Изобретение относится к получению новых бициклических бензоконденси- рованных соединений, которые обладают обезболивающим действием и могут быть использованы как средства про- тив поноса и как агенты для лечения и предотвращения имезиса и тошноты, особенно при введении лекарств против новообразований.
Целью изобретения является получение новых-соединений - производных бензопирана или тетралина, обладающих ценными биологически актив- фши свойствами.
Пример I. 4-Н-Ацетилкарбокс- имидо-5-окси-2,2-диметил-7-(1,1-ди- метилгептил)-3,4-дигидро- Н-бензопи- ран.
А. р-Нитрофенил-5-бензш1окси-2,2- диметил-7-(1,1-диметш1гептил)-3,4-ди- гидро-2Н-бензопиран-4-карбоксилат.
Смесь 3,3 г ( ммоль) 5-бензил- ,2-диметил-7-(1,I-диметнлгеп- тил)-3,4-дигидро-2Н-бензопиран-4- карбоновой кислоты, 5,0 г (21,3 ммоль п-нитрофенилтрифторацетата и 100 мл сухого пиридина в атмосфере азота перемешивают при комнатной температуре в течение трех часов. Пиридин выпари- вают под вакуумом, добавляют этиловый эфир к остатку и все это промьшают 1 н. гидроокисью натрия, водой, 10%- ной соляной кислотой, рассолом, высушивают MgS04H растворитель вьшарива- ют до получения 4,5 г сырого масла. Это масло берут в пентане и охлаждают до получения 3,38 г кристаллов, т.пл. 87-87,.
В. 4-Н-Ацетш1карбоксимидо-5-бен- зш10кси-2,2-диметил-7-(1,1-диметил- гептил)-3,4-дигидро-2Н-бензопиран.
К 301 мг (5,1 ммоль) ацетамида добавляют 35 мл сухого тетрагидрофуCH(CH,).j С(СН,),
во время добавления 103 мг гидрида натрия (99%) (4,3 ммоль). Полученную смесь перемешивают всю ночь в атмосфере азота, добавляют 400 мг (0,716 ммоль) сложного р-нитрофенил эфира, полученного в части А, и перемешивание продолжают при комнатной температуре в течение одного часа. Смесь выливают в лед-воду, подкисляют до рН 3 10%-ной соляной кислотой и экстрагируют этилацетатом. Экстрак промывают рассолом, насьш1енным раствором бикарбоната натрия, снова рассолом и высушивают MgS04. Выпаривани растворителя позволяет получить 416м сырого твердого вещества. Сырое вещество берут в метиленхлориде, промывают ра створом бикарбоната натрия, рассолом, высушивают (MgSO) и растворитель выпаривают, получая 331 мг пены. При добавлении гексана осаждаются кристаллы, 249 мг. Обработка их горячим гексаном дает 216 мг при охлаждении и фильтрации, т.пл. 157- 158 С.
Н-ЯМР(ООС1,)-, ч./млн,8 : 2,20 (с, ЗН), 3,80 (т, Ш), 5,0 (с, 2Н), 6,50 (с, 2Н), 7,30 (с, 5Н), 8,10 (с, 1Н).
С. 21,6 мг продукта части В, 45 мг 5%-ного палладия на угле и 25 мл этилацетата встряхивают в атмосфере водорода при атмосферном давлении в течение 2,5 ч. Фильтрацией и выпариванием фильтрата получают 158 мг продукта, т.пл. 146-147 С.
Повторяя методику части В с соответствующим амидом формулы R,,, CONH , .сульфонамидом формулы R,j,S02NH2 или мочевиной вместо ацетамида и гидрированием полученного продукта методом, изложенным в части С,.получают следующие имидосоединения формулы I, где Q - CONHCOR,, , CONHSOjR или
Т. пл. 143-148 С
Массовый спектр, м/е: М 431, основание 360
н-ЯМР (CDClj), ч./млн,& : 4,05 (т, 1Н), 6,50 (ri, 2Н), 7,1 (с, 1Н)
Се
CJ1,CH(CH,) (диастереомер А))
Cj,HyCH(CH,) (диастереомер В)
с,н,сн,
,
Q, CONHCONH-i
CONHSOjR, где R,, - СН,
Q-J V V MllCiy-J-Xpy,
Пример 2. 5-Окси-2,2-диме- тил-7-(1,1-диметилгептил)-3,4-дигидро-2Н-бинзопиран-4-гидроксамоваякислота.
A.Смесь 5,0.г 5-бензилокси-2,2- диметил-7-(1,1-диметилгептил)-3,4- дигидро-2Н-бензопиран-4-карбоновой кислоты, 500 мг 5%-иого катализатора - палладия на угле и I50 мл этил- ацетата гидрируют в течение 18 ч при давлении три атмосферы. После удаления катализатора и вьтаривания растворителя получают 4,25 г пены. Очистка хроматографией на силикаге- ле при элюировании 2:1 гексаном/эти- ловым эфиром позволяет получить 1,84 продукта, Т.пл. 147-148 С.
B.п-Нитрофенил-5-окси-2,2-диме- тил-7-(1,1-диметш1гептш1)-3,4-дигид- ро-2Н-бензопиран-4-карбоксилат.
Смесь 4,25 г (12,3 ммоль) продук- та части А, 8,67 г (37 ммоль) п-нит- рофенилтрифторацетата и 50 мл сухого пиридина перемешивают в течение 65 ч при комнатной температуре. Смесь вы13165634
Т.пл. 172-173 с
Т.пл. 141-146°С
Массовый спектр, м/е: М 479
Н-ЯМР (CDCl,), ч./млн, : 3,85 (т, 1Н), 4,3 (к, 1Н), 6,2 (м, 2Н), 7,2 (с, 5Н)
Т.пл. 144-149°С
Массовый спектр: м/е: М 479 основание 105
Н-ЯМР (CDClj), ч./мпн,К: 3,90 (т, Ш), 4,40 (к, 1Н), 6,35 (м, 2Н), 7,25 (с, 5Н)
Т.пл. 154-158 с
Массовый спектр, м/е: М 390 (M-NH) 373
Т.пл. 155-156 с
Массовый спектр, м/е: М 425, основание 303
5
0
5
Q
.паривают, чтобы удалить пиридин,и остаток обрабатывают так, как описано в в примере 1А, до получения 5,085 г (88%) требуемого сложного эфира. н-ЯМР (CDC1,), ч./млн., : 6,5 (м, 2Н), 6,7 (с, ОН), 7,03-7,3 (м, 2Н), 8,0-8,3 (м, 2Н).
С. Смесь 43 мг (1,066 ммоль) измельченной в порошок гидроокиси натрия в 10 мл пириди на в атмосфере азота перемешивают и нагревают для растворения, затем охлаждают до 0°С, добавляют 111 мг (1,6 ммоль) гидрохлорида гидроксиламина, и перемешивают 15 мин. Раствор 250 мг (0,533 ммоль) продукта части В в 3,0 мл пиридина добавляют, смеси.позволяют нагреться до комнатной температуры, оставляя ее с перемешиванием на всю ночь. Пиридин выпаривают, остаток берут в воду и экстрагируют этилацетатом. Объединенные экстракты промывают водой, насыщенным рассолом и высушивают MgSO. Выпаривание растворителя позволяет по-
5
лучить 260 мг сырого продукта, который загружают в колонку с 30 г сили- кагеля. Элюирование 1:1 гексаном/эти ловым эфиром для 10 фракций, затем этилацетатом - до элюирования требуемого продукта. Выпаривание растворителя (фракции 18-20) дает 158 мг целевого соединения.
Массовьй спектр (). н-ЯМР (CDClj,), ч./млн,8 : 6,20 (4Н, 2 ароматическое, NH, ОН), 10,0 (Н, обмены с DjO).
. Пример 3. Н-2-Пиридил-5-ок- си-2,2-диметил-7- (1,1 -диметштгеп- тил)-3,4-дигидро-2Н-бензопиранкарб- оксамид.
A.К-2-Пиридил-5-бензилокси-2,2- диметил-7-(1,1-диметилгептил)-3,4- дигидро-2Н-бензопиранкарбоксамид.
Смесь 1,118 г (2,0 ммоль) 4-нит- рофенш1-5-бензилокси-2,2-диметил-7- (1,2-диметилгептил)-3,4-дигидро-2Н- бе.нзопиранкарбоксилата, 376 мг (4,0 ммоль) 2-аминопиридина и 4 мл пиридина помещают в запаянную трубку и нагревают 18 ч при 155-157°С. После охлаждения трубку вскрывают, смесь концентрируют досуха под вакуумом, остаток растворяют в этиловом эфире, промывают 1 н. соляной кислотой (25 мл), 1 н. гидроокисью натрия (3 X 25 мл), водой (2 х 25 мл) и рассолом (25 мл), Промытый эфирный раствор высушивают (MgSO), растворитель вьтаривают до получения 966 мг масла. Масло очищают хроматографией на силикагелевой колонке, элюируя смесью гексан/метиленхлорид (1:4), затем 15%-Hbw этиловым эфиром в мети- ленхлориде. Фракции, содержащие продукт, объединяют и растворитель выпаривают под вакуумом до получения 823 мг (80%) требуемого амина. Ф
B.Смесь 691 мг каждого упомянутого амида и 10%-ного Pd/C катализатора, 1,08 г 1,4-циклогексадиена и 25 мл сухого этанола гидрируют по методике примера 2. После удаления растворителя под вакуумом получают 680 мг сырого дебензилированного продукта. Этот продукт очищают с по мощью колоночной хроматографии на силикагеле, элюируя метиленхлоридом затем метиленклоридрм, содержащим 10% этилового эфира, наконец, одним эфиром до получения 515 мг (90%) дебензилированного материала, который кристаллизуется из этилацетата/гек165636
сана, обеспечивая выход 398 мг (70%) продукта, т.пл. 166-167 с; массовый
спектр, м/е; 424 (молекулярный ион), 119 основание.
5 С. Применяя подходящий амин ArNH вместо 2-пиридиламина, получают с помощью упомянутой методики соединения, представленные в табл. 1.
Таблица 1
15
.
238,5-239 М 430
основание
Пример 4. 4-Нитрофенил-5бензилокси-2,2-диметил-7-(2-метилпропил)-3,4-дигидро-2Н-бензопиран-4карбоксилат.
А. 5-Бензилокси-2,2-диметил-7-(2- ме тшш ропил)-3,4-дигид ро б ен з опиран- 4- карбоновая кислота.
Смесь 7,8 г (22,2 ммоль) 5-бензил- окси-4-циано-2,2-диметш1-7(2-метилпропил)-3,4-дигидро-2Н-бензопираиа, 12,5 г таблеток КОН и 200 мл этилен- гликоля нагревают в азоте с верти- кальным холодильником в течение 18 ч и охлаждают до температуры окружающей среды. Смесь подкисляют до рН 3 концентрированной соляной кислотой, экстрагируют этилацетатом и экстракты высушивают MgS04. Выпаривание растворителя дает масло, которое бе713
ут в этиловый эфир, промьшают воой, рассолом, высушивают MgSO и астворитель выпаривают, обеспечивая олучение 8,25 г сырого продукта в виде кислоты, которую очищают с поощью хроматографии на силикагеле, элюируя этиловым эфиром/метиленхло- ридом в соотношении 1:4, просто эфиром и, наконец, метанолом/этиловым эфиром в соотношении 1:9, до получения 6,13 г выхода, который дает кристаллы из метиленхлррида/гексана, т.пл. 152-158°С.
В. Смесь 3,0 г (8,15 ммоль) кислоты, полученной в части А, 2,87 г (12,2 моль) п-нитрофенила трифтораце- тата и 40 мл сухого пиридина перемешивают при комнатной температуре в течение 60 ч. Пиридин выпаривают.под вакуумом, остаток промывают 1 н. соляной кислотой (3 X 25 мл), 1 н. гидроокисью натрия (4 X 25 мл), водой, рассолом и высушивают MgSO.Выпаривание растворителя обеспечивает получение 4,0 г сырого продукта в виде пены, который кристаллизуется из ме- тиленхлорида/гексана до получения 3,67 г целевого соединения, т.пл. 125-126°С.
Пример 5. 5-Окси-2,2-диме- тил-7-(2-метилпропил)-3,4-дигидро-2Н- бензопиран-4-карбоновая кислота, т.пл 185-187°С.
Получают каталитическим гидрирова- соответствующего 5-бензил эфира, используя описанные способы.
Пример 6. А. Используя способы примеров 1 и 3, но исходя из п- нитрофенил сложного эфира, полученного в примере 4, и подходящего амида или мочевины, получают таким же образом следующие соединения:
Q ОН
Т.пл., С
CONHCOCsHj. СОШ-Тетразол-5-ил
CONHCOC(CHj), CONHCOCHjCHCCH,) 10. CONHSO,jCH(CHj)2
CONHSO C H
Пример 7. A. Используя 3,3- диметил-6-(5-фенил-2-пентилокси)-8- бензилокси-1-тетралон) (получен в патенте США № 4188495), получают количественный выход соответствующего не- насьпценного нитрила - 8-бензилокси- 1-циано-3,З-диметил-6(5-фенил-2-пен- тилокси)-3,4-дигидронафталина в виде оранжевого масла.
Б. Масло, полученное в части А, гидрируют по методике примера 1C, получая соответствующий тетралин - 8-бензилокси-1-циано-3,3-диметил- 6-(5-фенил-2-пенталокси)тетралин с 89%-ным выходом в виде оранжевого масла.
0
5
0
5
0
5
C.Тетралиннитрил гидролизуют в этиленгликоле гидроокисью калия по методике примера 4А, получая соответствующую кислоту - 8-бензилокси-3,3- риметил-6-(5-фенил-2-пентилокси)- тетралин-1-карбоновую кислоту в виде белой пены с 39%-ным выходом.
D.Смесь 1,6 г продукта части С, 20 мл метанола и 320 мг 5%-ного палладия на угле (катализатора) гидрируют при давлении в три атмосферы в течение трех часов и продукт вьщеля- ют фильтрацией и выпариванием фильтрата. Получают 1,2 г бесцветной твердой пены, которая является на 92,5% чистой смесью диастереомеров по данным HPLC-анализа на Zobax Sil-колон- ке зарегистрированная торговая марка Е. I du Pont de Nemours and Co. I nc. Wilmington Del), 2%-ный изопро- пиловый спирт в гексане при 1 мл/мин.,
н-ЯМР (CDCl,), ч./млн.& : 0,8 (с, ЗН), 1,0 (с, ЗН), 1,2 (д, 4Н), 1,74 (м, 6Н), 2,5 (м, 4Н) 3,7 (м, 1Н), 4,16 (м, 1Н), 6,1 (с, 2Н), 7,1 (с, ЗН), 8,1( широкий с., 1Н), который согласуется со структурой для 8-окси-3,3-диметнл-6-(5-фенил-2-пен- токси)-тетралмн-1-карбоновой кислоты.
E,Реакция упомянутой кислоты (3,14 ммоль) с п-нитрофенилтрифтор- ацетатом (3,45 ммоль) в пиридине методом примера 2В дает 1,1 г (69%) п-нитрофенил-8-окси-З,З-диметил-6- (5-фенил-2-пёнтилокси)тетралин-1-кар боксилата в виде желтого масла.
F.Используя бензил-эфир, полученный в части С, по методике части
Е можно получить п-нитрофенш1-8-бен- зилокси-3,37Диметил-6-(5-фенил-2- пентилокси)-тетралин-1-карбоксилат в- виде масла с выходом 90%. TCX:R 0,68 с гексан/этилацетатом, 2:1 (рас т воритель).
И р и м е р 8. 8-Окси-З,З-диметил-6- ( 5-фенил-2-пентилокси тетралин 1-карбоксамид.
A.Реакция 2,3 г (3,9 ммоль) п-нитрофенил-8-бензилокси-З,3-диме- тил-6-(5-фeнил-2-пeнтшIOKcи)-тeтpa- лин-l-кapбoкcилaтa в избытке жидкого аммиака при -70°С в течение 30 мин
и выпаривание избытка аммиака дает желтую пасту, которая при силикаге- левой хроматографии, элюированная с помощью смеси 1:1 этилацетилат/гек сан, позволяет получить 730 мг амида R 0,15 при тех с системой растворителя 2:1 этилац -тат/гексан. Исходный материал также вьщеляют (1,15 г).
B.Гидрированием продукта части А в 50 мл метанола с 400 мг 5%-ного ка.тализатора Pd/C при давлении З.атм в течение 4,5 ч с применением обыч- ных способов можно получить сырой продукт, который очищают на сштика- гелевой колонке, используя в качестве растворителя зтилацетат/гексан (2:1), Получают 130 мг целевого со- единения в виде белого твердого вещества, т.пл. 155-157 с.
Н-ЯМР (CDCl,), ч./млн,Г: 0,8 (с, ЗН), 1,0 (с, ЗН), 1,16 (д, ЗН), 1,7 (м, 6Н), 2,47 (м, 4Н), 3,57 (м, 1Н), 4,16 (м, 1Н), 6,1 (д, ЗН), 7,2
(с, 5Н).
1
C.Проводят реакцию 0,55 г
(1,1 ммоль) п-нитрофенил-8-окси-3,3- диметш1-6-(5-фенш1-2-пентилокси)тет- рапин- 1-карбоксилата в 10 мл тетра- гидрофурана с избытком газообразного метиламина при комнатной температуре выливают полученную смесь в 10%-ную соляную кислоту, экстрагируют этил- ацетатом. Применяя обычные способы, получают 0,50 г N-метиламида - N-ме- ТИЛ-8-ОКСИ-3,З-диметил-6-(5-фенил-2- пентилокси)тетралин-I-карбоксамида в виде пены.
Н-ЯМР (CDC1,), ч./млн,б : 0,8 (с, ЗН), 1,0 (с, ЗН), 1,2 (д, 4Н), 1,7 (м, 6Н), 2,53 (м, 6Н), 3,6 (м, 1Н), 4,23 (м, NH), 6,2 (д, 2Н), 7,13 (с, 5Н).
Пример 9. 8-Окси-3,З-диметил-6- (5-фенил-2-пентилокси) -тетралин- I -карбонилмочевина .
С помощью реакции между 1,2 г (2 ммоль) п-нитрофенил-8-бензилокси- 3,З-диметил-6-(5-фенш1-2-пентилокси)- тетрапин-1-карбоксилата, 0,3 г (5 ммоль) мочевины и 0,248 г .(10 ммоль) гидрида натрия в 12 мл диметилсульфоксида при комнатной температуре в течение одного часа выделяют продукт методом примера 1В. Удаляя группу бензил гидрированием по методике примера 1C, можно получить чистое целевое соединение с общим выходом 36%.
Н-ЯМР (CDCl), ч./млн,У : 0,77 (с, ЗН), 1,1 (м, 8Н), 1,7 (м, 4Н),
2.5(м, 4Н), 3,6 (м, Ш), 4,16 (м, 1Н), 5,7 (с, 1Н), 6,1 (с, 2Н), 7,1 (с, 5Н), 8,2 (с, 2Н).
Пример 10. Реагирование 1,1 (1,9 ммоль) п-нитрофенил-8-окси-З,3- диметил-6-(5-фенил-2-пентилокси)тетралин- 1-карбоксилата, 1,0 г (17 ммоль ацетамида, 361 мг (15 ммоль) гидрида натрия в 70 мл тетрагидрофурана по ме методу примера 1, части В и С, позволяет заготовить 8-окси-3,3-диметил- 6-(5-фенил-2-пентилокси)-1-N-ацетил- карбоксимид с 55%-ным выходом продукта в виде пены.
н-ЯМР (CDC1,), ч./млч,5 : 0,7 (с, ЗН), 1,0 (с, ЗН), 1,16 (д, ЗН),
1.6(м, 6Н), 2,3 (с, ЗН), 2,5 (м, 4Н), 3,73 (м, 1Н), 4,13 (м, 1Н), 6,1 (с, 2Н), 7,1 (с, 5Н), 8,5 (NH).
I
Пример 11. 3- 8-Окси-З,З-диметил-6- (5-фенил-2-пентилокси) -тетралин- 1 -ил -3-оксопропионитрил.
К раствору 2,4 мл 2,1 М п-бутил- лития в 3,7 мл тетрагидрофурана при -78 С добавляют раствор 0,26 мл (5 ммоль) ацетонитрила в 3,7 мл ТГФ, и смесь перемешивают в течение одного часа при -78 с. Добавляют раствор 1,1 г (2,0 ммоль) п-«итрофенш1-8-бен- зилокси-3,З-диметил-6-(5-фенш1-2-пен- тилокси)тетралин-1-карбоксилата в
3.7мл ТГФ и продолжают перемешивать при -78 С в течение 30 мин. Реакционную смесь нагревают до комнатной температуры, гасят 7 мл 10%-ной соляной кислоты и экстрагируют этиловым эфиром. Вьщеление продукта, как и в предшествующем примере позволяет пол чить 1,13 г сырого бензил-эфира, котрый дает 500 мг очищенного промежуточного, продукта с помощью силикаге- левой хроматографии: массовый спектр молекулярный ион, 495.
Удаление группы бензила гидрированием по методу примера 1C дает чистое целевое соединение.
Н-ЯМР (CDClj), ч./млн,8 : 0,8 (с, ЗН), 1,06 (с, ЗН), 1,2 (м, 5Н), 1,7 (м, 4Н), 2,5 (м, 4Н), 3,5 (с, 2Н 4,0 (м, 2Н), 6,1 (с, 2Н), 7,1 (с, 5Н).
ОбезболиваюЕЦие свойства предлагаемых соединений определяются опытами использованием теплового ноцицептив- ного возбудителя, такого как удар по мьшиному хвосту, или химического но- цицептивного возбудителя, такого как измерение способности соединения по- давлять скрючивание у мьшей, вызванное фенилбензохиноновым раздражителем.
Опыты с использованием теплового ноцицептивного возбудителя.
Опыты по обезболиванию на мышах с использованием горячих пластин.
Используемый метод модифицирован. Контролируемое тепловое возбуждение прикладывают к лапе мьщ1и с помощью алюминиевой пластины толщиной 1/8 . Рефлектор инфракрасной тепловой лампы мощностью 250 В помещают под дно алюминиевой пластины. Терморегулятор со.единенный с термисторами на поверх нести пластины, задает программу лампам нагрева, с тем чтобы поддерживат температуру постоянной, (57 с). Каждая мышь опускается в стеклянный цилиндр (диаметр 6 1/2), находящийся на гррячей пластине, и отсчет времени начинается с того момента, когда лапка животного коснется пластины. Через 0,5 и 2 ч после лечения соединением, применяемым в эксперименте, мьш1ей наблюдают на первое ударное движение одной или обеих задних лапок или до тех пор, пока не пройдет 10 с без таких движений. Морфин имеет MPEjp .4-5,6 мг/кг/с (подкожное применение).
Обезболивающие эксперименты с мышами, подвергнутыми удару по хвосту.
Каждую мышь помещают в соответствующий металлический цилиндр, так чтобы хвост высовывался через один из концов. Цилиндр располагается таким образом, что хвост лежит прямо над скрытой тепловой лампой. При постановке эксперимента алюминиевый флажок, закрывающий лампу, отдергивается, позволяя световому пучку проходить через щель и фокусироваться на кончике хвоста. Одновременно включается секундомер. Мьш1и, которым не давали лекарства, обычно реагируют через 3-4 с после экспозиции лампы. Конечная точка для мьш1ей, подвергнутых лечению, - 10 с. Опыт с каждой мьш1ью проводят через 0,5 и 2 ч после принятия морфина и испытуемого соединения. Морфин имеет МРЕ 3,2-5,6 мг/ /кг (SC).
Метод погружения хвоста.
Метод представляет собой усовершенствованную процедуру с приемником предложенную Ben lassel et al. Самца белой мыщи (19-21 г) породы charles River CD-I взвешивают и помечают для идентификации. Обычно в каждой группе для лечения лекарством находится пять животных, каждому из которых соответствует контрольное животное.-С целью общей проверки новые опытные агенты назначают сначала в дозе 56 мг/кг внутрь брюшины или подкожным вливанием в объеме 10 мл/кг. Перед принятием лекарства и через 0,5 и 2 ч после него каждую мьш)ь помещают в цилиндр. Каждый цилиндр снабжен отверстиями для обеспечения адекватной вентиляции и закрывается круглой нейлоновой пробкой, через которую пропущен хвост животного. Цилиндр удерживают в вертикальном положении и хвост
полностью погружают в водяную ванну с постоянной температурой 56 С. Конечным моментом каждого испытания является энергичное судорожное подергивание хвоста в сочетании с двигательной реакцией. В некоторых случаях после принятия лекарства реакция может быть менее сильной. Чтобы предотвратить чрезмерное повреждение ткани, опыт завершают и хвост удаляют из ванны через 10 с. Ответное скрытое состояние записывается в секундах с точностью до 0,5 с. Контроль-носитель (без лекарства) и стандарт известной силы испытываются одновременно с кандидатами на скрининг. Ее1313
ли активность испытуемого агента не вернется к установленным величинам в течение 2-часового испытания, да- тентный период реакции определяется через 4 и 6 ч. Конечные измерения проводятся через 24 ч, если активность все еще наблюдается в конце экспериментального дня.
Опыт с использованием химического ноцицептивного возбуждения.
Подавление скрючивания, вызванного фенилбеизохиноновым раздражителем.
Группу ИЗ 5 мьшей Carworth Farm CF-I предварительно обрабатывают подкожно или орально физиологическим раствором, морфином, кодеином или опытным соединением. Двадцать минут (если обработка подкожная) или пятьдесят минут (если обработка оральная) спустя каждую группу обрабатывают внутрибрюшинной инъекцией фенил- бензохинона, известного раздражителя, вызывающего бркипинные сокращения.Через 5 мин мьшей наблюдают на наличие или отсутствие скрючивания в течение 5 мин после введения раздражителя. Устаноалено, что предварительная обработка лекарством MPEjo блокирует скрючивание.
Опыты с использованием давления как ноцицептивного возбуждения.
Эффект Haffner по методике защемления хвоста - модификация методики Haffner Experimentelle Prutung Schmer ztillender, Mittel Deutch - применяют для установления эффектов опытных соединений на.агрессивную атаку, вызванную в ответ на возбуждение защемлением хвоста.
Используют самцов белых крыс (50- ёО г)- породы Charls River (Sprague- Dawley) CD. Перед обработкой лекарством и спустя 0,5, 1,2 и 3 ч после обработки зажимом бульдбг (Johns Hopkins 2,5 дюйма)зажимают основание
7 МВЭ . 13 1 2йЗ Р20:Ё 2ё время - время выключения - контрольное время
Установлено, что соединения формулы I особенно полезны как противорвот- ные и противотошнотные агенты для млекопитающих для предотвращения рвоты и тошноты, вызванных введением антинео- пластических агентов.
Противорвотные свойства соединений формутая I определяют на неанестезиро656314
крысиного хвоста. Конечной точкой каждого испытания является явное агрессивное поведение по направлению к раздражающему возбудителю с латент-.
5 ностью атаки, выраженной в секундах. Зажим удаляется через 30 с, если ата- .ка больше не повторяется и латентный период реакции записывается как 30 с. Морфин имеет активность 17,8 мг/кг
to (i.p.).
Опыты с использованием электрического ноцицептивного возбуждения. Опыты раздирание-прьмски. Это усовершенствованная методика
15 раздирание - подпрыгивание. Применяется для определения болевых порогов. Используются самцы белых крыс (175-200 г) породы Charles Rever (Sprague-Dawley) CD. Перед получением
20 лекарства лапу каждой крысы погружают в 20%-ньй глицерин-солевой раствор. Животных затем помещают в камеру и подвергают последовательности 1-секундных ударов по лапе, которые
25 проводятся с повьппающейся интенсивностью с интервалами 30 с. Это разряды 0,26; 0,39; 0,52; 0,78; 1,05; 1,31; 1,58; 1,86; 2,13; 2,42; 2,72 и 3,04 мА. Поведение каждого животно30 го оценивается по наличию раздирания, писка, подпрыгивания или быстрого движения при ударной атаке.
Последовательность ударов с воз-. 35 растающей интенсивностью получает каждая крыса непосредственно до и через 0,5; 2,4 и 24 ч после принятия лекарства.
Результаты упомянутых опытов запи- 40 сываются как процент от максимально возможного эффекта (% МВЭ). % МВЭ каждой группы статистически сравнивается с % МВЭ стандарта и контрольной величиной, перед приемом . 45 лекарства. % МВЭ рассчитывается следующим образом:
ванных кошках согласно известной методике .
Антагонизм PGE (простагландин Е). Понос у мышей.
Противопоносная активность соединений формулы I определяется по усовершенствованной методике Dajani et al. European Jour. Pharmacol., 34, ,
151
105-113, 1975. Это метод надежного выявления поноса у не обработанных иным способом мышей в течение 15 мин Животные, у которых перед обработкой нет поноса, считаются защищенными. Запорные эффекты измеряются как ответ все или ничего, причем понос определяется как жидкий стул, очень отличающийся от нормального фекального вещества, которое состоит из хорошо сформированных щариков, крепких и относительно сухих.
Используют белых мышей, самцов, породы Charles River CD-К
Их помещают группами в клетки и, начиная с этого момента, эксперименты проводят в течение одной недели, Вес животных во время проведения опытов 20-25 г. Таблетированную крысиную пищу поставляют вдоволь. За 18 ч .до начала опытов пищу удаляют.
Животных взвешивают и метят для идентификации. Обычно используют до пяти животных в каждой группе, которая обрабатывается лекарством. Мышей весом 20-25 г помещают группами в клетки и держат голодными в течение ночи перед экспериментом. Воды дают вдоволь. Животным дают PGE о,32 мг/кг (i.p.) в 5%-ном этиловом спирте через один час после принятия лекарства и немедленно помещают отдельно в прозрачные акриловые ящики размером 15 х 18 см. Разового ис- прльзования картонный лист на дне ящика проверяют на наличие поноса через 15 мин. Группа, обрабатываемая растворителем +PGE, и группа, обрабатываемая растворителем, служат в качестве контрольных.
Данные анализируют, используя весовую линейную регрессию, с применением метода максимальной вероятное- ти. Компьютерная программа печатает результаты анализа формата линейной регрессии, включая степени свободы, сумму квадратов, среднеквадратичное и критические значения и пси/Chi квадрат. Если регрессия значительна, Е0„, EDso , , EDgoH затем 95%- ный интервал подсчитываются.
Соединения формулы I являются активными обезболивакицими, противопо- носными, противорвотными и противо6316
тошнотными соединениями, назначаемыми орально или парентерально в форме композиции. Композиции включают фармацевтический носитель, выбранный в зависимости от способа назначения и обычной фармацевтической практики, например, они могут быть назначены в форме таблеток, пилюль, порошков или гранул, содержащих та-7
кие эксципиенты, как крахмал, молочный сахар, определенные типы глин и т.д. Они могут быть назначены в капсулах, смесях, с теми же или эквивалентными эксципиентами или в виде оральной суспензии, растворов, эмульсий, сиропов и эликсиров, которые могут содержать вкусовые и цве- . товые агенты. Для орального назначения терапевтических агентов формулы
таблетки или капсулы, содержащие от около 0,01 до. около 100 мг, являются подходящими для большинства применений. I
Суспензии и растворы этих лекарств
обычно приготавливают непосредствен- но перед использованием для того, чтобы избежать их неустойчивости (окисления или осаждения) во время
хранения. Для этого используются в основном, сухие твердые композиции, которые перестроены для инъекционного назначения.
Врачи определяют дозу, которая йаиболее подходяща для пациента в каждом отдельном случае. Она колеблется в зависимости от возраста, веса и зависит от особенностей организма пациента и способа приема. Обычно начальная обезболивакщая доза так же, как и начальная доза для предотвращения или лечения тошноты для взрослых составляет 0,0)-5 00 кг в день в разовой или разделенной дозах. Во многих случаях нет необходимости превышать ежедневную дозу в 100 мг. Подходящая оральная доза составляет 0,01-50, парентеральная - 0,01-100 мг в день, предпочтительно 0,01-20 мг в день.
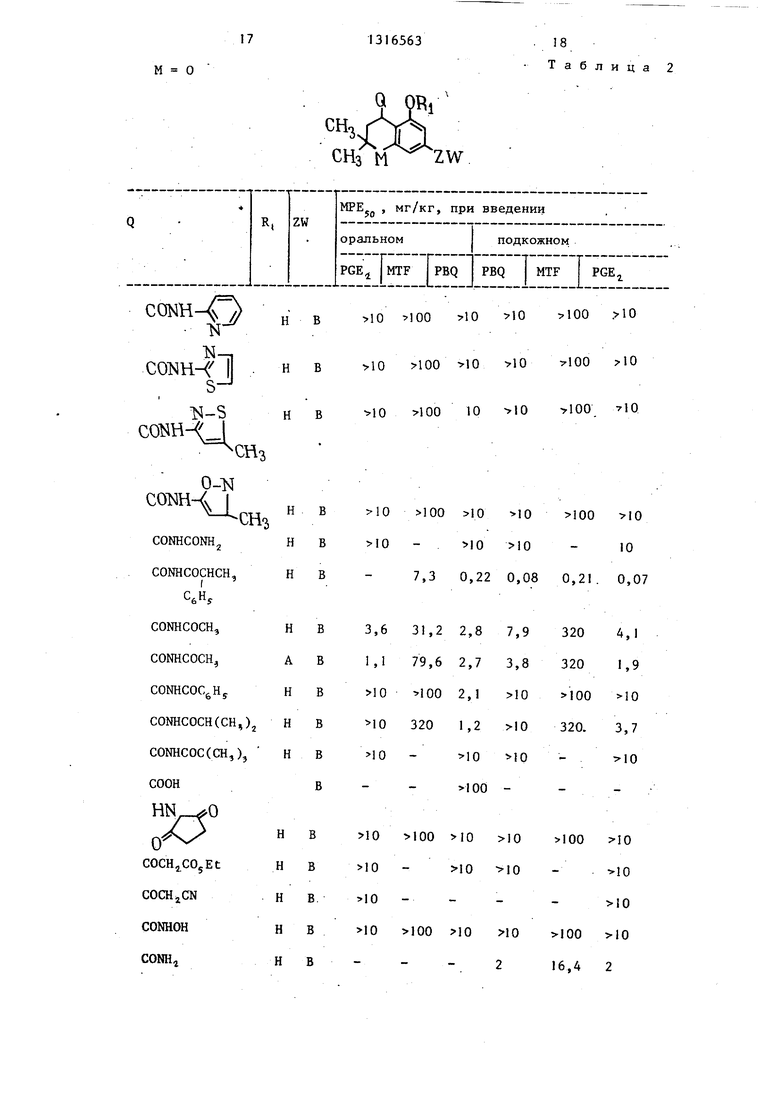
Результаты скрининговых обезболивающих и противопоносных испытаний соединений формулы I приведены в табл. 2.
17
М О
1316563
.18
Таблица 2
Q Q,
СНз М ZW
1
о ЯОО 10 10 100 .10
10
100 10 10 100 10
100 10 10 100 10
10 100 10 10 100 10 10 10-10
7,3 0,22 0,08 0,21. 0,07
19
Примечание. А- ОСНССН,) .
В - С(СН,),,(СН)5СН,;
С - CH2CH(CH,)j ;
PBQ - фенилбензохинон; .
PQEj - антагонизт простагландина
MTF - методика защемления хвоста.
Пример 13. d-1 -5-ОКСИ-4- (2-оксиэтил)-2,2-диметил-7-(1,1-ди- -метилгептил)-3,4-дигидро-2Н-бензопи- ран (100 мг) однородно смешивают и растворяют с 900 мл крахмала. Смесь затем загружают в телескопические желатиновые капсулы так, что каждая капсула содержит 10 мг лекарства и 90 мг крахмала.
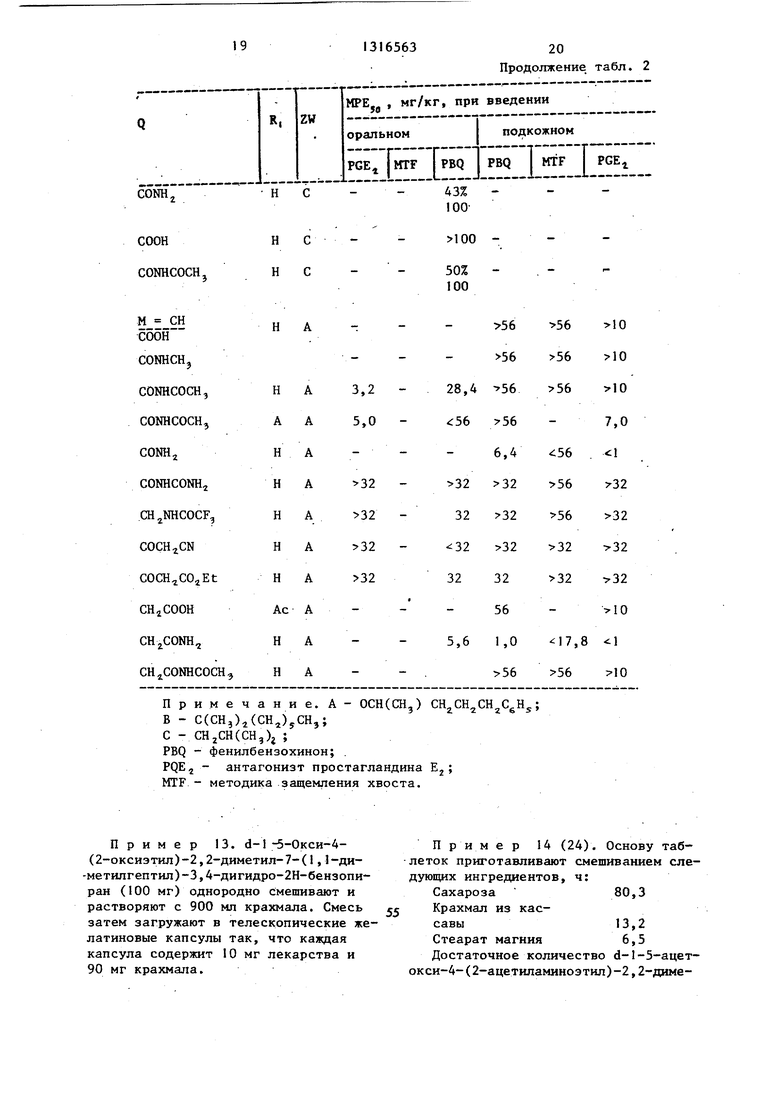
1316563
20 Продолжение табл. 2
Пример 14 (24). Основу таблеток приготавливают смешиванием следующих ингредиентов, ч:
Сахароза 80,3
Крахмал из кассавы13,2
Стеарат магния 6,5 Достаточное количество d-l-5-ацет- окси-4-(2-ацетнламиноэтил)-2,2-диме2113
тил-7-(1,1-диметилгептил)-3,4-дигид- ро-2Н-бензопиран смешивают с этой основой, чтобы получить таблетки, содержащие 0,1; 0,5; 1,5; 10 и 25 мг лекарства,
П р и м .8 р 15 (25). Суспензию d-1-5-окси-З-(3-оксипропил)-2,2-ди- .метил-7-(5-фенил-2-пентилокси)-3,4- дигидро-2Н-бензопирана получают добавлением достаточного количества лекарства к. 0,5% мети.пцеллюлозы для получения суспензий, содержащих 0,05 0,1; Oj5; 1,5 и 10 мг лекарства на 1 мл.
Из данных табл. 2 по флику мьппино- го хвоста следует, что при оральном введении мыши переносят дЬзы предлагаемых соединений от 7,3 до 320 мг/кг Предпочтительная доза для орального введения составляет 0,01-300 мг в день. Эта величина соответствует 0,0002-6,0 мг/кг в день для человека весом 50 кг. Таким образом, предлагаемые соединения достаточно безопасны.
Соединения формулы I не только обладают высоким уровнем активности как периферические обезболивающие.агенты, они достаточно селективны, так как обладают очень низким уровнем воздействия на центральную нервную систему и обезболивающей активностью, сравнимой с той, которой обладают структурно подобные соединения.
322
Опыт по дрожанию мышиного хвоста (MTF) является хорошо известным тестом для измерения активности агентов по отношению к центральной нервной
системе. Тест по определению раздражения и конвульсий, вызванных фенил- бензохиноном, является мерой выявления периферических обезболивающих эффектов.
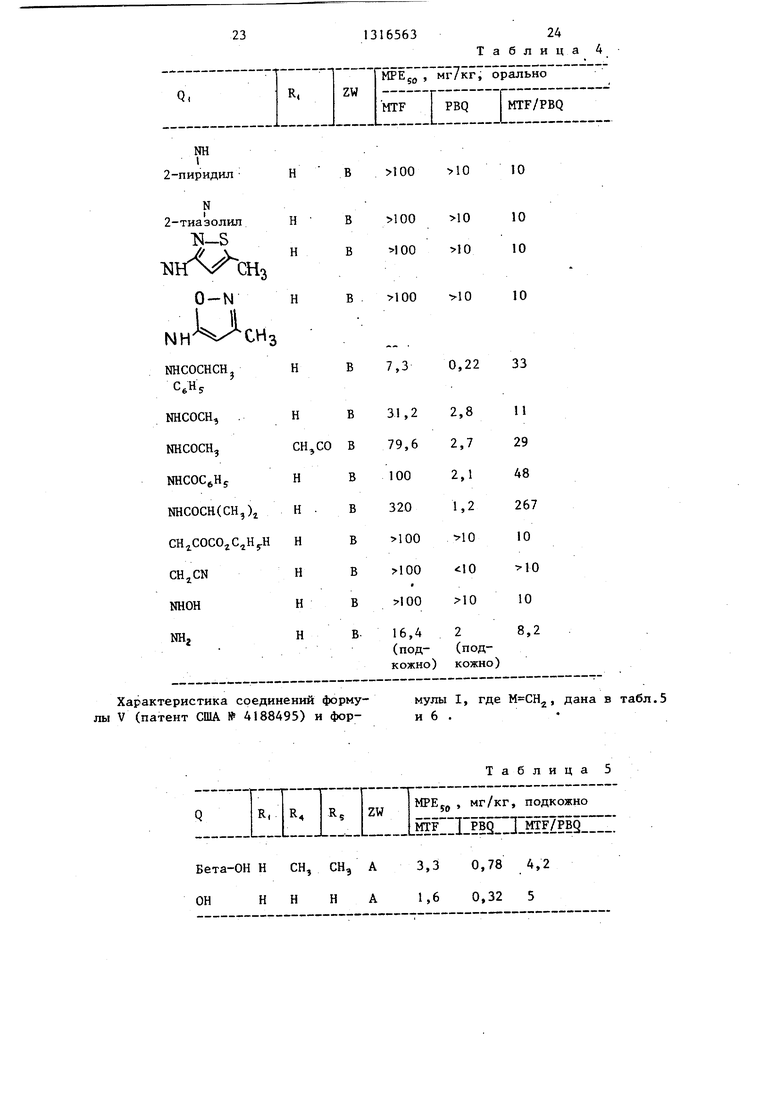
В качестве меры селективного периферического обезболивающего эффекта определяют соотношение MPEjg по значениям, полученным при проведении тестов MTF и PBQ (MTF/PBQ). Это соотношение для соединений формулы I и известных структурно подобных соединений, характеризуемьк .формулой V (описаны в патентах США № 4143139 () и № 4188495 (М-СН., приведено
в табл. 3-6:
COQl
OBi (I)
н
ZW Bi
снгм
30
в табл. 3 и 4 охарактеризованы соединения формулы V (патент США № 4143139) и формулы I соответствен но, где .
Бета-ОН СН, СН, А
Бета-ОН СН, СН, С
Бетан-ОН СИ, СН, D
Бетан-ОН Н
Морфин
СН, Е
Таблица 3
Ш 2-пиридш1
N 2-тиазолил
N-S
:NHrV CH3
O-N
н
NHCOCHCH,
Характеристика соединений формулы V (патент США № 4188495) и форБета-ОН Н СН, СН, А3,3 0,78 4,2
ОННИНА1,6 0,32 5
в 100 1010
в 100 1010
1010
в 100 10
10
7,3
0,22
33
мулы I, где , дана в табл.5 и 6 .
Таблица 5
в табл. 3-6 А - (CH)CH2CHiCHjC5H ; C(CH,)j (СНз)5СНз; С - OCHCCHOCH CH CgHj; D - СН(СН,)СН(СНз)СуН,, ; Ё - ,(СН,,СН.,СН2С5Н) .
На основании данных, представленных в табл. 3-6, можно сделать вывод что соединения формулы I характеризуются значительно большей селектив- ностью при их использовании в качестве периферических обезболивающих агентов по сравнению со.структурно им.подобными соединениями, являющимися известными и указанными в патентах США № 4143139 и № 4168495.
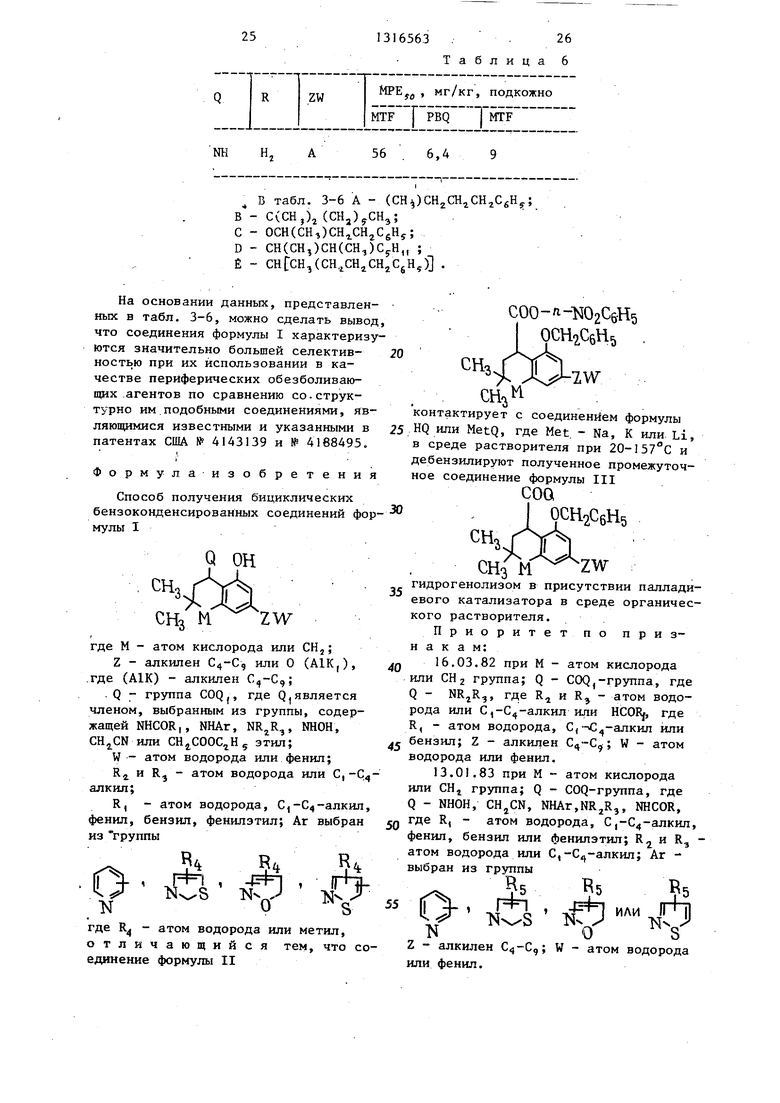
Формула-изобретения
Способ получения бициклических бензоконденсированных соединений фор- мулы I
Q ОН
СНз Н ZW
где М - атом кислорода или СН,,;
Z - алкилен С4-С, или О (А1К,), .где (А1К) - алкилен С,-С,;
. Q - группа COQj, где Q является членом, выбранным из группы, содержащей NHCOR,, NHAr, NR,jR,, NHOH, CHjCN или CHjCOOC H, зтил;
W - атом водорода или фенил;
R и R - атом водорода или С,-С алкил;
R, - атом водорода, С -С -алкил, фенил, бензил, фенилэтил; Аг выбран из группы
0
.
где R - атом водорода или метил.
отличающийся единение формулы II
тем, что СОО-л-1502СбН5
7.W
контактирует с соединением формулы HQ или MetQ, где Met. - Na, К или. Li, в среде растворителя при 20-157°С и дебензилируют полученное промежуточное соединение формулы III
COQ
ОСН2СбН5
5
0
5
0
5
СНз
ZW
гидрогенолизом в присутствии паллади- евого катализатора в среде органического растворителя.
Приоритетпо признакам:
16.03.82при М - атом кислорода или СН2 группа; Q - COQ,-группа, где Q - NRjR,, где R и R, - атом водорода или С -С -алкил или HCORj, где R, - атом водорода, С,- ; -алкил или бензил; Z - алкилен ,; W - атом водорода или фенил.
13.01.83при М - атом кислорода или СН группа; Q - COQ-группа, где Q - NHOH, CHjCN, NHAr,NR2R3, NHCOR, где R, - атом водорода, С,-С4-алкш1, фенил, бензнл или фенилэтил; R и R, - атом водорода или С,-С -алкил; Аг - выбран из группы
оВ.
или
5 Г
К Z - алкилен ,; W - атом водорода или фенил.
й
о
к








Изобретение касается бицикли- ческих соединений бензопиранового или тетралинового ряда, в частности веществ общей формулы (OHVC-CRtQ)-CH2 W-Z-C СН- С- Н- С-СЩ CH CH-CH CH-CH lvi CH-Nx, /Шо CH-S- . R/t-t- /S CH-f-T I C-CH2.-0 Й,-Н,СНз которые как обладающие обезболивающим действием могут быть использованы в медицине. Для выявления физиологической активности веществ бензопиранового или тетралинового ряда получены новые вещества I. Процесс ведут превращением группы при Б-угле- родном атоме: -C(0)-n-N02-CgH5- C(0)Q, и далее группы при А-углеродном атоме: -OH-CH2-CgH5., причем первое превращение ведут с использованием HQ, или Мет Q,, где Мет - Na, К, Li, в среде растворителя при 20-157 с, а второе - гидрогенолизом в присутствии Pd-катализатора в среде растворителя. Вещества (I) представляют собой в основном кристаллические продукты с т.пл. 143-240°С, но некоторые маслянистые жидкости. Обезболивающее действие веществ I лучше, чем известных аналогов. Также наблюдается противорвотный и противопоносный эффект. 6 табл. g СО :о У1 9д DO CM
| Вейганд-Хильгетаг | |||
| Методы эксперимента в органической химии | |||
| М.: Химия, 1965, с | |||
| ПРИСПОСОБЛЕНИЕ ДЛЯ АВТОМАТИЧЕСКОЙ БОКОВОЙ СТАБИЛИЗАЦИИ | 1921 |
|
SU445A1 |
