Изобретение относится к химии гетероциклических соединений, конкретно к способу получения фторированного производного у-бутиролактона- а-трифто- рацетил-у-бутиролактона формулы
,-J-CF, О °
который является одним из компонентов синтеза люминесциругащих красителей.
Известны способы получения производных у-бутиролактона- а-ацетилу -бутиро- лактона (V, сх. I, II). Например, способ, по которому а -ацетил- у-бутиролактон (V) с выходом 29% получают из зтилацетата (VI, сх. I) через стадию образования ацетоуксус- ного эфира (VII) с последующей обработкой окисью этилена (VIII) в присутствии этилата натрия (IX).
Схема I.
CH3-C-OC2H5- CHS-C-CH2-C-OC2HS+
о
VI
II
о
VII
II
о
о
СО
CH2-CH2 lM r-j-C-CH3
О VIII
о о
V
Известен также способ, по которому а- ацетил-у-бутиролактор (V) получают с выходом 60-70% из ацетоуксусного эфира и окиси этилена в-присутствии этилата натрия.
Схема II.
Снз-| сн2-с-ос2н5+ед0а1
C2H5ONa
О
о
сс°-сн
О
О
XI Ы
ю
ю
.
Известные способы не позволяют получить а-трифторацетил-у-бутмролактон (),
Наиболее близким к описываемому является способ получения кристаллогидрата а -трифторацетил- у-бутиролактона (схема III) из бутиролактона (Я) и трифторэтилаце- тата (VI) с применением в качестве конденсирующего агента гидрида натрия в эфире 3.
Схема III.
ГТ+СР.-С-ОСоН,
k Sл Ц 5 o.Sc.3
о
ее
О
О
О и
-C-CF3 О
.ЭФИР
НоО
К суспензии (0,75 моль) гидрида натрия абсолютном эфире в токе азота добавляют 0,035 моль) абсолютного этанола. Затем по аплям добавляют раствор смеси (0,7 моль) -бутиролактона и (0,7 моль) этилтрифтора- етата в 670 мл сухого эфира с такой скоротью, чтобы смесь кипела.
Реакционную массу перемешивают в ечение ночи, охлаждают и подкисляют 10%-ным раствором HCI до рН 5-6, Водный слой экстрагируют эфиром. Объединенные эфирные вытяжки промывают водой и упаривают..
Образовавшийся кристаллический осадок представляет собой гидрат (I). В известном способе для создания некоторой полярности среды добавляется в качестве затравки этанол в соотношении с гидридом натрия 1:20, что подтверждает тот факт, что в данном способе конденсирующим агентом является гидрид натрия,
Недостатком указанного способа является длительность процесса, использование взрывоопасных веществ - гидрида натрия м диэтилового эфира и связанная с этим необходимость проведения реакции в токе инертного газа, а также недостаточный выход целевого продукта (44% - в виде кри- сталлогидрата).
Целью изобретения является повышение выхода целевого продукта в индивидуальном виде, упрощение процесса и повышение его безопасности.
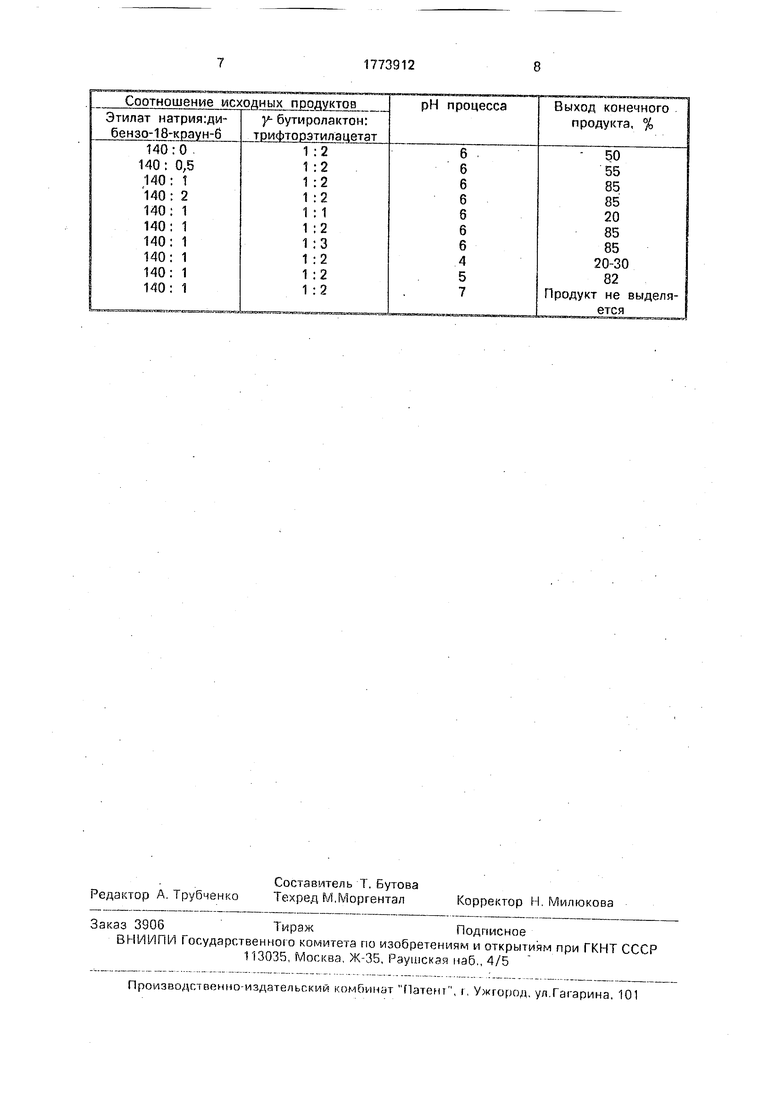
Поставленная цель достигается описываемым способом получения I взаимодействием у-бутиролактона с зтилфторацетатом в присутствии натрийсодержащего соединения с последующим подкислением реакционной смеси разбавленной соляной кислотой до рН 5-6, отделением органического слоя и экстракцией, отличием которого является использование в качестве натрийсодержащего соединения этилата натрия в смеси с дибензо-18-краун-6 при их мольном соотношении 1:140 и проведение процесса при мольном соотношении реагентов 1:2, при этом экстракцию проводят четыреххлористым углеродом, после чего продукт подвергают фракционированию. 0 Изучено влияние условий реакции на выход а -трифторацетил- у-бутиролактона: мольных соотношений и у-бутиролактона м этилфторацетата, катализатора краун-эфи- ра и этилата натрия. При эквимольных соот- 5 ношениях исходных веществ (1:1) выход не превышает 20%. Увеличение доли этилт- рифторацетата приводит к повышению выхода и достигает максимума .при соотношении 1:2 (в отсутствии краун-эфира 0 -50%, в присутствии -85% индивидуального продукта). Активация,этилата натрия кра- у.н-эфиром достигается благодаря тому, что последний комплексует катион натрия и способствует образованию этилат-иона, ко- 5 торый по существу является конденсирующим агентом.
Оптимальным условием выделения hpoдукта является слабокислая среда - рН 6,
т.к. в более кислой среде (рН 4) продукт
0 разлагается, а при рНвыше7 выделение его
невозможно.Изобретение иллюстрируется следующими примерами.
Пример 1, К смеси 13,6 г (0.2 моль) 5 этилата натрия и 0,5 г (0,0014 моль) дибен- зо-18-краун-б, что соответствует соотношению 140:1, в течение 15-20 мин прибавляют смесь 17,2 г(0,2 моль)у-бутиролактонаи63,2 г (0,4 моль) трифторэтилацетата (соотноше- 0 ние 1:2), при этом температура реакционной . смеси повышается до 75°С. Затем реакционную массу охлаждают до 25°С, обрабатывают 10%-ным раствором соляной кислоты до рН 6, отфильтровывают краун-эфмр и от- 5 деляют органический слой. Из водной части экстрагируют четыреххлористым углеродом. Экстракт объединяют с органическим слоем, упаривают, остаток фракционируют в вакууме. Длительность процесса 3,5 ч. 0 Выход а-трифторацетил-у-бутиролактона составляет 31 г (85%). Температура кипения 82-85°С/1б мм рт.ст.; пЬ° 1,419, о1о 1,456.
СеНбРзОз
Найдено. %: С 39,38; Н 2,60; F 31,15, Вычислено, %: С 39,56, Н 2,74, F 31,31. ИК-спектр: 1780 (ОО), 940 (С-О-С), лак- тон, 1150-200 (C-F) . Масс-спектр: М+ 182 (молекулярный ион, соответствующий молекулярной массе).
5
Пример 2. Опыт проведен в условиях, аналогичных примеру 1 (соотношение реагентов, температурный режим, метод выде- ления), за исключением обработки реакционной массы, которая ведется 10%- ным раствором HCI до рН 5. При этом выход составляет 82%.
Таким же образом были проведены эксперименты при запредельных соотношениях исходных продуктов.
Результаты экспериментов приведены в таблице.
Примеры получения а-трифторацетал- у-бутиролактона в кристаллогидратной форме.
Пример 3. К смеси 13,6 г (0,2 моль) этилата натрия и 0,5 г (0,0014 моль) дибен- зо-18-краун-6, что соответствует соотношению 140:1, в течение 15-20 мин прибавляют смесь 17,2 г (0,2 моль) у-бутиролактона и 56,8 г (0,4 моль) этилтрифторацетата (соотношение 1:2). При этом температура реакционной смеси не превышает 75°С. Затем реакционную массу охлаждают до 25°С, обрабатывают 10%-ным раствором соляной кислоты до рН 6, отфильтровывают краун- эфир и отделяют органический слой. Из водной части проводят экстракцию эфиром (4 30 мл). -Экстракт объединяют с органическим слоем, упаривают на водяной бане досуха. Образовавшийся кристаллогидрат а -трифторацетил- у-бутиролактона пере- кристаллизовывают из смеси гексан:эфир в соотношении 4:1.
Выход кристаллогидрата а-трифтора- цетил-у -бутиролактона составляет 29,7 г (75%). Т.пл. 97-98°С.
Пример 4. К 13,6 г (0,2 моль) зтилата натрия в течение 15-20 мин прибавляют смесь 17,2 г (0,2 моль) у-бутиролактона и 56,8 г (0,4 моль) этилтрифторацетата (соотношение 1:2). При этом температура реакционной смеси не превышает 75°С. Затем реакционную массу охлаждают до 25°С, обрабатывают 10%-ным раствором соляной кислоты до рН 6, отделяют органический слой. Из водной части проводят экстракцию
эфиром (4 30 мл). Экстракт объединяют с органическим слоем, упаривают на водяной бане досуха. Образовавшийся кристалло- 5 гидрат а -трифторацетил- у-бутиролактона перекристаллизовывают из смеси гек- сан:эфир(4:1).
Выход кристаллогидрата а-трифторацетил- у-бутиролактона составляет 23,8 г 0 (60%). Т.пл. 97-98°С, ИК-спектр: в области 1780-1760 () трифторацетил и лак- тон; в области 3450 (ОН)
Таким образом, предложенный способ в отличие от известного, дает возможность получить чистый индивидуальный а -трифторацетил- у-бутиролактон с высоким выходом (85%). Длительность процесса при этом сокращается до 3,5-4 ч. В известном способе длительность составляет 12-13 ч. Способ совершенно безопасен, т.к. в нем не используются взрывоопасные вещества - гидрид натрия и диэтиловый эфир.
Формула изобретения
Способ получения а -трифторацетил- у- бутиролактона формулы
5
0
5
С
о
г
х
о
II C-CF3
О
взаимодействием у-бутиролактона с этилт- рифторацетатом в присутствии нзтрийсо- держащего соединения с последующим подкислением реакционной смеси разбавленной соляной кислотой до рН 5-6 с последующим отделением органического слоя-и экстракцией, отличающийся тем, что, с целью повышения выхода целевого продукта, упрощения процесса и повышения его безопасности, в качестве натрийсодер- жащего соединения используют этилат натрия в смеси с ,дибензо-18-краун-6 при их молярном соотношении 1:140 и процесс ведут при молярном соотношении исходных реагентов 1:2, а экстракцию проводят четы- реххлористым углеродом, после чего продукт подвергают фракционированию.


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФТОРСОДЕРЖАЩИХ ТЕТРАКЕТОНОВ | 2010 |
|
RU2421442C1 |
| Способ получения динитропроизводных дифениловых и трифениловых эфиров | 2017 |
|
RU2671581C1 |
| СПОСОБ ПОЛУЧЕНИЯ {2-[4-(АЛЬФА-ФЕНИЛ-П-ХЛОРБЕНЗИЛ)ПИПЕРАЗИН-1-ИЛ]-ЭТОКСИ}-УКСУСНОЙ КИСЛОТЫ И НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2248974C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-МЕТИЛ-3-ФЕНИЛ-3-(4-ТРИФТОРМЕТИЛФЕНОКСИ)-ПРОПИЛАМИНА ГИДРОХЛОРИДА | 2007 |
|
RU2336264C1 |
| Способ получения низших С @ -С @ -алкиловых эфиров 2,2-диметил-3-(2,2-дихлорэтенил) циклопропанкарбоновой кислоты | 1991 |
|
SU1817771A3 |
| СПОСОБ ПОЛУЧЕНИЯ ДИГИДРОХЛОРИДА 2,7-БИС[2-(ДИЭТИЛАМИНО)ЭТОКСИ] ФЛУОРЕНОНА (АМИКСИНА) | 2006 |
|
RU2317974C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(1-АМИНОЭТИЛ)АДАМАНТАНА ГИДРОХЛОРИДА | 1997 |
|
RU2118313C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИДИВИНИЛФОСФИНОВОЙ КИСЛОТЫ ИЗ КРАСНОГО ФОСФОРА И АЦЕТИЛЕНА | 2015 |
|
RU2632816C2 |
| ИЗВЛЕЧЕНИЕ РАДИОНУКЛИДОВ ЭКСТРАГЕНТАМИ, СОДЕРЖАЩИМИ КРАУН-ЭФИРЫ | 2004 |
|
RU2318258C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНОГО СИНТЕТИЧЕСКОГО НИКОТИНА | 2021 |
|
RU2780283C1 |
Сущность изобретения: продукт а - трифторацетил-у -бутиролактон СбРбРзОз; т.кип/82-85°С/16 мм рт.ст. п&° 1,419. Реагент: у-бутиролактон. Реагент 2: этилтриф- торацетат. Условия реакции: при мольном соотношении 1:2 в присутствии CaHsONa в смеси с дибензо-18-краун-6 при их мольном соотношении 1:140 с последующим подкис- лением разбавленной HCI до рН 5-6, отдле- лением органического слоя, экстракцией ССЦ и фракционированием. 1 табл.
| Кожух паровых турбин | 1929 |
|
SU40813A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| Прибор для вычисления процентов | 1950 |
|
SU95116A1 |
| Кровля из глиняных обожженных плит с арматурой из проволочной сетки | 1921 |
|
SU120A1 |
