H3C-CH(OH)j-C(-HVHCRi-f|-5-А-О г
C(0)-ORi
где R - С ;,-С4 злкил; С -С -ал- кил, бензил или алкилкарбоксиметил; R- - Н, napa-нитробензильная защитная группа при наличии противоиона; А - -(СН)- или -(CHj)-; моно- или дизамещенные С,- С 4-алкилом группы
N-N
-@;4j; AJ)
V
N-N
Ч I
41
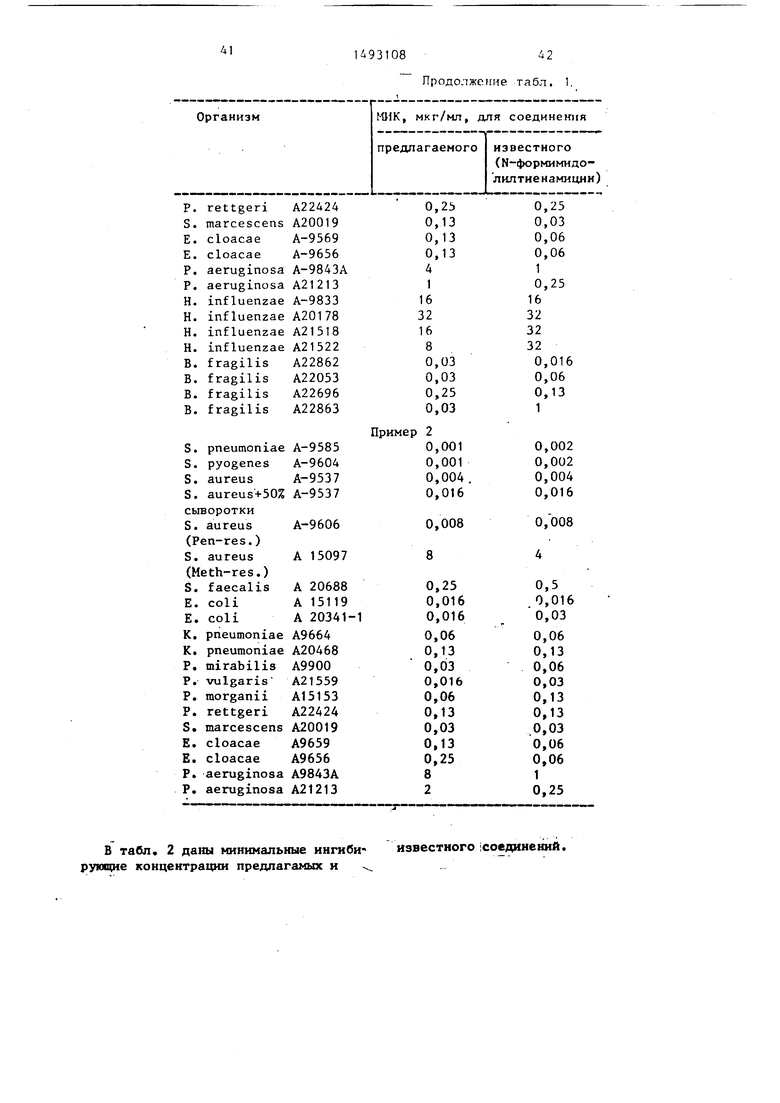
Nпричем указанное кольцо присоединено к А через углерод кольца и содержит азот, кватернизированный R., которые обладают антибактериальной активностью, что может быть использовано медицине. Цель - создание новых тивны( и малотоксичных веществ указанного класса. Синтез ведут реакцией соответствующего кетоэфира (вместо
в
VHCRi-f|-5-А-О г
я и
сой
в
C(0)-ORi
группы RIB ф-ле (I)
держится 0) дифенилхлорфосфатом в присутствии диизопропиламина в среде инертного органического основания с последующей обработкой производным
меркаптана формулы HS-A--r- N
см. вьше, в среде инертного
органического растворителя в присутствии диизопропилэтиламина и дальнейшим алкилированием соединение м ф-лы RJ - X, где X - галоген или группа сульфонатного сложного эфира; RI см. вьппе. Выделение целевого лродук- та ведут в свободном виде или при необходимости, когда R 3 - napa-нитробензильная группа, удалением карбок- сизащитной группы. В другом случае исходят из соединения, содержащего
группы -S-A-Q J , которое алкйлируют
о S
сл
4
со
СА9
00
сх
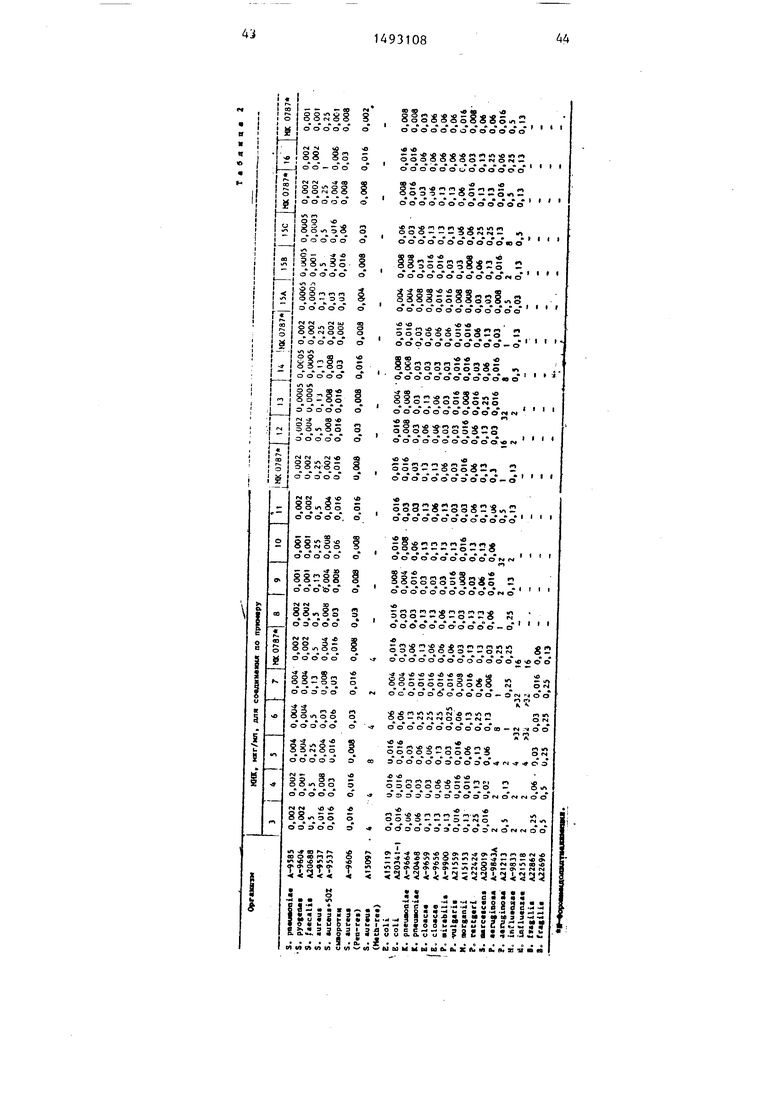
соеди)1еннсм ф-лы RjX с аналогичным выделением или удалением карбоксиза- щитной группы. Новые вещества имеют невысокую токсичность, например , , l-i мг/кг при активной дозе 5 мг/кг
(без признак токсич гости) в отношении к различным грамотрицате)1ьным и грамположительным бактериям. 2 с.п. ф-лы, 8 табл.




















Изобретение касается гетероциклических веществ в частности получения производных 7-оксо-1-азабицикло (3,2,0)гепт-2-ен-2-карбоновой кислоты общей ф-лы 1 @ , где R1-C1-C4-алкил
R2-C1-C4-алкил, бензил или алкилкарбоксиметил
R3-H, пара-нитробензильная защитная группа при наличии противоиона
A--(CH2)- или -(CH2)2-
- @ N+-моно- или дизамещенные C1-C4-алкилом группы @
@
@
@
@
@
@
@ , причем указанное кольцо присоединено к A через углерод кольца и содержит азот, кватернизированный R2, которые обладают антибактериальной активностью, что может быть использовано в медицине. Цель - создание новых активных и малотоксичных веществ указанного класса. Синтез ведут реакцией соответствующего кетоэфира (вместо группы S-A- @ N+-R2 в ф-ле 1 содержится "о")дифенилхлорфосфатом в присутствии диизопропиламина в среде инертного органического основания с последующей обработкой производным меркаптана формулы HS-A- @ N, где A и @ N- см.выше, в среде инертного органического растворителя в присутствии диизопропилэтиламина и дальнейшим алкилированием соединением ф-лы R2-X, где X- галоген или группа сульфонатного сложного эфира
R2-см.выше. Выделение целевого продукта ведут в свободном виде или при необходимости, когда R3-пара-нитробензильная группа, удалением карбоксизащитной группы. В другом случае исходят из соединения, содержащего группы -S-A- @ N, которое алкилируют соединением ф-лы R2X с аналогичным выделением или удалением карбоксизащитной группы. Новые вещества имеют невысокую токсичность, например, ЛД50= 14 мг/кг при активной дозе 5 мг/кг (без признаков токсичности) в отношении к различным грамотрицательным и грамположительным бактериям. 2 с.п. ф-лы, 7 табл.
Изобретение относится к области получения новых карбапенемных производных, а именно производных 7-оксо- 1-азабицикло(3,2,0)гепт-2-ен-2-карбо- новой кислоты общей формулы
ОН I
RI
HC S-A-C - R
0
де I водород или С -С -алкил;
С
R- - С -С -алкил, бензил или алкилкарбоксиметил;R - водород или пара-нитробензильная запцстная труппа при
наличии также и противоиона; А - группа (CHj)j,, где п 1
или 2; 1 -. моно- или дизамещенные
С -С4 алкилом группы:
O -fy
N К
1У-ТУ: 1
причем указанное кольцо присоединено к А через атом углерода кольца и содержит атом азота, который кватер- низирован группой R, обладающих антибактериальной активностью по отношению к различным грам- отрицательным и грамположительным бак териям.
Целью изобретения является разработка на основе известных методов способа получения новых соединений, обладающих ценным фармакологическим свойством при низкой токсичности.
Пример 1. Получение внутренней соли 1-метил-4-(2-карбокси)- 6о(- {t1-(R)-oкcиэтил -7-оксо-1-азаби5
0
5
0
5
0
5 /
цикло(3,2,0)гепт-2-ен-З -тиометилI- пиридиний гидроксида.
А. Раствор 673 мг (1,86 ммоль) па- ра-нитробензил-6о/- 1-(R)-OKCH3Tmi J- 3,7-диоксо-1-азабицикло(3,2,0)гепт- 2-ен-2-карбоксилата (соединение I) в 10 мл ацетонитрила охлаждают в атмосфере азота. Прибавляют раствор 245 мг (1,90 ммоль) диизопропилэтил- амина в 1 мл ацетонитрила, а затем по каплям в течение 2 мин прибавляют 510 мг (1,90 ммоль) дифенилхлорфосфа- та в 1 мл ацетонитрила. Результирующий раствор перемешивают при в течение 15 мин с получением пара-нит- робензилового эфира 3-(дифенилфосфо- рилокси)-6о(- 1-(R)-oкcиэтил J-7-OKCO- 1-азабицикло(3,2,0)гепт-2-ен-2-карбо- новой кислоты. К этому раствору прибавляют раствор 245 мг (1,90 ммоль) диизопропилэтиламина в 0,5 мл ацетонитрила, а затем раствор 270 мг (2,16 ммоль) 4-меркаптометилпиридина в 0,5 мл ацетонитрила. Реакционную смесь перемешивают при в течение 60 мин, и образовавшийся белый осадок собирают фильтрованием, промывают 5 мл охлажденного льдом ацетонитрила и получают 660 мг (выход 76%) П-нитробензил-3-(пиридин-4-ил-метан- тио)-6о(- 1 -(R)-oкcиэтил 3-7-оксо-1- азабицикло (3,2,0)гепт-2-ен-2-карбок- силата в виде белых-кристаллов, т.пл, 145°С.
Рассчитано: С 58,01; Н 4,56; N 9,23; S 7,04. ,N/)S
Найдено: С 57,74; Н 4,56; N 9,58; S 7,21 .
К раствору 660 мг (1,41 ммоль) промежуточного соединения 2 в 140 мп ацетона прибавляют 5 мл йодистого метила. Раствор реагентов перемешивают при 25 С в течение 8 ч. Растворитель вьтаривают под вакуумом и получают бледно-желтое твердое соединение, которое растирают с диэтиловым эфиром и получают 779 мг (выход 90%) п-нитробензил-3-(Ы-метилпиридин-4J 1Д
ил-метаитио)-Гхз(-Г1-(К)-оксиэтил 1-7- оксо-1 азабии 1кло(3 ,2 ,0) гепт-2-ен-2- карбоксилата в виде белого аморфного твердого соединения, т.пл. (г разложением).
Рассчитано: С 44,39; Н 4,22; N 6,82; S 5,20.
С2эН24МэОбЗ Е20
Найдено: С 44,66; Н 4,0; N6,84; S 5,64.
В. К раствору 779 мг (1 ,27 ммоль) соединения 3 в смеси тетрагидрофуран вода - диэтиловый эфир (80 мл - 80 мл - 100 ма) прибавляют 140 мг (1,4 ммоль) бикарбоната калия и 125мг (0,7 ммоль) дикалийфосфата. Затем прибавляют 700 мг 10%-ного палладия на угле и смесь гидрируют при 2,8 атм в течение 45 мин в шейк эре Парра. Затем смесь фильтруют и катализатор промывают водой (2x10 мл). Объединенные фильтрат и промывные воды экстрагируют диэти- ловым эфиром (150 мл), лиофилизуют и получают коричневьш порошок. Это сы- рое соединение очищают на обращенно- фазовой колонке .с 30 г C jBONDAPAK (Water Associates), элюируя водой при давлении 0,56 атм. Каяодую фракцию (20 мл) анализируют с помощью жидко- стной хроматографии высокого давления, фракции, поглощающие УФ 300 нм, собирают, лиофилизуют и получают 135 мг (выход 32%) указанного в заглавии соединения 4 в виде слабо-желтого твердого соединения.
Пример 2, Способ получения 3-(Н-метилпиридин- 4-ил-этантио) 1-(и)-оксиэтил }-7-оксо-1-азабицйкло- (3,2,0)гепт-2-ен-2-карбоксилата.
А. Суспензию 1,1 г (2,93 ммоль) Зо((К)-оксиэтш1 3-4 (3-диазо-З-П- нитробензилоксикарбонил-2-оксопропил) азетидин-2-она в 30 мл сухого бензола в течение 5 мин продувают азотом при комнатной температуре. Ее обрабатьгоа- ют 25 мг димера ацетат родия и смесь кипятят с обратным холодильником в течение 45 мин. Теплый раствор разбавляют этилацетатом (25 мл), фильтрованием удаляют катализатор, вьтаривают до сухого остатка и получают п-нитро- бензил-6- 1 -(R)-оксиэтил J-3,7-диоксо- 1-азабицикло-(3,2,0) гепт-2-ен-карбок силат.Это соединение растворяют в су; хом ацетонитриле (20 мл) и охлаждают до -10°С. К этому раствору при продувании азотом прибавляют 417 мг (3,2 ммоль)диизопропилэтиламина, а
Q
g п 5 о
0
5
0
08
затем 810 мг- (3,0 ммоль) дифенилх.:;( р фосфата, и реакционную смос перемешивают при в течение 20 , Затем реакционную смесь обрабатывают диизопрогшлэтиламином (420 пг, 3,2 ммоль) и 2-(4-пиридил)этантиолом (560 мг, 4,03 ммоль) в 2 мл ацето- нитрила. Реакционную смесь перемешивают при температуре от -5 до -10 С в течение 1 ч, затем разбавляют мети- ленхлоридом (100 мгг) и тщательно промывают смесью рассол - (1:1), 4%-ной фосфорной кислотой, 5%-ным бикарбонатом натрия, водой и рассолом. Органическую фазу сушат (над сульфатом магния), выпаривают и получают белое твердое соед1тнение. Это твердое соединение промывают смесью диэтиловый эфир - гексан (1:1), cyuiai над высоким вакуумом и получают 901 мг (63,9%) П -нитробензил-3- Iv-метилпири- дин-4-ил-этантио -bcf- (1-(R)-оксиэтилJ- 7-ОКСО-1-азабицикло(3,2,0)гепт-2-ен- 2-карбоксилата.
B.Суспензию полученного на стадии А карбапенема (890 мг, 1,85 ммоль)
и 7 мл иодометана в 200 мл сухого ацетона и 12 мл метиленхлорида пере- мешивают при 25 С в течение 24 ч. Реакционная смесь образует прозрачньп раствор за 18 ч. Растворитель удаляют при пониженном давлении, остаток промьшают диэтиловым эфиром и получают 920 мг (1,48 ММОЛЬ} 79,8%) 3-(N- метилпиридин- 4-Ш1-этантио)-6в(- f1-(R)- оксиэтилЗ-7-оксо-1-азабицикло(3,2,0)- гепт-2-о1;-2-карбоксилата в виде твердой пены.
C.Карбапенем со стадии В (920 г,
1,47 ммоль) растворяют в 90 мл тетра- гидрофурана, 90 мл диэтилового эфира и 90 мл воды и обрабатывают 265 г (1,51 ммоль) дикалийфосфата, 190 мг (1,9 ммоль) бикарбоната калия и 800 мг 10%-ного палладия на угле. Проводят гидрирование при 3,15 атм в течение 1 ч. Фильтрат отфильтровьшают на
. CELITE и промывают диэтиловым эфиром (3x25 мл). Водный слой лиофилизуют, получая коричневатое соединение, которое затем дважды хроматографически очищают на обращенно-фазовой колонке с 12 г С fi/С (jBONDAPAK (Waters
Associates) с использованием воды в качестве элюента, и получают 55 мг 3-(N-метилпиридин-4-ил -этантио- 6с Г1 - (R)-оксиэтил 1-7-ОКСО-1-азабицикло- (3,2,0)гепт-2-еи-карбоксилата
7
Пример 3. Получение 3-(N- метилпиридин-3-илметантио) 1- (R)- оксиэтил -7-оксо-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксилата.
A.Пара-нитробензиловый эфир 3- (пиридин-3-ил-метилтио)(К)-ок- сиэтнлJ-7-oкco-1-aзaбициклo(3,2,0)- гeпт-2-eн-2-кapбoнoвoй кислоты.
К охлажденному (О С) раствору 925 мг (2,66 ммоль) п-нитробензил- 3,7-диоксо- 1-(К)-оксиэтил 3 1-азаби- цикло(3,2,0)-гептан-2-карбоксилата в
14мл ацетонитрила прибавляют раствор 377 мг (2,9 ммоль) диизопропил- этиламина в 1 мл ацетонитрила, а затем раствор 786 мг (2,9 ммоль) дифе- нилхлорфосфоната в 1 мл ацетонитрила в атмосфере азота. Результирующий раствор перемешивают при 0°С в течение
15мин, а затем прибавляют раствор 377 мг (2,9 ммоль) 3-меркаптометилпи- ридина в 2 мл ацетонитрила. Реакционный раствор перемешивалот в течение
90 мин при . Осадок собирают филь- трованием, промьшают 20 мл ацетилаце- тата и получают 950 мг (выход 60%) указанного соединения в виде белых кристаллов.
Рассчитано: С 58,01; Н N 9,23; S 7,04.
.,
Найдено: С57,19;Н 5,19; N 8,76; S 7,08.
B.Получение 3-(Н-метилпиридин-3- ил-метантио)-6о{- 1-(К)-оксиэтил -7- оксо-1-азабицйкло(3,2,0)гепт-2-ен- карбоксилата.
К раствору 730 мг (1,56. ммоль) полученного соединения в 120 мл ацетона прибавляют 5 мл метилиодида и реакционную смесь перемешивают в течение 18 ч при комнатной температуре. Осадок собирают фильтрованием, промывают ацетоном (10 мл) и получают , 940 мг (выход 100%) кватернизованногЬ пиридина - натрий 3-(Н-метилпиридин- 3-ил-метантио-6о(- 1 - (R) -оксиэтил J-7- оксо-1-азабицйкло(3,2,0)гепт-2-ен-2- карбоксилата - в виде бледно-желтогб порошка.
Рассчитадо:С 46,24; Н 4,05; 7,03; S 5;37.
4,65;
N
q,H2,N,,I,
Найдено: С 45,82; Н4,11; N6,87; S 6,10.
К раствору 933 мг (1,6 ммоль) полученного соединения в 90 мл тетра- гидрофурана и 90 мп эфира прибавляют
10
20
25
493108
200 мг
8
KHCOj затем
30
45
50
55
130°С (сраз- Н 5,44; .
Н 349 мг KjHPO в 90 чп воды, а затем 1,0 г палладия на угле. Смесь гидрируют при 3,15 атм в шейке- ре Парра в течение 45 мин. Смесь отфильтровывают на слое CELITE и катализатор промывают водой (2x10 мл) . Объединенные фильтрат и промывные воды экстрагируют дизтиловым эфиром (2x100 мп) , лиофилизуют и получают желтое твердое соединение,которое очищают на обра- щенно-фазовой колонке с 8 г С., ВОШАРАК (Waters Associates) и элюируют 5%-ным раствором ацетонитрила в воде при дав- 15 лении 0,56 атм. Каждую фракцию по 15 МП анализируют с помощью жидкостной хроматографии высокого давления, фракции, поглощающие УФ при „ 300 им, собирают, лиофилизуют и получают 230 мг (выход 43%) указанного в заглавии соединения в виде бледно- желтых кристаллов, т.пл ложением).
Рассчитано: С 51,87; N 7,56.
C,,H,,,-H,jO
Найдено: С 51 ,95; Н 5,66; N 7,56.
Пример 4. Получение 3-(N-ме- тилпиридин-2-ил-метантио) -6 rit- 1 - (R) - оксиэтил J-7-оксо 1-азабицйкло(3,2,0)- гепт-2-ен-2-карбоксилата.
А. Пара-нитробензил-3-(пиридин-2- ил-метантио))-оксиэтил J-7- оксо-1-азабицйкло(3,2,0)гепт-2-ен-2- карбоксилат.
К охлаяоденному (0°С) раствору 925 мг (2,65 ммоль) п-нитробензил- 3,7-диоксо- 1-(К)-оксиэтилЗ-1-азабицйкло (3 ,2,0)гептан-2-карбоксилата в 15 мл ацетонитрила прибавляют раствор 377 мг (2,92 ммоль) диизопропил- этиламина в 1 мл ацетонитрила, а тем 786 мг (2,90 ммоль) дифенилхлор-- фосфата в 1 мл ацетонитрила в атмосфере азота. Полученный раствор перемешивают при в течение 15 мин, прибавляют раствор 377 мг (2,92 ммоль) диизопропилэтил- амина в 1 мл ацетонитрила, а затем раствор 350 мг (3,0 ммоль) 2-меркап- тометилпиридина в 1 мл ацетонитрипа. Реакционньй раствор перемешивают в течение 2 ч при -Ю с. Осадок собирают фильтрованием, промьтают 20 мп метштенхлорида и получают 650 мг Хвыход 54%) указанного соединения в виде желтого порошка.
Рассчитано: С 58,01; Н 4,65; N 9,23; S 7,04.
35
40
.,
Найдено: С 57,56 ; Н4,92;N8,94; S 7,03,
В. Получение 3-(Ы-метилпирндин-2- ил-метантио)-6о((К)оксизтил J-7- оксо-1-азабицикло(3,2,0)гепт-2-ен-2- карбоксилата.
К раствору 650 мг (1,39 ммоль) П-нитробензил-3-(пиридин-3 -ил-метан тио)-6о(- 1-(К)-оксиэтил J-7 Oкco-1- aзaбициклo (3,2,0) гепт-2-ен-2-карбок- силата в 100 мл ацетона прибавляют
4мл метилиодида. Реакционную смесь перемешивают в течение 3 дней при комнатной температуре. Осадок собирают фильтрованием, промывают ацетоном (10 мл) и получают 500 мг (выход 60%) кватернизованного пиридина- родиевая соль 3-(Ы-метилпиридин-2- ил-метантио )-без/- 1 - (R)-оксиэтил J-7- оксо-1-азабицикло(3,2,0)гепт-2-ен-2- карбоксилата - в виде бледно-желтого твердого соединения.
Рассчитано: С 46,24; Н 4,05; N 7,03; S 5,37.
.,,
Найдено: с 45,62; Н4,27; N6,80;
55,30.
К раствору 1,0 г (1,167 ммоль) полученного соединения в 90 мл тетра- гидрофурана и 90 мл диэтилового эфира прибавляют 215 мг (2,15 ммоль) KHCOj и 374 мг (2,1 ммоль) KjHPO B 90 мл воды, а затем 1 г 10%-ного палладия на угде. Смесь гидрируют при 3,15 атм в шейкере Парра в течение 45 мин. Смесь отфильтровывают через слой СБЫТЕ и катализатор промьшают водой (2x10 мл). Объединенные фильтрат и промывные воды экстрагируют ди этиловым эфиром (2x200 мл), лиофили- зуют и получают желтое твердое соединение, которое очищают на обращен- но-фазовой колонке с 10 г С,ВОШАРАК (Waters Associates) при давлении 0,56 атм с использованием в качестве элюента 5%-ного раствора ацетонитри- ла в воде. Каждую фракцию по 15 мл анализируют с помощью жидкостной хроматографии высокого давления, фракции, имеющие поглощение УФ при 300 нм, собирают, лиофилизуют и получают 390 мг (выход 44%) указанного продукта. Перекристаллизацией этого соединения из смеси вода - ацетон - этанол получают тонкие игольчатые кристаллы, Т-.ПЛ. 194-196 4: (с разложением) .
.
o
5
0
5
0
5
0
5
O
5
.Рассчитано: С 51,87 И 5,44; N 7,56.
CtsH2gNjO S,.2H
Найдено: С 51,37; Н 5,69; N7,37.
Пример 5. Получение 3-(N- метилпиридин-2-ил-этантио)-6о(-(, 1-(R) - оксиэтил J-7-OKCO-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксилата.
A.Пара-нитробензил-3-(пириднн-2- ил-этантио)-6о((К)-оксиэтнл J-7- оксо-1-азабицикло(3,2,0)гепт-ен-2- карбоксилат.
К oxлaждeннo fy раствору 1,78 г (5,0 ммоль) п-нитробензил-6с/ - 1-(R)- оксиэтил J-3,7-диоксо-1-азабицикло- (3,2,0)гепт-2-ен-2-карбоксилата в 25 мл ацетонитрила прибавляют 710 мг (5,5 ммоль) диизолропилэтиламина в 1 мл ацетонитрила, а затем 1,4 г (5,и ммоль) дифенилхлорфосфатав 1 мл ацетонитрила в атмосфере азота.
Полученный раствор перемешивают в течение 20 мин при 0°С, прибавляют раствор 710 мг (5,5 ммоль) диизопро- пилэтиламина в 1 мл ацетонитрила, а затем раствор 850 мг (6,1 ммоль) 2- меркаптоэтилпиридина в 2 мл ацетонитрила. Реакционную смесь перемешивают 60 мин при . Осадок собирают фильтрованием, промывают метиленхлоридом (20 мл) и получают 1,3 г (57%) указанного соединения в виде твердого желтого продукта.
B.Получение 3-(N-мeтилпиpидин-2- ил-этантио) 1г (К)-оксиэтил -7-oк- co-1-aзaбициклo(3 ,2 ,0)гепт-2-ен-2-кар- боксилата.
К суспендированному раствору 800 мг (1,7 ммоль) полученного соединения в 50 мл ацетона прибавляют 5 мл метилиодида. Реакционную смесь перемешивают в течение 48 ч при комнатной температуре. Осадок собирают фильтрованием, промьшают ацетонитрилом (15 мл) и получают 810 мг (выход 76%) кватернизованного пиридина - йодистая соль 3-(Н-метш1Пиридин-2-ил-этантио) 1-(R)-оксиэтил J-7-OKCO-1-аэабицикло- (3,2,0)гепт-2-ен-2-карбоксилата - в виде бледно-желтого порошка.
К раствору 790 мг (1,27 ммоль) полученного соединения в 100 мл тетра- гидрофурана и 100 мл эфира прибавляют 100 МП буферного раствора рН 7,0, а затем 1,0 г 10%-ного палладия на угле. Смесь гидрируют при 2,8 атм в шейкере Парра в течение 40 мин.Смесь отфильт-
n14
ровывают на слое CELITE и катализатор пром1лвают Бодой (2x10 мл).
Объединенные фильтрат и промывные воды экстрагируют эфиром (3x100 мл, лиофи.п изуют-и получают желтьш порошок который очищают на колонке с 30 г CjgBOiroAPAK (Waters Associates) при давлении 0,56 атм, используя вкачест- ве элюента 10%-ный раствор ацетонит- рила в воде.
Каждую фракцию по 15 мл анализируют с помощью жидкостной хроматографии высокого давления, фракции, имеющие поглощение УФ 300 им, соби- рают, лиОфилизуют и получают 65 мг (выход 15%) указанного соединения в виде желтого порошка.
Пример 6. Получение 3-(N-. проп1шпиридин-4-ил-метантио)-6о( - (К)-оксиэтилJ-7-oкco-1-aзa6ициклo- (3 , 2 ,0)гепт-2-ен-2-карбоксилата,
А. Получение пара-нитробензил-3- (пиридин-4-ил-метантио)(1- (R)-oK- сиэт1ш}-7-оксо-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксилата.
Раствор 673 мг (1,86 ммоль) пара- нитробензил-бс 1 -(К)-оксиэтил7-3,7- диоксо-1-азабицикло(3,2,0)гепт-2-ен- 2-карбоксилата в 10 мл ацетонитрила охлаждают до -10°С в атмосфере азота. Прибавляют раствор 245 мг (1,90 ммоль диизопропилэтиламина в 1 мл ацетонитрила, а затем в течение 2 мин по каплям прибавляют раствор 510 мг (1,90 ммоль) дифенилхлорфосфауа в 1 мл ацетонитрила. Результирую1ций раствор перемешивают при в течение 15 мин и получают пара-нитробен- зил-3- (дифенилфосфорилокси) 1 - (R) оксиэтил }-7-оксо-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксш1ат. К этому раствору прибавляют раствор 245 мг (1,90 ммоль) диизопропилэтиламина в Oj5 мл ацетонитрила, а затем раствор 270 мг (2,16 ммоль) А-меркаптометил- пиридина в 0,5 мл ацетонитрила. Реакционную смесь перемешивают при в течение 60 мин, образовавшийся белы осадок собирают фильтрованием, промывают 5 мл охлажденного льдом ацето- нитрила и получают 660 мг (выход 76%) продукта в виде белых кристаллов, Т.Ш1. 145 С.
Рассчитано: С 58,01; Н 4,36; N 9,23; S 7,04.
Найдено: С 57,74; Н 4,56; N9,58; S 7,21.
- д
j
0
;5
ЗО 0
5
5
081 2
B.Получение йодистой соли робензил-3-(1-аллилпиридин-4-ил-ме- тантио) -6о/- 1 -(К)-оксиэтил J-7-OKCO- 1-азабицикло(3,2,0)гепт-2-ен-3-2- карбоксилата.
К раствору 900 мг (2,13 ммоль) нитробензил-3-(пиридин-4-ил-мвтан- тио)-6о((К)-оксиэтил -7-оксо-1- азабицикло(3,2,0)-гепт-2-ен-2-карбок- силата в 150 мл ацетона прибавляют 2 мл аллилбромида и 380 мг йодистого натрия. Смесь перемешивают в течение 48 ч при комнатной температуре, растворитель выпаривают под вакуумом и получают желтое твердое соединение. Это соединение суспендируют в 120 мл ацетонитрила, фильтруют, вьшаривают под вакуумом и получают 1,0 г (выход 87%) указанного продукта в виде желтого твердого соединения.
Рассчитано: С 48,16; Н 4,21; N 6,74; S 5,15.
Czf Z bOiSib
Найдено: С 48,55; Н 4,46; N 6,69; S 5,15.
C.Получение 3-(1-пропилпиридин-4- ил-метантио)( 1-(R)-оксиэтил -7- оксо-1-азабицикло(3,2,0)гепт-2-ен-2- карбоксилата,
К раствору 1,27 г (2,Т5 ммоль) соединения стадии В в 100 мл тетрагид- рофурана и 100 мл эфира прибавляют 100 мл буфера 7, а затем 1,0 г 10%-ного палладия на угле. Смесь гидрируют при 2,8 атм в аппарате Парра в течение 40 мин. Смесь фильтруют на слое цеолита и катализатор промьшают водой (2x10 мл). Соединенные фильтрат и промьшные воды экстрагируют эфиром (3x100 мл), лиофилизуют и получают желтьа г порошок, который очищают на колонке с 40 г С ,g BONDAPAK (Waters Associates) при 0,56 атм с использо- ванием в качестве элюента 10%-ного раствора ацетонитрила в воде. Калщую фракцию по 15 мл анализируют с по- - мощью жидкостной хроматографии высокого давления, фракции, имеющие по глощение УФ при 1 300 нм, собирают , лиофилизуют и получают 48 мг (выход 6%) указанного соединения в виде желтого порошка.
Рассчитано: С 54,52; Н 6,10; N 7,07.
-
Найдено; С 54,32; Н 6,03; N 6,99.
Пример 7. Получение 3-(N- метил-3-метш1пиридин-2-ил-метантио)(К)-оксиэтил}-7-оксо-1-азаби- цикло(3,2,0)гепт-2-ен-2-карбоксилата.
А. Получение З-метил-2-меркапто- метилпиридина.
Раствор 2,45 г (17,0 ммоль) 2-хлор метил-3-метилпиридина и 1,37 г (18,0 ммоль) тиомочевины в 60 мл абсолютированного этанола кипятят с об- ратйым холодильником в течение 5ч. Выпариванием этанола и последующим прибавлением эфира получают 3,08 г (выход 72%) соли изотиомочевины, которую растворяют в 10 мл воды, содержащей 1,44 г (26 ммоль) гидроокиси натрия. Затем раствор нагревают при в течение 5 мин в атмосфере азота. Реакционную смесь охлаждают до 5°С, доводят до рН 6,4 уксусной кислотой и экстрагируют эфиром (4x50 мл) Объединенные эфирные экстракты промывают 5%-ным водным бикарбонатом натрия и рассолом. Осушенный сульфатом магния растворитель вьшаривают и получают 1,4 г (выход 83%) З-метил-2меркаптометилпиридина в виде желтого масла, которое используют на следующей стадии без дальнейшей очистки.
В. Получение пара-нитробензил-3- (З-метилпиридин-2-ил- метантио)-6о6- 1- (К)-оксиэтил -7-oкco-1-aзaбициклo- (3,2,0)гeпт-2-eн-2-кapбoкcилaтa.
К охлажденному (0°С) раствору 1,74 г (5,0 ммоль) п-нитробензил- 3,7-диоксо- 1-(К)-оксиэтил }-1-азаби- цикло(3,2,0)гептан-2-карбоксилата в ;25 мл ацетонитрила прибавляют 960 мг (5,8 ммоль) диизопропилэтиламина в 2 мл ацетонитрила, и затем 1,4 г (5,8 ммоль) дифенилхлорфосфата в 2 мл ацетонитрила в атмосфере азота. Результирующий раствор перемешивают в течение 20 мин при 0°С, прибавляют раствор 760 мг (5,8 ммоль) диизопропилэтиламина в 2 мл ацетонитрила, а затем раствор 810 мг З-метйл-2-мер- каптометилпиридина в 3 мл ацетонитрила. Реакционную смесь перемешивают при в течение 2 ч. Осадок собирают фильтрованием, промывают аце
тонитрилом и получают 1,36 г (выход 66%) указанного продукта в виде белого твердого соединения, т.пл. 145.
Рассчитано: С 47,91; Н 4,69; N 6,98; S 10,66.
, Ca4H7tNj03S,
Найдено: .С 47,72; Н 4,34; N 6,72; S 11,22.
,
O 5 05
0
г 0
0
5
С, Получение 3-(Х-мсти.ч-З-метилпи- ридин-2-ил-метантис)-бы-С1-(К)-оксн- этилJ-7-oкco-1 -азабицик.чо (3,2,0)гепт- 2-ен-2-карбоксилата.
К раствору 680 мг (1,45 ммоль) соединения стадии В в 120 мл метилен- хлорида прибавляют 270 мг (2,33 ммоль) метилфторсульфоната. Реакционною смесь перемешивают в течение 3 ч при комнатной температуре. Осадок собирают фильтрованием, npohn.iBaJOT метилен- хлоридом (5 мл) и получают 840 мг (выход 99%) кватернизованного пиридина в виде белых кристаллов.
Рассчитано: Г 49,14; Н 4,47; N 7,13; S 11 ,43.
Cj H NjOgG I
Найдено: С 49,56; Н 4,16; N7,26; S 11,03.
К раствору 810 мг (1,39 ммоль) полученного 3-(N-метил-З-метилпиридин- 2-ил-метантио) -(К)-оксиэтил J- 7-оксо-1-азабицикло(3,2,0)гепт-2-ен- 3-карбоксилат-фторсульфоната в 100мл тетрагидрофурана и 100 .ш эфира прибавляют раствор буфера рН7,0 (100мл), а затем 750 мг 10%-ного палладия на угле. Смесь гидрируют при 3,15 атм в аппарате Парра в течение 60 мин в холодной комнате (4-6 С). Смесь отфильтровывают на слое целита и катализатор промьшают эфиром (2x10 мл). Объединенные фильтрат и промывочньп растворитель экстрагируют эфиром (2x40 мл) лиофилизуют и получают желтое твердое соединение, которое очищают на колонке с 20 г С ,j ВОШАРАК (Waters Associates) при 0,56 атм, элюируя 5%-ным раствором ацетонитрила в воде. Каждую фракцию по 15 мл анализируют жидкостной хроматографией высокого „ давления, фракции, имеющие УФ-погло- щение 400 нм, собирают, лиофилизуют и получают 14 1 мг (выход 30%) указанного продукта в виде желтого твердого соединения.
Рассчитано: С 57,85; Н 5,85; N 7,94.
C.,X«Ni04Sv 1/4 HjO
Н айдено: С 58,60; Н 5,86; N 7,87.
Пример 8. Получение 3-(2- метил-М-метш1Тиазол-4-ил-метантио)- бвС-f 1 - (R) -оксиэтил J-7-OKCO-1 -азаби- .цикло(3,2,0)гептан-2-карбоксилата.
А. Получение пара-нитробензил-3- (2-метилтиазол-4-ил-метантио) -6о(- П (R)-оксиэтилJ-7-OKCO-1-азабицикло- (3,2,0)гепт-2-ен-2-карбоксилата
К охлажденному () раствору 1,4 г (4,0 ммоль) промежуточного нитробензил-3,7-дноксо- 1-(К)-окси- этил 3-1-азабицикло(3,2,0)гептан-2- г карбоксилата в 12 мл ацетонитрила прибавляют 0,83 МП (4,6 ммоль) диизопро- пиламина, а затем 1,16 г (4,3 ммоль) дифенилхлорфосфата в 2 мл ацетонитрила в атмосфере азота. Результирую- |о щкй pacfBop перемешивают при в течение 30 мин и получают пара-нитро- бензил-3-(дифенилфосфорилок1:и)-6с/- 1- (К)-оксиэтил -7-оксо-1-азабицикло- (3,2,0)гепт-2-ен-2-карбоксилат. К это-|5 му раствору прибавляют раствор 0,83 мл (4,6 ммоль) диизопропилэтиламина в 2 мл ацетонитрила, а затем раствор 0,62 г (4,2 ммоль) 2-метил-4-меркап- тометилтиазола в 3 мл ацетонитрила. 20 Реакционный раствор перемешивают при в течение 40 мин. Осадок собирают, промывают эфиром 30 мл и получают 943 мг указанного в заголовке стадии А соединения в виде белого твердого 25 продукта.
В. Получение 3-(2-метил-Н-метил- тиазол-4-ил-метантио) - (R) -окси- этил 1-7-оксо-1-азабицикло (3,2,0)гепт- 2-ен-2-карбоксилата.30
К раствору 525 мг (1,1 ммоль) соединения стадии А в 20 мл метиленхло- рида прибавляют 0,27 мл (3,3 ммоль) метилфторсульфоната. Реакционную смесь перемешивают в течение 90 мин j при комнатной температуре. Осадок собирают фильтрованием, промывают метиленхлоридом (50 мл) и получают 650 мг (выход 100%) кватернизованного тиазола, который используют в следую- 0 щей стадии без дальнейшей очистки.
К раствору фторсульфоната 3-(2- метил-Ы-метил-тиазол-4-ил-метантио)- 1 -(R)-оксиэтил J-7-OKCO-1 -азаби- цикло(3,2,0)гептан-2-карбоксилата в j 100 мл тетрагидрофураиа и 100 мл зфи- ра прибавляют 100 мл буферного раствора рН 7,0, а затем 500 мг 10%-ного палладия на угле. Смесь гидрируют при 2,45 атм в аппарате Парра в течение 45 мин. Смесь фильтруют через слой целита и катализатор промьшают водой
П
50
-.
COipNB
- г риро-ь) - |о ро- 1- то-|5 мл п- . 20 ри ааютии о 25
- си- пт- 30
оео-) j го
)- j и- ори й
50
-.
(2x10 мп). Соединенные фильтрат и npoMiifflHLie воды гжстрагируют эфиром (2x100 мл) , лиофилизутот и получают желтый порошок, Koropbrii очищают на обращенно-фазовой колонке с 8 г С ,gBONDАРАК (Waters Associates),элю- ируя 5%-ным раствором гзцетонитрила в воде при давлении 0,5б атм. Каждую фракцию по 15 мл лнализируют с помощью жидкостной хроматографии высокого давления, фракции, пог. УФ при/ д 300 им, собирают, лио- филизуют и получают 145 мг (выход, 48%) указанного н заголовке стадии В соединения в виДе блед);о-жс:лтого порошка..
Рассчитано: С 46, j; Н 3,64: N 7,17; S 16,41.
CyjH ,gN,,O S2-2H,0
Найдено: С 46,50; Н 5,26; N 7,13; S 16,20.
Пример 9. Получение 3-(N,N - диметилиьп1дазол-2-ил-метантио)-6о(- С1 -(R)-оксиэтил J-7-OKCO-1-азабицикло- (3,2,0) гепт-2-е1:-2-карбоксилата.
A.Получение 2-г.1еркаптометил-ме- тилимидазола.
К раствору 10,4 г (58 ммрль) 2- клор-метил-Ы-метилимидазола в 200 мл ацетонитрити г прибанляют 7,1 г (60 ммоль) N-ацетилтиомочевины и реакционную смесь кипятят с обратным холодильни .ом в течение 90 мин. Осадок отфильтровывают, промывают ацето- нитрилом (20 мл) и получают ичотиуро- ниевую соль, которую растворяют в 120 мл этанола и кипятят с обратньгм холодильником в течение 18 ч в атмосфере азота. Реакционную смесь охлаждают до комнатной температуры,концентрируют под до объема приблизительно 60 мл и осадок удаляют фильтрованием. Выпариванием фильтрата под вакуумом получают 2-меркаптоме- тил-метилимидазола в виде желтого масла, которое используют на следующей стадии без дополнительной очистки,
B.Получение пара-нитробензил-3- (Ы-метилимидазол-2-ил-метантио)-6с((К)-оксиэтил J-7-oкco-1-aзaбициклo- (3,2 ,О)гепт-2-ен-2-карбоксилата.
Q ip(o«V
HCt(И)
К охлажденному () раствору 7,24 г (20,3 ммоль) промежуточного (I -нитробензнл-3,7-диоксо- 1-(К)-окси- этил -1-азабицикло(3,2,0)гептан-2- карбоксилата в 35 мл ацетонитрила прибавляют раствор 2,8 г (21,3 ммоль) диизопропилэтиламина в 2 мл ацетонитрила, а затем 5,5 г (20,4 ммоль) ди- фепилхлорфосфата в 2 мл ацетонитрила 10 в атмосфере азота. Полученньп раствор перемешивают при в течение 15 мин и затем прибавляют раствор 4,1 г (3,0 ммоль) диизопропилэтиламина в 2 мл ацетонитрила, после чего 15 прибавляют 4,6 г (31,0 ммоль) тиола 32. Реакционную смесь оставляют перемешиваться в течение 60 мин при . Белый осадок собирают фильтрованием, промьшают метшгенхлоридом (20 мл) и 20 получают 6,6 г (выход 71%) указанного в заголовке продукта в виде белого твердого соединения, т.пл. 142 С.
Рассчитано: С 52,18; Н 4,79; N 11,59.
CiiH-j N O S,.
Найдено: С 52,22; Н 4,91; N 12,16.
С. Получение 3-(N,N -диметилимида25
эфиром (3x100 мл), лиофялизуют и получают желтмй аморфлый порошок, который очищают на колонке с 30 г С ВОШЛРАК (Waters Associates) при 0,56 атм с использовани м 10%-ного раствора ацетонитрила в воде в качестве элюента. Каждую фракцию по 20 мл анализируют с помощью жидкостной хроматографии высокого давления, фракции, имеющие поглощение УФ при 300 нм, собирают, лиофилизуют и получают 220 мг (выход 35%) указанного в заголовке стадии С продукта в виде желтого порошка.
Рассчитано: С 51,68; Н 5,67; N 12,06; S 9,50.
HjO
Найдено: С 49,93; Н 5,94; N 11,46; S 9,03.
Пример 10. Получение 3-(2,3, 4-триметилтиазол-5-ил-метантио)-6с/- 1-(К)-оксиэтил J-7-OKCo 1-азабицикло- (3 ,2,0)гепт-2-ен-2-карбоксялата.
А, Получение 2,4-димeтил-5-мepкaп- тoмeтил тиазола .
К раствору 4,8 г (26,0 ммоль) 5- хпорметил-2-метилтиазола в 50 мл абсолютированного этанола прибавляют
дильником в течение 18 ч. Осадок собирают фильтрованием, промывают эфиром (20 мл) и получают изотиурониевую соль, которую растворяют в 22 мл 1н. NaOH и нагревают при 100°С в течение i мин в атмосфере азота. Затем реакционную смесь охлаждают до комнатной температуры, доводят до рН 7,0 1н„
зол-2-ил-метантио)-6с/- 1 -(К)-оксиэтил J-7-oкco-1-aзaбициклo(3,2,0)гепт-30 2,4 г (30 ммоль) тиомочевины. Реакци2-ен-2-карбоксилата.онную смесь кипятят с обратным холоК суспендированному раствору 1 ,34 г (3,0 ммоль) соединения, полученного на стадии В, в 2/0 мл ацетона прибавляют 20 мл метилиодида. Реакцион- ную смесь перемешивают в течение 4 дней при комнатной температуре.
Осадок собирают фильтрованием,промывают ацетоном (20 мл) и получают 1,70 г (выход 96%) кватернизованного 40 экстрагируют эфиром (3x50 мл), имидазола - натрий 3-(Ы,М -диметил- Соединенные эфирные фазы промьшают
водой, раствором соли и сушат над сульфатом магния.
Выпариванием сухого растворителя д5 получают 780 мг (выход 49%) тиола в виде бесцветного масла, которое используют на следующей стадии без дальнейшей очистки.
в. Получение пара-нитробензил-3- (2,4-диметилтиазол-5-ил-метантиоХ-6о(- 1-(К)-оксиэтилJ-7-oкco-1-aзaбициклo- (3,2 ,0)гепт-2-ен-2-карбоксилата. К охлажденному (0°С) раствору 1 ,А г (4,0 ммоль) промежуточного ке- тосоединения (пример 9, стадия В); в 25 мл ацетонитрила прибавляют 610мг (4,7 ммоль) диизопропилэтиламина в
имидазол-2-ил-метантио) C1 -(R)- oкcиэтил J-7-oкco-1-aзaбициклo(3,2 ,0) гепт-2-ен-карбоксилата - в виде желтых кристаллов, т.пл. 175-177 С.
Рассчитано: С 43,08; Н 9,60; N 5,48.
.,
Найдено: С 43,02; Н 9,02; N 5,44.
К раствору 1,30 г (1,86 ммоль) полученного соединения в 120 мл тетра- гидрофурана и 120 мп эфира прибавляют 120 мл буфера рН 7,0, а затем 900 мг 30%-ного палладия на целите. Смесь гидрируют при 2,8 атм в аппарате Пар- ра в течение 40 мин. Смесь фильтруют на слое целита и катализатор промьша50
55
ют водой (2x1,5 мл). Соединенные фильтрат и промьшные воды экстрагируют
0 5 0
5
эфиром (3x100 мл), лиофялизуют и получают желтмй аморфлый порошок, который очищают на колонке с 30 г С ВОШЛРАК (Waters Associates) при 0,56 атм с использовани м 10%-ного раствора ацетонитрила в воде в качестве элюента. Каждую фракцию по 20 мл анализируют с помощью жидкостной хроматографии высокого давления, фракции, имеющие поглощение УФ при 300 нм, собирают, лиофилизуют и получают 220 мг (выход 35%) указанного в заголовке стадии С продукта в виде желтого порошка.
Рассчитано: С 51,68; Н 5,67; N 12,06; S 9,50.
HjO
Найдено: С 49,93; Н 5,94; N 11,46; S 9,03.
Пример 10. Получение 3-(2,3, 4-триметилтиазол-5-ил-метантио)-6с/- 1-(К)-оксиэтил J-7-OKCo 1-азабицикло- (3 ,2,0)гепт-2-ен-2-карбоксялата.
А, Получение 2,4-димeтил-5-мepкaп- тoмeтил тиазола .
К раствору 4,8 г (26,0 ммоль) 5- хпорметил-2-метилтиазола в 50 мл абсолютированного этанола прибавляют
дильником в течение 18 ч. Осадок собирают фильтрованием, промывают эфиром (20 мл) и получают изотиурониевую соль, которую растворяют в 22 мл 1н. NaOH и нагревают при 100°С в течение i мин в атмосфере азота. Затем реакционную смесь охлаждают до комнатной температуры, доводят до рН 7,0 1н„
экстрагируют эфиром (3x50 мл), Соединенные эфирные фазы промьшают
1 мл ацетонитрила, а затем 1,15 г (4,3 ммоль) дифенилхлорфосфатав 1 мл
ацетонитрила в а тмосфере азота. Результирующий раствор перемешивают в течение 20 мин при 0°С и прибавляют раствор 610 мг (4,7 ммолЬ) динзопрог- пилэтиламина в 1 мл ацетонитрила, после чего прибавляют 750 мг (4,7 ммоль) полученного тиола в 2 мл ацетонитрила. Реакционную смесь переме- шивают в течение 3 ч при . Осадок собирают фильтрованием, промывают 20 мл метиленхлорида и получают 1,14 г (вькод 61%) указанного соединения в виде белого твердого продукга.
Рассчитано: С 53,73: Н 4,71; N 8,57; S 13,44.
2
Найдено: С 53,97; Н 4,74; N8,58; S 13,10.
С. Получение 3-(2 ,3,4-триметилтиазол-5-ил-метантио) -6с/- С1 (R) -окси- этил jf-7-оксо-1 - азабицикло (2,2,0) гепт- 2-ен-2-карбоксилата.
К раствору 1,97 г (4,0 ммоль) соединения стадии В в 180 мл метиленхлорида прибавляют раствор 0,98 мл (13 ммоль) метилфторсульфонатав 2 мл метиленхлорида,. Реакционную смесь перемешивают в течение 70 мин при комнатной температуре. Реакционную смесь вьтивают в раствор эфира (400 мл и н-пентана (100 мл). Осадок собирают фильтрованием, промывают 20 мл эфира и получают 1,6 г (выход 65,5%) фтор- сульфонат 3-(2,3,4)-триметил-тиазол- 5-ш:-метантио)-6с(- С1-(К)-оксиэтил}-7- оксо-1-азабицикло(3,2,0)гепт-2-ен- карбоксилата (кватернизованного ти- азола) в виде белого аморфного порошка.
Рассчитано: С 45,09; Н 4,44; N 6,86.
Найдено: С 44,50; Н4,38; N 6,58.
К раствору 1,0 г (1,72 ммоль) полученного соединения в 100 мл тетра идрофурана и 100 мп эфира прибавляют 100 мл буферного раствора рН 7,0, а затем 1,0 г 10%-ного палладия на угле. Смесь гидрируют в аппарате Пар- ра при давлении 2,8 атм в течение 40 мин.Смесь,фильтруют на слое целита и катализатЪр промывают водой (2х х10 мл). Соединенные фильтрат и промывные воды экг-рагируют эфиром (Зх х100 мл), лиог.лизуют и получают желтый порошок, чоторый очищают на колонке с 40 г Cj BONDAPAK (Waters Associates) при давлении 0,56 атм, ис
,-
15
-,«
25 ) 35
5Ю
45
пользуя в качестве элюента 10%-1гый раствор ацетоннгрила в поде.
Каждую фракциро по 15 мл анализируют с помощью жидкостной хроматографии высокого давлештя и фракции, имеющие поглощение УФ 300 им, собирают, лиофилизуют и получают 31 5мг (выход 50%) указанного в заглгзвии соединения в виде желтого твердого продукта.
Рассчитано: С 48,25; Н 6,09; N 7,79.
С,уН ,з043 - 2Н20
Найдено: С 47,96; Н 5,83; N 7,89.
Пример 11. Получение 3-f2- (N-метилтиазолийзметилтио) ( (Е)-гидроксиэтил J-7-OKCO-1-азабицикло (3 ,2 ,0)гепт-2 ен-2-карбаксилата.
А. Получсргие 2-меркаптометилтиа- зола.
К раствору :-LnopncToro тионила 3,81 мл (0,052 ммоль) в хлороформе (30 мл) добавляют при комнатной температуре 3,60 г (0,026 ммоль) гидрок- симетилтиазолл, псзсле чего нагревают реакционную смесь в течение 2 ч при 50 С. Хлороформ отгоняют D вакууме, получая в результате коричневое твердое вещество, которое растворяют в 30 мл абсолютированного этилового спирта. Затем к полученному раствору добавляют 2,04 г тиомочевины. Реакционную смесь нагревают при кипении с обратным холодильником в течение 18 ч. Осадок собирают ф1шьтрованием промывают этанолом и эфиром и получают в результате 3,4 г (выход 55%) соли изотиоурония. Соль изотиоуро- ния растйоряют п 30 мл воды и продувают азотом полученный раствор в течение 20 мин. Затем к нему добавляют 1,10 г (0,027 ммоль) гидрата окиси натрия и нагрепают полученную смесь при 100°С п течение 2 мин, рН охлажденного до 0°С раствора доводят до 6,0 посредством уксусной кислоты, после чего осуществляют экстракцию двумя порциям ПО 35 Mil этилацетата. Органический слой сушат над сульфатом магния и разгоняют в вакууме, получая 0,75 г (выход 42%) 2-меркапто- метилтиазола в виде желтого масла, которое используют без специальной очистки.
в. Получение п-нитробензил- 3-(2- тиазолметилтио)-6о{- С1-(И)-гидрокси- этил J-7-OKCO-1-азабицикло(3,2 ,0) - гепт-2-ей-2-карбоксилата.
К ох.паждеииому до 0 С раствору 1,4 г (4,0 ммоль) кетопроизводного (.пример 9, стадия А) в 8 мл ацето- иитрила добавляют сначала 0,79 мл (4,4 ьмоль) диизопропилэтиламина, а затем 1,17 г (4,4 ммоль) дифенил- хлорфосфата в атмосфере азота. Полу- чеиньп раствор перемешивают при О С в течение 30 мин, получая в результа те р-нитробензил-3-(дифенилфосфорил- окси)-6о(- С1 -(К)-гидрооксиэтш1 3-7- оксо-1-азабицнкло(3,2,0)гепт-2-ен-2- карбоксилат. К этому раствору добавляют раствор 0,79 мл (4,4 ммоль) ди- изопропилэтиламина в 2 мл ацетонит- рила, после чего добавляют раствор 0,72 г тиола (полученного по стадии А) в 2 мл ацетонитрила. Реакционную смесь перемеипизают в течение 6 мин при , после чего разбавляют ее 50 мл эт11лацетата и промывают 30 мл воды, 20 мл 10%-ного водного раствора фосфорной кислоты и 30 мл рассола После отгонки раствора, высушенного няд сульфатом магния, получают крис- таллизующуся твердую фракцию, котору растирают с эфиром, получая 782 мг (выход 42%) указанного в заголовке продукта в виде белого кристалличес- кого продукта, т.пл. 158-160 С.
Рассчитано: с 52,05; Н 4,15; N 9,10; S 13,89.
С/гоН fgWjO Sj
Найдено: С 52,35; Н 4,40; N 8,72; S 13,90.
С. Получение 3- 2-(Ы-метилтиазо- лий)метилтио (-Cl -(R) -гидроксиэтил 7-ОКСО-1-азабицикло(3,2,0)гепт-2-ен- 2-карбоксилата.
К раствору 782 мл (1,36 ммоль) соединения 6 (стадия В) в 55 мл хлористого метилена добавляют 0,5 мл ме тилфторсульфоната и перемешивают полученную реакционную смесь в течение 90 мин при комнатной температуре.Осадок собирают фильтрованием и промывают хлористым метиленом (30 мл) и эфиром (20 мл), получая в результате 630 мг ЗЧ2-(Ы-метилтиазрлий)-метил- тиoJ-6o/- 1-(R)-oкcиэтил J-7-OKCO-1- азабицикло(3,2,0)гепт-2-ен-2-карбок- силат фторсульфоната (неочищенного кватернизованного тиазола), который используют на следующей стадии без дальнейшей очистки.
К раствору этого соединения в 140 тетрагидрофурана и 120 мл эфира добавляют 140 мл буферного раствора рН
Q 5 0 п
5
5
0
5
7,0, а затем 6jO -ц- ЮГ -мстч пллл; - дия на актиопрояаии(1Г у г-ле. Полученную спесь гидро ерп1зируют n;ut давлении водорода 30 ()у1П-/кп . лыим (2,11 кг/смМ на аппарате Парра в точение 35 мин. Затем смесь фильтруют и промывают катализатор двумя порциями по 10 мл воды. Объединенный фильтрат и промьшные воды экстрагируют двумя порциями эфира по 150 мл и лио- филизируют с получением желтого порошка. Неочищенный желтьй порошок очищают на колонке, запол)1снпой 7 г С« BONDAPAK (обратная фаза, фирма Уотерс Ассошиэйтс), используя для элюирования 5%-ный растгюр ацетонитрила в воде при давлении 8 фунт/кв . ллюйм (0,56 кг/см). Каждую из фракций по 15 мл исследуют методом жидкостной хроматографии под высоким давлением. Фракции, обладаютцие поглос;е- нием в ультрафиолетовой оботасти при лллкс - Ь собирают и лиофилизи- руют, получая 23 мг (выход 5%) ука-.; занного в заголовке соединения в виде аморфноготвердого вещества желтого цвета.
Пример 12. Получение (RS)-метил-Мметиппир1Вдин-3-ил-ме- . тантио -6о(-С1-(К)-гидроксиэт1и1 -7- оксо-1-азабицикло(3,2,0)гепт-2-ен- 2-карбоксилата.
А. Получение п-нитробензил-3- 1- (RS)-мeтилпиpидин-3-ил-мeтaнтиo -6o(- (1-(R)-гидpoкcиэтил }-7-оксо-1-азаби- цикло(3,2,0)гепт-2-ен-2-карбоксилата.
К охлажденному до О С раствору 1,85 г (5,3 ммоль) кетопроизводного (пример 9, стадия А) в 20 мл ацетонитрила добавляют 754 мг (5,8 ммоль) диизопропилэтиламина в 1 мл ацетонитрила, после чего вводят раствор 1,57 г (5,84 ммоль) дифенилхлорфосфата в
2мл ацетонитрила в атмосфере алоть. Полученный раствор перемешивают при 0°С в течение 15 мин, после чего добавляют раствор 754 мг (5,8 ммоль) диизопропилэтиламина в 1 мл ацетонитрила, а затем 814 мг (5,8 ммоль) 4- (1 -меркаптоэтил)-пиридина в 2 мл ацетонитрила. Полученную реакционную смесь перемешивают при 0°С в течение
3ч, после чего разбавляют реакционную смесь 200 МП этилацетата ипрогил- вают последовательно 200 мл охлажденного льдом рассола, 200 мл воды,
100 мл водного раствора бикарбоната.
и 100 мл рассола. После сушки над сульфатом магния и отгонки растворителя получают желтое масло, icoTopoe подвергают хроматографической очистке методом хроматографирования на колонке, заполненной силикагелем, используя д,пя элюирования смесь 50% ацетона и 50% хлористого метилена, получая в результате 1,65 г указанного в заголовке продукта в виде твердого вещества желтого цвета.
Рассчитано: С 58,83; Н 4,94; N 8,95; S 6,83.
С2зН,зН.,0,,5,
Найдено: С 57,15; N 5,04; N 8,26; S 6,78.
B.Получение 4-(l -меркаптоэтил)- пиридина.
К раствору 25 г 1-(4-пиридил)- этанола в 100 мл хлороформа добавляют 50 г хлористого тионила. Реакционную смесь нагревают с обратным холодильником в течение 2 ч. После отгонки растворителей в вакууме получают 1-(4-пиридил)-хлорэтан в виде полужидкой массы, которую используют на следующей стадии без дополнительной очистки. Затем к раствору этого соедннения в 160 мл этанола добавляют горячий раствор 14,4 г тио- мочевины в 75 мл этанола. Реакционную смесь нагревают при кипении с обратным холодильником в течение 18 ч. Этанол затем отгоняют и растворяют полученный остаток в 100 мл воды, до водя рН полученного раствора до значения, равного 10, посредством 2 н. гидрата окиси натрия. Полученную смесь перемешивают при комнатной температуре в тече{ше 90 мин, доводят р до 6,0 посредством введения 6 н. НС1 и экстрагируют эфиром (две порции по 200 мл). После сушки над сульфатом магния и отгонки растворителя получают желтое масло, которое разгоняют при давлении 5 мм рт.ст., и собирают фракцию, имеющую т.кип. 60-65 С, получая в результате 11,0 г (выход 38%) чистого тиола - 4-(1 -меркапто- этил)-пиридина в виде бесцветного масла.
C.Получение (К8)-метил-Н- метилпиридин-3-ил-метантио (R гидроксиэтил }-7-оксо-1-азабицикло- (3,2,0)гепт-2-ен-2-карбоксилата.
К раствору 1,1 г (2,34 ммоль) соединения стадии А в 100 мл ацетона добавляют 10 мл йодистого метила. Ре0
5
0
5
0
5
0
5
0
5
в 120 мл тетра- добавляют
акциоипую смесг, перемешивают в течение 18 ч при комнатной температуре. Осадок собирают фильтрованием и промывают 10 мл х-гтористого метилена, получая 1,4 г (выход 100%) п-нитробен- зил-3- 1 - (KjS) -мс тил-М-метилпиридин- 3-ил-ме тантио J-пoдид 6cl - С1-(К)-окси- 3TnnJ-7-oKco- 1 -а ч а бицикле ( 5,2,0)гепт- 2-ен-2-карбокс шата (кплтсрнизиро- ванное произво/;нс,-е пиридина) в виде желтого порошка.
Рассчитано: С 47,14; Н 4,29; N 6,87; S 5,24.
C24H24 jOiSJ,
Найдено: С 47,19; И 4,78; N 6,11; S 5,41 .
К раствору 1,45 г (2,37 ммоль) полученного соединения гидрофурана и 120 мл 120 МП буферного раствора рН 7,0, после чего вводят 1,5 г 10%-ного палладия на активированном угле. Полученную смесь гидрогениэируют при давлении 45 фунт/кв.дюйм (3,17кг/см2) в устройстве Парра н течение 60 мин. Затем смесь фильтруют через слой це- лита и пpo Iывaют катализатор водой (две порции по 15 мл). Объеди}1еннГ 1е фильтрат и промывные воды экстрагируют двумя порциями по 200 мл эфира и лиофилизируют, получая желтое твердое вещество, которое подвергают очистке на колонке с обратной фазой (С j, ВОШЛРАК, фирма Уотерс Лссошиэйтс, 50 г), используя для члюирования 57-ньгй раствор ацетонитрила в воде при давлении 8 фунт/кв.дюйм (0,56кг/см).
Каждую фрак1Ц1ю объемом 20 мл исследуют методом жидкостной хроматографии при высоком давлении, фракции, имеющие поглощение в области ультрафиолета с 00 нм, собирают и лиофилизируют, получая 200 мг (выход 24%) указанного в заголовке продукта в виде аморфного твердого вещества желтого цвета.
Рассчитано: С 54,38; Н 5,77; N 7,46.
1-1.5
НаГщено: С 54,39; Н 5,98; N7,68.
Пример 13. Получение 3-(N- метил-N -бензилимидазол-2-ил-метан- THo)-6o/-f 1-(R)-гидроксиэтил J-7-оксо- 1-азабицикло(3,2,0)гепт-2-ен-2-карб- оксилата.
А. Получение Н-бензил-2-меркапто- метилимидазола.
Найдено: С 49,39; Н 3,97; N 7,20; S 10,98.
К раствору 1,10 г (1,88 ммоль) полученного соединения в 80 мл тетра- гидрофурана и 80 мл эфира добавляют ВО МП буферного раствора рН 7,0, после чего вводят 800 мг IOZ-ного палладия на активированном угле. Реакционную смесь гидрогеиизируют при давлении 30 фунт/кв.дюйм (2,11 кг/смО в аппарате Flappa в течение 40 мин. Смесь фильтруют через слой целита и промывают катализатор водой (две порции по
10 мл). Объединенные фильтрат и про- 15 повышенного давления в результамывные воды экстрагируют эфиром (две порции по 100 мл) и лиофилизируют, получая желтый порошок, который очищают методом хроматографии на колонке (НР-20), используя для элюирова}1ия 20 вначале воду, а затем 5%-ный раствор ацетонитрила в воде. из фракций по 15 мл исследуют методо.м жидкостной хроматогфафии под высоким дав- /1ением, фракции, имеющие поглощение 25 в ультрафиолете 300 нм, собирают и лиофилизируют, получая 614мг (выход 42%) указанного в заголовке гтродукта в виде слабо-желтого порошка.
Рассчитано: С 55,73; Н 5,46; N 7,65; S 8,74.
С,7 2о гОл2 -НгО
HafuieHo: С 55,50; Н 6,05; N 7,74; S 8,68.
Пример 15. Получение 3-f4- (Ы,М-диметил-1,2,3-триазолил)метил- тио j-Cifi- Cl - (R) -гидроксиэтил J-7-OKCO- 1-аза бицикло(3,2,0)reпт-2-ен-2-карбоксилата.
А. Получение изомера А.
0,58 мл (5,16 ммоль) метилфторме- тансульфоната добавляют по каплям к охлажденному льдом раствору 590 мг (3,52 ммоль) 4-метантиолацетат-1-ме- тил-1,2,3-триазола в 2 мл сухого хлористого метилена при перемешивании в атмосфере азота. После проведения реакции в течение 0,5 ч баню убирают и по истечении 1 ч растворитель убирают посредством вытяжного вентилятора Остающееся при этом масло растворяют в нескольких миллилитрах воды и охлаждают полученный раствор на бане со льдом. Затем к нему добавляют охлажденный раствор гидрата окиси натрия (305 мг, 7,59 ммоль) в нескольки миллилитрах воды и оставляют реакиионную смесь на 0,73 ч при перемешивании. Раствор разбавляют 25 мл воды
и доводят его рИ ;i(i 7, : }i рг ;y;iiiT; Ti добанлепия Tin-|-;;uirii M lji oi и; ,р,гга замеценного фосф/пм иаптич. Erne s 1Д МП этого pacTHO,ia (чт; crsi пп-тст- вует примерно 1,9 ммо.;;;- Tpii- i з.л ттио. тл) вводят при nepeN L ;:iH ; HHH и о а1аждаа- мьш льдом раствор ано;1 ; ОС(;;,та (1,0 г, 1,72 ммоль) в 10 м 1 тетрагидрофурана (ТГФ). Реакционную смесь оставляют при перемеип1вании на 0,75 ч, причем часть кристал;1пчсс1:их проду -:тов (очевидно ) .;;ает в ходе i CaK- ции. Суспензию псрев( 1ят в реактор
0 5
0
5
те добавления к нему пе-болыиих количеств тетрагидро 11ура11 1 (20 MI) и БО- ды (20 мт). В тот же реактор вводя-.- 30 мл эфира и 1,0 г палладия на aKTHiinpoBiiHiioM уг.че и осуществляют гидрогено.чия реакционной смеси при давлении 40 фунт/кв .;1,юйм (2,8 кг/см) в течение 1 ч. Органическую фазу от;1сля т п npciNn iPahiT дву- мя порциями ПОД1.1 по 5 мп. Об1)е;;и1;ен- ные водные фазы фильтр ппт, а фильтрат концентри п ют в вакууме (примерно при 0,5 мм рт.ст. в т(чение 1,5ч). ПолученныГ же.птьп раство затем хро- матографируют (колонка средног-о давления с обратн(1Г фазой, 35x90 .чм, элюент - вода) , получал пос тс лиофи- лизации 395 мг клрбапенема, немного загрязненного иeбoлыl t и количествами неорганических примесей. Продукт подвергают очгютке методом жидкостной хроматографии под высоким
0
0
5
давлением (к(.)лонка, заполне1 ная С
те
Микробондапак, фирма Уотерс, 10х хЗОО мм, многократное введение, элюент - вода), получая в резуль- .тате 310 мг (57%) изомера А в виде рыжевато-коричневого порошка.
В. Получение изомера В и изомера С.
1,60 мл (14,0 ммоль) метилтрифтор- метансульфоната добавляют по каплям к охлажденному льдом раствору 1 ,20 г (7,02 ммоль) 4-метантиолацетат-2- метил-1,2,3-триазола в 6 мл сухого хлористого метилена в атмосфере азота. Реакционной смеси дают нагреться до комнатной температуры и оставляют при перемешивании на 16 ч. К ней добавляют дополнительное количество метилтрифторметансульфоната (0,40 мл, 3,56 ммоль) и после выдерживания ре- акционной смеси в течение 3 ч при комнатной температуре растворитель уда10
15
20
:)фи)е (60 мл). Реак- 25 2,47 г (0,065 ) литийалюмини1 1гидляют посредством вытяжного вентилятора. Остающееся ггри этом масло растирают с эфиром и получаемую при этом смолообра:;иую массу растпоряют з 5 Ш7 поды. Эту смесь охлаждают в бане со льдом и добавляют раствор 844 мг (21,1 ммоль) в 5 ш воды. После гтере- мептваиия п течение 0,75 ч раствор разба Г1Я от 60 мл воды и доводят рИ до значения, равного 8, посредством введения твердого монозамещениог-о фосфата калия. Затем 40 мл :: того раствора (что со(твстствует :римерно 4,7 ммоль смеси изомеров триазолтио- лов) добавляют в охлаждаем1 1Й льдом раствор енолфосфатп (2,00 г, 3,45 ммоль) в 60 мл тет;5агидрО(1:урана при неремеишпаини. Полученную смесь перемешивают при охлаждении в бане со Л1)Дом в течение 0,5 ч, после чего перенодят в реактор для польпиенного ддавлеипя, содержащи суспензию 10%-ного палладия на активированном угле (2,00 г)
ционн по смесь подверг ают гидрогеноли- зу при дагстснии 40 фунт/кв.дюйм (2,8 }U /CM ) 73 течение 1 ч. Органическую фазу отделяют и промывают двумя порп.иями по 10 мл вода. Об ьединен- ные водные |Ьазы фильтруют и фильтрат кондентрируют при высоком вакууме (примерно 0,5 мм рт.ст.) в течение 1,5 . Остающийся раствор хроматогра- фируют (колонка среднего давления с обратно фазой, 45x130 мм, вода в ка-- честве элюента), получая после лиофи- лизадии 595 мг смеси изомерных карба- пенемов, которая загрязнена небодыяи- ми коллчесгвами неорганических продуктов. Продукт реакции отделяют и очитцают методом жидкостной хроматографии под иысоким давлением (колонка, наполненная С и Микробондапак, фирма Уотерс, 10x300 мм, многократное введение, вода в качестве элюеи- та), получая и порядке элюирования изомер В, 153 мг (13Z).
Пример 16. (5R,6S)-6-(lR- гидроксиэт1ш)-3-(2-метил-1,2,3-тиади- азолий-4-ил-метилтио)-7-оксо-1-аза- бицикло (3,2 ,.0) гепт-2-ен-2-карбоксш1ат.
Л. Этил-,2,З-тиадиазол-4-ил-карб- оксилат.
Раствор этил- с -Н-карбэтоксигидра- зонпропионата (31,2 г, 0,154 моль) в 55 80 мл хлористого тиоиила перемешиваютют
Tbtfi тионил отгоняют и остаток растирают с гексаном ( reibijie порции по 30 мл) . Красное твердое вещество растворяют в 150 мл дихлорметана и промывают раствор насыщенным раствором бикарбоната натрия и водоГг. После сушки над сульфатом магния раст1зор концентрируют до К1зпстал.1П1зации соединения. После выдерживания при 23 С в течен11е некоторого промежутка времени образу- Ю1циеся кристаллы отфильтровывают, получая 16,8 г продукта (выход 69%), имеющего т.пл. 86°С. Фильтрат концеи-- трируют и очищают методом хроматографии на колонке, заполненной силика- гелем, используя в качестве раство 1И- теля-э.чюента д.ихлорметан, получая 3,17 г (13%) продукта, имеющего т.пл. 86°С.
В. 1 ,2,З-Тиадиазил-4-ил-метанол.
К суспензии 18,35 г (0,116 ммоль) этил-1,2,3-тиадиазол-4-ил-карбоксила- та в 400 мл эфира добавляют по частям
30
40
течение 3 ч при и нагрева- 70°С в течение 20 мин. Хлориспри
рида (в течение 1 ч), Реакционную смось перемеигинают при Р reqeirne 7 ч, после чего обрабатывают 2,47 г (0,065 моль) гийалюмипийгидрида . Перемешивание продолжают 24 ч, после чего вводят послодовательпо воду (7 wi), 15%-ный водный раствор гидря- та окиси натрия (7 м.т) и снова воду (21 мл). После перемешивания в течение 15 Mim эфирный раствор декантируют и смолообразньи остаток экстрагируют эфиром (5 раз по 100 м::) . Эфирные экстракты о 71единяют, с.ушат над сульфатом магния и концентрируют, получая 5,4 г продукта. Неочищенный продукт подвергают очистке на колонке, заполненной силикагелем (120 г, 4x16 см), используя для элюирования эфир, получая 1,3 г (7%) этт1-1,2,3- тиадиазол-4-пл-карбоксилата и 2,45 г (18%) 1,2,3-типдпазол-4-ил-метанола.
С. 1,2,3-Тпадиазол-4-ил-метанол- метансульфонат.
Раствор 1,2, З-тиадиазол-4-ил-ме- танола (0,75 г, 6,5 ммоль) в дихлор- метане (20 мл) охлаждают до 5°С в атмосфере азота и обрабатывают триэтил- амином (1,018 мл, 7,3 ммоль) и метан- сульфонилхлоридом (0,565 мл, 7,3 ммоль). Спустя 15 мин баню со льдом удаляют и перемешивают реакционную
смесь в течение 2 ч. Раствор промы- J
вают 1 н. раствором соляной кислоты (2 раза по 2 мл) и водой, сушат над
Tbtfi тионил отгоняют и остаток растирают с гексаном ( reibijie порции по 30 мл) . Красное твердое вещество растворяют в 150 мл дихлорметана и промывают раствор насыщенным раствором бикарбоната натрия и водоГг. После сушки над сульфатом магния раст1зор концентрируют до К1зпстал.1П1зации соединения. После выдерживания при 23 С в течен11е некоторого промежутка времени образу- Ю1циеся кристаллы отфильтровывают, получая 16,8 г продукта (выход 69%), имеющего т.пл. 86°С. Фильтрат концеи-- трируют и очищают методом хроматографии на колонке, заполненной силика- гелем, используя в качестве раство 1И- теля-э.чюента д.ихлорметан, получая 3,17 г (13%) продукта, имеющего т.пл. 86°С.
В. 1 ,2,З-Тиадиазил-4-ил-метанол.
К суспензии 18,35 г (0,116 ммоль) этил-1,2,3-тиадиазол-4-ил-карбоксила- та в 400 мл эфира добавляют по частям
5
0
0
рида (в течение 1 ч), Реакционную смось перемеигинают при Р reqeirne 7 ч, после чего обрабатывают 2,47 г (0,065 моль) гийалюмипийгидрида . Перемешивание продолжают 24 ч, после чего вводят послодовательпо воду (7 wi), 15%-ный водный раствор гидря- та окиси натрия (7 м.т) и снова воду (21 мл). После перемешивания в течение 15 Mim эфирный раствор декантируют и смолообразньи остаток экстрагируют эфиром (5 раз по 100 м::) . Эфирные экстракты о 71единяют, с.ушат над сульфатом магния и концентрируют, получая 5,4 г продукта. Неочищенный продукт подвергают очистке на колонке, заполненной силикагелем (120 г, 4x16 см), используя для элюирования эфир, получая 1,3 г (7%) этт1-1,2,3- тиадиазол-4-пл-карбоксилата и 2,45 г (18%) 1,2,3-типдпазол-4-ил-метанола.
С. 1,2,3-Тпадиазол-4-ил-метанол- метансульфонат.
Раствор 1,2, З-тиадиазол-4-ил-ме- танола (0,75 г, 6,5 ммоль) в дихлор- метане (20 мл) охлаждают до 5°С в атмосфере азота и обрабатывают триэтил- амином (1,018 мл, 7,3 ммоль) и метан- сульфонилхлоридом (0,565 мл, 7,3 ммоль). Спустя 15 мин баню со льдом удаляют и перемешивают реакционную
смесь в течение 2 ч. Раствор промы- J
вают 1 н. раствором соляной кислоты (2 раза по 2 мл) и водой, сушат над
К раствору 3,23 г (13,0 ммоль) хлор гидра та 4-бенэил-2-хлорметилнми- дазола в 80 мл ацетонитрила добавляю 1,72 г (14,5 ммо.чь) N-ацетилтиомоче- вины. Реакционную смесь нагревают в течение 3 ч при кипении с обратным холодильником. Осадок собирают фильтрованием и промывают ацетонитрилом (10 мл), получая соль изотиоурония, которую затем растворяют в 80 мл абсолютированного этилового спирта и нагревают в течение 18 ч при кипении с обратным холодильником в атмосфере азота. Реакционную смесь охлаждают до комнатной температуры, упаривают в вакууме до объема, равного примерно 30 мл, и удаляют фильтрованием образуюпшйся осадок. После разгонки фильтрата в вакууме получают 3,5 г (выход 97%) тиола, указанного в заголовке, в виде густого сиропа желтого цвета,
B,Получение П-нитробензил-3-(Пбензилимидазол-2-ил-метантио)-6с/-Г1 (R)-гидроксиэтил J-7-OKCO-1-азабицик- ло(3,2,0)гепт-2-ен-2-карбоксилата, К охлажденному до раствору 3,03 г (8,5 ммоль) кетопроизводного (пример 9, стадия А) в 70 мл ацето- нитрила добавляют 1,17 г (9,0 ммоль) диизопропилэтиламина в 2 мл ацетонитрила, а затем вводят 2,4 г (9,0 ммоль) дифенилхлорфосфата в 2 мп ацетонит- в атмосфере азота. Полученный раствор перемеип1вают в течение 20 .i. мин при , после чего к нему до- )Т раствор 1,17 г (9,0 ммоль) диизопропилэтиламина в 2 мл ацетонитрила и 4,8 г (15 ммоль) тиола (продукт стадии А), К реакционной смеси добавляют еще 1,93 г (15 ммоль) диизопропилэтиламина и оставляют перемешивать ее в течение еще 2 ч при , Осадок собирают фильтрованием и пром1.1вают 20 мп охлажденного хлористого метилена, получая 2,5 г (выход 55%) указанного в заголовке продукта в виде белого твердого вещества .
C.Получение 3-(Н-метил-М -бензил
и raдaзoл-2-ил-мeтaнтиo)-6of-C1(R) гидроксиэтил }-7-оксо-1-азабицикло (3,2,0)гепт-2-ен-2-карбоксилата.
К раствору 1,76 г (3,3 ммолъ) соединения полученного на стадии В в 1,1 л хлористого метилена добавляют 1,15 мл (13,4 ммоль) метилфторсульфо ната. Реакционную смесь перемепмвают
Q5 20 5д 50
55
35
45
в течение 2 ч при комнатной температуре, после чего концеИтцируют и вакууме до обт.ема, рапного примерно 15 мл. Осадок собирают фильтрованием и промывают хлористым метиленом (10 мп), получая в результате 1,58 г (выход 74%) л-нитро5ензил-3-(Н-ме- TiuT-N -бензилимидлзол-2-ил-метантио)- фторсульфонат-бо - 1-(К)-оксиэтил7-7- оксо-1-азабицикло(3,2,0)гепт-2-ен-2- карбоксилата (кватернизованного ими- дазола) в виде белого твердого вещества ,
Рассчитано: С 51,48; Н 4,47; N 8,67; S 10,20,
С 2 g Н g О 4 S 2 F
Найдено: С 51,84; Н 4,52; N 8,65; S 9,87,
К раствору 1,11 г- (1,71 ммоль) полученного соединения в 100 мл тет- рагидрофурана и 100 мл эфира добавляют 120 мл буферного раствора рН 7,0 после чего вводят 1,0 г 10%-пого палладия на активированном угле. Полученную смесь гидрогенизпруют при давлении 45 фунт/кв,дюйм (3 , I 7 кг/см ) в аппарате Парра в течение 45 мин. Реакционную фильтруют через слой целита и промывают катализатор водой (2 порции по 10 мл), Объединенные фильтрат и промывные воды экстрагируют 2 порциями эфира по 70 мл и лиофилизир тот, получая желтьп порошок, который подвергают очистке на колонке (С jjBONDAPAK, фирма Уотерс Ассошиэйтс, 40 г), используя для элюирования 10%-ный раствор ацетонитрила в воде при давлении 8 фунт/кв.дюйм (0,56 кг/смМ,
Каждую ия фракций объемом по 15 мл подвергают исследованию методом жидкостной хроматографии под высоким давлением, фракции, имеющие поглощение в ультрафиолете с. 300 нм, собирают и лиофилизируют, получал 305 мг (43%) указанного в заголовке продукта D виде слабо-желтого аморфного твердого вещества.
Рассчитано: С 57,25; Н 5,94; N 9,54; S 7,28.
C HjjNjO S ,1,5
Найдено: С 56,66; Н 5,70; N 9,49; S 8,30.
Пример 14. Получение 3-(2- метил-Ы-метилпиридин-З-ил-метантио)- 6of- О (R) -гидроксиэтил J-7-OKCO-1 - азабицикло(3,2,0)гепт-2-ен-2-карбок- силата.
А. Получение 2-метил-З-меркапто- метилпириднна.
К охлажденной до суспензии 2,86 г литййалгаминий гидрида в 50 мл сухбго тетрагидрофурана добавляют по каплям 6,23 г (0,038 моль) сложного эфира 2-метил-З-этилпиридинкарбокси- лата в 15 мл тетрагидрофурана в течение 15 мин. Полученную смесь перемешивают в течение 60 мин при , после чего добавляют к ней 50 мл этилацетата. Осадок отфильтровывают и промывают насьоценным водным раствором хлористого аммония. Органический слой сушат над сульфатом магния,фильтруют и отгоняют растворитель в ваку ие, получая в результате 3,2 г (выход 70%) 2-метил-З-гидроксиметилпири дина в виде желтого масла.
К охлажденному до О С раствору 4 мл хлористого тионила в 10 кш хлористого метилена добавляют по каплям раствор 3,2 г (0,026 моль) полученного спирта в 10 мл хлористого метилена за 15 мин в атмосфере азота. Охлажденную баню убирают и перемешивают реакционную смесь в течение 3 ч при комнатной температуре. Все растворители отгоняют в вакууме, получая в остатке 2-метил-З-хлорметилпиридин в виде коричневого твердого вецест- ва, которое используют на следующей стадии без очистки. Неочищенный продукт коричневого цвета растворяют в 30 мл абсолютированного этанола, добавляют 2,5 г (0,032 ммоль) тиомоче вины и нагревают реакционную смесь при 65-7b C в течение 18 ч, после чего указанную смесь охлаждают до комнатной температуры. Осадок собирают фильтрованием и промывают последовательно этанолом (20 мл) и эфиром (50 мл), получая 30 г соли изотио- урония. Эту соль растворяют в 10 мл воды и добавляют к полученному раствору раствор 640 мг (0,016 моль) гидрата окиси натрия в 10 мл воды в атмосфере азота. Реакционную смесь нагревают до в течение 2 мин, после чего охлаждают до ОС, доводят рН смеси, до значения, равного 6,0, посредством уксусной кислоты и экстрагируют двумя порциями хлороформа по 35 мл. После сушки над сульфатом магния и отгонки растворителя (хлороформа) получают 941 мг (выход 46%) тиола, указанного в заголовке в виде желтого масла.
B.Получение п-нитробензил-3-(2- метил пиридин-3-ил-ме тан тио)-6 с|( (К)-гидроксизтил -7-оксо-1-азабицикло(3,2,0)гепт-2-ен-2-карбоксилата. К охлажденному до 0°С раствору 1,52 г (4,37 ммоль) кетопроизводного (пример 9, стадия А) в 3 мл ацетонит- рила добавляют 0,86мл (4,80 ммоль)
диизопропилэтиламина, после чего вводят раствор 1,17 г (4,37 ммоль) дифенилхлорфосфата в 3 мл ацетонит- рила в атмосфере азота. Полученный при этом раствор перемешивают в течение 30 мин при 0°С, получая П-нитробензил-3-дифенилфос- форилокси-6о - С1 (R) -гидроксиэ ил -7- оксо-1-азабицикло(3,2,0) гепт-2-ен-2 карбоксилат. К этому раствору добавляют раствор 0,86 мл (4,80 ) диизопропилэтиламина п 2 мл ацетонитри- ла, после чего вводят раствор 940 мг (6,76 ммоль) тиола (продукта стадии А) в 2 мл ацетонитрила. Полученную
реакционную смесь перемешивают в течение 60 мин при . Осадок собирают фильтрованием и промывают эфиром (30 ил), получая в результате 1,12 г (выход 55%) указанного в заголовке продукта в виде светло-желтого твердого вещества, т.пл. 186-188°С (с разложением).
Рассчитано: С 58,83; Н 4,94; N 8,94; S 6,83.
CjjHj jO S Найдено: С 58,63; Н 4,99; N 9,06;
S 6,58.
C.Получение 3-(2-метил-Н-метил- пиридин-3-ил-метаитио)-bd- С1 -(R)- гндроксиэтилJ-7-oкco-1-aзaбициклo- (3 , 2 ,0) гепт-2-ен-2-карбоксилата.
К раствору 697 мг (1,19 ммоль) продукта стадии В в 100 МП хлористого метилена добавляют по каплям при ЮС 0,5 мл (6,18 ммоль) метилфторсульфо- ната в течение 10 мин. Смесь перемешивают в течение 2,5 ч при комнатной температуре, собирают осадок фильтрованием и промывают его 30 мл хлористого метилена, получая 777 мг (90%) п-н11Тробензил-3-(2-метилпиридин-3- ил-метантио)фторсульфонат-6 - 1-(К)- оксиэтнл1-7-оксо-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксилата (кватернизо- ванное производное пиридина) в виде желтого твердого вещества.
Рассчитано: С 48,91; Н 4,55; N 7,23; S 11,04.
3 ОэЗ
смесью сульфата и окиси магния и концеЕ1Трируют, Остаток подвергают очистке хроматографическим способом (колонка с силикагелем 1,5x21 см), используя в качестве элюирующего растворителя эфир, получая в результат 0,90 г (71%) 1,2,3-тиaдиaзoл-4-ил- мeтaнoлмeтaнcyльфoнaтa .
Рассчитано: С 24,73; Н 3,11; N 14,42; S 33,02.
C IbNjOjS
Найдено: С 24,78; Н 3,09;- N 14,66; S 31,94.
Кроме того, найдено 0,13 г (19%) ди-(1,2,З-тиадиазол-4-ил-метил)-эфира.
D. 4-Лцетилтиометил-1,2,3-тиадиа- зол.
К раствору 1 ,2 ,3-тиадиазол-4-ил- метаиолметансульфопата (0,90 г, 4,6 ммоль) в тетрагидрофуране (9 мл) добавляют водный раствор (2 мл) тиол ацетата натрия, полученный из тиол- yKcycHofi кислоты (0,38 мл, 5,3 ммоль и бикарбоната натрия (0,445 г, 5,3 ммсль) .Полученную реакционную смес перемешивают при 23 С в течение 1 ч и разбавляют 75 мл эфира, Оргаю чес- кий раствор промывают подои (3 раза по 3 мл), сушат над сульфатом магния и концентрируют. Сырую смесь очищают хроматографическим способом (колонка С С1шикагелем 1,4x19 см), используя качестве элюирующего растворителя
смесь 50% эфира и гексана, получая
в результате 0,60 г (75%) продукта.
Рассчитано: С 34,47; Н 3,47; N 16,08; S 36,80.
C,,H,N,OS,
Найдено: С 34,48; Н 3,83; N 16,28; S 36,80.
Е. 4-Ацетилтиометнл-2-метил-1,2,3- тиадиазолийтрифторметансульфонат и 4-ацетилтиометил-3-метил-1,2,3-тиа- диазолий трифторметансульфонат.
К раствору 4-ацетилтиометил-1,2,3- тиадиазола (0,60 г, 3,44 ммоль) в смеси эфира (4 мл) и дихлорметана (0,4 мл) добавляют несколько кристаллов указанного в заголовке соединения, а также 0,407 мг (3,6 ммоль) трифтор- метансульфоната в течение 5 мин. Реакционную смесь перемешивают при 23 С в атмосфере азота в течение 6ч. Белое твердое вещество, которое представляет собой смесь указанных в заголовке, двух соединений, отфильтровы-
5
0 5 Q
5
5
0
5
вают и промывают эфиром, пп.чучая
1,05 г продукта (90%).,
Рассчитано: С 20,27; Н 2,38; N 9,45; S 32,46
С7ПдЫ20 5,Гз
Найдено: С 24,61; Н 2,57; N 8,47; S 28,21.
F.4-Меркаптометил-2-метил-1,2,3- тиадиазолийтрифторметансульфонат и |4-меркаптометил-3-метил-1,2,3-тиадиазолийтрифторметансульфонат.
Раствор смеси 4-ацстилтиомети.т1-2- метшт-1,2,3-тиадиазолийтрифторметаН сульфоната и 4-ацети.пгиометил-З-ме- тил-1,2,3-тиалиазолийтрифторметил- сульфоната (1,05 г, 3,1 ммоль) в 6 Н. соляной кислоте (10 мп) нагревают при в атмосфере азота в течение 1,75 ч. Растворитель отгоняют при пониженном давлении, получая в остатке желтьй сироп в количестве 0,91 г. Это соединение используют на следующей стадии без очистки.
G.(5R,6S)-6-(1К-гидроксиэтил)-3- (2-метил-1,2,З-тиадиазолий-4-ил-ме- тилтио)-7-оксо-1-азабицикло(3,2,0)- гепт-2-енг2-карбоксилат.
Охлажденньп до 5° С раствор (5R,6S)- пара-нитробензил-6-(1R-гидроксиэтил)- З-дифенилофосфон-7-оксо-1-азабицикло- (3,2,0)гепт-2-ен-2-карбоксипата (1,7 г, 2,92 ммоль) в тетрагидрофуране (10 мл) обрабатывают раствором неочищенной смеси 4-меркаптометил-2- метил-1,2,3-тиадиазолийтрифторметан- сульфоната и 4-меркаптомет1ш-3 метил- 1,2,3 -тиадиазолийтрифторметансульфо- ната (0,9 г) в смеси с фосфатньм буфером (рН 7,2, 0,3м, 15 мл) и тет- рагидрофураном (5 мл). Реакг юнную смесь перемешивают в течение 1 ч,поддерживая значение рН 7,2 посредством
2и. раствора гидрата окиси натрия. Перемешивание продолжают в течение еще 1 ч, после чего добавляют 50 мл эфира и 1 г 10%-ного палладия на активированном угле. Полученную реакционную смесь гидрогенизируют при и давлении 45 фунт/кв.дюйм
(3,17 кг/см) в течение 2 ч, после чего фильтруют через слой целита.Органическую фазу отделяют, разбавляют эфиром (50 мп) и фосфатным буфером (рН 7,2, 0,ЗМ, 20 мп) и гидрогенизируют (используя 2 г 10%-ного палладия на активированном угле) в течение 2 ч при 50 фунт/кв.дюйк (3,5 кг/см). Водные фазы, получен10
15
ные при первой и второй гидрогенизации, объединяют, промывают эфиром и подвергают очистке методом хроматографии на Преп-Пак 500-С/18, используя- в качестве растворителя для элюи- рования воду, получая в результате 0,22 г неочищенного продукта. Его подвергают повторной очистке жидкостной хроматографией при высоком давлении-, используя для элюирования воду. Получено в результате 0, г (А%) указанного в заголовке соединения после лиофилизации его.
Пример 17. (5R, б5)--пара- нитробензил-6-(1К-гидроксиэтил-4Е- метил-З- f ( 1,2,З-тпадиазол-4-ил)- ;метилтиоJ-7-OKCO-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксилат.
Холодный (5°С) раствор (5R,6S)- пара-нитробензил-6-(1R-гидроксиэтил)- 3-(дифе1шлфосфоно)-4К-метил-7-оксо- 1-азабицикло(3,2 ,0) гепт-2-ен-2- карбоксилата (3,0 ммоль) в (12 ш) обрабатывают 4-меркаптоме- 25 тил-1 , 2 , 3-тиадиазолом (528 мг,4 ммоль) и диизопропилэтиламином (426 мг, 3,3 ммоль, 0,58 мл). После перемешивания в течение 1 ч раствор разбавляют EtOAc (35 Nm), дважды промывают холодной водой. Органический раствор, высушенньц г MgSO- упаривают и неочищенный продукт пропускают через слой SiO особой чистоты (40 г).Элю- иропание проводят (10x20 мл) и
30
16 мл), гидрируют 75 мин в присутствии Pd/C (1,0 I ) при давлении 45 psi. Затем реакционную смесь охлаждают на льду, фильтруют и фильт рат j- важды экст 1агируют EtjO. Водную фазу хроматографируют на обращенно- фазовом SiO, )люируя и затем 5%-ным в воде. Описанзюе гидри рование повторяют в тех же усттониях при тех же загрузках. Обе порции сое диняют и продукт под.вергают рехрома тографии на обращенно-фазовом Si02) элюируют водой (175 мл) и 5%-ным CH. в , соответс-твующие фракции соединяют и лиофи п- зуют, получая карба- пенем с выходом 164 мг (14,8%).
Пример 18. (1,3-Диметилими- дазолий-4-ил)метантиол трифторметан- сульфонат.
К раствору (1-метилимидазол-5-ил) метилтиоацетата (20,43 г, 0,12 ммоль) в смеси ацето}и1трил - эфир (1:2, 180 мл) прикапывают (0,5 ч) метил- трифдат (15,84 мг, 0,14 моль). В те чение 1 ч смесь перемеи01вают в атмосфере азота при и затем разбавляют эфиром (500 мл). Жидкость декан тируют, оставляя красный сироп, кото рьш дважды промывают эфиром (ЮО мл) перед добавлеии1 м соляной кислоты (200 мл, 6 н.). 1олучившийся водный раствор в течение 4 ч нагревают при 65 С на воздухе и упаривают. Следы соляной кислоты удаляют перегонкой
40
затем EtOAc (10x20 мт) , Соответств-уго- с водой, оставляя желтьп щие фракции упаривают и получают 1,18 г (82,5%) бледно-желтой пены.
(5R,6S)-6-(1R-гидроксиэтил)-4R- метил-3- СЗ-метил-1 ,2 ,3-тиадиазолий- 4-ил)метилтио J-7-оксо-1-азабицикло- (3,2,0)гепт-2-ен-2-карбоксилат (ВМУ- 26659).
К раствору (5R,б5)-пара-нитробен- зил-6-(1R-гидроксиэтил)-4R-мeтил-3- (1,2, 3-тиадиазол-4-ил)-метилтио-7- оксо-1-азабицикло(3,2,0)гепт-2-ен-2- карбоксилата (2,25 г, 4,72 ммоль) в CnjCl2(14 мл) и EtjO (9,5 г-ш) прикапывают метил трифторметансульфонат (775 г, 4,72 ммоль) при комнатной 5 температуре. Раствор мутнеет и выпадает масло.V После перемешивания 2 ч супернатант ,декантир пот, а масло растирают в , получая желтый поро45
сироп (32,4
92л). Часть сиро.ча очищают на колонке с обращенHoi j фазой водой и злюиру ющим растворителем, ИК (неразбавленный) .
(5R,6S)-3-C(1,З-диметилимидазо- лий- ил)-мeтилтиoJ-6-( IR-гидрокси- этил)-4R-мeтил-7-oкco-1-aзaбициклo- (3,2,0)гепт-2-он-2-карбоксилат (ВМУ- 27463).
К холодному (-20°С) раствору (5R, бЗ)-пара-нитробензил-3,7-диоксо-6- (1R-гидроксиэтил)-4R-мeтил-1-азаби- цикло(3,2,0)гептан-2К-карбоксилата (34,78 г, 0,096 моль) в сухом ацето- нитриле (150 мл), находящемуся в атмосфере азота, добавляют дифенилхлор фосфат (20,73 мл, 0,10 ммоль), диизо пропилэтиламин (17,42 мл, 0,10 моль) в течение 5 мин и 4-диметиламинопири
шок (2,08 г, 68,8%). Кватернизован- 55 дин (0,040 г). Охлаждающую баню за- ный карбапенем (1,0 г, 1,56 ммоль) растворяют в ТГФ (25 мл) ,Et20 (25мл) и фосфатном буфере (0,2 моль, рН 7,
меняют на баню с ледяной водой.Смесь перемешивают при в течение 1 ч, охлаждают до -20 С и последовательно
0
5
0
16 мл), гидрируют 75 мин в присутствии Pd/C (1,0 I ) при давлении 45 psi. Затем реакционную смесь охлаждают на льду, фильтруют и фильтрат j- важды экст 1агируют EtjO. Водную фазу хроматографируют на обращенно- фазовом SiO, )люируя и затем 5%-ным в воде. Описанзюе гидрирование повторяют в тех же усттониях при тех же загрузках. Обе порции соединяют и продукт под.вергают рехрома- тографии на обращенно-фазовом Si02) элюируют водой (175 мл) и 5%-ным CH.CN в , соответс-твующие фракции соединяют и лиофи п- зуют, получая карба- пенем с выходом 164 мг (14,8%).
Пример 18. (1,3-Диметилими- дазолий-4-ил)метантиол трифторметан- сульфонат.
К раствору (1-метилимидазол-5-ил)- метилтиоацетата (20,43 г, 0,12 ммоль) в смеси ацето}и1трил - эфир (1:2, 180 мл) прикапывают (0,5 ч) метил- трифдат (15,84 мг, 0,14 моль). В течение 1 ч смесь перемеи01вают в атмосфере азота при и затем разбавляют эфиром (500 мл). Жидкость декантируют, оставляя красный сироп, кото- рьш дважды промывают эфиром (ЮО мл) перед добавлеии1 м соляной кислоты (200 мл, 6 н.). 1олучившийся водный раствор в течение 4 ч нагревают при 65 С на воздухе и упаривают. Следы соляной кислоты удаляют перегонкой
с водой, оставляя желтьп
сироп (32,4 г
92л). Часть сиро.ча очищают на колонке с обращенHoi j фазой водой и злюиру- ющим растворителем, ИК (неразбавленный) .
(5R,6S)-3-C(1,З-диметилимидазо- лий- ил)-мeтилтиoJ-6-( IR-гидрокси- этил)-4R-мeтил-7-oкco-1-aзaбициклo- (3,2,0)гепт-2-он-2-карбоксилат (ВМУ- 27463).
К холодному (-20°С) раствору (5R, бЗ)-пара-нитробензил-3,7-диоксо-6- (1R-гидроксиэтил)-4R-мeтил-1-азаби- цикло(3,2,0)гептан-2К-карбоксилата (34,78 г, 0,096 моль) в сухом ацето- нитриле (150 мл), находящемуся в атмосфере азота, добавляют дифенилхлор- фосфат (20,73 мл, 0,10 ммоль), диизо- пропилэтиламин (17,42 мл, 0,10 моль) в течение 5 мин и 4-диметиламинопиридин (0,040 г). Охлаждающую баню за-
меняют на баню с ледяной водой.Смесь перемешивают при в течение 1 ч, охлаждают до -20 С и последовательно
обрабатывают раствором (1 , З-димет ш- имядазолий-4-цл)-метантнспа (32,4 г, 0,11 ноль) в ацетонитриле (130мл) и диизопропилэтиламином (19,16 мл, 0,11 моль) с такой скоростью, чтобы температура поддерживалась от -15 до -20 С (10 мин). Охлаждающую баню заменяют на баню с ледяной водой. Смесь перемешивают при в течение 1,5 ч, затем разбавляют водой (840мл Образующуюся смесь обрабатывают Dowex 1x8 50 СГ (200 кп) и выпивают на слой с обращенной фазой (14x14 см) В качестве элюирующего растворителя используют смесь (25-35%) ацетонит- рила и ноде. Фракции, содержащие чистый кар 3апенем, пропускают через колонку с Dowex 1x8 50 С1 (4х5 ск) со скоростью 3 мл/мин. Лиофилизаци- efi водного раствора получают желтый сухой остаток 27,6. (55%), который растворяют в буфере ( - NaOH, 0,3 и., 615 мл, рН 7,0). После добавления 10%-ного палладия на угле (20 г) , тетраг идрофурана (400 мл) и эфира (600 :-ш) смесь гидрируют при 23 С и 45 psi в течение 2 ч и фильтруют чорс слой броунмиллерита (Се- fite). OpгaничecкyкJ фазу отделяют и экстрагируют водой (2x50 NLT) . Боднуло фазу прог ывают эфирс- М (2x70 мл) , откачивают в высоком вакууме для того, чтобы удалить следы органических растворителей, и выливают па слой с обращенной фазой (14x13 см). Элюиро- вание проводят смесью (0-4%) ацето- нитрила в воде, после ;пюфилизации соответствую цих фракций получают жел- тьш порошок (14,6 г, 78,8%).
Пример 19. (5R,6S) 3-(3,4- диметилтиазолий-5-ил)метилтио -6- (1 К-гидроксиэтил)-4Рч.-мет1Ш-7-оксо- 1-азабицикло-(3,2,0)гепт-2-ен-2- карбоксилат (ВМУ-27061).
Раствор (5R, 63)-аллил-6-( 1 Рч.-гид- роксиэтил)-4К-метил-3-(4-метилти- азол-5-ил)метилтио -7-оксо-1-азаби- цикло(3,2,0)гепт-2-ен-2-карбоксилат (1,15 г, 3,34 ммоль) Б сухом дихлор- метане (25 мл) охлаждают до О С и обрабатывают прикапыванием (2 мин) ме- тилтрифторметансульфонатом (0,53 г, 3,24 ммоль). После выдерживания АО ми при 0°С добавляют трифенилфосфин (0,080 г) и тетракис(трифенилфосфин) палладий (0,080 г), затем добавляют 0,5м раствор 2-этилгексаната калия (6,7 мл, 3,33 ммоль) в этилацетате.
После выдерживания в течение 30 Ntiin при 0°С реакционной смеси позволяют нагреться до 22 С и с интервалами в 30 мин еще 3 раза добавляют когтлекг палладия и трифени.чфосфин (ка;кдого по 0,080 г). После выдерживания в течение 3 ч при 22 С томную реакционную смесь упаривают при пониженном давле0 НИИ и остаток разделяют между водой (15 мл) и эфиром (15 м,л). Органическую фазу опять экстрагируют водой (5 мл) и объединенные водные фазы очищают на колонке с обращенно-фазо5 вым силикагелем (2,5x12 см), используя в качестве элюирующего агента воду, а затем воду и ацето1П1трил (9:1). После повторно} хроматографии получают основной продукт в ниде белого
0 порошка после лиофилизации (0,248 г, 21%).
Пример 20. (5R, 65)- 1ара-нит- робензил-6-(1К-гидроксиэтил)-4К-ме- тил)-3-С(1-метил-1,2,З-триазол-4-ил)5 метилтиоJ-7-окео-1-азабицикло(3,2,0)- гепт-2-ен-2-карбоксилат.
Холодный (5°С) раствор (5R,6S)- пара-нитробензил-6-(1Я-гицроксиэтил)- 3-(дифенилфосфоно)-4К-метил-7-оксо-1- азабицикло(3,2,0)гепт-2-ен-2-карбокси- лата (4,0 ммоль) в CHjCN (20 мл) обрабатывают 4-меркаптомет1ш-1-метил- 1 ,2,3-триазолом (735 мл, 5,7 ьи-юль) и диизопропилэт1шаю1но 1 (646 мг, 5 ммоль, 0,87 мл). После перемешивания при 5 С в течение 15 мин и комнатной температуре в течание 1 ч реакционную смесь разбавляют EtOAc (75 мл), промывают холодпой водой, сушат MgS04 и упаривают. Оставшуюся пену хроматографируют на SiO особой чистоты (50 г), элюируя холодным EtOAc (200 мл) и затем CHjCN (200 ) Соответствуюш 1е фракции объединяют и упаривают, чтобы осталась твердая пена. Выход 1,1 г (58,1%).
(5R,6S)-6-(1R-гидpoкcиэтил)-4R- мeтил-3-(1 ,3-диметил-1,2,3-триазо- лий-4-ил)-метилтио -7-оксо-1-азаби- цикло(3,2,0)гепт-2 ен-2-карбоксилат
0 (ВМУ-26873).
К раствору (5R,6S)-пapa-нитpoбeн- зил-6-(1R-гидpoкcиэтил)-4R-мeтил)-3- f(1-иетил-1,2,3-триазол-4-ил)метил- тио }-7-оксо-1-азабицикло(3,2,0)гепт5 2-ен-2-карбоксилата (1,1 г, 2,32 ммоль) в (6 мл) и (5 мл) прика- пьшают при комнатной температуре три- фторметансульфонат (419мг, 2 ,55 ммоль,
0
5
0
5
0,29 Mji). Сразу же выпадает масло. После перемешивания в течение 1 ч су периатантную жидкость декантируют и масло растирают в Et20, при этом об- разовывается бледно-желтый порошок. Выход 1,31 г (88,6%). Кватернизован- ное соединение (1,02 г,.1,6 ммоль), растворенное в ТГФ (25 ил), (25 мл) и фосфатном буфере (1,2 М, рН 7,0, 16 мл) гидрируют в течение 60 мин в присутствии 10%-ного Pd/C (1,0 г) при давлении 40 p.s.i. Затем реакционную смесь охлаждают на льду, фильтруют и фильтрат экстрагируют (2x20 мл). Водный раствор хро- матографируют на обращение-фазовом SiOj (20 г), элю1груют холодной () водой (200 мп) и 57о-ным в (100 мп). Соответствующие фракции объединяют и лиофилизирутот. Выход 320 мг (56,9%).
S. pneumonia
S. pyogenes
S. aureus
S. aureus+50
сыворотки
S. aureus
(Pen-res.)
S. aureus
(Meth-res.)
S. faecalis
E. coli
(
E. coli
(10-3)
E. coli .
(10-O
E. coli
(10-)
E. coli
Ло-)
E. coli
(10-2)
)
K. K.
p. p.
p.
pneumonia
pneumonia
mirabilis
vulgaris
morganii
Полученные соединения (1) являются сильными антибиотиками, активны -ш по отношению к различным грамотрица- тельным и грамположительным бактериям.
Активность in vitro. Установлено, что образцы карбапенемньк соединений по примерам 1 и 2 после растворения в воде и разбавления питательным бульоном обнаруживают минимальные ин- гибирующие концентрации (МИК), выраженные в мкг/мл, по отношению к указанным микроорганизмам, что определено инкубированием при 37°С в течение ночи с разбавлением в пробирке.
I .....
В табл. 1 дана антибактериальная
активность in vitro карбапенемных производных по примерам 1 и 2.(N- формимидолилтиенамицин включен в честве соединения сравнения). Таблица 1
Пример 1
0,004 0,001 0,004 0,016
0,008
0,5 0,5
0,016
0,03
0,06
0,03
0,03
0,13 0,13 0,06 0,06 0,03 0,13
41
В табл. 2 даны минимальные ингиби рующие концентрации предпагамых и
1Д9310842
Продолжение табл.
известного соединений.
§8Я88
о О О о о
О ( о о о о
о о о о
ii: I сч fM I
2 I р о lA о ( и о о Г4 о (
I о о о о оо
|Л Г
§О 0
о - лm
о л о оо
о о о о оо
is,
I 5 о U1 5 о о
1
I о о о о о о
я. ч % я
о о о
ГМ (
Г4
о irt о оо
о о см о оо
о о о о оо
Л и
85 0 5 - 5 о о
о о о о о о
:П
1fT
S
I S
i
I Ш t l
I g§:-g
- о СЭ - о о
о о о о о
гч -
о о о о .
о о о о о
tvi см (N О
о S м о о
о о о о о
SS 32
о о ю о о о о сГ о сГ.
88Й§§
с о о о о
оSp fnnrt - О1Л Л о
5 - о5 о55оооооЗооо
оо оto о
Г4Г430
о9о
оо VIс5 а
оэ оо о
( л
оо ло о
о о оо о
§р «
5 -о э
о о ос о
о о о о о
« - р о о
о о о о о
N о о л о оо
о о о сГ ос
ГМ N чО vC-л
8 о «л 5 оо
I
(оо«чоо(( eooen о - %О в Л1ПО«л л(м - «-«Оо-« V4«NCw-4U- « о - .ОоО ЧвОи сО О
ТО«Ов«О« Ф ч ШO OO«9 O AMO0«0 - NN -.iaii i 5 5ai3iii5i5aai3i333
: S: S . газ:;
ft т ш tnч4-м 4 r4w4«ioaMH v
e« 4 ae ci -«- -«i.u3eidc- 4- f
iS :;H 3: ii55: S
5WU« HAttSb- - 94 | MW«OOM 4bUbGCi«w M4fl|
e.«93D.3i 9ЛооеС - - 49д«я««еа е &. 3040 iuuuo.a.vu в вСа«ч- «-Э м м
и - сЛ - |иЫМЫ1иЫЙмЬяё(.Ь93Ят«а
§qn4
О( Л4чэ -О4 Ф оооооооооош-
осГовоооеГовос
« «
( ооороооо - мом-
ООООООООООООО
(п . ° я . ч 1 я
оооооосГооооосГ
tCfn Oi f O tninci
°. ч ч .. . ч ч
о о о о сГ сГ о о о о о ю о
W3 «с
fn - - Ш (t (
э о о о о г
i ( - л о о -
..... ООООООООООО(ЧО
ОО 00 6 0
о о о о
о ЭОЛ
ооооооооооооо
о O« «
-«- ПъО д « - С ГЧ
ооооооэоо - о
оооооооооооо
«lO
((Л( IOOOOOOOOO ft
f V« в « о
- ОО - ОООООГ4О
ОООООООООООГ4ГЧ
ООООООООООО. «г
о «
. - . о о о о S 3 q S 2 3 3 S q «, г
оо оооо
f
о с
00000000000 04
ППМ П ППЛ(П «л ЭОО - N
ООдОООО ОООО - о
о
ООО - - OfNfV
ОООООООООООООФ ЛОО
оооо (м о Ы
о о о о о о о
ОСОООбоОООО -
.J..
1Л. оо - (Mc fMOO -fM -OIM
оо оо оо ооо ооев - м (-Т m
А л
«е«
«- - л 1 пт - р Л Оf «
ООООО - ООО - 5ОГЧ
э о о о о о о о о о о - N о Э
О lO
- ,ОО -«- пм г ф ООООООООО О - ОЛ
ЭОЭЭЭЭЭОООЭ МО(М(МОО
«П - 4гЧ(Пгч - т-Щ
ООЭО - - - о - гчо «л(Ntrt
осГо о сГсГэ сГсГо о сГ
51493108
Терапевтическая эффективность in мами, представлена в таОл. 3, где vivo соединения по примеру 1 и N- указана ЗДдо (доза выраженная в -формимидолилтиенамицина после внутри- мг/кг, требующаяся для защиты 50% мьпиечного введения мышам, экспериментально зараженньм различными организзараженных мьшей).
Примечание. За исключением Е. coli А15119 и К. pneumoniae А9964 лекарство вводили мышам внутримышечно через О и 2 ч после заражения. В случае Е. coli и К. pneumoniae обработку проводят спустя 1 и 3,5 ч после заражения. В каждом испытании при каждой дозе используют по 5 мьшей, Токсичность. Была определена токПродолже у
сичность соединения по примеру 1 после внутричерепного введения мьппам. Результаты представлены в табл. 4.
Таблица 4
По примеру 1 14
Бремя полуразло жения.
Площадь под кривой зависимости концентрации в крови от времени.
мами, представлена указана ЗДдо (доза мг/кг, требующаяся
зараженных мьшей).
15119 нутриЕ. coli ,5 ч озе исПродолжение табл.4 v у ----N-формимидо-лилтиенамицин
32
Среднее значение по 25 мышам.
Содержание в крови и время полу- разложения соединения по примеру 1 после внутримьшечного введения . 20 мг/кг мышам показаны в табл. 5, (соединения растворяют в 0,1 М фосфатном буфере при рН 7; данные по одному испытанию: по 4 мыши на соединение).
Таблица 5
7U93108
Выведение с мочой соединения по примеру 1 после внутрцмьшечпого введения 20 мг/кг мышам представлено в табл. 6 (соединения растворяют в
По примеру 1
N-формимидолилтиенамицин
В табл. 7 показано запщтное действие (3D50 ) при внутримышечном ввеВ табл. 8 дано содержание в крови и время полуразложения предлагаемьте соединений после внутримьшечного введения мьшам 20 мг/кг (соединения растворяют в О,1 М фосфатном буфере рН 7; значения основаны на одном испытании: по 4 мыши на соединение).
Таблица 8
Время полуразложения .Площадь под кривой зависимости концентрации в крови от времени. АЛ N-Форьтмидолилтиенамицип.
48
0,1 М фосфатном буфере рН 7; данные по одному испытанию: по 4 мыши на соединение).
26,1 0,5 12,1 0,1
0,1 26,7+6,7 0,1 12,2+3,6
дении заражен}1ьй мышам предлагаемых и известного соединений.
Твблнця 7
Формула изобретения
Ч
H3C-HC.S-A-OJ-R2
где R, - водород или С -С -алкил;
R.- - С,-С -алкил, бензил или алкилкарбоксиметил;R J - водород или пара-нитробен- зильная защитная группа при наличии также и противоиона; А - группа (СН), где п 1 4- или 2;
N моно- или дизамешенные С -алкилом группы
:
Н -N s.
хГХ / Г4-1 IN-1 з-,
- -9-
N
N
t,
г. т5,
N N (
причем указанное кольцо присоединено к А через атом углерода кольца и содержит атом азота, который кватер- низирован группой R2,
отлича-ющийся тем, что
кетоэфир общей формулы
ОН „RI
I п I
О СООРз
где R.и R имеют указанные значения, подвергают взаимодействию с дифенил- хлорфосфатом в присутствии диизопро- пилэтиламина в среде инертного органического основания, на полученное при этом соединение общей формулы
° н, НзС-саДЧ ь
Q - COOR;
где R и RJ имеют указанные значения; L - дифеноксифосфинилоксигруппа,
действуют производным меркаптана общей формулы20
HS-A-GN
где А и- ;; имеют ука: анные значения, в среде инертного органического раст-35 верителя в присутствии диизопропил- этиламина с последующим алкнлировани- ем образовавшегося соединения общей формулы
R ,. ° H3C-HC..
0 СООКз
где R, R, Аи имеют указанные
значения,
алкилирую1цим агентом общей формулы
Rj - X
где R имеет указанные значения;
X - галоген или группа сульфонатного сложного эфира,
и выделением целевого продукта или в случае необходимости, когда Rj- па- ра-нитробензильная группа, удалением карбоксизащитной группы.
ОН jjRi
НС-pfV QJ-N,-h
COORj
где R ( - водород или С -С -алкил; R-- Cf С.-алкил, бензил или
алкилкарбоксиметил; R - водород или пара-нитробен-
зильная защитная группа
при наличии также и противоиона;
группа (СН), где п - 1
или 2,
моно- или дизамещенные
С,- Сд-алкшюм группы
N-. N-N N-N &4)4J 1,4 1iJlА -CN-О
N
N
N
t
.-y-N. y-N N1 lx J-x N 3 N IJ
причем указанное кольцо присоединено к А через атом углерода кольца и содержит атом азота, который кватер- низирован группой R,
отличающийся, тем, что
соединение общей формулы
9 Б НС
R
S-A-Ol СООНз
где R, Rj, Аи имеют указанные
значения,
обрабатывают алкилирующим агентом
общей формулы
X
где R имеет указанные значения; I X - галоген или группа сульфонат- ного сложного эфира, с последукяцим выделением целевого продукта, или в случае необходимости, когда R j па- ра-нитробензильная группа, удалением карбоксизащитной группы.
Приоритет по признакам:

| СПОСОБ ПРИГОТОВЛЕНИЯ КОНЦЕНТРАТА ЛАКТАТА КАЛЬЦИЯ ИЗ МОЛОЧНОЙ СЫВОРОТКИ | 1996 |
|
RU2112391C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Патент США № 3950357, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
