Изобретение относится к новому способу получения нового промежуточного продукта, а именно 6/i-(1 R)- (трет-бутилдиметилсилилокси)-этил - ангидропенициллина формулы
где R.y - трет-бутилдиметилси-
лильная группа.
Цель изобретения разработка на I основе известных методов способа по- j лучения нового соединения, йвляющёго- 1 ся ключевым промежуточным продуктом I при синтезе карбапеяемовых илИ пене™ I новых антибиотиков.
I Изобретение иллюстрируется следу- :ющими примерами.
: П р И м е р 1. Получение {АК)-аце- токси-(ЗК)- (1 ,К)-{т| ет-бутилдиметклсилилоксн)этил}-2-азетидинона из ан- гкдро,-6,6-дибромпенициллина.
А, Ангидро-6,6-дибромпенициллин. Обрабатывают 20,00 г (55,5бммоль) 6,6-дибромленициллиновой кислоты, :растворенной в 200 мл метиленхлорида, |охлажденной на бане лед-метанол, по |каплям 58,4 ммоль (8 мл) триэтиламина 1и перемешивают 15 мин. К раствору |прибавляют по каплям 8 ,,40 мл 1(61,2 ммоль) трифторуксусной кислоты :Смесь перемешивают 30 мин, затем при- :бавляют по каплям 4,8 мл (6 1,2 ммоль) пиридина. Смесь перемешивают 30 мин йри -10°С, далее 18 ч при 5°С,
Смесь последовательно промывают 1 н. водной соляной кислотой, водой, 1 м раствором водным NaHCOs и рассолом, сушат над сульфатом магния. Остаток, полученный при выпаривании растворителя, снова растворяют в этилацетате (EtOAc) и обрабатывают активированным углем для полз чення целевого соединения: т.пл« 102 103°С (СНзОН), 16,, 1 г (47,2 ммоль), вы- код 85%.
Спектр ПМР (СДС1з, 80 МГц) f : 5,80 (1Н, с, Н-5); 2,21 (ЗН, с, CHj) 2,15 млн.д. (ЗН, с, СН j).
ИК-спектр (СНлгС) л) макс : 1800 (с, |5-лактам ) ; 1708 (с, лактон ) и 1640 (W, олефин).
+ 88,(с, 0,144, СНзОН). Вычислено, %: С 28,17; Н 2,07; N 4,10.
г
0
0
,.
35
40 45 СП
CjH NOjSBr.
Найдено, %: С 28,08; Н 1,98; N 4,06.
Б. Ангидро-6о( -бром-5/Ь-( Г R)-oK- сиэтилЗпенициллин.
Растворяют 15,02 г (44 ммоль) ан- гидро-6,6-дибромпенициллина в холодном (-78 С) ТГФ (450 мл), обрабатывают по каплям 18,0 мл (51,3 ммоль) 2,85 М раствора СНзМ§Вг в эфире и перемешивают 20 ютн при -78°С. Полу- ченньш в результате энолят магния улавливают избытком ацетальдегида (25 мл, 0,45 моль) и перемешивают 20 мин. Удаляют охлажденную баню и прибавляют к реакционной смеси 70 мл 1 н„ водной соляной кислоты и 300 мл эфира. Удаляют водную фазу и дважды экстрагируют по 200 мл эфира.Объединяют органические фазы, последова- тельно промьгоают 1 н. водной соляной кислотой, водой, 1 М водным раствором NaHCO и рассолом, сушат над сульфатом магния. Удаляют растворитель, получают целевое соединение (13,08 г, 42,7 ммоль), выход 97%, в виде масла.
ПМР-спектр (СДС1з) f : 5,61 (1Н, с, Н-5); 4,28 (1Н, квартет. ,0, Н-1 ); 2,21 (ЗН, с, СНР; 2,16 (ЗН, с, CHj); 1,64 (1Н, широкий синглет, ОН); 1,31 (ЗН, д, ,0 Гц, СНj) млн.д.
ИК-спектр (СН,С1з) V макс. : 3560 (м, ОН); 1785 (с, р-лактам СО); 1710 (с, лактон ) и 1635 см (м, олефин).
, + 83,8 (с, 0,128, МеОН).
Вычислено, % ,23; Н 3,95; N 4,57.
С,оН ,
Найдено, %: С 38,31; Н 4,63; N 4,61.
В. Ангидро-6с -бром-6 - f(1 R) - (трет-бутилдиметилсилилокси) -этилЗ пенициллин.
Прибавляют к 60 мл холодного (ледяная баня) раствора вещества в ме- тиленхлориде 6,0 г (.19,6 ммоль) сначала 4,50 мл (39 ммоль) 2,6-лутидина, затем по каплям 7,8 мл (34 ммоль) трет-бутш1днмет1-шсилилтрифталата. Смесь перемешивают 1 ч при 5°С, затем промывают 1 н. водной соляной кислотой, водой, 1 М водньм NaHCOj и рассо-. лом. Полученный органический слой сушат над сульфатом магния, разбавляют равным объемом смеси эфира - 140050Д
петролейного эфира (1:2) и обрабаты- целевое соединение 96,79%. Т,пл. 56- вают активированным углем. Твердый остаток после выпаривания растворителей снова растворяют в горячем гек- сане и дают выкристаллизоваться целевому соединению ,5,35 г. Концентрируют маточную жидкость, обрабатывают активированным углем и после кристал- лизаи 1и на холоде (5°С) получают 1,28 г. Объединение двух осадков дает 6,63 г защищенного по гидроксилу производного (15,7 ммоль), выход 80,6%. Т.пл. U6-U7°C (НеОН).
ПМР-спектр (СДС,) : 5,53 (1Н, с, Н-5); 4,27 (1Н, квартет, ,1 Гц, Н-1 ); 2,20 (ЗН, с, СН3); 2,13 (ЗН, с, СН,); 1,28 (ЗН, д, ,1 Гц, 0,91 (ЗН, с, трет-бутил); 0,09 (ЗН, с, CHj); 0,07 (ЗН, с, CH,) .
ИК-спектр () ч м,кс : 1785 (с, /i-пактам ); 1700 (с, лактон ) и 1635 см Чм, олефин) .
/O(/D + 119,6(9, 0,14 МеОН).
Вычислено, %: С 45,60; Н 6,45; N 3,32.
j NO SBrSi
Навдено, %: С 46,38; Н 6,02; N 3,23.
Г. Ангидро-6с -(1 К)-(трет-бутил- диметилсилилокси)этил -пенициллин.
К раствору 1,00 г (2,38 ммоль) ангид ро-бо -бром-б /3- (1 R) -(трет-бу7°С (МеОН).
rafP-спектр (C/ICIj) сГ : 5,29 (1Н, д, J 1,8 Гц, Н-5); 4,34 (1Н, двойной квартет, J 6,3 Гц, J 3,5 Гц Н-1 ); 3,52 (1Н, дд, J 3,5 Гц, J 1,8 Гц, Н-6); 2.17 (ЗН, с, СН,); 2,08 (ЗН, с, СНз); 1,25 СЗН, д, J 10 6,3 Гц, CHj); 0,89 (9Н, с, трет- бутил); 0,09 млн.д. (6Н, с, СНj).
ИК-спектр ( л «оке : 1775 (с, -лактам, ); 1695 (с, лактон ) и 1635 (м, олефин). в + 42,8°(с, 0,114 МеОН).
Вычислено, %: С 56,10; Н 8,24; N 4,09, S 9,38. C|f Н NOjSSi.
Найдено, %: С 56,65; Н 7,82; 20 N 4,07,- S 9,04.
Вьщеляют в виде масла соответствующий 6-р-(1 К)-изомер.
ГМР-спектр (СДС1з) сГ ; 5,35 (1Н, д, J 4,6 Гц, Н-5); 4,31 (1Н, двой- 25 ной KBapTeT,,0 Гц, ,4 Гц,Н-1 ); 3,83 (1Н,дд, ,6 Гц, ,4 Гц,Н-6); 2,18 (ЗН, с, СН); 2,08 (ЗН, с, CHj); 1,23 (ЗН, д, J 6,0 Гц, CHj); 0,89 (9Н, с, трет-бутил); 0,10 (ЗН, с,СНз); 30 0,06 млн.д. (ЗН, с, СИ,).
ИК-спектр (CH-,Cli) %)„акс 1785 (с, jj-лактам ); 1695 (с, лактон ) ; 1635 см- (м, олефин). +
+ 171,2 (с, 0,084 МеОН).
тилдиметилсилилокси)зтил -пеницилли- 35 Д.(3R)-(1 К)-(трет-бутилдиме- на в 25 мл 25%-ной смеси ТГФ-МеОН, тилсилилокси)(4К)-ацетокси-азетидион-1-ил р-метилкротоновая кислота.
охлажденному до -45 С, прибавляют Zn(Ag)(10 г),Цинково-серебряную пару готовят из 1 ч. ацетата серебра и 11,7 г цинка. Смесь перемешивают до тех пор, пока не израсходуются все исходные материалы (ТХС на пластине, RJ 0,4, 2% CHjCN/CH Cl,), а затем фильтруют через слой ЦЕЛИТА в холод40 Обрабатьшают раствор 5,00 г
(14,6 ммоль) ангидро-6о -(1 R)-(тpeт- бyтилдимeтилcилилoкcи)-этилJпeницил- линa в 75 мл уксусной кислоты при 22°С 14 г (44 ммоль) ацетата ртути .ном 1 М водном растворе . Фазы 45 перемешивают 24 ч. К смеси прибав- встряхивают, разделяют и экстрагируют ляют дополнительно 9,3 г (29 ммоль) водную фазу 3 раза по 10 мл эфира. ацетата ртути и продолжают перемеши- Объединяют органические фазы и после- вание еще 24 ч. Реакционную смесь довательно промывают 1 и. водной со- фильтруют через слой ЦЕЛИТА и твер5Q дый продукт промывают уксусной кислотой. Фильтрат разбавляют 150 мл воды и экстрагируют 5 раз по 40 мл эфира. Объединяют органические экстракты промывают 3 раза по 40 мл воды СЕ рассолом, сушат над сульфатом магния и обрабатывают активированным углем. При вьтаривании растворителя получаляной кислотой, водой, 1 М водным МаНСОз, рассолом, сушат над сульфатом магния. После выпаривания растворителя получают масло, которое кристаллизуется в вакууме (815 мг, 2,38 ммоль), выход 99,6%. Анализ с помощью высокоэффективной жидкостной хроматографии твердого продукта покацелевое соединение 96,79%. Т,пл. 56-
7°С (МеОН).
rafP-спектр (C/ICIj) сГ : 5,29 (1Н, д, J 1,8 Гц, Н-5); 4,34 (1Н, двойной квартет, J 6,3 Гц, J 3,5 Гц Н-1 ); 3,52 (1Н, дд, J 3,5 Гц, J 1,8 Гц, Н-6); 2.17 (ЗН, с, СН,); 2,08 (ЗН, с, СНз); 1,25 СЗН, д, J 6,3 Гц, CHj); 0,89 (9Н, с, трет- бутил); 0,09 млн.д. (6Н, с, СНj).
ИК-спектр ( л «оке : 1775 (с, -лактам, ); 1695 (с, лактон ) и 1635 (м, олефин). в + 42,8°(с, 0,114 МеОН).
Вычислено, %: С 56,10; Н 8,24; N 4,09, S 9,38. C|f Н NOjSSi.
Найдено, %: С 56,65; Н 7,82; N 4,07,- S 9,04.
Вьщеляют в виде масла соответствующий 6-р-(1 К)-изомер.
ГМР-спектр (СДС1з) сГ ; 5,35 (1Н, д, J 4,6 Гц, Н-5); 4,31 (1Н, двой- ной KBapTeT,,0 Гц, ,4 Гц,Н-1 ); 3,83 (1Н,дд, ,6 Гц, ,4 Гц,Н-6); 2,18 (ЗН, с, СН); 2,08 (ЗН, с, CHj); 1,23 (ЗН, д, J 6,0 Гц, CHj); 0,89 (9Н, с, трет-бутил); 0,10 (ЗН, с,СНз); 0,06 млн.д. (ЗН, с, СИ,).
ИК-спектр (CH-,Cli) %)„акс 1785 (с, jj-лактам ); 1695 (с, лактон ) ; 1635 см- (м, олефин). +
+ 171,2 (с, 0,084 МеОН).




Изобретение относится к гетероциклическим соединения, в частности к получению 6/1-( 1 К)-(трет-бутилди метилсилилокси)-этилJ-aнгидpoпeницил- лина (АП) общей формулы OKi .НН„ кШ I .0 тгде R - трет-бути.пдиметилсилильная группа, который может применяться в качестве промежуточного продукта при синтезе карбапенемовых или пенемовьк антибиотиков. Цель изобретения - разработка способа получения нового АП. Получение его ведут из 6,6-дибром- ангидропенициллина и в безводном инертном растворителе при (-15)-(-78)с с последую цим добавлением ацетальдегида. На образовавшийся цис-изомер действуют трет-бутил- диметилсилилтрифторметансульфонатом в присутствии органического основания при 0-5°С. Полученное при этом соединение восстанавливают в присутствии Zn-содержащего,катализатора. «о
зал следующее соотношение: исходный материал 0,70%; цис-изомер 2,51% и
ют масло, которое кристаллизуется в вакууме (5,38 г, 13,8 ммоль, выход
95%), т,пл. 119-120 С (CH-jCl,: петро лейный эфир 9:1),
ПМР-спектр (СДС1з) : 6,32 (1Н, д, J 1,4 Гц, Н-4); 4,24 (1Н, uentp из ПЯ.ТИ линий, J 6,0 Гц, Н-1 ) I 3,20 С1Н, дц, J 1,4 Гц, ,8 Гц, Нь-З); 2,24 (ЗН, с, СНз)| 2,05 (ЗН, с, СНзСО,),- 1,97 (ЗН, с, СНз); 1,29 (ЗН, д, I 6,3 Гц, СНз); 0,86 (ЗН, с, трет-бутил); 0,08 (ЗН, с, млн.д. (ЗН, с, СНэ).
ИК-спектр (,,) л)л,с«кс : 1770 (с, р-пактам ); 1745 (м, );
.це :, сн} и проводят озонолиз до полного исчезновения исходного материала (1,5 ч). Холодный (-78°с) раствор
g озонида восстанавливают (СНз)гЗ
(6 мл) и перемешивают при комнатной температуре 1,5 ч. Прибавляют 30 мл метанола, затем 1,2 мл 2,6-лутидина. Смесь перемешивают 2 ч при , раз10 бавляют эфиром, промывают 1 н, водной соляной кислотой, водой, 1М водным NaHCOj, рассолом, сушат над сульфатом магния При вьтаривании растворителя получают 1,31 г (4,56 ммоль) це1690 (м, ); 1620 см (м, олефин) „ 15 левого продукта, выход 95%, в виде
+ 18,9 (с, 0,088, МеОН).
: /с//
I Вычислено, %: С 56,07; Н 8,10;
,,бз.
i .
: Найдено, %: С 56,00; Н 8,25; ,73.
Е, (4Ю-Ацетокси-(ЗК)-(1 к)-(трет- бутилдиметилсилилокси)-этил j-2-азетиг дйонон.
К раствору г (5,2 ммоль) о {(,ЗR)( К)-(трет-бутилдиметилсилил окси) этил J-(4R)-elцeтoкcи-2-aзeтиди- 1 -ил - |,-метилкр-отоновой кислоты в 30 мл метиленхлорнда прибавляют при -15°С (баня лед-метанол) 1,63 г (6,20 ммоль) ЕЕДО. Удаляют охлаждаю- щу|ю баню и реакционную смесь переме- ши|вают 18 ч при . Ее последова- те|льно промывают 1 н. водной соляной ки слотой., водой, 1 М водным NaHCO, раюсолом, сушат над сульфатом магния, LpH выпаривании растворителя си1С|ланньй ангидрид (2,14 г выход 93:аЗ%) в виде производного, защищен- по карбоксильной функции
IMP-спектр (СДС1з) сГ : 6,23 (1Н, д, I 1,4 Гц, ); 4,32 (2Н, квартет, I 7,1 Гц, ); 4,05-4,39 (1Н, м, I 6,1 Гц, Н-1 )| 3,23 (1Н, дд, I 1,4 Гц, I . 6,1,. Н-3); 2,25 (ЗН, с, СНз)| 2,06 (ЗН, с, СНзСО ); 1,17 (ЗН, с, СНз); (ЗН, т, I 7,1 Гц, СНзСН) Ь29 (ЗН, д, I 6,1 Гц, СНз); 0,86 (9Н, с, третбелого твердого продукта. Т.пл. 104- 106°С, эфир : петролейный эфир 1:1 (литература: т.пл. 104-10б с) /cf/ + + 47,4° (с, 0,136 CHClg). Лит. дан20 ные: о1/ + 48,8 (с, 0,41 СНС1з).
И р и м е р 2. 6d -EpoM-6ft- (1 R)- оксиэтил ангидропенициллин и бо -бром- 6/3- (1 К)-(трет-бутилдиметилсилилок- си)-этилJaнrидpoпeнициллин - иллюст25 рапля реакции альдольной конденсации
при относительно высокой (-20 С) тем- .пературе.
Реагенты:ангидропенициллин 10,23 г (0,03 ммоль); CHjMgBr 12,21 мл
30 (0,0338 моль, 16% избытка, 2,85 М раствор в эфире, Альдрич); СНОСНО 6,6 г 8,4 мл (0,15 моль), of 0,788, Альдри-ч); ТГФ 150 мл (высушен над молекулярными ситами); TfOSic + 11,54 г 10,02 мл -(0,0436 моль, о( 1,151 пе- регн„)| 2,6-лутидин 6,42 г 6,98 мл (0,06 моль, 0/0,92 Альдрич); 100 мл (высушен-над молекулярными ситами).
40 К раствору 10,23 г ангидро-б5б-ди- бромпенициллина в 150 мл сухого ТГФ, охлажденному до , прибавляют по каплям 12,21 мл метилмагнийбромида в течение 10 мин, при этом температура поддерживается на уровне (-15) - (-20) С, Полученный в результате раствор переменшвают при в течение 10 мин, а затем прибавляют сначала по каплям в течение 5 мин 8,4мл
35
45
бутил); 0,08 (ЗН, с, СН,); 0,05млн„д.50 ацетальдегида, поддерживая температу(ЗН, с, СНз)„
ИК-спектр () „дс.кс : 1800 (GJ смешанньй ангидрид); 1775 (с, |}-лактам );; 1750 (с, ацетат ) | 1625 CM (W, олефин),
Растворяют 2,10 г (4,76-ммоль) полученного таким образом смешанного ангидрида в 30 мл метиленхлорида, ох- ла Кденного до -78 С (баня сухой ледру на уровне (-15)-(-20)С. К реакционной смеси прибавляют 10 мл насыщенного раствора хлористого аммония, затем 80 МП воды. Затем смесь экстраги- 55 РУют 150 мл и 50 мл этилацетата. Этил- ацетатный экстракт дважды пpo aIвaют по 100 МП рассола, сушат над безводным суль4/атом натрия и концентрируют, получая 8,9 г масла (97%). По данным
.це :, сн} и проводят озонолиз до полного исчезновения исходного материала (1,5 ч). Холодный (-78°с) раствор
озонида восстанавливают (СНз)гЗ
(6 мл) и перемешивают при комнатной температуре 1,5 ч. Прибавляют 30 мл метанола, затем 1,2 мл 2,6-лутидина. Смесь перемешивают 2 ч при , разбавляют эфиром, промывают 1 н, водной соляной кислотой, водой, 1М водным NaHCOj, рассолом, сушат над сульфатом магния При вьтаривании растворителя получают 1,31 г (4,56 ммоль) цебелого твердого продукта. Т.пл. 104- 106°С, эфир : петролейный эфир 1:1 (литература: т.пл. 104-10б с) /cf/ + + 47,4° (с, 0,136 CHClg). Лит. дан0 ные: о1/ + 48,8 (с, 0,41 СНС1з).
И р и м е р 2. 6d -EpoM-6ft- (1 R)- оксиэтил ангидропенициллин и бо -бром- 6/3- (1 К)-(трет-бутилдиметилсилилок- си)-этилJaнrидpoпeнициллин - иллюст5 рапля реакции альдольной конденсации
при относительно высокой (-20 С) тем- .пературе.
Реагенты:ангидропенициллин 10,23 г (0,03 ммоль); CHjMgBr 12,21 мл
0 (0,0338 моль, 16% избытка, 2,85 М раствор в эфире, Альдрич); СНОСНО 6,6 г 8,4 мл (0,15 моль), of 0,788, Альдри-ч); ТГФ 150 мл (высушен над молекулярными ситами); TfOSic + 11,54 г 10,02 мл -(0,0436 моль, о( 1,151 пе- регн„)| 2,6-лутидин 6,42 г 6,98 мл (0,06 моль, 0/0,92 Альдрич); 100 мл (высушен-над молекулярными ситами).
0 К раствору 10,23 г ангидро-б5б-ди- бромпенициллина в 150 мл сухого ТГФ, охлажденному до , прибавляют по каплям 12,21 мл метилмагнийбромида в течение 10 мин, при этом температура поддерживается на уровне (-15) - (-20) С, Полученный в результате раствор переменшвают при в течение 10 мин, а затем прибавляют сначала по каплям в течение 5 мин 8,4мл
5
5
ацетальдегида, поддерживая температуру на уровне (-15)-(-20)С. К реакционной смеси прибавляют 10 мл насыщенного раствора хлористого аммония, затем 80 МП воды. Затем смесь экстраги- РУют 150 мл и 50 мл этилацетата. Этил- ацетатный экстракт дважды пpo aIвaют по 100 МП рассола, сушат над безводным суль4/атом натрия и концентрируют, получая 8,9 г масла (97%). По данным
высоко эффективной жидкостной матографии имеется 92% цис-: не имеется транс-изомера, 8% примесей. В хроматографии используют: Колонка Порасил (Вотерс) 3% CHgC/CHjCl,
Растворитель Скорость потока
Детекция Разбавление
90 мл/ч УФ 275 нм 0,2
Указанное сырье-масло растворяют в 100 мл сухого метиленхлорида, охлажденного до 0 С. Прибавляют 6,98 м
2,6-лутидина, затем по каплям в тече-15 экстр акт промыва-от 2 раза по 300 -т
вне 20 мин прибавляют 10,02 мл трет- бутилдиметилсилилтрифторметансуль фоната, поддерживая температуру 0-5 С. Полученный в результате раствор перемешивают 1 ч при 0-5°С. Данные ТХС 20 (окись кремния, эфир : петролейный эфир 1:1,12) показывают, что реакция завершилась. Реакционную смесь промывают 100 мл 1 н. соляной кислоты, 100 мл насыщенного раствора NaHCO 5 и 100 мл рассола соответственно, сушат над безводным сульфатом натр1-ш и кондентрирутот, получая темное мас- -io, которое постепенно затвердевает. Масло растворяют в теплом петролейном 30 эфире, обрабатывают углем и концентрируют досуха (14 г). Сырой твердый продукт снова растворяют в 70 мл теплого йзопропанола, разбавляют 35 мл воды, пока раствор еще является теплым, охлаждают до 0°С и фильтруют. Осадок на фильтре промывают смесью
изопропанол : вода 2:1 и сушат в вакуумном эксикаторе. Выход 7,0 г (55% в двух стадиях).
Примерз. 6o -BpoM-6/i-(1 R)- (трет-бутилдиметилсилилокси)этил ангидропенидиллин - иллюстрация использования CH3MgCl в качестве реактива
Гриньяра.
35
40
Реагенты: ангидропенициллин 34, 1 г (0,1 моль); CHjMgCl 39,6 мл (0,116 моль, 16% избытка, 2,9 М раствор в ТГФ); CHjCHO 28 мл 22 г (0,5 моль) d 0,788); ТГФ 350 мл (высушен над молекулярйыми ситами); 2,6- лутидин 23,3 мл 21,4 г (0,2 моль, о1 0,92, высушен над КОН); TfOSix + 32 г 27,6 мл (0,12 моль, «t 1,151); толуол 800 мл.
Охлаждают раствор 34,1 г ангидро- 6,6-дибромпенициллина в 350 m сухого ТГФ др -45°С и прибавляют туда по
каплям 39,6 t-m м-егилмагн;.;;хлорида, поддерживая температуру Н1сже -40° С . Полученный в результате раствор перемешивают 10 мин при температуре в интервале (-45)-(-40)С и прибавляют сначала по каплям 28 мл ацетальдеги- да в течение 5 мин, поддерживая температуру ниже . Раствор перемешивают 15 мин при -40 С. К peaKijjiOH- ной смеси прибавляют 35 мл насьпцен- ного раствора хлористого аммония, затем 400 мл воды. Смесь зкстрагиру- ют 350 и 150 мл толуола, Толуольный
рассола, сушат н,я;; безводным сульфатом магния и кок;:,ентрируют до объема 100 мл. К концентрированному раствору прибавляют 300 мл толуола, затем продолжают концентрирование до примерно 300 мл.
Высоко эффективная жидкостная хроматография показывает 92% чистоты, нет транс-изомера.
ка
Порасил (Вотерс) 3% CHjCN/CH.Cl 90 мл/ч УФ 275 нм 0,2
Толуольный раствор охлаждают до G°C и прибавляют 23,3 мл 2,6-лутидина, затем по каплям прибавляют в течение 15 мин 27,6 мл трифлата. При этом поддерживается температура О
5 С. Полученный в результате раствор перемешивают при 0°С в течение 1 ч. ТСХ: силикагель, эфир : петролейный эфир 1:1,1 г. К реакционной смеси прибавляют 250 m воды и устанавливают рН 2,5-5 с помощью примерно 8 мл концентрированной соляной кислоты. Отделяют органический слой. К органическому слою прибавляют 250 мл воды и устанавливают рН 8,0 с помощью 10 мл 1%-ного гидроксида натрия. Органический слой промывают 2 раза по 250 МП рассола, обрабатывают 15 г угля и концентрируют до примерно 50 мл. К концентрату прибавляют 200мл
изопропанола и по каплям 100 мл воды при перемешивании. Удаляют при пониженном давлении 100 мл растворителя, получают пасту, которую ох.паждают до 0°С, перемешивают 0,5 ч и фильтруют.
Осадок на фильтре промывают охлажденной на льду смесью изопропанола и воды 2:1 (80 мл), сушат в вакуум-эксикаторе. Выход 29 г (69%), т.пл, 95 - 100°С, Сырой продукт перекристаллизолывают следуюишм образом. Растворнгот п 150 мл толуола, обрабатывают углем, но возможности больше концентрируют, прибавляют 200 мл изопропанола, затем по каплям прибавляют 100 мл воды при охлаждении льдом и перемешивании. ОтфильтровЕ,шают осажденный продукт, промывают охлажденной на льду смесью изопропанол : вода 2:1 и сугаат в ва- куум-эксикаторе. Выход 21,0 г (50), т.пл. 104-108°С.
П р и м е р 4. 6(У-Г( 1 R)-тpeт-бy тилдимeтилcилилoкcи) этил ангидропени- циллин - иллюстрация использования восстановления цинком (уксусной кислоты) ,
Реагенты: ангидропенициллин 42,0 г (0,1 моль)J цинковый порошок 42,0 г (0,65 моль, Анахемиа); уксусная кис- лота 11 мл 11,54 г (О, 19 моль, d- 1,049, ледяная); СНзОН 1000 мл,
В трехгорлую колбу емкостью 2 л, снабженную механической мешалкой, термометром, капельной воронкой и вводом азота и отводной трубкой, загружают и суспендируют 42 гангидро- 60f-6poM-6/b- f( l R)-oкcиэтнлJпeницилли- на в 100 мл метанола. Реакционную I смесь охлаждают до -20 С и прибавля- Iют туда 42 г цинкового порошка, затем iмедленно, прибавляют 11 мл уксусной кислоты. Полученную в результате - смесь перемешивают при температуре (-25)-(-15) С в течение 0,,5 ч. ТСХ I (окись кремния, эфир : петролейный |эфир 1:3,22 или раствор молибдата) показывает завершение реакции. Реакционную- смесь фильтруют на ЦЕЛИТЕ в И 00 мл насыщенного раствора хлористо- го аммония и промывают ЦЕЛИТ метилен- хлоридом.. Фильтрат разбавляют 500 мл воды и экстрагируют 1000 и 500 мл метилс-нхлорида. Метиленхлоридный экстракт промывают 1000 мл рассола, су- шат над безводным сульфатом магния и концентрируют для получения 34 г (100% сырого) масла, которое постепенно кристаллизуется. Анализ показывает 83,3% транс-, 5,7% цис- и 11% примесей.
П р и м е р 5. Получение ангидро- 6о(- и 6/i- f(l К)-оксиэтил пенициллина
Обрабатывают охлажденный на бане лед-метанол раствор 4,20 г (13,7 ммоль ангидро-6о(-бром-6/Ь- (1 К)-оксиэтил пенициллина в 40 мл метанола 4,2 г Zn(Ag) и перемеишвают 15 мин, Прибавляют еще 1,1 г Zn(Ag) и смесь переме
5
0
5 0 0 5 0
5
шивают 10 мин. Холодную суспензию фильтруют через ЦЕЛИТ в холодный концентрированный водный раствор хлористого аммония. Твердый продукт промывают эфиром и разделяют две фазы.Водную фазу экстрагируют 2 раза по 20 мл эфира. Эфирные экстракты объединяют и промывают последовательно 1 н. водной соляной кислотой, водой, 1 м водным КаНСОз и рассолом, затем сушат. Остаток после выпаривания растворителя тщательно растирают с холодной смесью петролейный эфир : эфир 9:1, получают 1,3 г (5,7 ммоль, выход 42%) в виде белого твердого продукта: т.лл. 174-174 С ( эфир 2:8).
ПМР-спектр (СДС1з) : 5,28 (1К, д, I 1,7,Н-5); 4,35 (1Н, квинтет, Г 6,2, Н-Г); 3,57 (1Н, дд,,2, I 1,7, Н-6); 2,19 (ЗН, с, СНд); 2,11 (ЗН, с, СНз); 1,72 (1Н, широкий синглет ОН); 1,40 млн.д. (ЗН, д, I 6,3, СНз).
ИК-спектр (.) : -иллпкс : 3500 (W, ОН); 1775 (с, ft -лактам )| 1695 (с, лактон ) | 1635 см Чм, олефин).
Вычислено, %: С 52,84; Н 5,76; N 6,16.
CioHijNO S.
Найдено, %: С 52,86; Н 5,72;N 6,08,
Вьшаривают холодную смесь эфир : : петролейный эфир 1:9 и вьщеляют чистый 6 -изомер с помощью препаративной ТСХ (2% CHgCN/CH Cl)-.
ПМР-спектр (СДС1з) (f : 5,40(1Н, д, I 4,6, Н-5); 4,36 (1Н, двойной квартет, I 6,1, I 9,0, Н-1); 3,74 (1Н, дд, I 9,0, ,6,Н-6); 2,19 (ЗН, с, СН,); 2,25-1,90 (1Н, широкий синглет, ОН); 2,09 (ЗН, с, СНз); 1,27 млн.д. (ЗН, д, ,6, CH,
ИК-спектр (CHjCl) )макс : 3580 (W, ОН); 1770 (с, р-пактам ); 1700 (с, лактон ); 1640 см (м, олефин).
Условия реакции не оптимизированы, при повторении при -50°С бьт получен выход 56,6% 6с|(-изомера. Формула изобретения
ORiiHH
-R-y-
0 -тя- «
о
S п
-hf f
Qsss -NЦподвергают взаимодействию с СНзМ§С1 в безводном инертном растворителе при температуре от -15 до -78°С с последукнцим добавлением ацетальдеги- да, на образовавшийся цис-изомер общей формулы
.действуют трет-бутилдиметилсилилтри- фторметансульфонатом в присутствии органического основания при ,полученное при этом соединение общей ORi.BrH
.S.,0
где R имеет указанные значения;
подвергают восстановлению в присутствии Zn-содержащего катализатора.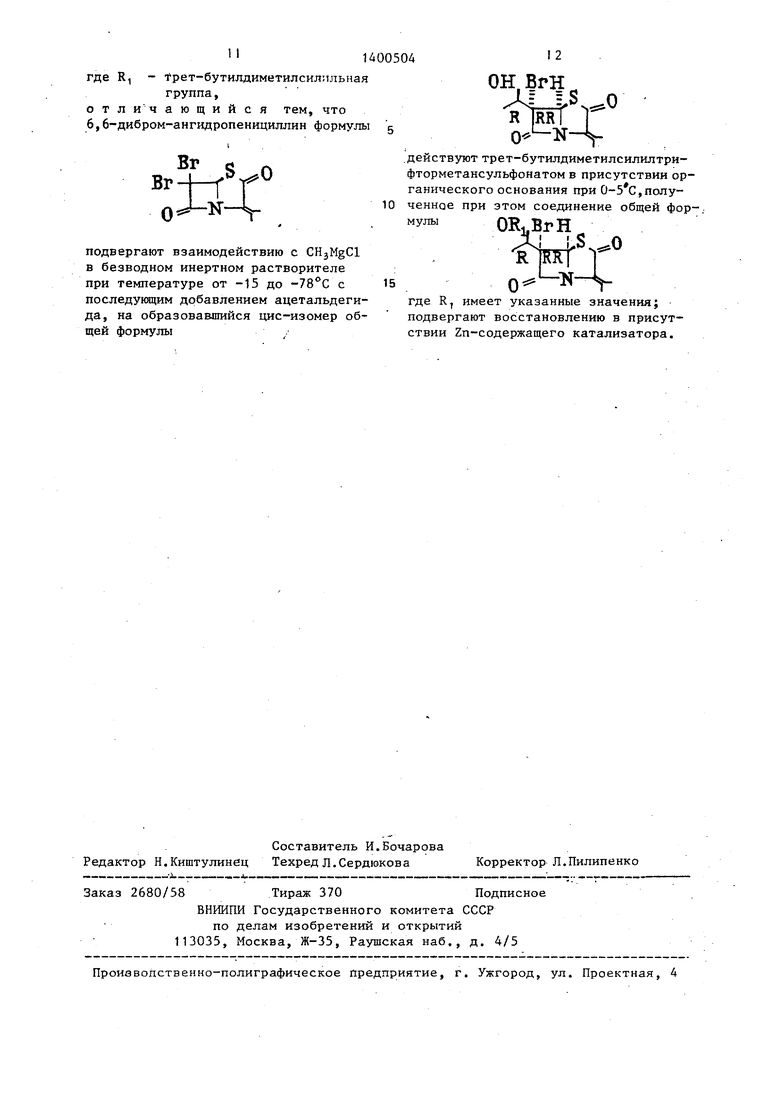
| Патент США № 3943123, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Tetrahedron Letters | |||
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
