(5) СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНОВ 2р-ХЛОРМЕТИЛ-2о6-МЕТИЛПЕНАМ-3 А-КАРБОНОВОЙ КИСЛОТЫ ИЛИ ЕЕ СЛОЖНЫХ ЭФИРОВ, ИЛИ ЕЕ СОЛЕЙ (ЕГО ВАРИАНТЫ)
12
Изобретение относится к способу получения новых пёнициллиновых соединений, а именно, сульфонов 20-хлорметил-2е(гметилпенам-3о -карбо- 5 новой кислоты или ее сложных эфиров или ее солей (его варианты). Эти соединения могут найти применение в медицине.
Известен способ получения пени-;- Q циллановой кислоты или ее сложных эфи.ров, или ее солей, заключающийся в том, что 6-бром-пенициллановую кислоту или ее соль подвергают восстановлению каталитическим гидрогенолизом, ,5 предпочтительно в инертном растворителе под давлением водорода от 1 до 5 кг/см2.или водородом в момент выделения и выделяют целевой продукт в виде свободной кислоты или ее соли jo или свободную кислоту или ее соль переводят в сложный эфир l }.
Известен способ получения сульфона ленициллановой кислоты или ее . .сложных эфиров или ее солей окислением сульфоксида ленициллановой кислоты или ее соли путем обработки таким окислителем, как перманганат металла, взятым в количестве от 0,5 до 5 мольных эквивалентов, предпочтительно 1 мольный эквивалент, в среде растворителя при температуре от -20 до 50°С, предпочтительно при , либо окислением путем обработки органической надкислотой, взятой в количестве от 1 до мольных эквивалентов, предпочтительно 1,2 мольных эквивалентов в среде органического растворителя при температуре от -20 до 50ЧС; предпочтительно при Целевой продукт выделяют в виде свободной кислоты, ее соли или сложного эфира.
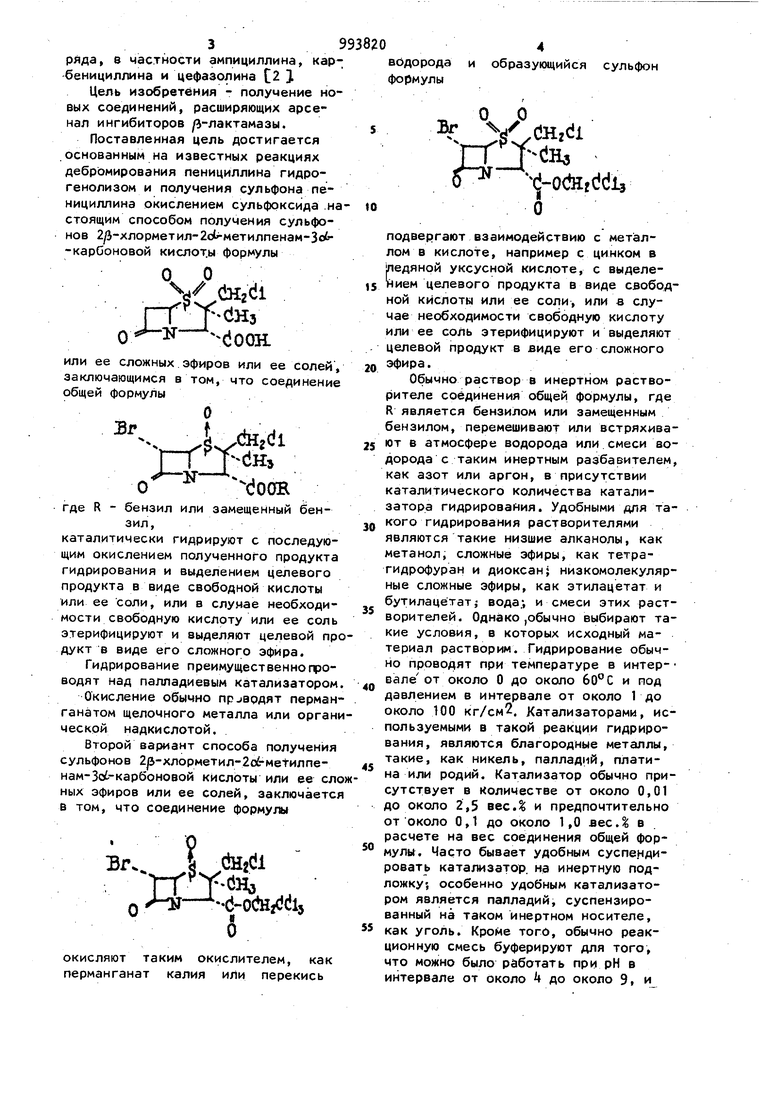
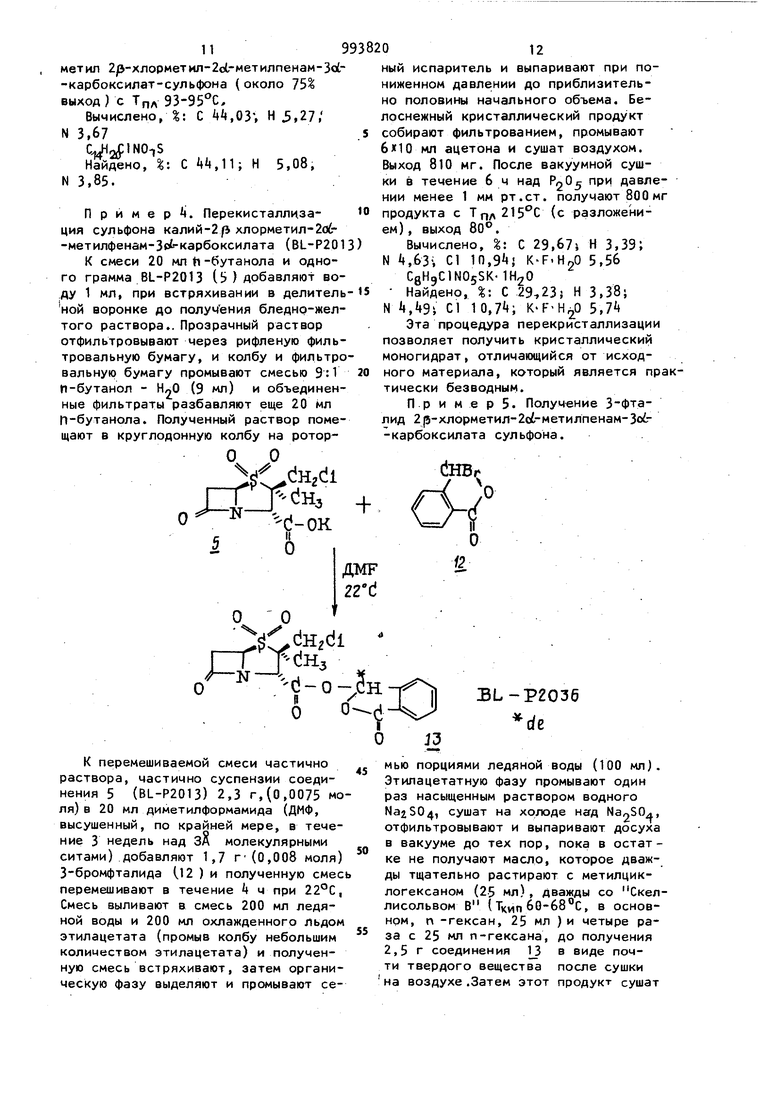
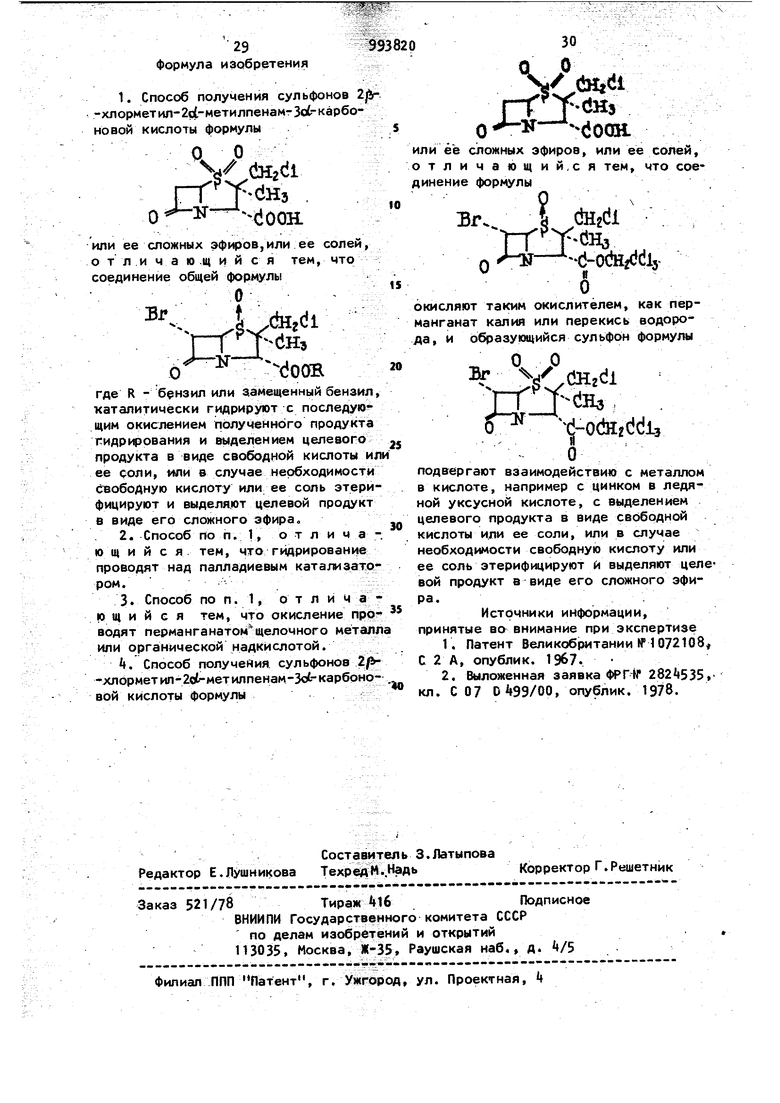
Эти соединения обладают антибактериальной активностью, а также способностью защищать от разрушения под действием |Ь-лактамазы антибиотиков цефалоспоринового и пенициллинового 3 ряда, в частности ампициллина, карбенициллина и цефазолина С2 J Цель изобретения - получение новых соединений, расширяющих арсенал ингибиторов /i-лактамазы. Поставленная цель достигается основанным на известных реакциях дебромирования пенициллина гидрогенолизом и получения сульфона пенициллина окислением сульфоксида .на стоящим способом получения сульфонов 2/1-хлорметил-2о1 -метилпенам-ЗоЬ-карОоновой кислоты формулы N/ (iHzcii енз 0 -CjOOK или ее сложных эфиров или ее Солей заключающимся в том, что соединение общей формулы V J(l,(l , kT« dOORR - бензил или замещенный бензил, каталитически гидрируют с последующим окислением полученного продукта гидрирования и выделением целевого продукта в виде свободной кислоты или ее соли, или в случае необходимости свободную кислоту или ее соль этерифицируют и выделяют целевой пр дукт в виде его сложного эфира. Гидрирование преимущественно проводят над палладиевым катализатором Окисление обычно пpJBOДят перман ганатом щелочного металла или орган ческой надкислотой. Второй вариант способа получения сульфонов 2р-хлорметил-2о6-меТилпенам-Зо -карбоновой кислоты или ее сл ных эфиров или ее солей, заключаетс В том, что соединение формулы I dHzCJi , - -cJ-ouizddis окисляют таким окислителем, как перманганат калия или перекись водорода и образующийся сульфон формулы подвергают взаимодействию с металлом в кислоте, например с цинком в Ледяной уксусной кислоте, с выделением целевого продукта в виде свободной кислоты или ее соли, или в случае необходимости свободную кислоту или ее соль этерифицируют и выделяют целевой продукт в виде его сложного эфира. Обычно раствор в инертном растворителе соединения общей формулы, где R является бензилом или замещенным бензилом, перемешивают или встряхивают в атмосфере водорода или смеси водорода с таким инертным разбавителем, как азот или аргон, в присутствии каталитического количества катализатора гидрирования. Удобными для такого гидрирования растворителями являются такие низшие алканолы, как метанол, сложные эфиры, как тетрагидрофуран и диоксан; низкомолекулярные сложные эфиры, как этилацетат и бутилацётат; вода;, и смеси этих растворителей. Однако ,обычно выбирают такие условия, в которых исходный материал растворим. Гидрирование обычно проводят при температуре в интер- валеот около О до около 60° С и под давлением в интервале от около Г до около 100 кг/см2. Катализаторами, используемыми в такой реакции гидрирования, являются благородные металлы, такие, как никель, палладий, платина или родий. Катализатор обычно присутствует в количестве от около 0,01 до около 2,5 вес.% и предпочтительно от около 0,1 до около 1,0 вес.% в расчете на вес соединения общей формулы. Часто бывает удобным суспендировать катализатор, на инертную подложку, особенно удобным катализатором является палладий, суспензированный на таком инертном носителе, как уголь. Кроме того, обычно реакционную смесь буферируют для того, что можно было работать при рН в интервале от около до около 9, и
предпочтительно, от 6 до 8. Обычно используют боратный или фосфатный буфер. Реакция протекает около одно го часа.
Предлагаемый способ иллюстрируётся приведенными ниже примерами.
Скеллисольв В является фракцией петролейного эфира с т.кип. 606В°С, состоящей глйвным образом, изп-гексана (Скеллисольв является торговой маркой Скелли Ойл Ко).
Пример, Получение калиеврй соли 2/3-хлорметил-2с6-метилпенам-Зо -карбоновой кислоты-сульфона (BL-P201.3).
а) Получение 5-сульфоксид 6с -бромпенициллановой кислоты )
Растворяют 30 г (37,5 моля) N ,N -дибензилэтилендиаминовой соли Bd-бромпенициллановой кислоты в 330 мл метиленхлорида. Перемешивают и охлаждают до 0°С.
Затем медленно добавляют 13 мл (156 ммоля)концентрированной соляной кислоты в растворе метиленхлорид-а. Осаждение НС1 соли дибензилэтилендиамина (ДБЭДНС) происходит в течение минуты, взвесь перемешивают при в течение 10 мин.
Для выделения осадка ДБЭДНС отфильтровывают через фильтр с диатомитовой землей (Дикалит) , промывают фильтровальную лепешку 150 мл метиленхлорида (фильтрование следует проводить как можно быстрее) избегают хранения кислого метиленхлоридного раствора в течение длительного времени, так как могут возникнуть некоторые трудности при фильтровании, связанные с природой осадка, поэтому полезно ускорить фильтрование взвеси. Промывают объединенные метиленхло- ридные фильтраты 60 мл холодной воды, перемешивают 5 мин и сливаю т водную фазу, рН промывки равен 2,0-2,3. Метиленхпоридный раствор, содержащий бо бромпенициллановую кислоту, концентрируют при пониженном давлении до объема 65-80 мл, охлаждают и перемешивают раствор при 5°С.
При интенсивном перемешивании осторожно добавляют 3 мл (86,9 ммоля) kQ% перуксусной кислоты в течение 30 мин, реакция экзотермична; поддерживают температуру между с помощью ледяной бани. Сульфоксид начинает кристаллизоваться после добавления 0 мл перуксусной кислоты.
его охлаждают и перемешивают взвесь при , в течение 2 ч.
Белоснежную фильтровальную лепешку отфильтровывают и промывают в следующей последовательности: 10 мл 5°С воды, затем 10 мл метиленхлорида, и наконец.. 15 мл геп тана. Сушат ({жльтровальную лепешку при 45°С в темростате до постоянного веса около 6-10 ч. Более длительное нагревание может привести к появлению розоватого цвета. Вес соединения составляет около 16,26 г, выход - 73,. Реакционную смесь и целевой продукт исследуют с помощью тонкослойной жидкостной хроматог:ра({н4И, используя в качестве системы растворите-ля смеси 15 толуол С ацетон) 1 уксусная кислота (НАС) или 8 ацетон (8 метанол) 3 толуол ( НАС). Конечный продукт анализируют с помощью ЯМР и ИК.
б) Получение паранитробензил 6-oi-бромпеницилланат-S -сульфоксида (2)
К раствору 12 г (0,0 моля) З-сулфоксида боС-бромпенициллановой кислоты в 100 мл ацетона добавляют 7,5 г (0, моля) калий-2-этилгексаноата. Полученную соль собирают фильтрованием, промывают холодным ацетоном и сушат на воздухе, в результате чего получают: 10 г вещества. Кристаллическую соль калия растворяют в 75 мл диметилацетамида.и 7,8 г (О,О моля) паранитробензилбромида добавляют к раствору. Этот раствор перемешивают при 23°С в течение . Полученную смесь разбавляют 500 мл воды и экстрагируют этилацетатом. Этилацетатный слой промывают четыре раза водой и сушат над безводным сульфатом магния. Растворитель выпаривают при (5 мм) до получения масла, которое кристаллизуется. Свет локоричные кристаллы соединения 2. размешивают до получения взвеси в эфире и отфильтровывают, в результате чего получают 9 г (70% выход) соединения с ТПА 12 -125° (с разложением) .
- Вычислено, %: С 1,98; Н N 6,52
Найдено,: СЛ2,00; Н 3, N 6,98
ИК (КВг) 1800 (с), (с), (ел), 1520 (с), (ср), . 1350 (с), 1060 (ср), (ср) см-
Н-ЯМР (60 МГц, ДМСО):сУЧ,22(с, ЗН/, 1,6 (с., ЗН), if,67 (с),
1Н (5,2) d - 1-5 Гц (с, 1Н), 5,45 (с-, 2Н), 5.68 (д. J-l-5 Гц, 1Н), 7,5-8,5 (м, АН),5
в ) Получение пара-нитробензил 2/V-хлорметил-2с6-мётил-6-бронпенам-3о -карбоксилат О ).
Раствор 5 г (0,012 моля) паранитробензил &5 бромпвницилланат-57сульфоксида (2 Гв 2Q мл безводного диоксана нагревают при температуре кипения с обратным холодильником в атмосфере азота в течение ч с 1,5 г (0,012 моля) хинолина и 1,6 г 5 (0,012 моля)бёнзойлхлорида. Раствор разбавляют 600 .мл воды и экстрагируют этилацетатом. Этилацетатный экстракт промывают 5 раствором бикарбонат натрия, 5 раствором фосфорной -20 кислоты и наконец водой. Органичес;кий слой сушат над безводным сульфатом магния и выпаривают до получения .масла при 35С (15 мм). Масло кристаллизуется, его промывают эфиром 25 и наконец холодным толуолом, в резулы тате чего получают соединение (3), 3,5 г (65° выход), 130-135°С с разложением. ,
Вычислено,: С 0,06; Н 3,1 30 N 6,23
C..H..C18rN2°5
Найдено, УО: С 40,19i Н 3,12;
N 6,75
ИК {КВг);1792 (с), (с), 35 1610 (ел), 1520 (с), 1353 (с), 1280 (ср), 1025 (ел), 990 (ел), 750(сл)см-1. ЯМР (60 МГц, ДМСО),45 (с, ЗН), 3,5-4,3 (м, 2Н), 5,05 (cj 1Н.), 5,42 (с, 2Н), 5,5 (d J-1,5 Гц, 1Н), 5,62 40 (d, J-1,5 Гц, UI), 7,5-8,5 (м, 4Н).
г). Получение пара-нитробензи.п 2 -хлорметил-2оЬ-метилпенам-6с6-карбоксилат-сульфоксида (4 X
Раствор 1 г (0,0022 моля) пара- нитробензил Зр хлорметил-2о(гметил- ; -6о6-бромпенам-ЗоЬ-карбоксилата t 3 , растворенного в 50 мл метиленхлорида, перемешивают с 473 мг (0,0022 моля) метахлорпербензойной кислоты. Раствор перемешивают при 23 С в течение 3ч. Метиленхлорид выпаривают до 20 мл при 15 мм и 33°С,, полученный концентрированный раствор разбавляют 50 мл гептана (Скеллисольв В). Раствори- тель декантируют, а остаток тщательно перемешивают до получения взвеси с эфиром и соединение (4 ) вскоре крисJталлизуется, в результате чего получают 250 мг (24 выход) Тр 13б-137° с разложением. .
Вычислено, %: С 38,68} Н 3,02j Ы 6,02
С.М..(,В
Найдено, I: С 39,14 Н 3,13) N5,96
ИК (KBг): 1800 (с), 1760 (с), 1520 (с), 1350 (с), 1200 (с), 1050(с 830 (ел), 740 (ел) см1
Н-ЯМР (60 мГц,ДМСО): сМ,32 (с, ЗН 3,8-4,5 (м, 2Н), 4,97 (с, 1Н), 5,25 (d-JM,5 Гц, 1Н), 5,45 (с, 2Н), 5,.6 (d, J-1,5 Гц, 1Н), 7,8-8,5 (м, 4Н).
д ) Получение 2р-хлорметил-2фметилпенам-Зсб-карбоксилат-сульфон калиевой соли (5), (В1.-Р2013).
К раствору 7 г (0,015 моля) паранитробензил 2р-хл6рмет 1Л-2о -метил-6с1-б0омпенам-3о -карбоксилат-0ульфоксида (4) в 150 мл этилацетата добавляют суспензию 4 г 301 палладия на диатомитовой земле (Целит) и2,8 J- бикарбоната натрия в 150 мл воды. Полученную смесь гидрируют 5 ттечение 3 у при 50 пси (3,5 кг/см ). Катализатор удаляют фильтрованием, а водный слой выделят и обрабатывают 1,5 г перманганата калия в 50 мл воды. Полученную смесь перемешивают в течение 1 ч, после чего добавляют 250 мг бисульфита натрия. Полученную смесь отфильтровывают, и рН полученного фильтрата устанавливают концентрированной соляной кислотой до 2. Раствор лиофилизируют до получения белого аморфного порошка. Твердую часть экстрагируют этилацетатом, выпаривают до объема 20 мл и разбавляют 100 мл гептана (Скеллисольв В). Собирают твердый, гигроскопичный 2 -Хлорметил-2о -метилпенам-3о -карбоновая кислота-сульфон. Кислоту растворяют в ацетоне, обрабатывают твердым калий-2-этилгексаноатом. После фильтрования получают белый кристаллический осадок соли. 170 мг соединения (5 )с с разложением.
Вычислено, %: С 28,27; Н 3,24; N 4;12 .
CgH ClKNOjSHjO
Найдено, %: С 28,27; Н 3,б9; N 3,84
ИК (КВг): 1790 (с), 1770 (ср), 1620 (с), 1460 (ср), 1370 (с). 1310 (с), 1200 (с), (с), 995(ср (ср) см-1 Н-ЯМР (100 МГц, Д-0),68 (с, З 3,2-3,9 (м,. Гц, J-- Гц, Гц 2н), Ц,Q-k, (м, 2Н), Ц,3 (.с, 1Н), 5,02 (двд, J Гц, Гц, 1Н). П р и мер 2, Получение пивалоилоксиметил 2-р-хлррметил-2с -метилпенам-Зо -карбоксйлат-суяьфона. 2/3-хлорметил-2 1 -«етилпенам-Зо1-карбоновая кислота-сульфон в диметилформамиде обрабатывает одним эквивалентом триэтиламина и переме шивают для получения раствора. добавляют бррмметйлпивалат (1 эк вивалент) в диметилформамиде, после чего полученную смесь переманивают при комнатной температуре. Затем смесь очищают фильтрованием, и полу% Х Нз-f f dHzdi + Na (306; tBZ.-P2013) K. перемешиваемой суспензии И,6 (0,0t87 моля) 8L-P20i3 (5) в 200 мл ацетона добавляют k мл 10 водного раствора иодида натрия, и полученную смесь доводят до Кипения- с обратным холодильником на паровой бэ не. К этой суспензии, кипящей с обратным холодильником, добавляют, 1,8 мл (0,1 моль) повторно перегнанного хлорметилпивалата ( при 7 мм рт.ст.) сразу. Полученную смесь перемешивают при кипении с об ратным холодильником в течение 3 ч, а затем охлаждают до комнатной температуры (22°С). Кристаллическую тв дую часть собирают фильтрованием, промывают 3X30 мл ацетона и объединенные фильтраты выпаривают до масла в вакууме при температуре менее 22°С. Затем полученное масло помеща ЮТ в 500 мл этилацетата и промывают один раз водой (200 мл), и один раз насыщенным при перемеши вании с2 г обесцвеченного угля и
993820
10 i+H20.+(iidH2-o-d-d()TTj ciHzdi 0 d02(iHj-o-b-ci{dH3)3 ченный фильтрат выливают в ледяную воду.. Выделившуюся твердую частьотфильтровывают, промывают.водой и сушат до получения указанного соединения (сложного эфира). . Соответствующие ацетоксиметиловый, метоксиметиловый, ацетониловый и фенациловый сложные эфиры той же самой кислоты получают, заменяя в вышеописанном способе бронметилпмвалат на эквивалентный вес хлорметилацетата, хлорметйпметиловбго простого эфира, хлорацетона и фенацилбромида, соответственно. Примерз. Получение пивалоилоксиметил-2о(гхлорметил-2в6-метилпенам-ЗсЬ-карбоксилат-сульфона {BLР202). ацетон. 0 0 X dT4 fjL-P2(fO) при охлаждении (ледяная баня). Спустя 20 мин смесь профильтровывают через диатомитовый фильтрующий материал (Дикалит)с подсосом, а затем этот фильтрующий материал промывают I.XIOO мл этилацетата. Объединенные фильтраты концентрируют в вакууме при 22°С до масла. Затем это масло концентрируют далее при температуре около 22°С и давлении менее 1мм рт.ст, для удаления большей части оставшегося хлорметилпивалата. Оставшееся масло дважды тщательно растирают с 50 мл порциями h -пентана и оставляют на два дня в холодной комнате(около 10°с) в п-пентане. Затем твердую кристаллическую массу размельчают в твердый порошок под 40 мл смеси 1:1 эфир -п-пентан. Полученный продукт отфильтровывают, промывают смесью эфир - пентан (1 :1), потом пентаном и сушат на воздухе. После сушки в вакууме в течение ч над Р205 получают 13,37 г пивалоилоксй119метил 2р-хлорметил-2о(гметилпенам-3о(.-карбоксилат-сульфона (около 75% выход Гс ТПА 93-95°С, Вычислено,: С 4,03, Н5,27/ N 3,67 CW afNOlS Найдено, %; С if.ll; Н 5,08, N 3,85. Пример. Перекисталлкзация сульфона калий-2/ хлорметил-2о6-метилфенам-3« карбоксилата (BL-P201 К смеси 20 мл tt -бутанола и одного грамма BL-P2013 (5 ) добавляют воду 1 мл, при встряхивании в делитель ной воронке до получения бледно-желтого раствора.. Прозрачный раствор отфильтровывают через рифленую фильтровальную бумагу, и колбу и фильтро вальную бумагу промывают смесью 9:1 П-бутанол - НлО (9 мл) и объединенные фильтраты разбавляют еще 20 мл П-бутанола. Полученный раствор помещают в круглодонную колбу на ротор0 о CJ-OK
о о
т
- Л-0-3
Ь 0 к перемешиваемой смеси частично раствора, частично суспензии соединения 5 (BL-P2013) 2,3 г,(о,0075 мо ля) в 20 мл диметилформамида (ДМФ, высушенный, по крайней мере, в течение 3 недель над ЗА молекулярными ситами) добавляют 1,7 Г(0,008 моля) 3-бромфталида (12 ) и полученную смес перемешивают в течение k ч при 22°С, Смесь выливают в смесь 200 мл ледяной воды и 200 мл охлажденного льдом этилацетата (промыв колбу небольшим количеством этилацетата) и полученную смесь встряхивают, затем органическую фазу выделяют и промывают сеБЬ-Р203б
de 0 ныи испаритель и выпаривают при пониженном давлении до приблизительно половины начального объема. Белоснежный кристаллический продукт собирают фильтрованием, промывают 6x10 мл ацетона и сушат воздухом. Выход 810 мг. После вакуумной сушки в течение 6 ч над давлении менее 1 мм рт.ст. получают 800 мг продукта с Tf,/ 215°С (с разложением) , выход 80°. Вычислено, % С 29,67 Н 3,39; N i,63-, С1 in,9Aj KF.HgO 5,56 CgHjClNOsSK-1Н20 Найдено, % С 29,23} Н 3,38; N i,it9i С1 10, K.F-H,O 5,7 Эта процедура перекристаллизации позволяет получить кристаллический моногидрат, отличающийся от исходного материала, который является практически безводным. П р и м е р 5. Получение Зфталид 2р-хлорметил-2о6-метилпенам-3о6-карбоксилата сульфона. мью порциями ледяной воды (100 мл). Этилацетатную фазу промывают один раз насыщенным раствором водного N32804, сушат на холоде нггд , отфильтровывают и выпаривают досуха в вакууме до тех пор, пока в остатке не получают масло, которое дважды тщательно растирают с метилциклогексаном (25 мл), Дважды со Скеллисольвом В (Ткип 60-68°С, в основном, П -гексан, 25 мл ) и четыре раза с 25 мл п-гексана, до получения 2,5 г соединения в виде почти твердого вещества после сушки на воздухе .Затем этот продукт сушат
над P давлении менее 1 мм рт. до получения 2,5 г соединения 3 с Т;,д 10А°С (с разложением) . Его чистота 85-95%;Вычислено, %: С 51,615 Н 3,79$ N 3,77i Cl 9,53
Найдено, %: С 52,59} Н ,б7; N 3,21, С1 7,73i КР.Н,0 0,27
П р и м е р 6о Получение пивалоилоксиметил 2|&-хлорметил-2о6-метилпенам- Зо1-карбоксилат- сул ьфона.
Смесь сульфонгидрата 2р-хлорметил-2о(.-мет илпенам-3о6-кар($оксилата, калиевой соли 1 г (0,0031 моля) и 1 г ЗА молекулярных сит перемешивают в 15 мл димет1 фармамида в течение 2 ч при 23°С. К этой смеси добавляют 70 мг (0,0031 моля) пива; лоилоксиметилхлорида и перемешивание продолжают в течение 18 ч. Молекулярные сита собирают, фильтрат разбавляют 100 мл воды и экстраn ySf
Чо.к
(ЗОб) к перемешиваемому раствору 500 м BL-P2013 (калиевая соль) в 15 мл HgO и 10 мл этилацетата добавляют 2 Н НС1 до получения рН 1 (осуществляют это в ледяной бане при интенсивном перемешивании). Затем-полученную смесь насыщают , водный слой выделяют, а органическую фазу быстро сушат на льду над фильтруют и добавляют по каплям 50 NaOH (натрий-2-этилгексаноат) в бе водном П-бутаноле до нейтральной реакции по влажной индикаторной бумаге. Продукт не кристаллизуется при поскребывании, и поэтому его концентрируют в вакууме до масла, которое растворяют в ацетоне (5 мл), опять скребят - нет кристаллов, добавляют эфир до точки помутнения - нет кристаллов. Концентрируют в вакууме на ро торном испарителе до марла, которое растворяют в этилацетате, добавили одну каплю Н20 - поскребли - нет кристаллов. Сконцентрировали в вакууме, а затем остаток тщательно растерли с 5 мл п-бутанола - получили
гируют этилацетатом. Этилацетат. промывают 9 раз водой и сушат над безводным сульфатом магния. Растворитель удаляют при 30°С (15 мм), после чего остается масло, которое хроматографируют на силикагеле, используя силикар СС-7 (метиленхлорид 8, Этилацетат 2), и получают 1 пятно с Rf 5. Остаток, полученный кристаллизацией из гептана Скелли -. cojibB В , составляет 100 мг ( Э 95°С) пивалоилоксиметил 2/3-хлоометил-2о -метилпенам- 3 -карбоксилат-сульфона.
Вычислено, %: С kk,Q3 Н 5,27, N 3,67
Найдено, %: С 4,20; Н 5,
N 3,63
Спектры ЯМР и ИК соответствуют
предложенной CTpyKtype.
П р и м е р 7. Получение 2/3-хлорметил-2о6-метилпенам-3оь-карбоксилат-сульфон-натриевой соли.
О О
-К - ОгНа
О
dJNOg a f 283.67} 200 мг аморфного белого порошка, промыли эфиром, высушили на воздухе высушили в вакууме над течение 24 ч, в результате получили 180 мг 2ргхлорметил-2 -метилпенам-ЗоС-карбоксилат-сульфон-натриевой соли, teMпература разложения более 100°С (неопределенная). Вычислено, %: С 33,10; Н 3,13; N k,B3 CgHgCIN05SNa Найдено, %: С 33,20; Н 3,69; N Ц, KT-HjO ,ОЦ П р и м е р 8 Получение 2/3-хлорметил-2о6-метил пенам-ЗоС-карбоксилат-сульфон, калиевой соли (BL-P2013) К 10 л, воды, 130 г (1,25 моля) кислого карбоната натрия и 200 г 10% Pd на BaSO добавляют 271 г (0,5б5-моля) пара-нитробензил-бс -бром-2 хлорметил-2-метилпенаМ-3-карбоксилат-сульфона, растворенного в 5 л этилацетата. Полученную смесь гидрируют при и давлении 1 кг. Спустя 5 ч потребление воорода становится очень медленным. 15 . 99 добавляют 200 г 101 Pd на BaS04 и полученную смерь гидрируют до дальнейшего заметного снижения поглощения водорода Взвесь фильтруют через диатомитовую землю (Целит), материал фильт-;; ра промывают водой и водную фазу промывают 3 л этипацетата. К водному раствору добавляют 3 л этилацетата и рН смеси устанавливают 1,5 150 мл 12 и НС при 10°С. Органическую фаЗУ выделяют и водный раствор насыщают и экстрагируют 2x1 л этилацетата. Объединенные экстракты сушат над сульфатом магния, Высушивающий агент удаляют и при 0°С добавляют 260 мл 2 н калий-2-этилгексановой кислоты в бутаноле. После перемешивания в течение 2 ч при0°С собирают 2р-хлорметил-2-метилпенам-3-карбоксилат-сульфон натриевую соль (BL-P2013) и сушат в еекууме при комнатной температуре. Выход 13,8 г (около 70%).
О о
ч,
rfScSf 2а
СНзj ; - J. , П л
К смеси 25 мл этипацетата и 10 мл воды добавляют 800 мг (О,00261 моля) BL-P2013 в виде калиевой соли. После того, как твердая часть полностью растворится, полученную смесь обрабатывают по каплям 5й% водной фосфорной кислотой при интенсивном встр хивании до тех пор, пока из водного слоя больше не происходит выделение осадка. Этилацетатный слой выделяют, затем промывают насыщенным раствором хлористого натрия и сушат над безвод ным сульфатом магния. Высушенный агент удаляют фильтрованием и промы вают 10 мл этилацетата (растворитель от промывки объединяют с исходным фильтратом). Затем к этилацетату добавляют Скеллисольв В до точки помутнения (приблизительно 10 мл). Полученную смесь обрабатывают 500 мг активированного угля (Дарко KB) Бг I dHzdi о« -%0о
о о - .dHzdi
-3
CJOzH 016 П р и м е р 9. Получение пара-нитробензил 6с(.-.бром-2р-хлорметил-2-метилпенам-3-карбоксилатсульфона. К 16 л уксусной кислоты добавляют 36,6 г (0,812 моля) пара-нитробензил 6о1 бром-2ргхлорметил-2метилпенам-3-карбоксилата. К полученному таким образом раствору, перемешива- . емому при комнатной температуре, по-; каплям за 3 ч добавляют раствор 282 г (1,7.8 моля) КМпОч в 26 л воды. Получанную смесь перемешивают при комнатной температуре в течение 1 ч и по каплям добавляют Н,0 (37%} до получения бесцветного раствора. Затем добавляют 30 л воды, полученную смесь перемешивают в течение 1 ч при комнатной температуре и собирают кристаллический осадок. который промывают л воды и 2X2 л этанола, и сушат в вакууме при комнатной температуре. Выход 297 г (7б%)о(.з) (0,5% + 75,9.Пример 10. Получение BL-P2013 в виде свободной кислоты. и фильтруют. Фильтрат разбавляют 15 мл Скеллисольва В, затем вводят затравку кристаллов BL-P2013 в виде свободной кислоты. Спустя приблизительно 3 ч при комнатной температуре кристаллический осадок свободной кислоты собирают и сушат в вакууме в течение 15 мин над РпОс до получе- -.-f 1 л С- .. ния 323 мг (k6%) продукта Тпл медленным разложением выше 100°С. Вычислено, %: С 35,89; Н 3,77} N 5,23v С1 13,25 CeHjoCINOsS Найдено, %: С 35,88, Н 3,91; N 5,+1 i С1 13,52 Этот продукт теряет стабильность при хранении при 23°С в течение 7 дней. Пример 11. Получение 2 -хлорметил-2-метилпенам-З-карбоксилат-сульфона, калиевая соль, (BL-P2013) О О .f dHzdi ШН, 1/. jJ-VW dHs о - dOrK 3fSL P20l3 MB-305-77
К 600 мл Н20 добавляют В г 30 Pd на Целите и 16 г (0,19 моля) бикарбоната натрия. Затем 32 г (0,69 моля) пара-нитробензил 6ot-бpoм-2(i-xлopмeтил-2-мeтилпeнaм-3-карбоксилатсульфрксида растворяют в kOQ мл этилацетата и добавляют к водной взвеси. Полуменную смесь гидрируют на аппарате Паара при давлении 50 пси (3V5 кг/см) при в течение k ч. Полученную взвесь фильтруют через целитовый фильтроральный материал на воронке из спече ного стекла, затем фильтровальный 1атериал промывают 2xSO мл Н20 и вод ный слой объединенных фильтратов и прокмвок отделяют. Водный слой промывают 200 мл диэтилового эфира, затем йхлаждают до 5°С и при перемешивании добавляют по каплям за 1/2 ч раствор 12 г (0,076 моля) 200 мл Н20, причем рН поддерживают между 7,5 и а,Оt добавляя kO% H-jPO. После появления розового цвета через 5 мин добавление КМпО4 прекращают. Реакционную смесь п емешивают с небольшим количеством (примерно 50 мг) бисульфита натрия в течение 1/2 ч, а затем взвесь фильтруют через целитовый фильтровальный материал. Этот фильтровальный материал промыва1ЮТ 2x50 мл . Объединенный фильтрат и промывки расслаивают 500 мл этиацетата и, при перемешивании рН. устанавливают 1,5 добавляя 2 н. НС . Слои разделяют и водный слой Хасыщ1ают сульфатом натрия. Его повторно экстрагируют 2)((iOO мл этилацетата, и затем объединенные экстракты этилацетата сушат над сульфатом натрия в течение 1/2 ч при 5°С. Сульфат натрия удаляют фильтрованием, и полученный фильтрат выпаривают при пониженном давлении до остатка. Этот остаток растворяют в 16 О МП ацетона и 1бО мл диэтилового Э( и 50% (по весу) калий-2-этилгексаноата в Н-бутаноле добавляют до нейтрализации раствора по влажной индикаторной бумаге. Калиевая соль BL-P2013 кристаллизуется, ее собирают фильтрованием промывают диэтиловым эфиром и сушат. В результате получают .16 г 2 -хлорметил-2о{гМетилпенам-Зо6-карбоксилат-сульфона калиевой соли (BL-P2013) . Выход 76% (с Тпл ).
ИК и ЯМР спектры соответствуют предложенной структуре.
Вычислено, %: С 31,2; Н 2,97} N 1,58
CdHqClKNO S
Найдено, %: С 31,18; Н 2,98; N it,51, HgO 0,93
П р и м е р 12. Пивалоилоксиметил-2/ -хлорметил-2-метилпенам-3-карбоксилатсульфона (BL-P202j) .
rY TCJHa .
О к перемешиваемой суспензии 14,6 г (0, моля) 2р-хлорметил-2-метилпенам-3-карбрксилат-сульфонат калиевой соли (BL-P20T3) в 200 мл ацетон добавляют мл 10% водного раствора йодида натрия, и полученную смесь до водят до кипения с обратным холодильником на паровой бане, К этой суспензии, кипящей с обратным холодильником, добавляют сразу Н, 8 мл (0,1 мо ля) перегнанного хлорметилпивалата КипЗ при 7 мм рт.ст.). Полученную смесь перемешивают при кипении с обратным ХОЛОДИЛЬНИКОМ в течение 3 ч, а затем рхлажда1от до комнатной
МБ 381.83 температуры (22°С). Твердые кристаллы собирают фильтрованием, промывают 3x30 мл ацетона и объединенные фильтраты выпаривают до масла при пониженном давлении при температуре менее 22°С. Затем масло помещают 8 500 мл этилацетата и промывают один раз водой (200 нп), и один раз насыщенным раствором NanSO (200 ш). Затем раствор быстро сушат над , причем его перемешивают с 2 г обес цвеченного угля при охлаждении (ледяная баня.) . Спустя 20 мин полученную смесь фильтруют через целитный фильтро9вальиый материал, и этот материал промывают мл этилацетата. Объединенные фильтраты .концентрируют пр пониженном давлении при до маела. Затем это масло концентрируют далее при температуре около 22°С и давлении менее 1 мм рт.ст. до Удаления большей части оставшегося хлор метилпивалата. Оставшееся масло затем тщательно дважды-растирают с 50 мл порциями п-гептана, и затем оставляют на уикэнд при температуре около под п-пентаном. Полученную твердую кристаллическую массу растирают затем в порошок под АО мл смеси диэтиловый эфир-п-пентан . Полученный продукт собирают фильтрованием, промывают смесью диэтиловый ,эфир П-пентан (1:1), а затем п-пен таном и сушат воздухом. После сушки при высоком вакууме в темени ч над Р2О5 получают 13,37 г пйвалоилоксиметил 2р-хлорметил-2-метил-пенам-3-карбоксилат-сульфона (BLv93-95°C. Р2024), около 75% с Тр;, Очистка BL-P202if. Приблизительно 3 г неочищенного BL-P202 (полученного описанным выше способом) растворяют в 5 мл этил ацетата, помещают в колонку с силикагелем размером ,5 0 см ( Наллинкродт СС7 и элюируют смесью СН2С12 этилацетат : , объемное отношение). Фракции, содержавшие отдельное пятно при RP 0, (тонкослойная жидкостная хроматография на пластинах силикагеля смесью l: СН2С12 этилацетат, Ij детектирование) объ единяют и концентрируют при понижен ном давлении до 1,38 г кристаллического твердого продукта. Часть эт го материала (900 мг) растворяют в 5 мл этилацетата. Полученный раствор фильтруют, разбавляют почти до точки помутнения п-етролейным эфиром ( Скеллисольв В ), а затем хран при комнатной температуре в течение 3 дней. Образовавшиеся кристаллы со бирают фильтрованием, промывают пет ролейным эфиром и сушат, в результа чего получают 5бО мг очищенного BL Р202 с ЮО-ЮТ С. Вычислено, %: С ,03; Н 5,27, N 3,67 C HjjTlNO-jS Найдено, %: С 44, П; Н 5,08j N 3,85 П р и м е р 17. Получение аммони евой соли BL-P2013. 020 1.BL-P2013 в форме свободной кислоты (250 мг), растворенную а 20 мл смеси ацетон-метанол (1:1 по объему), фильтруют до получения прозрачного раствора. 2.Получают безводный аммониевый раствор, добавив 1 мл гидроокиси аммония (30%, реагентной степени уистоты) к 10 мл растворителя-смеси ацетон - метанол (1:1 по объему), а затем 1 г безводного сульфата магния добавляют к этому раствору при слабом перемешивании, и полученную смесь фильтруют через фильтровальную бумагу. Полученный фильтрат обозначают как безводный аммониевый раствор. 3.К фильтрату, полученному по методу 1, постепенно добавляют приблизительно 2 мл безводного аммониевого раствора и хорошо перемешивают, k. Смесь, полученную по методу 3, смешивают с порцией в 100 мл диэтилрвого эфира для осаждения аммониевой соли В-Р2013. 5. Белую аммониевую соль выделяют из растворителя и промывают 2 порциями по 50вмл каждая диэтилового эфира. 6. Выделенный порошок сушат при 35 С в вакуумном шкафу в течение ночи. 7. Результаты анализов: Вычислено, %: С 33,7} Н 4,6, N 9,8 Найдено, °4: С 33,66; Н 4,63, N 10,12. Сушили KF. Микроскопическое исследование: кpиcтaллиt ecкoe вещество. П р и м е р 14. Получение негигроскопичной натриевой соли BL,-P2013. 1. Растворяют 50 мг свободной кислоты BL-P2013 в 4 мл смеси ацетонметанол (1:1 по объему), фильтруют до получения прозрачного раствора. 2о Готовят натрий-2-этилгексаноатный раствор, растворив 40 мг натрий-2-этилгексаноата в 10 мл смеси ацетЬн - метанол (1:1 по объему) 3.К фильтрату (метод 1) добавляют 10 мл раствора (метод 2) и хорошо перемешивают. 4.Порцию 10 мл диэтиЛового эфира смешивают со смесью, полученной методом 3, для осаждения натриевой соЛИ BL-P2013. 2199 5. Белую соль погружают в диэтиловый эфир на 1-2 ч, а затем выделяют из растворителя и промывают тремя порциями диэтилового эфира по 5 мл каждая. V/ н Hw di V;i« nepeH/Jucma/rw НгО . ЙОгК-НгО
kOQ мл BL-P2013 растворяют в минимальном количестве смеси ацетон Н20 (1:1 по объему) и разбавляют 10 мл ацетона, фильтруют, затем разбавляют ацетоном до объема около 25 мл, поскребли, и спустя 30 мин фильтрованием собирают кристаллический гидрат, хорошо промывают его ацетоном, потом сушат на воздухе, а затем в вакууме при давлении менее 1 мм рт.ст. в течение ночи. Выход 280 мг.
Вычислено, %: С 29,67i Н 3,39; N 4,63; С1 10, Н,0 5,55 C0H9C1NOSKH20
Найдено, %: С 29,32,- Н 3,32j Н Ц,ЦЦ. С1 11.31; Н20 5,90.
П р и м ер 16. Получение NiN -дибензилэтилендиаминовой соли BLР2013.
BL-P2013+1/2 Ы,М-дибензилэтилендиамин диацетата перекристаллизация
ацетон-эфир 820 6. Выделенный порошок сушат при 30°С в вакуумном шкафу в темение ночи. П р и м е р 15- Перекристаллизация BL-P2bl3. 0. о А PNf Н J-Jj C OfK-HzO
/(
ЙОгН-(бН5(Нгда-с Нг
о
зоб мг (0,001 моля BL-P2013 растворяют в 7 мл и добавляют к раст-35 вору 180 мг (.0,0005 моля) N, N-дибеизилэтилендиамин диацетата в 7 мл . Н-О. Полученную смесь перемешивают, и соль кристаллизуется, после перемешивания приблизгительно в течение о 10-15 мин соль собирают фильтрованием и сушат на воздухе, в результате чего получают N,N-дибензилэтилендиаминовую соль BL-P2013 (300 мг). Полученный материал перекристаллизовыва- 45
ют, растворив приблизительно в 10 мл кипящего ацетона и разбавив эфиром до точки помутнения. При этом получают 26О мг высушенного на воздухе и в вакууме материала.
Вычислено, %: С 5t,69i Н S,2; N 7,53) С1 9,55
Найдено, %: С 9,39; И 5,
N 7,05, С1 8,96i Н20 1,23 (KF).
Пример 17. Получение хлорметилового сложного эфира BL-P2013.











ASf
(JHtdi о
f3J6,J7)
7 К иитенсивно перемешиваемой смеси 15,25 г (O.OfI моля) BL-P2013 15), 15 г (0,15 моля) KHCOj и 1,7 г (0,005 моля) тетрабутиламмоний кислого сульфата (/(лдрич Кем.Ко) в смеси с 50 мл воды и 50 мл СН2С12.по каплям добавляют раствор 9,5 г (0,0575 моля) C1CH2-0-S02t;.l в 40 мл CHjClj. Температура повышается до 26°С, и после добавления (которое заняло приблизительно 15 мин) смесь перемешивают еще дополнительно 30 мин. Так как продукт кристаллизуется, добавляют еще (около 400 мл) до получения раствора. Выделенный слой CHoCI и 50 мл СН2С12промывок объединяют, сушат над сульфатом магния при перемешивании, и добавляют 2 г обесцвеченного угля (Дарко KB). Спустя около 30 мин смесь фильтруют, концентрируют до около 50 мл и добавляО, о
feedl
-odHzdi « о к перемешиваемой смеси г (0,0159 моля) хлорметилового сложного эфира BL-P2013 (7 ) в 25 мл ацетона добавляют 3 г (С,,02 моля) йодина натрия. Полученную взвесь перемешивают в течение 17 ч, а затем охлаж дают до 0°С. Добавляют две капли нас ценного водного КНСО и полученную смесь медленно разбавляют по каплям водой в течение более Ю мин до тех пор, пока не добавят 50 мл. Взвесв изменяет цвет с желтого на серый, затем на пурпурный и потом на черный и после этого кристаллы немедленно собирают фильтрованием и про
- J- tiHzdi
о/ -d-odHiddiy
50
ЛеЗлнм yftcyctfo ffc/f/fffma 99
,
Nai
d-odHzT о
8
+кмл04-кн:202
о dH,di
Cjl-odH ddi,
Ч о .2 ют 150 мл изопропилового спирта. Затем удаляют остаток при пониженном давлении. Полученный кристаллический осадок собирают фильтрованием, хорошо промывают изопропиловым спиртом и сушат на воздухе. После сушки в вакууме при давлении менее 1 мм рт. ст. получают 8,5 г хлорметил-2(3-хлор мет ил-2-мет илпенам-3 . .. . - /-карбоксилат-сульфона (7). Тал 116 С {разложение при температуре свыше 100°С, потемнение). Элементный анализ для Вычислено, %: С З, 18; Н 3,5i; N ,kЗ С1 22, Найдено, %: С 3,16 J Н 3,5,N ,47j С1 22,46; HgO 0,33 (KF) : Оценка чистоты: 90-95. П р и м е р 18. Получение йодометилового сложного эфира BL-P2313. мывают холодной смесью ацетон - вода (1:2), а затем изопропиловым спиртом (3X10 мл), а затем диэтиловым эфиром и наконец п-пентаном. После высушивания на воздухе получают 5,55 г (91 выход) йодметилового сложного эфира BL-P2013 V8 ). Тг7/ П8119 0 с разложением. Чистота продукта около 90. Пример 19. Получение N,N-дибензилэтилендиамин-2 -хлорметил-2о -метилпенам-2оС-карбоксилата сульфона. Стадия 1. Соединение 3 растворяют .в 1 л ле дяной уксусной кислоты и при переме шивании при 22С по каплям добавля ют насыщенный водный раствор КМп04, до появления розового цвета (т.е. капля, помещенная на-кусочек фильтр вальнрй бумаги дает розовую окраску). Затем при охлаждении добавляют N20 по каплям до тех пор, пока не получают п)р1ОЗрачный раствор (при .этом о(5разуется некоторое количество белого осадка). Полученньй раствор выливают в 2,5 л воды и про дукт i экстрапфуют )Pj мл этилац тата. Этилацетат промывают 5 водным p:act вором бикарбЬнаг натрия ЯО нейтрального (т.е„ более не происходит выделения пузырьков при добавлении) , сушат над суль.фатом натрия и выпаривают,; в результате чего получают в остатке соединение k. ЕГО оставляют при в течение одного дня, а затем тщательно расти рают со Скеллисольвом В до получения 9,1 г соединения. Выход сое тавляет 28 от теоретического. Стадий i3 4- -ф2п0укс(/гяоу (, . g ciHzdi 1:1оох 5 (Б1.-Р2013) Зг75 г цинкового, порошка смешивают до получения взвеси с 5 мл ледяной уксусной кислоты и охлаждают до 5°С. К этой добавляют 3 г раствор j (О,(Ю57 моля) в 15 мл димитилформамида и полученную взвесь перемешивают-при в течение 2,5 ч Затем циНк отфильтровывают и свет ложелтый раствор выливают в 80 мл 5 водной соляной кислоты. Полученную смесь экстрагируют 3x25 МП этилацетата. Объединенные экстракты этил ацетата экстрагируют 3X20 мл 5 водным раствором бика| боната натрия, сохранив после разделения этилацетат ную фазу. Бикарбонатные экстракты объединяют, помещают под слой этилацетата, устанавливают рН 1,5 , добавив 2 н. НС1 и насытив сульфатом натрия. Этилацетатный слой выделяют, и водную фазу экстрагируют 2x30 мл этм1ацетата. Всю этилацетатную ( объединяют, сушат над сульфатом натрия и выпаривают до масла (которое было сёободной кислотой BL-P2013), а затем растворяют а около 20 мл ацетсма, к которому добавляют 20 мл диэтилсаого эфи-. рае Потом добавляют 50 калййг2-этипгексаноат (КЭГ)., в сухОм п-бутеноле до нейтральности кристаллизуд тся продукт 5 (BL-P2013). После перемешива ния о75 ч при его собирают фильтрованием, в.результате чего получают 650 мг соединения 5 (выход З). Стадия 3.50 мг соединения 5 растворяют в й,5 мл воды и добавляют 20 мг N,N -дибензилэтилендиамин.(ДБЭД) диацетата. ДБЭД соль 5 кристаллизуется , и ее собирают фильтрованием, промывают водой и сушат над Р205 в вакууме, в результате чего получают N} N -дибензил тилендиамин-2р|-хлорметил-2Ы-метилпенам-3(Л-карбоксилат-. сульфон (ДБЭД соль свободной кислое-, ты 5). Другую часть соединения 5 мг) растворяют в 3 мл воды, к которой добавляют раствор 270 мг ДБЭД диаце- . тата в 2 мл Н20. При поскребывании Д6ЭД соль S кристаллизуется (430 мг). перекристаллизации Из около 5 мл кипящего ацетона пс лучают 270 мг. . Результаты биологических испытаний. Продукт примера 1, соединение 5 имеющее строение О о fV(iH,(ii 6Д5 . именуется BL-P2013. Хотя BL-P2013 сам по себе в лучшем случае является очень слабым антибактериальным агентом, он ингибирует р-лактамазы и защищает щефоранид и амоксициллин от разрушения бактериями, продуцирующими fb-лактамазы ifn vitro и In vivo, когда его используют в сочетан с этими двумя, агентами. Предлагаемые соединения являются полезными при оральном или парэнтеральном введений, для повышения эффективности |)-лактамовых антибиотиков против бактерий, продуцирующих р-лактамазы. В расчете на вес, их до9зировка должна составлять от одной пятой до пяти частей, и предпочти тельно должна быть равной весу 3-лак тамового антибиотика. Например, эти соединения при использовании в отношении t:1 демонстрируют заметное повышение активности цефорадйна и амок сициллина против штаммов анаэробных Bacteroides, продуцирующих/3-лак тамазы, например, В. fragilis, В. thetaiotaomicron и других штаммов этого рода, а также против устойчивых Staphylococus aureus. Соединения вводят либо в смеси или одновременно с ft-лактамовыми антибиотиками,. в дозах внутри указанного интервала соотношений, причем антибиотик при этом вводят в обычных дозах. Способность соединений повышает эффективность -лактамовых антибиотиков против продуцирующихj3-лакт;амазы бактерий, делает их ценными дл совместного введения с некоторыми в-лактамовыми антибиотиками при лечении бактериальных заболеваний у млекопитающихся, в частности, челове ка. При лечении бактериальных заболе ваний соединения можно совместно измельчать с р-лактамовыми антибиотиками, и таким образом вводить два агента одновременно. Кроме того, сое динения можно вводить как самостоятельные агенты на протяжении курса лечения р-лактамовыми .антибиотиками При использовании соединения или его соли для повышения антибактериальной активности р-лактамового анти биотика, его можно вводить отдельно или предпочтительно в составе со ста дартными фармацевтичекими носителя ми и разбавителями. Соединение, нахо дящееся в форме свободной кислоты или ее фармацевтически- приемлемой соли, можно вводить орально или парэнтерально. Соединение в форме слож ного эфира, который легко гидролизу ется in vivo, лучше вводить орально Парэнтеральное введение включает вн римышечные, подкожные, внутрибрюшин ные и внутривенные инъекции. Если соединение используют в соче тании с носителем или разбавителем, указанный носитель или разбавитель выбирают на основании предполагаемого способа введения Например, для орального введения, соединение можно использовать в форме таблеток 28 капсул,блоков, лепешек, порошков, сиропов, элексиров, водных растворов и суспензий, и тому подобное, в соответствии стандартной фармацевтической практикой. Пропорции активных ингредиентов и носителя будут, естественно, зависеть от химической природы, растворимости, стабильности и потенции ак тивных ингредиентов, также как и от необходимой дозировки. Однако эти фармацевтические композиции могут содержат.ь от около 5 до около 80 носителя. В случае таблеток для орального прием а носители обычно включают лактозу, цитр натрия, и соли фосфорной кислоты , Для таблеток обычно используют различные дезинтегранты, например крахмал, и такие смазочные агенты, как стеарат магния, натри и лау рил сульфат и тальк. Для орального приема в виде капсул пригодны такие разбавители как лактоза и высокомолекулярные полиэтиленгликоли. Если для орального приема необходимы суспензии, то активные ингредиенты сочетают с эмульгирующими и суспендирующими агентами. При желании можно добавить подслащивающие и/или одорирующие вещества. Для парэнтерального введения, которое включает внутримышечные, внутрибрюшинции, обычно приготавливают стерильные растворы активных ингредиентов, и рН растворов соответствующим образом устанавливают и буферируют. Для внутривенного применения полную концентрацию растворов следует контролировать для создания изотонических растворов. Хотяименно врач должен определять дозировку, необходимую пациенту, отношение дневной дозы соединения или его соли и /ь-лактамового антибиотика должно быть в интервале от около 1:5 до 5:1, и предпочтительно, 1:1. Далее, дневная доза орального приема каждого компонента будет в интервале от около 10 до около 200 мг на кг живого веса, а дневная парэнтеральная доза каждого компонента бу-дет от около 10 до около 100 мг на кг живого веса. Эти цифры,, однако, являются лишь иллюстративными, и в некоторых случаях могут понадобиться дозы, выходящие за эти пределы. 1. Способ получения сульфонов 2 -хлорметил-2|1-метилпенамгЗо6-карбоновой кислоты формулы .dHzdi ЙНэ , (iooH. или ее сложных эфиров,или ее солей, о тли ч а ю .щ и и с я тем, что соединение общей формулы :0-: xiHzdi trtf : -l3---. 0 dOOR где R - бензил или замещенный бензил каталитически гидрируют с последую щим окислением полученного продукта г;идр| ования и выделением целевого продукта в виде свободной кислоты ил ее соли, 1ли в случае необходимости Свободную кислоту или ее соль этерифицируют и выделяк т целевой продукт в виде его сложного эфира о 2.Способ по п. 1, о т л и ч а ю щ и и с я тем, что гидрирование проводят над палладиевым катализатором. 3.Способ по п. 1, отличающий с я тем, что окисление проводят перманганатом щелочного металл или органической надкислотой. k. Способ получения сульфонов 2fV -хлормет ил-2оЬ-мет илпенам-3о г карбоновой кислоты формулы. бНэ -(JoOtt или её сложных эфиров, или ее солей. отличающ и и,с я тем, что соеинение формулы -J dHjdi rnf-бнз X-OsF---I (J-ixJHiCldiy «у окисляют таким окислителем, как перманганат калия или перекись водороа, и образующийся сульфо н формулы вр VdHzdi , -e-odHzcidia - о подвергают взаимодействие с металлом в кислоте, например с цинком в ледяной уксусной кислоте, с выделением целевого продукта в виде свободной кислоты И.ЛИ ее соли, или в случае необходимости свободную кислоту или ее соль этерифицируют и выделяют целевой продукт а виде его сложного эфира. Источники информации, принятые во внимание при экспертизе 1.Патент Великобритании №1072108, С 2 А, опублик. 1967. 2.Выложенная заявка If 2B2kS3S, кл. С 07 0 199/00, опублик. 1978.
