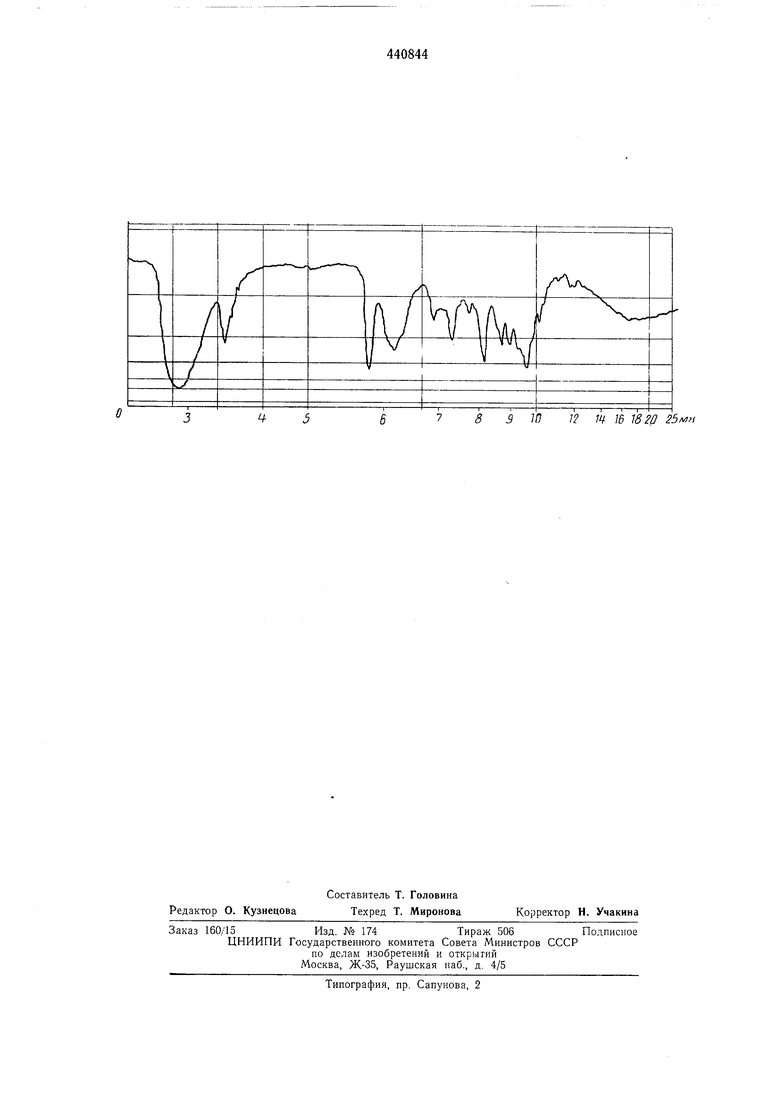
Изобретение относится к производству антибиотиков. Известен способ получения триацетильного производного спирамицина 1, заключающийся в том, что спирамицин 1 подвергают взаимодействию с уксусным ангидридом в те чение 24 час без нагревания. Реакционную смесь выливают в воду со льдом, доводят рН до 8,0, отфильтровывают осадок целевого продукта и перекристаллизовывают его из петролейного эфира. Целью изобретения является получение изомерного продукта, обладающего повышенной противобактериальной активностью. Поставленная цель достигается тем, что ацетилирование осуществляют в течение 5-70 час при 50-150°С, а образовавшееся тетраацетильное производное нагревают в водно-метанольной среде до удаления одной ацетильной группы. Триацетиловый спирамидин I соответствует формуле CsiHsaOigNs и обладает следующими физико-химическими свойствами: порошок кремового цвета, т. пл. 156-157С; dj - 97° (с-1, этанол); элементарный анализ, %: С 60,0-60,5; Н 8,2-8,4; О 28,3; N 2,6-2,65; СНзСО 12,5-12,9. УФ-спектр (определен в растворе этанола): максимальная абсорбция при 230 нм (EjcM 280). ИК-спектр (определен в таблетках в сочетании с КВг) приведен на чертеже. Пример 1. Приготовление тетраацетилового спирамицина 1. Раствор 1000 г спирамицина 1 выдерживают при 100°С в течение 24 час в 5400 г ацетилирующей смеси, полученной путем перемешивания 4752 г пиридина и 648 г уксусного ангидрида. После охлаждения реакционную смесь выливают на 10 кг ледяной воды, доводят значение рП раствора до 7,5 путем прибавления 800 см 10 я. едкого натра и экстрагируют хлороформом сначала по 2 л, а затем по 1 л. Хлороформные растворы сливают вместе и промывают 4 раза водой (по 4 л каждый раз), затем фильтруют и выпаривают досуха. Остаток высушивают при пониженном давлении (1 мм рт. ст.), затем вновь растворяют в 30 л циклогексапа. Полученный раствор фильтруют при 28°С, затем выпаривают досуха. Твердый остаток высушивают при пониженном давлении (1 ММ рт.ст.). Получают 1150 г тетрацетилового спирамицина I, имеющего титр 1185 ед/мг, т. пл. 148-152°С; -106° (С 2, этанол); СНзСО 17,0%. Приготовление триацетилового спирамнцина I.
Кипятят с обратным холодильником в течение 24 час 100 г тетраацетилового спирамнцина I, полученного описанным способом, в 9000 см смеси, состоящей из 7200 см метанола и 1800 см воды. После охлаждения добавляют 10 л воды и экстрагируют хлористЫМ метиленом по 4 л. После осветления жидкости ставят на испарение хлористый метилен и высушивают остаточный продукт под пониженным давлением (1 мм рт. ст.) при 60°С до постоянного веса. Получают 705 г триацетилового спирамицина I, имеющего титр 1670 ед/мг; т. нл. 148-150°С; 86°С (С 2, этанол), СНзСО 12,6%.
Пример 2. Очистка триацетилового спирамицина I кристаллизацией.
27,5 г полученного в примере 1 триацетилового спирамицина I растворяют в 50 см диизопропилового эфира. Образовавшийся раствор кристаллизуют в ледяной бане при встряхивании в течение 24 час. Кристаллический продукт разделяют фильтрованием, промывают три раза (по 10 см) диизопропиловым эфиром и высущивают при температуре 40°С и пониженном давлении (2 мм рт. ст.) в течение 12 час.
Получают 13,5 г кристаллов триацетилового спирамицина I, т. пл. 150-152°С.
Пример 3. Очистка триацетилового спирамицина I распределением против течения.
44,5 г триацетилового спирамицина I, полученного по примеру 1, очищают распределением против течения между разделенными фазами смеси: бензол + цикл огексан- -метанол + вода (45 : 55 : 80 : 20 по объему), устраивая 300 переносов в аппарате Крейга, где фракции имеют объем 50 см. Триацетиловый спирамицин I в таком случае находится во фракциях 140-210 (коэффициент распределения 1,38).
Содержимое фракций 164-196 собирают, концентрируют до 200 см при температуре
Активность относительно
25°С и пониженном давлении. Полученный водный концентрат экстрагируют при значении рН 8,5 два раза в одном объеме бензола. Собранные бензольные экстракты промывают 20% (от их объема) воды, высушивают на безводном сульфате натрия и концентрируют досуха при температуре 30°С и пониженном давлении (3 мм рт. ст.). Таким образом получают 15,6 г очищенного триацетилового спирамицина 1, d -89,2° (, этанол), титр равен 2300 ед/мг.
6,4 г этото продукта растворяют в горячем виде в 140 см диизопропилового эфира. Полученный раствор фильтруют и концентрируют до 50 см под пониженным давлением. Охлаждением в ледяной бане и взбалтыванием в течение 2 час получают бесцветные иголки, которые разделяют фильтрованием,
промывают в б см диизопропилового эфира и высушивают при температуре 40°С и пониженном давлении (3 мм рт. ст.) в течение 12 час. Получают 5,25 г кристаллического триацетилового спирамицина I с титром
2300 ед/мг, т. пл. 150-152°С.
i3,75 г этого кристаллического триацетилового спирамицина I вновь растворяют в горячем виде в 150 см диизопропилового эфира и полученный раствор фильтруют, затем концентрируют до 45 см при пониженном давлении. Кристаллизуя в холодной бане, получают 2,5 г рекристаллического триацетилового снирамицина I с титром 2300 ед/мг, т. пл. 156- 157°С, -97° (, этанол).
Полученный предлагаемым способом триацетилспирамицин I является изомером, в котором гидроксильный радикал микаминозы замещается ацетоксильным радикалом, а один
из ацетоксилов микарозы замещается гидроксилом, он обладает повышенной противобактериальной активностью.
Противобактериальная активность триацетилспирамицина I приведена в таблице.

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных фенотиазина или их солей | 1975 |
|
SU577994A3 |
| Способ получения производных цефалоспорина или их солей с металлами или азотсодержащими основаниями | 1974 |
|
SU566525A3 |
| Способ получения антибиотика | 1965 |
|
SU556732A3 |
| ЯДТЕНТНО- ТЕХНИЧЕСКАЯ БИБЛИОТЕКА | 1969 |
|
SU240564A1 |
| Способ получения производных 1,5,10, 10а-тетрагидротиазоло /3,4-в/изохинолина или их оптических изомеров или кислотно-аддитивных солей | 1977 |
|
SU635874A3 |
| Способ получения 2-(3-бензоил2-оксифенил)-пропионовой кислоты | 1974 |
|
SU517246A3 |
| ПРОИЗВОДНЫЕ ПЕРГИДРОИЗОИНДОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2127260C1 |
| Способ получения производных даунорубицина или их солей | 1976 |
|
SU659095A3 |
| Способ получения производных 1,2-дитиоланов | 1977 |
|
SU683622A3 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВОЗНИКАЮЩИХ ВСЛЕДСТВИЕ ДИСФУНКЦИИ НИКОТИНОВЫХ РЕЦЕПТОРОВ АЛЬФА7 | 2004 |
|
RU2376290C2 |
Предмет изобретения Способ получения триацетильного производного спирамицина I .путем обработки спирамицина I уксусным ангидридом, выделения 45 и очистки целевого продукта, отличающийся тем, что, с целью получения изомерного продукта с повыщенной противобактериальной активностью, адетилирование осуществляют в течение 5-70 час при 50-150°С, а образовавшееся тетраацетильное производное нагревают в водно-метанольной среде, 8 3 w 1 т J6 18 га 25мн
