1
Изобретение относится к получению новых гетероциклических соединений, в частности к получению производных .тиeнo-(2,3-d)-имидaзoлa общей формулы
К4
RZ
II н -„ ТОГ
K.
« (0)„ Re
где R, - водород или ацетрт;
R
R
R6и
и
водород;
независимо друг от друга
водород или С -С4-алкил;
водород или С -С -алкоксил
О или 1,
обладающих свойством понижать желудочную секрецию, т.е. антисекреторным действием.
Целью изобретения является создание на основе известных методов новых соединений - производных тиено-(2,3СХ)
CrJ
1456018
-6)-им1адазола, обладающих высокой антисекреторной, противоязвенной активностью и низкой токсичностью.
П р и м е р 1. 2(2-Пиридил)-метил- тиo-ЗH-тиeнo(2,3-d)имидaзoл (1, где R, по Rj - атомы водорода, ).
1,20 г (7,68 ммоль) 1,3-дигидро- тиено(2,3-й)имидазол-2-тиона(11,где R и R. представляют собой атомы водо- о рода) и 1,13 г (6,91 ммоль) гидрохлорида 2-хлорметилпиридина растворяют в 15 мл метилового спирта, после чего к приготовленному раствору при перемешивании прибавляют по каплям в is течение 20 мин 7,4 мл (14,8 ммоль) 2 н.раствора гидроокиси натрия. Затем реакционную смесь дополнительно перемешивают в течение 1,5 ч при комнатно перемешивают в течение 4 ч при комнатной температуре. Непосредственно- после этого реакционную смесь упаривают в вакууме, остаток растворяют в 50 мл воды, после чего посредством прибавления по каплям ледяной уксусной кислоты раствор подкисляют до рН 4,5. Образовавшийся осадок растворяют в 80 мл хлороформа, смесь энергично встряхивают и для разделения фаз фильтруют через вспомогательный филь -рующий материал. Фазы отделяют друг от друга. Водную фазу еще три раза экстрагируют хлороформом, применяя каждый раз по 40 мл последнего. Объединенные органические фазы сушат над сернокислым натрием и упаривают. Неочищенный продукт (5,8 г 82,2% от теоретически ной температуре и непосредственно по- 20 рассчитанного значения) растворяют
в 400 мл ацетонитрила при нагревании до температуры кипения, производят фильтрование в горячем состоянии в присутствии активированного угля, раствор упаривают до 150 мл и выдерживают с целью кристаллизации образовавшегося продукта.
еле этого упаривают на роторном испарителе. Остаток распределяют меяоду 60 МП воды и 80 мл хлористого метилена. Для разделения фаз сначала производят фильтрование через вспомогательный фильтрующий материал, причем затем водную фазу еще три раза экстрагируют хлористым метиленом, применяя каждьй раз по 70 мл последнего. Объединенные органические фазы сушат над сернокислым натрием и упаривают. , Неочищенный продукт (1,52 г окрашенного в коричневый цвет кристалли25
Выход: 4,34 г окрашенного в коричневый цвет кристаллического вещест- ва (61,8% от теоретически рассчитанного значения).
Т.ПЛ. ла). .
140-141 С (из ацетонитри- 3. 5-Ацетш1-2- (4Т.ПЛ. ла). .
Прим
140-141 С (из ацетонитри- 3. 5-Ацетш1-2- (4е р
ческого вещества, 92,4% от теоретиче- .. .. .. .
ски рассчитанного значения) перекрис- 35 -метокси-3,5-диметил-2-пиридил)-ме- таллизовывают из ацетонитрила при тилтио -ЗН-тиено(2,3-а)имидазол добавлении активированного угля. (j где R - СОСН, , R,
Выход;1,10 г бесцветного кристалли- водорода, RjH Ry -метильные радика- ческого вещества (64,3% от теорети- j R метоксильный остаток, ). чески рассчитанного значения). 40
атомы
5,64 г (28,4 ммоль) 5-ацетил- -1,3-дигидротиено(2,3-d)-имидазол- -2-тиона и 6,00 г (27,0 ммоль) гидрохлорида 2-хлорметил-4-метокси-3,5- -диметилпиридина суспендируют в
R 1 } у 250 мл метилового спирта, после чего к приготовленной суспензии при перемешивании прибавляют по каплям в течение 15 мин 28 мл 2 н.раствора гидроокиси натрия (56,0 ммоль) приТ.шт. 155-156 С (из ацетонитрила) .
Пример 2. 2- (4-Метокси-2- -пиридил)-метилтиоЗ-ЗН-тиено (2,3-d) имидазол (I, где атомы водорода; таток, ).
4,65 г (29,76 ммоль) 1,3-дигидротиено (2, 3-d) имидазол-2-тиона и 4,91 г
R - метоксильный ос(25,30 ммоль) гидрохлорида 2-хлорме- gg чем температура повышается при этом
тил-4-метоксипиридина суспендируют в 60 мл метилового спирта, после чего к приготовленной суспензии при перемешивании медленно прибавляют по каплям в течение 25 мин 27,6 мл : (55,2 ммоль) 2 н.раствора гидроокиси калия. При этом температура повьш1ает- ся до 32 С. После завершения прибав-с ления реакционную массу дополнительдо 27 С. Уяе через короткий промежуток времени из образовавшегося раствора начинает вьщеляться в осадок кристаллический продукт. 55 Реакционную смесь дополнительно перемешивают в течение 4 ч при комнатной температуре и непосредственно после этого полностью упаривают. Полученный остаток растворяют в
но перемешивают в течение 4 ч при комнатной температуре. Непосредственно- после этого реакционную смесь упаривают в вакууме, остаток растворяют в 50 мл воды, после чего посредством прибавления по каплям ледяной уксусной кислоты раствор подкисляют до рН 4,5. Образовавшийся осадок растворяют в 80 мл хлороформа, смесь энергично встряхивают и для разделения фаз фильтруют через вспомогательный филь -рующий материал Фазы отделяют друг от друга. Водную фазу еще три раза экстрагируют хлороформом, применяя каждый раз по 40 мл последнего. Объединенные органические фазы сушат над сернокислым натрием и упаривают. Неочищенный проВыход: 4,34 г окрашенного в коричневый цвет кристаллического вещест- ва (61,8% от теоретически рассчитанного значения).
Т.ПЛ. ла). .
Прим
140-141 С (из ацетонитри- 3. 5-Ацетш1-2- (4е р
.. .. .. .
-метокси-3,5-диметил-2-пиридил)-ме- тилтио -ЗН-тиено(2,3-а)имидазол (j где R - СОСН, , R,
)-ме- л
атомы
до 27 С. Уяе через короткий промежуток времени из образовавшегося раствора начинает вьщеляться в осадок кристаллический продукт. 55 Реакционную смесь дополнительно перемешивают в течение 4 ч при комнатной температуре и непосредственно после этого полностью упаривают. Полученный остаток растворяют в
200 мл воды, а затем посредством прибавления приблизительно 2 мл ледяной уксусной кислоты значение рИ раствора доводят до 4,5. Образовавшую- ,ся суспензию четыре раза экстрагируют хлороформом, причем суммарно применяют 250 мл последнего, объединенные органические фазы сушат над сернокислым натрием при добавлении активированного угля и затем упаривают
Полученное в виде остатка и окрашенное в темный цвет маслообразное вещество кристаллизуют из 80 мл аце- тонитрила, охлаждают, после чего оса док отфильтровывают. Продукт трижды промывают, после чего осадок отфильтровывают. Продукт трижды промывают холодным ацетонитрилом и сушат в вакууме при 40°С.
Выход: 8,92 г слегка окрашенного в желтоватый цвет кристаллического вещества (95,0% от теоретически рассчитанного значения).
Т.пл. 177-180°С (из ацетонитри- ла) .
Пример 4. 5-Ацетил-2 (Л-ме токси-2-пиридил)-метилтио -ЗН-тиено) (2,3-d)имидaзoл (1,где R - СОСН,., Rj,Rj, Ry и Rfe - атомы водорода, метоксильный остаток, ).
1,30 г (6,567 ммоль) 5-ацетил- -1,3-дигидротиено(2,3-ё)-имидазол-2- -тиона и 1,08 г (5,565 ммоль) гидрохлорида 2-хлорметил-4-метоксипириди на суспендируют в 15 мл метилового спирта, после чего к приготовленной суспензии при перемешивании прибавляют по каплям в течение 10 мин .. 6,2 мл 2 к. раствора гидроокиси натрия (12,4 ммоль), причем температура повьшается до 27 С. После этого реакционную смесь дополнительно перемешивают в течение 1,5ч при комнатной .температуре.
Непосредственно после этого реакционную смесь полностью упаривают, остаток растворяют в 50 мл воды, после чего раствор подкисляют до рН 4 посредством прибавления приблизительно 1 мл ледяной уксусной кислоты. Затем раствор трижды экстрагируют хлороформом, причем каждый раз применяют по 40 мл последнего, объединенные органические фазь сушат над сернокислым натрием и упаривают.
Неочищенньй продукт (1,52 г окрашенного в коричневый цвет маслообразного вещества) растирают с ацетонитрилом, а затем перекристаллизовывают
20
25
.j.
456П186
из ацетонитрила при добавлении активированного угля.
Выход: 0,76 г бесцветного кристал- лического вещества (42,8% от теоретически рассчитанного значения).
Т.пл. 192-194 е (из ацетонитрила).
Пример 5. 5-Ацетил-2-(2-пи- g ридил)-метилтио-ЗН-тиено(2,3-d)ими- дазол (I, где СОСН, R - R - атомы водорода, ).
1,50 г (7,565 ммоль) 5-ацетил-1,3- дигидротиено(2,3-d)-имидаз ол-2-тиона и 1,12 г (6,809 iмoль) гидрохлорида 2-хлорметилпиридина суспендируют в 60 мл метилового спирта, после чего к приготовленной суспензии при перемешивании прибавляют по каплям в течение 8 мин 7,2 мл 2 н. раствора гидроокиси натрия (14,4 ммоль), причем температура повьш1ается до 26 С. После этого реакционную смесь дополнит- тельно перемешивают в течение 2 ч при комнатной температуре.
Образовавшийся прозрачный раствор полностью упаривают, остаток раство- 1ряют в 80 мл воды, а затем получен- ньй раствор ПОДКИС.ЛЯЮТ до рН 4,5 посредством прибавления 1,2 мл ледяной уксусной кислоты. Раствор три раза экстрагируют, причем для этой цели суммарно применяли 150 мл хлороформа, органическую фазу сушат над сернокислым натрием и упаривают. Полу- 35 ченное в виде остатка кристаллическое вещество перекристаллизовывают из ацетонитрила.
Выход: 1,85 г бесцветного кристаллического вещества (84,5% от теоре- 40 тически рассчитанного значения).
Т.пл. 171-173 С (из ацетонитрила). Пример 6. 2-(2-Пиридил)- метилсульфинил-ЗН-тиено(2,3-d)ими- дазол (I, где R, - Rg -атомы водоро- 45 да, ).
0,45 г (1,82 ммоль) 2-(2-пиридил}- метилтио-ЗН-тиено(2,3-d)имидазола растворяют в 10 мл хлороформа, после чего к приготовленному раствору 50 при температуре, лежащей в интервале между -11 и , прибавляют по каплям при перемешивании раствор ,0,37 г (1,82 ммоль) 85%-ной 3-хлорнадбензойной кислоты в 5 Nm хлоро- 55 форма. Реакционную смесь перемешивают в течение 30 мин при температуре ниже О С, раствор разбавляют не- больпгим количеством хлористого метилена и два раза промывают насыщенным
30
7145
раствором кислого углекислого натрия, причем каждый раз применяют по 5 мл указанного раствора.
Водную фазу два раза экстрагиру- ют хлористым метиленом, причем каждый раз применяют по 5 мл последнего, объединенные органические растворы сушат над сернокислым натрием и упаривают.
Образовавшийся после растирания с ацетонитрилом кристаллический остаток (0,42 г, 87,7% от теоретически рассчитанного значения) перекрис- таллизовьшают из ацетонитрила при добавлении активированного угля.
Выход: 0,33 г бесцветного кристаллического вещества (68,9% от теоретически рассчитанного значения).
Т.пл. ISI-ISZ C (разл.) (из ацетонитрила) .
Пример 7. 2-(4-Мвтокси- -2-пиридил)-метилсульфинил1-ЗН-тиено (2,3-d)имидaзoл.(I, где R, Rj, Ry и Rg - атомы водорода, представляет собой метоксильный остаток, п 1).
4,30 г (15,50 ммоль) 2-(4-меток- си-2-пиридил) -метилтиб -ЗН-тиено(2,3 -ё)имидазола растворяют при комнатной температуре в 90 мл хлороформа, после чего приготовленный раствор охлаждают до -10°С. К охлажденному раствору при перемешивании и температуре, лежащей в интервале между и -8°С, прибавляют по каплям в течение 30 мин раствор 3,08 г (15,19 ммоль) 85%-ной 3-хлорнадбен- зойной кислоты в 35 мл хлороформа.. После этого реакционную смесь дополнительно перемешивают в течение 15 мин при С.
Образовавшийся раствор два раза экстрагируют насьш;е шым раствором кислого углекислого натрия, причем суммарно применяют 30 мл указанного раствора, водную фазу трижды экстрагируют хлороформом, применяя каждый раз по 20 мл последнего, после чего объединенные органические растворы сушат над сернокислым натрием и упа- ривают.
Неочищенный продукт (5,2 г окрашенного в красный цвет маслообразного вещества) растирают с неболыпим количеством ацетонитрила и образова шийся коричневый кристаллический прдукт растворяют при 80 С в 22 мл ди метилформамида. Раствор смешивают с активированным углем, производят
88
фильтрование в горячем состоянии, после чего фильтрат вводят в 130 мл нагретого до 60°С ацетонитрила. Смесь медленно охл аждают, а затем производят кристаллизацию в холодильнике в течение ночи.
Выход: 4,03 г бесцветного кристаллического вещества (88,6% от теоретически рассчитанного значения).
Т.пл. 157-159 С (разл.) (из ацетонитрила) .
Пример 8. 5-Ацетил-2-С(- метокси-3,5-диметил-2-пиридил)-метил- сульфинилЗ-ЗН-тиено(2,3-d)имидазол (I, где R, - ацетильный остаток, RZ и R - атомы водорода, R, и Rj. - ме- тильные радикалы, R4 метоксильный остаток, ).
4,5 г (12,95 ммоль) 5-ацетил-2- (4-метокси-3,5-диметил-2-пиридил)- метилтио -ЗНг-тиено( 2,3-d) имидазола почти полностью растворяют при комнатной температуре в 120 мл хлороформа, после чего приготовленный раствор охлаждают до . При температуре, лежащей в интервале между -8 С и , к охлажденному раствору в течение 25 мин прибавляют по капля при перемешивании раствор 2,58 г (12,69 ммоль) 85%-ной 3-хлорнадбен- зойной кислоты в 40 мл хлороформа.
Реакционную смесь затем дополнительно перемешивают в течение 10 мин при -100°С,после чего образовавшийся совершенно прозрачный раствор три раза экстраги луют насыщенным раствором кислого углекислого натрия, причем суммарно применяют 60 мл такого раствора. Водную фазу три раза промывают небольшим количеством хлорофор-г ма, объединенные органические растворы сушат над сернокислым натрием и упаривают.
Полученный маслообразный остаток (приблизительно 5 г окрашенного в красный цвет маслообразного вещества) растворяют при 80 С в 50 мл ди- метилформамида, после чего раствор фильтруют с применением активированного угля. Фильтрат вводят в 300 мл нагретого до ацетонитрила, медленно производят охлаждение, после чего в течение ночи в холодильнике происходит кристаллизация. Образовавшийся продукт отфильтровывают и три раза промывают его ацетонитрилом.
9U
Выход: 3,30 г бесцветного кристаллического вещества (70,1% от теоретически рассчитанного значения).
Т.пл. 190-191 с (из смеси диме- тилформамида и ацетонитрила).
Пример 9. 5-Ацетил-2- (4- -метокси-2-пиридил)-метилсульфинил - -ЗH-тиeнo(2,3-d)имидaзoл (I, где R, - ацетильный остаток, R, ЕЗ 5 RU атомы водорода, 4 токсильньт остаток, ),
0,53 г (1,628 ммоль) 5-ацетил-2-. ((4-метокси-2-пиридил) -метилтио - -ЗН-тиено(2,3-d)ш-овдаэола суспендируют в 15 мл хлороформа, после чего при температуре, лежащей в интервале между -12°С и , к приготовленной суспензии при перемешивании прибавляют по каплям в течение 7 мин раствор 0,33 г (1,628 ммоль) 85%-ной 3-хлорнадбензойной кислоты в 6 мл хлороформа, в результате чего образуется прозрачный раствор. Затем его дополнительно перемешивают в течение 10 мин при -10 С. Непосредственно после этого полученный раствор два раза экстрагируют насыщенным раствором кислого углекислого натрия, причем каждый раз применяют по 6 мл этого раствора, водную фазу дважды экстрагируют хлороформом, причем каждьй раз применяют по 5 мл последнего, объединенные органические растворы сушат над сернокислым натрием и упаривают. Неочищенный маслообразный продукт neper кристаллизовывают из ацетонитрила при добавлении активированного угля.
Выход: 0,37 г бесцЕвтиого кристаллического вещества (67,8% от теоретически рассчитанного значения).
Т.пл. 159-1б2°С (разл.) (из ацетонитрила).
Пример 10. 5-Ацетил-2- (4- -метокси-2-пиридил)-метилсульфинил |- -ЗН-тиено(2,3-d)имидaзoл (1,где R, - ацетильный остаток, Rj, R и Rg атомы водорода, R - метоксильный остаток, ).
0,53 г (1,628 ммоль) 5-ацетил-2 . (4-мвтокси-2-пиридш1) -метилтио} -ЗН- -тиено(2,3-d)имидaзoл растворяют в 15 мл ледяной уксусной кислоты,пос- ле чего приготовленный раствор при температуре от 5 до 10 С медленно смешивают со 181 мл (1,628 мммоль) перекиси водорода, примененной в ви- де 30%-ного раствора, в результате чего образуется прозрачный раствор.
1810
Этот раствор дополнительно перемешивают в течение 20 мин при комнатной температуре. Непосредственно после .этого раствор разбавляют прибавлением 100 мл воды, три раза производят экстрагирование хлороформом, причем каждый раз применяют по 20 мл последнего, и объединенные органические растворы промывают до нейтральной реакции раствором кислого углекислого натрия, сушат над сернокислым натрием и упаривают. Неочищенный маслообразный продукт перекристаллизовывают из ацетонитрила при добавлении акти-t вированного угля.
Выход: 0,28 г бесцветного кристаллического вещества (61,5% от теоретически рассчитанного значения) .
Т.пл. 159-1б2 с (разл.Хиз ацетонитрила) .
П р и м е р 11. 5-Ацетил-2-(2-пи- ридил)-метилсульфонил-ЗН-тиено(2,3- -d)ими ;aзoл (1, где Rf - ацетильный
остаток, Rjno R - атомы водорода,
t
) ,
1,0 г (3,46 ммоль) 5-ацетил-2- -(2-пиридил)-метилтио-ЗН-тиено(2,3- -d)имидaзoлa почти полностью растворяют при комнатной температуре в 15 мл хлороформа, после чего приготовленный раствор охлаждают до -12 С. При температуре, лежащей между -14 С и , к раствору прибавляют по каплям в течение 10 мин раствор 0,69 г (3,39 ммоль) 95%-ной 3-хлорнадбензойной кислоты в 10 мл хлороформа.
Затем реакционную смесь дополнит.; тельно перемешивают в течение 10 мин
при -10 С, причем в результате образуется совершенно прозрачньЕ раствор, который разбавляют небольшим количеством хлороформа, после чего дважды производят экстрагирование насьш5ённым раствором кислого углекислого натрия, применяя каждый раз п6 7 мл указанного раствора. Органический раствор сушат над сернокислым натрием и упаривают. Полученный остаток
кристаллизуют из ацетонитрила, а затем перекристаллизовывают из ацетонитрила при добавлении активированного угля.
Выход: 0,78 г бесцветного кристаллического вещества (73,9% от теоретически рассчитанного значения).
Т.пл. 194-197°С (из ацетонитрила) .
11145601
Пример 12. 2-(4-Метокси- 3,5-диметнл-2-пиридил)-метилтио - ЗН-тиено(2,3-d)имидазол.
10,0 г (64,0 ммоль) 1,3-дигидро- иено(2,3-6)-имидазол-2-тиона и 95 г (44,8 ммоль) гидрохлорида -хлорметил-4-метокси-3,5-диметш1- иридина суспендируют в 120 мл метиового спирта, после чего к.приго- ,д овленной суспензии медленно прибавяют по каплям 2 и. раствор гидроокии натрия (0,12 молб),
Смесь перемешивают в течение 4 ч при комнатной температуре. Поскольку д е было достигнуто полное прекращение, добавляют еще 0,50 г (2,24 ммоль) 2-хлорметил-4-метокси-3,5-диметш.пи- ридингидрохлорида и 1,5 мл 2 и. раствора NaOH. Затем реакционную смесь Q выпаривают в вакууме. Полученньй остаток .растворяют в 100 мл воды, а затем посредством прибавления ледяной уксусной кислоты значение рН раствора доводят до 4,5. Образовав- 35 шийся осадок растворяют в 160 мл хлороформа, смесь сильно встряхивают и отжимают, органическую фазу отделяют, водную фазу трижды промывают по 100 мл хлороформа. Объединен- ные органические фазы сушат над сернокислым натрием при добавлении активированного угля и затем упаривают. Получают 14,8 г коричневого сырого продукта. Выкристаллизовывают из ацётонитрила.
Выход: 9,94 г бесцветных кристаллов (69,2% от теорет.).
Т пл. 134°С (ацетонитрил). Пример 13. 2-(4-Метокси- -3,5-диметил-2-пиридил )етилсуль- финил -ЗН-тиено(2,3-d)имидазол.
9,40 г (30,8 ммоль) 2-(4-меток- си-3,5-диметил-2-пиридил)-метилтир - -ЗН-тиено(2,3-d)имидазола раство- ряют при комнатной температуре в 5 180 мл хлороформа, после чего приго- товленный раствор охлаждают до -10 С. К охлажденному раствору в течение 30 мин прибавляют по каплям при пе- ремешивании раствор 5,20 г (30,2ммольр 85%-ной 3-хлорнадбензойной кислоты в 80 мл хлороформа.
Реакционную смесь затем дополнительно перемешивают в течение 15 мин при -10°С. Поскольку превращение бы- 55 ло неполным, добавляют еще 0,08 г (0,40 ммоль) 3-х.т;орнадбензойной кислоты (85%). Через 15 мин двсяжды встряхивают с насыщенным раствором
ки су во 45 ни ки ро
во ря в ри ак вы л
5
кислого углекислого натрия, причем суммарно применяют 60 мл такого раствора. Водную фазу три раза промывают 45 мл хлороформа, объединенные органические растворы сушат над сернокислым натрием, обрабатывают активированным углем, фильтруют и упаривают
Получают 10,46 г светло-коричневого сырого продукта, который растворяют в 44 мл ДМФА при и вводят в 260 мл нагретого до 70 с ацётонитрила. Б горячем виде обрабатывают активированным углем, фильтруют и выкристаллизовывают в холодильнике. Выход: 6,7 г бесцветных кристаллов (69,0% от теорет,).
Т.пл. 175°С с разложением (СН2.0Н/ДМФА).
Для изучения фармакологических свойств применяют следующий метод исследования.
Соединение растворяют в 20%-ном диметилсульфоксиде, после чего 8 бодрствующим самкам крыс (штамм Charles River CD) в возрастающих дозах 125, 250 и 500 мкмоль/кг вводят через рот единый дозированный объем 20 мл/кг. Через 60 мин в животных вводят привратниковые лигатуры. Через 4 ч после введения при- вратниковой лигатуры в желудочном объеме животных определяют концентрацию кислоты и общую кислотность. Группы контрольных животных получают диметилсульфоксид в количестве 20 мл/кг, дистиллированную воду в количестве 20 мл/кг.
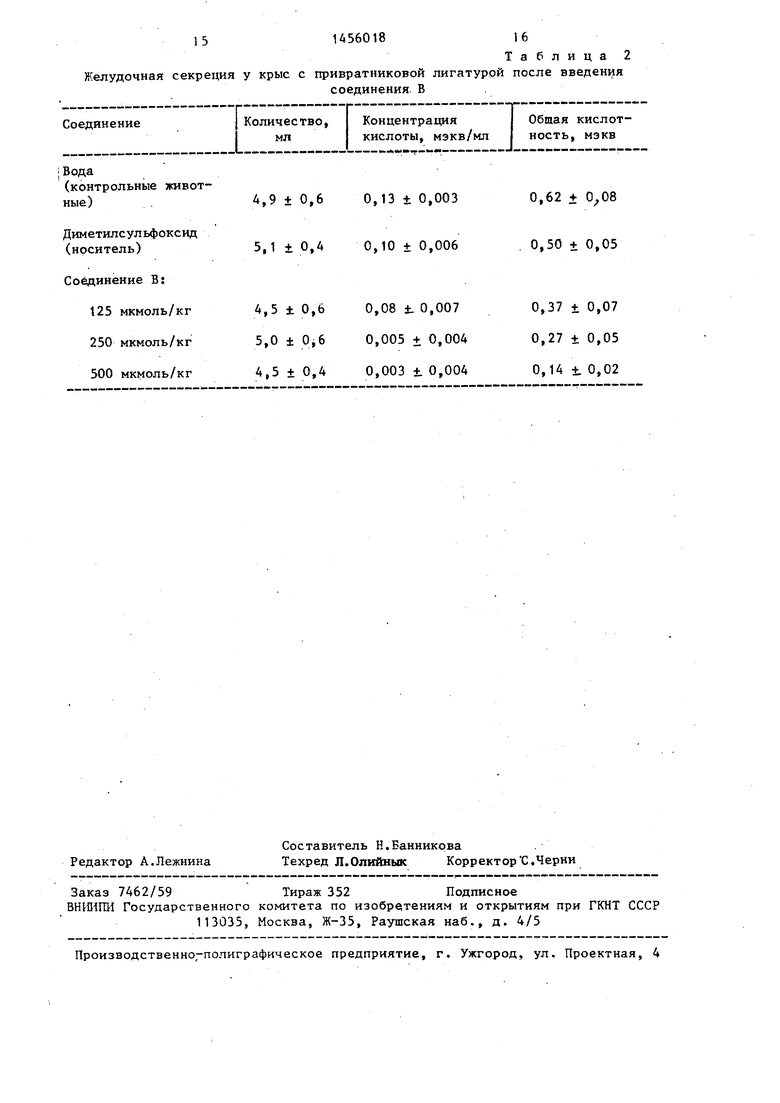
В этом стандартном тесте соединения общей формулы I, например 5-аце тил-2- ;(4-метокси-3,5-диметил-2- пиридил)-метилсульфинил -ЗН-тиено (2,3-d)имидазол (соединение А) или 2- (;4-метокси-2-пиридил)-метилсульфинил -ЗН-тиено (2,3-d) имидазол (соединение В), вызывали значительное и зависящее от дозы торможение желудочной секреции (табл. 1 и 2).
Соединения о€щей формулы I нетоксичны. Так, при введении соединения А в дозе 6000 мг/кг все подопытные животные остались живы. Следов побоных явлений не обнаружено.
Формула изобретения
1. Способ получения производных тиeнo-(2,3-d)-иl идaзoлa общей формулы I ,
13
R
R2
RI
,1
4
I JL
W ,
(oV Re
H
где Rf - водород или ацетил; Rj и Rg- водород;10
R, и R,- независимо друг от друга водород или С,-С4 алкил;
R - водород или С,-С4-алкоксил;
п - О или 1,
отличающийся тем, что, 15 соединение общей формулы II
R
2
УН
RAV-S
н
где R, и R имеют указанные значения,
подвергают взаимодействию с соединением общей формулы III
114
ciCH
Re
S
10
сил;
, 15
20
чене25
30
где R, .R., R5 H R имеют указанные
значения, в присутствии двух эквивалентов силь-ноге основания и вьщеляют целевой, продукт, либо в случае необходимости полученное соединение общей формулы I, где , окисляют эквивалентным количеством органической надкислоты или перекиси водорода в соединение общей- формулы I, где .
2.Способ по П.1, отличающийся тем, что взаимодействие соединений общих формул II и III осуществляют в низкокипящем спирте таком, как метиловый.
3.Способ по пп.1 и2, отличающийся тем, что соединение общей формулы II используют в избытке.
4.Способ по П.1, отличающийся тем, что окисление осуществляют эквивалентным количеством 3-хлорнадбензойной кислоты.
5.Способ по ПП.1 и 4, отличающийся тем, что окисление осуществляют в хлороформе или при температуре (.-6) - (-14) С.
6. Способ по П.1, отличающийся тем, что окисление осуществляют 30%-ной перекисью водорода в ледяной уксусной кислоте.






Изобретение относится к гетероциклическим соединениям, в частности к получению производных тиено-(2,3- -d)-ш«lидaзoлa формулы I (On)X N-C C-S-CR, CR, где X-ClReCh-C CHR,-CHR a R,-H. или ацетил; Rj HRj;-H; R, и Rj- - независимо друг от друга Н или С, алкил;К4 Н и С,-С-4-алкоксил;п-0 или 1, которые обладают антисекреторным действием. Цель - разработка способа по лучения новых соединений, обладающгас указанной активностью и низкой токсичностью. Получение их ведут из соединения формулы N-C(S)-NH-C C-S-CR. CRg предпочтительно в избытке и ClX HCl, где R,,R2. и X указано вьше, в присутствии двух эквивалентов сильного основания и вьщеляют. целевой продукт. Процесс предпочтительно ведут в низкокипящем спирте таком, как метиловьй. В случае необходимости полученное соединение формулы I, где , окисляют эквивалентным количеством органической надкислоты (предпочтительно 3-хлорнадбензойной кислоты) или перекиси водорода (предпочтительно 30%- ной перекисью водорода в ледяной уксусной кислоте) в соединение формулы I, где п 1. Предпочтительно окисление проводят в хлороформе при температуре (-6) - (-14) С. 5 .з.п. ф-лы, 2 табл. с ел 05
Таблица 1
Желудочная секреция у крыс с привратниковой лигатурой пос ле введения
соединения А
Вода
(контрольные живоные)
Диметилсульфоксид (носитель)
Соединение А: 125 мкмоль/кг
250 мкмоль/кг 500 мкмоль/кг
0,12 ± 0,005
0,56 ± 0,09
0,34 ± 0,07
0,37 ± 0,06 0,16 ± 0,03 0,13 ± 0,03
15145601816
Таблица 2
Желудочная секреция у крыс с привратниковой лигатурой после введения
соединения В
Соединение
Количество, мл
4,9 ± 0,60,13 ± 0,003
5,1 ± О,А0,10 ± 0,006
4,5 ± 0,60,08 t 0,007
5,0 ± 0,60,005 ± 0,004
4,5 ± 0,40,003 + 0,004
I I
Общая кислотность, мэкв
0,62 1 0,50 ± 0,05
0,37 + 0,07 0,27 t 0,05 0,14 +. 0,02
| Вейганд-Хильгетаг | |||
| М.: Химия, 1968, с | |||
| ПРИСПОСОБЛЕНИЕ ДЛЯ АВТОМАТИЧЕСКОЙ ПЕРЕСТАНОВКИ ЛЕНТЫ В УКАЗАТЕЛЯХ ОСТАНОВОК | 1914 |
|
SU584A1 |
