Изобретение относится к органической химии, а именно к новому способу получения как новых, так и известных производных 3-аминохинолина общей формулы I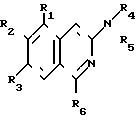 где R1 и R2 - низшая алкоксигруппа; R3 - атом водорода, R2 и R3 - низшая алкоксигруппа, а R1 - атом водорода или R2 и R3 вместе образуют метилендиоксигруппу;
где R1 и R2 - низшая алкоксигруппа; R3 - атом водорода, R2 и R3 - низшая алкоксигруппа, а R1 - атом водорода или R2 и R3 вместе образуют метилендиоксигруппу;
R4 и R5 имеют одинаковые или разные значения и представляют собой атомы водорода, низшие алкильные, ацильные группы, либо один из них представляет группу -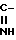 R7
R7
R6 и R7 имеют одинаковые или разные значения и представляют собой низший алкил, замещенный или незамещенный фенил или бензил.
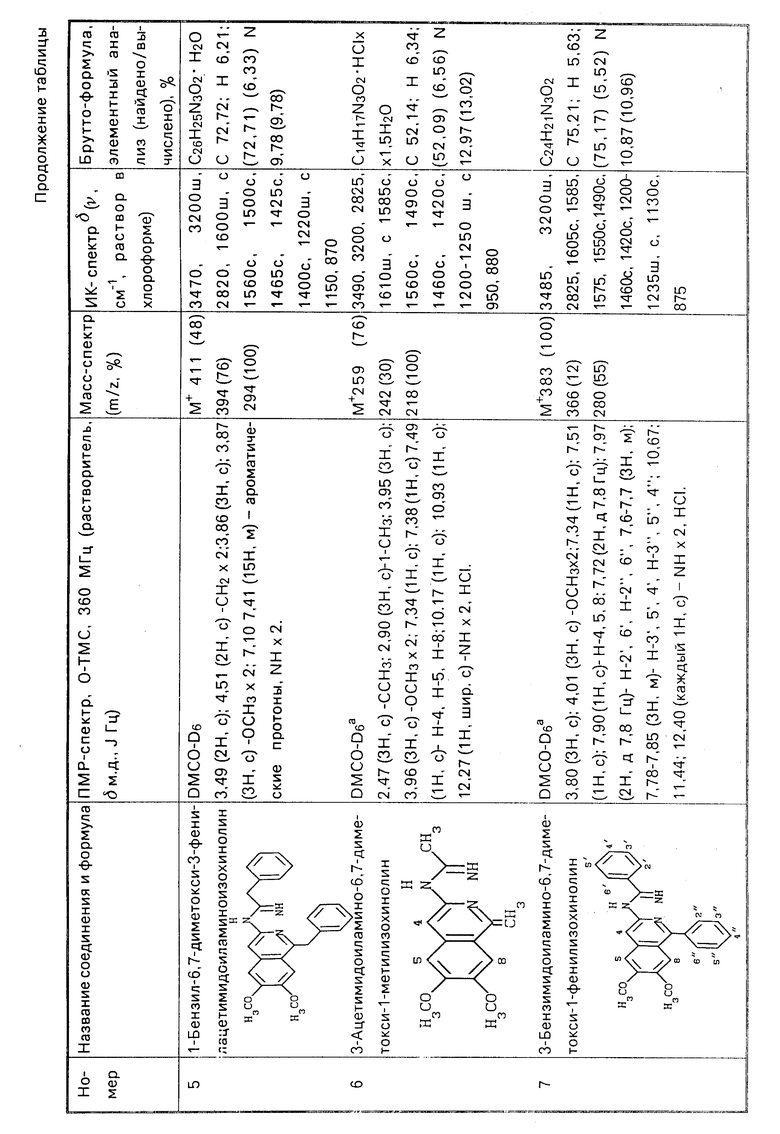
Соединения формулы I являются производными алкалоидов и могут найти применение в медицине или в качестве промежуточных продуктов в синтезе других биологически активных веществ.
Цель изобретения - увеличение выхода целевого продукта, упрощение способа получения и расширение ассортимента производных 3-аминоизохинолинов.
Поставленная цель достигается заявленным способом, заключающимся в том, что нитрилы фенилуксусных кислот формулы II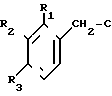


или их смесь с нитрилами общих формулы III и IV
R6-C ≡ N R7-C ≡ N
подвергают циклоди- или тримеризации под действием хлористого водорода с выделением целевого продукта или с гидролизом, ацилированием или алкилированием с целью получения соединений общей формулы I с соответствующими значениями радикалов.
П р и м е р 1. Через раствор 0,5 г (2,82 ммоль) 3,4-диметоксибензилцианида в 1 мл сухого диоксана пропускают ток безводного хлористого водорода сначала при комнатной температуре, затем при охлаждении до -21оС (баня из смеси льда и хлористого натрия) до насыщения. При этом поглощается 1 г (27,4 ммоль) хлористого водорода. Реакционную колбу плотно укупоривают, пробку привязывают, чтобы предотвратить улетучивание хлористого водорода, и оставляют стоять при комнатной температуре (18-21оС) в течение 2 суток. По прошествии этого времени реакционную колбу охлаждают и осторожно вскрывают. Растворитель вместе с избытком хлористого водорода упаривают в вакууме, из оставшейся кристаллической массы извлекают тремя порциями (по 10 мл) серного эфира 0,095 г непрореагировавшего исходного нитрила. Оставшийся после экстракции продукт перекристаллизовывают из 10 мл этанола и получают 0,344 г хлористоводородной соли 6,7-диметокси-1-(3,4-диметоксибензил)-3-(3,4- диметоксифенилацетимидоиламино)изохинолина (1˙ HCl) в виде бесцветных кристаллов с т. пл. 139-140оС. Маточник подщелачивают и получают еще 0,03 г основания 1, т.пл. 198-200оС (разл.). Общий выход 70,1% (или 86,5%, считая на вступивший в реакцию нитрил).
П р и м е р 2. Растворяют 0,25 г (1,41 ммоль) 3,4-диметоксибензилцианида в 0,5 мл сухого диоксана и проводят реакцию с 0,5 г (13,7 ммоль) хлористого водорода при 50оС в запаянной ампуле в течение 12 ч. Реакционную смесь обрабатывают, как это описано в примере 1, и получают 0,0623 г непрореагировавшего исходного нитрила и 0,177 г (66,2%) продукта 1 HCl, что составляет 88,1% в пересчете на вступивший в реакцию нитрил.
П р и м е р 3. Растворяют 0,25 г (1,41 ммоль) 3,4-диметоксибензилцианида в 0,5 мл сухого диоксана и проводят реакцию с 0,5 г (13,7 ммоль) хлористого водорода при +7оС в течение 5 суток, как это описано в примере 1. По прошествии этого времени колбу вскрывают, удаляют в вакууме растворитель и избыток хлористого водорода. Остаток обрабатывают двумя порциями серного эфира по 10 мл и извлекают 0,03 г непрореагировавшего исходного нитрила. Остаток перекристаллизовывают из 2%-ного раствора КОН в метаноле и получают 0,11 г основания 1. Маточник упаривают и хроматографируют на колонке с силикагелем L 40/100 (Chemapol) (20 г адсорбента, элюент - смесь хлороформа с метанолом 10:1). Получают дополнительные 0,026 г основания I (общий выход 54,4% ) и 0,045 г (18%) основания 3-амино-6,7-диметокси-1-(3,4-диметоксибен-зил)изохинолина (10) в виде бесцветных кристаллов с т.пл. 180-181оС (из метанола). Гидрохлорид (10 HCl) - желтые кристаллы; т.пл. 220-228оС (разложение из метанола).
П р и м е р 4. Растворяют 20,0 г (0,113 моль) 3,4-диметоксибензилцианида в 40 мл сухого хлороформа и проводят реакцию с 4,4 г (0,121 моль) хлористого водорода при комнатной температуре в течение 8 суток, как это указано в примере 1. Получают 11,59 г непрореагировавшего исходного нитрила и 7,37 г продукта 1 HCl, что составляет 34,5% (или 82,0% в пересчете на вступивший в реакцию нитрил).
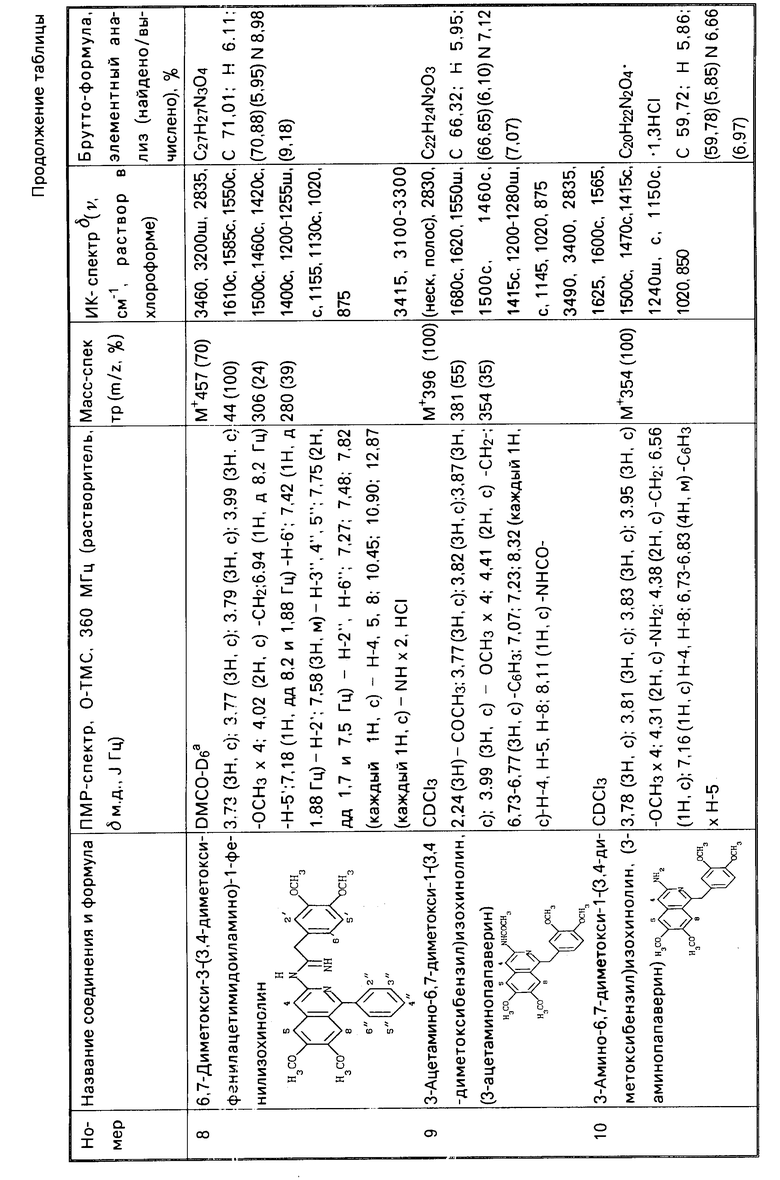
П р и м е р 5. Растворяют 0,8 г (4,52 ммоль) 3,4-диметоксибензилцианида в 5 мл сухого толуола и проводят реакцию с 0,5 г (13,7 ммоль) хлористого водорода в течение 11 суток при комнатной температуре, как это описано в примере 1. Получают 0,25 г непрореагировавшего исходного нитрила и 0,35 г (40,9% ) продукт 1 HCl, что составляет 59,5% в пересчете на вступивший в реакцию нитрил.
П р и м е р 6. Через раствор 3 г (16,9 ммоль) 3,4-диметоксибензилцианида в 4 мл хлорокиси фосфора пропускают ток хлористого водорода сначала при комнатной температуре, а затем при охлаждении до -10оС до насыщения. При этом поглощается 1,8 г (49,3 ммоль) хлористого водорода. Реакционную колбу укупоривают и оставляют стоять при комнатной температуре в течение 10 суток. По прошествии этого времени колбу вскрывают, упаривают в вакууме избыток хлорокиси фосфора и обрабатывают смесь 30 г льда. Образовавшийся маслянистый продукт промывают двумя порциями воды по 15 мл и растворяют в 15 мл горячего этанола. При охлаждении из раствора постепенно выпадает кристаллический осадок продукта 1 HCl. Выход 1,71 г. Маточник упаривают, хроматографируют на силикагеле L 40/100 (Сhemapol) (100 г адсорбента, элюент - хлороформ + метанол 100:3) и получают дополнительно 0,31 г продукта и 0,15 г не вступившего в реакцию исходного нитрила. Общий выход продукта 1 HCl 2,02 г (63,0%) или 66,3%, считая на вступивший в реакцию нитрил.
П р и м е р 7. Растворяют 0,5 г (2,44 ммоль) 3,4-диэтоксибензилцианида в 1,5 мл сухого диоксана и проводят реакцию с 1 г (27,4 ммоль) хлористого водорода в течение 8 сут при комнатной температуре, как это описано в примере 1. Получают 0,17 г непрореагировавшего исходного нитрила, 0,22 г гидрохлорида 6,7-диэтокси-1-(3,4-диэтоксибензил)-3-(3,4 -диэтоксифенилацетимидоиламино)изохинолина (2˙ HCl) и 0,04 г основания 2. Общий выход 49,1% (или 74,4% в пересчете на вступивший в реакцию нитрил).
Гидрохлорид (2˙ HCl) - бесцветные кристаллы, т.пл. 186-191оС (из этанола). Основание 2 - бесцветные кристаллы с двумя температурами плавления 129-313оС и 147-149оС (из этанола).
П р и м е р 8. Растворяют 1,66 г (10,3 ммоль) 3,4-метилендиоксибензилцианида в 2 мл сухого диоксана и проводят реакцию с 2 г (54,7 ммоль) хлористого водорода в течение 7 суток при комнатной температуре, как это указано в примере 1. Получают 0,015 г не вступившего в реакцию исходного нитрила, 1,26 г гидрохлорида 6,7-метилендиокси-1-(3,4-метилендиоксибензил)-3-(3,4- метилендиоксифенилацетимидоиламино)изохиноли- на (3˙ HCl), и 0,03 г основания 3. Общий выход 72,3% (или 72,9%, cчитая на вступивший в реакцию нитрил). Гидрохлорид (3 ˙HCl) - бесцветные кристаллы; т.пл. 287-293оС (разл. , из этанола). Основание 3 - бесцветные кристаллы, т.пл. 186-187оС (из метанола).
П р и м е р 9. Пропускают ток сухого хлористого водорода через 2 г (11,3 ммоль) 2,3-диметоксибензилцианида при -19оС до насыщения. При этом поглощается 1,05 г (28,8 ммоль) хлористого водорода. Колбу укупоривают и оставляют стоять при комнатной температуре в течение 7 суток. По прошествии этого времени колбу вскрывают и удаляют избыток хлористого водорода в вакууме. Остаток обрабатывают тремя порциями по 20 мл серного эфира и извлекают 0,12 г непрореагировавшего исходного нитрила. Маслянистый остаток растворяют в 2 мл метанола, добавляют 5 мл воды и оставляют на 12 ч при комнатной температуре. Затем смесь экстрагируют хлороформом, хлороформные вытяжки сушат, упаривают досуха и обрабатывают бензолом (3 порции по 10 мл). Бензольный экстракт отбрасывают, а маслянистый остаток перекристаллизовывают из 5%-ного этанольного раствора КОН и получают 0,23 г основания 5,6-диметокси-1-(2,3-диметоксибензил)-3-(2,3- диметоксифенилацетимидоиламино)изохинолина (4) - бесцветные кристаллы, т.пл. 141-142оС (из этанола). Из маточника методом колоночный хроматографии на силикагеле L 40/100 (Chemapol) (15 г адсорбента, элюент - хлороформ + метанол 10:1) получают дополнительно 0,05 г основания 4. Общий выход 14% (или 14,9%, считая на вступивший в реакцию нитрил).
П р и м е р 10. Через раствор 1,77 г (0,01 моль) 3,4-диметоксибензилцианида в 15,2 г (0,13 моль) бензилцианида пропускают ток сухого хлористого водорода сначала при комнатной температуре, а затем при охлаждении до -20оС до насыщения. При этом поглощается 11 г (0,3 моль) хлористого водорода. Реакционную колбу плотно укупоривают, пробку привязывают, чтобы предотвратить улетучивание хлористого водорода и оставляют стоять при температуре 35-40оС в течение 5 суток. По прошествии этого времени реакционную колбу вскрывают при охлаждении и избыток хлористого водорода удаляют в вакууме. Застывшую стеклообразную массу растирают с тремя порциями серного эфира по 70 мл каждая. Образовавшийся порошкообразный продукт высушивают в вакууме от остатков эфира, растворяют при нагревании в 40 мл метанола, добавляют 200 мл воды и оставляют на ночь. На следующий день отфильтровывают выпавший осадок, высушивают от остатков воды током горячего воздуха и обрабатывают тремя порциями по 70 мл горячего хлороформа, каждый раз отфильтровывая нерастворимый осадок. Осадок отбрасывают, а хлороформные вытяжки объединяют и упаривают досуха в вакууме. Полученный маслянистый остаток растворяют при нагревании в 20 мл этанола, добавляют 40 мл 10%-ного раствора едкого натра в этаноле и оставляют кристаллизоваться на холоде в течение 2 ч. Образовавшийся осадок отфильтровывают, промывают сначала этанолом, затем водой до нейтральной реакции, высушивают и получают 1,61 г основания 1-бензил-6,7-диметокси-3-фенилацетамидоиламиноизохинолина (5), бесцветные кристаллы, т. пл. 131-132оС (из этанола). Методом колоночной хроматографии на окиси алюминия (100 г адсорбента, элюент хлороформ) из спиртового маточника, а также из хлороформного экстракта, полученного из подщелочного водного фильтрата, выделяют дополнительно 0,12 г продукта 5 (общий выход 42,1%) и 0,09 г (3,1% ) основания 3-амино-1-бензил-6,7-диметоксиизохинолина (12), бесцветные кристаллы, т. пл. 155-156оС (из этанола). Гидрохлорид (12˙ HCl) - желтые кристаллы, т.пл. 129-132оС (из смеси этанолэфир).
П р и м е р 11. Проводят реакцию 1,77 г (0,01 моль) 3,4-диметоксибензилцианида с 11,5 г (0,28 моль) сухого ацетонитрила в присутствии 13 г (0,36 моль) хлористого водорода при 35оС в течение 5 суток, как это описано в примере 10. По прошествии этого времени реакционную колбу вскрывают и удаляют в вакууме избыток хлористого водорода и ацетонитрила. Оставшуюся полукристаллическую массу растворяют в 30 мл воды и нейтрализуют раствор, добавляя небольшими порциями порошкообразный гидрокарбонат натрия, до слабощелочной реакции. Выпавший осадок отфильтровывают и промывают двумя порциями воды по 15 мл. После высушивания получают 1,35 г (52,1%) основания 3-ацетимидоиламино-6,7-диметокси-1-метилизохинолина (6) в виде бесцветных кристаллов, т.пл. 289-290оС (разложение, из смеси этанол - вода). Гидрохлорид (6 ˙HCl) - бесцветные кристаллы; т.пл. 270-274оС (разложение из метанола).
П р и м е р 12. Проводят реакцию 1,77 г (0,01 моль) 3,4-диметоксибензилцианида с 15,28 г (0,148 моль) бензонитрила в присутствии 6,5 г (0,178 моль) хлористого водорода при 30оС в течение 7 суток, как это описано в примере 10. По прошествии этого времени реакционную колбу вскрывают и удаляют в вакууме избыток хлористого водорода. Из реакционной смеси экстрагируют тремя порциями по 30 мл серного эфира 9,2 г непрореагировавшего бензонитрила. Остаток растворяют в 10 мл метанола, добавляют 70 мл воды и оставляют на ночь. На следующий день отфильтровывают выпавший осадок, высушивают от остатков воды и обрабатывают тремя порциями бензола по 5 мл. Бензольные вытяжки отбрасывают, а остаток разделяют методом колоночной хроматографии на силикагеле L 40/100 (Chemapol) (100 г адсорбента, элюенты - последовательно бензол, хлороформ и 1%-ный раствор диэтиламина в хлороформе). Получают 1,69 г (44,1%) основания 3-бензимидоиламино-6,7-диметокси-1-фенилизохинолина (7) и 0,99 г (25,8%) основания 6,7-диметокси-3-(3,4-диметоксифенилацетимидоиламино)- 1-фенилизохинолина (8). Основание 7 - бесцветные кристаллы, т.пл. 96-97оС (из смеси этанол - вода). Основание 8 - бесцветные кристаллы, т.пл. 86-88оС (из этанола).
П р и м е р 13. Смешивают 0,1 г (0,513 ммоль) амида 3,4-диметоксифенилуксусной кислоты с 1 мл (10,95 ммоль) хлорокиси фосфора. Реакционный сосуд с частично растворившимся амидом укупоривают и оставляют при температуре 35-40оС в течение 7 сут. По прошествии этого времени реакционную смесь обрабатывают при охлаждении 10 г льда. Образовавшуюся вязкую массу промывают двумя порциями воды по 3 мл, удаляют остатки воды в токе горячего воздуха и растворяют в 3 мл горячего этанола. После охлаждения отфильтровывают выпавший осадок, промывают 2 мл этанола, высушивают и получают 0,05 г хлористоводородной соли 1. Маточник упаривают и хроматографируют на силикагеле L 40/100 (Chemapol) (10 г адсорбента, элюент - смесь хлороформа с метанолом 100: 3). Получают дополнительно 0,015 г продукта 1˙ HCl. Общий выход 67,01%. Полученный продукт по температуре плавления и спектральным характеристикам идентичен продукту, полученному в примере 1.
П р и м е р 14. Проводят реакцию 0,1 г (0,513 ммоль) 3,4-диметоксифенилацетальдоксима с 1 мл хлорокиси фосфора, как это указано в примере 13. Получают 0,055 г (56,7%) продукта 1 HCl, идентичного продукту, полученному в примере 1.
П р и м е р 15. Смешивают 1,7 г (3,00 ммоль) хлористоводородной соли 6,7-диметокси-1-(3,4-диметоксибензил)-3-(3,4- диметоксифенилацетимидоиламино)изохинолина (1˙ HCl), 0,5 г (3,68 ммоль) трехводного ацетата натрия и 10 мл уксусного ангидрида, нагревают смесь до кипения и кипятят 3 мин. К еще теплой смеси добавляют 30 мл бензола и отфильтровывают выпавший осадок. Осадок отбрасывают, а фильтрат упаривают в вакууме. Маслянистый остаток растворяют в 25 мл серного эфира и оставляют на ночь на холоде. На следующий день выпавший осадок отфильтровывают, промывают двумя порциями по 10 мл эфира, высушивают и получают 1,17 г (98,9%) 3-ацетамино-6,7-диметокси-1-(3,4-диметоксибензил)изохинолина (9), бесцветные кристаллы, т.пл. 160-161оС (из бензола). Гидрохлорид (9 ˙HCl) - бледно-желтые кристаллы, т.пл. 208-213оС (из смеси этанол - эфир).
П р и м е р 16. Раствор 1,2 г (3,03 ммоль) 3-ацетаминопапаверина (9) в 20 мл этанола смешивают с 10 мл 25%-ного водного раствора едкого калия и кипятят с обратным холодильником 2 ч. Затем раствор упаривают на три четверти первоначального объема и после охлаждения отфильтровывают выпавший осадок. Осадок промывают сначала тремя миллилитрами этанола, а затем водой до отсутствия щелочной реакции, высушивают и получают 0,85 г 3-амино-6,7-диметокси-1-(3,4-диметоксибензил)изохинолина (10). Из маточников методом колоночной хроматографии на силикагеле L 40/100 (Chemapol) (30 г адсорбента, элюент смесь хлороформа с метанолом 10:1) получают дополнительно 0,16 г продукта 10. Общий выход 94,2%. Полученный продукт по температуре плавления и спектральным характеристикам идентичен продукту, полученному в примере 3.
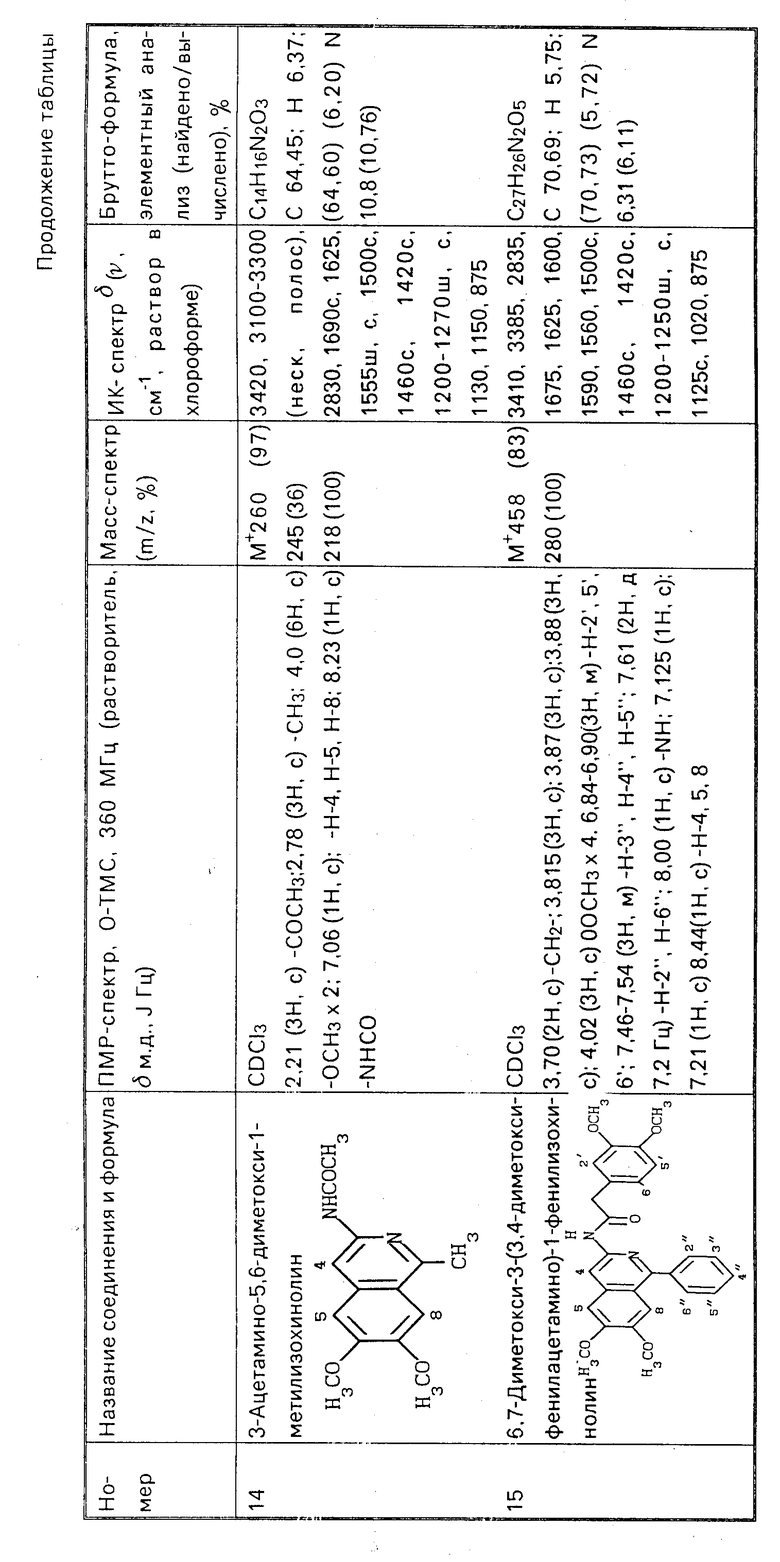
П р и м е р 17. Смесь 0,23 г (0,56 ммоль) 1-бензил-6,7-диметокси-3-фенилацетимидоиламиноизохинолина (5) и 0,7 мл уксусного ангидрида нагревают до кипения и кипятят 7 мин. Затем реакционную смесь охлаждают, смешивают с 15 мл серного эфира, выпавший осадок отфильтровывают, промывают двумя порциями эфира по 5 мл, высушивают и получают 0,105 г 3-ацетамино-1-бензил-6,7-диметоксиизохинолина (11), бесцветные кристаллы, т.пл. 180-181оС (из эфира). Из маточника методом колоночной хроматографии на силикагеле L 40/100 (Chemapol) (10 г адсорбента, элюентхлороформ) дополнительно получают 0,062 г продукта 11. Общий выход 88,8%.
П р и м е р 18. Проводят гидролиз 0,05 г (0,149 ммоль) 3-ацетамино-1-бензил-6,7-диметоксиизохинолина (II), как это описано в примере 16. Получают 0,042 г (96,0%) 3-амино-1-бензил-6,7-диметоксиизохинолина (12). Полученный продукт по температуре плавления и спектральным характеристикам идентичен продукту, полученному в примере 10.
П р и м е р 19. Смешивают 0,58 г (2,24 ммоль) 3-ацетимидоиламино-6,7-диметокси-1-метилизохинолина (6) с 15 мл 10%-ного спиртового раствора NaOH и кипятят с обратным холодильником в течение 2,5 ч. После этого реакционную смесь упаривают наполовину, охлаждают, выпавший осадок отфильтровывают, промывают сначала, небольшим количеством спирта, затем водой до нейтральной реакции. После высушивания осадка получают 0,37 г (75,8%) 3-амино-6,7-диметокси-1-метилизохинолина (13), бесцветные кристаллы, т.пл. 234-235оС (из этанола). Гидрохлорид (13˙ HCl) - желтые кристаллы, т.пл. 146-152оС (из смеси метанол - эфир).
П р и м е р 20. Раствор 0,11 г (0,5 ммоль) 3-амино-6,7-диметокси-1-метилизохинолина (13) в 1 мл уксусного ангидрида нагревают до кипения и кипятят 3 мин. После охлаждения к реакционной массе добавляют 20 мл бензола и отгоняют в вакууме смесь бензола и уксусного ангидрида. Остаток перекристаллизовывают из бензола и получают 0,12 г (91,5%) 3-ацетамино-6,7-диметокси-1-метилизохинолина (14); бесцветные кристаллы, т.пл. 211,5-212оС. Гидрохлорид (14 ˙HCl) - бесцветные кристаллы, т.пл. 228-230оС (из этанола).
П р и м е р 21. Раствор 0,016 г (0,035 ммоль) гидрохлорида 6,7-диметокси-3-(3,4-диметоксифенилацетимидоиламино)-1- фенилизохинолина (8 ˙HCl) в 3 мл воды кипятят с обратным холодильником в течение 5 ч. Отфильтровывают выпавший осадок, перекристаллизовывают из метанола и получают 0,0085 г (53,1% ) 6,7-диметокси-3-(3,4-диметоксифенилацетамино)-1-фенилизохинолина (15), бесцветные кристаллы, т.пл. 169-170оС.
П р и м е р 22. Растворяют при нагревании 0,88 г (2,48 ммоль) 3-амино-6,7-диметокси-1-(3,4-диметоксибензил)изохинолина в 30 мл диоксана, добавляют раствор 0,26 г (2,6 ммоль) янтарного ангидрида в 3 мл диоксана и кипятят смесь с обратным холодильником 30 мин. Реакционную смесь упаривают от диоксана наполовину в вакууме, добавляют 10 мл бензола и отфильтровывают выпавший осадок 6,7-диметокси-1-(3,4-диметоксибензил-3- (3- карбоксипропиониламино)изохинолина (1,1 г или 97,5%, т.пл. 210-212оС).
Растворяют 30 мг (0,066 ммоль) 6,7-диметокси-1-(3,4-диметоксибензил)-3-(3- карбоксипропиониламино)изохинолина в смеси 0,5 мл диметилформамида и 0,04 мл (0,3 ммоль) триэтиламина. К полученному раствору добавляют 0,02 мл (0,2 ммоль) этилхлорформиата и перемешивают при 0оС в течение 30 мин. Затем реакционную смесь разбавляют 2 мл воды, осадок отфильтровывают, промывают 2 мл воды, высушивают и получают 25 мг (86,8%) 6,7-диметокси-1-(3,4-диметоксибензил)-3- сукцинимидоизохинолина (16), бесцветные кристаллы; т.пл. 161-162оС (из смеси диоксан - бензол).
П р и м е р 23. Растворяют 0,093 г (0,37 ммоль) гидрохлорида 3-амино-6,7-диметокси-1-метилизохинолина в 1,5 мл диметилсульфоксида, добавляют 0,2 г карбоната калия и греют на паровой бане до исчезновения желтого окрашивания, затем добавляют 0,25 мл (4,0 ммоль) йодистого метила и греют при 90оС с обратным холодильником 15 мин. После этого добавляют еще 0,2 г карбоната калия и 0,5 мл (8,0 ммоль) йодистого метила и продолжают нагревание в течение 20 мин. После охлаждения обрабатывают смесь серным эфиром три раза порциями по 10 мл. Остаток перекристаллизовывают из 50%-ного водного этанола, подкисленного H1, и получают 0,085 г (60,0%) йодметилата 3-диметиламино-6,7-диметокси-1-метилизохинолина в виде бесцветных кристаллов; т.пл. 225оС (разл.) по литературным данным т.пл. 245оС (разложение).
ПМР-спектр полученного продукта: δ , м.д. (CDCl3/DMCO - D6 (1:1; o-TMC); 2,93 (3H, C) - ACH3; 3,69 (9H, c); N (CH3)3 4,02 (3H, c ); 4,04 (3H, c); OCH3·2; 7,43 (1H, c); 7,48 (1H, c); 8,15 (1H, c); H-4; H-5; H-8. Приведенные данные идентичны описанным в литературе.
В таблице приведены спектральные и физико-химические характеристики полученных соединений.
Таким образом, предложенный способ позволяет увеличить выход целевых продуктов и получить новые производные 3-аминоизохинолина, являющиеся ценными биологически активными веществами.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения анилинового производного или его фармацевтически приемлемой соли | 1982 |
|
SU1316560A3 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА | 1994 |
|
RU2124511C1 |
| Способ получения фенилэтиламинов или их солей | 1978 |
|
SU700061A3 |
| Способ получения производных изохинолина | 1976 |
|
SU718008A3 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И ХИНАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2130934C1 |
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2195454C2 |
| Способ получения фенилэтиламинов или их солей | 1978 |
|
SU688127A3 |
| Способ получения 6-замещенных гексагидроиндазолизохинолинов или их солей | 1982 |
|
SU1189349A3 |
| ТРИАЗОЛО[4,3-А][1,4]-БЕНЗОДИАЗЕПИНЫ И ТИЕНО[3,2-F]-[1,2,4]-ТРИАЗОЛО[4,3-А] [1,4]ДИАЗЕПИНЫ, В СЛУЧАЕ НАЛИЧИЯ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО АСИММЕТРИЧЕСКОГО ЦЕНТРА ИХ ЭНАНТИОМЕРЫ, РАЦЕМАТЫ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ФАКТОР АКТИВАЦИИ ТРОМБОЦИТОВ. | 1992 |
|
RU2094436C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2276140C2 |

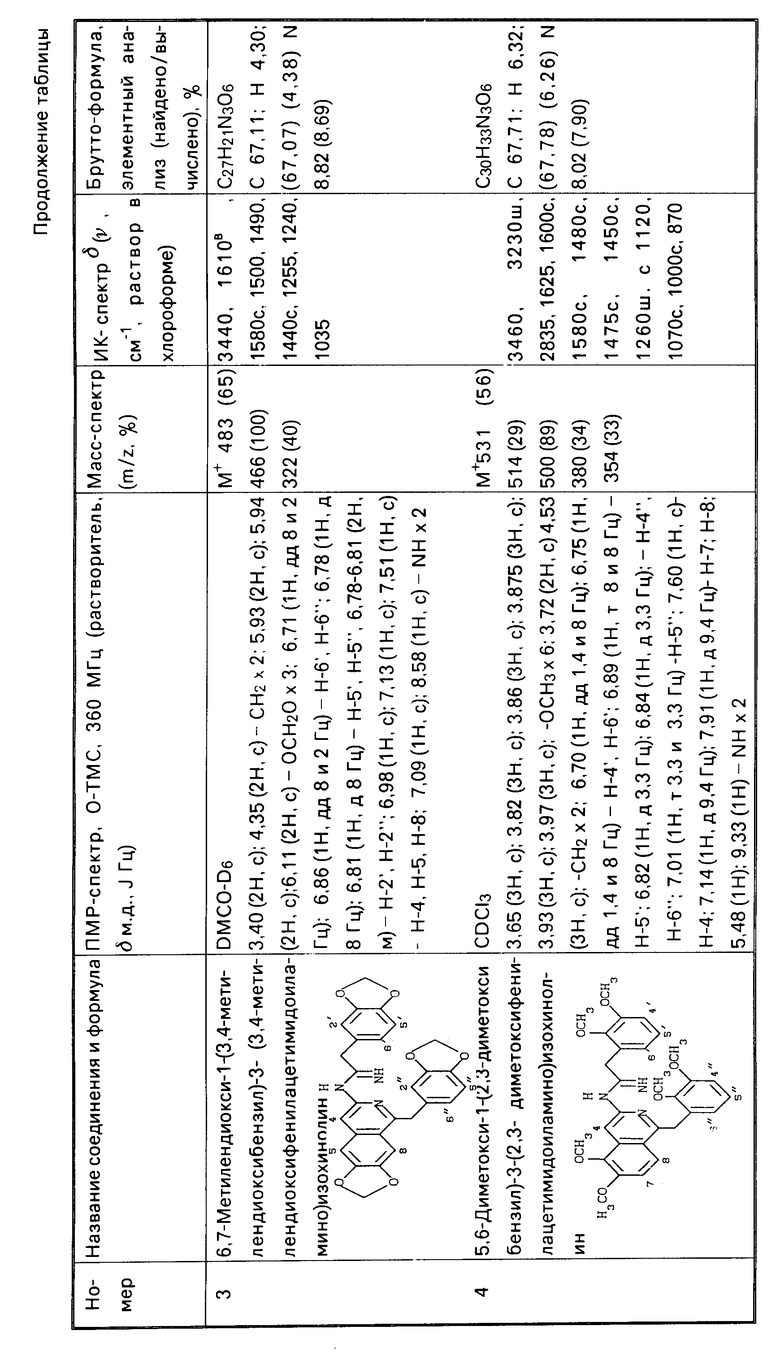


Изобретение относится к гетероциклическим веществам, в частности к способу получения производных 3-аминоизохинолина общей формулы I 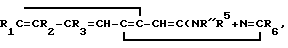 где R1 и R2 - низшая алкоксигруппа и R3= H , либо R1= H, R2 и R3 - низшая алкоксигруппа, либо R2 и R3 вместе образуют метилендиоксигруппу; R4 и R5 - одинаковые или различные, H, низший алкил, ацил, либо один из них представляет группу -C[NH]-R1; R6 и R7 - одинаковые или разные, низший алкил, замещенный или незамещенный фенил или бензил, являющихся физиологически активными веществами, может быть использовано в медицине. Цель - увеличение выхода и расширение ассортимента производных 3-аминоизохинолина. Синтез целевых веществ ведут реакцией циклоди- или тримеризации нитрилов фенилуксусных кислот общей формулы II
где R1 и R2 - низшая алкоксигруппа и R3= H , либо R1= H, R2 и R3 - низшая алкоксигруппа, либо R2 и R3 вместе образуют метилендиоксигруппу; R4 и R5 - одинаковые или различные, H, низший алкил, ацил, либо один из них представляет группу -C[NH]-R1; R6 и R7 - одинаковые или разные, низший алкил, замещенный или незамещенный фенил или бензил, являющихся физиологически активными веществами, может быть использовано в медицине. Цель - увеличение выхода и расширение ассортимента производных 3-аминоизохинолина. Синтез целевых веществ ведут реакцией циклоди- или тримеризации нитрилов фенилуксусных кислот общей формулы II 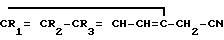 , или их смеси с нитрилами общих формул III и IV R6- C≡ N (III), R7-C≡ N (IV), где R6 и R7 см. выше, под действием HCl с последующим выделением целевого соединения, где R4 и R5 или один из них группа C[=NH]-R7 , или ацилированием и выделением соединения, где NR4R5 - ациламиногруппа, или гидролизом и выделением соединения, где NR4R5 - амино- или ациламиногруппа, или алкилированием и получением соединения, где R4 и R5 - низший алкил. Реакцию предпочтительно проводить в среде апротонного органического или неорганического растворителя (диоксана, хлороформа, хлорокиси фосфора), а необходимые нитрилы и галогеноводород могут быть получены in situ из амидов или альдоксимов под действием хлорокиси фосфора. Способ позволяет повысить выход целевых соединений с 72 до 87,5%. 2 з.п. ф-лы. 1 табл.
, или их смеси с нитрилами общих формул III и IV R6- C≡ N (III), R7-C≡ N (IV), где R6 и R7 см. выше, под действием HCl с последующим выделением целевого соединения, где R4 и R5 или один из них группа C[=NH]-R7 , или ацилированием и выделением соединения, где NR4R5 - ациламиногруппа, или гидролизом и выделением соединения, где NR4R5 - амино- или ациламиногруппа, или алкилированием и получением соединения, где R4 и R5 - низший алкил. Реакцию предпочтительно проводить в среде апротонного органического или неорганического растворителя (диоксана, хлороформа, хлорокиси фосфора), а необходимые нитрилы и галогеноводород могут быть получены in situ из амидов или альдоксимов под действием хлорокиси фосфора. Способ позволяет повысить выход целевых соединений с 72 до 87,5%. 2 з.п. ф-лы. 1 табл.
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-АМИНОИЗОХИНОЛИНА общей формулы 1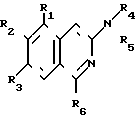
где R1 и R2 - низшая алкоксигруппа;
R3 - атом водорода либо R1 - атом водорода, а R2 и R3 - низшая алкоксигруппа, либо R2 и R3 вместе образуют метилендиоксигруппу;
R4 и R5 имеют одинаковые или разные значения и представляют собой атомы водорода, низшие алкильные, ацильные группы или один из них представляет группу R1
R1
R6 и R7 имеют одинаковые или разные значения и представляют собой низший алкил, замещенный или незамещенный фенил или бензил,
с использованием нитрилов, отличающийся тем, что, с целью увеличения выхода целевого продукта, упрощения способа получения и расширения ассортимента производных 3-аминоизохинолинов нитрилы фенилуксусных кислот общей формулы II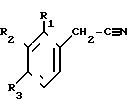
или их смесь с нитрилами общих формул III и IY
Rо-C≡N R7-C≡N
подвергают циклоди- или тримеризации под действием хлористого водорода с выделением целевого продукта, где R4 и R5 представляют собой атомы водорода либо один из них представляет группу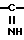 R7
R7
или с последующим ацилированием и получением соединений формулы I, где -NR4R5 представляет собой ациламиногруппу, либо с гидролизом и выделением соединений формулы I, где -NR4R5 представляет собой аминогруппу или ациламиногруппу, либо алкилированием и получением соединений формулы I, где R4 и R5 - низший алкил.
| Liepa A.I | |||
| Australian Yorn of Chem., N 35, 1391-1403 (1982). |
