Изобретение относится к способу получения новых производных 1, 2, 4-триазин:-5-онов, которые могут найти применение в сельском хозяйстве. ...-......,... Известные способы получения производных 1, 2, 4-триазин-5 нов не позволяют вводить любые радикалы в 3- и 6-ом положении триаэинового кольца. Известен способ получения 3,6-д.йзамещенных 1, 2, 4-триазин.-5-онов реакцией обменного разложения 1-циано-4-хлор-2,3 -диазабутадиенов с аммиаком и последующим гидролизом полученного 1,2,4-триазин-5-имина. Этот способ применим при условии, что оба заместителя одинаковы. В другом случае образуется смесь изомеров, которая либо вообще не может быть разделена, либо разделена с плохим выходом. Предлагается неизвестная модификация способа получения новых 1, 2, 4-триазин-5-онов обшей формулы N-Ri 2 R где f - водород незамещенный или ешенный алкил, алкенил, алкинил, циклкил, гидрокси-, axncoKCHjpjrana, арил, кил, или группы R Г приче 4 - водород или алкил с 1-4 атомами углерода; - водород, алкил, галогеналкил, фенил, алкилфенил, алкоксифенил, алкилмеркаптофенил и фенилалкил Ц. и ЯR вместе с соредним атомом 4о азота означает 5™б™ти членное гетерохшк личесхое холъцо; Бодород, незамещенный или замещенньпЕ алкил, алкенил, циклоал- кнл, арил, аралкил, или гетероциклический радикал; незамещенный или замещенный алкил, алкенил,. циклоалкил, арал кил или арил. Способ заключается в том, что диаза бутадиеиы общей формулы С-В2,(Е) где Ц и f{ имеют указанные значения;1 хлор или алкоксигруппа, подвергают реакции обменного разложе ния с аминосоединением, например первич ным амином, аммиаком, гидразином или гидроксиламином общей формулы (ш) где Ц имеет указанное значение, , при температуре С в прис.утстВии связываюшетх) кислоту средства. Реакх-шю проводят в присутствии разбавителя, в качестве которого используют полярные органические растворители, например спирты (метанол, этанол или изо-, пропанол), эфиры (тетрагидрофуран или диоксан), нитрилы (ацетонитрил), амиды кислот (диметилформамид), сульфоксиды (диметилсульфоксйд). В качестве связывающего кислоту средства применяют избыточное количество аминосоединения формулы 1И , обычно 2-5 молярное избыточное количество. В качестве связывающего кислоту сред ва можно также использовать карбонаты щелочных или щелочноземельных металлов Реакшпо предпочтительно проводят при 50-12О С при нормальном давлении, но может быть проведена и при повыщенном давлении Целевые продукты выделяют известными способами. По предлагаемому способу производные 1, 2, 4.триазин-5-она получают с высоки выходом и хорошей чистотой. В них заместители в 3 и 6-ом положениях триазинового кольца могут быть широко варьированы. Исходные диазабутадиены формулы |) могут быть получены реакцией обменного азложения 2-ацилгидразонов сложного эфира глиоксиловой кислоты формулы сбов7 ,И где и Но имеют указанные значения,.. Чу водород или алкил с 1-4 атомами углерода, с хлорирующими средствами, например пентагалогенндом фосфора, тионилхлоридом или фосгеном при О - 1ОО°С и при необходимости в растворителе. 2-А1Щлгидразонь1 эфиров глиоксиловой кислоты формулы IV могут быть получены реакцией обменного разложения сложного эфира глиоксиловой кислоты формулы СООК ,(г) где Ц„ и Б(„ имеют указанные значения, 3 7 с ацилгидразином формулы NH- NH, имеетуказанное значение, в присутствии органического растворителя при 5О-13О°С. Пример. 2-Этил-4-амино-6- -трет-бутил-1,2,4-триазин-5-он. А. 24,1 г (О,1О35 моля) 1-трет-бутил-1т-метоксикарбонил-4-хлор-4-этил-2,3-диазабутадиена при внешнем охлаждении :прикапьшают при размешивании в раствор :из 15,6 мл (0,32 моля) гищэазингидрата в 10О мл изопропанола таким образом, : что температура в реакционной массе. не превышает 4O°G. По окончании экзотерми- ческой реакции отгоняют растворитель, остаток обрабатывают водой и продукт реакпии отфильтровьшают. Получают 19,9 г i(94,4% от теоретического) 2-этил-4--амино-6-трет-бутил-1,2,4-триазин-5-она с т. пл. 149-151 С. После кипячения с 3 ч. циклогексана (находящиеся загрязнения растворены), после фильтрования при .15 Си высушивания при 5О С в вакууме т. пл. повышается до . Выход чи- стого триазинона составляет 18,4 г . (91% от теоретического). Б. 7,1 г (0,0021 моля) 92%-ного , сьфого продукта 1-трет-бутил-1-хлор-кар, бонил-4-хлор-4-этил-2,3-диазобутадиена при 20-ЗО С и размешивании прикапывают в раствор из 7,5 мл (0,14 моля) гидразингидрата в 5О мл изопропанола. Об{забатывают как указано в А. Получают 2,1 г (51,3% от теоретического) 2-этил -4 амино-6-трет-бутил-1,2,4-.триазин-5-он с т. пл. 154°С. П р и м е р 2. 3-Этинил-4-амино-6-фенил-1,2,4-триазин-5-он, В раствор нз 26,7 (0,1 моля) 1-феН1Ш-1 -этоксикарбонил-4-хлор-4-этил-2,3 -диазабутадиена в 100 мл диметилформамида при 5 - 1О°.. и размешивании прикапывают 10,0 г (0,2 моля) гидразингидрата, растворенного в 20 мл диметилформамида. После размешивания в течение 3 час реакционную смесь разбавляют 250 мл воды и оставляют стоять в течение ночи. Затем отсасывают от выпавшего твердого вещества, последний хорошо промывают водой и высушивают. Желтовато-белый сьфой продукт (17,4 г, 80% от теоретического) очищают переосаждением из изопропанола/воды. Получают 14,5 г (67,1% от теоретического) 3 этил-4-ам но-6-фенил-1,2,4-.триазин-5-он с т. пл. 164 С. П р и м е р 3. 3-Циклопропил-4-ами -6-фенил-1,2,4-триазин-5-он. В раствор из 53,0 г (0,2 моля) 1-ф нил- 1-этоксикарбонил-4-хлор-4-циклопро пил-2,3-диазабутадиена в 200 мл ацето- нитрила при О-5 С прикапывают 20 г (0,4 моля) гидразингидрата , растворенного в 100 мл а цетонитрила. Затем при комнатной температуре размешивают в течение одного часа, реакционньш раст вор сгущают до одной урети, разбавляю водой и отсасывают от образовавшегося осадка. После перекристаллизации из метанол/воды получают 26,5 г (58% от теоретического) З-циклопропил-4-амино-6-фенил-1,2,4-триазин-5-она с т. пл. 121°С. П р и м е р 4. 3-мeт л-4-амино-6- -(4-- метоксифенил)-1, 2, 4-триазин-5-он. В раствор из 7,5 г (0,23 моля) безводного гидразина в 1ОО мл ацетонитрила при температуре около 0°С прикапыв ют раствор иа 28,3 г (0,1 моля) 1-(4 -метокси-фенил) -1 -этоксикарбонил-4- -хлор-4-метил-2,3-диазабутадиена в 10О мл аценитрнла. Затем при ком натной температуре размешивают в течение 12 час. После отгонки растворителя осадок растворяют в эфире уксусной кис лоты и раствор промывают водой. Высу- раствор эфира освобождают рт растБорителя, оставшийся маслянистый оста кристаллизуют при растирании с эфиром. Получают IS г (65%. от теоретического ) З-метил-4-амино-6 - (4 -метокси-фенил)-1,2,4--триазин-5-она с т. пл. 203- 206°С, после перер ;азкдения из этанола с т. шт. 206°С. П р и м е р 5. 3-Этил-4-метил-6- -трет-бутил-1, 2, 4 триазин-5-он. В раствор из 9,3 г (0,3 моля) метиламина мл изопропанола при 1О20°С и размешивании прикапывают :,,,, 23,25 г (0,1 моля) 1-трет-бутил-1-метоксикарбонил-4-ХЛОР-4-ЭТИЛ-2,3-диазабутадиена. Затем при комнатной температуре размешивают в течение 3 час и оставляют реакционньй раствор стоять в течение ночи при комнатной температуре. После быстрого нагревания до 80 С растворитель отгоняют в вакууме, остаток поглощают водой и нерастворившийся в ней триазинон отфильтровывают. Получают 16,7 г сырого продукта с т. пл. 1О8112°С, который после повторного раствореш{я из промьшного бензина дает 15,1 г (77% от , теоретического) чистого 3-этил-4-метнл-6-трет-бутил-..1,2,4-триазин-5он с т. пл. 112-113°С. П р и м е р 6. 3-Этил-4-метил-6-фенил-1,2,4-триазин-5-он. В раствор из 26,7 (0,1 моля) 1-фенил-1-этоксикарбонил-4-хлор-4-этил-2,3-ди- азабутадиена а 100 мл ацетонитрила при С прикапывают раствор из 15 г (0,48 моля) метиламина в 100 мл аиетойитри- ла. Затем при комнатной температуре размешивают в течение 15 час и в течение : :6 час нагревают до кипения. По окончании i реакции растворитель отгоняют в вакууме, остаток растворяют в эфире уксусной кислоты и промывают водой. Раствор эфира уксусной кислоты высушивают и растворитель отгоняют в вакууме. Маслянистый . остаток кристаллизуется при растирании с эфиром. Получают 11,8 г (55% от теоретического) 3-этил-4-метил-6-(l)eнил-1,2,4-триазин-5-она в виде бесцветных кристаллов с т, пл. 179-18О С, которая после перерастворения из 20-кратного количества бензола повышается до . П р и м е р 7. 3-этил-4-аллил-6-(4.-метил.-фенил )-1,2,4-триазин-5-он. В раствор из 15 г (0,26 моля ) аллиламина в 100 мл ацетонитрила при 0-5 С прикапьшают раствор из 29 г (О,1 моля) 1-(4 метил-фенил)-1-этоксикарбонил-4-хлор-4-этил-2,3-диазабутади- бна в 10О мл ацетонитрила. После размешивания в течение 12 час при комнатной температуре реакционную смесь нагревают до кипения еще в течение 3 час, затем
7
растворитель отгоняют В вакууме, остаток растирают с водой и растворяют в эфире уксусной кислоты. Эфирный раствор высу шнвают и растворитель отгоняют в вакууме. Получе1шый маслянистый остаток кристаллизуется при растирании с эфиром. Получают 12 г (48%-, от теоретического) 3-эти п-. лил-6 - (4-метил-фени/1) -1,2, jlp триазин-5 она с . т. пл. 93-98 С, , рая после перерастворения из 30-кратного количества лигроина повышается до
101-102°С.
Пример 8. 3-Этил-4-гидроксц-6-фенил-1,2,4-триазин-5-он.
В раствор из 14,9 г (0,059 моля) 1-фенил-1-метокси-кар6онил-4-хлор-4 Этил-2,3-диазабутадиена в 5О мл изо-
8
пропанола прикапывают смесь из 6,2 г (0,089 моля) гидроксиламингидрохлорида и 15,0 г (0,148 моля) триэтиламина в
150 мл изопропанола. При этом температура повышается 20-35 . Затем в течение 2 час кипятят с обратным холодильником,, растворитель отгоняют в вакууме и остаток обрабатывают водой, причем последний крист лизуется. Получают 8,6 г (67% от теоретического) сьфого продукта с т. пл. 16О-165 С, а после перера.ст-., ворения из 7О мл метанола 5,2 г(40,от теоретического) чистого 3-этил-4-гидрокси-6-фенил-1,2,4-триазин-5-она с т. пл. 171-171,5 С. Аналогичным образом получают соединения общей формулы I, характеристика которых приведена в табл. 1.
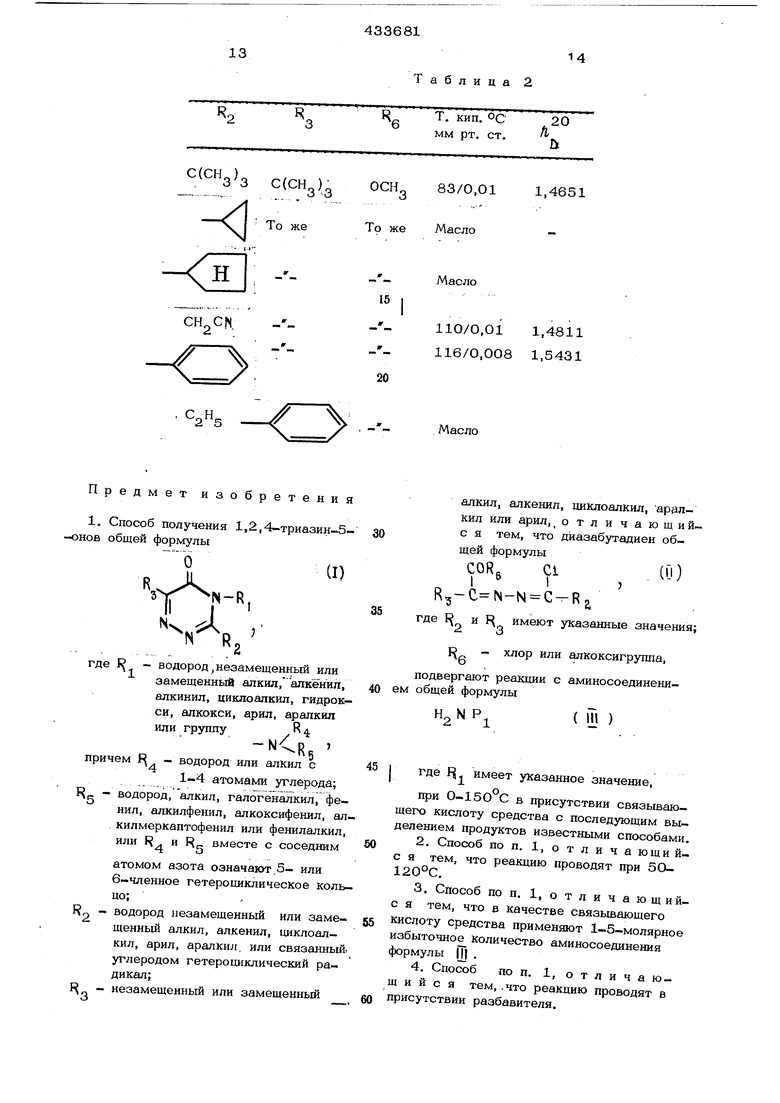
.Таблица 1 П p и м e p 9. Получение исходных материалов формулы П (диазабутадиены А, 1-.трет-бутил-1-метоксикарбонил-4 -хлор-4-4-этил-2,3-диазабутадиен. Раствор из 42,8 г (0,2 моля) 2.пропионилгидразона метилового эфира 1-треТ -бутилглиоксиловой кислоты в 50 мл ме тилеихлорида, размешивая при 20-30°С (наружное охлаждение необходимо, так ка реакция протекает экзотермически) при размешивании прикапывают в суспензию 41,7 г (О,2 моля) фосфорпентахлорида в 150 мл метиленхлорида. Когда фосфорпен тахлорид полностью переходит в раствор, реакционную смесь нагревают с обратным холодильником до окончания вьщеления га за. После этого отгоняют растворитель и образовавшийся фосфороксихлорид отгоня ют при давлении 0,015 мм. рт. ст. и температуре бани 30°С. Остается желтоватое масло, достаточно чистое для дальнейшего обменного разложения. При перегонке в вакууме получают 42,5 г (87% от теоретического) 1-.трет-бутил-1-метоксикарбонил-4-хлор-4-.этил-2,3 -диазабутадиена с т. кип. Кр. . 64с и роо.оив П. 1.46 5 4. Вариант способа. Через раствор из 42,8 г (0,2 моля) 2-пропионилгидразона метилового эфира 1-трет -бутил-глиоксиловой кислоты в 1ОО мл тетрахлоруглерода после добавления 2 мл диметилформамида пропускают в течение 2 час 30 мин при температуре реакции . 5О-6О С медленный поток фосгена. При этом наряду с неизрасходованным фосгеном из отводной трубы реакционного сосуда выходит много хлористого водорода. По окончании реакции нагревают до кипения и одновременно вводят азот для удаления оставшегося фосгена и хлористого водорода. Затем растворитель отгоняют в вакууме и остаток вьщерживают в течение нескольких минут при 50°С 0,1 мм рт. ст. Получают 44,7 г масла, содержащего наряду с 1-трет-бутил-1-метокси- карбонил-4-хлор-4-этил-2,3-диазабутади- еном небольшое количество исходного материала как побочный продукт. Определённый газохроматографический выход состав ляет 57,4% от теоретического. Необходимый как исходный материал 2-пропионил- гидразон метилового эфира 1-трет бутилглиоксиловой кислоты получают следующим образом: 1,1 л водного раствора соли калия 2,2-диметил-1-оксо-масляной , кислоты, содержащего 6,75 моля ки лоты, разбавляют 66 г (0,75 моля) I. гидразида пропионовой кислоты и ЗОО мл метиленхлорида. В течение 2 час при сильном размешивании прикапывают 93 мг концентрированной соляной кислоты, размешивают еще в течение 6 час, дают отстояться и разделяют обе фазы. Водную фазу дважды экстрагируют 200 мл метиленхлорида. Соединенные органические . экстракты высушивают над хлоридом кальция и отгоняют в вакууме. Получают масЛО; постепенно застывающее и дающее 135 г (90% от теоретического) 2-пропи- онилгидразона 1-трет-бутилглиоксиловой кислоты с т. пл. 103-105°С (из петролейного эфира). Свободный 2-пропионилгидразон-1-трет-бутил-глкоксиловой кислоты обработкой эквивалентным количеством карбоната каг-, ЛИЯ, переводят в его калиевую соль. 24 г (0,О77 моля) калия пропионилгидразон-трет-бутилглиоксиловой кислоты растворяют в 120 мл хлорбензола, разбавляют 12 мл (О,О9 моля) диметилсульфата и размешивают в течение 5 час при 80 С. После отсасывания образовавшейся соли калия и последующего промывания хлорбензолом растворитель отгоняют в вакууме. Оставшееся масло может быть очищено дистилляцией в высоком вакууме. Получают, 19,2 г (9О% от теоретического) 2-прошюнилгидразона метилового эфира 1-трет-бутилглиоксиловой кислоты с т. кип. Кр ,. 90-91°С и т. .пл. ,006 28-29°С. , Б. 1-Фенил-1-этоксикарбоннл-4-хлор.,-4-этил-.2,3-диазабутадиен. 37,2 г (0,145 моля) 1-фенил-1-хлоркарбони Л-4-Х ЛОР-4-ЭТИЛ-2,3-диазабутадиена при охлаждении льдом прикапывают в раствор этилата натрия из 6,9г(О,3 моля) натрия в 15О мл этанола. Затем реакционную смесь фильтруют, отгоняют растворитель и остаток поглощают в эфире. Эфирный раствор дважды промывают 1ОО мл воды, высушивают и выпаривают досуха. Получают 31,8 г( 90,3%,от теоретического) 1-фенил-1-этоксйкарбонил-4-хлор-4- этил-2,3-диазабутадиена в виде вязкого масла «24 . Применяемый в качестве исходного ма- териала 1-фенил-1-хлоркарбонил-4-хлор-4- -этил-2,3-диазабутадиен может быть получен следующим образом. Раствор из 15О г (1 моль) фенилглиоксиловой кислоты в ЗОО мл воды и раствор из 88 г (1 моль) пропирнилгидразина в lOO мл вЪда соединяоот вместе и охлаждают до + 1О°С. После 3- или 4-час стояния от- фильтровывают образовавшийся осадок. Получают 196 г (89% от теоретического) 2-.пропио11илгидразона фенилглиоксиловой кислоты с т. пл. . 44 г (0,2 моля) 2-пропионилгидразона фенилглиоксиловой кислоты суспендируют в 250 мл дихлорметана и порциями смешивают с 83,2 г (0,4 моля) фосфорпентахлорида. Смесь размешивают при j, комнатной температуре так долго, пока не образуется прозрачный раствор и не закан чивается вьщеление хлорводорода. Дистилляцией в вакууме получают 37,2 г (72% от теоретического) 1-фенил-1-хлоркарбо нил-4-хлор-4-этил-2,3-диазабутадиена в виде бесцветной жидкости с т. кип. 127 С. В. 1-фенил-1-этдксикарбонил-.4-хлор-4-циклопропил-диазабутадиен. 52,0 г (0,21 моля) 2-циклопропилкарбонилгидразона этилового эфира фенилглиоксиловой кислоты растворяют в 300 мл метиленхлорида и порциями смеши- . вают с 41,6 г (0,2 моля) фосфорпентахлорида. Размешивают до полного раство рения. Затем растворитель и образовавшийся фосфороксихлорид отгоняют в ваку- уме. Получают 53 г (94,6% от теоретического ) 1.-фенил-1-этоксикарбонил-4-хлор-4-циклопропил-2,3-диазабутадиен 24 1,5679. Исходный 4-циклопропилкарбонилгидра- зон этилового эфира фенилглиоксиловой кислоты получают следующим образом: 89,0 г (0,5 моля) этилового эфира февилглиоксиловой кислоты растворяют в 150 мл метанола и смешивают с 50 г (0,5 моля) гидразида циклопропанкарбоновой кислоты (т. пл. 97 С). Реакционную смесь нагревают до кипения с обратным холодильником в течение 30 мин, ох лаждают, оставляют стоять в течение нескольких часов и отсасывают от образова . шегося осадка. Сгущением фильтрата получают осадок. Соединенные остатки филь ра промьшают небольшим количеством холодного метанола и эфира и высушивают. Получают 120 г (92% от теоретического) 2-циклопропилкарбонилгидразона этилового э4)ира 1-фенилглиоксиловой кислоты с т. пл. 116-120°С. Г. 1-(4-Д етокси-фенил)-1-этоксикар бонил-4-хлор-4-метил-2,3-диазобутадиен. В раствор из 24 г (0,1 моля) 2-ацетилгидразона этилового эфира 1 (4-меток си1 -фенилглиоксиловой кислоты в 250 мл метиленхлорида добавляют порциями при r.2. разм ешивании 20,8 г (0,1 моля) фосфорпентахлорида. После размешивания в течение получаса при -5 С реакционную смесь выливают в 1 л ледяной воды, содержащей 100 г гидрогенкарбоната натрия. Органическую фазу отделяют и высушивают. После отдистиллированйя растворителя остается 24,5 г сырого (95% от теоретического) 1-(4-метокси)-фенил-1-зтоксикарбонил-4-.хлор-4-метил-2,3-диизабутадиена в виде масла ( м, 1,5545), Исходный продукт 2-ацетилгидразон этилового эфира 1-(4-метокси)-фенил-п -глиоксиловой кислоты получают реакцией обменного разложенияэтилового эфира (4-метокси)-фенилглиоксиловой кислоты и ацетилгидразина в кипящем этаноле с T. пл. 91°С. Д. 1-трет-Бутил-1-хлоркарбонил-4- -ХЛОР-4-.ЭТИЛ-2,3-Диазабутадиен. В суспензию из 1О4 г (0,5 моля) фосфорпентахлорида в 150 мл метиленхлорида при размешивании прикапывают раствор из 50 г (0,25 моля) пропионилгидразона 1-трет-бутилглиоксиловой кислоты в 50 мл метиленхлорида и реакционную смесь кипятят с обратным холодиль- НИКОМ до окончания выделения хлористого водорода. Затем растворитель и образовавшуюся хлорокись фосфора отгоняют в вакууме и остаток дистиллируют в вакууме. Получают 52 г (81,4% от теоретического) 1-трет-бутил-1-хлоркарбонил-4- -хлор-4-этил -2,3-диазабутадиена с т. кип. Кр 61-62 С. Для реакции замьпсания кольца в 3-алкил-1,2,4-триазин-5-она очистка посредством дистилляции не является необходи-мым, может быть применен сырой продукт, содержаший 92% диазабутадиена. Исходный продукт 2-пропионилгидразона 1-трет-бутил- глиоксиловой кислоты .как описано в примере А. Аналогично могут быть получены 4-.хлор-2,3-диазабутадие.. ны, приведенные в табл. 2,
Этоксим(уЩ1
62
NH,
64-67
13
R,
с{сн„)„
О
То же
14
Таблица 2
RТ, кип. ос20
мм рт. ст. j.
ОСН„ 83/0,011,4651
о
То же Масло
н
Масло
110/0,01 1,4811 116/0,008 1,5431
/
Масло







| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 4-амино-1,2,4триазин-5-онов | 1974 |
|
SU553933A3 |
| Способ получения производных 1,2,4-триазола или их солей | 1975 |
|
SU615857A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ФЕНИЛИМИДАЗОЛИЛАЛКАНОВ | 1971 |
|
SU415875A3 |
| Способ борьбы с грибками | 1973 |
|
SU648045A3 |
| Способ получения -(1-алкен-1-ил)амино-симмтриазинов | 1972 |
|
SU437296A1 |
| Способ получения 3,4-дигидро-1,2,4триазинов | 1974 |
|
SU540570A3 |
| Способ получения производных диазепина | 1973 |
|
SU482045A3 |
| СПОСОБ ПОЛУЧЕНИЯ 1,3,3-ТРЕХЗАМЕ1ЦЕННЫХ 3-АЗОЛИЛПРОПИНОВ | 1971 |
|
SU430551A1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ ПРИМЕНИМЫЕ В СЕЛЬСКОМ ХОЗЯЙСТВЕ СОЛИ И ГЕРБИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2097380C1 |
| Способ получения пиразолонов-5 или их солей | 1974 |
|
SU668600A3 |
Предмет изобретения 1. Способ получения 1,2,4-триааин-5- -онов общей формулы О R к.Д N R, где t - водород незамещенный или замешенный алкш1,алкенип, алкинил, циклоалкил, гидрокси, алкокси, арил, аралкил или группу R -NC D ) причем J - водород или алкил с 1-4 атомами углерода; Ч - водород, алкил, галогеналкил, фенил, алкилфенил, алкоксифенил, ал килмеркаптофенил или фенилалкил, или Нд и Яр, вместе с соседним атомом азота означают.5- или 6-членное гетероциклическое коль цо; - водород незамещенный или замещенный алкил, алкенил, циклоалкил, арил, аралкил, или связанный углеродом гетероциклический радикал;1 - незамещенный или замещенный алкил, алкенил, циклоалкил, аралкил или арил,о тл и чающийс я тем, что диазабутадиен общей формулы 1 и f имеют указанные значения; о5 хлор или алкоксигруппа. подвергают реакции с аминосоединеним общей формулы где FJ имеет указанное значение, при 0-15О С в присутствии связывающего кислоту средства с последующим выделением продуктов известными способами. 2.Способ по п. 1, о т л и ч а ющи йс я тем, что реакцию проводят при 50120°С. 3.Способ по п. 1, о т л и ч а ю щ ийс я тем, что е качестве связьшающего кислоту средства применяют 1-5-молярное избыточное количество аминосоединения формулы . 4.Способ по п. 1, отличающийся тем, .что реакцию проводят в присутствии разбавителя. 15 5. Способ по п. 1, о. т л и ч а ю щ и и с я тем, что в качестве разбавителей используют полярные органические J.6 растворители, например спирты, эфиры, нитрилы, диметилсульфоксид, диметилформамид.
