тилформамида, или этилацетата, или дихлорметана, или ацетона при температуре от комнатной до 80°С, предпочтительно при 25-50 С. В случае, когда X - бензилоксикарбониламиногруп- па, а Q - 1-метил-2-метоксикарбонилвиниламиногруппа, поапедние переводят в аминогруппу. Целевой продукт вьще- ляют в свободном виде или в виде фармацевтически приемлемых щелочных или четвертичных аммониевых солей, з.п, ф-лКл I табл.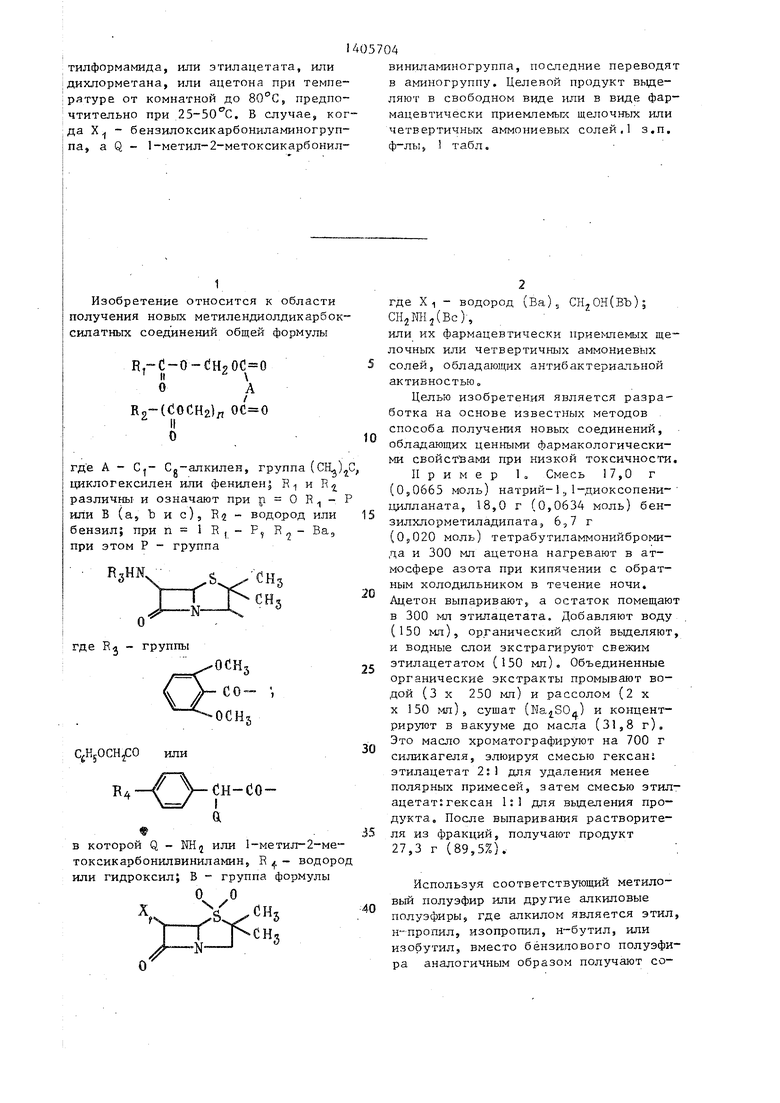














Изобретение относится к способу получения метилендиолдакарбоксилат- ных соединений фор{1улы В.-С-О-СНоОС О II I О А I B2-((JOCH2)/,OC«0 где А - Ci- Cg-алкилен, группа (, циклогексилен или фенилен; Е и R различные и чэзиачают: при п О R; - Р или В(а, Ъ и с); Rj - водород или бензил, при п 1 R,, -Р, Ва, при этом Р - группа где R - группы ОСН. С НуОСН СО- или сн-согде Q - NH или 1-метип-2-метокси- карбонклвиниламин; R }-- водород или гидроксил; В - группа формулы «N/ N CUs СНз где Х - водород (Ва), СН40Н(ВЪ), CHjNH(Bc), или их фармацевтически приемлемых щелочных ихш четвертичных аммониевых солей, обладающих антибактериальной активностью. Получение целевых соединений ведут из соедине- НИН формулы РСООМ или ВСООМ, где Р указан вьше, кроме Q - аминогруппа; В указан вьппе, когда X - Н, или CjHyCH OCGNHCMj; М - На, К или тетрабутиламмоний, и соединения формулы ХСН20(1 0 N I R2tClO)OCH2) где А, п, R указаны выше; X - галоген. Процесс ведут в среде TJ,N-AHMe4;: СП Ы
1
Изобретение относится к области получения новых метилендиолдикарбок- силатных соединений общей формулы
R,-C-O-{ H, и -
оА
К2(С10СН2)л О
где А - Cj- Cg-алкилен, группа (CIL)C, циклогексилен или фенилен; R и R различны и означают при п О R - Р или В (а5Ъис)5 R2- водород или 15 бензил; при ,-P, , при этом Р - группа
-b
N--Ч
СНз СНз
где Rj группы
qKjOCH O или
в которой Q - NHj или 1-метил-2-метоксикарбонилвиниламин, R - водоро или гидроксил; В - группа формулы
/°
А / (.н-
о
X Н УУ .Х.. I
г Т-СН;
/-N-J
0
5
5
0
5
0
где Xi - водород (Ва), СН20Н(ВЪ); (Ec}или их фармацевтически приемлемых щелочных или четвертичных аммониевых солей, обладающих антибактериальной активностью„
Целью изобретения является разработка на основе известных методов способа получения новых соединений, обладающих ценными фармакологическими свойствами при низкой токсичности.
Ц р и м е р К Смесь 17,0 г (0,0665 моль) натрий-1,, 1-диоксопени- цилланата, 18,0 г (0,0634 моль) бен- зилхлорметиладипата, 6,7 г (О5,020 моль) тетрабутиламмонийброми- да и 300 мл ацетона нагревают в атмосфере азота при кипячении с обратным холодильником в течение ночи. Ацетон выпаривак)Т5 а остаток помещают в 300 мл этилацетата. Добавляют воду (150 мл), органический слой вьщеляют, и водные слои экстрагируют свежим этилацетатом (150 мл). Объединенные органические экстракты промывают водой (3 X 250 мл) и рассолом (2 х X 150 мп), сушат () и концентрируют в вакууме до масла (31,8 г). Это масло хроматографируют на 700 г силикагеля, элюируя смесью гексанг этилацетат 2s для удаления менее полярных примесей, затем смесью этил- ацетат; гексан 1:1 для вьщеления продукта. После выпаривания растворителя из фракций, получа от продукт 27,3 г (89,5%).
Используя соответствующий метиловый полуэфир или другие алкиловые полуэфиры, где алкилом является этил, н- пропил, изопропил, н-бутил, или изобутил, вместо бёнзипового полуэфира аналогичным образом получают соответствующий алкил-1,1-диоксопени- цилланоилоксиметилдикарбоксилат.
Пример 2. Монобензиловый сложньй эфир цис-1,2-циклогександи- карбоновой кислоты.
К 15,4 г (0,10 моль) цис-1,2-цик- логександикарбонового ангидрида в 200 мл толуола по каплям добавляют
15
30 мин кристаллический продукт удаляют фильтрованием, промывают водой и сушат воздухом до получения 58,2 г указанной карбононой кислоты. После перекристаллизаци и из зтилах етата получают кристапличйский моногидрат, т. пл. 00-102 С.
Элементный анализ.
Вычислено, %: С 44,00; Н 5,66;
20 N 3,42,
С,Е,
С 43,93; Н 5,65;
раствор 10,8 г (о,10 моль) бензилово- ю воды. После перемешивания в течение го спирта в 50 МП толуола. Полученную смесь перемешивают при комнатной температуре В течение 1 ч, затем подогревают до и вьщерживают при этой температуре в течение 1 ч. Растворитель выпаривают до небольшого объема и целевой моноэфир получают после охлаждения и фильтрования твердого осадка (т, пл. 69-7l c),
В другом варианте реакционную смесь в толуоле обрабатывают эквимо- лярным количеством этанольной гидроокиси калия до получения калиевой соли монобензиловс1го сложного эфира. Натриевую соль получают, используя аналогичном образом метанолькую гидроокись натрия.
Соответствующие монобензиловые сложные эфиры или их натриевые или калиевые соли получают по описанному способу из следующих ангидридов ди- . карбоновых кислот: ангидрид янтарный, глутаровьш ангидрид (кипяче1-ше с обратным холодильником в течение ночи).
Пример 3. Кристаллический гидрат I,1-диоксопеницш1ланЬилокси- метиладипиновой кислоты.
К 400 мл ацетона добавляют 48,5 г (0,19 моль) натрий-1,1-диоксопеницилНайдено, /„ , N 3,42.
Кристалличность продукта пидтзерж- 25 дена кристаллографией рентгеновски1 ш лучами.
Пример 4. Натрий-1,1-диок- сопеницилланоилок симети-и-транс- , 4- -диклогександикарбоксилат. 30 А Бензштхлорметкл-траыс-1,4-цик- логександикарбоксилат.
К смеси 3,06 г (0,036 моль) бикарбоната натрия, 5,46-Г (0,018 моль) ,калийбензил-транс-1,4-диклогександи- gg карбоксилата, 50 мл воды и 500 мл хлороформа добавляют 6,17 г (0,018 моль) кислого сульфата тетра- бутиламмония. Полученного смесь переIмешивают при комнатной температуре в
. ланата, 48,0г (0,27 моль) беизилхлорме- до течение ночи. Слои разде пнют. Водный, тиладипатаи 19,3 г (0,06 моль) тетрабу- слой экстрагируют дважды хлороформом, тиламмонийбромида. Полученную смесь объединённые хлороформовые слои сушат нагревают при кипении с обратным хо- и выпаривают досуха. Пол ченную соль лодильником в течение ночи, фильтруют, тетрабутиламмония помещают в метилен- промывают ацетоном и фильтрат выпари- 45 хлорид (20 мл) и этот раствор прика- вают. Полученный остаток помещают в пывают к 20 мл бромхлорметана при 500 мл этилацетата, промывают попеременно рассолом и водой (порциями по 250 мл), снова рассолом и сушат над сульфатом магния. После выпаривания g растворителя в вакууме-получают 89,6 г сйетло-желтого масла. Это.масло помещают в 250 мл этилацетата.
ОС. Полученную смесь перемешивают
добавляют 20,0 г 10% Pd/C и полученную смесь гидрируют при давлении 3,52 кг/см в течение 1 ч. После добавления 15 г свежего катализатора гидрирование продолжают в течение 2,5 ч. Катализатор удаляют фильтрова55
при температуре окружающей среды в течение 70 ч, растворитель выпаривают и добавляют к остатку этилацетат. Выпавши в остаток тетрабутиламмоний- бромид удаляют фильтрованием, фильтрат сушат () и выпаривают в вакууме до получения 5 г (91%) неочищенного продукта. После очистки хро- матографированием на силикагеле, при элюировании смесью этиловый эфирггек- сан 1:3 получают 1,9 г (35%) целевого ггррдукта в виде масла.
нием, лепешку промывают ацетоном ,(1500 мл) и объединенные фильтрат и промывки выпаривают в вакууме до по- лу 1ения вязкого масла. Это масло помещают в 150 МП ацетона и медленно добавляют воду для начала кристаллизации, а затем продолжают до тех пор, пока всего не будет добавлено SCO мл
30 мин кристаллический продукт удаляют фильтрованием, промывают водой и сушат воздухом до получения 58,2 г указанной карбононой кислоты. После перекристаллизаци и из зтилах етата получают кристапличйский моногидрат, т. пл. 00-102 С.
Элементный анализ.
Вычислено, %: С 44,00; Н 5,66;
N 3,42,
воды. После перемешивания в течение
С,Е,
С 43,93; Н 5,65;
Найдено, /„ , N 3,42.
Кристалличность продукта пидтзерж- дена кристаллографией рентгеновски1 ш лучами.
Пример 4. Натрий-1,1-диок- сопеницилланоилок симети-и-транс- , 4- -диклогександикарбоксилат. А Бензштхлорметкл-траыс-1,4-цик- логександикарбоксилат.
К смеси 3,06 г (0,036 моль) бикарбоната натрия, 5,46-Г (0,018 моль) ,калийбензил-транс-1,4-диклогександи- карбоксилата, 50 мл воды и 500 мл хлороформа добавляют 6,17 г (0,018 моль) кислого сульфата тетра- бутиламмония. Полученного смесь перетечение ночи. Слои разде пнют. Водны слой экстрагируют дважды хлороформо объединённые хлороформовые слои суш и выпаривают досуха. Пол ченную сол тетрабутиламмония помещают в метиле хлорид (20 мл) и этот раствор прика пывают к 20 мл бромхлорметана при
ОС. Полученную смесь перемешивают
течение ночи. Слои разде пнют. Водный, слой экстрагируют дважды хлороформом, объединённые хлороформовые слои сушат и выпаривают досуха. Пол ченную соль тетрабутиламмония помещают в метилен- хлорид (20 мл) и этот раствор прика- пывают к 20 мл бромхлорметана при
при температуре окружающей среды в течение 70 ч, растворитель выпаривают и добавляют к остатку этилацетат. Выпавши в остаток тетрабутиламмоний- бромид удаляют фильтрованием, фильтрат сушат () и выпаривают в вакууме до получения 5 г (91%) неочищенного продукта. После очистки хро- матографированием на силикагеле, при элюировании смесью этиловый эфирггек- сан 1:3 получают 1,9 г (35%) целевого ггррдукта в виде масла.
1Н-ЯМР (CDCl.) еЛм.д.г 1,0-2,4 (м, lOlOl 5,1 (с, 2Н); 5,7 (с, 2Н); 7,3 (с, 5н),
В. Бензил-1,1-диоксопенициллано- шюксиметил-транс ,4 циклогександикарбоксилат.
Раствор 4,2 г (13,5 ммоль) бензил хлорметил-транс-1,4-циклогександикар боксилата, 3,63 г (14,2 гмоль.) натрий- , -диоксопеницилланата5 1545 г (4,5 ммоль) и 100 мл ацетона нагревают при кипячении с обратным холодильником в течение ночи. Ацетон выпаривают, добавляют этилацетат (100 мл), раствор промывают водой (трижды), рассолом и сушат над без- водным сульфатом натрия. Растворитель удаляют, выпаривая в вакууме до получения неочшценного продукта, который очищают хроматографически на колонке с силикагелем, элюируя смесь зтилацетат гексан 1:1 до получения 5,3 г (78%) очищенного продукта в виде масла, которое используют на следующей стадии,
. С К раствору 2j5 г (4,9 ммоль) сложного бензилового эфира, полученного в части В, в 50 нп этилацетата в атмосфере азота добавляют 1,5 г
30 К раствору 18,53 г (0,062 моль) калийбензилтерефталата в 300 мп вод добавляют 600 мл хлороформа, 10,38 (0,121 моль) бикарбоната натрия и 20,95 г (0,062 моль) кислотного сул
0% Pd/C катализатора. Полученную смесь гидрируют при давлении - 2 атм в течение около 20 мин. Катализатор удаляют фильтрованием, и к фильтрату добавляют 0,82 г (4,9 ммоль) gg фата тетрабутиламмония. Полученную натрий-2-этилгексаноатс1о. После пере-смесь перемешивают при комнатной
мешивания в течение 30 мин при комнатной температуре полз ченную смесь концентрируют до одной трети объема
температуре в течение 3 ч, органич кий слой вьщеляют, и годную фазу экстрагируют дважды хлороформом, Ор
и добавляют три объема этилового йфи- 40 ганические слои объединяют, сушат
ра„ Выпавшее в осадок соединение отфильтровывают, промывают эфиром и сушат в атмосфере азота до получения 1,7 г (79% выход стадии).
Пример 5 о Кристаллическая Ij -диоксопеницш1ланоилоксиметил- транс 194-циклогексанкарбоновая кислота.
(Na. и растворитель выпаривают до получения тетрабутиламмониевой соли бензилтерефталата. Ее помещают в 25 мл метиленхлорида и полученньй 45 раствор добавялют по каплям к 100 мл бромхлорметана при , Полученной смеси дают нагреться до комнатной температуры, перемешивают в течение ночи .и продукт выделяют и очищают
К раствору 6,07 г (12 ммоль) бензил- , 1-диоксопеницилланоилоксиметил- gQ по примеру 5 (часть А) до получения -транс-154-диклогександикарбоксилатауказанного сложного диэфира в виде
в 100 мл этилацетата в атмосфере азо- кристаллов, т.-пл, 64-6б с,
В. Бензил-,1-диоксопеницилланоилоксиметил тер ефталат.
Раствор 6,34 г (0,021 моль) бен- зилхлорметилтерефталата, 5,58 г (0,022 моль) натрий-,1-диоксопени- цилланата, 2,24 г (0,0069 моль) тетта добавляют 3,2 г 10% РбУС в качестве катализатора. Полученную смесь гидрируют в течение 45 мин, встряхи вая при 50 пси (3552 кг/см -). Смесь фильтруют, фильтрат концентрируют в вакууме до получения в остатке масла, которое кристаллизуется при стоянии.
55
рабутиламмонийбромида и 200 мл ацето
057046
Этот продукт перекристаллизовывают из смеси этилапетата и гексана в атмосфере азота до получения 2,35 г
g кристаллического продукта, который содержит.некоторое количество масла. Его помещают в этилацетат (100 мп) и добавляют эквивалентное количество натрий-2-этилгексаноата, Выпавшую
10 в осадок натриевую соль перемешивают в течение 45 мин, концентрируют до одной трети объема и добавляют этиловый эфир для завершения осаждения. Натриевую соль собирают фильтровани-
15 ем, промывают эфиром и сушат в атмосфере азота, затем помещают в воду (50 мл), подкисляют соляной кислотой и полученную смесь экстрагируют этил- ацетатом. Экстракты сушат (),
20 растворитель выпаривают в вакууме, остаток кристаллизуют из смеси этил- ацетата и гексана и сушат в атмосфере азота до получения 1,85 г (37%) кристаллического продукта, т, пл.
25 118,5-119°С,
Пример 6, 1,1-Диоксопени- -цилланоилоксиметилтерефталат и его натриевая соль,
А, Бензилхлорметилтерефталат,
30 К раствору 18,53 г (0,062 моль) калийбензилтерефталата в 300 мп воды добавляют 600 мл хлороформа, 10,38 г (0,121 моль) бикарбоната натрия и 20,95 г (0,062 моль) кислотного суль
) gg фата тетрабутиламмония. Полученную смесь перемешивают при комнатной
температуре в течение 3 ч, органичес-- кий слой вьщеляют, и годную фазу экстрагируют дважды хлороформом, Ор- ,
(Na. и растворитель выпаривают до получения тетрабутиламмониевой соли бензилтерефталата. Ее помещают в 25 мл метиленхлорида и полученньй раствор добавялют по каплям к 100 мл бромхлорметана при , Полученной смеси дают нагреться до комнатной температуры, перемешивают в течение ночи .и продукт выделяют и очищают
по примеру 5 (часть А) до получения указанного сложного диэфира в виде
рабутиламмонийбромида и 200 мл ацето
на перемешивают при температуре кипения с обратным холодильником в атмосфере азота в течение 18 ч. Затем ацетон выпаривают, остаток помещают в этилацетат, промывают водой три раза и сушат (). После выпарк вания растворителя получают 1I г неочищенного продукта, который очищают, пропуская через колонку с силикагелем (20x2 см), элюируя смесью ди- этилацетат:гексан 1:1. После выпаривания полученных фракций получают 10 г (96%) целевого бензилового сложного эфира в виде масла.
C.Раствор 9 г бензилового сложного эфира, полученный в части В в 50 МП этилацетата откачивают для удаления воздуха и заполняют сосуд азотом, К раствору добавляют 2,5 г 10% Pd/C катализатора и полученную смес гидрируют при давлении 3 атм в течение 20 мин. Полученную смесь фильтруют через фильтр, промьгоают этил- ацетатом, К фильтрату и промывкам добавляют 2,98 г натрий-2-этилгекса- ноата, полученную смесь перемешивают в течение 30 мин. Дополнительно по
50 мл зтилацетата и эткл ового эфира добавляют к полученной густой смеси, которую фильтруют и промывают этиловым эфиром. После суики в течение ночи получают 5,8 г (75%) кристаллической натриевой соли.
D.К раствору 1 г з азанной натриевой соли в 50 МП воды добавляют 50 мл 0,1 н.соляной кислоты. Полученную смесь экстрагируют 75 мп этил ацетата. Этилацетат концентрируют в вакууме до получения суспензии и добавляют достаточное количество этилацетата, чтобы растворить осадок Раствор перемешивают при медленном добавлении гексана при комнатной температуре до точки помутнения. Затем раствор подогревают на паровой бане для улучшения растворения, и добавляют несколько капель гексана, смесь охлаждают до комнатной температуры
и помещают в холодильник. Полученные кристаллы собирают фильтрованием и сушат в атмосфере азота до получения 900 мг (95%) указанной кислоты, т, пл 167-169 С (с разложением).
Пример 7. Натрий-1,1-диок- сопеницшшаноилоксиметилизофталат.
А. Бензилхлорметилизофталат.
По примеру 7 (часть А) 17,0 г (0,058 моль) калийбензшшзофталата в
45 мл воды и 500 мл хлороформа прев-; ращают в его тетрабутиламмонийную соль и ее подвергают взаимодействию с избытком бромхлорметана, .Полученный неочищенный продукт (15 г) помещают в этилацетатс его добавляют к 45 г . силикагеля,„полученную смесь суспендируют и растворитель выпаривают. Оставшийся скликагель в сухом состоянии загружают в 8-дюймовую колонку с силикагелем и элюируют смесью этиловый эфирггексан 1:3, Поспе выпаривания растворителя из фракции, содер- жащк-х продукт, получают целевой Д1-1эфир в виде масла,
B,Бензил-,i-диоксопенициллано- илоксимети.ткз офталат.
Смесь 12,22 г (0,04 моль) бензилхлорметилизофталата, 10,75 г
( моль) бензилхлорметилизофта- лата, 10,75 г (0,042 моль) натрий- 1S i-дис.ксопенкцилланата, 4,,31 г (0,0134 моль) тeтpaбyтилaммoшiйбpoмида и 400 мл aiteTOKa нагревают при
кипячении с обратным холодильником в . течение 30 ч. Ацетон выпаривают и заменяют этилацетатом. Раствор промь;--. вают трижды водой, рассоло.м и сушат
(Na.SO), По1:ле выпаривания растворителя и хроматографкрования остатка на силикагеле с элюированием смесью этилацетат:гексан.65:35 получают продукт в,виде масла, которое кристаллизуе ся при .стоянии, выход 41%,
C,Смесь 8,14 г (0,016 моль) сложного бензилового эфира, полученного
в части Б, 2,5 г 10% Pd/C катализатора и 50 мл этилацетата гидрируют
по примеру 7 (часть С), Полученную смесь фильтруют для удаления катализатора и добавляют 2,7 г (0,016 моль) натрий 2-этилгексаноата. После перемешивания в течение 20 мин вязкую
взвесь концентрирз т до треть объема и добавляют этиловый эфир для завершения осаждения. Полученные кристаллы, вьщеляют фильтрованием и сушат в атмосфере азота до получения 6,35 г
(90%) указанной натриевой соли.
Пример 8. 6-tD-(2-амино-2- -фенилацетамидо) пеницилланоилоксиме- тил-транс-1,4-циклогександикарбоновая кислоте, гидрохлорид.
55
А. Бензил-6-{1)( 1-метил-2-ме- таксикарбонилвиниламино)-2-фенилацет- амидо} пеницилланоилоксиметил-транс- -1,4-циклогександикарбоксилат.
Раствор 2,22 г (3,28 ммоль) тетра- б утил аммоний-6 D- 2- ( 1 -метил- 2-ме- токсикарбонилвиниламино)-2-фенилацет- aMHfloJjпеницилланата, 1,0 г (3,23 ммоль) бензилхлорметил-транс- 1,4-циклогександикарбоксилата и 100 мл ацетона перемешивают при комнатной температуре в течение ночи„ Ацетон выпаривают и заменяют этил- ацетатом. Раствор промывают водой, сушат (NanSOx ) и растворитель выпаривают в вакууме. Оставшийся неочищенный материал очищают хроматографи чески на силикагеле, элюируя смесью этилацетатггексан 40;60 до получени 1,5 г (53%),
В. Бензилрвый сложный эфир, полу- ченньй в части А, ,5 г (2,08 ммоль) растворяют в 25 мп ацетона и добавляют 20,1 мл 0,1 н,соляной кислоты, Полученную смесь перемершвают 10 мин затем добавляют дополнительно 2,0 мл 0,1 н,соляной кислоты и растворитель выпаривают, К остатку добавляют 75 мл воды, полученную смесь экстрагируют дважды этиловым эфиром, содер жащим небольшое количество этилацета та, К полученным экстрактам добавляю 0,75 г 10% Pd/C катализатораJ и полученную смесь встряхивают под давлением водорода 50 пси (3s52 кг/см ) в течение 30 мин. Катализатор удаляют фильтрованием, полученньй фильтрат сушат вымораживанием до получения 700 мг продукта.
Пример 9, 6- В- 2-aivMHO- -(4-оксифенил)ацетамидо пенициллано илоксиметил-транс-1,4-циклогександи- карбоновая кислота, гидрохлорид,
А, Бензил-6-(Г)( 1-метил-2-ме- токсикарбонилвиннлами,но )-2-(4-оксифенил) ацетамидо j пеници лланоилокси- Метил-транс-1,4-циклогександикарбйк силат.
Раствор 0,5 г (1-561 ммоль) бензил хлорметил-транс-1,4-циклогександикар боксилата и 1,14 г (ls61 ммоль) тет- рабутиламмоний-б-Г (1-метил-2- -метоксикарбонилвиниламино)-2(4-ок- сифенил)ацетам1що пеницилланата в 50 мл диметилформамида перемепшвают в течение ночи при комнатной температуре. Реакционную смесь разбавляют этилацетатом, трижды промывают водой, затем рассолом и сушат (NajSO.) Растворитель выпаривают в вакууме. К остатку добавляют этилаце- тат, полученную смесь снова промыва
0
5
ют водой, рассолом, сушат и выпаривают для удаления остатков диметилформамида. Остаток очищают на силикагеле с помощью хроматографии, элюируя смесью этилацетат:гексан 7:3 до получения 500 мг (42%) очищенного сложного диэфира,
В. К раствору 0,5 г (0,678 ммоль) очищенного сложного диэфира, полученного в части А, в 25 мл ацетона добавляют 6,8 мл 0,1 н,соляной кислоты. После перемешивания в течение 10 мин добавляют дополнительно 1,0 мл 0,1 н,соляной кислоты, и ацетон выпаривают в вакууме. Остаток разделяют между водой и этиловым эфиром, и водный слой промывают эфиром. К водной, фазе добавляют 0,35 г 10% Pd/C катализатора в атмосфере азота, и полученную смесь гидрируют при 50 пси (3,52 кг/см ) в течение ночи. Полученную смесь фильтруют для удаления катализатора, водный фильтрат сушат 5 вымораживанием до получения 200 мг (50%) указанного соединения.
Пример 10, Натрий-6-(2-фе- нок си ацетамидо) пеницил-паноилоксиметил- диметилмалонат,
0АО Бензил-6(2-феноксиацетамидо)
пеницилланоилоксиметилдиметилмалонате - К 50 мл диметилформамида добавляют 3,88 г (0,01 моль) калий-6-(2-феноксиацетамидо) пеницилланата, 2,7 г g ( моль) бензилхлорметилдиметил- малоната. Полученную смесь перемеши™ вают при комнатной температуре в те- чение 3 ч. Смесь выливают в 150 мл этилацетата, промывают водой (З х 0 X 50 мп), рассолом (1 х 50 мл), сушат () и выпаривают досуха в вакууме. Остаток помещают в небольшое количество этилацетата и переносят в колонку с силикагелем (200 г), Ко- 5 лонку элюируют смесью этилацетат:гексан 1:, 1 , полученные фракции с продуктом объединяют и концентрируют в вакууме до получения 2,0 г продукта в виде бесцветного масла.
В Смесь 2,0 г (3,4 ммоль) продукта из части А, 40 мп этилацетата и 2,0 г 10% Pd/C катализатора перемешивают в атмосфере водорода при давлении 50 пси в течение 45 raн, Добавляют еще 1 г катализатора и перемешивание продолжают в течение 30 мин, Полут-шнную смесь фильтруют, промывают фильтровальную лепешку этилацетатом. Фильтрат и промь вки , перемешивают, до0
55
бавляя 0,56 г (3,37 ммоль) натрий-2- -этилгексаноата. Перемешивание продолжают, добавляя равньй объем этилового эфира. Осевший твердый продукт гранулируют, перемешивая в течение 30 мин, фильтруют, промывают эфиром и сушат в атмосфере азота до получения 1,35 г (77%) указанной соли.
Пример П. Натрий-6-( 2-фе- Q ноксиацетамидо)пеницилланоилоксиметил глутаратат.
А, Бензил-6-(2-феноксиацетамидо) пеницилланоилоксиметилглутарат,
К 50 мл диметилформамида добавляют 15 соль осаждают этиловым эфиром и со- 3,88 г (0,01 моль) калий-6-(2-фенок- бирают фильтрованием до получения сиацетамидо)пеницилланата, 2,7 г 1,,95 г (63%/ указанного соединения, (0,01 моль) бензилхлорметилглутарата. Пример 13, Повторяют способ Полученную смесь перемешивают в те- примера 12 с натрий-6-(2,6-диметокси-; чение 3 ч, после чего добавляют 3,0 г 20 бензаь{идо)пеницилланатом и бензилхлор
очищают по примеру 11 до получения бензил-6-(2,6-диметоксибензамидо)пе- ницилланоилоксиметилдиметилмалоната с выходом 65%,
К 3,5 г (5,7 ммоль) этого сложного бензилового эфира в 50 мл этилацетата добавляют 2,5 г 10% Pd/C катализатора. Полученную смесь гидрируют при давлении 50 пси в течение 1 ч. После фильтрования для удаления катализатора к фильтрату добавляют эквимолярное количество натрий-2-этилгексаноата в этилацетате, Полученн по натриевую
(0,02 моль) иодида натрия. Перемешивание продолжают в течение ночи. Реакцию гасят, добавляя 150 мл этилацетата. Промывают реакционную смесь водой (Зх50 мл), рассолом (1x50 мл) и сушат Na SO . После выпаривания растворителя в вакууме получают 6,0 г масла, которое очищают на хроматометилглутаратом на 2,2 молярном масштабе до получения бензил-6-(2,6- -диметоксибензамидо)пенициллакоилокг симетилглутарата с количественным 25 выходом в виде масла.
Гидрирование 1,4 г (2,2 ммоль) описанного сложного бензилового эфи- . ра на катализаторе Pd/C по способу, использованному в г.редыдз цих приме- 30 paxj и превращение в натриевую соль натрий-2-этилгексаноатом приводят s к получению 0,87 г (72,5%) натрнй-6- . -(2,6-диметоксибензамидо)пениииллгно ииоксиметилглутарата.
графической колонке на силикагеле (300 г), элюируя смесью этиладетат: гексан 1:1. После концентрирования фракций, содержащих продукт, получают 5,0 г (85%) бесцветного масла.
В. Продукт, полученный в части А, 5,0 г (0,0085 моль), 50 мл этилацета- 35 та,и 5 г 10% Pd/C катализатора гидрируют при давлении 3 атм в течение 1 ч. Добавляют дополнительно 2,5 г катализатора и гидрирование продолжают еще 2 ч. Полученную смесь фильт- 40 руют через диатомовую земпю, промывают этилацетатом. Объединенные фильтрат и промывки 200 мл выливают в чистую колбу и добавляют 6,13 мл натрий-2-этилгексаноата в этилацетате (0,23 г/мл). После перемешивания в течение 30 мин полученную смесь разбавляют равным объемом этилового эфира и фильтруют до получения 2,25 г (51%) натриевой соли.
Пример 12. Натрий-6-(2,6- -диметоксибензамидо)пеницилланоилок- симетилдиметилмалонат.
Смесь 4,02 г (0,01 моль) натрийГидрирование 1,4 г (2,2 ммоль) описанного сложного бензилового эфи- . ра на катализаторе Pd/C по способу, использованному в г.редыдз цих приме- 30 paxj и превращение в натриевую соль натрий-2-этилгексаноатом приводят s к получению 0,87 г (72,5%) натрнй-6- . -(2,6-диметоксибензамидо)пениииллгно ииоксиметилглутарата.
Пример 14, 1,1-Диоксопени- цилланоилоксиметил-6- D- 2-(1-метил- -2-метоксикарбош-1лвиниламино)-2-фе- нилацетамидо пеницилланоилоксиметил- -транс-1,4-циклогександикарбоксилат. Смесь 2,33 г i3,97 ммоль) натрий- -1,1-диоксопеницилланоилоксиметнл- -транс-1,4-циклогександикарбоксилата, 1,72 г (3,97 ммоль) иодометил-6- D- 2-(1-метил-2-метоксикарбонилвинил- 45 амино)-2-фенилацетамидо1 пенициллана- та и 40 мл диметилформамида перемешивают при комнатной температуре в те- . чение 5 мин. Смесь разбавляют этил- ацетатом, трижды промывают небольшими порциями воды, один раз рассолом и . сушат (). После выпаривания растворителя в вакууме и хроматографиро- вания остатка на колонке с силикаге - лем при элюировании смесью этилацета50
-6-(2,6-диметоксибензамидо)пеницилла- 55 та и гексана получают 1,4 г (40%) це- ната, 3,3 г (0,01 моль) бензилхлор-левого енамина.
метилдиметилмалоната и 30 мл диметил-Пример 15. 1,1-Диоксрпениформамида перемешивают при 25 С в те- цилланоилоксиметил-6- 1 -( 2-амино-2- чение 60 ч, затем продукт выделяют и-фенилацетамидо) пеницилланоилоксиочищают по примеру 11 до получения бензил-6-(2,6-диметоксибензамидо)пе- ницилланоилоксиметилдиметилмалоната с выходом 65%,
К 3,5 г (5,7 ммоль) этого сложного бензилового эфира в 50 мл этилацетата добавляют 2,5 г 10% Pd/C катализатора. Полученную смесь гидрируют при давлении 50 пси в течение 1 ч. После фильтрования для удаления катализатора к фильтрату добавляют эквимолярное количество натрий-2-этилгексаноата в этилацетате, Полученн по натриевую
метилглутаратом на 2,2 молярном масштабе до получения бензил-6-(2,6- -диметоксибензамидо)пенициллакоилокг симетилглутарата с количественным выходом в виде масла.
Гидрирование 1,4 г (2,2 ммоль) описанного сложного бензилового эфи- ра на катализаторе Pd/C по способу, использованному в г.редыдз цих приме- paxj и превращение в натриевую соль натрий-2-этилгексаноатом приводят s к получению 0,87 г (72,5%) натрнй-6- -(2,6-диметоксибензамидо)пениииллгно ииоксиметилглутарата.
Пример 14, 1,1-Диоксопени- цилланоилоксиметил-6- D- 2-(1-метил- -2-метоксикарбош-1лвиниламино)-2-фе- нилацетамидо пеницилланоилоксиметил- -транс-1,4-циклогександикарбоксилат. Смесь 2,33 г i3,97 ммоль) натрий- -1,1-диоксопеницилланоилоксиметнл- -транс-1,4-циклогександикарбоксилата, 1,72 г (3,97 ммоль) иодометил-6- D- 2-(1-метил-2-метоксикарбонилвинил- амино)-2-фенилацетамидо1 пенициллана- та и 40 мл диметилформамида перемешивают при комнатной температуре в те- . чение 5 мин. Смесь разбавляют этил- ацетатом, трижды промывают небольшими порциями воды, один раз рассолом и . сушат (). После выпаривания растворителя в вакууме и хроматографиро- вания остатка на колонке с силикаге - лем при элюировании смесью этилацета
та и гексана получают 1,4 г (40%) це- левого енамина.
метил-транс-1,4-циклогександикарбок силат, гидрохлорид.
. К раствору 1,4 г (1,61 ммоль) 1,1-диоксопеницилланоилоксиметил 6- (1-метил-2-метоксикарбонилви- ниламино)-2-фенилацетамидо пеницил- ланоилоксиметил-транс-1,4-циклогек- сандикарбоксилата.в 150 мл ацетона добавляют 20 мл 0,1 щсоляной кислот ты. Раствор перемешивают в течение 5 мин. Растворитель выпаривают в вакууме, остаток разбавляют водой, водную фазу дважды промывают смесью этиловый эфир:этилацетат,,; 1, Водную фазу фильтруют и сушат выморахшвани- ем до получения 634 мг (48%) указанного соединения, т, пл, 155-170 ,С (.с разложением)
Пример 16, Бензилхлорметил себацинат, К смеси 48,67 г (о,155 моль) монобензилсебацината, г (0,301 моль) бикарбоната, натрия, 200 мл воды и 52,55 г (0,155 моль) кислого сульфата тетра- бутиламмония добавляют 100 мл хлороформа. После встряхивагшя органический слой выделяют, водную фазу снова экстрагируют хлороформом и объединенные хлороформовые слои сушат ( После выпаривания растворителя остаток помещают ,в 50 мя бромохлорметана и перемешивают в течение ночи при комнатной температуре. Смесь выпари™ вают в вакууме, остаток смешивают с этилацетатом, фильтруют, и фильтрат концентрируют в вакуумео Оставшийся сьфой продукт очищают в хроматографи- ческой колонке с силикагелем до получения 2 г очищенного сложного моно™ эфира в виде масла
Пример 17в 1,1 Диоксопени- цилланоилоксиметил-6-|D 2-(1-метил- 2 метоксикарбонилвиниламино)-2-фе- нилацетамидо пеницилланоилоксиметил себацинато
К раствору 0,59 г ( ммоль) иодометил-6- 1 - 2-(1-метил-2 метокси карбонилвиниламино)-2-феиилацетамидо пеницилланата в 10 мл диметилформами да добавляют 0,47 г (1,0 мгдаль) натрий- 1 , 1 Д4 оксопеницилх;анбш1оксимет1-ш себацината, полученную смесь переме- пшвают до получения раствора. Реакционную смесь подвергают мгновенному хроматографированию на колонке с си- ликагелем 23 см слоя, эл оируя смесью этилацетат;гаксаи 7:3 до получения 200 мг (22%) целевого енамина.
5 0 о
g
д g
5
Пример 18, 1,-Диоксопени- цилланоилоксиметил-6- 1)-( 2-амино-2- -фенилацетамидо) пеницилланоилоксиме- тилсебацинат, гидрохлорид.
К перемешиваемому раствору 200 мг (0,22 ммоль) пеницилланоилоксиметил- (1-метил-2-метоксикарбонил- виниламино)-2-фенилацетамидо пени- цилланоилоксиметилсебацината в 25 мл ацетона добавляют 3,2 мл О,1.н,соляной кислоты Полученную Смесь перемешивают несколько минут, добавляют еще 1,0 МП соляной кислоты и перемешивание продолжают еще 1 мин. Ацетон выпаривают, остаток разбавляют водой и дважды промывают смесью этилового эфира и этилацетата. Водный слой фильтруют и сушат вымораживанием до получения 110 мг (59%) продукта.
Пример 19, I,l-Диoкcoпeни- циллaнoилoкcимeтил-6- D-( 2-амино-2- -фенилацетамидо)j пеницилланоилокси- метилтерефталат, гидрохлорид.
A.1,}-Диoкcoпeнициллaнoилoкcимe- тил-6- D- 2-( 1-метил-2-метоксикарбо- нилвиниламино)-2-фенилацетамидо пе- 1-шцш1ланоилоксиметилтерефталат.
К раствору 0,59 г (1 ммоль) иодо- метил-6- - 2-(1-метил-2-метоксикар- бонилвиниламино)-2-фенилацетамидр} пеницилланата в 10 мл диметилформами- да добавляют 0,48 г (1,1 ммоль) нат- рий-1,1-диоксопеницилланоилоксиметил- терефталата. Полученную смесь перемешивают до получения раствора, который разбавляют этилацетатом и промывают небольшими порциями воды (трижды), один раз рассолом и сушат (), Растворитель вьтаривают в вакууме, и остаток помещают в небольшом количестве этилацетата и очищают на хро- матографической колонке с силикагелем, элюируя смесью этилацетат:гек- сан 614..фракции, содержащие лродук-- ты, выпаривают до получения енаминза- мещенного соединения 0,3 г (23%),
B,К перемешиваемому раствору
г (о,35 ммоль) описанного енамин- защищенного продукта в 25 мл ацетона добавляют 4,5 мп 0,1 н„соляной кислоты. Полученную смесь перемешивают в течение нескольких минут, растворитель выпаривают, и остаток разделяют между водой и этиловым эфиром. Затем водную фазу промывают смесью этиловый зфир;этилацетат 1:1, фильтруют и полученньш фильтрат сушат выморажи-
151405704 6
ванием до получения 222 мг (78%) ука- сопеницилланоилоксиметилглутарат, занной соли соляной кислоты,гидрохлорид.
Пример 20, 1,1-Диоксопени- Аналогично 2,94 г (5 ммоль) того цилланоилоксиметил-6- 1)-( 2-амино-2- же иодометилового сложного эфира ме- -фенилацетамидо) пеницилланоилокси- тилацетоацетатенаминзащищенного ам- метилпэофталат, гидрохлорид,пициллина и 3,0 г (7,5 ммоль) натA,1,1-Диоксопеницилланоилоксиме- рий-1,1-диоксопеницилланоилоксиметил- тил-6- 1 -С2-(1-метил-2-метоксикарбо- глутарата перемешивают в 20 мл диме- килвиниламино)-2-фенилацетамидо}1пе- ю тилформамида в течение 5 мин и га- ницилланоилоксиметилизофталат, сят реакцию 150 мл этилацетата. Полу-
К раствору 0,59 г (110 ммоль) иодо- ченную смесь промывают водой (3 х метил-б-ГГ)( 1-метил-2-метоксикар- х 50 мл), рассолом (50 мл), сушат бонилвиниламино)-2-фенилацетамидо Jne- () и выпаривают досуха в ваку- ницилланата в 10 мл диметилформамида js Уме, Остаток очищают на хроматогра- добавляют 0,43 г (1,0 ммоль) натрий- фической колонке с,силикагелем -1,1-диоксопеницшшаноилоксиметилизо- (100 г), элюируя смесью метиленхло- фталата, Пол.ученную смесь .перемешива- рид:зтилацетат 60:40 (по объему), ют при комнатной температуре до за- отбирая фракции каждые 60 с. Фракции вершения растворения. Реакционную 20 16-24 объединяют и растворитель вы- смесь обрабатывают по примеру 19 паривают в вакууме до получения 1,8 г (часть А) до получения 200 мг (23%) пены. Ее растворяют в 30 мл ацетона, объединенного енамина,добавляют 21,5 мл 0,1 н,соляной ;,кисB,Енаминзащищающую группу удаляют лоты и полученную смесь перемешивают и получают гидрохлорид по примеру 19 25 в течение 20 мин. Ацетон выпаривают (часть в) с выходом 94%, при пониженном давлении, водную фазу
Пример 21, (2-Амино- , зкстрагируют этиловым эфиром (30 мл) -2-фенилацетамидо) пеницилланоилокси- и смесью зтилацетат:этиловый эфир метил-1,-диоксопеницш1ланоилоксиме- 1:1, Водный слой фильтруют через ди- тилсукдинат, гидрохлорид,30 атомовую землю и полученный фильтрат
К раствору 5,9 г (0,01 моль) иодо- лиофилизируют до получения 1,45 г метил-6- 1)( 1-метил-2-метоксикар- (37%) целевого гидрохлорида, бонилвиниламино)-2-фенилацетамидо 5 По описанному способу получают пеницилланата в 30 мл.диметилформами- также другие соединения, да добавляют 5,5 г (0,014 ммоль) нат- g В, (2-Амино-2-фенилацетамк- рий-1,1 -диоксопеницшшаноилоксиметил- до )J пеницилланоилоксиметил-1,1 -диок- сукцината при перемешивании. Спустя- сопеницилланоилоксиметиладипат, гид- 20 мин добавляют 150 мл этилацетата, рохлорид.
Полученную смесь промывают водой С, (2-Амино-2-фенилацетами- (3x50 мл), рассолом (50 мл).водой 40 до)}пеницилланоилоксиметил-1,1-диок- (2x50 мп), рассолом (50 мл), сушат сопеницилланоилоксиметилдиметилмало- (Na,jS04) и концентрируют до 6,3 г жел- нат, гидрохлорид,
той пены, Полученньй продукт раство- D. (2-Амино-2-фенилацетами- ряют в 60 мл ацетона и гидролизуют . до)пеницилланоилоксиметил-1,1-диок- при перемешивании с 80 мл 0,1 н,со- ДБ сопеницилланоилоксиметилмалонат, гид- ляной кислоты в течение 15 мин, Аце- рохлорид,
тон удаляют в вакууме и водный оста- Пример 22, (2-Амино-2- ток экстрагируют этилацетатом 50 мл)1, -фенилацетамидо) пеницилланоилоксиме- смесью этилацетат:этиловый эфир тил-1,1-диоксопеницилланоилоксиметил- 1:1,75 мл и снова этилацетатом 50 глутарат, гидрохлорид, (50 мл). Водную фазу фильтруют до А, 6-(1)-(2-Азидо-2-фенилацетамит-, получения прозрачного раствора, ко- до) пеницилланоилоксиметил-1,1-диок- торый после сушки вымораживанием да- сопеницилланоилоксиметилглутарат, ет 2,95 г твердой смеси. После хрома- К смеси 1,18 г (0.0023 моль) иодо- тографирования на Сефадексе L Н-20 55 метил-6- Е1-( 2-aзидo-2-фeнилaцeтaми- (вoдa) получают 0,26 г (3%) чистого до) пеницилланата и 1,2 г (0,003 моль) гидрохлорида,натрий-1,1-диоксопеницилланоилоксиме-,
А, 6-{D-(2-Амино-2-фенилацетами- тилглутарата добавляют 15 мл диметил- до) пеницилланоилоксиметил-1,1-диок- формамида. Полученную смесь перемешивают до получения раствора. Спустя 1 ч добавляют еще 1,0 г натриевой соли, и полученный раствор перемешивают еще 30 мин, разбавляют этилацетатом (100 мл), промывают рассолом (2 х X 30 мл), водой (2x30 мл), рассолом (1x30 мл), сушат (NajSOA) и концентрируют в вакууме до получения пены. После хроматографирования на колонке с силикагелем (100 г) и элюирования сме- смесью этилацетат:гексан 7:3 получают 0,72 г (43%) очищенного азидосоедине- ния.
в. Получен1№1Й азид растворяют в 15 мл дх хлорметана и 3 5 мл изопропа- нола и объединяют с 0,5 г 0% Pd/C катализатора о Полученную гидри руют под давлением 50 пси (3,52хг/см) водорода в течение 45 мин. После до- бавления 0,25 г катализатора, гидрирование продолжают еще 30 мин. Катализатор удаляют фильтрованием, промыва ют смесью дихпорметана и изопропанола
и полученный фильтрат концентрируют 25 хлорметил-i, 1-ди оксопеницилланоилокв вакууме до получения 3 мл суспензии. После добавления 30 мл диэтилового эфира образуется осадок, который после перемешивания в течение 5 мин фильтруют, и получают 0,24 г (35%) свободного основания. Порцию оскова- ния 0,21 г растворяют в 2,8 мл Ojl н.соляной кислоты и сушат вымораживанием после фильтрования через диатомовую землю до получения 0,14 г гидрохлорида.
Пример 23, (2-АМИНО-2- -фенилацетамидо )J пеницилланоилоксиме- тил-1,1-дноксопеницилланоилоксиметил- адипат, гидрохлорид.
А. Используя натрий-1,1-диоксопе- ницшшаноилоксиметкладипат вместо натрий-,-диоксопеницилланоилоксиме- тилглутарата.по примеру 4 (часть А)
симетилглутарата в.2 мл ацетона добавляют 0,323 г (0,0005 моль) тетра- бутилам1 оний-6-{1 - 2 ( 1 метил-2--ме- токсикарбоннпвинш1амино)2-фенил-аце :
30 амидо пекицилланата при перемешивании. После перемегаивания в течение 20 ч при комнатной температуре раст . воритель удаляют в вакууме, а полу- ченньй остаток хроматографируют на
25 колонке с силикагелем, элюнруя смесью зтилацетат:гексан 7:3, до получения 0,18 г масла, К раствору полученного масла в 55 мл ацетона добавляют 2 мл 0,1 н,соляной кислоты.
40 а затем дополнительно 5 мл воды. По- лученьхук смесь (рН 1,2) .перемешивают 30 мин. Ацетон удаляют в вакууме, а водный остаток промывают этиловым эфиром (2x30 мл)5 фильтруют и вымора50
получают )(2.-азидо-2-фенилацет g живакием сушат до получения 0,12 г .амидо)пеницшшаноилоксиметил 1,1-ди- (75% в расчете на енамин) гидрохло- оксопеницилланоилоксиметиладипат с выходом 37,7%.
Пример 24. . Хлорметил-,I- -диоксопеницилланоилоксиметилглута- рат.
Раствор 3,9 г (0,0084 моль) бензил- 1 , 1-диоксопеницилланоилоксим.етшг- глутарата,в 50 мл тетрагидрофурана (ТГФ) гидрируют в присутствии 3,0 г gg диметилмапоната в 25 мл воды добавляп 10% Pd/C катализатора под Давлением ют 150 мл хлороформа, а затем 8,5 г 50 пси (3,52 кг/см ) водорода в аппа- (0,025 моль) кислого сульфата тетра- рате гидрирования Паара, Катализатор бутиламмония. рН водного слоя устанав- удаляют фильтрованием, фильтровальную ливают при перемешивании до 7,5, дорида.
Пример 26. Иодометил-1,1- -диоксоперицйлланоилоксиметилдиметил- малонат,
А. Хлорметил-1,1-диоксопеницилла- ноилоксиметилдиметилмалонат.
К раствору 10 г (0,025 моль) натрий- J1-диоксопеницилланоилоксиметилiлепешку промывают ТГФ и фильтраты кон концентрируют в вакууме до 3,5 г вязкого масла« Это масло растворяют в 25 мл хлороформа, добавляют 10 мл воды. Полученную смесь перемешивают и устанавливают рН 8,0, добавляя 40%-ную гидроокись тетрабутиламмония. Хлороформовык слой выделяют, а водный слой экстрагир зтс Т хлороформом (2 х X 30 мл). Объединенные хлороформовые слои сушат () и концентрируют в вакууме до получения 5,8 г мйсла, которое растворяют в 33 мл иодохлор - метана и перемешивают в течение 15 ч, После концентрирования в вакууме к хроматографи.рования ка сигшкагеле (смесь этилацетата и гексана) получают 0,20 г (б%) указанного соедингния.
Пример 25. (2-Амино-2-- -фенилацетамкдо} пеницшшаноилоксиме- тил- i,диоксопеницилланоилоксиметхш- глутарат, гидрох.порид.
К раствору 0,2 г (0,0005 моль)
симетилглутарата в.2 мл ацетона добавляют 0,323 г (0,0005 моль) тетра- бутилам1 оний-6-{1 - 2 ( 1 метил-2--ме- токсикарбоннпвинш1амино)2-фенил-аце :
амидо пекицилланата при перемешивании. После перемегаивания в течение 20 ч при комнатной температуре раст воритель удаляют в вакууме, а полу- ченньй остаток хроматографируют на
колонке с силикагелем, элюнруя смесью зтилацетат:гексан 7:3, до получения 0,18 г масла, К раствору полученного масла в 55 мл ацетона добавляют 2 мл 0,1 н,соляной кислоты.
а затем дополнительно 5 мл воды. По- лученьхук смесь (рН 1,2) .перемешивают 30 мин. Ацетон удаляют в вакууме, а водный остаток промывают этиловым эфиром (2x30 мл)5 фильтруют и вымораживакием сушат до получения 0,12 г (75% в расчете на енамин) гидрохло-
живакием сушат до получения 0,12 г (75% в расчете на енамин) гидрохло-
диметилмапоната в 25 мл воды добавляп ют 150 мл хлороформа, а затем 8,5 г (0,025 моль) кислого сульфата тетра- бутиламмония. рН водного слоя устанав- ливают при перемешивании до 7,5, дорида.
Пример 26. Иодометил-1,1- -диоксоперицйлланоилоксиметилдиметил- малонат,
А. Хлорметил-1,1-диоксопеницилла- ноилоксиметилдиметилмалонат.
К раствору 10 г (0,025 моль) натрий- J1-диоксопеницилланоилоксиметилбавляя бикарбонат натрия, Хлороформо вый слой вьщеляют и водную фазу экстрагируют хлороформом (1x100 мл). Объединенные хлороформовые слои сушат () и концентрируют в вакууме до получения вязкого масла все еще содержащего хлороформ. Это масло ра- створятот в 95 мл хлороиодметана и перемешивают в течение ночи. После концентрирования в вакууме и хромато графирования на 300 г силикагеля, элюируя смесью этилацетат:гексан 1:1 (по объему)5 получают 7,4 г (70%) хлорметилового сложного эфира в виде масла,
В, К раствору 7,4 г (0,0156 моль) хлорметил-1,1-диоксопеницилланоилок- симетилдиметилмалоната в 50 мл ацетона добавляют 11,75 г (0,78 моль) иодида натрия, Полученный раствор перемешивают в течение 20 ч. После концентрирования в вакууме получают маслянистое твердое вещество, которое разделяют между 50 мл воды и JOO мл этилацетата, Водньй слой вьщеляют, органический слой промывают водой (50 мл), рассолом (50 мл), сушат (Na2S04) и концентрируют в вакууме до желтого масла, В результате хроматографировакия на силикагеле (150 г), элюируя смесью этилацетат: гексан 1:1 (по объему) получают 8,3 г (100%) иодометилового сложного эфира в виде прозрачного вязкого масла.
Пример 27. 6-{г - 2-Амино- -2-(пара-оксифенил)ацетамидо пени- цилланоилоксиметил-,1-диоксопеницшт ланоилоксиметилдиметилмалонат, гидрохлорид,
А, (1-Метил-2-метоксикар боннлвиниламино)-2-(пара-оксифенил) ацетамидо пеницилланоилоксиметил- -1,1-диоксопеницилланоилоксиметилди- метилмалонат.
К смеси 1,83 г (0,0026 моль) тет- рабутиламмоний-6- 1)( 1-метил-2-ме- токсикарбонилвиниламино)-2-(пара-ок- сифенил)ацетамидо Iпеницилланата.и 1,35 г (0,0026 моль) иодометил-i,1- .-диоксопеницилланоилоксиметилдиметил- малоната добавляют 10 мл диметилформ- амида. После перемещивания в течение 15 мин раствор разбавляют 100 мл этилацетата, промывают рассолом (25 мл), водой (3x25 мл), рассолом (25 мп), сушат () и концентрируют до пены. Эту пену помещают в этилацетат и хроматографируют на
100 г силикагеля, элюируя смесью этилацетат:гексан 1:1 (по объему), до получения 1,2 г (54%) енаминзащи- щенного аддукта,
В, К oпиcaннo гy енамину (1,2 г), растворенному в 30 мл ацетона, добавляют 14 МП 0,1 н.соляной кислоты, спустя 20 мин ацетон удаляют в вакууме и ВОДНЬЙ остаток экстрагируют этиловьм эфиром (2x50 мл) и этил- ацетатом (30 мл). После сушки вымораживанием водной фазы получают 0,8 г (72%) указанного гидрохлорида.
Пример 28. 6- - 2-Амино- -2-( пара-ацетоксифе 5ил) ацетамадо Jj пе- ницилланоилоксиметил-1,1-диоксопени- дилланоилоксиметилдиметилмалонат,
A,6-{D- 2-(1-Метил-2-метоксикар бонилвинилам1-;но)-2-(пара-ацетоксифенил)-ацетамидо пеницилланоилоксиме- тил-1 , 1-диоксопенишшланоилоксиметил- диметилмалонат.
2-( 1-Метил-2-метоксикарбонил
виниламино)-2-(пара-оксифенил)-ацетамидо пеницилланоилоксиметил-1,1-ди .ок со пеницилл аноило к симе тилдиме тилма- лонат, полученный по примеру 27 (часть А), 2,55 г (0,003 моль) и 4диметиламинопиридина (0,366 г)
(0,003 моль) растворяют в 30 мп ди- метилхлорметана и добавляют 0,28 мл (ОуООЗ моль) уксуснот о ангидрида, Раствор перемешивают в течение 25 мин,
разбавляют до 100 мл, промывают водой (30 мп), рассолом (30 мл),-сушат и концентрируют до получения 2,1 г (78%) желтой пены.
B,Пену, полученную по способу
части А, 2,1 г растворяют в 50 мл ацетона и добавляют 23 мл 0,1 н,соляной кислоты. После перемешивания в течение 20 мин ацетон удаляют в вакууме и водный слой промывают этиловым эфи-
ром (2x30 мп), фильтруют через диатомовую землю и сушат вымораживанием до получения 1,77 (71%) указанного гидрохлорида.
Пример 29, А, Повторяя процедуру, используя пивалоилхлорид вместо уксусного ангидрида, получают не- очищенньй продукт, который хромато- графически очищают на 100 г силикагеля, при элюировании смесью метиленхлорид:этилацетат 60:40 (объем/объем) После концентрирования содержащих продукт фракций получают 2,3 г (82%) бесцветной пены, которая является 6- (1-метил-метоксикарбонилвинил21
амино)-2-(пара-пивалоилоксифенил) адетамидо пеницилланоилоксиметил- -1,l-диoкcoпeнициллaнoилoкcимeтиJЩи- метилмалонатом.
B.К 2,2 г (2,35 моль) енамина, полученного в части А, в 30 мл ацетона добавляют 24 мл О,1 н.соляной кислоты. Эту смесь перемешивают при температуре окружающей среды в течение 5 мин, ацетон выпаривают в вакууме и водньй остаток промывают этиловым эфиром (3x50 мл). Остаточный эфир удаляют из водного слоя, выпаривая его в вакууме. Водный раствор делают прозрачным путем фильтрования и сушат вымораживанием до получения
l,61 г (80%). )- 2-амино-2-(пара- пивалоилоксифенил)ацетамидо пеницил ланоилоксиметил-,1-диоксопеницилла- ноилоксиметилдиметилмалоната, гидрохлорид.
C,Используя их муравьиног-.уксусны ангидрид в качестве ацилирующего агента в части А и удаляя задитную группу по описанному способу, получают 6-|D- 2-aминo-2-(пapa-фopмилoкcи- фeнил)aцeтaмидo J пеницилланоилокси- метил-1,1-диоксопеницилланоилоксиме- тилдиметилмалонат, гидрохлорид.
Пример ЗОо Бензил-6- Г)-(2- -(-метил-2-метоксикарбонилвинилами- но)-2-фенш1ацетамидо пеницилланоило симетилглутарат.
1, Бензилхлорметилглутарат.
Смесь 1,5 г (3,75 ммоль) тетрабу- тиламмонийбензилглутарата и 20 мл хлоридметана перемешивают при комнатной температуре в течение 3 ч и концентрируют в вакууме до вязкого масла. Это масло помещают в 20 мл этил- ацетата и 30 мл гексана и фильтруют для удаления тетрабутиламмонийиодида Растворитель выпаривают в вакууме и
остаток очищают хроматографически на 45 Д° смолы. Смолу тщательно растирают
75 г силикагеля, элюируя смесью этил- ацетат: гексан 70:30 (по объему). Каждые мин отбирают фракции по 15 мл. Фракции, содержащие целевой продукт (фракции 8-11), объединяют и растворитель выпаривают в вакууме до получения 0,55 г (62,5%) целевого продукта.
В. Смесь 0,55 г (2 ммоль) бензил- хлорглутарата, 1,37 г (2 ммоль) тет- рабутиламмоний (1-метил-2- -метоксикарб онилвиниламино)-2-фенил- ацетамидо -пеницилланата и 20 мл ацетона перемешивают в течение ночи
05704
при комнатной температуре. Ацетон выпаривают и остаток очищают хромато- графически на силикагеле, элюируя смесью этилацетат:гексан 60:40 (по объему) до получения 1,2 г (88%) продукта в виде масла.
Пример 31. (2-Амино-2- -фенил ацетамидо ) пеницилланоилокси- 10 метил-1,1-диоксопеницилланоилоксиме- тилдиметилмалонат-пара-толуолсульфо- нат.
А, 6-{D- 2-(1-Метил-2-метоксикар- бонилвиниламино)-2-фенилацетамидо 15 пеницилланоилоксиметил-1,1-диоксопв- ницилланоилоксиметилдиметилмалонат.
К 4,0 г (0,01 моль) натрий-1,1-ди- оксопеницилланоилоксиметилдиметил- малонат и 6,0 г (0,01 моль) иодоме- 20 тил-б-(Г)- 2-( 1-метил-2-метоксикарбо- нилвиниламино)-2-фенилацетамидо jne- ницилланата добавляют 40 мл диметил- формамида. Полученную смесь перемешивают при комнатной температуре в течение 30 мин. Смесь выливают -в
300 мл этиладетата, промывают водой (4x100 мл), рассолом (1x100 мл), сушат Na.SOj и концентрируют в вакууме до получения 9,3 г пены. Пену очища - ют хроматографически на силикагеле (300 г), элюируя смесью этилацетатг гексан 60:40, отбирая фракции по. 25 мл.. Фракции 39-65 объединяют и выпаривают в вакууме до получения 4,3 г (51%) коричневатой пены.
В. К 30 МП этилацетата добавляют 0,836 г (1 ммоль) енамина, полученного в части А, и полученную смесь перемешивают до получения раствора. Раствор 0,19 г (I ммоль) гидрата
пара-толуолсульфокислоты в 5 мл этил- ацетата добавляют в смесь и перемешивают в течегше 15 мин, и растворитель выпаривают до получения твер
с 150 мл этилового эфира, перемешивают в течение ночи, фильтруют, промывают этиловым эфиром и сушат воздухом до получения 0,84 г (92%) тозилата.
Пример 32. Натрий-1, 1-дио1{- со-б-бета-оксиметилпеницилланоилок- симетиладипат.
А. Смесь натрий-1,1-диоксо-6-бета-оксиметилпеницилланата (285 мг, 1 ммоль) и эквимолярных количеств (по 1 ммоль) бензилхлорметиладипата и тетрабутиламмонийбромида в 5 мл ацетона нагревают в атмосфере азота
при кипячении с обратным холодильником в течение ночи. Ацетон выпарива- - ют, остаток помещают в этилацетат и дважды промывают водой и сушат (). После выпаривания растворителя в вакууме и тщательного растирания остатка с хлороформом получают сырой продукт, который очищают хрома- тографически на силикагеле, элюируя смесью метиленхлорид:этилацетат 4:1 до получения 410 мг (78%) бензилово- го сложного эфира,
Бензиловый сложньш эфир помещают в 40 мл этилацетата и гидрируют на 5% Pd/C катализаторе при давлении 3,5 кг/см водорода в течение 30 мин. Катализатор отфильтровывают, промы вают этилацетатом. Фильтрат концентрируют до небольшого объема и при быстром перемешивании добавляют 129 мг натрий-2-этилгексаноата в этиладетате. Выпавший в осадок продукт отфильтровывают, промывают этил- ацетатом и сушат на воздухе до получения 199 мг. указанного соединения в виде бесцветного твердого продукта.
Пример 33, Натрий-J,1-диок- со-6-бета-оксиметилпеницш1ланоилок- симетилглутарат.
Используя смесь 485 мг (1,7 ммоль) 1,1 диоксо-6-бета-оксиметилпеницилла- ната, 450 мг (1,66 ммоль) бензилхлор- метилглутарата и 548 мг (1,7 ммоль) тетрабутиламмонийбромида в 5 мл ацетона получают соответствующий сложный бензиловый эфир (664 мг) по способу, описанному в примере 32, После гидрирования на Pd/C катализаторе и превращения в натриевую соль также по описанному способу получают 315 мг указанного соединения.
Пример 34, 1,l-Диoкco-6- -альфа-аминометилпеницилланоилоксиметил-транс-1,4-циклогександикарбоно- 45 динения вводили орально (по 5 крыс
вая кислота,
А, Тетрабутиламмоний-1,1-диоксо- -6-альфа-(б ензило ксикарб онил аминоме- тил)пеницилланат,
К раствору 0,67 г (1,69 ммоль) 6- -альфа-(бензилоксикарбонш1аминометид) пенициллановой кислоты 1,1-диоксида в 50 мл метиленхлорида добавляют 100 мл воды и 0,142 г (1,69 ммоль) бикарбоната натрия, рН полученной смеси доводят до 8,0 1 н,гидроокисью натрия. Добавляют 0,573 г (1,69 ммоль) тетрабутиламмонийбисульфата и рН снова доводят до 8,0 1 н,гидроокисью
натрия . Полученную смесь перемешивают в течение 20 мин, слои разделяют и водную фазу экстрагируют 25 мл мети- ленхлорида. Объединенные органические слои промывают рассолом, сушат (КалБО) и растворитель выпаривают в вакууме до пол чения соли в виде белой пены 0,98 г,
В, Бензил-1,l-диoкco-6-aльфa-(бeн- зилoкcикapбoнилaминoмeтил)пeницшIлa- нoилoкcимeтил-транс-1 ,4-циклогексан- дик ар бок сил ат.
Продукт, полученный в части А
(0,98 г, 1,53 ммоль), и 0,476 г
(1,53 ммоль) бензилхлорметил-транс- - ,4 1 ;иклогександикарбоксилата перемешивают в 50 мл ацетона при комнатной температуре (в атмосфере азота)
в течение 17 ч. Полученную смесь концентрируют в вакууме до получения масла, которое очищают хроматографи- чески на 100 г силикагеля, элюируя смесью этилацетат:хлороформ 30:70 в
15 мл фракции. Фракции (10-13) объединяют и выпаривают до получения .бесцветной пены 0,45 г,
. С. Продукт, полученный в части В, 0,45 г объединяют с 0,5 г 10% Pd/C
катализатора в 20 мл воды и 20 мл тетрагидрофурана и гидрируют при 50 пси (3,52 кг/см ) в течение 15 мин Полученную смесь фильтруют, прО -1ыва- ют смесью тетрагидрофурана и воды и
полученньй фильтрат и промывки выпаривают в вакууме до небольшого объема. Оставшиеся кристаллы смеши- вают с тетрагидрофураном и сушат па воздухе до получения 0,22 г (73,5%)
указанного соединения.
Фармакокинетические исследования. Данные по фармакокинетике получили на крысах весом 80-100 г штамма аутбрид Spraque-Dawley,Тестовые соедля каждого соединения) в виде водной суспензии, содержащей 10 или 20 мг/кг препарата. Образцы крови отбирали в указанное время и подвергали дифференцированному бионализу для определения уровней ампициллина и салбактам пенициллановой кислоты 1,1-диоксида, В биоанализе ампициллина использовали Sarcina lutea (АТСС9341),
Который чувствителен к ампициллину, но нечувствителен к бета-лактамазно- му ингибитору в концентрациях столь высоких как 100 мгк/мл, так как он не содержит бета-лактамазы. Таким
пластиковые пластины. Пластины затем инкубируют при в течение 18ч, и полученные зоны измеряют.
Биоанализ амоксициллина и пенициллина V проводят таким же образом, используя стандартные кривые, полученные с соответствующим пеници.апи- ном.
Определение салбактама основано на нечувствительности Pasteurella histolytica (59В010) к высоким концентрациям либо ампициллина, либо салбактама отдельно. Однако, так как ее устойчивость осуществляется через бета-лактамазу, культура реа- рирует синергетически на сочетание ампициллина и салбактама. Стандартные кривые получают способом, аналогичным описанному для ампициллина. Пластины для анализа подготавливают, добавляя 1 мл ночной культуры Pasteurella. histolytica к 100 мл агара Моллер-Хинтона , и к этому добавляют 50 мкг/мп ампициллина и 5% стерильной бычьей крови. Пластины инкубируют при в течекне около 18ч, после чего измеряют зоны.
Определение ,1-диоксида-6-бета- оксиметилпенициллановой кислоты и I,1-диоксида-6-альфа-аминометилпени- циллановой кислоты осуществляют тем же способом.
Фармакологичные данные для крыс при оральном приеме тестовых соединений приведены в таблице (для Р значения AMP, АМОХ и РеЩ соответствуют продуктам ампициллину, амоксициллину и пенициллину V).
Из полученных данных следует, что при оральном приеме соединения, где п О, а R - В, создают заметно высо кие уровни в крови ингибитора соотве ствующей бета-лактамазы, ВСООН.
муль
К.-С-О-СНоОС О.
о
А I
К2-((ОСН2)
О
где А - л-алкилен, группа
(он 3)7 С, циклогексилен или- фенилен;
R и ч - различны и означают: при
п О К - Р или В (а, Ъ и с) i 1- водород или бей- зил; при п 1 RI, - Р, Hj
при этом
Ва, Р - группа
40
C HjOCHjCO
или
где Q - NHJ или 1-метил-2-метоксикарбониининиламин;R - водород или гидроксил; В - группа формулы
V
riCr ,
СН5
27
где X - водород (Ва), (ВЪ),
СН,№Ц(Вс),
или их фармацевтически приемлемых щелочных или четвертичных аммониевых солей, отличающий ся тем, что соединения общей формулы
Р СООМ или В СООМ,
где Р указан выше, исключая Q - аминогруппа;
В указан выше, когда X - водород CHjOH или C HjCHjOCOHHCH ;
М - натрий, калий или тетрабутил- аммоний,
подвергают взаимодействию с соединением общей формулы
ЛСНп
А Ftg(COCH2)
О
А, п и R,j X
указаны выше, галоген;
Р -СООСН ОСгО
П
R 4COCH)
о
,,,,
,(
6-. /
J
9
:CHj соон
0428
в среде К,К-диметилформамида или этилацетата, или -дихлорметана, или ацетона, при температуре от комнатной до , и в случае, когда.X, - бен- зилоксикарбониламиногруппа, а Q - 1-метил 2-метоксикарбонилвиниламино- группа, последние переводят в аминогруппу с последующим выделением целе- вого продукта в свободном виде или в виде фармацевтически приемлемых ще- лочньгх или четвертичных аммониевых солей.
Приоритет по признакам:
| Патент США № 2985648, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
| Патент США № 3192198, кл | |||
| Прибор для периодического прерывания электрической цепи в случае ее перегрузки | 1921 |
|
SU260A1 |
