Изобретение относится к новым селективным и автономно реплицирующимся векторам экспрессии, рекомбинантной ДНК, которые состоят из транскрибируемой и транслируемой, активированной нуклео- тидной последовательности, находятся в рамке считывания гена, который кодирует биоактивное производное бычьего гормона роста (6ГР), и репликона, который в индуцированных условиях утрачивает контроль числа копий.
Изобретение относится также к способу биосинтеза бычьего гормона роста с использованием указанных векторов.
Развитие и эксплуатация технологии рекомбинантной ДНК для получения 6ГР были сильно затруднены проблемами экспрессии
гена. Некоторые из этих проблем, детально описанные в заявке на Европейский патент КЬ 0075444, , включают транскрипцию функционально субоптимальной мРНК . Такая мРНК имеет вторичные конфигурации стержня и петли, что препятствует нормальному рибосомному связыванию и таким образом резко снижает или даже предотвращает 6ГР экспрессию. Изобретение разрешает эту проблему, обеспечивая векторы для высокого уровня экспрессии производных 6ГР.
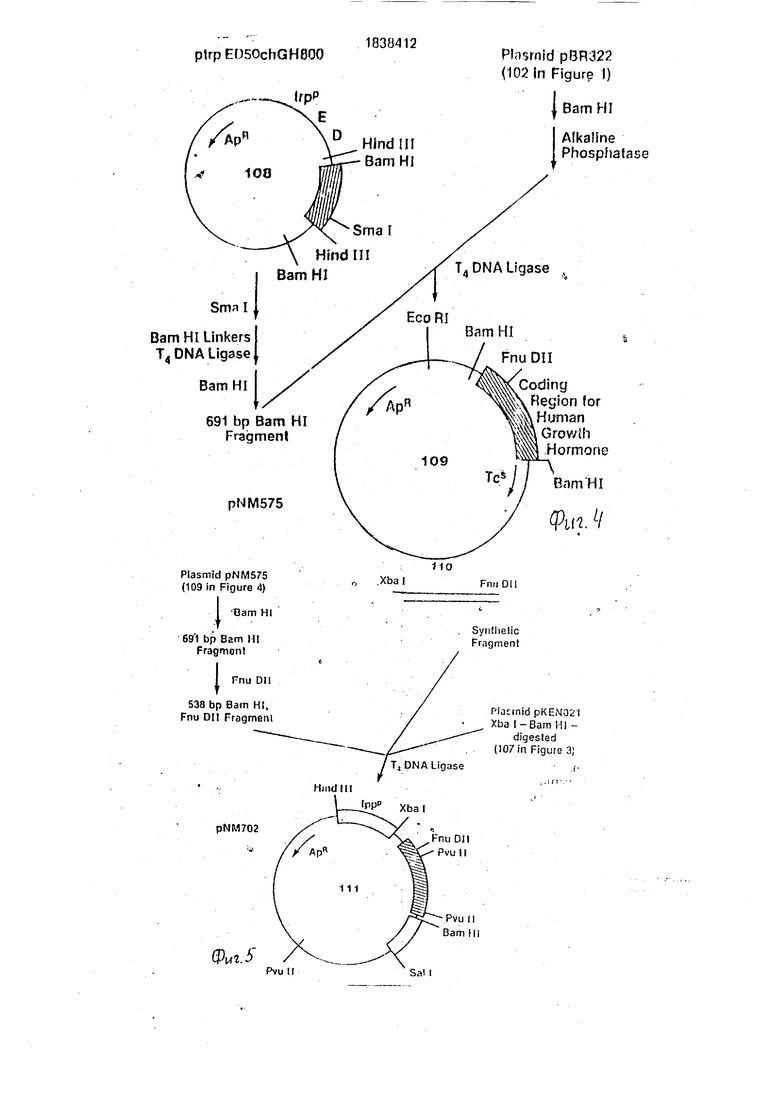
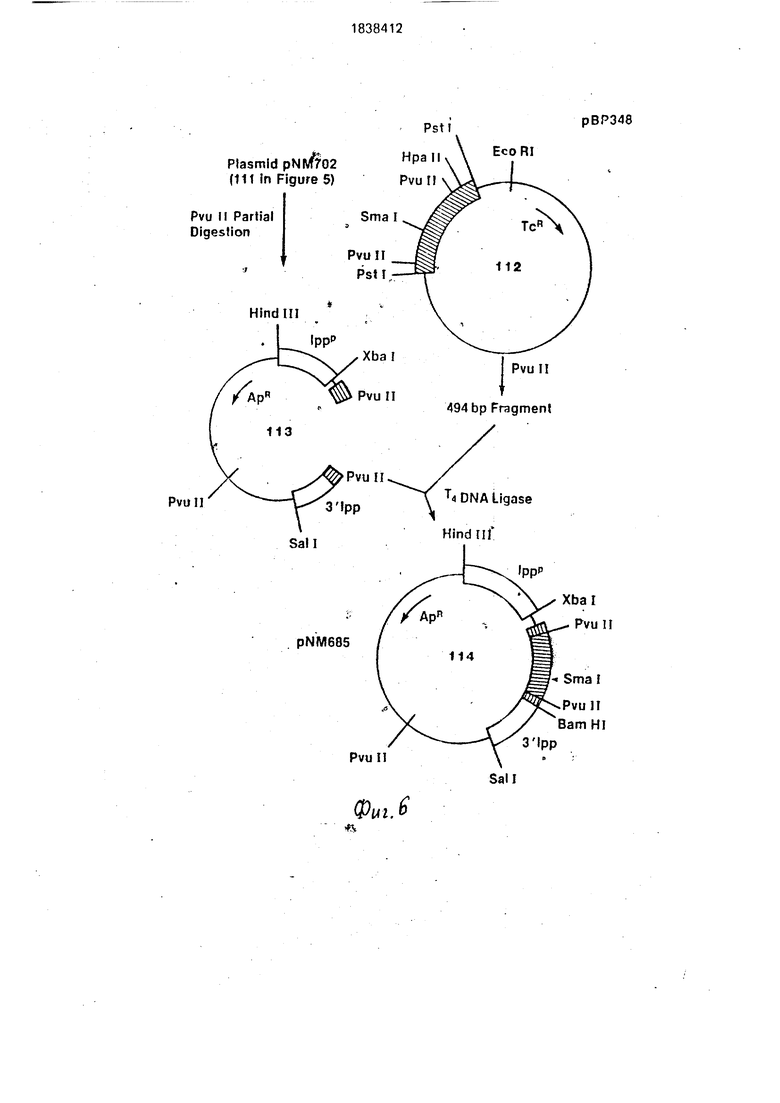
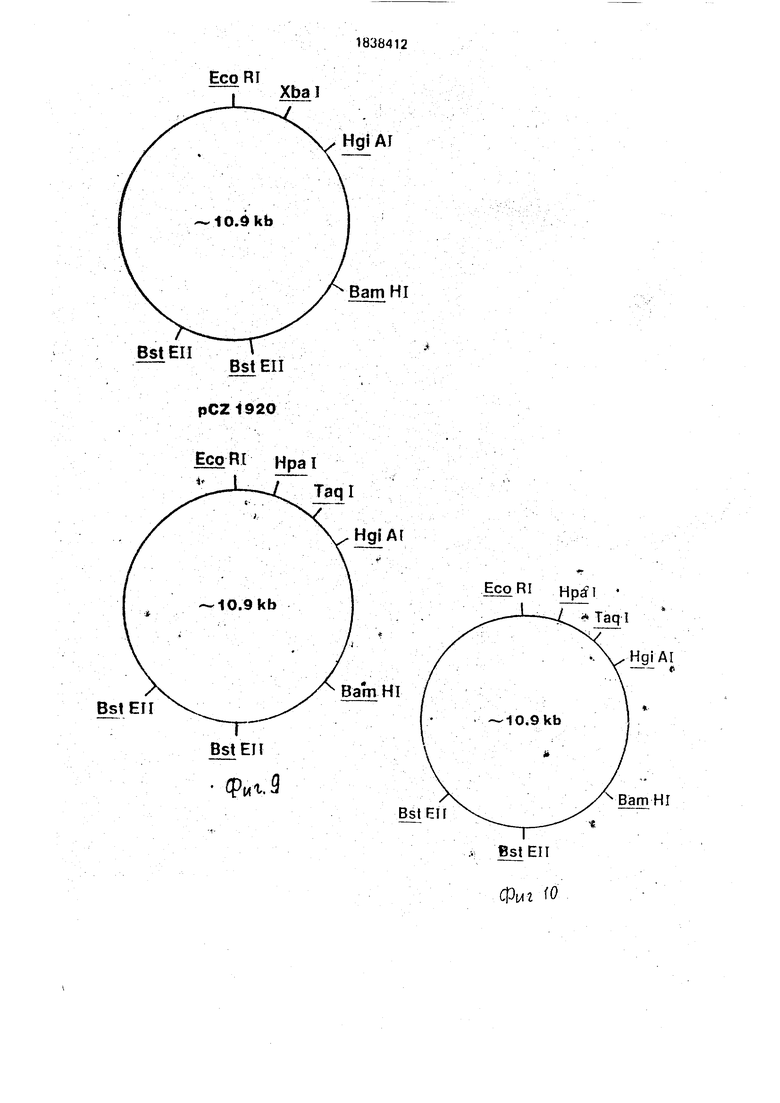
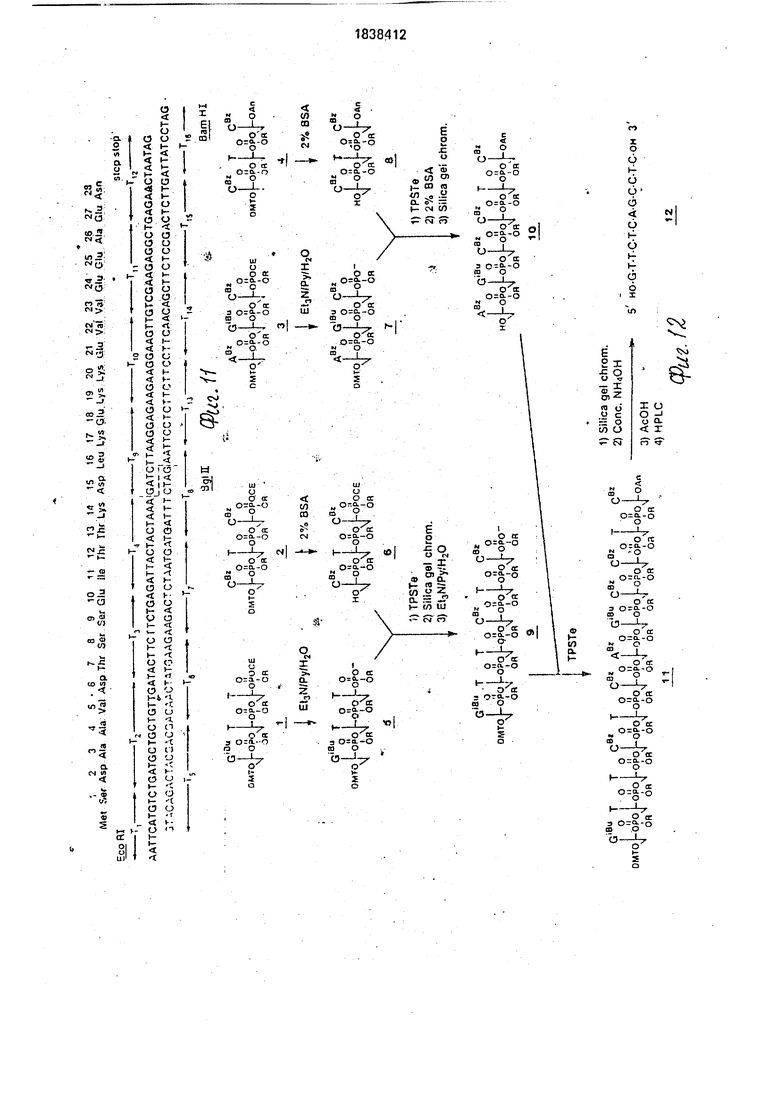
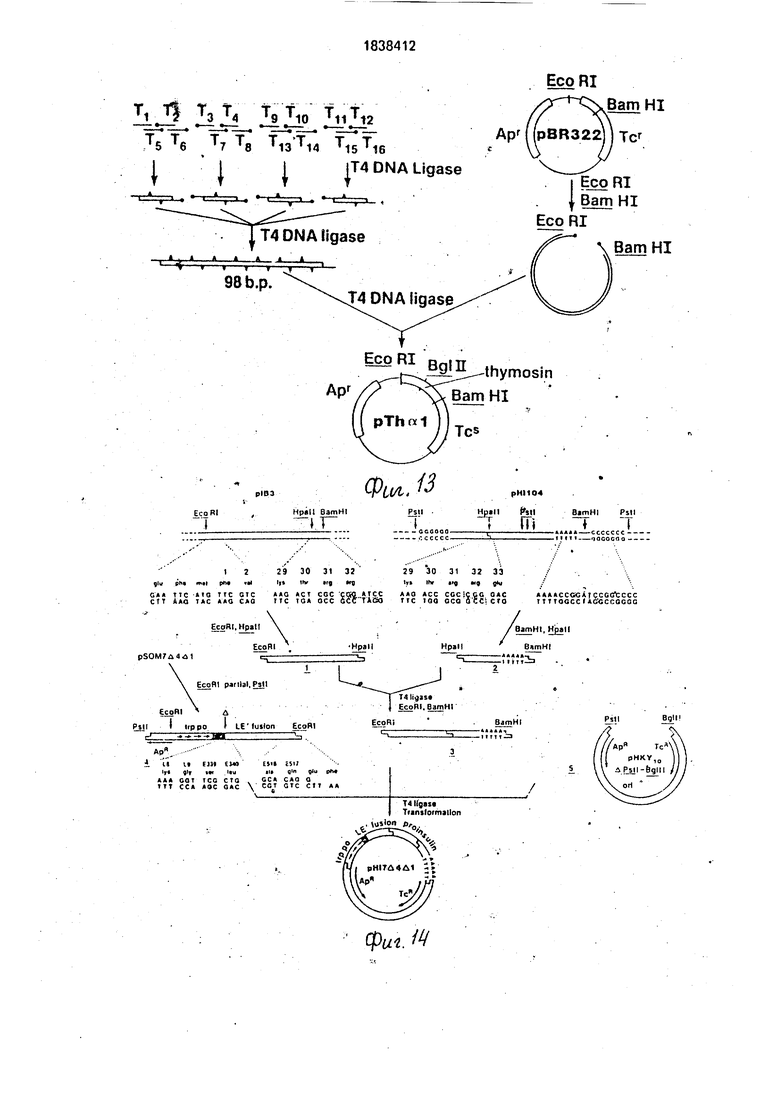
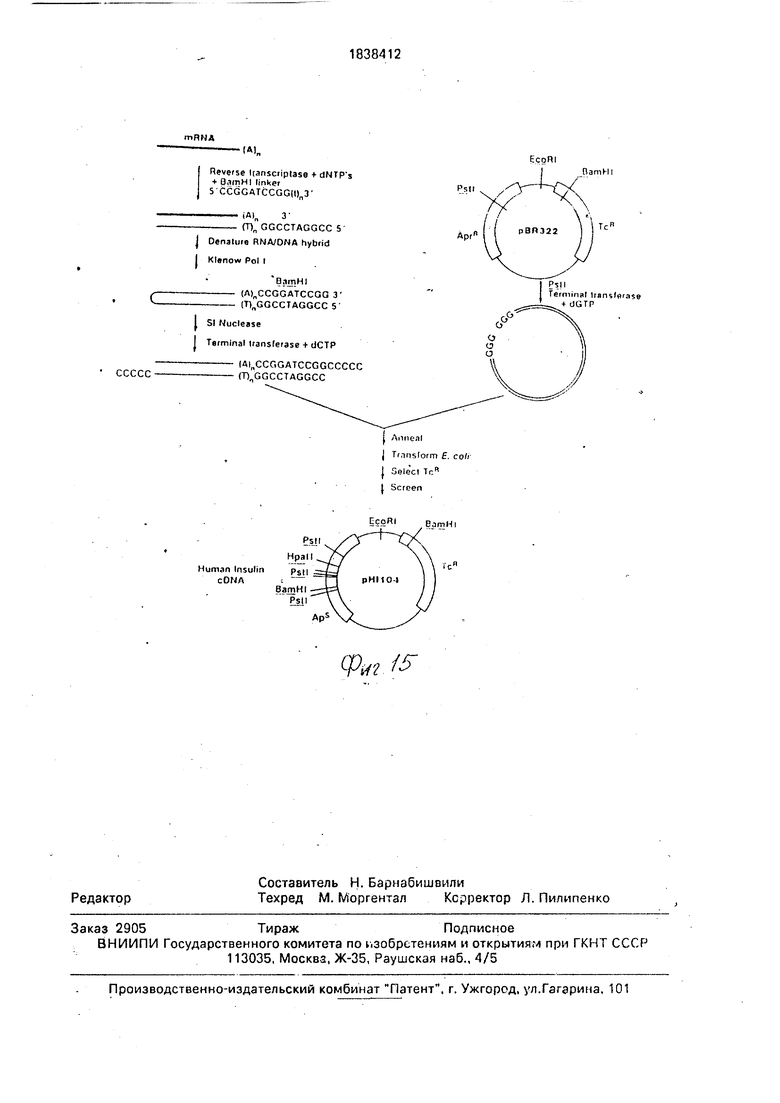
На фиг. 1-4 изображена схематическая иллюстрация протокола конструирования плазмиды pNM575; на фиг.5-8 - схематическая иллюстрация протокола конструирования плазмиды pNM789B: на фиг.9 - карта сайта рестрикции плазмид pCZ1920 и pJR1;
00
СА) 00
N
So
СА)
на фиг. 10 - карта сайта рестрикции плазмиды pCZ112; на фиг.11 - синтетический тем тимозин альфа 1; на фиг.12 - протокол синтеза нуклеотидного фрагмента Т15; ма фиг.13 - протокол конструирования плазмиды pHnal; на фиг.14- протокол конструирования плазмиды рН17Д4Д1. на фиг.15 - протокол конструирования плазмиды рН 1104.
(1 ) 5 АТС АЛА CGC ЛЛТ ТСТ АТС ССС ТТС 3 ТАС ТТТ ССС ТТЛ АСЛ ТАС CGG AAG
SССА GCC АТС ТСС ТТС ТСС GGC j GOT CGG ТЛС AGO ЛАС AGG CCG
( 2 ) 5 АТС GAT GAT АЛС ПТ CCG GCT АТС
Ю э. ТЛС СТА СТА ТТС ААЛ ССС ССА ТАС
3
13)
тст сто тсс ссс t
АСЛ ёАс Асе 5, АТС ТТТ ССА GCC АТС- GCT СТА ТЛС АЛА GGT ССС ТЛС ССЛ CAT
Селективный автономно реплицирующий рекомбинангную ДНК вектор экспре- сии в соответствии с изобретением состоит из неконтролируемого репликона и транскрибируемой и транслируемой активирующей последовательности, которая находится в рамке считывания гена, который кодирует биоактивное производное бычьего гормона роста, причем ген является
5 АТС CAT ТТТ CCG CCT АТС ТСТ 3 ТАС СТА АЛЛ ССС С(;Л ТАС ЛСЛ
3
СТО ТСС GGC ПАС ЛСК CCG
R-B
(Я.
АТС ТТТ ССА GCT АТС ТСТ СТА ТЛС АЛЛ C.GT ССА ТЛС АСА СЛТ
ТСТ CGT
3 5
(7)
АТС СТГ ТТТ ССО ССТ АТС ТСТ ТАС САЛ АЛЛ СОС СсА ТЛС АСА






















Использование: генетическая инженерия, биосинтез белков, в частности бычьего гормона роста. Сущность изобретения: ре- комбинантные плазмидные ДНК, кодирующие бычий гормон роста получают в результате лигйрования фрагмента BamHI-Xbal плазмиды pCZ101 размером 10,2 kb или BamHI-EcoRI фрагмента той же, плазмиды с Ipp промотором и репликоном, теряющим контроль числа копий при 30°С, с BamHI- Xbal .фрагментом плазмиды pCZ101 или pCZ106 размером 0,6 kb, кодирующим аминокислоты с 15 до 191 бычьего гормона роста и ДНК-линкером, содержащим нуклеотиды. кодирующие последовательность активации трансляции, стартовый ко- дон и кодон для N-конца производного бычьего гормона роста. Способ биосинтеза состоит в том, что культивируют штамм Escherlchia coli, трансформированный ре- комбинантной плазмидной ДНК pCZ110, или pCZ153. или pCZ154, культивирование трансформантов, выделение и очистку целевого продукта. 2 с.п. ф-лы, 2 табл., 15 ил. ел с
тст ест
;-R-R
ЛГ,Л ССЛ
АТС ТТС ССА GCT АТС ТСТ СТА
тле Але ест сел тле Асл CAT тст опт
АСЛ ССЛ
-R-R
где А является деоксиаденилом;
G - деоксигуанилом;
С - деоксицитидилом;
Т - тимидилом;
R - последовательностью ДНК, которая кодирует аминокислоты 9 (лейцин) по 1.91 (фенилаланин) бычьего гормона роста;
Векторы настоящего изобретения могут быть сконструированы путем независимого
а| 5 CTAGAGGGTATTAATA АТС ААЛ GCG ЛАТ ТСТ АТС ТССсАтАЛТТЛТ ТЛС TtT ССС ТТЛ AGA ТАС
ССС ТТС ССА ССС ATG ТСЙ ТТС ТСС ССС СТС CGC AAG GCT CGG ТАС AGG ЛАС AGO CCG GAC
Ь) 5 СТАСАСССТЛТТАЛТА, ЛТС ТТТ ССА GCC ATG GCT 1 ТСССАТЛЛТтАт тАс АЛЛ GGT CGG ТАС CGA
СТА ТСТ ССТ СТС ТТТ GCC AAC CCT GTGCT 3 CAT АсА ССЛ СЛС ААА CGG TTC CGA С 5
сто тсс сое
РАС AGG CCG
а) 5 АТС црт ттт ссс сет АТС тст
З1 ТАС ССЛ АЛЛ tJGC ССЛ ТАС АСА
сто.-тсс стс
Э 5
30
R1 является последовательностью ДНК, которая кодирует транслируемый стоп-сигнал, который представляет собой
ТАА АТТ
TGA ACT
TAG АТС
ИЛИ
г связывания следующих Xbal-H AI линкер- 40 ных последовательностей ДНК:
ттт ссс;ААС ест стсст ААА CGG ттс сс-А с
CTAGAGGCTATTAATA АТС ТТГ ССА ССТ АТС ТСТ
тсссАтААттАт тле ААЛ ест ссА ТАС АСА
СТА ТСТ GGT CTG ТТТ GCC AAC GCT CTGCT 3
CAT АсА сел сАс ААА CGC TTG CGA с 5
во фрагменты примерно 10,2 kb BamHI-Xbal и 0,6 kb BamHI-Hg AI плазмиды pCZ101. Полученные в результате плазмиды обозначают соответственно как плазмиды pCZ1920, pJRI и рАТ2, они содержат транслируемую и транскрибируемую активирующую последовательность липопро- теинового гена Е. coli (Nakamura and Inouye 1979, Cell 18:1109) при считывании каркаса кодирующей последовательности для био- активного производного бычьего гормона роста; соответственно размещенный транслируемый стоп-сигнал; и репликон неконтролируемогороста. Клетки, трансформированные плазмидами pCZ1920, pJR1 и рАТ2 соответственно, экс- прессируют MET-LIS-CLY-ASPN-SER-MET- ALA-бГР, MET-PHE-PRO-ALA-MET-ALA-R2 и МЕТ-бГР, где
МЕТ является метионином,
LIS - лизином,
л) V V
ССЛСС ЛТС СЛТ СЛТ ЛАС ТТТ CCG GCT АТС ТСТ TCG ТЛС СТЛ СТЛ ТТС ЛЛЛ GCC CGA ТЛС ЛСЛ
CTG ТСС GGC CTG ТТТ ССС АЛС GCT GTGCT СЛС ЛСС CCG СЛС ЛЛЛ CGO TTG ССЛ С
ы
з1
сслсл лтс; ттс сел ест лтс тет стл тст GGT
TGT ТЛС АЛС. GGT ССЛ ТАС ЛСЛ CAT АСЛ ССА
CTG ТТТ ССС ЛАС ССТ GTGCT СЛС АЛЛ CGG TTG ССЛ С
с)
CGACC АТС CAT ТТТ ССС ОСТ АТС ТСТ СТС ТСС
тсс тле стл Алл ссс сел тле лсл слс лсс
GGC CTG ТТТ GCC АЛС GCT GTCCT ССС..САС АЛЛ ССС TTG ССЛ С
ССАТС ЛТС GAT ТТТ ССС ОСТ АТС ТСТ СТС ТСС
тле тле стл Алл GCC сел ТАС лсл слс -лес
ССС СТС ТТТ ССС ААС GCT GTGCT 3 ССС СЛС АЛЛ ССС TTG ССА С 5
где А является деоксиаденилом,
G - деоксигуанилом,
С - деоксицитидилом,
Т - тимидилом,
во фрагменты примерно 10 kb EcoRI- BamHI и примерно 0,6 kb BamHI-Hgi AI плазмиды pCZ101 и фрагмент примерно 0,29 kb EcoRI-Clal плазмиды pNM608. Полученные в результате плазмиды. обозначен- ные соответственно как плазмиды pCZ112, рАТ1, pASP1, pASP2, pCZ154, PCZ155 и pCZ156, содержат транскрибируемую и транслируемую активирующую последовательность триптофанового гена Е. coli (Hallewell and Emtage, 1980, Gene 9:27) в рамке считывания кодирующей последовательности для биоактивного производного
GLY - глицином,
A.SPN - аспарагином,
SER - серином,
ALA - аланином,
РНЕ - фенилэланином,
PRO - пролином.
6ГР - природной аминокислотной последовательностью бычьего гормона роста, начинающейся с N-терминального (первого) фенилаланина.
R является природной аминокислотной последовательностью бычьего гормона роста, начинающейся с шестой аминокислоты (лейцина) от М-терминала.
Карта сайта рестрикции каждой из плаз- мид pCZ1920 и pJR1 представлена на фиг.9.
Дополнительные плазмиды, которые также входят в область настоящего изобретения, могут быть сконструированы независимо связыванием следующих линкерных последовательностей ДИК Tagl-Hgi Ai:
5 ССЛСС АТС GTT ТТТ CCG ССТ АТС 3 TCG ТАС САД ЛЛЛ GCC ССЛ ТЛС
ТСТ СТС ТСС ССС СТС ТТТ ССС ЛСЛ GAC ЛСС ССС СЛС ЛЛЛ ССС
ЛАС GCT GTGCT TTG ССЛ С
СС.ЛСС ЛТС ССТ ТГГ ССС ССТ АТС ТСС ТЛС ССЛ ЛЛЛ ОСС ССЛ ТАС
ТСТ CTG ТСС СТС СТС ТТТ ССС ЛСЛ GAC ACG СЛС (-ЛС ЛЛЛ ССС
ЛАС ССТ СТССГ
ттс сел с
5 51
ССЛТА АТС CAT ТТТ ССС С.СТ АТС ТАТ ТАС СТА АЛЛ CGC ССА ТЛС
ТСТ СТС ТСС GGC СТС ТТГ GCC АСА САС ЛСС CCG СЛС АЛЛ ССС
ААС ССТ GTCCT 3 TTG CGA С . 5
бычьего гормона роста; соответственно помещенный транслируемый стоп-сигнал и неконтролируемый репликон. Клетки, трансформированные плазмидами pCZ112, рАТ1, pASP1, pASP2, pCZ154, pCZ155 и pCZ156, соответственно экспрессируют MET-ASP-ASP-LYS-бГР. МЕТ-бГР, МЕТ- ASP-бГР, MET-ASP-бГР, MET-VAL-бГР, MET-ALA-PHE-PRO-ALA-MET-SER-IEU-SER- VAL-бТР и MET-ASP-бГР, где
МЕТ является метионином,
ASP - аспарагиновой кислотой,
LED - лейцином,
SER - серином.
PRO - фенилаланином,
LYS -лизином,
VAL - валином,
ALA - аланином,
б ГР является природной аминокислотной последовательностью бычьего гормона роста, начинающейся с аминокислоты 9, лейцина;
6ГР является природной аминокислотной последовательностью, начинающейся с N-терминального,(первого) фенилаланина. Карта сайта рестрикции иллюстративной плазмиды pCZ112 представлена на фиг. 10. Плазмида pCZ101 исходного материала содержит примерно 10,8 kb и сконструирована лигацией фрагмента примерно 0,6 kb Xbal-BamHI плазмиды pNM789B в подобным образом переваренную плазмиду р М-1 -АЗ. Последняя плазмида, которая содержит транскрибируемую и транслируемую активирующую последовательность липопротеинового гена Е. coli, а также и репликон неконтролируемого роста, может быть получена из Е. coli K12 RV308/plM-V- . A3, штамма, депонированного и составляющего часть постоянно хранящейся коллекции культур Северной региональной исследовательской лаборатории, Пеория, Иллинойс. Штамм является доступным как предпочтительный источник и хранилище плазмиды под регистрационным номером NRRLN В-15733. Исходный материал плазмида pNM789B происходит из плазмиды pKENUT в соответствии со стадиями, иллюстрированными и описанными на фиг.1- 8 и в примере 1 ниже. Плазмида рКЕШ может быть получена из Е, coli K12 CC620/pKEN111 штамма, депонированного и являющегося частью постоянно хранящейся коллекции куль тур Северной региональной исследовательской лаборатории. Пеория, Иллинойс. Штамм является доступным как предпочтительный источник и хранилище плазмиды под регистрационным номером NRRL 8-15011. Плазмида pNM789B также содержит транскрибируемую и транслируемую активирующую последовательность липопротеинового гена Е.соЧ и, кроме того, кодирующую последовательность, включающую соответственно расположенный .транслируемый стоп-сигнал, для конденсированного протеина, включающего 6ГР и девятичленный пол- ипептид у 6ГР N-терминала. Лигирование последовательности, кодирующей конденсированный протеин, содержащийся в фрагменте Xbal-BamHI, к соответственно расщепленной плазмиде р1М-1 -АЗ приводит в результате в вышеупомянутой плазмиде pCZ101 исходного материала.
Исходный материал плазмиды рМ608 содержит примерно 4,6 kb и сконструирована путем лигации фрагмента EcoRI-Clal плазмиды рН17 Д4Д1 в EcoRI-Clal - переваренную плазмиду рВ.322. Плазмида рН17 ДА Л1 может быть сконструирована в соответствии с методикой примера 5 ниже, Из-за множественности сайтов рестрикции
Та 1 в плазмиде рН 17 М Д1 желаемый фрагмент EcoRI-Tagl лучше всего может быть генерирован субклонированием фрагментов рН17 Д4 А1 EcoRI-Hpal и Hpal-Tagl, Плазмида р ММ608 содержит Е. coll триптофан транскрибируемую активирующую последовательность и, следовательно, является полезной для конструирования в изобретении.
Специалисты в данной области должны
знать, что различные линкеры ДНК, описанные выше, являются важными компонентами изобретения. Эти последовательности могут быть удобно синтезированы с помощью модифицированного фосфор-триэфирного метода, использующего полностью защищенные дидеоксирибонуклеотидные строительные блоки. Такие методы синтеза хорошо известны на данном уровне техники и могут быть осуществлены по существу в
соответствии с методикой Итакуры и др., 1977, Science, 198; 1056; и Криассотр., 1978, Pro, Nat, Acad, USA 75:5765. Кроме того, особенно предпочтительный способ описан в Hslung et al., .1983. Исследование нуклеиновых кислот 11:3227, и Narang eta., 1980. Методы вэнзимологии68:90. Линкеры коди-. руют транслируемую активирующую последовательность и также аминокислоты, составляющие первый участок (М-терминальную область) вышеупомянутых производных бГР. Остальная Кодирующая последовательность бГР (включая соответ- ственно расположенный транслируемый стоп-сигнал) и также транскрибируемая активирующая последовательность могут быть обеспечены лигацией соответствующего фрагмента плазмиды pCZ101. Такие лигации приводят в результате к иллюстра,- тивным плазмидам экспрессии производно.
го бычьего гормона роста изобретения. ,-,;
Деления фрагмента рестрикции при-/ мерно 900 пар оснований BstE l 1 плазмиды pCZ101 с последующей перециркуляриза-.
0 цйем приводит в результате в плазмиде pCZ103. BstE11 деления не оказывает воздействия на область кодирования бГР.в плазмиде pCZ101, Используя плазмиду pCZ103 вместо плазмиды pCZ101 в выше5 упомянутых конструированиях получают в результате подобные плазмиды, но меньше 900 пар оснований. Производные бГР, экс- прессируемые этими происходящими из pCZ103 плазмидами, следовательно, являются идентичными производным, экспрессируемым происходящими из pCZ101 их дубликатами.
Итак, используя фрагменты рестрикции плазмиды pCZ103 примерно 9.3 kb BamHI- Xpal и примерно 0,6 kbBamHI-Ho AI вместо фрагментов рестрикции плазмиды pCZ101 примерно 10,2 kb BamHI-Xbal и примерно kb BamHI-Hgi AI в вышеупомянутых кон- руированиях из плаэмид рЛ и рАТ2 получают в результате конструкцию производных плазмид pJR1.3 и рАТ2.3 соответственно. Используя фрагменты рестрикции плазмиды pCZ103 примерно 9,3 kb BfemHI-EcoRI и примерно 0,6 kb BamHI-Hgi А1 вместо фрагментов рестрикции плазмиды pCZ101 примерно 10,2 kb BamHI-EcoRI и npHMepHO.O,6kb BamHI-Hgi AI в вышеупомянутых конструированиях из плазмид рАТ1, pASP1, pASP2, pCZ154, pCZ155 и pCZ156 получают в результате конструкции производных плазмид рА И.З, pASP2.3, pCZ154.3, pCZ155.3 и pCZ156.3 соответственно,
Изобретение никоим образом не ограничивается использованием конкретной транскрибируемой активирующей последовательн остью, так как выбор определенной последовательности не является критическим для работоспособности изобретения.
Транскрибируемые активирующие последовательности, которые могут быть заменены ранее описанными липоиротеиновой и триптофановой активирующими последовательностями, включают, но не ограничиваются Е. coli лактозной (lac), бэктериофаговой АРгОг и бактериофаговой ApROR транскрибируемыми активирующими последовательностями. В дополнение к этому одна или несколько транскрибируемых активирующих последовательностей, или их частей, могут быть использованы совместно, такие как, например, trp и lac или tac транскрибируемые активирующие последовательности. Все вышеупомянутые последовательности были охарактеризованы ранее и могут быть сконструированы или синтетически или из известных плазмид.
В определенных вариантах, описанных здесь, репликация плазмиды определяется термоиндуцирующимся репликоном неконтролируемого роста, описанным как в британской патентной публикации № 1557774, так и в Uhlin et al., 1979, е е 6:91. При температурах ниже 30°С, в частности 25°С, репликон поддерживает относительно низкое число копий, примерно 10-15 копий на клетку. При повышении температуры до 37°С контроль числа копий теряется и плазмиды, содержащие репликон, амплифицируются до 1000-2000 копий на клетку. Конкретный репликон неконтролируемого роста, приведенный здесь в качестве примера, содержится в ранее описанной плазмиде рШ-11- АЗ исходном материале. Специалисты в данной области должны понимать, что изо- 5 бретение нерграничивается использованием какого-либо конкретного репликона неконтролируемого роста или мутанта числа копий. Другие репликоны, вызывающие безудержный рост или большое число ко- 0 пий, могут быть получены путем соответствующей селекции или могут быть сконструированы в соответствии с процедурой, описанной в Международной публикации № W082/02901. Такие репликоны могут 5 быть использованы для конструирования векторов экспрессии, которые также входят . в область изобретения.
Клонирование генов в векторы, содержащие репликон неконтролируемого роста,
0 приводит в результате, к индуцированию и потере контроля числа копий, к сильному увеличению скорости синтеза белка и сопутствующему образованию до настоящего времени неизвестных и неописанных видов
5 внутриклеточных белковоподобных гранул. Такие гранулы, которые также входят в изобретение, являются высоко гомогенными по их белковому составу, и, следовательно, отличимы от известных высокомолекулярных
0 агрегатов и инклюзий, которые иногда встречаются в клетках-хозяевах; содержащих рекомбинзнтную ДНК. Последние включения являются гетерогенными, обычно содержащими ассортимент клеточных
5 компонентов, таких как нуклеиновые кислоты, углеводы, липиды и пептиды, и содержат только 10-20% конкретного белкового продукта. Гранулы изобретения являются высоко однородными по целевому протеиновому
0 продукту, составляющему по крайней мере 50%, а чаще более 80% гранулы.
Настоящие новые виды гранул могут
с быть легко выделены из лизатов клеток, они
являются стабильными к промывке низкими
5 концентрациями мочевины или детерген - тов. Промывка удаляет белки, которые связаны не специфически в грануле, Выделение, которое генерирует высоко специфический активный материал, представ0 ляет собой полезную первую стадию в очистке чужеродных белков. Все последующие стадии очистки, следовательно, упрощаются. Особенно полезным является тот факт, что компоненты клеточных стенок мо5 гут быть легко отделены от гранул.
Полагаем, что образование гранул изобретения изолирует такой чужеродный белок, чтобы не происходило нарушения нормального клеточного метаболизма и
дреждевременной гибели клеток. Некоторые белки, такие как человеческий проинсу- лин, быстро деградируют в E.coli и не накапливаются. Однако, когда такие белки кодируются в векторах, содержащих репли- кон неконтролируемого роста, проинсули- новый белок образует нерастворимые гранулы, которые защищают первоначально нестабильный белок от протеолитическогоо разложения. Следовательно, образование настоящего вида гранул является полезным не только для накопления нестабильных белков, но также для упрощения процедур выделения и очистки генных продуктов.
Многие модификации и варианты насто- ящих иллюстративных последовательностей ДНК являются возможными. Например, вырожденность генетического кода допускает замещение нуклеотидов на всем протяжении областей, кодирующих полипептиды, а также замещения транслируемых стоп-сигналов TAG или TGA не спеАСТ . ACTциально подтвержденный примером транслируемый стоп-сигнал ТАА. СледоваАТГ.
тельно, как определено здесь выше, может быть одной из любых возможных последовательностей ДНК, которые кодируют аминокислоты 9 (лейцин) до 191 (фенилаланин) 6ГР. Такие последовательности могут быть выведены из.известной аминокислотной последовательности 6ГР и могут быть сконструированы по следующим традиционный синтетическим процедурам. Однако нуклео- тидные триплеты должны быть выбраны в соответствии с известными принципами и экстраполяциями, рассмотренными у Цуке- ра и Стейглера, 1981. Исследование нуклеи новых кислот 9(1):133, чтоВы избежать генерирования дополнительных оснований в мРНК. Водородное связывание междута-. кими комплиментарными основаниями приводит в результате к конфигурациям стержня и петли и свертыванию, которое снижает эффективность трансляции. Специалисты в данной области должны понимать, что все описанные выше модификации и вариации могут быть традиционно синтезированы в полном соответствии с цитированными .ранее синтетическими методиками. Следовательно, изобретение никоим образом не ограничивается специально представленными в примерах последовательностями ДНК и плазмидами. Векторы экспрессии и способ изобретения могут быть применены к широкому кругу организмов-хозяев, например, грамотрицател ьным прокариотным организмам, таким как Escherichia cotl. Е. сой К12 Е. соГсК12 RV308, E. coli K12 НВ101, Е.
coli K12 С600, Е. coli K12 С600 RirMic, E. coll К12 RRI, E. coli K12.MM294 и подобным. Хотя все варианты изобретения являются полезными, некоторые векторы и трансформанты
являются предпочтительными. Предпочти-, тельные векторы включают pCZ1920, pJR1, рЛ.З, pCZ1T2, pAT1, рАТ1.3, рАТ2, рАТ1.3, pASP1,pASP1,3,pCZ154, pCZ154.3, pCZ156, PCZ156.3. pASP2.3 и pASP2, а предпочтительные трансформанты включают Е. coli К12 RV 308/pJR1, E. coli K12 RV308/pCZ1920, E. coll К12 RV308/plRI, E. coll K12 RV308/pJRI.3, E. coli .К12 RC308/pCZ112, E,-co.ll K12 RV308/pAT1, E.
5 coli K12 RV308/pAT1.3, E, coli K12 RV308/pAT2, E. coli K12 RV3 Q8/pAT2.3, E. coli K12 RV308/pASP1, E. coli K12 RV308/pASP1.3, E.coli 312 RV308/pCZ154, E. COH.K12 RV308/pCZi54.3, E. coli K12
.0 RV308/pCZ156, E. coli K12 RV308/pCZ156.3, E. coli K12 RV308/QASP2.3- и E. coli K12 RV308/pASP2. Из этой предпочтит-ельной группы особенно предпочтительными являются плазмиды pCZ112, pCZ154, pCZ154.3,
5 pCZ156,pCZ156.3,pASP2.3npASP2HTpaHC- форманты E. coli K12 RV308/pCZ112, E. col K12 RV3087pCZ154, Eo. coli K12 RV308/pCZ154.3, E. coli K12 RV308/pCZ156 ; E. coli K12 RV308/pCZ156.3,.E. coli K12
0 RV308/pASP2.3 и E.coli K12 RV308/pAP2.
Специалисты в данной области должны знать, что векторы экспрессии изобретения используются для трансформации подходящих организмов-хозяев, таких, чтобы произ5 водное бычьего гормона роста
окспрессировалось при использовании к стандартных условий ферментации. Продукт, экспрессированный в виде высоко гомогенной гранулы и. выделенный
0 рутинными методами из полученного с результате лизата клеток-хозяев, является полезным для повышения молочной продуктивности и обычно для стимулирования роста крупного рогатого скота. Способы
5 использования, введения и подходящие дозировки для крупного рогатого скота известны иописаны в Публикации Европейского патентного ведомства N 0085036, страницы 7-9, приведенной здесь
0 в качестве уровня техники.-Следующие при- меры далее иллюстрируют изобретение, описанное здесь.
П р и м е р 1. Конструирование плазмиды pf JM789B.
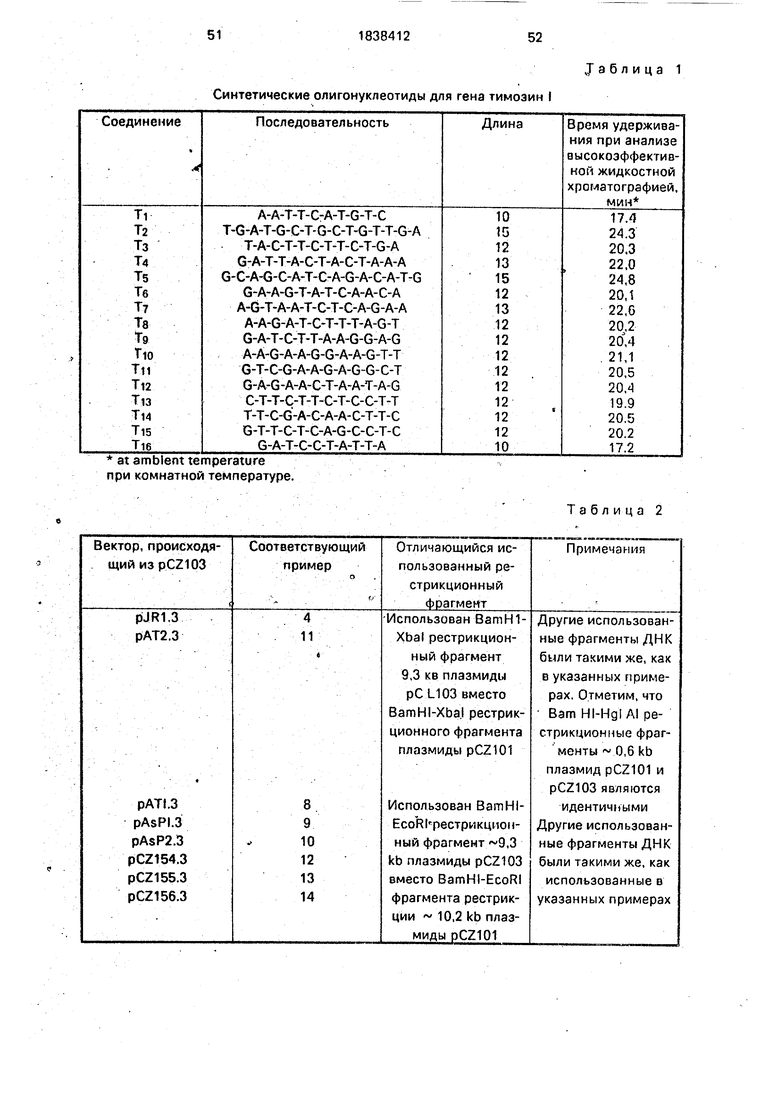
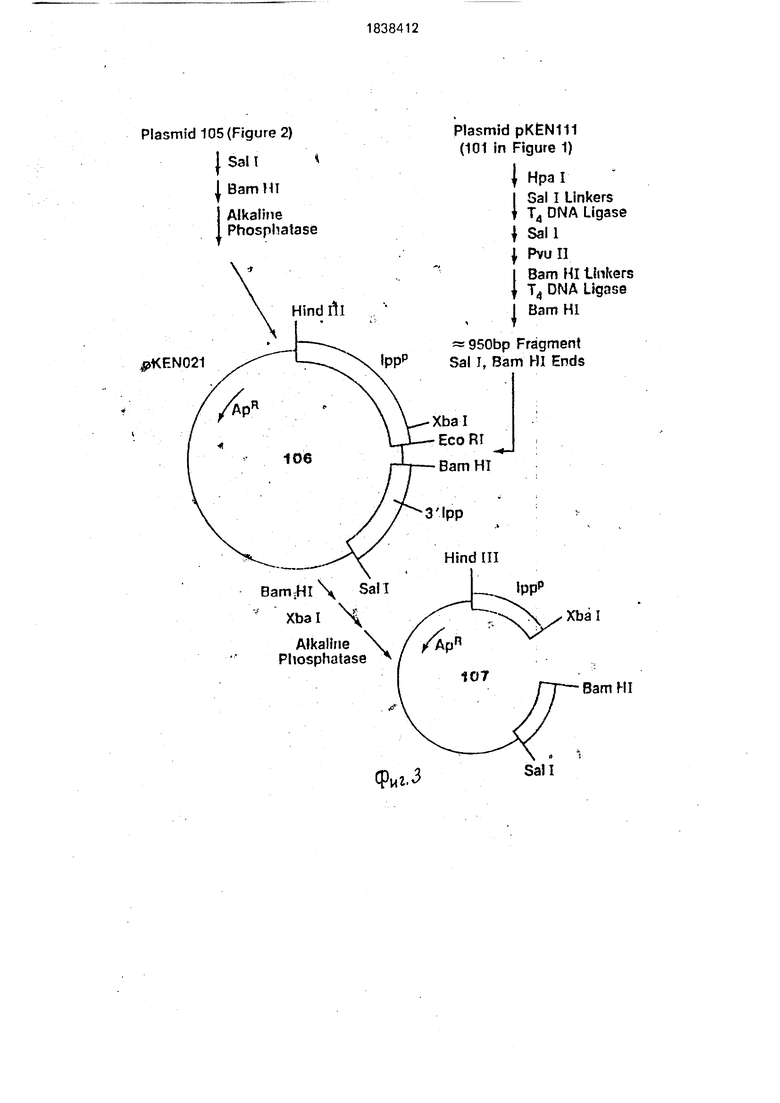
5 А. Конструирование плазмиды pKEN021 и ее фрагмента Xbal-BamHI.
В качестве исходного материала используют фрагмент 5,1 kb полученный путем Xbal-BamH расщепления плазмиды pKEN021(106 на фиг.З). Плазмида pKEN021
является производной pKEN111 (101 на фиг.1) и далее описана у Ли с сотр., 1981, g Bact. 1.46:861-866иуЦвибеляисотр,, 1981, J. Вас. 145: 654-656), которая внесена в Е. coli CC620 (NRRL номер хранения 15011) и которая имеет фрагмент примерно 2,8 kb, который содержит липопрстеиновый ген Е. coli. Описание этого фрагмента приведено НакамураиИноуе, 1979. Cell 18: 1109-1117. В pKEN021 последовательность из 650 пар оснований между единственными сайтами рестрикции EcoRI и Sail pB 322 была заменена последовательностями, взятыми из ли- попротеинового гена Е. coli. Последовательность липопротеинового гена (Накамура и Иноуе, 1979) включает фраг- мент462 пар оснований AI и I, идущий вверх от первого триплета (метионин) липопротеинового гена, который содержит промотор, 5 нётранслируемый участок и участок связывания рибосомы. Единственный сайт рестрикции Xbal локализован в сайте связывания рибосом 16 пар оснований перед метиониновым сигналом инициации трансляции. Pvull сайт рестрикции, локализующий 105 пар оснований вверх от концевого кодона трансляции структурального гева, был заменен нз сайт рестрикции BamHI при добавлении синтетического линкера ДНК (5 ССССАТССССЗ , полученного из-Коллаборатив Рисерч). Кодирующая последовательность для последних тридцати пяти аминокислот липопротеина, сигнал окончания трансляций, и последовательность, соответствующая 3 нетранслируемо- му участку информационной РНК, следуют за BamHI сайтом. Плазмида pKEN021 также включает некоторые периферические по- следовательности 850 пар оснований; не относящиеся к липопротеиновому гену и локализованные вниз от него в хромосоме Е.. coli. Эти последовательности включены как следствие способов и сайтов рестрикции ферментов, использованных при первоначальном выделении гена.
Ссылаясь на фиг.1, 2 и 3, плазмида pKEN021 получена из pKEN11 Т следующим образом: примерно 50 мкг рКЕМШ (101 на фиг.1) переваривают 25 ед. фермента рестрикции Hpall в 300 мкл буфера, содержащего 20 мМ Трис. HCI, рН 7,4, 10 мМ1ИдС 2 и 6 мМ / -меркаптоэтанола, при 37°С в течение 2 ч, Смесь дважды экстрагируют 300 мкл смеси 50:50 фенола и хлороформа и извлеченную водную фазу затем осаждают 2,5 объемами этанола и 0,1 объема ЗМ ацетата натрия. Шарик ДНК растворяют в 100 мкл электрофоретического буфера и фракционируют на 5%-ном полиакриламидном геле (акриламид: бис отношение равно 29:1 для
всех гелей, за исключением того, где отмечено). Гель окрашивают в растворе, содержащем 0,5 мкг/мл этидинийбромида и визуализируют полосы при длинноволно- 5 вом ультрафиолетовом облучении.Изолируют полосу 950 пар оснований и извлекают из геля электроэлюцией в мешок. После экстракции фенолом/СНС з и осаждения эта- нолом извлеченную ДНК (приблизительно
0 2,5 мк) растворяют в 25 мкл TEN (10 мМ NaCI, 10 мМ Трис. HCI, рН 7,4 и 1 мМ этилен- динитрилтетраацетата натрия (ЭДТА) рН 8.0).
Примерно 2 мкг фрагмента Hpall 950
5 пар оснований переваривают ферментом рестрикции Alu l в 200 мкл буфера, содержащего 50 мМ NaCI, 6 мМ Трис. HCI (рН 7,6), 6 мМ MgCl2 и 6 мМ Д-меркаптозтанола, в течение 2 ч при 37°С. ДНК фракционируют
0 на 6%-ном полиакриламидном геле и извлекают и очищают описанными ранее способами фрагмент Alul 462 пэр оснований. Фрагмент 462 пар оснований Alul (приблизительно 1 мкг) растворяют в 10 мкл Т4 ДНК
5 лигазного буфера (66 мМ Трис. HCI, рН 7,6, 10 мМ MgCl2, 10 мМ дитиотрейтола, 0,4 мМ АТР), содержащего 150 пикомолей фосфо- рилированного EcoRI линкера (S GGAATTCCS1 из Коллаборатив Рмсерч)и 2
0 ед. Т4 ДНК лигазы. После инкубации в течение 16 ч при 4°С смесь нагревают 10 мин при 65°С и разбавляют до 100 мкл добавлением EcoRI буфера (100 мМ Трис. HCI, рН 7,2, 50 мМ NaCI, TO мМ MgCla, 6 мМ / -мер5 каптоэтанола) и 40 ед. фермента EcoRI. После 2 ч при 37°С образец удобно экстрагируется фенолом ( и осаждается этанолом. Затем ДНК растворяют в 20 мкл Т4 ДНК лигазного буфера, содержащего
0 0,1 ед. Т4 ДНК лигазы и 0,1 мкг pBR322 (102 на фиг.1), которую затем линеаризуют EcoRi, а затем обрабатывают щелочной фос- фатазой. После лигации при 4°С в течение 16 ч полученную в результате ДНК исполь5 зуют., чтобы удобно трансформировать Е. , coli штамм К12 НВ101. Трансформанты отбирают на агаровых пластинах, содержащих 12 мкг/мл тетрациклина и изолируют плазмиды из устойчивых колоний путем 5ы0 строй щелочной экстракции, описанной у Бернбойма и Доли, 1979. Исследование нуклеиновых кислот 7: 1513:1523. Отбирают плазмиду (103 на фиг.1), содержащую 466 пар оснований фрагмента Xbal-BamHI, и ис5 пользуют в качестве исходного материала для следующей описанной стадии.
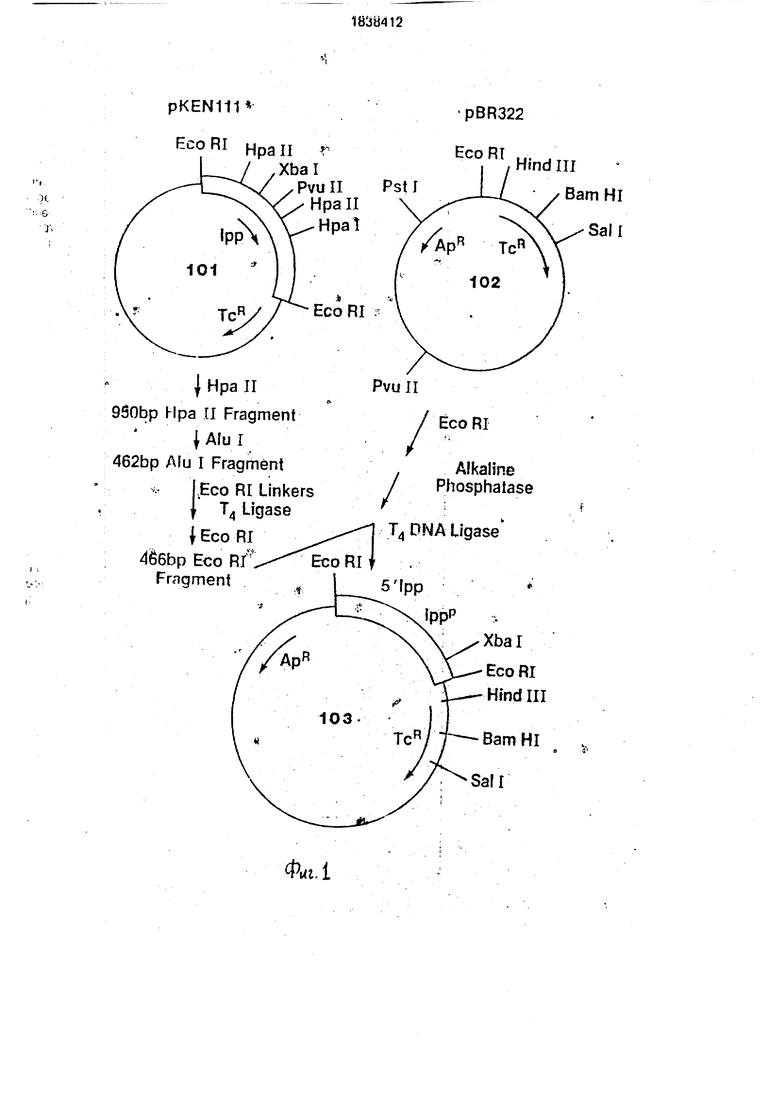
Около 2 мкг этой плазмиды (103 на фиг.2) переваривают 2 ед. фермента Hind III в 50 мл Hind HI буфера (60 мМ NaCI. 10 мМ
. HCI, рН 7,4. 10 мИ MgCl2 и 6 мМ
/3-меркаптоэтанола) в течение 1 ч при 37°С. После экстракции фенолом/СНС з и осаждения этзнолом ДНК растворяют в 200 мкл буфера, содержащего 300 мМ NaCI, 30 мМ ацетата натрия, рН 4,25, 1 мМ ZnCte и 200 ед. SI нуклеазы (Майлс Лебораториз). После 1 ч при 15°С реакцию останавливают экстракцией фенолом/СНС1з и осаждением этанолом. Полученную в результате ДНК растворяют в 10 мкл Т4 ДНК лигазного буфера, содержащего 20 пикомолей фосфори- лиров.анныхBamHI линкеров (5 CCGGATCCGG3 из Коллаборатив Рисерч) и 2 ед. Т4 ДНК лигазы. После 16 ч при 4°С реакционную смесь нагревают 10 мин при 65°С для инактивации лигазы, а затем разбавляют до 100 мкл в BamHI буфере(150 мМ NaCI, 20 мМ Трис. HCI, рН 8,0,10 мМ MgCla, 6 мМ /J-меркаптоэтанола), содержащем 20 ед. фермента BamHI. После 2 ч при 37°С смесь очищают на 1%-ном агарозном геле. Гель окрашивают и извлекают элюцией больший фрагмент (примерно 4,5 kb) после охлаждения, а затем очищают экстракцией фенолом/СНС з и осаждением этанолом, Извлеченный фрагмент с BamHI липкими концами растворяют в 20 мкл Т4 ДНК лигазного буфера, содержащего 0,1 ед. Т4 ДНК лигазы. После 16 ч при 4°С используют ДНК для трансформации E. coli HB101. Трансформанты отбирают по устойчивости к ам- пициллину (Арг) при 100 мкг/мл и скринируют на чувствительность к 10 мкг/мл тетрациклина (Тс). Несколько плаз- мид, полученных по описанной ранее процедуре Вирнбойма, из колоний, которые являются AprTcs, были исследованы на отсутствие Hindlll сайта и присутствие единственного BamHI сайта. Последовательный перевар EcoRI. Sail дает фрагмент 466 пар оснований и фрагмент 305 пар оснований. Отбирают плазмиду (104 на фиг.2) с этими характеристиками и затем модифицируют для превращения EcoRI сайта, локализованного выше Ipp промотора, в Hind 111 сайт рестрикции.
Переваривают 2 мкг плазмиды (104 на фиг.2) в 100 мкл EcoRI буфера с 0,2 ед. EcoRI о течение 10 мин при.37°С. Реакцию останавливают нагреванием в течение 10 мин при 65°С, а затем после экстракции фено- лом/СНС з осаждают ДНК этанолом, растворяют в 200 мкл SI нуклеазного буфера, содержащего 5 нуклеазу в количестве 1000 ед./мл и проводят реакцию при 12°С в течение 1 ч. Реакцию останавливают экстракцией фенолом/СНС1з и осаждением этзнолом. Полученную в результате ДНК ре- суспендируют в 10 мкл Т4 ДНК лигазного буфера, содержащего 20 пикомолей фосфорилированного Hind III линкера (S CCAAGCTTGGS1 из Коллаборатив Рисерч) и 2 ед. Т4 ДНК лигазы. После 16 ч при 4°С смесь нагревают 10 мин при 65°С, разбавляют до 150 мл в Hind III буфере, содержащем 10 ед. фермента Hind III, инкубируют 2 ч при 37°С, а затем фракционируют на 1%-ном агарозном геле. Самую большую полосу (эквивалентную единственному отрезку плазмиды) обычно извлекают и очищают, растворяют в 20 мкл Т4 лигазного буфера, содержащего 0,2 ед. лигазы, инкубируют при 4°С в течение 16 ч, а затем используют при трансформации в Е. coll
5 НВ101. Отбирают трансформанты по устойчивости к ампициллину, изолированные плазмиды анализируют с помощью рестрмк- ционного ферментного анализа. Отбирают плазмиду (105 на фиг.2) с фрагментом EcoRI0 Hind 111 из 500 пар оснований и используют как вектор клонирования для введения 3 участка Ipp гена.
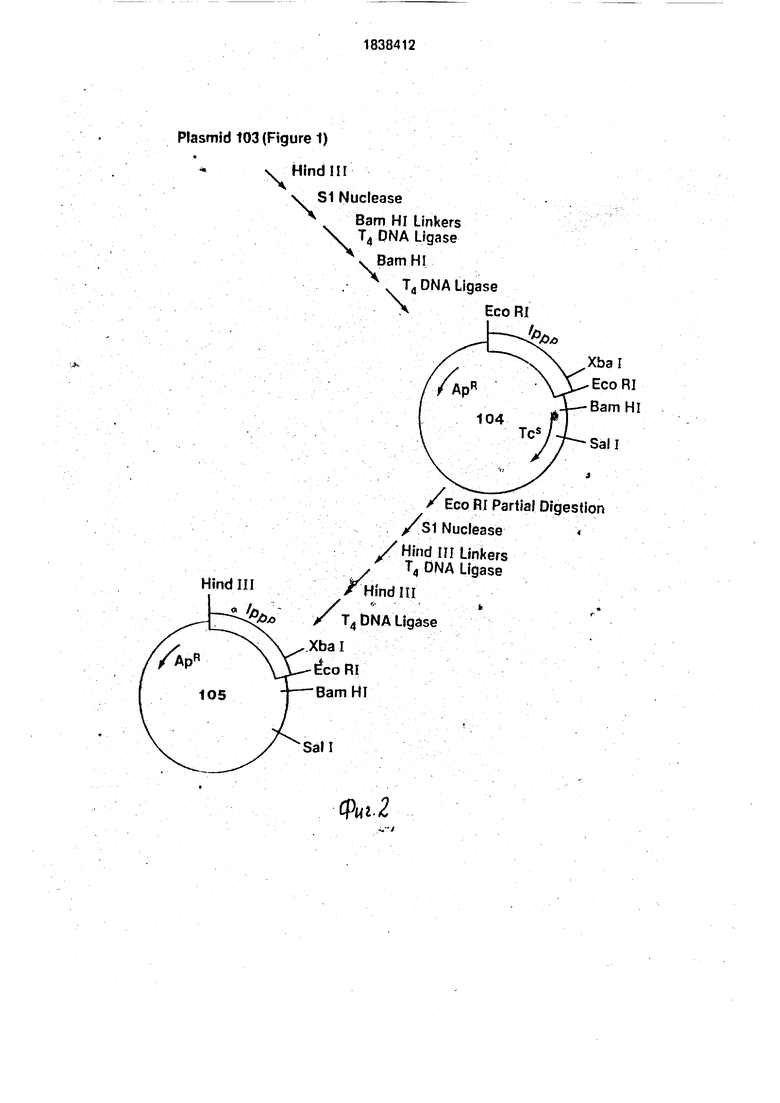
Переваривают около 2 мкг плазмиды (105 на фиг.З) в 50 мкл буфера Sail рестрик5 Ции (150 мМ NaCI, 6 мМ Трис. HCI, рН 7,9, 6 мМ , 6 мМ / -меркаптоэтанола) с 2 ед. Sail в течение 1 ч при(370С, а затек; разбавляют в 150 мкл буфера BamHI, содержащего 2 ед. BamHI. После 1ч при 37°С
0 прибавляют 2,5 ед. щелочной фосфатазы и продолжают инкубирование в течение 1 ч при 65°С. Материал экстрагируют фенолом/ , осаждают этанолом, растворяют в TEN и используют в качестве
5 клонирующего вектора для Ipp З -фрагмента. Для получения фрагмента, содержащего область Ipp 3, переваривают 10 мкг pKEN III (101 на фиг.З) в 200 мкл Нра буфера (20 мМ KCI, 10 мМ Трис. HCI, рН 7,4, 10 мМ
0 MgCl2 и 6 мМ / -меркэптоэтанола) с 10 ед. Нра в-течение 2 ч при 37°С. После экстракции фенолом/СНС1зи осаждения этанолом. растворяют ДНК в 10 мкл Т4 ДНК лигазного буфера, содержащего 20 пикомолей фосфо5 рилированногоSail линкера (5 GGTCGACC3 из Коллаборатив Рисерч) и 2 единицы Т4 ДНК лигазы, а затем инкубируют 16 н при4°С. Лигазу инактивиру ютпри нагревании при 65°С в течение 10 мин. Пол- 0 ученный в результате мате риал разбавляют в 100 мл.в Sail буфере, содержащем 10 ед. Sail, и инкубируют 1 ч при 37°С, затем разбавляют в 300 мл в Pvull буфере (60 мМ NaCI, 6 .мМ Трис. HCI, 7,5, 6 мМ MgCl2, 6 мМ
5 /3-меркаптоэтанола), содержаа1ем 10 ед. ре- стриктивного фермента Pvulll. После 1 ч при 37°С ДНК фракционируют на 5%-ном поли- акриламидном геле. Извлекают примерно 0,5 мкг фрагмента 950 г.-ар оснований, очи- щаюти растворяют вТЕ. РазбавляютО,2 мкг
фрагмента в 20 мкл Т4 ДНК лигазного буфера, содержащего 20 пикомолей фосфорили- рованногоBam HI линкера (5 CCGGATCCCG3, из Коллаборатив. Ри- серч) и 2 ед. Т 4 ДН К лигазы, а затем инкуби- руют 16 ч при 4°С. Полученную в результате ДНК затем нагревают в течение 10 мин при 65°С, разбавляют в 100 мкл BamHI буфера, содержащего 20 ед. BamHI, инкубируют при 37°С в течение 2 ч, а затем фракционируют на 5%-ном полиакриламидном геле, чтобы удалить избыток линкерных молекул. Полученный в результате фрагмент 950 пар оснований, имеющий-BamHI и Sail липкие концы, очищают традиционным способом и растворяют в 20 мкл Т4 ДНК лигазного буфера, содержащего как 0,2 мкг клонирующего вектора, описанного ранее, и 0,2 ед. Т4 ДНК лигазы. После инкубирования в течение 16 ч при 4°С используют ДНК длятранс- формации в Е. coli K12 НВ101. Получают плазмиды из устойчивых к ампициллину трансформантов и традиционно анализируют Sall-BamHI фрагмент. Описанную плаз- ми-ду (примерно 5,2 kb) обозначают PKEN021 (106 на фиг.З).
Переваривают Юмкг pKEN021. при37°С D 200 мкл Xbal/BamHI буфера (150 мМ NaCI, 10 мМ Трис. HCI, рН 8. 10 мМ MgCl2, 6 мМ / -меркаптоэтанола). используя 10 ед. BamHI, в течение 1 ч, затем 10 ед. Xbal в течение еще 1 ч при 37°С. Желаемую переваренную Xbal-BamHI ДНК затем обрабатывают 2.5 ед щелочной фосфатазы в течение 1,5 ч при 65°С экстрагируют фенолом /СИСЬ, собирают при осаждении этанолом и растворяют в 50 мкл TEN для дальнейшего использования (107 на фиг.З).
Б. Конструирование плазмиды pNM575.
В качестве источника фрагмента ДНК, содержащего кодирующую последовательность для участка гена человеческого гормона ро ста используют плазмиду pt rpED50chGH800 (108 на фиг.4) описанную Мартиалом с сотр. 1979, Science 205:602- 607, Этот фрагмент также может быть сконструирован синтетически (Итакура с сотр. 1988-и Кри с сотр. 1978) или может быть получен с использованием известных методик, описанных Гудманом с сотр. 1979; Me- тоды в Энзимологии 68:75-90, путем выделения мРНК, кодируюа ей человеческий гормон роста, из гипофиза человека. Участок гена человеческого гормона роста плазмиды ptrpED50chGH800 содержит единственный Smal сайт рестрикции 6 пар оснований ниже от кодона окончания трансляции гена. Этот сайт был заменен на сайт BamHI при использовании следующей процедуры: переваривают 6 мкг плазмиды 6 ед.
Smal в 200 мкл буфера Smal рестрикции (15 мМ Трис. HCI, рН 8,0, 6 мМ МдС1з. 16 мМ KCI и 6 мМ Д-меркаптоэтанола) в течение 1,5 ч при 37°С. После окончания переваривания проводят экстракции фенолом ( и извлекают ДНК осаждением этанолом, затем растворяют ее в 24 мкл TEN, Прибавляют 40 пикомолей фрагмента фосфорилированно- го BamHI адаптера (Коллаборатив Рисерч) к 0,5 мкг (0,2 пикомоля концов) переваренной выше плазмиды к 16 мкл лигазного буфера, содержащего 2 ед. Т4 ДНК лигазы. Смесь инкубируют 2 ч при 22°С, 16 ч при 4°С и затем 10 мин при 65°С. Генерируют BamHI липкие концы путем традиционного переваривания ВатН рестрикционным ферментом. Фермент расщепляет линкерную последовательность, а также BamHI сайт, локализованный у начала клонированной последовательности ДНК человеческого гормона роста. Это дает фрагмент 691 пар оснований с липкими BamHI концами, который выделяют на 6%-ном полиакриламидном геле, а затем извлекают обычным способом. Извлеченный фрагмент ДНК ли- гируют (используя 0,2 ед. Т4 ДНК лигазы в 20 мкл буфера в описанных ранее условиях) с 0,2 мкг переваренной BamHI и обработанной щелочной фосфатазой рВ 322 (102 на фиг.4). После 16ч при4°С материал используют для трансформации с E.coli штамм JA221/NRRL NB-15014 по существу в соответствии с процедурой трансформации Вен- синка с сотр. 1974, Cell 3:315-325. Отбирают трансформанты на агаровых пластинах, содержащих 100 мкг/мл ампициллина, а затем изолируют обычным способом плазмиды и идентифицируют их с помощью анализа ре- стрикционными ферментами и гель-электрофореза. Желаемые плазмиды, обозначенные как pNM575 (109 на фиг.4), содержат BamHI фрагмент приблизительно 700 пар оснований, и амплифицируют их обычным способом для дальнейшего использования.
В. Конструирование плазмиды pNM702.
Последовательность ДНК зрелого человеческого гормона роста содержит сайт РпиД11, который имеет 47 пар оснований от первого нуклеотида. Переваривают 25 мкг pNM-575 в 250 мкл BamHi буфера с 25 ед. BamHI при 37°С в течение 1 ч. Фрагмент591 пара оснований с BamHI липкими концами обычно выделяют на 6%-ном полилакрила- мидном геле и очищают. После очистки фрагмента одну треть его (эквивалентную 8 кг плазмиды) переваривают в 100 мкл РпиД11 буфера (6 мМ NaCI, 6 мМ Трис. HCI, рН 7,4, 6 мМ MgCla, 6 мМ / -меркаптоэтано- ла)с2,5 ед. РпиД11 в течение 1,5 ч при37°С.
Используют электрофорез на 6%-ном полиакриламидном геле и стандартные процедуры изилочения для выделения фрагмента 538 пар оснований ДНК, содержащего кодирующую последовательность для последних 175 аминокислот гена после транслируемого стоп сигнала.
Фрагмент двуиитевой ДНК (110 на фиг.5) был синтезирован фосфотриэфирным методом для соединения области Ipp промотора с участком, кодирующим человеческий гормон роста. Двунитевый фрагмент ДНК (110 на фиг.5) имеет следующую последовательность:
ZSXbjt 5 3
СТАОАСОСТЛГГАЛТЛЛТСЛТСССАТТССАЮАТСАТПАТЛАСТТСССАА- ТСССАТМТТЛТТАСАЛСССТААсёгАс ГАСТАСТАТГСАЛСССГГ30 ССЛТТСССТТЛТССЛССС7ТТГ1СЛСАЛСССТА1-ОСТССС 3 ССТЛЛГ.СГ.ЛАТАСОТСССАЛАААСТОТТССС-ЛТАССЛаСС S
Фрагмент б ыл получен известным фосфотриэфирным способом, по которому были получены следующие сегменты:
S 2)ТААТАЛТСТТСС
3)СЛТТССЛТСЛТ
4)GATGATAAGTTCC
5)САЛССАТТССС
6) TTATCCAGGC
LO 7)TTTTTGACAACG
О)CTATGCTCCG
9)СЛТТЛТТЛАТАСССТ
10)ЛТСССЛА :
11)СТТЛТСАТСЛТСАТССЛ «
15 12)GGTTGGGAA
13)GGATAAGGGAAT
14)GTCAAAAACCCT
15 )CGGAGCATAGCGTT
Используя приготовленные выше сегменты проводят реакции соединения, катализируемые Т4 лигазой, как описано ниже:
1) Соединяют 5 -нефосфорилировэнный сегмент 1 с 5 -фрсфорилированным сегментом 2 в присутствии 5 -фосфорилированного сегмента 9, используя Т4 лигазу, для образования дуплекса 1 ДНК(Броун с сотр., 1979, Методы в Энзимологии 68:109-151). Дуплекс выделяют с помощью препаратив- ного гель-электрофореза на 15%-ном поли- акриламидё,
2) Соединяют 5 -фосфорилированный сегмент 3 с 5 -фосфорилированным сегментом А в присутствии 5 -фосфорилировзннэго
сегмента 11, используя Т4 лигазу, для образования дуплекса 2 ДНК, который очищают с помощью электрофореза на 15%-ном полиакриламидном геле.
3) Соединяют 5 -фосфорилированный сегмент 5 с5 -фосфорилировзнным сегментом 6 в присутствии 5 -фосфорилированных сегментов 12 и 13, используя Т4 лигазу, для образования дуплекса 3 ДНК, который очи0 щают электрофорезом на 15%-ном полиак- риламидном геле.
4) Соединяют 5 -фосфорилироианный сегмент 7 с 5 -фосфорилированным сегментом 8 в присутствии Б -фосфорилированного
5 сегмента 14 и 5 -нефосфорилированного сегмента 15, используя Т лигазу, для образования дуплекса 4 ДНК, который очищают электрофорезом на 15%-ном полиакрила- мидном. геле.
0 5) Соединяют вместе дуплексы ДНК 2, 3 и 4 с помощью Т4 пигазы для образования дуплекса ДНК 5, который очищают электрофорезом на 15%-ном полиакриламидном геле.
5 6) Прибавляют 5 -фосфорилированный сегмент 10 и дуплекс 5 ДНК в присутствии Т4 лига зы к дуплексу 1 ДНК. Полученный в результате дуплекс ДНК (110 на фиг.5) очи: щают на 10%-ном полиакриламидном геле с
0 помощью электрофореза. Этот дуплекс ДНК затем зизиматически фосфорилируют, используя Т4 полинуклеотид киназу и (у- 32Р/ЛТР по следующей установленной процедуре.
5Конструируют плазмиду экспрессии pNM702 с помощью ферментного присоединения 0.1 гшкомоля (0,4 мкг) Xbal-BamHt фрамгента- плазмиды pKEN021 (107 на фиг.5). 0,025 пикомолей. синтетического
0 фрагмента ДНК (110 на фиг.5) и 0,3 пикомолей (0,08 мкг) фрагмента 538 пар оснований (из 109 на фиг.5) в 24 мкл лигационного буфера, используя 1,5 ед. Т4 ДНК лигазы. После инкубирования в течение 16 ч при 4°С
5 смесь используют для трансформации, в E.coli A211, как описано ранее. Отбирают трансформанты на агаровых пластинах, содержащих 100 мкг/мл ампициллина, и культивируют обычным образом как
0 предпочтительный источник желаемой плазмиды экспрессии.
Определяют экспрессию человеческого гормона роста по стандартной радиоимму- нологической процедуре (Твомей с сотр.,
5 1974 Ciin Chem 20:389-391) и определяют наличие по крайней мере 2 миллионов молекул на клетку.
Д. Конструирование плазмиды pNM789.
Используют плазмиду PNM702 (111 на
фиг.б). плазмиду экспрессии гормона чело10
веческого роста, в качестве исходного материала для конструирования плазмиды экспрессии Met-Phe-Pro-Leu-Asp-Asp-Asp- A$p-Lys-6fP.
Используют плазмиду рВР348 (112 на фиг.6), описанную Миллером с сотр., 1980, J. Blol. Chem, 255:7521-7524. в качестве источника двух фрагментов ДНК, содержащих кодирующую последовательность участка гена бычьего гормона роста. Плазмида содержит 831 пар оснований последовательности, кодирующей бычий гормон роста, клонированной в Psjl сайт рестрикции рЕ|5322. В качестве альтернативы методу, описанному Миллером с сотр., 1980, после- 15 до|вательность бычьего гормона роста мо- жфт быть сконструирована синтетически (И|гакура с сотр., 1977, и Кри с сотр., 1978) же может быть также получена из информационной РНК, выделенной из бычьих 20 гипофизов по уже рутинным процедурам, описанным Гудманом с сотр., 1979.
Последовательности; кодирующие человеческий гОрмон роста, и бычий гормон роста, являются очень похожими и показы- 25 вают много гомологии. Особенно полезными при конструировании плазмиды . экспрессии бычьего гормона роста являются фрагменты, генерированные при переваривании рестрикционным ферментом Pvull. 30 Размер продуцированных фрагментов составляет 497 пар оснований для человеческого гормона роста и 494 пар оснований для бычьего гормона роста, а соответствующие сайты рестрикции встречаются в одина- 35 ковых рамках считывания в обеих последовательностях.
Частично переваривают 10 мкг pNM702 (111 на фиг.6) с помощью 1 ед, Pvull в 200 MKjn буфера Pvuf рестрикции (60 мМ NaCI, 6 40 мМ.Трис. НС.. р Н 7,5, 6 мМ М , 6 мМ /3- еркаптоэтанола) в течение 10 мин при 37°С. После этого реакцию останавливают нагреванием при 65°С в течение 10 мин, ДНКобрабатывают щелочной фосфатазой и 45 отделяют фрагменты на 1%-ном агарозном геле. Линейный фрагмент ДНК (113 на фиг.6), размер которого соответствует ДНК с недостающим фрагментом Pvull 497 пар оснований (серия опытов несколько быст- 50 рее, чем единственный разрез плазмиды), был вырезан очищен и использован при конструировании промежуточной плазмиды (114 на фиг.6).
Готовят фрагмент 494 пар оснований 55 Pvull плазмиды рВР348 путем переваривания 10 мкг плазмиды в200мкл Pvull буфера, содержащего 10 ед. Pvull в течение 1 ч при 37°С. Выделяют фрагменты на 6%-ном по- лиакриламидном геле и визуализируют и
0
5
очищают обычным способом целевой фрагмент 494 пар оснований (из 112 на фиг.6).
Конструируют промежуточную плазмиду (1 14 на фиг.6) при взаимодействии 0,2 мкг плазмиды pNM702 Pvull фрагмента с 0,05 мкг фрагмента 494 пары оснований в 20 мкл Т4 ДНК лигазного буфера, содержащего 2 ед. Т4 ДНК лигазы, в течение 16 ч при 4°С. После трансформации и отбора трансформантов на устойчивость к ампициллину плазмиды подвергают обычному анализу на наличие и четкую ориентацию Pvull фрагмента 494 пар оснований. Плазмиды с фрагментом Pvull 494 пары оснований и фрагментом Xbal-Smal 440 пар оснований отбирают для использования при дальнейшем конструировании.
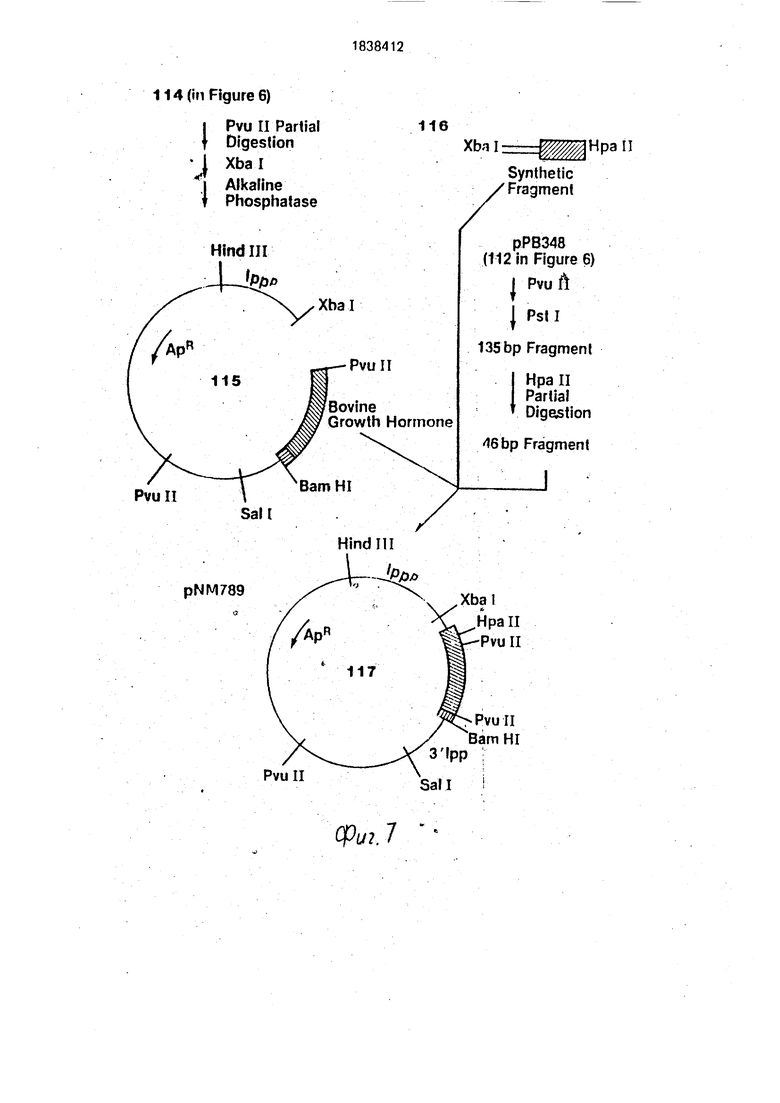
Переваривают 10 мкг промежуточной плазмиды (114 на фиг.7) с помощью 1 ед, Pvull в 200 мкл Pvull буфера в течение 5 мин при 37°С. После нагревания при 65°С.в течение 10 мин смесь наносят на 1 %-ный ага- розный гель и извлекают линейную ДНК, имеющую только один Pvull разрез на молекулу, очищают ее. Этот извлеченный материал (приблизительно 3 мкг) полностью переваривают 5 ед. Xbal и обрабатывают щелочной фосфатазой. Фрагменты наносят на 1 %-ный агарозный гель и извлекают самый большой фрагмент потерявший фрагмент 109 пар оснований между Xbal и первым сайтом Pvull у человеческого и бычьего гормона роста по традицией ной методике (1 15 на фиг.7).
Последовательность ДНК для первых 23 аминокислот (69 пар оснований) бычьего гормона роста до первого Pvull сайта содержит два-Hpall сайта рестрикции, первый из которых содержит 23 пары оснований от первого нуклеотида кодирующей последовательности. Фрагмент 63 пары оснований (116 на фиг.7) был синтезирован фосфорот- ризфирным методом. Этот фрагмент соответствует последовательности 19 пар оснований от Xbal сайта в сайте Ipp рибо- сомного связывания через АТС транслируемый старт-сигнал, после кодирующей последовательности для Phe-Pro-Leu-Asp- . Asp-Asp-Asp-Lys (24 пары оснований) и 20 нуклеотидов кодирующей последовательности бычьего гормона роста (от Ре до первого Hpall сайта). Фрагмент имеет следующую последовательность: Xbal
5 CTAGAGGGTATTAATAATGTTI I t t I -I I I I I I Г I I I I Г
,3 ТСССАТААТТАТТАСААСССATTGGATGATGATGATAAG-i i i i i i i i t i t I i t i i i i 10 GGGTAACCTACTACTACTATTC. TTCCCAGCCATGTCCTTGTC 3 ИИЁ.1 I
I I f 1 I I I I I I I I I t I Г ( t I
AAGGGTCGGTACAGGAACAGGC5
15 5
При получении фрагмента 63 пары осноаний готовят следующие девять сегментов:
1) CTAGAGGGTAT
2) TAATAATGTTCC 203) CATTGGATGAT 10
4) GATGATAAGTTCC
5) CAGCCATGTCCTTGTC
6) ATGGGAACATTATTAATACCCT
7) ТТАТСАТСАТСАТССА 258) ATGGCTGGGAAC 15
9) CGGACAAGGAC
Используя приготовленные выше сегменты, осуществляют реакции каталитического связывания Т4 лигазой, как описано выше:20
а) соединяют 5 -нефосфорилированный сегмент 1 с 5 -фосфорилированным сегментом 2 в присутствии 5 -фосфорилированно- го сегмента б с использованием Т4 лигазы для образования дуплекса 1 ДНК, который 25 очищают электрофорезом на 15%-номполи- акриламидном геле;
б) соединяет 5 -фосфорилированные сегменты 3, 4 и 5 в присутствии 5 -фосфори- лированных сегментов 7 и 8 и 5 -нефосфори- 30. лированного сегмента 9 с использованием Т4 лигазы для образования дуплекса 2 ДНК, который очищают электрофорезом на 15%- ном полиакриламидном геле;
в) соединяют затем дуплексы 1 и 2 с 35 помощью Т4 лигазы для образования дуплекса ДНК (116 на фиг.7), который очищают электрофорезом на 15%-ном полиакрила-: мидном геле. Этот дуплекс ДНК затем фер- ментативно фосфорилируют, используя Т4 40 полинуклеотид киназу и (у- Р/АТР в сост ветствии с установленной процедурой.
Фрагмент ДНК 46 пар оснований, который простирается от вышеуказанного Hpall сайта до Pvull сайта, может быть или скон- 45 струирован синтетически, или получен из первоначальной рВР348 плазмиды. Следовательно, переваривают 0,01 мкг плазмиды рВР348 в 400 мкл Pvull буфера с 50 единицами Pvul в течение 2 ч при 37°С. После 50 экстракции фенолом и осаждения этанолом растворяют ДНК в 400 мкл Pst буфера (50 мМ NaCI, 6 мМ Трис. HCif pH 7,4, 6 мМ MgCla, 6 мМ / -меркаптоэтанола) с 50 ед. pstl в течение 2 ч при37°С. Фрагменты ДНК 55 наносят на 6%-ный полиакрилэмидный гель и извлекают фрагмент 135 пар оснований, содержащий желаемую последовательность 46 пар основамий. очищают его па
стандартным методикам. Одну треть извлеченной ДНК (эквивалентно 33 мкг) подвергают ограниченному перевариванию 1 ед. Hpail рестрикционного фермента в 100 мкл Hpall буфера (20 мМ Трис. HCI, рН 7,4, 7 мМ MgCla, 6 мМ,- /3-меркаптоэтанола) в течение 40 мин при 37°С. После нагревания при 65°С в течение 10 мин фрагменты ДНК наносят на 5%-ный акриламидный гель (акри- ламид:бис соотношение 19:1) вместе с маркером соответствующего размера. Желаемый фрагмент 46 пар оснований, полученный при частичном переваривании Hpall фрагмента в 135 пар оснований (из 112 на фиг.7), очищают по стандартным методикам.
Инкубируют 0,2 мкг Xbaj-Pvull фрагмент плазмидного вектора (115 на фиг.7), 3,2 пикомоля синтетического фрагмента 63 пары оснований (116 на фиг.7), и 0,5 пикомоля фрагмента 46 пар оснований (из 112 на фиг.7), в 10 мкл лигационного буфера с 2 ед. Т4 ДНК лигазы в течение 16 ч при 4°С. Ли- гационную смесь используют для трансформации E.coli JA221, и отбирают полученные трансформанты, которые содержат, следовательно, желаемую плазмид у pNM789, на устойчивость к ампициллину. Идентичность плазмиды pNM789 (117 на фиг.7) была подтверждена традиционным скринингом в присутствии обоих фрагментов 494 пары оснований Pvuli и 109 пар оснований Xbal- Pvjjll.
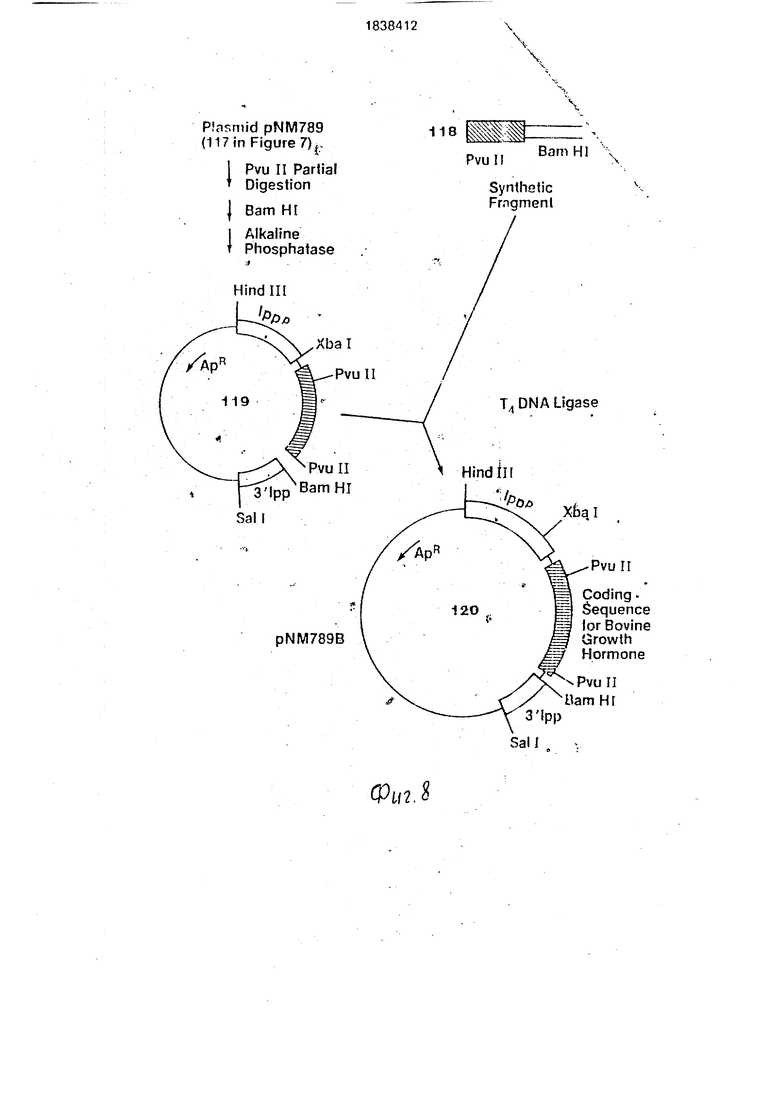
Д. Окончательное конструирование плазмиды pNM789B.
Для плазмиды pNM789 (117 на фиг.8) требуется изменение одного аминокислотного кодона для полного превращения в бычий гормон роста. Это осуществляется при удалении фрагмента 28 пар оснований от Pvull до. BamHI в pNM789 и замене синтетическим двунйтевым фрагментом, имеющим следующую последовательность (118 на фиг.8):
5 С ТС- Т& С. С Т Т С Т А Сг
I I I I I | | | , |
э(Ј А С А С О G А А с / г С СТА &s,
Переваривают 10 мкг pl IM789 1 ед. Pvuil в 200 мкл Pvull буфера в течение 5 мин при 37°С, После нагрева при 65°С в течение 10 мин смесь-разбавляют в 300 мкл добавлением BamHI буфера, переваривают полностью 10 ед. BamHI в течение 1 ч при 37°С, обрабатывают 5 ед. щелочной фосфатазы и инкубируют 1 ч при 65°С. Выделяют фрагменты ДНК на 1%-ном агарозном геле и традиционным образом очищают фрагмент ДНК(119 на фиг.8), примерно соответствую- щиГ; по размеру плазмиде pNM789 с единстоенным разрезом. Лигируют 0,2 мкг этого фрагмента 5 пикомолями синтетического фрагмента с использованием 2 ед. Т4 лига- эы в 20 мкл лигазного буфера. Лигацию про- врдят в течение ночи при 4°С. После трансформации выделяют несколько плаз- мйд и скринируют их на соответствующие фрагменты Pvull (494 пары оснований) и Xtyal-BamHI (628 пар оснований). Плазмиды, содержащие указанные выше фрагменты, составляют желаемую плазмиду pNM789B (120нафиг.8).
П р им е р 2. Конструирование плазмиды pCZ101 и E.coli K12 RV308/pCZ101. ; А. выделение плазмиды рШ-1 -АЗ.
Культивируют бактерии E.coli K12/p1 M-. Г-АЗ (NRRL № В-15733) в ТД-бульоне (1% трмптона, 0,5% дрожжевого экстракта, 0,5% хлористого натрия, рН 7,4) с 50 мкг/мл канамицина при 25°С в соответствии с традиционными микробиологическими процедурами. Затем культуру разбавляют в соотношении 1:10 свежим бульоном и после 3 4 инкубации при 37°С около 0,5 мл культу- ры переносят в трубку Эппендорфа 1,5 мл и центрифугируют примерно 15 с. Если нет других указаний, все манипуляции проводятся при комнатной температуре. Полученный в .результате надосадочный слой осторожно удаляют аспиратором с тонким наконечником, и повторно суспендируют клеточный шарик примерно п 100 мкл свежеприготовленного раствора лизбцима (2 мг/мл), который содержит 2 мг/мл лизоци- ма, 50 мМ глюкозы, 10 мМ ЭДТА (диамино- тетраацетата) и 25 мМ Трис. HCI, рН 8. Прибавляют примерно 200 мкл щелочного раствора 505(додецилсульфата натрия) (0,2 н. NaOH, 1% SDS), трубку осторожно пере- воЬачивают и затем хранят при 0°С до тех пор, пока не закончится лизис (примерно 5 ми-н). Затем прибавляют примерно 150 мкл З.М ацетата натрия и содержимое трубки осторожно перемешивают переворачиванием в течение нескольких секунд.
Трубку выдерживают при 0°С по крайней мере 60 мин, а затем центрифугируют 15 мин для получения почти прозрачного надосадочного слоя. Надосадочный слой. переносят во вторую трубку для центрифугирования, в которую добавляют 3 обьема холодного 100%-ногоэтанола. Затем трубку на 5 мин помещают на сухой лед-этанол, полученный в результате осадок собирают центрифугированием (5 мин), а надосздоч- ный слой удаляют отсасыванием. Собранный шарик растворяют в 100 мкл ТЕ (10 мМ Трис HCI, рН 8,0, 1 мМ ЭДТА), он представляет собой желаемую плазмиду р1М-1 -АЗ ДНК. .
Б. Xbal-BamH переваривание плазмиды pNM789B и генерация фрагмента примерно 0,6 kb Xbal-BamHI фрагмента.
Инкубируют примерно 5 мкг плазмиды 5 pNM789B ДНК в 50 мкл солевого буфера с 10 ед. каждого из рестрикционных ферментов BamHI и Xbal при 37°С в течение 1 ч. После добавления 5 мкл ЗМ ацетата натрия при рН 7,0 осаждают ДНК добавлением 2
0 объемов 100%-ного этанола. Целевой перевар ДНК растворяют в 100 мкл ТЕ буфера и хранят при 0°С для дальнейшего использования.
Солевой буфер обычно следующего со5 става:
100 мМ NaCI, 20 мМ Трис. HD, рН 8,0, 10 мМ MgCla, 5 мМ/3-меркаптоэтанола.
В. Xbal-BamHI переваривание плазмиды рШ-1 -АЗ.
0 Желаемое переваривание осуществляют по существу в соответствии с методикой примера 2,В, с тем исключением, что ис- . пользуют плазмиду р1М-Г-АЗ вместо плазмиды pNM789B, Растворяют .целевую ДНК
5 примерно в 100 мкл ТЕ буфера и хранят при 0°С для дальнейшего использования, Г. Лигирование и трансформация. Инкубируют примерно 1 мкг перевара плазмиды рШ-Г-АЗ, 1 мкг перевара плаз0 миды pl IM789B Xbal-BamHI, 40 мкг воды, 5
мкл (5 мМ) АТР, 5 мкл лигационной смеси и
. 5 ед. Т4 ДНК лигазы при 20°С о течение
примерно 2 ч. . Лигационная смесь может
быть приготовлена в следующем составе:
5 500 мМ Трис. HCI, рН 7,8,200мМ дитиотрей- тола, 100 мМ MgCl2. После инкубирования в течение 2 мин при 65°С и последующего охлаждения на льду полученную лигацион- ную смесь используют для трансформации
0 по существу в соответствии, с методикой трансформации Венсинка, 1974, Celi 3:315, E.coli K12 RV308 на ТД пластинах (1 % трип- тона, 0,5% дрожжевого экстракта, 0,5% хлористого натрия, 1,5% агара, рН 7,4),
5 содержащих 50 мкг/мл кзнзмицина. Бакте- - риальный штамм E.coli K12 RV308 был депонирован и является частью постоянно хранящейся коллекции культур Северной региональной исследовательской лаборато0 рии, Пеория, Иллинойс, из которой он является доступным для публики под регистрационным номером NRRL-B-15624. Некоторые из полученных в результате трансформантов, как показано электрофо5 резом на агарозном геле (Мзнийтис с сотр. 1982) и с помощью других тестов, содержат только целевую плазмиду примерно 10,8kb. Такой трансформант здесь обозначен как E.coli K12 RV308/PVC101, его отбирают, помещают на ТД агаровые пластины, содержащие соответствующие антибиотики, а затем культивируют, используя традиционные микробиологические методики. Полученные в результате клетки используют для выделения плазмиды pCZ101 по существу в соответствии с процедурой примера 2,А.
П р и м е р 3. Конструирование плазмиды pCZ1920u E.coli K12 RV308/pCZ1920.
А. Конструирование фрагмента BamHI- Xbal около 10,2 kb плазмиды pCZ1920.
Целевой фрагмент был сконструирован по существу в соответствии с мотодикой примера 2,Б, с тем исключением, что использовали плазмиду pCZ101, а не плазми- ду pNM789B, Целевые рестрикцмонные фрагменты BamHI-Xba примерно 10,2 kb традиционным образом отделяют и выделяют электрофорезом на агарозном геле (Ма- ниатис с сотр., 1982), а затем растворяют примерно в 100 мкл ТЕ-буфера и хранят при 0°С для дальнейшего использования.
Б, Конструирование фрагмента BamHI- HgiA примерно 0,6 kb плазмиды pCZ101.
.Целевой фрагмент был сконструирован по существу в соответствии с методикой примера 2,Б, с тем исключением, что были использованы плазмиды pCZ101 и рестрик- ционный фермент HgiAl вместо плазмиды pNM789B и рестрикционного фермента Xbal. Целевые фрагменты рестрикции BamHl-HgiAl примерно 0,6 kb были традиционным образом отделены и выделены электрофорезом на агарозном геле (Маниа- тис с сотр. 1982), а затем растворены в примерно 100 мкл ТЕ-буфера и хранились при 0°С для дальнейшего использования.
В. Конструирование линкерной последовательности ДНК.
51 CTAGAGGGTATTAATA АТС ТТТ ССА ССС ATG ОСТ СТА TCI GGT Э ТСССАТЛЛГТАТ ТЛС АДА GGT CGC TAG COA CAT A JA CCA 20СТП ТТТ GCC ААС GCT GTGCT З1 я GAC ААА CGG TTG CGA С. 5
Целевая линкерная последовательность была синтезирована удобным образомпо модифицированной фосфотриэфирной методике по существу в соответствии с методикой Итакура с сотр. 1977 и Кри с сотр. 1789. Вышеупомянутый способ синтеза также специально иллюстрирован в примере 1,
Г. Лигирование и трансформация.
Лигируют примерно 20:пикомолей линкера ДНК примера 3В, 1 мкг фрагмента BamHl-HgiAl примерно 0,6 kb плазмиды pCZ101, а полученную в результате плазмиду используют для трансформации :E. К12 RV308 по существу в соответствии с методикой примера 2Г.
Некоторые из полученных в результате трансформантов, как удобно показано электрофорезом на агарозном геле (Маниатис с сотр., 1982) и другими тестами, содержат
только целевую плазмиду примерно 10,8 kb. Такие траисформанты, обозначенные здесь как E.coli K12 RV308/pCZ1920, отбирают, наносят на пластины с ТД-агэром, содержащие соответствующие антибиотики, а затем
культивируют, используя традиционные микробиологические методики, Полученные в результате клетки показывают при SDS - гель-электрофореза, RIА и других тестах экспрессию вышеупомянутого MET-LYS-CLYASPN-SER-MET-ALA-бГР производного на высоких уровнях. Поскольку плазмида pCZ1920 содержит термоиндуцируемый репликой неконтролируемого роста, максимальная экспрессия желаемого продукта
производного 6ГР происходит при культивировании при температурах около 37°С.
П р и м е р 4, Конструирование плазмиды pJRI и E.coli K12 RV308/pJRI.
Получают желаемые конструкции по существу в соответствии с методикой примера 3, с тем исключением, что линкерная последовательность примера 3,В была заменена на линкерную последовательность ДНК:
S CTAGAGGGTAITAATA АТС Р.ААCGG ДАТ ТСТ AtG ССС Т7С ССА GCC
3 ТСССАГААГГАТ ТАС ТТТССС ТТЛ АСА ТЛС CGG GGI CGG
ATG ТСС TTG ТСС GGC СТС. ТТТGCC АЛС GCT CTGCT 3
ТАС АСС-, АЛС ACG CCG GAC АЛАCGG ТТС ССЛ С 5 .
Определенная выше линкерная последовательность может быть сконструирована по существу в.соответствии с традиционной методикой Итакура с сотр., 1977 и Кри с сотр. 1978. Вышеупомянутый способ синтеза также специально проиллюстрирован в
примере 1.
Целевые трансформанты, обозначенные здесь как E.coli K12 RV308/pJRI, помещают на ТД агаровые пластины, содержащие соответствующие антибиотики, а затем культивируют обычным образом для последующего продуцирования и выделения плазмиды pJRI. Трансформанты также были показаны SDS-гель-электро- форезом, R1A и другими тестами, для
экспрессии вышеопределеиного производного MET-PHE-PRO-ALA-VET-ALA-R2 бычьего гормона роста. Поскольку плазмида pJRI содержит термоиндуцируемый репликон неконтролируемого роста, максимальная
экспрессия целевого продукта производного 6ГР происходит при культивировании при температуре около 37°С.
П р и м е р 5. Конструирование плазмиды рН17Д4Д1. ;
Частичная схема конструирования плазмиды РН17 Л4Д1 представлена на фиг.15.
А. Конструирование плазмиды pBRHtrp.
Плазмида pGMl несет E.coli триптофа- 5 новый оперон, содержащий делецию ALE1413 (Миоззари с сотр. 1798, J. Bacteriology, 1457-1466) и, следовательно, экспрессирует плавленный протеин, состоящий из первых 6 аминокислот trp лидера и ю приблизительно последней трети trp E пол- ипептидз (здесь далее упоминается вместе ка|к LE }, а также trp D полипептид целиком, все под контролем системы trp промотор- оператор. E.coli K12 W3110-tna 2trp- 15 A102/pGMI был депонирован в Американской коллекции типов культур (АТСС № 31622), a pGMl может быть удобно удалена из штамма для использования в описанных ниже процедурах.20
Примерно 20 мкг плазмиды переваривают рестрикционным ферментом Pvull, который расщепляет плазмиду в пяти сайтах. Фрагменты генов затем объединяют EcoRI линкерами (состоящими из самокомплемен- 25 тарного олигонуклеотида последовательности: pCATGAATTCATG). обеспечивая EcoRI сайт расщепления для последующего кло- нирования в плазмиду, содержащую EcoRI сайт. Обр абатывают 20 мкг фрагментов 30 ДНК, полученных из pGMl, 10 ед. Т4 ДНК лигазы в присутствии 200 пикомолей 5 -фос- фарилированного синтетического нуклеоти- да pCATGAATTCATG и в 20 мкл Т4 ДНК лигазного1 буфера (20 мМ Трис, рН 7,6, 0,5 35 мМ АТР, 10 мМ MgCla, 5 мМ дитиотрейтола) при 4°С в течение ночи. Затем раствор нагревают в течение 10 мин при 70°С для остановки лигации. Линкеры расщепляют перевариванием EcoRI u разделяют фраг- 40 менты, имеющие теперь EcoRI концы, используя электрофорез на 5%-ном полиакриламидном геле (ЭПАГ). Выделяют три самых больших фрагмента из геля, сначала окрашивая этидинийбромидом, а за- 45 тем локализуя фрагменты в ультрафиолетовом свете и вырезая из геля интересующие участки, Каждый фрагмент геля с 300 мк.п 0,1хТЕВ помещают в ванну ля диализа и подвергают электрофорезу 50 при 100 в в течение 1 ч в 0,1хТВЕ буфере ТВЕ буфер содержит: 10,8 г Трис основания, 5,5 г борной кислоты, 0,09 г №2 ЭДТА в 1 л воды). Собирают водный раствор из иализной ванны, экстрагируют фенолом, 55 кстрагируют хлороформом, и делают его ,2 М по отношению к хлористому натр ию. атем извлекают ДНК из воды после осажения этанолом. Фрагменты, содержащие rp промотор/оператор с EcoRi липкими
концами, были дентифицированы путем вставки чувствительной к тетрациклину плазмиды, которая при вставке промотор/оператор становится устойчивой к тетрациклину. Все операции выделения фрагмента ДНК, описанные здесь и далее в этом примере, осуществляются с использованием ЭПАГ с последующей электроэлю- цией по описанному выше способу.
Б, Конструирование плазмиды pBRH trp, экспрессирующей устойчивость к тетрациклину под контролем trp промотора/оператора, и идентификация и амплификация фрагмента ДНК содержащего trp промотор/оператор, выделенного в А выше.
Плазмида pBRH1 (Родригес с сотр. 1979, Исследование нуклеиновых кислот 6, 3267-3287 и АТСС № 37070) экспрессирует устойчивость к ампициллину и содержит ген устойчивости к тетрациклину, но не будучи ассоциированной с промотором, не экспрессирует такую устойчивость. Клетки, содержащие плазмиду, следовательно, являются чувствительными к тетрациклину. При введении системы промотор/оператор в сайт EcoRI плазмида будет экспрессиро- вать устойчивость к тетрациклину.
Плазмиду pBRHI переваривают EcoRI. Фермент удаляют экстракцией фенолом с последующей экстракцией хлороформом, затем ДНК повторно суспендируют в воде после осаждения этанолом, Полученную в результате ДНК в отдельных реакционных смесях объединяют с каждым из трех фрагментов ДНК, полученных в примере 5,А, и лигируют Т4 ДНК лигазой, как описано выше. Имеющуюся ДНК в реакционной смеси используют для трансформации компетентного E.coli K12 293/NRRLB-15G25/ по стандартным методикам (Хершфилд с сотр. 1974, Proc. Nat. Acad. Set. USA 71:3455-3459) .затем бактерии помещают на LB-пластины (Ма- ниатис с сотр.,1982), содержащие 20 мкг/мл ампициллина и 5 мкг/мл тетрациклина.
Отбирают несколько устойчивых к тетрациклину колоний и выделяют плазмиду ДНК, обозначенную pBRH trp. Наличие желаемого фрагмента подтверждается фер- ментным рестрикционным анализом. Плазмида pBRHIrp экспрессирует Д-лакта- мазу, придающую устойчивость к ампициллину, и содержит фрагмент ДНК, который включает trp-промотор/оператор. Фрагмент ДНК также кодирует первый протеин (обозначенный LE ), состоящий из расплава первых шести аминокислот trp лидера и приблизительно последнюю треть trpE пол- ипептида, второй протеин (обозначенный Д ), соответствующий приблизительно первой половине trp Д полипептида. и третий
протеин, кодируемый, геном устойчивости к тетрациклину.
В. Конструирование плазмиды pSOM7 Д2.
Плазмиду pBRH trp переваривают ре- стрикционным ферментом EcoRI и полученный в результате фрагмент, выделенный ЭПАГ -и электроэлюцией, объединяют с плазмидой pSOM11, переваренной EcoRI (Итакура с сотр., 1977, 198:1056, патент США № 4 366 246, патент Великобритании № 2 007 676А). Смесь лигируют Т4 ДИК ли- гззойи полученную в результате ДНКтранс- формируют n E.coll E12 294, как описано ранее. Трансформированные бактерии отбирают на пластинах, содержащих ампи- циллим, а полученные в результате устойчивые к ампициллину колонии подвергают скринингу гибридизацией колоний (Грюнштейн с сотр. 1975 Procc. Nat. Acad. Sci. USA 72:3951-3965). Фрагмент, содержащий trp промотор-оператор, выделенный из pBRH trp, а затем радиоактивно меченый 32Р,-используют в качестве зонда в описанной выше процедуре. Было показано, что некоторые колонии являются положительными к гибридизации колоний и были отобраны. Изолируют плазмиду ДНК и определяют ориентацию оставленных фрагментов с помощью рестрикционного анализа, используя в двойном переваре ферменты Bglll и BamHi. Колонии, содержащие желаемую плазмиду с фрагментом trp промотор/оператор в точной ориентации, были выращены на LB среде, содержащей 10 мкг/мл ампициллина. Желаемую плазмиду обозначали pSOMT Д2 и используют для последующих конструировании, описанных выше.. . Г. Конструирование плазмиды ptrp24. 1. Конструирование фрагмента гена,Кодирующего дистальные области LE пол- ипептида с сайтами рестрикции Bglfl и BamHI соответственно на 5 и 3 концах кодирующего тяжа.
Плазмиду pSOM7 Д2 переваривают Hind III.после переваривания лямбда экзо- нуклеазой (5 до 3 экзонуютеаза) в условиях, выбранных таким образом, чтобы переварить выше сайта рестрикции Bglll в LE , кодирующем участке. Растворяют примерно 20 мкг Hind HI переваренной pSOM7 A2 га буфере(20 мМ глициновогб буфера, рН 9,6, 1 мМ MgCl2, 1 мМ /3-меркаптозтанола), Полученную смесь обрабатывают 5 ед. лямбда экзонуклеазы в течение 60 мин при комнатной температуре. Затем полученную греак- ционную смесь экстрагируют фенолом, экстрагируют хлороформом и осаждают эта4- нолом.
Для создания EcoRI остатка на дисталь- ном конце фрагмента LE гена синтезируют преймер32рССТСТССАТСАТ по усовершенствованному фосфотриэфирному методу
(Крис сотр., 1978) и гибридизируютнзодно- тяжевом конце фрагмента LE гена, полученного при переваре лямбда экзонуклеазой. Гибридизацию осуществляют при растворении 20 мкг обработанного лямбда экзонуклеазой продукта перевара Hind lit плазмиды pSOM7 Л2 в 20 мкл воды и объединения с 6 мкл раствора, содержащего приблизительно 80 пикомолей 5 -фосфорилированного олигонуклеотида, описанного выше. Синтетический фрагмент гибридизуют к З -концу LE кодирующей последовательности, а оставшуюся часть LE1 фрагмента наполняют полимеразой Клейноу 1, используя dATP,
dTTP, dGTP и dCTP. Полимераза Клейноу 1
представляет собой фрагмент, полученный протеолитическим расщеплением ДНК пол- имеразы 1. Он содержит 5 полимеризу- ющуюактивность, 3 экзонуклеотическую активность, но не 5
экзонуклеотическую активность родительского фермента (Корнберг, 1974, W.H. Freeman and Со, 98).
Реакционную смесь нагревают до 50°С и медленно охлаждают, до 10°С, после чего
прибавляют 4 мкл фермента Клейноу. После 15 мин инкубирования при комнатной температуре, последующего инкубирования о течение 30 мин при 37° прекращают реакцию добавлением 5 мкл 0,25 молярной ЭДТА. Реакционную смесь экстрагируют
фенолом, экстрагируют хлороформом,
.осаждают этанолом. ДНК последовательно
расщепляют рестрикционным ферментом
Bgl I и разделяют фрагменты с помощью
ЭПАГ. Ауторадиограмма получена для выде- .ленного из геля Р-меченого фрагмента ожидаемой длины приблизительно 470 пар оснований, его извлекают электроэлюцией, Как отмечалось, это фрагмент LE (d) имеет
терминал Bgl И и тупой конец, совпадающий с началом праймера.
5 N-терминал и 5 -концы были сконструированы с однотяжевым липким концом для облегчения присоединения к плазмидам, расщепленным EcoRI и BamHi. Как быть легко оценено, Bglll сайт в центре гена
способствует анализу рекомбинантных плазмид.
Олигодеоксирибонуклеотиды от Ti по Tie были синтезированы по модифицированному фосфотриэфирному методу Итаку- ра с сотр., 1977, и Кри с сотр., 1978. Различные Олигодеоксирибонуклеотиды показаны в табл.1.
Приведенный выше синтез является типичным при следовании процедуре для фрагмента Tis, как суммировано на фиг.12. Различные нуклеотидные фрагменты, которые использованы при синтезе Tis, обозначены номерами на фиг.12. Использованные сокращения являются следующими: ТР Те, 2,4,6-триизопролилбензолсульфонилтетра- зол; BSA, бензолсульфокислота ТСХ, тонкослойная хроматография; HPLC, высокоэффективная жидкостная хроматография; ДМТ 4,4 -диметокситритил; Се, 2-цианоэ- тил; R, n-хлорфенил; Bz, бензоил; An, анизо- ил; iBu, изобутил; Ру, пиридин; АсОН, уксусная кислота, EtaN, триэтиламин.
Деблокируют полностью защищенные тридеоксирибонуклеотиды 4 (85 мг, 0,05 мо- пя) и 2 (180 мг, 0,1 ммоля) у 5 -гидроксилов Путем обработки 2% ВАЗ в смеси.хлороформ/метанол 7:3 по объему (10 и 20 мл соответственно) в течение.10 мин при 0°С. Останавливают реакции добавлением насыщенного водного раствора бикарбоната аммония (2 мл), экстрагируют 25 мл хлороформа и промывают водой 2 раза по 10 мл. Органические слои сушат над сульфатом магния, концентрируют до малого объема (около 5 .мл) и осаждают добавлением петролейного эфира (фракция 35-60°С). Бесцветные осадки собирают центрифугированием и сушат в эксикаторе в вакууме, получают 6 и 8 соответственно, каждый гомогенен при ТСХ на силикагелз (Мерк 60 F2.54, хлороформ/метанол, 9:1).
Превращают тримеры 1 и 3 (270 мг, 0,15 ммоля, 145 мг, 0,075 ммоля) в.их фосфодиэ- фиры (5 и 7) путем обработки смесью триэ- тиламин/пиридин/вода/1:3:1, объем/объем, 10 мл/ в течение 25 мин при комнатной температуре. Удаляют реагенты на роторном испарителе и высушивают остатки при повторном испарении безводного пиридина (3x10 мл). Объединяют трймер 8 (0,05 ммоля) и трймер 7 с ТР Те (50 мг, 0,15 ммоля) в 3 мл безводного пиридина, реакционную смесь оставляют в вакууме при комнатной температуре на 2 ч. Анализ ТСХ показывает, что 95% тримера 8 превращается в гексамерный продукт (визуализиро- вано при детекции ДМТ группы при обрызгивании 10%-ной водной серной кислотой и нагревании при 60°С). Реакцию закаливают при добавлении 1 мл воды, растворитель выпаривают в вакууме. После удаления пиридина, испаряющегося совместно с толуолом, деблокируют гексамер в 5 поло- 5 жении с помощью 8 мл 2%-ной BSA, как описано выше для тримеров 4 и 2. Продукт 10 очищают на колонке с силикагелем (Мерк 60 Н, 3,5 х 5 см) при элюировании хлороформом/метанолом с градиентом от 98:2 до 95:5
10 по объему. Фракции, содержащие продукт 10.выпаривают досуха. Подобным образом трймер 5 сочетают с б и очищают полностью защищенный продукт непосредственно на силикагеле. Это последнее соединение бы15 ло деблокировано с З -конца смесью триэти- ламин/пиридин/вода, как описано ранее, для получения фрагмента 9.
Наконец, сочетают гексамеры 9 и 10 в 2 мл безводного пиридина с ТР Те (75 мг,
0 0,225 ммоля) в качестве конденсирующего агента. По окончании реакции (4 ч при комнатной температуре) смесь выпаривают на роторном испарителе, остаток хроматогра- фируют на силикагеле. Получают 160 мг про5 дукта 11 при осаждении петролейным эфиром, который оказался гомогенным по дачным ТСХ. Полностью деблокируют часть (20 мг) соединения 11 в 0,5 мл пиридина при обработке 7 мл концентрированной гидро0 окиси аммония (8 ч при 60°С) и последующей обработке в течение 15 мин 80%-ной уксусной кислотой при комнатной температуре. После выпаривания уксусной кислоты твердый остаток растроряют в 4 мл 4%-ной
5 гидроокиси аммония и экстрагируют 3 х 2 мл этилового эфира. Водную фазу концентрируют до 1-2 мл и часть наносят на HPLC для очистки 12. Объединяют фракции, соответствующие основному пику (примерно 2 А25-5
0 единицы)и концентрируют до примерно 5 мл. Обессоливают конечный продукт 12 на Вио-геле Р-2 (1,5 х 100 см) при злюции 20%-ным водным этанолом, отгоняют растворитель при пониженном давлении
5 досуха и снова суспендируют в 20 мкл воды, получают раствор А254 10. Подтверждают последовательность 12 двумерным анализом последовательности. Полный типозин альфа 1 ген был собран
0 из 16 синтетических олигонуклеотидов по описанным ранее методам, детализированным для соматостатина (Итакурэ с сотр., 1977), инсулина (Гоеддел с сотр., 1979) и гормона роста (Гоеддел с сотр., 1979,
5 281:544). Олигонуклеотиды Т2 по Tis в количеств по 10 мкгбыли количественно фосфо- рилированы (-р32/-АТР/Нью Ингленд Нюклеар) в присутствии Т4 полинуклеотид киказы (Гоеддел с сотр., 1979), чтобы пол- учить определенные активности прмблизитед1|ьно 1 кюри/ммоль. Радиомеченные фрагменты были очищены электрофорезом Интеле 20%, полиакриламида/7М мочеви- (ны, последовательности элюированных фрагментов были подтверждены двумер- ным электрофорезом/гомохроматографией (Джей с сотр., 1974 Nucleic Acids Res,1:331) частичных переваров змеиным ядом. Фрагментов Т.1 и Tie были оставлены нефосфори- лированными для сведения к минимуму нежелательной полимеризации во время последующих реакций лигирования. По 2 мкг каждого из этих нуклеотидов объединяют в четыре группы из четырех фрагментов (см, фиг. 13)с помощью Т4 ДНК лигазы, ис- пользованной в опубликованных процедурах (Гоеддел с сотр., 1979). Продукты реакции очищают гель-электрофорезом на 15%-ном полиакриламидном геле, содержащем 7 М мочевины (Мэксам и Джилберт, 1977, Proc. Nat, Acad. Sci. USA 71:3455). Четыре выделенных продукта были лигирова- ны вместе и реакционную смесь разделяют электрофорезом на 10%-ном полиакрила- мидком геле. При таком интервале разме- ров электроэлюируется ДНК гена тимозина альфа 1 (90-105 пар оснований).
Обрабатывают плазмиду рВ 322 (0,5 мкг) BamHI и EcoRI рестрикциоиными эндо- нуклеазами и разделяют фрагменты элект- рофорезом на полиакриламидном геле. Извлекают большой фрагмент из геля элек- троэлюцией и последовательно лигируют для получения объединенной синтетической ДНК(Гоеделл и др.. Nature 281:544, 1979). Эту смесь используют для трансформации E.coli К12 294. Помещают 5% трансформи- рованной смеси на LB пластины. Содержа- щие 20 мкг/мл ампициллйна. Четыре полученные устойчивые к ампициллину ко- лонии являются чувствительными к тетрациклину, предполагая вставку в ген устойчивости к тетрациклину. Анализ плаз- мид из этих четырех колоний показал, что в каждом случае плазмида, обозначенная pTha Т, содержит сайт Bglll. не найденный в самой pBR322, следовательно, указывает на наличие гена тимозин аяьфа 1, как показано на фиг.11, и фрагмент приблизительно 105 пар оснований, генерированный при расщеплении BamHI/EcoRI. Рутиноэ конструирование плазмиды pTh «1 (Не вытянута в масштабе) представлено на фиг. 13, где толстые точки указывают 5 -фосфатные группы.
Плазмида pTh a содержит ген, определяющий устойчивость к ампициллину. и структуральный ген, определяющий тимо- зин альфа«1, клонированный в его 5 кодирующий тяжевый конец в сайте EcoRI и в его З -конец в BamHI .сайте. Тимозиновый ген содержит также Bgl II сайт. Для создания плазмиды, способной принять LE (cl) фрагмент, полученный выше, pThAi переваривают EcoRI,r затем проводят реакцию прлимеразы Клейноу 1 с dTTP и dATP для тупых остатков EcoRI. Bg II переваривание полученного в результате продукта создает линейный фрагмент ДНК, содержащий ген устойчивости к ампициллину, а на его противоположных концах липкий Bgl II остаток и тупой конец. Полученный о результате продукт может, быть рециркулирован при взаимодействии с LE (d) фрагментом, содержащим Bgl II липкий конец и тупой конец, в присутствии Т4 лигазы для .образования плазмиды ptrp24. Действуя таким образом, повторно создается сайт EcoRI в положении, где имеется тупой конец лигации.
Д, Конструирование плазмиды рЗОМ7А2Д4.
Последовательное переваривание ptrp24 Bgl И и EcoRI и проведение затем ЭПАП и злектроэлюции дает фрагмент, имеющий кодоны для LE (d) полипептида с Bgl II липким концом и EcoRI липким концом вблизи его 3 кодирующего терминала.(й) фрагмент может быть клонирован в Bgl II сайт плазмиды pSOM7 A2 для образования плавленного протеина полипептид/сомато- статин, экспрессированного под контролем триптофанового промотора/операторе. Для этого требуется частичное переваривание EcoRI pSOM7 Д2 для того, чтобы расщепить дистальный сайт EcoRI от трифтофанового промотора/оператора, и точно выбрать последовательность праймера, чтобы точно сохранить рамку считывания кодона, и снова создать EcoRI сайт расщепления,
Итак, 16 мкг pSOM7 Д2 разбавляют в 200 мкл буфера, содержащего 20 мМ Трис, рН 7,5, 5 мМ MgCte, 0,02 % NP40 детергента и 100 мМ NaCI, и обрабатывают 0,5 ед. EcoRI. После 15 мин при 37°С реакционную смесь экстрагируют фенолом, экстрагируют хлороформом, осаждают этанолом, и потом переваривают Bgl П. Изолируют больший полученный в результате фрагмент с помощью ЭПАГ с последующей электроэлюцией. Этот фрагмент содержит кодоны..Е (р) для проксимального конца LE полипептида, а именно вверх от сайта Bgl.11, Этот фрагмент затем лигируют к указанному выше LE (p) фрагменту в присутствии Т4 ДНК лигазы для образования плазмиды pSOM7 A2 М.которая при трансформации в E.coii K12 294 эффективно продуцирует плавленный протеин, состоящий из полностью восстановленного LE полияептида и соматостатина под контролем триптофанового промотора/оператора.
Е. Конструирование линейной ДНК, имеющей Pstl остаток на З -конце и Bgl II остаток на его 5 -конце, связывающем ген, определяющий устойчивость к тетрациклину.
Плазмиду рВ322 Hind III переваривают и выступающие Hind III концы переваривают SI нуклеазой. Переваривание SI нуклеа- зой включает обработку 10 мкг Hind III переваренной рВ322 в 30 мкл SI буфера (0,3 М NsCI, 1 мМ ZnCl2, 25 мМ ацетата натрия, рН 4,5) 300 ед. SI нуклеазы в течение 30 мин при 15°С. Прекращают реакцию добавлени- ем 1 мкл 30 х 51 нуклеазного стоп-раствора (0,8 М трис основания. 50 мМ ЭДТА), Смесь экстрагируют фенолом, экстрагируют хлороформом, осаждают этанолом, а затем переваривают EcoRI, как описано выше, Полученный в результате фрагмент выделяют с помощью ЭПАГ с последующей электроэлюцией, он имеет EcoRI липкий конец и тупой конец, у которого кодирующий тяж начинается с тимидинового нуклеотида, SI- переваренный Hind III остаток, начинающийся с тимидина, может быть присоединен к переваренному Полимера- зой Клейноу 1 Bgl II остатку, чтобы восстановить ВдГИ сайт рестрикции при лигации.
Следовательно, плазмиду pSOM7 Д2, полученную в примере 5В, Bgl И переваривают и полученные Bglll липкие концы делают двутяжевыми обработкой полимеразой Клейноу 1, используя все четыре деоксинук- леотидных трифосфата. EcoRI расщепление полученного продукта с последующим ЭПАГ и электроэлюцией маленького фрагмента дает линейную часть ДНК, содержащую триптофановый промотор/оператор и кодоны LE проксимальной последовательности выше Bgl II сайта (LE (p))- Продукт имеет EcoRI конец и тупой конец, полученный в результате заполнения в Bgl II сайте. Однако Bgl II сайт восстанавлива- ется лигацией тупого конца к тупому концу указанного выше SI переваренного Hind III фрагмента. Итак, оба фрагмента лигируют в присутствии Ъ) ДНК лигазы для образования рециркулярированной плазмиды рНКУ10, которая размножается при транс- , формации в компетентные клетки E.coll K12 294. Отбирают клетки, устойчивые к тетрациклину, несущие рекомбинантную плазмиду рНКУЮ и экстрагируют ДНК плазмиды. Переваривание Bgl II и Pst I с последующим выделением с помощью ЭПАГ и электро- элюции большого фрагмента приводит к целевому линейному участку ДНК, имеющему Pst I и Bgl II липкие концы. Этот фрагмент
ДНК, полученный таким образом из рНКУЮ, содержит источник репликации, и, следовательно, является полезным в качестве компонента для конструирования плаэ- миды рН17 Д4 А1, у которой оба гена, кодирующих .плавленный протеин trpLE . полипептида и устойчивость к тетрациклину, контролируются trp промотором/оператором.
Ж. Конструирование линейной ДНК. имеющей trp промотор-оператор.
Плазмиду pSOM7 Д2 Д4, полученную в примере 5,Д, подвергают частичному EcoRI перевариванию с последующим Pst I перевариванием. Полученный в результате фрагмент, содержащий trp промотор/оператор, выделяют с помощью ЭПАГ с последующей электроэлюцией. Частичное EcoRI переваривание необходимо для получения фрагмента, который расщепляется вблизи 5 -конца соматостатинового гена, но не расщепляется у сайта EcoRI, имеющегося между геном устойчивости к ампициллину и trp промотором/оператором. Устойчивость к ампициллину теряется при Fst I разрез в гене устойчивости к ампициллину. она может быть восстановлена при лигации с готовым производным линейной ДНК рНКУЮ, полученным в примере 5,Е.
Синтетические олигонуклеотиды ддя человеческого проинсулина
Соединение Последовательность Н1AATTCATGTT Н2 CGTCAATCAGCA НЗ CCTTTGTGGTTC Н4 TCACCTCGTTGA Н5 TTGACGAACATG Нб CAAAGGTGCTGA Н7 AGGTGAGAACCA Н8 AGCTTCAACG
81AGCTTTGTAC
82CTTGTTTGCGGT
83GAACGTGGTTTC
84ТТСТАСАСТССТ В51 AAGACTCGCC
86AACAAGGTACAA
87ACGTTCACCGCA
88GTAGAAGAAACC
89AGTCTTAGGAGT В10 GATCCGGCG
П р и м е р 6. Конструирование плазмиды рМ608 и E.coll K12 RV308/PNM608.
А. Конструирование EcoRI-Hpal фрагмента примерно 253 пары оснований плазмиды рН17 А4 Д1.
Инкубируют при 37°С в течение примерно 1 ч около 5 мкг ДНК плэзмиды рН17 М А1,10 мкл (1 Ох)реакционного буфера, 80 мкл воды и 5 мкл (5 ед.) фермента рестрикции Hpal. Реакционный буфер (ЮХ)
для Hpal рестрикционнго фермента готовят следующего состава: 100 мМ Трис. HCI, рН 7.100 мМ MgCla, 60 мМ -меркаптоэтанола, 200 мМ KCI. Реакцию прекращают инкуби- рованием при 70°С в течение 5 мин. После охлаждения смеси на льду прибавляют примерно 12 мкл 1 fvl Трис. HCI, рН 7,2, 7 мкл воды и 1 мкл (10 ёд.) рестрищионного фермента. EcoRI. Полученную в результате смесь инкубируют сначала при 37°С в течение 1 ч, а затем 5 мин при 70°С. После охлаждения на льду экстрагируют полученную в результате ДНК каждым из фенолом и хлороформ:изоамиловый спирт (24:1), а затем осаждают этанолом. Целевой фрагмент EcoRI-Hpal примерно 253 пары оснований отделяют электрофорезом на полиакрила- мидном геле, извлекают электроэлюцией, осаждают этанолом, затем растворяют примерно в 10 мкл ТЕ буфера и хранят при 0°С для дальнейшего использования.
Б. Конструирование Hpal-Tagl фрагмента примерно 34 пары оснований плазмиды РН17Д4Д1.
Инкубируют при 37°С в течение 1 ч примерно 20 мкг ДНК плазмиды рН17 М Д1 в 32 мкл (5Х) EcoRI реакционного буфера, 124 мкл воды и 4 мкл рестрикционного фермента EcoRI (20 ед.). Реакционный буфер (5Х) для EcoRI рестрикционного фермента готовят следующего состава: 250 мМ NaCI, 500 мМ Трис. HCI, рН 7,2. 25 мМ MgCl2, 30 мМ / -меркаптоэтанола. Реакцию заканчивают инкубированием при 70°С в течение 5 ммн.. Полученный в результате EcoRI фрагмент 862 пары оснований выделяют электрофорезом на 6%-ном полиакриламидном геле с последующей электроэлюцией и осаждением этанолом. Затем фрагмент расуворяют примерно в 100 мкл Hpal буфера, содержащего 16ед. Нра рестрикционного фермента. После 1,5 ч при 37°С прибавляют 7,5 ед. Tag I рестрикционного фермента, продолжают инкубирование еще 1,5 ч и полученные в результате фрагменты отделяют на 7,5%- ном полиакриламидном геле. Извлекают целевой Hpal-Tagl фрагмент примерно 34 пары оснований электроэлюированием и осаждением этанолом, затем растворяют его в 5 мкл ТЕ буфера.
В, EcoRI-Clal переваривание плазмиды рВ322.
Инкубируют при 37°С в течение 1 ч примерно 5 мкг ДНК плазмиды рН322, 10 мкл Clal реакционной смеси, 77,5 мкл воды и 7,5 мкл (15 ед.) Clal рестрикционного фермента. Реакционную смесь (10Х) для Clal рестрикционного фермента готовят следующего состава: 100 мМ Трис. HCI. рН 7,4, 100 мМ MgCl2, 60 мМ /J-меркаптоэтанола. Реакцию
заканчивают инкубированием при 70°С в течение 5 мин. После охлаждения смеси на льду прибавляют примерно 12,5 мкл Трис. HCI, рН 7.2. 6,25 мкл 1 М NaCI, 2,5 мкл 0,1 М
MgCla и 1 мкл (10 ед.) EcoRI рестрикционного фермента. Полученную в результате смесь инкубируют 1 ч при 37°С, а затем 5 мин при 70°С. После охлаждения на льду полученную в результате ДНК подвергают
электрофорезу на 1%-ном агарозном геле. Большой фрагмент извлекают электрозлю- цией и осаждением этанолом, затем растворяют примерно в 20 мкл ТЕ буфера и хранят при 0°С для дальнейшего использования,
Г. Лигирование и трансформация.
Лигируют примерно 0,2 мкг EcoRI-Hpal фрагмента примера 6,А, 0,5 мкг Hpal-Tagl фрагмента примера 6,В и 0,1 мкг EcoRI-Clal перевара примера 6,В и используют для
трансформации E.coli К12 RV308 по существу по методике, описанной в примере 2Г.
Некоторые из полученных в результате трансформамтов, как показано электрофорезом на агарозном геле (Мэниатис с сотр.,
1982) и другими тестами, содержат только желаемую плазмиду примерно 4,6 kb. Отбирают такие трансформанты, обозначенные как E.coli K12 RV308/pNM608, помещают на пластины с ТД агаром, содержащим соответствующие антибиотики, и затем культивируют, используя традиционные микробиологические методики. Плазмида pNM508 представляет собой и является предпочтительным источником EcoRI-Clal
(TagI) фрагмента примерно 287 kb плазмиды РН17Д4Д1.
Пример. Конструирование плазмиды pCZ112 и E.coli K12 RV308/pCZ112.
A. EcoRI-Clat переваривание плазмиды
pNM608 и генерация EcoRI-Tagl фрагмента примерно 0,288 kb (такого же как EcoRI-Tagl фрагмент 0,288 kb плазмиды рН17Д4 Д1).
Инкубируют примерно 5 мкг ДНК плазмиды pNM608 в 50 мкл солевого буфера с 10
ед. каждого из EcoRI и Clal рестрикционных ферментов при 37°С в течение примерно 1 ч. Солевой буфер следующего состава: 50 мМ NaCI, 100 мМ Трис. HCI, рН 7,5, 6 мМ MgCl2, 2 мМ / -меркаптозт.анола. После до0 бавяения 5 мкл 3 М ацетата натрия, рН 7,0, осаждают ДНК 3 объемами 100%-ного эта- нола. Целевой перевар ДНК растворяют в 100 мкл ТЕ буфера и хранят при 0°С для дальнейшего использования.
5 Б. Конструирование линкерной последовательности ДНК.
CGACC ATG GAT GAT AAG TIT CCG GCT АТС ТСТ СТО TGG TAG СТА СТА ТТС ААА GGC CGA ТЛС AGA GAC
ТСС GG.C СТО ТТТ GCC ААС ССТ GTGCT AGO CCG GAC ДАЛ CGG TTG ССА С
Синтезируют желаемую линкерную последовательность модифицированным фос- фотриэфирным способом по существу в соответствии с Итакура с сотр., 1977 и Кри с сотр., 1978. Вышеупомянутый способ синтеза также был специально проиллюстрирован в примере 1.
В. Лигирование и трансформация.
Примерно 20 пикомолей линкерной ДНК примера 7,Б, 1 мкг перевара pNM608 примера 7,А, 0,5 мкг BamHI-EcoRI фрагмента примерно 10,2 kb плазмиды pCZ101 (полученного в соответствии с методикой примера 2,Б, с тем исключением, что ис- пользуют EcoRI рестрикционный фермент и соответствующий буфер, а ке Xbal рестрикционный фермент и буфер) и 1 мкг Ba mHI- HgiAl фрагмент примерно 0,6 kb плазмиды pCZ101 примерз 3,Б подвергают лигации, а полученную в результате плазмиду используют для трансформации E.coli K12 RV308 по --существу в соответствии с методикой примера 2,Г,
Не которые из полученных в результате трансформантов, как показано электрофорезом на агарозном пзле (Маниатис с сотр., 1982) и другими тестами, содержат только желаемую плэзмиду примерно 10,8 kb. Такие траисформанты, обозначенные здесь как E.coli K12 RV308/pCZ112, отбирают, по- мещают на ТД агаровые пластины, содержащие соответствующие антибиотики, и затем культивируют, используя традиционные микробиологические методики. Как показано электрофорезом на SDS-геле, RIA и другими тестами, полученные в результате клетки экспрессируют вышеупомянутое производное MET-ASP-ASP-LYS-бГР с высокими уровнями. Поскольку плазмида pCZ112 содержит репликон неконтролируемого роста, термоиндуцируемый, максимальная экспрессия желаемого продукта производного 6ГР происходит при культивировании при температуре около 37°С.
П р и м е р 8, Конструирование pATI плазмиды и E.coli K12 RV308/pATI.
Желаемое конструирование осуществляют по существу в соответствии с методикой примера 7 с тем исключением, что линкерную последовательность прим.ера.7 заменяют линкерной последовательность ) ДНК:
5 ССАСА АТС ТТС ССЛ GCT АТС ТСТ СТА ТСТ СОТ j. ЈДЈ Дс Ј Ј ,j,Ј . Јj Д ЈЈ
CTG ТТТ CCU ЛАС С,СТ CTCCT 3 ОЛС ААА CGG ТТС ССА С 5
Определенная выше линкерная последовательность может быть сконструирована по существу в соответствии с традиционной методикой Итакур с сотр.,
1977 и Кри с сотр., 1978. Вышеупомянутый метод синтеза также специально иллюстрирован в примере 1.
Желаемые трансформанты, обозначенные здесь как E.coli K12 RV308/pATI, помещают на пластины с ТД-агаром, содержащим соответствующие антибиотики, а затем культивируют для последующего продуцирования и выделения плазмиды
pASTI. Как показано SDS-гель-электрофоре- зом, RIA и другими тестами, трансформанты экспрессируют вышеупомянутое производное МЕТ 6ГР с высокими уровнями. Поскольку плазмида pATI содержит
термоиндуцируемый репликон неконтролируемого роста, максимальная экспрессия желаемого продукта производного 6ГР происходит при культивировании при температурах около 37°С.
П р и м е р 9. Конструирование плазмиды pASP1 и E.coli K12 RV308/pASP1.
Желаемые конструкции получают по существу по методике примера 7 с тем исключением, что линкерную последовательность
примера 7заменяют на последовательность ДНК: ,
10 5 -CGACC АТС GAT ТГТ ССС ССТ АТС ТСТ С ГС ТСС СОС СТО 3г ТСга ТАС СТА АЛЛ ССС CGA ТАС AGC GAC ЛСС cci GAC
ТТТ GCC ЛЛС fiCT СТ(Х1 3 15 АЛА CGC ТТС CGA С 5
Вышеуказа нная линкерная последовательность может быть сконструирована по существу в соответствии с традиционной
методикой. Итакура с сотр., 1977 и Кри с сотр., 1978, Вышеуказанный способ синтеза также.специально иллюстрирован в примере 1.
Желаемые трансформанты, обозначенные здесь как E.coit K12 RV308/pASP1, помещают на пластины с ТД-агаром, содержащим соответствующие антибиоти- ки, и затем культивируют для последующего
продуцирования и выделения плазмиды
pASP1. Как показано SDS-гель-электрофо- резом, RIA и другими тестами, трансформанты экспрессируют вышеопределенное производное MET-ASP-бГР с высокими уровнями. Поскольку плазмида ,pASP2 coдержит термоиндуцируемый репликон неконтролируемого роста, максимальная экспрессия желаемого производного 6ГР происходит при культивировании при температурах около 37°С.
ПримерЮ. Конструирование плазмиды pASP2 и E.coli K12 RV308/pASP2.
Желаемые конструкции получают по существу по методике примера 7, с тем исключением, что линкерную последовательность
примера 7 замеряют на последовательность ДНК:
ССАТС АТС CAT ГГТ ССС GCT ATG ТСТ СТО ТСС ССС СТО ТЛО ТАС СТА ЛАА ССС CGA ТАС АСА САС AGG CCG GAC
IS
ТТТ ССС ААС ОСТ СТССТ ААА CGG ТГС SCA С
Вышеуказанная линкерная последовательность может быть сконструирована по сущестоу о соответствии с традиционной методикой Итакура с сотр., 1977 и Кри с сотр., 1978. Вышеуказанный способ синтеза также специально иллюстрирован в примере 1.
Желаемые трансформанты, обозначенные здесь как E.coli K12 RV308/pAP2, помещают на пластины с ТД-агаром, содержащим соответствующие антибиотики, и затем культивируют для последующего продуцирования и выделения плазмиды pASP1. Как показано SDS-гель-электрофорезом, RIA и другими тестами, трансформанты зкспрессируют вышеопределенное производное MET-ASP-бГР с высокими уровнями. Поскольку плазмида pASP2 содержит термоиндуцируемый репликон неконтролируемого роста, максимальная экспрессия желаемого производного 6ГР происходит при культивировании при температурах около 37°С.
Л р и м е р 11. Конструирование плазмиды рАТ2 и E.coli K12 RV308/pAT2.
Осуществляют желаемые конструирования по существу по методике примера23, с тем исключением, что линкерная последо-, вательность примера З.В заменена линкер- ной последовательностью ДНК:
10 5 CTAGAGGGTATTAATA ATG ,ТТТ CCA GCT АТС ТСГ З1 ТСССАТААТТАТ ТАС ААА GGT CGA ТАС AGA CAT АСА
15
GCT СТО ТТТ GCC ААС GCT GTGCT CCA CAC AAA CGG TTG, CCA С
Определенная выше линкерная последовательность может быть сконструирована по существу в соответствии с традиционными, методиками Итакура с сотр.. 1977, и Кри с сотр., 1978. Вышеуказанный способ синтеза специально иллюстрирован в примере 1.
Желаемые трансформанты, обозначенные здесь как E.coli K12 RV308/pAT2, помещают на пластины с ТД-агаром, содержащим соответствующие антибиотики, а затем культивируют для последующего получения и выделения плазмиды рАТ2. Как было показано SDS-гель-электрофорезом, RIA и другими тестами, трансформанты зкспрессируют определенное выше производное МЕТ-бГР с высокими уровнями.
Поскольку плазмида рАТ2 содержит термоиндуцируемый репликон неконтролируемого роста, максимальна экспрессия желаемого производного происходит при
культивировании при температуре около 37°С.
П р и м е р 12, Конструирование плазмиды pCZ154 и E.coli K12 RV303/pCZ154.
Осуществляют желаемые конструирования по существу в соответствии с методикой примера 7, с тем исключением, что линкерную последовательность примера 7 заменяют на линкерную последовательность ДНК:
10 5 CGACC АТС СТГ ТТТ ССС ОСТ АТС ТСТ CTG ТСС GGC СТО 3 TGG TAG САД АЛА CGC CGA ТЛС АСА CAC AGG ССО СЛС
ТТГ ССС ААС GCT СТССТ /АА CGG TTG CCA С
Определенная выше линкерная последовательность может быть сконструирована по существу в соответствии с традиционной методикой. Итакура с сотр., 1977 и Кри с сотр., 1978. Вышеуказанный способ синтеза
был специально иллюстрирован о примере
Желаемые трансформанты, обозначенные здесь как E.coli K12 RV308/pCZ154, помещают на пластины с ТД агаром,
содержащим соответствующие антибиотики, и культивируют для последующего получения и выделения плазмиды pCZ150. Так же показано SDS-гель-электрофорезом, RIA и другими тестами, что трансформанты экспрессируют вышеупомянутое производное МЕТЛ/АЫэ Р с высоким уровнем. Поскольку плазмида pCZ154 содержит термоиндуцируемый репликон неконтролируемого роста, максимальная экспрессия желаемого
производного 6ГР происходит при культивировании при температуре около 37°С.
П р и м е р 13. Конструирование плазмиды pCZ155 и E.coli K12 RV308/pCZ155.
Желаемые .конструирования осуществля ют по существу в соответствии с методикой примера 7, с тем исключением, доо заменяют линкерную последовательность примера 7 линкерной последовательностью ДНК:
.-.
10 5 CGACC АТС ССТ ТТГ ССС GCT АТС ТСС СТО ТСС GTC СТО 3 ТСС ТАС СС-А ААА GCC CGA ТАС АСА GAC AGG CAG GAC
ТТТ ССС ААС GCT СГССТ 3 15 ААА CGG TTG CGA С $
Ыг
ty
ч/;
Определенная выше линкернял последовательность может быть сконструирована по существу в соответствии с традиционной методикой Итакура с сотр., 1977 и Кри с сотр., 1978. Указанный выше
способ был также иллюстрирован специально в примере 1.
Желаемые трансформанты, обозначенные здесь как E.coli K12 RV308/pCZ155, no- . мещают на пластины с ТД-агаром, 5 содержащим соответствующие антибиотики, а затем культивируют для последующего получения и выделения плазмиды pCZ155. Также было показано, SDS-гель-электрофо- резом, RIA и другими тестами, что трансфер- 10 манты экспрессируют указанное выше производное MET-ALA-PHE-PRO-ALA-MET- SER-LEU-SER-VAL-бТР с высокими уровнями. Поскольку плазмида pCZ155 содержит
термоиндуцируемый неконтроли- 15 руемого роста, максимальная экспрессия желаемого производного 6ГР происходит при культивировании при температуре око- ло 37°С.
П ри м е р 14. Конструирование плазми- 20
ды pCZ156 и E.coli K12 RV308/pCZ156.
Проводят желаемые конструирования по существу по методике примера 7 с тем исключением, что линкерную последовательность примера 7 заменяют на линкер- 26 ную последовательность ДНК:
S1 CGATA ЛТС CAT ITT CCG ССГ ЛТО ТСТ CTG ТСС GCC CTG 3 ТЛТ.ТАС СТА ЛАЛ GGC Срл ТАС ЛСЛ СЛС ACG CCG СЛС
ТТТ GCC АЛС GCT GTGCT 3 АЛЛ CGG TTG ССЛ С 5
Определенная выше линкерная последовательность может быть сконструирована по существу в соответствии с 35 традиционной методикой Итакура с сотр. 1977 и Кри с сотр., 1978. Указанный выше способ синтеза также был специально иллюстрирован в примере 1. - Желаемые трансформанты, обозначен- 40. .ные здесь как E.coli K12 RV308 /pCZ156, бы- ли. помещены на пластины с ТД-агаром, содержащим соответствующие антибиотики, затем проводят традиционное культивирование для последующего получения и 45 выделения плазмиды pCZ156. Также показано SDS-гель-электрофорезом, R1A и другими тестами, что трансформанты экспрессируют вышеупомянутое производное MET-ALP-бГР с высокими уровнями. По- 50 скольку плазмида pCZ156 содержит термоиндуцируемый репликон неконтролируемого роста, максимальная экспрессия желаемого производного 6ГР происходит при культивирова нии при температуре око- 55 ло 37°С.
П р и м е р 15. Конструирование плазмиды pCZ103 и E.coli K12 RV308/-pCZ103.
Растворяют 10 мкг плазмиды pCZ101 в Юмкл 10Х BstE11 реакционного буфера (1,5
. 5 10
15
0
6
0
5 0. 5 0 5
М NaCI, 60 мМ Трис. HCI, рН 7,9, 60 мМ MgCla, 60 мМ 2-меркаптоэтанола и 1 мг/мл BSA). и 88 мкл воды и 2 мкл ( 20 ед.) ре- стрикционного фермента BstEII, полученную в результате реакционную смесь инкубируют при 60°С в течение 2 ч. После экстракций фенолом и хлороформом и нескольких осаждений этанолом ДНК обрабатывают 0,25 М раствором ацетата натрия.
Суспендируют около 0,1 мкг ДНК переваренной BstE11 плазмиды pCZ101 в 100 мкл лигазного буфера. После добавления ЮОед. Т4 ДНК лигазы и инкубирования при 16°С в течение 2 .ч лигированную ДНК используют для традиционного трансформирования E.col K12 RV308, Отбирают трансформанты E.coli K12 RV308/pCZ103 на канамицине и идентифицируют рестрикци- онным ферментным анализом ДНК их плазмиды. Выделяют плазмиду pCZ103 из трансформантов по существу в соответствии с методикой примера 2,А.
Пример 16. Конструирование векторов, происходящих из плазмиды pCZ103 для экспрессии производных бычьего гормона роста.
Так как BstE11 делеция, сделанная при конструировании плазмиды pCZ103, не воздействует-на последовательность, кодирующую протеин 6П1, плазмиды, происходящие из плазмиды pCZ103 изобретения, зкспрес- сируюг то же самое произЕюдное 6ГР, что и их дубликаты, происходящие из плазмиды PCZ101.
Так как плазмиды изобретения, происходящие из pCZ1Q3. 6bmM сконструированы по существу в соответствии с методикой примеров, и которых были сконструированы их дубликаты, происходящие из pCZIOI, о табл.2 представлено кратко суммированное конструирование илазмид, происходящих из pCZ103. В габл.2 перечислены плазмиды, .происходящие из pCZ103, соответствующий пример, описывающий конструирование дубликата, происходящего из pCZI.01, и размер только отличающегося фрагмента ДНК, включенного и конструирование.
Плазмиды, происходящие из pCZ103, были использованы для.традиционной трансформации E.coli K12 RV308. Полученные в результате трансформанты экспрессируют вышеупомянутые производные 6ГР. Поскольку плазмиды, происходящие из pCZ103, содержат термоиндуцируемый репликон неконтролируемого роста, максимальная экспрессия происходит при культивировании при температурах около 37°С.
Формула изобретения 1. Способ получения рекомбинэнтной плазмидной ДНК, кодирующей бычий гормон роста, заключающийся в том, что осу: ществляют лигирование фрагмента BamHI-Xbaf пла.змиды pCZ101 размером 10,2 kb или BamHI EcoRI фрагмента той же плазмиды с Ipp промотором и репликоном, теряющим контроль числа копий при 37°С, с BamHI-Xbal-фрагментом плазмиды pCZ101 или pCZ103 размером 0,6 kb, кодирующим аминокислоты с 15 до 191 бычьего гормона роста и ДНК-линкером, содержащим нуклеотиды, кодирующие последовательность активации трансляции, стартовый кодон и кодон для N-конца производного бычьего гормона роста, при этом в случае, когда для лигирования берут BamHI-Xbaf-фрагмент плазмиды pCZ101 используют линкер с последовательностью нуклеотидов
Г1 - CTAGA аебтлттмтл
. . I IIIIII11 . ..
« - ТСССАТААТГАТ ГАС ТТТ ССС
ЛТО. АДА C6G ЛАТ -ТСТ ATU Off ТТС ССА 0((
шишшшуш
Атб тсс TTS тсс &&с ста ттт GCC ЛАС ист стаст-з
III Ml III HUM III III III ИМИ I тле лев АЛ f MS tct VAC MA ess We ш c-s1
и получают плазмиду pCZ1920, кодирующую MeT-Lys-Gly-Asp-1N-SER-MET-A a- bGH, или линкер с последовательностью нуклеотидов
амш
ССА ССГ лтв.ссТ СТА тггеет
HI ill ill nun
ТАС С6А &ATAUC
66 ТАС С6А 6АТЛСЛ ССА
СТС ТТТ СГС ААС ОСТ OTGCT-J III III III III III I CAC AAA C66 ГГО CdA
и получают плазмиду plR1, кодирующую MET-PHe-PRO-ALa-MET-Ala-bGH, или линкер с последовательностью нуклеотидов
Г-СШЛОССТАТТАЛТ АТС ТТТ ССЛ; ССТ АТС ТСТ СТА ТСТ ССТ
НМШШН HI 111 III- l III Ml HI III, 3-- сСс11АЛТТЛТ ТЛС AAA GOT CCA ТАС ЛОЛ 5ЛТ АСА ССЛ
сто ттт ccc ЛАС ест CTCCT-J
Ill III III 111 III I
CAC AAA CCS ПО CCA C-S
и получают плазмиду рАТ2, кодирующую Met bGH, в случае, когда для лигирования берут BamHI-Xbal-фрагмент плазмиды pCZ103, линкер имеет последовательность нуклеотидов
-ciACAGUGTATWTA АТС fri сел GCC АТС ест стл тст ест
minium iii iii ill ч) in 111 111 111 ill
Г-ТСССАТЛЛтТ TAC AM CST CCC TAG CGA CAT АСА ССА
Ct6 ТТТ «С ЛАС ССТ GTGCT-Г
ill III 111 III HI I CAC AAA CCC TTC CCA C-S1
5
0
и получают плазмиду р1Я1„3 кодирующего Met-PHE-PRO-Ala-BGH или линкер с последовательностью нуклеотидов
S -CTACAGGGTATTAATA АТС ТТТ ССА ПС Г АТС ТСТ СТА ТСТ ССТ
МИШИН Ill III III III III Ml Ml III III
З -ТСССАТААТТАТ ТЛС ЛЛА GGT CGA ТАС АСА CAT АСЛ CCA СТС ТТТ ССС ЛЛС GCT CTCCT-31
III III III IM Ml I
CAC AAA CCG TTG CCA C-5 -..
и получают плазмиду рАТ2,3, кодирующую MET-bGH, при этом в случае, когда для ли-, гирования берут BamHI-EcoRI-фрагмент плазмиды pCZIOI илирСгЮЗ, его дополнительно лигируют с ЕсоЯ1-С1а1-фрагментом плазмиды pNM 608 с ДНК-линкером, кодирующим последовательность активации трансляции, стартовый кодон и кодон для N-конца производного бычьего, гормона, причем в случае лигирования фрагмента плазмиды pCZ101 используют линкере нук- леотидной последовательностью
5 -ССАСС,ЛТО GAT GAT ЛЛО Г{Т ССС ССТ АТС ТСТ СТО
МММ III III III III III ill II Ill III
З -ТСС.ТАС СТЛ СТЛ TTC ЛА) GCC CGA ТАС ЛПА СЛС ТСС ССС СТС И Т ССС ЛЛС ССТ СТССТ-31
III II МИ 11 III I I.I III I
ACC CCG СЛС ЛЛЛ CGG ПГ. ССЛ C-S
о
5
при этом получают плазмиду pCZ112, кодирующую MET-Asp-Asp-Lys-bGH или линкер с последовательностью нуклеотидов
S -CGACA ЛТО ТТС ССЛ ССТ АТС.ТСТ СТЛ ТСТ GGT
Ml Ml Ml Ml III III, Ml IM Ml III тот тле ллс CGT спл тлс лсл слт лсл ссл
стстттссс ллс сст стсст-э .
. IIIIflIII III III I :
СЛСЛЛЛCCC TTO ССЛ С-5
при этом получают, плазмиду pAST.1, кодирующую MET-Asp-bGH, или линкер с последовательностью нуклеотидов
5 -ССЛСС ЛТС СЛТ ТТТ ССС ССТ ЛТС ТСТ СТС ТСС CGC СТС
III III Ml-1II III III-Ml III III I If Ml III
З -TGG 1ЛС СТЛ ЛЛЛ ССС ССЛ ТЛС ЛСС СЛС ЛСС ССО СЛС
Т1Т ССС ЛЛС ССТ GTGCT-3 . 111 11МII 11II
ЛЛЛ CCG TTG ССЛ С-5 :
при этом получают плазмиду pASTPI, кодирующую MET-bGH, или линкер с последовательностью нуклеотидов
55
S -CCAIC ЛТО ЫТ ТТГ СИ ССТ ЛТО ТСТ ПС ТСС ССС СЮ ПТ ССС ЛЛС ССТ Г1ССТ-Э
I I I III III |П III ill III III ill ill ill III I
TM ТЛС СТА.АЛД ССС СОЛ ТЛС.ЛГЛ СЛС. ЛГ.С ССС СДС : IM CC5 ItC CCA C-5
при этом получают плазмиду pASP2, кодирующую MET-ASP-bGH, или линкере последовательностью нуклеотидов
ДСС АТС UT TIT ССЙ ОСТ АТС ТСТ СТО Т« СВС СТС
HI III III III III II ill iii ill ill ill HI Э -TCG TAC СЛЛ AAA ССС СОЛ ТАС АСЛ GAC AGG CCG CAC
TITссс втост-з1
. IllIII ИМИ I V
АЛЛCCG ПС ССЛ C-5
при этом получают плазмиду pCZ154, кодирующую MET-VAL-bGH, или линкер с последовательностью нуклеотидов .-э
V-ССЛСС АТС ОСТ ТТТ CCG GCT.ATG ТСТ СТО ТСС GTC СТО
111 IIPIII III III III III III III III III III
З -TGC ТЛС ССЛ ЛАЛ CGC ССЛ GAG-ЛСС CrtG GAC ТТТ ССС АЛС ССГ GTCCT-3
Ill IN III III I . . .
АЛЛ CGO TTG ССЛ C-S )
при этом получают плазмиду pCZ155, кодирующую MET-ALA-PHE-PRO-A.LA-MET-SER- LEU-SER-VAL-bGH, или линкер с последовательностью нуклеотидов
5 -СПЛТЛ АТС СЛТ ТТТ ССО С.СТ ДТП ТСТ СТО ТСС Г,«С СТС
III III III III III III III III 111. III Ml III
Э -ТЛТ ТЛС СТА АЛА GGC ССЛ ТЛС ЛОЛ СЛС AGO IXG КЛС
ТТТ ССС ЛЛС ОСТ GTCCT-31 III ИМИ ИМ
. ЛЛА CGC TTG ССЛ С-5 ,
при этом получают плазмиду pCZ156, кодирующую MET-ASP-CH, в случае, когда для легирования берут BamHI-EcoRI-фрагмент плазмиды pCZ103 используют линкер с последовательностью нуклеотидов
5 -СОЛСА ATG ТТС ССЛ- ОСТ ЛТО ТСТ СТЛ ТОТ CGT
Ml ill ill III III III III III IN Hi
TGT TAC AAG GGT ССЛ 1ЛС АСЛ GAT ЛОА CCA СТО ТТТ ССС ЛЛС OCT. CTCCT -3
III III МММ Ml I
СЛС АЛЛ CGG-ПО-СОЛ C-5
при этом получают плэзмиду рАЛ.З, кодирующую MET-bGH, или линкер с последовательностью нуклеотидов
5 -ССАСС ЛТО СЛТ TiT CCG GCT ATG ТСТ СТО ТСС GCC СТО III III III III III 111 III III III III ||| Ills -TEG TAC СТА ЛЛА CGC ССЛ ТЛС ЛСС СЛС AGG CCG .СЛС
ТТТ ССС АЛС ССТ GTCCT-31
III III ИМИ I
ЛАЛ CGG TTG СОЛ С-5 ,
при этом получают плазмиду рА5Рг.З,кодй рующую MET-ASP-bGH, или линкере последовательностью нуклеотидов
ГДТС ATG СЛТ TIT CCU GCT ЛТС ТСТ CTG ТСС GUC СТС II 111 111 III III 111 111 III III III HI HI AC ТЛС СТА ЛЛЛ GGC ССЛ ТАС ЛСА СЛС ЛСО CCG.GAC
ТТТ ССС ЛАС .UCT СТССТ-3
III III III 111 I
ЛЛЛ CCG.ТТС CGA C-51 .
при этом получают плазмиду pAZP2.3, кодирующую МЕТ-а a-bGH, или линкер с последовательностью нуклеотидов
5 -ССЛСС ЛТО СТТ ТТТ CCG fiCT АТС ГСТ СТО ТСС ССС СГС
, III III III III III III III III III III III Ml
3 -TCG ТЛС СЛЛ АЛЛ CGC CGA TAC АСЛ СЛС ЛСС ССС СЛС ТТТ ССС ЛЛС ССТ GTCCT-3
III III III III I
АЛЛ CCG TTG ССЛ С-51
при этом получают плазмиду pCZ154.3, кодирующую MET-VAL-bGH, или линкер с последовательностью нуклеотидов
0
IK S -CGACO АТС ССТ ТТТ CCG ССТ ЛТО ТСТ СТО ТСС GTC СТС
111 ИМИ III III III III III III III III III
З -TGG ТЛС ССЛ ЛАА Г,ОС ССЛ ТЛС ЛСА СЛС AGO СЛС СЛС
ТТТ ССС АЛС ССТGTGCT-3
III ИМИ IIII
АЛЛ CGG TTG ССЛС-5
при этом получают плазмиду pCZ 155.3, кодирующую MET-ALA-PHE-PRO- ALA-MET- SE R-LEU-SER-VAL-bGH, или линкер с последовательностью нуклеождов
-5. -седт мо .ет пт кс ест лтс ict стс ice cc: Cfi . m ссс (.-с не OTCCT-J
И Л HI III HI III III III HI HI III III in III III MM
3 -T.M:IJC СТА M. CCC M ТЛС ДСЧ СГ.С AM CCO CAC ii., CCC ПС С Л C-S
5
0
5
0
при этом получают плозмиду pCZ156.3, кодирующую MET-ASP-bGH.
Приоритет по признакам: 06.03.84 поп.1; 26.07.84 по п.2.
Синтетические олигонуклеотиды для гена тимоэин I
at ambient temperature при комнатной температуре.
J а б л и ц а 1
PKEN111
EcoRl
-
Hpall Xbal PvuII Hpall
Hpal
EcoRl
PvuII
/
| Hpall 950bp Hpa II Fragment
jAful 462bp Alu I Fragment
iJEco RI Linkers/ T4 Ligase
EcoRl
4e6bp Eco Rf Fragment
Aii.l
pBR322 EcoRl
PstI
PvuII
E
Eco RI Alkaline
,-.c
Pnosphatas
Bam HI
Sail
Plasmid 103 (Figure 1)
Hindi It
S1 Nuclease
Bam HI Linkers T4DNALIgase
Bam HI
ind HI
У Есо RI PartialDigestion Xsi Nuclease«
XHindlH Linkers ./ T4ONALigase
A Hind III
/ :.-. ..,-.
У T DNALigase
4
Xbal co RI Bam Hi
Sail
ttoi.2
Bam HI
.T DNALigase VEcoRl
Plasmid 105 (Figure 2)
Sail 1 BamHT
Alkaline Phosphatase
Hind ill
#KEN021
Bam;HI Sail Xbal
Alkaline Phosphatase
Plasmid pKEN111 (101 in Figure 1)
Hpal
Sal I Linkers T4DNALigase
Sail Pvull
Bam HI tinkers T4 DNA Ligase
Bam HI
lppp
« 950bp Fragment Sal I, Bam HI Ends
Xbal EcoRI
Bam HI
IppP
Xbal
Bam HI
Фиг-3
Sail
Sma I
Hind III Bam HI
8тл I
Bam HI Linkers T4 ONA Ligase
Bam HI
691 bp Bam Hi Fragment
pNM575
Plasmid pNM575 (109 in Figure 4)
I Bam HI
691 bp Bam HI Fragment
I Fnu Dll
538 bp Bam HI, Fnu 01Г Fragment
pNM702
OU5
Pvu II
I Bam HI
I Alkaline
Phosphafase
Coding Region for Human Growth Hormone
Bam HI
Via. Ll
. Synflielic Fragment
Placmid pKEN021 Xba I-Bam Hi digested(107 in Figure 3)
Pvu il BamUl
Sa 1
Plasmid pNI/fo2 (111 in Figures)
Pvu II Partial Digestion
Pvu 11
Pvu II
Pst
pBP348
EcoRI
Pvu II Bam HI
3 lpp
VlHJ
114 (in Figure 6)
j Pvu II Partial f Digestion
JXbal
ЧAlkaline
tPhosphatase
Xbal
Bovine
Growth Hormone
Pvu II
Bam HI
Salt
pNM789
Pvu II
Фиг. 7
116
Xbal:
IHpall
Synthetic Fragment
Hormone
pPB348 (112 in Figure 6)
I Pvu ft j PstI 135bp Fragment
IHpall
IPartial
Digestion
Pvu II Bam HI
3 ipp ;
Sail
PJasmid pNM789 (117 in Figure 7)
l Pvu II Partial Digestion
| Bam HI
I Alkaline
т Phosphatase
Hind III
pNM789B
118
n ., Bam HI Pvu 11N..
Synthetic Frngmenl
Тд DNA Ligase
Pvu II
Coding Sequence lor Bovine Growth Hormone
Pvu II UamHI
Фм.«
EcoRI
Xbal
HgiAl
BstEII
BstEII
pCZ 1920
EcoRt
Hpa I
Фиг. 9
Bam HI
EcoRI Hpcfl
HgiAl
-- - i
BamHI
BstEII
Фиг fO
EcoRI
AATTCATGTCTGATGCTGCTGTTGATACTTCrTCTGAGATTACTACTAAAiGATCTTAAGGAGAAGAAGGAAGTTGTCGAAGAGGCTGAGAACTAATAG
SriCAGACTACGACGACA CTATGAAGAAGACTCTAATGATOATTTCTAGlAATTCCTCTTCTTCCTTCAAGAGCTTCTCCGACTCTTGATTATCCTAG
«iOu
I 00 I
IIII OM7O ЦОРО h-OPO (-
5Я OR
OPO ь-о
OMTO Ь-ОРО
L
0 II PO
OR
G Bu i о
TO bOPO OR4
Т Т Г8 Т Га Д8 r 8u rSz PQz т
DJoToYoToYoto(o(
nii и M il и и и м ii
PO kopo h-opo t-opo ЬОРО ЬОРО kopo V-OPO UOPO l-op J 4 J I J J J 4j I M I SI I I
то4 rvo no4 no no4 nn лс. Лг, Ar.N л
OR Ой
OR ОН ОЯ OR 11
OR OR
OPO
6a
step stop
BSIH CpLll.l1
Bam HI
0 I
II OPO (-
OR
OPO ь-оросе
OR
CBl Т
Too
OMTO hOPO l-OPO
N O
О | О
itit OPO ЬОР I I
он с
e
о
OPQCE
СЯ ,Ql
iBu ,Bz
ПМТО f-OPO I
1 on
О I О I О IIII II ЦОР
iv i i
on J оя о
о
-OPOCE OR
-BI
Т о С
az
OMTO
O | О I II I II I OPO НЭРО kOAn
OH I Ofl
Et3N/Py/H20
J&
2% BSA
J
Et3N/Py/H20
2% BSA
u
РО
000 II I II I II PO h-OPO OPOOR OR4 OR
.81
Bi
HO
с т с
j О0 i О ) ЦОРО ГОРО -ОРОСЕ N ncN ORN OF
ОН4 ORN
Ј
) TPSTe
2) Silica gel chrom.
3) Et3N/Py. H20
ОЯ
nSz
-IBu
Bl
OMTO t-OPO
N он4
U I О I О IIII II PO f-OPO HOPO
ORN ОПМ
OR
„at T rei I -S i ,9
HO h OPO - OPO (-О An
M A«M
I
PO h-OAl
v .
OH OR
1) TPSTe
2) 2% BSA
3) Silica gei chrom.
со со
CO
-t
КЗ
Gia .T
sBz
-8r
|O|0|0|O|0|O I n и и OMTO j-OPO kQPO HOPO h-OPO -OPO
OR4 OR4 OR Ofl OR
.81
iBu
.Bz
Bz
-3l
I ° I ° I ° I ° I ° I
nii и n и HO f-OPO hOPO Ь-ОРО Ц-ОРО KOPO f-OAn
ОН бн4 OH4 OR4
OH4 10
Т Га Д8 r 8u rSz PQz т
oToYoto(o(
M il и и и м ii
po ЬОРО ЬОРО kopo V-OPO UOPO l-op I J J J 4j I M I SI I I
no no4 nn лс. Лг, Ar.N л
OR ОН ОЯ OR 11
OR OR
OPO
6a
-Bz
OPOl-OAn OH
1) Silica gel chrom.
2) Cone. NH4OH
-----------------------г ----------„---
3) AcOH
4) HPLC
5 HO-G-T-T-C-T-C-A-G-C-C-T-C-OH 3
(fut. (2
12
T5 T6 T7 T8 T13T14 T15T16 I-I I |T4DNALigase
EcoRI I
Mpell BamHI
--Ц
t г
phi mil Ы
29 30 31 32
I V rg rg
TIC ia тгс otc АЛО ст сое сдо rcc лес ccciccc оде err Ало тле «a ттс тол осе Sec ТХйо ттс TOO cco5cc;cro
Р50М7Д4Л1
EcaRI.Hpall EcoRI
Ecqfll partial, Psll
iI
Pill
Јсой
irppo LE lmlon §S°R
-..- ма-ч
ESK ЕЯ;
II Qlf1 u P
x GCA CAQ О
COT QIC CtT AA
фи.М
EcoRI
EcoRI | Bam HI Eco RI
Bam HI
EcoRI
ME thymosin Bam HI
Tcs
ил. К
PSII
--- - сссоаа----r.CCCCCрИНО-4
Hp«ll fill
т rn
B«mHI Psll
-.«..-LceeccccL- t I f 1 1 --luGOGaa -- - 29 30 31 32 33
tyft Ihr rg мф glw
A CCUC ICCOCftCCC ITtTOGCCIiG GCCQCOC
BamHl, Hpall
Hpall
T4 EcoRI. BamHI
EcoRi
BamHI
Bg.ll
T4 llga Tcinjlotmjllon
.WiiЈ« .
Reverse Iransciipiase + сШТрч + Gam HI linker S CCGGATCCGG(l)n3
tAln3- (T)n GGCCTAGGCC 5
j Denature RNA/DNA hybrid I Klenow Pol
QamHI
(A)nCCGGATCCGG 3 (T)nGGCCTAGGCC 5
I SI Nucleate
| Terminal Iransferase + dCTP
ссссс
IAlnCCGGATCCGGCCCCC fO.GGCCTAGGCC
E-SJI Hpall Human Insulinр8ц cOfM
BamHl Psll
Aps
ЈT
Фиг К
Ecofll
BamHl
Psll
Pi I
Terminal Irsnslflr. + dGTP
( Anneal
j Trnnsform Ј. coli
} Select Tr.R
} Screen
EcoRI
BamHl
ЈT

| Способ приготовления солодовых вин | 1948 |
|
SU75444A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
