которые могут быть использованы в качестве неподвижной жидкой фазы для газожидкостной хроматографии.
Поставленная цель достигается свойствами предлагаемых соединений - олигомеров 1 - (2-оксиэтокси) -2,3-пропиленоксида общей формулы I
Н -оеНг Сн-З он йнг ОСН2 5Нг031
где п-6-8 и способом их получения.
Известен способ получения полноксиалкиленов путем полимеризации кислородсодержащих циклических соединений в присутствии гидроокисей щелочных металлов 2. Однако указанным способом получить предлагаемые соединения невозможно.,.9,
Применение в данном способе оптимального режима проведения процесса, соотиощение исходных реагентов и катализатора, а также дополнительная стадия гидролиза промежуточного продукта позволяют получать новые соединения, обладающие указанным положительным эффектом.
Способ получения олигомера формулы I заключается в олигомеризации винилглицидилового эфира этиленгликоля в присутствии основного катализатора, взятого в количестве 2-5% от веса мономера, при 70-90°С в течение 2-8 ч с последующим гидролизом полученного олигомера в присутствии 0,5-5% НС1 от массы гидролизата в водной или водно-диоксановой среде при мольном соотношении воды, диоксана и олигомера 10:0:1 - 10:1:1 при 50- 100°С в течение 1-2,5 ч.
Как правило, в качестве основного катализатора используют гидроокиси щелочных металлов, третичные амины или четвертичные аммониевые основания.
Очистку полученного олнгомера от мономера, не вступивщего в реакцию полимеризации, низкомолекулярных фракций олигомера и катализатора проводят методом переосаждения раствора олигомера (в одном из низкокипящих растворителей типа ацетон, диоксан).
Получение целевого продукта осуществляют по схеме
dK,-с;ненгОс;нгС1НгОен ЙНг /
О
Н -OCHr iH- OHНгО Н:
СНг .гОЙн СН2
f
PodHzCiH- j OH-f UHjC
Н.
енг оСНгСНгОН
где B NaOH, КОН, EtaN, четвертичное аммониевое основание, триэтилбензиламмонийгидроксид; л 6-8.
Строение целевого олигомера подтверждено ИК-спектрами, в которых отсутствуют характеристические полосы винилоксигруппы при 820, 960, 1200, 1320, 1620, 1640, 3080, 3100 см-Ч Наличие гидроксильных групп подтверждено интенсивным поглощением в области 3200-3600 см- с максимумом при 3400 см-.
Элементарный анализ (содержание С, Н) соответствует составу олигомера. Получеиные олигомеры обладают следующими преимуществами по сравнению с известными аналогами.
Высокая полярность, сравнимая с полярностью адипонитрила и р,р-оксидипропиоиитрила, обусловленная наличием полярных эфирных групп в основной цепн полимера и сильно полярных гидроксильных групп, расположенных на концах полимерной цепи, и в каждом боковом звеие. Полярность составляет 89,2 единицы по щкале Роршнайдера (за 100 единиц принята полярность р,р-оксидипропионитрила). Среди НЖФ, содержащих простые эфирные и гидроксильные группы, предлагаемая фаза по величине полярности не имеет близких аналогов. Полярность полигликолей находится в пределах , диглицерина 59. Столь высокая полярность нового олигомера обусловлена легкой доступностью гидроксильной группы для донорно-акцепторного взаимодействия с растворенным веществом.
Высокая термостойкость, превышающая термическую устойчивость подавляющего большинства полярных НЖФ. Унос 5% фазы наблюдается при ступенчатом нагревании с одновременной продувкой гелием со скоростью 30 мл/мин до достижения 270°С, что позволяет вести хроматографирование в изотермическом режиме до 240°С. В программированном режиме при скоростях повышения температуры 4-20°С/мин НЖФ устойчиво работает до 260°С.
Высокая симметричность пиков высокомолекулярных соединений (в том числе имеющих несколько гидроксильных групп в молекуле) при использовании щирокого круга диатомитовых носителей для ГЖХ. Асимметрия пиков спиртов Са-Се составляет всего 1,00-1,04 (80-130°С), этиленгликоля 1,2 (160°С), глицерина 1,4 (220°С). Элюирование глицерина в виде столь слабо асимметричного пика ни на одной из известных НЖФ до сих пор не удавалось
достигнуть.
Широкий диапазон рабочих температур. Новая НЖФ устойчиво обеспечивает высокую эффективность (в больщинстве случаев превосходящую известные аналоги) в
диапазоне температур 20-240°С.
В качестве высокополярной гидроксилсодержащей жидкой фазы предлагаемый объект изобретения представляет значительный интерес для исследовательских целей в области физической химии, конкретно в исследовании теории растворов.
В своей совокупности эти преимущества обеспечивают высококачественный анализ практически неограниченной области полярных органических веществ.
Исходный винилглицидиловый эфир этиленгликоля является в настоящее время доступным мономером, получаемым известным способом 3.
Олигомеризация
Пример I. 10 г винилглицидилового эфира этиленгликоля (винилокса), 0,05 г мелкораздробленного КОН перемешивают в течение 2 ч при 90°С. После смещения полученной олигомерной массы с 10 мл ацетона полученный раствор фильтруют для удаления катализатора. Затем раствор олигомера вводят при эффективном перемещивании в колбу, содержащую 100 мл петролейного эфира. По окончании осаждения олигомера осадитель сливают, а олигомер подвергают вакуумной сущке при 70-80°С. В результате проведенных операций получают 9,6 г олигомера винилокса. Выход олигомера составил 96%.
Пример 2. 10 г винилокса, 0,025 г NaOH перемешивают в течение 6 ч при 80°С. После проведения операции по переосаждению олигомера с использованием в качестве системы растворитель-осадитель бутилвинилового эфира и гексана получают 9,1 г олигомера винилокса. Выход 91%.
Пример 3. 10 г винилокса, 0,02 г триэтилбензиламмонийгидроксида
(С2Н5)зМСН2СбН510Н,
перемешивают в течение 3 ч при 90С. После обработки олигомера в условиях, ана.логичных примеру 1, исключив стадию (Ьильтрации, выделяют 9,7 г олигомера. Выход 97%.
Пример 4. 10 г винилокса, 0,05 г триэтиламина перемешивают в течение 8 ч при 70С. После проведения операции переосаждения аналогично примеру 3 получают 8,4 г олигомера винилокса. Выход 84%.
Анализ количества винилоксигрупп, проведенный методом гидролитического оксимироваиия, подтверждает присутствие винилоксигруппы в каждом звене олигомера винилокса. Молекулярная масса синтезированных олигомеров находится в интервале 900-1500 м. ед.
Гидролиз
Пример 5. В трехгорлую колбу с обратным холодильником и мешалкой загружают 18 мл Н2О, 14,4 г олигомера винилокса и 0,23 мл концентрированного раствора
НС1. После перемешивания в течение 2,5 ч при 100°С из реакционной массы с применением вакуума отгоняют воду и выделившийся в результате реакции уксусный альдегид. В результате получают 11,7 г олигомера с выходом 99,2%.
Пример 6. К 18 мл НгО и 2,3 мл концентрированной HCI при перемешивании в термостатируемую колбу при 50°С в течение 10 мин приливают раствор 14,4 г олигомера винилокса в 8,8 г диоксана. После введения всего раствора олигомера осуществляют барботаж газообразным азотом реакционной массы с целью удаления выделившегося в результате реакции уксус ного альдегида. Общее время реакции 1 ч. После вакуумного удаления растворителей гидролизат подвергают дополнительной вакуумной сущке, выделяют гидроксилсодержащего олигомера 11,6 г, что соответствует 98%-ному выходу.
Испытание олигомера в качестве
неподвижной жидкой фазы Пример 7. Готовят наполнитель: 10%
олигомера от веса носителя стерхомола зернением 0,,3 мм (наносят из раствора в этиловом спирте). Колонку А из нержавеющей стали диаметром 4 мм и длиной 3 м продувают при 150°С. На колонке
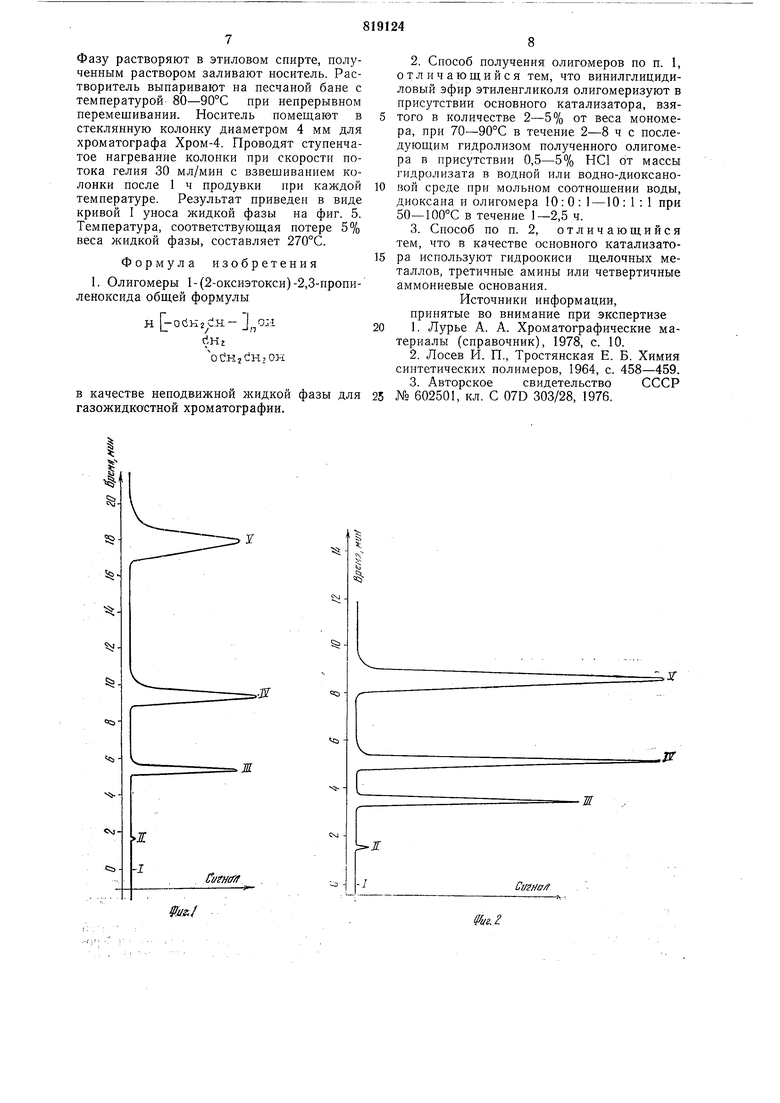
получают хроматограммы смесей нормальных спиртов €4-Сб - фиг. 1, пики 1П-V и нормальных углеводородов Сд-Сю-фиг. 2, пики П1-V. Температура термостата колонок и давление газа-носителя гелия на
входе в колонку при анализе смеси спиртов 100°С и 0,5 ати, для смеси углеводородов соответственно lOOC и 0,4 ати. Эффективность колонки А по углеводородам C9--Cio составила 1350, 1600, 1700 теоретических тарелок, по нормальным спиртам С4-Сб соответственно 1600, 1550, 1600 теоретических тарелок.
Для всех хроматограмм пик I соответствует запуску пробы, пик П несорбирующемуся компоненту - воздуху.
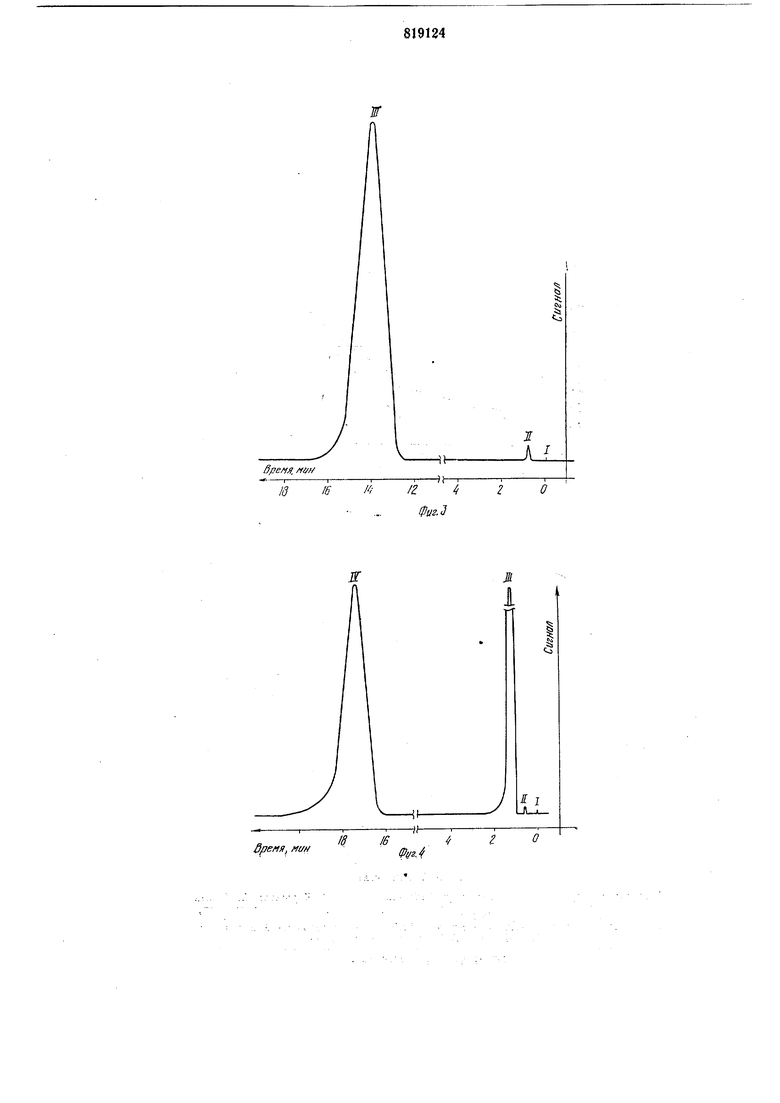
Пример 8. Готовят наполнитель: 15% олигомера от веса носителя хроматона-.М зернением 0,25-0,315 мм. Колонка В аналогична колонке А. Индекс удерживания
бензола при 80°С составляет 1099 ед., что соответствует полярности фазы 89,2 ед. по щкале Роршнейдера (за 100 ед. взят р,Р-оксидипропионитрил). На колонке В получают хроматограммы диметилформамида - фиг. 3, пик III, и диэтиленгликоля - фиг. 4, пик IV. Температура в термостате колонок при анализе диметилформамида 100°С, давление гелия на входе в колонку 0,4 ати. Анализ этиленгликоля осуществляют соответственно при 150°С и давлении 0,7 ати с использованием растворителя 1,4-диоксана, пик TII.
Пример 9. Берут 2,04 г исследуемой жидкой фазы (30% от веса носителя Хроматон N-AW зернением 0,25-0,315 мм).
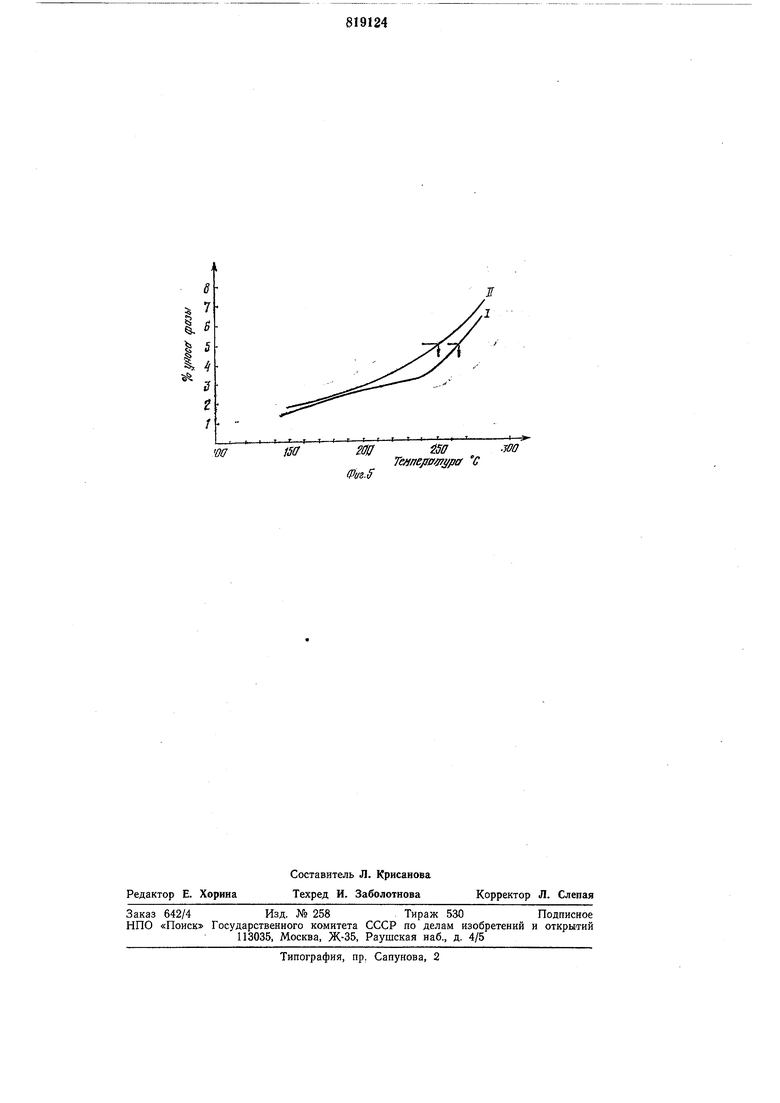
Фазу растворяют в этиловом спирте, полученным раствором заливают носитель. Растворитель выпаривают на песчаной бане с температурой 80-90°С при непрерывном перемешивании. Носитель помещают в стеклянную колонку диаметром 4 мм для хроматографа Хром-4. Проводят ступенчатое нагревание колонки при скорости потока гелия 30 мл/мин с взвешиванием колонки после 1 ч продувки при каждой температуре. Результат приведен в виде кривой I уноса жидкой фазы на фиг. 5. Температура, соответствующая потере 5% веса жидкой фазы, составляет 270°С.
Формула изобретения
1. Олигомеры 1-(2-оксиэтокси)-2,3-пропиленоксида общей формулы
н ,.o:i riHj
В качестве неподвижной жидкой фазы для газожидкостной хроматографии.
2.Способ получения олигомеров по п. 1, отличающийся тем, что винилглицидиловый эфир этиленгликоля олигомеризуют в присутствии основного катализатора, взятого в количестве 2-5% от веса мономера, при 70-90°С в течение 2-8 ч с последующим гидролизом полученного олигомера в присутствии 0,5-5% НС1 от массы гидролизата в водной или водно-диоксановой среде при мольном соотношении воды, диоксана и олигомера 10:0:1 -10:1:1 при 50-100°С в течение 1-2,5 ч.
3.Способ по п. 2, отличающийся тем, что в качестве основного катализатора используют гидроокиси щелочных металлов, третичные амины или четвертичные аммониевые основания.
Источники информации, принятые во внимание при экспертизе 1. Лурье А. А. Хроматографические материалы (справочник), 1978, с. 10.
2.Лосев И. П., Тростянская Е. Б. Химия синтетических полимеров, 1964, с. 458-459.
3.Авторское свидетельство СССР № 602501, кл. С 07D 303/28, 1976.


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения -алкоксиэтилглицидиловых эфиров гликолей | 1973 |
|
SU461924A1 |
| СОПОЛИМЕРЫ ВИНИЛХЛОРИДА, ВИНИЛГЛИЦИДИЛОВОГО ЭФИРА ЭТИЛЕНГЛИКОЛЯ И ВИНИЛОКСИЭТИЛОВОГО ЭФИРА ГЛИЦЕРИНА В КАЧЕСТВЕ ТЕПЛОСТОЙКИХ, ТЕРМОСТОЙКИХ, ХОРОШО РАСТВОРИМЫХ МАТЕРИАЛОВ С ВЫСОКОЙ ПРОЧНОСТЬЮ И АДГЕЗИЕЙ | 1995 |
|
RU2101298C1 |
| СОПОЛИМЕРЫ ВИНИЛХЛОРИДА, ВИНИЛГЛИЦИДИЛОВОГО ЭФИРА ЭТИЛЕНГЛИКОЛЯ, ВИНИЛОКСИЭТИЛОВОГО ЭФИРА ГЛИЦЕРИНА И ПРОСТЫХ АЛКИЛВИНИЛОВЫХ ЭФИРОВ, В КАЧЕСТВЕ ТЕРМОСТОЙКИХ, ХОРОШО РАСТВОРИМЫХ МАТЕРИАЛОВ С ВЫСОКОЙ ПРОЧНОСТЬЮ И АДГЕЗИЕЙ, СПОСОБНЫХ К РЕГУЛИРУЕМОМУ ОТВЕРЖДЕНИЮ | 1995 |
|
RU2100377C1 |
| Способ получения политиоацеталей | 1979 |
|
SU854945A1 |
| Способ получения дивиниловых эфиров уретановых тетраолов | 1980 |
|
SU988807A1 |
| НЕПОДВИЖНАЯ ФАЗА ДЛЯ ГАЗОВОЙ ХРОМАТОГРАФИИ И СПОСОБ ХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 1999 |
|
RU2155332C1 |
| Бис-(хелат)-бис(олигоэфир)-титанатыВ КАчЕСТВЕ КАТАлизАТОРОВ СиНТЕзАпОлиэфиРОВ | 1979 |
|
SU821452A1 |
| ПОРОШКООБРАЗНЫЙ ГИДРОФОБНЫЙ ПОЛИМЕР И КОЛОНКА ДЛЯ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ С ОБРАЩЕННОЙ ФАЗОЙ | 2002 |
|
RU2296137C2 |
| Способ получения самоотверждаемой эпоксидной смолы | 1980 |
|
SU952861A1 |
| Неподвижная жидкая фаза длягАзОВОй ХРОМАТОгРАфии | 1979 |
|
SU840733A1 |
со 0)
Ча
.ж
ем
Ж
L-J
CufHtf/f
9иг.1
Vus.Z.
, fioH
1
ЧЬ
Id IS A- Фиг.З
I I
18
Время, /iUH
-ih
16
Фуг, 4
1
I .
г
/
fjff20аЙЙ7 oa
т
Te/ineflffJjft/pcf °С
Фиг.5
