1
Изобретение касается получения (со)полимеров винилхлорида в микросуспензии, применяемых для производства листов, пленок, нитей, полых изделий, пеноматериалов, путем каландрирования, экструзии, литья под давлением, прессования и т. д.
Известен способ получения (со)полимеров винилхлорида путем (со) полимеризации винилхлорида в микросуспензии в присутствии радиального инициатора I.
Недостатком этого способа является то, что процесс (со)полимеризации имеет автоускоряемый характер, что влечет за собой, с одной стороны, затруднения, связанные с охлаждением, и вследствие этого плохое использование реактора, и, с другой стороны, неполное использование инициаторов, так как для обеспечения скорости, приемлемой в промышленном масштабе, необходимо, большое количество инициаторов. Кроме того, образуется много корок.
Ближайшим по технической сущности к изобретению является известный способ получения (со)полимеров винилхлорида путем (со)полимеризации винилхлорида в микросуспензии в присутствии содержащей радикальный инициатор дисперсии (со)полимера винилхлорида с частицами диаметром 0,05-2 мк 2 .
При осуществлении этого способа снижается коркообразование, частично замедляется автоускорение реакции, особенно при высоком содержании затравки, т. е. производительность процесса повышается. Однако этого недостаточно для полного использования способа в промышленном масштабе.
Цель изобретения - повыше;-;ие производительности процесса (со) полимеризации винилхлорида в микросуспензии. Эта цель достигается тем, что (со) полимеризацию проводят в присутствии водорастворимой соли уксусной или неорганической кислоты и металла, выбранного из группы, состоящей из железа, меди, кобальта, никеля, цинка, олова, титана, ванадия, марганца, хрома и серебра, и вводимого со скоростью 5-200 г/час комплексообразователя или в присутствии вводимого со скоростью 5-200 г/час предварительно полученного комплекса указанной водорастворимой соли и комплексообразователя.
Способ по изобретению позволяет избежать явления автоускорения, а также затруднений, которые связаны с этим явлением, дает возможность осуществлять полимеризацию или сополимеризацию винилхлорида в микросуспензии при обычных температурах со скоростью реакции, наиболее максимальной, которая возможна для данной аппаратуры, с лучшим использованием инициатора,следовательно, при использовании меньших количеств его, причем образование корок незначительное, а потому использование реактора оптимальное.
В качестве водорастворимых солей применяют сульфаты, хлориды, нитраты и а11етать в таком количестве, что молярное отношение соли металла к инициатору составляет от 0,1 до 10, предпочтительно от 0,1 до 2.
Эти сол:; можно вводить в реакционную среду до полимеризации или во время ее. 13 качестве комплексообразователей применяют соединения, способные переводить металл из его водорастворимой формы в форму, растворимую в винилхлориде, которая не оказывает ингибируюшего воздействия на полимеризацию и на активацию инициатора металлов. К таким соединениям относятся монокарбоновые кислоты, малорастворимые в воде, такие, как перфтормасляная, ос бромлауриновая, сульфосалициловая, гексагидробензойная, октановая, мвогоосновные (поликарбоновые) кислоты такие, как янтарная, винная, малеиновая, диоксималеиновая, и их соответгтвуюшие ангидриды; алкилфосфорные кислоты такие, как ди 2-этил -гексилфосфорная; лактоны такие, как аскорбиновая кислота и ее эфиры, JJ-бутиролактон; кетоны, содержащие в положениях ос-или/З-групы, активирующие карбонильную функцию, такие, как ацетилацетон, диокси-1,3-ацетон, бензоил, карбазоны, например дифенилтиокарбазон.
Количество применяемого комплексообразователя зависит от температуры полимеризации, охлаждающей способности реактора и чистоты реагентов. Оно может достигать молярных стехиометрических количеств по отношению к соли металла.
Этот комплексообразователь в присутствии соли металла дает растворимый в органической среде комплекс, который переводит металл в органическую фазу, где он оказывает активирующее действие на инициатор. Таким образом можно по желанию вызывать активирование инициатора, изменяя количество и момент введения комплексообразователя в реакционную среду, регулируя кинетику полимеризации, которая сильно активируется сначала, а затем замедляется. Благодаря оптимальному использованию охлаждающей способности реактора можно осуществлять полимеризацию за минимальное время. Однако этой цели не достигают, если сначала вводят весь комплексообразователь. В этом случае активация инициатора слишком быстрая с самого начала процесса полимеризации, инициатор быстро разлагается, и реакция не может закончиться из-за его недостатка.
Согласно одному из вариантов способа изобретения, инициатор активируют постепенным введение.м растворимого в органической среде металлического комплекса, предварительно полученного путем взаимодействия металлических солей и комплексообразователей.
Эти комплексы используют в таких пропорциях, что молярное отнощение металлический комплекс: инициатор составляет от 0,1 до 10.
Активацию инициатора ко.мплексом можно прервать в любой момент путем прекращения введения комплексообразователя или комплексов и/или снова переводят ион металла из его растворимой.в органической среде формы в его водорастворимую форму. Это легко достигается добавлением в реакционную среду агента образования внутрикомплексного соединения, выбираемого . среди щелочных солей кислот таких, как этилендиаминтетрауксусная кислота, нитрилтриуксусные кислоты, диэтилентриаминпентауксусная кислота, М-(2-оксиэтил)-этиленди.аминтриуксусная кислота. Агент образования внутрикомплексного соединения применяют в количествах, достигающих молярного стехиометрического по отношению к соли .металла. Затравку, необходимую для полимеризации, получают классическим способом полимеризации в микросуспензии. Например, используют воду, винилхлорид с сомономером или без сомономера, анионный эмульгатор и растворимый в органической среде инициатор. Мономер или мономеры тонко диспергируют в воде с помощью мощного механического средства такого, как, например, коллоидная мельница, ускоряющий насос, вибрационная мещалка, ультразвуковая аппаратура. Полученную мик5 росуспензию затем нагревают под автогенным давлением и приумеренном перемешивании при определенной температуре, в зависимости от молекулярного веса продукта, который хотят получить.
Затравка существует в форме дисперсии 0 частиц полимера или сополимера диаметром от 0,05 до 2 мк.
Для осуществлении с;гособа по изобретению частицы затравки должны содержать весь инициатор, необходимый для полимеризации. Это количество составляет от 0,1 до 5 вес. % по отношению к полимеру затравки и вводится перед полимеризацией указанного продукта.
Инициаторами, растворимыми в мономерах, являются органические перекиси такие, как диацетилперекиси, например перекиси лаурила, дед каноила, капроила.
При полимеризации, согласно изобретению, количество используемой затравки полимера в ней составляет 0,5-10°/о от веса мономера (ов), которые нужно полимеризовать, плюс полимер затравки. Количество присутствующего инициатора, по отношению к количеству полимеризуемого мономера,следовательно, незначительное по сравнению с количеством его по известному способу. Можно также применять большее количество затравки, но это нецелесообразно. Затравки, имеющейся в среде полимеризации, достаточно для обеспечения диспергирования мономера, поэтому нет необходимости прибегать к дополнительно.му энергичному диспергированию среды.
К мономерам, которые могут быть использо5 ваны для сополимеризации с винилхлоридом, относятся сложные виниловые эфиры моно- и поликарбоновых кислот, такие как винилацетат, винилпропионат, винилбензоат; сложные алифатические, циклоалифатические, .аромати ческие эфиры, амиды, нитрилы ненасыщенны ° Моно- или поликарбоновых кислот таких, как
акриловой, метакриловой, малеиновой, фумаровой кислот; алилгалогенид, винилгалогенид, винил иденгалогенид; алкилвиниловые простые эфиры; олефины. Количество сомономера может достигать 25% от веса сополимера.
Чтобы улучшить стабильность микросуспензии, можно выгодно добавлять перед и/или в процессе полимеризации анионный эмульгатор в количестве до 2 вес./о по отношению к мономеру (ам). Этот эмульгатор может быть таким же, как и при получении затравки. Его выбирают среди классических продуктов: мыла жирных кислот, алкилсульфаты, алкилсульфонаты, алкйларилсульфонаты, алкилсульфосукцинаты, алкилфосфаты.
Количество воды, используемое в полимеризации, согласно изобретению, должно быть таким, чтобы весовое отношение мономера (мономеров) + затравочный полимер к воде включая воду затравочного полимера) составляло от 0,3 до 1,5.
Температура полимеризации, в зависимости от количества полимера, которое желают получить, обычно составляет 30-70° С.
Способ по изобретению особенно пригоден для применения при непрерывной полимеризации. В этом случае введение комплексообразователя или комплекса позволяет поддерживать оптимальную кинетику реакции, обеспечивая постоянную и высокую степень конверсии.
Пример 1. В реактор емкостью 25 м, снабженный мешалкой, вводят 12000 кг воды, 1200 кг затравки, полученной путем полимеризации в микросуспензии, имеюшей концентрацию 33,3%, или 400 кг поливинилхлорида, содержашего 6 кг перекиси лаурила, 60 кг додецилбензолсульфанола натрия, 0,5 кг сульфата меди (11), 10000 кг винилхлорида. Средний диаметр частиц затравки 0,4 мк.
Реакционную массу нагревают до 52° С под автогенным давлением, поддерживая эту температуру в течение всей реакции. Как только температура среды достигнет 52° С, начинают вводить водный раствор аскорбиновой кислоты в количестве 4 г/л. Дебит по весу кислоты составляет 65 г/ч в течение 3 ч, затем 45 г/ч в течение 3 ч и, наконец, 20 г/ч до окончания реакции. Через 9 ч наблюдают понижение давления, характеризующее окончание реакции. Прекращают введение аскорбиновой кислоты и дегазируют непрореагировавший мономер.
Получают 22200 кг дисперсии полимера, концентрация которого составляет 42,2 вес.% (степень превращения 89,7 вес.% по отношению к используемому винилхлориду). Корка пленок полимера на стенках реактора составляет только 0,1 вес.°/о по отношению к взятому для реакции винилхлориду. Средний диаметр полученных частиц 1,1 мк.
После распыления и размалывания полученный полимер имеет вязкость 130 (измерена согласно французской норме Т 51-013).
Пластизоль, приготовленный путем смешивания 100 вес. ч. полученного поливинилхлорида и 60 вес. ч. диоктилфталата, имеет вязкость, измеренную в вискозиметре Брукфильда, 20 пз.
Для сравнения, с целью показать достоинства способа, осуществляют pa3vTH4Hbie опыты, аналогично описанному примеру 1.
А) Полимеризация в микросуспензии без затравки и без активации.
В тот же реактор, что и в примере 1, вводят (в кг): воды 12000; перекиси лаурила 8; додецилбензолсульфоната натрия 100; винилхлорида 10000.
Смесь гомогенизируют, чтобы получить микросуспензию, органическая фаза которой имеет средний гранулометрический состав 1 мк, затем нагревают до 52°С под автогенным давлением и поддерживают эту температуру в течение всей реакции. Скорость реакции не регулируется, очень медленная сначала она ускоряется, что вызывает необходимость в течение последних 2 ч максимально охлаждать реактор. Спустя 18 ч реакция заканчивается. Непрореагировавщий мономер дегазируют, и получают 20300 кг дисперсии с концентрацией полимера 41 вес.°/о. Это соответствует степени превращения 83 вес.% по отношению к используемому винилхлориду. Корка полимера на стенках реактора составляет 1 вес.% по отношению к используемому винилхлориду.
Таким образом, по сравнению со способом по изобретению, хотя количество взятого инициатора более высокое, время реакции в два раза выще, а степень конверсии незначительная и количество корок полимера на стенках реактора в 10 раз больше.
Б) Полимеризация в микросуспензии с затравкой, без активации.
Повторяют пример 1, но без введения сульфата меди и аскорбиновой кислоты. После 15 ч реакции получают 22000 кг дисперсии с концентрацией полимера 40,5 вес.% (или степень конверсии 85,1 вес.% по отношению к используемому винилхлориду).
Количество корок полимера на стенках реактора составляет 0,6% от используемого винилхлорида. Средний диаметр полученных частиц 1,08 мк.
По сравнению с примером 1, обнаруживают, что активация инициатора, согласно изобретению, позволяет осуществлять реакцию за 9 ч вместо 15 ч, увеличивает степень превращения на 4%, при этом образуется в 6 раз меньще корок.
В) Полимеризация в микросуспензии, без затравки и без активации в присутствии быстро разлагающегося инициатора.
В реактор емкостью 120 л, снабженный мешалкой, загружают (в кг): воды 60, перекиси лаурила 0,160, изопропилперкарбоната 0,120, додецилбензолсульфоната натрия 0,4, винилхлорида 40.
Смесь гомогенизируют, чтобы получить микросуспензию, органическая фаза которой имеет средний гранулометрический состав 1 мк, затем нагревают до 52° С под автогенным давлением и поддерживают эту температуру в течение всей реакции.
После 12 ч реакции получают грубую суспензию, содержащую большое количество коагулянта. На стенках реактора образуются 30% корок от используемого мономера.
Пример 2. Повторяют пример 1 с затравкой, частицы которой имеют средний диаметр 0,25 мк, и полимеризуют при 42° С. После 12 ч получают 22 100 кг дисперсии полимера с концеитрацией 42,4 вес.%. Степень превращения 89,7 вес.% по отношению к взятому винилхлориду. Средний диаметр полученных частиц 0,6 мк. Полимер имеет вязкость 180.
Пластизоль, приготовленный путем смешения 100 вес. ч. полученного поливинилхлорида и 60 вес. ч. диоктилфталата имеет вязкость 75 пз.
Пример 3. Повторяют пример 1, но вместо 10 000 кг вииилхлорида берут 9300 кг винилхлорида и 700 кг вииилацетата. После 9 ч 30 мин реакции получают 22400 кг содержащей 4,9% винилацетата дисперсии сополимера, концентрация которой 42,6 вес.%. Степень превращения 91,4 вес.% по отношению к исходным маномерам.
Средний диаметр полученных частиц 1,1 мк. Сополимер имеет вязкость 130. Пластизоль, приготовленный путем смешения 100 вес. ч. полученного сополимера и 60 вес. ч. диоктилфталата, имеет вязкость 30 пз.
Пример 4. В реактор, описанный в примере J. вводят (в кг): воды 12600, затравки с концентрацией полимера 39%, или 417,3 кг поливинилхлорида, содержашего 6.3 кг перекиси лаурила, 1070, додецилбензолсульфоната натрия 84, винилхлорида 8400 и сульфата меди 0,42. Средний диаметр частиц затравки 0,4 мк.
Реакционную массу нагревают до 52°С и постепенно вводят 840 г диоксималеиновой кислоты в течение 12 ч, или 105 г/ч в течение 4 ч, затем 65 г/ч в течение 4 ч и, наконец, 40 г/ч в течение 4 ч.
Получают 20 970 кг дисперсии с концентрацией полимера 36,9%. Степень превращения 97,1 вес.% по отношению к исходному мономеру. Со стенок реактора снимают тонкий слой полимера в количестве 0,12 вес.% от исходного винилхлорида. Средний диаметр полученных частиц 1,1 мк.
Пример 5. Повторяют пример 4, заменяя диоксималеиновую кислоту янтарной. После 14 ч
реакции получают 21 070 кг дисперсии с концентрацией полимера 37%. Степень преврашения 87,8%. Вес корок полимера со стенок реактора около 0,15% от веса используемого винилхлорида. Средний диаметр частиц 1,08 мк. Пример 6. Повторяют пример 4, заменяя
сульфат меди и диоксималеиновую кислоту 840 г ацетилацетоната ванадия. Последний постепенно вводят в среду , нагретую до 52° С, в течение 12 ч 30 мин, по 105 г/ч в течение 4 ч, затем 65 г/ч 4 ч и, наконец, 40 г/ч в течение 4 ч
30 мин.
Получают 20870 кг дисперсии с концентрацией полимера 36,5%, степень конверсии 85,5%. Количество корок полимера на стенках реактора 0,18% от веса используемого мономера. Средний диаметр частиц 1,09 мк.
Пример 7. Осуществляют несколько опытов .с различными агента.ми активации в следующих условиях.
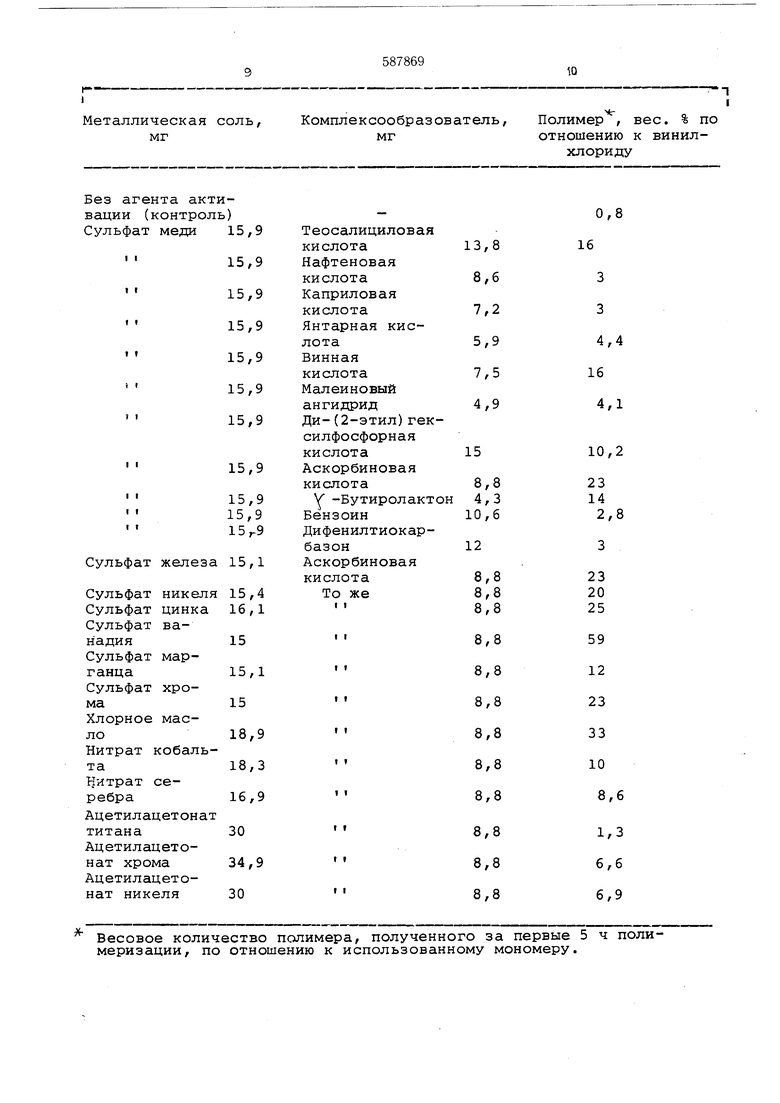
В стеклянную колбу емкостью 200 мл после создания в ней вакуума и заполнения газообразным винилхлоридом вводят 60 мл деионизировйнной воды, 10,5 г затравки, полученной путем полимеризации в микросуспензии, с концентрацией полимера 34,9%, или 3 г поливинилхлорида, содержащего 0,04 г перекиси лаурила, 0,18 г додецилбензолсульфоната натрия, 30 г винилхлорида и агент активации, образованный либо 10x10 моль соли металла и 5х10 моль комлексообразователя, либо 10x10 моль предварительно приготовленного металлического комплекса (см. таблицу). Средний диаметр частиц затравки 0,4 мк.
Колбу герметически закрывают, помещают
в термостатируемую при 52° С баню и перемешивают 5 ч, после чего охлаждают, дегазируют и воду выпаривают. Получают полимер. Весовое количество полимера, полученного за первые 5 ч полимеризации, по отношению к использованному мономеру.
Пример 8. В реактор объемом 25 м, снабженный мешалкой, вводят (в кг): воды 12000, затравочного вещества, полученного полимеризацией в микросуспензии с концентрацией полимера 33,3%, или 400 кг сополимера, образованного 95% винилхлорида и 5% винилацетата, содержащего 6 кг перекиси лаурила, 1200, додецилбензолсульфоната натрия 60, сульфата меди 0,5, винилхлорида 9300, винилацетата 700. Средний диаметр частиц затравки 0,4 мк.
Реакционную массу нагревают до 52°С при повысившемся давлении и эту температуру поддерживают в течение всей реакции. По достижении температуры 52° С начинают вводить водный раствор аскорбиновой кислоты из расчета 4 г/л. Весовой расход 65 г/ч в течение 3 ч, затем 45 г/ч в течение 3 ч и, наконец, 20 г/ч до конца реакции.
. После 9 ч 15 мин снимается давление, что характерно для конца реакции. Прекращают введение аскорбиновой кислоты и дегазируют непрореагировавший мономер.
Получают 22300 кг дисперсии полимера, концентрация которого 41,9 вес.%, степень превращения 89,4 вес.% по отношению к мономерам. Содержание винилацетата в сополимере 4,9 вес.%. Корка полимера на стенках реактора составляет только 0,1 вес.% по отношению к винилхлориду. Средний диаметр полученных частиц 1,1 мк.
Пример 9. В реактор объемом 25 м, снабженный мешалкой вводят (в кг): воды 12000; затравочного вещества, приготовленного полимеризацией в микросуспензии, имеющего концентрацию 33,3%, 1200, или 400 кг поливинилхлорида, содержащего 4 кг перекиси лаурила; додецилбензолсульфоната натрия 120; сульфата меди 0,5; винилхлорида 10000. Средний диаметр частиц затравки 0,06 мк.
Реакционную массу нагревают до 52° С при повысившемся давлении и поддерживают эту температуру в течение всей реакции. Как только температура среды достигнет 52° С, начинают вводить водный раствор аскорбиновой кислоты из расчета 4 г/л. Весовой расход кислоты 40 г/ч в течение 3 ч, 30 г/ч в течение 3 ч и, наконец, 15 г/ч до окончания реакции.
После 9 ч реакции давление снижается, что характерно для конца реакции. Введение аскорбиновой кислоты прекращают и непрореагировавший мономер дегазируют.
Получают 22000 кг дисперсии полимера, концентрация которого 41 Бес.%, или фактическая степень превращения 86 вес.% по отношению к винилхлориду. Средний диаметр полученных частиц 0,17 мк.
Пример 10. В реактор объемом 25 м , снабженный меша/1кой, вводят (в кг): воды 10800, затравочного вещества, приготовленного полимеризацией в микросуспензии, имеющего концентрацию полимера 33,3%, 2400, или 800 кг поливинилхлорида, содержащего 3 кг перекиси лаурила, додецилбензолсульфоната натрия 100, сульфата меди 0,5, винилхлорида 9000. Средний диаметр частиц затравки 1,8 мк.
Реакционную массу нагревают до 52° С при повысившемся давлении. Эту температуру поддерживают в течение всей реакции. Как только
температура среды достигнет 52° С, начинают вводить водный раствор аскорбиновой кислоты из расчета 4 г/л. Весовой расход кислоты составляет 65 F/4 в течение 3 ч, затем 45 г/ч 3 ч 5 и. наконец 20 г/ч до окончания реакции.
После 10 ч реакции давление снижается, что характерно для конца реакции. Введение аскорбиновой кислоты прекращают и непрореагировавщий мономер дегазируют.
Получают 21000 кг дисперсии полимера,
концентрация которого 40 вес.%, степень превращения 84,5 вес./о по отношению к винилхлориду. Корка полимера на стенках реактора составляет только 0,2 вес.°/о по отношению к винилхлориду. Средний диаметр полученных частиц 3,9 мк.
Пример 7/. В реактор объемом 25 м, снабженный мешалкой, вводят (в кг): воды 13000; затравочного вещества, приготовленного полимеризацией в микросуспензии, имеющего концентрацию 33,3%, 150, или 150 кг поли2 винилхлорида, содержащего 2,5 кг перекиси лаурила; додецилбензолсульфоната натрия 60; сульфата меди 0,5; винилхлорида 10000. Средний диаметр частиц затравки 0,12 мк.
Реакционную массу нагревают до 52° С при повышающемся давлении. Эту температуру поддерживают в течение всей реакции. Как только температура среды достигнет 52° С, начинают вводить водный раствор аскорбиновой кислоты из расчета 4 г/л. Весовой расход кислоты 65 г/ч в течение 3 ч, затем 45 г/ч в течение 3 ч 0 и, наконец, 20 г/ч до окончания реакции.
После 12 ч давление снижается, что характерно для конца реакции. Введение аскорбиновой кислоты прекращают и непрореагировавший мономер дегазируют.
5Получают 22000 кг дисперсии полимера,
концентрация которого 39,7 вес.%; степень превращения 86,7 вес. % по отношению к винилхлориду.
Тонкая пленка поли.мера на стенках реактора составляет только 0,1 вес.% по отношению к винилхлориду.
Средний диаметр полученных частиц 0,65 мк.
Таким образом, при осуществлении способа по изобретению значительно повыщается производительность процесса (уменьщается время реакции, увеличивается степень конверсии). Кроме того, коркообразование снижается.
Формула изобретения
Способ получения (со) полимеров винилхлорида путем (со) полимеризации винилхлорида в микросуспензии в присутствии содержащей радикальный инициатор дисперсии (со) полимера
винилхлорида с частицами диаметром0,05- 2 мк, отличающийся тем, что, с целью повыщения производительности процесса, (со)полимеризацию проводят в присутствии водорастворимой соли уксусной или неорганической кислоты и металла, выбранного из группы, состоящей из
железа, меди, кобальта, никеля, цинка, олова. 13 титана, ванадия, марганца, хрома и .серебра, и вводимого со скоростью 5-200 г/ч комплексообразователя или в присутствии вводимого со скоростью 5-200 г/ч предварительно полученного комплекса указанной водорастворимой со-5 ли и комплексообразователя. )Ч Источники информации, принятые во внимание при экспертизе: 1. Патент Франции № 1485547, кл. С 08 f, 1967. 2. Патент Франции № 91709, дополнительный к патенту 1485547, кл. С 08 f 1968.





| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения (со)полимеров винилхлорида | 1976 |
|
SU656531A3 |
| Способ получения поливинилхлорида | 1970 |
|
SU511017A3 |
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННОГО ПОЛИВИНИЛХЛОРИДА | 1972 |
|
SU357736A1 |
| Способ получения винилгалоид-НыХ пОлиМЕРОВ | 1977 |
|
SU799672A3 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРОВ | 1971 |
|
SU309528A1 |
| Способ получения винилхлоридныхпОлиМЕРОВ | 1978 |
|
SU841592A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРИВИТОГО СОПОЛИМЕРА | 1991 |
|
RU2021292C1 |
| Композиция для полимеризации полимера на основе винилхлорида и способ получения полимера на основе винилхлорида с использованием этой композиции | 2020 |
|
RU2819900C1 |
| Способ получения полимеров и сополимеров ненасыщенных соединений | 1971 |
|
SU452101A3 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНЫХ ДИСПЕРСИЙ ПОЛИМЕРОВ, ЭМУЛЬСИОННЫЕ ПОЛИМЕРЫ И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2435788C2 |
