(54) СПОСОБ ПОЛУЧЕНИЯ КАРБОСТИРИЛИЛИ 3,4-ДИГИДРОКАРБОСТИРИЛПРОИЗВОДНЫХ
ния, после чего перемешивают при в течение 20 ч. К реакционной смеси прибавляют 500 мл 10%-ного водного раствора соляной кислоты и удаляют нитробензол перегонкой с водяным паром. После охлаждения смеси высадившиеся кристаллы собирают фильтрованием, которые затем промывают 30 мл горячей вода. Перекристаллизацией из метанола получают 14,0 г 5-хлорацетил-3-оксикарбостирила с т.пл. 285-287°С (с разложением) в виде бледно-желтых кристаллов.
Пример 2. 20 г хрористого алюминия прибавляют к 5,0 г 8-оксикарбостирила и полученную смесь тщательно перемешивают. К смеси при охлаждении льдомпостепенно прибавляют 10 г хлорангидрида хлоруксусной кислоты. Смесь нагревают в течение 2 ч при 40-45°С для получения 8-хлорацетоксикарбостирила, после чего перемешивают в течение 3 ч при . После охлаждения высадившиеся кристаллы собирают фильтрованием, кристаллы промывают 300 мл воды, затем перекристаллизацией из метанола получают 2,6 г 5-хлорацетил-8-оксикарбосТирила в виде бледно-желтых кристаллов с т.пл. 285-287°С (с разложением) .
При этом часть образующегося 8-хлорацетоксикарбостирила отделяют и перекристаллизовывают из ацетона для получения бледно-желтых кристаллов. Температура плавления этих кристаллов 248 250°С (с разложением).
Пример 3. 1,5 г хлорангидрида хлоруксусной кислоты и 20 мл сероуглерода прибавляют к 0,5 г 8-оксикарбостирила и к полученному раствору при охлаждении льдом постепенно прибавляют 2 г хлористого алюминия. После тщательного перемешивания смесь постепенно нагревают и кипятят в течение 30 мин. После охлаждения непрореагировавший ;хлорангидрид хлоруксусной кислоты и сероуглерод удаляют и к полученному остатку для кристаллизации продукта прибавляют размельченный лед. Полученные кристаллы прогрызают водой, а затем перекристаллизовывают из ацетона и получают 0,45 г 8-хлорацетоксикарбостирила в виде бледно-желтых кристаллов с т.пл. 2 4 8-251° С (с разложением). Прймер4о20г хлористого алюминия и 10 г хлорангидрида хлоруксусной кислоты прибавляют к 10 г 8-хлорацетоксикарбостирила (см. пример 3) и полученную смесь нагревают при 75-85°С в течение 1 ч. Смесь в теплом виде выливают на размельченный лед и высадившиеся кристаллы фильтруют, промывают водой и перекристаллизовывают из метанола для получения 3,7 г 5-хлорацетил-8-рксикарбостирила в виде бледно-желтых кристсшлов с т.пл. 285-287°С (с разложением) .
Пример 5..7,3 г 8-оксикарбостирила и 12,5 г хлорангидрида хлоруксусной кислоты прибавляют к 70 мл нитробензола и к полученному раствору при перемешивании и охлаждении льдом постепенно добавляют 30г хлористого алюминия. Смесь перемешивают при 50-55 С в течение 6 ч, после чего сливают в ледяную воду. Высадившиеся кристаллы отфильтровывают, промывают метанолом, перекристаллизовывают из ацетона и получают 3,5 г 5-хлорацетил-8-хлорацетоксикарбостирилав виде бледно-желтых кристаллов с т.пл. 239-241С (с разложением) .
Примере. 1,7 г 5-хлор5 ацетил-8-хлорацетоксикарбостирила, полученного в примере 5, прибавляют к 50 мл 10%-ного водного раствора |СОляной кислоты, после чего перемешивают при 95 - 100с в течение 2 ч. После охлаждения высадившиеся кристаллы отфильтровывают, промывают водой, перекристаллизовывают из метаноЛа и получают 1,1 г 5-хлорацетил-8-оксикарбостирила в виде бледножелтых кристаллов с т.пл. 285-28б°С (с разложением).
Пример 7. 2,5 г 5-хлорацетил-8-хлорацетоксикарбостирила, полученного в примере 5, прибавляют к 30 мл 5%-ной водной гидроокиси калия.
после чего перемешивают при 20-25 С в течение 30 мин. Полученный раствор при охлаждении доводят до рН 2-3 разбавленной соляной кислотой. Высадившиеся кристаллы отфильтровывают, промывают водой, перекристаллизовывают из метанола и получают 1,7 г 5-хлорацетил-8-оксикарбостирила в виде бледно-желтых кристаллов с т.пл. 285-287°С (с разложением).
Пример8. К смеси 4,5 г 8-оксикарбостирила и 10 г хлорангидрида хлоруксусной кислоты постепенно добавляют при перемешивании и охлаждении льдом 220 г хлористого алюминия . Полученную смесь перемешивают при 55-60 С в течение 8 ч для образования 5-хлорацетил-8-хлорадетоксикарбостирила. Затем к реакционной смеси прибавляют 40 мл 5%-ной гидроокиси калия, после чего перемешивают при комнатной температуре в течение 30 мин. Кроме того, прибавляют 10%-ный водный раствор соляной кислоты для подкисления раствора и образования кристаллов. Высадившиеся кристалла отфильтровывают, промывают водой, затем перекристаллизовывают из метанола и получают 2,3 г 5-хлорацетил-8-оксикарбостирила в виде бледно-желтых кристаллов с т.пл. 285-286 С (с разложением).
Часть указанной реакционной смеси до прибавления 5%-ной гидроокиси калия отбирают и сливают в ледяную воду. Высадившиеся кристгшлы отфильтровывают, промывают метанолом, перекристаллизовывают из этилацетата и получают 5-хлорацетил-З-хлоацетоксикарбостирила в виде бледно-желтых кристаллов с т.пл. 238-241°С (с разложением) .
ПримерЭ. 12,б г 5-хлорацети-8-оксикарбостирила (см.пример 4 или 8), суспендируют в 130 мл изопропанола и к полученной суспензии при перемешивании по каплям прибавляют 2515 г изопропиламина. Затем реакционную смесь перемешивают при 55бО С в течение 3 ч. После охлаждения к смеси прибавляют концентрированную соляную кцслоту,чтобы довести смесь до рН 2-3, и высадившиеся кристаллы отфильтровывают, промывают ацетоном, перекристаллизовывают из смеси метанол-диметилформамид (1:1 по объему) и получают 6,5 г 5-изопропиламиноацетил-8-оксикарбостирилхлоргидрата в виде бледно-желтых кристаллов с т.пл. 28ь-288С (с разложением).
Пример 10. 8,О г 5-хлорацетил-8-оксикарбостирила (см. пример 4 или 8)- суспендируют в 100 мл этанола и при перемещивакии к полученной суспензии по каплям прибавляют 10 г трет-бутиламина, после чего перемешивают при 55-бО°С в течение 5 ч. После этого смесь концентрируют до половины объема и прибавляют затем концентрированную соляную кислоту, чтобы довести смесь до рН 2-3. Высадившиесн кристаллы отфильтровывают, промывают ацетоном, перекристализовывают из этанола и получают 4,1 г 5-трет-бутиламиноацетил-8 -оксикарбостирила в виде бледно-жел тых кристаллов с т.пл. 291-293 0 (с разложением).
Пример 11. 4,3 г-5-хлорацети-8-оксикарбостирила (см.пример 4 или 8) суспендируют в 50 мл этанола и к полученной суспензии при перемешивании по каплям прибавляют 5 г фторбутламина, после чего перемешивают при бО-бБ С в течение 5 ч. После охлаждения полученную смесь доводят до рН 3 концентрированной соляной кислотой. Высадившиеся кристаллы отфильтров|э1вают,промывают ацетоном,перекристаллиэовывают из смеси метанол-этанол (1:1 по объему) и получают 2,9 г 5-втор-бутиламиноацетил-8-оксикарбостирилхлоргидрата в виде бледно-желтых кристаллов с т.пл, 289-291°С (с разложением).
Пример 12. Юг 5-хлорацети-8-оксикарбостирила (см.пример 4 или 8) суспендируют в 30 мл бензола и к суспензии прибавляют 10 мл пиперидина, после чего смесь нагревают при перемешивании и температуре кипения с обратным холодильником в течение 6ч. Реакционную смесь отфильтровывают для вьоделения продукта реакции, который затем промывают бензолом и после этого 50 мл изопропанола. Полученное нерастворимое вещество .растворяют в 150 МП водной соляной кислоты. Раствор концентрируют досуха при пониженном давлении и полученный остаток перекристаллизовывают из этанола для получения 7,5 г белого аморфного полугидрата 5-пиперидино:ацетил-8-оксикарбостирилхлоргидратас т.пл. 239-242°С (с разложением).
Пример 13. Юг 5-хлорацетил.-8-оксикарбостирила (см.пример 4 или 8) суспендируют в. 60 мл бензола и к суспензии прибавляют 9 мл морфолина, после чего смесь нагревают при перемешивании и при температуре кипения с обратным холодильником в течение 4 ч. Реакционную смесь охлаждают и (Полученный осадок отфильтровывают. Осадок растворяют в 60 мл изопропанола и.раствор доводят до рН 2-3 с
концентрированной соляной кислотой. Полученный кислый раствор охлаждают
0 льдом и образующийся осадок отфильтровывают, после чего перекристаллизовывают из этанола. Осадок растворяют в 20 мл воды и раствор доводят до рН 7,5-8,0 бикарбонатом натрия
5 при охлаждении льдом. Осадок, образукадийся при охлаждении, отфильтровывают и перекристаллизовывают из этанола для получения 2,4 г белого аморфного 5-морфолиноацетил-8-окси0карбостирила с т.пл. 238-2 39, 5С (с разложением) , Полученный продукт идентифицирован ИК и ЯМР спектральными и элементными анализами.
Пример14, 120 мл бензиламина прибавляют к 15 г 5-хлорацетил 3-ок5сикарбостирила (см.пример 4 или 8) и смесь перемешивают при комнатной температуре в течение 1 ч. Осадок, который образуется при добавлении петролейного эфира к реакционной
0 смеси, растворяют добавлением разбавленной соляной кислоты и раствор отфильтровывают для удаления любого нерастворимого вещества. Солянокислотный слой затем концентрируют
5 досуха, а остаток перекристаллизовывают из метанола и получают 16,4 г вещества с т.пл. 274-279 С (с окра шиванием и разложеьшем), Полученный продукт идентифицирован ЯМР и ИК
0 спектральными и элементным анализами как моногидрат 5-бензиламиноацетил-8-оксикарбостирилхлоргидрата.
Пример15. Юг Ь-хлорацетил-8-оксикарбостирила растворяют в 50 мл этанола и к раствору по каплям
5 прибавляют 25 г 1-фенетиламина, после чего перемешивают при в течение 4 ч. Этанол затем отгоняют, а осадок промывают диэтиловым эфиром и растворяют в изопропаноле для удаления любо0го нерастворимого вещества.Изопропанольный слой концентрируют приблизительно до 1/3 от первоначального объема и доводят до рН 4 с концентрированной соляной кислотой. Образующий5ся осадок перекристаллизовывают из
смеси этанола и ацетона и получают 9,8 г вещества с т.пл. 246-249С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-(1-фенетиламиноацетил) -8-оксикарбостирилхлоргидрат,
Пример 16. 40 мл 1,1-диметилфенетиламина прибавляют к 5 г 5-хлорацвтил-8-оксикарбостирила и смесь перемешивают при комнатной температуре в темноте в течение 5 ч. Осадок, который образуется при добавлении петролеПного эфира к реакционной смеси, промывают диэтиловым эфиром и растворяют в метанольном растворе соляной кислоты для удаления любого нерастворимого вещества. Метанол затем отгоняют, а остаток перекристаллизовывают из метанола и получают 6,1 г вещества с т.пл. 246247 С (с окрашиванием и разложением) Полученный продукт идентифицирован ЯМР и Ж спектральными анализами как полугидрат 5-{1,1-диметилфенетиламиноацетил)-8-оксикарбостирилхлоргидрата.
П р и м е р 17. 24,3 г 8-окси-3,4-дигидрокарбостирила и ь8 г хлорангидрида хлоруксусной кислоты прибавляют к 130 мл сероуглерода и к смеси при охлаждении в ледяной воде и при перемешивании медленно добавляют 200 г хлористого алюминия. Смес перемешивают в течение 6 ч при 6070 С и отгоняют сероуглерод. Получающийся остаток сливают в ЬОО мл ледяной воды. Высадившиеся кристаллы отфильтровывают, промывают водой и перекристаллизовывают дважды ,из метанола для получения 8,0 г 5-хлорацетил-8-окси-З,4-дигидрокарбостирила в виде бледно-желтых кристаллов с т.пл. 189-191°С.
П р и м е р 18. 13 г 8-окси-3,4.-дигидрокарбостирила и 20 г хлорангидрида хлоруксусной кислоты растворяют- в 100 мл нит робензола и к смеси медленно добавляют 40 г хлористого алюминияя, после чего перемешивают в течение 15 ч при ТО-ТЗ С. Смесь затем подвергают перегонке с водяным паром для удаления нитробензола. После охлаждения смеси высадившиеся кристаллы отфильтровывают, промывают горячей водой, перекристаллизовывают из метанола и получают 7,2 г 5-хлорацетил 8-ойси-3,4-дигидрокарбостирила в ,виде светло; желтых кристаллов с т.пи. 189-190С.
Пример 19. 66 г хлорангидрида хлоруксусной кислоты хИ 30 мл нитробензола прибавляют к 17 г 8-метокс-3,4-дигидрокарбостирила и к полученной смеси при охлаждении льдом медленно добавляют 100 г хлористого алюминия, после чего перемешивают в течение 30 мин при комнатной температуре. После этого смесь оставляют еще на 30 мин, а затем сливают в 700 мл ледяной воды. Полученный осдок отфильтровывают, промывают этанолом, перекристаллизовывают из метанола и получают 20 г белых иглоподобных кристаллов с т.пл. 187-189С. Полученные кристаллы идентифицированы ЯМР и ИК спектральными и элементными анализами как 5-хлорацетил-8-метокси 3,4-дигидрокарбостирил.
Пример20. 33 г хлорангидрида хлоруксусной кислоты и 80 мл сероуглерода прибавляют к 8,5 г 8-метокси-3,4-дигидрокарбостирила и к полученной смеси при охлаждении льдом медленно добавляют 50 г хлористого алюминия, после чего перемешивают в течение 2 ч при комнатной температуре. Сероуглеродный слой удаляют декантацией и к оставшемуся раствору добавляют размельченный лед для кристаллизации продукта. Высадившиеся кристаллы, полученные таким образом,
0 отфильтровывают, промывают этанолом, перекристаллизовывают из метанола и получают 11 г белых иглоподобных кристаллов с т.пл. 187-188°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-хлорацетил-8-метокси-3,4-дигидрокарбостирил.
Пример 21, 24,3 г 8-окси-3,4-дигидрокарбостирила и 68 г хлорангидрида хлоруксусной кислоты прибавляют к 130 мл сероуглерода и к смеси при перемешивании и охлаждении ледяной воды медленно добавляют 200 г хлористого алюминия. Затем после 4-часовой реакции при охлаждении
5 льдом получают 8-хлорацетокси-З,4-дигидрокарбостирил. Реакционную смесь перемешивают в течение 2 ч при 60-70°С и отгоняют сероуглерод. Получающийся остаток сливают в 500 мл
0 ледяной воды и высадившиеся кристаллы отфильтровывают, промывают водой, перекристаллизовывают дважды из метанола и получают 8-,О г 5-хлорацетил-8-окси-З,4-дигидрокарбостирила в
с виде светло-желтых кристаллов с т.пл.
189-191°С. I
8-хлорацетокси-З,4-дигидрокарбостирил, полученный по указанной методике в качестве промежуточного
0 продукта, выделяют из аликвотной
части реакционной смеси и после перекристсшлизации из ацетона находят, что его т.пл. 183-186°С.
П р и м е р 22. 40 г хлористого
5 алюминия прибавляют к 11,5 г 8-окси-3,4-дигидрокарбостирила и смесь тщательно перемешивают. К смеси при охлаждении ледяной водой медленно, прибавляют 21 г хлорангидрида хлор0 уксусной кислоты иполучающуюся смесь перемешивают при 35-40°С в течение 2 ч, после чего перегонкой удешяют избыток хлорангидрида хлоруксусной кислоты. Полученный остаток выливгиот
на размельченный лед и высадившиеся кристаллы отфильтровывают, промывают 9 водой, перекристаллизовывают из аце тона и получают 5,6 г 8-хлорацетокс -3,4- дигидрокарбостирила в виде светло-желтых кристаллов с т.гш. 182-184°С. Пример 23. 13 г 8-окси-3,4-дигидрокарбостирила и 20 г хлорангилркда хлоруксусной кислоты раство ряют в 100 мл нитробензола и к смес медленно добавляют 40 г хлористого алюминия, после чего перемешивают пр 70-75°С в течение 15 ч. Нитробензол затем уд аляют перегонкой с водяным паром. После охлаждения смеси высадившиеся кристаллы отфильтровывают, промывают горячей водой, перекристаш лизовывают из метанола и получ-ают 7,2 г 5-хлорацетил-8-окси-3,4-дигидр карбостирила в виде светлс-желтых кристаллов с т.пл. 189-190°С. Пример 24. Къ,0г 8-окси-3,4-дигидрокарбостирила и 15,0 г хлорангидридг.а хлоруксусной кислоты медленно добавляют при перемешивании и охлаждении льдом 30 г хлористого алюминия, после чего перемешивают пр 55-60С в течение б ч и получают 5-хлорацетил-8-хлорацетокси-З,4-дигидрокарбостиркл. К реакционной сме си, прибавляют 50 мл 10%-ной водной соляной кислоты, а затем перемешиваю в течение 3 ч при 95-100 С, После охлаждения смеси высадившиеся криста лы отфильтровывают, промывают водой перекристаллизовывают из метанола и получают 2,7 г 5-хлорацетил-8-окси-3,4-дигидрокарбостирила в виде светло-желтых кристаллов с т.пл. 189-190°С. По указанной методике аликвотнук) часть реакционной смеси отбиргиот из реактора до ее гидролиза соляной кислотой и сливают в ледяную воду. Высадившиеся кристаллы отфильтровывают, промывают горячей водой, перекристаллизовывают из смеси диметилформамид-метанол (1:1 по объему) и получают 3-хлсрацетил-8-хлорацеток си-3 4-дигидрокарбостирил в виде светло-желтых кристаллов с т.пл. 206-207°С. Пример25. 4г 5-хлорацетил-8-окси-З,4-дигидрокарбостирила (см. пример 24), растворяют в 50 мл изопр панола и к раствору прибавляют 20 г изопропиламина,после чего перемешива ют, при 60°С в течение 3 ч. Реакционную смесь концентрируют до объема 1/3 - 1/4 от первоначального объема и насыщают высушенным газообразным хлористым водородом. После охлаждения смеси полученный осадок отфильтровывают, перекристаллизовывают из jэтанола и получают 3,5 г бесцветног9 аморфного вещества с т.пл. 274-275с Полученное вещество идентифицировано ЯМР и ИК спектральными и элемент Т1ЫМ анализами как Ь-изъпропиламиноацетил-8-окси-З,4-дигидрокарбостирилхлоргидрат. Пример 26, Зг 5-хлорацетил-8-окси-З,4-дигидрохарбостирила (см. пример 24) растворяют в 40 Ш изопропанола и к раствору при перемешивании прибавляют по каплям 3 г метиламина при в течение 30 мин. После завершения прибавления смесь перемешивают в течение еще 1 ч, нагревая при этой же температуре. Реакционную смесь затем концентрируют до 1/3 от первоначального объема и доволят до рН 1-2 с концентриг ванной соляной кислотой. Полученный осадок отфильтровывают, перекристаллизовывают из этанола и получают 1,5 г бесцветного аморфного вещества с т.пл, 254256 С. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как 5-метиламиноацетил-8-окси-З,4-дигидрокарбостирилхлоргидрат. Пример 27. 2,54 г 5-хлорацеТИЛ-8-ОКСИ-3,4-дигидрокарбостирила (см. пример 24) растворяют в 30 мл изопропанола и к раствору при перемешивании прибавляют по каплям 2,7 г этиламина при в течение 20 мин. После завершения прибавления смесь перемешивают в течение еще.40 мин, нагревая при этой же температуре. Затем реакционную смесь концентрируют до 1/3 от первоначального объема и доводят, до рН 1-2 концентрированной соляной кислотой, Образующийся осадок отфильтровывают,перекристаллизовывают из этанола и получают 1,7 г бесцветного аморфного вещества с т.пл. 258-260 с. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как 5-этиламиноацетил-8-окси-3,4- дигидрокарбостирилхлоргидрат. Пример 28. 4 г 5-хлорацетил-8-окси-3,4-дизгидрокарбостирила (см. пример 24) растворяют в 60 мл изопропанола и к раствору при перемешивании прибавляют по каплям 20 г трет-бутиламина при 60°С в течение 20 мин. После завершения прибавления смесь перемешивают в течение еще 40 мин, нагревая при этой же температуре. Затем реакционную смесь концентрируют до 1/2 от первоначального объема и доводят до рН 1-2 концентрированной соляной кислотой.Полученный оса,док отфильтровы вают,повторно перекристаллизовывают из смеси этанола и ацетона (1:2 по объему) и получают 2,0 г бесцветного аморфного вещества с т.пл. 253-25Ь°С. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным aнaлизaмli как 5-трет-бутиламиноацетил-8-окси-З,4-дигидрокарбостирилхлоргидрат. Пример 29, Зг 5-хлорацетил- . -8-окси-3,4-дигидрокарбостирила (см. пример 24) растворяют в 40 мл изопропанола и к раствору при перемешивании прибавляют.по каплям 10 г втор-бутиламина при ьО°С в течение 20 мин. Поеле завершения прибавления смесь перемешивают в течение еще 2,5 ч, нагревая при этой же температуре. Затем реакционную смесь концентрирую до 1/3 от первоначального объема и насыщают сухим газообразным хлористы водородом. Полученный осадок отфильт ровывают, перекристаллизовывают из этанола и получают 1,8 г бесцветного аморфного вещества с т.пл. 269-271С Полученное вещество идентифицировано ЯМР и ИК спектральным и элементным анализами как 5-втор-бутиламиноацеТИЛ-8-ОКСИ-3,4-дигилрокарбостирилхлоргидрат. Пример 30. 4 г 5-хлорацетил-8-метокси-3,4-дигидрокарбостирила (см.примеры 19 и 20) растворяют в ЬО мл изопропанола и к раствору прибавляют 20 г изопропиламина, после чего перемешивают в течение 3 ч при 60°С. Затем реакционную смесь концен трируют до 1/3-1/4 от первоначальног объема и насыщгиот сухим газообразным хлористым водородом. После охлаждени смеси полученный осадок отфильтровывают, пе1рекристаллизовывают из этанола и получают 3,5 г бесцветного иглоподобного кристаллического вещес ва с т.пл. 208-209°С. Полученное вещ ство идентифицировано ЯМР и ИК спект ральными и элементным ангшизами как 5-изопропиламиноацетил-8-метокси-3, -;9И ДРО Рбостирилхлоргидрат. пример 31. Зг 5-хлорацетил -8-метокси-З,4-дигидрокарбостирила (см.примеры 19 и 20) растворяют в 40 мл изопропанола и к раствору при перемешивании прибавляют по каплям 3 г метиламина при в течение 30 мин. После завершения прибавлени смесь перемешивгиот в течение еще 1 ч, нагревая при этой же температуре. Затем реакционную смесь концентрируют до 1/3 от первоначальног объема и доводят до рН 1-2 с концентрированной соляной кислотой. По лученный осадок отфильтровывают, перекристаллизовывают из этанола и получают 1,5 ,г бесцветного иглоподо ного кристаллического вещества с т.пл. 232-234 С. Полученное веществ идентифицировано ЯМР и ИК спекральными. и элементным анализами как 5-метиламиноацетил-8-метокси-3,4-дигидрокарбостирилхлоргидрат. П р и м е р 32, 2,54 г 5-хлораде -8-метокси-З,4-дигйдрокарбостирила (см.примеры 19 и 20) растворяют в 30 мл изопропанола и к раствору при перемешивании прибавляют по каплям 2,7 г этиламина при в течение 20 мин. После завершения прибавлени смесь перемешивают еще 40 мин, нагр вая при этой же температуре. Затем реакционную смесь концентрируют до 1/3 от первоначального объема и доводят до рН 1-2 концентрированной соляной кислотой. Полученный осадок отфильтровывают, перекристаллизоывают из этанола и получают 1,9 г есцветного иглоподобного вещества т.пл. 224-227С. Полученный проукт идентифицирован ЯМР и ИК спекральными и элементным анализами как -этиламиноацетил-8-метокси-3, 4-диидрокарбостирилхлоргидрат. Пример 33. 4г 5-хлорацетил-8-метокси-З,4-дигидрокарбостирила (см.примеры 19 и 20) растворяют в 60 МП изопропанола и к раствору при перемешивании прибавляют по капляк 20 г трет-бутиламйна при в течение 20 мин. После завершения прибавления смесь перемешивают в течение еще 40 мин, нагревая при этой же температуре. Затем реакционную смесь концентрируют до 1/2 от первоначального объема и доводят до рН 1-2 концентрированной соляной кислотой. Полученный осадок отфильтровывают, перекристаллизовывают из смеси этанола и ацетона (1:2 по объему) и получают 2,0 г бесцветного аморфного вещества с т.пл. 203-210°С. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как 5-трет-бутиламиноацетил-8-метокси-3,4ДигидЬокарбостирилхлоргидрат. Пример 34. Зг 5-хлорг1цетил-8-метокси-3,4-дигидрокарбостирила (см.примеры 19 и 20) растворяют в 40 мл изопропанола и к раствору при перемешивании прибавляют по каплям 10 г втор-бутиламина при 60°С в течение 20 мин. Затем реакционную смесь концентрируют до 1/3 первоначального объема и насыщают сухим газообразным .хлористым водородом. Полученный оса1док отфильтровывают, перекристгшлизовывают из этанола и получают 1,8 г бесцветных иглоподобных кристаллов с т.пл. 212-214 С. Полученное вещестIBO идентифицировано ЯМР и ИК спектральными и элементным анализами как 5-втор-бутиламиноацетил-8-метокси-3,4-дигидрокарбостирилхлоргидрат. ПримерЗЗ. 1г 5-изопропиламиноацетил-8-метокси-3,4-дигидрокарбостирилхлоргидрата (см.пример 30) растворяют в 10 мл 47%-ного водного раствора бромистого водорода и смесь кипятят с обратным холодильником при температуре бани 120-130 С в течение 2,5 ч. Затем к смеси прибавляют 20 мл воды и доводят рН смеси до 6,5-7,5 с помощью бикарбоната натрия. Образующийся при этом осадок отфильтровывают, промывают водой и высушивают. Полученное вещество растворяют в изопропаноле и раствор насыщают сухим газообразным хлористым водородом. Осадок отфильтровывают, перекристаллизовывают из этанола и получают 0,5 г бесцветного аморфного вещества с т.пл. 274-275°С. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как З-изопро шламиноацетил-Б-окси-3,4-дигидз1 окарбостирилхлоргидрат. 13 Пример 36. О,7г 5-метиламин ацетил-8-метокси-З,4-дигидрокарбостирилхлоргидрата (см.пример Л) рас .творяют в 10 мл 47%-ного водного рас твора бромистото водорода и смесь ки пятят с обратным холодильником при температуре бани 120-130С в течение 4 ч. Затем реакционную смесь обрабат вают аналогично примеру 35 и получаю 0,5 г бесцветного аморфного вещества с т.пл. 254-256®С. Полученное вещест во идентифицировано ЯМР и ИК спектральными и элементным анализами как 5-метиламиноацетил-8-окси-3,4-дигидрокарбостирилхлоргидрат. П р и м е р 37. 0,9 г 5-трет-бути ламиноацетил-8-метокси-З,4-дигидрокарбостирилхлоргидрата растворяют в 10 мл 47%-ного водного раствора бромистого водорода, а затем полученный раствор обрабатывают аналогично примеру 35 и получают 0,2 г светло-желтого аморфного вещества с т.пл. 253255°С. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как 5-трет-бути ламиноацетил-8-окси-З, 4-дигидрокар- бостирилхлоргидрат. Пример 38. О,5 г 5-втор-бути аминоацетил-8-метокси-З,4-дигидрокарбостирилхлоргидрата (см.пример 34 растворяют в 10 мл 47%-ного водного раствора бромистого водорода,а затем полученный раствор обрабатывают аналогично примеру 19 и получают 0,24 г бесцветного аморфного вещества с т.п 269-271 с. Полученное вещество идентифицировано ЯМР и ИК спектральными и элементньм анализами как 5-втор-бутиламиноацетил-8-окси-З,4-дигидро карбостирилхлоргидрат. Пример 39. 50 г бромангидрид -броммасляной кислоты (У) , 50 г безводного хлористого алюминия и 400 мл сероуглерода прибавляют к 20 г 8-оксикарбостирила (У1). Полученную смесь нагревают при 50°С в течение 13 ч и сероуглеродный слой удаляют декантацией. К остатку для кристаллизации продукта добавляют размельченный лед и образующиеся при этом кристаллы отфильтровывают, промывают водой, затем перекристаллизовывают из метанола и получают 27 г 5-(з --бpoмбyтиpил)-8-oкcикapбocтиpилa {1У) с т.пл. 218-219°С (с окргшшванием и разложением) . П р и м е р 40. 25 г хлорангидрида а. -броммасляной кислоты (У) и 25 г безводного хлористого алюминия прибавляют к 10 г 8-оксикарбостирила (У1) и полученную смесь нагревают пр в течение 4 ч с тщательным перемешиванием. К смеси для кристаллиЗёщии продукта добавляют после этого размельченный лед, образующиеся при этом кристаллы отфильтровывгиот, промывают водой, а затем перекристаллизовывают из метанола и получают 12,6 г 5- oi-бромбутирил)-8-оксикар19бостирила (1У) с т.пл. 213-219°С (с окрашиванием и разложением). Пример 41, 25 г бромангидрида 0 -броммасляной-кислоты (У) и 100 мл нитробензола прибавляют к 10 г 8-оксикарбостирила и к полученому раствору при охлаждении добавляют 25 г безводного хлористого алюм1 ния. Смесь нагревают при 70®С в течение 10 ч, после чего выливают на размельченный лед. Высадившиеся кристаллы отфильтровывают, промывают водой, перекристаллизовывают из метанола и получают 11,2 г 5-( а(.-бромбутирил)-8-оксикарбостирила с т.пл. 217218,5°С (с окрашиванием и разложением). Пример 42. 200 мл втор-бутиламина (Ш) прибавляют к 10 г 5-(оС-бромбутирил)-8-оксикарбостирила (1У) (см.примеры 39-41) и полученную смесь нагревают при в течение 20 ч, после чего концентрируют досуха. Кристаллы, которые образуются при добавлении воды, отфильтровывают, растворяют в 50 мл этанола и полученный раствор доводят до рН 1 с концентрированной соляной кислотой. Высадившиеся кристаллы отфильтровывают, перекристаллизовывают из метанола и получают 8,3 г 5-(с.-втор-бутиламинобутирил)-8-оксикарбостирилхлоргидрата (П) с т.пл, 212-214°С (с окрашиванием и разложением). Пример 43. 10 мл изопропиламина (И1) и 50 МГ1 метанола прибавляют к 5 г 5-(oi -бромбутирил)-8-оксикарбостирил (1У) (см.примеры 39-41). Полученную смесь нагревают при температуре кипения с обратным холодильником в течение 6 ч, после чего концентрируют досуха. В дальнейшем продукт реакции обрабатывают аналогично примеру 1 и получают 4,2 г метанольного сольвата 5(ai -изопропиламинобутирил)-8-оксикарбостирила (П) с т.пл. 136-137С (с разложением и вспениванием). ТП(ример44. 50 г бромангидрида ai -броммасляной кислоты (У) , 50 г безводного хлористого алюиминия и 400 мл сероуглерода прибавляют к 20 г 8-оксикарбостирила (У1). Полученную смесь нагревают при 50°С Р течение 13 ч и сероуглеродный слой удаляют декантацией. К остатку добавляют размельченный лед н высадившиеся кристаллы отфильтровывают, промывают водой и перекристаллизовывают из метанола для получения 27 г 5-(ot -бромбутирил)-8-оксикарбостирила (1У) с т.пл. 218-219°С (с окрашиванием и разложением). К 5 г полученного 5-(ot -бромбутирил)-В-оксигарбостирила (1У) прибавляют 100 мл изопропиламина (Ш) и смесь нагревают при 50°С в течение 4ч, после чего концентрируют досуха. Кристаллы, которые образуются при добавлении воды, отфильровывают, про1 ивают водой, а затем перекристаллизовывагат из метанола и получают 4,6 г метанольного сольвата 5- (oi -изопропкламинобутирил) -8-оксикарбостирила (И) с т.пл. 136-137°С (с разложением и вспениванием). Пример 45. 25 г 2-фенетилами на (14) прибавляют к 5 г 5-{о -бромбутирил)-8-оксикарбостирила (1У) (см примеры 39,40 или 41) и смесь перемешивают при 30°С в течение 8ч. Смесь диэтилового эфира и петролейного эфира прибавляют к реакционной смеси и высадившееся вещество растворяют в разбавленной соляной кислоте для удаления любого нерастворимого вещества. Солянокислотный слой концентрируют и осадок отфильтровывают и перекристаллизацией из этанол получают 5,3 г вещества с т.пл. 200-201°С (с окрашиванием и разло-жением). Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как дигидра.т (2-фенетиламино) бутирил -8-оксикарбостирилхлоргидрата (П). Пример 46. 5мп морфолина (III) прибавляют к 5 г 5-(оС -бромбут рил)-8-оксикарбостирила (1У) (см. примеры 39-41) и смесь перемешивают при 40°С в течение 4 ч. Затем реакционную смесь концентрируют при пониженном давлении к полученному остатку прибавляют 100 мл воды. Смесь перемешивают и отфильтровывают, после чего концентрируют отфильтрованный маточник при пониженном давлении Остаток растворяют в ацетоне и раствор отфильтровывают, чтобы удалить любое нерастворимое вещество. Фильт рат затем концентрируют при понижен ном Дсшлении, доводят до рН 2-3 с концентрированной соляной кислотой, а осадок, образующийся при охлаждени льдом, перекристаллизовывают из эта нола и получают 2,1 г белого аморфного моногидрата 5- (о(-морфолинобути рил)-В-оксикарбостирилхлоргидрата ( с т.пл. 179-182 С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами. П р и м е р 47. 17,1 хлорангидри да о -бромпропионовой кислоты, 27 г безводного хлористого алюминия и 8 нитробензола прибавляют к 8 г 8-мет сИ-3,4-дигидрокарбостирила и смесь при перемешивании нагревают при 50 60°С В;течение 1 ч. Затем реакционн смесь сливают в 200 мл ледяной водда и образующийся осадок отфильтровьюа ют и промывают водой. Затем осадок перекристаллизовывают из этанола и получают 11,5 г вещества с т.пл. 15 155°С. Полученный продукт идентифицирован ЯМР- и ИК спектральными и . элем2нтным анализами как . 5(оС-бром пропионил) -8-метокси-3, 4 дигидрокар бостирил. Пример 48, 26,4 г бромаыгид рида oi-броммасляной кислоты, 17,5 безводного хлористого алюьтания и 5 мл нитробензола прибавляют к 5 г 8-NKToкси-3,4-дигидрокарбостирила и смесь при перемешивании нагревают при 5060°С в течение 1 ч. Затем реакционную смесь сливают в 100 мл ледяной воды, а образующийся осадок отфильтровывают и промывают водой. Затем осадок перекристаллизовывают из этанола и получают 5 г вещества с т.пл. 151-152°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным ангшизами как 5-(ciбромбутирил)-8-метокси-З,4-дигидрокарбостирил. Пример 49. 2г 5-(о -бромпропионил)-8-метокси-З,4-дигидрокарбостирила (см.пример 47) суспендируют в 50 МП изопропанола и суспензию перемешивают в течение 2ч при 60°С. Затем отгоняют растворитель и получающийся остаток растворяют в 5 мл изопропанола. Раствор затем доводят до рН 2-3 с концентрированной соляной кислотой. Образующийся осадок отфильтровывают и смесь ацетона и диэтилового эфира прибавляют к фильтрату. Образующийся осадок отфильтровывают и перекристаллизацией из изо-пропанола получают 1,5 г белого аморфного вещества с т.пл, 172-174С (с разложением), Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как дигидрат 5- ( oi -изопропиламинопропионил)-8-метокси-3,4-дигидрокарбостирилхлоргидрата. Пример 50. 5 г 5-(ы: -бромпропионил)-8-метокси-З,4-дигидрокарбостирила (см.пример 47) суспендируют в 50 мл изопропанола и 10 г третбутиламина прибавляют к суспензии, после чего смесь перег йшивают при 60°С в течение 15 ч. Растворитель затем отгоняют и получающийся остаток растворяют в 10 мл изопропанола. Раствор затем доводят до рИ 2-3 концентрированной соляной кислотой. Образующийся осадок отфильтровывают и к фильтрату прибавляют ацетон. Образующийся осадок отфильтровывают и к фильтрату добавляют диэтиловый эфир. Образующийся осадок отфильтровьшают, перекристаллизовывгиот из смеси изопропанола и диэтилового эфира и получают 2,1 г бесцветного аморфного вещества с т.пл. 207-210 с (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как моногидрат 5- (о -трет-бутиламинопропионил) -8-метокси-З,4-дигидрокарбостирилхлоргидрата. Пример 51. 2 г 5-(о(-бромбутирил)6-метокси-З,4-дигидрокарбостирила (см.пример 48) суспендируют в 50 мл изопропанола и к суспензии прибавляют 5Г изопропиламина, после чего смесь перемешивают при в течение 4 ч. Затем отгоняют раствори тель и получающийся остаток растворя в 5 мл изопропанола. Раствор затем доводят до рН 2-3 с концентрированной соляной кислотой. Образующийся осадок отфильтровывают, перекристаллизовывгиот из смеси изопропанола и ацетона и получают 1,7 г бесцветного аморфного вещества с т.пл, 204-206 С (с разложением). Полученный продукт идентифицирован ЯМР и ИК -спектральными и элементным анализами как моногидрат 5- (oi-изопропиламинобутирил)-8-метокси-З,4-дигидрокарбостирилхлоргидрата, Пример 52. Зг 5-(oi-бромбутирил)-8-метокси-З,4-дигидрокарбостирила (см,пример 48) суспендируют в 50 мл изопропанола и к суспензии Прибавляют 9 г трет-бутилгинина, посл чего смесь перемешивают при в течение 19 ч. Затем отгоняют растворитель и получающийся остаток раство ряют в 5 мл изопропанола. Раствор затем доводят до рН 2-3 концентрированной соляной кислотой. Образующийс осадок отфильтровывают и к фильтрату добавляют ацетон. Образующийся осадо отфильтровывают и к фильтрату добавл ют диэтиловый эфир. Образуклцийся осадок отфильтровывают, перекристаллизовывают из изопропанола и получают 1,6 г бесцветного аморфного вещес ва с т.пл. 160-162°С (с разложением) Полученный продукт идентифицирован ЯМР и ИК спектрешьными и элементным aиaлизau lи как моногидрат 5-(о -трет-бутиламинобутирил)-8-метокси-З,4-дигидрокарбостирилхлоргидрата. Пример 53. 1,5г 5-(о -изопропиламинопропионил)-8-метокси--3,4-дигидрокарбостирила (см. пример 50) растворяют в 15 мл 47%-ного водного бромистого водорода и раствор кипятят в течение 15 ч, нагревая при 140С. Реакционную смесь концент рируют и для кристаллизации продукта к смеси прибавляют ацетон. Продукт затем перекристаллизовывают из смеси этанола и ацетона и получают 1,1 г. вещества с т.пл.223-226°С (с разложе нием) , Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-Сэ(-изопро пиламинопропионил)-8-окси-З,4-дигидрокарбостирилбромгидрат. Приме.р 54. 1,5 г 5-(с -изопропиламинобутирил)-8-метокси-З,4дигидрокарбостирила растворяют в 15 мл 47%-ной водной бромистоводородной кислоты и раствор кипятят в течение 15 ч, нагревая при 130-140 0. Реакционную смесь концентрируют и для кристсьплизации продукта к реакционной смеси прибавляют ацетон. Продукт затем перекристаллизовывают из смеси этанола и ацетона и получают 1,0 г вещества с т.пл. 165-Гб8 с. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как полугидрат 5-(о -изопропиламинобутирил) -8-окси-З,4-дигидрокарбостирилбромгидрат. пример 55. 1г 5-(с -трет-бутиламинопропионил)-8-метокси-З,4-дигидрокарбостирила, полученного из моногидратхлоргидрата, приготовленного аналогично примеру 50, растворяют в 15 мл 47%-ной водной бромистоводородной кислоты и раствор кипятят в течение 15 ч, нагревая при 140-150С. Реакционную смесь концентрируют и для кристаллизации продукта к реакционной смеси прибавляют ацетон. Продукт затем перекрис- . таллизовывают из смеси этанола и ацетона и получает 0,7 г вещества с т.пл. 224-227°С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как моногидрат 5-(Ы-трет-бутиламино -; пропионил)-8-окси-З,4-дигидрокарбостирилбромгидрата. Пример 56. 1,5г 5-(оС-трет-бутиламинобутирил)-8-метокси-З, 4-дигидрокарбостирила, полученного из моногидратхлоргидрата, приготовленного аналогично примеру 52, растворяют в 47%-ной водной бромистоводородной кислоте и раствор кипятят в течение 19 ч при 140-150° С. Реакционную смесь концентрируют и для кристаллизации продукта к реакционной смеси прибавляют ацетон. Продукт затем перекристаллизовывают из смеси ацетона и этанола и получают 1,1 г вещества с т.пл. 144-146°С (с разложением) . Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализс11.1и как дигидрат 5-(ы-трет-бутиламинобутирил)-8-окси-3,4-д11гидрокарбостирилбромгидрат. Пример 57. 5 г 5-(ot-6pOMпропионил)-8-окси-З,4-дигидрокарбостирила суспендируют в 30 мл бензола и к суспензии прибавляют 4,2 мл морфолина, после чего смесь нагревают при. температуре кипения с обратным холодильником в течение 4ч. Реакционную смесь отфильтровывают и фильтрат промывают водой, после чего концентрируют при пониженном давлении для полного удаления оставшейся воды. Получающийся остаток растворяют в. . 50 мл изопропанола и раствор доводят до рН 2-3 с концентриррванноП соляной кислотой. Образующийся при охлаждении льдом вязкий осадок разделяют и растворяют при нагревании в ацетоне . После охлаждения раствора образующийся осадок растворяют в 30 мл воды и доводят до рН 7,5-8 бикарбонатом натрия. Осадок, образующийся при охлаждении, отфильтровывают и растворяют в 10 мл 47%-ной водной бромистоводородной кислоты, после чего концентрируют при пониженном давлении. Полученный остаток промывгиот 10 мл этанола, перекристаллизовывают из этанола и получают 2,6 г белого аморф19кого моногидрата 5-(о(-морфолино)-проПИОНИЛ-8-ОКСИ-3,4-дигидрокарбостирил бромгидрата с т.пл. 23Ь-23бС (с разложением) . Пример 58. 30 мл нитробензола и 70 МП хлорангидрида хлоруксусной кислоты прибавляют к 40 г 1-метил -8-метоксикарбостирила и к смеси при охлаждении ледяной водой медленно добавляют 130 г хлористого гшюминия, после чего смесь перемешивают при в течение 4 ч. Затем реакционную смесь сливают в 1 л ледяной воды для осаждения продукта. Осадок от.фильтровывают, промывают диэтиловым эфиром, перекристаллизовывают из сме си хлороформа и этанола (2:5 по объему) и получают 32 г белого аморфного 1-метил-5-хлорацетил-8-метоксикарбостирила с т.пл. 204-205,5°С. Полученный продукт идентифицирован элементным анализом и ИК и ЯМР спект ральными анализами. Пример 59. 40 мл нитробензола и 12 мл хлорангидрида монохлоруксусной кислоты прибавляют к 7,4 г 1-метил-8-оксикарбостирила л к смеси при охлаждении ледяной водой добавляют медленно 20 г хлористого алюми ни я, после чего смесь перемешивают при 60°С в течение 18 ч. Затем реакционную смесь сливают в 500 мл ледяной воды для осаждения продукта. Оса- 30 док отфильтровывают, промывают диэ1;иловым эфиром, перекристаллизовывают из смеси этанола и диметилформамида (1:1 по объему) и получают 2,8 г белого аморфного 1-метил-5-хлорацетил-8-оксикарбостирила с т.пл. 287-289° (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и злементным анализами. ПримербО. 2,7 г 1-метил-5-хлорацетил-8-метоксикарбостирила, полученного аналогично примеру 58, растворяют в 40 мл изопропанола и к раствору при нагревании раствора при 55-60°С прибавляйт 9 г изопропиламина в течение 1,5 ч. После завершения прибавления смесь перемешивают в течение 1 ч.Реакционную смесь концент рируют при пониженном давлении и пол чающийся остаток растворяют в 40 мл изопропанола. Затем раствор отфильтровывают для полного удаления нераст воримы веществ и фильтрат доводят до рН 2,-3 с концентрированной соляной кислотой. Смесь охлаждают льдом и образующийся осадок отфильтровывают, перекристаллизовывают из этанола и получают 0,4 г белого аморфного 1-метил-5-изопропиламиноацетил-8-метоксикарбостирилхлоргидрата. Полученный продукт идентифицирован ИК спектральньол и элементным анализами. При мер 61.10 мл изопропаноламина прибавляют к 1/6 г 1-метил-5-хлорацетил-8-оксикарбостирила,,полученного аналогично примеру 59,и смес 9 концентрируют при пониженном давлении, а остаток подвеЬгают азеотропной перегонке с этанолом досуха. Получающийся остаток растворяют в 20 мл этанола и раствор отфильтровывают для извлечения нерастворимых веществ, которые затем растворяют в горячем этаноле. Раствор доводят до рН 2-3 с концентрированной соляной кислотой и охлаждают льдом. Образующийся осадок отфильтровывают и растворяют в 20 мл воды. Раствор доводят до рН 6,5-7,5 бикарбонатом натрия и образующийся осадок отфильтровывают,-, перекристаллизовывают из этанола и получают 1,1 г белого аморфного 1-метьл-5-изопропиламиноацетил-8-оксикарбо136-138°С (с разлосгирила с т.пл. жением). Пример 62. 2,0 г 1-метил-5-изопропиламиноацетил-8-метоксикарбостирилхлоргидрата (см.пример 60) растворяют в 30 мл 47%-ного водного раствора бромистоводородной кислоты и раствор нагревают на масляной бане при 120-130°С в течение 8 ч при кипячении с обратным холодильником. Затем к реакционной смеси прибавляют 10 мл воды, после чего концентрируют г ерегонкой. Заново к смеси прибавляют 10 мл воды, после чего концентрируют. После охлаждения смеси образующийся осадок отфильтровывают и расворяют в 60 мл воды при нагревании. олучающийся раствор доводят до рН 6,5-7,5 бикарбонатом натрия и образущийся осадок отфильтровывгиот. Перекристаллизацией осадка из этанола пoлyчaJOT 0,45 г белого аморфного 1-метил-5-иэопропиламиноацетил-8-оксикарбостирила с т.пл. 136-138 с (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анаьлизами. Пример 63. 1,0 г 5-изопропиламиноацетил-8-оксикарбостирилхлоргидрата (см. пример 9) растворяют в 40 мл воды и к полученному раствору добавляют 0,5 г пешладия на угле в качестве катализатора. Полученную смесь нагревают до 35-40С при перемешивании для поглощения водорода. После завершения реакции восстановления катализатор отфильтровывают, а фильтрат концентрируют досуха при пониженном давлении. К остатку прибавляют этанол, а затем заново концентрируют досуха для полного удаления воды. Остаток кристаллизуют из ацетона и перекристаллизовывают из смеси этанола и ацетона (1:1 по объему) для получения бледно-желтого аморфного 5-(1-окси-2-изопропиламино)-этил-8-оксикарбостирилхлоргидрата с т.пл. 210-212С (с разложением) . Пример 64. 2,О г свободного основания 5-втор-бутиламиноацетил-8-оксикарбо стирила ( см. пример 11) растворяют в lOO мл метанола и к полученному раствору при перемешива нии и охлаждении постепенно добавля ют 0,8 г боргидрида натрия, получаю щуюся смесь перемешивают при этой же температуре в течение 15 мин, после чего в дальнейшем перемешиваю при комнатной температуре в течение еще 1 ч. К смеси прибавляют концен рированную соляную кислоту для того чтобы довести смесь до рН 1,5-2, и перегонкой при пониженном давлении удаляют растворитель. К остатку при бавляют 30 мл этанола, а затем концентрируют смесь досуха при понижен ном давлении для удаления воды. Остаток растворяют в 50 МП абсолютног этанола и к полученному раствору пр бавляют этанольный раствор гидрооки натрия для того, чтобы довести раствор до рН 7-8,Ь. Образующиеся осад ки отфильтровывают, а фильтрат концентрируют досуха при пониженном давлении. Остаток затем экстрагирую с 50 мл абсолютного этанола и через экстракт пропускают хлористый водор экстракт концентрируют досуха при пониженном давлении, остаток перекристаллизовывают из изопропанола и получают 1,3 г бледно-желтого аморфного моногидрата 5-(1-окси-2-втор-бутиламино)-этил-8-оксикарбос тирилдихлоргидрата с т.пл. 143-144° (с разложением). Пример 65. 1,5 г 5-трет-бутиламиноацетил-8-оксикарбостирила(см.пример 10) растворяют в 100 мл метанола и к полученному раствору при перемешивании и охлаждении медленно добавляют 0,7 г боргидрида нат рия. Перемешивание в дальнейшем про должают при этой же температуре в те чение 15 мин и при комнатной темпер туре в течение еще 1 ч.Продукт реак ции обрабатывают аналогично примеру 2, а затем перекристаллизацией из этанола получают 0,9 г бледно-желтог аморфного 5-(l-oкcи-2-тpeт-бyтилaминo) -этил-8-оксикарбостирилхлоргидрата с т.пл. 242-244 С (с разложением) . Пример 66. 2,0 г 5-пиперидиноацетил-8-оксикарбостирилхлоргидрата (см. пример 12) растворяют в 200 мл метанола и к раствору при охлаждении льдом добавляют 2,0 г боргидрата натрия, после чего смесь перемешивают в течение 2 ч. Смесь дово дят до рН 2-3 с концентрированной соляной кислотой и смесь оставляют на1 ч при комнатной температуре, после чего фильтруют. Отфильтрованну реакционную смесь концентрируют при пониженом давлении, а остаток раство ряют в 30 мл этанола. Раствор отфильтровывают для полного удаления нерастворимых веществ, а фильтрат концентрируют при пониженном давлении. Эти операции (растворение в эта ноле, фильтрование и концентрирование) повторяют трижды. Получившийся остаток растворяют в ацетоне назреванием и образующийся при охлаждении осадок отфильтровывают, перекристаллизовывают иэ изопропанола и получают 0,6 г белого аморфного полуторного- гидрата 5-(1-окси-2-пиперидине этил-8-оксикарбостирилхлоргидрата с т.пл. 146-148 С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализс1ми. В соответствии со способом, описан;ным в примере 61, из соответствующих 5-(гетероциклических амино)этаноилкарбостириловых соединений можно получить следующие соединения: 5-(1-окси-2-пиперазино)-этил-8метоксикарбостирилдигидрохлорид, имеющий точку плавления (с разложением) 177-179 с (осаикдается из смеси водаметанол) . 5-(1-окси-2-пирролидино)этил-8метоксикарбостирилгидрохлорид, имеющий точку плавления (с разложением) 150-151С . (осаждается иэ смеси изопропил-ацетон). Пример 67. 1,0 г 5-морфолиноацетил-8-оксикарбостирила, полученного в примере 13, растворяют в 100 мл метанола и к раствору добавляют 1,2 г боргидрида натрия, после чего перемешивают смесь в течение 2 ч. Реакционную смесь доводят до рН 2-3 концентрированной соляной кислоаой и смесь оставляют на 1 ч при комнатной температуре. Затем реакционную смесь отфильтровывгиот и фильтрат концентрируют при пониженном давлении. Остаток растворяют в 20 МП этанола и раствор отфильтровывают для полного удаления нерастворимых веществ, после чего фильтрат концентрируют при пониженном давлении. Эти операции (растворение в эталоне, фильтрование и концентрирование) повторяют трижды и полученный осадок кристгшлизуют из ацетона. Высадившиеся кристаллы отфильтровывают и растирают в порошок с помощью 20 мл водного раствора бикарбоната натрия для полного удаления растворимых веществ фильтрованием, а нерастворимое вещество промывгиот водой и растворяют в этаноле при нагревании. Получающийся, раствор доводят до рН 2-3 концентрированной соляной кислотой и образующийся при охлаждении осадок отфильтровывают для получения 0,6 г белого аморфного полуторного гидрата 5-(1-окси-2-морфолино) -этил-8-оксикарбрстририл хлоргидрата с т.пл. 157-158, (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами. Пример 68. 50 мл метанола прибавляют к 5 г свободного основания 5-бензиламиноацетил-8-оксикарбостирила (см.пример 14) и к раствору при охлаждении льдом и перемешивании медленно добавляют 3 г боргидрида натрия, после чего смесь перемешиваю при комнаткой температуре в течение 1 ч. Полученную смесь доводят до рН 1 концентрированной соляной кислотой и образующийся осадок отфильтровываю Фильтрат концентрируют досуха и крис таллизуют из ацетона. Получающиеся кристаллы доводят до рН 8 водным раствором гидроокиси натрия, образующийся осадок отфильтровывают и пр мывают водой. Осадок доводят до рН 1 разбавленной соляной кислотой и концентрируют досуха. Полученный остато перекристаллизовываю из смеси метанола и ацетона и получают 4,2 г бе лого аморфного вещества с т.пл. 120-121°С. Полученный продукт иденти фицирован ЯМР и ИК спектральными и элементным анализаг и как дигидрат 5-(1-окси-2-бензиалмино)-этил-8 окси карбостирилхлоргидрата. Пример 69, 1г Ь-{1-фенетила миноацетил)-8-оксикарбостирилхлоргид та, полученного по примеру 15, растворяют в 50 мл метанола и раствор превращают в слабо щелочной раствор метанольным раствором гидроокиси натрия. Затем к смеси при охлаждении льдом добавляют медленно 0,5 г боргидрида натрия, после чего перемешивают смесь при комнатной температуре в течение 1 ч. Осадок, который образуется при прибавлении концентрированной соляной кислоты до рН 3, отфильтровывают, а фильтрат концентрируют досуха. Получающийся остаток растворяют в абсолютном этаноле и раствор доводят до рН 9 этанольным раствором гидроокиси натрия. Осадок отфильтровывают, а фильтрат концентрируют досуха. Остаток кристаллизуют из ацетона и промывают водой. Получающиеся кристаллы растворяют в изопропаноле и раствор насыщают газообразным хлористым водородом,после чего охлаждают. Образующийся осадок отфильтровывают, перекристаллизовывают из изопропанола и получают i 0,77 г белого аморфного вещества с т.пл. 1б2-1б4°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как диги рат 5- 1--ОКСИ-2-(1-фенетиламино) -этил-8-оксикарбостирилхлоргидоата. Пример 70. 0,1 г окиси пла тины и 50 мл воды прибавляют к 0,5 г моногидрата 5-бензиламиноацетил-8-оксикарбостирилхлоргидрата,получен ного по примеру 14,и смесь при встр хивании восстанавливают при комнатной температуре и атмосферном давле нии в атмосфере водорода в течение 24 ч. После завершения восстановлени катализатор отфильтровывают, а водную часть концентрируют досуха. Остаток перекристаллизовывают из смеси метанола и ацетона и получают 0,25 г белого аморфного 5(1-окси-2 амин6)-этил-8-оксикарбостирилхлоргидрата с т.пл. 261-262°С (с разло ением). Полученный продукт идентифиирован ЯМР и ИК спектральными и лементным анализами. Пример 71. 1,5 г 5-{1,1-димеилфенетиламиноацетил)-8-оксикарбостирила растворяют в 50 мл метанола к раствору при охлаждении ледяной водой и перемешивании медленно добавляют 1 г боргидрида натрия, после чего продолжают перемешивание при комнатной температуре в течение еще 1 ч. Затем полученную смесь доводят до рН 1 с концентрированной соляной кислотой и смесь концентрируют досуха. Остаток растворяют в этаноле и полностью удаляют фильтрованием нерастворимые вещества. Этанольный слой концентрируют досуха и остаток растворяют в изопропаноле. Изоприпанольный слой концентрируют и для кристаллизации продукта прибавляют ацетон. Перекристаллизацией продукта из смеси метанола и ацетона получают 1,4 г белого аморфного вещества с т.пл, 167-1б8 с (с окрашиванием и разложением). Полученное вещество идентифицировано ЯМР и ИК спектральными и элементным анализами как дигидрат (1,1-диметилфенетиламино)1-окси этил-8-оксикарбостирилхлоргидрата. Пример 72. 2,5 г хлоранила и 20 мл ксилола прибавляют к 2,2 г 5-(1-окси-2-амино-)-этил-8-окси-З,4дигидрокарбостирила и смесь нагревают при температуре кипения с обратным холодильником в течение 24 ч.. Затем реакционную смесь концентрируют досуха и остаток тщательно промывают четыреххлористым углеродом. Остаток затем растворяют в 30 мл метанола и раствор доводят до рН 1 пропусканием газообразного хлористого водорода в раствор, после чего охлаждают. Высадившиеся кристаллы отфильтровыВ 1ют, перекристаллизацией из метанола, получают 1,5 г вещества с т.пл, 261262 С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5- (1-окси-2-амино) -этил-8-оксикарбостирилхлоргидрат, Пример 73. 200 мл воды, 0,9 г гидроокиси натрия и 4,3 г никеля Ренея прибавляют к 4,3 г дигидрата (1,1-диметилфенетиламино)-1-окси -этил-8-окси-З,4-дигидрокарбостирилхлоргидрата и смесь нагревают при температуре кипения с обратным холодильником при в течение 15 ч. Затем реакционную смесь отфильтровывают для удаления катализатора, а фильтрат концентрируют. Полученные высадившиеся кристаллы перекристаллизовывают из воды и получают 2,6 г вещества с т.пл. 167-168с (с разложением) . Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как моногидрат 5-12-(i,1-диметилфенетиламино)-1-окси -этил-8-окси-карбостирилхлоргидрата. Аналогично примеру 10 получают 5-(2-изопропиламино-1-окси)-этил-8-оксикарбостирилхлоргидрат с т.пл. 210-212°С (с разложением), Пример 74. 2,О г 5-изопропиламиноацетил-8-окси-З,4-дигидрокарбо стирилхлоргидрата (см.пример 3L) рас творяют в 40 мл воды и к раствору прибавляют 0,5 г палладиевой черни в качестве катализатора. Смесь перемешивают при TO-VSC и атмосферном дав лении в присутствии газообразного водорода (для поглощения водорода). После завершения восстановления катализатор отфильтровывают, а фильтрат концентрируют досуха при пониженном давлении. Воду, которая оста ётся в получающемся остатке, полностью затем удаляют, используя этанол, далее к остатку прибавляют ацетон для кристаллизации продукта. Перекристаллизацией из смеси этанола и ацетона (1:2 по объему) получают 1,1 г бесцветного аморфного вещества с т.пл. 199-201С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-(1-окси-2-изопропиламино)-этил-8-окси-3,4-дигидрокарбостирилхлоргидрат. Пример 75. 1,5 г 5-трет-бутиламиноацетил-8-окси-З,4-дигидрокарбостирилхлоргидрата (см.пример 37 растворяют в 35 мл воды и к раствору добавляют 1,0 г палладия на угле в к честве катализатора.Смесь встряхиваю при 50-бО С под давлением 4-5 атм в присутствии газообразного водорода (для поглощения водорода). После завершения восстановления катгшизатор отфильтровывают и фильтрат концентрируют досуха при пониженном дав лении. К остатку для кристаллизации продукта прибавляют ацетон. Перекристаллизацией из смеси метанола и ацетона (1:2 по объему) получгиот 0,8 г бесцветного аморфного вещества с т.пл. 240-241°С. Полученный продук идентифицирован ЯМР и ИК спектргшьны ми и элементным анализами как 5-(1-окси-2-трет-бутиламино)-этил-8-окси .-3, 4-дигидрокарбостирилхлоргидрат. Пример 76.2,0г 5-втор-бутиламиноацетил-8-окси-З,4-дигидрокарбостирила (см. пример 29) растворяют в 100 мл метанола и к раствору при перемешивании и охлаждении льдом медленно добавляют 0,8 г боргидрййа натрия. Смесь затем перемешивают при этой же температуре в течение 15 мин а затем при комнатной температуре в течение 1 ч. Смесь доводят до рН 1,5 2 с концентрированной соляной кислотой, а затем при пониженном давлении отгоняют растворитель. Затем к остатку прибавляют 30 мл этанола и смесь заново концентрируют досуха при пониженном давлении для удаления воды. 926 К полученному остатку прибавляют 50 мл абсолютного этанолг и смесь доводят до рН 7-8,5. Полученный осадок отфильтровывают и фильтрат концентрируют досуха при пониженном давлении. Остаток экстрагируют с 50 мл абсолютного этанола и через экстракт пропускают газообразный хлористый войород. Затем экстракт концентрируют досуха при пониженном давлении и остаток перекристгшлизовывают из смеси метанола и ацетона (1:2 по объему) для получения 1,3 г бесцветного аморфного вещества с т.пл. 183-184°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-(2-втор-бутиламино-1-окси)-этил-8-окси-3,4-дигидрокарбостирилхлоргидрат. Пример 77. 0,7г палладиевой черни и 130 мл воды прибавляют к 1,5 г 8-окси-5-( -метилбензиламиноацетил)-карбостирилхлоргидрата и смесь встряхивают при 60°С под давлением водорода в 4 атм. После завершения реакции катализатор отфильтровывают и водный фильтрат концентрируют досуха. Для кристаллизации продукта к остатку прибавляют ацетон. Продукт затем промывают 100 мл этанола и перекристаллизацией из смеси метанола и этилацетата получают 0,8 г белого аморфного 5-(1-окси-2-бензиламино)-ЭТИЛ-8-ОКСИ-3,4-дигидрокарбостирилхлоргидрата. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами. Пример 78. 0,1 г палладиевой черни и 50 мл воды прибавляют к 0,4г 5-(1-окси-2-бензиламино)-этил-8-оксикарбостирилхлоргидратдигидратаИ смесь встряхивоют при комнатной температуре и атмосферном давлении в атмосфере водорода в течение 8 ч. После завершения реакции катализатор отфильтровывают и водный фильтрат концентрируют досуха. Полученный оста ок перекристаллизовывают из смеси метанола и ацетона и получают 0,2 г белого аморфного 5-(1-окси-2-амино)этил-8-оксикарбостирилхлоргидрата. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным ансшизами. Пример 79. 0,7 г паллс диевой черни и 100 мл воды прибавляют 1,4 г 5- (о{.-бензило№1иноацетил) -8-окси-3, 4-дигидрокарбостирилхлоргидрата и смесь восстанавливают при 60°С под давлением водорода в 4 атм. После завершения восстановления катализатор отфильтровывают и полученный водный фильтрат концентрируют досуха. Для кристаллизации продукта к остатку прибайлляют затем ацетон. Полученные крксталлы промывают 100 мл этанола и перекристаллизацией из смеси метанола и этилацетата получают 0,7 г белого аморфного 5-(1-окси-2-амино)-этил-8-окси-3,4-дигидрокарбостиилхлоргидрата. Полученный продукт дентифицирован ЯМР и ИК спектральныи и элементным анализами.
Пример 80. О,05 г палладиеой черни и 100 мл воды прибавляют 1 г 5-(1-окси-2-амино)-этил-8-оксикарбостирилх/Оргидрата и смесь при встряхивании восстанавливают под давлением водорода в 2 атм при температуре 50°С в течение 10 ч. После завершения восстановления катализатор отфильтровывают и водный фильтрат концентрируют досуха. Полученный осадок перекристаллизовывают из смеси метанола и ацетона и получают 0,9 г белого аморфного 5-(1-окси-2-амино)-. -ЭТИЛ-8-ОКСИ-3,4-дигидрокарбостирил-. хлоргидрата с т.пл. 270-272°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами.
Пример 81. О,5 г палладиевой черни и 50 мл воды прибавляют к 2 г 5-(1-окси-2-изопропиламино)этил-8-оксикарбостирила и смесь при встряхивании восстанавливают при атмосферном давлении в атмосфере водорода при 70°С в течение 8 ч. После завершения восстановления катализатор отфильтровывают и водный фильтрат концентрируют досуха. Полученный осадок перекристаллизовывают из смеси метанола и ацетона и получают 1,7 г белого аморфного 5-(1-окси-2-изопропиламино) -ЭТИЛ-8-ОКСИ-3,4-дигидрокар бостирилхлоргидрата. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами.
Пример 82. 0,1 г палладиевой черни и 30 мл воды прибавляют к 1 г 5-(1-окси-2-втор-бу иламино)-этил-8-оксикарбостирила и смесь при встряхивании восстанавливают под давлением водорода в 3 атм при в течение 10 ч. После завершения восстановления катализатор отфильтровывайт и водный ;;Фильтрат концентрируют досуха. Полученный осадок перекристаллизовывают из смеси метанола и ацетона и получают 0,6 г 5-(1-окси-2-втор-бутиламино)-этил-8-оксй-3,4-дигидрокарбостирилхлоргидрата. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами.
П ри мер 83. 0,5 г 10%-ного палладий на угле и 50 мл воды прибавляют к, 1,5 г 5-(l-oкcи-2-тpeт-бyтилaминd) -этил-8-окси кар бостирил хлоргидрата и смесь при встряхивании вое-, .станавливают под давлением водорода в 5 атм при в течение 16 ч. После завершения восстановления катализатор отфильтровывак1Т и водный фильтрат концентрируют досуха... Полученный осадок перекристаллизовывают из смеси ацетона и метанола и получают 1,1 г белого амсрфного 5-(1-окси-2-трет-бутиламино)-этил-8-окси-3,4-дигидрокарбостирила с т.пл. 2404lc. Полученный продукт идентифиирован ЯМР и ИК спектральными и лементньв. анализами.
Пример 84. 100 M7J палладиевой черни и 50 мл воды прибавляют к 300 мг моногидрата 5- 1-окси-2-(1,1-диметилфенетиламкно) -этиг5-8-оксикарбостиригхлоргидрата и смесь восстанавливают под давлением водорода в 2,5 атм при 45-50с в течение 8 ч. Катализатор отфильтровывают и фильтрат концентрируют досуха. Остаток перекристаллизовывают из воды и получают 260 мг аморфного вещества с т.пл. 120-121°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как дигидрат 5- 1-окси-2-(1,1-диметилфенетиламино)-этил -8-окси-З,4-дигидрокарбостирилхлоргидрата.
Пример 85. О,2 г окиси платины и 5р мл воды прибавляют к 1,О г полугидрата 5-(1,1-диметилфенетиламиноацетил)-8-оксикарбостирилхлоргидрата и смесь восстанавливают пол давлением водорода в 5 атм при 80С в течение 20 ч. Катализатор отфильтровывают и фильтрат концентрируют досуха. Получающийся остаток перекристаллизовывают из воды и получают 0,8 г белого аморфного вещества с т.пл. 120-121°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как дигидрат 5- 1-окси-2-(1,1-диметилфенетиламино)-этилУ-3,4-дигидрокарбостирилхлоргидрат а.
П р и Г4 е р 86. 1,5 г 5-пиперидиноацетил-8-оксикарбостирилхлоргидратарастворяют в 100 мл воды и к раствору прибавляют 0,5 г палладия на угле и 0,2 г палладиевой черни, после чего полученную смесь встряхивают для каталитического восстановления при с;тмосферном давлении в атмосфере водорода в течение 4 дней при.70°С. После завершения восстановления реакционную смесь отфильтровывают для удаления катализатора и фильтрат концентрируют досуха при пониженном давлении. Получающийся остаток растворяют при нагревании в ацетоне, после чего раствор охлаждают. Образующийся при охлаждении осадок перекристаллизовывают из изопропанола и получают 0,8 г белого с1морфного полугидрата 5-(2-пиперидино-1-окси)-этил-8-окси-З,4-дигидрокарбостирилхлоргидрата с т..пл. 13G-139°C (с разложением) . Полученный продукт идентифицирован ЙМР и ИК спектральными и элементным анализами.
Приме р.87. 10 мл 47%-ной водной бромистоводородной кислоты прибавляют к 1 г моногидрата 5-(1-окси-2-изопропиламино)-этил-8-метоксикарбостирилхлоргидрата и смесь нагревают при температуре кипения с обратным холодильником в течение 15 ч. 29 после чего концентрируют досуха. К остатку прибавляют ацетон для крист лизации продукта, который затем доводят до рН 8 разбавленным водным раствором гидроокиси натрия. Высадившиеся кристаллы отфильтровывают, промывают водой и растворяют в этаноле. Раствор доводят до рН 1 концентрированной соляной кислотой и концентрируют досуха. Полученный остаток перекристаллизовывают из см этанола и диэтилового эфира и получают 0,7 г вещества с т.пл. 210-212 (с разложением) . Получе.нный продукт идентифицирован ЯМР и ИК спектральн и элементным анализами как 5-(1-окс -2-изопропилэмино)-этил-8-оксикарбо стирилхлоргидрат. Аналогично примеру 72 получают из соответствующих 8-метоксисоедине ний следующие соединения: 5-(1-окси-2-изопропиламино)-этил -8-окси-З,4-дигидрокарбостирилхлорг рат с т.пл. 203-204 С (с разложением 5-(1-окси-2-трет-бутиламино)-эти i. -8-оксикарбостирил хлор гидрат-полу ги рат с т.пл. 244-246°С(с разложением 5-(1 окси-2-бензиламино)-этил-8-оксикарбостирилхлоргидрат-дигидра вещество с т.пл. 120-121°с 5- 1-окси-2-(1,1-диметилфенетилам но) -этил-8-оксикарбостирилхлоргидра -моногидрат с т.пл. 167-168°С (с раз ложением); 5-(1-окси-2-амино)-этил-8-окси-3, -дигидрокарбостирилхлоргидрат с т.пл 270-272,5 С(с разложением). ПриМер 88. 40 мл метанола прибавляют к 2 г 5-(о(-изопропиламинобутирил)-8-оксикарбостирила (П), полученного аналогично примеру 43 или 44, и к полученному раствору при перемешивании и охлаждении льдом добавляют по частям 2,5 г боргидрата натрия, после чего перемешивают при комнатной температуре в течение еще 1 ч. Для доведения рН до 1 в реакционную смесь прибавляют концентрированную соляную кислоту, а затем смесь концентрируют досуха. Осадки промывают ацетоном, растворяют в воде, а затем доводят до рН 8 водным раствором гидроокиси натрия для того, чтобы высадить кристаллы. Полученные кристаллы собирают фильтрованием, перекристаллизовывают из эта нола и получают 1,8 г моногидрата 5- (1-окси-2-изопропилс1мино) -бутил-8-оксикарбостирила с т.пл. 141-142С (с окрашиванием и разложением). Пример 89.30 мл метанола прибавляют к 1,5 г 5-(о1-втор-бутиламинобутирил)-8-оксикарбостирила (IIj см. пример 42) и к полученному раствору при перемешивании и охлаждении льдом добавляют по частям 1,5 г боргидрида натрия. В дальнейшем перё i мешивание продолжают вести при комнатной температуре в течение еще 1 ч. Реакционную смесь затем доводят 9 до рН 1 с концентрированной соляной кислотой, после чего концентрируют досуха. Осадки -отфильтровывают, промывают ацетоном, растворяют в воде, а затем доводят до рН 8 с водной гидроокисью натрия. Высадившиеся кристсшлы отфильтровывают, промывают водой и снова растворяют в разбавленной соляной кислоте. Полученный раствор концентрируют досуха, а осадки перекристаллиэовывают из этанола и получают 1,3 г моногидрата 5-{1-ркси-2-втор-бутиламино)-бутил-8-оксикарбостирилхлоргидрата (1) с т.пл. 182-183с (с разложением и вспениванием). Пример 90. 20 мл тетрагидфофурана прибавляют к 1 г 5-(.-изопропиламинобутирил)-8-оксикарбостирилхлоргидрата (П; см.пример 43 или 44) и полученную смесь по каплям прибавляют к суспензии 0,12 г литийалюминийгидрида в 10 мл тетрагидрофурана, перемешивая при комнатной, температу- ре. После завершения прибавления к реакционной смеси для полного разложения избытка литийалюминийгидрида прибавляют незначительное количество воды. Реакционную смесь затем сливают в 50 мл ледяной воды и водный слой полученного раствора разделяют и концентрируют досуха. Высадившиеся кристаллы отфильтровывают, промывают ацетоном и растворяют в воде. Раствор доводят до рН 8 водной гидроокисью натрия для того, чтобы высадить кристаллы, которые затем отфильтровывают. перекристаллизовывают из этанола и получают 0,8 г моногидрата 5-{1окси-2-изопропиламино)-бутил-8-оксикарбостирила (1) с т.пл. 141-142°С (с окрашиванием и разложением). При мер 91. 1,5 г свободного основания (2-фенетиламино)-бутирил)-8-оксикарбостирила (П1см. пример 45) растворяют в 150 мл метанола и к раствору при охлаждении ледяной водой и перемешивании медленно добавляют 2 г боргидрида натрия, после чего продолжгиот преемешивание при комнатной температуре в течение еще 1ч. Полученную смесь затем доводят до рН 1 концентрированной соляной кислотой и для удаления образующегося осадка смесь отфильтровывают . Фильтрат концентрируют досуха и остаток кристаллизуют добавлением ацетона. Затем к кристаллам прибавляют водный раствор гидроокиси натрия, чтобы довести рН до 8. Образующийся осадок отфильтровывают, перекристаллизовывают из этанола и получают 1,3 г белого аморфного вещества с т.пл. 147-«148С (с разложением и вспениванием). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как тригидрат 5- 1-окси-2-(2фeнэтилa o нo)-бyтилJ-8-oкcикapбo:тирила (I). Пример 92. 2,5г хлоранила и 20 мл ксилола прибавляют к 2,2 г 5-(1-окси-2-иэопропиламино)-пропил-8-окси-З,4-дигидрокарбостирила (УП и смесь нагревают при температуре кипения с обратным холодильником в течение 24 ч. Реакционную смесь концентрируют досуха и остаток тщательно промывеиот 50 мл четыреххлористого углерода.Остаток затем раст воряют в 30 мл метанола и в раствор пропускают газообразный хлористый водород до рН 1, после чего охлаждают. Высадившиеся кристгшлы отфиль ровывают, перекристаллизовывают из метанола и получают 1,5 г вещества с т.пл. 164-166С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как моногидрат 5-(1-окси-2-изопропиламино)-пропил-8-окси-кар бостирилхлоргидрат (I). Аналогично примеру 13 получгиот следующие соединения (I) из соответствующих 3,4-дигидрокарбостириль ных соединений (УП): 5-(1-окси-2-втор-бутиламино)-бутил-8-оксикарбостирилхлоргидрат-моногидрат с т.пл. 182-183С (с ра ложением) ; 5-(1-окси-2-этиламино)-бутил-8-оксикарбостирилхлоргидрат с т.пл, 174-177,5°С (с разложением). Пример 93. 10 мл 47%-ной водной бромистоводородной кислоты прибавляют к- 1 г моногидрата Ь-(1-окси-2-изопропиламино)-пропил-8.-метоксикарбостирилхлоргидрата ( И смесь нагревают при температуре кипения с обратным холодильником в течение 15 ч, после чего концент рируют досуха. К полученной смеси кристаллизации продукта прибавляют ацетон, а затем доводят до рН 8 разбавленным водным раствором гидроокиси натрия. Высадившиеся крист лы отфильтровывшот, промывают водо и растворяют в этаноле. Раствор до водят до рН 1 концентрированной со ляной кислотой и ко.нцентрируют дос ха. Полученный остаток перекристал лизовывают из смеси этанола и диэтилового эфира и получают 0,7 г ве ства с т.пл. 164-16б°С (с разложением) . Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-(1-окси -2-изопропиламино)-пропил-8-оксика бостириЛхлоргидрат (I) . Аналогично примеру 14 получают следующее соединение (I) из соот|Ветствующего 8-метоксисоединения JYIII): 5-(1-окси-2-изопропиламино -бутил-8-оксикарбостирилхлоргидрат с т.пл. 213-214 С (с разложением), П р и м е р 94 . 1,0 г , 5-(оС-изопропиламинопропионил)-8-окси-З,4дигидрокарбостирила, полученного из бромгидратполугидрата, приготов ленного аналогично примеру 54, рас 32 воряют в 50 мл метанола и к раствору при охлаждении льдом медленно добавляют 0,3 г боргидрида натрия, после чего смесь пере1 шивают в течение еще 1 ч при комнатной температуре. Метанол, который насыщен газообразным хлористым водсфодом,прибавляют затем к смеси,чтобы довести рН смеси до 1-2. Образующийся осадок отфильтровывают и фильтрат концентрируют досуха. Затем к остатку для доведения рН до 7,5-8 прибавляют 1 н. водный раствор гидроокиси натрия и образующийся осадок отфильтровывают, растворяют в этаноле и в раствор пропускают газообразный хлористый водород. Образующийся осадок отфильтровывыают, перекристаллизовывают из этанола и получают 0,8 г вещества с т.пл. 211-213С. Полученный продукт идентифицирован ЯМР и ЮС спектральными и элементным анализами как полугидрат 5-(1-окси-2-изопропиламино)-пропил-8-окси-З,4-дигидрокарбостирилхлоргидрата. Пример 95. К 1,0 г 5-(ы-изопропиламинобутирил)-8-окси-3,4-дигидрокарбостирила прибавляют 20 мл этанола и 0,05 г окиси платины и смесь восстанавливают под давлением водорода в 2 атм при 60°С в течение 10 ч. После завершения восстановления катализатор удаляют фильтрованием и . фильтрат доводят до рН 1 с концентрированной соляной кислотой и концентрируют досуха. Полученный остаток перекристаллизовывают из этанола и получают 0,9 г вещества с т.пл. 196198°С. Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным ансшизами как моногидрат . 5-(1-окси-З-изопропиламино)-бутил-8-окси-З,4-дигидрокарбостирилхлоргидрата. Пример96. К1,0г 5-(о -трет-бутиламинопропионил).-8-окси-3, 4 -дигидрокарбостирилбромгидрата, полученного из соответствующего моногид рата, приготовленного по примеру 55, прибавляют 50 мл воды и 0,2 г палладиевой черни и смесь восстанавливают при атмосферном давлении при в течение 20 ч. После завершения восстановления катализатор отфильтровывают и фильтрат концентрируют досуха. Полученный остаток перекристаллизовывают из этанола и получают 0,75 г вещества с т.пл. 198-199®С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как моногидрат 5-{1-окси-2-трет-бутиламино)-пропил-8-окси-З,4-дигидрокарбостирилбромгидрата. Пример 97. К1,0 г 5-(о(-трет-бутиламинобутирил)-8-окси-З,4-дигидрокарбостирилбромгидрата полученного из соответствующего дигидрата, приготовленного по примеру 56, прибавляют 50 мл воды и 0,2 г палладиевой черни и смесь восстанавливают при атмосферном давлении .при 80°С в течение 10 дней. После завершения востановления катализатор отфильтровывают и фильтрат концентрируют досуха. Полученный остаток перекристаллизовывают из этанола и получают 0,7 г вещества с т.пл. 164-166 С (с разложением) . Полученный продукт идентифицирован ЯМР и И спектральными и злементным анализам как 5-(1-окси-2-трет-бутиламино)-бутил-8-окси-З,4-дигидрокарбостири бром идрат (в виде зтанольного соль вата) . Пример98. К2 г 5-W-изопр пиламинобутирил)-8-оксикарбостирила прибавляют 0,1 г палладиевой черни и 50 мл этанола и при перемешивании смесь восстанавливают под давлением водорода в 35 атм при 75°С в течени 15 ч. После завершения востановлени катализатор отфильтровывают и фильт рат доводят до рН 1 концентрированной соляной кислотой. Смесь затем концентрируют досуха и получающийся остаток перекристаллизовывают из этанола и получают 1,7 г моногидрата 5-(1-окси 2-изопропиламино)-бутил-8 -окси-3,4-дигидрокарбостирилхлоргид рата с т.пл. 196-198°С. Пример 99. К1г 5-и-изопропиламинобутирил)-8-оксикарбостирилхлоргидрата прибавляют 0,05 г платиновой черни и 100 мл воды и смесь при перемешивании восстанавли вают под давлением водорода в 40 ат при в течение 18 ч. После завершения восстановления катализатор отфильтровывают и фильтрат концентрируют досуха. Образующийся осадок затем перекристаллизовывают из изопропанола и получают 0,85 г моногидрата 5-(1-окси-2-изопропиламино) -бутил-8-окси-З,4-дигидрокарбостирнл хлоргидрата с т.пл. 196-198°С. Пример 100. К 0,5 г 5-(«t-втор-бутиламинобутирил)-8-оксикарбо стирила прибавляют 0,0025 г палладиевой черни и 15 мл этанола и смес восстанавливают при стряхивании под давлением водорода в 50 атм при в течение 15 ч. После завершения востановления катализатор отфильтро вывают и фильтрат доводят до рН 1 концентрированной соляной кислотой и л онцентрируют досуха. Образующийся осадок перекристаллизовывают затем и этанола и получают 0,45 г моногидрата 5-(1-окси-2-втор-бутиламино)-бути -8-окси-З,4-дигидрокарбостирилхлоргидрата с т.пл. 205-207 с. Пример 101. 15 мл 47%-ной водной бромистоводородной кислоты прибавляют к 1,5 г 5-(1-окси-2-трет-бутиламино)-пропил-8-метокси-З,4-дигидрокарбостирила и смесь нагре вают при температуре 1 ипения с обрат ным холодильником в течение 15 ч. После этого к реакционной смеси для 93 кристаллизации продукта прибавляют ацетон и получающиеся кристаллы перекристаллизовывают из смеси этанола и ацетона и получают 1,4 г вещества с т.пл. 198-199С (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализaNM как моногидрат 5-(1-окси-2-трет-бутиламино)-пропил-8-окси-3,4-дигидрокарбостирилбромгидрата. Аналогично примеру 75, получают следующие соединения: 5- 1-окси-2-(-2-фенетиламино)-бутил -8-окси-3,4-дигидрокарбостирилхлоргидрат с т.пл. 127-129 С (с разложением); 5-(1-окси-2-морфолинобутил)-8-окси-З,4-дигидрокарбостирилбромгидрат с т.пл. 183-185 С (с разложением) . Пример 102. К 1,0 г 5(о( трет-бутиламинобутирил)-8-окси-3,4-дигидрокарбостирилбромгидрата прибавляют 50 мл воды и 0,2 г палладиевой черни и смесь восстанавливгиот при атмосферном давлении при температуре в течение 10 дней. После завершения восстановления катализатор отфильтровывают и фильтрат, концентрируют досуха. Получающийся остаток перекристаллизовывают из этанола и получают 0,7 г вещества с т.пл. 164166°С Сс разложением). Полученный продукт идентифицирован Я1-1Р и ИК спектральными и элементным анализами как 5-(1-окси-2-трет-бутиламино)-. -бутил-8-окси-З,4-дигидрокарбостирилбромгидрат (в виде этанольного сольвата). Пример 103. 1,0 г 5-(2-морфолинобутирил)-8-окси-З,4-дигидрок ар бо ст ирил бромгидрат а р а ст в ор яют в 70 мл воды и к раствору прибавляют 0,2 г палла,дия на угле и 0,3 г пал- : ладиевой черни, после чего смесь при встряхивании подвергают каталити-у ческому восстановлению при атмосфере водород. в течение 10 дней при 70°С. завершения восстановления реакционную смесь отфильтровывают для удаления катализатора и фильтрат концентрируют досуха при пониженном давлении. Получающийся остаток растворяют в ацетоне нагреванием после чего раствор охлаждают. Образующийся при охлаждении осадок перекристаллизовывают из этанола и получают 0,7 г белого аморфного полугидрата 5-(1-окси-2-морфолино)-бутил-8-окси-З,4-дигидрокарбостирилбромгидрата с т.пл. 183-185С (с разложением) . Полученный продукт идентифицирован ЯМР и ИК спектральными и элементныманализами. Пример 104. 0,45 г свободного основания 1-метил-5-изопропилс1Миноацетил-8-оксикарбостирила (см. пример 60) растворяют в 50 мл метанола и к раствору при охлаждении льдом и перемешивайии медленно добавляют
0,2 г боргидрита натрия. В дальнейше продолжают перемешивать в течение еще 1 ч и полученную смесь доводят до рН 2-3 концентрированной соляной кислотой. Затем образую11;ийся осадок удаляют фильтрованием. Фильтрат концентрируют при пониженном давлении и остаток растворяют в 20 мл этанола Для полного удаления нерастворимых веществ раствор отфильтровывают. Фильтрат концентрируют при пониженно Давлении, и затем прибавляют 20 мл этанола, после чего фильтруют для полного удаления нерастворимых веществ. Фильтрат концентрируют при пониженном давлении досуха и к остатку прибавляют 40 мл этанола для полного удаления растворимых в этаноле веществ, остающихся в остатке. Нерастворимое в этаноле вещество промывают дважды порциями по 20 мл холодной воды, перекристаллизовывают из этанола и получают 0,3 г белого аморфного 1-метил-5-(1-окси-2-изопропиламино)-этил-8-окси-карбостирилхлогидрата с т.пл„ 202-203,5°С (с разложением) о Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами о
Пример 105, 1 г 1-метил-5.изoпpoпилa Шнoaцeтил-8-oкcикapбocтирилхлоргидрат а суспендируют в 100 мл воды и к суспензии добавляют 0,1 г палладиевой черни и 0,1 г палладия нЪ. угле, после чего смесь подвергают каталитическому восстановлению водородом при атмосферном давлении и в течение 25 ч. После завершения восстановления реакционную смесь отфильтровывают и фильтрат концентрируют досуха при пониженном давлении. Получающийся остаток кристаллизуют из ацетона и затем перекристаллизовывают из этанола и получают 0,3 г белого аморфного 1--мет -ш-5- (1-окси 2-изопропиламино) этил-8 окси--, 4-дигидрокарбостирила с т.пл. 196-197°С с(с разложением) . Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами.
Пример 106. 1,0г 5-изо пропиламиноацетил-8-метоксикарбости
рила растворяют в 50 мл метанола и к раствору при охлаждении льдом и перемешивании медленно добавляют 0,6 г боргидрата натрия, после чего смесь перемешившот прикомнатной температуре в течение еще 1 ч. Реакционную смесь доводят до рН 2-3 с концентрированной соляной кислотой и образующийся осадок отфильтровывают. Фильтрат концентрируют досуха и кристаллизуют из ацетона. Перекристаллизацией продукта из этанола получают 0,8 г вещества с т.пл 230-231 0 (с разложением). Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как моноги,црат 5-(1-окси-2-изопропиламино)-этил-8-метоксикарбостирилхлоргидрата.
Пример 107. 2 г 5-изопропиламиноацетил-8-метокси-З,4-ди- . . гидрокарбостирйла растворяют в 70 мл метанола и к раствору при озслаждении 5 ледяной водой медленно добавляют 1 г боргидрида натрия, пбсле чего перемешивают при комнатной температуре в течение еще 1 ч. Реакционную смесь доводят до рН 1 с концентрированной соляной кислотой и образующийся осадок отфильтровывают. Фильтрат концентрируют досуха и получающийся остаток перекристаллизовывают из этанола для получег ния 1,5 г вещества с т.пл. 206208 С (с разложением) . Полученный продукт идентифицирован ЯМР и ИК спектральными и элементным анализами как 5-(l-oкcи-2-изoпpoпилaминo)-эткл-8-мeтoкcи-3,4-дигидpoкapбoстирилхлоргидрат.
При мер. 108. 3 г 5-(1-окси-2-изопропиламино)-этил-8-оксикар5 ростирилхлоргидрата растворяют в 20 МП воды и к раствору добавляют 0,9 г гидроокиси натрия. Затем к раствору при охлаждении льдом и перемешивании по каплям прибавляют Q 1,3 г диметилсульфата в течение
1ч. Смесь перемешивают в течение
2ч при 40-50°С и реакционную смесь экстрагируют хлороформом. Хлороформный экстракт промывают водой и высу5 шивают, а затем в хлороформный экстракт пропускают газообразный хлористый водород. Высадившиеся кристсшлы перекристаллизовывают из смеси этанола и ацетона и получают 2,3 г вещества с т.пл.. 235-237С (с разложением) . Полученный продукт идентифицирован ЯМР и КК спектральными и элементным анализами как моногидрат 5-(1-окси-2-изопропиламино)-этил-8-метоксикарбостнрилхлоргидрата.
Пример 109. 2,6 г 5-(1-окси-2-изопропиламино)-этил-8-окси-З,4-дигидрокарбостирйла растворяют в 30 МП воды и к раствору добавляют
0 0,45 г боргидрида натрия. Затем к раствору при охлаждении льдом и перемешивании в течение 1 ч прибавляют по каплям 1,3 г диметилсульфата. Реакционную смесь перемешивгиот
5 в течение 2 ч при 40-50С и экстрагируют хлороформом. Хлороформный экстракт про Фавают водой и высушивают, а затем в хлороформный экстракт пропускают газообразный хлористый
0 водород. Высадившиеся кристаллы перекристаллизовывают из. этанола и получают 2,2 г вещества с т.пл. 206208°С (с разложением). Полученный продукт идентифицирован ЯМР и ИК
5 спектральными и элементным анализами как 5-(1-окси-2-изопропиламино)-этил-8-метокси 3,4-дигидрокарбостирилхлоргидрат.
Пример 110. 10 мл 47%-ной водной бромистоводородной кислоты прибавляют к I г моногидрата 1-метил-5-(1-окси-2-изопропиламино)-этил-8-метокси-З, 4-д1.гидрокарбостирилхлоргидрата и смесь нагревают при температуре кипения с обратным холодильником в течение 15 ч, после чего концентрируют досуха. К полученнок у остатку прибавляют ацетон для кристаллизации продукта, которы затем доводят до рН 8 разбавленным водным раствором гидроокиси натрия. Высадившиеся кристаллы отфильтровывают, промывают и растворяют в этаноле. Раствор доводят до рН 1 концентрированной соляной кислотой и концентрируют досуха. Полученный остаток перекристаллизовывают из смеси этанола и диэтилового эфира и получают 0,7 г вещества с т.пл. 198-200°С ( с разложением). Полученный продукт идентифицирован ЯМР и ИК (спектральными и элементным анализами как 1-метил-5-{1-окси-2-изопропиламино)-ЭТИЛ-8-ОКСИ-3,4-дигидрокарбостирилхлоргидрат.
Формула изобретения
Способ получения карбостирил-или 3,4-дигидрокарбостирил-производных общей формулы J
да, алкильную группу , аралкильную группу, содержащую линейную или разветвленную алкильную группу Cj-Ci,., или циклоалкильную группу Ci,,-С или
RZ и Rj вместе с атомом азота, к которому они присоединены, образуют морфолино-, пиперидиво-, пиперазиноили пирролидиногруппы,
отличающийся тем, что 5-алкиламиноалканоилкарбостирил- или 5-алкиламиноалканоил-З,4 дигидрокар0бостирилпрокзводные общей формулы II
15
20
гле R .v .vj .4
R, R, R. и R
являются указанными группами,
подвергают восстановлению в присутствии катализатора или с помощью восстанавливающего агента с выделе25 .нием целевого продукта.
30 с.н-снмС -о щ ky где ,R4 и Ну, каждый, представ ляет -собой атом водорода-или алкил ную группу С(-Сц, причем по крайней мере один из Нц и Rj является атомом водорода, а Ra и R, которые могут быть одинаковыми или различными,каж дый представляют собой атом водороПриорите4192/1974; 12953/ 1974; 15116/ /19,74; 23604/1974 58315/1974; 58317/1974; 60Ъ04/19 74 ; 67823/ /1974; 67824/1974; 97660/1974/ 97661/1974; 97663/1974; 129381/1974; 101260/1974 129384/1974; 129382/1974 130718/1974; 130717/1974; 130721/1974; 130719/1974; 13.0722/1974; 130724/1974; 130725/1974; 130726/1974; 140339/1974; 140340/1974. м:




















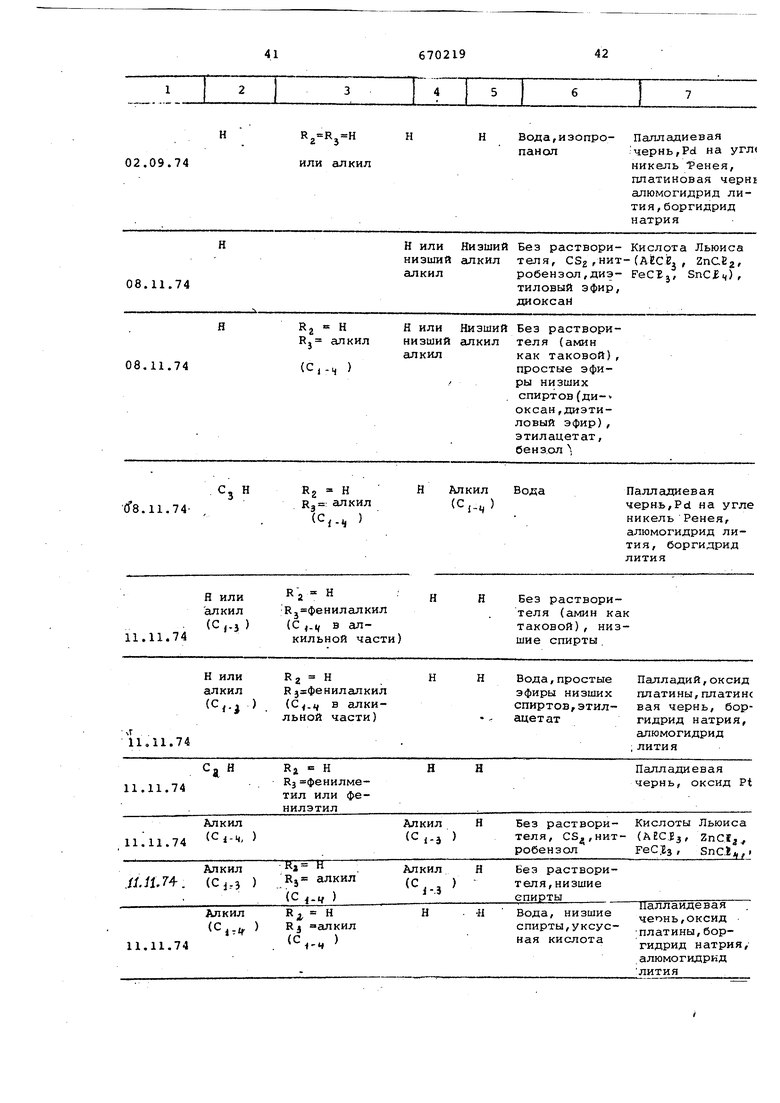
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения карбостирильных производных или их фармацевтически приемлемых солей с кислотами | 1979 |
|
SU1140687A3 |
| Способ получения карбостирильных производных | 1981 |
|
SU1367857A3 |
| Способ получения производных карбостирила или их фармакологически приемлемых кислотно-аддитивных или четвертичных солей | 1980 |
|
SU1091857A3 |
| Способ получения 3,4-дигидрокарбостирильных производных или их солей | 1973 |
|
SU580835A3 |
| Способ получения карбостирильных соединений или их фармацевтически приемлемых солей | 1984 |
|
SU1331430A3 |
| Способ получения карбостирильных производных или их фармацевтически приемлемых солей | 1982 |
|
SU1215621A3 |
| Способ получения карбостирильных производных | 1981 |
|
SU1169535A3 |
| Способ получения производных карбостирила | 1982 |
|
SU1331426A3 |
| Способ получения производных карбостирила | 1982 |
|
SU1356962A3 |
| Способ получения карбостирильных производных | 1982 |
|
SU1395140A3 |
диоксан
Rg RS Н или алкил
Н Rg Rj Н или алкил
Алкил R Н ( ) к, алкил (С,. )
27,Ю5.74
АлкилR 2 Н
(С ji. J )R J алкил
13 „Об. 74(i-Ч
Алкил
(С j. )Ri Н.
Rj алкил
(i-H ) Метил или 26.OS.74 этил
Н
R2 Н
К} алкил )
Без растворителя, метанол , этанол,изопропанол,диоксан, диэтиловый iэфир,этилаце:тат,бензол,
толуол,ксиол, ацетонитрил
Вода,метанол, Папладиевая
этанол,изопро- чернь,Pd на угле панол никель Ренея,
алюмогидрид лития, боргидрид натри я
Метанол,этанол ,изопропанол,алкиламины (т.е.без растворителя)
Без раствориКислоты Льюиса (А.Ссез,гпс.Еа, теля,CSa,нитFeCjtaробензол ,диэтиловый эфир, диоксан.
Н .Вода,этилаце-1Б.оргидрид наттатрия, алюмогидрид лития
Вода,изопро- Палладиевая панол этил- чернь,Pd на угле, ацетатплатиновая чернь,
никель Ренея
Без растворителя, (амин как таковой),изопропанол,диоксан ,диметиловый эфир,этила« цатат,бензол Н Без раствори-Кислоты Льюиса теля, (AtCEj, ZnC., робензол,диэ- FeCEj, БпСВц) тиловый эфир, диоксан
Источники информации, принятые внимание при экспертизе Редактор Т.Загребельная Заказ 3814/58
Филиал ППП Патент, г.Ужгород, ул.Проектная,4
1, СА, 62, 16212е, 1965. Составитель Г.Жукова Техред 3. Фант Корректор Е. Папп Тираж 512 Подписное ЦНИИПИ Государственного комитета СССР по делам изобретений и открытий 113035, Москва, Ж-35, Раушская наб., д.4/5
