(54) СПОСОБ ЭКСТРАКЦИИ СОЕДИНЕНИЙ МЕТАЛЛОВ ИЗ ВОДОЫХ РАСТВОРОВ где R - ал1 силеновый радикал, содержащий 1-20 атомов углерода, р 0-1, А моио- или полициютический радикал, имеющий кольцо или кольца, состоящие из 5-6 членов q - целое число 1 -5 г 0-2 R - алкильньга или алкенильный радикал, причем общее число атомов углерода в(Пн)д. Ч составляет по крайней мере 5 при условии, что когда q 2 то по крайней мере один из R радикалов содержит 5 или более атомов углерода, а когда q 3, то по крайней мере один из радикалов R содержит 3 или более атомов углерода, R - СЕ -Вг, -NOj или OR, где R - углеводорощгый радикал с числом углеродных атомов 1-20, пит равны О или I, или 3, П и R углеводородные или углеводородоксирадикаяы содержаише 1-20 атомов углерода, -С -Вг .или NOj с последующим отделением органической фазы, содержащей экстрагированные соединения металлов и извлечением их. По изобретению применяются 8-сульфЬнамидохинолины, которые имеют растворимость в водонесмешивающихся органических растворителях не менее 2 вес.%, и чьи металлические комплексы имеют такую же растворимость. Основная часть молекулы 8-сульфонамвдохинолина имеет строение WH OZпричем растворимость в водонесмещивающем ся органическом растворителе достигается за счет заместителей в хинолиновом кольце и/или радикалов, завершающих группу NHSO лучше последних. Как указано, Необходимо, чтобы этн 8-сульфонамидохйнолины имели требующуюся растворимость. Требуется также, чтобы азот хинолинового кольца и груп па NHSOj оставались активными, поскольку комплексирование металла происходит благо даря взаимодействию этих групп. Значительн количество наиболее подходящих 8-сульфонамидохинолинов приведено в примерах. Предпочтительная группа 8-сульфонамидохинолинов, применяемых по изобретению, пр ставляет собой соединения формуль где R - )тлеводородный радикал. Например R может быть алкил или алкенил с числом углеродных атомов 5-8 или более. Лучше, .если эти алкИлы и алкенилы содержат мене 20 углеродных атомов и являются разветвленными. Но предпочтительнее, чтобы R была группа формулы (А} Если р 1, то R - алкилен с 1-20 углеродными атомами, лучше 1-2 углеродными атомами; А - моно- или полициклический радикал с пяти или шестичленным кольцом или кольцами. Хотя эти моно- и полициклические радикалы могут быть насыщенными или ненасыщенными лучше, чтобы они были ненасыщенньсми и щестичленными, при, А - фенил или нафтил. В этих аралкильных, алкарильных или аЛкенарильных соединениях q - целое число 1-5, R,- алкил или алкенил, так чтобы общее число углеродных атомов в (R)q было не менее S, при условии что, если q 2, то не менее, чем в одном радикале R содержится 5 и более углеродных атомов, а если q 3 и более, то не менее, чем в одном радикале R содержится 3 и более углеродных атомов. Лучше,чтобы общее число углеродных атомов в (R)q составляло 8 и больше. Кроме того, лучше, чтобы R бьто алкил, а q 1-3. Отдельные группы R могут содержать 20 и более углеродных атомов, но такие группы не имеют преимуществ, поскольку они увеличивают суммарный молекулярный вес сульфонамидохинолинов без увеличения их зкстракционной способности. В наиболее интересных алкарильных и алкениларильных соединениях г 0-2, R - СЕ Вг, нитро или -OR, где R - алкил, алкенил, арил, аралкил, алка-. рил или алкенарил с 1-20 углеродными атомами. Лучще, чтобы г бьшо равно О или 1. Если. А - фенил, то q + г 5 и меньше. R и R в наиболее интересных соединениях по изобретению могут представлять собой алкил, алкеиил, арил, аралкил, алкарил или алкеиарил с 1-20 углеродными атомами, эфирные группы, -OR, как указано выше, или Ct, Вг или нитро группу. В этом случае лучше, чтобы пит были равны 0-3 и чтобы R и R были бы алкильные группы с 1-3 углеродными атомами, Ct, Вг или нитрогру ппы. В лучших соединениях m и п - эт О или 1. Если m или п 2 и R или В - это алифатические группы, то две такие группы могут при желании образовать дополнительное конденсированное кольцо при основном хинолиновом. Как видно из примеров, длина цепи алкила и апкешша и/или их разветвлеиность цепей в радикале R в наиболее подходящих соединениях (включая в аралкил, алкарил и алкениларильных соединениях) может определять указанную растворимость. Таким обр зом, наиболее подходящие соединения имеют радикал R с достаточной длиной цепи и/или разветвленность и тип цепи в алкильной и алкенильной группах, обеспечиваюидие минимальную растворимость в указшшьгх раствор телях. 8-Сульфоиамидохинолиновые соединения можно получать по реакции соответствующего 8-аминохинолина с соответствующим сульфонилхлоридом. В оптимальном варианте 8-аминохйнолин или замещенный 8-аминохинолин сначала растворяют в органическом основании или растворе органического основания в органическом растворителе. Затем полученный раствор охлаждают до О-10° С и медленно добавляют нужный сульфонилхлоРИД при перемешивании и температуре 0-20° По окончании добавления сульфонилхлорида реакционной смеси дают нагреться до ком натной температуры при перемешивании в те;Чение 1-3 ч. Затем реакционную смесь нагре вают до 80-100° С в течение 30 мин, добавляют воду и реакционную смесь при 75- 95° С перемешивают еще в течение 30 мин, Затем ее выливают в воду (отношение 250 м на 1 л) и вьщеляют сульфонамидохинолин или I) экстракцией органическим растворите1лсм, например бензолом, хлороформом и т.п или II) фильтрованием в случае получения твердых сульфонамидохинолинов. Если сульфонамидохинолины вьщеляют экстракцией, to органический экстракт лучше промыть три раза 2-5%-ным раствором бикарбоната натрия в 20-30%-ном водном метаноле, затем три раза водной серной кислотой с концентрацией 25 г/л и снова - раствором бикарбоната натрия. Наконец органическую фазу можно промыть рассолом и сушить над сульфатом натрия и фильтровать. Фильтрат можно выпарить в вакууме. По изобретению сульфонамвдохинолиновые соединения для экстракции растворяю в органическом растворителе, почти несмешивающимся с водой, затем этот раствор контактируют с содержащей металл водной фазой. Пр этом часть металла зкстрагируется органической фазой. Затем эти фазы разделяют и металлы можно извлечь из органической фазы с помощью воды В способе извлечения металлов по изобретению можно применять различные не смещивающиеся с водой органические раствортели, как алифатические так и ароматические, например керосины, бензол, толуол и ксилол. Выбор органического растворителя при промыщленнрм извлечении эавнст от ряда фак торов, в том числе от конструкции установки сопьвентной экстракции,цет1ости извлека,емого металла, удаления сточных вод и т.п. Способ по изобретению особенно хорощ при экстракционном вьщелении основных цветных :металлов: меди, никеля, цинка, кобальта (II), ;кадмия, ртути и серебра (I) и свинца, как зто более полно описано ниже. Почти во всех основных установках по выделению зтих металлов (особенно при выделении меди) применяют смесители-отстойники для. ;больщих количеств органических растворите;лей, позтому происходят неизбежные потери растворителя вследствие испарения, увлечения водной фазой и т.д. При этих услови,ях лучшими растворителями для выделения , металлов по изобретению являются алифатические и ароматические углеводороды с температурой вспыщки 65, и вьпне и растворимостью в воде менее 0,1 вес.%. Эти растворители почти нетоксичны, хилгачески инертны и сравнительно недороги. При экстракции по изобретенюо S-суль|фонамидохинолины растворяют в не смеишрающемся с водой органическом растворителе. Содержание 8-сульфонамидохинолинов в . растворе для прямого использовагшя в экстракционном способе 2-50 вес.%. В не которых случаях можно использовать концентри рованные растворы 8-сульфонамидохинолинов, .например 25-75%-ные, что удобно при транспорт1фовк-е и работе; кощентраЦию Перед использованием можно менять по необходимости. В процессе экстракции соотношение органической и подпой фаз может широко колебаться, поскольку контактирование любого количества сульфонамидохинолинового раст.вора с металлсодержащим водным раствором приводит к некоторому извлечению соединения металла в органическую фазу. В промышленном производстве соотношение органической и водной фаз предпочтительно 5:1-1:5. Практ1гчески экстракщ1ю и обратное извлечение обычно ведут при комнатной температуре и обычном давлении, хотя при желании можно применять более высокие температуры и давления. Весь процесс экстракции можно вести непрерьгВЕгб, яспользуя Очищенный органический растворитель ля обработки других, металлсодерж-ащнх расторов. Способ вьщеления металлов из их водных астворов по изобрете шю применим для ледующих металлов: Си, , Со, n-«-+, , Cd++, Hg++ и . Все эти еталлы, кроме , являются переходиыи металлами групп 1В, IIB и VIII периодиеской системы. Экстрагирование этих раз- , wiffbix металлов Из йгх в6дн1ь1х раствКров зависит от ряда факторов в том числе, например, от концентрации ионов металла, присутствующего аниона и рН и/или концентрации аммиака в водном растворе и концентрации данного сульфонамидохинолина в органической фазе. Для каждого йодногб раствора металла и раствора реагента сульфонамидохинолина существует оптимальный предел условий экстракции. Это также справедливо для операции обратного извлечегпш. Под обратным извлечением подразумевается to, что по крайней мере часть металла из органической аэы переходит в водную извлекаемую фазу. Затем металлы из зтой водной фазы можно вьщелять обычными способами, лучще электролизом. Отношение органической фазы к водной извлекающей фазе может иметь широкие пределы. Обычно в извлекающем водном растворе металл находится в более вы-; сокой концен:фацин, чем в исходном металлсодержащем растворе. Оптимальное отношение органического загруженного раствора к водной извлекающей фазе 1:1 - 10:1.
На основании многочисленньгх данных для сульфонамидохинолинов, приведенных в примерах 1 и 2, ниже приведены некоторые тфедпочтительные условия операций извлечения и обратной отмьшки для ионов разных металлов. Установлено, что легко извлекается при рН 0,5-7,0. Медь легко извлекается из аммиачных растворов с. концент-; рацией аммиака 10-150 г/л. Из органической фазы легко извлекается водным раствором 25-250 г/л HjSOa. Zn+, N +, и легко извлекаются из аммиачных растворов, как и Си+ . Для этих металлов лучшим интервалом рН являются: для Zn-H- 4-6, для N1+ 4,5-7,0, для 5-7 и для Cd ; 4-7. Все эти металлы легко извлекаются из органической фазы, в К1.1Орой они соДержат ся, водными кислыйи средами с содержанием 25-250 г/л серной кислоты. Свинец (РЬ) лучще извлекается при рН более 5,0, причем извлекается из органической фазы водным кислым раствором с 100-150 г/л азотной кислоты ( свинец плохо растворяется в водной H2SO4). РЬ++ не образует растворимого аммиачного комплекса. по имеющимся ограниченным данным лучще извлекается из ее водных растворов при рН 0,5-6,0. Для нее лучшей извлекающей средой является соляная кислота с концентрацией 20-50 г/л. АЭ экстрагируется из его аммиачного раствора при концентрации аммиака. 10 г/л. Извлекающим водньпл раствором для серебра в органической фазе является азотная кислота с концентрацией 63 г/л соляная с концентрацией 37 г/л и серная с концентрацией 150 г/л. Эти вьюоды сделаны на основании данных экстрагирования и водного извлечения, приведенных в примерах. Как указано вьппе, для каждого исходного металлсодержащего водного раствора существуют оптимальные услов.ия.
В первой rpjmne приведенных примеров (1-28) показано получение по изобретению наиболее интересных сульфонамидохинолийов, а во второй группе примеров -(29-59) извлечение металлов по изобретению. Пример 1А.
К раствору 43,2 г (0,3 моля) 8-аминрхинолина в 100 мл пиридина и 200 мл толуол медленно добавляют 103 г (0,3 моля) доде;цилбензолсульфонилхлорида, представляющего собой изомерную смесь, где додецильная груп,па находится больщей частью в п-положении. Реакционную смесь перемещивают в течение ночи, затем ее нагревают с обратным холодильником 1 ч и добавляют 500 мл дистиллированной воды. Перемещивание ведут еще 1 ч при нагревании, после чего реакционную смесь выливают в делительную воронку. Фазы разделяют и добавляют 1 л гептана ст. кип. 88-100° С. Затем органическую фазу два раза промывают поршями по 100 мл водной серной кислоты с концентрацией 25 г/л, четьфе раза свежеприготовленным 5%-ным раствором NaHCOs в 40%-ном водном метаноле (порциями по 200 мл), еще два раза раствором серной кислоты порциями по 200 мл, еще один раз раствором бикарбоната натрия и затем рассолом. Реакционную смесь сущат над сульфатом натрия, фильтруют и выпаривают досуха в вакууме. Получают 115,9 г продукта (выход 85%) в виде масла, который представляет собой 8- (додецилбензолсульфонамидо) хинолин формулы
Строение подтверждено в этом и последующих примерах ИК- и ЯМР-анализами.
Пример 1В. К раствору 12,9 г (0,09 моля) 8-аминохинолина в 150 мл пиридина добавляют 31.0 г (0,09 моля) додещшбензолсульфошшхлорида в 100 мл гептана при 0°С. Сульфонилхлорид был получен из додецилбензолсульфокислоты, где додецильная группа находится большей частью
в пара-положении. Реакционную смесь перемеишвают 1 ч при , затем перемешивают при комнатной температуре в течение ночи, затем ее нагревают до 70°С к выливют в 600 мл ледяной воды. Водную смесь экстрагируют гептаном, экстракт промывают четыре раза 5%-ным раствором МНСОз в 40%-ном водном метаноле и сушат над сульфатом натрия, фильтруют, нaqзeвaют до кипения и добавляют 10 г обесвдечивающего угля. Раствор фильтруют через целит и выпаривают в вакууме до светло-желтого масла (32,3 г). Полученный 8-додецйлбензолсульфонамидохинолин имеет строение, как в примере 1А, где додецильная группа находится в том же положении, что и в исходном додецилбензоле.
Пример 1C. Повторяют пример 1В, но в качестве исходного продукта используют, додецилбензол, представляющий собой синтетический алкилбензол, в котором боковая цепь разветвлена (твердый алкилат) и содержит в среднем 12 углероднь1х атомов.
Полученный сульфонилхлорид и 8-додецилбензолсульфонамидохинолин представляют собой изомернуго смесь, где додецильная группа находится в том же положении, что и в исходном додецилбензоле (в этом и последующих примерах алкильные группы в кольце находятся в тех же положениях, что. и в исходном алкилбензоле или алкилбе золсульфонилхлоридах, а сульфонамидохинолины являются обычно смесями изомеров).
Пример 2. В круглодонную колбу емкостью 5 л, снабженную воздушной мешалкой, термометром, капельной воронкой и ледяной баней помещают 365,7 г (2,54 моля) 8-аминохинолина и 2 л пиридина. Затем туда медленно подают 83§ г (2,54 моля) децилметилбензолсульфонилхлорида, так чтобы температура держалась 9-13°С, (время добавления 45 мин полученного из децилметилбензола. По окончании добавления сульфоннлхлорида, реакционную смесь нагревают до комнатной температуры и перемешивают; 3ч. Затем ее нагревают до 85° С вьщерживают при 80-С 45 мин и доба.вляют 1 л воды. Температуру снова под}|имают до 80° С и выдерживают при Этой температ)фе 30 мин. Смесь переносят в делительную воронку на 6 л и добавляют 1 л воды и 2 л гептана. После выстаивашш в течение ночи фазы разделяют, к водной фазе добавляют 2 л воды, экстрагируют гептаном и экстракт отделяют. Органические фазы соединяют и промывают следущим образом: 3 раза 4%-ным раствором NaHCOa в 25%-ном растворе метанол-во да, 3 раза водной серной кислотой 25 г/л, еше
2 раза раствором МаНСОз, еще 2 раза раствором H2S04 и 1 раз рассолом. Затем раствор сушат над сульфатом натрия, гептпн отгоняют и получают 1066,9 г светло-KopiNневого масла - 8-дешшметилбензолсульфонамидохшголина (90%-ной чистоты) строения
Сн,
10
Пример 3. Повторяют пример 2, но берут 196 мл пиридина, 36,0 г (0,25 моля) 8-аминохинолина и 86,5 г (0,25 моля) деЦилзтилбензолсульфонилхлорида. Этот сульфонилхлорид получен из деидлэтилбензола. Получают 94 г темного масла - 8- (деципзтилбензолсульфонамидо) хинолина формулы
20
СгКз
25
И р и м е р 4. Пример 2 повторяют, но применяют 120 мл пиридина, 23 г (0,16 моля) 8-аминохннолнна и 56 г (0,16 моля) диалкилбензолсульфонш1хлорида. Выход 83%, продукт - телтое масло, имеет формулу
СНз
5
(Jjy (ijjj- аянчп П р и м е р 5. Октилтолуол (50 г, 0,245 моля) мед;1енно добавляют при 5-10° С и перемешивании в течение 0,5 ч (экзотермическая реакция) к 81 г (0,69 моля) хлорсульфоновой кислоты загруженной в круглодонную колбу на 250 мл, снабженную воздушной мешалкой, термометром, капельной ворон кой, обратным холодильником, скруббером и ледяной баней. Реакциошгую смесь перемеигивают 3 ч при 25-30° С, затем она стоит в течение ночи. Ее выливают в 900 г льда, добавляют 500 мл ДНЭ-1ИЛОВОГО эфира и перемешивают до расплавления льда. Органическую фазу промьшают водой, 30%-ным водным Ыа2СОз, снова водой, сушат над. N32804 и растворитель отгоняют. Получают 39 г октилметнлбепзолсульфонилхлорида. При 14-18°С добавляют 20,4 г (0,067. моля) этого продукта к 14,4 г (0,1 моля) 8-амшюхгаюлина в смеси с 14,4 г (0,15 моля) триэтиламина и 25 мл бензола. Смесь перемеишвагот 2 ч при ком11натной температуре, затем нагревают до 80 С 1 ч. К реаквдоиной смеси добавляют 250 мл воды и 250 гептана, фазам дают разделиться в течение ночи. OpraHWiecKjTo фазу промывают так же, как в примере 2, сушат над N82804, отгоняют растворитель и получают 26,7 г темного масла - 8-октилметилбензолсульфонамидохинолина следующей формулы Пример 6. Повторяют пример 2, но применяют 23,04 г (0,16 моля) 8-аминохинолина, 100 мл пиридина и 49,5 г (0,ТЙ моля) нонилметилбензолсульфонилхлорида, который был получен из нонилтолуола с разветвленной ноннльной группой, который в свою очередь был получен из трипропилена. Получают 55,1 г темного масла, представляющего собой 8-нонилметилбензолсульфонимидохинолин следующего строения: CigHij; Пример. Повторяют пример 2, но применяют 20 г (0,139 моля) 8-аминохиноли на, 120 мл пиридина и 50 г (0,139 .моля) Жциййзопр6пилбен:з6лсульфонилхлорида (называется также децилкумолсульфонилхлорид) .Суп фонилхлорид был получен из децилкумола. Получают 50 г темного, вязкого масла.являющегося 8-децилизопропилбензолсульфонамидо хинояинрм следующего строения: Нз(5 .(JHj
П р им е р 8. Диамилбензол сульфонилхлорид получают из даамилбензола, который получают следующим образом. К суспензии 175,2 г (1,29 моля) АЕСРз в 660 мл четыреххлористого углерода добавляют 155,8 г (1,29 моля) хлорангидрида валериановой кислоты с такой скоростью, чтобы не повышать температуру, бани со льтом и солью выше 5°С (время добавления 20 мин). По окончании добавления смесь охлаждают до 0°С и начинают добавлять 159,2 г (1,07 мо692542
- Srnop.tfnn
П р И м е р 9. Повторяют пример 2, но применяют 28,8 г (0,2 моля) 8-аминохинолнна, 150 мл пиридина и 49,5 г (0,2 моля) 4-втор-амилбензолсульфонилхлорвда. Получают 50 г вязкого масла, представляющего собой ля) егор амилбензола (добавление ведут при в течение 3,5 ч). Реакдиокной смеси дают нагреться до 10° С в течение 1 ч, затем ее выливают в смесь льда и HCI и перемешивают в течение ночи. Фазам дают разделиться, водную фазу экстрагируют четыреххлористым углеродом и водную фазу выбрасьшают. Органические фазы соединяют вместе и промывают следующим образом: 2 раза водной 7%-ной HCt 2 раза 10%-ным водным Naj СОз, 1 раз водой и 2 раза рассолом. Продукт сушат над сульфатом натрия, отгоняют растворитель и перегоняют, получая фракщ1и, большей частью представляющие собой «-вгор-валерофенон с примесью орго-изомера. 104,8 г этого продукта смешивают с 86,3 г КОН, 61 мл 98-100%-кого jO и 500 мл диэтиленгликоля и нагревают с обратным холодильником в течение ночи; затем нагревают от 140 до 155°С, отгоняя выделившуюся воду. Реакционную смесь нагревают при 195° С 1 ч при слабом кипячении с обратным холодильником и собирают 50 мл дистиллята; его охлаждают и выливают в 50мл воды и 250 мл растворителя, который состоит в основном из н-гексана с т. кип. 60-71 С. Фазы разделяют, органическую промьгеают 2 раза 10%-ной HCfc сушат над сульфатом натрия, фильтруют и выпаривают до масла. Получают 83,1 г продукта, который перегоняют в вакзоме и по- лучают 49,8 г фракдии (температура в колбе 125-145 С, температура погона 105-ПО С), представляющей собой диамилбензол строения (еНз (СНг)д- ЬН- ((1Нг) J - СН 3 где а / б 2. Полученный диамилбензолсульфонилхлорид (25,5 г, 0,081 моля), 8-аминохинолин (12,4г, 0,085 моля) и 75 мл пиридина подвергают реакции, как в примере 2, и получают 33,5 г 8-диамилбензолсульфонамидохинолина следующего строения: 13 8- (вгор-амилобензол)сульфоиамидохинолин следующего строения: ;нэ (CHijV . ,Сн KfHzJB Tjjft а + в - 2. П р и м е р 10. ДинонилнафтаЛинсульфонилхлорид (125 г, 0,26 моля), .полученньга, как указано выше, растворяют в 150 мл то луола и добавляют к раствору 37 г (0,26 м ля) 8-аминохинолю1а в 100 мл пиридина при температре 10-20° С, при этом наблюдается .небольшое выделение тепла. Реакционную смесь перемешивают при комнатной температ)фе в тече ние ночи, затем ее нагревают до 80° С 30 ми потом добавляют 25 мл концентрированного NHa и перемешивают при 80°С 20 мин. Потом смесь выливают в 500 мл гептана и 300 мл воды, фазы разделяют, органическую фазу промывают 5%-ным NaHCOa (40%-ньш метанол в воде) до полного разделения фаз. Затем органическую фазу промывают серной кислотой 25 г/л до полного разделения фаз. После этих промывок, реакционную смесь нагревают до кипения, обрабатьшают 5 г обесцвечивающего угля, сущат над сульфатом натрия, фильтруют и выпаривают досуха в вакууме: Получают 152,2 г темного масла. Получают 8-динонилнафталинсульфонамидохннолин следующего строения: (J}fti4
Пример 11. Повторяют пример 2, но применяют 46,4 (0,32 моля) 8-аминохинолина, 180 мл пиридина и 82,35 г (0,3 моля) гептилбензолсульфонилхлорида, который был получен из гептилбензола. Получают 112,6 г (выход 97,35%) 8-гептилбензолсульфонамидохинолина следующего строения:
Пример 15. Пример 2 повторяют, но применяют 28,8 г (0,2 моля) 8.-амш охинол1 на, 75 мл пиридгага и 60,4 г (0,2 моля) 2,4,6-триизопропилбензолсуяьфонилхлорида. Получают 71,4 г пурпурно-белого твердого вещества, которое является 8-(2,4,6-триизопропилбензолсульфонамидохинолином следующего строения: 2 на, 50 мл пиридина и 74,9 г (0,2 моля) пентадецилбензолсульфонилхлорщда. Получают 79,1 г 8-пентадец|шбензолсульфонамидохинолина следующего строетшя: .Пример 13. Повторяют пример 2, но применяют 36 г (0,25 моля) 8-аминохинолина, 100 мл пиридина и 100 г (0,25 моля) и-«-гексадецилбензолсульфонилхлорида. Получают 24,6 г (около 2/3 реакционной смеси было потеряно прН размещении в делительной воронке) желтого масла, которое кристаллизовалось. Продукт .представляет собой 8-л-н-гексадецилбензолсульфонамидохинолии следующего строения Н-(,бН П р и м е р 14, Повторяют пример 13/ о применяют 22,68 г (0,167 моля) 8-ами- . охшюлина, 75 мл пиридина и 63 г (0,157 моля) гексадецилбензолсульфониллорида. Получают 49,25 г (выход 62%) зоотистого масла, представлянзщего собой 8гексадецилбенэолсульфонамидохннолйн следущего строения
где а V- в 4.
Пример 12. Повторяют пример 2, HOI применяют 28,8 г (0,2 моля) 8-аминохинолиНзс; енз Сн
СИз
П р и м е р 16. А. Получение 2-метил-8-аминох1шолкна. К охлаждаемому раствору 560 г метабисул фита натрия в 1 л воды добавляют 290 мл гидроокиси аммония. Смесь помещают в реактор из нержавеющей стали на 2 л и добав ляют 200 г 8-оксихинальдина и смеси дают стоять в течение ночи. Реактор закрьгоают и нагревают до 150°С. Затем реакционную смес перемещивают при 150° С 7 ч. за это время давление поднимается до 3,5 атм. Реакщ1онную смесь при охлаждении перемешивают в течение ночи, а затем реакторнагревают до 80 С. При этой температуре содержимое реактора сливают и реактор промывают I л бензола при 70-80° С. Бензольный раствор добавляют к реакционной смеси. Смесь фильтруют и фазы разделяют. Органическую фазу промьюают разбавленным водным раствором NaOH, затем рассолом, сушат над N82804, отгоняют растворитель и получают 84 г сырого продукта. Его перегоняют в вакууме и получают 50 г желтого твердого ве щества - 2-метил-8 -аминохинолина(называет также 8-аминохинальдин). В. Получение 8-додецилбензолсульфонамидо-2-мётилхинолинаПовторяют пример 2, но применяют 79,8 (0,505 моля) 2-метил-8-аминохинолина, полученного по примеру 16А, 100 мл пиридина в смеси с 200 мл толуола и 173,7 г (0,505 моля) додецилбензолсульфонилхлорида. Продуктом реакции является 8-додещшбеизолсульфонамидо-2-метилхинолин следующего строения: б «Has Пример 17. Повторяют пример 1бВ Яо применяют 25 г (0,158 моля) 2-метнл-8-амииохИнолийа,Л25 мл пиридина и 52,3 г (0,158 моля) деципббЛзолсульфонййШбрйй Получают 63,7 г 8-децилметилбензолсульфош амидо-2-мётилхинолина следующего строения Пример JB. А. Получение 8-амино6-метилхинолина. 30 г 8-нитро-6-мётилхинолина растворяют в 30 мл этилацетата,. 50 мл абсолютного эта нола и 50 мл этилового эфира. Раствор делят на две части, и к каждой добавляют по 0,4 rPtOj, обе части гидрируют. Затем их соединяют и перегоняют при температуре в. колбе 110-190°С при 0,45 мм рт. ст. Получают 20,7 высокочистого 8-амино-6-метилхинолина. В. Получение 8-децилметилбензолсульфонамидо-6-мётилхинолина. Повторяют пример 2, но применяют 19,4 г (0,123 моля) 8-амино-6-метш1хинолина, полученного по примеру 18А, 100 мл пиридина и 41,3 г (0,125 моля) децилметилбензол-. сульфонилхлорида. Получают 51,4 г свет.пого масла - 8-децилл1етилбензолсульфонамидо-6-метилхинолина следующего строения С,оН2, Пример 19. Повторяют пример 18В, но применяют 25 г (0,14 моля) 8-амино-6-метоксихинолина, 100 мл пиридина и 47,3 г (0,14 моля) децилметилбензолсульфонилхлорида. Получают, 58,6 г темного масла - 8децилметилбензолсульфониламидо-6-метоксихинолина следующего строения: (io% П р и м е р 20. А. Получение 8-амино-5-нитрохинолина. ; В круглодонную колбу на 5 л снабженную воздущной мещанкой, холодильником, капельной воронкой, термометром и баней с горячей водой, загружают 40 г (0,23 моля) 5-нитрохинолина в 100 г (1,44 моля) хлоргидрата гидроксиламина. Затем добавляют 1950 мл 95%-ного этилового спирта, твердое вещество растворяется, после чего добавляют 200 г КОН в 1200.мл метилового спирта в течение 50 мин при 54-57° С. Смесь перемешивают при 55° С еще 1 ч, затем ее выливают в. 10 л водь1, дают охладиться и фильтруют. Из 95%-ного этанола вьпфисталлизовьша ется оранжевый осадок 8-a,шю-5-нитрохинолина..;. . , В. Получение В-децииметилбензолсульфонамидо-5-ш1трохинолина. Повторяют пример 2, ио применяют 18,9 г (0,1 моля) 8-амшю-5-нитрохинолина, 60 мл пиридина и 36 г (0,1 г) децилметилбензолсульфршшхлорцда. Дополнительно реакциошо смесь нагревают при 80-85°С в течение 22- 24 ч, в отличие от более кратковременного |нагрсвания в примере 2. Получают 13 г тем ного масла, представляющего собой 8-децил метилбензолсульфонамвдо-5-нитрохинопина формулы П ff И м е р 21. А. Получение 8-амино5,7-дихлорхинолина Через раствор 10 г 8-аминохинолина в 50 мл ледяной уксусной кислоты барботируют газообразный хлор при охлаждении при 40-50°С. Ток хлора прекращают по окончании экзотермической реакции (всего подают 16,5 г хлора). Тфасный осадок отфильтровывают и суспендируют в 100 мл водного 2%-ногр МаОН и 300 мл этилового эфира. Смесь, фильтруют и разделяют фазы. Эфирную фазу промьшают рассолом, сушат над сульфатом натрия, фильтруют, выпаривают досуха и получают 5,6 г сырого продукта, который затем перекристаллизовывают -из смеси эфир-гептан. Получают 5,1 г коричневых игл (т.пл. 121-123°С), представляющзих собой 8-амино-5,7-дихлорхинолин. В. Получение 8-децш1метилбензолсульфонамидо-5,7-дихлорхинолина. Повторяют пример 20В, но применяют 7,2 г (0,034 моля) 8-амино-5,7-дихлорхиноли на,полученного по примеру 21 А, 25 мл пиридина и 11,9 г (0,036 моля) дещшметилбен золсульфошшхлорида. Получают 12,2 г красноватого масла - 8-децилметилбензолсульфон амидо-5,7тдихлорхинолина следующего строения/(iloM2) П р и м е р 22. А. Получение «-додецил фенилметансульфонилхлорида. Смесь 147 г (0,5 моля) додецилбензил- хлорида (с разветвленной додецильной группой твердого алкилатного типа) 79 г (0,5 мо ля) безводного тиосульфата натрия, 250 мл метанола и 250 мл дистиллированной водьг нагревают с обратным холодильником 3 ч при перемешивании. Летучие (около 75 мл) отгоняют при давлении водоструйного насоса до интенсивного вспенивания. Реакшюнную смесь переносят в колбу на 2 л, снабженную конденсатором с сухим льдом, термометром, механической мешалкой и газораспределительной трубкой. Колбу охлаждают до 0°С на ледяной бане и добавляют 250 мл ледяной уксусной кислоты и 500 г льда. Барботируют газообразный хлор с минимальной скоростью для поддержания минимального количества Cgj в колбе. Температуру держат 10° С и меньше ( барботируют 1 ч). Затем добавляют 500 мл гептана, реакгшонную смесь перемешивают и разделяют фазы. Органическую фазу промывают. 500 мл 50%-ного водного раствора NaHCOa, затем рассолом, сушат над сульфатом натрия, выпаривают и получают золотистое масло. Этот продукт частично очищают молекулярной перегонкой и получают «-додецилфеннлметансульфонилхлорвд (чистота ОКОЛО 50%). в. Получение 8-додецилфеннлметансульфонамидох1гаолш(а. Сырой сульфонипххлорид, полученный по примеру 22А, добавляют непосредственно к перемеипгоаемому раствору 8-амшгохга олина (0,064 моля) и тризтиламкна (0,07 моля) в 25 мл 1,1,2-трихлорэтана при-5-10 С, затем смеси дают нагреться до комнатной температуры. После перемешшания при комнатной температзфе в течение 2 ч реакционную смесь нагревают до 60° С и выливают смесь в 200 мл воды и 300 мл гептана. После встряхивания фазы разделяют, оргшгичсскую фазу промьшают три раза по 100 мл 5%-ной NaHCOa в 30%-ном водном растворе метанола, три раза по 100 мл серной кислоты 25 г/л,снова раствором бикарбогшта натрия, 5атем рассолом, затем ее сушат над сульфатом натрия и вьтар1шают досуха в вакууме. Полученное красноватое масло (50,4 г,. 30 - 50% сульфонамида по ИК-спектру) очищают молекулярной перегонкой и хроматографией на силикагеле и пол}чаюг вязкое масло (8,2г,75% сульфонамида) .Формула этрго .ешш... (JH2 Пример 23. Повторяют пример 2, но применяют 22,1 г (0,154 моля) 8-аминохинолина, 75 мл пирид1ша и 50 г (0,154 моля) гексадекансульфоннлхлорида. Получают 60,7 г .8-н-гексадекансульфонамидохинолина слеующего строения: Ю1$б2-((111г1,
19 Пример 24. A. Получение 2-этилгексан-1-сульфо1гилхяорида. Смесь 57,9 г (0,3 моля) 2-этилгексил-1-бромида, 22,8 г (0,3 моля) тиомочевт1ы и 75 мл абсолютного этанола перемеигивают с обратным холодильником 20 ч. После охлаждения 1з течение ночи этанол отгоняют в вакууме и получают белое воскообразное вещество: lero pacTBopmoT в 250 мл) воды при 80°С и добавляют 40% NaOH в воде до прекращения помутнения. Маслянистый продукт отделяют и растворяют в 75 мл уксусной кислоты и 25 мл воды. Раствор охлаждают до о С и барботируют Ctj до окончания реакции окисления (всего расходуют 80,2 г CBj). Полученное бесцветное масло является 2-этилгексан- 1-сульфонилхлоридом. В. Получение 8-2-этилгексансульфонамидо-. хинолина., Повторяют пример 2, но применяют 43,2 г (0,3 моля) 8-амин6хшюл1ша, 200 мл ттириДина и весь сульфонилхлорид, полученный в ча ти А этого примера. Получают 20,2 г 8-2-этилгексансульфонамидохинолина следующего строения: |Нз . . Нг }}Н Ог- Н2-Сн-(йНг)з-СНз П р и м е р 25. А. Получение изодецилб МИДа. 196 г (0,72 моля) РВгз медленно добавляют при перемешивании к 316 г (2,0 м ля) изодеканола (смесь изомерных спиртов 10 углеродными атомами) при температуре ниже о С. По окончании добавления РВгз реакционной смеси дают нагреться до комнат ной температуры при перемешивании, затем ее держат в течение ночи с осушительной трубкой. Сьфой продукт перегоняют при 60-65° С/0,45 мм рт. ст., два раза промьшают холодной H2SO4 (уд вес. 1,84), два раза 50%-ной смесью метанола и аммиака и один раз рассолом, затем сушат над СаС. Продукт еще раз перегоняют и получают 244,1 г фракции изодецилбромида, полученной при температуре в колбе 70° С, давлении 0,45 мм рт.-ст. и температуре в погоне 48°С. ;- - - :;--:;-- - ---В. Получение изодекансульфонилхлорида. Смесь ПО г (0,5 моля) изодецилбромида полученного а разделе А этого примера, 38 г (0,5 моля) тир мочевины и 250 мп 95,%-ного этанола нагревают с обратным хол дильником 8 ч, затем реакционную смесь ох лаждают и перемешивают в течение 1 сут. Отгоняют 125 мл этанола и добавляют раствор. NaOH (30 г) в 200 мл воды. Затем
692542
20 еакционную смесь нагревают с обратным хоодильником 3 ч, вьшивают в 300 мл воды экстрагируют 200 мл диэтилового эфира. кстракт сушат над сульфатом натрия, фильтруют и выпаривают до появления розоатого масла, которое растворяют в 250 мл едяной уксусной кислоты, добавляют 50 мл воды, охлаждают до 0°С и начинают пропускание газообразного хлора, которое ведут при ОС очень медленно во избежание изишнего вьоделения тепла. Хлор добавляют в течение 1 ч, всего расходуют 137 г хлора. Избыток хлора удаляют продувкой газообразного азота в раствор NaHSOj. Реакци- онную смесь выливают в 500 мл воды и экстрагируют гексаном; экстракт два раза промывают 5%-ным водным раствором NaHSpj и один раз рассолом, сушат над . сульфатом натрия, фильтруют и выпаривают в вакууме до образования белого масла. Ползд1ают 107 г изодекансульфонилхлорида. С. Полу1енне 8-(иэодекансульфонамидо)хинолина. Повторяют пример 2, но применяют 43,2 г (0,3 моля) 8-амйнохинолина, 200 мл пиридина и 72 г (0,3 моля) изодекансульфонилхлорида, полученного по методике примера 25В. Получают 92,0 г 8-чзодекансульфонш1амидохинолина след)тощего строения - iHz tlHzi J8Hi7) где изодецильная группа установлена по ЯМР-спектру и группа представляет собой смесь алкильных групп с разветвленной цепью. П р и м е р 26. А. Получение алкенютсульфонхлорида. Смесь 84,3 г (0,405 моля) РС8 и 96,7 г (0,324 моля) С|4-1б-злкенилсульфоната загружают в трехгорлую круглодонную колбу на 500 мл, снабженную холодильником и механической мешалкой. Смесь нагревают паром 2 ч при перемешивании. Вначале реакция протекает рчеяь бурно, экзотермически. После добавления 50 мл гептана реакционную смесь перегоняют в вакууме водоструйного насоса при обогреве паром. Остаток растворяют в 300 мл гептана и полученный раствор фильтруют. Затем его вьшаривают в вакууме до образования масла (62,5 г), которое применяют в части В этого примера. В. Получение 8-(С,4-1б-алкенилсульфонамидо)-хинолина. Сульфоннлхлорид из А этого примера мед;ленно добавляют к перемешиваемому раство2 py 30,6 г (0,212 моля) 8-аминохинолииа в 100 мл пиридина при 10-20° С. Реакционную смесь перемешивают в течение ночи при ком натной температуре, затем ее нагревают до 80° С, добавляют 200 мл воды и через 30 мин добавляют 28%-ный раствор водного аммиака. Смесь выливают в 300 мл воды и 500 ма гептана. Фазы разделяют, органическую фазу промывают метанольным рас вором бикарбоната натрия, затем серной ки лотой (25 г/л). В результате кислотной про мьшки образуется эмульсия, которой дают расслоиться в течение I сут. Органическую фазу промывают метанольным раствором би карбоната натрия до получения хорошо раз-, деляемых фаз. Затем органическую фазу сушат над безводным сульфатом натрия, филь руют, обрабатывают 5 г норита, фильтруют, выпаривают до масла, которое пропускают через колонку с силикагелем (100 г) с помощью гептана (1 л). Гептан выпаривают в вакууме и получают 41,8 г масла, которое очищают молекулярной перегонкой. Во времл перегонки наблюдается небольшое разлож ние. Получают 10,1 г масла, в котором по ИК- и ЯМР-спектрам обнаруживают 60-65% сульфонамида. Сульфонамидохинолиновая активная часть продукта имеет строение R где R - С14Мб-алкенильная группа. П р и м ё р 27. Повторяют пример 2, но применяют 21,6 г (0,15 моля) 8-аминох1шолина, 100 мл пиридина и 31,8 г (0,15 моля) к-октансульфонилхлорида.. Получают 39,2 г желтого масля, представляющего собой 8-(н-октансульфонамидо)хинолин .следующего строения: ( П р и м е р 28. Повторяют пример 2, но применяют 45 г (0,26 моля) сьфого н-пентансульфонилхлорвда, 37,4 г (0,26 моля) 8-аминохинолина и 150 мл пиридина. Получают 35,6 г 8- (пентансульфонамидо)-хинолина следующего строения: l H Oz-ltHzj/ C-Hj Далее приведены примеры методик применения сульфонамидохинолшгов для извле2че1шя металлов из их водных растворов по изобретению. Если нет особых указаний, то извлечение ведут по методикам 1-4. Методика 1. Сначала готовят 0,1 Л/ раствор сульфонамидохинолина в указанном растворителе, не смешивающемся с водой. Применяют пять водных растворов следующего состава: Си++ 0,05 М CuS04 (3,2 г/л Си++), 0,4 Л/ NHj и 0,1 М (NN4)2804 Ni++ 0,05 М NiS04 (2,9 г/л Ni++), 0,4 М NHj и 0,1 Л/ (NN4)2804 Zn++ 0,05 Af ZnS04 (3,2 г/л Zn++), 0,4 М NHs и 0,1 Л/ (NN4)2804 to++ 0,025 М CoS04 (1,5 г/л Со++), 1,7 М NHj и 0,1 М (NN4)2804 (полуген nf)H необходимости п атмосфере азота), Со+++ 0,025 Л/ CoSO4 (1,5 г/л Со++), 1,7 ЛГ NNa и 0,1 .W (NN4)2003 (окислено воздухом до Со+++). Порции органического раствора взба гтывают с различными водными растворами при ;отношении фаз органическая .водная 1:1 в течение 1 ч при комнатной температуре; затем орган1{ческую фазу анализируют на содержание металла. Если имеется .третья фаза, то органическую и подводную фазы осветляют и анализируют. Методика. 2. Целью этой методики являет.ся определение степени извлечения различных ионов металлов как функции рН 1-6. Как и в методике 1, готовят 0,1 М раствор суль({юнамидохшюлшга в задашюм не смспшвающемся с водой органическом растворителе. Полученный раствор смешивают с вощгым раствором в отношении 1:1 при встряхивании в течение 1 при комнатной температуре. Водную фазу составляют из равных объемов двух компонентов: компонент А -- 0,2 М раствор сульфата металла в воде, компонент В - вода, или водный раствор серной кислоты или едкого натра 0,005-0,5 М. Производят , несколько раз извлечения при разных значениях рН. В первой экстракции в качестве компонента В берут воду. После определения раффинатного рН выбирают компонент В, так чтобы рафф}шатное значение рН было 1-6 (целые числа). Анализируя орга гическую фазу на извлечение металла и водную фазу на рН, получают данные о степени извлечения металла как функцию рН для дагпгой исследуемой системы. . Методика 3. Целью этой методики является определение степегш извлечение разных ионов металлов как функции суммарной концентращо аммиака в водной фазе. Органические растворы сульфонамидохинолина готовят так же как в предьщущей методи(Kej контактирование производят при встря-.. хивании при отношении органической и водной фаз 1:1 в течение 1 при комнатной
температуре. Концентрации водных растворов сульфатов металлов, аммиака и сульфата аммония приведены в табл. 1.
Таблица














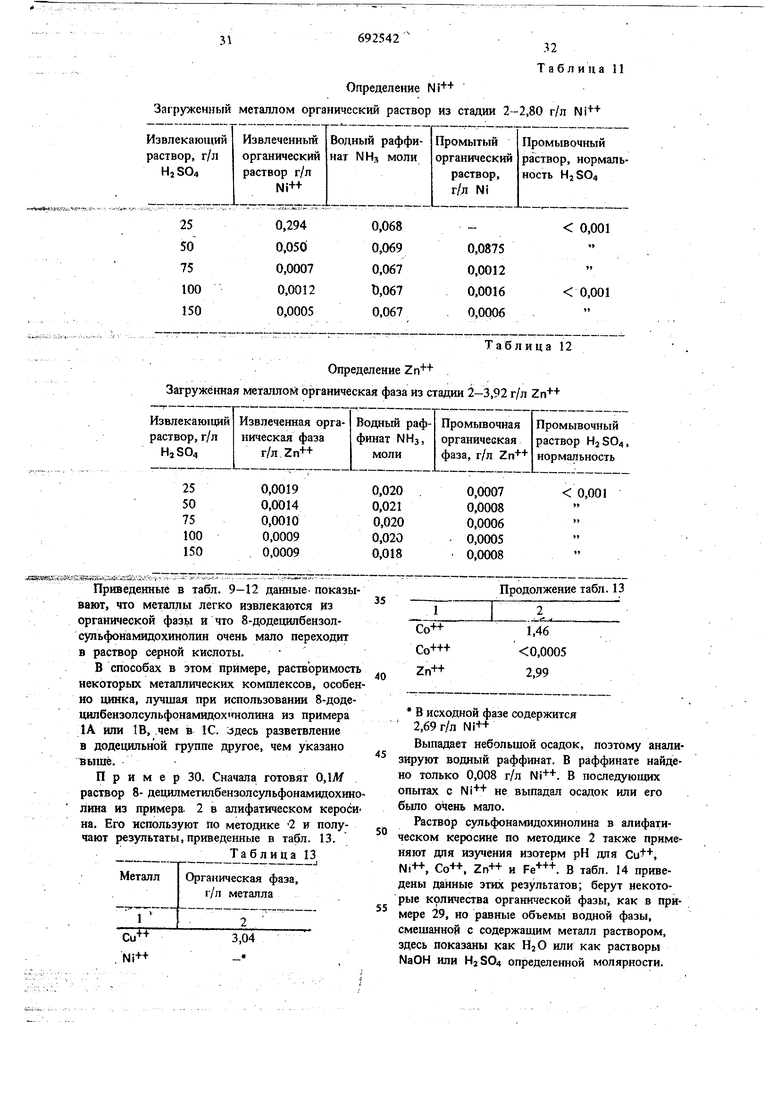
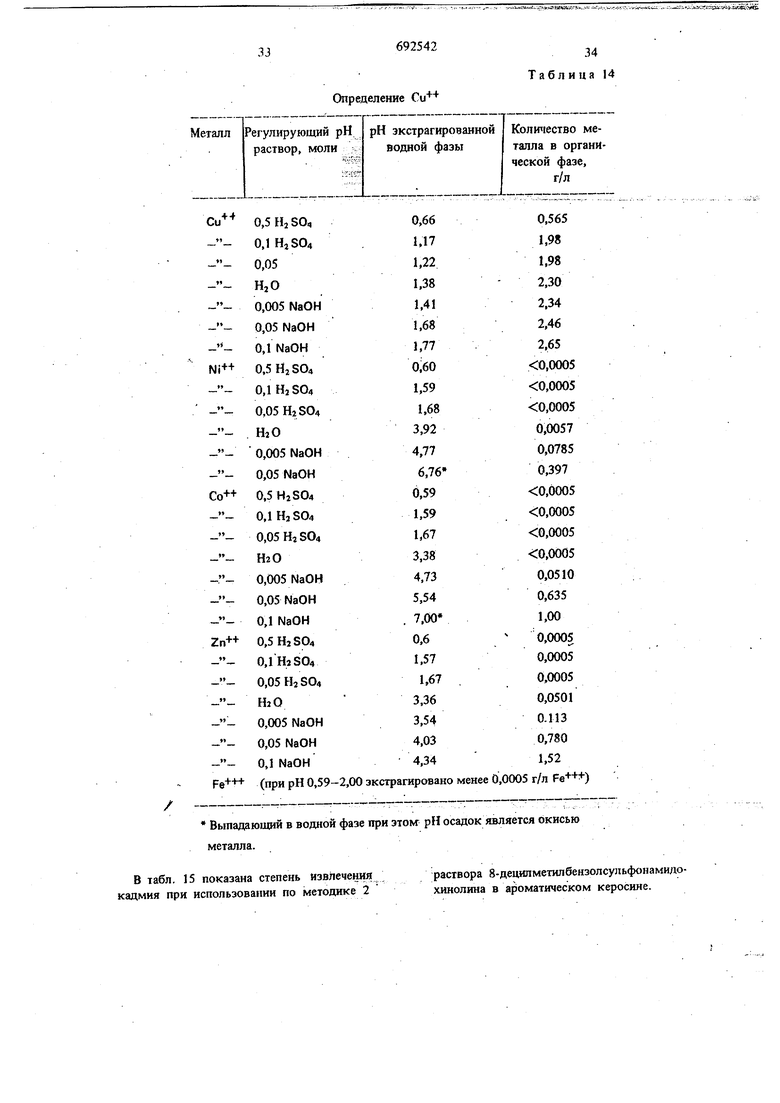
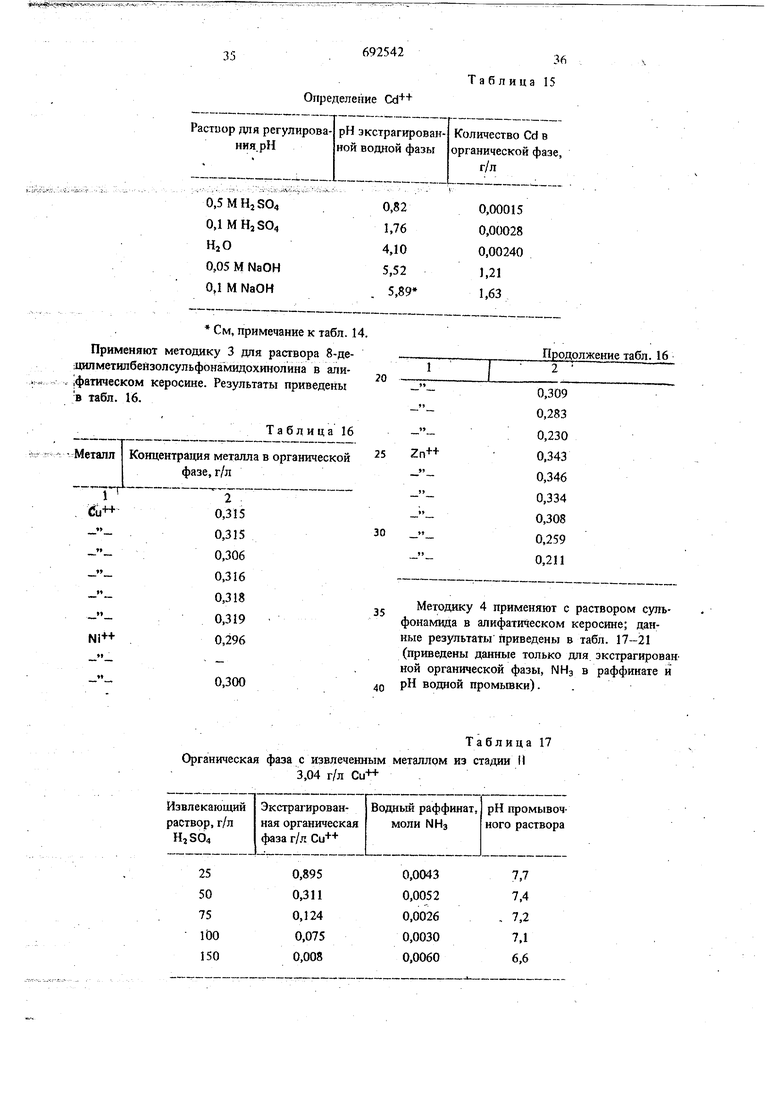
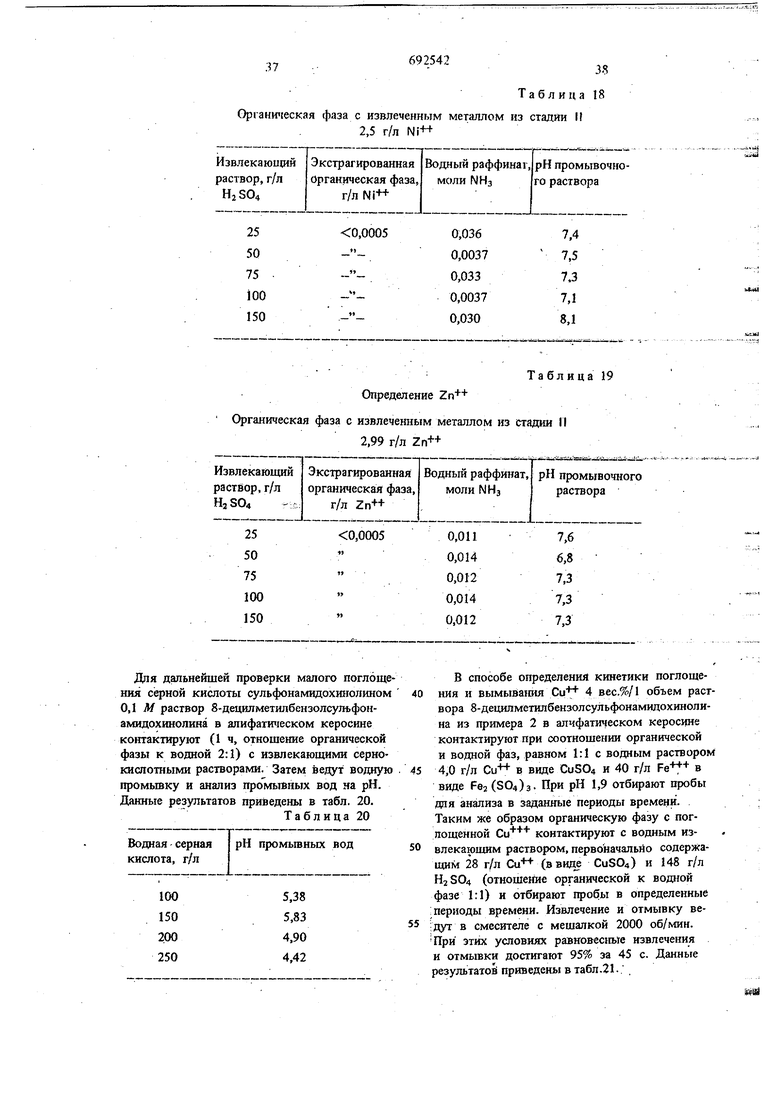











Разделенные органическую и водную фазы анализируют на содержание металла. Таким образом определяют влияние увеличения : концентрации аммиака на стеиень извлечения металла.
Методика 4. Целью методики являются: 1) определение степени извлечение металла из органической фазы в зависимости от когщентрации кислоты; 2) определение степени захвата аммиака во время извлечения и 3) определение степени захвата кислоты при извлечении из органической фазы. Органические растворы готовят так же, как и в ранее приведённых методиках. Кроме того, готовят следующие водные растворы:
A.0,1 М сульфат металла, 0,6 Л/МНэ и 0,15 Af(NH4)2S04.
B.Пять растворов, содержащих 25, 50, 75, 100 и 150 г/л серной кисЛоты, соответственно..
В первой стадии сульфонамидохинолиновый раствор контактируют с водным раствором А при отношении органической к водной фазе 1:2 при встряхивании-в течение 2 ч при комнатной температуре. Фазы разделяют и органическую фазу снова контактируют со свежим водным раствором А, как указано вьпие. Полученную отделенную органическую фа:зу анализируют на концентрацию метаял а. Ее делят на 5 порций, каждую из KOTOpbix встряхивают с одним из пяти водных растворов В (отношение органической к водной фазе 1:1, время контакта 1 ч). Фазы разделяют и в Органической фазе определяют содержание металла, а в водной фазе содержание МНз. Отмытую органическую фазу промывают водой при отношении .органической к водной фазе 1:1 (время
контакта 1 ч). Водный промывной раствор анализирую на содержание H2SO4.
П р и м е р 29. Сначала готовят 0,1 Л/ раствор 8-додецш1бензолсульфона1Сяодохинолипга из примера 1В в ароматическом керосине. . Его применяют по методике 1 и получают следуюицие результаты:
.
Металл
Содержание металла в органической фазе,
г/л
0,0170 3,06 . 2,75 3,87
При работе с р створом такого сульфонамидохинолина в алифатическом керосине
получают хорошее извлечение (1,35 г/л), но образуются ocaдкйV в случае Си и , {. в случае Zn органическая фаза загущается. Таким обраЬом, для зтого сульфонамидозсинолина более ароматический растворитель дает лучшие результаты.
По методике 2 для изучения изотерм рН для Си++, Zn++, Со++, Fe+++ и Ni++ применяют раствор 8-додецш1беизолсульфоиамидохинолина в ароматическом керосине. РезультаТЬ1 приведены и табл. 2-6, Во всех случаях применяют 10 МП сульфонамидохинолинового раствора и 5 мл 0,2 ЛГ раствора сульфата металла, при различных количествах воды и раствора NaOH и/или H2S04 (мл).
Маблюдается выпадение слабого осадка
27
Определение Со
о о
О 5 О О
о о о
5 О
23О
о о о
о о о о
1 о о о
3
1
Наблюдается вьшадение слабого осадка
Определение
Наблюдается вьшадение осадка.
Определение Ni
692542
28 Таблица 4
3,48
0,0028
1,29 0,0002
7,25 2,72
5,41 0,860
4,94 0,275
1,70 0,0008 0,0002.
2,18
Таблица 5
Таблица 6
29
Из данных приведенных в табл. 2--б видна степень извлечения Си , , Zn и Ni и незначительная степень извлечения , селективность по Си по сравнению с при сравнительно малых значениях
рН.
Методику 3 также применяют с раствором 8-додецилбензолсульфонамидохинолина в ароматическом керосине. В табл. 7-9 приведены данные этих результатов.
Таблица 7 Определение Си
Концентрация металла, г/л
Таблица 8
Определение Концентрация металла, г/л Органическая фаза из стадии 2-2,99 г/л Си
1,15 0,563 0,267 0,144 S0,05
. 30
Продолжение табл. 8
Определение N i Концентрация металл,а г/л
Т а б л и ц а 9 Определение
Концентрация металла г/л
Водный раффинат
Органическая фаза
0,0005
0,456 0,0005 0,445 0,448 0,0029 0,0150 0,424 0,0346 0,414 0,379 0,0710
Методику 4 также применяли для раствора ;8-додецилбензОлсульфоиамидохинолина в ароматическом керосине для Си++, N1++ и . Данные / результатов, приведены в табл. 10-12.
Таблица 10
Определение Си
0,001
1,15
0,560
0,253
0,143
0,0580 Загруженный металлом органический раствор из стадии
,.-,v.
Определение Zn Загружё1тая металлом органическая фаза из стадии 2-3,92 г/л
aiSfcS.V.VS rii- tvS-Ii...i/.i.-:-:.
Приведенные в табл. 9-12 данные-показывают, что металлы легко извлекаются из органической фазы и что В-додецилбензолсульфонамидохинолин очень мало переходит в раствор серной кислоты.
В способах в зтом примере, растворимость некоторых металлических комплексов, особенно цинка, лучшая при использовании 8-додецилбензолсульфрнамидохтнолина из примера 1А или IB, .чем в 1C. адесь разветвление в додецильной группе другое, чем указано выше.
П р и м е р 30. Сначала готовят 0,Ш раствор 8- децилметилбензолсульфонамидохино лина из примера- 2 в алифатическом керосина. Его используют По методике 2 и получают результаты, приведенные в табл. 13.
Таблица 13 Определение
Таблица 12
Продолжение табл. 13
1
1,46
0,0005 2,99
В исходной фазе содержится 2,69 г/л Ni-HВыпадает небольшой осадок, поэтому анализируют водный раффинат. В раффинате найдено только 0,008 г/л . В последующих опытах с N не выпадал осадок или его было очень мало.
Раствор сульфонамидохинолина в алифатическом керосине по методике 2 также применяют для изучения изотерм рН для Си, , Со-«-+, Zrt++ и . В табл. 14 приведены данные этих результатов; берут некоторые количества органической фазы, как в примере 29, но равные объемы водной фазы, смешанной с содержащим металл раствором, здесь показаны как или как растворы NaOH или HjSO4 определенной молярности. Таблица 11 2-2,80 г/л Ni
33
/
Выпадающий в водной фазе при этом рН осадок является окисью металла.
В табл. 15 показана степень извлечения кадмия при использовании по методике 2
692542
34 Таблица 14
Определение
раствора 8-децилметилбензолсульфонамидохинолина в ароматическом керосине.
35 Определение PacTUop для регулирова- рН экс ния.рНной во .,лг ..:.: : .,,...,.,. ,.ч 0,5MH2S04 0,lMHjS04 HjO 0,05MNaOH 0,1 MNaOH CM, примечание к табл. 14. Применяют методику 3 для раствора 8-де;цилметш1бейзолсульфонамидохинолина в али,фатическом керосине. Результаты приведены в табл. 16. „„-„JAP Металл Концентршшя металла в органической фазе, г/л . ,315 ,315 0,306 ,316 - -0318 ,319 iyjj+ц-0296 , ,300
Органическая фаза с извлеченным металлом из стадии 11 3,04 г/л Си++
Экстршированная органическая фаза Г/.1 Си++
0,895 0,311 0,124 0,075 0,008
692542
36
Таблица 15
Таблица 17
рН промывочВодный раффинат, моли ЫНз ного раствора
7,7 7,4 7,2 7,1 6,6 трагирован- Количество Cd в ной фазы органической фазе, - - . 0,820,00015 1,760,00028 4,100,00240 5,521,21 5,,63 - -Продолжение табл. 16 12 0,283 0,230 0,343 ,346 ,, ,308 30 ,211 -. ,, Методику 4 применяют с раствором сульфонамида в алифатическом керосине; результаты приведены в табл. 17-21 (приведены данные только для. экстрагирован ной органической фазы, NHa в раффикате и 40 Р водной промьюки).
я фаза с извлеченнглм металлом из стадии И
Экстрагированная Водный раффинаг, рН промывочноОрганическая фаза, моли МНзго раствора
r/nW
0,00050,0367.4
,0037 7,5
,0337,3
,00377,1
,0308,1
фаза с извлеченным металлом из стадии II
Экстрагированная Водный раффинат, рН нромывочного органическая фаза, моли МНзраствора
г/л ,
0,00050,0117,6
0,0146,8
.0,0127,3
0,0147,3
0,0127,3
Для дальнейшей проверки малого поглощения С(3рной кислоты сульфонамидохююлшюм 0,1 М раствор 8-децилметилбензолсульфонамидохинолина в алифатшюском керосине контактируют (1ч, отношение органической фазы к водной 2:1) с извлекающими сернокислотными растворами. Затем ведут водную промьтку и анализ промывных вод на рН. Данные результатов приведены в табл. 20,
Таблица 20
Водная серная
рН промьгоных вод кислота, г/л
5,38 5,83 4,90 4,42
692542
3 Таблица 18 2,5 г/л Ni-t- Таблица 19 Определение Zn
2,99 г/л Zn++
В способе определения кинетики поглощения и вымывагом 4 вес.%/1 объем раствора 8-децилметШ1бензолсульфонамидохинолина из примера 2 в алифатическом керосине контактируют при соотношении органической и водной фаз, равном 1:1 с водным раствором
4,0 г/л в виде CuS04 и 40 г/л Fe++ в виде Рв2 (304)3. При рН 1,9 отбирают пробы дня анализа в заданные периоды времениТаким же образом органическую фазу с поглощенной контактируют с водным извлекающим раствором, первоначально содержащим 28 г/л ( CuSO4) и 148 г/л H2SO4 (отношение органической к водной фазе 1:1) и отбирают пробы в определенные ; периоды времени. Извлечение и отмывку ве: дут в смесителе с мешалкой 2000 об/мин. При зтих условиях равновес1Ш1е извлечения и отмывки достигают 95% за 45 с. Данные результатов приведены в табл.21. . Предел определяемости Fe легко выделяется электролизом из водного отмывочног раствора с чистотой 99 П р н м е р 31. Повторяют методику 1 для извлечения и , но применяют 8-децилэтш1бензолсульфонамвдохинолш1 из пp мера 3 в растворе в алифатическом керосине с концентрациями 5, 10 и 15 вес.%/объем Эти растворы контактируют с водными металлсодержаищми растворами два раза по 15-20 мин, каждый раз для обеспечения максимального извлечения. Данные результатов приведены в табл. 22. Таблица 22 Концентрация рецКоличество металла гента, вес.% в органической фазе, 1$%-ную органическую фазу с извлёченньш цинком один раз промьшают при соотношении органической и водной фаз 1:1 в течение 15 мин 1 Л/ раствором (NN4)2564. рН водного промывочного раствора изменяется от 5,7 до 8,1, содержание в ней 0,193 г/л. Затем органическую фазу Контактируют с водным раствором 112864 100 г/л для извлечения из Нее цинка. В извлеченной органической фазе содержание Zn 0,0005 г/л, в водном извлекающем растворе содержание NHj 0,026 молей. Соответствующий 8-децилэтилсульфонамидохинолрн, полученный из децилэтилбензола при
Таблица 21 алкипированки, выполненном при О-5° С (См, табл. 2), дает комплекс , вызывающий желатинирование в случае алифатического керосина, но он хорошо растворяется в ароматическом керосине. П р и м е р 32. Повторяют пример 31, но применяют йульфонамидохинолин из примера 4. Полученный комплекс Си вызывает желатинизацию керосинового раствора. Комплекс дает мутную органическую фазу, в ней содержится 3,4 и 7,05 г/л. Zn при концентрациях 5 и 10 вес.%/объем, соответственно. Реагент и его комплексы с растворяют в ароматическом керосине; по методике 1 получают отделенную органическую фазу с 3,06 г/л без осадка. П р и м е р 33. Частич1го повторяют пример 3, но применяют 8-октилметш1бензолсульфонамидохинолин из примера 5. При 15 вес.%, /объем в Ароматике 150 реагент максимально извлек 9,8 г/л и 10,3 г/л . В. алифатическом керосине во время извлечения, образуются хлопья, что указывает на неполное растворение мет;аплического комплекса. Пример 34. Применяют методику 1 с 8-нонш1метилбензолсульфонамидохинолшюм Из примера 6 в ароматическом керосине. Получают результаты, приведенные в табл. 23. Количество металла в органической фазе, 2,61. NiHСо- 1,76 ,09 . При анализе возникают затруднения с эмульсией, ПОЭТОМУ псюбы перед анализом фазы центрифугируют.
41
П р и м е р 35. В 10 вес.%/объем растворе 8-децилизопропилбензолсульфонамидохинолин из примера 7 в алифатическом керосине максимально извлекаю: Си, как в примере 31. В органической фазе окаэываетП р и м е р 36. Методику 1 выполняют с 8-диамилбензолсульфонамидохиноли-Металл
Си++
Ni++
Co++t
Со++
Zn++ .Методику 2 также нением раствора в аро при смешении 0,2 М
692542
42
ся 6,25 г/л Си. Применяют также методику 2 с применением 0,1 М раствора сульфонамидохинолина в алифатическом керосине. 0,2 М водный раствор CuS04 смешивают с регулирующим рН раствором,как указано в табл. 24.
Таблица 24
ном из примера 8,растворенным в ароматическом керосине .Результаты приведены в табл. 25.
20
Таблица 25
Количество металла в органической фазе, г/л
2,90
2,47
0,0090
1,74
3,31
Таблица 26 Если 5%-ный раствор сульфонамидохинолина в Ароматике 150 дважды контактируют с водным раствором Си++, то в органической фазе содержится 3,62 г/л Си-Н-, но вьшадает небольшой осадок. Осадок также вы. падает если вместо ароматического, применяют алифатический керосин. Если в качестве растворителя в методике 1 применяют бензол, то при однократном контакте с раствором Си++ в выделенной органической фазе содержится 2,99 г/л Си++без осадка. существляют .с приме- с регулирующим тическом керосине j одного раствора CuSO4; рН раствором, приведены в |НЫе результатов табл. 26. 43 , Пример 37. Применяют методику Т с 0,1W раствором 8-вгор-амйЛбензолсульфонамидохйнолина из примера 9 в бензоле и содержа1вдм Си водный раствор. В полученной органической фазе содержание 3,58 г/л. При повторении методики 1 с соответствующим раствором сульфонамкдохинолина из примера 9 в ароматическом ке pocfffle образуется осадок и осадок (в отфильтрованной органической фазе содержится 1,01 г/л и 0,396 г/л , соответственно), а в случае Zn образуется эмульсия (в органической фазе 0,367 г/л , не дает осадка, когда в органической фазе было 1,70 г/л Со++. П р и м е р 38. Повторяют методику с применением 8-(динонилнафталинсульфонамидо) хинолина из примера 10, растворенПример 39. Применяют. меюдику 1 0,1 Л/ раствором 8- (гептилбензблсульфонмидо) хинолином из примера 11 в бензоле с содержащим водным раствором. В полученной органической фазе содержится 3,28 г/л Си+ при вьшадвнии слабого осадка, образование которого можно отнести за счет примесей. При повторении с 0,1 ЛГ раствором сульфонамидохинолина из примера И в ароматическом керосине (два контакта с содержащим Си++ водным раствором) выпадает небольшое количество гранулированного осадка из органической фазы при стоянии в течение ночи, в органической фазе содержится 1,66 г/л Си++.
Пример 40. Применяют методику 1 с 8- (пентадецилбензолсульфонамидо) хинолином из примера 12, растворенным в ароматиfliecKOM кёросше. рёзуль веденные в табл. 29..
Т а б л и ц а 29
Количество металла в
40 Металл органической фазе,
г/л
в других опьггах, проводимых по методике 1, с водным раствором Си++ образуется осадок, если реагент из примера 12 растворяют в алифатическом керосине при 5 вес.%/ /объем. Но если применяют растворы 10 вес. /объем в смесях 50:50 или 75:25 по объему алифатического и ароматического керосина, то осадка не образуется и органическая и него в алифатическом керосине при концентрации 0,1 М. Получают результаты, данные которых приведены в табл. 27; ,,... Металл Количество металла в органической фазе, г/л ---- По методике 2 применяют 0,15 Л/ раствор реагента из примера 10 в алифатическом кеpociffle с 0,2 М водным раствором CuS04 в смеси с регулирующим рН раствором, как указано в табл. 28.- «водная фаза хорошо разделяются после контактирования дая извлечения.
Как и в предыдущих примерах, методику 2 осуществляют с применением 0,1 М раствоП р и м е р 41. 8-(гексадецилбензолс 71ьфонамидо)хинолин из примера 13 растворяют в бензоле с концентрацией 0,1 Л/ и контактируют с раствором, содержащим , по методике 1. В отделеннойОрганической фазе содержится 2,1 г/л , при извлечении образуется небольшой осадок.
П р и м е р 42. 8-(гексадецилбензолсульфонамидо)хинолин из примера 14 растворяют в ароматическом керосине в количестве 15 вес.% на 1 объем и контакт фу1от с водным раствором Си и Zrf по методике1. В органических фазах содержится 10,3 г/л Си--- и 8,8 г/л .
П р и м е р 43. Повторяют методику 1, но применяют 8-(триизонропилбензолсульфонамидо) хинолин из примера 15 в ароматическом керосине. Данные результатов приведены в табл. 31.
Таблица 31
Металл I Количество металла в органической фазе, г/л Выпадает небольшой осадок П р и м е р 44. Получают 5%-ньш раствор 8- (додедилбензолсульфонамидо) -2-метилхинолина из примера 16 в ароматическом керосине. Его применяют в методике 1 цпя водных pacTBopoJB и Zn-. В полученном растворе комплекса Си содержи-сся 3,03 г/л;
ра 8- (пентадецилбензолсульфоиамидо) хинолина в ароматическом керосине; данные результатов опытов приведены в табл. 30.
Таблица 30
Си- -; он имеет яркое сине-зеленое окрашивание (небольшой осадок удаляют фильтрованием). При извлечении цинка образуется обильный осадок, который растворяют при добавлении равного количества бензола. Реагент как таковой не растворяется в алифатическом керосине.
П р и м е р 45. Применяют методику 1 дня 8-(децилметилбензолсульфонамидо)-2-метилхинолина из примера 17 как в алифатическом, так и в ароматическом керосине. Данные результатов приведены в табл. 32.
Т а б л и ц а 32
35 Растворитель и металл
Количество металла в органической фазе,
г/л
40 55 Пример 45а.. Методику 2 осуществляют с раствором 8- (дедилмегилбензолсульфонамидо)-2-метилхинолина из пpeдыдyLЦIтx примеров в. ароматическом керосине. Данные результатов приведеньг и табл. 33. .-69 47 Раствор для регулирования рН водн Р , 0,5 М Н, S04 0,2MH2S04 0,lMHjS04 НаО: 0,05MNaOH 0,lMNaOH Органическая фаза с извлеченны 3,14 г/л Извлекающий раствор. Экстр г/л HI SO4орган вор г 100 150 200 ..:..., 250 В методике 3 применяют раствор 8-(децилметилбензолсульфонамидй)-2-метилхинолкна в ароматическом керосине для и . Данные результатов приведены в.табл. 35. : .. Таблица 35 Т Металл Концентрация металла в -, органической фазе, г/л , 1 2 . 0,315 0.300 ,,0149 0,0416 ,п т t л - - U,U114 ,.. ,0045 Zn 0,351. . 0,342 ,322 .,218 542 48 Таблица 33 го раффината Количество в органической фазе, г/л 0,63 0,0005 1,33 0,0019 1,65 0,0068 . 2,54 0,226 2,89 0,930 3,37 1,52 Таблица 34 металлом из стадии II; гированный рН промывного ческий раст- раствора/ л 1,083,84 1,455,45 1,203,99 0,3783,84 , Продолжение табл. J5 j, --:----- ,115 j bо 5531 40 i П р и м е р 46. 8-(Децилметилбензолсульфонамидо)-6-метилхкнолин из примера 10 .растворяют в ароматическом керосине с концентрацкей 0,1 М н применяют по методикам 45 данные результатов, которых приведены табл. 36-39 соответственно. - Таблица 36 Металл Количество металла в ....органической фазе, 50 - I : ,08 NiH-2,63 €0-+1,80 ,0007 .98
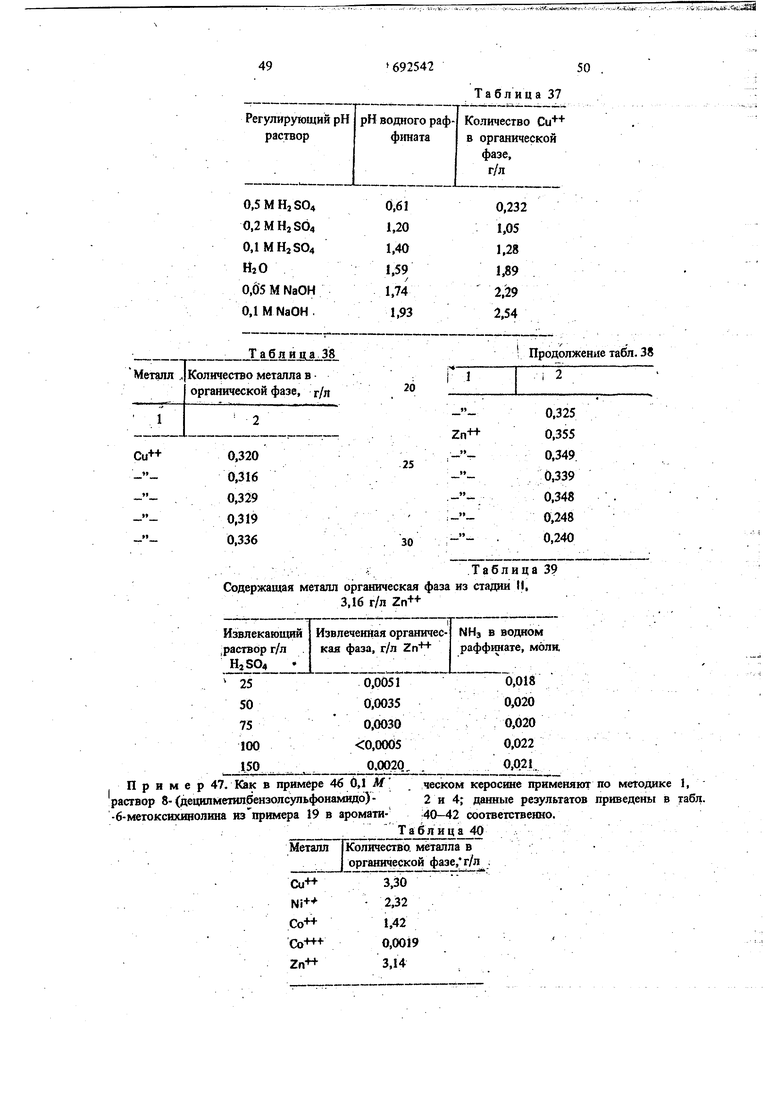
Содержащая металл органическая фаза из стадии 1|, 3,16 г/л Zn- -6-мегоксихинолииа из примера 19 в Металл Ni+Со++Со- - Zn-HТаблица 37
VТаблица 39
NN38 водном раффинате, моли, аромати- 40-42 соогветственно. Псолнчество. металла в I органической фазе, г/л 0,0019 3,14 0,021. ческом керосине применяют по методике , 2 и 4; данные результатов приведены в табл. Таблица 40 3.30 - 2.32 1.42 . -, . .П р и м е р ют 8-(децилметил хинолин из приме матическом керо ив бензоле. Дан в табл. 43. ; Растворител$ и ме Ароматик 150 Cц+
Раствор для регулирования , рН
0,5MH2S04 0,2MHjSO4 0,1 MH2SO4 HI О 0,05 М NaOH
Н водного раффиКоличество в орнатаганической фазе,
г/л
0,147
0,60 0,391 1,13 0,530 1,39 0,945
1,83 1,05 1,82 - /.t 514ii%jiE«jJft 5:;iK-ft:...-ftv- - -....t,;/;Vvi--- ..:....-.j:.:;..,,. ,.,,.,7.,,,,..,,,, .. , ..-- . . 5169254252 Раствор для регули- рН водного раф- Количество в рования рНфииатаорганической фазе, 0,5MHzS040,540,220 0,2MHjS041,010,765 0,1MH2S041,251,05 HjO1,591,36 0,005 MNaOH. 1,601,57 ---------- :j::Органическая фаза с извлеченным металлом из стадии II 3,24 г/л Извлекающий раствор, Экстрагированный органический г/л HI SO4раствор г/л 1001.05 1500,358 2500,0395 , Извлеченную органическую фазу промывают водой. рН воды до промывки 5,7, поела промывки 4,6. 48. По методике 1 примени- Продолжение табл. 43 бензолсульфонамидо)-5-нитро-: -..,--. ра 20, растворенный в аро-; сине с концентрацией 0,1 ные результатов1ЯрйВедены -., 35 Таблица 43Бензолталл Количество металла вСи 2,73 органической фазе, г/л ,72 2- 40 .-.--:...-:.Таблица4}г/л Таблица 42 По методике 2 д;1я раствора в Ароматике 150 получили данные, приведенные в табл. 44. Т а б л и ц.а 44 При максимальном извлечении меди ароматического керосина (2,8 г/л) и его отмывке водной серной кислотой (250 г/л) получают отмытый органический раствор, содержащий 0,055 г/л Си, при отмывке водной серной 5 кислотой (150 г/л) получают органический, раствор, содержащий 0,523 г/л . П р и м е р 49. Методики 1 и 2 применяют с 0,1 М раствором 8-(децилметнлбензол- Ю сульфонам1здо)-5,7-дихлорхинолина из примера 21 в ароматическом керосине.
При растворении в алифатическом керосине с концентрацией 0,1 Л/ и контактировании с водным раствором Си+ из методики 1, соединение из примера 22 дает желтую эмульсию,которая желатинизируется при стоянки
Таблица 46
П р и м е р 51. Применяют методику 1 с 0,1 М раствором 8- (гексадекансульфон23. Данамидо) хинолина из
примера
в приведены ные результатов табл. 49. Данные результатов приведены к табл. 45 методика 1) и в табл. 46 (методика 2). Таблица 45
Таблица 49
Кол№1ество металла в органической
Металл фазе, г/л
3,07 2,65 1,76
П р и м е р 53. 0,1 М раствор 8-(к-октансульфонамидо) хинолина из примера 27 в ароматическом керосине два раза контактируют гфи отношении органической к водной фазе 1:1 в течение 1 ч, каждый раз с водным раствором из методики I. Максимальное извлечение и органический раствор 2,20 г/л .. Осадок не обнаружен.
цекансульфонамвдо)хинолина в бензоле, то. ,в отделенной органической фазе находят 1,59 г/л и небольшое количество осадка.
Пример 52. Применяют методики 1, 2 и 4 для 0,1 ЛГ раствора 8-(2-этш1гексансульфонамвдо)хинолина из примера 24 в ароматическом керосине. Данные результатов приведены в табл. 50 (методика 1) 51 (мето- ; дика 2) и 52 (методика 4).
Таблица 50
П р и м е р 54. Методику 1 применяют для 0,1 М раствора 8-(пентансульфонамидо) хинолина из примера 28 в бензоле и водного раствора . В отделенной органической фазе 3,44 г/л . Однако, при попытке максимально извлечь в 0,1 М раствор 8-(пентилсульфонамидо) хинолина в ароматическом керосине, как в примере 53, выВыпадает небольшой осадок.фазе, г/л Вьтадает обильный осадок ,поэтомуорганическукИ 5i фазу не анализируют..,85 Когда методику 1 повторяют для водногоNi 2,42 створа и 0,1 М раствора 8-(н-гекса-CQ++2,80 Раствор для регулированиярН водного раффината Колич. в рНорганической фазе, г/л f 0,5MH2SO40,740,164 0,2MH2SO41,160,930 0,1 MHz SO,1,324,27 НгО1,551,72 0,05MNaOH1,752,00 ;; ..,,; Органический раствор из стадии II, около 2,80 г/л Извлекающий раст- Экстрагированная Промьгеочный раствор, вор, г/л H2SO4органическая фаза,рН г/л Си 1000,1005,36 1500,00255.36 2000,00253,93 250-4,8 . Исходный органический раствор не анализируют, содержание Си только оценивают. Металл Количество металла в органической Таблица 5 Таблица 52 пал уме Бывают. растворе П р ют такж хинолин вора в данные 56. по Металл ; 1 ,f - -
75 100 150 200
3,33 3,39 5,85 4,52 57692542 58 ренный осадок, который отфильтро- ZfJ Jl L В отфильтрованном органическомМеталл I Количество металла в органической содержится 0,86 г/л .фазе, г/л и м е р 55. Методики 1-4 применя- 5 е для 8-(изодекансульфонамвдо)-Ni2, а из примера 25. Для 0,1 М ряст-Со 1,80 ароматическом керосине получаютр ..0 ooQg результатов, приведенные в габл. 53методикам 1-4 соответственно. .-, -, --Раствор регулирования рН водного раф- Количество Си рНфинатав органической 0,5 М Нз S040,490,206 0,2MHjS041,061,14 0,lMHjS041,231,50 HjO1,612,25 0,05 М NaOH1,622,13 0,lMNaOH1,872,39 . , Продолже1ше табл. 55 Таблица 55-i;,,. ,. ,. , ;..I 2 Количество металла в органическойзо ,314 фазе, г/лZn-- 0,356 ....-, 340 .0,306,,f. , 03123S v,fJ ... 0,318 0,316- -,0,171 л -5 i т -., -, I, ,.U,jl - Органический раствор из стадии И 2,87 г/л Извлекающий раствор. Экстрагированная рН промьшного г/л H2S04органическая фа-раствора Таблица 54 фазе, г/л Таблица 56 за г/л
Приме р 56. 8%-ный раствор 8 (Ci 4-1 б алкенилсульфонамидо)хйнолина из примера 26 в алифатическом керосине контактируют по -методике 1. В полученном органическом растворе содержится 3,66 г/л Си++.
П р и м е р 57. Готовят 0,05 М раствор Ад, растворив 0,84 г AgNOs и 13,2 г (N44)2804 в 20 мл 2 М NH4 ОН и разбавив до 100 мл водой,. Затем с этим раствором Ад контактируют в течение 1 ч при встряхивании 0,1 Л/ раствор (З-децилметилбензолсульфонамидо) хйнолииа из примера 2 в ароматическом керосине при отношении органической к водной фазе 1:1. После разделения фаз в орагнической фазе содержится 2,96 г/л Ад. Затем порции этой фазы контактируют при встряхивании 1 ч с равным объемом различных водных растворов; для извлечения из него Ад. Данные результатов приведены в табл. 58.
Таблица 58
Извлекающий водный раствор
0,01 0,01
0,04
П р и м е р 58. Повторяют пример 57 но исходный водный раствор получают растворением 1,71 гНд(ЫОз)2 в 100 мл воды. Почти весь Нд(МОз)2 растворяется, ocтaвший ся осадок отфильтровывают и получают раствор с концентрацией Нд++ 0,05 М (рН 2,02).
Т аб л ица 57
При перемешивании с 1,0 М НСС. в органическом растворе содержится 0,93 г/л.
П р и м е р 59. 5,5%-ный раствор 8-(децилметилбензолсульфонамидо)хинолина из примера 2 в алифатическом керосине контактируют
при перемешивании и отношении органической к водной фазе 1:4 с водным раствором 2,5 г/л РЬ++ из РЬ(ЫОз)2 в воде (рН во время извлечения доводят до 7,1). Длительность контакта 2 мин. В отделенной органической фазе
содержится 9,56 г/л P/J+ . Органический раствор отмывают водной iHNOs (150 г/л) при отношении органической к водной фазе 6;1 и получают органическую фазу, не содержащую металла. В водном о1мывающем растворе наблюдается небольшой осадок Pb(N03)2.
Формул1а изобретения Способ экстракции соединений металлов из водных растворов, содержащих соединения меди, никеля, кобальта, цинка, кадмия, ртути,
серебра и/или свинца, путем контактирования указанных водных растворов с органической фазой, содержащей раствор сульфонамидохинолина в органическом растворителе, с последующим отделением органической фазы, содержащей экстрагированные соединения металлов и извлечением их, отличаю щийся тем, что, с целью повышения степени извлечения соединений металлов, в качестве органического растворителя используют практически
не смешивающийся с водой алифатический или ароматический углеводород юга их смесь, с 1емпературой вспышки 65-90,6°С, а в качестве сульфонамидохинолина используют 8-сульфонамидохинолин общей формулы
где R - алкильный или алкенильный радикал, содержащий по крайней мере 5 атомов углерода или группу, имейщую формулу
где R - алкиленовый радикал, содержащий 1-20 атомов углерода, р 0-1, А - моноили полициклический радикал, имеющий кольцо или кольца, состоящие из 5-6 членов, q - целое число 1-5, п 0-2, R - алкиль |й или алкенильиый радикал, причем общее 1ЧИСЛО атомов углерода в (R) q составляет по крайней мере 5, при условии, что когда q 2, то по крайней мере один из R
радикалов содержит 5 или более атомов углерода, а когда q 3, то по крайней мере один из радикалов R содержит 3 или более атомов углерода, R - CR-Br.-NO, или OR, где R - углеводородный радикал с числол углеродных атомов 1-20, пит равны О или 1, 2 или 3, R и R - углеводородные или углеводородоксирадикалы, содержащие 1-20 атомов углерода, -СИ -Вг. или NOj. Источники информации,
принятые во внимание при зкспертизе
