Изобретение относится к микробиологической промышленности и касается получения аит)иотика-макролида с макроциклическим лактониым кольцом.
Известен способ получен1тя антибиотика ти- ; лозина, обладающего оппеделенным антимикробным спектром 1}.
Однако известный антибиотик обладает ниченным спектром антимикробного действия.
Целью изобретенил является получение антиби тика, подавляющего ткроорганиз1уш, патогенные для животных, в частности обладающего активностью в отнощении грамгполо жителыП)1х микроорганизмов и микоштазм.
Эта цель достигается созданием способа получения антибиотика-макролида общей форму; где Q--CHO, юш CHgOH, или ацкльный слож Hbdi зфир, при зтом, когда в положении 3 в молекуле О-ацетильная группа, то в положени 4 отсутствует 0-изовалерианорая группа, в . KOTOpiSM штамм Streptomyces fradiae NRRL 12170 выращивают в глубинных азробных условиях в питательной среде, содержащей ис точники азота и углерода и минерашьные сол выделяют целевой продукт, где Q-CHO и пр необходимости переводят его химическим вос становлением в соединение, где и/или переводят путем этерификании в ациль 1Ш1Й сложный эфир. Для осуществпешш способа используютчнтамм Streptomyces fracJiae, который хранится в коллекции NRRL под номером 12170 и по-, лучен с помощью мутагенеза. Культурально морфологические признаки. Спорофозы-RA, цепи спор ; 10, поверхность спор гладкая, форма сферическая., Бульон с триптон-дрожжевым экстрактом N 2 (JSP). ; Рост хороший, обратная сторона колоний хселтая, в6здуцшь1й мипелий хорошо развит, .бельш растворимый пигмент осутствует. 3SP N 3. Рост плохой, обратная CTOpoira колоний белая, воздушный мицелий - следы растворимый пигмент отсутствует. OSP N 4:. РОСТ хороший, обратная сторона колоний желтая, воздушный мицелий плохо развит, растворимый пигмент отсутствует. Л8Р N 5. Рост хороший, обратная сторона колоний желтая, воздущный мицелий и расгворимый 1шгмент отсутствуют. 3SP N 7. Рост хороший, обратная сторона колоний желтая, воздушный мицелий хорошо развит, растворимый пигмент светлокоричневый, серый. Са-малат. Рост плохой, обратная сторона колоний белая, воздушный мицелий и расторимый пигмент отсутствуют. Среда Чапека.Рост значительный, обратная сторона колоний желтая, воздушный мицелий - следы, растворимый 1П1гмент отсутствует. Томатная паста - овсяная мука. Рост . хороший, обратная сторона колоний желтая. Физиолог о-биохимические свойства. Усваивает глюкозу, t-арабинозу, Д-фрукто-; зу, Д-,галактозу, йзо-инозий, Д-маннит, сахар, Д-ксилозу, Д-рамнозу. усваивает раффинозу, салицин. Жела- тин разжижает, молоки не сбраживает, крахмал гидролизует, каталаза положительная, устойчивость к хлориду натрия - 4%, фосфатаза положительная, уреаза отрицательная. Диапазон рН роста 6,1-7,8. Диапазон температур роста 10-30 С. Чувствительность к антибиотикам, хлорам феникол, цефалоридин, тетраш клин, нанкамипин - 30 мкг, стрептомицин 10 мкг - чувствителен, слабочувствителен -- эритромишш 15 мкг, полимиксии Б - 300 ед: нечувствителен к линкалицину. Антагонистические свойства. Обладает антимикробной активностью в отношении: Организммкг/мл Streptococcusруо- genes С-203о,25 Streptococcuspneumoniae Napu Io,13 Streptococcusbug (rpD) 2820,5 Staphylococcus aureus 1,0 3055 Staphylococcus aureus 209P Pasteurella multocide Pastoirella hemolytica Mycoplasma galliscpticum Mycoplasma hyopneumoniae Mycoplasma hyorhinis Способ получения антибиотика 23 демнци зилтипозина (ДМТ) и его црои.чнодных шшю рируется следующими примерами. Пример 1. Ферментация ЛМТ во встряхиваемой колбе. Лиофилизованные таблетки Streptomyces fradiae NRRL 12170 диспергируют в J -2 мл стерилизованной воды. Часть зтого раствора (0,5 мл) используют для инокуляций растительной среды (150 мл), имеющей следующ состав,%: Зерновая настойка1,0 Экстракт дрожжей0,5 Соевая мука грубого помола0,5 Карбонат калыщя0,3 Соевое масло (неочищенное)0,45 Обессоленная вода97,25 С другой стороны, растительную культуру Streptbmyces fradiae NRRL 12170,.. сохраняем в 1-миллилитровых объемах в жидком азоте быстро оттаивают и используют для инокуля ции растительной среды. Инокулированную р тительную среду выдерживают в 500 мл кол Эрленмейера при 29С и в течение 48 ч он находится в закрытой трясучке, совершающе 300 об/мин. Эту инкубированную растителыгую среду (0,5 мл) используют для инокуляции 7 мл проду1хирующей среды, имеющей следуюиетй состав,%; Свекольная патока2,0 Зерновая мука1,5 Рыбная мука0,9 Пшеничная клейковина0,9 Хлорид натрия0,1 (NHi,)tHro4 0,04 Карбонат кальция гСоевое масло (неочищенное) 3,0 рбессоле1гная вода91,36 Инокулированную ферментативную среду инкубируют в 50 мл бутылочках при в течение примерно 6 дней в закрытой трясучке, совершающей 300 об/мин. Ферментация ДМТ в ферментерах. Чтобы получить большой объем йнокулята берут 1200 мл инкубированной среды и шгокули руют 946,4 л (250 галлонов) растительной среды второй стадии, имеющей следующий состав, %: 84 Зерновуя настойка1,0 (оевая маслянап мука0,5 Дрожжевой экстряк 0,5 Карбопат кальция0,3 Соевое масло (неочищенное)0,5 Линетин (неочищенный)0,015 Вода97,185 рН среды ДОВОДЯ /ш 8,5 добавлением 51 г-ного раствора едкого натра. Эту растительную среду второй стадий шжубируют в ферментере емкостью 1325 л (350 галлонов в течение примерно 48 ч при 28 С при умеренном аэрировании и перемеишпании. Приготовленную инкубированную среду второй стадли (545 л, т. е. 144 гал.чона) используют для инокуляции 3785,4 л (1000 галлонов) стерильной ферменгапио1той средь, имсЕощей следуюисий состав,%: Рыбная мука0,875 Зерновая мука1,5 Зерновая клейковина0,875 Карбпнат кальция 0,2 Хлорид натрия0,1 (N),04 Свекольная патока2.0 Соевое масло (неочищенное)3,0 Лепитин0,09 Вода91,32 рН регулируют до 7,2 добавлением 50%ного раствора едкого натра. Инокулированиая продуцирутощая среда сбраживается в 6057 -литровом ферментере (1600 галлонов) в течение 8-9 дней при 28 С. Бродящ ю среду аэрируют стериальным воздухом, поддерживая концентращ1Ю растворенного кислорода в пределах примерно 30-50%, и перемешивают ставдартными мешалками со скоростью вращения 25.0 об/мин. Пример 2.. Вьщеление ДМТ. КультуральЕ1Л1р жидкость (3800 л) полученн)ю в соответствии с примером 1, фильтруют, используя мелкий порошок. Мицелиалъиую лепешку промывают водой, а эт1т водные промывки обавляют к фильтрату. рН фильтрата доводят до 9,2, используя;.. 0%-ный раствор едкого натра .(0,5 п) фил-трат затем экстрагируют 2000 л зттшацетата. этилацетатному экстракту добавляют 450 г бессоленной воды и 6,4 кг одноосновного осфата натрия и получешплй раствор тпвтепьо перемеишвают. рН раствора регулируют римерно от 6,0 до 4,35, используя раствор осфорной кислоты (3300 мл, 2 ч воды на 1 ч фосфорной кислоты). Водную фазу отдеяют. рН обогащенной водной фазы доводят до 6,5, используя 5W(}ibm водный раствор едкого натра (700 мл). Полученный раствор концентрируют до объема примерно 225 л в вакууме. рИ этого KOHueHtpHpoBaHHOro раствора довод гт до 9,2, добавляя 10%-н лй водный едкий натр -(26л)., Полученный щелочной раствор выдерж вают в течение восьми часов. Выделяюпдиеся кристаллы отфильтровывают, промывают обессоленной водой (50 л) и сушат, получая при мерно 8,6 кг продукта. Часть полученного продукта (3 кг| подвергают перекристаллизации из смеси ацетона - воды и получают гфимерно 2,07 кг ДМТ свободного основакия. 132°C ДМТ размягчается i примерно при 150° С. и медлеН1Ю плавится примерно до А 1ализом установлено, что ДМТ приблизитель но имеет следующий состав,%: углерод 61, водород 8,5, азот 2, кислород 28, следующую эмпирическую формулу: а также молекулярную массу примерно 742 ( по данным масс спектрометрии). Максимумы поглощения приходятся на сле дующие |а:стоты (см): 3634 (очень малый) 3559 (с уступом), 3423 (ишрокий), 2955 (иптенсивный), 2907 (интенсивный), 1710 (интенсивный), 1676 (с Уступом), 1588 (интенсивный), 1447 (с уступом), 1396 (с уступом), 1359 (малый), 1309 (очень малый 1178 (с уступом), 1156 (интенсивный), 1111 (с уступом), 1073 (с уступом), 1048 (интенсивный), 1013 (с уступом), 984 (с ус тупом), 926 (очень малый), 898 (очень, малый) и 833 (очень мальш). У-ф-спектр поглощения ДМТ в нейтральном этиловом спирте обнаруживает максимум , поглощения при 283 им ( 22296:- Е/. 300,9). ДМТ-свобод11ое основание имеет следующее удельное вращение; ot Itf -53,5(с 1,СНз-ОН Электрометрическое титрование ДМТ в 66%-ном водаом диметилформамиде указыва ют на наличие титруемой группы с рК при мерно 7,25. ДМТ-основалие является растворимым в роде и в больщиистве полярных органически .растворителях, таких как ацетон, метанол, . этанол, диметилформамвд, хлороформ, диметилсульфоксид. ДМТ-соли-ад1гукты кислотл чш растворяются в воде, чем ДМТ-основание. .Пример 3. Получение ОМТ(5-0-микоминозилтилонолида). ДМТ, приготовле1пП)Ш в соответствии с методикой примера 2, растворяют в разбавленной соляной кислоте (соляную кислоту добавляли к воде до рН раствора 1,0), Г1олученнйй раствор выдерживают в течение 24ч при комнатной тгмперагуре и затем рН регулируют до 9,0 добавлением едкого натра. Этот щелочной раствор экстрагируют этилацетатом, д 1хлорметаном ил.и хлороформом. Экстракт сушат под вакуумом и получают. Пример 4. Получение ОМТ. ДМТ (500 г) приготовленный в соответствии с примером 2, измельчают в ступе и добавляют к обессоленной воде (1,5 л); рН поддерживают в пределах 3,5-7,5 при необходимости добавлением серной кислоты. Чере:з 30 М1ш весь ДМТ растворялся, рН растврра затем доводят до 1,4 и выдерживают раствор в течение 42 ч при . После стандартной обработки получают 366 г ОМТ, идентичность которого была подтверждена . сопоставлением с тонкослойной хроматографией известной пробы. Приме р 5. Получение дигидро-ДМТ (20-дигидро- 23- демиципозилтилозина). 50мг ДМТ, приготовлепной в соответствии с примером 2, растворяют в водиом растворе изопрошшового спирта, (примерно 40%, 25мл). В 30%-ном водном, растворе изопро-, шшового спирта растворяют 20 мг борогидри-; да натрия. 1 мл раствора борогидрида . натрия добавляют к раствору, содержащую . ДД1Т. Полученную смесь перемешивают в течение 5 мин, рН регулируют до 7,5, добавляя фосфорную кислоту, и затем концентрируют в вакууме для удаления изопропилового спирта. -Затем к йог/ученному водному концентрату добавляют воду до объема 25 мл, а затем добавляют 50 мл хлороформа. рН:.; водной фазы регулируют до 7,5. После экстракции отделяют хлороформ и выпаривают в вакууме, в результате чего получают дигидро-ДМТ. Пример 6. Получение дигидро-ОМТ (20-дигидро-5-0-микалиноэтилти лонолида). Дигидро-ДМТ, полученный в соответствии с примером 5, обрабатывают способом, описанным и примере 3, и получают дигидроОМТ. Пример 7. .Альтернативное получение ОМТ. ОМТ получают из ДМТ путем обработки ДМТ в ферментативном бульоне, в котором его готовили в присутствии слабой кислоты в соответствии с примером 3. Выделение ОМТ достига.пось процедура, аналогичной той, которая описана для ДМТ в примере 2. Пример 8. 2-0-Лцетил-ДМТ. Ш г ДМТ (13,5 ммоль) растворяют в . 260 мл ацетона и обрабатьшагот jTccycHbiM .атпидридом (1,6 мл, 5,7 ммоль), добавляя по каплям при перемешивании при комнатной температ фе. После перемешивания в течение 7 18 ч растворитель вьтаривают при пош женном давлении. Остаток растворяют в 200 мл этилацетата и экстрагируют насыщен1 ым раст вором бикарбоната натрия(2 х 200 мл). Органический раствор сушат над сульфатом натрия, фильтруют и выпаривают. Остаток растворяют .в малом объеме этилацетата, загружают в колонку с силикагелем (Вотерс Преп 500) и элюируют этил ацетатом (4л). Фракции, содержащие целевой продукт, идентифиилруют методом тонкослойной хроматог фии, объединяют и выпаривают досуха,, получая 6,5 г (61%) 2-о-ацетил-ДМТ. Пример 9. 2-0-Пропионил-ДМТ. Аналогичным способом 6,0 г (8,1 ммоль) ДМТ в 120 мл ацетона обрабатывают 1,2 мл (9,2 ммоль) ангидрида йронионовой кислоты. После обработки и хроматографирования выделяют 3,7 г (57%) 2 -О-пропионил-ДМТ. Пример 10. 2, 23-Ди-О-ацетил-ДМ 3 г ДМТ (4,05 ммоль) растворяют в 90 мл хлористого метилена и 7,8 мл гафидина и обрабатьтают 1,4 мл (13,7 ммоль) ангидрида уксусной кислоты, добавляемого по каплям при перемеишвании при коляитной температуре. После перемеш1гваиия в тече}ше 17 ч раствор разбавляют 15 мп толуола и растворители выпаривают при пониже1шом давлении. Остаток растворяют в малом объеме этилацетатом, загружают в колонку с силика гелем. (Вотерс Преп 500) и элюируют четырь мя литрами этилацетата, получая 2,1 г сырого продукта. Этот материал растворяют в мтимальном объеме толуола, загружают в хроматографическую колонну с силикагелем (300 мл) и элюируют (а) 3:1 толуола к этилацётату (300 мл), (б) 5:4 толуола к этилацетату (600 мл) и (в) 1:1 толуола к этиладетату . (1000 мл). Фракции, содержащие целевой продукт, анализируют тонко:спойной хроматографией, объединяют и выпаривают досуха, получая 1,6 г (64%) 2 ,23 дигидро-0-ацетил-ДМТ. II. 2,23-Ди-О-пропионил. Пример 1Щ1 . В 90 мл. метиленхлорида и 7,8 мл пиридяна растворяют 3 т ДМТ (4,05 ммоль) и обрабатывают 1,8 мл (13,8 ммоль) ангидридй пропионовой кислоты, добавляемого по каплям при перемешивании при комнатной температуре. После перемешилания в течение ночи добавляют 15 мл толуола, и раствор выпаривают досуха про пониже1шом давлении Остаток растворяют в 20 мл толуола, загружают в хроматографическую колонку с сили кагелем (Е.Мерк 60,500 мд) и элюируют (а) 3:1 толуола к этилацетату (300 мл). 18 (б) 5:4 толуола к этилацетату (300 мл) и (в) 1:1 толуола к этилацегату (1000 мл). Получают 2,5 г (72%) 2, 23-ди-О-пропионил-ДМТ. П р и мер 12. 2- О-Ацетил-23-О-пропионил-ДМТ. В 180 МП метиленхлорида в 15 мл пиридина растворяют 6 г (7,7 ммоль) 2 -О- ;. ацетил-ДМТ и обрабатывают 1,2 мл (9,2 ммоль) ангидрида прогпюновой. кислоты, добавляемого по каплям при комнатной .. темггературе. После перемешивания в течение 17 ч раствор разбавляют 300 мл толуола и выпаривают досуха при пониженном давлении. Остаток растворяют в толуоле и экстраги- руют пасьпценным раствором бикарбоната натрия. Толуольный слой сушат над сульфатом . натрия, фильтруют и вьшаривают. Остаток растеоряют в малом объеме толуола, загру- . жают в хроматографическ}то колонку с силикагелем (300 мл), заполненную толуоломэтиляцетатом (1:1) и элюируют од1О1м литром засазанного растворителя. Фракции, содержащие целевой продукт, идентифшцфуют методом тонкослойной хроматографии, объедя 1яют, выпаривают при пониженном давдещш и получают 4,5 г (70%) 2-0-ацетид-23-0-Про-1 ПИОНИЛ-Д.МТ. Пример 13. 2-0-Пропио1™л-23-0-ацет1Ш-ДМТ. Аналогичиьгм образом обрабатывают 2 -Or -пропиопил-ДМТ (1,5 г, 2 ммоль) в хлористом метелене (45 мл) и 3,9 мл пиридина с помощью 0,3 мл (2,9 ммоль ангидрида уксусной кисяоть). После хроматографирования. на спликагеле (Вотерс Преп. 500) выделяют 0,81 г (51%). 2-0-прошюнил 23-О-ацетил-ДМТ. Пример 14. 2-0-Ацетил-23-0-фенилацетил-ДМТ. 2-О-Ацетил-ДМТ (26,5 г, 3,5 ммоль) растворяют в 75 мл хлористого метилена и 0,8 мл пиридшш и обрабатывают раствором 0,56 мл (3,5 ммоль) фенплацетилхлорида и , 13 мл хлористого метилена, добавляемых по аплям при перемешивании при комнатной емпературе. Спустя 1,5 ч смесь обрабатывают опопннтельным количеством фенилацетилхло-; ида (0,56 мл) в хлористом метилене (13 мй). пустя еще 3,5 ч исходный материал был зрасходован (установлено тонкослойной хроатографией). Затем раствор выпзривадат доуха при понижешюм давлении, остаток расторяют в хлористом метилене и экстрагирую асыщенным раствором бикарбоната натрия. рганический слой сушат над. сульфатом натия, фильтруют и вьтаривают досуха.. Остаок растворяют в толуоле и хроматограф1фу-. т на силикагельной колонке (Вотерс ПреН 500), : кггра|-ир)(ание проводят при линейном градн.яге толуола (4 л) и этилацетата (4 л) при соопюшепим 1:1. Фракшш, содержащие пеленой продукт, объеднпжот и выпаривают, получая 1,1 с (.15%) 2-0-Aцeтил-2: 0-фeнилaucrJl.т-ДMГ. Из П1«д111сствуго111их фракций так же было получено 0,86 г (24%) 2 -0-ацетил- 23,4-д,и-0-фенила11етил-ДМТ. Пример 15. 2-0-Ацетип-23-Г1р011иошш-ДМТ (1,6 г, ,9 ммоль) растворяют в 9. метаноле (80 мл) и перемешивают ггри комнатной температуре в течение 42 ч. Раствор выпаривают досуха при пониженном ллвлении. Остаток растворяют в малом объеме толуола, загружают в хроматографическую колонку с силикагслем (Е. Мерк 60, 2.00 мл) и элюируют 2 л толуола-этилацетата (1:1), Фракции, содержащие целевой продукт, идентифицируют методом тонкослойной хрома тографии, объединяют и выпаривают при тюни жеином давлении, получая 1,2 г (79%) 23-0-пропионил-ДМТ. Пример 16. 23-0-Фенилацетил-ДМТ. 0,6 г (0,67 ммоль) 2-0-ацетил-23-0-фенилацетил-ДМТ раст)5Оряют в 80 о-пом водном растворе (50 мл) и энергично кипятят с обратным холодильником в течение 4,5 ч. Раствор охлаждают до комнатной температуры и выпаривают досуха при пониженном давле НИИ, полу1ая 0,39 г (68%) 23-0-фенилацетил-ДМТ. „ Пример 17. 23,4 -ди-О-Фенилацетил-ДМТ. 2 - О- Ацетил-23,4 -ди-О-фенилацетил-ДМТ (0,36 г, 0,36 ммоль) растворяют в 80%-ном водном метаноле (30 мл) и кипятят с обрат ным холодильником в течение 4,5 ч. Раствор охлаждают до комнатной температуры и выпониженном давлении, пяривают досуха при 0,29 г (84%) 23,4 -ди-О-фениланетилполучая-ДМТ. Пример 18. 2 -О-Ацетил-3,23,4 -три -0-пропионил-ДМТ. 2-0- Аиетил-ДМТ (2 г, 2,6 ммоль) раство ряют в 40 мл ацетона и 8 мл пиридина и обрабатывают раствором 40 мл ангидрида Лропионовой кислоты в 20 мл ацетона, добавляя его .по каплям при перемецшвапии и комнатной температуре. Перемешивание осуществляют в течение 5 дней, смесь выпаривают при пониженном давлении. Остаточное мас растворяют в этилацетате и экстрагируют насы щенным раствором бикарбоната натрия. Органический слой сушат над сульфатом натрия, фильтруют и вьшаривают. Остаток растворяют н малом объеме- толуола и хроматотрафирую на силикягеле (Вотерс, Преп 500). Колонку злсирун)т при линейном градиенте толуола (4 л) и :)тила11етата (4 л). Фракции, содержа-, шие целевой продукт, объединяют, выпаривают и получают 1,4 г (57%) 2-0-анетил-3,23,4-0- . пропионил-ДМТ. Пример 19. 3,23,4 -Три-0-нропионил-ДМТ. 2:0-Ацегш1-3,2Л,4 -три-О-пропионил-ДМТ (0,7 г, Oi74 ммоль) растворяют в 80%-ном водном метаноле (55 мл) и кипятят с обратным холодильником в течение 5 ч. Раствор охлаждают и затем выпаривают до водного при пониженном давлении. Продукт экстрагируют хлористым метютеном, органический слой отделяют, сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении досуха, получают 0,38 г (56%) 3,23,4 -три-0-пропионил-ДМТ. Пример 20. 2,4 ,23-Tpи-0-aцeтил-ДMT)0,0 г ДМТ (13,5 ммоль) растворяют в 150 мл пиридина и обрабатывают 5,8 мл (60,7 ммоль) уксусного ангидрида при перемепшвании при комнатной температуре. После перемешивания в течение ночи растворитель выпаривают при потшженном давлении. Остаточное масло растворяют в дихлорметане и экстрагируют насыщенным бикарбонатом натрия; органический слой отделяют, сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Остаточный материал хроматофафируют на силикагеле (Вотерм Преп, 500), элюируют при линейном градиенте смеси толуола (4 л) и этилацетате и этилацетата (4 л) при соотношении 3:1. Фракции, содержащие целевой продукт, идентифицируют тонкослойной хроматографией, объединяют и выпаривают при пониженном давлении, получают 4,5 г 2,4-23-три-О-ацетил-ДМТ. Пример 21. 2 - О-Ацетил-23-0-фепоксиацетил-ДМТ. 2 -0-Ацетил-ДМТ (2,75 г, 3,5 ммоль) растворяют в 75 мл дихлорметана и 0,8 мл пиридина и обрабатывают раствором 1,2 мд (88 ммоль) феноксиацетилхлорида в 25 мл Дихлорметана, добавляя их по каплям при перемешивании и комнатной температуре. . Через один час реакционную смесь выливают в насыщенный раствор бикарбоната натрия (200 мл) и отделяют органический слой, сушат над сульфатом натрия, фильтруют и вьшаривают при пониженном давлении. Остаточную твердую пену загружают в хроматографическую колонку с силикагелем, продукты разделяют и элюируют используя толуолэтидацетат (1:1) в качестве растворителя. 1. 1,5 г 2 -О-ацетил-23-О-феноксиацетил-ДМТ.выделяют вместе с 0,55 г 2 -0-aцeтил-23,f -ди-0-фeнoкcиaцeтил-ДMT и 0,03 г 2 -0-ацетид- 3,23-аи- О- феноксиацети л- ДМТ.
П p и e p 22. 2-0-Ацетил-23-0-(napaхлорфенчлацетил) - ДМТ.
Пара -хлорфенилуксусную кислоту (4,3 г, 25 ммоль) и 1-оксибензотриазол (5,4 г, 25 ммопь) растворяют в 150 мл тетрагидрофурана. Раствор охлаждают в ледяной бане и обрабатывают 5,2 г (25,3 ммоль) дициклогексилкарбодимида. Реакционную смесь перемеишвают при О С в течение 3 ч и затем помешают в холодильник на ночь. Смесь фильтруют, и фильтрат выпаривают при пониже шм давлении. Остаток растворяют в ацетоне (75 мл), фильтруют и обрабатывают 10 г (12,8 ммоль) 2-0-ацетил-ДМТ и 0,87 г (12,8 ммоль) имидазола. Добавляют ацетон до объема 1235 мп и затем добавляют ,87 мл (12,8 ммоль) тризтиламина. После перемешивания в течение 20 ч при комнатной температуре растворитель выпаривают при пониженном давлении, остаток загружают в Kortoraiy с силикагелем и злюир)тот при градиенте толуола-этилацетата (4:1) и одного этилацетата. Получают 4,75 г 2-0-ацетил23-0- (пара-хлорфенилацетил)-ДМТ.
П р и м е р 23. 2 -О-Ацетил-4 -0-проПИО1ШЛ-ДМТ..
2-0-Ацетил-ДМТ (1,0 г, 1,3 ммоль) растворяют в 30 мл пиридина и раствор обрабатывают 0,6 МП (4,6 ммоль) пропиопового ангидрида, добавляемоо по каплям при перемешивании и комнатной температуре. Через 20 ч добавляют 3,0 мл (23 ммоль) пропионового ангидрида и реакцуюнную массу пере. мешивают в течение последующих 26. ч при KONtaaTHOu температуре. Смесь разбавляют 30 мл толуола и при пониженном давлении выпаривают растворители. Остаточное масло растворяют в 50 мл дихлорметане и экстрагируют насыще1шым раствором бикарбоната натрия, фильтруют я вьшаривают. Остаток загружают в колонну с силнкагелем (Вотерс Преп 500) и злюируют при линейном традиенте толуола (4 л) и этилэнетата (4 л).. Огачала элюируют 305 мг 2-0-ацетил-ЗЛЛ-. -ди-0-прошгонил-ДМТ, а затем 286 мг 2 -0-аце0, тил-4 -0-пропионил-ДМТ.
Предлагаемый способ позволяет получить , а 1тибиотик, обладаюцдай определенным спектром антимикробного действия, и может быть использован для лечения сельскохозяйственных и других жвиотных.;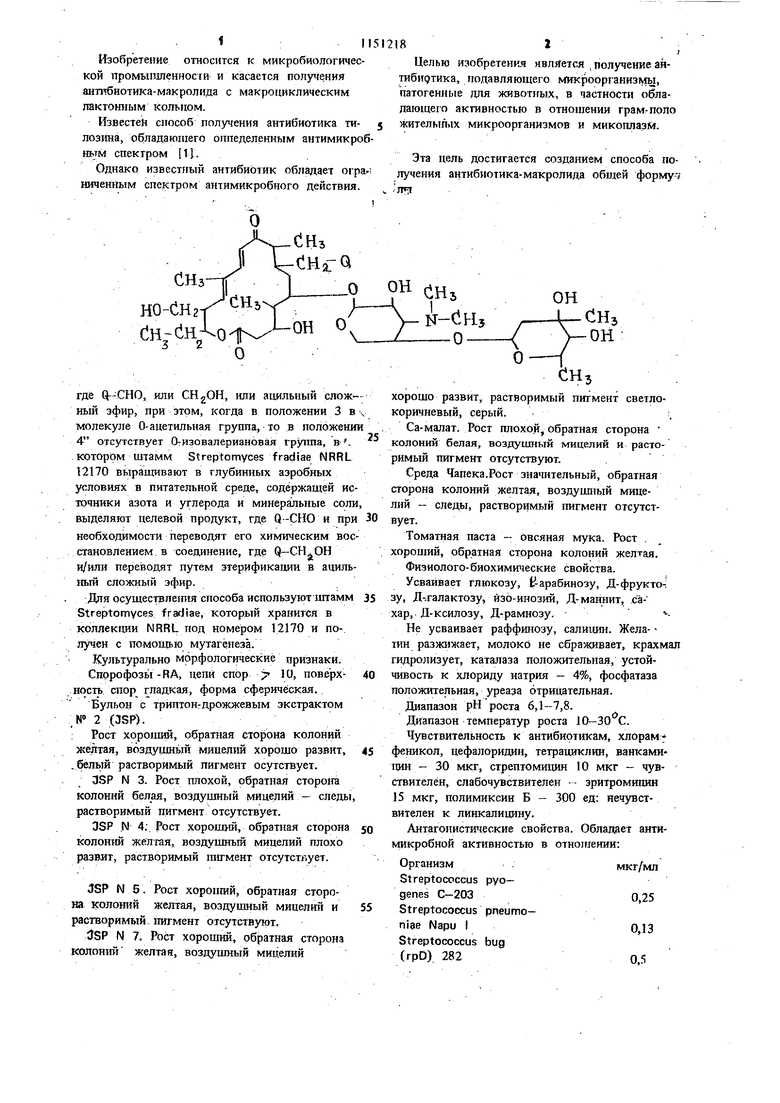





СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА-МАКГОЛИДА общей формулы СНз ОН N-СНз СНз (Л i ООН с dH источники азота и углерода и минеральные соли, выделяют целевой продукт, где Q СНО и при необходимости переводят его химичег КИМ восстановлением в соединение, 01 где GJ-CHgCffl и/или переводят этерификадш в ащшьный сложный 1 эфир. knM QO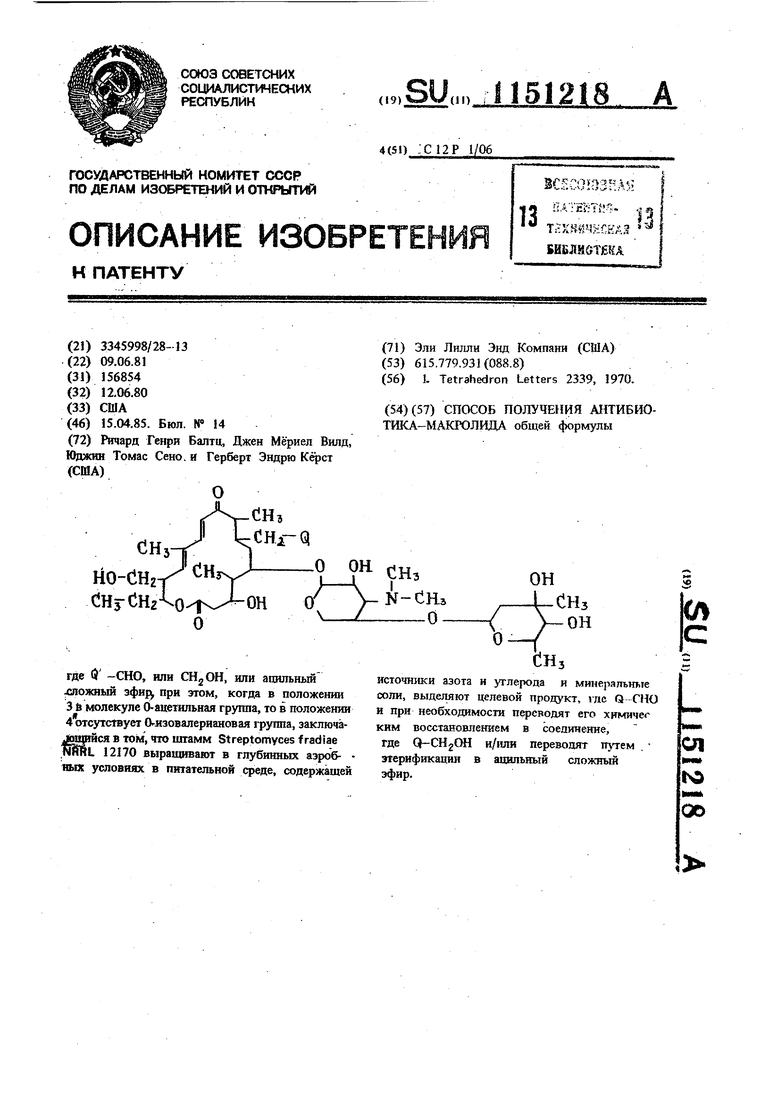
| Мокрый вакуум-насос | 1925 |
|
SU2339A1 |
