№ 15
(71)Хиноин Дьедьсер Еш Ведьесети Термекек Дьяра РТ (HU)
(72)Иштван Секели, Шандор Ботар, Марианна Ловас, Кристина Долгот, Габор Ковач, Шандор Вираг, Тамаш Сюч, Иштван Ракоци, Карой Тиханьи, Петер Кёрмеци, Пал Хадхази, Иштван Штадлер, Дьёрдь Блашко и Бела Косеги (HU)
(53) 547.514.71.07(088.8)
(56)Патент ФРГ № 2945781, кл. С 07 D 31 1/94, 1980.
(54)СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИНТЕ Р-м-ФЕПИЛЕНПРОСТАЦИКЛИНОВ
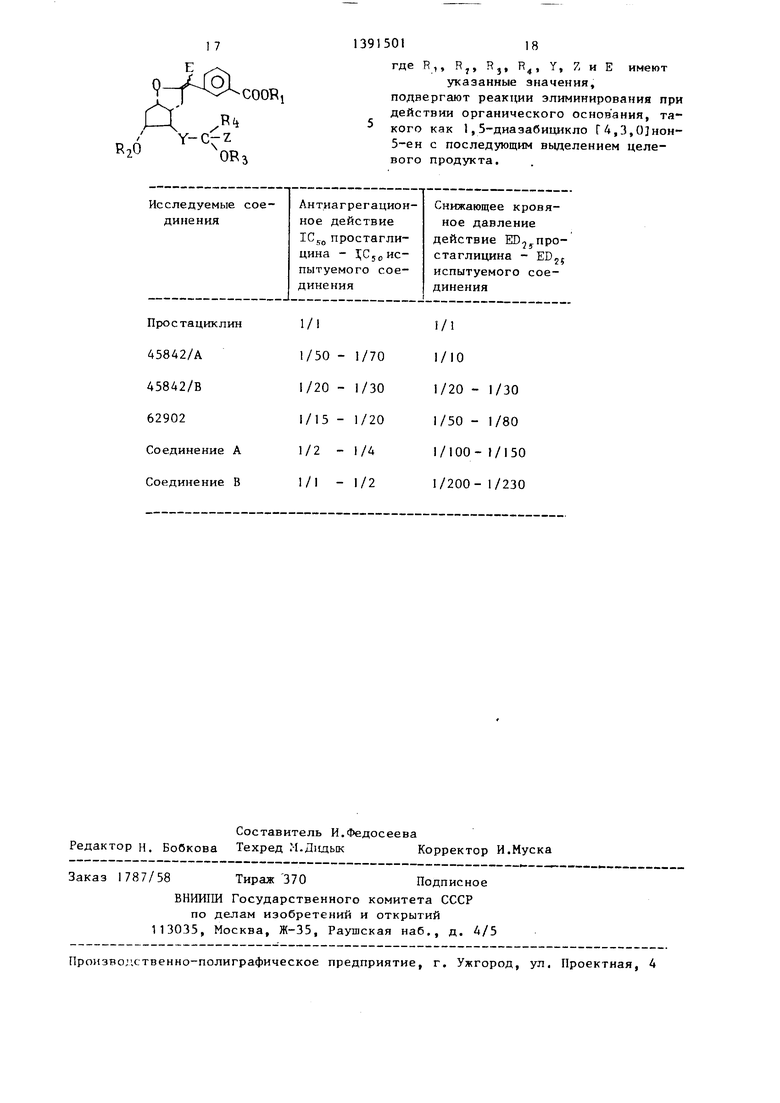
(57)Изобретение относится к способу получения производных интер-м-фени- ленпростациклинов формулы
jg)
tt
RzO
COORi
OR,
где R, - H, Cj- С -алкил, щелочной металл или С ,-С -тетраалкил аммоний; Н, тетрагидропиранил-2, алкил трет-бутилсилил, ацетил; Rj-H, тетрагидропиранил-2, С,-С -диалкил трет-бутилсилил, ацетил; R,, Н, С,-С,-алкил; Y - или транс-СН CW, где W - С1, Вг, F; Z - углеводородная цепочка с 6-7 атомами С,при этом в 16 положении простациклиновой структуры имеются такие заместители, как феноксиметил или трифторметил- феноксиметил, обладающих антиагрега- тизным действием. Получение целевых соединений ведут из соответствующего лактола и фосфорана. Полученные ПГФ - галогенпроизводные подвергают взаимодействию с электрофильными реагентами, такими как иод-М-бромсукцинимид, пербромид пиперидиния, фенилселенйл- хлорид. Образующееся производное про- стациклина подвергают реакции элиминирования при действии органического основания, такого как 1,5-диaзoбицик- лoГ4,3,OJнoн-5-eн с последующим выделением целевого продукта. 1 табл.
g
сл
с
со со
СП







| название | год | авторы | номер документа |
|---|---|---|---|
| Производные 2,3,4-тринор- 1,5-интер-м-фениленпростациклина, обладающие цитозащитными свойствами | 1983 |
|
SU1382834A1 |
| Способ получения производных 2,3,4-тринор- @ -интер-фениленпростагландина | 1982 |
|
SU1138020A3 |
| Способ получения производных интерфураниленпростациклинов | 1986 |
|
SU1470189A3 |
| Производные интерфураниленпростациклина, обладающие гипотензивными свойствами и способностью подавлять агрегацию тромбоцитов | 1986 |
|
SU1467056A1 |
| Способ получения производных 7-оксо-простациклина или их солей | 1985 |
|
SU1376939A3 |
| Оптически активные производные 7-оксопростациклина,обладающие антиагрегатным и гипотензивным действием | 1985 |
|
SU1421741A1 |
| Способ получения солей эфедрина с аналогами 7-оксо-PGJ @ | 1986 |
|
SU1454250A3 |
| Способ получения производных 7-оксопростациклина | 1986 |
|
SU1424735A3 |
| Соли аналогов 7-оксо-PGJ @ с эфедрином,проявляющие тормозящее свертываемость крови действие и снижающие кровяное давление | 1986 |
|
SU1447823A1 |
| Способ получения производных простациклина или их эпимеров | 1981 |
|
SU1072801A3 |

СМ
Изобретение касается способа получения новых аналогов интер-м-фенилен- простациклинов общей формулы
где R, - водород, С)-С4-алкил, щелочный металл или С -С тетраалкил аммоний; R, - водород, тетрагидропиранилС ,-С -диалки.п трет-бутилсилил, ацетил; R - водород, тетрагидропиранилС,-С -диалкил трет-бутилсилил, ацетил; I
R4 - водород, С,-С4-алкил;
Y - С ; С- или транс - ,
где W - хлор, бром, фтор; Z - углеводородная цепочка с
6-7 атомами углерода, при этом в 16 положении простацикли- новой структуры имеются такие заместители, как феноксиметил или трифтор метилфенокс.иметил, обладающих анти- агрегативным действием.
Цель изобретения - получение новых производных интер-м-фениленпро- стациклина, обладающих более селективным действием в сравнении с природным простациклином,
Пример. Метиловый эфир 2,3,4-тринор-1,5-интер-м-фенш1ен-1 А- бром-20-метил-РСГjp,
В круглодонную колбу объемом I00 мм в атмосфере азота помещают 722 мг (30,2 ммоль) гидрида натрия и 35 мл безводного диметилсульфоксида. Суспензию перемешивают сначала 30 ми при 65 - 70°С, затем 30 мин при 70 - . Предприняты меры для отвода получающегося водорода, например, при применении перегонной трубки с ртуть К полученному раствору в атмосфере азота добавляют 7,2 г (15,1 ммоль) трифенил-м-карбоксибензилфосфоний- бромида. Полученную суспензию перемешивают при в течение 30 мин.Полученный таким образом красный, вязкий раствор фосфорана добавляют при комнатной температуре в течение 10 мин к раствору 1,37 г (3,77 ммоль 3, Зар4, 3,6,6ар)-гексагидро-2-гидр
0
0
5
О
0
5
5
окси-А -(2-бром-З(S)-гидрокси-I-ноне- нил)-3-гидрокси-2Н-1и1клопента(Ь)фура- на в I мл безводного тетрагидрофура- на. Реакционную смесь перемешивают при 20 мин.
Затем реакционную смесь выпивают в смесь 100 мл воды и 30 г льда. Устанавливают значение рН 3 - 4 добавлением серной кислоты и раствор экстрагируют мл этилацетата. Соединенные органические фазы экстрагируют 3-15 мл 1н. едким натром. Объединяют щелочные экстракты и добавлением I н.серной кислоты устанавливают значение рН 3,5 - 4, раствор смешивают с 80 мл диэтилового эфира и непосредственно после этого с 10 мл 1 М эфирного раствора диазометана. Эфирную фазу отделяют, промывают насыщенным раствором поваренной соли, высушивают и затем испаряют. Получают 1,78 г сырого продукта, который хроматографируют через колонку с си- ликагелем (элюирующая жидкость - этилацетат:бензол 3:I).Фракции, соответствующие значению ,3l (мерк силикагель арт.5713, э Do фyющaя жидкость - как указано), собирают и испаряют. Получают 929 мг (50%) целевого соединения. Тонкослойная хроматография Rf 0,31 .
ПНР (СОС1з),с/ : 7,3-8,1 (4Н); 6,2-6,7 (транс-Н-3, Н-6 олефин.протоны) ; 5,7-5,93 (цис-Н-5, Н-6, Н-13 олефин.протоны); 4,0-4,35 (ЗН, Н-9, Н-11, Н-15); 3,92 (ЗН, метиловый эфир - CHj).
П р и м е р 2. Метиловый эфир 2,3,4-тринор-1,З-интер-м-фенилен-13, 14-дидвгидро-20-метил-РСР .
532 мг (1,08 ммоль) 2,3,4-тринор- 1,5-интер-м-фенилен-14-бром-20-метил- PGF метилового эфира растворяют в 12 мл диметилсульфоксида, раствор смешивают с 1,22 г
(10,8 ммоль) калий трет-бутоксида. Смесь перемешивают н течение 5 мин и затем разбавляют 20 мл насыщенного раствора поваренной соли, 20 мл воды и 80 мл этилацетата. Водную фазу подкисляют 1 н. раствором кислого сульфата натрия до значения рН 3. После отделения органической фазы ее экстрагируют 2-10 мл этилацетата. Органические фазы объединяют, охлаждают до и смешивают с I О - 20 мл 1 М эфирного раствора диазометана. Органическую фазу высушивают над безводным сульфатом натрия и после фильтрования испаряют при пониженном давлении.
Получают А74 г сырого продукта, который очищают, как описано в примере I, хроматографированием через колонку. Фракции со значением ,35 собирают и при пониженном давлении испаряют растворитель. Получают 2А7 г (59,8%) целевого соединения.
Тонкослойная хроматография ,3 (этилацетат:бензол 3:1).
ПМР (CDC1O,: 7,25-8,1 (4Н, аромат.протоны) ; 6,3-6,58 (2Н, Н-5, Н-6) 4,0-4,5 (ЗН, Н-9, Н-11, Н-15); 3,95 (S, ЗН, эфир сложный - СНз).
П р и м е р 3. Метиловый эфир 2,3,4-тринор-1,5-интер-м-фенилен-5- иод-14-бром-20-метилпростациклина.
577 мг (1,17 ммоль) метилового эфира 2,3,4-тринор-1,5-интер-м-фени- лен-14-бром-20-метил-РСР у растворяю в 2 мл метиленхлорида. При перемешивании добавляют 11,6 мл (11, 6 ммоль) раствора кислого карбоната натрия концентрации 1 ммоль/мл. К полученно двухфазной смеси добавляют 23,4 мл (2,34 ммоль)приготовленного с мети- ленхлоридом раствора иода концентрации 0,1 ммоль/мл. Реакционную смесь при комнатной температуре интенсивно перемешивают в течение 1 ч и затем разбавляют 100 мл этилацетата. Избыточный иод удаляют 5%-ным раствором тиосульфата натрия. Органические фазы отделяют и водные фазы экстрагируют 2x15 мл этилацетата. Органические фазы объединяют, промьшают насыщенным раствором поваренной соли, высушивают над сульфатом натрия безводным, фильтруют и фильтрат испаряют при пониженном давлении, освобождая от растворителя. Получают 720 мг целевого соединения.
Тонкослойная хроматография Rf 0,6 и 0,65 (этилацетат:бензол 3:1, двухразовое элюирование).
П р и м е р 4. Метиловый эфир 2,3,4-тринор-1,5-интер-м-фенилен-5иод-13, 14-дидегидро-20-метилпроста- циклина.
Работают, как описано в примере 3 с тем различием, что в качестве исходного вещества применяют 483 г (1,17 ммоль) метилового эфира 2,3,4- тринор-1,5-интер-м-фенилен-13,14-ди0
5
0
0
5
0
5
0
5
дегидро-20-метил-РСР2. Получают 603 мг продукта.
Тонкослойная хроматография Rf 0,67 и 0,60 (этилацетат:бензол 3:1).
П р и м е р 5. Метиловый эфир 2,3,4-тринор-1 , 5-интер-м -фенилен- 1 4 бром-20-метилпростациклина.
689 г (1,11 ммоль) метилового эфира 2,3,4-тринор-1,5-интер-м-фенилен- 5-иод-14-бром-20-метилпростациклина загружают в крутлодонную колбу объемом 10 мл. Объем колбы несколько раз продувают азотом. В атмосфере азота добавляют 1 мл 1,5-диaзaбициклo(4,3,0)- нoн-5-eнa. Реакционную смесь перемешивают при в течение 2 ч, затем охлаждают до комнатной температуры, разбавляют 50 мл эфира, эфирную фазу промьшают 3x5 мл водой, высушивают над безводным сульфатом натрия и после фильтрования испаряют при понижен - ном давлении. Получают 543 г сырого продукта. Его очищают с помощью хро- матографирования через колонку с си- ликагелем, элюирующей смесью дихлор- метан-ацетон в соотношении 5:1. Фракции, соответствующие Rf 0,73 ц 0,69, собирают и отделяют друг от друга испарением. Из фракции с Rf 0,73 получают 216 мг (39,5%) продукта, который имеет 5,6-двойную связь в (Z)- положении, в то время как фракция с Rf 0,69 дает 206 мг продукта с (Е)-изомерией.
Тонкослойная хроматография Rf 0,73 и 0,69 (пластина силикагеля мерк.арт. 5715; дихлорметан:ацетон 3:1, элюируют дважды).
ПМР (СВС1з),: 7,3-8,2 (т, 4Н, аромат, про тоны)-, 5,85 (d, 1Н, Н-19); 5,3 (S, 1Н, Н-5); 4,83 (т, 1Н, Н-9); 3.95-4,2 (т, 2Н, Н-11, Н-15); 3,94 (S, ЗН, эфир-СНз-).
П р и м е р 6. Метиловый эфир 2,3,4-тринор-1,5-интер-м-фенилен-13, 14-дидегидро-20-метилпростациклина.
Работают, как описано в примере 5, с тем различием, что в качестве исходного вещества применяют 599 мг (1, 1 1 ммоль) метилового эфира 2,3,4- тринор-1,5-интер-м-фенилен-5-иод-13, I4-дидегидро-20-метилпростациклина. Получают 218 г (52,8%) целевого соединения .
Тонкослойная хроматография Rf 0,50 (бензол:этилацетат 3:1).
5
ПНР (СОС1з)Х:7,3-8, 1 (m, 4Н,
аромат.протоны); 5,95 и 5,3 (S, 1Н,
Е- или Z-H-5); 4,05-4,5 (2Н, Н-11 и
Н-15);4,75 (dt, 1Н, Н-9); 4,95 (ЗН,
эфир-СНз) .
Пример. Метиловый эфир 2,3,4-тринор-I,5-интер-м-фенилен- 13,14-дидегидро-20-метилпростацикли- на.
494 мг (1 ммсль) метилового эфира 2,3,4-тринор-1,5-интер-м-фенилен-14- бром-20-метилпростациклина растворяют в 5 мл сухого диметилсульфоксида. К раствору при комнатной температуре добавляют 550 мг (5 ммоль) калий- трет-бутоксида. Суспензию перемешивают 5-10 мин и затем разбавляют водой Устанавливают значение рН 4 добавлением водного раствора 1 н. щавелевой кислоты. Затем раствор экстрагируют 30 мл, затем 2x20 мл этилацетата при . Органические фазы объединяют, высушивают над безводным сульфатом натрия и после фильтрации смешивают с 10 мл эфирного раствора диазомета- на концентрации 4 моль/л. Растворитель отгоняют при пониженном давлении и сырой продукт очищают на колонке с силикагелем хроматографически (дихлормета 1:ацетон 5:1). Фракции, соответствующие значениям Rf 0,5 (бензол:этилацетат 3:1), собирают и испаряют при пониженном давлении. Получают 255,6 мг (62%) целевого соединения. Физические константы соответствуют константам продукта, полученного согласно примеру 6.
Если в качестве исходного продукта применяют 5,6 (Z)-продукт, полу- ченный по примеру 5, то получают (г)-изомеры целевого соединения. Соответственно, если исходят из продукта 5,6-(Е), полученного по примеру 5, то получают (Е)-изомеры.
Примере. Натриевая соль 2,3,4-тринор-1,5-интер-м-фенилен-14 бром-2-метияпростациклина,
988 мг (2 ммоль) метилового эфира 2,3,4-тринор-I,5-интер-м-фенилен-14- бром-20-метилпростациклина растворяют в 1 мл метанола. К раствору при комнатной температуре добавляют 1 мл (10 ммоль) 5 н.водного раствора едкого натра. Реакционную смесь перемешивают в течение 1 ч при комнатной температуре, затем отгоняют метанол при пониженном давлении и остаток разбавляют смесью 5 мл насыщенного
c 0 5 Q
Q ,
5
5
5016
раствора поваренной соли и 5 мл воды. Раствор экстрагируют 35 мл эфира. Водную фазу охлазидают до , смешивают с 20 мл этилацетата и при интенсивном перемешивании устанавливают значение рН 4-4,5. Затем фазы отделяют друг от друга и водную фазу экстрагируют 3V5 мл этилацетата. Органические фазы объединяют: высушивают над безводным сульфатом натрия и после фильтрювания испаряют. Получают 841 мг сьфой кислоты. Продукт растворяют в 0,5 мл метанола и раствор смешивают с 1,75 мл 1 н. мета- нольного раствора едкого натра. Метанол при пониженном давлении испаряют. Сырой продукт суспендируют в 1 мл сухого тетрагидрофурана и затем фильтруют. Получают 878,5 мг (87,5%) целевого соединения.
Тонкослойная хроматография Rf 0,54 (мерк силикагелевые пластины, арт. 5715, бензол:диоксан:уксусная кислота 20:20:1).
П р и м е р 9. Натриевая соль 2,3,4-тринор-1,5-интер-м-фенилен-13, 14-дидегидро-20-метилпростациклина.
Работают, как описано в примере 8, с тем различием, что в качестве исходного вещества применяют Метиловый эфир 824 мг (2 ммоль) 2,3,4-тринор- 1,5-интер-м-фенилен-13,14-дидегидро- 20-метилпростацкклина. Получают 691,6 мг (82,3%) целевого соединения.
Тонкослойная хроматография Rf О,53 (мерк силикагелевые пластины, арт. 5719, бензол:диоксан:уксусная кислота 20:10:1).
П р и м е р 10. Метиловый эфир 2,3,4-тринор-I,5-интер-м-фенилен-20- метил-13,14-дидег идро-6а-карбапро- стагландина 1 ч.
Раствор 20 ммоль натрййметилсульЛ финилметида в 20 мл диметилсульфоксида (получен из 480 мг гидрида натрия и 20 мл сухого диметилсульфоксида) охлаждают до 15-20 С и затем смешивают с 4,77 г do ммоль) трифенил-3- карбоксибензилфосфонийбромида. Полученный красный раствор медленно перемешивают при 35 С 30 мин, затем охлаждают до комнатной температуры и смешивают с раствором 1,34 г (3 ммоль) 7о -(тетрагидропи11ан-2-Ш1-окси)-6 (ЗЯ)-тетрагидропиран-2-ил-окси- (1-ноне нил)-бицикло (3 . 3 ,0)октан-3-она в 1 мл сухого тетрагидрофурана. Реак7
ционную смесь перемешивают при 40 С в течение 48 ч, затем охлаждают до комнатной температуры и разбавляют 10 мл воды. Смесь нейтрализуют 1 г раствором кислого сульфата натрия и затем экстрагируют мл этилаце- тата. Органические экстракты объединяют, промывают сначала 3X10 мл водой и 1V10 мл насьпценным раствором поваренной соли, высушивают над безводным сульфатом натрия и затем фильруют. Фильтрат охлаждают до , смешивают с 10 мл эфирного раствора ди- азометана концентрацией 1 ммоль/л и затем при пониженном давлении отгоняют растворитель. Полученный сырой продукт (2 г) очищают хроматографи- чески через колонку с силикагелем смесью бензола и этилацетона в соот- ношении 4:1. Фракции, соответствую- щие значениям Rf 0,48 и 0,43, собирают и отделяют друг от друга испарением. (Rf : элюирующая жидкость - бензол : этилацетат 3:1). ,48 644,5 мг (37%) 5(г)-изоме ,43 679,3мг (39%) 5(Е)-изомер Полученные продукты отделяют друг от друга и растворяют в соответственно в 20 мл смеси из уксусной кислоты воды и тетрагидрофурана в соотношении 3:1:1,5. Раствор при перемешивают в течение 3 ч, затем охлаждают до комнатной температуры и смешивают с 40 мл насыщенного раствора соляной кислоты и 40 мл этилацетата. Органическую фазу отделяют и водную фазу экстрагируют 2X5 мл этилацетата Органические фазы соединяют и промывают до нейтральной реакции 2X1 о мл насьденного раствора гидрокарбоната натрия. Органическую фазу высушивают над сульфатом натрия, фильтруют и растворитель испаряют при пониженном давлении. Сырой продукт очищают хро- матографически на колонке с силикагелем этилацетатом. Получают в виде 5(Z)- или 5(Е)-изомера 380,4 мг (31,9%) или 387,6 мг (32,5%). Rf в этилацетате 0,28 или О,25.(мерк си- ликагелевые пластины арт.5715,этил- ацетат) .
ПМР (CDCl,,), : 7,25-8,0 (4Н); 5,6 (1Н); 3,5-4,25 (5Н).
Пример II. Натриевая соль 55 2,3,4-тринор-1,5-интер-м-фенилен-20- метил-13,14-дидегидро-6а-карбо- простагландина.
Q .20 25 -, зо д .,
0
5
5018
Работает, как описано в примере , с тем различием, что в качестве исходного вещества применяют 385,6 мг (0,97 ммоль) метилового эфира 2,3,4- тринор-1,5-интер-м-фенилен-20-метил- 13, 14-дидегидро-6а-карбапростаглан- дина-Ь . Получают 352,8 г (87%) целевого соединения.
Тонкослойная хрюматография Rf 0,6 (мерк силикагелевые пластины арт.5715, бензол:диоксан:уксусная кислота 20:10:1).
Указанным в примерах 1-11 образом из соответствующих исходных соединений получены следующие соединения (в том случае, если не указаны дополнительные данные, значение Rf on ределяют на силикагеле мерк.арт. 5719 смесью бензол: диоксан:уксусная кислота 20:10:1):
натриевая соль 2,3,4-тринор-1,5- интер-м-фенилен-13,14-дидегидро-20- этилпростациклина, ,57;
натриевая соль 2,3,4,17,18,19,20- гептанор-1,5-интер-м-фенилен-13,14- дидегидро-1б-(З-трифторметилфенокси) простациклина, Rf 0,62;
натриевая соль 2,3,4-тринор-1,5- интер-м-фенилен-14-бром-2О-этилпростациклина, Rf 0,57;
натриевая соль 2,3,4,17,18,19,20- гептанор-1,5-интер-м-фенилен-16-фе- нил-13,14-дидегидрюпростациклина, Rf 0,69;
метиловый эфир 2,3,4-тринор-1,5- интер-м-фенилен-11,15-бис(тетрагидро- пиран-2-ил-окси)-13,14-дидегидро-20- метилпростациклина, Rf 0,87 (бензол : этипацетат 3:1);
метиловый эфир 2,3,4-тринор-1,5- интер-м-фенилен-1t,15-бис (диметил- трет-бутилсилокси)-13,14-дидегидро- 20-метилпростациклина, Rf 0,95 (бензол : этилацетат 3 : 1 ).
Пример 12. 2,3,4-Тринор-1,5- интер-м-метилен-14-хлор-20-мeтил- PGF2o(мeтилoвый эфир получают аналогично примеру 1 с той разницей, что в качестве исходного материала используют 3,3o(|i, 4,5,6,6c/fl-гeкcaгидpo- 2-oкcи-4 /1- (2-бром-З ( S ) -окси-1 -ноне- яил)-5 1-окси-2Н-циклопента (Ь) фу- ран. Rf 0,33 (на пластине с силикагелем арт.7515, подвижная система: смесь этилацетата и бензола в соотношении 3:1).
Таким же образом получают 2,3,4- тринор-1,5-интер-м-фенилен-I4-фтор20-метил-РСР -метиловый эфир с той разницей, что в качестве исходного материала используют З. , 5, 6, 6e//i- гeкcaгидpo-2-oкcи-Д/ г-(2-фтop-3(S)- oкcи- 1 -ноненил) -5о -окси-2Н-циклопен- та(Ь) фуран. Последний получают путем взаимодействия 3,, 4,5,6, гексагидро-2-оксо-4/ |-(3-оксо-1 -ноненил) -5о -окси-2Н-циклопента (Ь)фурана с перхлоридфторидом в среде метанола. Образующийся в результате 3,3« /5,4,5, 6,6с/р-гексагидро-2-оксо-4р-(2-фтор-1- метокси-3-оксо-1 -нонил)-5/ -окси-2Н10
териала используют 2,3,4-тринор-1,5- интер-м-фенилен-5-иод - 14-хлор-20-ме- тил-PGI,-метиловый и 2,3,4-тринор- 1,5-интер-м-фенилен-5-иод-I4-фтор- 20-метил-РС11-метиловый эфиры.
Rf (1А-хлор-РС1,-производное) 0,76 и 0,73 (на пластине с силИкаге- лем арт.5715, после повторной хроматографии с использованием в качестве подвижной фазы смеси дихлорметана с ацетоном в соотношении 3:1).
Rf (14-фтор-РС71 -производное) 0,79 и 0,77 (на пластине с силикагециклопента(Ь)фуран защищают тетрагид-15 арт.5715, после повторной хроматографии с использованием в качества подвижной фазы смеси дихлорметана с ацетоном в соотношении 3:1).
ропиранильной группой и подвергают реакции элиминирования. Образующийся при этом 3,3о,4, 5, 6, 6o / l-гeкcaгидpo- 2-oкco-4/ -(2-фтop-l-мeтoкcи-3-oкco- l-нoнeнил)-5c -тeтpaгидpoпиpaншIOкcи- 2Н-циклопента(Ь)фуран используют для получения исходного материала по методу Corey (J.A.C.S 9,397, 1970). ,35 (на пластине с силикагелем
тографии с использованием в качеств подвижной фазы смеси дихлорметана с ацетоном в соотношении 3:1).
П р и м е р 15. 2,3,4-Тринор-1,5 20 интер-м-фенилен-14-бром-20-метил- РС1|2-тетрабутиламмониевая соль.
Проводят те же операции, что и в примере 8, с той разницей, что вмес то 1,75 мл I н. раствора гидроокис
арт.5715, подвижная фаза: смесь этил-25 натрия к кислоте добавляют 3,5 мл ацетата с бензилом в соотношении 3:1), метанольного раствора гидроП р и м е р 13. 2,3,4-Тринор-1,5- интер-м-фенилен-5-иод-I4-хлор-20-ме- тил-PGF, -метиловый эфир, соответст30
40
венно 2,3,4-тринор-1,5-интер-м-фени- лен-5-иод-14-фтор-20-метил-РСР, -метиловый эфир получают таким же образом, как это описано в примере 3, с той разницей, что в качестве исходного материала используют 2,3,4-тринор-35 I,5-интер-м-фенилен-14-хлор-20-метил- PGI дрметиловый эфир, соответственно 2,3,4-тринор-1,5-интер-м-фенилен14-фтор-20-метил-РС1 7о1 метиловый эфир.
Rf ()4-хлор-Р01 -производное) 0,63 и 0,67 (на пластине с силикагелем арт.5715, после повторной хроматографии с использованием в качестве подвижной фазы смеси этилацетата с бензолом в соотношении 3:1).
Rf (14-фтор-Р01 -производное) 0,67 и 0,7 (на пластине с силикагелем арт.5715, после повторной хроматографии с использованием в качестве подвижной фазы смеси этилацетата с бензолом в соотношении 3:1).
П р и м е р 14. 2,3,4-Тринор-1,5- интер-м-фенилен-14-хлор-20-метил- PGI,-метиловый и 2,3,4-тринор-1,5-интер-м-фенилен- 14-фтop-20-мeтил-PGI - метиловый эфир получают таким же образом, как и в примере 5, с той разницей, что в качестве исходного ма45
50
окиси тетрабутиламмония.
,5 (на пластине с силикагелем арт.5715, подвижная фаза: смесь бензола, диоксана и уксусной кислоты в соотношении 20:20:1),
П р и м е р 16.
A.2,3,4-Тринор-1,5-интер-м-фени- лeн-5-бpoм-PGI ,-1 метиловый эфир.
508 мг (1,26 мысль) 2,3,4-тринор- 1 ,5-интep-м-фeнилeн-PGFly(-мeтилoвoгo эфира растворяют в IО мл безводной смеси хлороформа и тетрагидрофурана взятых в соотношении 1:1, и охлажда ют раствор до в смеси сухого льда с ацетоном. Добавляют к раство ру 337 мг (1,86 ммоль) N-бромсукцин мида и перемешивают смесь сначала в течение 10 мин при -78°С, а затем в течение 30 мин при комнатной температуре.
Реакционную смесь промывают 50мл хлороформа, а затем трижды 20 мл во ды. Органическую фазу высушивают над сульфатом натрия и фильтруют. Раств ритель отгоняют в вакууме.
Вес полученного продукта 431 мг (выход 71 /).
Rf 0,5,1; 0,57 (подвижная фаза: смесь дихлорметана с ацетоном в соотношении 3:1).
B.2,3,4-Тринор-1,5-интер-м-фе- нилен-5-бром-15- метил-PGI,-метиловый эфир.
териала используют 2,3,4-тринор-1,5- интер-м-фенилен-5-иод - 14-хлор-20-ме- тил-PGI,-метиловый и 2,3,4-тринор- 1,5-интер-м-фенилен-5-иод-I4-фтор- 20-метил-РС11-метиловый эфиры.
Rf (1А-хлор-РС1,-производное) 0,76 и 0,73 (на пластине с силИкаге- лем арт.5715, после повторной хроматографии с использованием в качестве подвижной фазы смеси дихлорметана с ацетоном в соотношении 3:1).
Rf (14-фтор-РС71 -производное) 0,79 и 0,77 (на пластине с силикаге арт.5715, после повторной хроматографии с использованием в качества подвижной фазы смеси дихлорметана с ацетоном в соотношении 3:1).
П р и м е р 15. 2,3,4-Тринор-1,5- интер-м-фенилен-14-бром-20-метил- РС1|2-тетрабутиламмониевая соль.
Проводят те же операции, что и в примере 8, с той разницей, что вместо 1,75 мл I н. раствора гидроокиси
5 натрия к кислоте добавляют 3,5 мл метанольного раствора гидро0
0
5
5
0
окиси тетрабутиламмония.
,5 (на пластине с силикагелем арт.5715, подвижная фаза: смесь бензола, диоксана и уксусной кислоты в соотношении 20:20:1),
П р и м е р 16.
A.2,3,4-Тринор-1,5-интер-м-фени- лeн-5-бpoм-PGI ,-1 метиловый эфир.
508 мг (1,26 мысль) 2,3,4-тринор- 1 ,5-интep-м-фeнилeн-PGFly(-мeтилoвoгo эфира растворяют в IО мл безводной смеси хлороформа и тетрагидрофурана, взятых в соотношении 1:1, и охлаждают раствор до в смеси сухого льда с ацетоном. Добавляют к раствору 337 мг (1,86 ммоль) N-бромсукцини - мида и перемешивают смесь сначала в течение 10 мин при -78°С, а затем в течение 30 мин при комнатной температуре.
Реакционную смесь промывают 50мл хлороформа, а затем трижды 20 мл воды. Органическую фазу высушивают над сульфатом натрия и фильтруют. Растворитель отгоняют в вакууме.
Вес полученного продукта 431 мг (выход 71 /).
Rf 0,5,1; 0,57 (подвижная фаза: смесь дихлорметана с ацетоном в соотношении 3:1).
B.2,3,4-Тринор-1,5-интер-м-фе- нилен-5-бром-15- метил-PGI,-метиловый эфир.
n
Процесс проводят аналогично части А, с той разницей, что вместо 2,3,4- тринор-1,5-интер-м-фенилен-РСГ,-метилового эфира добавляют 525 мг (1,26 ммоль) 2,3,4-тринор-1,5-интер м-фенилен-15-метил-РСРэ -метилового эфира. Вес полученного продукта 430 мг (выход 69%).
-
Rf 0,49; 0,52 (подвижная фаза: смесь дихлорметана с ацетоном в соотношении 5:2).
C.2,3,4,17,18,19,20-Гептанор-1,5 интер-м-фенилен-5-бром-16-фенокси- РСГ -метиловьй эфир.
Процесс проводят аналогично части А, с той разницей, что в качестве исходного соединения вместо 2,3,4-три- нор-1,5-интер-м-фенилен-РСР -метилового эфира используют 551,8 мг (1,26 ммоль) 2,3,4,17,16,19,20-гепта нор-I,5-интер-м-фенилен-16-фенокси- PGF тилов о г о эфир а.
Вес полученного продукта 443 мг (выход 68%) .
Rg 0,48 и 0,45 (подвижная фаза; этилацетат .
D.2,3,4,17,18,19,20-Гептанор-1,5- интер-м-фенилен-5-бром-16-(3-трифтор- метилфенокси)-РС1,-метиловый эфир.
Процесс проводят аналогино части А, с той разницей, что в качестве исходного соединения вместо 2,3,4- тринор-1, 5-интер-м-фенилен-РСР тилового эфира используют 637,5 мг (1,26 ммоль) 2,3,4,17,18,19,20-геп- танор-1,5-интер-м-фенилен-16-(3-триф- торметилфенокси)-РСРг(у -метилового эфира.
Вес полученного продукта 505,7 мг (выход 68,5%).
Rg 0,46 и 0,49 (подвижная фаза : этилацетат).
74 мг (0,184 ммоль) 2,3,4-тринор- I,5-интер-м-фенилен-РСР -метилового эфира растворяют в 3 мл безводного дихлорметана, добавляют к полученному раствору 25,2 мл (0,184 ммоль) триэтиламина и охлаждают реакционную смесь с помо1цью смеси сухого льда и ацетона до -78°С. Затем к ней добавляют по каплям 356,5 мл (0,5677 ммоль /мл) раствора фенилселенилхлорида в дихлорметане и перемешивают смесь в течение 1 ч. Раствор оставляют стоять пока он не нагревается до ком
391
5
10
20
30
-
j,
35
р.
/55
45
50
50112
натйой температуры, после чего из него проводят экстракцию хлороформом, экстракт фильтруют и отгоняют из него растворитель.
Сырой продукт подвергают хроматографии на силикагеле, используя в качестве подвижной фазы смесь дихлорметана с ацетоном в соотношении 2:1.
Вес полученного продукта 45 мг (выход 44%).
Rg 0,57 (подвижная фаза : смесь ацетона с дихлорметаном в соотношении 1:2).
F.2,3,4,17,18,19,20-Гептанор-1,5- интер-м-фенилен-5-бром-11-15-диаце- токси-1б-фенокси-PGI,-метиловый эфир.
Прюцесс проводят аналогично части А, с той разницей, что вместо 2,3,4- тринор-1,5-интер-м-фенилен-РСР -ме- тилового эфира в качестве исходного соединения используют 657 мг (1,26 ммоль) 2,3,4,17,18,19,20-гепта- нор-1,5-интер-м-фенш1ен-11-15-диаце- токси-1б-фенокси-PGF -метилового эфира.
Вес полученного продукта 447 мг (выход 59%).
Rg - 0,69 и 0,71 (подвижная фаза : смесь бензола и этилацетата в соотношении l:l)i.
G.2,3,4,17,18,19,20-Гeптaнop-l,5- интep-м-фeнилeн-5-бpoм-l б-фенокси- PGI, -метиловый эфир.
518 мг (1 ммоль) 2,3,4,17,18,19,. 20-гептанор-1,5-интер-м-фенилен-5- бром-1б-фенокси-PGI,-метилового эфира растворяют в 5 мл перегнанного 1,5-диазо-бицикло-(4,3,0)-нон-5 и раствор перемешивают в течение 30 мин при комнатной температуре.
Полученную реакционную смесь разбавляют 50 мл эфира. Эфирную фазу высушивают над безводным сульфатом натрия и после фильтрации отгоняют из нее эфир. В результате получают 490 мг сырого продукта.
Сырой продукт подвергают очистке с помощью хроматографии на силикагеле (арт.7734, размер частш 0,03- 0,2 мм), используя в качестве подвижной фазы смесь эфира и ацетона в соотношении 3:1.
Вес полученного продукта 380,25мг (выход 85,4%).
Rg 0,49 и 0,51 (подвижная фаза : : этилацетат).
Таким же образом мо,--ут быть получены следующие соединения:
2,3,4-тринор-I,5-интер-м-фенилен- 15-метил-РС1j-метиловый эфир, Rg - 0,53 и 0,5А (подвижная фаза : смес дихлорметана и ацетона в соотноше- НИИ 5:2);
2,3,4,17,18,19,20-гептанор-1,5- интер-м-фенилен-16(3-трифторметил- фенокси)-РС1,-метиловый эфир, Rg - 0,51 (подвижная фаза: этилацетат).
Н. 2,3,Д-Тринор-1,5-интер-м-фени- лен-5-бром-РС1,-метиловый эфир.
Процесс проводят аналогично части А, с той разницей, что вместо N-бром сукцинимида в качестве исходного сое динения используют 487,5 мг (1,9 ммоль) N-дибромгидантоина.
Вес полученного продукта 405 мг (66,7%).
Rg 0,51 и 0,57 (подвижная фаза: смесь дихлорметанй и ацетона в соотношении 3:1).
I. 2,3,4-Тринор-1,5-интер-м-фени- лен-5-бром-РС1,-метиловый эфир.
Процесс проводят аналогично части А, с тон разницей, что вместо N-бром сукцинимида добавляют 451,5 мг (1,89 ммоль) пербромида пиперидиния.
Вес полученного продукта 394,6 мг (выход 65%).
,51 и 0,57 (подвижная фаза: смесь дихлорметана и ацетона в соотношении 3:1).
Обнаружено, что в случае соединений в соответствии с изобретением анти- агрегатное действие больше, одновременно снижающее кровяное давление действия может быть еще уменьшено, однако стабильность остается.
Для сравнения антиагрегатного и снижающего кровяное давление действия проведены сравнительные опыты. Испытаны известный простациклин, кроме того,
5(Z)-2,3,4-тpинop-l,5-интер-м-
фенилен-13,I4-дидегидропростациклин (45842/В);
5(Z)-2,3,4-тринор-I,5-интер-м-фе- нилен-20-метилпростациклин (62902);
5(Z)-2,3,4-тринор-1,5-интер-м-фе- нилен-13,14-дидегидро-20-метилпроста циклин (соединение А);
5(Z)-1,3,4-Тринор-1,5-интер-м-фе- нилен-13,14-дидегидро-20-этилпроста- циклин (соединение В).
Соединения используют в виде натриевых солей.
Агрегаты тромбоцитов измерены в двухканальном агрегометре по Иайтону в объеме крови 0,5 мл. Необ содимая для измерений кровь взята у пациентов, которые по крайней мере в течение двух недель перед взятием крови не принимали лекарственных препаратов, оказьюающих влияние на агрегаты тромбоцитов и не имели заболеваний почек и печени. Кубитальную венозную кровь разбавляют в соотношении 9:1 3,8%-ным раствором цитрата натрия и затем для получения богатой тромбоцитами плазмы центрифугируют в течение 5 мин с 230 г, в то время как бедная тромбоцитами плазма получена после 10-минутного центрифугирования. Количество тромбоцитов установлено на значении 2-3VI О на миллилит Исследования проведены от отбора пробы до расчета в течение 1ч. Перед измерением контролируют чув- , ствительность простациклина соответствующей плазмы. В случае богатой тромбоцитами плазмы для простациклина получается значение ,6 - 1,0 нг/мл.
Агрегация тромбоцитов вызвана 2f4 М AIP. Исследуемые соединения растворяли в О,1М растворе три-НС1 со значением рН 7,8 и выдерживают при . Некоторые соединения через определенное время снова замеряют и до этого момента выдерживают при 0-4°С. Из полученных результатов графически определены значения соответствующие ICjo .
Снижающее кровяное давление действие исследовано на наркотизированных крысах. Из результатов опытов рассчитаны дозы (ED) необходимые для снижения кровяного давления на 25 мм рт.ст. (3,33 кПа).
В таблице указаны относительные, считая на простациклин, значения тормозящего агрегацию и снижающего крю- вяное давление действия.
Из таблицы ясно видно, что соединения А и В по своим побочным действиям, учитывая силу эффективности, значительно выгоднее, чем известные соединения. Особенно это касается соединения В, полезное действие которого, т.е. антиагрегатное действие в основном достигает действия простациклина, в то время как нежелательное побочное действие - снижение
кровяного давления - составляет лишь 1/200 часть снижающего кровяное давление действия простациклина. Для стабильности соединений в соответст- ВИИ с изобретением характерно, что в сильно кислой среде период полураспада его разложения сохраняется еще в течение нескольких дней; из соединений в связи с этим могут быть приго- товлены также препараты для орального введения,
Соединения в соответствии с изобретением, кроме большой химической стабильности, сравнимой со стабиль- ностью простациклина, и сильного антиагрегатного и дезагрегатного действия имеют еще то преимущество, что их продолжительность действия превосходит во много раз продолжительность действия простациклина. Метаболизм природного простациклина осуществляется очень быстро. Для определения продуктов метаболизма физико-химическим путем можно доказать, что соеди- нения расщепляются либо в результате химического гидролиза, либо в результате /3 -окисления внешних цепей, либо в результате окисления 15-гидро ксильной группы до 15-кетогруппы.
В случае соединений в соответствии с изобретением эти очаги осуществления метаболизма заторможены. Ароматическое ядро, встроенное во внешнюю цепь, сразу же исключает алифати ческое /з-окисление , Критическая в отношении эффективности длина цепи не изменяется также в результате других метаболистических реакций фениль ной группы, например гидроксилиро- вания.
1C jo простациклина/1С;(, метилового эфира 2,3,4-три-1,5-м-интерфеннлен- 14-бром-20-метил PG1, 1/5
ЕД,5 простациклина/ЕД ,j метилового эфира 2,3,А-три-1,5-м-интерфени- лен-14-бром-20-метил PGI, 1-/50,
Острая токсичность для натриевой соли 2,3,4-тринор-1,5-интер-м-фени- лен-13, 14-дидегидро-20-метил-РС1/5 ((5Z),(5E) изомер 2:1) составляет i.V, 100 мг/кг .
Формула изобретения
Способ получения производных ин- тер-м-фениленпростациклинов общей формулы
М
COOKv
ГЧ Дч
/ Y-C-Z
«26 -Jp,
де R, - водород, С,-С,-алкил, щелоч- ный металл или С1-С4-тетра- алкил аммоний;
R1 - водород, тетрагидропиранил- -2, С -С -диалкил трет-бутил- си1|ил, ацетил;
R, - водород, тетрагидропиранил-2, С -С -диалкил трет- бутилси ил, ацетил;
R - водород, С,-С4-алкил;
Y - С С- или транс- СН CW, где W - хлор, бром, фтор;
Z - углеводородная цепочка с
6-7 атомами углерода, при этом в 16 полбжении проста- циклиновой структуры имеются такие заместители, как феноксиметил или трнфторме- тнлфеноксиметил,
тличающийся тем, что ак тол общей формулы
°т г:
ОН
R
OR,
где R,, R-i, R, Y и Z имеют указанные
значения,
подвергают взаимодействию с фосфора- ном общей формулы
(СбН5)
где R, имеет указанные значения; и полученные ПГФ .-производные общей формулы
ОП -х
J О1 Jx -x - C/ COOPt
L-X .Rt
R/ -C- OR
где R,, R, Rj, R , Y и Z имеют указанные значения; E - галоген;. ,
подвергают взаимодействию с электро- фильными реагентами такими, как йод . 1-бромсукци1 имид, парбромид пипери- линия, феь илселенилхлорид, и полученное производное простациклина общей формулы
1 7
9
О
COORi
139150118
где R,, Rj, Rj, R, Y, 7, и E имеют
указанные значения, подвергают реак1ц и элиминирования при
