Изобретение относится к способам получения новых химических соединений т моно-окса-бензоацетамидных соединений или их метансульфонатной или -бензамидных соединений, обладающих анальгетическим действием и некоторым успокаивающим действием на центральную нервную систему (ЦНС).
Цель изобретения - получение но- вых соединений, обладающих ценными знальгетическими свойствами.
Пример I. {+)-(5 d, 7d, 8ft) -3,А-Дихлор-М-метил-М- 7-( -пирроли- динил)-1-оксапиро(4,5) дец-8-ил бен- золацетамид.
Стадия А. 8-ГЗ-(1-Этоксиэтокси) пропил -1,4-диоксаспиро(4,5)декан-3- -ол.
В нагретую в печи двухлитровую колбу (трехгорлую, круглодонную), оснащенную питательной воронкой, термометром для низких температур, магнитной мешалкой, впуском для азота для положительного давления и емкостью для сыворотки, помещают 12,03 г (|,79 г-атом) лития в виде литиевой проволоки в 1/8 дюйма, содержащей 0,6% натрия и нарезанной на куски величиной около 1 дюйма, под потоком азота в 1 л сухого диэтилового эфира Применяют свежие емкости со свежим безводным этиловым эфиром без дополнительной сушки. Затем добавляют 161 г (0,76 моль) этил-3-бромпропил- ацетальдегидацетала следующим образом, добавляют порцию 25 мл (чистого бромида, регулируя температуру реакционной смеси, после достижения 28 С реакционную колбу погружают в охлади тельную ванну из сухого льда/тетра- хлорметана (около -20 с) , а скорость добавления остального бромида регулируют так, чтобы реакционная температура составляла от -5 до 0°С. По окончании добавления бромида реакционную смесь перемешивают примерно пр -20 С в течение 1 ч, затем подают с помощью шприца или .иглы в нагретую 6 печи трехгорлую трехлитровую круг- лодонную колбу, и извне охлаждают на водно-ледяной бане с целью получени и сохранения этоксиэтоксипропиллитие вого промежуточного продукта, в который по каплям добавляют раствор 5,8 (0,55 моль) 8-оксо-1,4-диоксаспиро (4,5)декана в 1 л сухого диэтилового эфира (из свежих емкостей) в течение приблизительно 2,5 ч. Полученная реакционная смег(, мед.менно нагренается до комиат1и1Й температуры, затем ее выливают в два литра ледяншч) полуна- сьш1еиного хлорис 1 ог(1 аммония н йодном растворе. Фачы отделяют, водную фазу зкстраг ируют 1 л диэтилоного эфира. Эфирные слои соединяют и промывают раствором рассола, сущат сульфатом натрия, затем удаляют эфирный растворитель при пониженном давлении и сушат при 45 с (0,25 мм рт.ст.) в течение 14 ч.
Получают 142,15 г декан-8-олацета- левого промежуточного продукта в виде бледно-желтого масла, частоты которого достаточно для использования его на следующей стадии. Небольшую,часть этого масла перегоняют при пониженном давлении, получая более чистый продукт ст.кип. 135-14CfC (0,01 ммрт.ст.) ЯМР-спектр (в хлороформе) соответствует .
Стадия Б. 1 ,4 , 9-Триоксаспиро (4,2,4,2)тетрадекан.
В раствор 142 г (0,52 моль) сырого декан-8-ол-ацеталя стадии А в i л метанола добавляют 50 г кислой смолы Dowex 50W-X8 (200-400 меш), затем смесь перемешивают при комнатной тем- пераутре 2,5 ч. Реакционную смесь фильтруют через целит, фильтрат упаривают, сушат при 55°С (О,25 мм рт.ст. в течение 16 ч.
Получают 108,1 г (выход 96%) сырого диолового промежуточного продукта, чистота которого достаточна для использования его на следующей стадии. ЯМР-спектр соответствует.
В раствор 108 г (0,5 моль) сырого диола, смешанного с 116,3 г (1,15 моль) триэгиламина в 2 л хлористого метилена, охлажденного до 0 С на ледяной бане, по каплям в течение 2 ч добавляют раствор 65,8 г (0,575 моль) метансульфонилхлорида. По завершении добавления реакционную смесь перемешивают 16 ч (без пополнения льдом), а затем один раз промывают водой и сущат сульфатом магния. Растворитель удаляют в вакууме. Сьфой остаток от продукта перегоняют при пониженном давлении.
Получают 77,6 г (выход 78%) без- цветной жидкости, которая кристаллизуется при выдержке при комнатной температуре, т.кип. 84-87°С ( 0,7 мм рт. ст. т. пл . С, ЯМР-спектр соответствует.
Стадия В. 1-Оксаспиро-(А,5)декан- -8-он.
В раствор 75,5 г (0,38 моль) ке- тальтетрадекана стадии Б в ДОО мл тетрагидрофурана добавляют 300 мл 5%- ный водный раствор хлорной кислоты, затем смесь нагревают 18 ч до . Полученную реакционную смесь охлаждают до комнатной температуры и рас- пределяют между 1,5 л насыщенного водного раствора бикарбоната натрия и 1 л диэтилового эфира. Жидкие фазы разделяют, водную фазу однократно экстрагируют этиловым эфиром. Эфирны фазы соединяют, сушат сульфатом магния после промывки раствором рассола Растворитель удаляют в вакууме.
Получают в качестве остатка 53,9 г (выход §2%) бесцветной жидкости, ко- торая содержит согласно газовой хроматографии 9% исходного кеталя. ЯМР- спектр соответствует, но показывает примеси кеталя при 3,93 ррт. Сьфой кетон/кеталь применяют для дапьнейше го превращения без дополнительной очистки.
Аликвоту этого сырого кетона/ке- таля перегоняют при пониженном давлении т.кип. 77-84°С (0,8 мм рт.ст.).
Стадия Г. 1-Оксаспиро (4,5)декан- -8-ОЛ-4-метилбензолсульфонат.
Во взвесь 4,7 г (0,116 моль) ли- тийалюминийгидрида в 600 мл сухого диэтилового эфира по каплям добавля- ют раствор 53,9 г (0,35 моль) 1-окса спиро (4,5) декан-8-она стадии В в 225 мл сухого этилового эфира так, чтобы сохранялась легкая обратная дефлегмация. По окончании добавления реакционную смесь перемешивают 0,5 ч при комнатной температуре и затем резко охлаждают, добавляя по каплям 10 мл этилацетата, затем 4,7 мл воды 4,7 мл 15%-ного водного раствора гид роокиси натрия и 14,1 мл воды. Полученную смесь фильтруют. Жмых промывают диэтиловым эфиром. Растворитель удаляют в вакууме.
Получают 52,3 г бледно-желтой жид кости (восстановленный кетон, сырой спирт), которую применяют без дополнительной очистки. ЯМР-спектр соответствует восстановленной кетоновой структуре.
В раствор 51,3 г (0,32 моль) сырого спиртового продукт а восстановления в 700 мл сухого пиридина, охлажденного до -17°С, добавляют раствор
69 г (0,36 моль) п-толуолсуфонилхло- рида в 400 мл сухого пиридина (раствор охлажден до ). Полученную смесь хранят 110 ч при -17°С. Массу пиридина удаляют в вакууме, остаток распределяют между этиловым эфиром и водой. Водную фазу дважды экстрагируют этиловым эфиром, объединенные жидкие этилэфирные фазы экстрагируют дважды ледяной 5%-ной водной хлористоводородной кислотой, один раз насыщенным водным бикарбонатом натрия, сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают 79 г (выход 80%) указанного соединения в виде твердого белого продукта.
Небольщую часть этого сульфата пе- рекристаллизовывают дважды из диэтилового эфира (т.пл. 82-84°с). ЯМР- спектр твердого продукта соответствует.
Вычислено, %: С 61,91; Н 7,14; S 10,33.
Си Наг 0,5
Найдено, %: С 61,64; Н 7,35; S 10,24.
Стадия Д. 1-Оксаспиро (4,5)дец- -7-ен.
Смесь 79,0 г (0,27 моль) тозилата стадии Г и 50 г (0,33 моль) ДБУ нагревают 8 ч до 100°С. Полученную реакционную смесь охлаждают до комнатной температуры и распределяют между 600 мл воды и 200 мл диэтилового эфира. Жидкие фазы разделяют, водную фазу дважды экстрагируют порциями по 200 мл этилового эфира, эфирные фазы соединяют, дважды промывают 10%-ной водной хлористоводородной кислотой, один раз насьщенным водным бикарбонатом натрия, один раз раствором расео- ла, сушат сульфатом натрия. Растворитель удаляют на паровой бане при атмосферном давлении через колонну Виг- ре в 12 дюймов. Полученный сьфой продукт перегоняют при пониженном давлении.
Получают 23,8 г (выход 64%) дец-7- -енового соединения, т.кип. 62-63 С (3,25 мм рт.ст.). ЯМР-cnekTp соответствует.
Вычислено, %: С 78,21; Н 10,21.
С, Найдено, %: С 77,80; Н 10,21.
Стадия Е. ()-(Гэ, З /з, б о/)-Ди- гидроспиро/фуран-2(ЗН), 3-(7)-окса- бицикло(4,1.0)гептан изомер Б, цис).
(-t-) - ( 1 а, 3 « , h n J -,. i 11 и; 1111 : 11 и p 1 ран-2 (ЗН) , J -(7) -.кг,-1пицик-р I , ,i) гептан (n:ioMep Л, ipaiii;) ,
В раствор 20,0 г (i),l 4) мм-п) 1--ь-, саспиро (4 , 5) дец-7-f H.I н . )) мп ;uixii;4 метана по к.шлям д(1Пап | ;к1 при -к рни и течение 2,5 ч jiacTHiip ), i
(0,16 моль) M-X. U)pIU lih4 nr)r Ч |
кислоты (80-90А Aldcich) н ЬОО -.i.- ; и-- ,,., хлорметана. Полученнун) р аьншчшум смесь 1 ч п рем1;МШ1иан1Т iiini комиатниц температуре, д)Г)анляь)т 100 ми
IO%- HOГC5 водного Гч I y/n.l}j,4T И КЛТрИИ.
По перемепшнании нол ученной pv aKiui ш- ,, ной смес .и н ч-шчии - 1 . гм(м:ь чг вает отрицате; Ы1|,Г| pt) учь г.гг njni тсгтг на крахмальный йод. I r а каць Ч1Н vid I Mc; дважды нром,|цаи1т HO|IHJIHMH н I чмпу - насыщенного nojiHuMi pn ii i ;ii I liiapoo- ната }1а-грия, цпрнч г,а i. х дааиш и вакууме нри 1о ---1М1Ном i . .ai(-)ia ; v;i: .
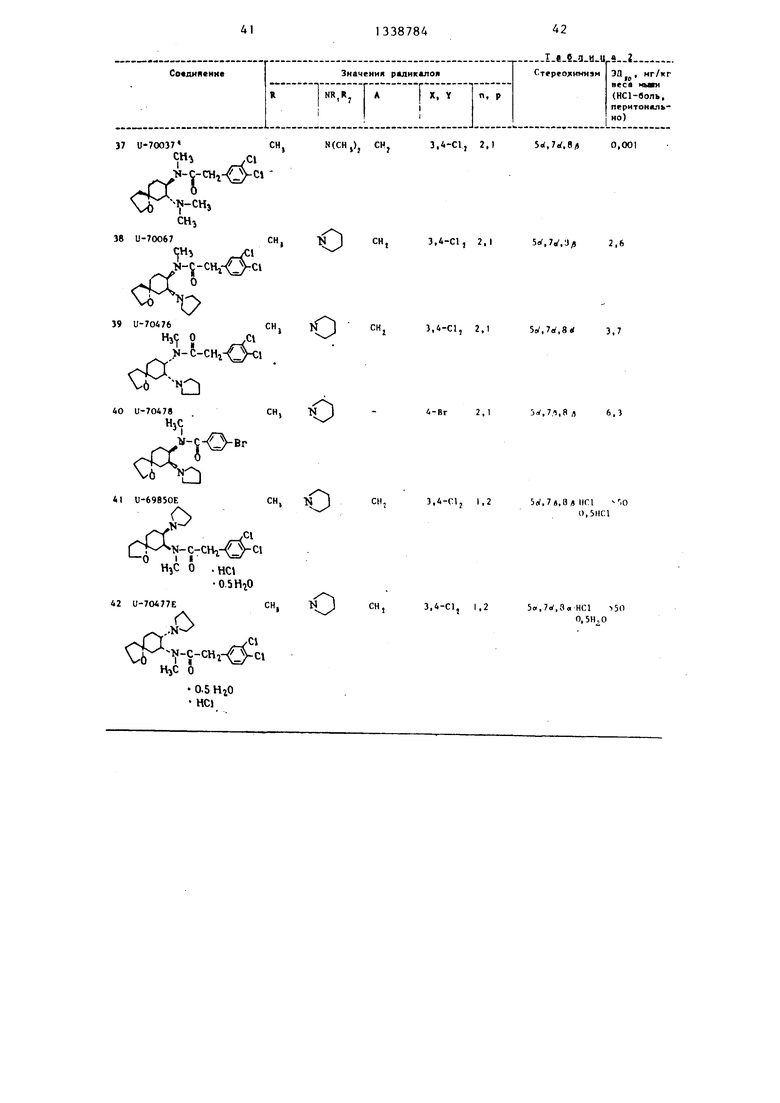
Получаиуг 21,0 i MaL nio-:-м:.гг а: .липкости., с: llONioiahl i ; а,чм и;;К T llion хроматографии уI/IM IN аомаю, что. :;
это смесь ичомсров Л и 1 соотно иен 1и ASrSS 1Т( шч . ГЖХ проводят на колонне ина 3/- SC-(i. Ичомер А ян.лиется тра не-Ч11ч- (а|д( м , а изомер Ii )поь 1:идог-1 го кгн(}1И гу1)а ii; НИИ на о ;ч1о1 /-}1ии .( 1 : а i ; ju } ;i/i- щения в ар Л 11 иа i HI IM i taiei:ХИМИЗМОМ.
I- I ,0 I aiai , o;i ..TUTII v;i;- ичяют, I lriai i. ; 11 r- .4i aim ч.лч vHiiiot x j, xMNni i Г КИ .. ;ч , I Ч 1 I: iiiv г a M . ; ь HOI очисть и.
1,0 I алик ); I Ы . ; -; i - . рафн p ;, ИЛ I 0 j ,a iiin-,a- г;:к i lO . ; .., .Ь-ы., 1 ,
ЧЛЫИрурГГ , Mall --; ia:.| . |1;--; I Mi : ,
ПТИ ; ;; i a ч h . .-i, i ,
Ilo -yaai : 0 v j -a-1- I a ; и ;: Л и 220 г aa ; . и .. ч : : . i ;а- . tii;e смг :i а а i: ;и : ;. i -: ш „
Ичомгр Л. ;li- I -;aii 1. 1 : :. i;. чч ; i-у- j, ет ,
(, ,, I i.i-n : а ,: ч а . .1,1 ; а 1 S/. , ; - 1и;.. . ,i; iH. .
Иа.ч-Ч Р К, -,ча Л- . , 1и 1 rv- ет,;.(
Для я : гпкли Нч t i . - па а
I 5. ,094.; найагна 1 i , :: - Н ,
Ста;;лч Ж. ( + )-(, , -1 - - ; Метил (ilje4iii:r -a л ал I ч-. И1-| i ; , а гни- г, г, о (4 , 5 ) Д1 :;-/ -н 1 I iai рро аи.ча и (i чомс) А j . ( + )- ( 5 , 7 , 8 О - - f 7- К- Л :. (фонпл- е гил) at-stiii 1 - 1 -.:;., 1 а i i;p il , i .,-Н- ил аи и ii (а -i. ,) J
I н I HI i;;. и 1 , (1 1 ( I l ) (анч и ниг- и ipaH -1-: 1 , I iHipi 1 I j ,)) )1-: } (},. i г I алии , 17, ч .: г
11 , i i ) t laiMH a (-ii : 11 a ) ами Ha и (i M: НОДЫ iiatpiaiai O I Я ч при П) С,. Ч1 чнуи Р оакнимннуin га-ичл выдерживают а ; X чаж;11ч ;ия JP кo нaтнoй ( РГ I и а(л:р1 д 10. -н|,м iuv4- Hi-i iai-THiip. a-i 1илрч окиси натрии и ;щ- :-;л, рм(; г а)1м ,1. Ф рачде.аяют. Водный (юй чкс ра I ир sail : один pa i /uixaop- Mi TanoN). Op I .-ашлес hiie жидкие слои .чче;щняы : , cvniai гvJn.itia roM магния. 1 агт1 о|пгу laib -д1чянгг в п.акууме. Полу- а 1ан1ый 1 Ы1)ой -ь.идкиГ остаток HI pei o- 1ИП1Г1 ajiii HoHn.iaiHoM давлении.
Иолучаьч 9,) 1 смеси аминогниртов нил1- , 4-iaiН1 | Ч-, 11 11 в 1чко1 о Mai- aa, I . ьпп . 1 )(-- 1Ь(; л,: (о ,1) мм 1)т. ст. ) .
И раствор 9,1) ) ( 1ч ммо.Л)) амино- laaipioBoii (ч-кча и ,1 г ( 57, 5 -iMoJib) I ри 41 н I . S a.a /шхлормет ана , Х.(ЧН1(; го ас- ()(;, но кан.лям доОав- ач 1 Т расгв р ч, 1 г (37,3 ммод1л) ме- I анс у abijii Ч1илхаорид.- в )() мл днхдор- I i.ina. Но окончании добавления полу- ч, смесь перемешивают 1 ч при и ) 111-м ч.--да1 11 в р,ччде1П1те.11ьную i-;i i-.-ii 41 |-,у . 11- лу aiainyi-i (а-ичл один раз
IH .--ч п-мнчт Ц11 :ии, суша г сульфатом м,1Г- ;;а:1. Г,-,: ми-рн-п ль -. да.чяшт в } акууме ар Н I-,-: - -41 а 1 Ml 41 i ( мнг|1 а гур с ,
1Ь ауч1Н1 1 i, Н ; c .ii oro ме|аисульфо мной ,т.л --aiioro ч шдукт а (мечи.|,:;ь ai па- -л,ч-- i oi ipanaTMiaiioT
, I I I) ,.- . -41 1-1-1 аи| ро аи л.ина и
1 .. ; ;1лм. |м, v4i-;-4iy:H: i-vaa a. нагрева- и , Ii р- iiiMM Xi I а: ;ai-4 ьнн |. ii)M f)3 С | . ., : а ii-л очН чч: Л рр; ;л1;и на
-I I : а 1 ч , .HHoii pi-ак ци 4;Hiui ччц н ч р| I I а;:. Ч1Н; а-1 Hi-iiNipHoM аннараМ , : I а : . а-, ра- а; i а -л Hi i r между 10%- iii-a i-41 aioi : 11 apt , a-aiia.i.i на Г1)Ия и ди- ;: p -ii i iHo--. - - aiiiH nioi i чкстрагируют один p/;-; аи x l , . Op г-чничес КИ(: чщ . ч а-гаи i;-.-a-i . - -iiiai сульфатом маг- , Рас I HI 1|-.1| I I 11- а1аля11:1Т р, вакууме.
11олуч,на , 1 ,с гмчч м.асла. Г И мощы: 1 чи о;- а- I lHi ч | хром а ТО гр афии r;i ia a-4 a yi i aHi-njicHo/ наличие по г(-1|1- :ца --к PC i ч.иамино(ых изоме(-ыроа i:p civi-i хром.пчн рафируют на i Ь1 спаи I-, а; с аи (ч()-63 мкм. Мерк), 1 чI 1И 11 Iа-11 ч а -I а к а / ме та11o:i а / эт а- Hicia (О ,М : 7 , 7 : . ; п-ч оГгьсму). Фр.чкции, - , а р,-,аг:н I ai- ;-ii а i a-icpi i, :аи-диняют.
71338784
получая 2,1 г изомера Л 2,5 г изомера В и 1,5 г смеси изомеров А, Б и В.
Изомер А. ЯМР-спектр соответствует. Масс-спектральный анализ (т/е): 328 (М), 24Д (мЧНг - ССН СН,, СН ) , 166 (CHj)jN СНСН C(Cflj)jO), 160 ((CH3) CHCjHy).
Изомер В. ЯМР-спектр соответствует. Масс-спектральный анализ, т/е 328 (М ), 216 (CtHjCHjtUCH) СНСН C(CHj) 0), 10 (CHj СНСН N(CHj)jCHj).
Стадия 3: .( + )-(5я , 7, 8)-3,4-/biЯМР- и ИК-спектры соответствуют, Вычислено, %: С 62,11; Н 7,11; С1 16,67; N 6,59. С„Н,„С1,М,0,
Найдено, %: С 62,06; Н 7,19; С1 16,58; N 6,39.
Рентгеновское определение кристаллической структуры соединения подверж- 10 дают регио- и стереохимизм.
Моногидробромид соединения получают путем добавления диэтилового эфира бромистоводородной кислоты в свободное основание в диэтиловом эфире. Поэтиловым эфиром, получая бедые микрокристаллы (с т.пл. 209-210°С) моно- г-идробромида указанного соединения.
Вычислено, %: С 52,18; Н 6,17; N 5,53; С1 14,00; Вг 15,78.
C,,,11,,N,0,C1,B
Найдено, %: С 51,91; Н 6,20; N 5,51; С1 13,92; Вг 15,69.
Соединение по примеру 1, стадия 3, Зе получают следующим образом; эпок- сидирование 1-оксаспиро(4,5)дец-7-ена осуществляют с-применением м-хлорпе- роксибензойной кислоты в диэтиловом
хлор-М-метил-М- 7-(1-пирролидинил)- 15 лученный осадок собирают и перекрис-1-оксаспиро(4,5)дец-8-ил бензолацет- таллизовывают из смеси метанола с диамид.
В раствор 2,0 г (6,1 ммоль) изоме-
ра А стадии Ж в 100 мл абсолютного
этанола добавляют 2,0 г 10%-ного пал- 20
ладия на угле, смесь встряхивают на
аппарате Парра при давлении водорода
35 атм с целью удаления N-бензиловой
защитной группы. После прекраш,ения
поглощения водорода реакционную смесь 25
с|ильтруют через целит, затем целит
промывают с применением нескольких
порций этанола, фильтрат выпаривают
в вакууме и сушат в глубоком вакууме.
Получают 1,3 г дебензилированного зо 3( в качестве растворителя, полуаминового промежуточного продукта чая смесь эпоксидов, которыми обогаЫ-(5с, , 8)-М-метил-7-(1-пирро- щен Г о, З /з, б а )-изомер; эту смесь
лидинил)-1-оксаспиро(4,5)декан-8-ами- эпоксидов подвергают взаимодействию
на в виде бесцветного масла.с пирролидином, получая пирролидиниВ раствор 0,75 г (362 ммоль) 3,4- 35 спирт, который превращают в ме-дихлорфенилуксусной кислоты в 25 мл зилат, который затем подвергают взасухого тетрагидрофурана добавляют (в имодействию с бензил(метил)амином; повиде одной порции) 0,59 г 3,62 ммоль лученную диаминовуюсмесь дебензилируют
1, -карбонилдиимидазола, полученную и ацилируют; продукт очищают перекрисмесь перемешивают 1 ч при комнатной 40 сталлизацией из ацетонитрила,
температуре. Затем по каплям добавля- Пример 2, ( + )-5е/, 7а/, 8/9ют раствор 0,75 г (3,15 ммоля) дебензилированного амина в 20 мл сухого
ТГФ. Полученную смесь перемешивают
18 ч при комнатной температуре. Массу 45
ТГФ удаляют в ротационном испарителе.
Остаток распределяют между этиловым
эфиром и водой. Жидкие фазы разделяют.
Эфирную жидкую фазу промывают один
раз водой, один раз раствором рассо- 50 ( ммоль) 4-бромбензоилхлорида в
ла, сушат сульфатом магния. Раствори- 25 мл сухого диэтилового эфира. После
окончания добавления реакционную смесь перемешивают при комнатной температуре -3 ч, затем распределяют меж- 55 ДУ водой и этилацетатом. Фазы отделяют, органическую фазу промывают один раз водой, один раз раствором рассола, сушат сульфатом магния, растворитель удаляют в вакууме.
-4-Бpoм-N-мeтил-N- 7-(1-пирролидннил)- -1-оксаспиро(4,5)дец-8-ил бензамид.
В раствор 0,54 г (2,28 ммоль) де- бензилированного диамииа стадии 3 примера 1 и 0,28 г триэтиламина в 35 МП сухого диэтилового эфира по каплям добавляют раствор 0,55 г
тель удаляют-в вакууме.
Получают 1,25 г белого воскообразного твердого продукта.
Этот, сырой аминоамидный твердый продукт дважды кристаллизуют из аце- тонитрнла.
Получают 0,8 г (выход 54%) указанного соединения, т.пл. 105,5-106,5 С.
ЯМР- и ИК-спектры соответствуют, Вычислено, %: С 62,11; Н 7,11; С1 16,67; N 6,59. С„Н,„С1,М,0,
Найдено, %: С 62,06; Н 7,19; С1 16,58; N 6,39.
Рентгеновское определение кристаллической структуры соединения подверж- дают регио- и стереохимизм.
Моногидробромид соединения получают путем добавления диэтилового эфира бромистоводородной кислоты в свободное основание в диэтиловом эфире. Поэтиловым эфиром, получая бедые микрокристаллы (с т.пл. 209-210°С) моно- г-идробромида указанного соединения.
Вычислено, %: С 52,18; Н 6,17; N 5,53; С1 14,00; Вг 15,78.
C,,,11,,N,0,C1,B
Найдено, %: С 51,91; Н 6,20; N 5,51; С1 13,92; Вг 15,69.
Соединение по примеру 1, стадия 3, Зе получают следующим образом; эпок- сидирование 1-оксаспиро(4,5)дец-7-ена осуществляют с-применением м-хлорпе- роксибензойной кислоты в диэтиловом
Пример 2, ( + )-5е/, 7а/, 8/9( ммоль) 4-бромбензоилхлорида в
-4-Бpoм-N-мeтил-N- 7-(1-пирролидннил)- -1-оксаспиро(4,5)дец-8-ил бензамид.
В раствор 0,54 г (2,28 ммоль) де- бензилированного диамииа стадии 3 примера 1 и 0,28 г триэтиламина в 35 МП сухого диэтилового эфира по каплям добавляют раствор 0,55 г
9I
Получают 1,0 I белого порошка, который перекристаллизовывают игз ацето нитрила, получая 0,7 г (выход 74%) названного аминоамнда в виде белых кристаллов, т.пл. IBl-lBS C (с разл ЯМР-, ИК- и масс-спектры соответствуют.
Вычислено, %: С 59,85; Н 6,94; N 6,65; Вг 18,96.
С,, Hj, BrNjO
Найдено, %: С 59,77; Н 7,00; N 6,69; Вг 19,10.
Пример 3. (±)-(5.i. Id, 9, -3,4-Диxлop-N-мeтил-N- 8-(1-пирроли- динил)-1-оксаспиро(4,5)дец-7-ил беи- золацетамид и его моногидрохлоридмета ноловын сольват.
В раствор 2,4 г (7,31 ммоль) диамина (изомер В, стадия Ж примера l) в 100 МП абсолютного этанола добавля ют 2,4 г 10%-ного палладия на угла. Полученную смесь встряхивают на аппарате Парра при 35 psig давления водорода с целью дебензилирования амина. После окончания поглощения водорода полученную реакционную смесь фильтруют. Жмых тщательно промывают абсолют ным этанолом. Фильтрат выпаривают в вакууме.
Получают 1 ,6 г дебензилированного амина (изомер А) в виде бесцветного масла.
В раствор 0,8 г (3,86 ммоль) 3,4- дихлорфенилуксусной кислоты в сухом ТГФ добавляют 0,63 г (3,86 ммоль) 1 , -карбонилдиимидазола. Полученную смесь перемешивают при комнатной температуре 1 ч. Добавляют по каплям раствор 0,8 г (3,36 ммоль) дебензи- лированного диамина (изомер А) в 20 мл сухого ТГФ. Полученный раствор перемешивают при комнатной температуре 18 ч. ТГФ удаляют на ротационном испарителе. Остаток распределяют между этиловым эфиром и водой. Жидкие фазы разделяют, эфирную фазу концентрируют в вакууме.
Получают 1,3 г сырого названного продукта в виде масла.
Полученный таким образом эфирный раствор сырого продукта обрабатывают эфирной хлористоводородной кислотой. Полученную гидрохлоридную соль амино амидного осадка собирают и дважды пе рекристаллизонывают из метанола/этил ацетата (50:50 по объему).
Получают 0,6 г указанной аминоами ной гидрохлоридной соли, т.пл. 203 - 210 С.
.5 N ;,
25
10
15
О 3540 45
50 55
30
Вычислено, N 6,8Ь; CI 22,8,
С,Н,„ С1, N,0,
1а11ден(5, ,,96; С1 22,99.
Прим е р 4. ()-(, Ь d, 7/3)- -3, 4-/ljix.nop-N-MeTH i-N- /- ( 1 -пирроли- диш)л ) - 1 -оксаспиро (4 , 5 ) Д(. ц-6-ил Оен- золацетамид.
Стл;и1я Л. I - 3- ( I-Этокситгокси ) пропил щклогекс,1Н- 1 -ола ,
В )1агретую н печи 3-горловую круг- лодонную колбу емкостью н 250 мл, ос- нащен)1ун) магнитной мешалкой, ничкотем- пературным термометром, перегородкой и уравниваюпич длн.чение тгитающей воронкой, соединеннее с источником азота, помещают 2,0 г (0,3 моль) .питиевой проволоки диаметром в 1/8 дюйма с содержанием 0,6% натрия, нарезанной на куски в потоке азота и 125 мл безводного диэтилового эфира. Затем при перемешивании по каплям добавляют около 3 мл этил-3-бромпропилацетальдегида в атмосфере азота. Когда температура реак1-1ионной смеси достигает и литиевая проволока становится блестящей реакционную смесь охлаждают до -5 С в бане из смеси сухого льда/тетрахлор- метана. Температуру смеси выдерживают от 5 и 04; путем добавления по каплям остатка 30,2 г (0,14 моль) этил-3- -бромпропилаце ал1)Де1 идацеталя. После окончания добавления полученную смесь перемешивают при температуре около -78°С помощью сухого, льда/тетрахлор- метана 2 ч, понерхность остаточной ли- тевой проволоки становится тусклой.
Литий органическое промежуточное соединение, полученное в ходе этой реакции, П1)дают с помощью в нагретую в печи круглодонную колбу и охлаждают до на водно-ледяной бане. Питающую воронку загружают 9,95 г (0,10 моль) 2и 1клогексен-1-она и 1 00 мл безвод- Hoi o этилового эфира. В холодное перемешиваемое литийорганическое промежуточное соединение по каплям добавляют циклогексановый раствор в атмосфере азота в течение 1,5 ч. Полученную смесь перемешивают 1 ч при 0°С, затем ей дают нагреться до комнатной температуры медленно путем перемешивания в течение 18 ч без пополнения бани льдом. Реакционную смесь выливают н 250 мл ледяного полунасыщенного ВОД1ГОГО раствора хлористого аммония. Жидкие фазы разделяют. Водную фаЗУ днажды экстрагируют порциями по 200 MJ1 диэтилового эфира. Соедлпенные жидкие эфирные фазы промывают раствором рассола, сушат сульфатом нагрия и концентрируют в вакууме.
Получают 21,4 г (выход 93:) промежуточного циклогекс-2-ен- -оловсто продукта в виде бесцнетног о масла, которое используют без обработки на следующей стадии. ЯМГ-спектр соответствует .
Ста;шя Б. I-Оксасгшро(4 ,5) бец-6- -ена.
Смесь 21 г (0,10 моль) ацеталя,
полученного на стадии Л, 10 г катион- обменноГ смолы Dowex 50W-X8 (200 - 400 меш) и 200 мл MeiaHojia перемешивают 4 ч при комнатной температуре для обеспече(шя полноты реакг ии , за- тем фильтруют через целит, который промывают дважды 50 мл метанола. Растворитель соединенных фильтрата и промывочной жидкости удаляют н вакууме. Получают в качестве остатка 7,2 г (выход 60Z) указанного -дец-6- -ена iiocjTe перегонки, т.кип. 90 - 96°С (25 мм рт.ст.). ЯМР-спектр соответствует этому соединен 1ю.
Масс-спектральны; 1 анализ т/е 138 (М).
Стадия В. (jb)-(l c/, 2 Л, б о/)-Ди- гидроспиро/фуран-2-(ЗН)-2 -(7)окса- бицикло (4,1,0).
( + )-( о/, 2V, 6 о/)-Дигидроспиро/ /фуран-2(5Н)-2 -(7)-оксабицикло (4,I,0)гептана (изомер А перемещается быстрее, изомер Б перемещается медленнее
В перемешиваемую порцию 36 г
(0,094 моль) 1-оксаспиро(4,5)дец-6- -ена, 150 мл этиленхлорида добавляют по каплям 23 г 80-90%-ной м-хлорпер- бензойной кислоты в 500 мл Хлористого метилена в течение 2 ч. Реакционную смесь перемешивают при комнатной температуре 18ч. В полученный раство .цобавляют 180 мл 10%-ного водного раствора бисульфита натрия (по объему), после чего полученную двухфазную сие- тему (жидкую) быстро перемешивают 1 ч до получения отрицательного результата в опыте на крахмал/йодид. Затем медленно добавляют в смесь 5 г бикарбоната натрия в виде нескольких пор- ций при перемешивании. Полученную смесь дважды промывают полунасыщенным раствором бикарбоната натрия, один раз насыщенным раствором хлористого
натрии и суша г сульфатом натрия (карбонатом калия. Растворитель удаляют. Остаток хроматографируют на силика- гс ;е, элюируя с применением гексан- I илацетатной смеси (1:I по объему).
Получают 9,5 г более быстро перемещающегося эпоксида (изомера А) и 4,64 г более медленно перемещающегося эпоксида (изомера В). ЯМР-спектры каждого изомера соответствуют, но относительный стереохимизм не определен. Масс-спектр каждого из изомеров А и Б т/е 154 (м ) .
Стадия Г. (:|)-(5f, 60;, 7/5) - Метил(фенилметил)-амино -1-оксаспи- ро(4,5)дец-7-ил пирролидина.
Смесь 3,65 г (0,024 моль) 5,6-эпок си-1-оксаспиро(4,5)декана (изомер А), 4,37 г (0,36 моль) бензил(метил)амина и 2 мл воды нагревают 18 ч при 90 С до завершения реакции. Реакционную смесь охлаждают на ледяной бане до , после чего добавляют при перемешивании 4 мл 25%-ного раствора гиД)- (1КИСИ натрия. Полученную основную смесь дважды экстрагируют 100 мл хлористого метилена. Соединенные жидкие органические фазы сушат сульфатом магния. Удалением растворителя в вакууме получают 4,9 г промежуточного аминовс го спирта ( + )-(5, 6о/, 7/з)-7- метил (фенилметилТамино -1-оксаспиро(4,5) цекан-6-ол.
ЯМР- и масс-спектры соответствуют, Полученный таким образом промежуточный аминовый спирт, 1,98 г (0,018 моль) триэтиламина и 60 мл хлористого метилена помещают в нагре- туют в печи круглодонную колбу емкостью 250 мл, оснащенную магнитной мешалкой и уравнивающей давление питающей воронкой, соединенной с источником азота. В эту воронку загружают 2,02 г (0,018 моль) хлористого метилена. Систему очищают азотом и охлаждают до 0°С на ледяной бане. Метан- сульфонилхлоридный (мезилхлоридный) раствор по каплям добавляют в течение 1 ч в перемешиваемую аминоспиртовую реакционную смесь. Полученную смесь после добавления перемешивают при еще 1 ч с целью обеспечения полноты реакции. Реакционную жидкую фазу отделяют, сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают остаток в 5,6 г сырого мезилатного промежуточного продукта.
13
К полученному таким обр.гном сырому меэилату добавляют 20 мл iiupptuin/uina и 6 мл водм. Смесь н.чгренают с riL pi-- мешиванием при 90 С в течение 18 ч с целью обеспечения полноты реакции. Избыток пирролидина уд.чляют в вакууме полученный сырой остаток распределяют между 100 мл хлористого метилена и 100 мл 10%-ного раствора гидроокиси натрия. Водную фазу один раз экстрагируют 50 мл хлористого метилена. Соединенные органические фазы сушат сульфатом магния. Растворитель удаляют в вакууме .
Получают 5,Ь г (выход 95%) пирроли диндиаминового произнодного после перекачки в глубоком вакууме (0,1 мм рт.ст.) в Te4eittie 20 ч. Диами
новое производное применяют в таком виде на следующей стадии.ЯМР- и масс- спектры соответствуют.
Стадия Д. (+ )-(3, 6о(, 7/з) -Метиламино-1-оксаспиро(4,5)дец-7-ил1 пирролидина.
Смесь 5,3 г (0,016 моль) 6-бензил (метил )-амино-7-пирроли;у1 НИЛ- -окса- спиро(А,5)декана стадии Г, 5 г 10%-но го палладия на угле и 150 мл абсолютного этанола гидрируют при АО psig в течение 4 дней в аппарате Парра с целью обеспечения полноты реакгщи.
Гидрированную смесь фильтруют через целит. Жмых дважды промывают 50 мл абсолютного этанола. Удалением растворителя в вакууме получают 3,11 г (выход 82%) промежуточного пирролиди- нилдиамина в виде бесцветного масла, которое применяют на следующей стадии ЯМР- и масс-спектры образцов этого ди- аминового масла соответствуют.
Стадия Е. (j)-(5, 60/, 7/})-3,4-Ди- хлор-М-метйл-М- 7-(1-1и рролидинил)- 1 - -оксаспиро(4,5)дец-6-ил бензоацетамид
I
В нагретую в печи круглодонную кол-45 ( ) и лево (-) вращающих энантиомеров
отдельных соединений по примеру 1. Получение ( + )-(5d, во1, 8|%)-3,4- -Дихлор-М-метил-К- 7(1-пирролиди- нил)- 1-оксаспиро(4,5)дец-8-ил бензо- (-)-(5с/, 7о/, 8)-3,4-ди- xлop-N-мeтил-N- 7-(1-пирролидинил)- - 1-оксаспиро(4,5)дец-8-ил бензолацет- амида.
В раствор 10,0 г (23,5 ммоль) 1,13 г (7,0 ммоль) I, l -карбонилдиими- бензолацетамида, полученного по при- дазола в течение 15 мин. Затем промы- меру 1, в 250 мл метанола добавляют вают азотом и получе гную смесь пере-9,1 г (23,5 ммолтО дц-п-толуолправомешивают 1 ч при ком)1атной температу-вращающей винной кислоты. Полученную
ре.смесь выпаривсШтт досуха в вакууме.
бу емкостью 50 мл, оснащенную магнитной мешалкой и питательной воронкой для уравновещивания давления, подключенной к источнику азота, помещают
Г,44 г (7,0 ммоль) 3,4-дихлорфенилук- 50 ацетамида сусной кислоты и 15 мл ТГФ (высушенного над молекулярными ситами ЗА). В перемешиваемую смесь вводят три порции в виде твердого продукта
5
, ю 15
- 20
30
Г)87ЯД1/,
R ак 1 инщюн.и nbiii таким («Пра и м paci Flop ),4-;и1х.( Ни.чукс;усН(Ч1 кислоты по канлям доОаиляют pai i пор 1,5 г, (h , } з ммоль) Ь- .11П11Л-лмино-7-( I-пир- ролиллпил)-1-оксасниро-(4,5)декана, полученного на стадии Д, в 10 мл су- Х1)го ТГФ « тече Ше 20 мин. После окончания добавления реакционную смесь 3 ч перемешивают при комнатной температуре с цел1)Ю обеспечения полноты реакции. ТГФ удаляют и вакууме, остаток распределяют между 100 мл дц- этилового эфира и 100 мл воды. Эфирную фазу промывают 50 мл раствора рассола, сушат сульфатом магния. Растворитель удаляют в вакууме хроматог ра- фией остатка на си;шкагеле с элюиро- ванием 2,5%-ным метлнолом (содержащим 10% аммиака/этилацетата).
Получают 2,1 г (выход 78%) гтродук- та.
Полученнын таким образом эфирный раствор свободного основания обраба- 5 тывают диэтилэфировым раствором хлористого водорода. Полученную в виде осадка аминогидрохлоридную соль пе- рекристаллизовывают из смеси метано- ла/диэтилового эфира (1:1 по объему), получая аминовь продукт в виде гид- рохлоридной соли, т . пл . 248-251°с. ЯМР- и масс-спектры соответствуют.
Вычислено, %: С 56,12; Н 6,85; 5,93; CI 22,59.
21 С1 -О
Найдено, % : С 56,30; Н 6,85; 6,15; С1 22,20.
Соединение, гтолученное по этому примеру, является отдельным единичным эппмером с неизвестным стереохи35
N
N
40
мизмом при С,, /1ругой Cj-эпимер име- 135-Г37 С и приведен в приет т.пл.
мере 14.
При
ер 5. Разделение право
Tii()i;iJiiii OejiMii продукт растиоряют в мл luiMiUibHOM nfibt Me кипящего метанола. Добавляют диэтилопый эфир в ко;1ичест- пе, достаточном для обеспечения легкого помут}1ения смеси. Слегка помутневшему раствору дают 18 ч стоять при комнатной температуре. Полученные кристаллы собирают.
Получают 9,8 г белог о кристалли- ческого продукта, который дважды пе- рекристаллияовывают из смеси (I:1 по объему) метанола/диэтилового эфира, получая 5,0 г соли. Эту правовращающую соль винной кислоты распределяют между диэтиловым эфиром и 10%-ным водным раствором гидроокиси натрия, полученные жидкие фазы разделяют. Эфирную фазу промывают рассолом, су- щат сульфатом магния. Растворитель удаляют.
Получают 3,0 г белого твердого продукта, который перекристаллигютзы- вают из диэтилового эфира/гексана (1:1 по объему), получая 2,3 г в основном чистого левовращающего (-)- изомера, т.пл. 106-107,5°С; (с/) -6,136; (rf)43t -1 ,38 (С 1,13, в этилацетате). ЯМР-спектр соответствует.
Вычислено, %: С 62,11; Н 7,11; N 6,59; С1 16,67
C..G,
Найдено, %: С 61 ,92; 1 7, 16; N 6,42; Ci 16,83.
Маточные растворы от кристаллизации выпаривают досуха в вакууме. Остаток распределяют между 10%-ным водным раствором гидроокиси натрия и диэтиловым эфиром. Жидкие фазы разделяют. Эфирную фазу промывают раствором рассола, сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают 4,1 г (+) свободного основания в виде белого твердого продукта, который растворяют в 150 мл метанола, в который добавляют 3,9 г (9,6 ммоль) ди-г-толуол-1-винной кислоты. Полученный раствор выпаривают досуха в вакууме. Полученный твердый остаток нерекристаллизовывают из смеси метанола/диэтилового эфира (1:1 по объему) три раза, получая 3,2 г соли Обработав эту левовращающую соль винной кислоты так же, как и ее правовращающую сЬпъ получают 2,0 г свободного левовращающего (-) основания, ко которое перекристаллизовывают из ди878416
этилового эфира/1-ексана (1:1 по объему) .
Получают 1,35 г левовращающего (-) н-юмерного свободного основания, .пл. 106.5-107, (с/) +6,18; -t-14,51. HNff-cneKTp соответст- ьует правовращающему (+) изомеру.
Вычислено, %: С 62,11; Н 7,11; N 6,59; С1 16,67
0
C,,H,CI,NjO,
N
Найдено, %: С 62,02; Н 7,17; 7,17; С1 16,60. Пример 6. Общий способ аце5 лирования (+)-(, 7о1, 8р)-М-метил- -7-(1-пирролидинил)-1-оксаспиро(4,5) декан-8-амина
Получение ()-(5o/, 7 d, 8p)-N- -метил-N- 7-(1-пирролидинил)-1-окса0 спиро(4,5)дец-8-ил бензолацетамида. В высушенный раствор, оснащенный смесительным устройством и питающий воронкой для уравновешивания давления, соединенной с источником азота,
5 помещают 0,48 г (3,5 ммоль) фенилук- сусной кислоты (или ее эквивалента выбранной замещенной в кольце фенил- уксусной, бензойной или фенилалкано- вой кислоты) и 5 мл ТГФ (высушенного
0 с применением молекулярных сит ЗА ). В перемешиваемую смесь добавляют в две порции в виде твердого продукта 0,56 г (3,5 ммоль) 1,1 -карбонилдиими- дазола. Смесь перемешивают 45 мин
с при комнатной температуре. В полученную перемешиваемую активированную кислотную смесь по каплям добавляют 0,75 г (3,15 ммоль) выбранного диамина, например декан-8-амина примера 1,
Q-стадия 3, Зе в 10 мл сухого ТГФ в течение 10 мин. Полученную смесь 18 ч перемешивают при комнатной температуре до полного завершения реакции. ТГФ удаляют в вакууме. Остаток распределяg ют между 30 мл этилацетата и 20 мл воды. Органическую жидкую фазу отделяют, сушат сульфатом магния, растворитель удаляют в вакууме. Получают ацилиро- ванный продукт, например (+)-(5о, 7о, 8 ft) -N-метил- 7-(1-пирролидинил)-1-ок- саспиро(4,5)дец-8-ил бенэолацетамид в виде порошка, который перекристаллизовывают, например, из ацетонитрила, получая бензолацетамид или бензамид,
е например, 800 мг (выход 71%) указанного аминоамида с т.пл. 123-125°С.
Пример 7. Получение 1-окса- спиро(4,5)дец-7-ена согласно стадии Д примера 1.
0
17,
В нагретую в печи трехг орлую круглодонную колбу, оснащенную двумя конденсаторами типа Dewar, покрытой стеклом магнитной мегиллкой и положительным впуском для аяота, конденсируют 1,75 л безводного аммиака (колбу погружают в баню из сухого льда/изо- пропанола во время конденсации аммиака) . Затем добавляют раствор 136.19 г (1 моль) 3-фенилпропанола в 200 мл сухого диэтилового эфира (охлажденного до и затем раствор 107,3 г (2,33 моль) абсолютного этанола в 200 мл диэтилового эфира (охлажденного до -78 с). В этот раствор в виде небольших порций добавляют в течение 2 ч 69,0 г (3 г-атом) металлического натрия в виде кусков (не содержащего минерального масла за счет промывки пентаном). Первые куски вызывают бурную экзотермическую реакцию, так что скорость добавления следует тщательно регулировать. По окончании добавления темно синюю реакционную смесь перемешивают без пополнения хладагентом в течение 18 ч.
Колбу, содержащую белый твердый продукт, охлаждают на водно-ледяной бане, затем добавляют 800 мл ледяной воды и затем 800 мл диэтилового эфира По растворении всего твердого продукта реакционную смесь подают в разделительную воронку. Фазы разделяют. Эфирную фазу трижды промывают 800 мл и один раз раствором рассола и сушат сульфитом магния. Растворитель удаляют.
Получают 143 г сырого 3-(1,4-цикло гексадиенил)-пропан-1-ола, который разделяют на две порции. Каждую по отдельности обрабатывают 2,5 г п-то- луолсульфоновой кислотой. Давление снижают до 12 мм.рт.ст., емкость погружают в масляную ванну, нагретую до 115 С. После примерно 3 ч начинается перегонка через короткую колонку Виг ре с вакуумными двойными стенками при пониженном давлении и с постоянной скоростью во время продолжения циклизации.
Получают 105,4 г (выход 76%) 1-ок саспиро(4,5)дец-7-ена в виде бесцветной жидкости, т.кип. 79-81°С (12 мм рт.ст.). ЯМР-спектр идентичен спектру соедине ия по примеру 1 , стадия Д.
П р и м е р 8. ( + )-(5о(, 7с/, 8/5)- -4-XJIop-N-мeтил-N- 7-Tl -пирролидинил)
i878418
- I -оксаспир ( 4 , ) ;и ii-8-M/ij (п ИчшыцетЛналогично прим(.ч)у 6, но испольчуя 4-хлорфенилуксуг11ую кислоту, получают аминоамид с т.кчи. 109-1lO C, выход
89%.
Вычисленс), 7,: С 67,59; 11 7,99; N 7,17; С1 9,07. C,,H,,CINjO.
Найдено, С 67,48; П 7,98; N 7,05; С1 9,11.
Пример 9. (f)-(5o/, 7а , 8/})- -4-BpoM-N-MeTHji-N- 7- (1 -пирролидинил) - 5 -|-оксаспиро(4, 5) Д1 ц-8-ил бeнзo.тIaцeт- aмид.
Аналогично примеру 6, но испол1)зуя 4-бромфенилуксусную кислоту, получают аминоамид с т.пл. 120-122 С, выход 0 93%.
Вычисле)1о, %: С 60,69; И 7.18; N 6,43; Вг 18,35
C,,H3,BrN,0,
Найдено, С 60 , 7 I ; И 7,16; N 6,35; Вг 18,31.
Пример 10. ( + )-(5d, 7с, 8 р)- -4-Метокси-М-метил-М- 7-(1-пирролидинил) - 1-оксаспиро(4,5)дец-8-ил бензол- ацетамид.
Аналогично примеру 6, но используя 5-метоксифенилуксусную кислоту получают аминоамид с т.пл. 110-111 С, выход 73%.
Вычислено, %: С 71,47; Н 8,87;
N 7,25.
С„Нз,М,Оз
Найдено, %: С 71,26; Н 8,97; N 7,J7.
Пример 11. { + )-{5ci, 7, 8)-N, 4-Диметил-М- 7-(1-пирролидинил)- - 1 -оксаспиро (4, 5)-дец-8-ил -бензолацет- амид.
Аналогично примеру 6, но используя 4-метилфенилуксусную кислоту, получают аминоамид, с, т.пл. 95-97°С, выход 74%.
Вычислено, %; С 74,55; Н 9,25; N 7,56.
Найдено, %: С 74,66; Н 9,22.
N 7,68.
Пример 12. ( + )-(5с/. Id, 8/4)- -З-Хлор-М-метил-М- 7-Т 1-пирролидинил)- -1-оксаспиро(4,5)дец-8-ил бензолацет- амид.
Аналогично примеру 6, но используя 3-хлорукСусную кислоту, получают аминоамид с т.пл. 91-92°С, выход 70%.
Вычислено, %: С f-7,59; И 7,99; N 7,17; С1 9.07.
,, N,0,C1
Найдено, %: С 67,11; Н 7,98; N 7,07; С1 8,98.
Пример 13. ()-(5а , 7о(, 8л)- -М-Метил-А-нитро-М- 7-(1-пирролиди- нил)-1-пксаспиро(А,5)дец-8-ил бензол- ацетамид,
Аналогично примеру 6, но используя 4-нитрофе1шлуксусную кислоту получают аминоамидное соединение (хлоргидрат, гидрат) с т.пл. ISe-fSO C, выход 71%.
Вычислено, %: С 60,33; Н 7,36; N 9,60; С1 8,06.
Cj.Hj.NjO.-HCl- HjO
Найдено, %: С 59,86; Н 7,27; N 9,61; С1 Я,10.
Пример 14. ( + )-(5, (id, 7/}) -3,4-Дихлор-М-метил-М- 7-(1-пирроли- динил)-1-оксаспиро(4,5)дец-6-ил бен- золацетамид.
Подвергая взаимодействию 6,7-эпок- си-1-оксаспиро(4,5)декановый изомер Б по примеру 4, стадия В, как описано для изомера А в примере 4, стадии Г. Д и Е, получают соединение с т.пл 135-137°С, которое является спироэпи- мером соединения, полученного по примеру 4, стадия Е.
Вычислено, %: С 62,11; Н 7,11; N 6,59; CI 16,67.
Ч,Нз„М,С1,0,
Найдено, %: С 62,06; Н 6,54; N 6,54; С1 16,60.
Пример 15. ( + )-(5с/. Id, -3,4-Дихлор-М-метил-М-Т8- 1-пирроли- динил -1-оксаспиро(4,5)дeц-7-ил бeн- зoлaцeтaмид и его многогидробромид -хлороформ-сольват.
Стадия А. ( + )-(5., 7о, 8;9) -(Метиламино)-Т-оксаспиро(4,5)дец-8- ил пирролидин (изомер А).
((5d, 7/i, 8о/)(Метилами- iHo)-1-оксаспиро(4,5)дец-8-ил пирроли- дин (изомер Б).
В круглодонную колбу емкостью 25 мл, оснащенную обратным холодильником и магнитной мешалкой, помешают 3,4 г (22,0 моль) эпоксидного изомера А по примеру 1, стадия Е, 1,72 г (24,0 ммоль) пирролидина и 1 мл воды Смесь нагревают с перемешиванием в атмосфере азота при 70°С в течение 2 ч. Реакционную смесь охлаждают до О С на водно-ледяной бане, затем добавляют 3,5 мл 25%-ной гидроокиси натрия. Смесь перемешивают в течение
878420
15 мин. Избыток пирролидина удаляют в вакууме. Водный остаток экстрагируют дважды 50 мл хлористого метилена. Соединенные органические продукты 5 -.ромывают 100 мл рассола и сушат
сульфатом магния. Растворитель удаляют в вакууме. Остаток перегоняют мо- лекулярно при пониженном давлении.
Q Получают 4,3 г (87%) промежуточного аминового спирта т.пл. 100-102 с (0,05 мм рт.ст.),
В нагретой в печи круглодонной колбе емкостью 100 мл, оснащенной
5 питательной уравнивающей давление воронкой и магнитной мешалкой помещают 4,3 г аминового спирта, 2,43 (24,0 ммоль) триэтиламина и 25 мл хлористого метилена, затем смесь охQ лаждают до 0°С на водно-ледяной бане. В перемешиваемую смесь по каплям в течение 30 мин добавляют 2,7 г (24,0 моль) метансульфонилхлорида в 20 мл хлористого метилена. Смесь пе5 ремешивают при этой температуре еще 30 мин, затем ее промывают 50 мл воды. Органическую фазу сушат сульфатом маг- магния. Растворитель удаляют в вакууме.
0 Получают 6,0 г сьфого мезилата.
Сырой мезилат помещают в кругло- донную колбу емкостью 100 мл, оснащенную обратным холодильником и магнитной мешалкой, затем добавляют 40 мл 40%-ного водного метиламина. Смесь нагревают 2 ч при 70 С. Избыточный метиламин удаляют в вакууме, остаток распределяют между 150 мл , 10%-ной NaOH и 150 мл хлористого мер тилена. Водную фазу экстрагируют
150 мл хлористого метилена. Соединенные органические продукты сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают 4,3 г сырого продукта. ГХ установлено, наличие смеси (2:1) изомеров А и Б, Хроматографией половины сьфого продукта на силикагеле R Р-2 с элюированием 2 o6.%, (50%-ной воднЬй), 10 об.% воды и 88 об.% CHjCN получают 1,6 г изомера А и 0,63 г изомера Б (95% в пересчете на регенерацию сырого продукта).
5
Н-ЯМР-спектр изомера А соответствует.
Н-ЯМР-спектр изомера Б соответствует.
35
21133
Масс-спектр изомера Б, гл/е: 238 (,7%), 194 (78,5%),126 (15,2 /;), 110 (100%) , 97 (61 ,9%) .
Стадия Б. (j)-(So/, 7/3, 8с1)-3,Д-ди- xлop-N-мeтил-N- 8-(1-пирролидиннл)- 1 - -оксаспнро(4,5)дец-7-ил бензолацет- амид и его моногидробромид.
В нагретую в печи круглодонную колбу емкостью 50 мл, оснащенную пи- тающей воронкой, уравновешивающей давление, и магнитной мешалкой, помещают 0,59 г (2,9 ммоль) 3,4-дихлорфе- нилуксусной кислоты и 10 мл ТГФ (высушенного с применением мгзлекулярных сит ЗА ). В перемешиваемую смесь добавляют в двух 0,5 г (2,9 ммоль) 1,1 -карбонилдиимидазола. Полученную смесь перемешивают при комнатной температуре в течение I ч, по каплям добавляют 0,63 г (2,6 ммоль) диаминового изомера В стадии Л в 5 мл ТГФ в течение 15 мин. Реак1щон- ную смесь перемешивают 18 ч. ТГФ удаляют в вакууме. Остаток разделяют на 30 мл этилового эфира и 30 мл воды. Эфирную фазу промывают 2 раза 30 мл воды. I раз 30 мл рассола и сушат на MgSO. Растворитель удаляют я вакууме Хроматограс1я1ей на силикагеле с элюи- рованием 3%-ного метанола (содержащего 10% аммиака и 97% этилацетата) получают 0,8 г (75%) аминоамида. Продук растворяют в этиловом эфире и обрабатывают раствором (эфирным) бpo иcтoI o водорода. Полученный ос:адок перекрис- таллизовывают из хлороформа/гексана.
Получают гидробромидную соль с т.пл. 212-215 С.
Масс-спектр высокой степени разрешения: вычислено 424,16926, найдено 424,1648.
Вычислено, %: С 50,95; Н 6,22; N 5,39; С1 14,37; В г 15,40.
CjjHj Nj OjClj- HBr-0,S-Н,0 X
X 0,034 СПС1 .
Найдено, %: С 50,94; Н 6,13; N 5,45; С1 14,37; Вг 16,20.
Н-ЯМР (80 МГп, СОС1з1 вободное ос
нование) соответствует аминоамиду. с,о : 1640 см
ИК (нуйол)
Масс-спектр т/е: М+ 424,426 (7,5; 5,1%); 314,316 (14,2; 9,3%), 207 (30,7%), 1 10 (100%), 97 (55,5%).
Пример 16. (j)-(5, 60, 7/1)- -3,4-Дихлор-М- 7-(диметиламино)-1-ок- саспиро(4,5)-дец-6-ил метилбенэолацет- амид.
д t,
Стадия Л . (+ ) - ( S ,
7 ,9)-М - Фе0Ь jo
5
0
0
5
нилметил-М , N -тримг ил-1-оксаспиро (4 , 5 )декан-6 , 7-;и)лмин .
В ()Нмун1 колбу емкостью 100 мл, оснащенную конденсатором и маг нитной меша.чкой, помещают 3,8 i- (25 ммоль) 5 , 5-эпокси-1-оксасггиро (4,5) декана (изомер f примера 4, ста- ди.л в), 4,5 г (37,5 ммоль) бензил(ме- тил) амина и 3 мл ноды. Смесь Hai pena- ют с перемеппиьчнием 3 дня при 90(;. Pea. шюнную смесь охлаждают до на водно-ледяной бане. К холодной смеси добавляют 4 мл 25%-ного раствора NaOH. Основную смесь дважды экстрагируют 75 мл хлористого метилена. Соединенные органические вещества сушат сульфатом магния, растворитель удаляют в вакууме. Избыток бензил(метил)амина удаляют путем tcarpeBa при 40 С. в глубоком вакууме.
Получают 6,32 г (92%) сырого амино- вого спирта.
ЯМР-спектр соответствует.
В круглодонпую ксхпбу емкостью 250 мл, оснащенную питающей уравновешивающей давление воронкой, магнитной мешалкой и положительным впуском для азота, помещают 4,0 г (14,5 ммоль) аминового спирта, 1076 г (16,0 ммоль) триэтиламина и 40 мл хлористого метилена. Смесь охлаждают на водно-ледяной бане до . затем по каплям в течение 30 мин добавляют 1,83 г (16,0 моль) метансульфонилхлорида в 30 мл хлористого метилена, Реак1Ц1ОН- ную смесь перемешивают в холоде 30 мин. Раствор промывают 70 мп воды, сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают 5,4 г сырого промежуточного мезилата.
Сырой мезилат помещают в круглодонную колбу емкостью 100 мл, оснащенную магнитной мешалкой и конденсатором, вместе с 40 мл 25%-ного водного диме- тиламина. Смесь наз ревают с перемешиванием 2 дня при 90°С. Затем ее охлаждают до в водно-ледяной бане и обрабатывают 2 мл 25%-ного водного раствора NaOH. Основную смесь дважды экстрагируют 50 мп хлористого метилена. Соединенные орг-анические продук ты сушат сульфатом магния, растворитель удаляют в вакууме. Хроматографией на силикагеле с элюированием 3% ме- метанола (с содержанием 10% NHj) и
2Т
97. :jTn:iniieT;n а гю.пучают 2,4 г (55%) чамещсиногсз лиамииа.
11-ЯМР (80 , CDClj) и мясс-спок тры (;п 1тнстс:тиук т.
(:та;и1я Б. ()-(5f, 60/, 7л)-м ,М N -Трнметнл- 1-окгас11Лро(4,5)-6, 7-ди- амл и.
Смесь 2,4 г (7,95 ммоль) диамина стадии Л, 2,4 г 20Z-iioro палладия на угле и 100 мл абсолютного этанола гидрируют иа аппарате Парра при 40 psig в течение 4 ч. Реакционную смесь фильтруют .через целит. Жмых тщательно промывают этанолом, соединенные фильтрат и промывочную жидкость концентрируют в вакууме.
Получают 1,38 г (82%), названного диамина, который используют без дополнительной очистки.
Стадия В. ()-(), 6с/, 7А)-3,4-ди xjiop-N- 7-(диметиламино- I -оксаспиро (4,5)дец-6-ил -N-метилбензолацетамид
В нагретую в печи круглодоиную колбу емкостью 100 мл, оснащен1гую питающей уравнивающей давление воронкой, магнитной мешалкой и положительным впуском для азота, помещают 800 мг (3,88 моль) 3,4-дихлорфенил- уксусной кислоты и 5 мл ТГФ (высу- щенного над молекулярными ситами ЗА ) В смесь добавляют в две порции 630 мг (3,88 ммоль)1,Г -карбонилди- имидазола, смесь перемащивают 1 ч пр при комнатной температуре. Систему очищают азотом, после чего в течение 15 мин по каплям добавляют 750 мг (3,53 ммоль) диамина стадии Б в 5 мл ТГФ. Полученную смесь 18 ч переме- щивают при комнатной температуре. ТГ удаляют в вакууме, остаток распределяют между 30 мл этилацетата и 20 мл воды. Органическую фазу сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают твердый продукт, который перекристаллизовывают из этилацетата получая 1,15 г (82%) аминоамида, т.пл. 131-133 С.
Н-ЯМР (80 МГц CDClj) , 1,2-1,95 209 (с 6Н): 2,89 (с, ЗН); 3,55-4,00 (м, 4Н); 4,75 (д, 2Н); 7,0-7,35 (м, ЗН).
ИК (нуйол)с-о : 1634 cm .
Масс-спектр, т/е: 398,400 (5,5; 3,6% М); 181(33%); 168 (21%); 98 (18%); 84 (100%).
Вычислено, %: С 60,15; Н 7,07; N 7,01; С1 17,76.
0
878424
C,,,0
Найдено, 7,: С 59,92; Н 7,08; N 6,93; С1 17,77.
Получен)1ый таким образом амино- амид представляет собой С -эпимер соединеьшя, гюлучен}{ого по примеру 17, стадии А-В.
Пример 17. ()-(5, Ь, 7ft)- -3,4-Дихлор-М- 7-(диметиламино)- 1-оксаспиро (4,5) дец-6-6ил-- -N-метилбензолацетамид и его монохлоридгидрат.
Стадия А. (:)-(5f, бе/, 7/э)-М -Фе- нилметил-N , N , N -триметил-1-окса- g спиро(4,5)декан-6,7-диамин,
В круглодонную колбу емкостью 250 мл, оснащенную конденсатором и магнитной мешалкой, помещают 9,8 г (64 ммоль) 5,6-ЭПОКСИ-1-оксаспиро (4,5)декана (изомер А по примеру 4, стадия в), 11,57 г (95 ммоль) бензил (метил) амина и 6 мл воды. Смесь нагревают с перемещиванием при 70°С в течение 18 ч, затем ее охлаждают 5 ДО О С на водно-ледяной бане. К холодной перемешиваемой смеси добавляют одноразово 6 мл 25%-ного раствора NaOH. Основную,смесь дважды экстрагируют 100 мл хлористого метилена, Сое- Q диненные органические фазы суиат сульфатом магния, растворитель удаляют в вакууме. Избыток бензил(метил)амина удаляют нагревом при в глубоком вакууме. Продукт перегоняют молекуляр- но при пониженном давлении. Получают 15,7 г (90%) промежуточного аминового спирта с т.кип. 154 - (0,02 мм рт.ст.) в виде жидкости, которая кристаллизуется при кпм- натной температуре, т.пл. 63-65 С.
Вычислено, %: С 74,14; Н 9,15; N 4,09.
,
Найдено, %: С 74,34; Н 9,03; N 5,09.
ЯМР- и масс-спектры соответствуют.
В нагретую в печи круглодонную колбу емкостью 100 мл, оснащенную магнитной мешалкой и питающей уравнивающей
Q давление воронкой, помещают 3,0 г
(10,9 ммоль) аминового спирта, 1,32 г (12,0 ммоль) триэтиламина и 40 мл хлористого метилена. Колбу охлаждают до О С на ледяной бане в атмосфере
55 азота. В холодный перемещиваемый раствор по каплям в течение 15 мин 1,38 г (12,0 ммоль) метансульфонилхЛо- рида в 20 мл хлористого метилена. Реакционную смесь еще 30 мин перемешива0
5
ют на холоде и промывают 50 мл воды. Органическую фазу отделяют, сушат сульфатом магния. Растворитель удаляют в вакууме.
Получают 4,2 г сырого промежуточного мезилата.
К половине сырого мезилата добавляют 15 мл 25 -ного водного диметил- амила. Смесь нагревают и перемешива- ют 24 ч в герметичной колбе из нержавеющей стали при . Реакционную смесь Охлаждают до комнатной температуры и обрабатывают 1,5 мл 25%-но ного раствора NaOH. Основной продукт дваящы экстрагируют 50 мл хлористого метилена. Соединенные органические продукты сушат сульфатом магния. Растворитель удаляют Б вакууме.
Остальной мезилат обрабатывают аналогично, получая 3,3 г сырого продукта. Соединенные продукты хромато- графируют на силикагеле, элюируя 1,5% метанола (содержащим 10% NN3) и 98,5% этилацетата, получая 1,35 г (41%) диамина.
ЯМР-спектр соответствует.
Стадия Б. (+.)-(5f, 60/, 7)-N ,гГ, N -Триметил-1-оксаспиро(4,5)-декан- -6,7-диамин.
Смесь 1,35 г (А,5 ммоль) диамина стадии А, 1,35 г 10%-ного палладия на угле и 100 мл абсолютного этанола гидрируют при 40 psig в течение 18ч, Реакционную смесь фильтруют через це- лит. Жмых тщательно промывают зтано- лом. Соединенные фильтрат и промывочную жидкость концентрируют в вакууме.
Получают 1 г (100%) дебензилирован ного диаминового продукта, который используют без дополнительной очистки
Стадия В. {+)-(5, 6а , 7/з)-3,4-Ди- хлор-N- 7- (д11метиламино) -оксаспиро (4,5)дец-6-илJ-N-метилбензолацетамид и его гидрат моног-идрохлорида.
В нагретой в печи колбе емкостью 50 мл, оснащенной магнитной мешалкой и питающей воронкой для выравнивания давления, помещают 0,53 г (2,6 ммоль) 3,4-дихлорфенилуксусной кислоты и
5 мл тетрагидрофурана (высушенного над молекулярными ситами ЗА ). В перемешиваемую смесь в две порции добавляют твердый 1,1 -карбонилдиимидазол (0,42 г, 2,6 ммоль) промывают азотом, затем при комнатной температуре 45 мин перемешивают в течение 15 мин по каплям добавляют 0,3 г (2,4 ммоль) диамина стадии Б в 5 мл тетрагидрофурана. Реакционную смесь 4 ч перемешивают при комнатной температуре. ТГФ удаляют в вакууме, остаток распределяют между 30 мл этилацетата и 20 мл воды. Органическую фазу сушат сульфатом магния. Растворитель отгоняют в вакууме. Затем хроматографируют на силикагеле, элюируя 2% метанола (содержащего 10% NH ) и 98% этилацетата
Получают 0,6 г (63%) свободного основания в виде бесцветного масла. Продукт растворяют в эфире и обрабатывают эфирным раствором хлористого водорода. Остаток собирают и пере- кристаллизовывают из метанола/эфира. Получают хлористоводородную соль, т.пл. 190-194°С.
ЯМР (80 мГц, СОСЦ, свободное основание), cf: 1,2-1,90 (м, юн); 2,18 (с, 6Н); 2,98 (с, ЗН); 3,55- 3,75 (м, 4Н); 4,5 (д, 2Н); 7,0-7,4 (м, ЗН).
Вычислено, %: С 54,00; Н 6,46; N 6,29; С1 23,91.
Cl,NjOj- HCl 0,5
Найдено, %: С 54,16; Н 6,69; N 6,39; С1 24,00.
ИК () 1645 см Масс-спектр, т/е: 398,400 (,16%); 181 (81%) 168 (42%); 84 (100%).
Полученный аминоамид представляют собой Cj-эпимер по примеру 16, стадии А-В.
Пример 18. ( + )-(5d, 7с/, В л) -4-Бром-3-метокси-М-метил-М- 7-(1- -пирролидинил)-1-оксаспиро(4,5)дец- -8-ил бензамид.
Дебензилированный амин по примеру 1, стадия 3, Зе ацилируют З-бром-4- -метоксибензойной кислотой, применяя способ ацилирования по примеру 6.
Получают аминоамид с т.пл. 156- 158°С, выход 60%.
Вычислено, %: С 58,67; Н 6,71; N 6,22; Вг 17,44
C.
Найдено, %: С 58,49; Н 6,71; N 6,71; Вг 17,38.
Пример 19. ( + )-(5d. Id, 80) -Бром-М-метил-М- 7-(1-пирролидинил)- -1-оксаспиро(4,5)дец-8-ил бензолацет- амид.
Аналогично примеру 6, но используя 3-бромфенилуксусную кислоту, получаю аминоамид с т.пл. 93,5-97°С.
Вычислено, %: С 60,69; Н 7,18; N 6,43; Вг 18,35.
C..O,
27Г
Нлид. ни, С 60,48; М 7,33; N Ь,30; В г 18,10.
II р 11 м е р 20. ()-(5o(, Id, 8/i)- -N-MoTHJi-N- 7- ( 1 -luippojiHAUHHji) -окса- ciiiipo (4,5) дец-8-ил -3- (трифторметил) бензолацстамид.
Аналогично примеру 6, но используя 3-(трифторметил)-фенилуксусную кислоту, получают амииоамид в виде полу- гидрата моногидробромида, т.пл. 198- 20 Г с;.
Вычислено, Z: С 53,70; Н 6,47; N 5,43; Вг ,
Cj5 Н,, N,jO,,FjHBr 0,5FijO
Найдено, /;: с 53,70; Н 6,33; N 5,28; Вг 15,77.
Пример 21 . (-f)-(5o, 7с/, 8/з)- -М-Метил-2-1штро-М- 7-(1-пирролиди- нил)-1-оксаспиро(4,5)дец-8-ил бен- золацетамид.
Аналогично примеру 6, но используя 2-нитрофенилуксусную кислоту, получают ам и1оамид с т.пл. 124-127 С (из этилацетата), выход 63%.
Вычислено, %: С 65,81; Н 7,78; N 10,47.
С2,Нз,МзО,
Найдено, %: С 65,52; Н 7,82; N 10,40.
Пример 22. ()-(5, 70/, 8/5)- -М-Метил-3-нктро-М-С7-(1-пирролиди- нил)-1-оксаспиро(4,5)дец-8-ил1бензол- ацетамид.
Аналогично примеру 6, но используя 3-}штрофенилуксусную кислоту, получают аминоамид в виде полугидрата мо- ногидрохлорида, т.пл. 186-189°С.
Вычислено, %: С 59,12; Н 7,44; N 9,40; С1 8, 16.
С.Н,, N, -0,5 H.jO
НаГщёно, %: С 59,17; Н 7,39; N 9,23; С1 8,.07.
Пример 23. (+)-(5о/, 7о/, 8/i)- -М-Метил-4-нитро-К- 7-(l-пиppoлиди- нил)-l-oкcacпиpo (4,5)дец-8-ил бензол- ацетамид и его моногидрохлорид.
Аналогично примеру 6, но используя 4-нитрофенилуксусную кислоту, получают аминоамид с т.пл. 168-17l C из диэтилового эфира/метанола
Прим е ip 24. (5о, 7о(, f)-()- -3,4-Дихлор-М-метил- 7-(1-пирролиди- нил) -1 -оксаспиро (4,5) дец-8-ил .1бензол- ацетамидметансульфонат.
Стадия А. В раствор 5,0 г (11,7 ммоль) бензолацетамида в 100 мл сухого метанола, охлажденного на водно-ледяной бане, по каплям добавляют
538784
28
11,7 мл (11,7 ммоль) 1,0 М раствора метансульфоновой кислоты в метаноле. После окончашш добавления растворитель удаляют в вакууме, получая белую пену. Сырой продукт перекристаллизо- вывают из метанола/диэтилового эфира, получая 5,1 г (выход 83%) белых кристаллов соли с т.пл. 210-214 С.
Вычислено, %: С 52,97; Н 6,57; N 5,37; С 13,60; S 6,15. С„Н,,С1,,5
Пайдено, %: С 52,62; Н 6,62; N 5,41; С 13,64; S 6,08.
Стадия Б. Порцию в 1130 г
(2,66 моль) (5о,, 7с, 8/з)-(+)-3,4-Ди- хлор-М-метил-М- 7-(1-пирролидикил)- -1-оксаспиро(4,5)дец-8-ил бензолацетамида растворяют в 5 л хлористого метилена и фильтруют в чистый сухой реактор емкостью 12 л, оснащенный механической мешалкой, впуском для азота, термометром и капельной воронкой (1 л) Поскольку свободное основание загрязнено неорганическим твердым продуктом, используют 1230 г продукта, из которых ИЗО г представляют собой свободное основание, неорганический твердый продукт отфильтровывают.
Раствор охлаждают примерно до 15 С на водно-ледяной бане, затем в течение примерно 1 ч добавляют раствор 256 г (173 мл), 2,66 М метансульфоновой кислоты в 500 мл хлористого метилена, после чего приблизительно 30 мин перемешивают. Показатель рН должен быть равен от 5 до 7. Если метансульфоновая кислота добавлена в большом избытке на стадии концентрирова1шя может появиться сложность с разложением. Питательную воронку меняют вакуумным перегоночным аппаратом, большее количество хлористого метилена удаляют под вакуумным давлением примерно при . Получают конечный объем около 2 л остаточного материала при некоторой степени кри- сталлизации.
Затем к остатку добавляют 600 мл метанола и перемеш1вают до полного растворения. Полученный раствор охлаждают до 20 С и разбавляют медленным добавлением (2ч) 6л диэтилового
эфира. Полученную смесь перемешивают в течение ночи при комнатной температуре в атмосфере азота, поскольку метансульфокислотная соль аминоамида кристаллизуется медленно.
Полученную смесь фильтруют, тнер- дьи; продукть Г1рс1№,пают 2 л дичтилопо- го эфирл. Фильтрованные кристаллы мо- тапсульфонатной соли сушат на воздухе в течеш1е 3 ч, а чатем в вакууме при 50 С в течение 20 ч. Если в целевых твердых продуктах имеется метиленхло- рид (что видно по ЯМР-амализу), может оказаться необходимой перекристаллизация из метанола/диэтилового эфира путем растворения кристаллического продукта в 3-4 мг/л метанола, разбавления полученного раствора 25 мл/г диэтиловым эфиром, перемешивания смеси в течение нескольких часов, фильтрации и сушки.
Испытание соединения на аналитическую активность.
Еди1шчная доза - величина, используемая в качестве доз-единиц для млекопитающих, причем каждая единица содержит в качестве активного действующего начала заданное количество соединения с необходимыми фармацевтическими свойствами для достижения положительного действия у человека и животных. В качестве единичных доз согласно предлагаемому способу используются таблетки, капсулы, орально вводимые жидкие препаративные формы на соотнетствуюиа1х жидких носителях, стерильные препараты на соответствующих жидких носителях, стерильные , препараты на соответствующих жидких носителях для внутримышечного и внутривенного введения, суппозитории и стерильные сухие препаративные формы для получения стерильных инъекцируе- мых препаратов на соответствующем жидком носителе, Соответствуюш 1е твердые разбавители или носители для получения твердых фармацевтических единичных доз для орального введения выбраны из группы, включающей липиды, углеводы, белки и минеральные твердые продукты, например крахмал, сахароза, лактоза, каолин, ,цикальцийфосфат, желатина, камедь, кукурузный сироп, кукурузный крахмал, тальк и т.п. Капсулы, как твердые, так и мягкие, наполняют составами на основе этих амино- амидных активных ингредиентов в сочетании с соответс вующими разбавителями и носителями, например съедобйыми маслами, тальком, карбонатом кальция и т.п., а также стеаратом кальция. Жидкие препаративные формы для орального введения получают на воде или
водгалх косупс лях, KOTOI LIO прсдио пи- тельно содержат ичппииианищк .И енты, например мсти.чцеллюлозу, камс Д,, по- ливинилнирролидон, поливиниловый спирт и т.п. И((ъекиируемая форма должна быть стерильно и жидкой, удобно при наполнении питрица. Такого рода препаративные формы должп. быть устойчивыми при изготовлении и хранении и обычно дополнительно сс держат консерванты типа бактериостатических и фунгостатических средсти, 1гапример парабенс, хлороформ, бешиловьпЧ
спирт, фенол, тимерозал и т.н. В некоторых случаях предпочтительно включать агенты осмотического действия, например сахар или хлористый натрий в изотонических концонтра щях. К носителям и наполнителям следует отнести растительные масла, этанол, поли- олы, например глицерин, пропиленгли- коль, жидкий пoJп этилeнгликoль и т.п. Любые твердые препаративные формы
стерилизуют стерилизуюиц1м газом, например, окисью этилена. Носители, наполнители, разбавители, растворители, консерванты, изотонические агенты и т.п. представляют собой фармацевтические средства, содействующие системному введению препаративных форм.
Одна фармацевтическая единичная доза соединений формулы (I) должна
содержать от 0,5 до 350 мг действующего начала. Количество основного действующего начала в каждой единичной дозе таково, чтобы оно было достаточным для достижения анальгетического действия в действенном не токсичном диапазоне, т.е. при системном применении количество основного активного ингредиента составляет примерно от 0,01 до 5 мг/кг веса тела
пациента. Предпочтительные дозы для большинства случаев применения составляют от 0,05 до 2,0 кг/кг веса тела, В полутвердых мазях местного назначения действующее начало может
содержаться в количестве 0,2-10%, предпочтительно 0,5-5% на носителе (фармакологический крем),
3/Ij(j соединений формулы (I) составляет примерно менее 75 мг/кг при подкожном введении в стандартных лабораторных анальгетических опытах на подопытных животных (легкие удары по хвосту, , судороги, вследствие введения хлористоводородной кислоты
31
или 1К)). а 6(jjiee cnjibni,ie из сое динеши обладают ЭД менее 10 мг/кг 11)И 1:одкожиом ипеденли в этих опытах н то рремя как при введении в ходе О1и,1та naxolone jumping fest они показывают довольно высокие показатели LDj (выше 250 MT/KI; подкожно), спе- ;| )11лтельно, о}1и имеют низкую токсичность и мало зависят от физического состояния подопытных животных по сравнению со стандартными контрольными средствами.
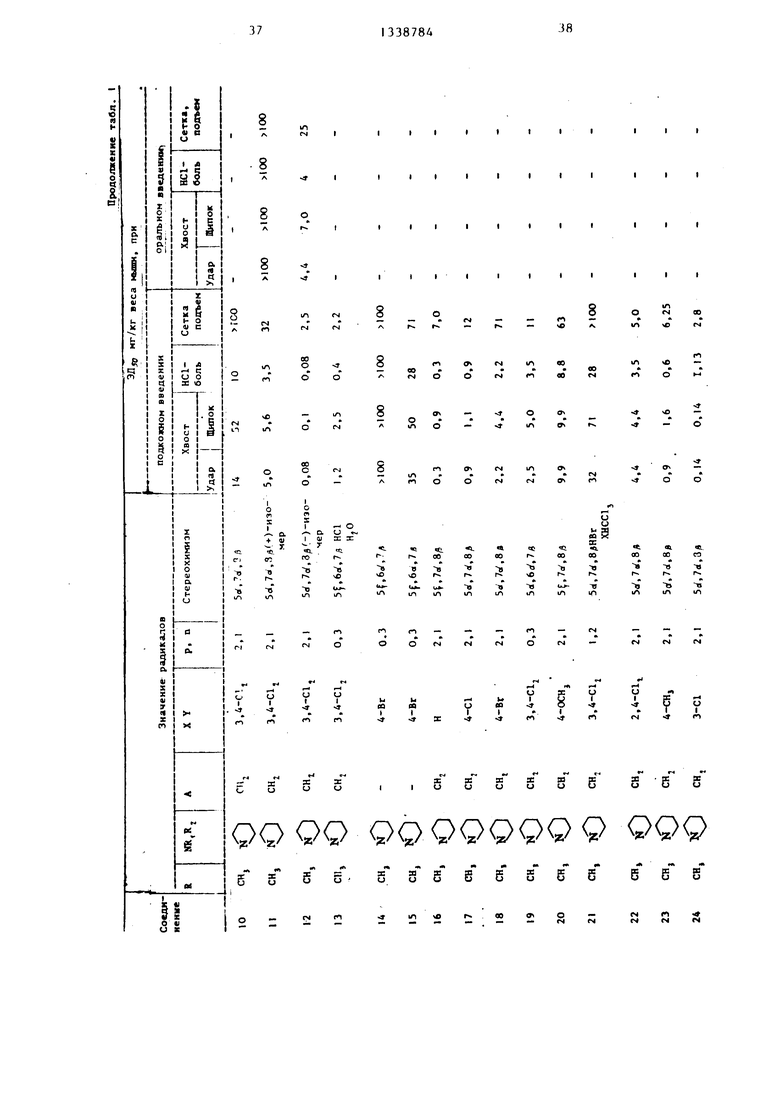
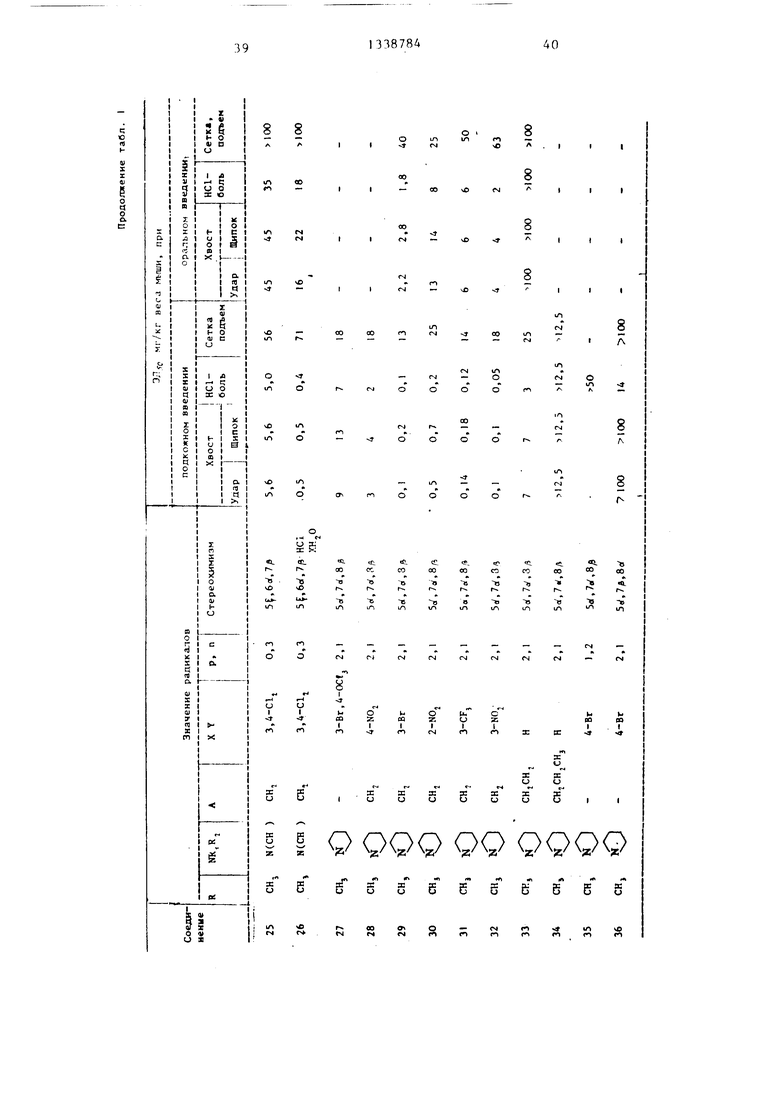
В табл. I - 3 приведены биологические данные при подкожном или оральном введении в организм мышей предлагаемых соеди}1ет1Й 1-36, за исключением соединения 22, которое приводится с ограничением, а также соединений .37-42, введенных перито- нально в сраннении с известными соединениями .
А - Ж. Лнальгетическое действие оценивается при ударе по хвосту, щипке в хвост и НС1-болевом опыте. Опыт с взлезанием вверх по сетке является показателем успокоительного и анальгетического действий испытуемых соединений.
При подкожном введении соединение 26 превосходит соединение Б.
Соединение 12 (-)-изомера соединения {I) более действенно при оральном введении, чем соединение F,
соединение 8 превосходит по действию соединение Ж. Поскольку оральная дача анальгетических средств, как правило, является наиболее приемлемым и удобным видом введения в организм, более сильное оральное действие предлагаемых соединений по сравнению в известными является наизначительнейшим фактором. Кроме того, в табл. 1 и 2 прийедены данные действия цис-соединений, полученные на подопытных животных (анальгети- ческий тест, болевое НС1 - испытание) , при этом три из них являются активными в качестве анальгетических лекарственных препаратов, а два не оказывают действия в этом испытании при максимальной испытанной дозе (50 мг/кг).
Формула изобретения
Способ получения моно-окса-бензо- ацетамидных или -бензамидных соеди- нений общей формулы (I)
32
R О
О (
(CHoA-N/Ri
Кг
5
0
5
0
0
5
где волнистые соединительные линии указывают цис- или транс-отношение двух азотсодержащих групп в положениях 1
ца; Р
и 2 ц 1клоалифатического коль5
целое число, равное 0; 1 или 2;
п - целое число, равное 1; 2
или 3, так что образующееся циклоалифатическое кольцо, содержащее их, имеет шесть атомов углерода;
А - (CHj)q (где q - целое число, равное 0; 1; 2 или 3;
X иУ -независимо друг от друга водород или галоген, три- фторметил, метоксигруппа, нитрогруппа или кил;
R - С -Cj-алкил; R, и Rj - ( -Cj-алкил или вместе с
атомом азота, с которым они связапы образуют пирролиди- новое кольцо, кроме соединений формулы (I), при р 1 и п 2, в 1и с-кон4игура- ции при условии, что, если р 2 и г, 1, А - (CH2)q при q I, К - метил, R и R,j вместе с атомом азота, с которым они связаны, образуют пирролидиновое кольцо и относительный стерео- химизм (5(5/, УС/, 8|5), то X и Y не могут означать атом хлора в положении 2 и 4 фе- нильного кольца или метан- сульфонатной соли моно-окса- бензоацетамидных соединений, отличающийся тем, что соединение формулы (II)
R
I
0
О (CH)p.JH L-(
Р.
55 где п. р, R, R и R имеют указанные
значения,
подвергают взаимодействию с соединением формулы (III)
О
R -C-CCHilq-xQ
в среде орг а)шческого растворителя в течет1е BpeMetm, достаточного для образования соединения формулы (I). где q, X и Y имеют указанные значения;5 выделяют целевой продукт в свободном виде или в виде метансульфонат- ной соли моно-окса-бензоацетамидных соединений.
R. - атом хлора, гидроксигрупна
141
или ,, группа,
u«
8 8
II-00 О N
о 1
. II
8
л
8
U-70037
СН.
i.
снг,ci
-с-снг- -с
-i;j-CHj
N(CH,)j CHj
CHa
U-70067
(jHl -5-CHi-(Cl
CH.
о
CH,
U-70476
CH,
PHjCf оci
N- -CH -O-Ct
6
U-7O478
CH,
HjC
о
о
3,4-Cl, 2,15o(,7rf,,001
CH,3,4-Cl, 2,15rf,7,,6
CH
j3,4-Cl, 2,15trf,7rf,8«(3,7
4-Br2, 1j,7,4,S /)6,3
,i -C-f3-Br
CH
41U-69B50E
.0 .rf
S N-C-CH,-O-C1 -0 II
0 .HC
O.
42U-70477ECH
c, Cr H,c 5
0.5 НгО
HCl
CH,3,4-Cl, 1,2
5(,7,Be IICl -- ,0 0,5MC1
CH,
3,4-Cl, 1,2
5с., 7rf,0 HCl 15CI 0, 5H,0
Л
УдарПилок
.
l.. ° Оч О
-9-сНг-( Vci t -Hct
сн.
с
1,1
II j4 J
В OXO..-N-C- Q-BP32
Uv 0
n,N-C-CHi- VCl
о
Us «
N-CH -HCl
CHi
сн,
1,40,93540454071
4028
-HBP
too
D- -P
18181863
ВГ
Cl-fVcVii-C-N H I Cl0 CH
С1чрьсн,-5Cl 0
CH,
10
.160.160,14 8.8
NvJ
0 Br-fVC- N-лХ I ° ° в 0.6 7.0
1 .AJ
Составитель И, Дьяченко Редактор Л. Пчолинская Техред И.Попович Корректор М. Пожо
Заказ 4150/59Тираж 371 Подписное
ВНИИПИ Государственного комитета СССР
по делам изобретений и открытий 113035, Москва, Ж-35, Раушская наб., д. А/5
Производственно-полнгра(1ическое предприятие, г. Ужгород, ул. Проектная, 4
т вл|1п« 2
УЯ«Р Ннпок
too
10
14139,9100
100 100 100)100

















Изобретение касается замещенных бензамидных соединений, в частности соединений общей формулы (I) О ) - ,)Л,„ где волнисто1е линии указывают цис- или транс-отношение двух азотсодержащих групп в положениях I или 2 цик- лоалифатического кольца; р 0; I или 2; п 1; 2 или 3, так что образующееся циклоалифатическое кольцо имеет 6 атомов углерода; А - (CHj),j, лы (I), где р 1 и п 2 в цис-кон- фигурации при условии, что если р 2 и п 1 , А - (CHjj (при q 1 , R, CHj), Rj + R с атомом азота образуют пирролидиноное кольцо и относительный стереохимизм (5в(, Id, 8/3), то X и Y - не могут быть хлором в положениях 2 и 4 фенильного кольца, или метансульфонатной соли моноокса- бензоацетамидных соединений, которые обладают анальгетическим и некоторым успокаивающим действием на центральную нервную систему и поэтому могут быть использованы в медицине. Цель - создание новых более активных веществ указанного класса. Получение новых соединений ведут из соответствующего диамина и ароматического соединения в среде растворителя с последующим выделением целевого продукта в свободном виде или в виде необходимой соли. Испытания новых веществ показывают, что они более активны (в 8 раз), чем ближайщие аналоги анальгетическо- го действия. 3 табл. СО со со 00 00 см

| Патент CUJA № 4212878, кл | |||
| Способ приготовления хлебного вина | 1925 |
|
SU424A1 |
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
