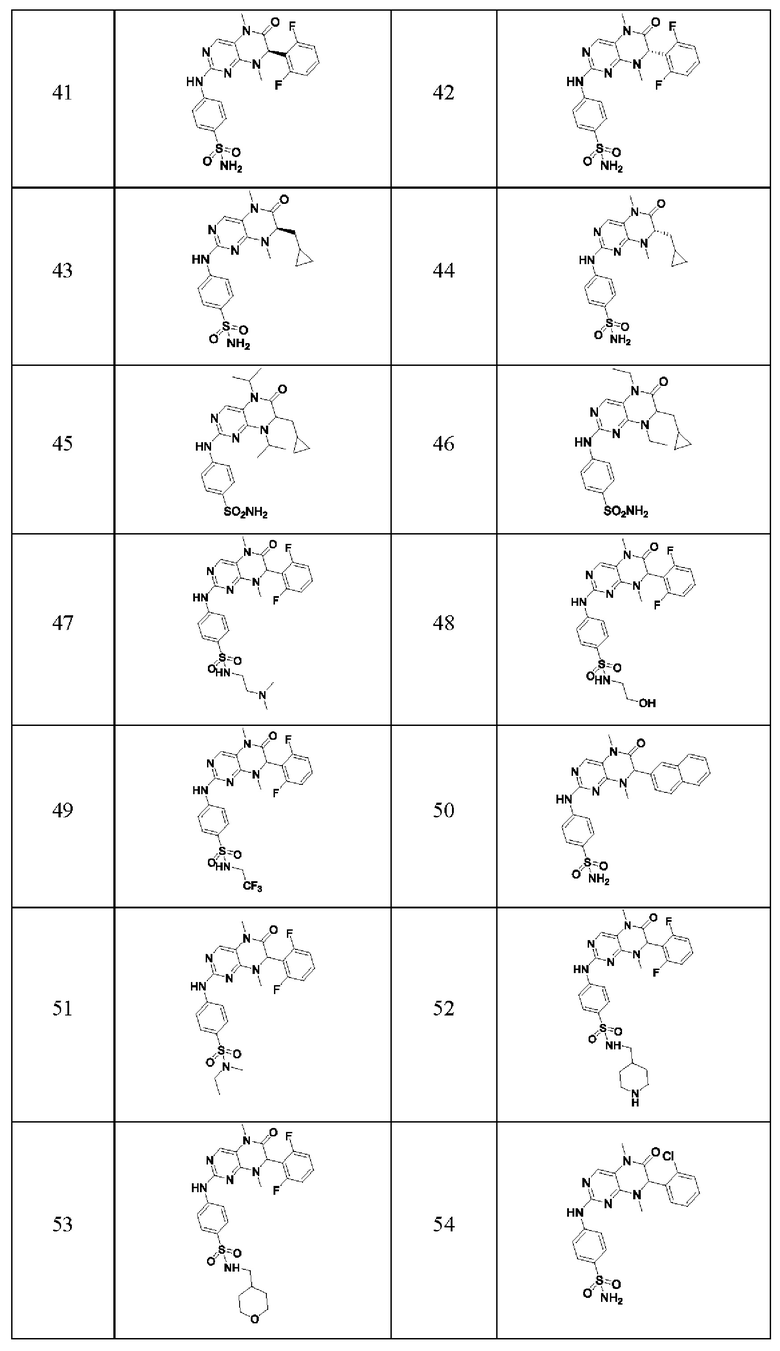

Наиболее предпочтительно, ингибитор киназы MST1/2 согласно настоящему изобретению представляет собой соединение 1.
В таком варианте реализации настоящего изобретения количество ингибитора киназы MST1/2 в культуральной среде обычно составляет от 1,25 мкМ до 10 мкМ, предпочтительно от 2,5 мкМ до 10 мкМ, более предпочтительно 5 мкМ.
Культуральная среда для первичных клеток согласно настоящему изобретению предпочтительно также содержит один или более из следующих факторов: инсулиноподобный фактор роста 1 (IGF-1); фактор роста фибробластов 7 (FGF7); комплекс инсулин-трансферрин-селен (ITS); фактор роста гепатоцитов (HGF); ингибитор киназы ROCK, выбранный из по меньшей мере одного из Y27632, фасудила и Н-1152; и ингибитор рецептора TGFβ типа I, выбранный из по меньшей мере одного из А83-01, SB431542, Repsox, SB505124, SB525334, SD208, LY36494 и SJN2511.
В предпочтительном варианте реализации количество фактора роста фибробластов 7 в культуральной среде предпочтительно составляет от 5 до 80 нг/мл, более предпочтительно от 20 до 80 нг/мл; соответствующие количества инсулина/трансферрина/селенита натрия в комплексе инсулин-трансферрин-селен составляют от 5 до 20 мкг/мл, от 2,5 до 10 мкг/мл, от 2,5 до 10 нг/мл, соответственно, и предпочтительно от 10 до 20 мкг/мл, от 5 до 10 мкг/мл, от 5 до 10 нг/мл, соответственно; количество инсулино подобно го фактора роста 1 предпочтительно составляет от 10 до 80 нг/мл, более предпочтительно от 10 от 40 нг/мл; количество фактора роста гепатоцитов предпочтительно составляет от 10 до 80 нг/мл, более предпочтительно от 10 до 40 нг/мл; ингибитор киназы ROCK предпочтительно представляет собой Y27632, при этом количество ингибитора киназы ROCK предпочтительно составляет от 1,25 до 20 мкМ, более предпочтительно от 2,5 до 10 мкМ; ингибитор рецептора TGFβ типа I предпочтительно представляет собой А83-01, при этом количество ингибитора рецептора TGFβ типа I предпочтительно составляет от 125 до 1000 нМ, более предпочтительно от 125 до 500 нМ.
По сравнению со средой для условного перепрограммирования клеток и средой для выращивания органоидов в состав предложенной среды добавлен ингибитор киназы MST1/2, но указанная среда не содержит неопределенные компоненты, такие как сыворотка, экстракт гипофиза быка или т.п., узкофункциональные факторы, необходимые для культивирования органоидов, такие как агонисты Wnt, белки семейства R-спондин, ингибиторы BMP (костный морфогенетический белок) или т.п., а также не содержит никотинамид и N-ацетилцистеин, что, тем самым, значительно снижает стоимость среды, упрощает технологический процесс приготовления среды и проведение культивирования in vitro эпителиальных клеток первичного рака гортани с регулируемой стоимостью и посредством удобной технологической операции.
Согласно настоящему изобретению эпителиальные клетки первичного рака гортани можно выбрать из клеток рака гортани, нормальных эпителиальных клеток рака гортани и эпителиальных стволовых клеток рака гортани.
В одном из аспектов настоящего изобретения предложен способ культивирования эпителиальных клеток первичного рака гортани, включающий следующие этапы:
(1) Приготовление культуральной среды для первичных клеток согласно настоящему изобретению в соответствии с приведенным выше составом.
(2) Предварительное нанесение на контейнер для культивирования облученных трофобластных клеток.
В частности, трофобластные клетки могут представлять собой, например, облученные клетки NIH-3T3, при этом источник облучения представляет собой рентгеновские лучи или γ-лучи, предпочтительно γ-лучи, при дозе облучения от 30 до 50 Гр, предпочтительно 35 Гр. В частности, облученные клетки NIH-3T3 инокулируют в контейнер для культивирования, такой как 48-луночный планшет, 24-луночный планшет, 12-луночный планшет, 6-луночный планшет или колба для культивирования клеток Т25, при плотности 2×104 клеток/см2 и сохраняют для применения после прилипания к стенке.
(3) Выделение эпителиальных клеток первичного рака гортани из ткани рака гортани.
Эпителиальные клетки первичного рака гортани можно получить, например, из образцов ткани рака гортани и образцов параканкрозной ткани. Например, образцы ткани рака гортани получают из раковой ткани информированного и давшего свое согласие пациента с раком гортани путем хирургической резекции, а образцы параканкрозной ткани собирают из ткани рака гортани, расположенной на расстоянии по меньшей мере 5 см от ткани гортани. Сбор вышеупомянутых образцов ткани осуществляют в течение получаса после хирургического вмешательства или биопсии. Более конкретно, в стерильной среде вырезают образец ткани из не затронутых некрозом участков, при этом объем образца составляет более 0,5 см3, после чего образец ткани помещают в предварительно охлажденную среду DMEM (минимальная эссенциальная среда Игла, модифицированная по способу Дульбекко)/F12 объемом от 3 до 5 мл, содержащуюся в пластиковой стерильной центрифужной пробирке с крышкой, и транспортируют в лабораторию на льду. При этом среда DMEM/F12 содержит от 1 до 2 об. % пенициллина/стрептомицина и/или от 0,2 до 0,4 об. % примоцина (в дальнейшем называемая жидкостью для переноса ткани). В случае применения стрептомицина/пенициллина диапазон концентраций стрептомицина составляет от 25 до 400 мкг/мл, предпочтительно от 50 до 200 мкг/мл, более предпочтительно 200 мкг/мл, диапазон концентраций пенициллина составляет от 25 до 400 Ед/мл, предпочтительно от 50 до 200 Ед/мл, более предпочтительно 200 Ед/мл; и в случае применения примоцина диапазон концентраций составляет от 25 до 400 мкг/мл, предпочтительно от 50 до 200 мкг/мл, более предпочтительно 100 мкг/мл.
В боксе биологической безопасности образец ткани переносят в чашку для культивирования клеток, которую затем промывают жидкостью для переноса и смывают клетки крови на поверхности образца ткани. Промытый образец ткани переносят в другую новую чашку для культивирования, добавляя от 1 до 3 мл жидкости для переноса, и разделяют образец ткани на фрагменты ткани объемом менее 3 мм3 с помощью стерильных лезвия скальпеля и пинцета.
Фрагменты образцов тканей переносят в центрифужную пробирку, которую центрифугируют со скоростью от 1000 до 3000 об/мин в течение от 3 до 5 минут в настольной центрифуге (Sigma, 3-18K); после удаления супернатанта добавляют жидкость для переноса ткани и раствор для разложения ткани в соотношении 1:1 (доза составляет примерно 5 мл раствора для разложения ткани на 10 мг ткани; способ приготовления раствора для разложения ткани включает: растворение от 1 до 2 мг/мл коллагеназы II, от 1 до 2 мг/мл коллагеназы IV, от 50 до 100 Ед/мл дезоксирибонуклеиновой кислоты I, от 0,5 до 1 мг/мл гиалуронидазы, от 0,1 до 0,5 мг/мл хлорида кальция, от 5 до 10 мг/мл бычьего сывороточного альбумина в HBSS (сбалансированный соляной раствор Хэнка) и RPMI-1640 в объемном соотношении 1:1); затем образец нумеруют и герметично закрывают с помощью герметизирующей пленки, после чего подвергают разложению в аппарате для встряхивания с постоянной температурой (Zhichu Instrument ZQLY-180N) при 37°С, число оборотов от 200 до 300, определяют, завершено ли разложение, путем наблюдения каждый 1 час, при отсутствии явного тканевого блока разложение можно прекратить; в противном случае продолжают разложение до тех пор, пока оно не станет достаточным, при этом время разложения составляет от 4 до 8 часов. После разложения неразложившиеся тканевые блоки фильтруют через сетчатый фильтр для клеток (размер ячейки фильтра для клеток составляет, например, 70 мкм); тканевые блоки на сетчатом фильтре промывают жидкостью для переноса ткани; остаточные клетки смывают в центрифужную пробирку и центрифугируют с помощью настольной центрифуги со скоростью от 1000 до 3000 об/мин в течение от 3 до 5 минут. После удаления супернатанта исследуют оставшиеся скопления клеток для определения, остались ли клетки крови; при наличии клеток крови добавляют от 1 до 5 мл лизата клеток крови (приобретенного в компании Sigma), который затем хорошо перемешивают, лизируют при 4°С в течение от 10 до 20 минут при встряхивании и тщательном перемешивании один раз каждые 5 минут; после лизиса полученный образец извлекают и центрифугируют со скоростью от 1000 до 3000 об/мин в течение от 3 до 5 минут. После удаления супернатанта добавляют культуральную среду для первичных клеток согласно настоящему изобретению для повторного суспендирования. Общее количество клеток получают путем подсчета с применением счетчика с визуализацией потока (JIМВIO FIL, Jiangsu Jimbio Technology Co., Ltd.).
(4) Инокуляцию эпителиальных клеток первичного рака гортани, выделенных на этапе (3), в сосуде для культивирования, предварительно инокулированном трофобластными клетками, и культивирование с применением культуральной среды для первичных клеток, полученной на этапе (1).
Более конкретно, клетки NIH-3T3, облученные с применением гамма-излучения при дозе облучения 35 Гр, предварительно инокулируют при плотности от 2×104 до 4×104 клеток/см2 (например, 2×104 клеток/см2) в одной лунке многолуночного планшета, при этом после прилипания к стенке клетки первичного рака гортани инокулируют при плотности от 2×104 до 8×104 клеток/см2 (например, 4×104 клеток/см2); в каждую лунку добавляют от 0,5 до 2 мл культуральной среды для первичных эпителиальных клеток и затем культивируют планшет в инкубаторе для клеток в течение от 8 до 16 дней, например, в условиях 37°С, 5% СО2; во время культивирования каждые 4 дня используют для замены свежую культуральную среду для первичных клеток; разложение и пассирование осуществляют при разрастании эпителиальных клеток первичного рака гортани до плотности клеток, составляющей от примерно 80% до 90% от площади дна многолуночного планшета.
В отличие от технологии выращивания органоидов на данном этапе не требуется непрерывно смешивать первичные клетки с матриксным гелем на льду для получения капель геля и ждать затвердевания указанных капель геля перед добавлением среды. Кроме того, предложенный способ позволяет экономить количество дорогостоящего геля внеклеточного матрикса и упрощает этапы технологической операции.
Необязательно, после культивирования инокулированных эпителиальных клеток первичного рака гортани в течение от 8 до 16 дней, когда клеточные клоны, образовавшиеся в контейнере для культивирования сливаются и покрывают 80% площади дна, удаляют супернатант и добавляют от 1 до 2 мл 0,25% трипсина (приобретенного в компании Thermo Fisher) для разложения в течение 1 минуты; после удаления 0,25% трипсина снова добавляют от 1 до 2 мл 0,05% трипсина для разложения клеток, после чего инкубируют при комнатной температуре в течение от 5 до 20 мин. Разложившиеся клетки повторно суспендируют в от 2 до 4 мл культуральной жидкости, содержащей, например, 5% (об./об.) фетальной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина, и центрифугируют со скоростью от 1000 до 3000 об/мин в течение от 3 до 5 минут. Разложившиеся одиночные клетки повторно суспендируют с применением культуральной среды для первичных клеток согласно настоящему изобретению и помещают полученную клеточную суспензию в колбу для культивирования клеток Т25, на которую предварительно нанесены трофобластные клетки для непрерывного культивирования. Процесс предварительной обработки колбы для культивирования клеток Т25 аналогичен процессу на этапе (2).
Размножаемые эпителиальные клетки рака гортани выращивают в двухмерной конфигурации, что позволяет избежать неравномерных размеров органоидов и внутреннего некроза в чрезмерно выросших органоидах, который может возникать при размножении с применением технологии выращивания органоидов.
В другом аспекте эпителиальные клетки рака гортани, в частности, клетки рака гортани, культивированные с применением способа культивирования эпителиальных клеток первичного рака гортани согласно настоящему изобретению, можно использовать для оценки эффективности лекарственных средств и скрининга лекарственных средств, включающих следующие этапы:
(1) Получение эпителиальных клеток первичного рака гортани, более предпочтительно, получение образцов раковой ткани или образцов биопсии раковой ткани, полученных от пациентов с раком гортани; выделение эпителиальных клеток первичного рака гортани и культивирование и размножение эпителиальных клеток первичного рака гортани (в частности, клеток первичного рака гортани) в соответствии со способом культивирования эпителиальных клеток первичного рака гортани, описанным выше, до обеспечения количества клеток, составляющего по меньшей мере 105, предпочтительно, по меньшей мере 106.
(2) Выбор лекарственного средства для тестирования.
(3) Основываясь на максимальной концентрации в плазме Cmax лекарственного средства в качестве эталона, применение концентрации, превышающей Cmax в 2-5 раз, в качестве начальной концентрации и разведение указанного лекарственного средства с получением различных градиентов концентраций лекарственного средства, например, градиентов концентраций лекарственного средства, составляющих от 5 до 10, предпочтительно градиентов концентраций лекарственного средства, составляющих от 6 до 8.
(4) Разложение эпителиальных клеток рака гортани, культивированных на этапе (1), с получением суспензии одиночных клеток, подсчет количества клеток с применением счетчика с визуализацией потока, разведение суспензии одиночных клеток с применением культуральной среды для первичных клеток согласно настоящему изобретению, равномерное добавление разведенной клеточной суспензии в многолуночный планшет при плотности от 2000 до 4000 клеток на лунку, например, 50 мкл разведенного раствора клеток на лунку, и выдерживание для прилипания в течение ночи.
Такой этап позволяет избежать проблемы, свойственной способу перепрограммирования клеток, состоящей в том, что присутствие питающих клеток может влиять на результат подсчета первичных клеток и последующий анализ жизнеспособности первичных клеток, и устраняет необходимость в трудоемком этапе смешивания с матриксным гелем, встраивания в него и последующего посева клеточной суспензии с матриксным гелем на льду, как в способе выращивания органоидов, что значительно упрощает технологический процесс и улучшает функциональность и реализуемость предложенной технологии. Поскольку инокулированные клетки представляют собой суспензии одиночных клеток, а не трехмерные структуры, такие как органоид, указанный способ может обеспечивать более равномерные количества высеваемых клеток и меньшую изменчивость количества клеток в лунках по сравнению со способом выращивания органоидов, что делает его более подходящим для последующих операций высокопроизводительного скрининга лекарственных средств.
(5) Добавление выбранных потенциальных лекарственных средств, таких как традиционные химиотерапевтические препараты, лекарственные средства направленного действия, лекарственные средства на основе антител или их комбинация, при градиентных разведениях к адгезивным клеткам, полученным на этапе (4), с применением высокопроизводительной автоматизированной установки.
(6) Через несколько часов после добавления лекарственного средства, например, 72 часа, определение уровня выживаемости эпителиальных клеток рака гортани с применением набора для люминесцентного определения жизнеспособности клеток Cell-Titer Glo (приобретенного в компании Promega) для скрининга активности лекарственного средства.
В частности, в каждую лунку добавляют, например, 10 мкл реагента Cell Titer-Glo (приобретенного в компании Promega) и после равномерного встряхивания измеряют интенсивность хемилюминесценции каждой лунки с помощью флюоресцентного микропланшетного ридера. Откладывая концентрацию лекарственного средства по оси абсцисс и интенсивность флуоресценции по оси ординат, применяют программное обеспечение GraphPad Prism для построения кривой зависимости «доза лекарственного средства-эффект» на основе измеренных значений и рассчитывают интенсивность ингибирования пролиферации тестируемых клеток под действием лекарственных средств.
При применении клеток первичного рака гортани согласно настоящему изобретению при скрининге лекарственных средств и тестировании чувствительности in vitro к лекарственным средствам взаимодействие с лекарственным средством происходит быстрее вследствие двухмерного роста клеток по сравнению с продолжительностью тестирования лекарственных средств в технологии выращивания органоидов (среднее время введения в технологии выращивания органоидов составляет 6 дней).
Положительные эффекты настоящего изобретения также включают:
(1) уровень успеха при культивировании эпителиальных клеток первичного рака гортани может быть улучшен и достигать более 85%;
(2) можно обеспечить воспроизводство первичными эпителиальными клетками рака гортани, культивированными in vitro, патологического фенотипа и гетерогенности пациента-хозяина первичных клеток;
(3) состав среды не содержит сыворотку и, соответственно, на указанную среду не влияет качество и количество сыворотки из разных партий;
(4) эпителиальные клетки рака гортани можно размножать с высокой эффективностью, при этом эпителиальные клетки рака гортани при начальном количестве клеток на уровне 104 можно успешно размножить до величины 106 в течение примерно двух недель, при этом размножаемые эпителиальные клетки рака гортани обладают способностью к непрерывному пассированию;
(5) нет необходимости в проведении операций на льду и диссоциации матриксного геля на этапе пассирования, при этом разложение и пассирование клеток можно завершить в течение от 10 до 15 минут;
(6) стоимость культивирования можно регулировать, поскольку культуральная среда для клеток первичного рака гортани не требует применения дорогостоящих агонистов Wnt, белков семейства R-спондин, ингибиторов BMP и подобных факторов, при этом стоимость культивирования клеток может быть снижена;
(7) технологические операции являются удобными: по сравнению с технологией выращивания органоидов нет необходимости встраивать клетки в матриксный гель, как в технологии выращивания органоидов, и этапы операций являются простыми и легкими;
(9) указанная технология позволяет культивировать и получать эпителиальные клетки рака гортани в больших количествах и с высокой однородностью, что подходит для высокопроизводительного скрининга новых потенциальных соединений и высокопроизводительных функциональных тестов in vitro на чувствительность к лекарственным средствам у пациентов.
Применение культуральной среды для клеток согласно такому варианту реализации позволяет культивировать эпителиальные клетки рака гортани, полученные от людей или других млекопитающих, в том числе клетки рака гортани, нормальные эпителиальные клетки рака гортани, эпителиальные стволовые клетки рака гортани или ткани, содержащие по меньшей мере какие-либо из перечисленных клеток. В тоже время культуральную среду согласно настоящему изобретению также можно использовать для разработки набора для размножения и культивирования in vitro клеток первичного рака гортани.
Кроме того, клетки, полученные с применением способа культивирования согласно такому варианту реализации, можно использовать в регенеративной медицине, фундаментальных медицинских исследованиях эпителиальных клеток рака гортани, при скрининге реакций на лекарственные средства и при разработке новых лекарственных средств для заболеваний, связанных с ESCC (плоскоклеточный рак пищевода), и т.п.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1A-1F показано влияние различных факторов в культуральной среде на пролиферацию клеток первичного рака гортани.
На рис. 2 показано влияние усиливающих факторов в культуральной среде на пролиферацию клеток первичного рака гортани.
На фиг. 3A-3G показано влияние концентрации каждого фактора на пролиферацию клеток первичного рака гортани.
Фиг. 4А и 4В представляют собой полученные под инвертированным микроскопом фотографии клеток рака гортани, которые культивировали из клеток, выделенных из одного клинического образца ткани рака гортани (№0S0003), с применением культуральной среды SCM согласно настоящему изобретению на день 4 и день 12, соответственно.
На фиг. 5 представлена сравнительная диаграмма, на которой показано общее количество клеток, которые были собраны после 7 дней культивирования в 6 различных культуральных средах из клеток, выделенных из одного хирургически удаленного образца рака гортани (№0S0006).
Фиг. 6 представляет собой сравнительную диаграмму, на которой показаны клеточные пролиферативные эффекты, полученные при культивировании клеток, выделенных из шести хирургически удаленных образцов рака гортани (№0S0011, 0S0012, 0S0013, 0S0014, 0S0015, 0S0016), в течение 7 дней в условиях применения 6 различных культуральных сред.
Фиг. 7 представляет собой сравнительную диаграмму, на которой показаны кривые роста клеток, полученные при культивировании клеток, выделенных из одного клинического образца ткани рака гортани (№0S0004), в условиях применения 6 различных культуральных сред.
На фиг. 8А и 8В представлены изображения клеток рака гортани, которые были получены путем культивирования клеток, выделенных из одного хирургически удаленного образца рака гортани (№0S0015), с применением культуральной среды SCM согласно настоящему изобретению, а затем окрашены с помощью неспецифического красителя DAPI для ядер и антитела р63, специфичного к раку гортани, соответственно.
На фиг. 9 приведено сравнение результатов иммуногистохимического анализа клеток исходной ткани одного хирургически удаленного образца рака гортани (№0S0001) и клеток рака гортани, полученных путем культивирования клеток с применением культуральной среды SCM согласно настоящему изобретению.
На фиг. 10А и 10В показаны кривые зависимости «доза-эффект» клеток рака гортани при применении различных химиотерапевтических препаратов и лекарственных средств направленного действия, при этом указанные клетки рака гортани получали путем культивирования хирургически удаленных образцов раковой ткани двух разных пациентов с раком гортани (№0S0020 и №0S0022) с применением культуральной среды SCM согласно настоящему изобретению, соответственно.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем описании эпителиальные клетки включают дифференцированные эпителиальные клетки и эпителиальные стволовые клетки, полученные из эпителиальных тканей. «Эпителиальные стволовые клетки» относятся к клеткам с долговременной способностью к самообновлению, которые могут дифференцироваться с образованием эпителиальных клеток, и стволовым клеткам, происходящим из эпителиальных тканей. Примеры эпителиальных тканей включают роговицу, слизистую оболочку полости рта, кожу, конъюнктиву, мочевой пузырь, почечный каналец, почки, пищеварительные органы (пищевод, желудок, двенадцатиперстную кишку, тонкую кишку (в том числе тощую кишку и подвздошную кишку), толстый кишечник (в том числе толстую кишку)), печень, поджелудочную железу, молочную железу, слюнную железу, слезную железу, предстательную железу, корень волоса, трахею, легкие и т.д. Наряду с прочим, культуральная среда для клеток согласно данному варианту реализации предпочтительно представляет собой культуральную среду для эпителиальных клеток, возникающих при раке гортани.
Кроме того, в настоящем описании «эпителиальные опухолевые клетки» относятся к клеткам, полученным при онкогенезе клеток, происходящих из указанных выше эпителиальных тканей.
В настоящем описании термин «органоид» относится к трехмерной, органоподобной клеточной ткани, образованной путем спонтанной организации и агрегации клеток в регулируемом пространстве при высокой плотности.
[Примеры получения ингибиторов киназы MST1/2]
В настоящем описании ингибитор киназы MST1/2 относится к любому ингибитору, который прямо или косвенно отрицательно регулирует передачу сигналов MST1/2. В общем случае, ингибиторы киназы MST1/2 уменьшают активность киназы MST1/2, например, путем связывания с нею. Поскольку MST1 и MST2 имеют похожие структуры, ингибиторы киназы MST1/2 могут представлять собой, например, соединения, которые связываются с MST1 или MST2 и снижают их активность.
1. Получение соединения 1, представляющего собой ингибитор киназы MST1/2
4-((7-(2,6-дифторфенил)-5,8-диметил-6-оксо-5,6,7,8-тетрагидроптеридин-2-ил)амино)бензсульфамид 1

Метил-2-амино-2-(2,6-дифторфенил)ацетат (A2): в круглодонную колбу помещали 2-амино-2-(2,6-дифторфенил)уксусную кислоту (2,0 г) и затем метанол (30 мл), после чего добавляли по каплям тионилхлорид (1,2 мл) на ледяной бани. Реакционная система взаимодействовала в течение ночи при 85°С. После завершения реакции систему выпаривали при пониженном давлении для высушивания растворителя и использовали полученное твердое вещество белого цвета непосредственно на следующей стадии.
Метил-2-((2-хлор-5-нитропиримидин-4-ил)амино)-2-(2,6-дифторфенил)ацетат (A3): в круглодонную колбу помещали метил-2-амино-2-(2,6-дифторфенил)ацетат (2 г) и затем ацетон (30 мл) и карбонат калия (2,2 г) и далее охлаждали систему до -10°С на ледяной солевой бане, после чего медленно добавляли раствор 2,4-дихлор-5-нитропиримидина (3,1 г) в ацетоне. Реакционную систему перемешивали в течение ночи при комнатной температуре. После завершения реакции реакционную смесь фильтровали, удаляли растворитель из фильтрата при пониженном давлении и очищали остаток с помощью колоночной хроматографии на силикагеле под давлением с получением соединения A3. ЖХ/МС: М+Н 359,0.
2-Хлор-7-(2,6-дифторфенил)-7,8-дигидроптеридин-6(5Н)-он (А4): в круглодонную колбу помещали метил-2-((2-хлор-5-нитропиримидин-4-ил)амино)-2-(2,6-дифторфенил)ацетат (2,5 г) и затем уксусную кислоту (50 мл) и железный порошок (3,9 г). Реакционную систему перемешивали при 60°С в течение двух часов. После завершения реакции реакционную систему выпаривали при пониженном давлении для высушивания растворителя, после чего полученный продукт нейтрализовали до щелочного состояния с помощью насыщенного раствора бикарбоната натрия и экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным солевым раствором и высушивали с применением безводного сульфата натрия. Органическую фазу фильтровали и выпаривали досуха при пониженном давлении с получением неочищенного продукта. Неочищенный продукт промывали диэтиловым эфиром с получением соединения А4. ЖХ/МС: М+Н 297,0.
2-Хлор-7-(2,6-дифторфенил)-5,8-диметил-7,8-дигидроптеридин-6-(5Н)-он (А5): в круглодонную колбу помещали 2-хлор-7-(2,6-дифторфенил)-7,8-дигидроптеридин-6-(5Н)-он (2 г) и N,N-диметилацетамид (10 мл) и охлаждали до -35°С, после чего добавляли йодметан (0,9 мл) и затем гидрид натрия (615 мг) и перемешивали реакционную систему в течение двух часов. После завершения реакции реакционную смесь гасили водой и экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным солевым раствором, соответственно, и высушивали с применением безводного сульфата натрия. Органическую фазу фильтровали и выпаривали досуха при пониженном давлении с получением неочищенного продукта. Неочищенный продукт промывали диэтиловым эфиром с получением соединения А5. ЖХ/МС: М+Н 325,0.
4-((7-(2,6-Дифторфенил)-5,8-диметил-6-оксо-5,6,7,8-тетрагидроптеридин-2-ил)амино)бензсульфамид (1): в круглодонную колбу помещали 2-хлор-7-(2,6-дифторфенил)-5,8-диметил-7,8-дигидроптеридин-6-(5Н)-он (100 мг), сульфаниламид (53 мг), п-толуолсульфоновую кислоту (53 мг) и втор-бутанол (5 мл). Реакционную систему перемешивали при 120°С в течение ночи. После завершения реакции реакционную смесь фильтровали и промывали метанолом и диэтиловым эфиром с получением соединения 1. ЖХ/МС: М+Н 461,1.
2. Получение других соединений согласно настоящему изобретению, представляющих собой ингибитор MST1/2.
Другие соединения согласно настоящему изобретению, представляющие собой ингибитор MST1/2, синтезировали посредством способа, аналогичного способу получения соединения 1, и их структуры и данные масс-спектров приведены в следующей таблице.






[Пример 1]
Выделение человеческих эпителиальных клеток первичного рака гортани
Образцы ткани рака гортани получали из раковых тканей трех информированных и давших согласие пациентов с раком гортани путем хирургической резекции, а именно образцы №0S0020, 0S0021 и 0S0022. Один из образцов (№0S0020) описан ниже.
Вышеупомянутые образцы ткани собирали в течение получаса после хирургического вмешательства или биопсии. Более конкретно, в стерильной среде вырезали образцы ткани из не затронутых некрозом участков объемом более 0,5 см3 и помещали в 4 мл предварительно охлажденной жидкости для переноса ткани (конкретный состав показан в таблице 1). Жидкость для переноса помещали в пластиковую стерильную пробирку для криоконсервации объемом 5 мл с крышкой (приобретенную в компании Guangzhou Jet Bio-Filtration Co., Ltd.) и транспортировали в лабораторию с помощью холодовой цепи (0-10°С).


В боксе биологической безопасности переносили образец ткани (№0S0020) в 100 мм чашку для культивирования клеток (приобретенную в компании NEST). Образец ткани промывали жидкостью для переноса ткани. Остаточную кровь на поверхности образца ткани смывали. Удаляли избыточные ткани, такие как жир на поверхности образца ткани. Промытый образец ткани переносили в другую новую 100 мм чашку для культивирования; добавляли 2 мл жидкости для переноса и использовали стерильные лезвие скальпеля и пинцет для разделения образца ткани на фрагменты ткани объемом менее 3 мм3.
Фрагменты образца ткани переносили в центрифужную пробирку объемом 15 мл и центрифугировали со скоростью 1500 об/мин в течение 4 минут в настольной центрифуге (Sigma, 3-18К); после удаления супернатанта добавляли жидкость для переноса ткани и раствор для разложения ткани в соотношении 1:1 (доза составляла примерно 5 мл раствора для разложения ткани на 10 мг ткани; конкретный состав был показан в таблице 2); затем образец нумеровали и герметично закрывали с помощью герметизирующей пленки, после чего подвергали разложению в аппарате для встряхивания с постоянной температурой (Zhichu Instrument ZQLY-180N) при 37°С, число оборотов 300; завершено ли разложение, определяли путем наблюдения каждый 1 час.
После разложения неразложившиеся тканевые блоки фильтровали через 70 мкм сетчатый фильтр; тканевые блоки на сетчатом фильтре промывали с применением жидкости для переноса ткани; остаточные клетки смывали в центрифужную пробирку и центрифугировали со скоростью 1500 об/мин в течение 4 минут.
После удаления супернатанта исследовали оставшиеся скопления клеток для определения, остались ли клетки крови; при наличии клеток крови добавляли 3 мл лизата клеток крови (приобретенного в компании Sigma), который затем хорошо перемешивали, лизировали при 4°С в течение 15 минут при встряхивании и тщательном перемешивании один раз каждые 5 минут; после лизиса полученный образец извлекали и центрифугировали со скоростью 1500 об/мин в течение 4 минут. Удаляли супернатант с получением разложившихся и выделенных клеток первичного рака гортани, в которые добавляли базальную среду (ВМ) для повторного суспендирования. Базальную среду получали путем добавления 0,2 об. % примоцина (приобретенного в компании Invivogen, с концентрацией 50 мг/мл) к коммерческой среде DMEM/F-12 с обеспечением конечной концентрации 100 мкг/мл. Общее количество клеток составляло 2080000, как было подсчитано с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.).
Другие два образца ткани рака гортани выделяли в соответствии с тем же способом, описанным выше, при этом общее количество клеток составляло 1970000 (0S0021) и 2320000 (0F0022), соответственно.
[Пример 2]
Оптимизация культуральной среды для эпителиальной клетки первичного рака гортани
1) Влияние различных факторов
Культивированные клетки NIH-3T3 (приобретенные в компании АТСС и культивированные в культуральной растворе DMEM, содержащем 10% фетальной бычьей сыворотки) подвергали разложению с применением 0,25% трипсина (приобретенного в компании Thermo Fisher), и прекращали разложение путем применения культур ал ьно го раствора DMEM (приобретенного в компании Corning), содержащего 5% (об./об.) фетальной бычьей сыворотки (приобретенной в компании ExCell Biotech Co., Ltd.), 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина; полученный образец собирали в центрифужную пробирку объемом 15 мл и центрифугировали со скоростью 1500 об/мин в течение 4 минут, а затем удаляли супернатант. Центрифугированные клеточные отложения повторно суспендировали в упомянутом выше культуральном растворе DMEM, содержащем 10% фетальной бычьей сыворотки, и подсчитывали с применением счетчика с визуализацией потока (ЛМВIO FIL, Jiangsu Jimbio Technology Co., Ltd.). Клетки облучали с помощью у-лучей при дозе облучения 35 Гр, а затем инокулировали в сосуде для культивирования при плотности 2×104 клеток/см2. Клетки инкубировали в инкубаторе при 37°С до тех пор, пока они не прилипли к стенке. Перед инокуляцией первичных клеток из сосуда для культивирования удаляли культуральную среду.
Приготовление базальной среды (сокращенно ВМ): ВМ получали путем добавления 0,2 об. % примоцина (приобретенного в компании Invivogen, с концентрацией 50 мг/мл) к коммерческой среде DMEM/F-12 с обеспечением конечной концентрации 100 мкг/мл.
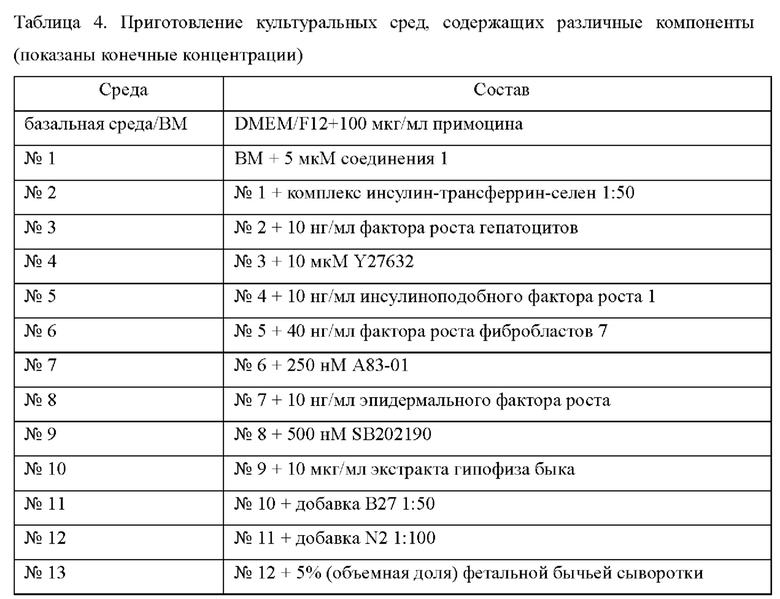
Далее для получения культуральных сред для эпителиальных клеток рака гортани, содержащих различные дополнительные компоненты, в базальную среду (ВМ) добавляли разные виды и концентрации аддитивных факторов (таблица 3).


Культуральные среды с различными компонентами добавляли в объеме 500 мкл/лунку в 48-луночные планшеты, на которые были предварительно нанесены облученные γ-лучами клетки NIH-3T3. Клетки рака гортани (№0S0064), выделенные из ткани рака гортани в соответствии с тем же способом, описанным в примере 1, инокулировали при количестве клеток 4×104 клеток/лунку в вышеупомянутых 48-луночных культуральных планшетах, на которые были предварительно нанесены облученные γ-лучами клетки NIH-3T3. После поверхностной дезинфекции планшеты помещали в инкубатор при 37°С, 5% СО2 (приобретенный в компании Thermo Fisher) с тем, чтобы равные количества свежевыделенных клеток рака гортани (№0S0064) культивировались в различных составах среды. Культуральной среды заменяли и пополняли трофобластные клетки каждые 4 дня после начала культивирования. После 10 дней культивирования осуществляли подсчет клеток. В качестве экспериментального контроля использовали базальную среду (ВМ) без добавления какой-либо добавки. Результаты представлены на фиг. 1A-1F.
Ордината на приведенных фигурах показывает отношение количества клеток, полученных после культивирования в различных средах, к количеству клеток, полученных после культивирования в базальной среде ВМ. Как показано на указанных фигурах, добавление к ВМ разных концентраций различных факторов в соответствии с таблицей 3 приведет к разным эффектам на пролиферацию клеток. Наряду с прочим, добавка В27, добавка N2, комплекс инсулин-трансферрин-селен, фактор роста гепатоцитов, инсулиноподобный фактор роста 1, фактор роста фибробластов 7, соединение 1, Y27632 и А83-01 обеспечивали определенные стимулирующие эффекты на пролиферацию клеток в конкретных диапазонах концентраций.
(2) Влияние усиливающих факторов в культуральных средах на пролиферацию клеток первичного рака гортани, полученных способом согласно настоящему изобретению
Для получения культуральных сред для эпителиальных клеток рака гортани, содержащих различные дополнительные компоненты, в базальную среду ВМ последовательно добавляли разные малые молекулы, добавки и факторы роста (таблица 4), соответственно.
Культуральные среды с различными компонентами добавляли в объеме 500 мкл/лунку в 48-луночные планшеты, на которые были предварительно нанесены облученные γ-лучами клетки NIH-3T3, и одновременно в качестве экспериментального контроля использовали среду ВМ. Клетки рака гортани (№0S0065), выделенные из ткани рака гортани в соответствии со способом, описанным в примере 1, инокупировали при количестве клеток 4×104 клеток/лунку в вышеупомянутых 48-луночных культуральных планшетах, на которые были предварительно нанесены облученные γ-лучами клетки NIH-3Т3. После поверхностной дезинфекции планшет помещали в инкубатор при 37°С, 5% CO2 (приобретенный в компании Thermo Fisher) с тем, чтобы равные количества свежевыделенных клеток рака гортани (№0S0065) культивировались в различных составах среды. После 7 дней культивирования осуществляли подсчет клеток. Результаты показаны на фиг. 2.
Как показано на указанной фигуре, было установлено, что в настоящем патенте среда №7 является наиболее предпочтительной культуральной средой для культивирования и размножения клеток первичного рака гортани (далее сокращенно SCM). Исходя из этого, дальнейшее добавление некоторых факторов, ингибиторов, представляющих собой малые молекулы, или определенной концентрации сыворотки и заменителя сыворотки, таких как добавка N2 и добавка В27, не оказывало значительного влияния на стимулирование пролиферации клеток.
(3) Влияние различных концентраций аддитивных факторов на пролиферацию клеток первичного рака гортани, полученных в настоящем изобретении
Приготовление культуральной среды для эпителиальных клеток первичного рака гортани согласно настоящему изобретению (сокращенно SCM): в базальную среду (ВМ) добавляли фактор роста фибробластов 7 (FGF-7) в конечной концентрации 40 нг/мл, инсулиноподобный фактор роста 1 (IGF-1) в конечной концентрации 10 нг/мл, фактор роста гепатоцитов (HGF) в конечной концентрации 10 нг/мл, исходный раствор комплекса инсулин-трансферрин-селен (ITS) при уровне разведения 1:50 (инсулин в конечной концентрации 10 мкг/мл, трансферрин в конечной концентрации 5 мкг/мл и селенит натрия в конечной концентрации 5 нг/мл в среде SCM), соединение 1 в конечной концентрации 5 мкМ, Y27632 в конечной концентрации 10 мкМ и TGFβ1-ингибитор А83-01 в конечной концентрации 250 нМ и получали культуральную среду для эпителиальных клеток первичного рака гортани.
Эпителиальные клетки рака гортани, происходящие из раковой ткани, выделяли и получали из раковой ткани пациента с раком гортани (образец №0S0005) с применением того же способа, что и в примере 1. Далее для получения общего количества клеток подсчитывали клетки рака гортани, происходящие из раковой ткани, с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.). Затем указанные клетки инокулировали в 12-лун очном планшете, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 при плотности 4×104 клеток/см2. 2 мл приготовленной культуральной среды SCM для эпителиальных клеток первичного рака гортани добавляли в 12-луночный планшет, который затем помещали в инкубатор при 37°С, 5% СО2 (приобретенный в компании Thermo Fisher) для культивирования. Когда клетки в культуральной планшете разрастались и покрывали примерно 80% площади дна, удаляли супернатант среды в 12-луночном планшете и добавляли 0,5 мл 0,25% трипсина (приобретенного в компании Thermo Fisher) для разложения в течение 1 минуты; после удаления 0,25% трипсина снова добавляли 0,5 мл 0,05% трипсина для разложения клеток, после чего инкубировали при комнатной температуре в течение от 5 до 20 минут до полного разложения клеток, которое наблюдали под микроскопом (EVOS М500, Invitrogen); затем разложение прекращали путем применения 1 мл культурального раствора DMEM/F12, содержащего 5% (об./об.) фетальной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл пенициллина; полученный образец собирали в центрифужную пробирку объемом 15 мл и центрифугировали со скоростью 1500 об/мин в течение 4 минут, а затем удаляли супернатант. Центрифугированные клеточные отложения повторно суспендировали в базальной среде ВМ и подсчитывали с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) с получением общего количества клеток. Полученные клетки использовали в следующих экспериментах по культивированию.
Далее для проведения экспериментов приготавливали следующие 7 культуральных сред с различными составами:
Состав 1: Состав среды SCM без фактора роста фибробластов 7;
Состав 2: Состав среды SCM без комплекса инсулин-трансферрин-селен;
Состав 3: Состав среды SCM без инсулиноподобного фактора роста 1;
Состав 4: Состав среды SCM без фактора роста гепатоцитов;
Состав 5: Состав среды SCM без Y27632;
Состав 6: Состав среды SCM без соединения 1;
Состав 7: Состав среды SCM без А83-01.
Суспензию разложившихся клеток разводили с применением приведенных выше составов 1-7, соответственно, и высевали в 48-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3, в количестве 10000 клеток и объеме 250 мкл на лунку.
При применении среды с составом 1 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученный фактор роста фибробластов 7 в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации фактора роста фибробластов 7 составляли 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл, 5 нг/мл, 2,5 нг/мл и 1,25 нг/мл, соответственно; среду с составом 1 использовали в качестве контрольной лунки (ВС).
При применении среды с составом 2 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученный комплекс инсулин-трансферрин-селен в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации исходного раствора комплекса инсулин-трансферрин-селен составляли 1:1600, 1:800, 1:400, 1:200, 1:100, 1:50 и 1:25, соответственно (что соответствует конечным концентрациям инсулина/трансферрина/селенита натрия 0,3125 мкг/мл, 0,15625 мкг/мл, 0,15625 нг/мл; 0,625 мкг/мл, 0,3125 мкг/мл, 0,3125 нг/мл; 1,25 мкг/мл, 0,625 мкг/мл, 0,625 нг/мл; 2,5 мкг/мл, 1,25 мкг/мл, 1,25 нг/мл; 5 мкг/мл, 2,5 мкг/мл, 2,5 нг/мл; 10 мкг/мл, 5 мкг/мл, 5 нг/мл; и 20 мкг/мл, 10 мкг/мл, 10 нг/мл, соответственно); среду с составом 2 использовали в качестве контрольной лунки (ВС).
При применении среды с составом 3 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученный инсулиноподобный фактор роста 1 в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации инсулиноподобного фактора роста 1 составляли 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл, 5 нг/мл, 2,5 нг/мл и 1,25 нг/мл, соответственно; среду с составом 3 использовали в качестве контрольной лунки (ВС).
При применении среды с составом 4 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученный фактор роста гепатоцитов в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации фактора роста гепатоцитов составляли 80 нг/мл, 40 нг/мл, 20 нг/мл, 10 нг/мл, 5 нг/мл, 2,5 нг/мл и 1,25 нг/мл, соответственно; среду с составом 4 использовали в качестве контрольной лунки (ВС).
При применении среды с составом 5 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученный Y27632 в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации Y27632 составляли 40 мкМ, 20 мкМ, 10 мкМ, 5 мкМ, 2,5 мкМ, 1,25 мкМ и 0,625 мкМ, соответственно; среду с составом 5 использовали в качестве контрольной лунки (ВС).
При применении среды с составом 6 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученное соединение 1 в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации соединения 1 составляли 40 мкМ, 20 мкМ, 10 мкМ, 5 мкМ, 2,5 мкМ, 1,25 мкМ и 0,625 мкМ, соответственно; среду с составом 6 использовали в качестве контрольной лунки (ВС).
При применении среды с составом 7 в 48-луночный планшет, инокулированный первичными клетками, добавляли полученный А83-01 в количестве 250 мкл на лунку таким образом, чтобы конечные концентрации А83-01 составляли 4000 нМ, 2000 нМ, 1000 нМ, 500 нМ, 250 нМ, 125 нМ и 62,5 нМ, соответственно; среду с составом 7 использовали в качестве контрольной лунки (ВС).
После размножения клеток до площади, составляющей примерно 85% от площади 48 лунок, клетки подвергали разложению и подсчитывали, при этом указанные отношения рассчитывали с учетом количества клеток в контрольной лунке (ВС), полученные результаты показаны на фиг. 3A-3G. На каждой из фиг. 3A-3G приведенное отношение представляет собой отношение количества клеток при первом пассировании, культивируемых с применением каждой культуральной среды, к количеству клеток при первом пассировании, культивируемых с применением соответствующей контрольной лунки. Если такое отношение больше 1, это указывает на то, что стимулирующий пролиферацию эффект полученной среды, содержащей различные концентрации факторов или низкомолекулярных соединений, является предпочтительным по сравнению с эффектом среды в контрольной лунке; если такое отношение меньше 1, это указывает на то, что стимулирующий пролиферацию эффект полученной среды, содержащей различные концентрации факторов или низкомолекулярных соединений, меньше, чем эффект среды в контрольной лунке.
Согласно результатам, показанным на фиг. 3А-3G, количество фактора роста фибробластов 7 в культуральной среде предпочтительно составляет от 5 нг/мл до 80 нг/мл, более предпочтительно от 20 нг/мл до 80 нг/мл; объемная концентрация комплекса инсулин-трансферрин-селен предпочтительно составляет от 1:25 до 1:100, более предпочтительно от 1:25 до 1:150 (что соответствует конечным концентрациям инсулина/трансферрина/селенита натрия от 5 до 20 мкг/мл, от 2,5 до 10 мкг/мл, от 2,5 до 10 нг/мл, соответственно, и более предпочтительно от 10 до 20 мкг/мл, от 5 до 10 мкг/мл, от 5 до 10 нг/мл, соответственно); количество инсулиноподобного фактора роста 1 предпочтительно составляет от 10 нг/мл до 80 нг/мл, более предпочтительно от 10 нг/мл до 40 нг/мл; количество фактора роста гепатоцитов предпочтительно составляет от 10 нг/мл до 80 нг/мл, более предпочтительно от 10 нг/мл до 40 нг/мл; количество Y27632 предпочтительно составляет от 1,25 мкМ до 20 мкМ и более предпочтительно от 2,5 мкМ до 10 мкМ; количество соединения 1 предпочтительно составляет от 1,25 мкМ до 10 мкМ и более предпочтительно от 2,5 мкМ до 10 мкМ; количество А83-01 предпочтительно составляет от 125 нМ до 1000 нМ и более предпочтительно от 125 нМ до 500 нМ.
[Пример 3]
Культивирование клеток первичного рака гортани, полученных из ткани рака гортани человека
Эпителиальные клетки рака гортани, происходящие из раковой ткани, выделяли и получали из раковой ткани пациента с раком гортани (образец №0S0003) с применением того же способа, что и в примере 1. Далее для получения общего количества клеток клетки рака гортани, происходящие из раковой ткани, подсчитывали с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.). Затем указанные клетки инокупировали в 12-луночном планшете, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 при плотности 4×104 клеток/см2. 2 мл приготовленной культуральной среды SCM для эпителиальных клеток первичного рака гортани добавляли в 12-луночный планшет, который затем помещали в инкубатор при 37°С, 5% CO2 (приобретенный в компании Thermo Fisher) для культивирования.
На фиг. 4А показано микроскопическое изображение (сфотографированное с помощью 100× инвертированного фазово-контрастного микроскопа) клеток, которые культивировали до дня 4 после инокуляции при плотности 4×104 клеток/см2 в 12-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 согласно данному примеру. Микроскопическое исследование показало, что культивированные клетки первичного рака гортани, полученные из раковой ткани, образовали крупные клоны. На фиг. 4В приведено микроскопическое изображение (сфотографированное с помощью 100× инвертированного фазово-контрастного микроскопа) клеток, которые культивировали до дня 12 после инокуляции в соответствии с данным примером, которое показало, что поле зрения было заполнено клетками. На фиг. 4А и 4В можно видеть, что формирование заметного клона можно наблюдать под микроскопом, когда выделенные клетки первичного рака гортани культивировали in vitro в течение 4 дней, при этом количество клеток значительно увеличивалось после 12 дней размножения, что предполагает, что технология согласно настоящему изобретению представляет собой эффективную технологию с точки зрения размножения in vitro эпителиальных клеток рака гортани.
[Пример 4]
Влияние различных культуральных сред на стимулирование пролиферации клеток первичного рака гортани, полученных из ткани рака гортани
(1) Сравнение влияния различных культуральных сред на клонообразование первичных клеток и пролиферативные эффекты первичных клеток
Культуральную среду SCM для эпителиальной клетки первичного рака гортани получали тем же способом, что и в примере 2, и приготавливали базальную среду ВМ в качестве контроля. В качестве другого контроля дополнительно приготавливали культуральную среду FM, применяемую в технологии условного перепрограммирования клеток. Описание этапов приготовления сред можно найти в Liu et al., Nat Protoc, 12(2): 439-451, 2017. Состав указанной культуральной среды приведен в таблице 5. Одновременно в качестве дополнительных контрольных условий приготавливали культуральные среды SCM-1 и SCM-2 для клетки первичного рака гортани, при этом составы указанных культуральных сред получали путем замены комплекса инсулин-трансферрин-селен в среде SCM на добавку В27 при объемном отношении 1:50 и добавку N2 при объемном отношении 1:100, соответственно. Кроме того, в качестве дополнительного контроля в компании Gibco приобретали коммерческую среду «Defined Keratinocyte SFM» (бессывороточную среду, оптимизированную для выделения и размножения кератиноцитов человека) (в дальнейшем также называемую «средой KSFM»), состав указанной культуральной среды для приведен в таблице 6.


Путем применения того же способа, что и в примере 1, получали клетки первичного рака гортани (№0S0006), полученные из тканей рака гортани. Далее указанные клетки культивировали при одинаковой плотности (4×104 клеток/см2) при следующих 6 условиях культивирования:
A. Технология согласно настоящему изобретению: клетки первичного рака гортани инокулировали в 24-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 (приобретенные в компании АТСС) при плотности 4×104 клеток/см2, культивировали с применением 1 мл культуральной среды SCM для эпителиальных клеток первичного рака гортани согласно настоящему изобретению.
B. Технология условного перепрограммирования клеток: клетки первичного рака гортани инокулировали в 24-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 (приобретенные в компании АТСС) при плотности 4×104 клеток/см2, культивировали в 24-луночном планшете с применением 1 мл среды FM для условного перепрограммирования клеток (подробное описание этапов см. в: Liu et al., Nat. Protoc., 12(2): 439-451, 2017).
C. Клетки первичного рака гортани инокулировали в 24-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 (приобретенные в компании АТСС) при плотности 4×104 клеток/см2, и культивировали в 24-луночном планшете с применением 1 мл культуральной среды SCM-1.
D. Клетки первичного рака гортани инокулировали в 24-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 (приобретенные в компании АТСС) при плотности 4×104 клеток/см2, и культивировали в 24-луночном планшете с применением 1 мл культуральной среды SCM-2.
E. Клетки первичного рака гортани инокулировали в 24-луночный планшет при плотности 4×104 клеток/см2 и культивировали в 24-луночном планшете с применением 1 мл коммерческой среды KSFM.
F. Клетки первичного рака гортани инокулировали в 24-луночный планшет, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 (приобретенные в компании АТСС) при плотности 4×104 клеток/см2, и культивировали в 24-луночном планшете с применением 2 мл базальной среды ВМ.
В описанных выше шести культурах клетки культивировали при шести условиях культивирования, при этом среды обновляли каждые 5 дней. Одновременно исследовали образование клеточных клонов и статус пролиферации клеток при культивировании в каждой среде в 24-луночном планшете, при этом статус роста клеток фиксировали путем фотографирования с помощью микроскопа (EVOS М500, Invitrogen).
Что касается первичных раковых клеток рака гортани (№0S0006), культивированных с применением технологии согласно настоящему изобретению, когда указанные клетки в культуральном планшете разрастались и покрывали примерно 80% площади дна, супернатант среды в 24-луночном планшете удаляли и добавляли 0,5 мл 0,25% трипсина (приобретенного в компании Thermo Fisher) для разложения в течение 1 минуты; после удаления 0,25% трипсина снова добавляли 0,5 мл 0,05% трипсина для разложения клеток, после чего инкубировали при 37°С в течение 10 минут до полного разложения клеток, которое наблюдали под микроскопом (EVOS М500, Invitrogen); затем разложение прекращали путем применения 1 мл культурального раствора DMEM/F12, содержащего 5% (об./об.) фетальной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл пенициллина; полученный образец собирали в центрифужную пробирку объемом 15 мл и центрифугировали со скоростью 1500 об/мин в течение 4 минут, а затем удаляли супернатант. Центрифугированные клеточные отложения повторно суспендировали в культуральной среде согласно настоящему изобретению и подсчитывали с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) с получением общего количества клеток, составляющего 317000. Клетки, культивированные при других пяти условиях культивирования, подвергали разложению и подсчитывали с применением того же технологического процесса, описанного выше. Общее количество клеток, культивированных с применением сред FM, SCM-1, SCM-2, KSFM и ВМ составляло 166700, 266100, 277200, 95600 и 35900, соответственно.
Фиг. 5 представляет собой график общего количества клеток, амплифицированных из клеток №0S0006 при различных условиях.
Фиг. 6 представляет собой сравнительную диаграмму, на которой показаны клеточные пролиферативные эффекты, полученные при культивировании клеток первичного рака гортани, выделенных из шести образцов пациентов с раком гортани (№0S0011, 0S0012, 0S0013, 0S0014, 0S0015, 0S0016), в соответствии с примером 1 в течение 7 дней в условиях применения шести различных культуральных сред, где √ означает умеренные клонообразующую способность и эффект, стимулирующий пролиферацию, √√ означает значительные клонообразующую способность и эффект, стимулирующий пролиферацию, √√√ означает чрезвычайно значительные клонообразующую способность и эффект, стимулирующий пролиферацию и × означает отсутствие клонообразования. Указанная фигура позволяет подтвердить, что культуральная среда SCM имеет значительное превосходство над другими пятью условиями культивирования с точки зрения клонообразующей способности и эффекта, стимулирующего пролиферацию клеток, при культивировании первичной клетки, происходящей из ткани рака гортани.
(2) Кривая непрерывного культивирования и роста клеток первичного рака гортани в различных культуральных средах
Культуральную среду SCM для эпителиальной клетки первичного рака гортани и культуральные среды FM, SCM-1, SCM-2, KSFM и ВМ в качестве контрольных сред получали с применением того же способа, что и в пункте (1) настоящего примера.
Ткань рака гортани, происходящую из клеток первичного рака гортани (№0S0004), культивировали в шести культуральных средах, а затем подвергали разложению, пассировали и подсчитывали с применением того же способа, что и в пункте (1) настоящего примера.
Когда пересеваемые клетки разрастались в культуральном планшете и снова покрывали примерно 80% площади дна планшета, указанные культивированные клетки подвергали разложению, собирали и подсчитывали в соответствии с описанным выше рабочим способом. Клетки повторно инокулировали при плотности 4×104 клеток/лунку и осуществляли непрерывное культивирование.
Ниже приведена формула для расчета количества эпителиальных клеток первичного рака гортани при удвоении клеточной популяции при различных условиях культивирования:
Удвоение популяции (PD)=3,32 × log10 (общее количество разложившихся клеток/количество клеток при начальной инокуляции), см. Chapman et al., Stem Cell Research & Therapy 2014, 5: 60.
На фиг. 7 показаны кривые роста клеток 0S0004 при шести различных условиях культивирования, полученные с помощью программного обеспечения Graphpad Prism. По оси абсцисс показаны дни, в которые осуществляли культивирование клеток, а по оси ординат показана величина, кратная уровню кумулятивной пролиферации клеток, то есть величина, кратная уровню размножения клеток в период культивирования. Чем больше указанное значение, тем больше величины, кратные уровню размножения клеток за определенный период, то есть, чем больше размножается клеток. Наклон представляет собой скорость размножения клеток. Указанная фигура позволяет подтвердить, что скорости пролиферации эпителиальных клеток рака гортани, культивированных с применением культуральной среды SCM согласно настоящему изобретению и культуральных сред SCM-1, SCM-2, превосходили другие три условия культивирования, а также позволяет подтвердить, что при применении технологии согласно настоящему изобретению можно непрерывно культивировать эпителиальные клетки первичного рака гортани.
[Пример 5]
Идентификация клеток первичного рака гортани, полученных из раковых тканей (1) Иммунофлуоресцентная идентификация тканей первичного рака гортани и клеток рака гортани после культивирования с применением пассирования
Эпителиальные клетки рака гортани, происходящие из раковой ткани, выделяли и получали из раковой ткани пациента с раком гортани (образец №0S0015) с применением того же способа, что и в примере 1. Далее для получения общего количества клеток клетки рака гортани, происходящие из раковой ткани, подсчитывали с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.). Затем указанные клетки инокулировали в 24-луночном планшете, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 при плотности 4×104 клеток/см2. Одновременно в 24-луночный планшет предварительно помещали круглые предметные стекла (приобретенные в компании Thermo Fisher) для иммунофлуоресцентного окрашивания. 1 мл приготовленной культуральной среды SCM для эпителиальных клеток первичного рака гортани добавляли в 24-луночный планшет, который затем помещали в инкубатор при 37°С, 5% СО2 (приобретенный в компании Thermo Fisher) для культивирования.
Когда клетки в 24-луночном планшете размножились и покрыли 80% площади дна, культуральный раствор удаляли и применяли 4% формальдегид для фиксации клеток на льду в течение 30 минут. Полученный образец промывали с помощью PBS (фосфатно-солевой буфер) (приобретенного в компании Shanghai Sangon Biotech) в течение 5 минут × 3 раза. После удаления PBS добавляли проникающую жидкость и в течение 30 минут встряхивали полученный образец (со скоростью примерно 100 об/мин) без воздействия света для разрушения мембраны, а затем промывали с применением PBS в течение 5 минут × 3 раза. После этого использовали PBS+0,3% Тритон Х-100 (приобретенный в компании Shanghai Sangong Biotech) для получения 5 об. % раствора BSA (бычий сывороточный альбумин) (приобретенного в компании Shanghai Sangong Biotech) для блокирования и осуществляли блокирование при 37°С в течение 30 минут.
Заранее приготавливали PBS+0,3% Тритон Х-100 для разведения антитела, при этом антитело р63, специфичное к плоскоклеточной карциноме (приобретенное в компании CST), разводили в соотношении 1:50. Блокирующий раствор удаляли и добавляли приготовленный разбавитель для первичного антитела. Полученный образец инкубировали в холодильнике при 4°С в течение ночи. Полученный образец вынимали при 4°С, доводили до комнатной температуры, продолжали инкубирование при 37°С в течение 1 часа, а затем промывали с помощью PBS в течение 5 минут × 3 раза.
Заранее приготавливали PBS+0,3% Тритон Х-100 для разведения вторичного антитела, в котором кроличье флуоресцентное вторичное антитело (приобретенное в компании Thermo Fisher) со светом возбуждения 488 нм разводили в соотношении 1:1000. Полученный образец инкубировали при комнатной температуре в темноте в течение 1 часа, а затем промывали с применением PBS в течение 5 минут × 3 раза.
Неспецифический флуоресцентный краситель DAPI (4,6-диамидино-2-фенилиндол дигидрохлорид) (приобретенный в компании Sigma) разводили с применением PBS в объемном соотношении 1:1000, а затем использовали для окрашивания при комнатной температуре в темноте в течение 5 минут. Полученный образец промывали с помощью PBS в течение 5 минут × 3 раза. Фотографии делали под микроскопом (EVOS М500, Invitrogen) для записи.
На фиг. 8А и 8В приведены флуоресцентные фотографии, сделанные в различных полях под объективом 10×, соответственно; при этом на фиг. 8А показана картина окрашивания ядер неспецифическим флуоресцентным красителем DAPI, а на фиг. 8В показана картина окрашивания антителом р63, специфичным к раку гортани (расположенным в ядрах). Как показано на указанных фигурах, положения, отмеченные ядрами на фиг. 8А, были все окрашены в зеленый цвет на фиг. 8В, что указывает на то, что культивируемые клетки представляют собой клетки плоскоклеточной карциномы гортани, что согласуется с клиническим патоморфологическим диагнозом.
(2) Иммуногистохимическая идентификация тканей первичного рака гортани и клеток рака гортани после культивирования с применением пассирования
Раковую ткань (образец №0S0001) размером примерно с боб мунг извлекали из удаленного путем хирургической резекции образца у пациента с раком гортани и погружали в 1 мл 4% параформальдегида. Эпителиальные клетки рака гортани (образец №0S0001) получали из оставшейся раковой ткани тем же способом, что и в примере 1. Образец 0S0001 непрерывно культивировали до шестого пассирования с применением культуральной среды SCM согласно настоящему изобретению в соответствии со способом, описанным в примере 3.
Для определения экспрессии важных связанных с раком гортани биомаркеров в исходной ткани образца 0S0001 и первичных клетках, полученных путем непрерывного культивирования до шестого пассирования, использовали иммуногистохимический анализ. Ткань фиксировали с применением 4% параформальдегида, заливали в парафин и разрезали с помощью микротома с получением тканевых срезом толщиной 4 мкм. Затем проводили обычный иммуногистохимический анализ (подробное описание этапов см. в Li et al., Nature Communication, (2018) 9: 2983). В качестве первичных антител использовали цитокератин (pCK) (приобретенный в компании CST), антитело р63 (приобретенное в компании CST) и антитело Ki67 (приобретенное в компании R&D).
На фиг. 9 приведено сравнение результатов иммуногистохимического анализа клеток исходной ткани и клеток рака гортани, полученных путем культивирования указанных клеток с применением культуральной среды SCM согласно настоящему изобретению. Фиг. 9 подтверждает, что экспрессия связанных с раком гортани биомаркеров на клетках, которые культивировали из клеток рака гортани (образец №0S0001) с применением технологии согласно настоящему изобретению до 6-го пассирования, соответствовала экспрессии маркеров на срезе исходной ткани, из которого были получены указанные клетки. Из этого можно сделать вывод, что клетки, культивированные с применением технологии согласно настоящему изобретению, сохраняют исходные патологические характеристики раковых тканей пациентов с раком гортани.
[Пример 6]
Эксперименты по ксенотрансплантатному онкогенезу клеток первичного рака гортани, полученных из раковой ткани, у мышей
Клетки рака гортани (№0S0003) выделяли и получали из раковых тканей одного пациента с патоморфологически диагностированным раком гортани с применением того же способа, что и в примере 1. Образец 0S0003 культивировали с применением культуральной среды SCM согласно настоящему изобретению в соответствии со способом, описанным в примере 3, и когда количество клеток рака гортани достигало 1×107, указанные клетки рака гортани подвергали разложению и собирали с применением того же способа, что и в примере 4. Культуральную среду SCM для клеток рака гортани согласно настоящему изобретению и Matrigel® (приобретенный в компании BD Biosciences) смешивали в соотношении 1:1 и использовали 100 мкл указанной культуральной среды, смешанной с Matrigel, для повторного суспендирования 5хЮб клеток рака гортани, после чего полученный образец вводили путем инъекции в жировую подушку рака гортани и подмышечную область правой передней конечности 6-недельных самок мышей с высоким иммунодефицитом (NCG) (приобретенных в компании Nanjing Model Animal Research Center), соответственно. Фиксировали объем и скорость роста опухолей, образующихся из раковых клеток гортани, у мышей и делали фотографии каждые три дня.
На 21-й день после инокуляции опухолевых клеток можно было наблюдать образование опухоли в обоих из двух мест инокуляции опухолевых клеток у мышей. С 21-го дня по 40-й день пролиферация опухоли у мышей была заметной. Это означает, что у мышей полученные из раковой ткани клетки рака гортани, культивированные с применением способа культивирования согласно настоящему изобретению, обладают онкогенностью.
[Пример 7]
Функциональный тест на чувствительность к лекарственным средствам клеток рака гортани, полученных из раковой ткани
При применении в качестве примера полученный путем хирургической резекции образец от пациента с раком гортани было подтверждено, что клетки рака гортани, культивированные из полученных от пациента образцов опухоли гортани, можно использовать для тестирования чувствительности опухолевых клеток пациента к разным лекарственным средствам.
1. Посев клеток первичного рака гортани: Клеточные суспензии выделенных клеток рака гортани (№0S0020 и №0S0022), полученных в соответствии со способом, описанным в примере 1, инокулировали в 12-луночном планшете, на который были предварительно нанесены облученные γ-лучами клетки NIH-3T3 при плотности 4×104 клеток/см2. 2 мл приготовленной культуральной среды SCM для эпителиальных клеток первичного рака гортани добавляли в 12-луночный планшет, который затем помещали в инкубатор при 37°С, 5% СО2 (приобретенный в компании Thermo Fisher) для культивирования. Когда клетки в культуральном планшете разрастались и покрывали примерно 80% площади дна, супернатант среды в 12-луночном планшете удаляли и добавляли 0,5 мл 0,25% трипсина (приобретенного в компании Thermo Fisher) для разложения в течение 1 минуты; после удаления 0,25% трипсина снова добавляли 0,5 мл 0,05% трипсина для разложения клеток; клетки инкубировали при 37°С в течение 10 минут до полного разложения клеток, которое наблюдали под микроскопом (EVOS М500, Invitrogen); затем разложение прекращали путем применения 1 мл культурального раствора DMEM/F12, содержащего 5% (об./об.) фетальной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл пенициллина; полученный образец собирали в центрифужную пробирку объемом 15 мл и центрифугировали со скоростью 1500 об/мин в течение 4 минут, а затем удаляли супернатант. Центрифугированные клеточные отложения повторно суспендировали в культуральной среде SCM и подсчитывали с применением счетчика с визуализацией потока (JIMBIO FIL, Jiangsu Jimbio Technology Co., Ltd.) с получением общего количества клеток, которое составляло 830000 и 768000, соответственно. Клетки инкубировали в 384-луночном планшете при плотности от 2000 до 4000 клеток/лунку и оставляли указанные клетки прилипать в течение ночи.
2. Эксперименты с градиентами концентраций лекарственных средств:
(1) Приготавливали планшеты для хранения лекарственных средств с применением метода градиентного разведения: 40 мкл 10 мкМ исходных растворов лекарственных средств, предназначенных для тестирования, применяли, соответственно, в качестве максимальных концентраций, и из них отбирали пипеткой 10 мкл растворов, соответственно, и добавляли в ЕР-пробирки объемом 0,5 мл, содержащие 20 мкл ДМСО; 10 мкл растворов из вышеупомянутых ЕР-пробирок снова отбирали пипеткой во вторые ЕР-пробирки объемом 0,5 мл, содержащие 20 мкл ДМСО, то есть разводили лекарственные средства в соотношении 1:3. Описанный выше способ повторяли для постепенного разведения и получали 7 концентраций, необходимых для дозирования. В 384-луночные планшеты для хранения лекарственных средств добавляли разные концентрации лекарственных средств. В каждую лунку группы для контроля растворителем добавляли равный объем ДМСО в качестве контроля. В указанном примере в качестве лекарственных средств, подлежащих тестированию, использовали бортезомиб (приобретенный в компании МСЕ), дисульфирам (приобретенный в компании МСЕ), гефитиниб (приобретенный в компании МСЕ) и эрлотиниб (приобретенный в компании МСЕ).
(2) Путем применения высокопроизводительной автоматизированной установки (JANUS, Perkin Elmer) группы с разными концентрациями лекарственных средств и контрольные группы с растворителем в 384-луночных планшетах для хранения лекарственных средств добавляли в 384-луночные планшеты для культивирования клеток, на которые были высеяны раковые клетки гортани. Все группы с лекарственными средствами и контрольные группы с растворителем помещали в 3 дублирующие лунки. Объем лекарственных средств, добавленных в каждую лунку, составлял 100 нл.
(3) Тест на жизнеспособность клеток: через 72 часа после введения использовали набор для анализа Cell Titer-Glo (приобретенный в компании Promega) для определения величины хемилюминесценции культивированных клеток после введения лекарственного средства. Значение величины хемилюминесценции отражает жизнеспособность клеток и влияние лекарственного средства на жизнеспособность клеток. В каждую лунку добавляли 10 мкл полученного раствора для обнаружения Cell Titer-Glo и использовали микропланшетный ридер (Envision, Perkin Elmer) для определения величины хемилюминесценции после смешивания.
(4) Тест на жизнеспособность клеток: В соответствии с формулой уровень выживаемости клеток (%) = величина хемилюминесценции лунки с лекарственным средством/величина хемилюминесценции контрольной лунки × 100% рассчитывали уровни выживаемости клеток, обработанных различными лекарственными средствами. С помощью программного обеспечения Graphpad Prism строили графики и рассчитывали показатели полумаксимального ингибирования IC50 и одновременно рассчитывали уровни выживаемости клеток при максимальных концентрациях Cmax различных лекарственных средств в крови в организме человека.
(5) Результаты тестирования на чувствительность к лекарственным средствам показаны на фиг. 10.
На фиг. 10А и 10В, соответственно, представлена чувствительность клеток рака гортани, культивированных из хирургически удаленных образцов раковой ткани двух разных пациентов с раком гортани (образец №0S0020 и образец №0S0022), к двум химиотерапевтическим препаратам бортезомибу и дисульфираму и к лекарственным средствам направленного действия гефитинибу и эрлотинибу. В частности, на фиг. 10А показаны результаты определения чувствительности клеток рака гортани, культивированных из образца №0S0020, к указанным четырем лекарственным средствам; на фиг. 14В показаны результаты определения чувствительности клеток рака гортани, культивированных из образца №0S0022, к указанным четырем лекарственным средствам. На приведенных фигурах концентрации по оси абсцисс, соответствующие пунктирным линиям, представляют собой максимальные концентрации Cmax указанных четырех препаратов в крови в организме человека. Полученные результаты показали, что клетки от одного и того же пациента имеют разную чувствительность к разным лекарственным средствам при их соответствующих максимальных концентрациях в крови в организме человека, при этом клетки от разных пациентов также имеют разную чувствительность к лекарственным средствам при максимальных концентрациях в крови в организме человека. Согласно полученным результатам можно определять эффективность лекарственных средств при клиническом применении у больных раком гортани.
Промышленная применимость
В настоящем изобретении предложены культуральная среда и способ культивирования или размножения in vitro эпителиальных клеток первичного рака гортани. Потомство клеток и органоид, культивированные с применением культуральной среды и способа культивирования согласно настоящему изобретению, можно использовать для оценки эффективности и скрининга лекарственных средств, определения токсичности и в регенеративной медицине. Следовательно, настоящее изобретение применимо в промышленности.
Хотя настоящее изобретение было подробно описано в данном документе, настоящее изобретение не ограничено таким описанием, и специалисты в данной области техники могут внести изменение в соответствии с принципом настоящего изобретения. Следовательно, все модификации, выполненные в соответствии с принципом настоящего изобретения, следует интерпретировать как находящиеся в пределах объема охраны настоящего изобретения.




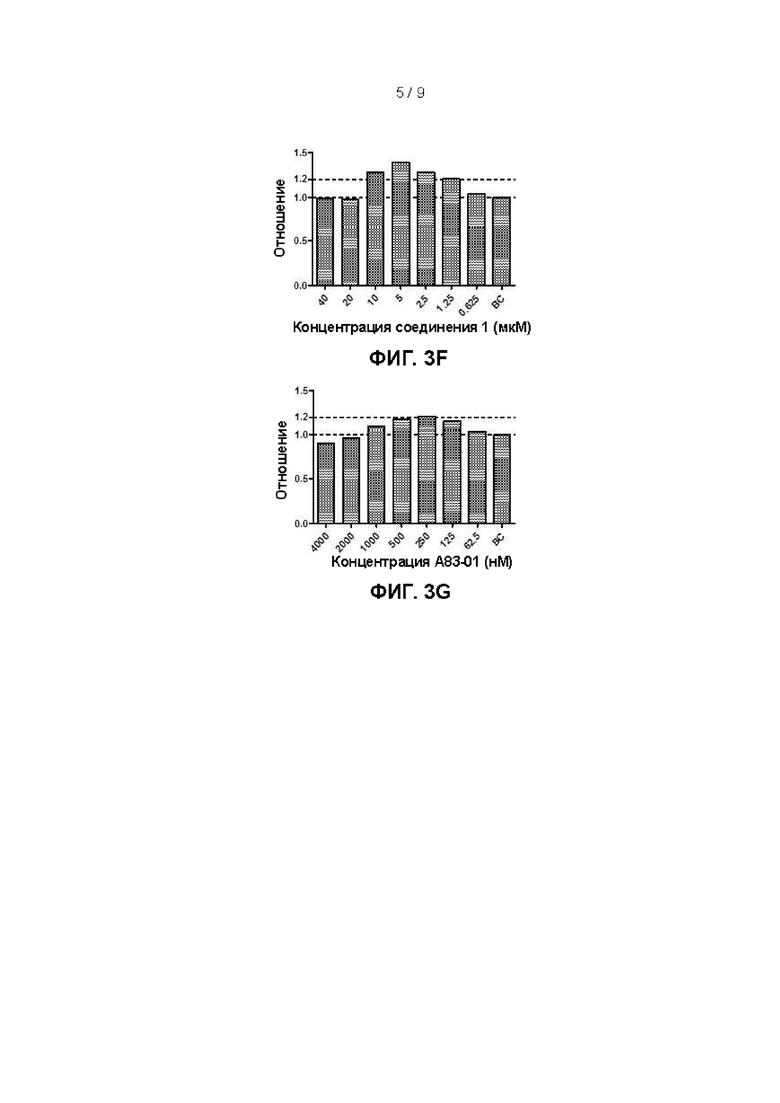

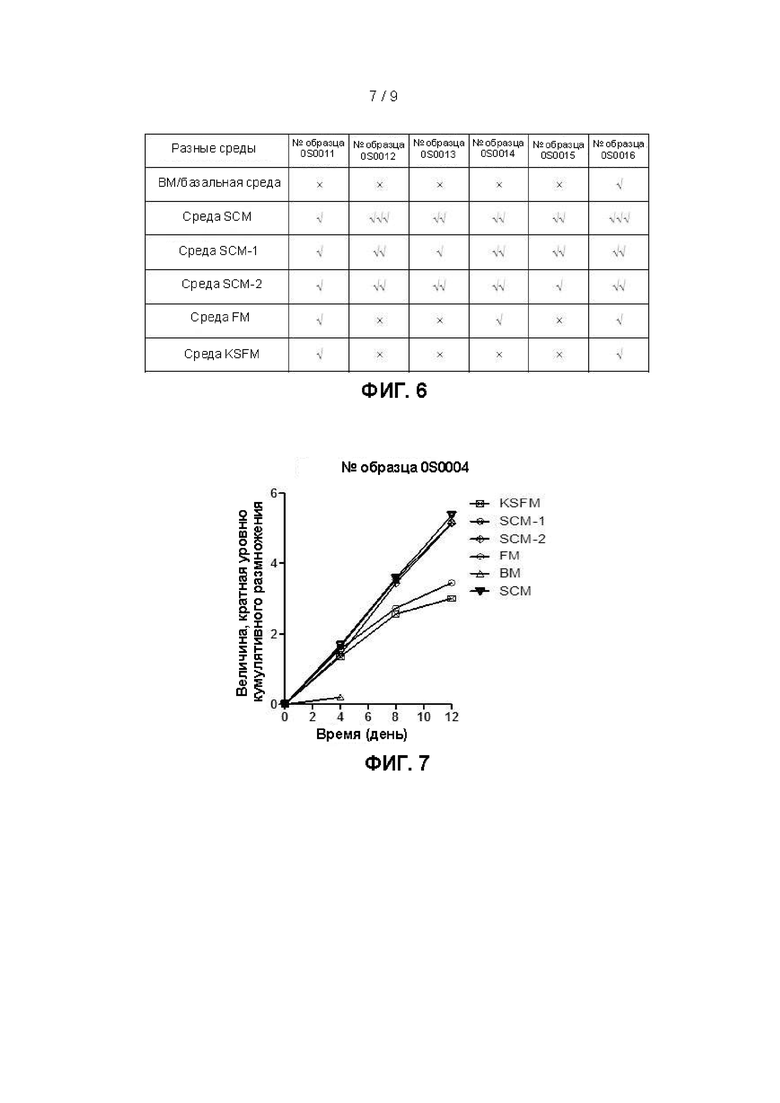


Настоящее изобретение относится к области биотехнологии. Изобретение раскрывает новую культуральную среду, содержащую ингибитор киназы MST1/2 и обеспечивающую более короткий период культивирования клеток в сравнении с известными аналогами. Изобретение может быть применимо для культивирования или размножения in vitro эпителиальных клеток первичного рака гортани. Клеточная модель, полученная с применением культуральной среды для первичных клеток согласно настоящему изобретению, может быть использована для оценки эффективности и скрининга лекарственных средств, подходящих для лечения рака гортани. 3 н. и 8 з.п. ф-лы, 10 ил., 6 табл., 7 пр.
1. Культуральная среда для первичных клеток для культивирования эпителиальных клеток рака гортани, характеризующаяся тем, что:
содержит ингибитор киназы MST1/2, при этом указанный ингибитор киназы MST1/2 включает соединение формулы (I) или его фармацевтически приемлемую соль или сольват,
 Формула (I)
Формула (I)
где
R1 выбран из C1-C6 алкила, C3-C6 циклоалкила, C2-C6 спироциклоалкила и фенила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6, и тиенила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6;
R2 и R3 каждый независимо выбраны из C1-C3 алкила;
R4 и R5 каждый независимо выбраны из водорода, C1-C6 алкила, C3-C6 циклоалкила и C4-C8 циклоалкилалкила;
R6 выбран из галогена, C1-C6 алкила и C1-C6 галогеналкила.
2. Культуральная среда для первичных клеток по п. 1, в которой ингибитор киназы MST1/2 включает соединение формулы (Ia) или его фармацевтически приемлемую соль или сольват,
 Формула (Ia)
Формула (Ia)
где
R1 выбран из C1-C6 алкила, фенила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6, и тиенила, необязательно независимо содержащего в качестве заместителя от 1 до 2 R6;
R5 выбран из водорода, C1-C6 алкила и C3-C6 циклоалкила;
R6 независимо выбран из галогена, C1-C6 алкила и C1-C6 галогеналкила.
3. Культуральная среда для первичных клеток по п. 2, в которой
R1 представляет собой фенил, необязательно независимо содержащий в качестве заместителя от 1 до 2 R6;
R5 представляет собой водород;
R6 представляет собой фтор, метил или трифторметил.
4. Культуральная среда для первичных клеток по п. 1, в которой ингибитор киназы MST1/2 представляет собой по меньшей мере соединение, выбранное из следующих соединений, или его фармацевтически приемлемую соль:

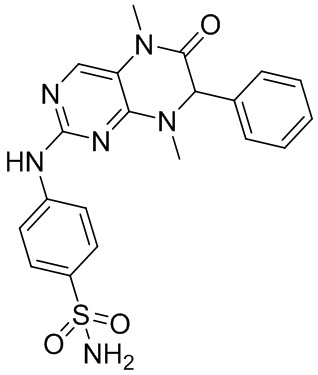
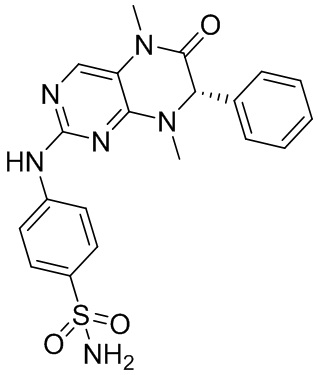



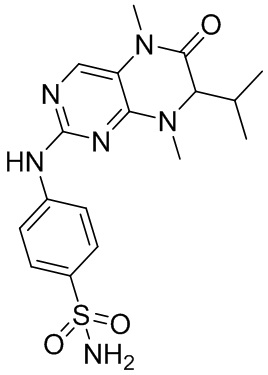
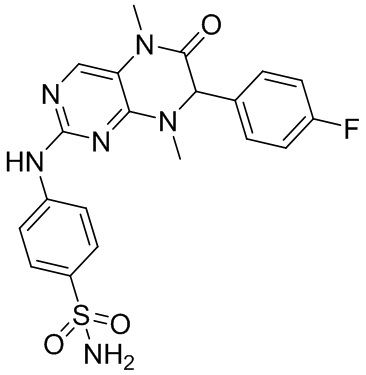



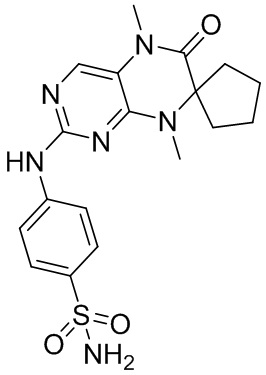






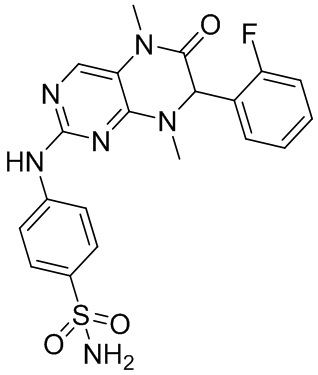


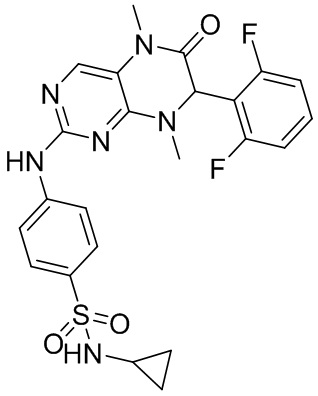

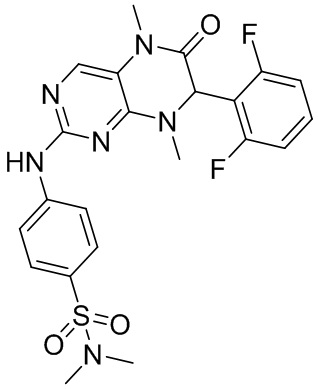





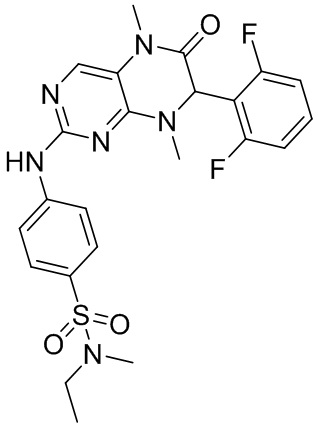
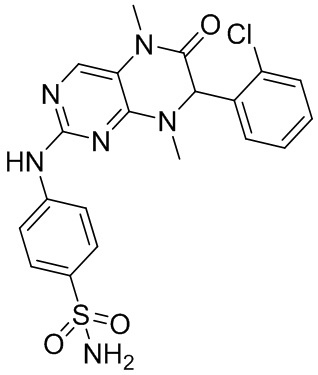
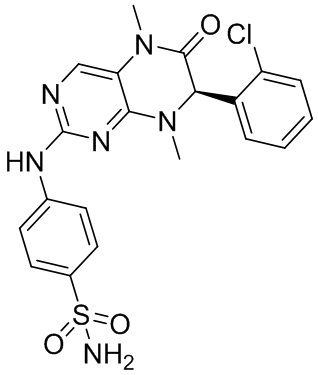
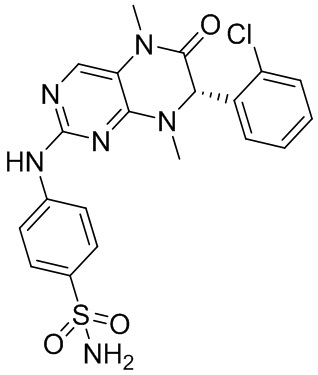

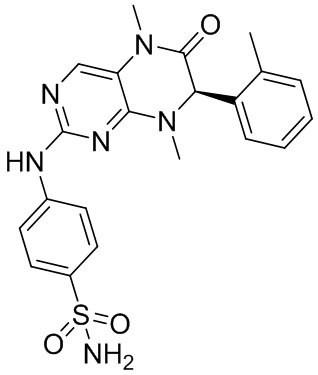
 .
.
5. Культуральная среда для первичных клеток по любому из пп. 1-4, характеризующаяся тем, что:
количество ингибитора киназы MST1/2 составляет от 1,25 до 10 мкМ, предпочтительно от 2,5 до 10 мкМ.
6. Культуральная среда для первичных клеток по любому из пп. 1-4, характеризующаяся тем, что:
дополнительно содержит один или более из следующих факторов: инсулиноподобный фактор роста 1; фактор роста фибробластов 7; комплекс инсулин-трансферрин-селен; фактор роста гепатоцитов; ингибитор киназы ROCK, выбранный из по меньшей мере одного из Y27632, фасудила и H-1152; и ингибитор рецептора TGFβ типа I, выбранный из по меньшей мере одного из A83-01, SB431542, Repsox, SB505124, SB525334, SD208, LY36494 и SJN2511.
7. Культуральная среда для первичных клеток по п. 6, характеризующаяся тем, что:
количество инсулиноподобного фактора роста 1 составляет от 10 до 80 нг/мл, более предпочтительно от 10 от 40 нг/мл;
количество фактора роста фибробластов 7 составляет от 5 до 80 нг/мл, более предпочтительно от 20 до 80 нг/мл;
соответствующие количества инсулина/трансферрина/селенита натрия в комплексе инсулин-трансферрин-селен составляют от 5 до 20 мкг/мл, от 2,5 до 10 мкг/мл, от 2,5 до 10 нг/мл, соответственно, и более предпочтительно от 10 до 20 мкг/мл, от 5 до 10 мкг/мл, от 5 до 10 нг/мл, соответственно;
количество фактора роста гепатоцитов составляет от 10 до 80 нг/мл, более предпочтительно от 10 от 40 нг/мл;
количество ингибитора киназы ROCK составляет от 1,25 до 20 мкМ, более предпочтительно от 2,5 до 10 мкМ;
количество ингибитора рецептора TGFβ типа I составляет от 125 до 1000 нМ, более предпочтительно от 125 до 500 нМ.
8. Культуральная среда для первичных клеток по любому из пп. 1-4, характеризующаяся тем, что:
не содержит сыворотку, экстракт гипофиза быка, агонисты Wnt, белки семейства R-спондин, ингибиторы BMP, никотинамид или N-ацетилцистеин.
9. Культуральная среда для первичных клеток по любому из пп. 1-4, характеризующаяся тем, что:
эпителиальные клетки рака гортани выбраны из клеток рака гортани, нормальных эпителиальных клеток рака гортани и эпителиальных стволовых клеток рака гортани.
10. Способ культивирования эпителиальных клеток рака гортани, характеризующийся тем, что указанный способ включает следующие этапы:
(1) приготовление культуральной среды для первичных клеток по любому из пп. 1-9;
(2) предварительное нанесение на сосуд для культивирования трофобластных клеток, облученных с применением рентгеновских лучей или γ-лучей;
(3) инокуляцию эпителиальных клеток первичного рака гортани, выделенных из тканей рака гортани, в сосуде для культивирования, на который предварительно нанесены трофобластные клетки, и культивирование с применением культуральной среды для первичных клеток, полученной на этапе (1).
11. Способ скрининга лекарственного средства для лечения раковых заболеваний гортани, характеризующийся тем, что указанный способ включает следующие этапы:
(1) культивирование эпителиальных клеток рака гортани с применением способа культивирования по п. 10;
(2) выбор лекарственного средства, подлежащего тестированию, и разведение с получением разных градиентов концентрации лекарственного средства;
(3) добавление лекарственного средства, разбавленного до градиентов, к эпителиальным клеткам рака гортани, полученным на этапе (1), и определение жизнеспособности клеток.
| TURUNEN S.P | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| WO 2020073906 A1, 16.04.2023 | |||
| CN 105801582 A, 27.07.2016 | |||
| WO 2008050096 A1, 02.05.2008 | |||
| RU 2018124591 A1, 06.04.2020. | |||

