Изобретение относится к области получения Ы,0-бмс-ацилпроизводных хинониминоксимов общей формулы
N-О-Z,
где Аг- 1,4-хиноидный остаток; Z - ацильный радикал.
Указанные продукты относятся к классу соединений, применяемых в промышленности при вулканизации каучука и для синтеза красителей.
Известен способ получения М,О-бис-ацилхинониминоксимов, заключающийся во взаимодействии хинониминоксимов с хлорангидридами кислот в водном растворе щелочи. (О. Fischer, «Justus Liebigs Annalen der Chemie, 186, 145, 1895).
Указанный способ не является универсальным и имеет следующие недостатки:
1.Ввиду применения едкой щелочи нроцесс сопровождается побочными реакциями омыления хлорангидрида кислоты и гидролиза хинониминоксима в хиноноксим, что приводит
к сильному загрязнению конечного продукта и к необходимости применения избытка хлорангидрида.
2.Ввиду того, что реакционная масса представляет собой гетерогенную систему, реакция идет сравнительно медленно, что еще более увеличивает выход побочных продуктов
и соответственно уменьшает выход основного продукта.
3. Способ практически непригоден для ацилирования твердыми хлорангидридами, которые в условиях гетерогенного процесса почти нацело омыляются.
Цель изобретения - изыскание универсального способа получения Ы,О-бис-ацилхинониминоксимов, свободного от указанных недостатков и обеспечивающего получение продуктов с повышенной степенью чистоты.
Получение продуктов с повышенной степенью чистоты имеет чрезвычайно большое значение для последующего синтеза красителей и для вулканизации каучука, так как присутствие примесей ухудшает качество красителей и полимерных материалов.
Предлагаемый сиособ заключается в том, что хинониминоксимы в среде органического растворителя обрабатывают хлорангидридами кислот в присутствии водного раствора бикарбоната натрия. Образующийся продукт выпадает в осадок или выделяется выливанием реакционной массы в воду. Растворитель может быть регенерирован отгонкой и снова использован в процессе синтеза.
генной системе. Кроме того, вместо едкой щелочи используется соль - бикарбонат натрия, создающая меньший рН среды.
Это обеспечивает способу следующие большие преимущества:
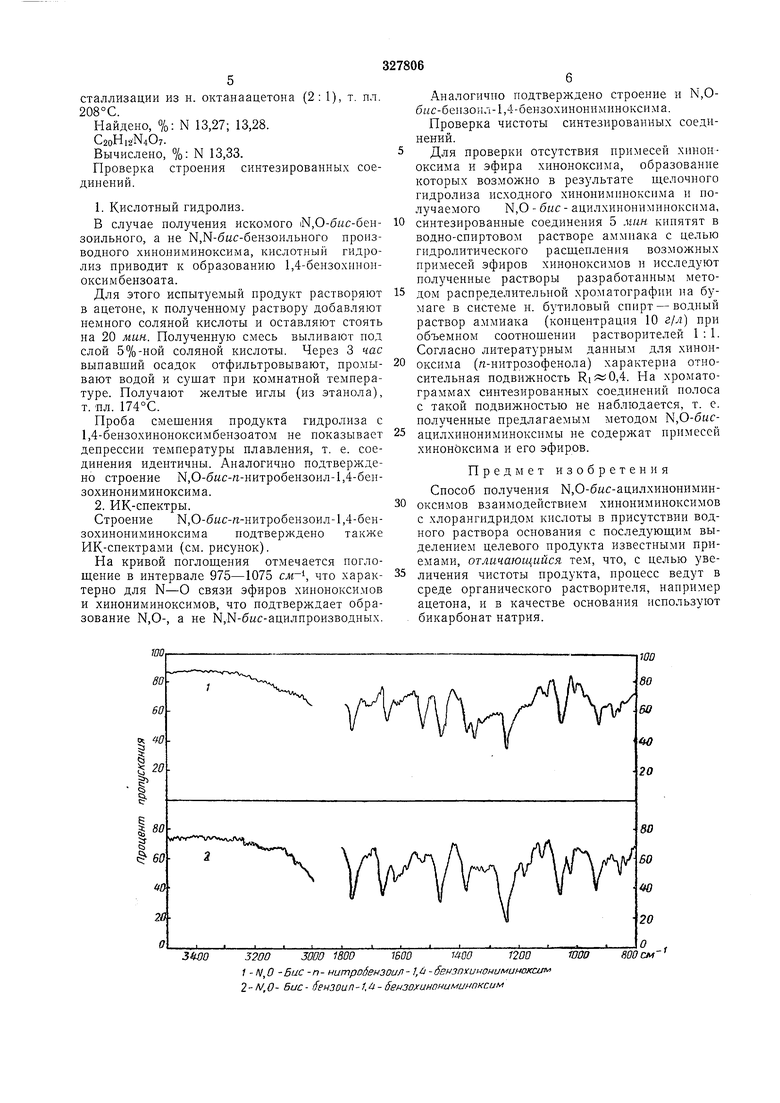
1.Ввиду того, что побочные реакции омыления хлорангидрида кислоты и гидролиза хинониминоксима практически не имеют места, |М,О-быс-ацилхиноннминоксимы получаются повышенной степени чистоты. Так, нанример, К,0-быс-бензоил-1,4-бензохинониминоксим (быс-бензоил-п-нитрозоанилин), полученный предлагаемым способом (см. пример 1), даже без дополнительной очистки перекристаллизацией имеет ту же температуру плавления, что и продукт, полученный известным снособом и подвергнутый перекристаллизации. Перекристаллизация Ы,0-быс-бензоил-1,4-бензохинониминоксима, синтезированного описываемым способом, обеснечивает получение продукта с т. пл. 149°С, т. е. на 7,5°С выше, чем известный способ {142°С).
2.Способ обеспечивает получение продуктов с высокими выходами (80-96%).
3.В отличие от известного способа новый способ не требует иснользования избытка хлорангидрида кислоты, а применяемые для синтеза исходные нродукты практически не подвергаются гидролизу, т. е. расходные коэффициенты по сырью для нредлагаемого способа значительно ниже.
4.Способ пригоден и для твердых хлорангидридов кислот (см. нрнмер 2), т. е. в отлнчие от известного способа является универсальным.
5.При проведении процесса требуется меньшее охлаждение реакционной массы, так как экзотермическая побочная реакция омыления хлорангидрида почти не наблюдается и процесс хорошо идет и при обычной температуре. Благодаря этому в производственных условиях может быть достигнута экономия хладагента и электроэнергии на его циркуляцию.
6.Способ, предусматривающий использование органических растворителей, обеспечивает протекание реакции не в водной суспензии, а в гомогенной системе, вследствие чего реакция идет быстрее и производительность аппаратуры в единицу времени значительно увеличивается.
7.Указанное выше обстоятельство дает возможность оформить процесс как непрерывный, т. е. облегчает механизацию и автоматизацию его в производственных условиях.
Предлагаемый способ технологически прост и может быть реализован не только в лабораторном, но и в производственном масштабе.
Пример 1. Получение Ы,0-бис-бензоил-1, 4-бензохинониминоксима.
онный раствор охлаждают до 5-10°С и при перемешивании постепенно добавляют раствор 13,44 г (0,16 г-моль бикарбоната натрия в 140 мл воды. Размешивание продолжают при комнатной температуре в течение 1 час. Образующийся коричневато-желтый кристаллический осадок отфильтровывают, промывают ацетоном для удаления следов примесей органических соединений, затем водои для удаления примесей неорганических соединений и сушат при 50°С. Выход 16 г, т. пл. 142°С.
Ацетоновый фильтрат и ацетоновый раствор после промывки осадка выливают нри
механическом перемешивании под слой холодной воды (3,5-4 л). Размешивание продолжают еще 3 час. Выпавший светло-коричневый осадок отфильтровывают, промывают водой и сушат при 50°С. Выход 9,4 г, т. нл.
139°С.
Общий выход 25,4 г (96,2%). После перекристаллизации из этанола получаются мелкие блестящие желтые пластинки с т. пл. 149,5°С.
В целях упрощения процесса можно также выделять продукт выливанием всей реакционной массы носле ацилнрования под слой воды (3,5-4 л). Выход тот же, т. пл. 141 °С. После перекристаллизации из этанола т. пл. 149,5°С.
По литературным данным (О. Fisher, «Justus Liebigs Annalen der Chemie, 286, 153, 1895) -дибензоилнитрозоанилин химически чистый продукт, плавится при 142°С.
Проба смешения продуктов, полученных
обоими методами, не показывает депрессии температуры плавления, что подтверждает идентичность обоих продуктов.
Количественный анализ подтверждает получение Н,0-быс-бензоил-1,4-бензохинониминоксима (старое название - дибензол-п-нитрозоанилин).
Пайдено, %: N 8,37; 8,44. СгоНиМаОз. Вычислено, %: N 8,48.
При м ер 2. Получение К,О-быс-и-нитробензоил-1,4-бензохинониминоксима.
К раствору 1,4 г (0,0115 г-моль} 1,4-бензохинониминоксима в 130 мл ацетона .при механическом перемешивании и комнатной темнературе постепенно приливают раствор 4,27 г (0,023 г моль п-нитробензоилхлорида в 70 мл ацетона и раствор 1,95 г (0,23 г-моль) бикарбоната натрия в 20 мл воды. Перемешивание продолжают еще 1,5 час, после чего выпавший осадок отфильтровывают, промывают ацетоном, затем водой и сушат при 50°С. Выход 2,4 3, т. пл. 202°С.
Дополнительное количество продукта получают выливанием ацетонового фильтра под
слой холодной воды (во избежание осмоления) или постененным добавлением воды к ацетоновому фильтрату. Выпавший осадок отфильтровывают, промывают водой и сушат при 50°С. Выход 0,96 г, т. пл. 197°С.

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ ХИНОНДИОКСИМОВ | 1967 |
|
SU201368A1 |
| СПОСОБ ПОЛУЧЕНИЯ N-МЕТИЛПИПЕРАЗИНА | 1962 |
|
SU146314A1 |
| ВСЕСОИЭЗИАЯБИЬЯии | 1971 |
|
SU321994A1 |
| Способ получения п-нитрозоанилинов | 1976 |
|
SU644780A1 |
| Способ получения производных -(бензтиазолил-2)-оксаминовой кислоты, или ее эфиров, или ее солей | 1977 |
|
SU680647A3 |
| СПОСОБ ПОЛУЧЕНИЯ 3- | 1970 |
|
SU433677A3 |
| Способ получения @ -ситостерина | 1983 |
|
SU1167187A1 |
| Способ получения 4- -(2метокси-5-хлорбензамидо)этил/бензолсульфонамида | 1976 |
|
SU662549A1 |
| СПОСОБ ПОЛУЧЕНИЯ б«с-ХРОМОНИЛОВЫХ СОЛЕЙ | 1972 |
|
SU341232A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АМИДИНОПЕНИЦИЛЛАНОВОЙ КИСЛОТЫ | 1973 |
|
SU406362A1 |
