4, 4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индол (А) является правовращающей, более высокоактивны, чем соединения, у которых указанная часть (А) является левовращающей.
Пример 1. Хлористоводороный ё{-транс-8-фтор-5-(п-фторфенил)-2- 4-окси-4-(п-фторфенил)бутил -2,3,4,4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индол и хлористоводородный транс-8-фтор-5-(п-фторфенил)(п-фторфенил)-3-бутенил -2,3,4,4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индол.
В реакционный сосуд емкостью 1000 мл, оборудованный магнитной мешалкой и капельной воронкой и поддерживаемый в атмосфере азота., помещают 177 мл 0,94 М борана и в тетрагидрофуране. Раствор охлаждают на ледяной бане и к холодному раствору добавляют в течение 30 ми раствор.25 г (0,0555 моль) 8-фтор-5-(п-фторфенил)-2- 4-окси-4-(п-фторфенил) бутил|-2,3,4,5-тетрагидропиридо(4,3-Ь) индола в 295 мл тетрагидрофурана. Полученную смесь перемешивают при температуре окружающей среды в течение 20 мин, затем нагревают с обратным холодильником в течение 2 ч. Реакционную смесь охлаждают, упаривают в вакууме и получают жидкий остаток. К нему добавляют смесь 50 мл уксусной кислоты и 50 мл 5 н. соляной кислоты, после чего происходит энергичное ввделение газа. Смесь нагревают с обратным холодильником в течение 1 ч, охлаждают до комнатной температуры и фильтруют. Фильтрат охлаждают на льду и подщелачивают путем добавления 50%-ного (вес/вес) раствора гидроксида натрия. Основную смесь экстрагируют дважды 150 мл порциями хлороформа, соединенные органические слои высушивают над сульфатом магния и выпаривают досуха в вакууме, получают желтое твердое вспененное вещество, 25 г. При помощи тонкослойной хроматографии на силикагеле с применением системы растворителя гексан/этилацетат 1:1 (по объему) установлено наличие двух продуктов. Вспененное твердое вещество хроматографируют на колонке с силикагелем и элюируют смесью
1680944
гексан/этилацетат 1:1 (объем) с контролем фракций при помощи -тех. Фракции, содержащие только более подвижньшпродукт, т.е. 8-фтор5 -5- (п-4/горфенил)(п-фторфенил)-З-бутенил -2,3,4,4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индол, выпаривают досуха, растворяют в ацетоне и превращают в хлористоводородную солЬ О путем добавления безводного хлористого водорода в ацетоне. Полученное более твердое вещество отделяют фильтрованием, высушивают, получают 1,5г 3-бутенил.соединения. Т.пл. 270 15 273С.
Фракции, содержащие только менее подвижный 8-фтор-5-(п-фторфенил)-2- 4-ОКСИ-4-(п-фторфенил)бутил -2,3, 4,4а,5,9Ь-гексагидро-1Н-пиридо20 (4,3-Ь) индол, выпаривают, растворяют в этиловом эфире и превращают в хлористоводородную соль путем добавления безводного хлористого водорода. Получают 10,8 г этого продукта в виде смеси двух, диастереомеров, Т.пл. 241-245 С.
Содержание более подвижного 3-бутенил соединения возрастает до 100% при соответствующих повышении кислот30 кости и увеличении продолжительности нагрева с обратным холодильником в смеси уксусная кислота/соляная кислота.
Пример 2. Процесс проводят 3 аналогично примеру 3, но с применением в качестве исходного вещества 8-фтор-5-(о-фторфенил)(п-фторфенил) бутил -2,3,4,5-тетрагидропи)ридо(4,3-Ь) индола.Быстрее движущий4Q ся через силикагель при хроматографии компонент идентифицирован как транс-8-СФтор-5-(о-фторфенил)-2С4-(п-фторфенил)-3-бутенил -2,3,4,4а, 5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) 45 индол. Т. пл. 141-142 0.
Медленнее движущийся компонент идентифицирован как транс-8-фтор-5-(о-фторфенил)-2- 4-окси-4-(п-фторфенил)бутилД-2,3,4,4а,5,9Ь0 -гексагидро-1Н-пиридо(4,3-Ь) индол. Т.пл. 195-197с. Каждый продукт представляет собой диастереомер.
Пример 3. Хлористоводородньй а1-транс-5-фенил-2 3-(п-фторбензо5 ил)пропил}-3,4,4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индол.
В реакционный сосуд емкостью 25 мл, оборудованный магнитной мешалкой и Поддерживаемый в атмосфере азота, помещают 0,828 мл (8,0 г, 10,3 ммоль) безводного пиридина и 10 мл дихлорметана. К раствору доба ляют 517 мг (5,17 ммоль) триоксида хрома и полученную темно-красную суспензию перемешивают в течение 15 мин при комнатной температуре. Добавляют одной порцией раствор 359 мг (0,862. ммоль) свободного основания d -тpaнc-5-фeнил-2- 4-оксиi 4- (п-фторфенил) бутил -2,3,4,4а,5,9Ь -гексагидро-1Н-пиридо(4,3-Ь) индола в 5 мл дихлорметана. Реакционная смесь быстро превращается в коричне вую суспензию. Ее перемешивают при комнатной температуре в течение 30 мин. Нерастворимое вещество отделяют фильтрованием, промывают дихлорметаном и соединенные фильтраты и промьюные воды экстрагируют 20 мл 10%-ного раствора гидроксида натрия. Органический слой сушат над сульфатом магния и выпаривают досух в вакууме, получают смолу.. Смолу очищают методом колоночной хроматографии на силикагеле, элюируя смесью 1:1 (по объему) гексан/этилацетат. Фракции, содержащие требуемый про.дуКт, соединяют, вьтаривают до полу чения желтой смолы, смолу растворяю в этиловом эфире и обрабатывают без водным хлористым водородом. Получен ную суспензию выпаривают досуха, суспендируют в 3 мл холодного дихло метана. Образуется бесцветное твердое вещество, которое отделяют филь рованием и сушат, получают 20 мг Т. пл. 244указанного соединения. 246,5°С. Пример 4. Хлористоводород,ньй d5-тpaнc-8-фтop-5-(п-фторфенил) -2-СЗ- (п-фторбензоил) пропилЗ-2,3,4, 4 5,9Ь-гексагидро-1Н-пиридо(4,3-Ь)инд В колбу емкостью 100 мл, содержа щую 20 мл дихлорметана и 1,76 мл (21,9 ммоль) пиридина добавляют 1,09 г триоксида хрома и полученную темную суспензию перемешивают в теч ние 15 мин. Затем добавляют одной порцией раствор 824 мг{1,82 ммоль) свободного основания df-TpaHC-8-фтор-5-(п-фторфенил)-2- 4-окси-4-(п-фторфенил)бутил -2,3,4,4а,5,9ЬГ8Ксагидро-1Н-пиридо(4,3-Ь) индола (полученного из хлористоводородной соли путем подщелачивания водного раствора гидрокскдом натрия, акстра гирования дихлорметаном и выпаривания экстракта досуха) в 10 мл дихлорметана. Полученную красно-коричневую суспензию перемешивают при комнатной температуре в течение 1 ч и обрабатывают по методике, примененной в примере 10, получают 25 мг требуемого продукта. Т.пл. 260-263 0. Пример 5. Разделение диастереомеров df.-TpaHc-8-фтор-5-(п-фторфенил)-2- 4-окси-4-(п-фторфенил)бутил -2,3,4,4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индола. А. 5 г смеси диастереомеров хлористоводородного с1 -транс-8-фтор-5-(п-фторфенил)-2- 4-окси-4-(п-фторфенил)бутил -2,3,4,4а,5,9Ь-гексагидро-1)-пиридо(4,3-Ь) индола, полученного в примере 3, превращают в свободное основание путем распределения между метиленхлоридом и 1.0%-ным водным раствором гидроксида натрия. Органическую фазу сушат (Na2 SO) и выпаривают до образоввния пены,которую растворяют в 12,5 мл этилацетата и 45 мл гексана при температуре кипения смеси. После охлаждения в течение ночи вьтавший в осадок продукт отделяют фильтрованием и получают 2,24 г продукта. Т.пл,126129°С. Его перекристаллизовывают трижды из смеси зтилацетат/гексан и получают 1,22 г одного диастереомера. Т.пл. 132-134с. Это свободное основание превращают в хлористоводородную саль путем добавления эфирного раствора хлористого водорода к раствору свободного основания в метаноле и получают 1,30 г, Т.пл. 259-260°С. Жидкостный кроматографический анализ при высоком давлении показал, что данньй продукт является сС/З -диастереомером 99%-ной чистоты. В. Маточньш раствор после первой кристаллизации выпаривают до.получения смолы, растворяют в этиловом эфире и превращают в хлористоводородную соль путем добавления зфирного раствора хлористого водорода. Оставшееся твердое кристаллическое вещество перекристаллизовывают трижды из смеси ацетонитрил/метанол и получают 1,03 г - второго диастереомера, обозначенного как у о -диастереомер. Т.пл. 237-239°С. Жидкостный хроматрграфический анализ при высоком давлении показал, что данный продукт содержит около 95 вес.% чис того уо -диастереомера. Пример 6. Разделение диаст реомеров d -тpaнc-8-фтop-5-(n-фтopфенил) -2г 4-окси-„4- (п-фторфенил) бутилJ-2,3,4,4а,5,9Ь-гексагидро-1Н-пи ридо(4,3-Ь)индола. А. Разделение ОС | -диастереомера на сх; -энантиомер и |3-этантиомер. Раствор 2,40 г (5,3 ммоль) рацемического iji, а -диастереомера, полу ченного как описано, и 2,0 г. (7,5 ммоль) N-трет-бутоксикарбонил-L-фенилаланина в 80 мл хлороформа охлаждают на ледяной бане в атмосфе ре азота. К перемешиваемому раствору добавляют 1,55 г (7,5 ммоль) дициклогексаилкарбодиимида и полученную смесь перемешивают в течение 1 при О С и еще в течение 1 ч при ком натной температуре. Выпавшее в осад твердое вещество (мочевину) отделяю фильтрованием и промывают метиленхлоридом. Фильтрат и промывные воды вьтаривают в вакууме и остаток хром тографируют на силикагеле, элюируя смесью 5:1 (по объему) метиленхлорид этилацетат. Фракции, содержащие эфи Ы-трет-бутоксикарбонил-1гфенилаланин соединяют и выпаривают в вакууме и получают 2,5 г белой аморфной пены. К этой пене добавляют 30 мл безводной трифторуксусной кислоты при и смесь перемешивают на ледяной бане в течение 30 мин, за это время происходит растворение. Трифторуксусную кислоту удаляют выпариванием в вакууме и вращающемся испарителе без внешнего подогрева колбы. Оставшееся твердое вещество растворяют в холодном метиленхлориде и промывают холодным 1%-ным (вес/вес) водным раствором бикарбоната натрия до появления нейтральной реакции на индикаторной рН-бумаге. Нейтральный органический слой сушат (MgSO,.) , растворитель вьшаривают и получают 1,6 г бледножелтой смолы. Смолу очищают хроматографией на 40 г силикагеля Me сК 230-400 меш, элюируя смесью 35:1 (объем) этилацетат/метанол. Фракции, содержащие L-фенилаланиновый эфир СС-энантиомера, -и фракции, содержащие L-фенилаланиновый эфир -этантирмера, разделяют, выпаривают досуха в вакууме и получают 636 и 474 мг соответственно. Перемешиваемый раствор 625 мг L-фенилаланинового эфира d, -энантиомера в 10 мл метанола при комнатной теьшературе Ьбрабатывают 10%-ным водным раствором гидроксида натрия до помутнения, затем перемешивают в течение 30 мин при комнатной температуре. Удаляют метанол выпариванием при пониженном давлении, затем добавляют 10 мл воды. Водную суспенйзию экстрагируют метиленхлоридом и соединенные органические слои сушат над сульфатом магния. После выпаривания растворителя получают бледно-желтую смолу, которую растворяют в ацетоне (5 мл), обрабатывают избытком эфирного раствора хлористого водорода, из которого кристаллизуется в виде пластинок хлористоводородная соль правовращающего об -энантиомера, 380 мг Т.пл. 251-255°C; oi +32,2 (С 1,67 в метаноле). В результате гидролиза L-фенилаланинового эфира -этантиомера (474 мг полученного продукта) аналогичным образом получают левовращающий /3 -энантиомер хлористоводородного 8-фтор-5-(п-фторфенил)-2- 4-окси-4- (п-фторфенил) -2,3,4, 4а,5,9Ь-гексагидро-1Н-пиридо(4,3-Ь) индола, Т,пл, 252-255с.. 33,0°(С 1,67 в метаноле). Анализ методом жидкостной хроматографии при .высоком давлении показал, что чистота оС -энантиомера и |3-энантиомера составляет 99% и более. В. Разделение УS -диастереомера на У и 6 -энантиомеры, 0 -Диастереомер транс-8-фтор-5-(п-фторфенил)-2-С4-ОКСИ-4-(п-фторфенил ) бутил -2 ,3,4,4а,5,9Ь-гексагидро-1Н-(4,3-Ь) индола взаимодействует с Ы-трет-бутоксикарбонил-Ьфенилаланином, затем полученный эфир -бок-Ь-фенилаланина взаимодействует трифторуксусной кислотой для удалеия аминозащитной (t-бок) группы,Затем фиры аминокислоты хроматографируют ля разделения L-фенилаланиновых эфиов jf -энантиомера и -энантиомера ак описано в части А примера 6. Затем азделенные у - и S -эфиры гидролиуют отдельно и очищают с целью полуения правовращакяцего У -энантиомера
зывания допамина взаимосвязано с их относительной фармакологической способностью действовать на поведение, вероятно, посредством допаминевых рецепторов. Лучшее исследование связьшания нейролептических рецепторов проведено Лей-, зономс применением Н-спироперидола (спиперона) в качестве меченого лиганда. Была применена еледующая методика.
Крысы-самцы 250-300 г были обезглавлены, и мозг немедленно препарировали на охлажденной льдом стеклянной пластинке для удаления полосатого -тела ( л- ЮО мг/мозг). Ткань гомогенизировали в 40 объемах (1 г + 40 мл) охлажденного льдом 50 мМ Трис(трис-оксиметиламинометана); буфер , рН 4,7. Гомогенат центрифугировали дважды при 50000 г (20000 об/мин) в течение 10 мин с повторной гомогенизацией промежуточного осадка в свежем буфере ТНАМ (таком же объеме). Конечный осадок осторожно суспендировали в 90 объемах холодного приготовленного (срок хранния менее одной недели) 50 мМ буфера Tris - рН 7,6, содержащего 120 мМ хлорида натрия (7,014 г/л), 5мМ хлорвда калия (0,3728 г/л), 2 мМ хлорида кальция (0,222 г/л), 1 Мм хлорида магния (0,204 г/л), 0,1% аскорбиново кислоты (1 мг/мл) и 10 мкМ паргилина (100 мкл смеси/100 мл буфера; исходный раствор 15 мг/10 мл бидистиллированной воды). Аскорбиновую кислоту и паргилин добавили свежеприготовленными. Суспензию ткани поместили в водную баню при 37 С на 5 мин для обеспечения дезактивации моноаминоксидазы ткани и затем до употребле;ния держали на льду. .Инкубационная смесь состояла из 0,02 мл раствора
:ингибитора, 1,0 мл гомогената ткани и 0,10 мл меченого индикатора (Н-спироперидол. New England nuclear
23,6 кюри/ммоль), приготовленного так, получить 0,5 нМ в конечной инкубационной сред (обычно разбавля с 2,5 мкл исходного раствора 17 мл бидистиллированной воды). Пробирки инкубировали в течение 10 мин при 37°С партиями по три, после че-го 0,9 мл от каждой инкубационной пробирки фильтровали через фильтры Whatman GF/B с применением высоковакуумного насоса. Каждый фильтр поместили в сдинтилляционную ампулу в нее добавили 10 мл жидкого сцинТШ1ЛЯЦИОННОГО фтора и каждую ампулу подвергли энергичному завихрению в течение приблизительно 5 с. Пробы оставлены на ночь, чтобы фильтры стали прозрачными, затем они снова были подвергнуты завихрению, и затем в течение 1,0 мин подсчитали радиоактивность. Связываниерассчитывали как фентамоли (10 моль) Н-спироперидола, связанные 1 мг пр теина. Опыты с контрольными растворами (носителе или 1-бутакламол, М; 4,4 мг растворили в 200 мкл ледяной уксусной кислоты, затем разбавили до 2,0 мл бидистиллированной водой для получения ЮМ исходного раствора и хранили в холодильнике) , растворами слепых проб (d-бутакламол, 10 М; j - на 10 М исходного раствора, так же, как для 1-бутакламола) и растворами ингибиторов повторяли по три раза. Концентрацию, при которой связывание уменьшалось на 50% (), оценивали на полулогарифмической бумаг Нерастворимые лекарства растворяли в 50%-ном этаноле (инкубация в 1%-ном этаноле).
Результаты, полученные с различными формами хлористоводородного транс-8-фтор-5-(п-фторфенил)-2- 4-окси-4-(п-фторфенил)бутш1 -2,3,4, 4,а,5,9Ь-гексагидро-1Н-пиридо (4,3-Ь)индола, представлены в табл.5
Таблица 1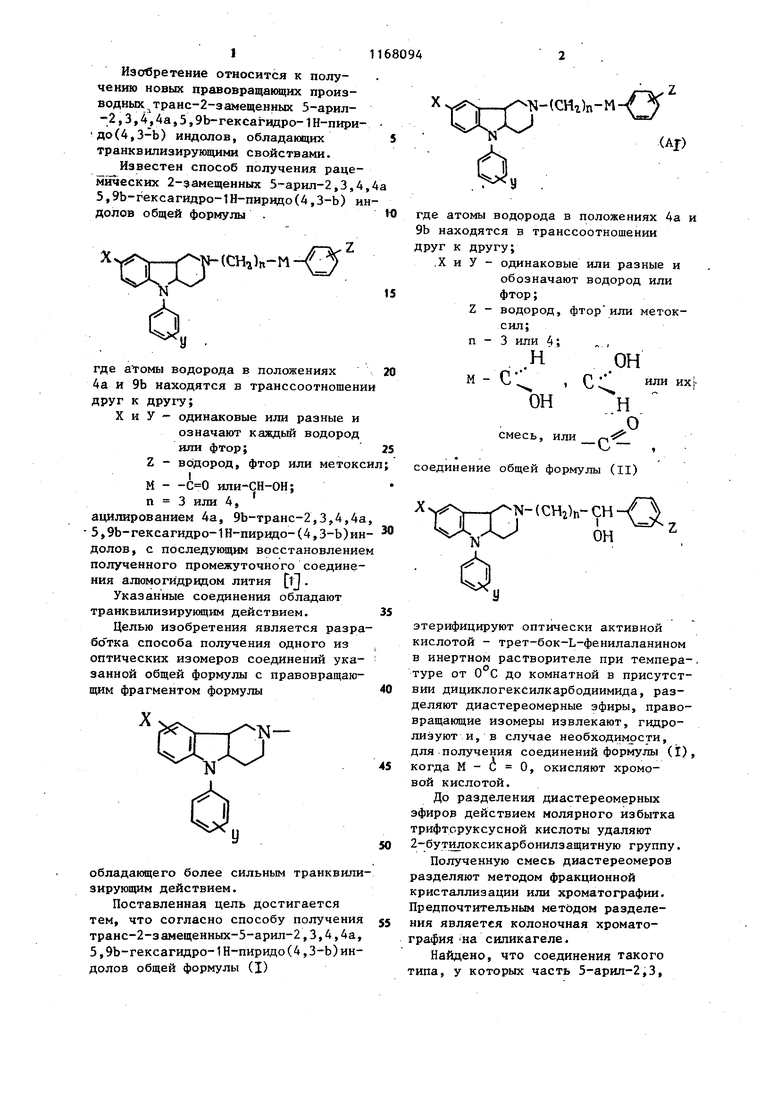








1. СПОСОБ ПОЛУЧЕНИЯ ТРАНС-2-ЗАМЕЩЕННЫХ 5-АРШ1-2,3,4,4а, 3,9Ъ-ГЕКСАгадРО-1Н-ПИРИДО
С,Н.СН-(СН,), О 5 tл л
ои
n-FCjH 9 ОН
n-CHjOC H CH-CCH;),- ь ч
он
n-FCgH C-CCHj),0
(СН2)зОН
n-FC l CH-CCHj),Oh(|;Н-(СН2)з ОНn-CHjOC H CH-CCHj,) ОНn-FCj H C -(С%)з
H/b) Н
C.Hj H/b/ Н
,H/b/ HC HjCCHj), ЦИС-9- з-(4-метил-1-пиперазинил)пропилиден -2-(диметилсудьфонамидо)-тиоксантен
HaifeeHO, что для соответствующего времени наблюдения 1 час. . Патент США 3 991 199 Патент США 3 310 553.
0,032-0,10,1-0,32
0,1-0,320,1-0,32
0,1-0,32лГО
0,1-0,,32
0,1-0,,32
0,0,32 0,32
0,32 ,032-0,1 0,032-0,1 0,032-0,1
0,1-0,32 0,1 0,32 1,0 1,0
1,0 3,2
3,2-32 32 3,2 ,0
Не испытан л.10 3,2-10 3,2-32 3,2
3,2-10
10
0,32-1,0
10
32
аналога 4а,9Ь-цис ED 56 мг/кг при
Соединение
Подавление связывания Н-спиропериодола, мП (HKjj)
Смеванные в с /1 - и у о -диастереомеры по i примеру 1
«,й-Диастереомер по примеру 5
Правовращаюпщй о -энантиомер по примеру 6
Леворращающий /3 -энантиомер по примеру 6
-Диастереомер по примеру 5
Правовращающий | -энантиомер - по примеру 6
Левовращающие &--энантиомеры jK примеру 6
Т а б
лица. 5
21
23
22 1800
23 25 30
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Топливная суспензия | 2023 |
|
RU2822465C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
