Изобретение относится к способу получения новых 4-пиридонтпроизводных, обладающих антибактериальным действием.
Целью изобретения является получение новых антибактериально более эффективных 4-пиридон-производных, чем известные структурные аналоги того же действия.
Пример 1. 0,42 г (1,60 ммоль) 7 хлор-6-фтор-1,4-дигидрометиламино-4 оксо-хинолин-3-карбоновой кислоты,
ы
31551248
0,60 г (4,8 ммоль) 8-метил 3,8-диаза- бицикло (3.2.1) октана и 1 мл диметил- сульфоксида (ДМСО) перемешивают в течение 3 ч при 140°С. После охлаждения 5 выпавшие в осадок кристаллы отсасывают, промывают эфиром и кристаллизуют из 2Н соляной кислоты. Получают гид- рохлорид 1 ,4-дигидро-6 фтор-1-метил- ами но-7(8-метил 3 8-диа заби цикло 3, 1 о 2.1 окт-3 ил)-4-оксо-хинолин-3 карбо- новой кислоты в виде желтоватых кристаллов с т.пл. 291 - 294°С (при разложении) Выход 0,2|j г (43 %).
П р и м е р 2. 0,5 г (1,4 ммоль) J5 ангидрида 6,/-дифтор-1 - (4-фторфенил) - -1 ,4-дигидро-4-оксо-хинолин-3 карбо- новой кислоты с дифторборной кислотой, полученной одночасовым нагреванием Ь,7-дифтор-1-(4-фторфенил)-1,4-дигид- 20 ро-4-оксо-хинолин-З-карбоновой кислоты с бортрифторид-этератом в ацетонит- риле, полученном после дистилляции
летучих компонентов, и 0, г (2,0 ммоль) дигидрохлорида 8-метил-3 8-диазабицик-25 ло(3.2.1)-октана перемешивают в 20 мл абсолютного ДМСО с 0,60 г (6,0 ммоль) триэтиламина в течение 2k ч при 40 С. Затем упаривают в вакууме для просушивания и остаток нагревают вместе с зо 3.0 мл 1Н NaOH в течение 1 ч при обратном потоке. После добавки активированного угля фильтруют, фильтрат подкисляют 2Н соляной кислотой. Образовавшийся осадок отфильтровывают и , кристаллизуют из смеси вода - диметил- формамид. Получают 0,28 г дигидрохлорида 6-фтор-1-(4-фторфенил)-1,4-дигид- ро-у-(8-метил-3,8-диазабицикло 3.2.1 -ОКТ 3 ИЛ) -4-ОКСО-ХИНОЛИН-3 КарбОНО- дд
вой кислоты в виде бесцветных кристаллов с т.пл. 280 С (разложение). Выход 0,27 г (40 % теоретического).
П р и м е р 3 Аналогично примеру 2, однако с 0,.19 г (1,68 ммоль) 2-ме- 45 тил-2,5-Диазабицикло 2. гептаном в качестве исходного продукта, получают 0,25 г гидрохлорид 6-фтор-1-(4- -фторфенил) -1, 4 -ди гидр о- }- (5 метил-2,
|5-диазабицикло 2.2.1 гепт-2-ил) -4-ок- 5о to-хинолин-З-карбоновой кислоты в ви-1 де бесцветных кристаллов. Т.пл. 26jj°C (разложение). Выход 0,48 г (56 % теоретического).
П р и м е р 4. К раствору 1,5 г ее (k ммоль) смешанного ангидрида 6, фтор-1-(-фторфенил)-Ч ,4-дигидро- -ок- со-хинолин-3 карбоновой кислоты и дифторборной кцслоты в 10 мл диметилсульфоксида добавляют 0,9 г (,78 ммол 2-(фенилметил)-2,5 диазабицикло 2.2. 1 гептана и 0, г ( ммоль) триэтиламина и перемешивают в течение k ч при комнатной температуре. Затем реакционный раствор разбавляют 150 мл эфира. Образовавшийся осадок отфильтровывают, диспергируют в смеси 60 мл диоксана и 9) мл 1,5 Н раствора NaOH и нагревают в течение 1 ч при 100°С. Раствор фильтруют, чтобы удалить нерастворившийся материал. Затем нейтрализуют концентрированной соляной кислотой. Полученный осадок отфильтровывают, промывают дистиллированной водой и сушат на воздухе. Получают 1,23 г 6-фтор-1-(4-фторфенил)--1,4-ди- гидро-4-оксо-7-(5 Фенилметил-2,5-Ди- азабициклоС2.2.1 гепт-2-ил)хинолин- -3-карбоновой кислоты. Т.пл. 275 - 28l)°C (из ДМСО) при разложении. Выход 1,23 г (63 % теоретического).
Пример5. У,ОУг (2,2 ммоль) 6-фтор-1-(4-фторфенил)-1,4-дигидро-4- -оксо-у-(2-фенилметил-2,5-Диаза-би- цикло 2.2.1 гепт-2-ил)-хинолин-3 кар- боновой кислоты растворяют в 70 мл абсолютного этанола, содержащего 7 г соляной кислоты. Добавляют 1 г катализатора гидрирования (порошкообразный активированный уголь с 10 % палладия), и полученная смесь гидрируется при 15 пси и 60 С в течение 90 мин Катализатор отфильтровываю фильтрат выпаривают из смеси на роторе и вновь помещают в 30 мл 1 Н раствора NaOH. Взвешенный осадок отфильтровывают и водный раствор нейтрализуют добавкой концентрированной соляной кислоты. После добавления 50 мл ди- метилформамида раствор нагревают до 100°С до тех пор пока гелеобраз- ный продукт вновь растворится. После охлаждения выпадает кристаллический осадок 0,68 г 7(2,5-Диазабицикло 2. 2,1 гепт-2-ил)-6-фтор-1-(4-фторфе- нил)-1,4-дигидро-4-оксо 3-хинолинкар- боновой кислоты в качестве моногидрата, т„пл. 290 - 309°С (разложение). Выход: 0,69 г (7,4 % теоретического)
П р и м е р 6. К 1,2 г (3 ммоль) 7(2,5-Диазабицикло 2.2.1 гепт-6-фтор -1-(4-фторфенил)-1,4-дигидро-4-оксо- -хинолин 3 карбоновой кислоты в 10 мл муравьиной кислоты добавляют 2,64 г (30 ммоль) смешанного ангидрида из уксусной и муравьиной кислоты. Смесь перемешивают 6 ч, затем выпаривают и
полученный осадок кристаллизуют из этанола. Получают 0,4 г 7 (5 формил- -2,5-ДиазабициклоГ2.2.1 гепт-2-ил)-6- фтор-1-(4-фторфенил)-1,4-дигидро-4-ок со-хинолин 3 кар(юновой кислоты, т.пл. 27k - 276°С. Выход 0,4 г (31 % теоретического).
П р и м е р / 15,0 г (37,3& ммоль смешанного ангидрида У хлор-б-фтор-(2,4-дифторфенил)-1,4-дигидро-4-ок- со-хинолин-3-карбоновой кислоты и ди- фторборной кислоты, полученной путем нагревания 7 хлор-6-фтор-1-(2,4-ди- фторфенил)-1,4-дигидро-4-оксо-хино лин-3-карбоновой кислоты в 60 %-ном водном растворе фторборной кислоты, 15,36 г (56 ммоль) дигидробромида г 2-метил-2,5 Диазабицикло 2.2.11 гептана и 31 мл (0,224 ммоль) триэтилами- на добавляют к 200 мл сухого диметил- сульфоксида и перемешивают 6 дней при комнатной температуре. Затем реакционную смесь выливают в 800 мл холодной воды. Образовавшийся осадок отфильтровывают, промывают водой и затем нагревают в смеси из 10 мл диок- сана и 150 мл 3 Н- натронной щелочи в течение 1 ч при возвратном потоке. После охлаждения при перемешивании добавляют 500 мл концентрированной соляной кислоты. Дополнительно перемешивают в течение 1 ч, отфильтровывают нерастворившуюся часть и желтый фильтрат экстрагируют еще несколько раз метиленхлоридом. Водную фазу при охлаждении льдом твердым гидроксидом натрия доводят до рИ , после чего проводится экстракция хлороформом.
-
) -
на и 5 мл сухого диметилсульфоксида, 2,73 г 6-фтор-1-(2,4-дифторфенил)-1, 4-дигидро-4-оксо-7-(5 Фенилметил-2, 5-Ana3a6niiHKnof2.2.1 -гепт-2-ил) -3-хи- нолинкарбоновой кислоты. Т.пл. 233 238°С (разложение) после кристаллизации из толуола. Выход 2,73 г (39 % теоретического).
ПримерЗ. 2,57 г (5,08 ммоль) 6-фтор-1-(2,4-дифторфенил)-1,4-дигид- ро-4-оксо-7-(5 фенилметил-2,5 диаза- бицикло 2.2.1Згепт-2-ил)-хинолин-3 карбоновой кислоты растворяют в 170 мл
15 абсолютного этанола, содержащего 18 г соляной кислоты, и замещают 2,1 г 10 %-ного активированного палладия на угле. Гидрируют 2 ч при 15 пси и 60°С. После удаления катализатора вы2о паривают при пониженном давлении. Полученное твердое вещество помещают в 80 мл воды и устанавливают рН 7. Полученную суспензию нагревают 20 мин до 60°С. После охлаждения осадок отфильтровывают. После кристаллизации из диметилформамида получают 1,53 г 7-(2,5-Диазабицикло 2.2.1 гепт-2-ил)- -6-фтор-1 -(2,4-дифторфенил)-1,4-ди- гидро-4-оксо-хинолин-3 карбоновой кислоты в виде гидрата. Т.пл. 198-204 С (разложение). Выход 1,53 г (69,5 % теоретического) .
Пример 10. 0,5 г (1,2 ммоль) 7(2,5 Диазабицикло 2.2.1Згепт-2-ил)- -6-фтор-1-(2,4-дифторфенил)-1,4-ди- гидро-4-оксо-хинолин 3 карбоновой кислоты суспендируют в 20 мл диметилформамида и замещают 3 мл смешанного ангидрида из уксусной и муравьиной кис25
30
35




Изобретение относится к гетероциклическим соединениям, в частности к получению 4-пиридон-производных ф-лы 1, CH=CY-CF=CH-C=C- N(R1) -CH=C-COOX-C(O), где R1 - 2,4-дифторфенил
X - H
Y - основание ф-лы II, III: @ где R2 - H, низший алкил, формил или ацетонил, или их фармацевтически приемлемых аддитивных солей с неорганическими или органическими кислотами или основаниями, или их гидратов. Цель - выявление более активных соединений. Получение ведут реакцией соединения ф-лы 1, где R1 - указано выше
Y - нуклеофильная отщепляемая группа такая, как CL или F
X - H, низший алкил или борный комплекс -B(R4R5), где R4 и R5 - одинаковые и равны F или остаток -O(CO)-низший алкил, с соединением ф-лы III, где R2 - низший алкил или означает защитную аминогруппу, в инертном растворителе таком, как диметил-формамид или диметилсульфоксид, с выделением соединения ф-лы 1а , где R1, X принимают указанные значения, Y1 - радикал ф-лы II, где R2 принимают указанные значения, или в случае, когда X - низший алкил или группа B (R4R5), соединение 1а действием водной щелочью или органическим основанием переводят в соединение 1, где X - H, или в случае, когда в соединении 1а в положении R2 - защитная группа, ее отщепляют, или при необходимости полученное соединение 1, где R2 - H, метилируют, формилируют или замещают бромацетоном, с выделением целевого продукта в свободном виде или в виде фармацевтически приемлемых аддитивных солей с кислотой или основанием, или их гидратами. 1 з.п. ф-лы, 2 табл.
Фазу хлороформа сушат сульфатом нат- 4Q лот. После 90 мин перемешивания при рия и хлороформ отфильтровывают в- ва- комнатной температуре растворитель кууме. Полученное таким образом темно- отдистиллировывают при пониженном дав- коричневое масло дает после кристал-лении. Маслянистый осадок после пере- лизации из 450 мл этанола 3,15 гмешивания с эфиром становится твер- 6-фтор-1-(2,4-дифторфенил)-1,4-дигид- 4g дым и может быть отфильтрован. Крис- -,.(,-. ,-с. , п таллизация из смеси 20 мл диметилформамида и 5 мл лигроина дает 0,24 г 7(5 Формил-2,5-Диазабицикло 2.2.1 гепт-2-ил)-6-фтор-1-(2,4-дифторфенил)- -1,4-дигидро-4-оксо-хинолин-3-карбо- новой кислоты. Т.пл. 252 - 258°С
(5 метил-2,5-Диазабицикло 2.2.1J гепт-2-ил)-4-оксо-хинолин-3 кар6оно- вой кислоты в качестве гидрата. Т.пл. 215 220°С (разложение). Выход 3,15 г (18,8% теоретического).
Примерв. Аналогично примеру 6 получают при применении г (13,85 ммоль) смешанного ангидрида 7 хлор-6-фтор-1 -(2,4-дифторфенил)-1, 4-дигидро-4-оксо-хинолин-3 карбоновой кислоты и дифторборной кислоты, г (16,62 ммоль) 2-(фенилметил)-2, азабицикло .2.1 гептан дигидробро- мида, 11,58 мл (83 ммоль) триэтилами50
(разложение). Выход 0,24 г (45,3 % теоретического).
П ри мер 11. К раствору 0,6 г 5 (1,5 ммоль) 7(2,5-Диазабицикло 2.2. 13-гепт-2-ил)-6-фтор-1-(4-фторфенил)- -1,4-дигидро-4-оксо-хинолин-3 карбо- новой кислоты в 60 мл диметилформамида прибавляют 0,23 г (1,65 ммоль)
4Q 4g
50
(разложение). Выход 0,24 г (45,3 % теоретического).
П ри мер 11. К раствору 0,6 г (1,5 ммоль) 7(2,5-Диазабицикло 2.2. 13-гепт-2-ил)-6-фтор-1-(4-фторфенил)- -1,4-дигидро-4-оксо-хинолин-3 карбо- новой кислоты в 60 мл диметилформамида прибавляют 0,23 г (1,65 ммоль)
карбоната калия и 0,82 г (6 ммоль) бромацетона и реакционную смесь перемешивают 6 ч при комнатной температуре. Затем избыточный бромацетон и ди- метилформамид отделяют при пониженном давлении, остаток помещают в 1 Н NaOH, фильтруют и доводят рН фильтра 4Н соляной кислотой до 6,5. Полученный оста ток отфильтровывают, помещают в метанол и хроматографируют через силикагель (растворитель бензол:ме- танол 5:4). Получают 0,20 г 7-( тонил-2,5-Диазабицикло 2.2.13-гепт-2- -ил)-6-фтор-1-(4-фторфенил)-1,4-ди- гидро-4-оксо-хинолин-З-карбоновой кислоты. Т.пл. 20U - 205°С (разложение). Выход 0,20 г (29,4 % теоретического) .
П р и м е р 12. 188 мг (0,434 ммоль) моногидрата 7(2,5 Диазабицикло 2.2. О-гепт-2-ил)-6-фтор-1(2,4-дифторфе- нил)-1,4-дигидро-4-оксо-хинолин-3-кар боновой кислоты (пример 9) суспендируют в 20 мл сухого диметилформамида, разлагают с 96 мг (0,85 ммоль) безводного бикарбоната калия и ,92 мг (0,48 ммоль) 4 бром-метил-5 метил-1, З-Диоксолен-2-она и перемешивают 10 ч при комнатной температуре. Затем вы-
Strepc.
pneumonia1
Aeinetobaccer
calcoauecicus
Baeteroidfs
Јragilis
Escherichia .
coli
Branharaella
cacarrhalis
Klebsiella
spp,
Scaphylococcus
aureus
Scaphylococcus
epidermidis
Scaphylococcus
spp.
Enterobacuer
0
ливают на смесь из 150 мл этилацета- та и 50 мл воды и разделяют фазы. Органическую фазу промывают пять раз ho 30 мл воды, сушат над сульфатом натрия, фильтруют и сгущают при пониженном давлении при комнатной температуре. Перекристаллизация из этилацетата дает 63 мг 1-(2,4-дифторфенил)-6-фтор- -1,4-дигидро-7-(5 С(5-метил-2-оксо-1, З-дикосолен-4-ил)метилJ-2,5 Диазаби- цикло 2.2 13гепт-2-ил }-4-оксо хино- лин-3-карбоновой кислоты. Т.пл. 133 138 С (разложение). Выход 63 мг (27,5 % теоретического).
Определение антибактериальных свойств в пробирке.
Антибактериальные свойства соединения примера 7 (соединение А) определены в МНК-тесте (минимальный тест концентрации замедления) по методу агар-разбавления по отношению к различным грамположительным и анаэробным штаммам бактерий и сравнены со структурным аналогом - 6-фтор-1-(4-фторфе- нил)-1,4-дигидро-7-(5-метил-2,5-Диаза- биц|ф«ло- 2.2.1 гепт-2-ил) -4-оксо-3 хи- нолин карбоновой кислоты (соединение В). Результаты помещены в табл. 1.
Таблица 1
0,25 1,OS 0,06 0,12 2,00 4,00 0,03 0,12
меньше 0,015 0,03
Л,), 06 0,06 0,12 0,12 0,12 0,25 0,25 0,50 0,12
0,50 1,00 0,25 0,50 2,00 4,00 0,06 0,12
о,оз о,оз
0,06 0,12 0,12 0,12 0,25 0,25 9,25 0,50 0,12
0,50 2,00 2,00 4,00 2,00 4,00 0,06 0,12
0,03 0,06 0,12 0,25 0,12 0,25 0,25 0,25 0,50 0,50 0,25
Определение антибактериальных свойств in vitro.
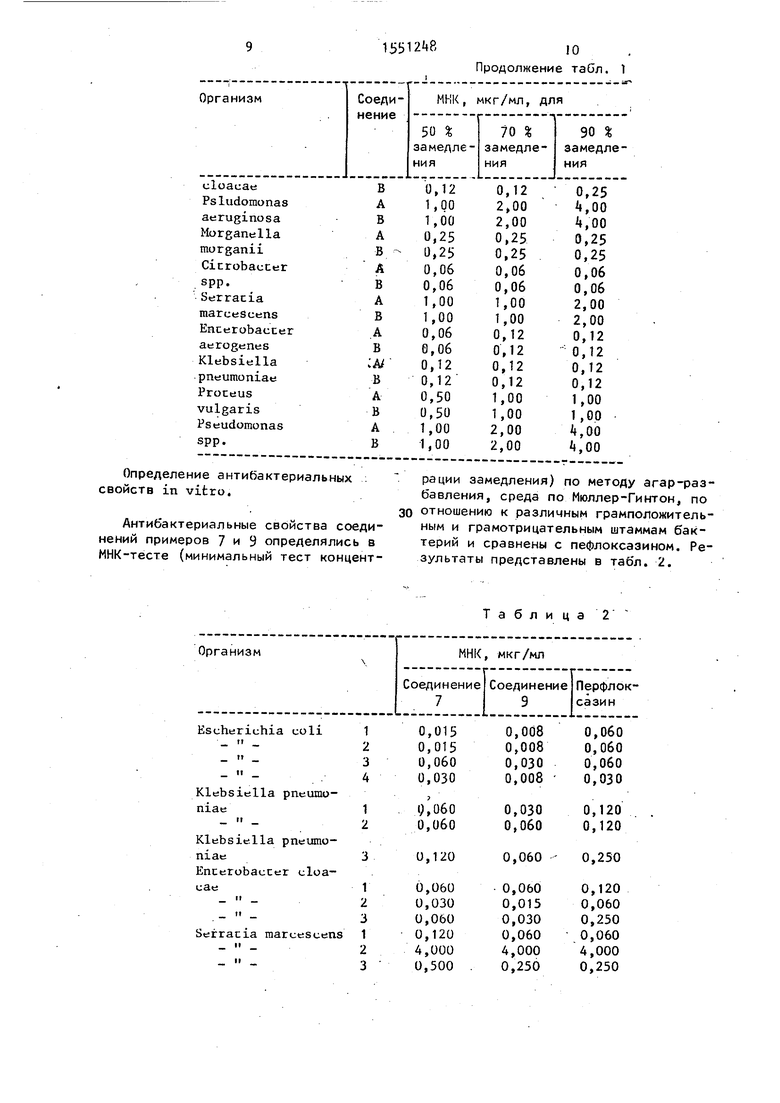
Антибактериальные свойства соединений примеров 7 и 9 определялись в МНК-тесте (минимальный тест концентEscherichia coli
Klebsiella pneumoniae
n
Klbbsiella pneumo- niae
Encerobaccer cloacae
к
it
Serracia marcescens ii
15512Wю
Продолжение табл. 1
рации замедления) по методу агар-разбавления, среда по Мюллер-Гинтон, по отношению к различным грамположитель- ным и грамотрицательным штаммам бактерий и сравнены с пефлоксазином. Результаты представлены в табл. 2.
Таблица 2
0,120
0,060
0,250
11
$5 2kQ12
i ,Продолжение таСл. 2
Как видно из габл. 1 и 2, предлагаемые соединения превосходят известные структурные аналоги по антибактериальному действию против различных штаммов бактерий.
Испытания на крысах соединения по примеру 7 показали, что соединения от носятся к категории малотоксичных.
Формула изобретения
F JLCOOX
N
i
Ri
где К,- 2,4-дифторфенил; X - водород; Y - основание общей формулы II
Rf $Nгде Ra - водород, низший алкил, фор- 55где кг низший алкил или защитная
мил или ацетонил,аминогруппа, в инертном растворителе,
или их.Фармацевтически приемлемыхтаком как-диметилформамид или димеаддитивных солей с неорганическимитилсульфоксид, с вь елением соединеили органическими кислотами или осно-Ния of-лцей формулы 1а
ваниями, или их гидратов, отличающийся тем, что соединение общей формулы III
О
ггЛСООКз i/4Ai
i
где К L
R.
имеет указанное значение; нуклеофильная отщепляемая группа, такая как хлор или фтор;
водород, низший алкил или борный комплекс - B(R4R5), где R и R5 являются одинаковыми и представляют собой фтор или остаток - 0(СО) - низший алкил, подвергают взаимодействию с соединением общей формулы IV
)N-H
где К1, RJ имеют указанные значения; Y -радикал общей формулы IVa
где R 2 имеет указанные значения; или в случае, когда RJ- низший ал- 15 кил или группа В (R4R), соединение 1а действием водной щелочью или органическим основанием переводят в соединение I, где X - водород;
10
15 2481
или в случае, когда в соединении 1а в положении R2 - защитная группа, ее отщепляют;
или при необходимости полученное соединение формулы I, где R2 - водород, метилируют, формилируют или замещают бромацетоном с выделением целевого продукта в свободном виде или в виде фармацевтически приемлемых аддитивных солей с кислотой или основанием, или их гидратами.

| Способ получения замещенных диазабициклоалкилхинолон карбоновых кислот с мостиковой связью или их фармацевтически приемлемых аддитивных солей | 1986 |
|
SU1482531A3 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
