1
Изобретение относится к способу получения производных пиридазина - новых биологически активных соединений, которые могут найти применение в медицине.
Цель изобретения - получение новых производных пиридазина, обладающих более высокой противосудорожной активностью, а также гипнотической .и успокаивающей активностью при сохранении низкой токсичности.
Пример 1. 3-(4-Оксипипери- дин)-6-(2-нитрофенил)-пиридазин (SR 41673).
Смешивают 3 г З-хлор-6- 2-нитрофенил) -пиридазина с 3,86 г 4-оксипи- перидина в 60 мл н. бутанола и нагревают при перемешивании при 100 С в течение 5ч.
Реакционную смесь концентрируют в вакууме досуха, затем остаток растворяют в воде и экстрагируют в этил- ацетате. Органическую фазу промывают водой, сушат на сульфате натрия и концентрируют досуха. Полученное масло хроматографируют на силикагеле, используя смесь хлороформ-метанол (90:10) в качестве элюента. После выпаривания растворителей оставшееся масло кристаллизуют. Повторную кристаллизацию проводят в ацетонитриле с получением 2,6 г целевого соединения,т.пл. 138-139°С, выход 68%.
П р и м е р 2. 3-(4-Оксипипери- дин)-6-(2-хлорфенил)-пиридазин (СМ 40907).
5 г 3-хлор-6-(2-хлорфенил)-пири- дазина растворяют в 120 мл н. бутанола и смешивают с 6,74 г 4-оксипипе ридина, затем нагревают при рефлюксе. В соответствии с условиями, ука- занными Б примере 1, получают после рекристаллизации в абсолютном этаноле 5 г целевого продукта, т.пл. 154- 155 С.
П р и м е р 3. 3-(4-Бутироилоксипиперидин )-6- (2-хлорфенил)-пиридазин (SR 41172).
2,9 г соединения, полученного в примере 2, растворяют в 100 мл -тетра гидрофурана, затем прибавляют 3,5 мл триэтиламина и 2,6 г бутирилхлорида, после чего смесь нагревают при рефлюсе в течение 3 дн. Прибавляют 0,7 мл триэтиламина и 0,52 мл бутирилхлорида и нагревают смесь снова при рефлюсе в течение 24 ч. Реакционную смесь затем концентрируют насухо, а оста
Q
15
20
25Q
35
, 45
50 55152
ток растворяют в 1 н. соляной кислоте, после чего промывают в простом эфире.
После добавления карбоната натрия экстрагируют в этилацетате. Органическую фазу сушат на сульфате натрия и концентрируют насухо. Получившееся масло подвергают хроматографии на си- ликагеле, используя в качестве элю- ента этнпацетат. После концентрирования и рекристаллизации в простом изопропиловом эфире получают 2,7 г SR 41172, т.пл. 89-90°С.
П р и м е р 4. 3-(4-Метилкарбамо- илок сипиперидин)-6-(2-хлорфенил)-пи- ридазин, (SR 41820). ,
Растворяют 2,7 г соединения, полученного в примере 2, в 50. мл тетра- гидрофурана. Нагревают раствор при рефлюксе в течение 48 ч в присутствии 1,65 мл метилизоцианата, после чего прибавляют еще 1,65 мл метилизоцианата, нагревание ведут еще в течение 48 ч.
Реакционную смесь концентрируют в вакууме, остаток хроматографируют на силикагеле, используя в качестве элюента этилацетат. После концентрирования соединение рекристаллизуют в этилацетате. Получают 1 г SR 41820, т.пл. 134-136°С.
Пример 5. 4-(4-Диметилкарба- моилок сипиперидин)-6-(2,4-дихлорфе- нил)-пиридаз ин (SR 42432).
A.3-(4-Оксипиперидин)-6-(2,4-ди- хлорфенил)-пиридазин (SR 41378).
Действзгют аналогично примеру 1, используя при этом как исходное соединение 3-хлор-6-(2,4-дихлорфенил)- пиридазин и 4-оксипиперидин.
Таким же образом получают целевое соединение, т.пл. 15 -153°С (этанол), выход 77,6%.
B.SR 42432.
4,7 г полученного вьше соединения растворяют в 60 мл диоксана, затем прибавляют 6,3 мл триэтиламина и 4,7 мл диметилкарбамоилхлорида. Проводят нагревание при рефлюксе в течение 7 дн, после чего выпаривают растворитель досуха -под вакуумом. Остаток растворяют в водном растворе (10%-1Н ом) карбоната натрия и экстрагируют в этилацетате. Органическую фазу отделяют, сушат на сульфате натрия и выпаривают растворитель досуха в вакууме. Остаток подвергают хроматографии на силикагеле, применяя в качестве элюента этилацетат.
Полученное вещество кристаллизуют. После повторной кристаллизации в эти ацетате получают целевой продукт, т.пл. Г96-198 С; выход 2,8 г.
П р и м е р 6. ,4-Диокса-8- аза-спиродек (4,5) -8-ил -6- (2-хлорфе- нш1)-пиридазин (SE 42487).
Нагревают при рефлюксе в течение 18 ч смесь 4,5 г З-хлор-6-(2-хлорфе- нил)-пиридазина и 8,2 г 1,4-диокса- 8-аза-спиродекан (4,5) в 100 мл бу- танола. Выпаривают бутанол досуха в вакууме и растворяют остаток в воде. Экстрагируют с этилацетатом и высушивают органический раствор на сульфате натрия. Растворитель выпаривают досуха в вакууме и рекристаллизуют остаток в этилацетате. Получают целевой продукт, т.пл. 156-158 С, вы- ход 5,2 г.
Пример. 3-(4-Оксопипери- дин)-6-(2-хлорфенил)-пиридазин (SR 42488).
Нагревание ведут при рефлюксе в течение 4 ч 4 г соединения, полученного в примере 6, растворенного в смси 40 мл муравьиной кислоты и 60 мл воды. Реакционную смесь выливают в раствор едкого натра, перенасыщенно го льдом, экстрагируют с метиленхло - ридом, отделяют органический слой и сушат на сульфате натрия.
Растворитель выпаривают досуха в вакууме, после чего проводят рекрис- таллизацию остатка в абсолютном этаноле. Получают целевой продукт, т.пл 158-160°С, выход 3,2 г.
Используя аналогичные методы, получают соединения, описанные в табл.
Биологические испытания.
Определение гипногенного и успокаивающего эффекта соединений.
Эффект соединений на самопроизволную подвижность.
Успокаивающий эффект проявляется в снижении самопроизвольной подвижности животных. Этот эффект определялся с помощью актиметрического теста, разработанного Буасье и Симон. Тест вьшолняли с помощью двух актиметричес ких клеток типа Apelat (26-21,5 10 см), просвечиваемых двумя световыми лучами, которые фиксируются фотоэлементом. Партия подопытных жн- вотных состояла из 12 самок мьшей Charles River CDl весом по 20-24 г. Животные были помещены индивидуально в клетки на 45 мин после введения
орально исследуемых соединений в дозе 250 или 100 мг/кг. Каждое пересечение светового пучка фиксировалос индивидуальным счетчиком. Соответствующее количество передвижений животных регистрировалось в течение 10 мин и сравнивалось с количеством перемещений, отмеченных у контрольны животных, которые бьши обработаны только эксципиентом(0,1 Н HCl).
Результаты приведены в табл. 2.
После введения орально в дозе 100 или 250 мг/кг предлагаемых соединени наблюдают мощное успокоительное . действие этих соединений, проявляющееся в значительном снижении подвижности животных.
С другой стороны, доза в 100 мг/к соединения СМ 41378 вызьшает потерю рефлекса характерного переворачивания от индуктивного эффекта у 60% обработанных животных;
В. Способность к усилению наркоза достигаемого пентобарбиталом.
Дпя уточнения гипногенного дай-, ствия предлагаемых соединений была изучена их способность к усилению эффекта, достигаемого одной субнар- конической дозой пентобарбитала, на примере мышей. Партия подопытных животных состояла из 10 мьш1ей Charles River CDl весом по 20-24 г. Пентобар битал (20 мг/кг, в.б.) бьш введен через 60 мин после введения мыщам орально предлагаемого соединения. Критерием отмечаемого засыпания была потеря рефлекса поворотов.
Способность к усилению действия пентобарбитала, мг/кг:
СМ 40907 96
SR 4115520
SR 4137821
SR 420957,4
SR 4248867
Предлагаемые соединения способны усилить наркотическое действие пен- тобарбитала; это свойство является предикативным гипногенному действию.
Оценка противосудорожного действия соединений.
Противосудорожное действие предлагаемых соединений на мышей было оценено на модели судорог, вызванных электрошоком, и на модели судорог, вызванных химическим веществом бикукулином.
Антагонизм судорог, вызванных электрошоком,
Этот тест был несколько изменен по сравнению с тестом Свингярдаи Азами. Использовали импульсный генератор Каста, снабженный 2 глазными электродами, дающий ток в 12,5 В в течение 0,3 с. Партия животных состояла из 10 мышей Charles Rivers GDI весом по 20-24 г. Соединения были введены животным орально за 60 мин до электрошока. Животные, не проявля- ющие тонических выпрямлений задних конечностей, расценивались как защищенные от судорожного криза.
Антагонизм судорог, вызванных би- кукулином.
Партия животных состояла из 10 мышей Charles Rivers GDI весом по 20- 24 г. Соединения были введены орально за 60 мин до введения бикукулина (0,8 мг/кг, в.в.). Появление тоничес- ких судорог было замечено в течение 60 мин после введения бикукулина.
Результаты приведены в табл. 3.
После орального введения мьш1ам предлагаемых соединений проявляются про- тивосудорожные свойства этих соединений по отношению к электрошоку и действию бикукулина.
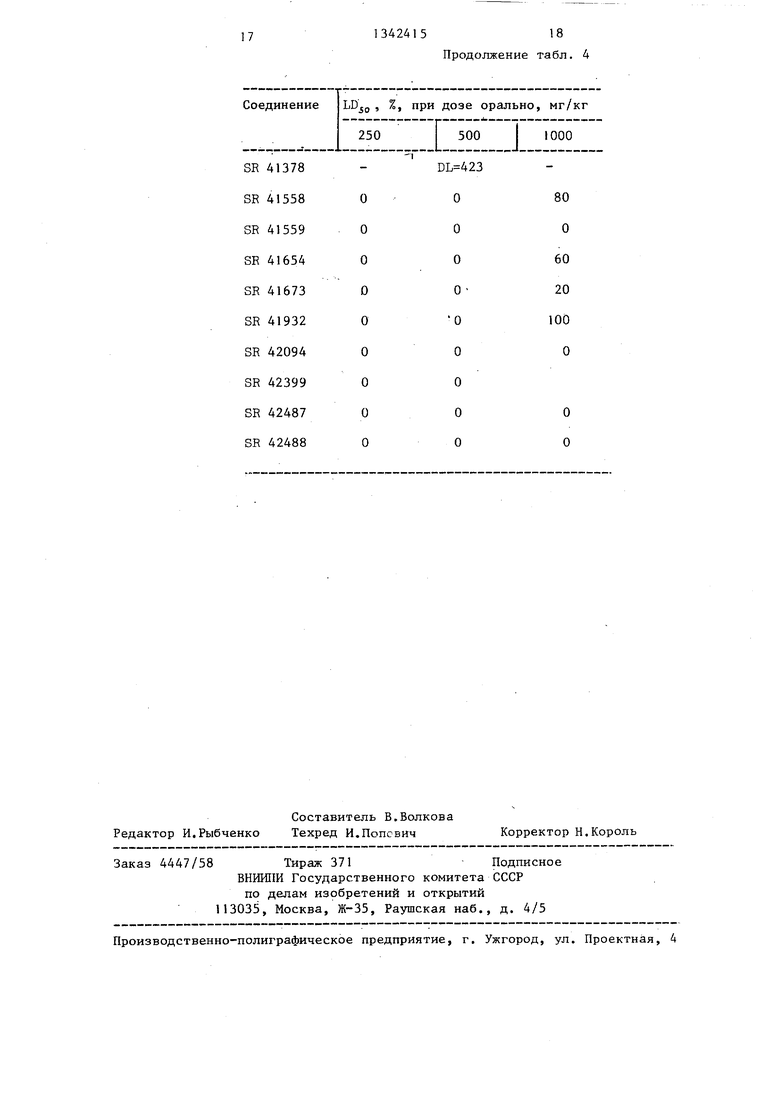
Определение летальной дозы дця мыши после скорого введения соединений
Соединения вводили орально партии животных из 5 самок мышей Charles Rivers GDI весом 20-24 г в виде раствора в 0,1 N соляной кислоты.
Токсичность проявлялась в течение 72 ч после введения соединений. Летальная доза (I Ljo была рассчитана для двух соединений.
Результаты (в процентах),показывающие количество животных, которые погибли в течение 71 часа после введения орально предлагаемых соединений, приведены в табл. 4.
Таким образом проведенные опыты показывают, что предлагаемые соеди- нения имеют низкую токсичность, обладают более высокой противосудорож- ной активностью, чем вальпроат натрия и, кроме того, проявляют успокаивающую и гипнотическую активность.
Формула изобретения
Способ получения производных пиридазина общей формулы I
Хг.
где Аг - группа
,«3 R
R, - водород, галоген, низшая алкильная группа, низшая алкоксилгруппа, группа три- фторметил, оксигруппа, нит- рогруппа или цианогруппа; Кд - водород или галоген,
Аг - незамещенная нафтилгруппа;
X.
де R, Ra
/Rr
- группа -С-NV
5.
и
составляют с атомом азота, с которым они связаны, одну из следуюш 1х групп пиперидина или пирролина;
.
о
RI
причем группа пиперидина замещена в 3- или 4-положении группой R, означающей -ОН; ,-ОН; .,
о
В
-0-И -0-C-N
RB
R
9
II О
где Rg и
R - независимо друг от друга
атом водород или низший алкил,
а в 4-м положении оксогруппой или группой 1,3-спиро-диоксолан 2-ил, пр этом группа пирролина замещена в 4- положении группой -ОН или -СН20Н; отличающийся тем, что подвергают взаимодействию соединение формулы II
С1
где Аг имеет указанные значения с амином формулы
/КГч
R.
де R, и R образуют с атомом азота, с которым они связаны, либо группу пиперидина, замещенную в 3- или 4-положении группой ( СН2)-ОН,, где или 2, или в 4- положении группой 1,37 . 134
спиродиоксолан-2-ил, либо группу пирролина, замещенную в 4-положении оксигруппой или CHjOH; в спирте при температуре кипения смеси и в случае необходимости превращают полученное соединение общей формулы I, /KI
где X, - группа-C-NС где В,
т
R
R
образуют с атомом азота, с которым ойи связаны, группу пиперидина, замещенную в 3- или в 4-положении группой ОН;
в сложный эфир действием хлорангид- рида кислоты в присутствии третичного амина в апротонном растворителе, в N-однозамещенный карбамат воздействием алкилизоцианата, нагревая
58
смесь в апротонном растворителе, В Н,М-дизамещенный карбамат воздействием хлористого карбамила в растворителе при температуре кипения смеси и в присутствии акцептора водородной кислоты, превращают полученное соединение .общей формулы 1, где Xj - группа
/Кг ч -C--N )
Rf
где R, и Rj составляют с атомом азота, с которым они связаны, группу пиперидина, замещенную в 4-положении группой спиродиоксолана, путем гидролиза в кислой среде в соответствующее оксопроизводное.
Таблица 1
1 /«2
V
.
АГ




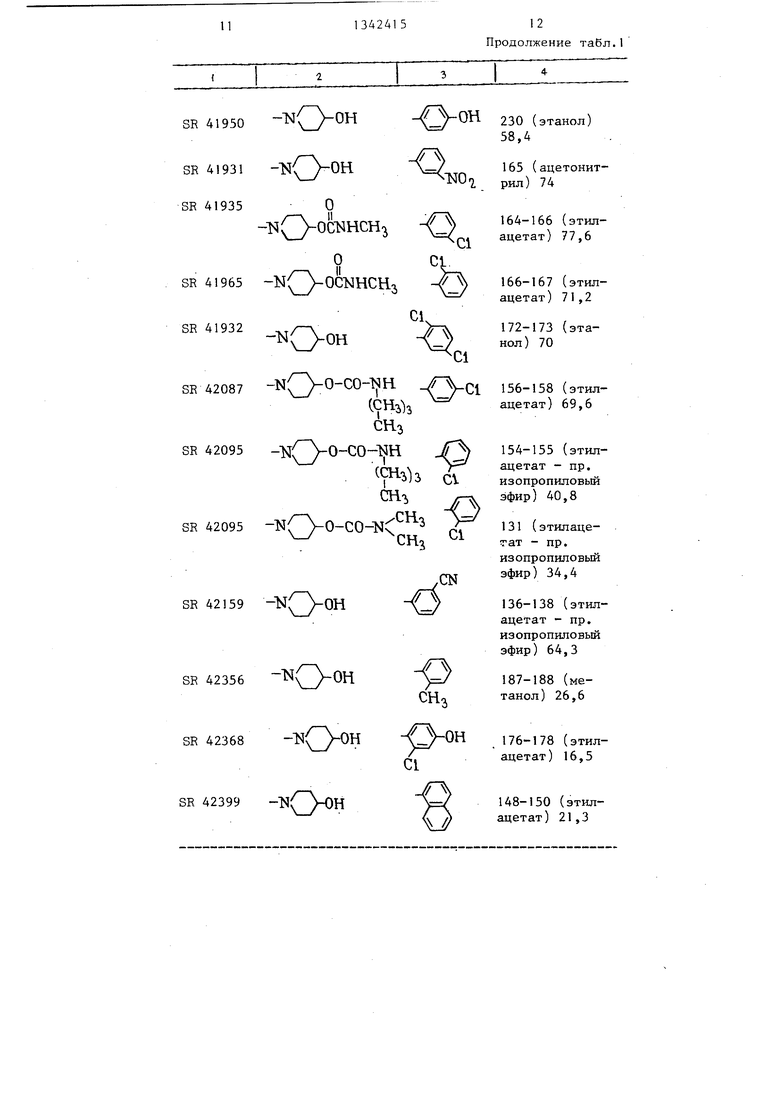


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения замещенных производных пиридазина | 1982 |
|
SU1140685A3 |
| Способ получения 4-цианопиридазинов или их фармацевтически совместимых солей | 1984 |
|
SU1274623A3 |
| Способ получения производных 4-фенилхиназолина или их солей | 1982 |
|
SU1299511A3 |
| Способ получения производных метилендифосфоновых кислот или их солей | 1985 |
|
SU1382402A3 |
| Способ получения производных пиридазина | 1982 |
|
SU1356960A3 |
| Способ получения пирролидинонов | 1984 |
|
SU1391497A3 |
| Способ получения производных метилендифосфоновой кислоты | 1983 |
|
SU1333240A3 |
| Способ получения аминопроизводных пиридазина или их солей с кислотами | 1984 |
|
SU1189345A3 |
| ПРОИЗВОДНЫЕ ГЛИЦИНАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2130923C1 |
| Способ получения аминопроизводных пиридазина | 1982 |
|
SU1138024A3 |
Изобретение касается азотсодержащих гетероциклических соединений, в частности производных пиридазина (ПРИ) обп;ей формулы I Х-М-М 6н-(Аг)- , где Аг - СбН,-...Е,К4; К, - галои)а;, низший алкил, низший алкок- сил, Н, CF,, N0, CN; R - водород, галоид, или Аг - незамещенный наф- тил; , причем R, и Rj азотом образуют замещенный R,, в 3- положении пиперидин (СН); -ОС(о)СзНт; ОС(0)-СН-( OC(0)NRgR,; ,-независимо равны Н, низшему алкилу или в 4-положении оксо- или 1,3-спиродиоксолан-2-ил, или образуют замещенный в 4-положении пирролин ( или ), которые обладают гипнотической или бронхорасширяющей активностью и могут найти применение в медицине. Цель изобретения - выявление более активных соединений указанного класса. Синтез ПРИ ведут из соединений фор- мул II и III СС1 СН-СН...CAr-N N (II); NHR,RI (III), где Аг - указано вьше, R, и Н с атомом азота образуют группу пиперидина, замещенную в 3- или 4-положении группой (СН2)ОН, где или 2, или в 4-положении 1,3-спиродиоксолан-2-илом, либо образуют группу пирролина, замещенную в 4-положении ОН или . Процесс ведут в среде спирта при температуре кипения смеси. В случае необходимости полученное соединение ПРИ, где R, и Rj с атомом азота образуют группу пиперидина, замещенную в 3- или 4-положении группой ОН, превращают: а) в сложный эфир обработкой хлорангидридом кислоты в присутствии третичного амина в апротон- ном растворителе; б) в N-однозаме- щенный карбамат обработкой алкилизо- цианатом при нагревании в апротон- ном растворителе; в) в ТТ,Н-дизамещен- ньш карбамат обработкой хлористым- карбамилом в растворителе при кипении и в присутствии акцептора водородной кислоты. Когда в ПРП R, и :R2 с атомом азота - группа пиперидина, замещенная в 4- положении спиродиоксо- ланом, то проводят гидролиз в кислой среде. Испытания производных пиридазина показьшают, что они малотоксичны и обладают более высокой противо- судорожной активностью, чем вальпро- ат натрия, и дополнительно проявляют успокаивающую и гипнотическую активность. 4 табл. СО со 4 Ю СП сн
см 41096
ОН
см 41127 SR 41155 SR 41171
154 (этилаце- тат) 56,9
174-176 (абсолют . э т анол) 62,9
150-152 (этил- ацетат) 80,2
150 (ацетонит- рил) 47,9
134241510
Продолжение табл.1
тонитрил) 70,7
42087
42095
42095
Cl
-N(O-CO-TJIH. Ус1 ССНз)з СНз
-ъг(о-со- н jQ Ь d
CH-i .т/СНз СН
-NQ O-CO-N - X. . С1
SR 42159
-К(УОН
SR 42356
нол) 70
156-158 (этил- ацетат) 69,6
154-155 (этил- ацетат - пр. изопропиловый эфир) 40,8
131 (этиладе- тат - пр. изопропнловый эфир) 34,4
136-138 (этил- ацетат - пр. изопропиловый эфир) 64,3
187-188 (метанол) 26,6
SR 42368
SR 42399
176-178 (этил- ацетат) 16,5
148-150 (этил- ацетат) 21,3
Соединение
ЬВд, , %, при дозе орально, мг/кг 250 Г 500 I 1000
СМ 40907 СМ 41127 SR 41155 SR 41171 SR 41172 SR 41184 SR 41185 SR 41188 SR 41254
Таблица 4
7
100 100 100 100
о
60 10
Редактор И.Рыбченко
Составитель В.Волкова Техред И.Попсвич
Заказ 4447/58 Тираж 371Подписное
ВНИШШ Государственного комитета СССР
по делам изобретений и открытий 113035, Москва, Ж-35, Раушская наб., д. 4/5
Производственно-полиграфическое предприятие, г. Ужгород, ул. Проектная, 4
Корректор Н.Король
| Вейганд-Хильгетаг | |||
| Методы эксперимента в органической химии | |||
| М | |||
| : Химия, 1967, с | |||
| СТАНОК ДЛЯ ИЗГОТОВЛЕНИЯ ГАЛЕЙ | 1923 |
|
SU413A1 |
