Изобретение относится к способу по чения нового производного соясапоген ла р ; именно 3-D-(D- глюкуронпирано зил)-соясапогенола В формулы ОН СНг,ОН обладающего антикомплементарной активностью. Цель изобретения - разработка сп соба получения нового соединения 3-0- (Я-В-глюкуронпиранозил)-соясапо генола В , обладающего преимуществами в фармакологической активности перед глицирризином. П р и м. е р 1. А. Соясапонин В (500 г) растворя ют в 30 мл метанола.. После добавле ния 1,5 мл 15 н.серной кислоты раствор нагревают с обратным холодильником в течение 3,5 ч. К реакционной смеси добавляют холодную воду для образования осадка который соби рают и промывают обильно водой. Пре ципитаты адсорбируют па к.олопке силикагеля и проявляют смесью хлрроформа и этанола поочередно (20:1 по объему 200 мл 10:1 по объему 150мл 5:1 по объему 200 мл и 2:1 по объему 200 мл) для получения 71 мг 3-0-(6-0-метил-В-р-глюкуронпиранозил)-соясапонина В. Полученное соединение растворяют в 2 мл метанола и 2 мл 1 н. водного раствора едкого натра добавляют к первому раствору с последующим нагре ванием с обратным холодильником в те чение 2 ч. После этого реакционную смесь смешивают.с холодной водой, .значение рН устанавливают равным 1 с помощью Т н. водного раствора солянрй кислоты и экстрагируют н-бутанолом. Фракцию н-бутанола концентрируют досуха при пониженном давлении. Кристаллизацией остатка из смеси хлороформа и ацетона (в отноше- НИИ 1:1 по объему) получают 50 мг 3-р-(В-глюкуронпиранозил)-соясапоген лаВ, т.пл. 231-232°С (с разл.). Вычислено,%: С 68,11, Н 9,21. ,Найдено,%: С 67,85, Н 9,08 . Тонкослойная хроматография на силикагеле с использованием Kiesel GebЕ (коммерческое название для силикагеля компании Merck). 1.Хлороформ-метанол-вода (при объемном отношении 65:35:8). R.j 0,38. 2.Изопропанол-2 н. водный раствор аммиака (при объемном отношении 100:15) R 0,21. Растворимость: хорошо растворим в метаноле, этаноле, н-пропаноле, н-бутаноле, водных растворах щелочей, пиридине, диметилсульфоксиде и диметилформамиде растворим в ацетоне, этилацетате и метилэтилкетоне; слабо растворим в бензоле, хлороформе,,- диэтиловом эфире, н-гексане ипетродейном эфире. Б. Соясапонин В (500 мг) растворяют в 30 мл этанола, насыщенного хлористым водородом, и раствор кипятят с обратным холодильником в течение 3 ч. К реакционной смеси добавляют холодную воду.для осаждения прёципитата, который собирают и промывают водой. Затем осадок очищают на колонке силикагеля методом хроматографин (растворитель для элюирования: смесь хлороформа и этанола в объемном отношении 20:1,10:1, 5:1,2:О и получают 75 мг 3-0-(6-0-этил-В-глюкуронпиранозил)-соясапогенола В. Полученное соединение смешивают с 2 мл этанола и 2 мл 1 н. водного .раств.ора едкого кали и смесь кипятят с обратным холодильником в течение 2 ч. Реакционную смесь смешивают с холодной водой, устанавливают рН около 1 с помощью 1 н. водного раствора соляной кислоты с последующим экстрагированием н-бутанолом. Фракцию н-бутанола выпаривают досуха в вакууме и кристаллизуют остаток из смеси хлороформа и ацетона (в объемном отношении 1:1). Получают 45 мг 3-0-(0-В-глюкуронпиранозил)-соясапонина В, т.пл. 231-232°С (с разл.). . Пример 2. Соясапонин В (Г г) растворяют в 70 мл дистиллированной воды .и раствор смешивают с 5 мл 15 н. водного раствора соляной кислоты, с последующим кипячением с обратным холодильником в те3
чение 2,5 ч. Реакционную смесь экстрагируют н-бутанолом. Фракцию н-бутанола выпаривают до.суха под вакуумом. Остаток адсорбируют на колонке силикагеля и проявляют смесью хлороформа и этанола (в объемном отношении 20:1, .10:1, 5:1, 2:1). Получают .350 мг З-О- -В-глюкуронпиранозил)-соясапонина В , т.пл.231232С (разлагается). -Пример 3. 3-0-(6-0-Метил- -В-глюкуронпиранозил)-соясапогенол В (70 мг) добавляют к 2 мл диоксана и к смеси добавляют 1 ,-5 мл 15 н.Н 80. Результирующую смесь нагревшот с обратным холодильником в течение 2,5 ч. .Реакционную смесь экстрагируют нг-бутанолом и н-бутанольную фракцию концентрируют до сухого остатка при пониженном давлении.. Остаток, адсорбируют в колонке .с силикагелем и проявляют смесью хлороформэтанол в последовательности 20:110:1-5:1-2:t (по объему) с получением 24 мг 3-0-(/ -В-глюкуронпиранолиз) соясапогенола В, т.пл. 231232°С. (с разл.).
П р и мер 4. Раствор 1 г соясапонина В в 70 мл дистиллирован|Ной воды c seшивaют с 5 мл водного jpacTBOpa, в котором содержится 3,6 г трифторуксусной кислоты, и смесь нагревают- с обратным холодильником в течение 2 ч. Реакционную смесь экстрагируют н-бутанол6м„ Н-бутанольную фракцию 1 онцентрируют до сухого остатка пониженном давлении. Остаток адсорбируют в колонке с силикагелем И:проя вляют смесью хлороформ - этанол в последовательности 1-10:1 -5:1-2:1. (по объему) с поо1учением 175 мг 3-0-(-ЕН глюкуронопиранозил)-соясапогенола В, т.пл. 231-232С (с разл.). .
П р им е р 5. 3-0-(6-0-Метил-р-D-глюкуронпиранозил)-соясапогенрл В (70 мг) добавляют к 2 мл воды и к смеси добавляют 1,5 мл концентрированной со 1яной кислоты. Полученную смесь нагревают при 50°С te течение 6 ч. Реакционную смесь экстрагируют н-бутанолом и фракцию- н-бутанола концентрируют досуха при пониженном давлении. Остаток адсорбируют на силикагеле в колонке «проявляют смесьюхлороформ - этанол последовательно составами (20:1, 10:1, 5:1, 2:1.по объему) с получением 22 мг 3-0-(
909894
-О-глюкуронпиранозши-тсоясапогенола В, т.пл. 231-232 0 (разложение).
Пример 6. 3-0-(6-0-Метил-fJ-D-глюкуронпиранозил) -соясапогенол 5 В (70 кг) добавляют к 2 мл диметилсульфоксида и к смеси добавляют 2 мл 1 н. водной гидроокиси натрия. Полученную смесь нагревают при 110 С в. течение 2 ч. После смешения реакционной
)0 смеси с холодной водой и доведения значения рН до 1 с помощью 1 н. водной соляной кислоты,, реакционную смесь экстрагируют н-бутанолом. и. фракцию н-бутанола концентрируют до-
15 суха при пониженном давлении. Остаток адсорбируют на колонке с силикагелем и проявляют смесью хороформэтанола последовательно составами (20:1, 10:1,5:1,2:1 по объему) с
2Q получением 3-0- (jb -D-глюкуронпирано-. зил)-соясапогенола В, т.пл.231-232 С (разложение).
Пример 7. 3-0-(6-0-Метил-/ -D-глюкуронпиранозил) -соясапогенол В
25 (70 мг) добавляют к 2 мл диметилформамида и к смеси добавляют 3 мл 1 н. бикарбоната натрия. Полученную смесь нагревают при 90 С в течение 3,5 ч. После смешения реакционной
Зб смеси с холодной водой и доведения значения рН до 1 с помощью 1 н. водного раствора соляной кислоты, реакгщонную смесь экстрагируют н-бутанолом и фракцию н-бутанола концент-
, рируют досуха при пониженном давлении. Остаток адсорбируют на колонке с силикагелем и проявляют смесью хлороформ-этанод последовательно составами (20:1, 10:1, 5:1, 2:1 по объему) с получением 27 мг 3-0-(Д-В-гл окуронпйранозил)-соясапогенола В , т.пл. 231-232С (разложение .
Примере. 3-0-(6-0-Метил-/1-D-глюкуронпиранозил)-соясапогенол
5 В (70 мг) добавляют к 2. мл тетрагидрофурана и к смеси добавляют 45 мл 1 н. карбоната калия. Полученную смесь кипятят с обратным холодильн.иком в течение 1 ч. После смед шения реакционной смеси с холодной
водой и доведения значение рН до 1 . . с помощью 1 н. водного раствора соляной кислоты, реакционную смесь экстрагируют н-бутанолом и фракцию 5 н-бутанола концентрируют досуха .при пониженном давлении. Остаток адсорбируют на колонке с силикагелем и проявляют смесью хлороформ- этанол последовательно составами (20-:.1, 10-: 1, 5:1, 2:1 по объему) с получение 19 мг 3-0-(/ -В-глюкуро-нпиранозил)-соясапогенола В , имеющего температуру плавления 231 2-32 С (разложение). . . Предлагаемые-, соединения обладаю .антиксплементарной активностью и используются в качестве, лечебных средств при аутоиммунных заболеваниях. . . . Фармакологические испытания. Испытываемые соединения: А. ЗгО-ф-В-глюкуронпиранозил)-соясапогенол- 8. Б. Глицирризйн (соединение для сравнения).. . . Антикомплементарная активность. Испытания заключаются в следующем, В пробирку . загружают по 0,5м воднойдисперсии каждого из испыты ваемых соединений, по О,5 мл сенси билизнрованнЬгх эритроцитов (СЭ),нр этом .культура .содержала 1x10 клеток/мл, 1 мл пятикратно разбавленного буферного раствора веронала, содержащего изотонический желатин (Этот 5-кратно разб-авленньй раство для крдткости назван ЖВБ) и по 0,5-мл комплемента сыворотки (комплемент морских свинок) разбавленного Т50 раз- ЖВД. Смесь вьщерживают при 37°С в течение 60 мин. Затем к ней добавляют 5 мл ледяного физиологического раствора и смесъ центрифугировали. Спектральная поглощательная способность отделен ного верхнего слоя определялась при ,и измерялась степень инги бирования гемолиза сенсибилизирова ных эритроцитов испытываемыми соед нениями. 50% значение ингибирующей гемолиз активности ( У /мл), из ренное предлагаемым методом, показ но в табл. 1 для каждого испытывае мого соединения. Острая токсичность. Острая токсичность (ЛДс,мг/кг веса) испытываемых соединений опре делена на мышах привнутрибрюшин. ном введении в случае соединения А и при внутривенном введении соединения В. . Полученные результаты показаны в табл. 1. Таблица 1 Как видно из результатов, представленны.х. в. табл. .1, значение острой токсичности (ЛДур) предлагаемого соединения такого порядка,- который позволяет применять его в качестве лечебного препарата. Терапевтическое действие на нефротоксин, вызывающий нефрит. Нефротоксин-крыс (для краткости НТ) получают следующим образом. Кара надпочечника крысы равномерно смешиваяась с равным количеством физиологического раствора. Гомогенизированн-ую смесь .смешивали с полным стимуля.тором Фрейнда (продукт компании Difco)-в- объемном отношении 1:1 ,2. мл полученной смеси инъекцировали внутримышечно кролику (весом 3100 г) для иммунизации кролика. Через полтора месяца; брали .кровь из сердца кролик-а и получали сьгооротку. Полученн-ую .сыворотку инактивировали при-56 С Втечение 30 мин, затем высаливали 40%-ным насьщенным водным раствором сульфата амомния и фракционировали. Фракции Y-глобулина (ИУ-Г) собирали для получения НФ. Оценку терапевтического -действия проводили на самцах крыс линии Вистар с весом тела 150-160 г при трех повторениях для каждого испытываемого - . соединения. Испытываемое соединение вводили внутрибрюшинно один раз в день в течение 7 дней. Через 1 ч после введения испытьшаемого соединения, на 3-й день вводили НТ (нефротоксин) . НТ инъекцировали внутривенно . в количестве 1- мл. в вену хвоста каждой крысе. Физиологический раствор соли использовали в контрольной группе.- ; . Уровень протеинурии (общее количество выделившейся мочи .в течение 24-часового периода) измеряли методом белкового помутнения с использованием альбумина бычьей сыворотки.
как эталона, с помощью сульфосалициловой кислоты.
Полученные результаты представлены в табл. 2.
г
Таблица 2



| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения тетразолилалкоксикарбостирилов | 1979 |
|
SU1064868A3 |
| Способ получения производных карбостирила | 1982 |
|
SU1331426A3 |
| Способ получения карбостирильных производных | 1981 |
|
SU1367857A3 |
| Способ получения производных карбостирила или их фармакологически приемлемых кислотно-аддитивных или четвертичных солей | 1980 |
|
SU1091857A3 |
| Способ получения производных тетразола или их фармакологически приемлемых кислотно-аддитивных солей | 1981 |
|
SU1212324A3 |
| Способ получения сесквитерпеновых производных или их солей | 1980 |
|
SU1056901A3 |
| Способ получения 3-0/ @ - @ -глукуронпиранозил/-соясапогенола | 1979 |
|
SU1074408A3 |
| Способ получения карбостирильных производных | 1982 |
|
SU1395140A3 |
| Способ получения карбостирильных производных | 1981 |
|
SU1169535A3 |
| Способ получения карбостириловых производных (его вариант) | 1982 |
|
SU1779249A3 |
СПОСОБ ПОЛУЧЕНИЯ 3-0-(/3-I)-ГЛЮКУРОНПИРАНОЗИЛ)-СОЯСАПОГЕНОЛА 5 формулы I отличающийся тем, что соединение формулы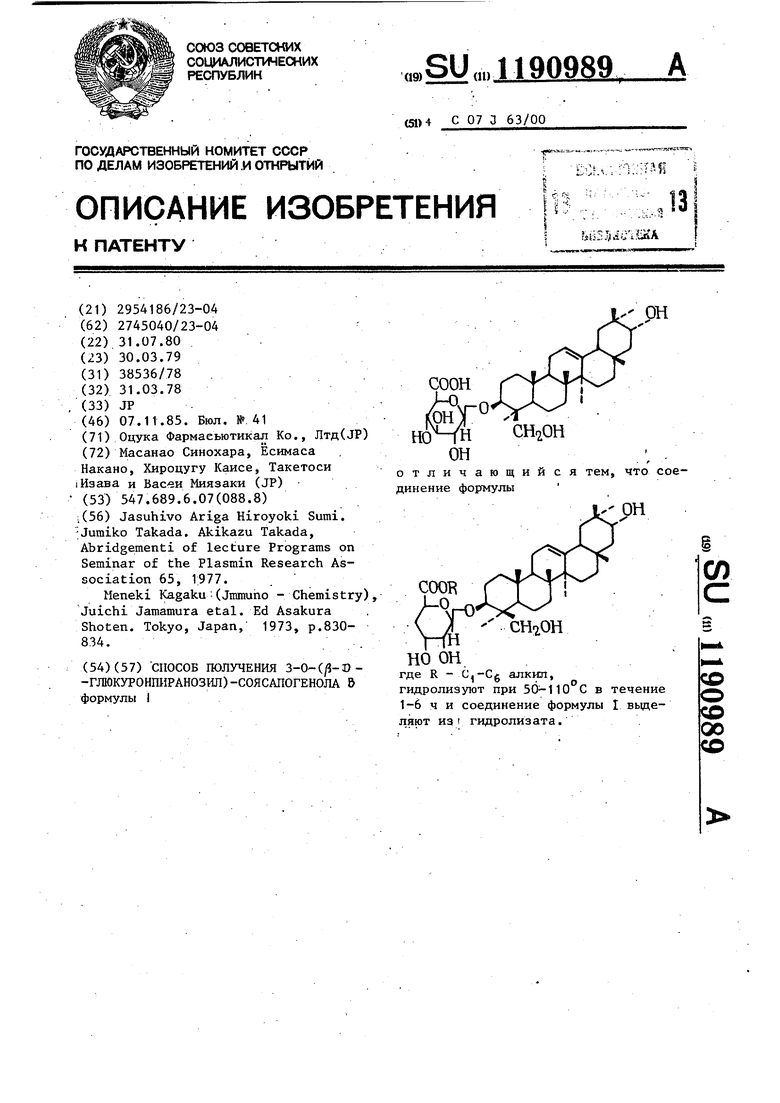
Среднее значение
Предлагаемое соединение-.
(3 мг/тело)
Номер дня отсчитывается со времени введения испытываемого соединения, которое происходит за час до введения нефротоксина.
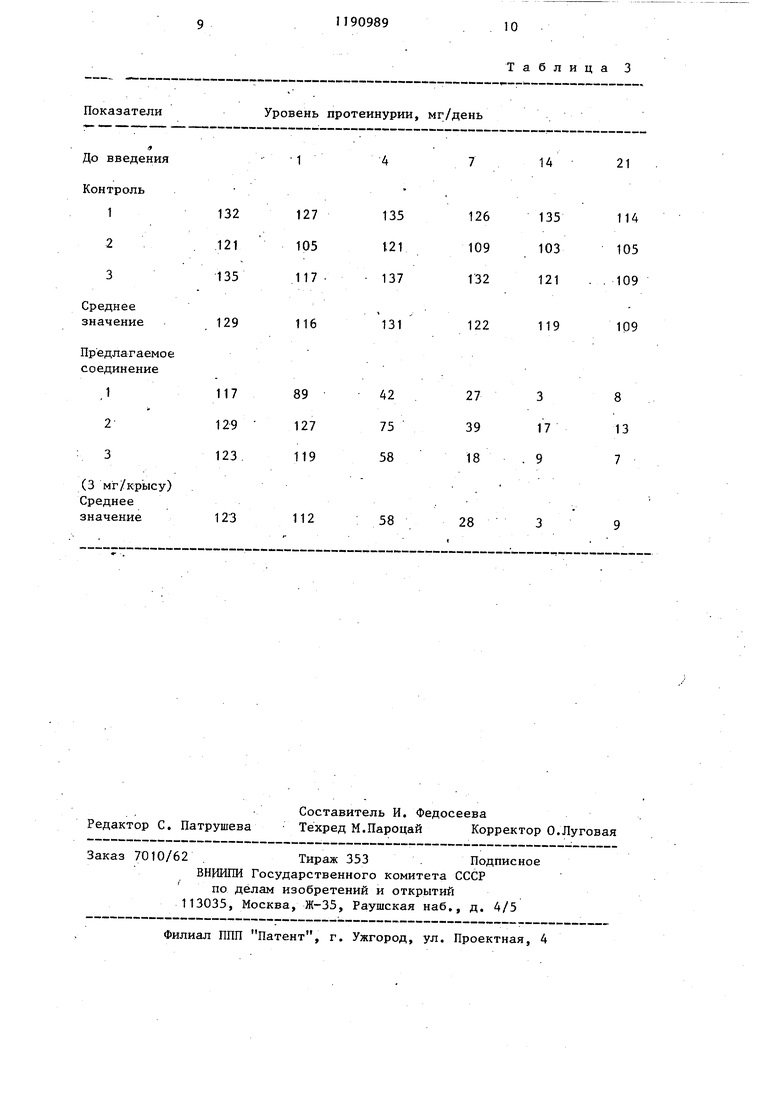
Уровень протеинурии у- здоровых крыс колеблется 0,5-5 мг/день. Если уровень протеинурии превьшает эти пределы, особенно если уровень протеинурии выше 10 мг/день, то можно с уверенностью говорить о том, что имеет место нефрит. Как можно видеть из результатов, табл. 2, нефрит появился в контрольной группе, а в случае применения испытываемых предлагаемых соединений уровень протеинурии со Времени введения нефротоксина до 10 дней после введения в значительной мере такой же, что и у здоровых крыс. Таким образом, введение предлагаемых соединений ингибирует первичные и вторичные иммунные реакции.
Терапевтическое действие при нефрите Хеймана.
В эксперименте использовали крыс линии Вистер самцов с весом 180-200 г. Экстрагировали кору над23
20
33
0,9
7
2,9 1,8
13 2,7
9 1,1
почечников крыс и гомогенизировали с равным количеством физиологического раствора по объему. Гомогенат центрифугировали при 1500 в течение 1 ч. Верхний слой жидкости очищали по известному методу и смешивали с полным стимулятором Фрейнда 37. Ra (выпускаемого компанией Difco в отношении 0,4:1 по объему.
Полученную смесь внутрибрюшинно инъецировали изологическим крысам в количестве 0,5 мг на крысу. Затем такое же количество смеси вводили каждые две недели до. тех пор, пока протеинурия достигала уровня, превышающего 100 мг в день. Этрт период составлял 6-8 недель.
Каждое из испытываемых соединени внутрибрюшинно вводили крысам, больным нефритом Хеймана (весом 300350 г) один раз в день в течение 7 дней и затем измеряли уровень протеинурии (мг/день) описанным способом. Физиологический раствор использовали для контроля. Полученные результаты представлены в табл. 3.
Уровень протеинурии, мг/день
Показатели
1
132
127
121 105
135 117
129
116
Таблица 3
21
14
126
135
114 103 109 105 109 132 121
122
119
109
| Jasuhivo Ariga Hiroyoki Sumi | |||
| -Jumiko Takada | |||
| Akikazu Takada, Abridgement of lecture Programs on Seminar of the Plasmin Research Association 65, 1977 | |||
| Meneki Kagaku :(Jmmuno - Chemistry), Juichi Jamamura etal | |||
| Ed Asakura Shoten | |||
| Tokyo, Japan, 1973, p.830834. |
