Изобретение относится к способу получения новых замещенных циклогексилиденпростагландинов общей формулы денпростагландины указанной обшей формулы 1 и 2. Цель изобретения - расширение класса аналогов природных простанглащцгаов, обладающих ценными фармакологическими свойствами. Цель достигается описываемым способом, заключающимся в том, что соецинение формулы
где У - водород или оксигруппа; К - карбонил или оксигруппа; L - этилен;
Q - водород или алкил Cj
один из R), R и Нз - алкил Ci-C, а два других - водорйд,
обладающих денными фармакологическими свойствами.
Использование известных в химии простаглан динов методом.взаимодействия замешенного циклопентенона с литиокупратом и гидролиза сложных эфиров позволяетчюлучить не описанные в литературе замещенные циклогексилиCOOQ
(П)
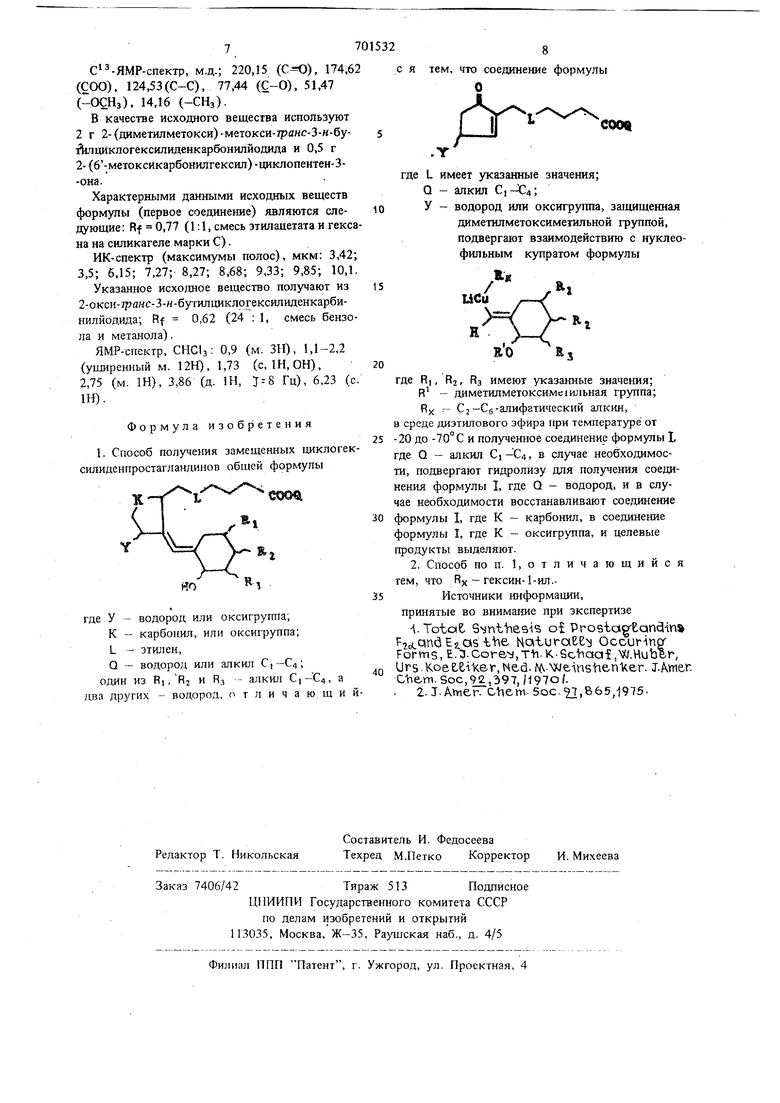
где L имеет указанные значения: Q - алкил Ci-C4;
У - водород или оксигруипа, чашишсиная диметилметоксимстилыюй группой. подвергают взаимодействию с нуклеофильным купратом формулы / RI LiCu Ч , RORSгде R, Ra, RS имеют указанные значения, R - диметилметоксиметил; RX - С2-Сб-алифатический алкия, предпочтительно гексин- 1-ил в среде диэтилового эфира при температуре от -20 до и полученное соединение формулы I, где Q алкил С)-€4, в случае необходимости подвер гают гидролизу для получения соединения формулы 1, где Q - водород, и в случае нео ходимости восстанавливают соединение форму лы I, где К - карбонил, в соединение форму лы Т, где К - оксигрушш, после чего целевые продукты выделяют известными методами. Пример. Раствор ,155 г 2-(диметил метокси)-мстокси-гр 7нс-3-метилциклогекс11лиденкарбонилбромида и 14 мл безводного эфи ра охлаждают в атмосфере аргона до -70 С, после чего добавляют 4,5 мл 1,8 М раствора трст-бутиллития в пентане. Образуется желтокоричневый раствор. После перемешивания в течение 3 ч по каплям прибавляют раствор ,262 г Си-С-::С-СцН, 2 Р (N Ме2)з и 5м эфира. Коричневато-красный раствор перемешивают 1ри в течение 40 мин. После этого г рибавляют раствор 0,5 г 2-(6-метокси карбонилгексил)-циклопентен-З-она в 12 м.ч безводного эфира и реакщтонную смесь перемешивают при -70°С в течение 1 ч, при -24°С в течение 2 ч и без охлаждения в течение 1 ч. Реакционную смесь сливают в смесь из 50 мл воды, 10 г сульфата аммония и 10 мл серной кислоты. Реакционную смесь перемешивают в течение 30 мин, в naainee в осадок желтое соединение меди отфильтровывают, промывают зфиром, соединен ные эфирные растворы отделяют от водной фазы, промывают последовательно дважды по 25 мл воды, дважды но 10 MJJ 5%-ного раствора гидроокиси аммония, 5 раз по 25 мл воды и 20 мл насьнценного раствора хлористого натрия, высушивают пад безводным суль фатом магния, фильтруют и удаляют раствори тель. Остаюшееся смолоподобное масло (1,35 г) подвергают хроматографии на колон ке (диаметр 20 мм, щгина 450 мм), содержа щей 60 г силикагеля (размер частиц 0,05-0,2 мм), и элюируют 1400 мл бензола, содержащего 19(- метанола. Элюат (1000-1100 м содержит 0,538 г 2-(6-карбоксигексил)-3-(2окси-7ранс-3-метилциклогексилиден)-карбонилциклопентанона. Согласно данным тонкослойной хроматографии продукт является од нородным. Rf 0,4 (силикагель, смесь бензола и метанола 10:1); Rf 0,54 (смесь зтилацетата и гексана 1:1). ИК-спектр, мкм: 2,8 (ОН), 3,4 и 3,5 (С-Н), 5,75 (С-0), 8,6 и 9,45 (С-О). Н - ЯМР-спектр, М.Д.; 6,7 (дублет 1Н, Т 10 Гц); 4,55 (д. Ш), 4,57 (синглет, ЗН), 3,4-2,94 (мультигшет, 26Н), 1,1 и 1,4 (2 д. ЗН). С3-ЯМР-спектр, М.Д.: 220,57 (), 174,65 (С-00-), 14,27 (дублет) (С ), 123,39 (), 122,67 (), 78,48 (Q-OH), 55,66 (j: ), 51,51 (ОСНз), 41,47 - 24,88 Н. (CHjCHj), 18,73 (дублет) (С-СНз). Исходное вещество может быть получено следующим образом. 8,6 г 50/г-ной дисперсии гидрида натрия в минеральном масле суспендируют в 300 мл безводного эфира, охлажденного до 10° С, обрабатывают с 1 мл этанола и перемешивают в теченне 15 мин. После этого прибавляют по каплям в течение 1,5 ч смесь 18,5 г 2-метилвдклогексанона и 15 г этилформиата. Реакционную смесь перемешивают в течение 6 ч, в результату чего образуется густая паста. Соль енола растворяют при охлаждении со льдом путем осторожного прикапывания 35 мл водь. Водный раствор отделяют от эфирного слоя и экстрагируют 50 мл эфира. Водный раствор подкисляют до значений рН 4 с помошью 6н. соляной кислоты, экстрагируют дважды по 100 мл эфиром, эфирный раствор высушивают над безводным сульфатом магния, фильтруют, растворитель удаляют и остаток перегоняют. Подустают 18 г светло-желтой жидкости; т. кип. 69-70°С/3 мм рт.ст.; выход 78%. Жидкость может хранитьс:я в холодильнике в течение нескольких недель без разложения. 25,6 г трифенилфосфина растворяют в 300 мл безводного бензола, после чего раствор охлаждают до 12-14°С при перемешивании и по каплям в течение 1,5 ч прибавляют 15,7 г брома в 75 мл безводного бензола. К образующейся белой суспензии прибавляют в течение 15 мин раствор 14,1 г дизтиланилина и 40 мл бензола и смесь перемешивают в течение 15 мин. После этого 13,17 г 2-формил-6-иетилциклогексанона, полученного по указанному способу, растворяют в 30 мл безводного бензола и раствор по каплям сливают в течение 30 мин в смесь, содержащую бромирующий агент, при энергичном перемещивании. Реакционную смесь перемешивают при 25°С
в течение часа, при 40°С в течение 3 ч, охлаждают до комнатной температуры и выпавший осадок кристаллического бромгидрата диэтиланилина отфильтровывают. Бензольный раствор концентрируют на роторном испарителе при 25° С/15-20 мм рт.ст. до объема 80-90 мл и остаточный бензольный раствор разбавляют 30-50 мл пентана в нескольких порциях. Кристаллический трифенилфосфиноксид немедленно выпадает в осадок. Смесь оставляют стоять при 0°С в течение нескольких часов, трифенилфосфиноксид отфильтровывают, растворитель полностью удаляют на роторном испарителе, остаточную кристаллическую пасту экстрагируют с несколькими порциями пентана,раствор отделяют от кристаллического трифенилфосфиноксида декантацией и фильтрованием. Следы трифенилфосфиноксида удаляют охлаждением до -14° С в течение 15-24 ч и фильтрованием. Пентан удаляют на роторном испарителе.
Таким образом получают 17,2 г 2-оксо-З-метилциклогексилиден-карбинил-бромида с выходом 90%. ИК-спектр, мкм: 3,23 (Н), 5,9 (, сопр.).
3,2 г литийалюминийгидрида суспендируют в 80 мл безводного эфира, суспензию охлаждают до 0°С, после чего по каплям прибавляют в течение 1 ч при перемешивании раствор из 17,2 г 2-оксо-З-метилциклогексилиденкарбинилбромида и 60 мл эфира. Реакционную смесь перемешивают при 5°С в течение 1,5 ч, после чего ее обрабатывают 25 мл 5%-ного водного раствора серной кислоты. Эфирный раствор отделяют от водного слоя, промывают водой до нейтральной реакции, высушивают над безводным сульфатом магния и выпаривают.
Таким образом получают 17-17,2 г коричневато-желтого масла, которое состоит из смеси двух изомеров; согласно данным тонкослойной хроматографии на пластинке с силикагелем Rf 0,47 и 0,56 соответственно в смеси хлороформа и метанола состава 70 : 1. Изомеры могут быть разделены на колонке с силикагелем путем злюирования бензолом. Сначала элюируется трансизомер, который может быть извлечен из пентана охлаждением до -18°С. Температура плавления перекристаллизованного продукта достигает 66°С:
ИК-спектр, мкм: 3,0 (широкая полоса ОН), 3,4 и 3,5 (СНз, СНг), 6,1 (С-С).
Н-ЯМР-спектр, М.Д.: 7,75 (дублет, Ш), 4,3 (д. 1Н, J-9 Гц), 5,1 (мультиплет, 1Н), 2-2,9 (м, 7Н), 2,95 (ЗН).
Тонкослойная хроматография: Rf 0,56 (хлороформ - метанол 70:1).
Продукт является 2-окси-3-7ран :-метилциклогексилиденкарбинилбромидом.
К раствору 1,79 г по.т1учеииого таким образом 2-окси-3-трйнс-3-метилш(клогексилидснкарбинилбромида и 4,2 мл свежеперегнанного изопропенилметилового эфира в 4 мл безводно5 го бензола прибавляют одну каплю хлорокиси фосфора и смесь оставляют стоять при комнатной температуре в течение 20 ч в условиях, исключающих попадание влаги. Хлорокись фосфора нейтрализуется тремя каплямитрйзтшг0 амина, растворитель удаляют на роторном испарителе при 25° С, Остаток растворяют в 20 мл безводного пентана и фильтруют.
После удаления пентапа получают 2,396 г 2- (диметилметокси)-меток си -З-трянс-мётил5 циклогексилвденкарбонилбромида в виде бесцветной жидкости.
Выход 99%.
Согласно данным тонкослойной хроматографии продукт является однородным; Rf 0 0,68 (хлороформ - метанол 70 : 1).
ИК-спектр, мкм: 3,25 (СН), 3,4 и 3,5 (CHj. CHj, 6,1 (), 9,3, 9,45 и 9,8 ().
Пример 2., 0,538 г 2-(6-метоксикарбонилгексип)-транс-З-(2 -окси-7ранс-3 - метилшж5логексилиденкарбинил) -циклопентанона перемешивают в растворе 400 мг гидроокиси натрия в 20 мл (3 : 1) смеси метанола и воды при комнатной температуре в течение 15 ч. после чего метанол отгоняют. К водному слою прибав0ляют 5 мл воды и его экстрагируют эфиром. Водный слой подкисляют соляной кислотой и снова экстрагируют эфиром. Соединенные . эфирные экстракты высуишвают над безводным сульфатом магния и выпаривают. 5 Таким образом получают 320 мг 2-(6-карбоксигексил) -транс-З- (2 -окси-:гр(2нс-3 -метилциклогексилиденкарбинил)-Ш1клопентанона в виде масла, которое является однородным по данным тонкослойной хроматографии.
0
ИК-спектр, мкм: 2,7 - 4,1 (широкая полоса ОН,-СООН); 3,4 и 3,5 (СН, СИ,); 5,75 (); 5,8 (СООН).
Rf 0,25 (бензол - метанол - уксусная кислота, 35:4:1).
Rf 0,50 (этилацетат - гексан - уксусная кислота, 25 : 10 : 4).
ПримерЗ:По примеру 1 получают 0,53 г 2-(6 -мeтoкcикapбoнил)-гeкcил -:Ipaнc-3-(2 -oкcи-7pш(c-3 н-бyтилциклoгeкcилидeнкapбинш)-Ш1клопентанона в виде продукта, который однороден согласно данным .тонкослойной хроматографии.
55 Тонкослойная хро.матография: Rf 0,24 (гексан-этилацетат-метанол, 30:10:1,2).
ИК-спектр, мкм: 2,85 (ОН). 3,4, 3,45 и 5,75 )-, 6,85, 8,55 и 10,2. С13-ЯМР-спектр, М.Д.; 220,15 (С), 174,6 (COO), 124,53(С-С), 77,44 (С-0), 51,47 (-ОСНз), 14,16 (-СНз). В качестве исходного вещества используют 2 г 2-(диметилметокси)Метокси-7ранс-3-к-буАлциклогексилиденкарбонилйодида и 0,5 г 2- (б-метоксикарбонилгексил) -циклопентен-3-она. Характерными данными исходных веществ формулы (первое соединение) являются следующие: Rf 0,77 (1:1, смесь этилацетата и гекс на на силикагеле марки С). ИК-спектр (максимумы полос), мкм: 3,42 3,5; 6,15; 7,27; 8,27; 8,68; 9,33; 9,85; 10,1 Указанное исходное вещество получают из 2-окси-7раис-3-н-бугш1Циклргексилиденкарбинилйодида; Rf 0,62 (24 : 1, смесь бензо ла и метанола). ЯМР-спектр, СНС1з: 0,9 (м. ЗН), 1,1-2,2 (уширенный м. 12Н), 1,73 (с, 1Н, ОН), 2,75 (м. Ш), 3,86 (д. Ш, JrS Гц), 6,23 (с 1Н). Формула изобретения 1. Способ получения замещенных Щ1клогек силиденпростагландинов общей формулы У - водород или оксигруппа; К - карбонил, или оксигруппа; L - этилен, Q - водород или алкил Cj-С4; один из RI/RJ и Нз алкил Ci-C4, а два других - водород, отличаюши 8 с я тем, что соединение формулы имеет зтсазанные значения; -алкил Ci--C4; -водород или оксигруппа, затдищенная диметилметоксиметильной группой, подвергают взаимодействию с нуклеофильным купратом формулы UK Ч VR, .-. R2, РЗ имеют указанные значения; - диметилметоксиме1Ш1Ьнал группа; RX -- С2-Cg-алифатический алкин, в среде диэтилового эфира при температуре от -20 до -70° С и полученное соединение формулы Ь где Q - алкил Cj . в случае необходимости, подвергают гидролизу для получения соединения формулы I, где О - водород, и в случае необходимости восстанавливают соединение формулы 1, где К - карбонил, в соединение формулы I, где К - оксигруппа, и делевые продукты выделяют. 2. Способ по п. 1, отличающийся тем, что RX - гексин-1-ил.. Источники информации, принятые во внимание при зкспертизе -l.Totoi. S ntliee-is oi Prosiag Ecjndin 4i QndEias NaturaE Occur-inor Forms, E.3.Core-y,Tti. K-Schaaf ,W.Hub6, иг5.КоеШ1 ег,Мед./А- «е- П511еп1 ег. J.,r. C-hem. 500,92,397, Л97О/. 2. J.AvYier-Cliem.Soc.,865,-1975.


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения оптически активных или рацемических 17-аза-производных простагландинов пгф | 1978 |
|
SU730297A3 |
| Способ получения производных 7-оксо-простациклина или их солей | 1985 |
|
SU1376939A3 |
| Способ получения 4-тиа-или 4-сульфинил- @ производных | 1981 |
|
SU1053746A3 |
| Оптически активные производные 7-оксопростациклина,обладающие антиагрегатным и гипотензивным действием | 1985 |
|
SU1421741A1 |
| Способ получения производных интер- @ -фениленпростациклинов | 1982 |
|
SU1391501A3 |
| Способ получения производных 7-оксопростациклина | 1986 |
|
SU1424735A3 |
| Способ получения производных 7-оксопростациклина | 1980 |
|
SU1056899A3 |
| Способ получения оптически активных или рацемических сложноэфирных производных хризантемовой кислоты | 1979 |
|
SU1088661A3 |
| Способ получения производных 2,3,4-тринор- @ -интер-фениленпростагландина | 1982 |
|
SU1138020A3 |
| Производные 2,3,4-тринор- 1,5-интер-м-фениленпростациклина, обладающие цитозащитными свойствами | 1983 |
|
SU1382834A1 |
