где R - низшая апкилькая группа;
R - или атом водорода, или вместе с Н образует кар6о101клическое кольцо и R - инэший алкил, фенил или низшая алкоксигруппа, с соединением формуль
8ч.хН
в органическок; растворителе, таком как ацетсщ. этилацетат или их смеси, при температуре 0-25° С, причем символы Аи В в формулах II и IV означают: А - водород или солеобразующнй , а В - сжсигруппа. алкилсульфонилоксигрушга, аршюупьфшоикжснгрушга или атом галоида, либо А - ацильный радикал, а В - оксигруппа, алкнпсульфонил(жснгруш1а или арнлсульфогашсжснгруппа.
Продукты выделяют в свободном виде или в виде аддитивной соли.
Предпочтительной аддитивной солью соедииеиия является хлористоводородная, но могут быть получены и соли с органическими кислотами, в особенности те, которые применяются для образования солей с самой 6 - (D) (а - аминофенилацетамидо) - пенициллаиовой кислотсж.
Группа X в формуле И может быть свободнш аминогруппш, но реакция проходит лучше, когда соединение II имеет защищениую аминогруппу (енаминовую группу формулы III).
В таких случаях проще действовать натриевой или калиевой солью N-зaulишeниoгo соединения 11 и соединения III, где R и R вместе означают карбоциклическое кольцо, а В - атом галоида, в частности бром или хлор.
Когда группа А является водородом или солеобразующим ионом, а группа В - оксигруппой, то сое,(ннение фактически представляет равновесную систему
НСО
НООС
и может оыть изомером, который и вступает в реакхщю. Предпочтительно использовать алкнлсульфониловый сложный эфир или арилсульфоииловый
(в особенности л толуолсульфоиат) для обеспечения более гладкого протекания реакции. При этом для достижения высоких выходов обычно требуется присутствие основания.
В случае, когда группа А является органической
ащшьной группой, ясно, что соединение II просто смешанный ангидрид, и ацильиая группа может быть одной из разнообразных алифатических или ароматических ацильиых групп, но обычно удовлетворительны алкоксикарбонильиые группы (например, группа CjHsCKIO). Когда в качестве реагеита II применяется смешанный ангидрид, то группа В в реагенте IV может быть оксигруппой, алкилсульфонилоксигруппой или арилсульфонилоксигруппой.
Другим реакционноспособным этерифицирующим производным соединением 11 является галондангидрид кислоты, в частности хлорангидрид. Это соединеиие может реагировать с гидроксисоединением IV или с алкилсульфоииловым сложным эфиром его (иапример, п - толуолсульфонатом) в присутствии связывающего кислоту агента с получением целевого фталидного эфира.
Пример. А. 3- Бромофталид - (3 - бром - I
-(ЗН) - изобензофуранен).
Фгалид (10,0 г, 0,75 моль) и N - бромсукциниМвд кипятят с обратным холодильником в 200 мл сухого четыреххлористого углерода 3-4 ч, в присутствии каталитического количества а - азоизобутироиитрила. Конец реакции определяют исчезновением N - бромсукцинимида со дна реакционного сосуда и накоплением его наверху. Сукцинимид удаляют фильтрованием и фильтрат концентрируют в вакууме до 15-20 мл. После охлаждения этого концентрата отфильтровывают 13,0 г неочищенного 3 - бромфталида (выход ) с тлл. 75-80С в внде белого кристаллического вещества. .
Продукт перекристаллизовывают из циклогексана и получают в виде бесцветных пластинок с Т.ПЛ. 78-80° С. Выход 95%.
ЯМР (CCl4)Br 5: 7,67 (4Нм., ароматика); 7,38 (CH.SCH-)Хлористоводородный D (-) а - аминобензилпенициллинфталидный сложный эфир.
Безводный D (-) а - аминобензилпеницшшин (17,5мл, 0,05 моль) и триэтиламин (7,10мл,
1 зкв) смешивают с ацетоном, содержащим 1% воды (350мл). Через 1/2 ч добавляют бикарбонат
(5 г) и 3 - бромфталид (10,65 г, 0,05 моль) и смесь перемешиваюпг арк комиатной температуре 4 ч. После фильтрования фильтрат концентрируют в вакууме до- 75 МЛ( добавляют этилацетат (500 мл) и полученный раствор промывают 2%-иым раствором
бикарбоната иатрия (2x100мл), затем водой ( 100 мл). К этилацетатиому раствору добавляют воду (150мл) и при сильном перемешивании по каплям добавляют 1 и. раствор соляной кислоты до получения в водной фазе рН2,5. Этнлацетатиый
слой отделяют и высушивают над безводным сульфатом магния. Затем к прозрачному желтому этцлацетатному фильтрату добавляют эфир до прекращения образования осаждавшегося белого аморфного твердого вещества и получают 7,8 г продукта (28,8%). Дальнейщий продукт (0,8г,3,0%) получают из водной фазы следующим образом. К водному слою добавляют н.-бутанол и полученную смесь испаряют в вакууме до удаления всей воды. Получившийся бутанольный раствор выливают в эфир (2000 мл)| выделившийся аморфный осадок отделяют. Общий выход составляет 31,8%.
УФ-спектр (КВч) содержит следующие сильные полосы при: 1778, 1682, 1500, 1285, 1149,978,752, 697см-.
ЯМР (CH3)jSO/D20; 5:7,88 (4Нм., ароматика, фталид) i
7,48 (бНм., ароматика); 5,50 (2Н м., р-лактамы); 5,16 (iHc., а-протон); 4,54 (1Нс., Сз-протон); 1,45, (6H,d сдв. диметилы).
Испытание на чистоту с гидрОксиЛаминрм и цнстеином дает соответственно 92,4 и 86,5%.
Вьпислено, %: С 55,65; Н 4,67; N 8,11; S 6,19;
06,84.
C24H2405N3Cli
Найдено. %: С 54,49; Н4,67; N7,83; S 6,20; С1 5,18..
И р Н м е р 2. Хлористоводородш 1й фталид 6- (D
.а - аминофенилацетамида) - пеницилланат.
Тонкую суспензию калиевой соли енаминзащищенного ампициллина (25,18 г, 0,05 моль) и 3 бромфталида (10,65 г, 0,05 моль) обрабатьшают в смеси ацетон - этилацетат (1:2, 1500мл) в течение 24 ч. После фильтрования органический слой дважды промьшают порциями по 250 мл 1 н. бикарбоната натрия, высушивают над сульфатом магния и концентрируют в вакууме. Добавление эфира вызьтает кристаллизацию фталида енаминзащищенного а - амии офени лацетамидопенииялланата с выходом 85%. ЯМР-спектр (СОз)25О : 7,86 (4Нм., ароматика, фталид); 7,60 (1Нс. СООСН); 7.35 (5Н с., ароматика); 5,30-5,65 (ЗН м., (J-лактамы и а-протон); 4,53 (Шс., С - 3 - протон); 4,50 (Шс.Н); 3,56 (ЗНс. ОСНз); 1,78 (ЗНс. СНз.); 1,50 (6Н м., сдв. диметилы).
Вычислено. %; С 59,26; Н5,11; N7,40; 55,68
СзвНзвЫзОа
Найдено, %: С 58,83; Н 5,00; N 6,89; S 5,34. Однокапельное пятно на биохроматограммё при R 0,95. Енаминозашлщенную группу удаляют из родукта растворением 10 г в водном ацетоне (250 мл воды, 250 мл ацетона) и сильным перемеиванием этого раствора при рН 2,5 в течение 1 ч. Ацетон удаляют в вакууме и сложньш эфир высалиают из водной фазы в виде липкой желтой смолы, растворяют в этилацетате (200 мл) и дважды промывают порциями по 200 мл 1 н. бикарбоната натрия и высушивают над сульфатом магния. Осторожое добавление сухого эфира (около 50 мл) к ухому этилацетатному слою дает хлористоводородный ампициллин-фталидный сложный эфир в
виде TOHKorcf белого аморфного твердого вещества с выходом 80%.
ЯМР-спектр подтверждает структуру полученного продукта. Чистота до шдроксиламиновому испытанию 110,3%. Одаокапельное пятно на биохроматограммё при Rf p,8S.
Вычислено, %: С 55,65; Н4.67; N8.11; S 6,19.
Cj4H,4NjOeSCI Найдено, %: С 54,60; Н 4.70; N 7,92; S 6,40.
Смесь ацетона (250мл), натрий (D) - (N
- метокснкарбоншшропан - 2 - ил - а - аминофенилацетата (30,51 г), этилхлорформината (10,9мл) и
N - метилморфолина (4-6 капель) пере1ыешивают
10-15 мин при температуре от -20 до -ЗСгС
К этому раствору добавляют сразу всю 6 -аминопенициллановую кислоту (25,4 г), растворенную в воде (50 мл) с помощью триэтнламина, затем разбавляют ацетоном (150мл) и охлаждают до -20 С.
Реакционную смесь перемешивают 45 мин без
дальнейшего охлаждения и сразу добавляют раствор 3 - бромфталида (25 г) в ацетоне (100мл), после чего перемешивание продолжают еще 5 ч, в течение которых температура поднялась до комнатной (23°С).
После первого осветления фильтрованием ацетон удаляют в вакууме и к остатку добавляют зтилацетат (375 мл) и 2%-ный раствор бикарбоиата натрия (200 мл). После короткого перемешивания,
во время которого фазы разделились, органический слой промывают опять 2%-ным раствором бикарбоната натрия (200 мл).
К полученному этилацетатному раствору добавляют воду (375 мл) и 2 н. HCI (60 мл) и эту смесь
перемешивают при комнатной температуре (23° С) 45 мин. Затем добавляют бензол (600 мл) и пооте короткого перемешивания фазы разделяют. Органический слой отбрасывают, а водный слой фильт. руют с небольшим количеством обесцвечивающего
. древесного угля. Затем к фильтрату добавляют хлористый натрий в количестве, достаточном для насыщения, и после нескольких минут Перемешивания осевшее масло экстрагируют хлористым метиленом (1x400 мл, 1 100мл). Эти экстракты
объединяют, высушивают над сульфатом магния, отфильтровывают и испаряют под уменьшенным давле1шем до объема 100 мл. Затем быстро добавляют эфир (500 мл) при перемешивании и получившийся осадок перемешивают 30 мин при окружающей температуре. Продукт отфильтровьшают с отсасьшанием, промывают эфиром (2 к 50 мл) и сушат 3 ч в шкафу с принудительной циркуляцией воздуха при 35-40° С. Продукт - идентичный с заведомым образцом фталида 6 - (D) - в -аминофекилацетамидо)- пеницилланата.
П р и м е р 3. 6 - Аминспенициллансяая кислота (18,5 г. 0,085 моль) и бикарбонат натрия (21 г, 0,25 моль) растворяют в 250 мл воды и 100 мл ацетона. К этому раствору, охлажденному льдом, добавляют 16.6 г (0,085 моль) а - азндофенил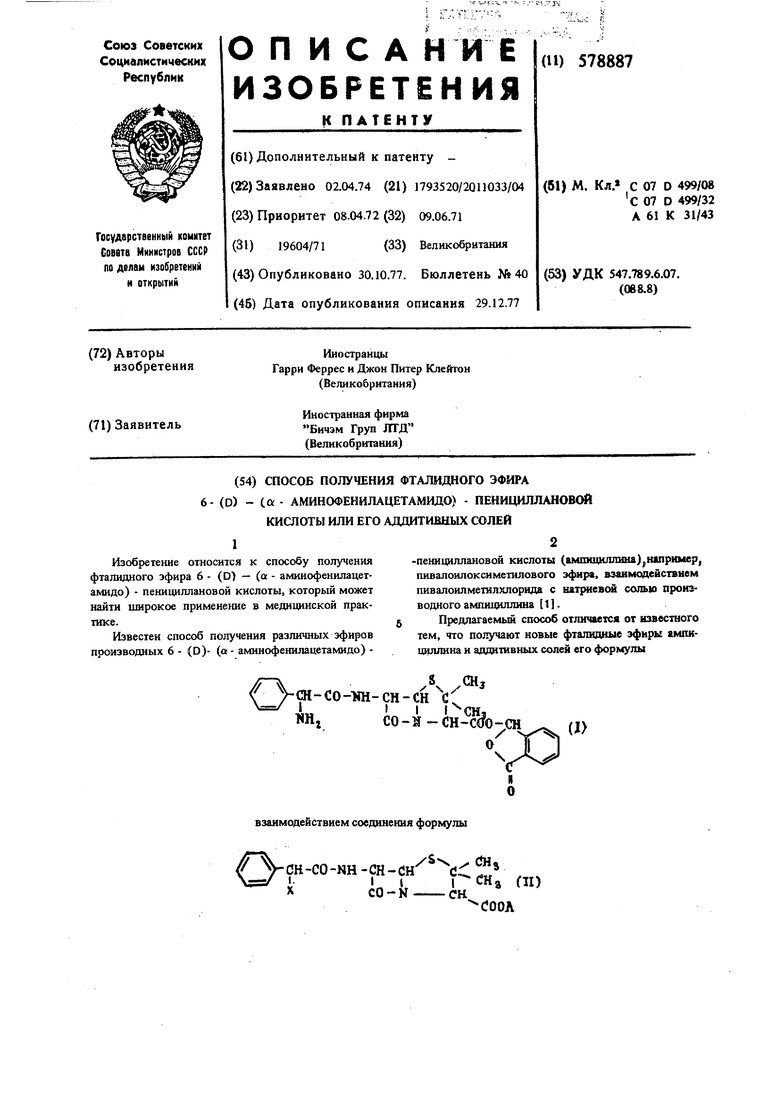


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения фталидного эфира 6-/д/- -аминофенилацетамидо/ пенициллановой кислоты | 1972 |
|
SU507244A3 |
| Способ получения сложных эфиров @ -аминопенициллинов или их кислотно-аддитивных солей | 1973 |
|
SU1153830A3 |
| Способ получения производных @ -фталидил-5-фторурацила | 1982 |
|
SU1258325A3 |
| Способ получения 6-метокси- карбоксипенициллинов или их солей | 1976 |
|
SU656524A3 |
| Способ получения 1,1-диоксидов пенициллановой кислоты или ее эфиров или ее солей | 1978 |
|
SU860706A1 |
| Способ получения производных бензодипиранов или их солей | 1973 |
|
SU553934A3 |
| Способ получения производных простанкарбоновой кислоты | 1971 |
|
SU439962A1 |
| Способ получения сложных эфиров -аминопенициллина | 1971 |
|
SU553936A3 |
| Способ получения 3-(4-аминоэтоксибензоил) бензо( @ )тиофенов или их солей | 1982 |
|
SU1155157A3 |
| Способ получения 7-замещенных 3-винилцефалоспоринов или их аддитивных солей с кислотами | 1983 |
|
SU1309911A3 |
